Способ получения ретиналя или аддукта (полностью-е)-ретиналя с гидрохиноном и промежуточные продукты - RU2318805C2
Код документа: RU2318805C2
Описание
Настоящее изобретение относится к способу получения альдегида витамина А (ретиналя), ценного промежуточного соединения в синтезе других производных витамина А (ретиноидов). Известно, что ретиноиды, особенно витамин А-спирт (ретинол), являются ценными веществами, которые способствуют здоровью человека, среди прочего в отношении зрения, иммунной системы и роста и по этой причине часто применяются в качестве компонентов поливитаминных препаратов и как добавки для некоторых пищевых продуктов и кормов. Настоящее изобретение относится далее к новым исходным веществам для упомянутого выше способа и, кроме того, к этапам способа, приводящим к (полностью-Е)-ацетату витамина А.
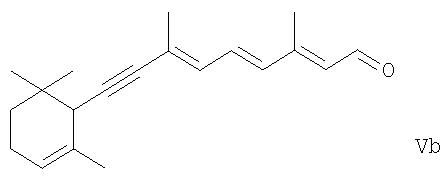
В соответствии с настоящим изобретением обеспечивается способ получения ретиналя формулы

который включает взаимодействие производного 5-(2,6,6-триметилциклогекс-1-енил)-1,4-пентадиена общей формулы
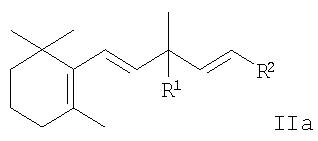
или производного 5-(2,6,6-триметилциклогекс-2-енил)-1,4-пентадиена общей формулы

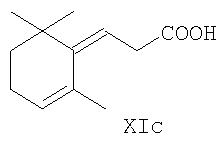
или производного 5-(2,6,6-триметил-2-циклогексен-1-илиден)-1-пентена общей формулы

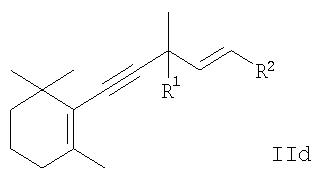
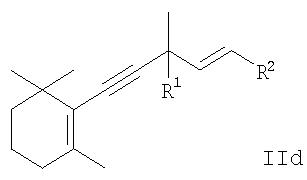
или производного 5-(2,6,6-триметилциклогекс-1-енил)пента-1-ен-4-ина общей формулы

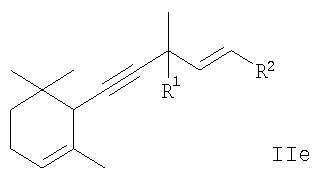
или производного 5-(2,6,6-триметилциклогекс-2-енил)пента-1-ен-4-ина общей формулы

где R1 означает гидроксил или группу OR3,
R2 означает хлор, бром, (С1-С6)алкокси, (С1-С6)алкилтио, арилокси, арилтио, (С1-С6)алкилкарбонилокси, ароилокси, три(С1-С6)алкилсилилокси, ди(С1-С6)алкилфосфонилокси, (С1-С6)алкилсульфонилокси, (С1-С6)алкилсульфонил, арилсульфонил, ди(С1 -С6)алкиламино, N-арил(С1-С6)алкиламино или диариламино, и
R3 означает (С1-С6)алкил, (С1-С6 )алкилкарбонил, ароил, (С1-С6)алкоксикарбонил, три(С1-С6)алкилсилил, ди(С1-С6)алкилфосфонил, диарилфосфонил, (С1-С6)алкилсульфонил или арилсульфонил, с производным 1,3-бутадиена общей формулы
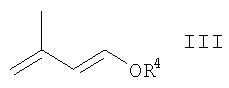
где R4 означает (С1-С6)алкил, (С1-С6)алкилкарбонил или три(С1-С6)алкилсилил, в присутствии кислоты Льюиса или Бренстеда и подвергая полученное таким образом соединение общей формулы

(исходя из производного 5-замещенного 1,4-пентадиена формулы IIa) или

(исходя из производного 5-замещенного 1,4-пентадиена формулы IIb), или

(исходя из производного 5-замещенного 1-пентена формулы IIc), или

(исходя из производного 5-замещенного пента-1-ен-4-ина формулы IId), или

(исходя из производного 5-замещенного пента-1-ен-4-ина формулы IIe) действию основных или кислотных условий, чтобы элиминировать из них фрагмент R2Н, и таким путем получить из соединения формулы IVa ретиналь формулы I или из соединения формулы IVb соединение формулы,

или из соединения формулы IVc - соединение формулы

или из соединения формулы IVd - соединение формулы

или из соединения формулы IVe - соединение формулы
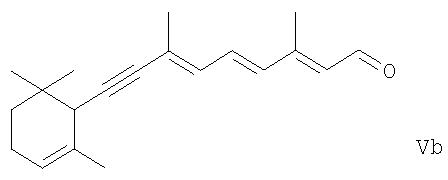
и где соединение формулы Va или Vb было получено, гидрирование его, чтобы получить ретиналь формулы I или соединение формулы I' соответственно, и в каждом случае, когда соединение формулы I' или I" было получено, изомеризация его в основных или кислых условиях или в присутствии металлического катализатора в требуемый ретиналь формулы I.
В приведенных выше обозначениях термин "(С1-С6)алкил" охватывает (от С3) линейные или разветвленные алкильные группы до шести атомов углерода, такие, как метил, этил, изопропил, трет-бутил, неопентил и н-гексил. Это применимо в равной мере к (С1-С6)алкильной части таких групп, как "(С1-С6)алкокси" и "(С1-С6)алкилтио", и ко всем, взятым в скобки, группам (C1-С6)алкил и (С1-С6)алкокси. Где более чем одна (С1-С6)алкильная группа присутствует в заместителях, обозначенных R2, R3 или R4, например, в таких группах, как "три(С1 -С6)алкилсилилокси", "ди(С1-С6)алкиламино", "ди(С1-С6)алкилфосфонил" или "три(С1-С6)алкилсилил", такие (С1-С6)алкильные группы могут быть одинаковыми или разными. "Арил" как таковой или как арильная (или "ар") часть "арилокси", "арилтио", "ароилокси", "арилсульфонилокси", "арилсульфонил", "N-арил(С1-С6)алкиламино", "диариламино", "ароил" или "диарилфосфонил" означает фенил, 1-нафтил или 2-нафтил, или обычно замещенную такую группу, например толил.
Приведенные выше формулы I, I', I", IIa, IIb, IIc, IId, IIe, III, IVa, IVb, IVc, IVd, IVe, Va и Vb охватывают в каждом случае изомерные формы, например, оптически активные или неактивные E/Z-изомеры, а также их смеси, если специально не заявлено иначе. Что касается E/Z-изомерии, то хотя (полностью-E)-изомерная форма промежуточных соединений формул IVa, IVb, IVc, IVd, IVe, Va, Vb, I' или I" и продукта, ретиналя, формулы I является в каждом случае предпочтительной, каждое соединение обычно присутствует или получается, если уместно, в виде смеси Е- и Z-изомеров.
На первом этапе способа в соответствии с изобретением, т.е. при взаимодействии производного формулы IIa, IIb, IIc, IId или IIe [впредь обозначаемых для краткости, как производное "5-замещенного пентадиена", "пентадиена", "пентена", "пента-1-ен-4-ина" или "пента-1-ен-4-ина" соответственно, или все вместе, как "5-замещенный пент(ади)ен(ин)"] с производным 1,3-бутадиена в кислых условиях происходит избирательная атака первого производного по γ-положению производного 1,3-бутадиена.
Первый этап процесса обычно осуществляется взаимодействием производного 5-замещенного пен(ади)ен(ин)а формулы IIa, IIb, IIc, IId или IIe с производным 1,3-бутадиена формулы III в органическом растворителе при температуре в диапазоне примерно -70°С до примерно +60°C, предпочтительно в температурном диапазоне примерно -30°С до комнатной температуры и в присутствии в качестве катализатора кислоты Льюиса или Бренстеда. Как, правило, соответствующими органическими растворителями являются полярные или неполярные апротонные растворители. Такими растворителями являются, например, низшие галоидированные алифатические углеводороды, например, метиленхлорид и хлороформ; низшие алифатические и циклические простые эфиры, например, диэтиловый эфир, трет-бутилметиловый эфир и тетрагидрофуран; низшие алифатические нитрилы, например ацетонитрил; низшие алифатические сложные эфиры, например этилацетат; низшие алифатические углеводороды, например, пентан и гексан; а также ароматические углеводороды, например толуол. Предпочтительным растворителем является ацетонитрил, необязательно в комбинации с другими упомянутыми выше растворителями. Там, где используется смесь ацетонитрила с другим растворителем, объемное соотношение ацетонитрила и такого другого растворителя составляет предпочтительно примерно 1:1 до примерно 1:0,5. Примерами кислот Льюиса, которые могут быть использованы, являются хлорид цинка, диэфират хлорида цинка, бромид цинка, ди(трифторметансульфонат) цинка, тетрахлорид титана, тетрахлорид олова, эфират трехфтористого бора, хлорид железа(III), триметилсилилтрифлат, а также перхлорат лития, а примерами кислот Бренстеда, которые могут быть использованы, являются n-толуолсульфоновая кислота, метансульфоновая кислота, трифторметансульфоновая кислота, серная кислота, а также трифторуксусная кислота. Обычно предпочтительными являются кислоты Льюиса, особенно упомянутые выше соли цинка, эфират трехфтористого бора и хлорид железа(III). Кислотные катализаторы обычно применяются в каталитических (ниже стехиометрических) количествах, удобно в количестве, которое составляет 0,5-30 молярных процентов в расчете на количество используемого производного 5-замещенного пент(ади)ен(ин)а, причем диапазон молярных процентов составляет 1-15%. Когда используется производное 5-замещенного пент(ади)ен(ин)а формулы IIa, IIb, IIc, IId или IIe, в которой R2 означает основную группу, особенно ди(С1-С6)алкиламино, N-арил(С1-С6)алкиламино или диариламино, безусловно потребуется большее относительное количество кислотного катализатора, обычно по меньшей мере один молярный эквивалент. Кроме того, удобно использовать 1,1-2,5 эквивалента, предпочтительно 1,1-1,8 эквивалента производного 1,3-бутадиена на эквивалент производного 5-замещенного пент(ади)ен(ин)а. Более того, реакцию удобно проводить при нормальном давлении. В большинстве случаев давление не является решающим.
При подготовке к выделению продукта формулы IVa, IVb, IVc, IVd или IVe, если уместно, к реакционной смеси может быть добавлена кислота, предпочтительно слегка разбавленная водная уксусная кислота, например, характерное объемное соотношение уксусная кислота:вода примерно 9:1, и смесь затем перемешивается в течение периода, например, около 30 мин до примерно 2 ч, удобно при интервале температур от примерно -10°С до примерно 30°С. Включенный в это этап подкисления/гидролиза гарантирует, что требуемое производное формулы IVa, IVb, IVc, IVd или IVe в конце концов получается по реакции производного пент(ади)ен(ин)а формулы IIa, IIb, IIc, IId или IIe с производным 1,3-бутадиена формулы III. Однако этап подкисления/гидролиза обычно является ненужным, когда в последнем производном R4 представлен как три(С1 -С6)алкилсилил. В любом случае этап подкисления/гидролиза является полезным для удаления избытка производного бутадиена из смеси после реакции и перед выделением продукта формулы IVa, IVb, IVc, IVd или IVe.
Продукт формулы IVa, IVb, IVc, IVd или IVe может быть затем выделен из реакционной смеси и, если необходимо, очищен известным по сути способом. Обычно смесь соединяют с водой, и все экстрагируют не смешивающимся с водой органическим растворителем, таким, как, например, низший алкан, диалкиловый эфир или алифатический сложный эфир, например, гексан, трет-бутилметиловый эфир или этилацетат, соответственно органическую фазу промывают водой и/или раствором бикарбоната натрия и/или насыщенным водным раствором хлорида натрия, сушат и концентрируют. Выделенный таким образом и по меньшей мере до некоторой степени промытый неочищенный продукт затем может быть очищен, если необходимо, например, колоночной хроматографией, например, используя силикагель в качестве стационарной фазы и элюирующий агент, такой, как гексан, этилацетат, толуол или смесь одного или нескольких из них.
В отношении дальнейшего (второго) этапа способа, т.е. элиминирования соединения R2Н из соединения формулы IVa, IVb, IVc, IVd или IVe, это может быть осуществлено с основанием или кислотой. Элиминирование алканола из β -алкоксиальдегидов или δ-алкокси-α,β-ненасыщенных альдегидов с образованием соответствующих α,β-ненасыщенных альдегидов известно в химической литературе и может быть осуществлено в различных условиях, и такая методология может быть применена в настоящем случае. Например, в области известных индуцированных основанием элиминирований 1,8-диазабицикло[5.4.0]ундец-7-ен очень часто используется как основание в количестве примерно 1-2 эквивалентов на эквивалент используемого альдегида. Такие условия применяются при известном получении каротиноидов [среди прочего смотри Bull. Chem. Soc. Japan, 50, 1161 и послед. (1977); там же, 51, 2077 и послед. (1978); Chem. Lett. 1975, 1201 и послед.; и German Offenlegungsschrift 2701489] и витамина А [смотри среди прочего Chem. Lett. 1975, 1201 и послед.; и J. Gen. Chem. USSR, 32, 63 и послед. (1962)]. Другая методология, использующая алканоат натрия в качестве основания, описана, например, в ЕР 814078816334 и 978508. В качестве примеров расщеплений, индуцированных кислотой, ссылка снова делается на Bull. Chem. Soc. Japan, 50, 1161 и послед. (1977) и на J. Gen. Chem. USSR, 30, 3875 и послед. (1960), в которых n-толуолсульфоновая кислота или 85% фосфорная кислота применяется в качестве кислотного катализатора. Буферная система ацетат натрия/уксусная кислота [Helv. Chem. Acta, 39, 249 и послед, и 463 и послед. (1956) и патенты US 2827481 и 2827482] была использована для такого расщепления, особенно при получении каротиноидов. В случае соответствующих алкоксикетонов (β-алкоксикетонов или δ-алкокси-α,β-ненасыщенных кетонов) отщепление алканола также происходило очень хорошо: в этом отношении смотри Synthesis, 1986, 1004 и послед, или J. Org. Chem., 49, 3604 и послед. (1984). Принимая во внимание эту и другую подходящую литературу, у специалиста в этой области не будет трудностей в нахождении условий реакций для успешного осуществления второго этапа способа в соответствии с настоящим изобретением.
Кроме того, элиминирование соединения R2Н может быть также осуществлено с несколькими эквивалентными количествами основания на каждый эквивалент соединения формулы IVa, IVb, IVc, IVd или IVe. Так, этап способа в этом случае удобно осуществлять, подвергая соединение формулы IVa, IVb, IVc, IVd или IVe, растворенное в соответствующем органическом растворителе, обработке основанием с элиминированием соединения R2Н. Соответствующими органическими растворителями в большинстве случаев являются протонные или апротонные растворители или их смеси, такие, как, например, спирты, например, этанол и изопропанол, и смеси спиртов; низшие галоидированные, преимущественно хлорированные, алифатические углеводороды, например, метиленхлорид и хлороформ; и ароматические углеводороды, например толуол. Основание может быть неорганическим или органическим, и это в большинстве случаев соответственно сильные основания, особенно те алкоголяты щелочных металлов, которые являются более сильными основаниями, например этилат натрия, и азотсодержащие основания, такие, как 1,8-диазабицикло[5.4.0]ундец-7-ен, триалкиламины, например, триэтиламин, и пиридин. Как указано выше, удобно использовать по меньшей мере один эквивалент основания на эквивалент соединения формулы IVa, IVb, IVc, IVd или IVe, предпочтительно примерно 1 до примерно 2 эквивалентов основания.
Когда алкоголят щелочного металла применяется в качестве основания, раствор алкоксида натрия в алканоле или приготавливается заранее, или этот раствор является свежеприготовленным из металлического натрия и алканола. Объединение алканольного раствора алкоксида натрия с раствором или суспензией соединения формулы IVa, IVb, IVc, IVd или IVe в (том же) алканоле, предпочтительно также приготовленном заранее, может осуществляться в любой из двух последовательностей. Реакционную смесь затем перемешивают соответственно в температурном интервале от примерно - 20°С до примерно 120°С, предпочтительно при температурах от примерно 0°С до примерно 70°С. В зависимости от точки кипения растворителя реакцию удобно проводить при нормальном давлении или при слегка избыточном давлении (чтобы достичь требуемой температуры). Однако в большинстве случаев давление не является решающим. В этих условиях реакция элиминирования обычно завершается в пределах нескольких часов, особенно после примерно 30 мин до примерно 4 ч.
В случае элиминирования соединения R2Н, индуцированного кислотой, соответствующими кислотами в большинстве случаев являются сильные минеральные кислоты, такие, как, например, хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота и перхлорная кислота, и сульфоновые кислоты, такие, как, например, метансульфоновая кислота, трифторметансульфоновая кислота и n-толуолсульфоновая кислота. Минеральные кислоты могут быть водными и в зависимости от природы кислоты могут иметь концентрацию от примерно 10 до примерно 50%. Хлористоводородная кислота (особенно примерно 10-37%), бромистоводородная кислота (особенно примерно 25-63%) или йодистоводородная кислота (например, 47%) является наиболее подходящей. В этом случае требуется только каталитическое количество, т.е. до самое большее 1 эквивалент на эквивалент соединения формулы IVa, IVb, IVc, IVd или IVe, предпочтительно примерно 0,1-1 эквивалент. Кроме того, индуцированное кислотой элиминирование осуществляется в растворителе, в котором соединение формулы IVa, IVb, IVc, IVd или IVe имеет хорошую растворимость и находится в растворенном состоянии (так называемое, "гомогенное расщепление"), или в растворителе, в котором это не происходит, т.е. вместо растворения соединение формулы IVa, IVb, IVc, IVd или IVe находится в суспензии ("гетерогенное расщепление"). Однако в обоих случаях кислотный катализатор необязательно должен быть полностью растворим. Соответствующими растворителями для гомогенного расщепления являются особенно галоидированные алифатические углеводороды, например, метиленхлорид, хлороформ и 1,2-дихлорэтан, и ароматические углеводороды, например, бензол или толуол. Подходящими растворителями (дисперсионной средой) для гетерогенного расщепления являются низшие алифатические нитрилы, кетоны и карбоновые кислоты, например, ацетонитрил, ацетон и уксусная кислота, соответственно предпочтительно ацетонитрил и ацетон. В обоих случаях элиминирование удобно проводить в температурном интервале от примерно -20°С до примерно +50°С, предпочтительно в интервале от примерно 0°С до комнатной температуры. Время реакции зависит в каждом случае от температуры реакции и может доходить до нескольких часов, причем реакция элиминирования обычно завершается самое позднее после примерно 5 ч.
Независимо от выбранного способа этапа элиминирования R2Н продукт формулы I, I', I", Va или Vb как подходящий может быть выделен из реакционной смеси известным по своей сути способом, обычно охлаждением реакционной смеси, удобно до комнатной температуры или даже до примерно 0°С, необязательным добавлением воды и экстракцией с органическим растворителем, не смешивающимся с водой. В качестве такого растворителя соответственно используется низший алифатический углеводород, например пентан или гексан; ароматический углеводород, например толуол; или низший алифатический сложный эфир, например этилацетат. После своего выделения продукт может быть дополнительно очищен, если необходимо или требуется, колоночной хроматографией или другим общеизвестным способом очистки ретиналя или его изомера, или его производного.
В случае, когда одним исходным веществом в способе по настоящему изобретению является производное 5-замещенного пента-1-ен-4-ина формулы IId или IIe, продукт на этом этапе способа, т.е. после элиминирования остатка R2Н из соединения формулы IVd или IVe как подходящий, является соединением формулы IVa или IVb соответственно. Следующим этапом является гидрирование для получения ретиналя или соединения формулы I' соответственно. Упомянутое гидрирование может проводиться при каталитических условиях, известных по своей сути, например, в каталитических условиях, применяемых в аналогичных способах, описанных в Houben-Weyl, Methoden der Organischen Chemie, том IV/1c ("Reduction", часть 1), 105 и послед., Thime Stuttgart 1980 и патенте UK 722911. Катализатор является подходяще "отравленным", типа катализатора Линдлара, предпочтительно 5% палладием на карбонате кальция, отравленным 3,5% свинцом, а температура и давление являются подходящими в интервале примерно 5-50°С и примерно 1-5 бар (примерно 0, 1-0,5 МПа), соответственно. В качестве растворителя, пригодного для использования, является органический растворитель относительно низкой полярности, например, алканол или алифатический сложный эфир, предпочтительно этилацетат. Кроме того, (отравленный) катализатор может быть выгодно модифицирован добавлением азотсодержащего соединения, такого, как хинолин.
После этапа элиминирования R2H или упомянутого выше случая дополнительного этапа гидрирования соответствующий выделенный и необязательно очищенный продукт, являющийся ретиналем или продуктом формулы I' или I" как подходящей, обычно присутствует в виде изомерной смеси четырех изомеров по двойным связям в положении 9 и 13, причем упомянутая изомерная смесь традиционно обозначается как (9 E/Z, 13 E/Z)-изомер.
В случае, когда получается соединение формулы I' (исходя из производного 5-замещенного 1,4-пентадиена или пента-1-ен-4-ина формулы IIb или IIe, через соединение формулы IVb или соединения формул IVe и IVb, соответственно) или альтернативно, где получается соединение формулы I" (исходя из производного 5-замещенного 1-пентена формулы IIc, через соединение формулы IVc), соответственный продукт затем изомеризуется в требуемый ретиналь формулы I. Условия для соответствующей изомеризации с основанием, кислотой или металлическим катализатором известны по своей сути и описаны, например, в J. Am. Che. Soc. 108. 2090 и послед. (1986) и Chem. Ber. 118, 348 и послед. (1985) с использованием 1,8-диазабицикло[5.4.0]ундец-7-ена и оксида алюминия в эфире, соответственно, в качестве основания; в ЕР 647624 и J. Chem. Res. Synop. 296 и послед. (1987) в отношении кислых условий; и в J. Chem. Soc. Perkin Trans. 1, 1593 и послед. (1984) и Tetr. Lett. 1979, 1499 и послед., для изомеризации в присутствии нонакарбонильного комплекса с двумя атомами железа [Fe2(CO)9; в бензоле] и три(трифенилфосфинил)родийхлоридного комплекса [Rh(PPh3)3Cl], соответственно в качестве металлического катализатора.
Если требуется, образуемый таким образом ретиналь, преимущественно в (9 E/Z, 13 E/Z)-изомерной форме затем изомеризуют в требуемую, как правило, изомерную форму ретиналя (полностью-E)-ретиналь. Изомеризация может проводиться в обычных изомеризующих условиях, и особенно предпочтительный способ включает катализируемое кислотой образование аддукта (преимущественно) (полностью-Е)-ретиналя с гидрохиноном из ретиналя, полученного, как определено и описано выше, и первоначально по существу в (9 E/Z, 13 E/Z)-изомерной форме, и гидрохинона в органическом растворителе, в котором сам аддукт умеренно растворим, тем самым, позволяя требуемому аддукту (полностыо-Е)-ретиналь-гидрохинон находиться в кристаллической форме. Это образование аддукта представляет дальнейший аспект по настоящему изобретению. В качестве кислотного катализатора соответственно применяется хлористоводородная кислота (особенно примерно 37%), бромистоводородная кислота (48%) или йодистоводородная кислота (47%), n-толуолсульфоновая кислота или элементарный йод. Молярное соотношение исходный ретиналь:гидрохинон составляет соответственно примерно 1:0,5, и два компонента обычно соединяют вместе в органическом растворителе, в котором сам аддукт умеренно растворим, таком, как низший алифатический углеводород, например, пентан или гексан; или низший алифатический простой эфир, например, диэтиловый эфир. Предпочтительным растворителем является диэтиловый эфир или смесь упомянутого простого эфира и гексана. При нахождении двух компонентов аддукта в таком растворителе со следовым количеством кислотного катализатора при температуре окружающей среды, как правило, происходит кристаллизация аддукта (полностью-Е)-ретиналь-гидрохинон после относительно короткого времени, например, примерно одного часа. Кристаллический аддукт может быть затем легко удален из жидкой среды путем фильтрации и, если требуется, промыт, например, растворителем, применяемым для его образования, как упомянуто выше, и высушен, предпочтительно при комнатной температуре при пониженном давлении. Такая общепринятая методология образования и выделения аддукта (полностью-E)-ретиналь-гидрохинон описана, например, в патенте US 3013081, патентах Франции 1288975 и 1291622.
В качестве альтернативы предшествующему выделению и необязательной очистке ретиналя, образованного в охарактеризованном/описанном выше двух- или трехэтапном способе (не трех- или четырехэтапном способе, исходя из производного 5-(2,6,6-триметилциклогекс-1-енил)пента-1-ен-4-ина формулы IId или IIe и включая дополнительный этап гидрирования) по настоящему изобретению, в тех случаях, где элиминирование R2Н (второй этап способа) индуцируется кислотой, и предполагается описанное выше катализируемое кислотой образование аддукта ретиналь-гидрохинон, упомянутый второй этап способа и этап образования аддукта ретиналь-гидрохинон могут быть объединены, избегая таким образом выделения промежуточного вещества и необязательной очистки ретиналя перед образованием аддукта. При такой альтернативе растворитель, использованный для индуцированной кислотой реакции элиминирования R2Н, если он не тот, в котором аддукт был бы легко растворим, в таком, как галоидированный алифатический углеводород, должен быть заменен по завершении реакции элиминирования на растворитель, в котором аддукт нерастворим или умеренно растворим. Таким заменяющим растворителем соответственно является низший алифатический углеводород, например, пентан или гексан, или низший алифатический простой эфир, например, диэтиловый эфир, т.е. растворитель, указанный выше в качестве пригодного для "отдельного" образования аддукта. Замена растворителя соответственно осуществляется выпариванием "непригодного" растворителя при пониженном давлении и добавлением подходящего растворителя. Затем прибавляют гидрохинон, и способ кристаллизации и выделения образующегося кристаллического аддукта (полностыо-Е)-ретиналь-гидрохинон осуществляют, как описано выше для "отдельного" образования аддукта.
Полученный таким образом аддукт (полностью-E)-ретиналь-гидрохинон может быть превращен в используемый далее продукт, витамин А (ретинол) в преимущественно (полностью-E)-изомерной форме способами, известными по сути, например прямым каталитическим восстановлением с использованием рутениевого катализатора, как описано в J. Molec. Catalysis 79, 117-131 (1993), там же 66, L 27-L 31 (1991) и патенте US 4906795. Это восстановление может быть также осуществлено с боргидридом натрия, как описано в Chem. Lett. 1975, 1201 и послед., где также указано, как ретинол может быть превращен в ацетат витамина А путем ацетилирования уксусным ангидридом в пиридине.
За исключением соединений формулы IIa, где одновременно R1 означает гидроксил, и R2 означает арилсульфонил, исходные производные 5-замещенного пент(ади)ен(ин)а формул IIa, IIb, IIc, IId и IIe являются новыми соединениями и составляют еще дополнительный аспект по настоящему изобретению.
Эти новые исходные вещества формул IIa, IIb, IIc, IId и IIe могут быть получены с использованием принципов уровня техники, подходящих для присоединения по тройным связям, применяя в качестве исходных соединений соответствующие ацетиленовые соединения формул


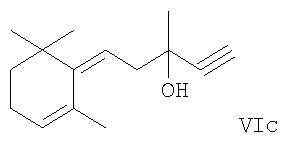
(для получающихся в конечном счете исходных соединений формул IIa, IIb и IIc, соответственно, с характерной особенностью R1 как гидроксила) или, как уместно, соответствующие ацетиленовые соединения, в которых группа OR3 присутствует вместо гидроксильной группы. Соответственно последние соединения имеют следующие формулы VIa′, VIb′ и VIe′:

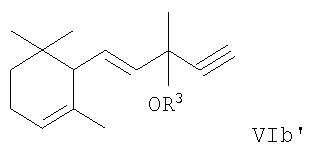

где R3 имеет соответствующее значение, приведенное выше, т.е. означает (C1-С6)алкил, (C1-С6)алкилкарбонил, ароил, (C1-С6)алкоксикарбонил, три(C1-С6)алкилсилил, ди(C1-С6)алкилфосфонил, диарилфосфонил, (C1-С6)алкилсульфонил или арилсульфонил.
Исходное ацетиленовое соединение формулы VIa, VIb, VIc, VIa′, VIb′ или VIc′ реагирует в каждом случае с соединением, несущим остаток R2, особенно с соединением формулы R2H. Условия реакции в каждом случае, как правило, те, которые используются для реакций с металлоорганическими соединениями, т.е. применение в основном эфирного растворителя, например низшего алифатического простого эфира, например диэтилового эфира или диметоксиэтана, или циклического простого эфира, например тетрагидрофурана, и низких температур реакции, например, примерно -100°С до примерно 0°С, особенно примерно -70°С до примерно -40°С (смотри Organometallics in Synthesis, A Manual, ред. М. Schlosser, с. 126, 1994, John Wiley и Sons Ltd.).
Примерами специфических литературных ссылок, обеспечивающих принципы осуществления соответствующих реакций присоединения такого исходного ацетиленового соединения к соединению формулы R2Н, распределенных по категориям в соответствии с природой группы R2, являются следующие:
R2 означает (C1-С6)алкокси: ЕР 274600; Tetr. Lett. 40, 6193-6195 (1999), Synlett. 7, 880 и послед. (1996) и J. Org. Chem. USSR (Engl.) 6, 903 и послед. (1970);
R2 означает (C1-С6)алкилтио или арилтио: Tetr. Lett. 24, 61 и послед. (1983) и Tetr. Lett. 25, 189 и послед. (1984);
R2 означает (C1-С6)алкилкарбонилокси или ароилокси:: Bull. Soc. Chim. Fr. 133, 939-944 (1996), Organomet. 15, 3998-4004 (1996) и J. Organomet. Chem. 551, 151-157 (1998);
R2 означает арилсульфонил: Tetrahedron. 46, 7197-7206 (1990);
R2 означает хлор или бром: Org. Reactions. 13, 150 и послед. (1963) и Tetr. Lett. 32, 5861-5864(1991).
Эти новые производные 5-замещенных пент(ади)енов формул IIa, IIb и IIc,
где R1 означает гидроксил, и
R2 означает одну из ранее приведенных (выбранных) групп R2 или означает арилокси, три(C1-С6 )алкилсилилокси, ди(C1-С6)алкилфосфонилокси, (C1-С6)сульфонилокси, арилсульфонилокси, (C1-С6)алкилсульфонил, ди(C1-С6)алкиламино, N-арил(C1-С6)алкиламино или диариламино, могут (также) быть получены по двухэтапной методике, представленной схематически следующим образом:
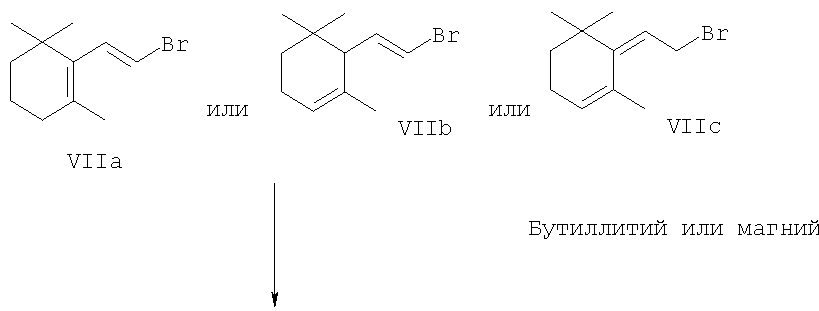

В каждом случае условия реакций обычно те, которые использованы для такого же типа реакции, как описано в упомянутой выше публикации Organomellics in Synthesis, ред. М. Schlosser, а также в J. Org. Chem. 43, 1595 и послед. (1978).
Те производные 5-замещенного пент(ади)ена формул IIa, IIb и IIc, где R2 означает хлор или бром, могут также быть получены "непрямым" присоединением HCl или HBr, соответственно по тройной связи соответствующих исходных ацетиленовых соединений формул VIa, VIb, VIc, VIa′, VIb′ и VIc′ путем первоначального присоединения органического соединения бора [смотри, например, J. Org. Chem. 54, 6068 и послед. (1989), Tetr. Lett. 39, 3103-3106 (1998), Synth. Commun. 11, 247 и послед. (1981), Synth. Commun. 1983, 1027 и послед, и J. Chem. Soc. Perkin Trans. 1, 2725-2726 (1992)], гидрида алкилалюминия [Tetr. Lett. 29, 6243 и послед. (1988)], гидрида олова [Synth. 1986, 453 и послед., J. Am. Chem. Soc. 106, 5734 и послед. (1984), Tetr. Lett. 39, 6099 и послед. (1998) и Helv. Chim. Acta 66, 1018 и послед. (1983)] или циркониевого соединения [J. Org. Chem. 56. 2590 и послед. (1991), Synth. 1993, 377-379 и Tetr. Lett. 31, 7257 и послед. (1990)], затем галоидированием соответствующего промежуточного вещества галоидирующим агентом, таким, как элементарный хлор или N-хлорсукцинимид, или элементарный бром, или N-бромсукцинимид соответственно.
Альтернативный способ получения этих производных 5-замещенного пент(ади)ена формул IIa, IIb и IIc,
где R1 означает гидроксил, и
R2 означает (C1-С6)алкокси, также основан на принципах, описанных в такой литературе, как Organometallics in Synthesis, A Manual, ред. М. Schlosser, с. 126, 1994, John Wiley и Sons Ltd., а также в J. Org. Chem. 23, 1063 и послед. (1958) и J. Org. Chem. 43, 1595 и послед. (1978). Согласно способу β-ионон, α -ионон или ретро-ионон формулы Xa, Xb или Хс соответственно,
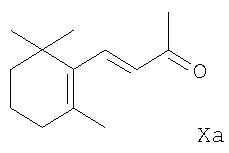

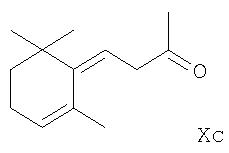
реагирует с соответствующим цис-1-литио-2-алкоксиэтиленом [цис-LiCH=СНО(С1-С6)алкилом] в эфирном растворителе при температуре приблизительно 100°С до примерно 0°С. В качестве эфирного растворителя в основном используются низшие алифатические простые эфиры, например, диэтиловый эфир или диметоксиэтан, или простой циклический эфир, например, тетрагидрофуран. Реакцию предпочтительно осуществляют при температуре -70°С до примерно -40°С. Кроме того, исходный цис-1-литио-2-алкоксиэтилен удобно получается in situ из соответствующего цис-1-бром-2-алкоксиэтилена [цис-BrCH=СНО(С1-С6)алкила] и н-бутиллития или втор-бутиллития (примерно эквимолярные количества двух реагентов), или трет-бутиллития (примерно в двойном молярном количестве относительно количества используемого производного этилена), с использованием того же растворителя и температуры реакции, которые приведены выше для последующей реакции с полученным таким образом 1-литио-2-алкоксиэтиленом. Полученное в конечном счете производное 5-замещенного пент(ади)ена после завершения реакции удобно выделяется из смеси добавлением к ней воды и экстракцией органическим растворителем, не смешивающимся с водой, особенно апротонным растворителем, таким, как низший алифатический углеводород, например, гексан; ароматическим углеводородом, например, толуолом; низшим галоидированным алифатическим углеводородом, например, метиленхлоридом или хлороформом; низшим алифатическим или циклическим простым эфиром, например, диэтиловым эфиром, тетрагидрофураном или диоксаном; или галоидированным ароматическим углеводородом, например, хлорбензолом. После удаления экстрагирующего растворителя соответственно при слегка повышенной температуре и пониженном давлении соответствующий требуемый 5-замещенный 1-алкокси-3-метил-3-гидроксипент(ади)ен формулы IIa, IIb или IIc получается с хорошим показателем чистоты и практически количественным выходом. Обычно он не должен далее подвергаться очистке перед применением в способе по настоящему изобретению, включающему реакцию с производным 1,3-бутадиена формулы III.
Предшественники формул VIa, VIb и VIc, как также и предшественники формул Ха, Xb и Хс, являются известными соединениями. Литературные ссылки, касающиеся этих предшественников, включают, например, в отношении предшественника VIa: патент Германии (DE) 2731284; предшественника VIb: Can. J. Chem. 46, 3041 и послед. (1968); и предшественников VIc и Хс: WO 00/002854 и Bull. Soc. Chim. Fr.132, 696 и послед. (1995). Предшественники формул Ха и Xb доступны в продаже.
Только некоторые предшественники формул VIa', VIb' и VIc' являются известными соединениями, например, соединения формулы VIa', где R3 означает метил или этил (см. патенты DE 2321141 и US 4035425); формулы VIb′, где R3 означает ацетил (см. WO 00/002854 А1); формулы VIa′, где R3 означает бензоил [см J. Am. Chem. Soc. 102, 6355 и послед. (1980), там же, 104, 6115 и послед. (1982), там же, 107, 1028 и послед. (1985) и там же. 107. 1034 и послед. (1985)]; формулы VIb′, где R3 означает метоксикарбоил (см. ЕР 647624); и формулы VIa′, где R3 означает триэтилсилил [см. J. Org. Chem. 63, 8704 и послед. (1998)].
Оставшиеся предшественники формул VIa′, VIb′ и VIc′ могут быть получены из соответствующих предшественников формул VIa, VIb и VIc следующими способами в соответствии с природой группы R3:
R3 означает (С1-С6)алкил: как описано в патентах DE 2321141 и US 4035425 (R3' означает метил или этил: см. выше), или аналогично соответствующему способу;
R3 означает (С1-С6)алкилкарбонил: как описано в WO 00/002854 А1 или J. Org. Chem. 56, 5349 и послед. (1991), или аналогично подходящему способу путем алканоилирования соответствующего соединения формулы VIa, VIb или VIc соответствующим ангидридом карбоновой кислоты, например уксусным ангидридом в присутствии основания;
R3 означает ароил: как описано в J. Am. Chem. Soc. 102, 6355 и послед. (1980), там же, 104, 6115 и послед. (1982), там же, 107, 1028 и послед. (1985) или там же, 107, 1034 и послед. (1985), или аналогично подходящему способу путем ароилирования соответствующего соединения формулы VIa, VIb или VIc с соответствующим ангидридом ароматической кислоты в присутствии основания;
R3 означает (С1-С6)алкоксикарбонил: как описано в ЕР 647624, или аналогично подходящему способу путем взаимодействия соответствующего соединения формулы VIa, VIb или VIc с соответствующим алкоксикарбонилхлоридом в присутствии основания;
R3 означает три(С1-С6)алкилсилил: как описано в J. Org. Chem. 63, 8704 и послед. (1998), или аналогично подходящему способу путем взаимодействия соответствующего соединения формулы VIa, VIb или VIc с соответствующим триалкилсилилхлоридом в присутствии основания, например, триэтиламина или диметиламинопиридина;
R3 означает ди(С1-С6)алкилфосфонил или диарилфосфонил: аналогично подходящему способу, описанному в J. Org. Chem. 54, 627 и послед. (1989), взаимодействием соответствующего соединения формулы VIa, VIb или VIc с соответствующим диалкилхлорфосфитом и последующим окислением полученного таким образом фосфита в фосфат, или аналогично подходящему способу, описанному в Tetr. Lett. 1984, 4195 и послед., взаимодействием соответствующего соединения формулы VIa, VIb или VIc с соответствующим диалкил- или диарилхлорфосфатом в присутствии основания;
R3 означает (С1-С6)алкилсульфонил или арилсульфонил: аналогично подходящему способу, описанному в Synlett. 1053 и послед. (1999) или Tetrahedron 55, 6387 и послед. (1999), взаимодействием соответствующего соединения формулы VIa, VIb или VIc с соответствующим хлорангидридом алкансульфоновой кислоты или хлорангидридом арилсульфоновой кислоты в присутствии основания, например, триэтиламина или пиридина. Предшественники формул VIa, VIb или VIe могут быть получены из β-ионона, α-ионона и ретро-ионона (соединений формул Ха, Xb и Хс, соответственно приведенных выше) путем реакции деградации трехгалоидных производных метана, особенно с бромом в водном гидроксиде натрия, в соответствии со способом, описанном на стр.325-326 в Carotenoids, том 2: Synthesis, ред. G. Britton, S. Liaaen-Jensen и Н. Pfander, Birkhäuser Veriag, Basel, Boston, Berlin, 1996, давая соответствующую β -замещенную акриловую кислоту формулы XIa или XIb или γ-замещенную винилуксусную кислоту формулы XIc, соответственно,


затем по реакции Хунсдиккера, например, с N-бромсукцинимидом, в соответствии со способом, описанном в Tetrahedron 43, 4601-4608 (1987), давая соединение формулы VIIa, VIIb или VIIc соответственно.
Предшественники формулы IX в некоторых случаях являются известными соединениями, а в других случаях могут быть получены аналогично известным соединениям; соответствующая литература для различных типов группы R2 приводится далее:
R2 означает хлор или бром: J. Chem. Soc. Perkin Trans. 1990, 3317-3319;
R2 означает (С1-С6)алкокси: Collect. Czech. Chem. Commun. 1992, 1072-1080, где описано получение предшественника формулы IX, где R2 означает этокси; соответствующее соединение с R2, означающим метокси, доступно в продаже.
R2 означает (С1-С6)алкилтио или арилтио: J. Org. Chem. 46, 235 и послед. (1981);
R2 означает арилокси: Fette, Seifen, Anstrichmittel 82, 82-86 (1980);
R2 означает (С1-С6 )алкилкарбонилокси или арилокси: J. Org. Chem. 50, 1955-1959(1985);
R2 означает три(С1-С6)алкилсилилокси: Liebigs Ann. Chem. 12, 2352 и послед. (1985);
R2 означает ди(С1-С6)алкилфосфонилокси: патент Швейцарии 490016;
R2 означает (С1-С6)алкилсульфонил или арилсульфонил: Aust. J. Chem. 41, 881 и послед. (1988);
R2 означает ди(С1-С6)алкиламино, N-арил(С1-С6)алкиламино или диариламино: Org. Prep. Proced. Int. 16, 31-36 (1984).
В случае предшественника формулы IX, где R2 означает (С1-С6)алкилсульфонилокси или арилсульфонилокси, соединения являются новыми, и никакие аналогичные способы их получения неизвестны. Однако они могут быть получены взаимодействием 3-кетобутиральдегида с соответствующим алкан- или арилсульфонилхлоридом в таком растворителе, как тетрагидрофуран и в присутствии основания, например, триэтиламина. J. Heterocyclic Chem. 1991, 885-890 и J. Chem. Soc. Perkin Trans. 1, 1992, 2855-2861, соответственно, обеспечивают подходящую информацию, касающуюся методологии.
Альтернативные новые исходные вещества для способа по настоящему изобретению, т.е. производные 5-замещенного пента-1-ен-4-ина формул IId и IIe, могут быть получены из известных 1-этинил-2,6,6-триметилциклогекс-1-ена или -2-ена, соответственно, формулы


[(см. патент US 2775626 и Tetrahedron 55, 15071-15098 (1999), патент СН 651287, соответственно, для получения каждого в отдельности 1-этинил-2,6,6-триметилциклогексена] путем реакции конденсации при депротонировании сильным основанием в безводных условиях в инертном органическом растворителе с кетоном формулы IX, приведенным выше. В качестве сильного основания соответственно применяют основание, которое является обычным для депротонирования ацетиленов, особенно основания, содержащие литий, натрий или магний. Примерами таких оснований служат литийорганические соединения, такие, как метиллитий, бутиллитий или фениллитий, реактивы Гриньяра, такие, как алкилмагнийгалоиды и диалкилмагний, амиды, такие, как амид лития и амид натрия, и гидриды, такие, как гидрид лития и гидрид натрия, предпочтительным основанием является алкиллитий или алкилмагнийхлорид. В качестве растворителя соответственно используются простые алифатические или циклические эфиры, например, диэтиловый эфир или, соответственно, тетрагидрофуран или диоксан; или алифатический или ароматический углеводород, например, гексан или петролейный эфир, или бензол, толуол либо ксилол соответственно. Предпочтительным растворителем для реакции является тетрагидрофуран. Кетон формулы IX предпочтительно используется в эквивалентном количестве или с легким избытком, например, 1,1-1,3 эквивалента на эквивалент 1-этинил-2,6,6-триметилциклогексена формулы XIIa или XIIb. Однако больший избыток необязательно вреден для выхода реакции конденсации. Более того, легкий избыток основания относительно количества соединения формулы XIIa или XIIb, например, 1, 1-1,3 экв. соответственно используется. Кроме того, возможно осуществить реакцию конденсации при дополнительном присутствии неорганической соли лития или церия, которые, как известно, немного повышают выход реакции. Примерами таких солей являются галоидные производные лития, галоидные производные церия и тетрафторборат лития, причем трихлорид церия и бромид лития являются преимущественными солями. Количество соли лития или церия не является определяющим и может составлять, например, примерно 0,5-1,0 экв. на эквивалент количества соединения формулы XIIa или XIIb. Независимо от используемых особых условий, указанных выше, конденсация соответственно осуществляется при температурах от примерно -80°С до примерно 10°С, предпочтительно от примерно -70°С до примерно -50°С. Реакция конденсации дает те производные 5-замещенного пента-1-ен-4-ина формул IId и IIe, где R1 означает гидроксил. Остающиеся производные 5-замещенного пента-1-ен-4-ина формул IId и IIe, где R1 означает группу OR3, могут быть получены из предшествующих производных по аналогии со способами получения предшественников формул VIa′, VIb′ и VIc′ из соответствующих предшественников формул VIa, VIb и VIc, как указано выше при упоминании подходящих ссылок для трансформации гидроксила в группу OR3.
Эта реакция конденсации, как указано выше, дает исходное производное 5-замещенного пента-1-ен-4-ила формулы IId или IIe для непосредственного применения по настоящему изобретению, т.е. для реакции с производным 1,3-бутадиена формулы III. Однако вместо такого непосредственного применения производное может быть превращено в другое используемое исходное вещество, а именно в производное 5-замещенного 1,4-пентадиена формулы IIa или IIb, соответственно, путем избирательного восстановления комплексным гидридом металла, предпочтительно гидридоалюминатом, наиболее предпочтительно бис(2-метоксиэтокси)алюмонатрий-гидридом, в условиях, описанных в литературе, например, аналогично методикам, описанным в Helv. Chim. Acta 73, 868-873 (1990) и патентах US 4952716 и 5227507. Восстановление удобно проводить в инертном органическом растворителе, примерами таковых являются те растворители, которые приведены выше в связи с реакцией 1-этинил-2,6,6-триметилциклогексена формулы XIIa или XIIb с кетоном формулы IX, особенно растворители, упомянутые там, как являющиеся предпочтительными. Температура и давление, при которых осуществляется избирательное восстановление, не являются определяющими. Поскольку восстановление протекает быстро, его осуществляют предпочтительно при температурах от примерно -50°С до примерно 30°С, более предпочтительно при температурах от примерно 10°С до примерно 0°С и при атмосферном давлении. Восстанавливающий агент может быть использован в почти эквивалентном количестве или предпочтительно в избыточном количестве относительно количества исходного производного 5-замещенного пента-1-ен-4-ина формулы IId или IIe. Количество, которое составляет по меньшей мере примерно 1,1 экв., например, 1,2-1,4 экв. на эквивалент упомянутого производного, является предпочтительным. Однако больший избыток необязательно вреден для результата избирательного восстановления.
Гидролиз алюминиевого комплекса, образованного в качестве промежуточного вещества при описанном выше избирательном восстановлении, в требуемое производное 5-замещенного 1,4-пентадиена формулы IIa или IIb, может быть осуществлен известным по сути способом, например, обработкой водой в присутствии органической или неорганической кислоты, такой, как n-толуолсульфокислота, или более предпочтительно в присутствии щелочи, такой, как раствор гидроксида натрия. Температура и давление не являются определяющими. Однако, как правило, гидролиз осуществляют при атмосферном давлении и комнатной температуре или более низкой температуре, предпочтительно при температурах от примерно 0°С до комнатной температуры.
Производные 1,3-бутадиена формулы III, использованные в качестве исходных веществ на первом этапе способа по настоящему изобретению, являются или известными соединениями, или могут быть получены по аналогии с известными соединениями, например, согласно методикам, описанным в Bull. Soc. Chim. Fr. 130 (2), 200-205 (1993), Tetr. Lett. 22 (29), 2833-2836 (1989) и там же, 26 (47), 8591-8594 (1995).
Изобретение иллюстрируется следующими примерами.
Пример 1
Получение (1Z,4E)-1-этокси-3-метил-3-гидрокси-5-(2,6,6-триметилциклогекс-1-енил)-1,4-пентадиена
К 750 мл тетрагидрофурана в колбе на 750 мл, снабженной магнитной мешалкой и приспособлениями для пуска аргона, прибавляли в атмосфере аргона 17,6 мл (приблизительно 25 г, 0,16 моля) цис-1-бром-2-этоксиэтилена, и затем смесь охлаждали до -70°С. Затем к перемешиваемой смеси медленно прибавляли 210 мл (0,32 моля) трет-бутиллития (1,5 М раствор в гексане) при поддерживании температуры от -70°С до -60°С, добавление завершалось в пределах примерно 30 мин. После перемешивания смеси в течение дальнейших 30 мин при -70°С медленно прибавляли раствор 20,2 г (0,103 моля) β-ионона (приблизительно 98% чистоты) в 60 мл тетрагидрофурана, поддерживая температуру смеси при -70°С до -60°С. Реакционную смесь затем перемешивали при -70°С в течение 3 ч, после чего по данным ВЭЖХ реакция была завершена.
Охлаждение убирали, и к содержимому колбы прибавляли 200 мл смеси лед/вода в течение 10 мин. После того, как температура смеси достигла 0°С, ее выливали в 250 мл гексана, и все вместе промывали последовательно 3×200 мл воды и 2×200 мл насыщенного раствора хлористого натрия. Затем объединенные водные фазы экстрагировали 250 мл гексана. Объединенные гексановые экстракты сушили над безводным сульфатом натрия и после удаления высушивающего агента фильтрацией раствор концентрировали при 35°С при пониженном давлении 100-200 мбар (10-20 кПа), затем путем концентрации при комнатной температуре в высоком вакууме удаляли последние следы растворителя. Получали 30,25 г (выход количественный) (1Z,4E)-1-этокси-3-метил-3-гидрокси-5-(2, 6,6-триметилциклогекс-1-енил)-1,4-пентадиена в виде бледно-желтого масла.
1H-ЯМР (400 МГц, CDCl3): 6,05 (d, J=16 Гц, 1Н), 5,99 (d=8 Гц, 1Н), 5,54 (d, J=16 Гц, 1Н), 4,60 (d, J=8 Гц, 1Н), 3,84 (q, J ˜ 8 Гц, 2Н), 1,95 (t, J ˜ 8 Гц, 2Н), 1,65 (s, 3Н), 1,6 (m, 2Н), 1,45 (m, 2Н), 1,25 (t, J ˜ 8 Гц, 3Н), 0,98 (ls, 3Н);
ИК (пленка, см-1): 1659 (С=O), 1102 (С-О-С);
МС: 264,3 (M+).
Пример 2
Получение (1Z,4E)-1-этокси -3-метил-5-(2,6, 6-триметилциклогекс-2-енил)-пента-1,4-диен-3-ола
Используя методику, аналогичную той, которая описана в примере 1, получали 14,32 г (выход 92%) заглавного соединения из 10,10 г (50 ммолей) α-ионона.
1H-ЯМР (400 МГц, CDCl3): 5,96 (m, 1Н), 5,59 (d, J=12 Гц, 1Н), 5,45 (d=11 Гц, 1Н), 5,39 (m, 1Н), 4,57 (d, J=8 Гц, 1Н), 3,82 (q, J ˜ 8 Гц, 2Н), 2,13 (d, J ˜ 9 Гц, 1Н), 1,98 (s, 2Н), 1,53 (m, 2Н), 1,36 (m, 3Н), 1,27 (t, J ˜ 8 Гц, 3Н), 0,93 (s, 3Н), 0,84 (s, 3Н);
ИК (пленка, см-1): 1658 (С=С), 1103 (С-О-С);
МС: 264,3 (М+).
Пример 3
Получение 2-(5-этокси-3-метокси-3-метил-(1E,4Z)-пента-1,4-диенил)-1,3,3-триметилдиклогексена
Промывали три раза 0,8 мл (приблизительно 5,6 ммоля) гидрида калия (35% суспензия) в атмосфере аргона в трубке Шленка с перемешивающей пластинкой гексаном и декантировали. После прибавления 0,72 мл (7,5 ммоля) диметилсульфата при температуре окружающей среды по каплям прибавляли растворенные в 3 мл сухого тетрагидрофурана 1,43 г (5 ммолей) (1Z,4E)-1-этокси-3-метил-5-(2,6, 6-триметилциклогекс-2-енил)-пента-1,4-диен-3-ола.
После того, как смесь перемешивали в течение 18 ч при температуре окружающей среды, реакцию прекращали добавлением 1,5 мл 25% аммиака и перемешивали в течение дальнейших 30 мин. Затем раствор разбавляли 50 мл гексана и экстрагировали 3×25 мл воды и 2×25 мл насыщенного солевого раствора. Водные фазы снова экстрагировали 50 мл гексана. Объединенные органические фазы концентрировали при пониженном давлении, получая 1,498 г (выход 98%) 2-(5-этокси-3-метокси-3-метил-(1E,4Z)-пента-1,4-диенил)-1,3,3-триметилциклогексена в виде желтоватого масла. Оно было достаточно чистым для дальнейшей реакции.
1H-ЯМР (400 МГц, CDCl3): 6,05 (d, J=16 Гц, 1Н), 5,99 (d, J=8 Гц, 1Н), 5,48 (d, J=16 Гц, 1Н), 4,61 (d, J=7 Гц, 1Н), 3,81 (q, J=7 Гц, 2Н), 3,23 (s, 3Н), 1,97 (t, J ˜ 7 Гц, 2Н), 1,68 (s, 3Н), 1,6 (m, 2Н), 1,51 (s, 3Н), 1,45 (m, 2Н), 1,23 (t, J ˜ 8 Гц, 3Н), 0,98 (s, 6Н);
ИК (пленка, см-1): 1660 (С=С), 1105 (С-О-С);
МС: 278,2 (М+).
Пример 4
Получение 3-метокси-3-метил-5-(2,6, 6-триметилдиклогекс-1-енил)-(1Z,4Е)-пента-1,4-диенилбензоата
В двухгорлую колбу, снабженную мешалкой, резиновой перегородкой и впуском для аргона, вводили 3,60 г (15 ммолей) 2-(3-метокси-3-метил-(E)-пент1-ен-4-инил)-1,3,3-триметилциклогексена, 2,76 г бензойной кислоты, 20 мл толуола и 90 мг (0,15 ммолей) комплекса бис(2-метилаллил)этилен-бис-дифенилфосфинрутений(II) в атмосфере аргона. После перемешивания в течение 12 ч при температуре окружающей среды реакционную смесь нагревали до 45°С в течение 4 ч для завершения реакции.
Впоследствии все летучие вещества удаляли при пониженном давлении 100-150 мбар (10-15 кПа), и полученное коричневое масло очищали колоночной хроматографией, используя 50 г силикагеля (0,04-0,063 мм) в качестве стационарной фазы и смесь 9:1 (об./об.) гексана и этилацетата в качестве элюирующего агента. Таким способом получали 2,67 г (выход приблизительно 50%) 3-метокси-3-метил-5-(2,6, 6-триметилциклогекс-1-енил)-(1Z, 4E)-пента-1,4-диенилбензоата.
1H-ЯМР (400 МГц, CDCl3): 8,09 (d, J=7 Гц, 2Н), 7,60 (t, J=7 Гц, 1Н), 7,46 (t, J=8 Гц, 2Н), 7,38 (d, J=7 Гц, 1Н), 6,14 (d, J=16 Гц, 1Н), 5,57 (d, J = 16 Гц, 1Н), 5,11 (d, J=7 Гц, 1Н), 3,28 (s, 3Н), 1,96 (t, J ˜ 6 Гц, 2Н), 1,65 (s, 3Н), 1,63 (s, 3Н), 1,57 (m, 2Н), 1,43 (m, 2Н), 0,97 (s, 3Н), 0, 96 (s, 3Н);
ИК (пленка, см-1): 1737 (С=O), 1667 (С=С), 1095 (С-О-С);
МС: 354,2 (M+).
Пример 5
Получение 3-гидрокси-3-метил-5-(2,6,6-триметилциклогекс-1-енил)-(1Z,4E)-пента-1,4-диенилбензоата
Используя методику, аналогичную той, которая описана в примере 4, получали 9,75 г (выход 95%) 3-гидрокси-3-метил-5-(2,6,6-триметилциклогекс-1-енил)-(1Z,4E)-пента-1,4-диенилбензоата из 6,60 г (30 ммолей) (Е)-3-метил-1-(2,6,6-триметилциклогекс-1-енил)пент-1-ен-4-ин-3-ола в виде неочищенного продукта, который был достаточно чистым для дальнейшей реакции.
1Н-ЯМР (400 МГц, CDCl3): 8,07 (d, J=7 Гц, 2 Н), 7,61 (t, J=7 Гц, 1Н), 7,47 (t, J=8 Гц, 2Н), 7,31 (d, J=7 Гц, 1Н), 6,19 (d, J=16 Гц, 1Н), 5,53 (d, J=16 Гц, 1Н), 5,22 (d, J=7 Гц, 1Н), 1,96 (t, J ˜ 6 Гц, 2Н), 1,65 (s, 6Н), 1,59 (m, 2Н), 1,45 (m, 2Н), 0,98 (s, 6Н);
ИК (пленка, см-1): 1738 (С=O), 1666 (С=С);
МС: 340,2 (M+).
Пример 6
Получение 1-этокси-3-метил-5-(2,6, 6-триметилциклогекс-2-енилиден)-(Z)-пент-1-ен-3-ола
Используя методику, аналогичную той, которая описана в примере 1, получали 2,64 г (выход 43%) заглавного вещества из 4,67 г (23,1 ммоля) ретро-ионона после хроматографической очистки, используя 30 г силикагеля (0,04-0,063 мм) в качестве стационарной фазы и смесь 98:2 (об./об.) гексана и этилацетата в качестве элюирующего агента.
1Н-ЯМР (400 МГц, CDCl3): 5,96 (d, J=7 Гц, 1Н), 5,61 (уш. s, 1Н), 5,56 (t, J=7 Гц, 1Н), 4,51 (d, J=7 Гц, 1Н), 3,84 (q, J=8 Гц, 2Н), 2,70 (dd, J=18 Гц, J=7 Гц, 1Н), 2,59 (dd, J=18 Гц, J=7 Гц, 1Н), 2,06 (уш. s, 2Н), 1,83 (s, 3Н), 1,45 (m, 2Н), 1,36 (s, 3Н), 1,28 (t, J ˜ 8 Гц, 2Н), 1,19 (s, 6Н);
ИК (пленка, см-1): 1659 (С=С), 1100 (С-O-С);
МС: 247,2 (М+-ОН).
Пример 7
Получение 2-(5-этокси-3-метил-3-триметилсилилокси-(1Е,4Z)-пента-1,4-диенил)-1,3, 3-триметилциклогесена
В трубке Шленка, снабженной мешалкой и резиновой перегородкой, растворяли 2,95 г (10 ммолей) 1-этокси-3-метил-5-(2,6,6-триметилциклогекс-1-енил)-(1Z,4Е)-пента-1, 4-диен-3-ола в смеси 50 мл сухого диметилформамида, 0,50 г (0,4 ммоля) 4-диметиламинопиридина и 7,0 мл (50 ммолей) триэтиламина. Смесь охлаждали до 0°С, и по каплям вводили 5,0 мл (40 ммолей) триметилхлорсилана.
После того, как смесь перемешивали в течение 14 ч при температуре окружающей среды, ее разбавляли 250 мл гексана и экстрагировали 3×250 мл воды, 2×250 мл насыщенного раствора бикарбоната натрия и 2×250 мл насыщенного солевого раствора. Объединенные органические фазы сушили над сульфатом натрия и концентрировали при пониженном давлении, получая 3,49 г (выход 93%) 2-(5-этокси-3-метил-3-триметилсилилокси-(1E,4Z)-пента-1,4-диенил)-1,3,3-триметилциклогесена в виде желтоватого масла. Оно было достаточно чистым для дальнейшей реакции.
1Н-ЯМР (400 МГц, CDCl3): 6,02 (d, J=16 Гц, 1Н), 5,81 (d, J=7 Гц, 1Н), 5,64 (d, J=16 Гц, 1Н), 4,45 (d, J=7 Гц, 1Н), 3,75 (q, J=7 Гц, 2Н), 1,97 (t, J ˜ 6 Гц, 2Н), 1,66 (s, 3 Н), 1,6 (m, 2Н), 1,55 (s, 3Н), 1,40 (m, 2Н), 1,23 (t, J ˜ 7 Гц, 3H),0,99(s, 6H), 0,14 (s, 9H);
ИК (пленка, см-1): 1660 (C=С), 1105 (С-О-С);
МС: 336,3 (М+).
Пример 8
Получение транс-1-хлор-3-метил-5-(2,6,6-триметилциклогекс-1-енил)пент-1-ен-4-ин-3-ола
Раствор 1,48 г (10 ммолей) 2-этинил-1,3,3-триметилциклогексена в 90 мл сухого тетрагидрофурана в трехгорлой реакционной колбе на 250 мл, снабженной магнитной мешалкой, резиновой перегородкой и приспособлениями для ввода аргона, охлаждали в бане с сухим льдом в атмосфере аргона. Раствор обрабатывали по каплям 7,5 мл (12 ммолей) 1,6 М раствором н-бутиллития в гексане с такой скоростью, чтобы поддерживать температуру при -70°С до -60°С. Прибавление было завершено в течение 10 мин. После того, как реакционную смесь перемешивали в течение дополнительных 30 мин при -70°С, медленно прибавляли раствор 1,045 г (10 ммолей) 4-хлорбутен-2-она в 10 мл сухого тетрагидрофурана. После дальнейшего перемешивания в течение 3 ч при -70°С реакционную смесь выливали в 250 мл смеси льда с водой и экстрагировали 3×70 мл гексана. Объединенные органические фазы промывали 3×100 мл воды и 50 мл насыщенного солевого раствора, сушили над безводным сульфатом магния. Все летучие вещества удаляли при пониженном давлении 100-200 мбар (10-20 кПа), и получали 2,57 г неочищенного продукта. Хроматография на 60 г силикагеля (0,04-0,063 мм) со смесью 95:5 (об./об.) гексана и этилацетата дала 1,19 г (4,7 ммоля, выход 47%) чистого вещества.
1Н-ЯМР (400 МГц, CDCl3): 6,56 (d, J=13 Гц, 1Н), 6,11 (d=13 Гц, 1Н), 2,02 (t, J ˜ 6 Гц, 2Н), 1,86 (s, 3Н), 1, 63 (s, 3Н), 1,60 (m, 2Н), 1,46 (m, 2Н), 1,09 (s, 6Н);
ИК (пленка, см-1): 3373 (ОН), 2210

МС: 252,2, 254,2 (М+).
Пример 9
Получение (1E,4E)-1-хлор-3-метил-5-(2,6,6-триметилциклогекс-1-енил)пента-1,4-диен-3-ола
Растворяли 1,96 г (7,5 ммоля) 1-хлор-3-метил-5-(2,6,6-триметилциклогекс-1-енил)пент-1-ен-4-ин-3-ола в 100 мл сухого тетрагидрофурана в трубке Шленка на 250 мл, снабженной мешалкой и резиновой перегородкой, в атмосфере аргона и охлаждали до -10°С, с использованием шприца прибавляли по каплям 2,71 мл (9,5 ммоля) натрийдигидридо-бис(2-метоксиэтокси)алюмината (3,5 М в толуоле). После перемешивания в течение 4 ч при -10°С реакционную смесь "гасили" добавлением 5 мл 40% раствора этанола в гексане при 0-5°С.
Для обработки раствор подвергали действию 13 мл 28% водного раствора гидроксида натрия при 0-5°С в течение 10 мин. Образующуюся эмульсию разбавляли 110 мл воды, экстрагировали дважды по 50 мл гексана. Объединенные органические фазы экстрагировали 5×50 мл воды и 50 мл насыщенного солевого раствора, сушили над сульфатом магния. После удаления всех летучих веществ при пониженном давлении 100-200 мбар (10-20 кПа) получали неочищенный продукт, который содержал 94% (1E,4E)-1-хлор-3-метил-5-(2,6,6-триметилциклогекс-1-енил)пента-1,4-диен-3-ола и был достаточно чистым для дальнейшей реакции.
1H-ЯМР (400 МГц, CDCl3): 6,28 (d, J=13 Гц, 1H), 6,11 (d, J=16 Гц, 1Н), 6,09 (d=13 Гц, 1Н), 5,51 (d, J=16 Гц, 1Н), 1,97 (t, J ˜ 7 Гц, 2Н), 1,65 (s, 3H), 1,60 (m, 2H), 1,45 (m, 2H), 1,44 (s, 3 H), 0,98 (s, 6H);
МС: 254,2, 256,2 (M+).
Пример 10
Получение (9E/Z,13E/Z)-11,12-дигидро-11-этоксиретиналя
В двухгорлую колбу на 100 мл, снабженную магнитной мешалкой и приспособлениями для ввода аргона, вводили 12,3 г (приблизительно 42 ммоля) цис-1-этокси-3-метил-3-гидрокси-5-(2,6, 6-триметилциклогекс-1-енил)-1,4-пентадиена (приблизительно 90% чистоты) и 12,5 г (80 ммолей) 1-триметилсилилокси-3-метил-1,3-бутадиена и 60 мл ацетонитрила. Смесь охлаждали до -30°С и прибавляли 550 мг (приблизительно 4 ммоля) безводного хлорида цинка при перемешивании в атмосфере аргона. Реакционную смесь перемешивали последовательно в течение 1 ч при -30°С, 1 ч при 0°С и 1 ч при комнатной температуре, после чего было установлено с помощью ВЭЖХ, что реакция завершена примерно на 90%.
С целью обработки к перемешиваемой смеси при комнатной температуре прибавляли 10 мл смеси 9:1 ледяной уксусной кислоты и воды, и смесь перемешивали при данной температуре в течение дальнейших 30 мин. Полученный раствор прибавляли к 100 мл воды, и водный раствор экстрагировали 2×200 мл гексана, объединенные отделенные органические фазы промывали последовательно 3×100 мл воды и 3×100 мл насыщенного раствора хлорида натрия. Отделенную гексановую фазу сушили над безводным сульфатом натрия и после удаления фильтрацией осушающего агента концентрировали при 35°С при пониженном давлении 100-200 мбар (10-20 кПа). Получали 15,6 г неочищенного продукта в виде масла, которое затем очищали колоночной хроматографией, используя 400 г силикагеля (0,04-0,063 мм) в качестве стационарной фазы и смесь 9:1 (об./об.) гексана и этилацетата в качестве элюирующего агента. Таким путем получали 8,84 г (выход приблизительно 64%) чистого (9E/Z,13E/Z)-11,12-дигидро-11-этоксиретиналя в виде желтого масла.
ВЭЖХ: изомерный состав ретиналя: 4,8% (9,13-ди-цис), 7,4% (13-цис), 28,0% (9-цис) и 56,7% (полностью-E) [общая площадь в процентах 96,9 (%)];
1Н-ЯМР (400 МГц, CDCl3): среди прочего 4 дублета (СНО) примерно при 9,85-10 (1˜8 Гц);
МС: 247,2 (М+);
ИК (пленка, см-1): 1676 (СН=O), 1633 (С=С), 1082 (С-О-С); УФ (циклогексан): 232 нм (∑=49100, log ∑=4,69).
Пример 11
Получение (2E/Z,8E/Z)-5-этокси-3,7-диметил-9-(2,6,6-триметилциклогекс-2-енил)нона-2,6, 8-триеналя
Используя методику, аналогичную той, которая описана в примере 10, получали (2E/Z,8E/Z)-5-этокси-3,7-диметил-9-(2,6,6-триметилциклогекс-2-енил)нона-2,6,8-триеналя из 2,145 г (7,7 ммоля) (1Z,4E)-1-этокси-3-метил-5-(2,6,6-триметилциклогекс-2-енил)пента-1,4-диен-3-ола.
ВЭЖХ: состав изомеров ретиналя: 4,7% (9,13-ди-цис), 7,9% (13-цис), 23,9% (9-цис) и 61,7% (полностью-E) [общая площадь в процентах 95,2 (%)];
1Н-ЯМР (400 МГц, CDCl3): среди прочего 4 дублета (СНО) приблизительно при 9,85-10 (J˜ 8 Гц);
МС: 330,2 (М+);
ИК (пленка, см-1): 1676 (СН=O), 1633 (С=С), 1082 (С-О-С);
Пример 12
Получение (9E/Z,13E/Z)-11, 12-дигидро-11-бензоилоксиретиналя
Используя методику, аналогичную той, которая описана в примере 10, получали 970 мг (2,4 ммоля, выход 48%) (9E/Z,13E/Z)-11, 12-дигидро-11-бензоилоксиретиналя из 1,87 г (5 ммолей) 3-метокси-3-метил-5-(2,6,6-триметилциклогекс-1-енил)-(1Z,4Е)-пента-1,4-диенилбензоата.
ВЭЖХ: состав изомеров ретиналя: 4,3% (9, 13-ди-цис), 8,5% (13-цис), 34,2% (9-цис) и 46,8% (полностью-Е);
1Н-ЯМР (400 МГц, CDCl3): среди прочего 4 дублета (СНО) приблизительно при 9,85-10 (J˜ 8 Гц);
МС: 406,3 (М+);
ИК (пленка, см-1): 1718 (СО=O), 1677 (СН=O).
(9E/Z,13E/Z)-11,12-Дигидро-11-бензоилоксиретиналь (266 мг, выход 52%) получали также из 935 мг (2,5 ммоля) 3-гидрокси-3-метил-5-(2,6,6-триметилциклогекс-1-енил)-(1Z,4E)-пента-1,4-диенилбензоата и 380 мг (3,75 ммоля) 1-метокси-3-метил-1,3-бутадиена в аналогичных условиях.
ВЭЖХ: состав изомеров ретиналя: 3,9% (9,13-ди-цис), 2,2% (13-цис), 28,0% (9-цис) и 54,4% (полностью-Е).
Пример 13
Получение 5-этокси-3, 7-диметил-9-(2,6,6-триметилциклогекс-2-енилиден)-(2E/Z, 6E/Z)-нона-2,6-диеналя
Используя методику, аналогичную той, которая описана в примере 10, получали 1,404 г (4,3 ммоля, выход 47, 2%) 5-этокси-3,7-диметил-9-(2,6,6-триметилциклогекс-2-енилиден)-(2E/Z, 6Е/Z)-нона-2,6-диеналя из 2,380 г (9,0 ммолей) 1-этокси-3-метил-5-(2,6,6-триметилциклогекс-2-енилиден)-(Z)-пент-1-ен-3-ола.
ВЭЖХ: состав изомеров: 5,1%, 9,8%, 23,2% и 52,7% (полностью-E) [общая площадь в процентах 95,1 (%)];
1Н-ЯМР (400 МГц, CDCl3): среди прочего 4 дублета (СНО) приблизительно при 9,85-10 (J˜ 8 Гц);
МС: 330,5 (М+);
ИК (пленка, см-1): 1675 (СН=О), 1633 (С=С), 1083 (С-О-С).
Пример 14
Получение (9Е,13E)-5-хлор-3,7-диметил-9-(2,6,6-триметилциклогекс-1-енил)-нона-2,6-диен-8-иналя
Используя методику, аналогичную той, которая описана в примере 10, получали 325 мг (1,0 ммоль, выход 78%) (9Е,13E)-5-хлор-3,7-диметил-9-(2,6,6-триметилциклогекс-1-енил)нона-2,6-диен-8-иналя из 330 мг (1,3 ммоля) 1-хлор-3-метил-5-(2,6, 6-триметилциклогекс-1-енил)пент-1-ен-4-ин-3-ола.
1H-ЯМР (400 МГц, CDCl3): 9,99 (d, J=8 Гц, 1Н), 6,39 (d, J=13 Гц, 1Н), 5,93 (d=8 Гц, 1Н), 5,87 (d, J=13 Гц, 1Н), 2, 49 (d, J=13 Гц, 1Н), 2,41 (d, J=13 Гц, 1Н), 2,26 (s, 3Н), 2,00 (t, J ˜ 6 Гц, 2Н), 1,82 (s, 3Н), 1,59 (m, 2Н), 1,45 (m, 2H), 1,43 (s, 3H), 1,06 (s, 6H);
ИК (пленка, см-1): 2211
МС: 318,2, 320,2 (М+).
Пример 15
Получение (9E/Z,13E/Z)-11,12-дигидро-11-хлорретиналя
Используя методику, аналогичную той, которая описана в примере 10, получали 303 мг (0,9 ммоля, выход 24,4%) (9E/Z, 13E/Z)-11,12-дигидро-11-хлорретиналя из 1,21 г (3,9 ммоля) (1Е,4E)-1-хлор-3-метил-5-(2,6,6-триметилциклогекс-1-енил)пента-1,4-диен-3-ола.
1Н-ЯМР (400 МГц, CDCl3): среди прочего 4 дублета (СНО) приблизительно при 9,85-10 (J˜8 Гц);
МС: 320,2 (М+).
Пример 16
Получение (9E/Z,13E/Z)-ретиналя
К 2,48 г (7,5 ммолей) 11,12-дигидро-11-этоксиретиналя в 22 мл толуола в двухгорлой реакционной колбе на 25 мл, снабженной магнитной мешалкой и приспособлением для ввода аргона, прибавляли 2, 40 г (приблизительно 15 ммолей) 1,8-диазабицикло[5.4.0]ундец-7-ена, и реакционную смесь перемешивали в течение 3 ч при 70°С (согласно анализу ВЭЖХ и тонкослойной хроматографией элиминирование этанола завершилось на этом этапе). Образующийся раствор затем выливали в 50 мл 10% водной серной кислоты, и всю смесь экстрагировали 2×100 мл гексана, объединенные органические фазы промывали последовательно 50 мл 10% водной серной кислоты, 3×100 мл воды, 2×50 мл насыщенного раствора бикарбоната натрия и, наконец, 2×50 мл раствора хлористого натрия. После высушивания отделенной органической фазы над безводным сульфатом натрия и концентрации при 35°С при пониженном давлении 100-200 мбар (10-20 кПа) получали 2,42 г неочищенного ретиналя в виде смеси E/Z-изомеров. Хроматография на 60 г силикагеля (0,04-0,063 мм) со смесью 9:1 (об./об.) гексана и этилацетата дала 1,81 г (выход 85%) чистого ретиналя (смеси E/Z-изомеров) в виде вязкого красного масла.
ВЭЖХ: состав изомеров ретиналя: 6,8% (9,13-ди-цис), 19,5% (13-цис), 20,1% (9-цис) и 52,7% (полностью-Е) (всего 99,1%);
1H-ЯМР (400 МГц, CDCl3) среди прочих 4 дублета (СНО) при 10,1-10,2 (J ˜ 8 Гц);
ИК (пленка, см-1): 1661 (СН=O), 1580 (С=С сопряженная);
МС: 284,2 (М+); УФ (циклогексан): 367 (∑=69400, log ∑=4,84).
Пример 17
Получение (9E/Z,13E/Z)-ретиналя из 5-этокси-3,7-диметил-9-(2,6,6-триметилциклогекс-2-енилиден)-(2E/Z, 6E/Z)-нона-2,6-диеналя
К раствору 350 мг (1,0 ммоля) 5-этокси-3,7-диметил-9-(2,6,6-триметилциклогекс-2-енилиден)-(2E/Z, 6E/Z)-нона-2,6-диеналя в 6 мл ацетона в атмосфере аргона в трубке Шленка на 10 мл прибавляли 0,05 мл 48% бромистоводородной кислоты при -10°С. После того, как реакционную смесь перемешивали в течение 1 ч при температуре окружающей среды, ее переносили в делительную воронку, разбавляли 50 мл гексана и экстрагировали 4×25 мл воды и 2×25 мл насыщенного солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали при пониженном давлении. Очищали 456 мг неочищенного продукта колоночной хроматографией на 8 г силикагеля (0,04-0,063 мм) со смесью 98:2 (об./об.) гексана и этилацетата и получали 139 мг (выход 49%) чистого ретиналя (смеси E/Z-изомеров) в виде вязкого красного масла.
Пример18
Получение аддукта (полностью-E)-ретиналь-гидрохинон
В реакционную колбу на 50 мл, снабженную магнитной мешалкой и приспособлением для ввода аргона, вводили 2,60 г (9,14 ммоля) (9E/Z,13E/Z)-ретиналя (очищенного вещества из примера 16) и 0,514 г (4,57 ммоля) гидрохинона в 6 мл диэтилового эфира. К реакционной смеси под аргоном прибавляли следовое количество, т.е. приблизительно 5-10 мкл 55% водной йодной кислоты. После примерно 1 ч происходила кристаллизация образовавшегося аддукта, так что содержимое колбы представляло собой не поддающуюся перемешиванию кристаллическую массу. Примерно через 16 ч в атмосфере аргона к кристаллической массе прибавляли 25 мл гексана, и образующуюся суспензию перемешивали при комнатной температуре в течение двух часов. Затем кристаллы собирали фильтрацией, промывали гексаном и сушили в высоком вакууме при комнатной температуре, получая 2,27 г (выход 84%) цвета охры аддукта (полностью-Е)-ретиналь-гидрохинон с приблизительным соотношением ретиналя к гидрохинону 8:1 (по данным1Н-ЯМР).
1 Н-ЯМР (400 МГц, CDCl3): 10,10 (doublet; J ˜ 8 Гц, 1H, CHO);
ВЭЖХ: состав изомеров ретиналя: 3,0% (13-цис), 95,3% (полностью-Е) (гидрохинон не включен; всего 98, 3%).
Пример 19
Получение аддукта (полностью-Е)-ретиналь-гидрохинон из (9E/Z,13E/Z)-11,12-дигидро-11-этоксиретиналя
В реакционную колбу на 10 мл, снабженную магнитной мешалкой и приспособлением для ввода аргона, вводили 992 мг (3 ммоля) (9E/Z,13E/Z)-11,12-дигидро-11-этоксиретиналя в 5 мл метиленхлорида. После добавления следового количества, т.е. приблизительно 5-10 мкл 55% водной йодистоводородной кислоты реакционную смесь нагревали до 40°С в течение 1 ч. По истечении этого времени по данным ВЭЖХ было установлено, что элиминирование этанола завершено.
Растворитель метиленхлорид затем удаляли упариванием при пониженном давлении и заменяли 1 мл диэтилового эфира. Затем прибавляли 170 мг (приблизительно 1,5 ммоля) гидрохинона, и смесь перемешивали при комнатной температуре. После этого прибавляли несколько кристаллов требуемого аддукта из партии, полученной ранее, затем 7 мл гексана вводили медленно в темный раствор/суспензию для активации кристаллизации. Полученный осадок отделяли фильтрованием, промывали гексаном и сушили в высоком вакууме. Таким путем получали 348 мг (выход 38%) цвета охры аддукт (полностью-Е)-ретиналь-гидрохинон с соотношением ретиналя и гидрохинона примерно 4:1.
Состав изомеров ретиналя: 3,1% (13-цис), 95,3% (полностью-Е) (всего 98, 4%).
Реферат
Изобретение относится к способу получения ретиналя формулы

возможно в виде его аддукта (полностью-Е)-ретиналя с гидрохиноном, который включает взаимодействие производного 5-(2,6,6-триметилциклогекс-1-енил)-1,4-пентадиена или производного 5-(2,6,6-триметилциклогекс-2-енил)-1,4-пентадиена, или производного 5-(2, 6,6-триметил-2-циклогексен-1-илиден)-1-пентена, или производного 5-(2,6,6-триметилциклогекс-1-енил)пента-1-ен-4-ина, или производного 5-(2,6,6-триметилциклогекс-2-енил)пента-1-ен-4-ина с производным 1, 3-бутадиена в присутствии кислоты Льюиса или Бренстеда, и полученное соединение подвергают обработке основанием или кислотой в среде растворителя, чтобы элиминировать группу R2H, и в случае необходимости гидрируют и подвергают изомеризации в основных или кислых условиях или в присутствии металлического катализатора в требуемый ретиналь формулы I, при этом ретиналь, полученный в виде смеси (9E/Z,13E/Z) изомеров при необходимости изомеризуется в (полностью-Е)-ретиналь путем образования аддукта (полностью-Е)-ретиналя с гидрохиноном. Изобретение также относится к новым производным 5-замещенного пент(ади)ен(ин)а, используемым в качестве промежуточных продуктов. 3 н. и 10 з.п. ф-лы.
Формула













Комментарии