Способ получения производных изосерина - RU2557546C2
Код документа: RU2557546C2
Описание
Область техники, к которой относится изобретение
Данная заявка относится к "one pot" способу получения производных изосерина с высокой диастереоселективностью.
Уровень техники
α-Гидрокси-β-аминокислоты (изосерины) являются важными целевыми соединениями, т.к. данные аминокислоты входят в состав молекул, представляющих большой биологический интерес, например, новый липопептидный сидерофор, именуемый орникорругатин (ornicorrugatin)[1]; KRI-1314[2] - мощный полипептидный ингибитор ренина человека; амастатин[3] - тетрапептид, обладающий иммунорегуляторной, противоопухолевой и антибактериальной активностью; микрогинин (microginin)[4]; трео-β-бензилоксиаспартат (TBOA)[5], первый непереносимый блокатор всех подтипов EAAT; и чаще всего производные таксана[6].
С точки зрения стерических особенностей биологические требования к упомянутым выше аминокислотам четко определены и обычно активность проявляют трео-изомеры (2R, 3S), которые, таким образом, являются предпочтительными.
Известные синтетические подходы к получению изосериновых соединений посредством образования связи C-2/C-3 основаны на реакции сульфинилимина с литиевым енолятом защищенного α-гидрокси сложного эфира [7a, b] или обычного имина с литиевым енолятом сложного эфира и последующей стадией окисления[7c]. Основными недостатками упомянутых выше реакций являются: в первом случае[7a, b] - использование дорогих сульфинилиминов, синтезировать которые непросто. Во втором случае достигаются только умеренные выходы, а также требуется стадия окисления с использованием дорогого реагента (оксазиридина) для введения α-гидроксильной группы. В соответствии с другим синтетическим подходом [7d] производные простых иминов вводят в реакцию с силилацеталем α-метоксикетена в кислых условиях. Соединения изосерина, функционализированные α-метокси группой, образуются с умеренным выходом и диастереоселективностью. Кроме того, на отдельной стадии необходимо удалить защитную α-метокси группу. Наконец, известна конденсация N-арилиминов с опасными арилдиазоацетатами, используемыми в большом стехиометрическом избытке, и катализируемая дорогим родиевым катализатором[7e]. В зависимости от механизма замещения производные изосерина получаются с различной диастереоселективностью и с умеренными выходами. Основная трудность, которая затрагивает все известные методы синтеза, заключается в получении чистого энантиомера, т.к. во всех методах, за исключением приведенного в ссылке [7d], используется хиральный вспомогательный фрагмент в исходном реагенте(-ах), так что в процессе конденсации образуются четыре диастереомера.
Описание изобретения
Настоящее изобретение относится к способу получения соединений общей формулы 1, в которой R1 представляет собой линейную или разветвленную алкильную группу Cl-C6, незамещенную или замещенную арильную или гетероарильную группу, R2 представляет собой линейную или разветвленную алкильную группу Cl-C6, арилалкильную группу; R3 представляет собой Н, алкильную группу Cl-C4. Все стереоизомеры включены в формулу 1.
Предпочтительными соединениями формулы 1 являются соединения, в которых R1 представляет собой изо-Bu, арил, тиенил; R2 является PhCH2, н-Bu; а R3 является Н, алкильной группой С1-С4.
Предпочтительны трео-диастереомеры (2R*,3S*) формулы 1.

Способ, описанный в данном изобретении, представлен на схеме 1, где группы R1, R2 и R3 определены выше.
Способ заключается в реакции приблизительно эквимолекулярных количеств силильного эфира енола общей формулы 3, в которой R4 представляет собой Me, Et, а R5 представляет собой Me, Et и имина общей формулы 4, в которой R1 является линейной или разветвленной алкильной группой Cl-C6, арилалкильной группой, незамещенной или замещенной арильной группой, гетероарильной группой, R2 является линейной или разветвленной алкильной группой Cl-C6 или арилалкильной группой, приводя к промежуточным соединениям общей формулы 5 (3R*,5S*,6S*,1'S*) и 6 (3R*,5S*,6S*,1'R*), в которых R1 является линейной или разветвленной алкильной группой Cl-C6, арилалкильной группой, незамещенной или замещенной арильной группой, гетероарильной группой; R2 является линейной или разветвленной алкильной группой Cl-C6, арилалкильной группой; R4 представляет собой Me или Et. При алкоголизе промежуточные соединения 5 и 6 непосредственно превращаются в производные изосерина общей формулы 1 (2R*,3S*) и 1' (2R*,3R*), в которой R3 является Н, или в производные изосерина общей формулы 1 (2R*,3S*) и 1' (2R*,3R*), в которой R3 является алкильной группой С1-С4.

Реакция конденсации между соединениями 3 и 4 катализируется как протонными кислотами, так и кислотами Льюиса (0,1-1 экв.).
Типичными представителями катализаторов Льюиса являются ZnCl2, CoCl2, InCl3, NbCl5, Eu(OTf)3, PdCl2, SnCl2 и MgBr2. Было показано, что распределение продуктов 5 и 6 реакции, а следовательно, и продуктов 1 (2R*,3S*) и 1' (2R*,3R*), зависит от типа кислоты Льюиса.
Кислоты Льюиса, такие как InCl3, SnCl2 и MgBr2 приводят и к лучшей трео-диастереоселективности (т.е. к увеличению образования предпочтительного диастереомера 1 (2R*,3S*)), и к более высокому выходу продукта реакции. Следовательно, InCl3, SnCl2 и MgBr2 являются предпочтительными.
Реакция может быть осуществлена в широком диапазоне температур, например, от -70°C до 25°C. Лучшие результаты были получены при температуре от -40°C до -30°C. Этот диапазон температур, таким образом, является предпочтительным.
Реакция протекает предпочтительно в полярных апротонных растворителях, например, в диметилформамиде, ацетонитриле, дихлорметане, хлороформе, тетрагидрофуране. Причем ацетонитрил и дихлорметан являются предпочтительными.
При необходимости все продукты реакции могут быть очищены с использованием стандартных процедур.
Алкоголиз осуществляется с использованием подходящего спирта R3OH в присутствии триметилсилилхлорида.
Согласно предпочтительному варианту данного изобретения, приводящему к лучшим результатам с точки зрения выхода и диастереоселективности, реакцию осуществляют как "однореакторную реакцию", что позволяет избежать выделения всех промежуточных соединений и приводит после стадии простой кристаллизации к чистому конечному трео-диастереомеру.
В соответствии с этим способом, сначала в ацетонитриле в присутствии молекулярных сит при комнатной температуре или путем азеотропной отгонки смеси ацетонитрил/вода из альдегида (R1CHO) и амина (R2NH2, предпочтительнее PhCH2NH2) in situ получают имин 4. Температуру реакции понижают до -30°C и добавляют силильное производное 3. Затем добавляют катализатор и реакционную смесь перемешивают в течение 1 ч. Неочищенную реакционную смесь сразу обрабатывают раствором триметилсилилхлорида в спирте и после кристаллизации выделяют чистый диастереомер 1 (2R*,3S*) (где R3 представляет собой алкильную группу С1-С4).
По желанию способ, описываемый в настоящем изобретении, может непосредственно приводить к энантиомерно чистым соединениям (таким образом, избегая этапа разделения), исходя из энантиомерно чистого соединения 3 (R4=Me, R5=Et).
Соединения 3 получают из соответствующих лактонов 2 в соответствии со схемой 2.
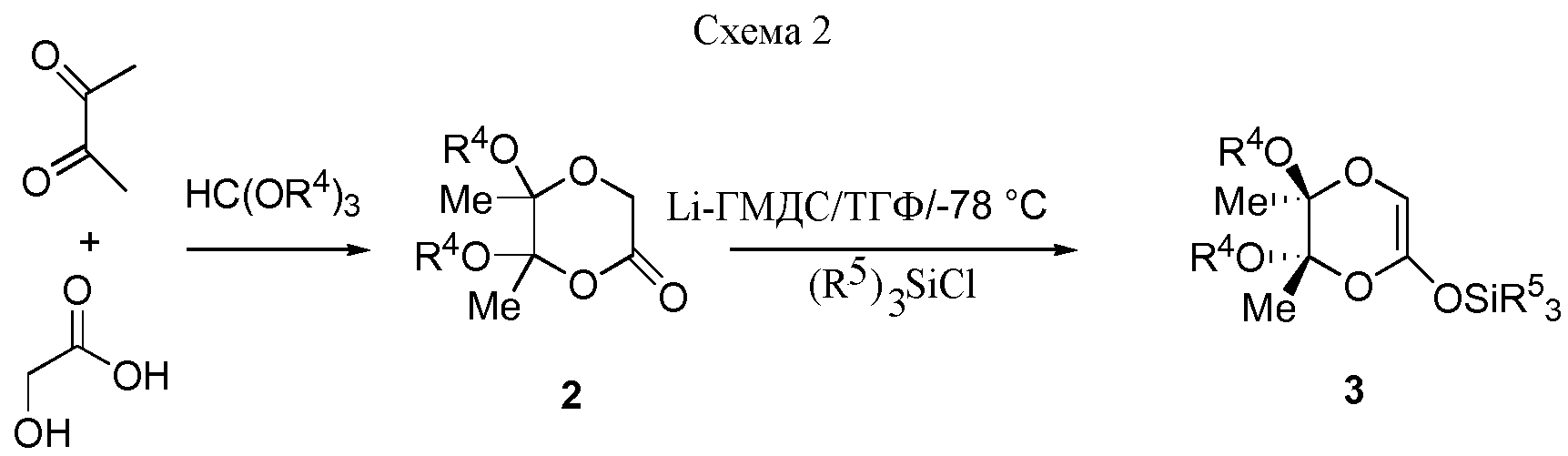
Соединение 2 (R4=Me) является известным соединением, которое может быть получено обычным способом [8c]. Соединения 2 (R4=Et), 3 (R4=Me, R5=Et), 3 (R4=R5=Et) являются новыми соединениями, которые вместе со способами их получения являются также частью настоящего изобретения. Новый лактон 2 (R4=Et) получают в соответствии с очень эффективной "однореакторной" методикой, исходя непосредственно из смеси 2,3-бутандиона, гликолевой кислоты и этилортоформиата в присутствии каталитических количеств H2SO4. Соединения 3 получают с превосходными выходами из соединений 2, используя гексаметилендисилиламид лития (LiHMDS) в ТГФ при -78°C и Et3SiCl в качестве силилирующего реагента (схема 2). Преимуществом является короткое время реакции.

В соответствии с предпочтительным признаком настоящего изобретения, имины 4 получают in situ из соответствующего альдегида и амина, и реакционную смесь немедленно вводят в реакцию с соединениями 3, таким образом, избегая выделения иминов, которое является сложным и понижающим выход реакции по причине того, что имины, как известно, являются достаточно нестабильными соединениями.
Преимущества данного изобретения
Способ, описанный в настоящем изобретении, является лучшим благодаря простоте методики, которая не требует выделения промежуточных соединений и сложной хроматографической очистки соединений.
Мягкие условия необходимы для снятия защиты как с гидроксильной, так и с карбоксильной групп. Это важно, потому что полностью удается избежать риска рацемизации. Хорошие выходы реакций и высокая диастереоселективность являются существенными дополнительными преимуществами.
Данный способ приводит к рацемической смеси производных изосерина, которые могут быть разделены с использованием обычных методов. Однако при желании можно легко избежать стадии разделения и при использовании энантиомерно чистой защищенной глицидиловой кислоты [10] непосредственно получить энантиомерно чистые соединения.
Примеры
Предварительно InCl3 был обезвожен нагреванием в вакууме при 200°C в течение 2 ч.
5,6-Диэтокси-5,6-диметил-[1,4]-диоксан-2-он. 2,3-Бутандион (4,6 мл, 46,22 ммоль) и гликолевую кислоту (3,07 г, 40,40 ммоль) растворяют в этилортоформиате (40 мл) и добавляют каталитическое количество H2SO4. Реакционную смесь оставляют при 25°С на 1 ч. Прибавляют насыщенный раствор NaHCO3 (10 мл) и полученную смесь экстрагируют этилацетатом (3×30 мл). Органическую фазу сушат над Na2SO4 и неочищенную реакционную смесь очищают методом хроматографии на силикагеле (циклогексан/этилацетат, 10:1), получая после кристаллизации чистое соединение 2 (R4=Et, 52%). Белое твердое вещество, т.пл. 37°С (CH2Cl2/пентан, 0°С). ИК (NaCl) Vmax 1752 см-1;1Н ЯМР (CDCl3) δ 4,32, 4,23 (AB система, J 16,6, 2H), 3,79-3,69 (м, 2H), 3,58 (кв, J 7,2, 2H), 1,53 (с, 3H), 1,41 (с, 3H), 1,21 (т, J 6,9, 3H), 1,99 (т, J 7,0, 3H);13C ЯМР (CDCl3) δ 168,1, 105,2, 97,8, 60,5, 58,8, 57,3, 18,8, 17,9, 15,9, 15,7, 15,3. Масс-спектр (ЭС) m/z 341,4 [M+23]+; Рассчитано для C10H18O5: C, 55,03; H, 8,31; найдено C, 55,12; H, 8,38.
5,6-диметокси-5,6-диметил-[1,4]диоксен-2-илокси)триэтилсилан. Лактон 2 (R4=Me) (3,23 г, 17 ммоль) растворяют в безводном ТГФ (35 мл) и полученную смесь охлаждают в атмосфере N2 до -78°C. Растворяют Li-ГМДС (4,52 г, 27 ммоль) в ТГФ (10 мл) и затем добавляют по каплям к полученной выше смеси. После добавления реакционную смесь перемешивают в течение еще 10 минут, после чего добавляют триэтилсилилхлорид (4 мл, 26 ммоль). Затем полученному раствору дают нагреться до 25°С и продолжают перемешивание в течение ночи. После чего удаляют ТГФ при пониженном давлении и к остатку добавляют пентан (50 мл). Полученную суспензию фильтруют через целит, а растворитель удаляют в вакууме, получая неочищенное соединение, которое перегоняют в вакууме (120°C, 0,8 мм рт.ст.). Чистое соединение 3 (R4=Me, R5=Et) получают (4,26 г, 82%) в виде бесцветного масла. ИК (NaCl) vmax 1719, 1149, 740 см-1;1H ЯМР (CDCl3) δ 5,52 (с, 1H), 3,37 (с, 1H), 3,22 (с, 1H), 1,45 (с, 3H), 1,38 (с, 3H), 1,06-0,94 (м, 9H), 0,75-0,67 (м, 6H);13C ЯМР (CDCl3) δ 143,9, 104,3, 96,6, 90,4, 49,5, 48,6, 17,6, 17,1, 6,6, 4,8. Масс-спектр (ЭС) m/z 218,9; Рассчитано для C14H28O5Si: C, 55,23; H, 9,27; найдено C, 55,11; H, 9,15.
5,6-Диэтокси-5,6-диметил-[1,4]-диоксин-2-илокси)триэтилсилан. Силильное производное 3 (R4=R5=Et) (4,8 г, 85%) получают в соответствии с вышеописанной синтетической методикой, но к смеси 2 (R4=Et) (3,7 г, 17 ммоль) и триэтилсилилхлорида добавляют основание. Бесцветное масло (130°C, 0,8 мм рт.ст.). ИК (NaCl) vmax 1721, 1149, 744 см-1;1H ЯМР (CDCl3) δ 5,50 (с, 1H), 3,68-3,63 (м, 2H), 3,51 (кв, J 7,0, 2H), 1,46 (с, 3H), 1,40 (с, 3H), 1,22-1,03 (м, 6H), 1,02-0,88 (м, 9H), 0,75-0,63 (м, 4H), 0,55 (кв, J 10,8, 2H);13C ЯМР (CDCl3) δ 143,8, 104,3, 100,2, 96,4, 57,4, 56,6, 18,4, 17,9, 15,9, 15,7, 6,6, 4,9. Масс-спектр (ЭС) m/z 355,2 [M+23]+; Рассчитано для C16H32O5Si: C, 57,79; H, 9,70; найдено C, 57,60; H, 9,58.
Получение соединений 5 и 6. МетодА. В атмосфере азота при перемешивании растворяют имин 4 (0,66 ммоль) в сухом MeCN (1,2 мл). Затем реакционную смесь охлаждают до -30°C и одной порцией добавляют безводный InCl3 (73 мг, 0,33 ммоль). После перемешивания при этой температуре в течение 10 мин по каплям добавляют раствор силильного производного 3 (0,66 ммоль) в сухом MeCN (1 мл). Реакционную смесь перемешивают в течение 1 ч, а затем обрабатывают насыщенным раствором NaHCO3 (1 мл). Неочищенное вещество экстрагируют этилацетатом (3×5 мл) и органическую фазу промывают рассолом, сушат над Na2SO4, фильтруют и в вакууме удаляют растворитель. Неочищенный продукт реакции очищают методом флэш-хроматографии на силикагеле. Соединения 5/6 (R4=Et) очищают методом картриджной флэш-хроматографии (SiO2; н-гексан/Et2O, 7:2; скорость элюирования: 30 мл/мин). В последних случаях были выделены только изомеры 5. Метод Б. В двугорлой круглодонной колбе, снабженной магнитной мешалкой и устройством для ввода азота, растворяют альдегид (бензальдегид: 60 мкл, 0,59 ммоль; изовалериановый альдегид: 63 мкл, 0,59 ммоль) и амин (бензиламин: 79 мкл, 0,59 ммоль) в MeCN (1,5 мл) в присутствии молекулярных сит (60 мг, активированных в вакууме при 200°С в течение 2 ч). По истечении 1 ч реакционную смесь охлаждают до -30°C и одной порцией добавляют безводный InCl3 (65,3 мг, 0,29 ммоль). После перемешивания при этой температуре в течение 10 мин по каплям добавляют раствор силильного эфира енола 3 (R4=Me, R5=Et) (180,2 мг, 0,59 ммоль) в сухом MeCN (1 мл). Реакционную смесь перемешивают в течение 1 ч. Реакционную смесь обрабатывают как описано в методике выше и выделяют соединения 5/6 (R1=Ph, R2=Bn, R4=Me) или 5/6 (R1=Me2CHCH2, R2=Bn, R4=Me) соответственно.
(3R*,5S*,6S*)-3-(1'-N-бензиламино-1'-фенил-метил)-5,6-диметокси-5,6-диметил-[1,4]-диоксан-2-он (5,6: R1=Ph, R2=Bn, R4=Me). Колоночная хроматография: этилацетат/циклогексан, 1:5; МетодА: 65% (4: 1), 5: 52%,6: 13%. Метод Б: 5: 71%, 6: 16%.
1'S*-5 (R1=Ph, R2=Bn, R4=Me): 108°C (н-пентан/CH2Cl2). ИК (KBr) Vmax 3372, 1744 см-1;lH ЯМР (CDCl3) δ 7,40-7,22 (м, 10H), 4,31, 4,28 (AB система, J 2,9, 2H), 3,60, 3,51 (AM система, J 13,2, 2H), 3,21 (с, 3H), 3,14 (с, 3H), 3,00-2,00 (шир, 1H, обмен), 1,47 (с, 3H), 1,37 (с, 3H);13C ЯМР (CDCl3) δ 169,2, 140,8, 139,7, 128,7, 128,6, 128,5, 127,2, 105,1, 98,6, 75,7, 63,6, 51,1, 50,1, 49,5, 18,4, 17,2. Масс-спектр (ЭС) m/z 386,0 [M+1]+; Рассчитано для C22H27NO5: C, 68,55; H, 7,06; N, 3,63; найдено C, 68,43; H, 7,14; N, 3,56.
1'R*-6 (R1=Ph, R2=Bn, R4=Me): бледно-желтое масло.
ИК (NaCl) vmax3318, 1748 см-1;1H ЯМР (CDCl3) δ 7,41-7,26 (м, 10H), 4,54, 4,30 (AB система, J 2,7, 2H), 3,75, 3,56 (AM система, J 13,2, 2H), 3,33 (с, 3H), 2,77 (с, 3H), 2,76 (уш.с, 1H, обмен), 1,37 (с, 3H), 1,36 (с, 3H);13C ЯМР (CDCl3) δ 168,1, 140,5, 138,4, 129,7, 128,7, 128,2, 127,8, 127,4, 105,1, 98,6, 74,7, 63,4, 51,5, 49,6, 49,5, 18,3, 17,2. Масс-спектр (ЭС) m/z 386,2 [M+1]+; Рассчитано для C22H27NO5: C, 68,55; H, 7,06; N, 3,63; найдено C, 68,40; H, 7,15; N, 3,51.
(3R*,5S*,6S*)-3-(1'-N-Бензиламино-3'-метил-бутил)-5,6-диметокси-5,6-диметил-[1,4]-диоксан-2-он (5,6: R1=Me2CHCH2, R2=Bn, R4=Me). Колоночная хроматография: соединения 5 и 6 нестабильны при хроматографировании на силикагеле (этилацетат/циклогексан, 1:5) с невысокой скоростью с целью разделения двух изомеров, и наблюдается существенное уменьшение выхода продукта реакции. По этой причине только основной изомер 5 (R1=Me2CHCH2, R2=Bn, R4=Me) был выделен в чистом виде. Альтернативно, при хроматографировании неочищенной реакционной смеси на нейтральном оксиде алюминия (циклогексан/Et2O, 7:1) с хорошим выходом была получена смесь соединений. Метод A: 74%.
1'S*-5 (R1=Me2CHCH2, R2=Bn, R4=Me): бледно-желтое масло. ИК (NaCl) vmax 3339, 1747 см-1;1H ЯМР (CDCl3) δ 7,37-7,19 (м, 5H), 4,17 (д, J 2,6, 1H), 3,78 (с, 2H), 3,28-3,24 (м, 1H), 3,28 (с, 3H), 3,24 (с, 3H), 1,90 (уш.с, 1H, обмен), 1,80-1,60 (м, 1H), 1,50-1,38 (м, 2H), 1,46 (с, 3H), 1,37 (с, 3H), 0,91 (д, J 6,6, 3H), 0,86 (д, J 6,3, 3H);13C ЯМР (CDCl3) δ 170,1, 141,2, 128,5, 128,3, 126,9, 105,0, 98,3, 72,5, 57,5, 51,8, 50,1, 49,3, 40,8, 25,4, 23,0, 22,98, 18,2, 17,1. Рассчитано для C20H31NO5: C, 65,73; H, 8,55; N, 3,83; найдено C, 65,60; H, 8,70; N, 3,71.
(3R*,5S*,6S*)-3-(1'-N-н-бутил-1'-фенил-метил)-5,6-диметокси-5,6-диметил-[1,4]-диоксан-2-он (5,6: R1=Ph, R2=Me(CH2)3, R4=Me). Колоночная хроматография: этилацетат/циклогексан, 1:5; Метод A: 68% (3,2:1), 5: 52%, 6: 16%.
1'S*-5 (R1=Ph, R2=Me(CH2)3, R4=Me): бледно-желтое масло. ИК (NaCl) vmax 3343, 1755 см-1;1H ЯМР (CDCl3) δ 7,33-7,21 (м, 5H), 4,26, 4,22 (AB система, J 2,7, 2H), 3,31 (с, 3H), 3,11 (с, 3H), 2,55-2,20 (м, 3H, 1H обмен), 1,55-1,20 (м, 4H), 1,44 (с, 3H), 1,35 (с, 3H), 0,83 (т, J 7,0, 3H);13C ЯМР (CDCl3) δ 169,2, 139,9, 128,3, 128,2, 127,3, 104,9, 98,4, 75,5, 63,9, 49,9, 49,3, 46,7, 32,6, 20,5, 18,1, 17,0, 14,2. Масс-спектр (ЭС) m/z 352,1 [M+1]+; Рассчитано для C19H29NO5: C, 64,93; H, 8,32; N, 3,99; найдено C, 64,80; H, 8,47; N, 3,87.
1'R*-6 (R1=Ph, R2=Me(CH2)3, R4=Me): бледно-желтое масло.
ИК (NaCl) Vmax 3323, 1747 см-1;1H ЯМР (CDCl3) δ 7,37-7,24 (м, 5H), 4,53, 4,24 (AB система, J 3,5, 2H), 3,32 (с, 3H), 2,75 (с, 3H), 2,75-2,40 (м, 3H, 1H обмен), 1,60-1,20 (м, 4H), 1,35 (с, 3H), 1,33 (с, 3H), 0,86 (т, J 7,1, 3H);13C ЯМР (CDCl3) δ 168,1, 138,4, 129,4, 127,5, 127,4, 104,9, 98,5, 74,0, 63,7, 49,4, 49,3, 47,2, 32,3, 20,6, 18,1, 17,0, 14,2. Масс-спектр (ЭС) m/z 352,1 [M+1]+; Рассчитано для C19H29NO5: C, 64,93; H, 8,32; N, 3,99; найдено C, 64,78; H, 8,43; N, 3,88.
(3R*,5S*,6S*,1'S*)-3-(1'-N-Бензиламино-1'-фенил-метил)-5,6-диметокси-5,6-диметил-[1,4]-диоксан-2-он (5: R1=Ph, R2=Bn, R4=Et). Метод A: 74%; т.пл. 154°C (с разл.), (CH2Cl2/Et2O).
ИК 3344, 1749 см-1;1H ЯМР (CDCl3) δ 7,38-7,17 (м, 10H), 4,30, 4,23 (AB система, J 2,5, 2H), 3,66, 3,43 (AM система, J 13,2, 2H), 3,73-3,19 (м, 4H), 3,00-2,00 (уш, 1H, обмен), 1,49 (с, 3H), 1,38 (с, 3H), 1,06 (т, J 6,9, 3H), 0,87 (т, J 6,9, 3H);13C ЯМР (CDCl3) δ 169,2, 140,5, 139,7, 128,6, 128,5, 128,4, 128,3, 127,5, 127,0, 105,0, 98,2, 75,5, 63,2, 58,3, 57,3, 50,8, 18,9, 17,7, 15,2, 15,1. Масс-спектр (ЭС) m/z 414,0 [M+1]+; Рассчитано для C24H31NO5: C, 69,71; H, 7,56; N, 3,39; найдено C, 69,58; H, 7,68; N,3,27.
(3R*,5S*,6S*,1'S*)-3-(1'-N-Бензиламино-1'-(3,4-метилендиокси)фенил-метил)-5,6-диэтокси-5,6-диметил-[1,4]-диоксан-2-он (5: R1=Ph, R2=3,4-OCH2O-Ph, R4=Et). 87%; т.пл. 112°C (пентан/CH2Cl2). ИК (NaCl) vmax 3459, 1736 см-1;1H ЯМР (CDCl3) δ 7,26-7,22 (м, 5H), 6,94 (с, 1H), 6,77 (с, 2H), 5,98 (с, 2H), 4,18 (уш.с, 2H), 3,65, 3,42 (AM система, J 13,2, 2H), 3,78-3,20 (м, 4H), 2,55-1,80 (уш, 1H, обмен), 1,49 (с, 3H), 1,40 (с, 3H), 1,09 (т, J 7,0, 3H), 0,88 (т, J 7,0, 3H);13C ЯМР (CDCl3) δ 169,0, 147,9, 147,0, 140,5, 133,9, 128,6, 128,3, 127,0, 122,0, 108,6, 108,1, 104,9, 101,1, 98,2, 75,6, 62,9, 58,3, 57,4, 50,6, 18,9, 17,8, 15,2, 15,0. Масс-спектр (ЭС) m/z 458,1 [M+1]+; Рассчитано для C25H31NO7: С, 65,63; H, 6,83; N, 3,06; найдено С, 65,45; H, 7,00; N, 2,96.
Общая методика метанолиза соединений5 и 6: Соединение 5 (R1=Ph, R2=Bn, R4=Me; R1=Ph, R2=Bn, R4=Et) или 6 (R1=Ph, R2=Bn, R4=Me) (0,225 ммоль) растворяют в течение 10 мин при перемешивании в 0,5 М растворе триметилсилилхлорида в MeOH (1,0 мл, 0,5 ммоль) при 25°C. После удаления растворителя остаток кристаллизуют, что приводит к чистому соединению 1 или 1' (R1=Ph, R2=Bn) [1: 97% из соединения 5 (R1=Ph, R2=Bn, R4=Me); 1: 95% из соединения 5 (R1=Ph, R2=Bn, R4=Et), 1': 80% из соединения 6 (R1=Ph, R2=Bn, R4=Me).
"One pot" получение производных метил-3-(амино)-2-гидрокси-пропионата 1/1' МетодC: реакцию между 4 (R1=Ph, R2=Bn) и 3 (R4=R5=Et) проводят в соответствии с общей методикой.1H ЯМР анализ неочищенной реакционной смеси показал наличие диастереомера 5 (R1=Ph, R2=Bn, R4=Et), и только следовых количеств диастереомера 6 (R1=Ph, R2=Bn, R4=Et) (ВЭЖХ: «Ascentis SI» 150x4.6 мм, 3 мкм, 0,8 мл/мин, λ=210 нм, н-гексан /изо-PrOH, 98:2; 92:8). В соответствии с вышеописанной методикой, неочищенную реакционную смесь обрабатывают MeOH/TMSCl и после кристаллизации выделяют производные метилового эфира 1 (R1=Ph, R2=Bn, R3=Me) (60%). Дополнительную порцию соединения 1 (R1=Ph, R2=Bn, R3=Me; 10%) выделяют после очистки методом колоночной хроматографии на силикагеле (циклогексан/этилацетат, 4:1). МетодD: В атмосфере азота в присутствии молекулярных сит (60 мг, активированные в вакууме при 200°С в течение 2 ч) растворяют подходящие альдегид (0,59 ммоль) и бензиламин (79 мкл, 0,59 ммоль) в MeCN (1,5 мл) и полученную смесь перемешивают в течение 1 ч. Альтернативно, получают имин 4 в MeCN, после чего отгоняют азеотропную смесь MeCN/H2O. Добавляют MeCN (2 мл), смесь перемешивают в течение 10 мин, растворитель упаривают. Эту процедуру повторяют еще и, в конце, добавляют MeCN (1,5 мл). Реакционную смесь охлаждают до -30°C и добавляют соединение 3 (R4=R5=Et) (0,59 ммоль) и катализатор (InCl3 или MgBr2, 0,29 ммоль), перемешивание продолжают в течение 1 часа. Нечищеную реакционную смесь обрабатывают MeOH/TMSCl в соответствии с вышеописанной методикой метанолиза и после кристаллизации или после колоночной флэш-хроматографии на силикагеле выделяют производные метилового эфира 1.
Метил-3-(бензиламино)-2-гидрокси-3-фенил-пропионат: диастереомерный избыток (d.e.) 83% (колоночная хроматография: этилацетат/циклогексан, 1:4). (2R*,3S*): 74% т.пл. 107°C (н-пентан/Et2O), (107-108°C)[9].1H ЯМР (CDCl3) δ 7,41-7,22 (м, 10H), 4,26, 3,95 (AX система, J 4,1, 2H), 3,77, 3,49 (AM система, J 13,2, 2H), 3,70 (с, 3H), 3,00-2,00 (уш, 2H, обмен), (2R*,3R*): 10%. Т.пл. 99°C (н-пентан/CH2Cl2), (98-99°C)[9].1H ЯМР (CDCl3) δ 7,38-7,22 (м, 10H), 4,54, 4,06 (AX система, J 4,0, 2H), 3,78, 3,61 (AM система, J 12,8, 2H), 3,60 (с, 3H), 3,00-2,00 (уш, 2H, обмен).
Метил-3-(бензиламино)-2-гидрокси-3-(4-нитрофенил)-пропионат: диастереомерный избыток (d.e.) 83% (колоночная хроматография: этилацетат/циклогексан, 1:3). (2R*,3S*): 74%. Т.пл. 147-149°C (н-гексан/CH2Cl2). ИК (KBr) vmax 3491, 1729 см-1;1H ЯМР (CDCl3) δ 8,23, 7,53 (AA'XX' система, J 8,8, 4H), 7,38-7,10 (м, 5H), 4,25, 4,06 (AM система, J 3,3, 2H), 3,76, 3,43 (AM система, J 13,5, 2H), 3,75 (с, 3H), 3,20-2,00 (уш.с, 2H, обмен).13C ЯМР (CDCl3) δ 173,4, 147,9, 147,6, 139,6, 128,9, 128,5, 128,3, 127,4, 123,9, 74,6, 62,9, 52,8, 50,8. Масс-спектр (ЭС, катионы) m/z 353,1.
Метил-3-(бензиламино)-2-гидрокси-3-(4-метоксифенил)-пропионат: диастереомерный избыток (d.e.) 83%. Колоночная хроматография (этилацетат/циклогексан, 1:3). (2R*,3S*): 72%. ИК (KBr) vmax 3491, 1733 см-1;1H ЯМР (CDCl3) δ 7,40-7,18 (м, 7H), 6,90 (д, J 8,8, 2H), 4,22, 3,89 (AM система, J 4,1, 2H), 3,82 (с, 3H), 3,75, 3,47 (AM система, J 13,2, 2H), 3,70 (с, 3H), 2,80-1,90 (уш.с, 2H, обмен).
Метил-3-(бензиламино)-2-гидрокси-3-(4-хлорфенил)-пропионат: диастереомерный избыток (d.e.) 82%. Колоночная хроматография (этилацетат/циклогексан, 1:4). (2R*,3S*): 73%. Т.пл. 106-108°C (н-гексан/CH2Cl2). ИК (KBr) vmax 3491, 1729 см-1;1H ЯМР (CDCl3) δ 7,40-7,10 (м, 9H), 4,21, 3,91 (AM система, J 3,6, 2H), 3,71 (с, 3H), 3,75, 3,45 (AM система, J 14,5, 2H), 3,70 (с, 3H), 2,80-1,90 (уш.с, 2H, обмен).
Метил-3-(бензиламино)-2-гидрокси-5-метил-гексаноат: диастереомерный избыток (d.e.) 87% (колоночная хроматография: Et2O/н-гексан, 1:4). (2R*,3S*): 68%. Т.пл. 89-90°C. ИК (NaCl) vmax 3467, 1739 см-1;lH ЯМР (CDCl3) δ 7,40-7,20 (м, 5H), 4,05 (д, J 2,2, 2H), 3,80-3,60 (2H, перекрыв.), 3,73 (с, 3H), 3,03 (дт, J 7,0, 1,8, 1H), 2,50-2,05 (уш, 2H, обмен), 1,80-1,60 (м, 1H), 1,50-1,20 (м, 2H), 0,92 (д, J 1,8, 3H), 0,86 (д, J 1,8, 3H);13C ЯМР (CDCl3) δ 175,3, 140,5, 128,5, 128,4, 127,3, 72,0, 57,4, 52,3, 52,0, 41,6, 25,3, 23,0, 22,7. Масс-спектр (ЭС, катионы) m/z 266,1.
Метил-3-(бензиламино)-2-гидрокси-3-тиофенил-пропионат: диастереомерный избыток (d.e.) 79% (колоночная хроматография: этилацетат/циклогексан, 1:4). (2R*,3S*): 73%. Масло. ИК (NaCl) vmax 3467, 1739 см-1;lH ЯМР (CDCl3) δ 7,38-7,20 (м, 6H), 7,03-6,95 (м, 2H), 4,33, 4,22 (AB система, J 3,7, 2H), 3,83, 3,55 (AM система, J 13,5, 2H), 3,74 (с, 3H), 3,00-2,00 (уш, 2H, обмен);13C ЯМР (CDCl3) δ 173,6, 143,6, 140,0, 128,5, 128,4, 127,3, 126,6, 126,0, 125,3, 75,2, 59,2, 52,7, 50,8. Масс-спектр (ЭС, катионы) m/z 314,1.
Список литературы


Реферат
Изобретение относится к способу получения соединений общей формулы 1, в которой Rпредставляет собой линейную или разветвленную C-Cалкильную группу, незамещенную или замещенную арильную или гетероарильную группу; Rпредставляет собой линейную или разветвленную C-Cалкильную группу, арилалкильную группу; Rпредставляет собой Н, C-Cалкильную группу. Способ осуществляют в соответствии с приведенной ниже схемой. Способ включает реакцию силильного эфира енола общей формулы 3, в которой Rпредставляет собой Me, Et, а Rпредставляет собой Me, Et, и имина общей формулы 4, в которой Rявляется линейной или разветвленной алкильной группой C-C, арилалкильной группой, незамещенной или замещенной арильной группой, гетероарильной группой, Rявляется линейной или разветвленной алкильной группой C-Cили арилалкильной группой. Далее способ включает алкоголиз или гидролиз полученных промежуточных соединений общей формулы 5 (3R*,5S*,6S*,1'S*) и 6 (3R*,5S*,6S*,1'R*), в которых Rпредставляет собой линейную или разветвленную C-Cалкильную группу, арилалкильную группу, незамещенную или замещенную арильную группу, гетероарильную группу; Rпредставляет собой линейную или разветвленную C-Cалкильную группу, арилалкильную группу; Rпредставляет собой Me или Et, с получением соединений формулы 1. Предлагаемый способ позволяет получать производные изосерина с хорошими выходами и высокой диастереоселективностью без выделения промежуточных соединений. 7 з.п. ф-лы, 22 пр.
Формула
в которой R1 представляет собой линейную или разветвленную Cl-C6 алкильную группу, незамещенную или замещенную арильную или гетероарильную группу; R2 представляет собой линейную или разветвленную Cl-C6 алкильную группу, арилалкильную группу; R3 представляет собой Н, Cl-C4 алкильную группу;
включающий:
- реакцию силильного эфира енола общей формулы 3, в которой R4 представляет собой Me, Et, а R5 представляет собой Me, Et, и имина общей формулы 4, в которой R1 является линейной или разветвленной алкильной группой Cl-C6, арилалкильной группой, незамещенной или замещенной арильной группой, гетероарильной группой, R2 является линейной или разветвленной алкильной группой Cl-C6 или арилалкильной группой;
- алкоголиз или гидролиз полученных промежуточных соединений общей формулы 5 (3R*,5S*,6S*,1'S*) и 6 (3R*,5S*,6S*,1'R*), в которых R1 представляет собой линейную или разветвленную Cl-C6алкильную группу, арилалкильную группу, незамещенную или замещенную арильную группу, гетероарильную группу; R2 представляет собой линейную или разветвленную Cl-C6 алкильную группу, арилалкильную группу; R4 представляет собой Me или Et

- получение in situ имина 4 из альдегида (R1CHO) и амина (R2NH2, предпочтительнее PhCH2NH2) в ацетонитриле в присутствии молекулярных сит при комнатной температуре или путем отгонки азеотропной смеси ацетонитрил/вода;
- добавление силильного производного 3 при -30°C с последующим добавлением катализатора;
- обработку неочищенной реакционной смеси раствором триметилсилилхлорида в спирте и выделение после кристаллизации чистого диастереомера 1 (2R*,3S*) (где R3 представляет собой С1-С4 алкильную группу).
Комментарии