Способ получения простых ариловых эфиров - RU2330012C2
Код документа: RU2330012C2
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к усовершенствованному способу получения некоторых простых ариловых эфиров. Изобретение также относится к промежуточным соединениям, используемым в способе, и к способам получения таких промежуточных соединений.
Предпосылки изобретения
Патент США 4229449 раскрывает соединения формулы (А)

в которой
n и n1 независимо равны 1, 2 или 3;
каждая из групп R и R1, которые могут быть одинаковыми или различными, представляет собой атом водорода; атом галогена; галоген-С1-С6 алкил; гидрокси; С1-С6 алкокси; С1-С6 алкил, необязательно замещенный; арил-С1-С6алкил, необязательно замещенный; арил-С1-С6 алкокси, необязательно замещенный; -NO2; NR5R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил, или две соседние группы R или две соседние группы R1, взятые вместе, образуют -О-СН2-О-радикал;
R2 представляет собой атом водорода; С1-С12 алкил, необязательно замещенный, или арил-С1-С6 алкил;
каждая из групп R3 и R4, которые могут быть одинаковыми или различными, представляет собой атом водорода, С1-С6 алкил, необязательно замещенный, С2-С4 алкенил, С2-С4 алкинил, арил-С1-С4 алкил, необязательно замещенный, С3-С7 циклоалкил, необязательно замещенный, или R3 и R4 с атомом азота, к которому они присоединены, образуют пятичленный или шестичленный насыщенный или ненасыщенный, необязательно замещенный, гетероциклический радикал, необязательно содержащий другие гетероатомы, принадлежащие к классу O, S и N;
или R2 и R4, взятые вместе, образуют -СН2-СН2-радикал;
и их фармацевтически приемлемые соли.
Раскрыто, что соединения проявляют активность антидепрессантов.
В частности, патент Соединенных Штатов 4229449 раскрывает соединение: 2-[α-(2-этоксифенокси)бензил]морфолин:

и его фармацевтически приемлемые соли, которые обладают ценными антидепрессантными свойствами. Это соединение также известно как ребоксетин.
Ребоксетин ведет себя не так, как большинство антидепрессантов. В отличие от трициклических антидепрессантов и даже отдельных ингибиторов повторного захвата серотонина (SSRis), ребоксетин оказывается неэффективным в 8-ОН-DPAT гипотермическом тесте, что свидетельствует о том, что ребоксетин не является SSRi. Смотри публикацию Brian E. Leonard, "Noradrenaline in basic models of depression", European-Neuropsychopharmacol, 7 Suppl., 1 pp. S11-6 and S71-3 (April 1997), включенную в настоящее описание во всей ее полноте в качестве ссылки. Ребоксетин представляет собой селективный ингибитор обратного захвата норэпинефрина только с ингибирующим действием только по отношению к минимальному количеству серотонина, но не к допамину. Ребоксетин не проявляет никакой холиноблокирующей активности в различных животных моделях и по существу лишен ингибирующей активности к моноаминоксидазе (МАО).
Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. В описании оптических активных соединений используют приставки R и S для обозначения абсолютной конфигурации молекулы относительно ее хирального центра(ов). Приставки D и L или (+) и (-) обозначают направление вращения плоскополяризованного света соединением, где L или (-) означают, что соединение является левовращающим. В отличие от этого, соединение с приставкой D или (+) является правовращающим. Не существует корреляции между номенклатурой для абсолютной стереохимии и для вращения энантиомера. Таким образом, D-молочная кислота представляет то же, что и (-)-молочная кислота, и L-молочная кислота представляет то же, что (+)-молочная кислота. Для данной химической структуры каждая пара энантиомеров идентична, за исключением того, что они являются не налагаемыми зеркальными отражениями одно другого. Отдельный стереоизомер также может быть рассмотрен как энантиомер, и смесь таких изомеров часто называют энантиомерной или рацемической смесью.
Когда в молекуле существует два хиральных центра, то возможны четыре стереоизомера: (R,R), (S,S), (R,S) и (S,R). Из них (R,R) и (S,S) являются примером пары энантиомеров (зеркальные отражения одно другого), которые обычно делят химические свойства и точки плавления, как любые другие энантиомерные пары. Зеркальные отражения (R,R) и (S,S), однако, не являются взаимоналагаемыми с (R,S) и (S,R) конфигурациями. Эта зависимость называется диастереомерной, и (S,S) молекула является диастереомером молекуле (R,S), тогда как (R,R) молекула является диастереомером молекулы (S,R).
С химической точки зрения ребоксетин имеет два хиральных центра и поэтому существует в двух энантиомерных парах диастереомеров, (R,R) и (S,S) энантиомерной паре и (R,S) и (S,R) энантиомерной паре. В настоящее время ребоксетин коммерчески доступен только в виде рацемической смеси энантиомеров, (R,R) и (S,S) в отношении 1:1, и ссылка в настоящем описании на общее название «ребоксетин» относится к данной энантиомерной или рацемической смеси. Ребоксетин коммерчески продается под торговыми названиями EDRONAX™, PROLIFT™, VESTRA™ и NOREBOX™.
Известно (смотри WO 01/01973, включенную во всей полноте в качестве ссылки), что (S, S)-энантиомер ребоксетина обладает существенно улучшенной селективностью к обратному захвату норэпинефрина по сравнению с повторным захватом серотонина. Соответственно, WO 01/01973 раскрывает способ селективно ингибируемого захвата норэпинефрина, способ, включающий стадию введения терапевтически эффективного количества композиции человеку, композиция включает соединение, обладающее фармакологической селективностью серотонин (Ki)/норэпинефрин (Кi), по меньшей мере, примерно 5000. В документе дополнительно раскрыт ряд новых областей использования (S, S)-ребоксетина, включая использование (S,S)-ребоксетина для лечения хронических болей, периферической нейропатии, недержания (включая стрессовое недержание, генуинное стрессовое недержание и смешанное недержание), фибромиалгии и других соматических нарушений и мигреней.
Патенты US 5068433 и 5391735, а также GB-A-2162517 раскрывают способы получения и промежуточные соединения, используемые для получения единых диастереомеров соединений формулы VIb:

где R представляет С1-С6 алкокси или тригалогенметил. Данные диастереомеры описаны как важные промежуточные соединения для получения соединений формулы А, включая ребоксетин. Способы, раскрытые в упомянутых патентах, и патенте США 4229449, однако, неэффективны и дают низкий суммарный выход соединений формулы А при осуществлении в промышленном масштабе. Кроме того, способы требуют использования дорогих реагентов и продолжительных времен производства. Таким образом, не экономично получать соединения формулы А в промышленном масштабе с использованием способов, раскрытых в данных патентах.
WO 00/39072 относится к способу получения амина формулы VIIa:

включающему:
а) окисление необязательно замещенного транс-коричного спирта с образованием промежуточного эпоксида формулы Ia:

b) взаимодействие эпоксида с необязательно замещенным фенолом с образованием диола формулы IIa:

с) взаимодействие диола с силилирующим агентом с получением спирта формулы IIIa:

где Р представляет соединенный с силилом радикал;
d) взаимодействие спирта формулы IIIa с реакционноспособным производным сульфоновой кислоты с получением соединения формулы IVa:

где Ra представляет остаток сульфоновой кислоты;
е) удаление Р из соединения формулы IVa с получением спирта формулы Va:
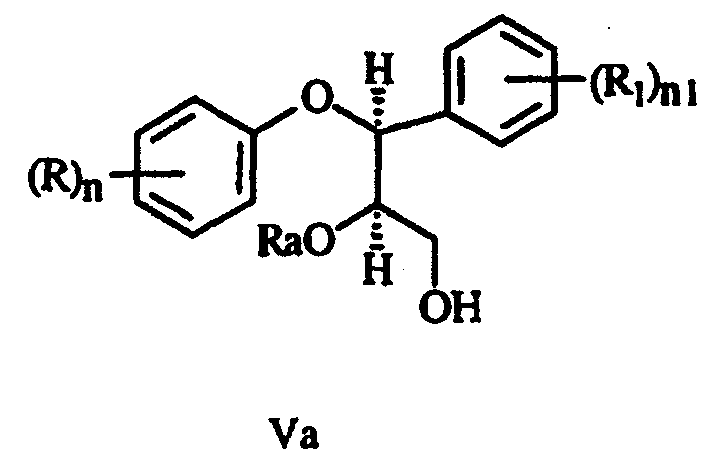
f) замещение сульфонилоксигруппы с получением эпоксида формулы VIa:

и
g) взаимодействие эпоксида с аммиаком с получением соединения формулы VIIa.
WO 00/39072 также относится к способу получения рацемического ребоксетина из вышеуказанного амина, включающему:
h) взаимодействие соединения формулы VIIa:

с карбоновой кислотой формулы НООССН2L или ее реакционноспособным производным, где L означает отщепляемую группу, с получением амида формулы VIIIa;

i) взаимодействие соединения формулы VIIIa с получением соединения формулы IXa:

и
j) восстановление соединения формулы IXa с образованием соответствующего соединения следующей формулы:

в вышепредставленных формулах R, R1, n и n1 имеют значения, определенные в патенте США 4229449, упомянутом выше.
Все упомянутые выше публикации раскрывают способы получения ребоксетина и родственных соединений в форме рацемической смеси (R,R)- и (S,S)-энантиомеров. Необходима дополнительная стадия разрешения для выделения более активного (S,S)-энантиомера.
Альтернативный синтез (S,S)-ребоксетина раскрыт в GB-A-2167407. Этот документ раскрывает хиральный синтез (S,S)-ребоксетина, исходя из хиральной фенилглицидиловой кислоты. Однако не существует никакого адекватного хирального синтеза фенилглицидиловой кислоты, так что хиральная кислота должна быть получена разделением, что неэффективно. Последующее восстановление до фенилглицидола дает низкий выход. После этой стадии синтез, описанный в GB-A-2167407, идет параллельно рацемическому синтезу: как отмечено выше, синтез характеризуется низкой селективностью и низким выходом и является неэффективным.
Было бы желательно разработать энантиоселективный синтез (S,S)-ариловых сложных эфиров, используемых в производстве (S,S)-ребоксетина, который бы исключал получение нежелательного (R,R)-энантиомера и позволил бы более эффективно получить (S,S)-ребоксетин, с большим выходом и чистотой, чем при синтезе согласно известным техническим решениям.
Авторы изобретения неожиданно обнаружили, что новые системы согласно настоящему изобретению дают возможность получения промежуточных соединений, используемых в синтезе (S,S)-ребоксетина, энантиоселективно, эффективно, с высоким выходом и чистотой.
Краткое изложение сущности изобретения
В одном аспекте настоящее изобретение относится к способу получения соединения формулы (IV):

в которой
n и m независимо равны 0 или целому числу от 1 до 5;
каждая из групп R и R1, которые могут быть одинаковыми или различными, представляет собой атом галогена; галоген-С1-С6 алкил; гидрокси; С1-С6 алкокси; С1-С6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из атома галогена, гидрокси, С1 -С6 алкокси, NR5R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил, или -CONR5R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил; арил-С1-С6 алкил, где арильное кольцо необязательно замещено одним или несколькими заместителями, выбранными из С1-С6 алкила, атома галогена, галоген-С1-С6 алкила, гидрокси, С1-С6 алкокси и NR5R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил; арил-С1-С6 алкокси, где арильное кольцо необязательно замещено одним или несколькими заместителями, выбранными из С1-С6 алкила, атома галогена, галоген-С1-С6 алкила, гидрокси, С1-С6 алкокси и NR5R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил; -NO2; NR5R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил; или две соседние группы Rили две соседние группы R1, взятые вместе, образуют -О-СН2-О- радикал;
включающий:
(а) асимметричное эпоксидирование соединения формулы (I):

где R1 и m имеют значения, указанные выше, агентом окисления в присутствии оптически активного соединения с образованием соединения формулы (II):

где R1 и m имеют значения, указанные выше, с последующей
(b) реакцией соединения формулы (II) с соединением формулы (III):

где R и n имеют значения, указанные выше, в присутствии основания,
при этом способ осуществляют без выделения соединения формулы (II).
Неожиданно авторы изобретения установили, что проведение стадий (а) и (b) последовательно, без выделения соединения формулы (II) позволяет получить соединение формулы (IV) энантиоселективно и с хорошим выходом и чистотой. Это находится в противоречии с тем, что ожидалось, так как предполагалось, что необходимо отделять соединение формулы (II) от агентов, использованных при его синтезе и обработке; ожидалось, что присутствие этих агентов будет препятствовать синтезу и очистке соединения формулы (IV).
Изобретение дополнительно относится к способу получения соединений формулы (IX):

где R, R1, n и m имеют значения, указанные выше, включающему:
(а) получение соединения формулы (IV) в соответствии с описанным выше способом;
(b) реакцию соединения формулы (IV) с силилирующим агентом с образованием соединения формулы (V):

где R, R1, n и m имеют значения, указанные выше, и Р представляет собой силильную защитную группу;
(с) реакцию соединения формулы (V) с агентом сульфонилирования формулы R′SO2X, где R′ представляет собой остаток сульфоновой кислоты и Х представляет отщепляемую группу, с получением соединения формулы (VI):

где R, R1, n и m имеют значения, указанные выше, Р имеет значения, указанные выше, и R′ представляет остаток сульфоновой кислоты;
(d) удаление силильной защитной группы с получением соединения формулы (VII):

где R, R1, n и m имеют значения, указанные выше, и R′ имеет значение, указанное выше;
(е) замещение сульфонилоксигруппы с получением соединения формулы (VIII):

где R, R1, n и m имеют значения, указанные выше, и
(f) реакцию соединения формулы (VIII) с аммиаком или соединением аммония с получением соединения формулы (IX).
Предпочтительно способ осуществляют без выделения соединений формул (V), (VI), (VII) и (VIII).
В другом аспекте изобретение относится к соединениям формулы (V):

где R, R1, n и m имеют значения, указанные выше, Р представляет собой силильную защитную группу.
В другом аспекте изобретение относится к соединению формулы (VI):

где R, R1, n, m, Р и R′ имеют значения, указанные выше.
Подробное описание предпочтительных вариантов осуществления изобретения
В соединениях формул с (I) по (IX) каждая из групп R и R1, которые могут быть одинаковыми или различными, представляет собой атом галогена; галоген-С1-С6 алкил; гидрокси; С1-С6 алкокси; С1-С6 алкил, необязательно замещенный одним или несколькими заместителями, выбранными из атома галогена, гидрокси, С1-С6 алкокси, NR5R6 , где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил, или -CONR5R6, где R5 и R6 независимо представляют атом водорода или С1-С6 алкил; арил-С1-С6 алкил, где арильное кольцо необязательно замещено одним или несколькими заместителями, выбранными из числа таких групп, как С1-С6 алкил, атом галогена, галоген-С1-С6 алкил, гидрокси, С1-С6 алкокси, и NR5R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил, арил-С1-С6 алкокси, где арильное кольцо необязательно замещено одним или несколькими заместителями, выбранными из числа таких групп, как С1-С6 алкил, атом галогена, галоген-С1-С6 алкил, гидрокси, С1-С6 алкокси, и NR5R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил; -NO2; NR5 R6, где R5 и R6 независимо представляют собой атом водорода или С1-С6 алкил, или две соседние R группы или две соседние R1 группы, взятые вместе, образуют -О-СН2-О-радикал; и n и m независимо означают 0 или целое число от 1 до 5.
Термин «алкил» означает насыщенную углеводородную группу линейной или разветвленной цепи, содержащую от 1 до 6 атомов углерода.
Термин «галогеналкил» означает алкильную группу, определенную выше, которая замещена одним или несколькими атомами галогена.
Термин «алкокси» означает «алкил-О-», где алкильная группа определена выше.
Термин «арил» означает фенильную или нафтильную группу.
В подходящем случае группы R и R1, которые могут быть одинаковыми или различными, выбраны из числа таких групп, как гидрокси или С1-С6 алкокси. Предпочтительно R означает метокси или этокси, более предпочтительно этокси.
Предпочтительно n равно 1 или 2, более предпочтительно 1.
Предпочтительно m равно 0 или 1, более предпочтительно 0.
В особенно предпочтительном варианте осуществления n равно 1, m равно 0 и R представляет этоксигруппу в положении 2 фенильного кольца.
Стадия 1
Способ настоящего изобретения начинается с асимметричного эпоксидирования соединения формулы (I) агентом окисления в присутствии оптически активного соединения с образованием (R,R) энантиомера соединения формулы (II).
Асимметричное эпоксидирование соединения формулы (I) достигается при использовании агента окисления в присутствии оптически активного соединения. Точная природа агента окисления и оптически активного соединения не являются критическими факторами, при условии, что они могут обеспечивать эпоксидирование олефина асимметричным образом с образованием (R,R) энантиомера соединения формулы (II). Агент окисления сам по себе может быть оптически активным, в этом случае отпадает необходимость в использовании отдельного оптически активного соединения.
Примеры подходящих агентов окисления включают такие гидропероксиды, как трет-бутилгидропероксд, гидропероксид кумола, α,α -диметилгептилгидропероксид, бис-диизобутил-2,5-дигидропероксид, 1-метилциклогексилгидропероксид, циклогексилгидропероксид или диоксираны, особенно хиральные диоксираны типа, описанного Yian Shi et al., J.Am. Chem. Soc. 1997, 119, 11224-11235. В случае хиральных диоксиранов отпадает необходимость в использовании отдельного оптически активного соединения, поскольку сам диоксиран является хиральным.
Примеры подходящих оптически активных соединений включают соли и сложные эфиры оптически активных органических кислот, особенно сложных эфиров винной кислоты. Особенно предпочтительным примером оптически активного соединения является (-)-диизопропилтартрат.
Реакция необязательно может быть осуществлена в присутствии дополнительных реагентов, примеры которых включают алкоксиды титана, такие как метоксид титана, этоксид, н-пропоксид, изопропоксид, н-, втор-, изо- или трет-бутоксид. Изопропоксид титана является предпочтительным.
Особенно предпочтительно, чтобы асимметричное эпоксидирование проводилось в условиях, описанных в J. Am. Chem. Soc., 1980, 102, 5974-5976 ("Sharpless asymmetric epoxidation"), где окислителем является трет-бутилгидропероксид, оптически активным соединением является (-)-диизопропилтартрат и реакцию осуществляют в присутствии изопропоксида титана. Дополнительную информацию о методике можно найти в патенте США 4471130 и в публикации K.B. Sharpless et al., J. Am. Chem. Soc. 1987, 110, 5765-5780.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя, природа которого не является особенно важной, при условии, что он инертен к реакционной смеси и может растворять реагенты, по меньшей мере, в какой-то степени. Примеры подходящих растворителей включают алифатические углеводороды, такие как пентаны, гексаны, гептаны и октаны, нонаны или деканы, ароматические углеводороды, такие как бензол, толуол и ксилолы, галогенированные углеводороды, такие как метиленхлорид, хлороформ и 1,2-дихлорэтан, простые эфиры, такие как метил трет-бутиловый простой эфир, и их смеси. Предпочтительно, чтобы растворитель представлял смесь метиленхлорида и алифатических углеводородов.
Температура реакции зависит от различных факторов, таких как природа реагентов и растворителя. Однако обычно она составляет от -50°С до комнатной температуры и предпочтительно от -30°С до 0°С.
Продолжительность реакции зависит от различных факторов, таких как природа реагентов, растворитель и температура. Однако обычно она составляет от 10 минут до 24 часов, предпочтительно от 30 минут до 12 часов и более предпочтительно от 1 до 6 часов.
Реакцию предпочтительно проводят в безводных условиях. Для поглощения любого количества содержащейся воды в реакционной смеси предпочтительно присутствуют молекулярные сита.
После завершения реакции обычно добавляют агент обрыва реакции для того, чтобы нейтрализовать любой избыток содержащегося окислителя. Природа агента обрыва реакции важна, поскольку наиболее удобные агенты обрыва реакции являются либо неэффективными, либо в их присутствии продукт является нестабильным. Предпочтительными агентами обрыва реакции являются (С1-С6)алкилфосфиты, такие как триметилфосфит и триэтилфосфит. Триэтилфосфит является особенно предпочтительным.
Соединения формулы (II) не выделяют из реакционной смеси. Наоборот, на следующей стадии проводят непосредственное взаимодействие реакционной смеси, содержащей соединение формулы (II) и агент обрыва реакции, с соединением формулы (III).
Стадия 2
На следующей стадии осуществляют взаимодействие соединения формулы (II) с соединением формулы (III) в присутствии основания.
Природа основания не является особенно важным фактором при условии, что оно способно действовать как основание. Примеры подходящих оснований включают гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид рубидия и гидроксид цезия, гидроксиды тетра(С1-С6)алкиламмония, такие как гидроксид тетраметиламмония и гидроксид тетраэтиламмония, карбонаты щелочных металлов, такие как карбонат лития, карбонат натрия, карбонат калия, карбонат рубидия и карбонат цезия, и органические амины, такие как триметиламин и триэтиламин. Гидроксиды щелочных металлов являются предпочтительными, а особенно предпочтительным является гидроксид натрия.
Реакцию обычно и предпочтительно проводят в присутствии растворителя, природа которого не является особенно важной при условии, что он инертен к реакционной смеси и способен растворять реагенты, по меньшей мере, в некоторой степени. Примеры подходящих растворителей включают воду, амиды, такие как диметилформамид, ДМА и НМФ, алифатические углеводороды, такие как пентаны, гексаны, гептаны и октаны, ароматические углеводороды, такие как бензол, толуол и ксилолы, галогенированные углеводороды, такие как метиленхлорид, хлороформ и 1,2-дихлорэтан, простые эфиры, такие как тетрагидрофуран, метил трет-бутиловый эфир, диоксан, диэтиловый эфир, диизопропиловый эфир и диметоксиэтан, и их смеси. Предпочтительно, чтобы растворитель представлял собой двухфазную смесь метиленхлорида и воды.
Реакцию предпочтительно проводят в присутствии катализатора межфазного переноса, действие которого заключается в переносе аниона основания, такого как гидроксид-ион, в органический слой. Примеры подходящих катализаторов перехода фаз включают соли тетра(С1-С6)алкиламмония и бензилтри(С1-С6)алкиламмония, такие как тетра-н-бутиламмонийхлорид и бензилтриэтиламмоний хлорид. Тетра-н-бутиламмонийхлорид является предпочтительным.
Температура реакции зависит от различных факторов, таких как природа реагента, основания и растворителя. Однако обычно она составляет от комнатной температуры до 80°С и более предпочтительно от 35°С до 70°С.
Продолжительность реакции зависит от различных факторов, таких как природа реагентов, растворитель и температура. Однако обычно она составляет от 10 минут до 24 часов, предпочтительно от 30 минут до 12 часов, и более предпочтительно от 1 до 6 часов.
После завершения реакции соединение формулы (IV) выделяют из реакционной смеси обычным методом. Например, соединение может быть экстрагировано органическим растворителем, органический слой может быть промыт водным раствором, таким как вода, гидроксид натрия или хлорид натрия для того, чтобы удалить любые содержащиеся ионные фрагменты, отфильтрован для удаления любого твердого материала и сконцентрирован для удаления растворителя. Продукт может быть дополнительно очищен обычными методами, такими как перекристаллизация или колоночная хроматография.
Стадия 3
На этой стадии простой ариловый эфир формулы (IV) силилируют с получением соединения формулы (V). Превращение достигается за счет осуществления реакции с агентом силилирования в присутствии основания.
Точная природа силильной защитной группы Р, присоединяемой на этой стадии, не является критической для осуществления настоящего изобретения при условии, что она обычно способна защитить гидроксильную группу. Предпочтительно силильная защитная группа способна защитить первичную гидроксильную группу в присутствии вторичной гидроксильной группы. Примеры подходящих силильных групп включают триметилсилильную, трет-бутилдиметилсилильную, триэтилсилильную, триизопропилсилильную и трет-бутилдифенилсилильную группы, из которых триметилсилильная группа является предпочтительной.
Агентом силилирования обычно является силилгалогенид, предпочтительно хлорид.
Природа основания не является особенно важным параметром, при условии, что она способна действовать как основание. Примеры подходящих оснований включают гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид рубидия и гидроксид цезия, такие гидроксиды тетра(С1-С6) алкиламмония, как гидроксид тетраметиламмония и гидроксид тетраэтиламмония, карбонаты щелочных металлов, такие как карбонат лития, карбонат натрия, карбонат калия, карбонат рубидия и карбонат цезия, такие органические амины, как триметиламин, триэтиламин, пиридин, метилдиэтиламин, диметилэтиламин, три-н-пропиламин, триизопропиламин, диизопропилэтиламин, трибутиламины и высшие триалкиламины, пиколины, лутидины и колидины, 2-метил-5-этилпиридин (лонза пиридин), 2,6-ди-трет-бутилпиридин, 2,6-ди-трет-бутил-4-метилпиридин и алкилхинолины и изохинолины, N-метилпиперидин, N-метилпирролидин, N-метилморфолин и высшие алкиловые аналоги данных соединений. Органические амины являются предпочтительными, а триэтиламин является особенно предпочтительным.
Реакцию обычно и предпочтительно проводят в присутствии растворителя, природа которого не особенно важна, при условии, что он является инертным по отношению к реакционной смеси и способен растворять реагенты, по меньшей мере, в некоторой степени. Примеры подходящих растворителей включают амиды, такие как диметилформамид, алифатические углеводороды, такие как пентаны, гексаны, гептаны и октаны, ароматические углеводороды, такие как бензол, толуол и ксилолы, галогенированные углеводороды, такие как метиленхлорид, хлороформ и 1,2-дихлорэтан, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран, диизопропиловый эфир, диметоксиэтан и диоксан, такие сложные эфиры, как этилацетат, и их смеси. Предпочтительно, чтобы растворителем являлся галогенированный углеводород, особенно метиленхлорид.
Температура реакции зависит от различных факторов, таких как природа агента силилирования, основания и растворителя. Однако обычно она составляет от -78°С до комнатной температуры и предпочтительно составляет от -50°С до -10°С.
Продолжительность реакции зависит от различных факторов, таких как природа реагентов, растворитель и температура. Однако обычно она составляет от 5 минут до 2 часов, предпочтительно от 10 минут до 1 часа.
Соединения формулы (V) являются новыми и поэтому составляют часть настоящего изобретения.
Соединения формулы (V) обычно не выделяют из реакционной смеси. Наоборот, взаимодействие осуществляют между раствором, содержащим соединение формулы (V), непосредственно с агентом силилирования на следующей стадии.
Стадия 4
На этой стадии защищенное силильными группами соединение формулы (V) подвергают сульфонилированию с получением соединения формулы (VI). Данное превращение достигается за счет осуществления реакции соединения формулы (V) с агентом сульфонилирования формулы R′SO2X в присутствии основания.
Природа агента сульфонилирования не является особенно важной для осуществления настоящего изобретения при условии, что он используется для сульфонилирования гидроксильной группы. Примеры остатков сульфоновых кислот R′ включают (C1-C6) алкильные группы, галоген-(С1-С6) алкильные группы и фенильные группы, необязательно замещенные 1-3 (С1-С6) алкильными группами или атомами галогенов, и предпочтительные примеры включают метил, этил, трифторметил, фенил и п-толил. Примеры отщепляемой группы Х включают атомы галогенов и сульфонилоксигруппы формулы R′O, где R′ представляет один остаток сульфоновой кислоты, упомянутый выше, и предпочтительными являются атомы галогенов. Предпочтительные примеры агентов сульфонилирования включают метансульфонилхлорид, бензолсульфонилхлорид, п-толуолсульфонилхлорид и ангидрид п-толуолсульфоновой кислоты, и особенно предпочтительным является метансульфонилхлорид.
Природа основания не является особенно важным параметром, при условии, что оно способно действовать как основание. Примеры подходящих оснований включают органические амины, такие как триметиламин, триэтиламин, пиридин, метилдиэтиламин, диметилэтиламин, три-н-пропиламин, триизопропиламин, диизопропилэтиламин, трибутиламины и высшие триалкиламины, пиколины, лутидины и коллидины, 2-метил-5-этилпиридин (лонза пиридин), 2,6-ди-трет-бутилпиридин, 2,6-ди-трет-бутил-4-метилпиридин и алкилхинолины и изохинолины, N-метилпиперидин, N-метилпирролидин, N-метилморфолин и высшие алкиловые аналоги данных соединений, и триэтиламин является особенно предпочтительным.
Реакцию обычно и предпочтительно проводят в присутствии растворителя, природа которого не особенно важна, при условии, что он является инертным по отношению к реакционной смеси и способен растворять реагенты, по меньшей мере, в некоторой степени. Примеры подходящих растворителей включают амиды, такие как диметилформамид, ДМА и НМФ, алифатические углеводороды, такие как пентаны, гексаны, гептаны и октаны, ароматические углеводороды, такие как бензол, толуол и ксилолы, галогенированные углеводороды, такие как метиленхлорид, хлороформ и 1,2-дихлорэтан, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран, диизопропиловый эфир, диметоксиэтан и диоксан, такие сложные эфиры, как этилацетат, и их смеси. Предпочтительно, чтобы растворителем являлся галогенированный углеводород, особенно метиленхлорид.
Температура реакции зависит от различных факторов, таких как природа агента сульфонилирования, основания и растворителя. Однако обычно она составляет от -78°С до комнатной температуры и предпочтительно составляет от -50°С до -10°С.
Продолжительность реакции зависит от различных факторов, таких как природа реагентов, растворитель и температура. Однако обычно она составляет от 5 минут до 2 часов, предпочтительно от 10 минут до 1 часа.
Соединения формулы (VI) являются новыми и поэтому составляют часть настоящего изобретения.
Соединения формулы (VI) обычно не выделяют из реакционной смеси. Наоборот, взаимодействие осуществляют между раствором, содержащим соединение формулы (VI), непосредственно с агентом удаления защитных групп на следующей стадии.
Стадия 5
На этой стадии силильные защитные группы удаляют с получением соединения формулы (VII). Данная стадия реализуется за счет осуществления реакции с агентом удаления защитных групп.
На этой стадии точная природа агента удаления защитных групп не является критической для осуществления настоящего изобретения при условии, что она обычно может быть использована для удаления силильных защитных групп из гидроксильных групп. Примеры подходящих агентов удаления защитных групп включают кислоты, такие как соляная кислота, бромистоводородная кислота, серная кислота и уксусная кислота, и источники ионов фтора, такие как фторид калия и тетра-н-бутиламмонийфторид. Кислоты являются предпочтительными, и особенно предпочтительной является соляная кислота.
Реакцию обычно и предпочтительно проводят в присутствии растворителя, природа которого не особенно важна, при условии, что он является инертным по отношению к реакционной смеси и способен растворять реагенты, по меньшей мере, в некоторой степени. Примеры подходящих растворителей включают воду, спирты, такие как метанол и этанол, амиды, такие как диметилформамид, алифатические углеводороды, такие как пентаны, гексаны, гептаны и октаны, ароматические углеводороды, такие как бензол, толуол и ксилолы, галогенированные углеводороды, такие как метиленхлорид, хлороформ и 1,2-дихлорэтан, простые эфиры, такие как диэтиловый эфир, метил-трет-бутиловый эфир, тетрагидрофуран, диизопропиловый эфир, диметоксиэтан и диоксан, и их смеси. Предпочтительно, чтобы растворителем являлся галогенированный углеводород, особенно метиленхлорид, и вода.
Температура реакции зависит от различных факторов, таких как природа агента удаления защитных групп, основания и растворителя. Однако обычно она составляет от 0°С до 50°С и предпочтительно равна комнатной температуре.
Продолжительность реакции зависит от различных факторов, таких как природа реагентов, растворитель и температура. Однако обычно она составляет от 2 до 48 часов, предпочтительно от 6 до 24 часов и более предпочтительно от 8 до 16 часов.
После завершения реакции раствор, содержащий соединение формулы (VII), может быть обработан традиционным методом. Например, раствор может быть промыт водным раствором, таким как вода, бикарбонат натрия или хлорид натрия, для удаления любых присутствующих ионных соединений, или профильтрован для удаления любого твердого вещества. Однако соединение формулы (VII) не выделяют из раствора. Наоборот, осуществляют немедленное взаимодействие раствора, содержащего соединение формулы (VII), для замещения сульфонилоксигрупп на следующей стадии.
Стадия 6
На этой стадии соединение формулы (VII) претерпевает внутримолекулярное замещение сульфонилоксигрупп с образованием соединения формулы (VIII). Предпочтительно это достигается осуществлением реакции соединения формулы (VII) с основанием.
Природа основания не является особенно важным параметром, при условии, что оно способно действовать как основание. Примеры подходящих оснований включают гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид рубидия и гидроксид цезия, гидроксиды тетра(С1-С6)алкиламмония, такие как гидроксид тетраметиламмония и гидроксид тетраэтиламмония, и карбонаты щелочных металлов, такие как карбонат лития, карбонат натрия, карбонат калия, карбонат рубидия и карбонат цезия. Гидроксиды щелочных металлов являются предпочтительными, и особенно предпочтительным является гидроксид натрия.
Реакцию обычно и предпочтительно проводят в присутствии растворителя, природа которого не особенно важна, при условии, что он является инертным по отношению к реакционной смеси и способен растворять реагенты, по меньшей мере, в некоторой степени. Примеры подходящих растворителей включают воду, спирты, такие как метанол и этанол, амиды, такие как диметилформамид, сульфоксиды, такие как диметилсульфоксид, алифатические углеводороды, такие как пентаны, гексаны, гептаны и октаны, ароматические углеводороды, такие как бензол, толуол и ксилолы, галогенированные углеводороды, такие как метиленхлорид, хлороформ и 1,2-дихлорэтан, простые эфиры, такие как диметоксиэтан, диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан, и их смеси. Предпочтительно, чтобы растворитель представлял двухфазную смесь метиленхлорида и воды.
Реакцию предпочтительно проводят в присутствии катализатора межфазного переноса, действие которого заключается в переносе аниона основания, такого как гидроксид-ион, в органический слой. Примеры подходящих катализаторов перехода фаз включают соли тетра(С1-С6) алкиламмония и бензилтри(С1-С6) алкиламмония, такие как тетра-н-бутиламмонийхлорид и бензилтриэтиламмонийхлорид. Тетра-н-бутиламмонийхлорид является предпочтительным.
Температура реакции зависит от различных факторов, таких как природа реагента, основания и растворителя. Однако обычно она составляет от 0°С до точки кипения растворителя, предпочтительно от 10°С до 40°С, более предпочтительно она равна комнатной температуре.
Продолжительность реакции зависит от различных факторов, таких как природа реагентов, растворитель и температура. Однако обычно она составляет от 5 минут до 12 часов, предпочтительно от 10 минут до 6 часов и более предпочтительно от 30 минут до 3 часов.
После завершения реакции раствор, содержащий соединение формулы (VIII), может быть обработан традиционным методом. Например, раствор может быть промыт водным раствором, таким как вода или хлорид натрия, для удаления любых присутствующих ионных соединений, профильтрован для удаления любого твердого вещества или повторно растворен в другом подходящем растворителе, примеры которого даны выше. Однако соединение формулы (VIII) не выделяют из раствора. Наоборот, осуществляют немедленное взаимодействие раствора, содержащего соединение формулы (VIII), с аммиаком или соединением аммония на следующей стадии.
Стадия 7
На этой стадии осуществляют взаимодействие эпоксида формулы (VIII) с аммиаком или соединением аммония с получением соединения формулы (IX).
Превращение достигается за счет взаимодействия с аммиаком, точный источник не особенно важен. Аммиак может быть газообразным или растворенным в растворителе (таком как вода, метанол или этанол). В альтернативном варианте аммиак может быть образован in situ взаимодействием соли аммония (такой как хлорид аммония или ацетат аммония) с основанием (примеры которого даны на стадиях 2 и 6).
Реакцию обычно и предпочтительно проводят в присутствии растворителя, природа которого не особенно важна, при условии, что он является инертным по отношению к реакционной смеси и способен растворять реагенты, по меньшей мере, в некоторой степени. Примеры подходящих растворителей включают спирты, такие как метанол, этанол и изопропанол, амиды, такие как диметилформамид, ДМА и НМФ, алифатические углеводороды, такие как пентаны, гексаны, гептаны и октаны, ароматические углеводороды, такие как бензол, толуол и ксилолы, простые эфиры, такие как диметоксиэтан, диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан, и их смеси. Предпочтительно, чтобы растворителем являлся спирт, особенно метанол.
Температура реакции зависит от различных факторов, таких как природа реагента, основания и растворителя. Однако обычно она составляет от комнатной температуры до точки кипения растворителя и предпочтительно равна от 30°С до 50°С.
Продолжительность реакции зависит от различных факторов, таких как природа реагентов, растворитель и температура. Однако обычно она составляет от 10 минут до 12 часов, предпочтительно от 30 минут до 6 часов и более предпочтительно от 2 до 4 часов.
После завершения реакции соединение формулы (IX) выделяют из реакционной смеси традиционным методом. Например, соединение может быть экстрагировано с использованием органического растворителя и сконцентрировано для удаления растворителя. Соединение может быть экстрагировано водой в виде аддитивной соли кислоты при добавлении кислоты, такой как соляная кислота, нейтрализовано добавлением основания, такого как гидроксид натрия, а затем экстрагировано органическим растворителем и концентрировано для удаления растворителя. Продукт может быть дополнительно очищен традиционными методами, такими как кристаллизация или колоночная хроматография.
Примеры
Далее способ настоящего изобретения будет дополнительно описан со ссылкой на следующие примеры. Данные примеры предназначены для пояснения, а не для ограничения объема настоящего изобретения.
Пример 1. (2R,3R)-2, 3-эпокси-3-фенилпропанол

Коричный спирт (150 г), порошкообразные молекулярные сита 4А (60 г) и D-диизопропилтартрат (39,3 г) перемешивали с метиленхлоридом (2,25 л) и смесь охлаждали до температуры от -15 до -20°С. Добавляли Ti(OiPr)4 (33,2 мл) и реакционную смесь перемешивали при температуре от -20 до -25°С в течение 30 минут. Сухой раствор трет-бутилгидропероксида в изооктане (500 мл, 4,47 М, KF менее 0,1%) добавляли в течение промежутка времени, превышающего 1 час, поддерживая температуру менее чем -20°С. После завершения введения реагента смесь перемешивали в течение 3-х часов при температуре примерно -20°С. После окончания реакции медленно, при охлаждении добавляли триэтилфосфит (210 мл), поддерживая температуру менее +20°С. Затем смесь фильтровали через Celite™ и получали раствор указанного в заголовке соединения.
Пример 2. (2R,3S)-3-(2-этоксифенокси)-2-гидрокси-3-фенилпропанол

Гидроксид натрия (49,2 г), тетра-н-бутиламмонийхлорид (15,5 г) и 2-этоксифенол (169,9 г) растворяли в воде (1080 мл). Добавляли раствор (2R,3R)-2,3-эпокси-3-фенилпропанола из примера 1 и смесь нагревали до внутренней температуры примерно 40°С и метиленхлорид отгоняли. Внутреннюю температуру постепенно повышали до 65°С по мере отгонки метиленхлорида и нагревание осуществляли в течение трех часов после завершения удаления метиленхлорида. Реакционную смесь охлаждали до 30°С. Добавляли метил-трет-бутиловый простой эфир (1,5 л) и смесь перемешивали в течение 30 минут. Фазам давали возможность разделиться и водную фазу удаляли. Органический слой промывали 1М NaOH (2 × 1 л), водой (1 л) и насыщенным водным раствором NaCl (1 л). Органический раствор осушали кипячением в колбе с обратным холодильником с ловушкой Дина-Старка до тех пор, пока содержание KF не составило 0,9%. Горячий раствор фильтровали через фильтр калибром 2 мкм. Метил-трет-бутиловый простой эфир (950 мл) отгоняли из фильтрата и добавляли изооксан (200 мл). Дополнительно отгоняли 200 мл растворителя. Добавляли изооктан (200 мл) и раствор охлаждали, пока не начинали образовываться кристаллы. Как только кристаллизация начиналась, добавляли изооктан (700 мл) тремя порциями в течение примерно 1 часа. Суспензию перемешивали в течение 15 минут, затем охлаждали до -20°С и поддерживали при -20°С в течение 1 часа. Маточный раствор удаляли через колоночный фильтр и остаток промывали изооктаном (500 мл). Промывную жидкость удаляли через колоночный фильтр. К остатку добавляли метил-трет-бутиловый простой эфир (300 мл) и смесь нагревали до растворения осадка. Раствор охлаждали до температуры примерно 20-25°С и добавляли изооктан (400 мл) порциями по 50 мл в течение примерно 30 минут. Затем суспензию медленно охлаждали до -15°С. Суспензию перемешивали при -15°С в течение одного часа и фильтровали. Кристаллы промывали изооктаном (300 мл), а затем сушили в токе азота, после чего получили 206 г указанного в заголовке соединения. По данным анализа данный материал содержал 97%-ный энантиомерный избыток.
Анализ методом хиральной ЖХВР:
Колонка: Chiracel OD-H
Подвижная фаза: 90:10 гексаны: изопропанол
Расход: 1 мл/минута
Детектор: 215 нм
Время опыта: 20 минут
Времена удерживания:
(2S,3R)-3-(2-этоксифенокси-2-гидрокси-3-фенилпропанол: 8,7 минут
(2R,3S)-3-(2-этоксифенокси-2-гидрокси-3-фенилпропанол: 10,8 минут
Температура плавления: 91,9-93,4°С
[α]20D(с = 10) + 8,33°
1Н ЯМР (400, 13 МГц, CDCl3): δ 1,51 (т, J=6,6 Гц, 3Н), 3,24 (дд, J=3,5 Гц, 9,5 Гц, 1Н), 3,33 (д, J=8 Гц, 1Н), 3,67 (м, 1Н), 3,96 (м, 1Н), 3,96 (д, J=3,5 Гц, 1Н), 4,12 (кв, J=6,6 Гц, 2Н), 5,28 (д, J=3,5 Гц, 1Н), 6,59-6,65 (м, 1Н), 6,67-6,75 (м, 1Н), 6,86-6,94 (м, 2Н), 7,26-7,41 (м, 5Н).
13С ЯМР (100,62 МГц, CDCl3): δ 14,76, 62,02, 74,28, 86,30, 112,37, 116,09, 120,72, 122,34, 126,33, 128,12, 128,75, 137,85.
Примеры 3 и 4

Пример 3. (2R,3S)-3-(2-этоксифенокси)-2-мезилокси-3-фенил-1-гидроксипропан
(2R, 3S)-3-(2-этоксифенокси)-2-гидрокси-3-фенилпропанол (40,00 мг) и триэтиламин (23,4 мл) растворяли в метиленхлориде (400 мл) и раствор охлаждали до примерно -20°С. Добавляли раствор триметилсилилхлорида (18,7 мл) в CH2Cl2 (28 мл), поддерживая температуру реакционной массы менее -15°С. После завершения введения реагента смесь перемешивали примерно в течение 15 минут при температуре менее -15°С. К раствору добавляли метансульфонилхлорид (13,2 мл), поддерживая температуру между -20 и -15°С, затем триэтиламин (19,5 мл), вновь поддерживая температуру реакционной смеси между -20 и -15°С. Смесь перемешивали в течение 15 минут после завершения добавления триэтиламина. К реакционной смеси добавляли соляную кислоту (1М, 140,6 мл). Смесь нагревали до 20-25°С и перемешивали в течение 12 ч. Фазы разделяли и органический раствор промывали 5%-ным водным раствором бикарбоната натрия (131 мл). Водную фазу отделяли и получали раствор указанного в заголовке соединения.
Пример 4. (2S,3S)-1,2-эпокси-3-(2-этоксифенокси)-3-фенилпропан
Гидроксид натрия (25 г), тетра-н-бутиламмонийхлорид (1,92 г) и воду (100 мл) перемешивали до растворения твердых веществ. Добавляли раствор (2R,3S)-3-(2-этоксифенокси)-2-мезилокси-3-фенил-1-гидроксипропана из примера 3 в метиленхлориде и смесь перемешивали с высокой скоростью до завершения реакции. Фазы разделяли и водную фазу экстрагировали метиленхлоридом (100 мл). Объединенные органические фазы промывали насыщенным водным раствором NaCl (76 мл). Органический раствор концентрировали под вакуумом до 60 мл. Добавляли метанол (300 мл) и раствор концентрировали до 60 мл. Добавляли метанол (300 мл) и смесь отгоняли до объема 60 мл, в результате чего получали раствор указанного в заголовке соединения.
Пример 5. (2S,3S)-3-(2-этоксифенокси)-2-гидрокси-3-фенилпропиламин

Раствор (2S,3S)-1,2-эпокси-3(2-этоксифенокси)-3-фенилпропана из примера 4, метанол (280 мл) и концентрированный гидроксид аммония (450 мл) загружали в 1л колбу высокого давления, снабженную магнитной мешалкой. Колбу закрывали и нагревали при 40°С в течение трех часов. Смесь охлаждали и добавляли метиленхлорид (340 мл). Фазы разделяли и водную фазу экстрагировали метиленхлоридом (2 × 150 мл). Органические фазы объединяли и перегоняли под вакуумом до объема 450 мл. К раствору продукта вновь добавляли метиленхлорид (250 мл). Органическую фазу промывали водой (375 мл), водную фазу экстрагировали метиленхлоридом (150 мл) и растворы в метиленхлориде объединяли. Добавляли соляную кислоту (0,5 М, 375 мл) и смесь перемешивали, а затем оставляли осадиться. Фазы разделяли и органическую фазу промывали водой (375 мл). Водные фазы объединяли и промывали метиленхлоридом (70 мл). Органическую фазу отделяли и отбрасывали. Добавляли метиленхлорид (220 мл), а затем 50%-ный раствор NaOH до достижения рН более 12. Фазы разделяли и водную фазу экстрагировали метиленхлоридом (100 мл). Органические фазы объединяли и перегоняли до твердого остатка. Добавляли этилацетат (2 × 250 мл) и перегоняли. Добавляли этилацетат (116 мл) и гептан (116 мл). Смесь нагревали до растворения твердого вещества. Раствор медленно охлаждали до 20-25°С при быстром перемешивании. Когда температура суспензии достигала 20-25°С, добавляли гептан (116 мл) и суспензию охлаждали до -15°С и поддерживали при 15°С в течение 1 часа. Твердое вещество отфильтровывали и сушили в токе азота, в результате чего получали 25 г указанного в заголовке соединения.
Тпл. 98,5-99,9°С
[α]20D (c = 10) + 37,04°
1Н ЯМР (400, 13 МГЦ, CDCl3): δ 1,51 (т, J=7,1 Гц, 3Н), 2,5-2,7 (м, 4Н), 3,94 (м, 1Н), 4,11 (кв, J=7,1 Гц, 2Н), 4,75 (д, J=8,1 Гц, 1Н), 6,62-6,86 (м, 4Н), 7,23-7,40 (м, 5Н).
13С ЯМР (100,62 МГц, CDCl3): δ 14,83, 43,11, 64,21, 87,77, 112,84, 119,58, 120,82, 123,32, 127,25, 128,60, 148,06.
Реферат
Изобретение относится к способу получения соединений формулы (IV), включающему асимметричное эпоксидирование соединения формулы (I) агентом окисления в присутствии оптически активного соединения с образованием соединения формулы (II); добавление агента обрыва реакции, чтобы погасить любой избыток присутствующего агента окисления, где агентом обрыва реакции является три(С1-С6)алкилфосфит; без выделения соединения формулы (II) взаимодействие реакционной смеси, включающей соединение формулы (II) и окисленный агент обрыва реакции, с соединением формулы (III) в присутствии основания и очистку соединения формулы (IV) кристаллизацией. Изобретение также относится к способу получения соединения формулы (IX), включающему реакцию соединения формулы (IV) с силилирующим агентом с образованием соединения формулы (V); реакцию соединения формулы (V) с агентом сульфонилирования формулы R'SO2X, где R' представляет собой остаток сульфоновой кислоты (С1-С6)алкил и X представляет отщепляемую группу, с получением соединения формулы (VI); удаление силильной защитной группы с получением соединения формулы (VII); замещение сульфонилоксигруппы с получением соединения формулы (VIII) и реакцию соединения формулы (VIII) с аммиаком или соединением аммония с получением соединения формулы (IX). Изобретение также относится к промежуточным соединениям (V) и (VI). Соединение формулы (IX) может быть использовано для получения биологически активного вещества - (S,S)-ребоксетина.

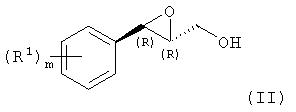







В приведенных структурных формулах n и m независимо равны 0 или целому числу от 1 до 5; каждая из групп R и R1, которые могут быть одинаковыми или различными, представляет собой С1-С6алкокси или С1-С6алкил; Р представляет собой силильную защитную группу; R' представляет собой остаток сульфоновой кислоты (С1-С6)алкил. 4 н. и 10 з.п. ф-лы.
Формула









Документы, цитированные в отчёте о поиске
Способ получения 3-арилокси-3-замещенных пропанаминов или их фармакологически приемлемых кислотно-аддитивных солей
Комментарии