Кобальтовые катализаторы - RU2252072C2
Код документа: RU2252072C2
Чертежи








Описание
Настоящее изобретение имеет отношение к созданию кобальтовых катализаторов. Более конкретно, оно имеет отношение к созданию предшественника кобальтового катализатора, способа приготовления предшественника кобальтового катализатора и способа приготовления кобальтового катализатора.
Способы приготовления кобальтовых катализаторов широко известны. Например, в патенте США No.5733839 описан способ приготовления пропитанного катализатора Фишера-Тропша, который имеет носитель из оксида алюминия и активный компонент, выбранный из группы, в которую входят кобальт, железо и их смеси.
Задачей настоящего изобретения является создание поддерживаемого кобальтового катализатора (кобальтового катализатора на носителе), который имеет более высокую производительность, чем известные кобальтовые катализаторы.
В соответствии с первым аспектом настоящего изобретения предлагается предшественник кобальтового катализатора, который содержит носитель катализатора, пропитанный кобальтом, вместе со всем восстановимым кобальтом, который присутствует в носителе как поддерживаемый оксид кобальта в соответствии с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0.
Другими словами, в соответствии с первым аспектом настоящего изобретения, предлагается предшественник кобальтового катализатора, который содержит носитель катализатора, который был пропитан кобальтом и прокален, таким образом, что весь присутствующий в нем восстановимый кобальт, то есть кобальт, который объединен с такими элементами, как водород и кислород, в отсутствие поддерживаемого кобальтом взаимодействия, такого как образование алюминатов кобальта или силикатов кобальта, которые могли бы снизить его способность к восстановлению, присутствует в носителе как поддерживаемый оксид кобальта в соответствии с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0.
Таким образом, например, весь восстановимый кобальт может присутствовать в виде Со2О3·Н2 О или СоО(ОН), то есть при а=2 и b=1. Однако вместо этого весь восстановимый кобальт может также присутствовать, например, в виде смеси Со3O4 с СоО(ОН) или Со2О3·Н2О, при 45% восстановимого кобальта в предшественнике катализатора, который присутствует как Co3O4, и 55% восстановимого кобальта, который присутствует как СоО(ОН) или Со2О3·Н2О. Это могло бы привести к тому, что в предшественнике катализатора весь восстановимый кобальт присутствует как поддерживаемый оксид кобальта в соответствии с блочной формулой СоОaНb, в которой а=1,7 и b=0,55. Другим примером является смесь Со2О3 с СоО(ОН) или Co2O3·H2O, с 60% восстановимого кобальта, который присутствует как Со2О3, и с 40% восстановимого кобальта, который присутствует как СоО(ОН) или Co2O3·H2O. Это могло бы привести к тому, что в предшественнике катализатора весь восстановимый кобальт присутствует как поддерживаемый оксид кобальта в соответствии с блочной формулой СоОaНb, в которой а=1,7 и b=0,4.
Предшественник катализатора может содержать от 5 г Со на 100 г носителя до 70 г Со на 100 г носителя, преимущественно от 20 г Со на 100 г носителя до 50 г Со на 100 г носителя, а еще лучше от 25 г Со на 100 г носителя до 40 г Со на 100 г носителя.
В соответствии со вторым аспектом настоящего изобретения предлагается способ приготовления предшественника кобальтового катализатора, который включает в себя следующие операции:
пропитка порошкового пористого носителя катализатора солью кобальта и частичная сушка пропитанного носителя и
прокаливание частично высушенного пропитанного носителя для получения предшественника кобальтового катализатора, причем прокаливание производят при условиях прокаливания, выбранных таким образом, что весь восстановимый кобальт присутствует в носителе как поддерживаемый оксид кобальта в соответствии с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0.
Прокаливание ведут путем нагревания носителя и пропускания горячего воздуха до температуры от 95° С до 400° С, причем управляют скоростью нагревания так, чтобы вначале удалялась остаточная влага, а затем происходило разложение соли кобальта на продукты, содержащие оксиды и любую воду гидратации.
Соль кобальта представляет собой нитрат кобальта, так что оксиды, которые образуются в качестве продуктов разложения, представляют собой оксиды азота, причем прокаливание проводят в кальцинаторе с псевдоожиженным слоем, при этом после прокаливания концентрация азота в предшественнике катализатора составляет менее чем 1,0 вес. %.
Объемная скорость воздуха должна составлять по меньшей мере 1,0 м3n на 1 кг Со(NO3)2·6Н2О в час, причем скорость нагревания соответствует следующим критериям: когда объемная скорость воздуха равна 1,0 м3n на 1 кг Со(NО3)2·6Н2О в час, то скорость нагревания составляет ≤ 1° С /мин; однако когда объемная скорость воздух превышает 1,0 м3n на 1 кг Со(NО3)2·6Н2О в час, то допустимая скорость нагревания возрастает до х° С /мин, где x≥ 1.
На стадии прокаливания может осуществляться первоначальное нагревание пропитанного носителя, пока он не достигнет температуры прокаливания Тc, после чего поддерживают его при температуре прокаливания Тc в течение периода времени tc. Скорость нагревания до температуры прокаливания Тc является нелинейной. Период времени tc, в течение которого проводят изотермическое прокаливание при температуре прокаливания Тc, составляет от 0,1 до 20 часов.
Частично высушенный пропитанный носитель из стадии пропитки носителя не хранят, а также не нагревают или не охлаждают ранее последующей стадии прокаливания с псевдоожиженным слоем, так что его сразу же направляют на стадию прокаливания с псевдоожиженным слоем.
В соответствии с настоящим изобретением могут быть использованы любые имеющиеся в продаже пористые оксидные носители катализатора, такие как оксид алюминия (Аl2О3), диоксид кремния (SiO2), диоксид титана (TiO2), оксид магния (MgO) и смесь диоксида кремния с оксидом алюминия. Носитель преимущественно имеет средний диаметр пор от 8 до 50 нм, а преимущественно от 10 до 15 нм. Объем пор носителя может составлять от 0,1 до 1,0 мл/г, а преимущественно от 0,3 до 0,9 мл/г. Средний размер частиц составляет от 1 до 500 мкм, преимущественно от 10 до 250 мкм, а еще лучше от 45 до 200 мкм.
В качестве носителя может быть использован защищенный модифицированный носитель катализатора, который содержит, например, кремний в качестве модифицирующего компонента, как это показано в публикации WO 99/42214.
Пропитка носителя катализатора в принципе может быть осуществлена при помощи любого известного способа или процедуры пропитки, такой как пропитка с зачаточной влажностью или пропитка в фазе суспензии. Однако стадия пропитки может, в частности, предусматривать использование такого способа, который описан в публикации WO 00/20116. При этом стадия пропитки носителя может предусматривать использование способа пропитки в фазе суспензии с двумя операциями, характеристики которого зависят от требования желательной загрузки кобальта и от объема пор носителя катализатора.
В ходе любой из двух операций пропитки в фазе суспензии может быть добавлен растворимый в воде предшественник соли палладия (Pd), платины (Pt), рутения (Ru) или их смеси, в качестве легирующей примеси, способной усилить восстановимость кобальта. Массовая пропорция палладия, платины, рутения или комбинированной смеси указанных металлов, если она используется, к металлическому кобальту может составлять от 0,01:100 до 0, 3:100.
Пропитку и сушку носителя обычно проводят в конической вакуумной сушилке с вращающимся шнеком или в поворотной вакуумной сушилке.
На стадии прокаливания прокаливание может предусматривать пропускание горячего воздуха над частично высушенным носителем и вокруг него, что приводит к дополнительной сушке пропитанного носителя за счет удаления присутствующей в нем остаточной влаги; и прокаливание полученного в результате сухого пропитанного носителя, за счет чего происходит разложение соли кобальта на продукты разложения, содержащие оксид(ы) и любую воду гидратации, причем продукты разложения выделяются в виде пара, что способствует формированию поддерживаемого предшественника катализатора, который содержит поддерживаемый оксид кобальта в соответствии с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0.
В частности, так как соль кобальта представляет собой нитрат кобальта, оксид(ы) представляют собой диоксид азота, при равновесии между NO2 и N2O4, а также оксид азота (NO).
Процесс может предусматривать разбавление продуктов разложения, которые получают в ходе прокаливания. Другими словами, образование поддерживаемого оксида кобальта в соответствии с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0, усиливается за счет разбавления продуктов разложения в ходе прокаливания.
Присутствие фазы поддерживаемого оксида кобальта в соответствии с блочной формулой CoOaHb, в которой а≥ 1,7 и b>0, может быть задано за счет использования программируемого снижения температуры (ПСТ) в качестве определяющей техники.
Минимальной температурой, при которой проводят прокаливание, является такая температура, при которой начинается разложение предшественника кобальта, то есть соли кобальта, в то время как максимальной температурой прокаливания является температура, при которой фаза преимущественного поддерживаемого оксида кобальта в соответствии с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0, преобразуется в нежелательную фазу шпинели Со3О4.
Прокаливание может быть проведено при помощи любого известного оборудования для прокаливания, такого как кальцинатор с псевдоожиженным слоем, барабанная печь, а также кальцинатор или печь с насыпным слоем.
В частности, прокаливание может быть проведено в кальцинаторе с псевдоожиженным слоем, причем преимущественно прокаливание осуществляют на воздухе, при температурах от 95° С до 400° С. Минимальной температурой прокаливания является такая температура, при которой начинается разложение нитрата, то есть температура около 120° С, в то время как максимальной температурой прокаливания является такая температура, при которой преимущественный оксид кобальта в соответствии с блочной формулой СоОaНb преобразуется в нежелательную фазу шпинели Со3О4. Максимальная температура прокаливания обычно составляет от 200° С до 300° С. После прокаливания концентрация азота в предшественнике катализатора преимущественно составляет менее 1,0 вес. %.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения проводят удаление оксида (оксидов) азота и воды в ходе прокаливания в воздухе для усиления стабилизации фазы поддерживаемого оксида кобальта в соответствии с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0.
В ходе прокаливания в псевдоожиженном слое можно управлять скоростью нагревания пропитанного и высушенного носителя и объемной скоростью воздуха таким образом, чтобы в первую очередь удалялась остаточная влага в носителе, а затем происходило разложение нитрата кобальта.
Преимущественно используют минимальную объемную скорость воздуха, чтобы достичь максимальной относительной внутренней активности кобальтового катализатора по отношению к полученному в результате катализатору; однако с минимальной объемной скоростью объединяют максимальную скорость нагревания. В частности, заявитель обнаружил, что для конкретного пропитанного нитратом кобальта носителя существует минимальная объемная скорость воздуха 1,0 м3n на 1 кг Со(NO3)2·6Н2О в час, которая должна быть использована для того, чтобы достичь максимальной относительной внутренней активности кобальтового катализатора. Если использована минимальная объемная скорость воздуха менее 1,0 м3n на 1 кг Co(NO3)2·6Н2О в час, то не удается достичь максимальной относительной внутренней активности результирующего кобальтового катализатора. Однако заявитель также обнаружил, что тогда максимальная скорость нагрева 1° С/мин, преимущественно 0,5° С/мин, а обычно от 0,1 до 0,5° С/мин, может быть использована в сочетании с минимальной объемной скоростью воздуха, равной 1,0 м3n на 1 кг Со (NO3)2·6Н2О в час. Другими словами, если скорость нагрева носителя превышает 1°С/мин при поддержании минимальной объемной скорости, равной 1,0 м3n на 1 кг Со (NO3)2·6Н2О в час, то это может оказывать вредное влияние на начальную активность катализатора.
Однако если используют объемную скорость, превышающую 1,0 м3n на 1 кг Со(NО3)2·6Н2О в час, например объемную скорость 100 м3n на 1 кг Со(NO3O)2·6Н2О в час, то при этом может быть использована объединенная с ней более высокая максимальная или пороговая скорость нагрева, а именно 100° С/мин. Таким образом, может быть применено мгновенное прокаливание при использовании достаточно высоких объемных скоростей.
Способ может включать в себя на стадии прокаливания первоначальное нагревание пропитанного носителя, пока он не достигнет температуры прокаливания Тc. Таким образом, нагревание носителя до температуры прокаливания Тc может быть осуществлено при одновременном поддержании объемной скорости по меньшей мере 1,0 м3n на 1 кг Со (NO3)2·6H2O в час и соответствующей скорости нагревания слоя, объединенной с объемной скоростью, как это было описано ранее. Способ может также включать в себя после этого поддержание температуры прокаливания носителя Тc в течение периода времени tc.
Управление скоростью нагревания может осуществляться за счет управления предварительными нагревателями питающего газа (воздуха), через которые проходит воздух, необходимый для флюидизации (создания псевдоожиженного слоя) и прокаливания, и/или за счет управления температурой стенки кальцинатора. В то время как нагревание до температуры Тc идет линейно (с постоянной скоростью), можно полагать, что усиление активности может быть достигнуто при нелинейной скорости нагревания, что позволяет подстроиться к профилям освобождения (выхода) оксида (оксидов) азота и воды.
Период времени tc, в течение которого проводят изотермическое прокаливание при температуре прокаливания Тc, может составлять от 0,1 до 20 часов, при условии, что содержание азота в прокаливаемом катализаторе составляет менее 1,0 вес. %.
Частично высушенный пропитанный носитель из стадии пропитки носителя преимущественно не хранят, а также не нагревают или не охлаждают ранее последующей стадии прокаливания с псевдоожиженным слоем, так что его (сразу же) направляют на стадию прокаливания с псевдоожиженным слоем, главным образом при такой же температуре, при которой он выходит из стадии пропитки носителя. Таким образом, частично высушенный пропитанный носитель выходит из стадии пропитки носителя при температуре от 60° С до 95° С, а обычно около 75° С, и поступает на стадию прокаливания с псевдоожиженным слоем ориентировочно при такой же температуре, без хранения носителя между двумя стадиями. Стадию прокаливания с псевдоожиженным слоем преимущественно проводят при помощи кальцинатора с псевдоожиженным слоем, который непосредственно подключен к вакуумной сушилке.
Псевдоожиженная среда, которую используют в кальцинаторе, представляет собой, воздух, который требуется для прокаливания, причем линейная скорость воздуха через кальцинатор, естественно, должна быть достаточной для обеспечения надлежащей флюидизации.
Настоящее изобретение имеет также отношение к созданию катализатора, который получен при помощи способа в соответствии с третьим аспектом настоящего изобретения или получен за счет восстановления предшественника катализатора в соответствии с первым аспектом настоящего изобретения или предшественника катализатора, полученного при помощи способа в соответствии со вторым аспектом настоящего изобретения.
Предложенный способ особенно хорошо подходит для приготовления кобальтового катализатора Фишера-Тропша в фазе суспензии, то есть такого катализатора, который подходит для ускорения конверсии синтез-газа, содержащего оксид углерода и водород, в углеводородные продукты, проводимой при повышенных температуре и давлении.
Указанные ранее и другие характеристики изобретения будут более ясны из последующего детального описания примеров, не имеющего ограничительного характера и приведенного со ссылкой на сопроводительные чертежи.
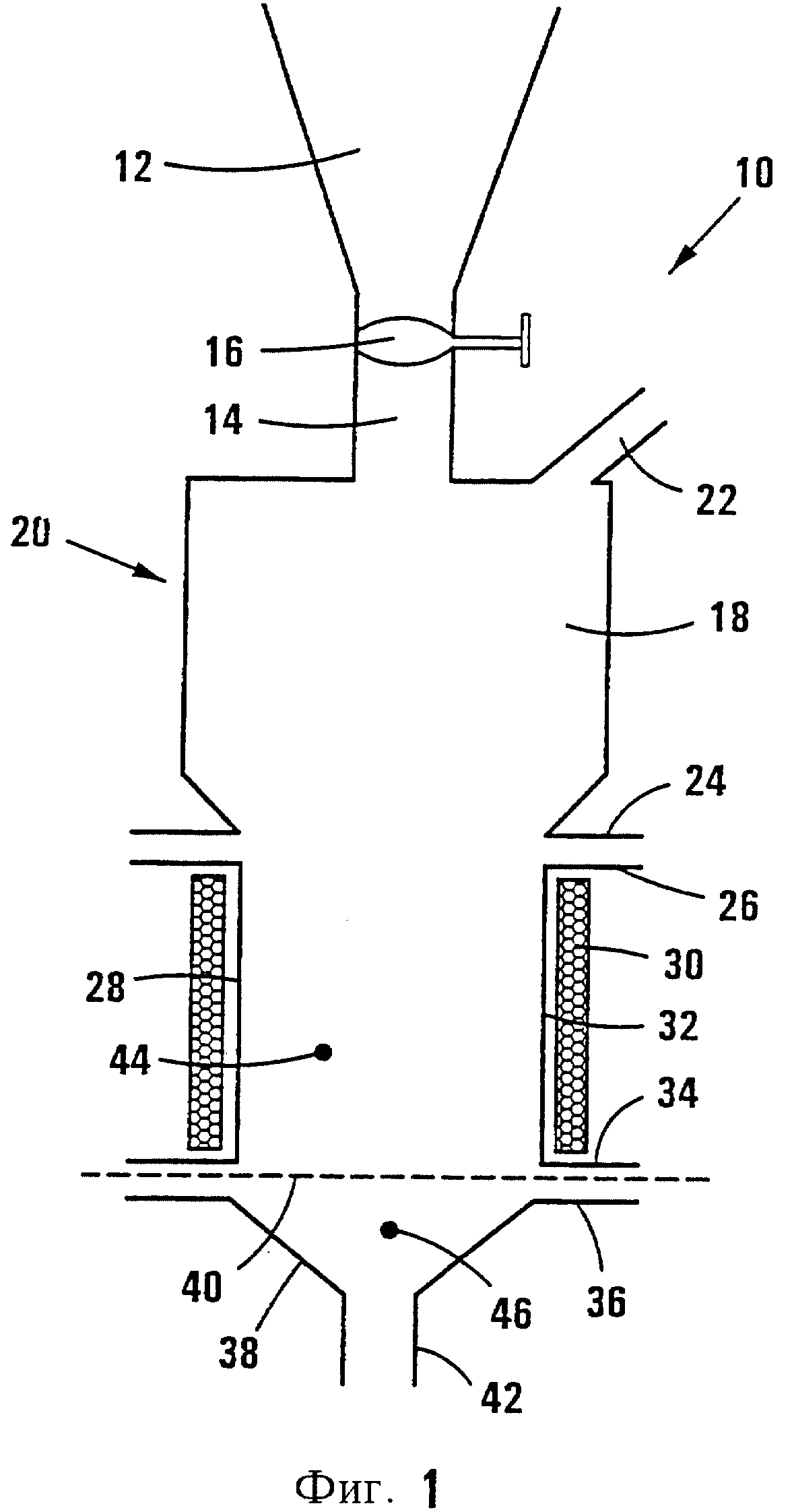
На фиг.1 показана схематически установка, которую используют для приготовления катализатора Примера 1, на стадии опытного производства.
На фиг.2 показана картина программируемого снижения температуры (ПСТ) катализатора Р. ПСТ эксперимент был осуществлен с использованием скорости нагревания 10° С/мин и смеси водорода с аргоном, содержащей 10% водорода.
На фиг.3 показана картина программируемого снижения температуры катализатора Н. ПСТ эксперимент был осуществлен с использованием скорости нагревания 10° С/мин и смеси водорода с аргоном, содержащей 10% водорода.
На фиг.4 показаны относительные внутренние специфические активности катализаторов Фишера-Тропша в соответствии с Примерами 1, 2 и 8, в функции объемной скорости, в ходе их прокаливания в псевдоожиженном слое.
На фиг.5 показаны относительные внутренние специфические активности катализаторов Фишера-Тропша в соответствии с Примерами 1, 2 и 8, в функции скорости нагревания, в ходе их прокаливания в псевдоожиженном слое.
На фиг.6 показаны области производительности нежелательного, предпочтительного и наиболее предпочтительного катализаторов Фишера-Тропша, в функции объемной скорости воздуха и скорости нагревания, в ходе прокаливания в псевдоожиженном слое.
На фиг.7 и 8 показаны результаты термогравиметрических анализов (TGA) катализатора В Примера 1, после пропитки и вакуумной сушки носителя, но перед прокаливанием на воздухе, причем:
на фиг.7 показаны потери в массе катализатора В в функции времени, при использовании скорости нагревания 0,40° С/мин и времени выдержки при температуре 250° С, составляющем 6 часов;
на фиг.8 показаны потери в массе катализатора В в функции времени, при использовании скорости нагревания 5° С/мин от 30° С до 75° С и скорости нагревания 0,4° С/мин от 75° С до 130° С, а затем времени выдержки 4 часа при температуре 130° С, скорости нагревания 0,17° С/мин от 130° С до 160° С и скорости нагревания 0, 7° С/мин от 160° С до 250° С, а затем времени выдержки 4 часа при температуре 250° С.
На фиг.9 показана картина программируемого снижения температуры катализатора Q. ПСТ эксперимент был осуществлен с использованием скорости нагревания 10° С/мин и смеси водорода с аргоном, содержащей 10% водорода.
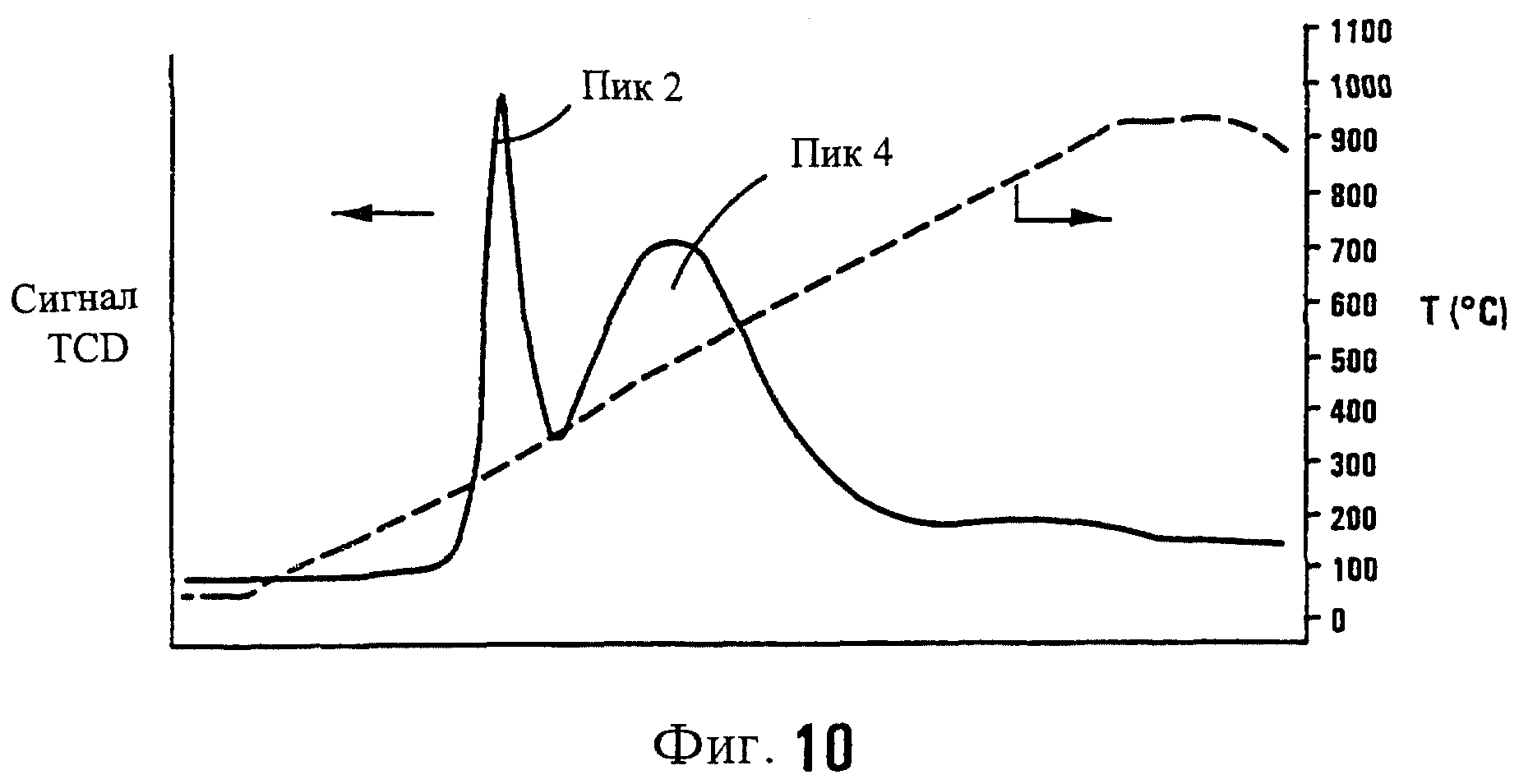
На фиг.10 показана картина программируемого снижения температуры катализатора R. ПСТ эксперимент был осуществлен с использованием скорости нагревания 10° С/мин и смеси водорода с аргоном, содержащей 10% водорода.
ПРИМЕР 1
На опытной установке, показанной на фиг.1, была приготовлена серия кобальтовых на базе оксида алюминия катализаторов Фишера-Тропша, названных А, В, С, D, Е, G, Н и I. Указанные катализаторы были приготовлены с использованием кальцинатора с псевдоожиженным слоем. Катализаторы D, Е, G, Н и I выполнены в соответствии с настоящим изобретением, а катализаторы А, В и С приведены для сравнения.
На фиг.1 позицией 10 обозначена в целом опытная установка, которая была использована для приготовления катализаторов Фишера-Тропша. Установка 10 содержит коническую вакуумную сушилку 12 объемом 100 дм3, имеющую канал загрузки 14, снабженный клапаном 16. Указанный канал 14 непосредственно соединен с секцией отделения (разделения) 18 кальцинатора с псевдоожиженным слоем, который обозначен в целом позицией 20.
Кальцинатор 20 содержит вентиляционный канал 22, ведущий из секции 18 в скруббер (газоочиститель) (не показан). Секция 18 соединена при помощи фланцев 24, 26 с трубчатым компонентом 28, который образует цилиндрическую стенку кальцинатора с внутренним диаметром 350 мм и высотой 655 мм. Нагревательный элемент 30 охватывает компонент 28, причем предусмотрен датчик и контроллер температуры 32, установленный между элементом 30 и компонентом 28.
У своего нижнего конца компонент 28 имеет фланец 34, соединенный с фланцем 36 компонента в виде воронки 38. Между фланцами 34, 36 установлен фильтр 40, который перекрывает выход компонента 28. Компонент 38 имеет впускной воздушный канал 42, через который подогретый воздух поступает в кальцинатор.
Датчик температуры 44, который установлен в канале компонента 28, служит для измерения температуры внутри псевдоожиженного слоя (не показан) сухого порошкового пропитанного материала носителя, который образован на фильтре 40. Датчик температуры 44 установлен над фильтром 40 на расстоянии около 35 мм. Аналогичный датчик температуры 46 установлен под фильтром 40.
В установке 10 вакуумная сушилка 12, которая использована для пропитки и вакуумной сушки носителя, непосредственно соединена с кальцинатором с псевдоожиженным слоем 20.
Катализаторы были приготовлены при помощи процедуры пропитки в фазе суспензии и вакуумной сушки. Эта процедура была идентичной для всех катализаторов в этом Примере.
В каждом случае, в первой операции пропитки и прокаливания, производят перемешивание раствора 17,4 кг Со(NO3)2·6H2O, 9,6 г (NH3)4 Pt(NO3)2 и 11 кг дистиллированной воды с 20,0 кг носителя из γ оксида алюминия (Puralox SCCa 5/150, объем пор 0,48 мл/г, производство фирмы Condea Chemie GmbH Uberseering 40, 22297 Hamburg, Germany) путем добавления носителя в раствор. Полученную в результате
суспензию вводили в коническую вакуумную сушилку 12 и непрерывно перемешивали. Температуру указанной суспензии повышали до 60° С и затем создавали вакуум 20 kПа(а). В течение первых 3 часов операции сушки температура медленно возрастала и достигала 95° С через 3 часа. Через 3 часа вакуум снижали до 3-15 kПa(a). Пропитанный носитель катализатора сушили в течение 9 часов, после чего указанный пропитанный носитель катализатора был немедленно и непосредственно загружен в кальцинатор с псевдоожиженным слоем 20. Температура сухого пропитанного носителя катализатора в момент загрузки в кальцинатор составляла около 75° С. Время загрузки составляет ориентировочно 1-2 минуты, поэтому температура внутри кальцинатора сохранялась равной точке уставки и составляла около 75° С. Для получения катализатора с содержанием кобальта 30 г Со на 100 г Al2O3 проводили вторую операцию пропитки и сушки. Для этого перемешивали раствор 9,4 кг Со(NO3)2·6Н2О, 15,7 г (NH3)4 Pt(NO3)2 и 15,1 кг дистиллированной воды с 20,0 кг пропитанного и прокаленного материала из первой операции пропитки и прокаливания путем добавления этого твердого промежуточного материала в раствор. Полученную в результате суспензию вводили в коническую вакуумную сушилку и непрерывно перемешивали. Температуру указанной суспензии повышали до 60° С и затем создавали вакуум 20 kПа(а). В течение первых 3 часов операции сушки температура медленно возрастала и достигала 95° С через 3 часа. Через 3 часа вакуум снижали до 3-15 kПa(a). Полученный в результате пропитанный носитель катализатора сушили в течение 9 часов, после чего указанный пропитанный носитель катализатора был немедленно и непосредственно загружен в кальцинатор с псевдоожиженным слоем. Температура сухого пропитанного носителя катализатора в момент загрузки в кальцинатор составляла около 75° С. Время загрузки составляет ориентировочно 1-2 минуты, поэтому температура внутри кальцинатора сохранялась равной его точке уставки и составляла около 75° С.
Объемную скорость воздуха в кальцинаторе и скорость нагревания пропитанного и высушенного материала в диапазоне от 75° С до 250° С изменяли в попытке получения катализатора с максимальной относительной начальной внутренней активностью. Время выдержки 6 часов при температуре 250° С сохраняли постоянным для всех операций приготовления катализаторов. Температуру псевдоожиженного слоя контролировали (компонент 44 на фиг.1) в течение каждого цикла прокаливания.
Условия, использованные в ходе прямого прокаливания в псевдоожиженном слое пропитанного и высушенного материала, указаны в Таблице 1.

Содержание азота в каждом из приготовленных на опытной установке катализаторов было меньше или равно 0,5 вес. %, как это показано в Таблице 2.
ПРИМЕР 2
На лабораторной установке была приготовлена серия кобальтовых на базе оксида алюминия катализаторов Фишера-Тропша, названных К, L, М и О, при использовании таких же процедур, как и при приготовлении катализаторов на опытной установке, описанном в Примере 1, но начиная с 50 г носителя вместо 20 кг. Количество других химикалий также было понижено в отношении 0,05/20. Указанные катализаторы были также приготовлены с использованием прокаливания в псевдоожиженном слое. Приготовление указанных катализаторов производили на лабораторной установке, аналогичной установке 10 фиг.1. Катализаторы L и М выполнены в соответствии с настоящим изобретением, а катализаторы К и О приведены для сравнения.
Условия, использованные в ходе прокаливания в псевдоожиженном слое носителей катализаторов, приготовленных на лабораторной установке, указаны в Таблице 3.

ПРИМЕР 3
При помощи пропитки суспензии был приготовлен кобальтовый катализатор Р, который не соответствует настоящему изобретению. В первой операции пропитки и прокаливания перемешивали раствор 43,68 г Co(NO3)2·6Н2О, 0,024 г (NH3)4 Pt(NO3)2 и 50 мл дистиллированной воды с 50 г носителя из γ оксида алюминия (Puralox SCCa 5/150, объем пор 0,48 мл/г, производство фирмы Condea Chemie GmbH Uberseering 40, 22297 Hamburg, Germany) путем добавления носителя в раствор. Полученную в результате суспензию вводили во вращательный испаритель (rotarvap) и непрерывно перемешивали. Температуру указанной суспензии повышали до 60° С и затем создавали вакуум 25 kПa(a). В течение первых 3 часов операции сушки температура медленно возрастала и достигала 95° С через 3 часа. Через 3 часа вакуум снижали до 5 kПа(а). Пропитанный носитель катализатора сушили в течение 9 часов, после чего указанный носитель катализатора был немедленно прокален. Прокаливание было проведено в статической обжиговой печи при температуре 250° С, без протекания воздуха над образцом. Для получения катализатора с содержанием кобальта 30 г Со на 100 г Al2O3 была проведена вторая операция пропитки и прокаливания. Раствор 23,51 кг Co(NO3)2·6Н2О, 0,039 г (NH3)4 Pt(NO3)2 и 50 мл дистиллированной воды перемешивали с 50 г пропитанного и прокаленного материала из первой операции пропитки и прокаливания путем добавления этого твердого промежуточного материала в раствор. Полученную суспензию вводили во вращательный испаритель и непрерывно перемешивали.
Температуру указанной суспензии повышали до 60° С и затем создавали вакуум 25 kПа(а). В течение первых 3 часов операции сушки температура медленно возрастала и достигала 95° С через 3 часа. Через 3 часа вакуум снижали до 5 kПа(а). Полученный в результате пропитанный носитель катализатора сушили в течение 9 часов, после чего указанный носитель катализатора был немедленно прокален. Прокаливание было проведено в статической обжиговой печи при температуре 250° С, без протекания воздуха над образцом.
ПРИМЕР 4
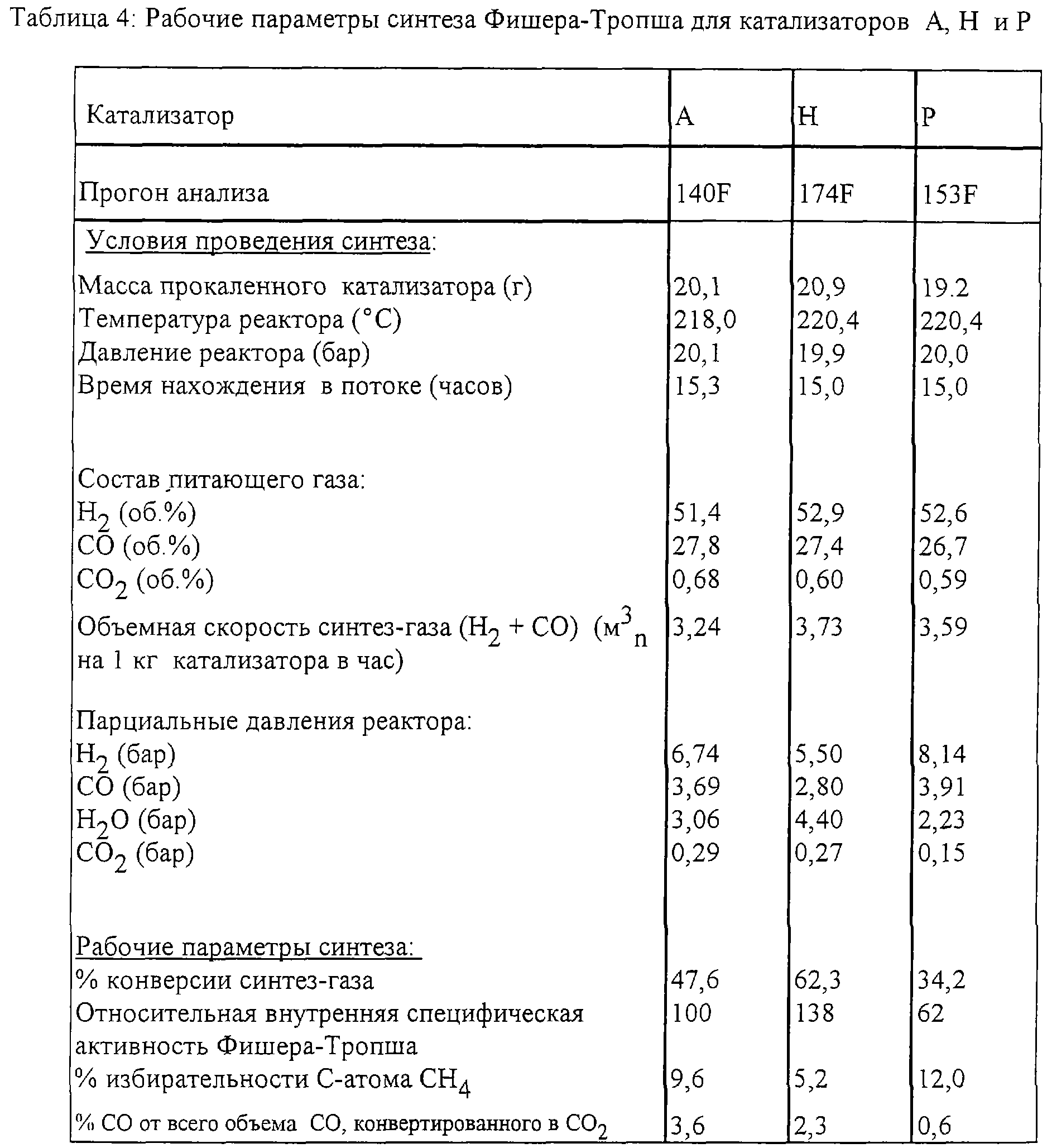
Катализаторы А, Н и Р были испытаны при проведении синтеза Фишера-Тропша. Экспериментальные данные, а также условия проведения синтеза Фишера-Тропша приведены в Таблице 4.
Ранее проведения синтеза в лабораторном микрореакторе суспензии Фишера-Тропша при реальных условиях проведения синтеза Фишера-Тропша прокаленные предшественники катализаторов были восстановлены в лаборатории с использованием стандартной лабораторной методики восстановления (скорость нагревания 1° С/мин от 25 до 425° С, выдержка 16 часов при 425° С, GHSV 200 млn водорода на 1 г катализатора в час, 1 бар чистого водорода), после чего проводилась выгрузка и покрытие воском Фишера-Тропша.
При применении известного кинетического выражения Фишера-Тропша к кобальту:
rFT=(kFTPH2PCO )/(1+KPСО)2
получим производный предварительный экспоненциальный коэффициент Аррениуса AFT (то есть АFT=kFT/(е-Ea|RT)) для каждого из проведенных прогонов.
Относительная внутренняя специфическая активность Фишера-Тропша может быть определена как ((предварительный экспоненциальный коэффициент катализатора х) / (предварительный экспоненциальный коэффициент катализатора А))* 100, где катализатор х представляет собой катализатор специфических примеров, то есть катализаторы Н и Р.
Эксперименты на Программируемое Снижение Температуры (ПСТ) были проведены на катализаторах Н и Р (фиг.2 и 3) с использованием скорости нагревания 10° С/мин и смеси водорода с аргоном, содержащей 10 об. % водорода.
Результаты синтеза Фишера-Тропша показывают, что катализатор Н является намного более активным (так как он имеет относительный внутренний коэффициент FT, равный 138), чем катализатор Р (который имеет относительный внутренний коэффициент FT, равный 62). Картины ПСТ также показывают четкие различия между указанными двумя катализаторами. Эти различия можно типизировать по следующим параметрам:
1. Отношение высоты пиков, для пика 2 и пика 4.
2. Ширина пика 2 на половине высоты.
3. Присутствие или отсутствие пика 3.
Картина ПСТ катализатора Н является типичной для поддерживаемого кобальтового катализатора, который содержит восстановимый оксид кобальта в соответствии с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0, в то время как ПСТ картина катализатора Р говорит о присутствии нежелательной фазы шпинели Со3О4.
Отсюда можно прийти к выводу о том, что катализатор, который был надлежащим образом прокален, будет иметь ПСТ картину, типичную для желательной фазы оксида кобальта с блочной формулой СоОaНb, в которой а≥ 1,7 и b>0, а также желательную высокую относительную внутреннюю активность Фишера-Тропша.
ПРИМЕР 5
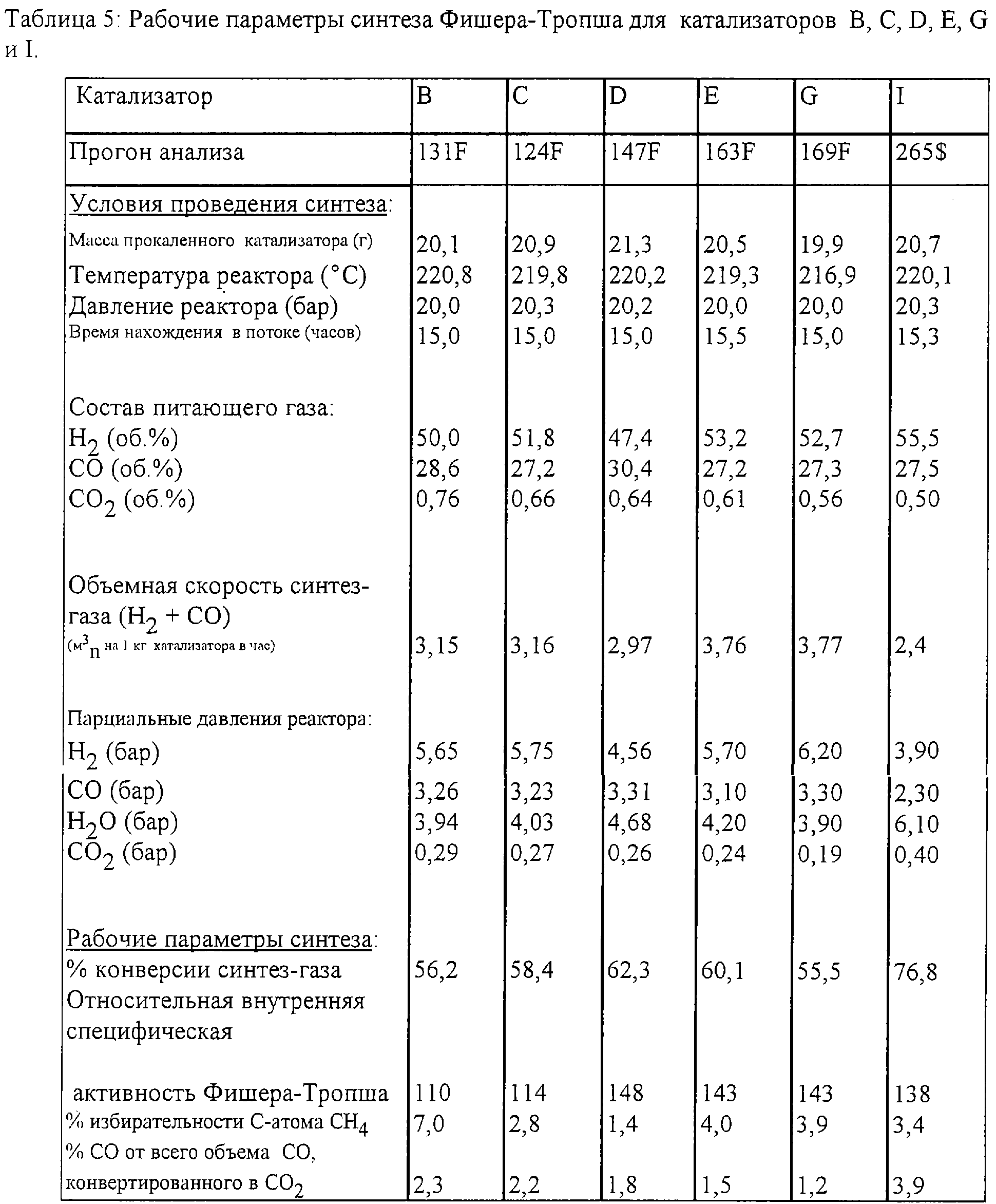
Все катализаторы А, В, С, D, E, G, Н, К, L, М и О, которые были приготовлены с использованием прокаливания в псевдоожиженном слое, были испытаны при проведении синтеза Фишера-Тропша. Экспериментальные данные, а также условия проведения синтеза Фишера-Тропша приведены в Таблицах 5 и 6.
Катализаторы были восстановлены и испытаны аналогично описанному в Примере 4. Относительные внутренние коэффициенты активности Фишера-Тропша также были вычислены в соответствии с описанным в Примере 4.
Относительные внутренние специфические активности Фишера-Тропша всех катализаторов были нанесены на график в функции объемной скорости в ходе прокаливания в псевдоожиженном слое (фиг.4). Активности катализаторов А, В, С, D, Е, G, Н, К, L и М возрастают линейно до относительной внутренней специфической активности Фишера-Тропша, составляющей около 140, при объемной скорости 1,02 м3n на 1 кг Со (NO3)2·6Н2О в час, после чего можно ожидать сохранения уровня относительной внутренней активности Фишера-Тропша около 140.
Можно полагать, что положительное влияние объемной скорости на наличие предпочтительной прокаленной фазы оксида кобальта и на желательные параметры катализатора вызвано тем фактом, что при повышенных объемных скоростях снижаются уровни максимальной концентрации NO и воды вокруг частиц катализатора в ходе прокаливания.
Относительные внутренние специфические активности Фишера-Тропша для катализаторов А, В, С, D, Е, G, Н, I, К, L, М и О также были нанесены на график в функции скорости нагревания в ходе прокаливания в псевдоожиженном слое (фиг.5). Можно прийти к заключению о том, что при постоянной объемной скорости воздуха внутренняя активность катализатора Фишера-Тропша не зависит от вариаций скорости нагревания в диапазоне от 0,1° С до 0,5° С/мин. Оказалось, что при более высоких скоростях нагревания начальная внутренняя активность может падать (например, для катализатора О), как это показано штриховыми экстраполяциями. Можно полагать, что это вызвано возрастанием концентраций NOx и воды вокруг частиц катализатора за счет быстрого разложения нитратов кобальта при высоких скоростях нагревания. На фиг.5 показана начальная (через 15 часов нахождения в потоке при реальных условиях синтеза) внутренняя FT активность катализатора I. Катализатор I показывает, что если объемная скорость является очень высокой, например 1000 м3n на 1 кг Со (NO3)2·6Н2О в час, то можно допустить мгновенное прокаливание, то есть скорость нагревания 100° С/мин.
Как это показано на фиг.6, правильный выбор сочетания скоростей нагревания с объемными скоростями воздуха позволяет получить катализатор с желательной внутренней активностью Фишера-Тропша.

ПРИМЕР 6
Разложение объема нитрата кобальта, то есть освобождение объема NOx, происходит при температуре от 130° С до 160° С, как это показано на фиг.7. Для подавления максимального пика NOx, то есть для усиления начальной активности, была проведена подстройка к данному профилю освобождения NOx, то есть была исследована возможность использования нелинейной скорости нагревания, целями которого являются максимальное удаление остаточной влаги и кристаллизационной воды ранее достижения ориентировочно 130° С, с последующим сглаживанием выделения NOx в диапазоне от 130° С до 160° С. Имитация указанного была проведена при помощи TGA эксперимента с использованием выдержки при 130° С, низкой скорости нагревания от 130° С до 160° С и высокой скорости нагревания от 160° С до 250° С, как это показано на фиг.8. Нашли, что в этом случае выделение NOx продолжается в течение значительно более длительного периода времени, за счет чего максимальная концентрация NOx вокруг катализатора снижается.
ПРИМЕР 7
Катализатор Q был приготовлен аналогично катализатору I Примера 1. ПСТ картина для катализатора Q показана на фиг.9.
Часть образца этого катализатора Q была дополнительно прокалена при 450° С, то есть при более высокой температуре, чем предлагается в соответствии с настоящим изобретением. Это было сделано для того, чтобы убедиться в наличии Со3О4 в этом конкретном образце катализатора R. ПСТ картина для катализатора R показана на фиг.10.
Из относительного потребления водорода в стадии восстановления, которое рассчитано из площадей под пиками 1, 2 и 4 фиг.9 и 10, можно прийти к заключению о том, что катализатор R, который был приготовлен не в соответствии с настоящим изобретением, главным образом содержит Со3О4. В отличие от этого катализатор Q, который был приготовлен в соответствии с настоящим изобретением, преимущественно содержит СоО(ОН). Это видно также из Таблицы 7.
ПРИМЕР 8
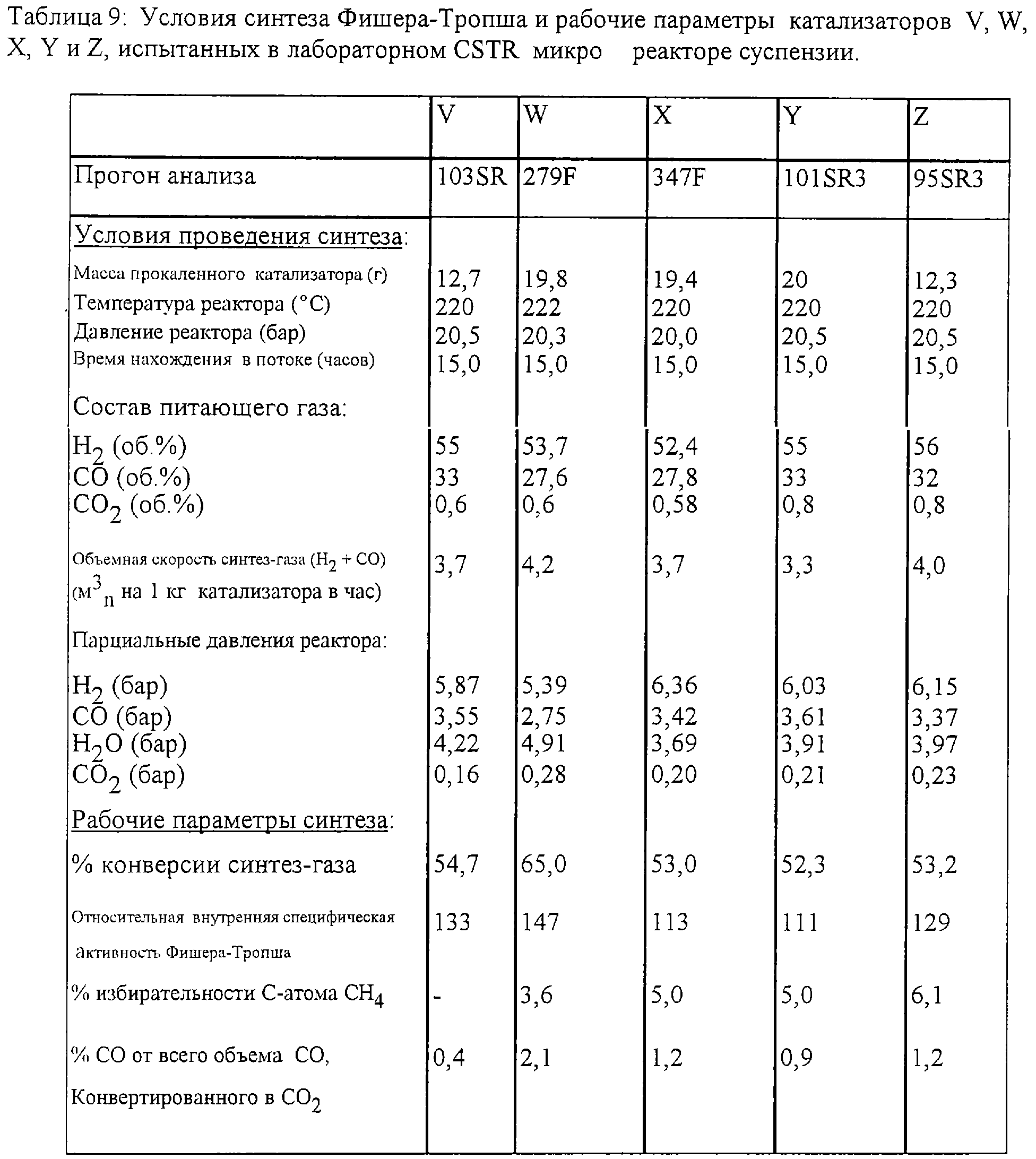
На лабораторной установке была приготовлена серия кобальтовых на базе оксида алюминия катализаторов Фишера-Тропша при использовании таких же процедур, как и использованные в Примере 2. Примененные условия прокаливания приведены в Таблице 8.
Катализаторы V, W и Z приготовлены в соответствии с настоящим изобретением, а катализаторы Х и Y приведены для сравнения.
Все катализаторы были восстановлены и испытаны на рабочие параметры синтеза Фишера-Тропша, как это описано в Примере 4. Были также рассчитаны относительные внутренние коэффициенты активности Фишера-Тропша, как это описано в Примере 4. Полученные данные представлены в Таблице 9, а также показаны на фиг.4-6.
ПРИМЕР 9
Два катализатора (АА и АВ) были приготовлены при помощи пропитки с зачаточной влажностью, при которой 50 г оксида алюминия с объемом пор 0,48 мл/г было пропитано 24 мл водного раствора нитрата кобальта, который также содержит нитрат аммония и платины. После пропитки полученный промежуточный продукт был высушен и прокален в блоке прокаливания с псевдоожиженным слоем до температуры 250° С. Точные условия прокаливания приведены в Таблице 10. После прокаливания промежуточный продукт был пропитан второй раз при помощи пропитки с зачаточной влажностью и прокален аналогично первой операции пропитки и прокаливания. После второй операции пропитки и прокаливания получили катализатор, который имеет следующий состав: 30 Со/ 0,075 Pt/ 100 Al2O3.
Катализатор АА выполнен в соответствии с настоящим изобретением, а катализатор АВ приведен для сравнения.
Все катализаторы были восстановлены и испытаны на рабочие параметры синтеза Фишера-Тропша, как это описано в Примере 4. Были также рассчитаны относительные внутренние коэффициенты активности Фишера-Тропша, как это описано в Примере 4. Полученные данные представлены в Таблице 11.

Полученные результаты показывают, что кобальтовые катализаторы, приготовленные при помощи процедур пропитки с зачаточной влажностью, прокаливание которых проведено в условиях в соответствии с настоящим изобретением, могут быть использованы для получения катализатора с улучшенными рабочими параметрами для синтеза Фишера-Тропша.
ПРИМЕР 10
Термогравиметрический анализ (TGA) на воздухе катализатора Q, проведенный после второй операции его пропитки и прокаливания, показывает, что потери в массе в диапазоне от 250° С до 550° С составляют 3,5-4,0 вес. %. В предположении, что в ходе TGA образуется соединение кобальта Со3О4, был проведен расчет для нахождения истинного соединения кобальта, присутствующего после стандартного прокаливания катализатора при 250° С. Возможными кандидатами являются соединения СоО, СоООН или Co2O3·H2O, Co(NO3)2 и/или Со2О3. Ожидаемые в ходе конверсии указанных соединений в Co3O4 изменения массы представлены в Таблице 12. Из рассмотрения этих данных можно прийти к заключению о том, что соединением кобальта, которое присутствует после прокаливания при 250° С, проведенного с использованием условий в соответствии с настоящим изобретением, является главным образом СоООН или Со2О3·Н2О.
Реферат
Изобретение относится к области каталитической химии. В изобретении предложен предшественник кобальтового катализатора, который содержит носитель катализатора, пропитанный кобальтом. Весь восстановимый кобальт присутствует в носителе в виде поддерживаемого оксида кобальта в соответствии с блочной формулой СоОаНb, в которой а≥1,7 и b>0. В изобретении предложены также варианты способа приготовления предшественника кобальтового катализатора. Технический результат: получен катализатор с более высокой активностью. 3 н. и 17 з.п. ф-лы, 12 табл., 10 ил.
Комментарии