Терапевтические соединения - RU2470908C2
Код документа: RU2470908C2
Чертежи



Описание
Область техники, к которой относится изобретение
Наcтоящее изобретение имеет отношение к терапевтическим соединениям, пригодным в качестве анестетиков.
Уровень техники
Пропофол (2,6-диизопропилфенол) является внутривенным седативным/снотворным средством, широко используемым для индуцирования и поддержания общей анестезии (общего наркоза), обезболивания серьезно больных пациентов и обезболивания при процедурах (например, эндоскопии) (Langly, M.S. и Heel, R.C. Drugs, 1988, 35, 334-372). Пропофол только умеренно растворяется в воде и в настоящее время продается в 10% соевом масле на основе липидной эмульсии, подобно композициям для парентерального питания.
Пропофол представляет собой агонист GАВАA (гамма-аминомасляной кислоты, ГАМК), активирующий разные подтипы рецепторов GАВАA, которые являются ионными каналами, переносящими анионы хлора через клеточные мембраны в центральной нервной системе. Несмотря на то, что пропофол является ахиральным, рацемические смеси ряда диалкилфенолов являются известными агонистами рецептора GАВАA (James et al., J. Med. Chem. 23, 1350, 1980; Krasowski et al., J. Pharmacol. & Exp. Therapeutics 297, 338, 2001). James et al., сообщает об открытии факта, что пропофол является преобладающим в общем профиле по отношению к другим оцененным аналогам.
Многие клиницисты предпочитают пропофол из-за его прекрасного фармакокинетического и фармакодинамического профилей, профилей выхода из наркоза и восстановления сознания после наркоза. Однако нежелательные побочные действия (например, угнетение дыхания, синдром ICU, инъекционная боль и гемодинамические эффекты), вызываемые терапевтической дозой или близкой к ней дозой, значительно ограничивают его пользу в разнообразных клинических ситуациях. Это, в частности, относится к гемодинамическим эффектам. Введение пропофола, особенно в болюсной форме часто вызывает снижение кровяного давления без компенсаторного увеличения частоты сердечных сокращений. Некоторые клинические состояния являются несовместимыми с применением пропофола из-за нежелательных и потенциально вредных гемодинамических последствий. Примеры таких состояний включают сердечно-сосудистую болезнь, такую как болезнь коронарных артерий, кардиомиопатию, ишемическую болезнь сердца, порок клапана сердца и врожденный порок сердца. Хроническая гипертония, цереброваскулярная болезнь, травма головного мозга и пожилой возраст могут сделать применение пропофола затрудненным или проблематичным из-за его гемодинамических свойств. Пациенты с острой потерей крови, обезвоживанием или тяжелой инфекцией, включая пациентов с геморрагическим шоком, гиповолемическим шоком или септическим шоком могут быть подвергнуты чрезмерному риску при применении пропофола. Гемодинамические свойства пропофола могут ограничивать его использование у пациентов, получающих другие медикаменты или виды лечения, такие как спинальная анестезия, эпидуральная анестезия или вазоактивные медикаменты.
Раскрытие изобретения
Изобретение предоставляет терапевтические соединения, которые демонстрируют сходную или улучшенную фармакологическую активность по сравнению с пропофолом наряду с улучшенным гемодинамическим профилем. Соответственно в одном варианте осуществления изобретение предоставляет (-)-стереоизомер формулы (I):
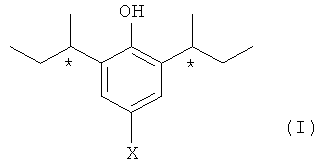
в которой Х представляет собой Н или F, или его соль, или пролекарство.
Изобретение также предоставляет фармацевтическую композицию, содержащую
(-)-стереоизомер формулы (I) или его фармацевтически приемлемую соль или пролекарство и фармацевтически приемлемый носитель.
Изобретение также предоставляет способ лечения тошноты, рвоты, мигрени, нейродегенеративных состояний нервной системы (например, болезни Фридриха, болезни Паркинсона, болезни Альцгеймера, болезни Хантингтона, бокового амиотрофического склероза (ALS), рассеянного склероза (MS), болезни Пика и т.д.), травм центральной нервной системы (например, перелома черепа и происходящего в результате этого отека, сотрясения, контузии, тяжелого кровоизлияния в мозг, резаных повреждений, субдурального и эпидурального кровоизлияния и повреждения спинного мозга (например, механического повреждения вследствие сжатия или изгибания позвоночника)), конвульсий (например, эпилептических припадков) или болезни, связанной со свободными радикалами (например, ишемического реперфузионного повреждения, воспалительного заболевания, системной красной волчанки, инфаркта миокарда, инсульта, травматической геморрагии, образования катаракты, увеита, эмфиземы, язвы желудка, неоплазии, лучевой болезни и т.д.) у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I) или его фармацевтически приемлемой соли, или пролекарства животному.
Изобретение также предоставляет способ индуцирования или поддержания общей анестезии у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I) или его фармацевтически приемлемой соли, или его пролекарства животному.
Изобретение также предоставляет способ промотирования обезболивания у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства животному.
Изобретение также предоставляет способ лечения мигрени у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства животному.
Изобретение также предоставляет способ лечения бессонницы у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства животному.
Изобретение также предоставляет способ промотирования анксиолитического эффекта у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства животному.
Изобретение также предоставляет способ лечения синдрома отмены зависимости у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства животному.
Изобретение также предоставляет способ промотирования антиэметического эффекта у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства животному.
Изобретение также предоставляет способ агонизирования рецептора GABA, включающий соприкосновение рецептора (in vitro или in vivo) с эффективным количеством (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства.
Изобретение также предоставляет способ агонизирования рецептора GABA у животного, включающий введение эффективного количества (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства животному.
Изобретение также предоставляет (-)-стереоизомер формулы (I), или его фармацевтически приемлемую соль, или его пролекарство для применения в лекарственной терапии.
Изобретение также предоставляет применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента для лечения тошноты, рвоты, мигрени, нейродегенеративных состояний нервной системы (например, болезни Фридриха, болезни Паркинсона, болезни Альцгеймера, болезни Хантингтона, бокового амиотрофического склероза (ALS), рассеянного склероза (MS), болезни Пика и т.д.), травм центральной нервной системы (например, перелома черепа и происходящего в результате этого отека, сотрясения, контузии, тяжелого кровоизлияния в мозг, резаных повреждений, субдурального и эпидурального кровоизлияния и повреждения спинного мозга (например, механического повреждения вследствие сжатия или изгибания позвоночника)), конвульсий (например, эпилептических конвульсий) или болезни, связанной со свободными радикалами (например, ишемического реперфузионного повреждения, воспалительного заболевания, системной красной волчанки, инфаркта миокарда, инсульта, травматической геморрагии, образования катаракты, увеита, эмфиземы, язвы желудка, неоплазии, лучевой болезни и т.д.) у животного.
Изобретение также обеспечивает применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента для индуцирования или поддержания общей анестезии у животного.
Изобретение также обеспечивает применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента для промотирования обезболивания у животного.
Изобретение также обеспечивает применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента для лечения мигрени у животного.
Изобретение также обеспечивает применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента для лечения бессонницы у животного.
Изобретение также обеспечивает применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента для промотирования анксиолитического эффекта у животного.
Изобретение также обеспечивает применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента для лечения синдрома отмены зависимости у животного.
Изобретение также обеспечивает применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента промотирования антиэметического эффекта у животного.
Изобретение также обеспечивает применение (-)-стереоизомера формулы (I), или его фармацевтически приемлемой соли, или его пролекарства для приготовления медикамента для агонизирования рецептора GABA у животного.
Изобретение также предоставляет способ синтеза и промежуточные соединения, раскрытые в данном описании, которые являются пригодными для получения (-)-стереоизомера формулы (I), или его соли, или его пролекарства
Краткое описание чертежей
Фигура 1 показывает эффект от действия на среднее артериальное кровяное давление (мм рт.ст.) (-) стереоизомера формулы (I), в которой Х представляет собой Н, у свиней после внутривенного (ВВ) вливания, по сравнению с пропофолом.
Фигура 2 показывает эффект от действия на сердечный ритм (ударов в мин) (-) стереоизомера формулы (I), в которой Х представляет собой Н, у свиней после внутривенного (ВВ) вливания, по сравнению с пропофолом.
Фигура 3 показывает эффект от действия на минутный сердечный выброс (литров в минуту, или л/мин) (-)стереоизомера формулы (I), в которой Х представляет собой Н, у свиней после внутривенного (ВВ) вливания, по сравнению с пропофолом.
Осуществление изобретения
Настоящее изобретение предоставляет (-)стереоизомер формулы (I) или его соль, или его пролекарство, как определено выше.
Абсолютная конфигурация такого стереоизомера определена как (R, R).
В одном варианте Х является Н. Когда Х является Н, стереоизомер также может называться (R,R)-2,6-ди-втор-бутилфенолом.
Было обнаружено, что, по сравнению с пропофолом, (R,R)-2,6-ди-втор-бутилфенол демонстрирует неожиданно улучшенный общий профиль активности в качестве анестетика. В частности, было найдено, что соединение оказывает более сильное анестезирующее действие, показывает более высокий терапевтический индекс и сохраняет сравнимый фармакокинетический профиль, например демонстрирует сходную скорость выведения из организма. Также соединение может оказывать менее сильное воздействие на артериальное давление, сердечный ритм и/или минутный сердечный выброс. Более того, полагают, что клинические испытания продемонстрируют, что соединение вызывает меньше боли при инъекции, чем пропофол. Боль при инъекции пропофола коррелировала с концентрацией пропофола в его липидном эмульсионном носителе. Было обнаружено, что при включении в состав в идентичной липидной эмульсии концентрация водной фазы (R,R)-2,6-ди-втор-бутилфенола значительно уменьшается (до более чем 90%) по сравнению с пропофолом.
Также неожиданно было обнаружено, что два других изомера 2,6-ди-втор-бутилфенола, (S,S) или (+) и (мезо) стереоизомера демонстрируют улучшенный гемодинамический профиль наряду со сходной или улучшенной фармакологической активностью по сравнению с пропофолом. Однако было обнаружено, что улучшенный суммарный профиль активности как анестетика (R,R)-2,6-ди-втор-бутилфенола является исключительным для этого изомера данного диалкилфенола.
Соответственно соединения согласно изобретению являются, в частности, пригодными для индуцирования или поддержания общей анестезии или промотирования обезболивания у пациента. Они особенно являются пригодными для анестезирования пациентов, имеющих повышенную чувствительность к гемодинамическим эффектам. Такие пациенты включают пациентов, страдающих от сердечно-сосудистой болезни, такой как болезнь коронарных артерий, кардиомиопатия, ишемическая болезнь сердца, порок клапана сердца и врожденный порок сердца; пациентов, страдающих от хронической гипертонии, цереброваскулярной болезни или травмы головного мозга; пациентов пожилого возраста (например, старше 50, 60, 70 или 80-летнего возраста); пациентов с острой потерей крови, обезвоживанием или тяжелой инфекцией, включая пациентов с геморрагическим шоком, гиповолемическим шоком или септическим шоком; и пациентов, получающих спинальную анестезию, эпидуральную анестезию или вазоактивные медикаменты (см., например, Reich DL et al, 2005, Anesth Analg 101, 622). Например, пациентом может быть человек, физическое состояние которого по классификации Американского общества анестезиологов (ASA) составляет, по меньшей мере, 3. Настоящее изобретение также рассматривает введение соединения согласно изобретению пациентам, которые не могут подвергаться предварительному обезболиванию при инъекции.
При использовании здесь термин "фармацевтически приемлемый носитель" включает растворители, адъюванты, эксципиенты или наполнители.
Термин "животное" включает млекопитающих таких как, например, люди, домашние животные, зоологические животные и крупный рогатый скот.
Термин "лечение" болезни или расстройства включает: 1) улучшение болезни или расстройства (т.е., остановку или уменьшение развития болезни или расстройства или, по меньшей мере, одного из его клинических симптомов); 2) улучшение, по меньшей мере, одного физического параметра, который может быть незаметным для пациента; 3) ингибирование болезни или расстройства, которое может быть выражено или физически (например, стабилизация заметного симптома), или физиологически (например, стабилизация физического параметра) или и тем, и другим, или 4) задержку начала болезни или расстройства.
Стереоизомерная чистота соединений и пролекарств, описанных здесь, может быть установлена с помощью обычных аналитических методов, хорошо известных специалистам в данной области техники. Например, для установления стереохимической чистоты отдельного стереоизомера можно использовать применение хиральных ЯМР шифт-реагентов, газохроматографический анализ на хиральных колонках, высокоэффективную жидкостную хроматографию с использованием хиральных колонок, поляриметрию, изотопное разбавление, калориметрию, ферментные методы, капиллярный электрофорез на хиральных гелях, образование диастереоизомерных производных посредством реакции с хиральными реактивами и стандартный способ анализа с помощью принятых аналитических методов. Альтернативно для установления стереохимической чистоты описанного здесь соединения можно использовать синтез с использованием исходных материалов известного стереохимического обогащения. В данной области известны другие аналитические способы для демонстрации стереохимической однородности.
Настоящее изобретение предоставляет стереоизомер формулы (I) или его соль, или его пролекарство в нерацемической (т.е. энантиомерно обогащенной) форме в центрах, отмеченных "*" в формуле (I). Таким образом, изобретение включает стереоизомер формулы (I) в обогащенной смеси, которая содержит не больше чем 45% другого энантиомера того соединения формулы (I), которое показано, или его соли, или пролекарства. (-)-Энантиомер, выделенный в примере 1 ниже, является специфическим стереоизомером изобретения. В некоторых вариантах изобретения обогащенная смесь содержит не больше чем около 40%, 35%, 30%, 25%, 20%, 15%, 10%, 5%, 4%, 3%, 2% или 1% других энантиомеров или диастереоизомеров соединения формулы (I), или его соли, или его пролекарства. В другом варианте осуществления изобретения обогащенная смесь содержит меньше чем около 1% других энантиомеров, или диастереоизомеров соединения формулы (I), или его соли, или его пролекарства.
Способы получения соединения формулы (I)
Как правило, соединения формулы (I) можно получить с помощью, по меньшей мере, трех разных подходов. В одном подходе рацемическую и/или диастереомерную смесь получают, используя обычные способы органического синтеза, или приобретают из коммерческих источников, а смесь разделяют с помощью методов, известных специалистам в данной области техники, таких как, например, фракционная кристаллизация, разделение на хиральных колонках (см. пример 1 ниже), образование и разделение производных или их кинетическое оптическое расщепление, и т.д., для того чтобы обеспечить по существу чистые стереоизомеры формулы (I) или стереоизомерно обогащенные смеси соединений формулы (I). Альтернативно для получения соединения формулы (I) можно использовать асимметричный синтез. Для получения по существу чистых стереоизомеров формулы (I) или стереоизомерно обогащенных смесей соединений формулы (I) можно использовать известные хиральные предшественники, применяя известные методы. Другие способы включают получение хиральных промежуточных продуктов, используя, например, энантиоселективную гидрогенизацию, энантиоселективное восстановление и/или энантиоселективное образование углерод-углеродной связи, ферментативное расщепление рацемических ацетатов и т.д., с последующим превращением в соединение формулы (I), используя обычные методы органического синтеза.
В одном способе стереоизомер формулы (I) можно получить с помощью хирального изоцианата чтобы образовать смесь карбаматных диастереомеров, которые можно разделить с получением искомого диастереомера формулы (I) после гидролиза карбаматного остатка.
Следовательно, согласно другому аспекту настоящее изобретение представляет способ получения (-) стереоизомера формулы (I) или его соли, или его пролекарства, который включает проведение гидролиза диастереизомера (-)-2,6-ди-втор-бутилфенилового эфира карбаминовой кислоты формулы

в которой R1 представляет хиральную аминогруппу с последующим, если необходимо, образованием свободного фенола, или его соли (такой, как фармацевтически приемлемая соль), или его пролекарства.
Гидролиз может быть проведен посредством реакции карбамата с основанием, например гидроксидом щелочного металла, таким как гидрокисид калия или натрия, которая дает соль (-)стереоизомера формулы (I), такую как соль щелочного металла. Свободный фенол можно получить обработкой этой соли кислотой, такой как соляная кислота. Хиральная аминогруппа может быть, например, хиральной 1-арилэтиламиногруппой, например (R)-1-арилэтиламиногруппой, такой как (R)-l-фенилэтиламиногруппа.
Карбаматный исходный материал можно получить путем реагирования рацемической смеси соответствующего 2,6-ди-втор-бутилфенола с хиральным изоцианатом с получением смеси диастереоизомеров, содержащей диастереоизомер (-)-2,6-ди-втор-бутилфенилового эфира карбаминовой кислоты; и отделения соответствующего диастереоизомера (-)-2-втор-бутилфенилового эфира карбаминовой кислоты формулы (II).
Хиральный изоцианат может быть, например, хиральным 1-арилэтилизоцианатом, например (R)-1-арилэтилизоцианатом, таким как (R)-(+)-1-фенилэтилизоцианат. Получающийся в результате продукт является смесью соответствующих диастереоизомеров 2-втор-бутил-6-изопропилфенилового эфира 1-арилэтилкарбаминовой кислоты. Искомый диастереоизомер можно отделить с помощью хроматографии с использованием, например, кремния в качестве стационарной фазы или посредством кристаллизации.
Неожиданно было обнаружено, что использование (R)-(+)-1-фенилэтилизоцианата в вышеописанном способе обеспечивает исключительно хорошее разделение стереоизомеров 2,6-ди-втор-бутилфенола по сравнению с использованием других хиральных ацилирующих или сульфонирующих реагентов, таких как хиральные карбоксильные кислоты или хиральные сульфоновые кислоты.
Способы получения стереоизомера формулы (I) или его соли предоставлены как дополнительные варианты осуществления изобретения.
Соли
В случаях, когда соединения являются достаточно кислыми, соль соединения формулы (I) может быть использована как промежуточное соединение для отделения и очистки соединения формулы (I) или его обогащенной смеси. Кроме того, подходящим может быть введение соединения формулы (I) в виде фармацевтически приемлемой соли. Примеры фармацевтически приемлемых солей включают соли, которые получены с помощью обычных методик, хорошо известных в данной области техники, например посредством реагирования достаточно кислого соединения формулы (I) с подходящим основанием, дающим физиологически приемлемый катион. Например, можно получить соли щелочного металла (например, натрия, калия или лития) или щелочноземельного металла (например, кальция).
Фармацевтические композиции
Фармацевтические композиции, раскрытые здесь, содержат соединение формулы (I), раскрытое здесь, с подходящим количеством фармацевтически приемлемого носителя, для того, чтобы обеспечить форму для надлежащего введения пациенту. Соединения формулы (I) могут быть включены в состав фармацевтических композиций и введены пациенту в целом ряде форм, приспособленных для выбранного способа введения, т.е. орально, парентерально, внутривенно, внутримышечно, местно или подкожно.
Таким образом, соединения формулы (I) могут быть введены системно в комбинации с фармацевтически пригодными носителями, такими как инертные растворители или съедобные носители. Такие композиции и препараты могут содержать, по меньшей мере, 0,1% активного соединения. Несомненно, процентное содержание композиций и препаратов может изменяться и может условно находиться в интервале между от около 0,1% до около 60% от массы данной стандартной лекарственной формы. Количество активного соединения в таких терапевтически пригодных композициях является таким, чтобы получался эффективный уровень дозировки.
Соединения формулы (I), описанные здесь, обычно включают в состав фармацевтических композиций, пригодных для внутривенного введения. Соединения формулы (I) могут быть относительно нерастворимыми в воде. Таким образом, для внутривенного введения соединения формулы (I) обычно включают в состав в водных средах, используя один или больше растворителей, несмешивающихся с водой, и один или больше эмульгаторов или поверхностно-активных веществ. Отдельные композиции могут включать один или больше дополнительных компонентов, таких как стабилизирующие вещества, модификаторы тоничности, основания или кислоты для регулирования рН и солюбилизаторы. Композиции также могут необязательно содержать консерванты, такие как, например, этилендиаминтетрауксусная кислота (EDTA) или метабисульфит натрия. Пригодные эмульсии масло-в-воде, содержащие консервант, такой как EDTA, который можно использовать вместе с соединениями, описанными здесь, охарактеризованы в Патентах США №5,908,869, 5,714,520, 5,731,356 и 5,731,355.
В описанных здесь фармацевтических композициях можно использовать широкий диапазон несмешивающихся с водой растворителей. Несмешивающийся с водой растворитель может быть растительным маслом, таким как, например, соевое, сафлоровое, хлопковое, кукурузное, подсолнечное, арахисовое, касторовое или оливковое масло. Альтернативно несмешивающийся с водой растворитель может быть сложным эфиром средне- или длинноцепочечной жирной кислоты, такой как, например, моно-, ди- или триглицерид, сложным эфиром комбинации средне- и длинноцепочечной жирной кислоты или химически модифицированным или синтетическим веществом, таким как этилолеат, изопропилмиристат, изопропилпальмират, сложный эфир глицерина, полиоксил или гидрогенизированное касторовое масло. Несмешивающийся с водой растворитель также может быть маслом морской рыбы, таким как, например, печень трески или другое масло из рыбы. Другие подходящие растворители включают фракционированные масла, такие как, например, фракционированное кокосовое масло или модифицированное масло соевых бобов. Несмешивающийся с водой растворитель может включать "структурированные липиды" (см., например, Lipid Biotechnology, T.M.Kuo and H.W.Gardner (eds.). Marcel Dekker, Inc. New York, NY). Многие структурированные липиды доступны от частных поставщиков, таких как Danisco A/S, Copenhagen Denmark и S&J Lipids, Ostrander, Огайо.
Фармацевтические составы, описанные здесь, также могут содержать эмульгатор. Подходящие эмульгаторы включают синтетические неионные эмульгаторы, такие как, например, этоксилированные эфиры, полиоксипропилен-полиоксиэтилен блок-сополимеры и фосфолипиды. Также можно использовать природные фосфолипиды, такие как фосфолипиды яиц или сои, и модифицированные или искусственно созданные фосфолипиды или их смеси. В некоторых вариантах осуществления эмульгаторы являются фосфолипидами яиц и фосфолипидами сои. Фосфолипиды яичного желтка включают фосфатидилхолин, лецитин и фосфатидилэтаноламин.
Фармацевтические составы, описанные здесь, могут включать липидную эмульсию, содержащую от около 0,1% до около 5% (масс./масс.) соединения формулы (I), от около 5 до около 25% (масс./масс.) несмешивающегося с водой растворителя и от около 40% до около 90% (масс./масс.) воды. Предпочтительная композиция содержит от около 0,5% до около 2% (масс./масс.) соединения формулы (I). В одном варианте осуществления фармацевтическая композиция содержит от около 0,5% до около 5% (масс./масс.) соединения формулы (I) и от около 0% до около 50% (масс./масс.) несмешивающегося с водой растворителя.
Фармацевтические составы, описанные здесь, также могут включать стабилизирующие вещества. Анионные стабилизаторы включают, например, фосфатидилэтаноламины, конъюгированные с полиэтиленгликолем (PEG-PE), и фосфатидилглицерины, отдельным примером которых является димиристоил фосфатидилглицерин (DMPG). Дополнительные стабилизаторы включают, но не ограничиваются этим, олеиновую кислоту и ее натриевую соль, холевую кислоту и дезоксихолевую кислоту и их соответствующие соли, катионные липиды, такие как стеариламин и олеиламин, и 3|3-[N-(N',N'-диметиламиноэтан)карбамоил]холестерин (DC-Chol).
Фармацевтические составы, описанные здесь, можно сделать изотоническими с кровью включением подходящего модификатора тоничности. Наиболее часто в качестве модификатора тоничности используется глицерин. Альтернативные модифицирующие тоничность агенты включают ксилитол, маннитол и сорбитол. Фармацевтические композиции обычно составляют так, чтобы они имели физиологически нейтральное значение рН, обычно в пределах 6,0-8,5. Значение рН можно отрегулировать добавлением основания, например NaOH или NаНСО3, или в некоторых случаях кислоты, такой как НСl.
Соединения формулы (I) могут быть заключены в состав с фармацевтически безопасными эмульсиями типа масло-вода, содержащими растительное масло, фосфатидный эмульгатор, обычно лецитин яиц или лецитин соевых бобов, и модификатор тоничности, такой как, например, Liposyn® II и Liposyn® III (Abbott Laboratories, Норт-Чикаго, Иллинойс) и Intralipid® (Fresenius Kabi AB, Упсала, Швеция), или другими подобными эмульсиями типа масло-вода.
Соединения формулы (I) также могут быть включены в состав с триглицеридом, содержащим сложные эфиры, по меньшей мере, одной жирной кислоты со средней длиной цепи (С6-С12). В некоторых вариантах осуществления триглицериды являются сложными эфирами C8-С10 жирной кислоты. Триглицериды, подходящие для включения в состав соединений формулы (I), включают, но не ограничиваются этим, Miglyol® (Condea Chemie GmbH (Witten, Германия)). Например, Miglyol® 810 или 812 (каприловый(С10)/каприновый (С8) глицерид) является пригодным для состава соединений формулы (I).
Кроме того, соединения формулы (I), описанные здесь, могут быть включены в состав аналогично фармацевтическим композициям пропофола, описанным, например, в Патентах США №4,056,635, 4,452,817 и 4,798,846.
Другие подходящие составы для применения в настоящем изобретении можно найти, например, в Remington's Pharmaceutical Sciences, Philadelphia, Pa., 19th ed. (1995).
Терапевтическое/профилактическое введение и дозы
Соединение формулы (I) и/или его фармацевтические композиции можно вводить отдельно или в комбинации с другими фармацевтическими средствами, включая соединения, раскрытые здесь, и/или их фармацевтические композиции. Соединения, раскрытые здесь, можно вводить или применять сами по себе или как фармацевтические композиции. Определенная фармацевтическая композиция зависит от желательного способа введения, как хорошо известно специалисту в данной области техники.
Соединения, раскрытые здесь, и/или их фармацевтические композиции могут быть введены субъекту посредством внутривенной болюсной инъекции, непрерывного внутривенного вливания, в виде таблетки для перорального приема, капсулы для перорального приема, раствора для перорального приема, внутримышечной инъекции, подкожной инъекции, трансдермального всасывания, посредством буккального (щечного) всасывания, внутриносового всасывания, ингаляции, подъязычно, интрацеребрально, интравагинально, ректально, местно, в частности в уши, нос, глаза или на кожу или любым другим удобным способом, известным специалистам в данной области техники. В некоторых вариантах осуществления соединения, раскрытые здесь, и/или их фармацевтические композиции, доставляются посредством лекарственных форм с замедленным высвобождением, включая лекарственные формы с замедленным высвобождением для приема внутрь. Введение может быть системным или местным. Известны разные системы доставки (например, инкапсулирование в липосомы, микрочастицы, микрокапсулы, капсулы, системы доставки лекарственного средства "аналгезия, управляемая пациентом", и т.д.), которые можно использовать для доставки соединений, раскрытых здесь и/или их фармацевтических композиций.
Количество соединений, раскрытых здесь, и/или их фармацевтических композиций, которое будет эффективно, может быть определено с помощью стандартных клинических методик, известных в данной области техники. Введенное количество соединений, раскрытых здесь, и/или их фармацевтических композиций будет конечно зависеть, в числе других факторов, от субъекта, которого лечат, веса субъекта, возраста субъекта, состояния субъекта, ожидаемого действия соединения, способа введения и мнения лечащего врача. Например, уровень дозировки (R, R) или (-) стереоизомера формулы I для получения общей анестезии может находиться в пределах от около 1 до около 10 мг/кг. Предпочтительные дозы для индуцирования анестезии находятся в пределах от около 1 до около 2,5 мг/кг. Предпочтительные поддерживающие дозы находятся в пределах от около 1 до около 15 мг/кг/час. Предпочтительные дозы для получения седативного эффекта находятся в пределах от около 0,3 до около 6 мг/кг/час.
Комбинированная терапия
В определенных вариантах осуществления соединения, раскрытые здесь, и/или их фармацевтические композиции можно использовать в комбинированной терапии, по меньшей мере, с одним терапевтическим средством. Соединения, раскрытые здесь, и/или их фармацевтические композиции и терапевтическое средство могут действовать аддитивно или, более предпочтительно, синергично. В некоторых вариантах осуществления соединения, раскрытые здесь, и/или их фармацевтические композиции вводят одновременно с введением другого терапевтического средства, такого как, например, другие седативные снотворные средства (например, этомидат, тиопентал, мидазолам, дексмедетомидин, кетамин), анестетики (например, десфлюран, севофлюран, изофлюран, закись азота), анальгетики (например, опиоид, такой как ремифентанил, морфий, меперидин, гидроморфон, метадон, фентанил, сулфентанил или алфентанил, или неопиоидный анальгетик, такой как, кеторолак, гапапентин, лидокаин или кетамин), паралитические средства, такие как рокурониум, цис-атракуриум, векурониум или панкурониум бромид, противорвотные средства (например, ондансетрон, доласетрон, дроперидол), сердечно-сосудистые средства (например, метопролол, пропранолол, эсмолол, клонидин, фенилэфрин, эфедрин, эпинефрин, норэпинефрин, допамин, дилтиазем, атропин, гликопирролат, лизиноприл, нитроглицерин, натрия нитропруссид, дигоксин, милринон), стероиды (например, дексаметазон, гидрокортозон, метилпреднизолон), антисептики (например, цефазолин, ванкомицин), мочегонные средства (например, фуросемид, гидрохлортиазид, спиронолактон), изменяющие настроение средства (например, флуоксетин, арипипразол) или возбуждающие средства, такие как никотин или цитизин.
Например, соединения, раскрытые здесь, и/или их фармацевтические композиции можно вводить вместе с другими терапевтическими средствами. В других вариантах осуществления соединения, раскрытые здесь, и/или их фармацевтические композиции вводят до или после введения других терапевтических средств.
Пролекарства
Термин "пролекарство" при использовании здесь относится к соединению, которое может метаболизироваться или превращаться in vivo для предоставления соединения формулы (I). Обычно пролекарства включают соединения, которые получаются при модификации фенольной группы в соединении формулы (I) с получением соответствующего соединения, которое может быть метаболизировано или превращено in vivo для того, чтобы предоставить соответствующее соединение формулы (I). О пролекарствах из фенольных соединений, а также способах их получения уже сообщалось. Например, см. публикации Патентных Заявок США №20070015716, 20060287525, 20060205969, 20060041011, 20050239725 и 20050107385.
Другие пригодные группы пролекарств обсуждаются в следующих публикациях Международных Патентных Заявок и публикациях Патентных Заявок США: WO 2005023204; US 2005107385; US 2005004381; WO 2004092187; WO 2004032971; US 2006100163; WO 2006033911; WO 2004033424; US 2005267169; WO 2003086413; US 2002370213; WO 2003057153; US 2001342755; US 2002099013; WO 2002034237; US 2004127397; WO 2002013810; WO 2000048572; US 2006166903; WO 200008033; US 2001025035; WO 9958555 и US 199875356; и в других последующих публикациях: Krasowski, M.D. Current Opinion in Investigational Drugs (Thompson Scientific) (2005) 6(1), 90-98; Fechner, J. et aL, Anesthesiology, 2004, 101, 3, 626-639; Altomare C. et al., European Journal of Pharmaceutical Sciences; 2003, 20, 1, 17-26; Sagara, Y. et al., Journal of Neurochemistry; 1999; 73, 6, 2524-2530, и Trapani, G., et al., International Journal of Pharmaceuticals, 1998, 775, 2, 195-204.
Как описано выше, было обнаружено, что другие два изомера 2,6-ди-втор-бутилфенола, (S,S) или (+) или (мезо) изомеры формулы (I) демонстрируют улучшенный гемодинамический профиль наряду с подобной или улучшенной фармакологической активностью по сравнению с пропофолом. Соответственно настоящее изобретение также обеспечивает каждый из этих изомеров, их пара-фтор производные и их фармацевтически приемлемые соли и их пролекарства, и их фармацевтические композиции для применения в качестве анестетиков.
Каждый из (S,S) или (+) или (мезо) стереоизомеров формулы (I), их соли и их пролекарства могут быть получены, следуя общим способам, описанным для получения соответствующих (R,R) или (-) стереоизомеров. Например, стереоизомеры можно отделить от рацемического соединения с помощью хиральной фазовой хроматографии, например, как описано в примере 2 здесь. Было обнаружено, что (S,S) или (+) стереоизомер 2,6-ди-втор-бутилфенола можно легко получить при реагировании рацемической смеси соответствующего 2,6-ди-втор-бутилфенола с ацилгалидом (например, ароилгалидом, таким как бензоилхлорид) с получением смеси карбонатных диастереомеров, которые можно разделить с получением искомого диастереомера формулы (I) после гидролиза карбонатного остатка. Пример такого способа описан в примере 5а в дальнейшем.
(S,S) или (+) и (мезо) стереоизомеры формулы (I) могут существовать, быть включены в состав и введены пациентам, как описано и приведено в пример здесь для (R,R) или (-) стереоизомеров. Для (S,S) или (+) стереоизомеров, уровень дозировки для получения общей анестезии может находиться в пределах от около 1 до около 12 мг/кг. Предпочтительные дозы для индуцирования анестезии находятся в пределах от около 1,2 до около 4 мг/кг. Предпочтительные поддерживающие дозы находятся в пределах от около 1,5 до около 30 мг/кг/час. Предпочтительные дозы для получения седативного эффекта находятся в пределах от около 0,5 до около 12 мг/кг/час. Для (мезо) стереоизомеров уровень дозировки для получения общей анестезии может находиться в пределах от около 1 до около 10 мг/кг. Предпочтительные дозы для индуцирования анестезии находятся в пределах от около 1 до около 3 мг/кг. Предпочтительные поддерживающие дозы находятся в пределах от около 1 до около 20 мг/кг/час. Предпочтительные дозы для получения седативного эффекта находятся в пределах от около 0,3 до около 8 мг/кг/час.
Способность соединения изобретения оказывать седативное или снотворное действие может быть установлена с помощью стандартных фармакологических моделей, которые хорошо известны в данной области техники. Гемодинамический профиль соединения изобретения можно определить, используя стандартные фармакологические модели, которые хорошо известны в данной области техники.
Изобретение теперь будет проиллюстрировано следующими неограничивающими примерами.
Пример 1
Отделение стереоизомера соединения формулы (I) посредством ВЭЖХ разделения диастереомерных карбаматов 2,6-ди-втор-бутилфенола.

где:
racemic-2,6-di-sec-butylphenol - рацемический 2,6-ди-втор-бутилфенол;
(R)-(1-isocyanatoethyl)benzene - (R)-(1-изоцианатоэтил)бензол;
pyridine - пиридин;
HPLC-ВЭЖХ;
1b. diastereomerically enriched carbamatc - диастереомерно-обогащенный карбамат;
dioxane - диоксан;
2. stereoisomer of Formula (I) (-)-2,6-di-sec-butylphenol - стереоизомер формулы (I) (-)-2,6-ди-втор-бутилфенол
Синтез R-(+) - (1-фенил-этил)-карбаминовой кислоты 2,6-ди-втор-бутилфенилового эфира (I):
Смесь 2,6-ди-втор-бутилфенола (2,06 г; 10 ммоль), R-(+)1-фенилэтилизоцианата (1,47 г; 10 ммоль) и 4-(диметиламино)пиридина (0,06 г; 0,5 ммоль) нагревали при 100°С в 10 мл безводного пиридина в течение ночи. Реакционную смесь выпаривали и полученный осадок обрабатывали этилацетатом (75 мл) и 1М HCl aq (100 мл). Органический слой дважды промывали 1М НСl aq (2×100 мл), рассолом (100 мл) и высушивали над безводным MgSO4. Выпаривание растворителя предоставляло карбамат (1) (3 г,85%).
Разделение диастереоизомеров R-(+) - (1-фенил-этил)-карбаминовой кислоты 2,6-ди-втор-бутилфенилового эфира (1b):
Разделение с помощью ВЭЖХ выполняли на ВЭЖХ колонке с силикагелем (250×41,5 мм), сорбент Si-60A 10 мм. Градиент: гексаны-этилацетат 0-10% за 72 мин; скорость потока 50 мл/мин; загрузка 1 г (1) в 10 мл гексана. Фракцию с искомым изомером карбамата (1b) собирали и высушивали (0,18 г, 72%).
Исследование оптической чистоты с помощью хиральной хроматографии
Исследования 2,6-ди-втор-бутилфенолов выполняли на колонке CHIRALCEL OD-H (4,6×250 мм) в изократическом режиме, мобильная фаза - н-гексаны, скорость потока 1 мл/мин, 20 мин, обнаружение 270 нм. Образцы растворяли в гексанах. Карбаматы предварительно гидролизовали до 2,6-ди-втор-бутилфенолов при 100°С в течение 1-2 минут в 1:1 смеси диоксан: 1М NaOH aq. 2,6-ди-втор-бутилфенолы экстрагировали эфиром. Эфирный слой выпаривали и остаточное масло растворяли в н-гексанах.
Синтез (-)-2,6-ди-втор-бутилфенола (2): R-(+) - (1-Фенил-этил)-карбаминовой кислоты (-)-2,6-ди-втор-бутилфеноловый эфир (1b) (4,1 г; 11,6 ммоль) растворили в 100 мл 1:1 смеси диоксан: 1М NaOH aq. Реакционную смесь перемешивали при 70°С в течение 15 мин. Летучие компоненты удаляли при пониженном давлении до объема 50-70 мл. Значение рН доводили до 3-4 с помощью 1М НСl. Фенол экстрагировали эфиром (3×50 мл), промывали 1 М НСl, рассолом и высушивали над безводным MgSO4. Выпаривание давало сырой продукт в виде желтого масла (2,4 г, ~100%). Выполняли вакуумную перегонку (120-125°С/~5 мм) (2,1 г, 89%). Оптическое вращение:

Пример 2. Прямое разделение стереоизомеров 2,6-ди-втор-бутилфенола
Разделение смеси стереоизомеров 2,6-ди-втор-бутилфенола выполняли с помощью хиральной ВЭЖХ. 2,6-ди-втор-бутилфенол (1 мг/мл в ВЭЖХ градиенте н-гексана) вводили в хиральную ВЭЖХ колонку (Daicel Inc., CHIRALCEL OD-H 20×250 мм, 5 мкм). Разделение выполняли с помощью изократического градиента, используя ВЭЖХ градиент н-гексана в качестве мобильной фазы при скорости потока 10 мл/минуту при температуре окружающей среды. Пик обнаружения был при 270 мм. 2,6-ди-втор-бутилфенол показал три пика в отношении 1:2:1, соответствующих энантиомеру 1 (искомый стереоизомер), (мезо)-2,6-ди-втор-бутилфенол и энантиомер 2. Отделенный энантиомер 1 (1 мг/мл) растворяли в ВЭЖХ градиенте н-гексана и вводили в хиральную ВЭЖХ колонку (Daicel, Inc., 5 CHIRALCEL OD-H 4,6×250 мм, 5 мкм), прогоняли с изократическим градиентом, используя ВЭЖХ градиент н-гексана в качестве мобильной фазы при скорости потока 0,7 мл/минуту при температуре окружающей среды. Пик обнаружения был при 270 мм, показал время удержания 17,1 минут и чистоту изомера >%99. Оптическое вращение:
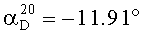
Пример 3. Состав
Следующая таблица иллюстрирует репрезентативную лекарственную форму, содержащую соединение формулы (I) для терапевтического применения.
Пример 4. Состав
Следующая таблица иллюстрирует репрезентативную лекарственную форму, содержащую соединение формулы (I) для терапевтического применения.
Пример 5. Получение (R,R)-ди-втор-бутилфенола с помощью хроматографии для разделения карбаматных диастереоизомеров
a) (R)-(+) - (1-фенил-этил)-карбаминовой кислоты 2,6-ди-втор-бутилфениловый эфир
Ди-втор-бутилфенол (доступный от компании Acros & АК Scientific) (5 грамм (г);
21,1 миллимоль (ммоль)) азеотропно высушивали на роторном испарителе (55°С, 48 Торр), используя 5 миллилитров (мл) толуола, затем загружали в 100-мл трехгорлую колбу, снабженную магнитной мешалкой, обратным холодильником, термопарой и входом для азота (N2). Добавили толуол (10 мл) и 4-диметиламинопиридин (0,085 г; 0,7 ммоль). Последним вводили (R)-(+)-(1-фенилэтилизоцианат (3,5 г; 3,65 мл; 23,63 ммоль). Получающуюся прозрачную желтую смесь нагревали под N2 при 90°С, используя колбонагреватель, и продолжали перемешивать при этой температуре, в то же время контролируя развитие реакции с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). После завершения реакции (18-24 часа (час)), судя по ВЭЖХ, реакционную смесь концентрировали на роторном испарителе (50-55°С/45-50 Торр) до получения полутвердого вещества (~9,4 г), которое растворяли в горячем 2-пропаноле (18 мл). Раствору позволяли достичь температуры окружающей среды, затравляли чистым (R)-(+) - (1-фенил-этил)-карбаминовой кислоты-2,6-ди-втор-бутилфениловым эфиром и помещали в холодильник (4°С) на 24-36 часов для медленной кристаллизации. Осажденные желтые твердые вещества фильтровали охлажденными и высушивали на фильтровальной воронке в течение 1-2 час. Первая порция продукта весила 2,8 г (выход 37,5%), как было обнаружено с помощью ВЭЖХ анализа ее чистота была больше чем (>) 95 процентов площади (А%). Материнскую жидкость концентрировали на роторном испарителе до ~2/3 исходного объема (отгоняли 4 мл пропанола) и затем охлаждали до 0-5°С в течение 6-8 час. Вторую порцию продукта фильтровали охлажденным, высушивали на фильтровальной воронке с получением дополнительных 2,6 г (выход 34,9%) продукта, который, как было обнаружено с помощью ВЭЖХ, был ~88 А%.
b) (R,R,R)-1-фенилэтилкарбаминовой кислоты-2,6-ди-втор-бутилфениловый эфир
В систему для ВЭЖХ Agilent, снабженную детектором на диодной матрице и колонкой Silica Column KROMASIL 0,46 см ID × 25 см длиной 10 мм, загрузили 714 мг рацемического R-(+) - (1-фенил-этил)-карбаминовой кмслоты-2,6-ди-втор-бутилфенилового эфира, растворенного в 10 мл гексан/этилацетата (98:2), чтобы получить 71,4 г/л подающий (загрузочный) раствор. Образец элюировали гексан/этилацетатом (98:2) при скорости 2 мл/мин при 25°С. Фракции, содержащие (R,R,R)-1-фенилэтилкарбаминовой кислоты 2,6-ди-втор-бутилфениловый эфир собирали и выпаривали под уменьшенным давлением при <55°С. При максимальной нагрузке (R,R,R)-стереоизомер собирали с хиральной чистотой 98,7% диастереомерного избытка (de) и суммарным выходом 53%.
с) (R,R)-ди-втор-бутилфенол
В 100-мл трехгорлую колбу, снабженную магнитной мешалкой, обратным холодильником, термопарой и входом для азота (N2), добавили тетрагидрофуран (ТГФ) (9 мл), (R,R,R)-1-фенилэтилкарбаминовой кислоты-2,6-ди-втор-бутилфениловый эфир (1 г; 2,8 ммоль) и 1,0 М гидроксид натрия (11,4 мл; 11,4 ммоль). Полученную прозрачную смесь нагревали под N2 при 55-60°С, используя колбонагреватель, и продолжали перемешивать при этой температуре, одновременно контролируя развитие реакции с помощью ВЭЖХ. После завершения реакции (6-8 час), судя по ВЭЖХ, реакционную смесь охлаждали до 15°С и фильтровали, чтобы удалить осажденную мочевину. Осадок на фильтре промывали холодным ТГФ (5 мл). Фильтрат и смыв объединяли, подкисляли до рН 2-3 3,0 М соляной кислотой (HCl) (3,5 мл). После перемешивания в течение 10 минут (мин) добавляли эфир (10 мл), затем полученную смесь энергично перемешивали в течение 15 мин, после чего отделяли слои. Органический слой промывали 3,0 М HCl (3 мл), рассолом (5 мл), высушивали сульфатом магния (MgSO4), фильтровали, чтобы удалить сушильный агент, затем концентрировали на роторном испарителе с получением полутвердого желтого осадка, который перемешивали с метилтретбутиловым эфиром (МТВЕ) (3 мл) в течение 15 мин и затем фильтровали. Осадок на фильтре промывали МТВЕ (2 мл). Фильтрат и смыв объединяли и затем концентрировали на роторном испарителе с получением титульного соединения в виде желтого масла (0,6 г, 100% сырой выход), которое, как было найдено с помощью ВЭЖХ, имело чистоту больше чем 93 А%.1Н ЯМР (DMSO-d6), как было обнаружено, согласуется с данной структурой.
Пример 5а. Получение (S,S)-ди-втор-бутилфенола с помощью хроматографии для отделения карбонатных диастереоизомеров
а) 2,6-ди-втор-бутилфенолбензоиловый эфир
Ди-втор-бутилфенол (доступный от компании Acros & АK Scientific) высушили на роторном испарителе (55°С, 48 Торр), используя толуол, затем загрузили в 100-миллилитровую (мл) трехгорлую колбу, снабженную магнитной мешалкой, обратным холодильником, термопарой и входом для азота (N2). Добавили толуол и 4-диметиламинопиридин, а затем бензоилхлорид. Полученную смесь нагревали под N2 при 90°С, используя колбонагреватель, и продолжали перемешивать при этой температуре, одновременно контролируя развитие реакции с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). После завершения реакции, судя по ВЭЖХ, реакционную смесь концентрировали на роторном испарителе (50-55°С/45-50 Торр) с получением полутвердого вещества.
b) (S,S)-2,6-ди-втор-бутилфенолбензоиловый эфир
В систему для ВЭЖХ Agilent, снабженную детектором на диодной матрице и колонкой Silica Column KROMASIL 0,46 см ID × 25 см длиной 10 мм, загрузили 2,6-ди-втор-бутилфенолбензоиловый эфир, растворенный в гексан/этилацетате (98:2), чтобы получить подающий (загрузочный) раствор. Образец элюировали гексан/этилацетатом (98:2) при 25°С. Фракции, содержащие (S,S)-2,6-ди-втор-бутилфенолбензоиловый эфир, собирали и выпаривали под уменьшенным давлением при <55°С с получением маловязкого масла.
c) (S,S)-ди-втор-бутилфенол
В 100-миллилитровую (мл) трехгорлую колбу, снабженную магнитной мешалкой, обратным холодильником, термопарой и входом для азота (N2), поместили тетрагидрофуран (ТГФ), (S,S)-2,6-ди-втор-бутилфенолбензоиловый эфир и 1,0 М гидроксид натрия. Полученную смесь нагревали под N2 при 55-60°С, используя колбонагреватель, и продолжали перемешивать при этой температуре, одновременно контролируя развитие реакции с помощью ВЭЖХ. После завершения реакции, судя по ВЭЖХ, реакционную смесь охлаждали до 15°С и фильтровали, чтобы удалить осажденную мочевину. Осадок на фильтре промывали холодным ТГФ. Фильтрат и смыв объединяли, подкисляли до рН 2-3 3,0 М соляной кислотой (НСl) (3,5 мл). После перемешивания в течение 10 минут (мин) добавляли эфир (10 мл), затем полученную смесь энергично перемешивали в течение 15 мин, после чего отделяли слои. Органический слой промывали 3,0 М НСl, рассолом, высушивали сульфатом магния (MgSO4), фильтровали, чтобы удалить сушильный агент, и затем концентрировали на роторном испарителе с получением осадка, который перемешивали с метилтретбутиловым эфиром (МТВЕ) в течение 15 мин и затем фильтровали. Осадок на фильтре промывали МТВЕ. Фильтрат и смыв объединили и затем концентрировали на роторном испарителе с получением титульного соединения.
Биологические тесты
Фармакологический профиль (R,R)-ди-втор-бутилфенола оценивали в сравнении с пропофолом в тестах, описанных в следующих примерах. В этих примерах (R,R)-ди-втор-бутилфенол называется соединением 1.
Пример 6. Исследование срезов гиппокампа мозга крыс
Способность соединения 1 и пропофола усиливать действие агонистов на рецепторе g-аминомасляной кислоты подтипа А (GАВАA рецептор) проверяли и сравнивали в электрофизиологическом исследовании срезов гиппокампа мозга крыс.
Соединение 1, полученное, как описано в примере 5, и пропофол тестировали в пяти концентрациях: 0,1; 1, 3, 10 и 30 микромолей (мМ). Стоковые растворы 100 миллимолярного (мМ) пропофола и 100 мМ соединения 1, каждый в ДМСО, разводили в солевом растворе, чтобы достигнуть соответствующих концентраций; 30 мкМ образцы содержали 0,03% ДМСО; растворы, содержащие до 0,1% ДМСО, не оказывают значительного влияния на исследование срезов. Значения ЕС50 и ЕС20 определяли с помощью методики, сходной с методикой, описанной в Casasola et al, 2002, Epilepsy Research 47, 257, с изменениями как указано ниже.
Срезы гиппокампа мозга крысы получали, как указано далее: Самцов крыс линии Wistar (100-125 г) обезболивали изофлураном и декапитировали, мозг быстро удаляли, собирали, замораживали и резали с применением прибора вибратом (OTS-4000, Electron Microscope Sciences) на поперечные срезы толщиной 400 микрон (мкм). Срезы переносили в подогретую (33°С), погруженную (в жидкость) регистрирующую камеру, перфузируемую при скорости 2,5-3 мл/мин модифицированной искусственной спинномозговой жидкостью (120 мМ хлорида натрия, 3,5 мМ хлорида калия, 2,5 мМ хлорида кальция, 1,3 мМ хлорида магния, 1,25 мМ фосфата натрия, 26 мМ карбоната натрия, 10 мМ глюкозы, насыщенной на 95% кислородом, рН 7,4). Срезы гиппокампа приводили в равновесие в регистрирующей камере в течение, по меньшей мере, 1 часа.
Электрофизиологическое исследование выполняли следующим образом:
Стеклянный стержневой электрод (диаметр кончика 1-2 мкм) заполняли 3М хлоридом натрия (NaCl) и помещали в слой пирамидальных клеток СА1 срезов гиппокампа. Концентрический биполярный стимулирующий электрод 25 мкМ (SNE-100, Rhodes Medical Supply) помещали в stratum radiatum области СА1, чтобы стимулировать коллатеральный/комиссуральный путь Шеффера. Ответы популяции пирамидальных клеток СА1 регистрировали на приборе Axoprobe-1A (Axon Instruments, Molecular Devices, Sunnyvale, Калифорния). Для получения данных использовали pCLAMP 8.2 (Axon Instruments), и для анализов использовали Clampfit (Axon Instruments). Стимулирование состояло из одного прямоугольного импульса (продолжительность 0,3 миллисекунды (мсек)) от стимулятора Grass SI 1 Stimulator (Grass Medical Instruments), который доставлялся каждые 20 сек на протяжении всего экспериментального периода. Интенсивность стимула была отрегулирована так, чтобы вызывать ответ 80-90% максимума. Размах амплитуды ответа популяции от каждого стимула измеряли как показатель возбудимости клеток.
Соединение 1 и пропофол, каждое в присутствии ЕС20 мускимола (2 мкМ), каждое перфузировали последовательно, начиная от самой низкой до самой высокой концентрации, в модифицированной искусственной спинномозговой жидкости соответствующих срезов гиппокампа. Действия каждой концентрации измеряли от 4 до 7 мин после применения соединения 1 или пропофола, соответственно, причем было найдено, что в это время изменения в ответе являлись устойчивыми. Мускимол (10 мкМ) применяли после применения соединения 1 или пропофола, чтобы проверить чувствительность препарата, только если соединение 1 или пропофол не вызывал соответствующего ингибирования амплитуды спайка (суммарного ответа) популяции СА1 (<90% ингибирование). Антагонист GАВАA рецепторного канала пикротоксин (50 мкМ) применяли в конце снятия показаний, чтобы подтвердить, что ответ был опосредован GABAA рецептором.
Данные собирали и анализировали с применением программ Clampfit и Excel (Microsoft) и представляли как средние и отдельные значения. Степень популяционного эффекта (%) получали с помощью измерения амплитуды спайка популяции СА1 до (контроль) и после совместного применения мускимола (ЕС20) и соединения 1 или пропофола (различие нормализовали к контролю и умножали на 100 для получения процента эффекта).
Данные показали, что соединение 1 было сильным потенцирующим средством действия агонистов на рецепторе GАВАA в срезах гиппокампа мозга крыс со значением ЕС50 2,5 мкМ. Для пропофола значение ЕС50 составляло 4,8 мкМ. Таким образом, соединение 1 ведет себя подобно пропофолу в исследовании срезов гиппокампа мозга и действительно усиливает мускимол-опосредованный ответ на рецепторе GABAA.
Пример 7. Исследования специфичности
Проверили и сравнили способность соединения 1 и пропофола взаимодействовать с целым рядом биологических мишеней.
Фармакологические анализы соединения 1, полученного как описано в примере 5, и пропофола были выполнены компанией Cerep, Inc. (Redmond, WA, США) в их системе «Профиль разнесения» ("Diversity Profile"), стандартный профиль 71 рецептора (59 пептидов, непептидов или ядерных рецепторов; 7 ионных каналов; 5 аминных транспортеров) и 16 ферментов. И соединение 1, и пропофол были проверены в дозе 10 мкМ (терапевтически релевантная концентрация).
Результаты показали, что соединение 1 ведет себя подобно пропофолу в отношении проверенных 71 рецептора и 16 ферментов. Например, соединение 1 и пропофол (каждый из них) показали самый большой эффект (больше чем 30% ингибирования контрольного связывания) в исследовании по измерению связывания пикротоксинина (активное соединение пикротоксина) на хлорном канале, выделенном из коры головного мозга крысы. Этот лиганд-зависимый ионный канал g-аминомасляной кислоты (GABA) является главной мишенью действия пропофола. Более того, и соединение 1, и пропофол (каждый из них) показали больше чем 20% ингибирования контрольного связывания относительно только одного из 16 проверенных ферментов: фосфодиэстеразы 2 (PDE2). Не наблюдалось значительных эффектов для альфа2, NMDA, РСР, бензодиазепиновых или опиоидных рецепторов.
Пример 8. Боль при инъекции - Концентрация водной фазы
Полагают, что боль при инъекции, общая проблема введения пропофола, вызывается пропофолом, присутствующим в водной фазе липидной эмульсии (см., например, Klement W et al, 1991, Br J Anaesth 67, 281). В некоторых исследованиях сообщалось о значительном снижении боли при инъекции, когда концентрацию водной фазы пропофола уменьшали по сравнению с количеством пропофола в водной фазе дипривана (см., например, Doenicke AW et al, 1996, Anesth Analg 82, 472; Ueki R et al, 2007, J Anesth 21, 325).
Была определена концентрация соединения 1 в водной фазе (концентрация водной фазы) липидного эмульсионного состава. Эта концентрация водной фазы была сравнима с концентрацией водной фазы пропофола, включенного в состав такой же композиции, и с концентрацией водной фазы дипривана® (AstraZeneca, Wilmington, DE, USA).
Была составлена однопроцентная (1%) композиция соединения 1 в соответствии с примером 4, соединение 1 получили, как описано в примере 5. Композиция 1% пропофола была составлена таким же образом. Использовали диприван (эмульсия для инъекции 1% пропофола) от компании AstraZeneca.
Концентрации водных фаз соединения 1 и пропофола определяли с помощью метода ультрафильтрации, описанного в Teagarden DL at al., 1988, Pharmaceutical Research 5, 482. Коротко, четыре образца композиции (0,4 мл) с 1% соединения 1, четыре образца композиции (0,4 мл) с 1% пропофола и два образца (0,4 мл) дипривана поместили на микроцентрифужные фильтры Ultrafree®-MC (Millipore, Billerica, MA) и отделили водные фазы от жидких фаз с помощью микроцентрифугирования в течение 15 мин при 5000 об/мин. Концентрации соединения 1 и пропофола в соответствующих водных фазах количественно определили с помощью метода жидкостной хроматографии/тандемной масс-спектрометрии (LC/MS/MS) по отношению к стандартным кривым соединения 1 и пропофола с использованием тимола в качестве внутреннего стандарта (анализы были выполнены Alturas Analytics, Inc., Москва, ID).
Концентрация водной фазы соединения 1 в композиции с 1% соединения 1 составляла 0,38±0,02 мкг/мл. Концентрация водной фазы пропофола в композиции с 1% пропофола составляла 6,28±0,41 мкг/мл. Концентрация водной фазы пропофола в диприване составляла 4,1 мкг/мл.
Эти результаты показали уменьшение на 94% в концентрации водной фазы соединения 1 по сравнению с концентрацией водной фазы пропофола в идентичных композициях и уменьшение на 91% в концентрации водной фазы соединения 1 по сравнению с концентрацией водной фазы пропофола в диприване.
Пример 9. Фармакокинетические исследования
Фармакокинетические (РК) исследования проводили на домашних свиньях, чтобы оценить фармакодинамические эффекты соединения 1 и сравнить их с фармакодинамическими эффектами пропофола.
Композицию с 1% соединения 1, полученную как описано в примере 5 и составленную в соответствии с примером 4, вводили 6 свиньям с помощью 20-мин внутривенного (ВВ) вливания при скорости 0,380 мг/кг/мин (общая доза 7,6 мг/кг) и одной свинье при 0,456 мг/кг/мин (общая доза 9,12 мг/кг). Концентрации в плазме соединения 1 сравнивали с литературными данными по пропофолу со сходным протоколом, в котором композицию 1% пропофола, составленную таким же образом, как композиция соединения 1, вводили 5 свиньям с помощью 10-мин внутривенного (ВВ) вливания при скорости 0,750 мг/кг/мин (общая доза 7,5 мг/кг).
По данным этого исследования соединение 1 показало сходный фармакокинетический профиль относительно пропофола на модели на свиньях. Трехчастевая модель лучше всего описала соединение 1 и данные для пропофола. Клиренс соединения 1 превышал предполагаемый печеночный кровоток, подобно пропофолу. Также соединение 1 показало сходный метаболический путь пропофола у свиней и у людей: глюкуронидация в 1-положении вместе с 4-положением подвергается гидроксилированию с последующей конъюгацией глюкуронида и сульфата. Исследование на собаке с повышением дозировки показало сходные концентрации в плазме при выведении соединения 1 и пропофола, также указывая на сходные уровни клиренса в этих видах.
Пример 10. Анестетические эффекты у крыс
Исследовали ответ на анестезирующую дозу болюсной ВВ инъекции соединения 1 по сравнению с пропофолом у крыс.
Для определения начала и продолжительности анестезии использовали утвержденную модель общей анестезии на грызунах (см. Hill-Venning С et al., 1996, Neuropharmacology 35, 1209; Lingamaneni R et al., 2001, Anesthesiology 94, 1050), как показано с помощью потери рефлекса выпрямления (LORR) и времени восстановления (временной интервал от возвращения рефлекса выпрямления до тех пор, пока крыса не будет способна цепляться и влезать на стальной каркас и нормально переходить с места на место). Также оценивали минимальную дозу для достижения LORR и максимально переносимую дозу (MTD).
Композицию 1% соединения 1, полученную, как описано в примере 5, и составленную в соответствии с примером 4, или диприван вводили посредством болюсной внутривенной (ВВ) инъекции 2,5 мл/мин 6 самцам крыс Sprague-Dawley (200-300 г) на дозовую группу за время, необходимое для введения доз, описанных ниже. Относительную активность оценивали путем установления дозы, необходимой для того, чтобы вызвать у 50% крыс потерю рефлекса выпрямления (HD50), и дозы, необходимой для того, чтобы вызвать 7-минутную анестезию (НD7 мин). Были исследованы дозы 1,9; 2,3; 3,0; 7,0; 13,7; 14,0 и 15,2 мг/кг для соединения 1 и 3,5; 4,0; 7,0 и 14,0 мг/кг - для дипривана.
Результаты показали, что болюсное внутривенное (ВВ) введение соединения вызывало дозозависимую продолжительность анестезии у крыс. Время наступления (начала) LORR составляло меньше чем 15 сек, когда вводили соответствующие лекарственные средства в дозе, по меньшей мере, 3 мг/кг для соединения 1 и в дозе, по меньшей мере, 7,0 мг/кг для пропофола. Соединение 1 не вызывало LORR при дозе 1,9 мг/кг, но вызывало LORR при всех других испытанных дозах. Пропофол не вызывал LORR у 4 из 6 крыс, проверенных в дозе 3,5 мг/кг, но вызывал LORR при всех других проверенных дозах. Таблица 1 сравнивает результаты по HD50, НD7 мин, MTD и терапевтический индекс (TI; определенный здесь как отношение MTD к HD7 мин) для соединения 1 и пропофола. Одна крыса погибла при введении 14 мг/кг дипривана. Две крысы погибли при введении 15,2 мг/кг соединения 1. Время восстановления показало незначительную взаимосвязь с дозой, за исключением высоких доз соединения 1, которые также вызывали продолжительную LORR.
Итак, соединение 1 показало эффективность при более низких дозах, чем пропофол, и также показало более высокую MTD и улучшенный TI по сравнению с пропофолом.
Также в этих тестах был оценен (S,S)-2,6-ди-втор-бутилфенол, полученный в соответствии с примером 2, в дозах 2, 3, 4, 5, 6, 28, 35, 42, 49 и 56 мг/кг. Таблица la показывает результаты по HD50, HD7мин, MTD и TI для этого соединения. Одна из шести крыс погибла при введении 49 мг/кг (S,S)-2,6-ди-втор-бутилфенола.
В отдельном исследовании крысам вводили 7 мг/кг соединения 1 в кремафоре или пропофол, (S,S)-2,6-ди-втор-бутилфенол или (мезо)-2,6-ди-втор-бутилфенол (полученный в соответствии с примером 2) в таких же дозах и композициях. Результаты показаны в таблице Ib. Одна из 6 крыс погибла при введении 21 мг/кг 1% (мезо)-2,6-ди-втор-бутилфенола, включенного в кремафор; однако остальные 5 крыс показали продолжительность анестезии 34 мин.
Итак, активность (S,S)-2,6-ди-втор-бутилфенола была сходна с пропофолом. Активность (мезо)-2,6-ди-втор-бутилфенола была улучшена по сравнению с пропофолом. Оба стереоизомера показали улучшенные MTD и терапевтические индексы по сравнению с пропофолом.
Пример 11. Анестетический и гемодинамический эффекты у собак породы бигль.
Исследование с повышением дозировки проводили на собаках, чтобы продемонстрировать анестетический и гемодинамический эффекты болюсного внутривенного (ВВ) введения соединения 1 по сравнению с пропофолом.
Конечными точками в этом исследовании была зависимость от дозы индуцирования, продолжительности, глубины и качества анестезии и гемодинамических эффектов болюсного внутривенного (ВВ) введения соединения 1 или пропофола. Использовали композицию 1% соединения 1, полученного как описано в примере 5 и составленную в соответствии с примером 4, и композицию 1% пропофола, составленную таким же образом.
Электроэнцефалографическое (ЭЭГ) измерение глубины анестезии проводили с помощью биспектрального индекса (BIS), который является одной из нескольких систем, используемых для измерения эффектов анестетических лекарственных средств на мозг, и для того, чтобы следить за изменениями уровня обезболивания или анестезии. BIS представляет собой математический алгоритм, который анализирует данные от ЭЭГ, и результатом является одно число от 100 (полное сознание) до 0 (изоэлектрическая ЭЭГ). Другие оценки включали шкалу седативного эффекта, клинические наблюдения, кровяное давление, электрокардиограмму (ЭКГ) и насыщение кислородом.
Собакам породы бигль (самцы, 2-4-летнего возраста, 8-10 кг) имплантировали порты для сосудистого доступа. Во время хирургической операции по вживлению портов головы собак брили, размечали для размещения ЭЭГ электродов и вводили ВОТОХ® (Allergan, Inc., Irvine, CA; очищенный комплекс ботулических токсинов типа А): всего вводили 40 единиц на собаку за 5 внутримышечных (IM) инъекций через бровь. Инъекции предназначались для подавления мышечных движений и электромиографическую (EMG) интерференцию с сигналом BIS.
Исследование было перекрестным. Каждая собака получала от 2 до 4 возрастающих болюсных внутривенных (ВВ) доз (введенных в течение 60 секунд) соединения 1 или пропофола, отделенных, по меньшей мере, 30 минутами (или временем, пока собака не просыпалась) до тех пор, пока не будет достигнута MTD. MTD была определена как доза, которая уменьшала среднее артериальное давление крови (MAP) на 50% или до значения меньше чем 50 миллиметров ртутного столба (мм рт.ст.). Все животные получали дополнительно кислород и, если необходимо, вентиляторную поддержку после 4 минут апноэ.
Глубину анестезии определяли посредством оценки наличия или отсутствия lash (махать хвостом) рефлекса, ответа на легкий удар между бровями или слуховой раздражитель, сжатие пальца (toe pinch), и дыхания. Наличие каждого признака считали как 1 и отсутствие каждого как 0. Это давало возможность подсчитать совокупный счет седативного эффекта во множестве временных точек в течение 30 мин между дозами (5=пробуждение, 0=апноэ/глубокая анестезия). Качество анестезии оценивали, отмечая плавность индуцирования (состояния анестезии), с помощью качественной оценки мышечного тонуса и по наличию непроизвольного движения. Случаи непроизвольных движений (например, во время «выхода») подсчитывали, как присутствие или отсутствие на всем протяжении наблюдаемого периода для каждой дозы. BIS и гемодинамические эффекты анализировали с помощью 2-факторного дисперсионного метода анализа ANOVA с последующим t-тестом с поправкой Бонферрони для множественных сравнений действия времени и дозы.
А. Анестетические эффекты
Способность соединения 1 и пропофола, введенных с помощью болюсного внутривенного (ВВ) вливания, чтобы вызвать дозозависимую анестезию самопроизвольно дышащих собак породы бигль без премедикации (3,3-30 мг/кг/дозу; 1-10 собак на дозу), показаны в таблице 2. Две из 3 собак, которым вводили 15 мг/кг пропофола, достигли MTD при 15 мг/кг. Следовательно, только 1 собаке дали дозу 30 мг/кг пропофола.
Также данные показали, что анестезия была вызвана в течение 1 минуты при всех дозах соединения 1 и пропофола. Продолжительность анестезии, измеренная временем сна, была длиннее с соединением 1, чем с пропофолом во всех дозах. Совокупный счет седативного эффекта показал приблизительно равносильную глубину анестезии обоих препаратов, и пропофола, и соединения 1, в дозе выше 5 мг/кг. Не наблюдалось значительного различия между значениями BIS у собак, которым вводили соединение 1 в дозе 10 мг/кг либо пропофол в дозе 10 мг/кг или 15 мг/кг. Соединение 1 вызывало большее действие на BIS в дозах, по меньшей мере, 15 мг/кг, но эти дозы являются очень высокими, и потенциально не уместными клинически. Качество анестезии (плавность индуцирования анестезии, качественная оценка мышечного тонуса, наличие непроизвольного движения) соединения 1 было сходно с пропофолом.
Также в этом тесте были оценены (S,S)-2,6-ди-втор-бутилфенол и (мезо)2,6-ди-втор-бутилфенол, полученные в соответствии с примером 2. Таблица 2а показывает дозозависимую продолжительность анестезии (время сна) для этих соединений.
Также данные показали, что анестезия была вызвана в течение 1 мин при всех дозах (S,S)-2,6-ди-втор-бутилфенола и (мезо)2,6-ди-втор-бутилфенола. Продолжительность анестезии, измеренная временем сна, была сходна с пропофолом для (S,S)-2,6-ди-втор-бутилфенола и длиннее для (мезо)2,6-ди-втор-бутилфенола. Качество анестезии (S,S)-2,6-ди-втор-бутилфенола было сходно с пропофолом, но было хуже для (мезо)2,6-ди-втор-бутилфенола.
В.Гемодинамические эффекты: кровяное давление
Гемодинамические данные, такие как среднее артериальное давление (MAP), регистрировали на базовом уровне 1, 2, 4, 8, 15, 20 и 30 мин. Соединение 1 вводили в дозах 5, 10, 15 и 30 мг/кг соответственно 3, 6, 6 и 3 собакам. Пропофол вводили в тех же самых дозах 3, 5, 5 и 1 собакам, соответственно. Только 1 собака получила 30 мг/кг пропофола, так как критерий MTD был достигнут при дозе 15 мг/кг у двух животных. Данные анализировали с помощью 2-факторного дисперсионного метода анализа ANOVA с последующим t-тестом с поправкой Бонферрони для множественных сравнений.
Сравнение данных показало, что пропофол вызывал значительно более сильное действие на MAP, чем соединение 1. Таблица 3 представляет пример, в котором процент изменений среднего артериального давления (MAP %) от базового уровня 4 мин после болюсного внутривенного (ВВ) введения 10, 15 или 30 мг/кг соединения 1 сравнивается с изменениями MAP %, вызванными некоторыми дозами пропофола.
Также в этом тесте оценили (S,S)-2,6-ди-втор-бутилфенол и (мезо)2,6-ди-втор-бутилфенол, полученные в соответствии с примером 2. Сравнение данных показало, что пропофол оказывал значительно большее действие на MAP, чем (S,S)-2,6-ди-втор-бутилфенол и (мезо)2,6-ди-втор-бутилфенол. Таблица 3а предоставляет пример, сравнивающий изменения MAP % от базового значения 4 мин.
Пример 12. Анестетический и гемодинамический эффекты на собаках монгрелах
Это исследование сравнивало эффект общей внутривенной анестезии на хронически оснащенных измерительной аппаратурой собаках монгрелах, которым вводили соединение 1 или пропофол. Оценки включали анализ гемодинамических параметров, таких как кровяное давление, сердечный ритм, минутный сердечный выброс, а также клинические биохимические параметры и ЭЭГ исследования.
Композицию 1% соединения 1, полученного как описано в примере 5 и составленную в соответствии с примером 4, и диприван (инъецируемая эмульсия 1% пропофола) сравнивали на взрослых (по меньшей мере, 9-месячного возраста; приблизительно 20-40 кг) собаках монгрелах.
Общую анестезию вызывали у собак внутривенным (ВВ) введением 7 мг/кг дипривана, собак трахеально инкубировали и механически вентилировали. Общую анестезию поддерживали с использованием end-tidal (в конце спокойного выдоха) 2,2% севофлурана в кислороде. Торакотомия была выполнена в пятом межреберном пространстве слева, причем наполненные гепарином катетеры помещали в проксимальную нисходящую грудную аорту (датчик кровяного давления Р50 Gould, Окснард, Калифорния) и в правую, и левую артерии, чтобы обеспечить ВВ доступ. Ультразвуковой датчик времени прохождения потока (Т108, Transonic Systems, Итака, Нью-Йорк) был помещен вокруг восходящей грудной аорты. 20 кГц допплеровский потоковый зонд (Model HDP-20-3.5, Triton Surgical Technologies, Сан-Диего, Калифорния) был помещен вокруг левой передней нисходящей коронарной артерии. Шесть сономикрометрических кристаллов МГц (Hartley, Хьюстон, Техас) были имплантированы в субэндокард. Высокоточный микроманометр (Р7, Konigsberg Instruments, Пасадена, Калифорния) был вставлен в левый желудочек. Гидравлический сосудистый обтуратор (In Vivo Metric Systems, Хилдсбург, Калифорния) был расположен вокруг грудной нижней полой вены. Управление техническими средствами было выведено на поверхность, грудную стенку закрыли послойно, а пневмоторакс был эвакуирован. Собакам предоставляли период восстановления минимум 7 дней до проведения опытов и давали возможность приспособиться к стоянию в поддерживающей повязке в течение восстановительного периода.
Собаки голодали в течение ночи. Бодрствующих собак помещали в поддерживающую повязку и вставляли игольчатые электроды для регистрирования ЭКГ в отведении II. Электроды для отведений с черепа размещали для записи ЭЭГ (МР150, Biopac Systems, Голета, Калифорния) в трех биполярных записывающих конфигурациях, которые замеряли лобную, височную, теменную и затылочную области. Затем собаки получали ВВ болюс 500 мл нормального солевого раствора, после чего внутривенное (ВВ) вливание нормального солевого раствора устанавливали со скоростью 3 мл/кг/час (60-120 мл/час на собаку) в продолжение эксперимента. Собакам давали возможность стабилизироваться в течение 30 минут. ЭЭГ записывали непрерывно в течение эксперимента. Анализы крови на газы и кислотность и химические анализы включали рН, рO2, sO2, pCO2, tCO2, карбонат, калий, натрий и избыток оснований, и измеряли сразу после отбора крови с помощью анализатора оценки газового и электролитного состава крови (ABL-505, Radiometer, Копенгаген). Клинические биохимические анализы крови включали альбумин, отношение альбумин/глобулин, щелочную фосфатазу, аланинаминотрансферазу (SGPT), аспартатаминотрансферазу (SGOT), бикарбонат, прямой билирубин, азот мочевины крови BUN, отношение BUN/креатинин, кальций, хлориды, холестерин, СК, креатинин, глобулин, глюкозу, фосфор, калий, натрий, отношение натрий/калий и общий белок. После стабилизации регистрировали измерения ЭЭГ, показатели гемодинамики, ЭКГ и газы крови. Извлекали образцы крови для исследования параметров РК и клинической химии и вызывали петли давление-объем, данные регистрировали.
Сразу после базовых измерений собаки получали 4 мг/кг (1 собака) или 5 мг/кг (6 собак) внутривенную (ВВ) болюсную дозу соединения 1 или 7 мг/кг внутривенную (ВВ) болюсную дозу пропофола (7 собак) в течение 1 минуты, чтобы вызвать общую анестезию. После введения в состояние анестезии собак инкубировали трахеально и механически вентилировали, используя 50% кислород в азоте на всем протяжении вливания лекарственного средства и периодов восстановления. Начиная с 4 минут после окончания болюсной дозы, собакам, которые получили болюс соединения 1, вводили серию из четырех 15-минутных внутривенных (ВВ) вливаний со скоростью 0,25; 0,5; 1,0 и 2,0 мг/кг/мин соединения 1 поэтапно (in a stepwise crossover fashion); такой же протокол использовали для собак, получивших болюс пропофола, за исключением того, что пропофол вливали с указанной скоростью и временем (введения). MAP контролировали непрерывно, и дозирование немедленно прекращали, если MAP уменьшалось ниже 50 мм рт.ст. в любое время, или если сердечный ритм превышал 200 ударов в минуту. Дозирование прекратили у одной собаки в конце периода вливания соединения 1 в дозе 1,0 мг/кг/мин и у двух других собак в течение периода вливания соединения 1 в дозе 2,0 мг/кг/мин. В конце каждого 15-минутного вливания регистрировали измерения ЭЭГ, показатели гемодинамики, ЭКГ и газы крови, образцы крови отбирали для исследования РК и вызывали петли давление-объем, данные регистрировали. После дозирования собакам давали возможность восстановиться. Когда субъективное определение клинических параметров указывало на достаточное восстановление от общей анестезии, вентилирование останавливали, трахею экстубировали. Отмечали время экстубирования трахеи. Через 30 минут после завершающего вливания регистрировали ЭЭГ, параметры гемодинамики, ЭКГ и результаты анализов крови на газы и кислотность, отбирали образцы крови для РК и вызывали петли давление-объем, данные регистрировали.
Были определены концентрации соединения 1 и пропофола в плазме собак и были оценены концентрации 5 метаболитов (1 окислительного, 3 глюкуронид-конъюгированных и 1 сульфат-конъюгированного), с помощью жидкостной хроматографии (LC) и тандемной масс-спектрометрии (MS/MS) (выполнена при Alturas Analytics).
Результаты показали, что данные анализов крови на газы и кислотность и клинические биохимические данные были устойчивы. Исследования ЭЭГ показали дозозависимый седативно-наркотический эффект и не было подтверждения судорожной или предсудорожной активности. Все собаки приходили в себя (восстанавливались) после общей анестезии с похожей скоростью, независимо от того, вводили им соединение 1 или пропофол. Соединение 1 и метаболиты глюкуронида и в 1-положении и в 4-положении были обнаружены в плазме. Концентрации в плазме были сопоставимы с режимом введения лекарственного средства.
На этой модели при терапевтически релевантных дозировках результаты ЭЭГ показали более сильный анестетический эффект соединения 1 по сравнению с пропофолом. Не было статистически значимых различий между показателями MAP и сердечного ритма для соединения 1 и пропофола. Минутный сердечный выброс у собак, получавших пропофол, был значительно снижен от базового; в отличие от этого собаки, получавшие соединение 1, не показали статистически значимого снижения минутного сердечного выброса.
Пример 13. Анестетический и гемодинамический эффекты у свиней
Анестетический и гемодинамический эффекты соединения 1 и пропофола сравнивали на анестезированных вентилируемых свиньях, которым внутривенно (ВВ) вливали композицию с 0,5% соединения 1, полученную как описано в примере 2 и составленную в соответствии с примером 6, или диприван (эмульсия 1% пропофола для инъекции). Оценки включали ЭЭГ измерения глубины анестезии с использованием BIS, показатели фармакокинетики, кровяное давление, ЭКГ, сердечный ритм, минутный сердечный выброс, температуру тела и насыщение кислородом.
Эксперименты проводили на коммерческих свиньях фермерского разведения того и другого пола (средняя масса 33,6 кг). Анестезию вызывали изофлураном. Внутрисосудистый доступ получали через ушную вену. Каждую свинью инкубировали и механически вентилировали. Насыщение кислородом ткани контролировали с помощью непрерывной пульсовой оксиметрии с размещением на языке. За вентилированием следили с помощью анализатора вдыхаемого/выдыхаемого воздуха, который измерял концентрации кислорода, углекислого газа и сильного ингаляционного анестетика. Установочные параметры вентилирования при необходимости регулировали для поддержания устойчивого состояния.
Непрерывный уровень анестезии достигался с помощью изофлурана и вливания панкурониума (10 мг/час). ЭКГ контролировали на всем протяжении исследования. Артериальное давление крови контролировали через катетер в левой бедренной артерии. MAP, систолическое и диастолическое артериальное давление и сердечный ритм регистрировали каждые 5 секунд. Во внутреннюю яремную вену был помещен легочный артериальный катетер для термодилюционной оценки минутного сердечного выброса и температуры крови. Поддерживали температуру тела 37°С. Для проведения мониторинга ЭЭГ с помощью приборов применяли липкие электроды в лобно-затылочных областях (Aspect Medical, Норвуд, Массачусетс, США).
План эксперимента включал 30-минутный период стабилизации, с последующим внутривенным (ВВ) вливанием соединения 1 (0,384 мг/кг/мин × 20 мин) или пропофола (0,750 мг/кг/мин × 10 мин). За соответствующим вливанием следовал 180-минутный период вымывания. Образцы крови для гемодинамических измерений и для фармакокинетических исследований отбирали перед вливанием, каждые 2 мин в течение вливания соединения 1 или пропофола и с повторяющимися интервалами в течение периода вымывания. Время и скорости вымывания соединения 1 и пропофола были установлены заранее, чтобы вызвать максимальное снижение BIS (<10) в течение периода вливания. Образцы артериальной крови для определения рН, рO2, рСO2, глюкозы, калия и лактата измеряли на базовом уровне до вливания соединения 1 или пропофола, во время вливания и ежечасно после вливания.
А. Анестетические эффекты
Соединение 1 и пропофол вызывали максимальное подавление BIS (<10) при внутривенных (ВВ) вливаниях 0,384 мкг соединения 1 в течение 14,7±3,8 мин (на кг в мин) и 0,750 мкг пропофола в течение 9,4±1,9 мин (на кг в мин), соответственно. Действие на ЭЭГ было обратимо, и возвращение к базовому уровню происходило в течение 60 минут. Площадь под кривой (AUC) соединения 1, необходимая для достижения максимального фармакодинамического эффекта (Emax), была значительно меньше, чем для пропофола (51,5+15,5 против 108,7+24,3 мкг-мин/мл, соответственно). В заключение, данные показали, что соединение 1 было более эффективно, чем пропофол.
B. Гемодинамические эффекты
Среднее артериальное давление и сердечный ритм измеряли с интервалами на всем протяжении внутривенного (ВВ) вливания и «вымывания» соединения 1 (0,384 мг/кг/мин, 5 свиней) и пропофола (0,750 мг/кг/мин, 6 свиней). Результаты показаны на фигурах 1 и 2 соответственно. Фигура 3 сравнивает минутный сердечный выброс, вызываемый соединением 1 по сравнению с пропофолом. У свиней, которым вводили соединение 1, брали образцы артериальной крови для анализа крови на газы и кислотность и биохимического анализа крови; средние значения представлены в таблице 4.
АВЕс относится к кислотно-основному избытку, исправленному. Базовые значения MAP и HR не различались между соединением 1 и пропофолом. Оба соединения уменьшали MAP, но пропофол вызывал значительно большее снижение MAP (66±4), чем соединение 1 (106±3) (р<0,001). Самое низкое значение HR, измеренное для пропофола (88+6 ударов в мин), было значительно ниже, чем самое низкое значение HR измеренное для соединения 1 (129±6 ударов в мин) (р<0,5). Оба значения и MAP, и HR возвращались к базовому значению после прекращения вливания соединения 1 или пропофола. Не наблюдалось значительного различия в уменьшении минутного сердечного выброса, вызываемого соединением 1 по сравнению с пропофолом.
Таблица 4 показывает, что все значения анализа крови на газы и кислотность и биохимического анализа крови находились в пределах нормы: соединение 1 не вызывало значительных метаболических изменений, таких как метаболический ацидоз или повышенный лактат.
Пример 14. Антиэметическая активность
Соединение 1 проверили на антиэметическую активность на хорьках и сравнивали с активностью пропофола.
Самцов descented (без запаха) хорьков весом 1,0-1,5 кг с портами для сосудистого доступа в яремной вене содержали при цикле день/ночь 12 час/12 час при контролируемой температуре с неограниченным доступом к воде и пище (ad libitum). Каждый день на протяжении исследования хорьков кормили за один час до введения дозы вещества. Непосредственно перед введением дозы вещества воду и пищу убирали. Композицию 1% соединения 1, приготовленную как описано в примере 2 и составленную в соответствии с примером 5, или диприван вводили хорькам с помощью внутривенного (ВВ) вливания; см. Wyim RL et al, 1993, Eur J Pharmacol 241, 42 re DIPRBBAN administration in ferrets. После введения соединения 1 или дипривана животных помещали в чистые прозрачные клетки (с крышками) и оставляли свободными в течение 45-мин периода наблюдения (наблюдение проводили «вслепую» относительно конкретно примененного воздействия).
Рвота у хорьков отличается ритмичными сокращениями брюшных мышц, которые связаны или с оральным выталкиванием твердого или жидкого вещества из желудочно-кишечного тракта (т.е., рвотой), или с движениями, которые не включают прохождение вещества (т.е., позывы на рвоту). Случаи позывов и/или рвоты рассматривались как отдельные случаи, когда интервал между позывами и/или рвотой достигал 5 сек.
Проэметическую активность соединения 1 или пропофола исследовали на 6 хорьках (на одно лекарственное средство) как указано далее: хорькам давали наркоз посредством ингаляции изофлураном. Соединение 1 или пропофол вводили внутривенным (ВВ) вливанием в течение 15 мин при скорости 1 мг/кг/мин. После завершения вливания за хорьками наблюдали непрерывно в течение 45 мин, подсчитывая количество случаев рвоты и позывов на рвоту.
Антиэметическую активность соединения 1 или пропофола исследовали на 6 хорьках (на одно лекарственное средство) как указано далее: хорькам давали наркоз изофлураном, вводили соединение 1 или пропофол посредством 15 мин внутривенного (ВВ) вливания при скорости 1 мг/кг/мин. После завершения вливания подкожно вводили 0,5 мг/кг морфина сульфата, за хорьками наблюдали в течение 45 минут, как описано выше. Еще шести хорькам вводили только 0,5 мг/кг морфина сульфата подкожно.
Один морфин сульфат (0,5 мг/кг) обладал проэметической активностью у хорьков, приводя к 15 случаям рвоты и 157 случаям позывов на рвоту. Соединение 1 не вызывало какого-либо количества случаев рвоты или позывов на рвоту, когда его вводили отдельно или в присутствии морфина. У хорьков, получивших пропофол и морфина сульфат, насчитали 3 случая рвоты и 47 случаев позывов на рвоту. Следовательно, и соединение 1, и пропофол уменьшали долю случаев рвоты и позывов в присутствии морфина.
Все публикации, патенты и патентные документы включены в данное описание путем отсылки, как-будто включены в данное описание по отдельности. Изобретение описано со ссылкой на различные особые и предпочтительные варианты осуществления и методики. Однако должно быть понятно, что можно сделать много изменений и модификаций, но и оставаться в духе и рамках изобретения.
Реферат
Настоящее изобретение относится к новому (-)-стереизомеру формулы (I), где X является Н, или к его фармацевтически приемлемой соли, которые агонизируют рецептор GABA, к фармацевтической композиции на основе предложенного соединения, к способу получения (-)-стереизомера формулы (I) или его фармацевтически приемлемой соли, к способу индуцирования или поддержания общей анастезии, к способу промотирования обезболивания и к способу прототирования антиэметической активности с использованием предложенного (-)-стереоизомера или его фармацевтически приемлемой соли, а также к новому диастереизомеру (-)-2,6-ди-втор-бутилфенилового эфира карбаминовой кислоты формулы (II), где Rпредставляет собой хиральную аминогруппу и X является Н, или к его фармацевтически приемлемой соли. 8 н. и 6 з.п. ф-лы, 15 пр., 10 табл., 3 ил.
Формула

где X является Н; или его фармацевтически приемлемая соль.
где X является Н; или его фармацевтически приемлемой соли.
где X является Н; или его фармацевтически приемлемой соли.

где R1 представляет хиральную аминогруппу, и где X является Н, с последующим, если необходимо, образованием свободного фенола или его фармацевтически приемлемой соли.

где R1 представляет хиральную аминогруппу, и где X является Н.
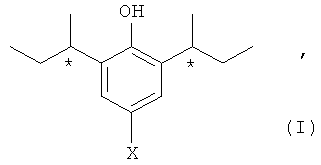
где X является Н; или его фармацевтически приемлемой соли.
где X является Н; или его фармацевтически приемлемой соли.
Комментарии