Способ тионирования и тионирующий реагент - RU2605412C2
Код документа: RU2605412C2
Чертежи


Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к способу тионирования. Более конкретно, настоящее изобретение относится к способу превращения оксогруппы (>C=O) в соединении в тиогруппу (>C=S) или ее таутомерную форму указанной тиогруппы.
Уровень техники
В 1951 году Klingsberg1 и др. описали использование P4S10, растворенного в пиридине, в качестве тионирующего реагента. Пиридин и P4S10 легко взаимодействуют с образованием цвиттер-ионных соединений без запаха, состав которых P2S5·2C5H5N, изучен еще в 1967-1968 гг. немецкими неорганиками2,3, которые предложили структуру, установленную с помощью31P ЯМР данных4, а также относительно родственных молекул.
Несмотря на работу Klingsberg и др., преимущественно используемым реагентом в реакции тионирования соединений, содержащих оксогруппу, был так называемый реагент Лавессона (IUPAC название: 2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дитион), далее обозначаемый LR. LR был введен в органическую химию в 1968 и использовался со значительным количеством реагентов, таких как кетоны и амиды, которые были тионированы с хорошим выходом. Однако LR в качестве тионирующего реагента имеет ряд недостатков. Например, его термическая стабильность посредственная, даже сообщалось, что LR начинает разлагаться выше 110°C5,6. Кроме того, LR имеет в основном низкую растворимость, что нередко обусловливает необходимость использования гексаметилфосфорамида (ГМФА) в качестве растворителя. Предполагается, что ГМФА является канцерогенным для человека, и его использование запрещено во многих странах. Дополнительным недостатком LR является сильный, неприятный запах у самого соединения и то, что в ходе реакции существует тенденция к формированию зловонных побочных продуктов, которые трудно отделить от целевого продукта реакции (часто требуется колоночная хроматография).
По всей видимости, сохраняется потребность в разработке усовершенствованного способа тионирования соединения, содержащего оксогруппу, а также улучшенного тионирующего реагента для использования в таком процессе.
Раскрытие изобретения
В соответствии с первым аспектом предложен способ превращения группы >C=O (I) в соединении в группу >C=S (II) или таутомерную форму группы (II) по реакции, дающей тионированный продукт реакции, при использовании кристаллического P2S5·2C5H5N в качестве тионирующего реагента.
В соответствии с другим аспектом предложен тионирующий реагент, который является кристаллическим P2S5·2C5H5N.
Краткое описание чертежей
Фиг.1 представляет (А) молекулярную структуру и (Б) кристаллическую структуру P2S5·2C5H5N.
Фиг.2 представляет (А) молекулярную структуру и (Б) кристаллическую структуру дигидромонотиофосфата пиридиния.
Осуществление изобретения
Авторы настоящего изобретения определили кристаллическую структуру P2S5·2C5H5N рентгеноструктурным анализом, детали которого приведены в экспериментальной части. Ortep-представление молекулярной структуры соединения показано на фиг.1. Молекулы связаны друг с другом несколькими Ван-дер-ваальсовыми связями. Самая короткая Ван-дер-Ваальсова связь (C-H…S) связывает молекулы вместе в бесконечную цепочку вдоль оси с. Коэффициент упаковки (процент заполнения пространства элементарной ячейки Ван-дер-Ваальсовыми взаимодействиями) составляет 67,7%, что свидетельствует о молекулярной каркасной структуре в твердом состоянии. Упаковке молекул способствует π-стэкинг ароматических колец. Расстояние между плоскостями двух соседних ароматических групп составляет около 3,5 Å.
Как было указано выше, настоящее изобретение предлагает тионирующий реагент, состоящий из кристаллического P2S5·2C5H5N. Более преимущественным является то, что этот реагент может храниться в течение длительного периода времени и, кроме того, свободен от примесей, обычно содержащихся в тионирующем реагенте, поскольку эти примеси (из P4S10) удаляются с маточным пиридиновым раствором.
Улучшенная чистота будет приводить к более чистым продуктам тионирования и более легкому выделению продукта. Особым преимуществом является то, что тионирующий реагент может быть переведен в растворители, такие как ацетонитрил и диметилсульфон. Действительно, цвиттерионное кристаллическое соединение имеет хорошую растворимость в горячем ацетонитриле и в горячем пиридине. Оно также имеет хорошую растворимость в циклических сульфонах или в низших алкилсульфонах, таких как диметилсульфон.
В одном осуществлении способа изобретения тионирующий реагент и соединение, подлежащее тионированию, взаимодействуют в жидкой для соединения и тионирующего реагента среде растворителя. Другими словами, тионирующий реагент используют растворенным в жидкой среде растворителя.
В одном осуществлении способа изобретения тионирующий реагент, используемый в виде расплава, смешивают с тионируемым соединением. В этом осуществлении тионирующий реагент нагрет до его температуры плавления (167-169°C) и тионируемое соединение смешивают с тионирующим реагентом до, после или во время нагревания.
Среда растворителя должна быть выбрана из апротонных растворителей. В одном осуществлении жидкая среда растворителя является органическим растворителем, который является жидкостью при комнатной температуре и который может быть нагрет до соответствующей температуры реакции, например до температуры 60-200°C, например 60-100°C, таким как ацетонитрил, который является жидким при комнатной температуре (температура плавления -42°C) и имеет температуру кипения 82°C. В этом случае кристаллический P2S5·2C5H5N и тионируемое соединение, оба, растворены в органическом растворителе, который необязательно нагревают, например, с обратным холодильником.
В одном осуществлении кристаллический P2S5·2C5H5N смешивают со средой растворителя при температуре ниже точки плавления среды растворителя и кристаллического P2S5·2C5H5N и смесь нагревают для получения жидкого раствора, содержащего P2S5·2C5H5N, растворенного в жидкой среде растворителя.
Тионируемое соединение может быть смешано с другими компонентами реакционной смеси в любой точке процесса, например, до или после плавления и/или растворения.
Например, температура плавления диметилсульфона составляет 107-109°C. В случае использования расплавленного диметилсульфона в качестве жидкой реакционной среды растворителя кристаллический P2S5·2C5H5N и твердый диметилсульфон могут быть смешаны, например, при комнатной температуре и нагреты до температуры, по меньшей мере, около 109°C, после чего получается раствор P2S5·2C5H5N в жидком диметилсульфоне. В этой реакционной среде может быть выполнено тионирование соединения, содержащего оксогруппу.
Предпочтительным признаком P2S5·2C5H5N является его термостойкость, которая позволяет выполнять реакцию тионирования при температурах свыше 100°C, например при температуре 100-200°C или 115-180°C, или при температуре 150-175°C, в частности при температуре 165-175°C, хотя и более низкие температуры могут быть использованы, например 60-100°C. В некоторых осуществления реакцию проводят при температуре кипения растворителя.
В настоящее время не ясно или P2S5·2C5H5N как таковой после растворения в жидкой среде растворителя тионирует соединение, или же реакция протекает через диссоциацию до некоторой другой промежуточной реакционно-способной формы. Для целей настоящего изобретения, однако, точный механизм реакции не является существенным, и указание на то, что растворенный P2S5·2C5H5N вступает в реакцию с растворенным соединением, означает реакцию, протекающую по любому возможному промежуточному пути, ведущему к целевому тионированному продукту.
В присутствии воды или протонного растворителя, такого как низший спирт, например метанол или этанол, P2S5·2C5H5N быстро подвергается интенсивному разложению. Например, добавление воды к горячему водному раствору/суспензии P2S5·2C5H5N в ацетонитриле быстро приводит к прозрачному раствору соли пиридина и тиофосфорной кислоты, а именно дигидромонотиофосфату пиридиния, формулы

Эта соль легко растворяется в воде, и ее легкое образование и высокую растворимость можно с успехом использовать для выделения тионированного продукта реакции изобретения, например тиоамидов. Таким образом, в типичной реакции изобретения четыре эквивалента амида нагревают с 1,1 эквивалента кристаллического P2S5·2C5H5N в сухом ацетонитриле и при выделении продукта весь остающийся тионирующий реагент легко удаляется добавлением воды.
P2S5·2C5H5N также будет разлагаться при обработке спиртами; например, обработка P2S5·2C5H5N этанолом дает O,O-диэтилдитиофосфонат пиридиния, формулы

Таким образом, одним преимуществом настоящего изобретения является то, что целевой тионированный продукт легко отделяется от остающегося тионирующего реагента P2S5·2C5H5N обработкой протонным растворителем, таким как вода или низший спирт, например этанол.
Таким образом, в одном осуществлении изобретения предложен способ превращения группы >C=O (I) в соединении в группу >C=S (II) или таутомерную форму группы (II) путем приведения в контакт соединения с P2S5 2C5H5N так, чтобы получить тионированный продукт реакции, включающий смешивание кристаллического P2S5·2C5H5N с указанным соединением в жидкой среде растворителя, так, чтобы получить жидкий раствор соединения и P2S5·2C5H5N, и проведение реакции между P2S5·2C5H5N и соединением в растворе с последующим добавлением протонного растворителя к раствору.
После добавления протонного растворителя к раствору соль, образующаяся при разложении всего оставшегося P2S5·2C5H5N, легко будет отделить от тионированного соединения, например, экстракцией водным раствором или водой. В некоторых осуществлениях добавление протонного растворителя, такого как вода, приведет к осаждению тионированного продукта реакции, который затем может быть отделен от водной фазы, например, простой фильтрацией. Дальнейшая очистка продукта реакции необязательно может быть выполнена, например, перекристаллизацией.
Группа >С=O (I), которую нужно превратить в группу >C=S (II), может присутствовать, например, в виде кетонной или амидной функциональной группы и может присутствовать в соединении, включающем одну или несколько функциональных групп, и в этом случае может быть достигнуто селективное тионирование, как будет показано в примерах ниже.
В одном осуществлении группа (I) присутствует в виде амидной функциональной группы -C(O)-N<, например, в соединении
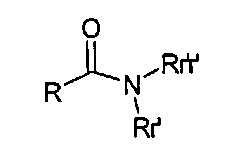
где R, например, может быть выбран из C1-C12 гидрокарбилов и R′ и R″ могут быть независимо выбраны из H и C1-C12 гидрокарбилов или где R и R′ и/или R′ и R″ могут быть соединены друг с другом с образованием вместе с углеродом и/или азотом амида, с которым они связаны, моно- или полициклического кольца, например моно- или полициклического 5-20-членного кольца, необязательно включающего один или несколько дополнительных гетероатомов, например один или несколько гетероатомов, выбранных из О, N и S, причем кольцо может быть насыщенным или ненасыщенным и ароматическим или неароматическим.
В одном осуществлении соединение представляет собой пептид, олигопептид или полипептид, например пептид, содержащий 1-10 групп (I) в основной цепи, или 1-5 оксогрупп (I).
В одном осуществлении группа (I) присутствует в виде кетонной группы, например, в соединении

где R и R′, например, могут быть независимо выбраны из H и C1-C12 гидрокарбилов, или могут быть соединены друг с другом с образованием вместе с углеродом кетона моно- или полициклического кольца, например моно- или полициклического 5-20-членного кольца, необязательно содержащего один или несколько гетероатомов, например один или несколько гетероатомов, выбранных из О, N и S, причем кольцо может быть насыщенным или ненасыщенным и ароматическим или неароматическим.
Группы R, R′ и R″ могут быть необязательно и независимо замещены одним или несколькими заместителями, например одной или несколькими дополнительными оксогруппами или одной или несколькими другими функциональными группами.
Когда группа (I) присутствует в виде кетонной группы, предпочтительно должна присутствовать, по меньшей мере, одна электронодонорная группа в соединении, что приводит к увеличению электронной плотности на группе (I). Такая электронодонорная группа (EDG) может быть, например, группой с неподеленной парой электронов, которая может повышать электронную плотность кетонной группы делокализацией указанной пары электронов по одной или нескольким двойным связям, находящихся между EDG и кетонной группой. Электронная плотность на кетонной группе также может быть повышена индуктивным эффектом.
Продукт реакции тионирования изобретения является тионированным соединением, содержащим группу >C=S (II) или ее таутомер, например группу >C=C(SH)-.
Кристаллический P2S5·2C5H5N предпочтительно смешивают в мольном отношении к превращаемой группе (I) 1 моль P2S5·2C5H5N на 1-4 моля группы (I), например 1 моль P2S5·2C5H5N на 2-4 моля группы (I), в частности 1 моль P2S5·2C5H5N на 3-4 моля группы (I).
Таким образом, в случае, если соединение содержит более одной группы (I), превращаемой в группу (II), мольное отношение P2S5·2C5H5N к соединению будет соответственно выше. Например, в случае, если соединение содержит две группы (I), превращаемые в две группы (II), кристаллический P2S5·2C5H5N предпочтительно смешивают с тионируемым соединением в мольном соотношении 1 моль P2S5·2C5H5N на 0,5-2 моля соединения, например 1 моль P2S5·2C5H5N на 1-2 моля соединения или 1 моль P2S5·2C5H5N на 1,5-2 моля соединения.
Как правило, для соединения, содержащего n функциональных групп, выбранных из, например, кетонных и амидных функциональных групп, например n амидных функциональных групп, мольное отношение P2S5·2C5H5N к соединению может составлять n/4-n, или n/4-n/2, например n/4-n/3.
Предпочтительным признаком P2S5·2C5H5N в качестве тионирующего реагента является его селективность. Так, например, сложноэфирные функциональные группы обычно не реагируют с P2S5·2C5H5N и, следовательно, настоящее изобретение также предлагает способ селективного тионирования, например, амидных или кетонных групп в соединении, также содержащем сложноэфирную группу.
Изобретение далее будет описано в следующих не ограничивающих примерах.
Пример 1
Кристаллический P2S5·2C5H5N
Декасульфид тетрафосфора (P4S10, 44,5 г, 0,1 моль) добавляют порциями к сухому пиридину (560 мл) при 80°C при перемешивании мешалкой. После кипячения с обратным холодильником (1 ч) получают прозрачный желтый раствор, из которого осаждаются светло-желтые кристаллы при охлаждении раствора. Через 2 часа кристаллы собирают, промывают сухим ацетонитрилом и, наконец, переносят в эксикатор (с емкостью с конц. серной кислотой) для удаления всего избытка пиридина, выход 62,3 г (84%), т.пл.: 167-169°C, ИК vмакс: 3088, 3040, 1608, 1451, 1197, 1044, 723, 668 см-1, см. фиг.1.
Дигидромонотиофосфат пиридиния
Кристаллический P2S5·2C5H5N (3,80 г, 10 ммоль) кипятят с обратным холодильником в ацетонитриле (35 мл), содержащем воду (1,0 мл). Прозрачный раствор (полученный в течение 3 мин), концентрируют и продукт оставляют кристаллизоваться, 3,15 г (79%). Кристаллы пригодны для рентгеноструктурного анализа, т.пл. 110-120°C разл. с выделением H2S;
1Н ЯМР (300 МГц, ДМСО-d6) δ 7,51 (м, 2H, 3-H), 7,95 (дд, 1H, 4-H), 8,63 (д, 2H, 2-H), 9,7 (шир. с, 3H);13С-ЯМР (75,5 МГц, ДМСО-d6) δ 124,7 (д), 138,5 (д), 147,8 (д); см. фиг.2.
О,O-Диэтилдитиофосфонат пиридиния
Кристаллический P2S5·2C5H5N (1,0 г) нагревают с обратным холодильником в этаноле (5 мл) в течение 5 мин, прозрачный раствор упаривают с получением масла, которое быстро затвердевает (100%).
ИК vmax: 2976,2891, 1630, 1600, 1526, 1479, 1383, 1020, 920, 748, 681 см-1
1Н-ЯМР (300 МГц, ДМСО-d6) δ 1,08 (т, J=7,1 Гц, 6H), 3,79 (м, 4H), 8,09 (м, 2H), 8,62 (м, 1H), 8,97 (м, 2H);13С-ЯМР (75,5 МГц, ДМСО-d6) δ 16,1 (кв,3JC-P=8,8 Гц), 59,8 (т,2JC-P=7,1 Гц), 127,2 (д), 142,5 (д), 146,0 (д).
Пример 2
(S)-11-Тиоксо-2,3,11,11а-тетрагидро-1H-бензо[е]пирроло[1,2-а][1,4]диазепин-5-(10H)-он (таблица 1, строка 17).
К раствору (200 мл) 2,3-дигидро-1H-бензо[е]пирроло[1,2-а][1,4]диазепин-5,11(10H,11аH)-диона (4,0 г, 20 ммоль) в MeCN добавляют кристаллический P2S5 2C5H5N (2,3 г, 6 ммоль) и нагревают до 60°C 3 ч, в течение которых образуется желтый осадок. Реакционную смесь оставляют при комнатной температуре на ночь для полного осаждения. Продукт фильтруют в вакууме и промывают небольшим количеством холодного MeCN с получением указанного в заголовке соединения (3,9 г, 85%) в виде бледно-желтого твердого вещества, т.пл. 268-270°C; (с 0,16, МеОН);
ИК vmax: 3170, 2979, 1616, 1602, 1477, 1374, 1271, 1141, 831, 813,752 см-1;
1H-ЯМР (300 МГц, ДМСО-d6) δ 1,89-1,94 (м, 1H), 1,99-2,16 (м, 2H), 2,84-2,94 (м, 1H), 3,40-3,50 (м, 1H), 3,53-3,60 (м, 1H), 4,27 (д, J=6,11 Гц, 1H), 7,22-7,27 (м, 1H), 7,30-7,37 (м, 1H), 7,55-7,60 (м, 1H), 7,80-7,85 (м, 1H), 12,46 (шир. с, 1H);13С-ЯМР (75,5 МГц, ДМСО-d6) δ 22,7 (т), 29,0 (т), 46,8 (т), 59,8 (д), 121,8 (д), 125,7 (д), 127,8 (с), 130,2 (д), 132,2 (д), 136,5 (с), 164,2 (с), 201,9 (с).
Пример 3
2,5-Пиперазиндитион из глицина (табл.2, строка 1).
Глицин (1,50 г, 20 ммоль), кристаллический P2S5·2C5H5N (9,12 г, 28 ммоль) и диметилсульфон (8,0 г) нагревают при 165-170°C в течение 1 ч, после чего реакционную смесь (после охлаждения) обрабатывают кипящей водой в течение 30 мин. Полученное коричневатое твердое вещество перекристаллизовывают из смеси этанол/ДМФ, 1,85 г (63%), т.пл. 284°C;1H-ЯМР (300 МГц, ДМСО-d6) δ 4,19 (с), 10,7 (с);13C-ЯМР (75,5 МГц, ДМСО-d6) δ 54,4 (кв), 191,9 (с).
Пример 4
2,5-Пиперазиндитион из 2,5-пиперазиндиона (табл.2, строка 2).
2,5-Пиперазиндион (2,28 г, 20 ммоль) и кристаллический P2S5·2C5H5N (2,28 г, 8 ммоль) нагревают с обратным холодильником в ацетонитриле (50 мл) в течение 2 ч, когда смесь концентрируется, добавляют воду. Образовавшееся твердое вещество собирают после перемешивания в течение 1 ч, 2,63 г (90%).
Температура плавления и спектр ЯМР идентичны данным, полученным выше для 2,5-пиперазиндитиона из глицина (табл.2, строка 1).
S,S′-1,4-Диацетил-2,5-бис-ацетилтиоло-1,4-дигидропиразин, 35.
Вышеуказанный 2,5-пиперазиндитион (1,46 г, 10 ммоль) нагревают при температуре кипения в уксусном ангидриде (20 мл) в течение 2 ч, после чего реакционную смесь концентрируют и обрабатывают диизопропиловым эфиром, 2,06 г (93%), т.пл. 190-192°C;1H-ЯМР (300 МГц, ДМСО-d6) δ 2,17 (с, 6H), 2,45 (с, 6H), 6,99 (с, 2H);13C-ЯМР (75,5 МГц, ДМСО-d6) δ 22,2 (кв), 29,4 (кв), 117,0 (с), 131,6 (д), 166,3 (с), 193,7 (с); Элементный анализ, вычислено для C12H14N2O4S2, C 45,75, H 4,48, N 8,88. Найдено: C 45,90, H 4,32, N 8,71.
Восстановительное расщепление тетрасульфида, 25.
3,3′-Дииндолил-2,2′-тетрасульфид 25 (3,58 г, 10 ммоль), растворяют в ТГФ, 50 мл, и добавляют к смеси NaBH4 (1,50 г, 40 ммоль) в ТГФ (75 мл). Происходит выделение газов, содержащих H2S, и реакционную смесь перемешивают в течение 3 ч при 40-45°C в токе аргона. Этот чувствительный к воздуху раствор, содержащий дианион 26, не хранят, а непосредственно используют в реакции, описанной ниже.
2,2′-Бис(метилтио)-1H,1′H,H-3,3′-бииндол.
Диметилсульфат (1,51 г, 12 ммоль), растворенный в MeOH (15 мл), добавляют по каплям к раствору, полученному восстановительным расщеплением тетрасульфида 25 (5 ммоль) при 25°C. После перемешивания (1 ч) раствор выпаривают и обрабатывают водой. Неочищенное твердое вещество кристаллизуют из смеси метанол-вода с получением желтого твердого вещества (0,45 г, 57%), т.пл. 184-186°C;1H-ЯМР (300 МГц, ДМСО-d6) δ 2,44 (с, 6H), 6,95-6,99 (м, 2H), 7,10-7,22 (м, 4H), 7,36-7,45 (м, 2H), 11,55 (с, 2H);13C-ЯМР (75,5 МГц, ДМСО-d6) δ 18,0 (кв), 110,8 (с), 110,9 (д), 119,0 (д), 119,2 (д), 121,5 (д), 128,0 (с), 129,1 (с), 137,0 (с).
Синтез циклодисульфида, 23.
Раствор, полученный восстановительным расщеплением тетрасульфида 25, после добавления воды (50 мл), перемешивают в течение 24 ч при контакте с воздухом. Желтое твердое вещество собирают и перекристаллизовывают из ацетонитрил-ДМФ 4:1 с получением 2,20 г (77%) твердого вещества, все еще содержащего ДМФ, который удаляют сушкой при пониженном давлении, т.пл. >227-228°C.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7,04-7,08 (м, 1H), 7,28-7,31 (м, 2H), 7,33-7,51 (м, 1H), 12,16 (с, 1H);13C-ЯМР (75,5 МГц, ДМСО-d6) δ 136,3 (с), 127,0 (с), 124,9 (с), 124,6 (д), 120,3 (д), 120,2 (д), 119,3 (с), 112,2 (д).
Пример 5
Циклодисульфид 23, получаемый тионированием оксиндола при 160°C (табл.3, строка 13).
Оксиндол (1,33 г, 10 ммоль) и кристаллический P2S5·2C5H5N (1,52 г, 4 ммоль) нагревают с диметилсульфоном (4,0 г) и затем нагревают при 160°C в течение 5 мин. Расплав охлаждают и затем нагревают с водой. Полученное твердое вещество кристаллизуют из ацетонитрил-ДМФ 4:1 с получением 1,37 г (92%), т.пл. >227-228°C. Этот материал идентичен полученному восстановительным расщеплением тетрасульфида 25.
3,3′-Битио-оксиндол, 27.
Раствор, полученный восстановительным расщеплением тетрасульфида 25, подкисляют AcOH, что приводит к образованию указанного в заголовке соединения в виде желтого осадка, 2,52 г (85%). Которое перекристаллизовывают из ацетонитрила, т.пл. 180°C разл. Эта молекула является чувствительной к окислению воздухом.
1H-ЯМР (300 МГц, ДМСО-d6) δ 4,66 (с, 2H), 6,85-6,91 (м, 4H), 6,96-6,98 (м, 2H), 7,07, 7,13 (м, 2H), 13,06 (с, 2H);13C-ЯМР (75,5 МГц, ДМСО-d6) δ 60,8 (д), 110,4 (д), 123,0 (д), 123,4 (д), 128,6 (д), 130,2 (с), 144,2 (с), 204,3 (с). Элементный анализ, вычислено для C16H12N2S2; C, 64,60, H, 4,08, N, 9,43. Найдено: C, 64,26, H, 3,99, N, 9,31.
Пример 6
Метил 5-меркапто-4-(2-метокси-2-оксоэтил)-2-метил-1Н-пиррол-3-карбоксилат, 34b.
Диэфир 33а (2,13 г, 10 ммоль) и кристаллический P2S5·2C5H5N (1,14 г, 4 ммоль) кипятят с обратным холодильником в ацетонитриле (50 мл) 1 ч. После концентрирования до 25 мл добавляют воду и образовавшееся твердое вещество собирают и кристаллизуют из 2-пропанола, 1,85 г (81%), т.пл. 185-187°C; ИК vmax: 3273, 2954, 1742, 1724, 1707, 1681, 1562, 1440, 1341, 1269, 1200, 1173, 1171, 1080, 1003, 782 см-1;1H-ЯМР (300 МГц, ДМСО-d6) δ 2,43 (с, 3H, CH3), 3,17 (с, 1H, SH), 3,49 (с, 3H, OCH3), 3,64 (с, 3H, OCH3), 11,90 (с, 1H, H);13C-ЯМР (75,5 МГц, ДМСО-d6) δ 13,4 (д), 30,6 (кв), 50,4 (кв), 51,4 (кв), 111,2 (с), 117,1 (с), 126,9 (с), 139,9 (с), 164,4 (с), 171,1 (с). Элементный анализ, вычислено для C10H13NO4S; C, 49,37, H, 5,38, N 5,75. Найдено: C, 49,25, H, 5,46, N, 5,61.
Пример 7
3-(1H-Индол-3-ил)-3,3′-бииндолин-2-тион (табл.3, строка 9).
3-(1H-Индол-3-ил)-3,3′-бииндолин-2-он (728 мг, 2 ммоль), кристаллический P2S5·2C5H5N (228 мг, 0,6 ммоль) и диметилсульфон (3,05 г) нагревают (165-170°C) в течение 20 мин. Расплав охлаждают и затем нагревают в воде в течение 10 мин. Образовавшееся твердое вещество собирают, 766 мг (94%), т.пл. >260°C.1H-ЯМР (300 МГц, ДМСО-d6) δ 7,09-7,15 (м, 2H), 7,18-7,20 (м, 5H), 7,24-7,30 (м, 7H), 13,00 (с, 1H);13C-ЯМР (75,5 МГц, ДМСО-d6) δ 72,7 (с), 111,2 (д), 124,4 (д), 126,5 (д), 127,5 (д), 128,6 (с), 128,7 (с), 129,0 (д), 129,1 (д), 129,1 (д), 139,2 (с), 143,0 (с), 143,5 (с), 145,3 (с, 2C), 208,4 (с). Элементный анализ, вычислено для C24H17N3S; С, 75,96, Н, 4,51, N, 11,07; Найдено: C, 76,10, H, 4,46, N, 11,00.
Результаты ряда реакций тионирования в соответствии с изобретением, используя кристаллический P2S5·2C5H5N, растворенный в горячем ацетонитриле, перечислены в таблице 1. В приведенных в качестве примера реакциях отношение кристаллический P2S5·2C5H5N к тионируемому соединению составляет 1,1:4. В некоторых случаях проводят прямое сравнение с LR. Например, ε-капролактам и P2S5·2C5H5N дает соответствующий тиоамид в течение 5 мин, a LR тионирует еще быстрее.
Действительно, суспензию LR в горячем ацетонитриле можно титровать добавлением ε-капролактама. Преимущества тионирующего реагента по изобретению по сравнению LR, прежде всего, состоят в том, что тионирующий реагент изобретения проще в приготовлении, без запаха (при достаточной чистоте), и в том, что тионированные продукты являются очень чистыми. В описанных в заявке примерах образование нитрилов из первичных амидов никогда не было проблемой. Этот тип побочной реакции иногда может быть проблематичным, когда используемым тионирующим реагентом является LR7,8. Тионирование в качестве примера кетонов с помощью P2S5·2C5H5N проходит хорошо (табл.2, строки 3 и 4). Кетопроизводные 20а и 21а могут быть превращены в 20b и 21b, соответственно, когда тионирующий реагент по изобретению используют в горячем пиридине или в виде расплава, и еще лучше - при нагревании вместе с диметилсульфоном (табл.1, строка 20, и в таблице 3, строка 3).
В то время как тионирование 3,3-диметилоксиндола (строка 7, таблица 1) дало превосходный выход, исходное соединение оксиндол (строка 6, таблица 1) дало неприемлемо низкие выходы (~10%). В этом случае образование комплексов с низкой растворимостью, по-видимому, вызывает проблему. Синтез 3,3-дииндолилиндолин-2-тион также неудачен, но может быть осуществлен с диметилсульфоном в качестве растворителя (см. таблицу 3). Тионирование 3-гидрокси-2-пиридона проходит хорошо без осложнений с получением интересного класса 3-гидрокси-2-(1Н)-пиридинтиона, который в некоторых типах комплексов металлов (например, Zn2+), как сообщалось, имеет некоторые перспективы в лечении сахарного диабета.
Селективность может быть достигнута в случаях, когда более чем одна карбонильная группа присутствует в исходных материалах. Таким образом, монотионированные молекулы (табл.1, строки 12, 16 и 17) могут быть получены с хорошими выходами. Тионирование пиперидин-2,6-диона дает монотионированный продукт в горячем ацетонитриле, в то время как с избытком тионирующего реагента в горячем пиридине может быть получен полностью тионированный продукт.


Тионирование Gly-Gly, а также пиперазин-2,5-диона дает хорошие выходы целевого дитионированного продукта (таблица 2, строки 1 и 2). Для дополнительной характеристики нерастворимого продукта его ацетилируют в горячем уксусном ангидриде, что дает тетраацетилированный продукт 35, который легко дает хорошие спектры ЯМР.

Тионирование при достаточно высоких температурах (165-175°C) может быть осуществлено, например, с P2S5·2C5H5N, растворенным в диметилсульфоне (т.пл. 107-109°C, т.кип. 238°C). Результаты некоторых примеров реакций изобретения перечислены в таблице 3. В одном случае (таблица 3, строка 6) продукт частично превращается в дисульфид 22 с очень низкой растворимостью. Аналогичные наблюдения были опубликованы, например, Stoyanov9 и Hino et al10. В последней работе установлено, что ряд производных 3-замещенных индол-2-тионов легко могут быть окислены до соответствующих дисульфидов. Формирования продуктов окисления можно избежать проведением реакции в атмосфере аргона.
Ранее бензальдегид был тионирован много раз11-16 и продукт неизменно выделяют в виде тримера (29) неустойчивого первичного продукта 30, и тример 29 действительно был продуктом при реакции тионирующего реагента по изобретению с бензольдегидом в диметилсульфоне.

Сложноэфирная группа, как правило, не затрагивается P2S5·2C5H5N, как можно продемонстрировать тионированием (табл.3, строка 10) моноацетата койевой кислоты (31), которое селективно дает тион 32 (табл.1, строка 17). Тионирование диэфира 33а дает еще один пример, а именно производное пиррол-2-тиола 34b.


Исходный материал полностью (ЯМР данные) находится в виде таутомера 33а, тогда как продукт полностью находится в виде таутомера тиола 34b. Но что более важно, две сложноэфирные функциональные группы остаются неизменными.
Из-за низкой растворимости и высокой температуры плавления трудно охарактеризовать 2,5-пиперазиндитион (табл.3, строка 12), поэтому получен легко растворимый тетраацетат 35.


В свете вышеприведенного общего описания и с помощью иллюстративных примеров, специалисты в данной области техники вполне способны осуществить изобретение во всем объеме формулы изобретения, с использованием рутинного экспериментирования при необходимости выбора подходящих условий реакции, например, в зависимости от функциональных групп, которые могут присутствовать в тионируемом соединении. Например, реакцию можно проводить при нормальной атмосфере или в атмосфере инертного газа, например аргона или азота. Другие параметры, которые могут быть оптимизированы или изменены, например среда растворителя, температура реакции и время реакции, и все такие модификации и вариации рассматриваются как входящие в объем притязаний настоящего изобретения.
Ссылки
(1) Klingsberg, Е.; Papa, D.J. Am. Chem. Soc. 1951, 73, 4988-4989.
(2) Meisel, M.; Grunze, H.Z. Anorg. Allg. Chemie, 1967, 360, 277-283.
(3) Fluck, E.; Binder, H.Z. Anorg. Allg. Chemie 1967, 354, 113-129.
(4) Brunel, E.; Monzur, J.; Retuert, J.J. Chem. Res (M) 1981, 3437-3445.
(5) Jesberger, M.; Davis, T.P.; Berner, L. Synthesis 2003, 1929-1958.
(6) a) Ozturk, Т.; Erdal, E.; Olcay, M. Chem. Rev. 2007, 107, 5210-5278.
b) Ozturk, Т.; Erdal, E.; Olcay, M. Chem. Rev. 2010, 110, 3419-3478.
(7) Scheibye, S.; Shabana, R.; Lawesson, S.O.; Römming, C. Tetrahedron 1982, 38, 993-1001.
(8) Ley, S.V.; Leach, A.G.; Storer, R.I. J. Chem. Soc., Perkin Trans 1 2001, 358-361.
(9) Stoyanov, S.; Petkov, I.; Antonov, L.; T. Stoyanova; Karagiannidis, P.; Aslanidis, P. Can. J. Chem. 1990, 68, 1482-1489.
(10) Hino, Т.; Suzuki, Т.; Nakagawa, M. Chem. Pharm. Bull 1974, 22, 1053-1060.
(11) Baumann, E.; From, E. Ber. 1889, 22, 2600-2609.
(12) Stanfield, J.A.; Reynolds, L.B. J. Am. Chem. Soc. 1952, 74, 2878-2880.
(13) Böttcher, В.; Bauer, F. Liebigs Ann. Chem. 1951, 574, 218-226.
(14) Takikawa, Y.; Shimoda, K.; Makabe, Т.; Takizawa, S. Chem. Lett. 1983, 1503-1506.
(15) Sekido, K.; Hirokawa, S. Acta. Cryst. C 41 1985, 379-400.
(16) Bonini, B.F.; Mazzanti, G.; Zani, P.; Maccagani, G.; Foresti, E. J. Chem. Soc., Perkin Trans 1, 1988, 1499-1502.
Реферат
Изобретение относится к способу превращения группы >C=O (I) в соединении в группу >C=S (II) или в таутомерную форму группы (II) в реакции, дающей тионированный продукт реакции, при использовании кристаллического PS·2CHN в качестве тионирующего реагента, а также к тионирующему агенту, который является кристаллическим PS·2CHN и имеет температуру плавления 167-169°С. Технический результат: предложен новый способ превращения группы >C=O (I) в соединении в группу >C=S (II). 2 н. и 20 з.п. ф-лы, 3 табл., 4 ил., 7 пр.
Формула
Документы, цитированные в отчёте о поиске
Замещенные иминоазины, хлоразиниевые соединения, соединения пиридинона и гербицидное средство на ихоснове
Комментарии