Новые оптически-активные соединения фенилэтаноламинов и способ их получения - RU2416597C2
Код документа: RU2416597C2
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым оптически активным соединениям фенилэтаноламинов, обладающих эффектом агониста β2-рецептора, которые особо полезны для лечения астмы или бронхита. Изобретение также относится к способам получения указанных соединений, фармацевтическим композициям, содержащим их и их применению.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Астма и бронхит являются общими заболеваниями. В большинстве случаев для лечения астмы и бронхита применяют антибиотики, которые не очень эффективны и обладают некоторыми побочными эффектами при длительном применении. Агонисты β2-рецептора хорошо известны в качестве средств против астмы. Однако эти лекарственные средства и их физические и химические свойства все еще недостаточно эффективны.
Китайский патент № 01128234.7 раскрывает новые соединения фенилэтаноламинов, обладающие хорошим эффектом агониста β2-рецептора. Однако в этом патенте не дано никакой информации об оптически активных изомерах соединений фенилэтаноламинов.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является предоставление новых оптически активных соединений фенилэтаноламинов, которые показывают более высокую активность агониста β2-рецептора и более низкую токсичность, чем их рацемическая смесь.
Настоящее изобретение предоставляет соединения формулы (I), имеющие (-)- или (+)-конфигурацию или их фармацевтически приемлемые соли

где
R1 представляет H или галоген; R2 представляет CF3, CN или галоген; R3 представляет собой линейный или разветвленный алкил, имеющий от 1 до 6 атомов углерода, или циклоалкил, имеющий от 3 до 6 атомов углерода,
или их фармацевтически приемлемые соли.
Предпочтительно, в формуле I R1 представляет собой Cl или Br; R2 представляет собой CF3, CN, или F; и R3 представляет собой линейный или разветвленный алкил, имеющий от 3 до 6 атомов углерода.
В предпочтительном варианте осуществления настоящего изобретения соединения формулы (I) имеют (-)-конфигурацию.
Более предпочтительно, соединения согласно настоящему изобретению выбраны из группы, состоящей из:
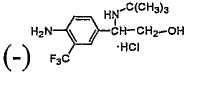
гидрохлорида (-)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола,
гидрохлорида (+)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола,
гидрохлорида (-)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола,
гидрохлорида (+)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола,
гидрохлорида (-)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола,
гидрохлорида (+)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола,
гидрохлорида (-)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола и
гидрохлорида (+)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола.
Используемый в настоящем описании термин «фармацевтически приемлемая соль» относится к традиционным кислотно-аддитивным солям, которые сохраняют биологическую эффективность и свойства соединений формулы (I) и которые образованы из подходящих нетоксичных органических или неорганических кислот. Примеры кислотно-аддитивных солей включают такие соли, которые получены из неорганических кислот, таких как хлористоводородная кислота, бромистоводородая кислота, иодистоводородная кислота, серная кислота, фосфорная кислота и азотная кислота, такие соли, которые получены из органических кислот, таких как уксусная кислота, винная кислота, салициловая кислота, метансульфоновая кислота, бутандиоевая кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота и т.п. В частности, фармацевтически приемлемыми солями соединений формулы (I) предпочтительно являются гидрохлорид и гидробромид.
Оба указанных выше оптически активных изомера, имеющих (+)- и (-)-конфигурацию, соответственно, обладают активностью агониста β2-рецептора, причем изомер, имеющий (-)-конфигурацию, обладает активностью агониста β2-рецептора, в 3-7 раз большей, чем активность изомера, имеющего (+)-конфигурацию, и в 2-5 раз большей, чем активность рацемической смеси. Кроме того, токсичность изомера, имеющего (-)-конфигурацию, меньше токсичности рацемической смеси и изомера, имеющего (+)-конфигурацию.
Настоящее изобретение также предоставляет способ получения соединений формулы (I) посредством разделения, включающего в себя реакцию рацемической смеси формулы (II)

где R1, R2 и R3 являются такими, как определено выше,
с соединением, выбранным из группы, состоящей из: D(-)-винной кислоты, L(+)-винной кислоты, дибензоил-D-винной кислоты, дибензоил-L-винной кислоты, (+)(-)камфор-10-сульфоновой кислоты, L(-)-яблочной кислоты, L(+)-миндальной кислоты, d-α-бромкамфорсульфоновой кислоты и 1-хининовой кислоты, в безводных условиях с образованием соли; и кристаллизации этой соли два или более раз для разделения на соединения формулы 1 согласно п.1 формулы изобретения, имеющие оптическую активность.
Предпочтительно, в вышеуказанном разделении кристаллизацию проводят от 2 до 5 раз. Более предпочтительно, кристаллизацию проводят два или три раза.
В вышеуказанном разделении реакционным растворителем предпочтительно является какой-либо спирт (например, абсолютный этиловый спирт), простой эфир (например, абсолютный этиловый эфир) или углеводород (например, петролейный эфир). Реакцию предпочтительно проводят при температуре между комнатной температурой и температурой кипячения с обратным холодильником. Выход продукта обычно составляет 50-60%.
Настоящее изобретение также предоставляет фармацевтическую композицию, включающую в себя соединения формулы (I), имеющие (+)- или (-)-конфигурацию, и фармацевтически приемлемые эксципиенты.
Термин «фармацевтически приемлемые эксципиенты» означает эксципиент, который можно применять в фармацевтике, который является полностью безопасным, нетоксичным и не является нежелательным в биологическом или каком-либо ином смысле. Эти эксципиенты включают лактозу, крахмал, воду, спирт и т.п.
Фармацевтическая композиция согласно настоящему изобретению может также включать пропелленты, антисептики, солюбилизаторы, стабилизаторы, увлажнители, эмульгаторы, подсластители, красители, отдушки, соли для регулирования осмотического давления, буфер, кроющие средства, антиоксиданты и т.п. Фармацевтическая композиция согласно настоящему изобретению может также включать другие терапевтически ценные вещества, например, другие активные ингредиенты, отличные от соединения формулы (I).
Фармацевтическая композиция согласно настоящему изобретению может быть получена в виде таблеток, капсул, растворов, спреев, инъекций и т.п. Ее можно вводить в пероральной, парентеральной, аэрозольной, ингаляционной формах, через полость рта или носа или в других формах.
Соединения согласно настоящему изобретению обладают эффектом агониста β2-рецептора и их можно применять для лечения астмы и бронхита. Соответственно, настоящее изобретение также относится к применению соединений формулы (I), имеющих (+)- или (-)-конфигурацию, для получения лекарственного средства, обладающего эффектом агониста β2-рецептора. Настоящее изобретение также относится к применению соединения формулы (I), имеющего (+)- или (-)-конфигурацию, для получения лекарственного средства для лечения астмы и бронхита.
Соединения согласно настоящему изобретению можно вводить в терапевтически эффективном количестве. Термин «терапевтически эффективное количество» означает количество, которое эффективно предупреждает, ослабляет, облегчает состояния заболеваний. Такое «терапевтически эффективное количество» могут определить специалисты в данной области.
Терапевтически эффективное количество или дозу можно менять в широком диапазоне и они могут быть установлены согласно требованию конкретного случая. Обычно для взрослых с массой тела около 70 кг, предпочтительно, доза составляет, примерно от 10 мкг до 20 мг/день, более предпочтительно, от 50 мкг до 10 мг/день в пероральной или парентеральной форме. Когда требуется, можно выйти за верхний предел и нижний предел дозы. Дневные дозы можно вводить одной дозой или дозами, разделенными на несколько раз.
Следующие примеры иллюстрируют способ синтеза этих соединений.
ПРИМЕР 1
Гидрохлорид (-)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола и
гидрохлорид (+)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола
a) Гидрохлорид (-)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола
4,45 г (0,0143 моль) 2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола растворяли в 53,4 мл абсолютного этанола. По каплям добавляли раствор 2,57 г (0,00717 моль) дибензоил-D-винной кислоты в 25,7 мл абсолютного этанола, затем добавляли 188 мл петролейного эфира (т. кип.60-90°C). После перемешивания в течение 1 часа смесь фильтровали и сушили, получая дибензоил-D-тартрат 2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола (2,9 г). Выход: 82,6%.
2,9 г дибензоил-D-тартрата 2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола добавляли к 60 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до pH 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования этиловый эфир удаляли при пониженном давлении, что давало 1,7 г (-)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола. ee%=92,2%. Выход=76,4%.
Второе разделение проводили с дибензоил-D-винной кислотой, и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле до достижения рН 2. После фильтрования и сушки получали 1,43 г гидрохлорида (-)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола, ee%=99,0%, т.пл.: 209-210,7°C (десятичн.). Общий выход: 57,4%.
[α]D=-18,8° (c=0,5; абсолютный метанол)
b) Гидрохлорид (+)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола
4,45 г (0,0143 моль) 2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола растворяли в 53,4 мл абсолютного этанола. По каплям добавляли раствор 2,57 г (0,00717 моль) дибензоил-L-винной кислоты в 25,7 мл абсолютного этанола, затем добавляли 188 мл петролейного эфира (т. кип.60-90°C). После перемешивания в течение 1 часа смесь фильтровали и сушили, получая дибензоил-L-тартрат 2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола (3,2 г). Выход: 91,2%.
3,2 г дибензоил-L-тартрата 2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола добавляли к 65 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до pH 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования удаляли этиловый эфир при пониженном давлении, что давало 1,9 г (+)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола. ee%=94,4%. Выход=85,4%.
Второе разделение проводили с дибензоил-L-винной кислотой, и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле до достижения рН 2. После фильтрования и сушки получали 1,52 г гидрохлорида (+)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола. ee%=99,0%, т.пл.: 209,6-211,0°C (десятичн.). Общий выход: 61,0%.
[α]D=+18,5° (c=0,5; абсолютный метанол)
ПРИМЕР 2
Гидрохлорид (-)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола и гидрохлорид (+)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола
a) Гидрохлорид (-)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола
4,5 г (0,0163 моль) 2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола растворяли в 65 мл изопропанола. Затем по каплям добавляли раствор 1,23 г (0,0082 моль) L-винной кислоты в 25 мл изопропанола. После перемешивания в течение 2 часов смесь фильтровали и сушили, получая L-тартрат 2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола (2,24 г). Выход: 78,1%.
2,24 г L-тартрата 2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола добавляли к 50 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до pH 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования этиловый эфир удаляли при пониженном давлении, что давало 1,67 г (-)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола. ee%=90,0%. Выход=74,2%.
Второе разделение проводили с L-винной кислотой, и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле до достижения рН 2. После фильтрования и сушки получали 1,38 г гидрохлорида (-)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола. ee%=95,0%. Общий выход: 54,0%.
b) Гидрохлорид (+)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола
4,5 г (0,0163 моль) 2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола растворяли в 65 мл изопропанола. Затем по каплям добавляли раствор 1,23 г (0,0082 моль) D-винной кислоты в 25 мл изопропанола. После перемешивания в течение 2 часов смесь фильтровали и сушили, получая D-тартрат 2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола (2,3 г). Выход: 80,2%.
2,3 г D-тартрата 2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола добавляли к 52 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до pH 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования удаляли этиловый эфир при пониженном давлении, что давало 1,63 г (+)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола. ee%=90,6%. Выход=72,4%.
Второе разделение проводили с D-винной кислотой, и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле до достижения рН 2. После фильтрования и сушки получали 1,31 г гидрохлорида (+)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола. ee%=94,6%. Общий выход: 51,5%.
ПРИМЕР 3
Гидрохлорид (-)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола и гидрохлорид d-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола
a) Гидрохлорид (-)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола
2,5 г (0,0093 моль) 2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола растворяли 50 мл безводного этанола. По каплям добавляли раствор 1,67 г (0,0047 моль) дибензоил-D-винной кислоты в 16,7 мл безводного этанола, затем добавляли 200 мл безводного этилового эфира. После перемешивания в течение 1 часа смесь фильтровали и сушили, получая дибензоил-D-тартрат 2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола (1,58 г), Выход: 75,6%.
1,58 г дибензоил-D-тартрата 2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола добавляли к 32 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до рН 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования удаляли этиловый эфир при пониженном давлении, что давало 0,9 г (-)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола. ee%=89,7%. Выход=72,0%.
Второе разделение проводили с дибензоил-D-винной кислотой, и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле до достижения рН 2. После фильтрования и сушки получали 0,77 г гидрохлорида (-)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанол. еe%=97,0%. Общий выход: 54,4%.
[α]D=-19,3° (c=0,5; абсолютный метанол)
b) Гидрохлорид (+)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола
2,5 г (0,0093 моль) 2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола растворяли в 50 мл безводного этанола. По каплям добавляли раствор 1,67 г (0,0047 моль) дибензоил-L-винной кислоты в 16,7 мл безводного этанола, затем добавляли 200 мл безводного этилового эфира. После перемешивания в течение 1 часа смесь фильтровали и сушили, получая дибензоил-L-тартрат 2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола (1,67 г). Выход: 80,2%.
1,67 г дибензоил-L-тартрата 2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанол добавляли к 33 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до рН 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования удаляли этиловый эфир при пониженном давлении, что давало 0,94 г (+)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола. ee%=89,4%. Выход=75,9%.
Второе разделение проводили с дибензоил-L-винной кислотой, и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле до достижения рН 2. После фильтрования и сушки получали 0,82 г гидрохлорида (+)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанол. ee%=96,0%. Общий выход: 58,0%.
[α]D=+18,5° (c=0,5; абсолютный метанол)
ПРИМЕР 4
Гидрохлорид (-)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола и гидрохлорид (+)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола
a) Гидрохлорид (-)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола
2,5 г (0,008 моль) 2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола растворяли 50 мл безводного этанола. По каплям добавляли раствор 1,43 г (0,004 моль) дибензоил-L-винной кислоты в 14,3 мл безводного этанола, затем добавляли 193 мл безводного этилового эфира. После перемешивания в течение 1 часа смесь фильтровали и сушили, получая дибензоил-L-тартрат 2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола (1,61 г). Выход: 82,0%.
1,61 г дибензоил-L-тартрата 2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола добавляли к 32 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до рН 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования удаляли этиловый эфир при пониженном давлении, что давало 0,96 г (-)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола. ee%=90,2%, Выход=76,6%.
Второе разделение проводили с дибензоил-L-винной кислотой, и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле до достижения рН 2. После фильтрования и сушки получали 0,75 г гидрохлорида (-)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола. ee%=95,0%. Общий выход: 54,0%.
b) Гидрохлорид (+)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола
2,5 г (0,008 моль) 2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола растворяли в 50 мл безводного этанола. По каплям добавляли раствор 1,43 г (0,004 моль) дибензоил-D-винной кислоты в 25,7 мл безводного этанола. Затем добавляли 193 мл безводного этилового эфира. После перемешивания в течение 1 часа смесь фильтровали и сушили, получая дибензоил-D-тартрат 2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола (1,59 г). Выход: 81,0%.
1,59 г дибензоил-D-тартрата 2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола добавляли к 32 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до рН 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования удаляли этиловый эфир при пониженном давлении, что давало 0,97 г (+)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола. ee%=89,4%. Выход=77,8%.
Второе разделение проводили с дибензоил-D-винной кислотой, и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле до достижения рН 2. После фильтрования и сушки получали 0,78 г гидрохлорида (+)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола. ee%=95,5%. Общий выход: 56,0%.
ПРИМЕР 5. Основные таблетки
1) Рецептура препарата
2) Способ получения
Вышеуказанное количество соединения согласно настоящему изобретению отвешивали и растворяли в подходящем количестве этанола вместе с PVP. Смешивали лактозу и крахмал и добавляли к раствору для однородного смачивания. Влажные частицы получали, применяя сито с отверстиями размером 1,5 мм, и аэрационно сушили при 50°C. Для гранулирования применяли сито с отверстиями размером 1,0 мм. Гранулят однородно перемешивали со стеаратом магния и спрессовывали. 120 мг на таблетку. Применяли плоскую насечку 7 мм.
ПРИМЕР 6. Таблетки, покрытые пленкой
1) Рецептура препарата
2) Способ получения
Вышеуказанное количество соединения согласно настоящему изобретению отвешивали и растворяли в подходящем количестве этанола вместе с PVP. Смешивали лактозу и крахмал и добавляли к раствору для однородного смачивания. Влажные частицы получали, применяя сито с отверстиями размером 1,5 мм, и аэрационно сушили при 50°C. Для гранулирования применяли сито с отверстиями размером 1,0 мм. Гранулят однородно перемешивали со стеаратом магния и спрессовывали. 120 мг на таблетку. Применяли глубокую зубчатую насечку 7 мм. Таблетку покрывали сахарной пленкой. 200 мг на таблетку, покрытую пленкой.
ПРИМЕР 7. Капсулы
1) Рецептура препарата
2) Способ получения
Вышеуказанное количество соединения согласно настоящему изобретению отвешивали, однородно смешивали с лактозой и крахмалом и вносили в капсулы. Содержимое каждой капсулы имело массу 120 мг.
ПРИМЕР 8. Инъекционные растворы
1) Рецептура препарата
2) Способ получения
Вышеуказанное количество соединения согласно настоящему изобретению отвешивали и добавляли к 1000 мл воды для инъекций. Затем к раствору при перемешивании добавляли и полностью растворяли вышеуказанные количества лимонной кислоты, Na2HPO4 и NaCl. Добавляли активированный уголь и раствор перемешивали при 80°С в течение 20 минут. Активированный уголь затем удаляли фильтрованием через мембраны с микропорами размером 0,2 мкм. Добавляли дополнительно 1000 мл воды для инъекций. Измеряли содержание соединения и рН раствора. Раствор разливали в 2-мл ампулы и стерилизовали при 120°С в течение 20 мин.
ПРИМЕР 9. Суппозитории
1) Рецептура препарата
2) Способ получения
Вышеуказанное количество соединения согласно настоящему изобретению сплавляли с основой при 40°С и помещали в форму. Суппозитории получали после охлаждения до температуры ниже 37°С.
ПРИМЕР 10. Сиропы
1) Рецептура препарата
2) Способ получения
60 мл дистиллированной воды нагревали до 80°C и в ней в достаточной степени диспергировали вышеуказанные количества бензойной кислоты, яблочной кислоты, соединения согласно настоящему изобретению, красного пигмента и сахарозы. Добавляли ароматическую апельсиновую эссенцию и полное количество воды. Сироп получали после фильтрования.
ПРИМЕР 11. Порошки
1) Рецептура препарата
2) Способ получения
Вышеуказанное количество соединения согласно настоящему изобретению отвешивали, помещали в псевдоожиженный слой и пульверизовали в сверхзвуковом потоке воздуха для получения ультрамикроскопического сухого порошка с размером частиц от 1 мкм до 5 мкм. Лактозный эксципиент с размером частиц менее 200 мкм получали высокоскоростным дроблением и помолом. Ультрамикроскопический сухой порошок и лактозный эксципиент однородно смешивали способом «увеличения равных количеств» и помещали в капсулы размера 3.
ПРИМЕР 12. Ингаляционные лекарственные формы
1) Рецептура препарата
2) Способ получения
Соединение согласно настоящему изобретению, гидроксипропилметилцеллюлозу, микрокристаллическую целлюлозу и лактозу пульверизовали и просеивали через 100 меш. 0,1 г гидроксипропилметилцеллюлозы и 50%-ный (по объему) раствор этанола в воде смешивали для получения 1%-ного (масса к объему) раствора гидроксипропилметилцеллюлозы в водном растворе этанола. Вышеуказанное количество просеянного соединения согласно настоящему изобретению растворяли в 8 мл раствора гидроксипропилметилцеллюлозы в водном растворе этанола. 0,2 г просеянной гидроксипропилметилцеллюлозы, 1,5 г просеянной микрокристаллической целлюлозы и 9 г просеянной лактозы однородно смешивали и затем получали мягкий материал с полученным ранее раствором гидроксипропилметилцеллюлозы в водном растворе этанола, содержащим соединение согласно настоящему изобретению. Этот мягкий материал гранулировали с помощью сита (30 меш), сушили при 60°С в печи и затем гранулировали с помощью двух сит (100 меш и 400 меш соответственно). Отбирали грануляты между 100 меш и 400 меш и ими заполняли пузырьки для получения порошкового ингалятора пузырькового типа.
ПРИМЕР 13. Компрессы (припарки)
1) Рецептура препарата
2) Способ получения
Отвешивали вышеуказанное количество желатина и давали достаточно набухнуть в воде. Для ускорения растворения применяли водяную баню при 60°С. К раствору желатина при перемешивании добавляли натрий-карбоксиметилцеллюлозу, получая первый раствор. Однородно смешивали вышеуказанные количества полиакрилата натрия, AlCl3, лаурокапрама, каолина, лимонной кислоты и соединения согласно настоящему изобретению, получая второй раствор. Затем первый раствор смешивали со вторым раствором и добавляли глицерин и олеиновую кислоту в указанных количествах. После тщательного перемешивания смесь наносили на нетканую материю (250×300 см2), которую покрывали несклеивающимися слоями (5 см×6 см), получая 50 штук припарок.
ПРИМЕР 14. Пластыри
1) Рецептура
2) Способ получения
К смеси 40,0 г блок-сополимера стирол-изопрен-стирол, 34,5 г терпеновой смолы и 10,0 г алифатической углеводородной смолы добавляли 2,0 г соединения согласно настоящему изобретению в форме 20%-ного водного раствора, 5,0 г N-метил-2-пирролидона, 2,5 г α-моноизостеарилового эфира глицерина, 5,0 г изопропилмиристата и 1,0 г смеси сложных эфиров сорбитана и жирных кислот. Добавляли подходящее количество этилацетата и однородно перемешивали, придавая форму пластыря. Пластырь однородно наносили на отделяемые пленки и сушили теплым воздухом. Пленки подложки затем наклеивали на отделяемые пленки и прессовали с вращением для получения клейкого пластыря.
ПРИМЕР 15. Исследование эффекта соединений согласно настоящему изобретению на β2-рецептор
Экспериментальные животные
Использовали морских свинок (Hartley, закупленных в Experimental Animal Center of Shenyang Pharmaceutical University. Сертификат разрешения: SCXK (Liao) - 2003-011) обоего пола, с массой тела 400-500 г.
Реагент
Образец: Все испытуемые соединения были в виде 10-6 M раствора.
Раствор фосфата гистамина: Shanghai LiZhu Biotechnology Co., Ltd. Партия № 1703
Гидрохлорид изопреналина, ISO: Shanghai Harvest Pharmaceutical Co., Ltd. Партия № 20040901
Инструменты
Термостатическая водяная циркуляционная баня типа S-501-A: Liaoyang Boda Scientific Instrument Co., Ltd.
Многоканальный самописец RM-6240: Chengdu Instrument Co., Ltd.
Электронные аналитические весы FA1004: Shanghai Jingke Industrial Co., Ltd.
Экспериментальные методики и результаты:
Антагонистические эффекты соединений согласно настоящему изобретению на сокращение трахеи, индуцированное гистамином.
Морских свинок забивали и на вентральной стороне шеи разрезали кожу и подкожные ткани. Удаляли трахею и срезали щитовидный хрящ до бифуркации трахеи, затем помещали в охлажденный льдом насыщенный кислородом раствор Кребса-Хенсляйта. Срезали соединительные ткани вокруг трахеи. Один конец трахеи удерживали щипцами и по спирали вырезали полоски трахеи (2 см×3 мм). Затем образцы помещали в баню, содержавшую 20 мл раствора Кребса-Хенсляйта. Нижний конец полосок трахеи закрепляли крючком, а верхний конец соединяли с датчиком напряжения, таким образом измеряя рекордером изменения натяжения. Баню контролировали при 37°С и непрерывно подавали газообразный кислород. Предварительная нагрузка на образцы составляла 2 г. Образцы уравновешивали в питательном растворе в течение 1 часа и раствор освежали каждые 20 мин.
Когда натяжение образцов достигало стабильного значения, в баню добавляли различные концентрации гистамина (1×10-10-3×10-4 моль·л-1). Для определения концентрации гистамина, при которой достигалось 50% максимального сокращения, строили кривую «доза-эффект». После освежения раствора и уравновешивания образцов в течение 60 мин, в баню добавляли некоторое количество гистамина. Когда натяжение полоски трахеи достигало 50% максимального сокращения, в баню добавляли испытуемые соединения и рассчитывали антагонистический эффект (представленный как степень расслабления) данных соединений на сокращение изолированной трахеи, индуцированное гистамином, как показано ниже:
Степень расслабления (%)=[(интенсивность сокращения после добавления гистамина-интенсивность сокращения после добавления испытуемых соединений)/интенсивность сокращения после добавления испытуемых соединений]×100%
Степень расслабления для каждого соединения представлена в Таблице 1.











Дополнительные примеры, где R3 представляет циклоалкил, имеющий от 3 до 6 атомов углерода



Гидрохлорид (-)-2-(3-бром-4-амино-5-трифторметилфенил)-2-циклопентиламиноэтанола
5,0 г (0,0136 мол) 2-(3-бром-4-амино-5-трифторметилфенил)-2-циклопентиламиноэтанола растворяли в 50 мл абсолютного этанола. По каплям добавляли раствор 1,02 г (0,0068 мол) L-винной кислоты в 25 мл абсолютного спирта и затем добавляли 150 мл этилового спирта. После перемешивания в течение 2 часов смесь фильтровали и сушили, получая 2-(3-бром-4-амино-5-трифторметилфенил)-2-циклопентиламиноэтанол L-тартрата (2,44 г). Выход: 81,0%.
2,44 г 2-(3-бром-4-амино-5-трифторметилфенил)-2-циклопентиламиноэтанол L-тартрата добавляли к 50 мл воды. После перемешивания добавляли 20%-ный раствор NaOH для доведения смеси до рН 10. Смесь экстрагировали этиловым эфиром и сушили над безводным сульфатом натрия. После фильтрования удаляли этиловый эфир при пониженном давлении и получали 1,89 г of (-)-2-(3-бром-4-амино-5-трифторметилфенил)-2-циклопентиламино-этанола. ее%=91.2%. Выход = 75,6%.
Второе разделение проводили с L-винной кислотой и процедура была такой же, как описанная выше. К полученному эфирному раствору добавляли раствор HCl в изопропаноле для достижения рН 2. После фильтрования и сушки получали 1,41 г гидрохлорида (-)-2-(3-бром-4-амино-5-трифторметилфенил)-2-циклопентиламиноэтанола. ее%=95,3%. Выход: 51,4%.
Реферат
Изобретение относится к новым оптически активным соединениям фенилэтаноаминов формулы (I), имеющим (-)-конфигурацию, или их фармацевтически приемлемым солям, которые обладают эффектом агониста β2-рецептора и могут использоваться для лечения астмы или бронхита. В формуле (I) ! ! R1 представляет собой Н или галоген; R2 представляет собой CF3, CN или галоген; R3 представляет собой линейный или разветвленный алкил, имеющий от 1 до 6 атомов углерода, или циклоалкил, имеющий от 3 до 6 атомов углерода. Изобретение также относится к способам получения указанных соединений и к композиции, их содержащей. 5 н. и 4 з.п. ф-лы, 1 табл.
Формула

где R1 представляет собой Н или галоген; R2 представляет собой CF3, CN или галоген; R3 представляет собой линейный или разветвленный алкил, имеющий от 1 до 6 атомов углерода, или циклоалкил, имеющий от 3 до 6 атомов углерода,
или его фармацевтически приемлемые соли.
гидрохлорида (-)-2-(3-хлор-4-амино-5-трифторметилфенил)-2-трет-бутиламиноэтанола,
гидрохлорида (-)-2-(3-трифторметил-4-аминофенил)-2-трет-бутиламиноэтанола,
гидрохлорида (-)-2-(3-хлор-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола,
гидрохлорида (-)-2-(3-бром-4-амино-5-цианофенил)-2-трет-бутиламиноэтанола.

где R1, R2 и R3 являются такими, как определено в п.1,
с соединением, выбранным из группы, состоящей из: D(-)-винной кислоты, L(+)-винной кислоты, дибензоил-D-винной кислоты, дибензоил-L-винной кислоты, (+)(-)-камфор-10-сульфоновой кислоты, L(-)-яблочной кислоты, L(+)-миндальной кислоты, d-α-бромкамфорсульфоновой кислоты и 1-хининовой кислоты, в безводных условиях с образованием соли; и
кристаллизацию соли дополнительно два или три раза для разделения ее на соединения формулы (I) по п.1, имеющие оптическую активность.
Комментарии