Циклические пептиды, радиофармацевтический препарат, циклические пептиды, имеющие радиоактивную метку - RU2145608C1
Код документа: RU2145608C1
Чертежи
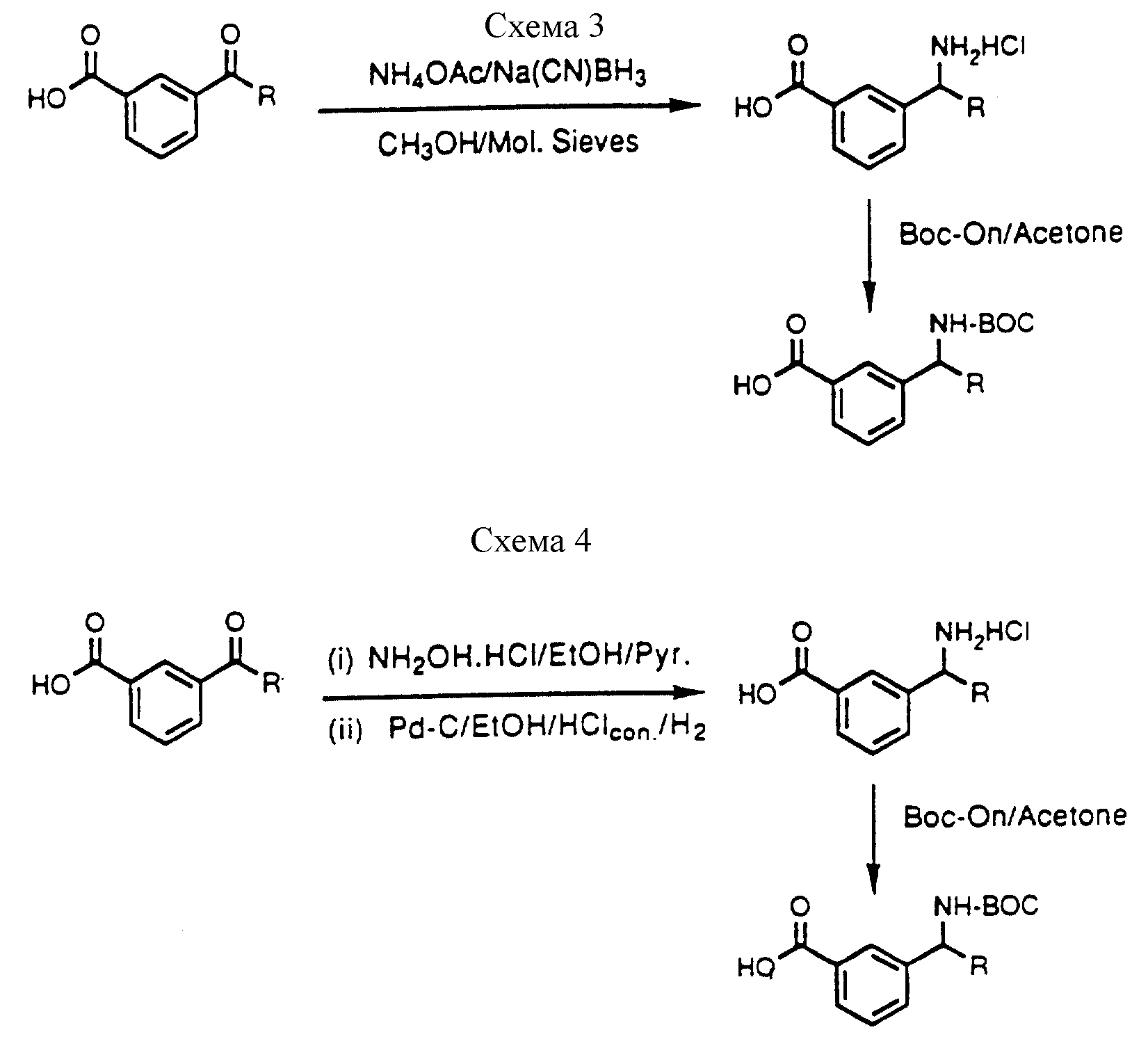
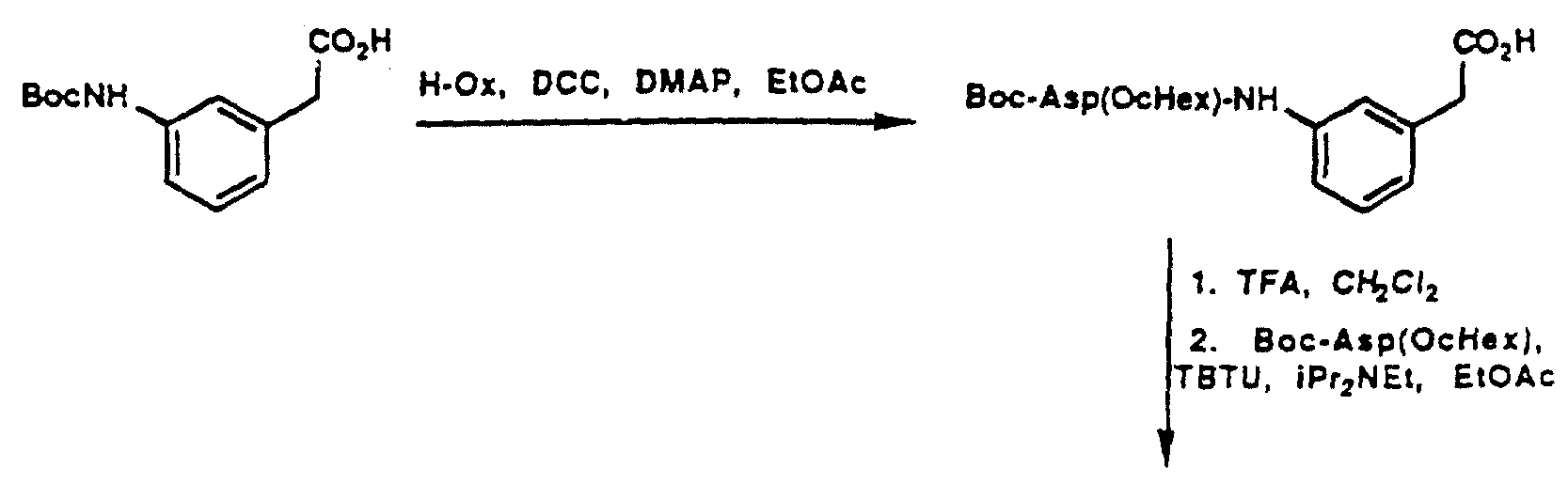

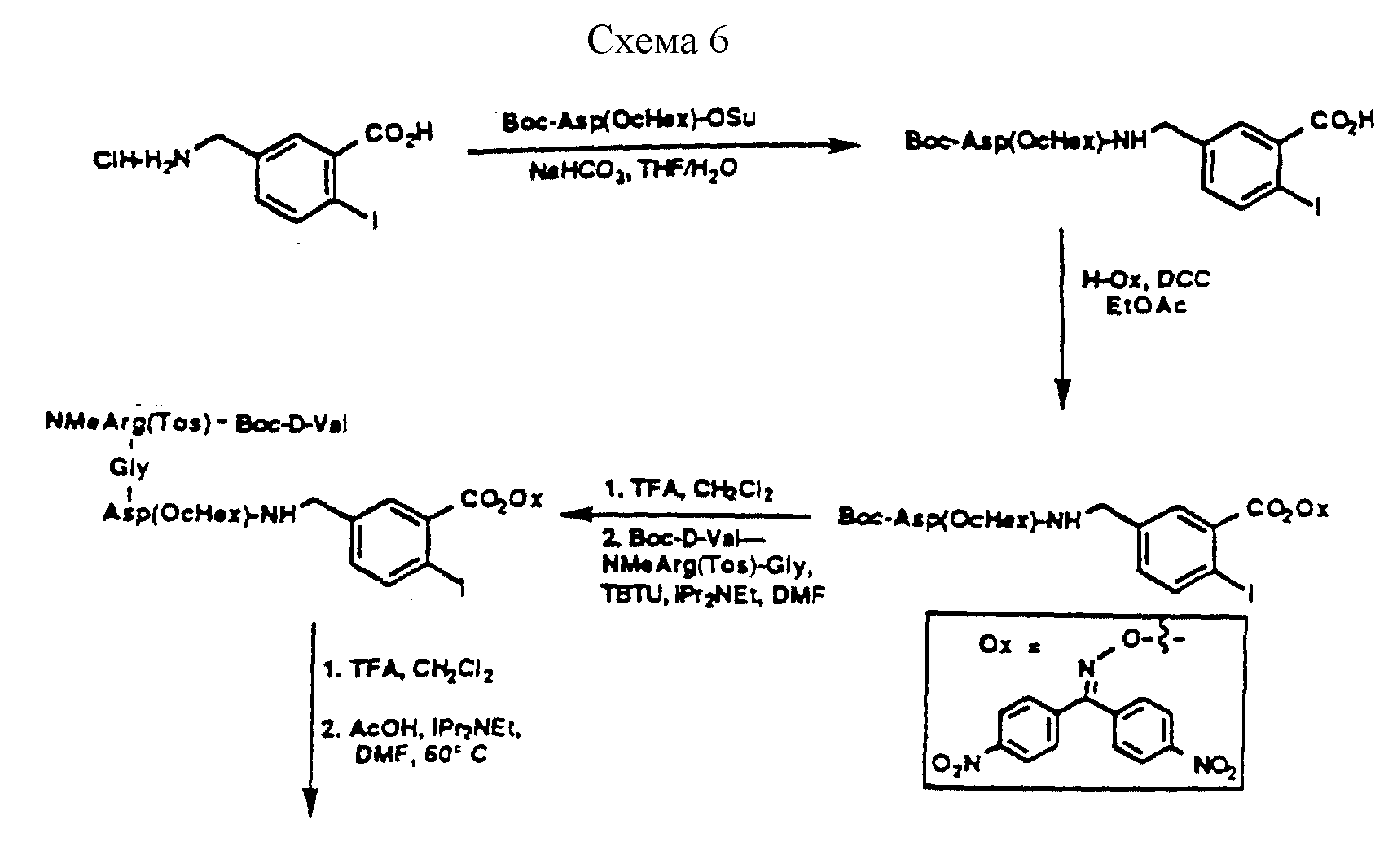
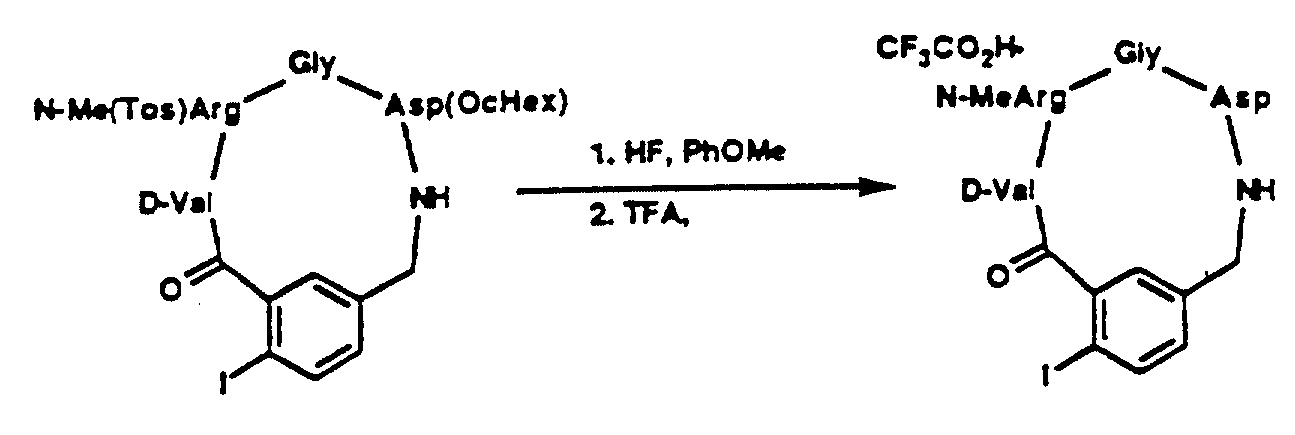

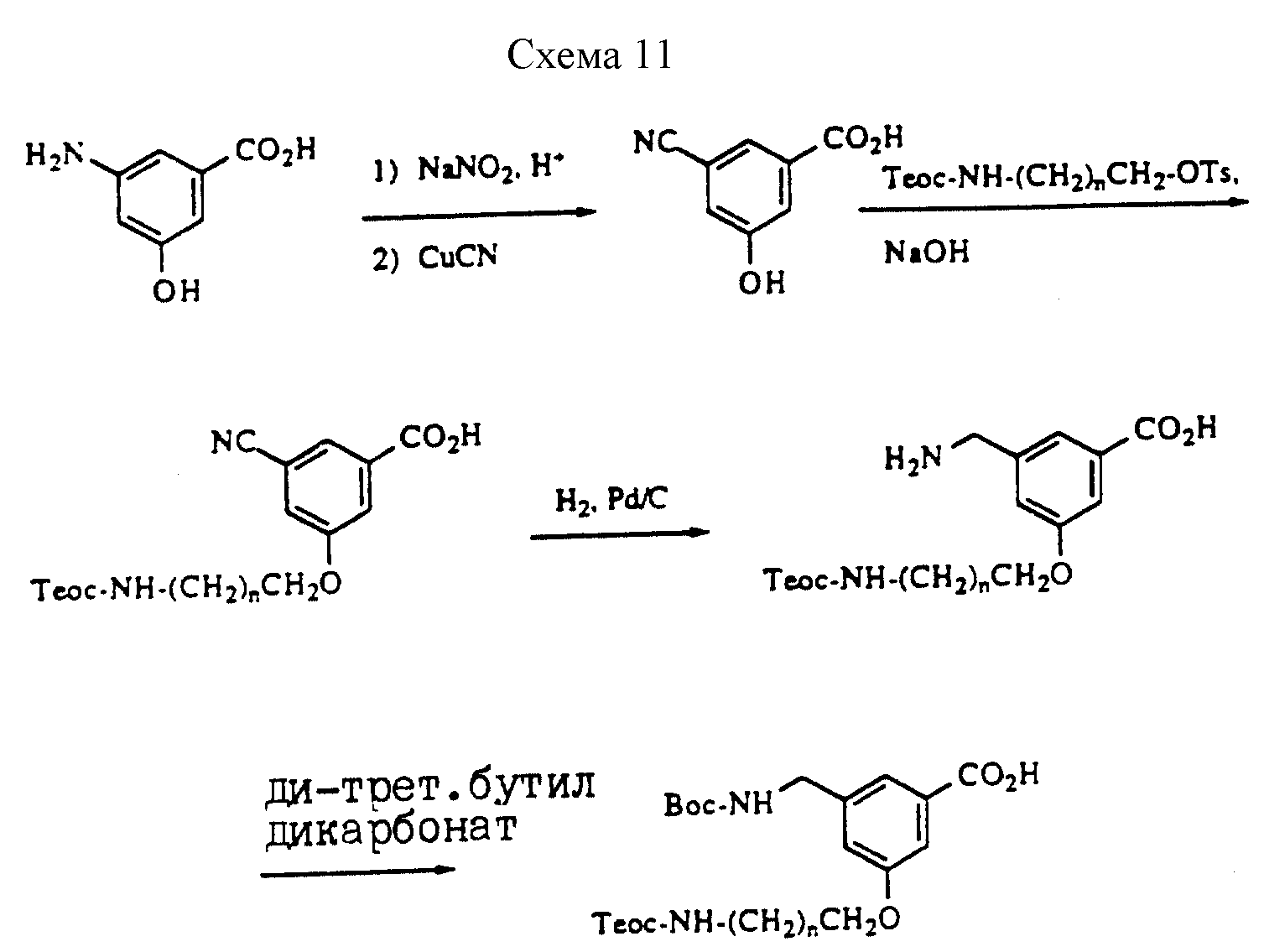
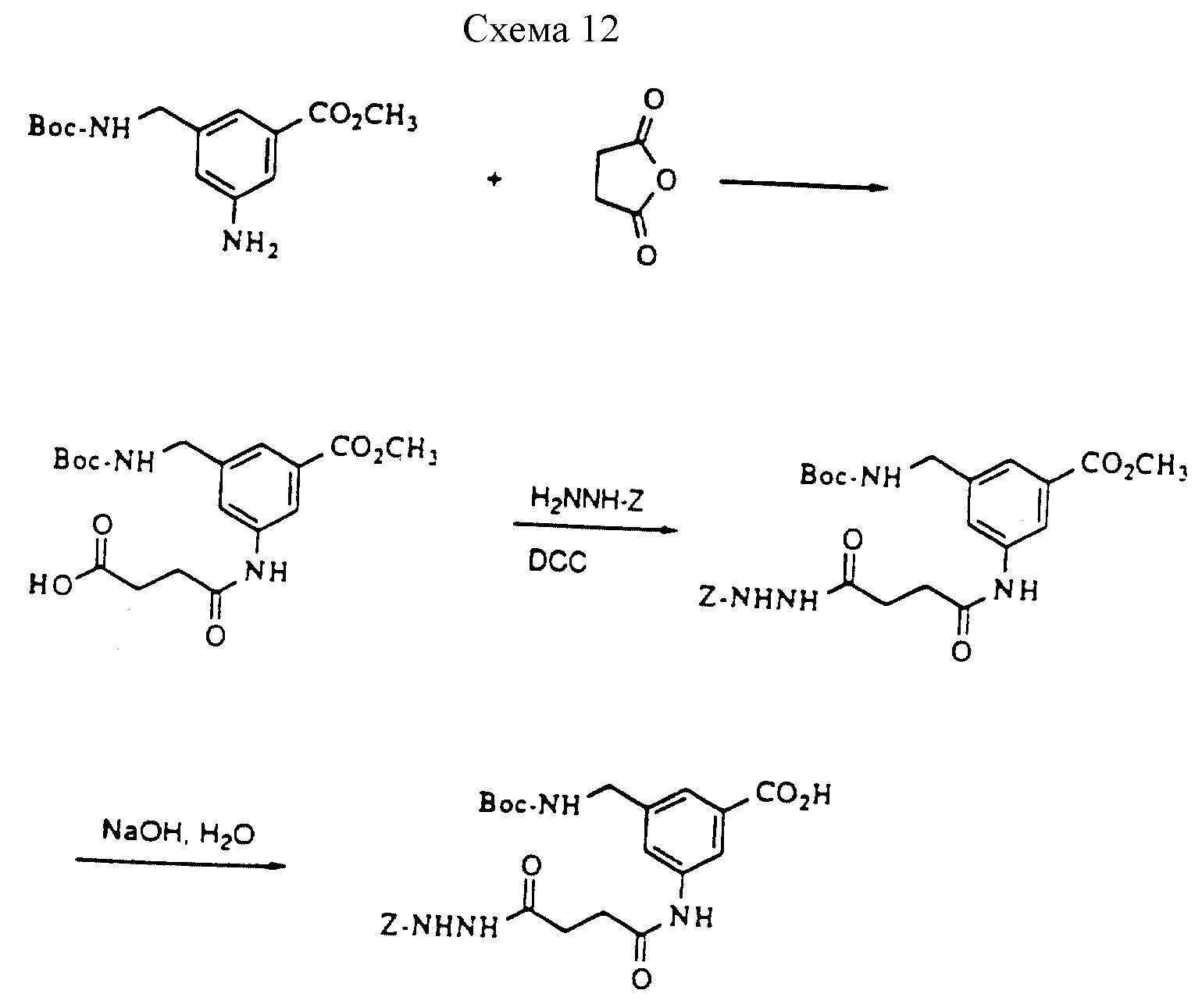
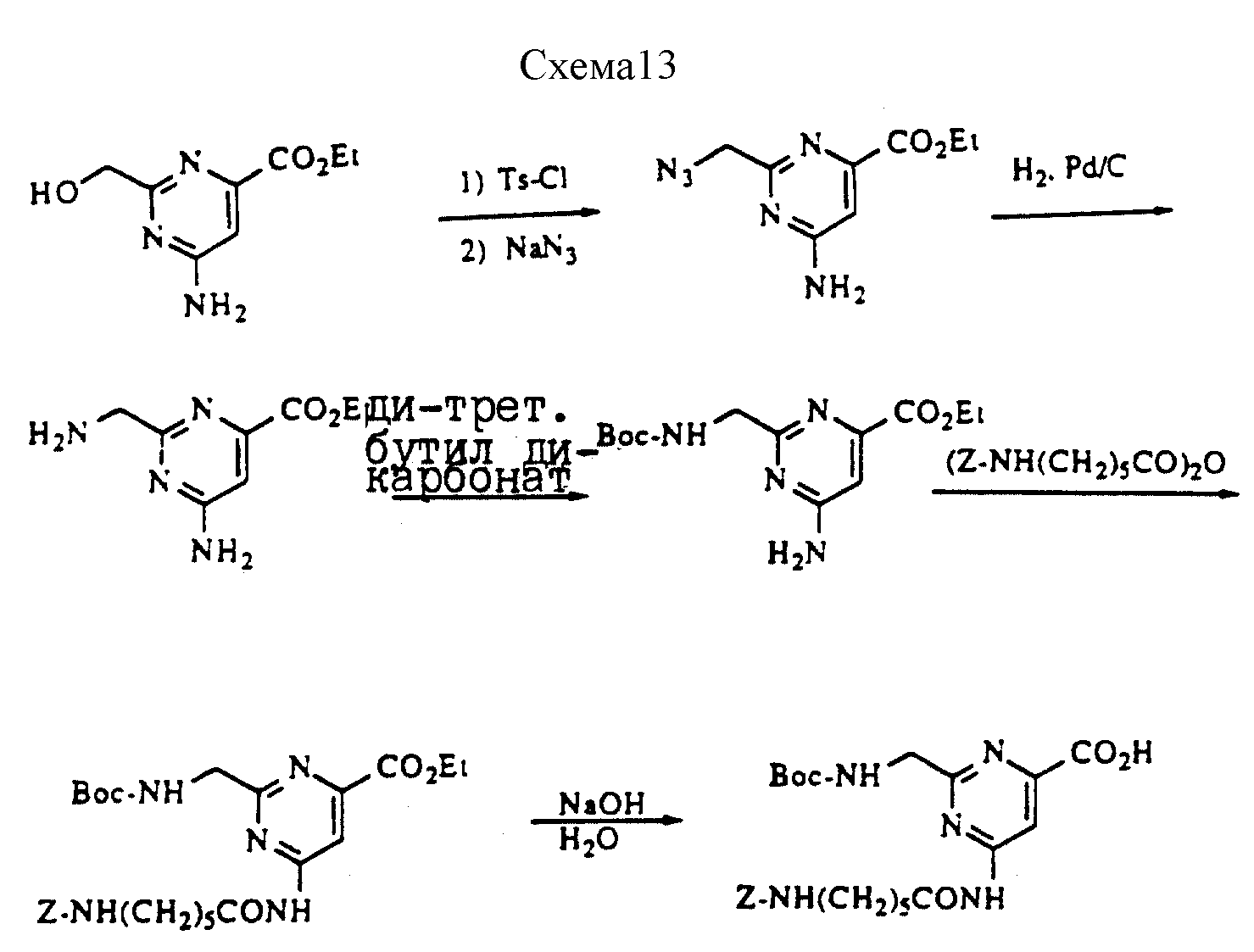
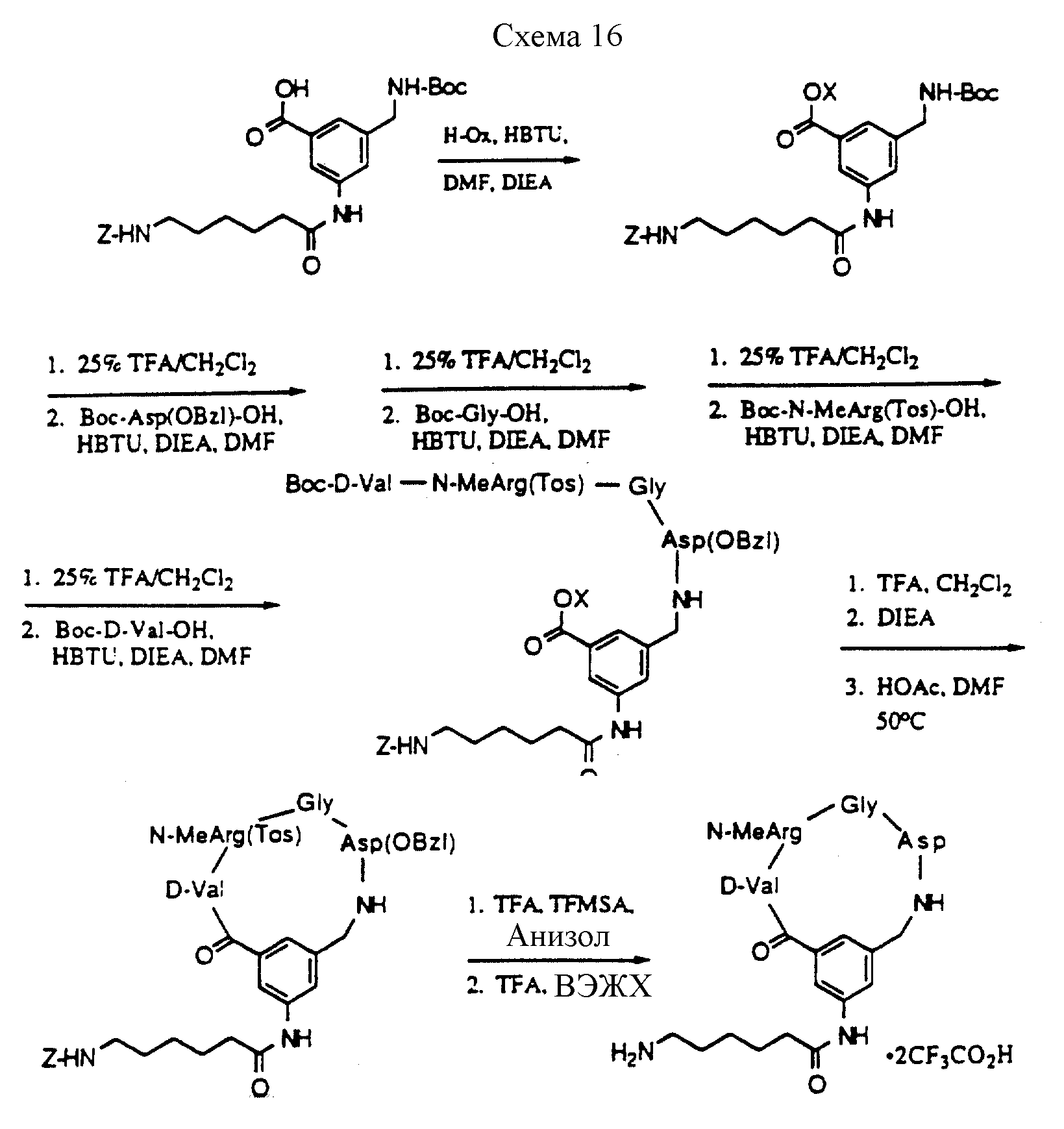

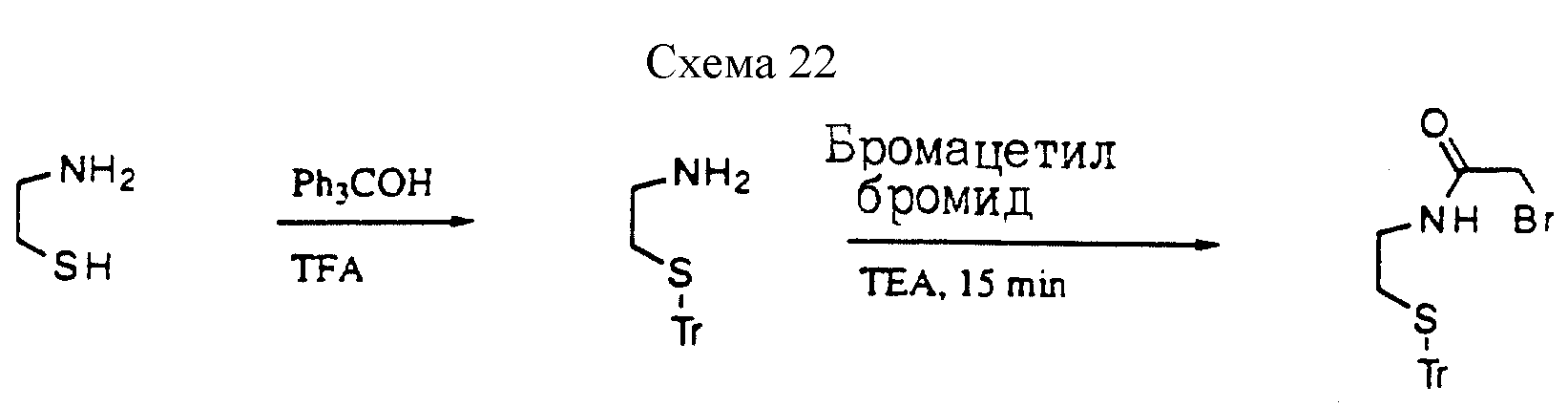
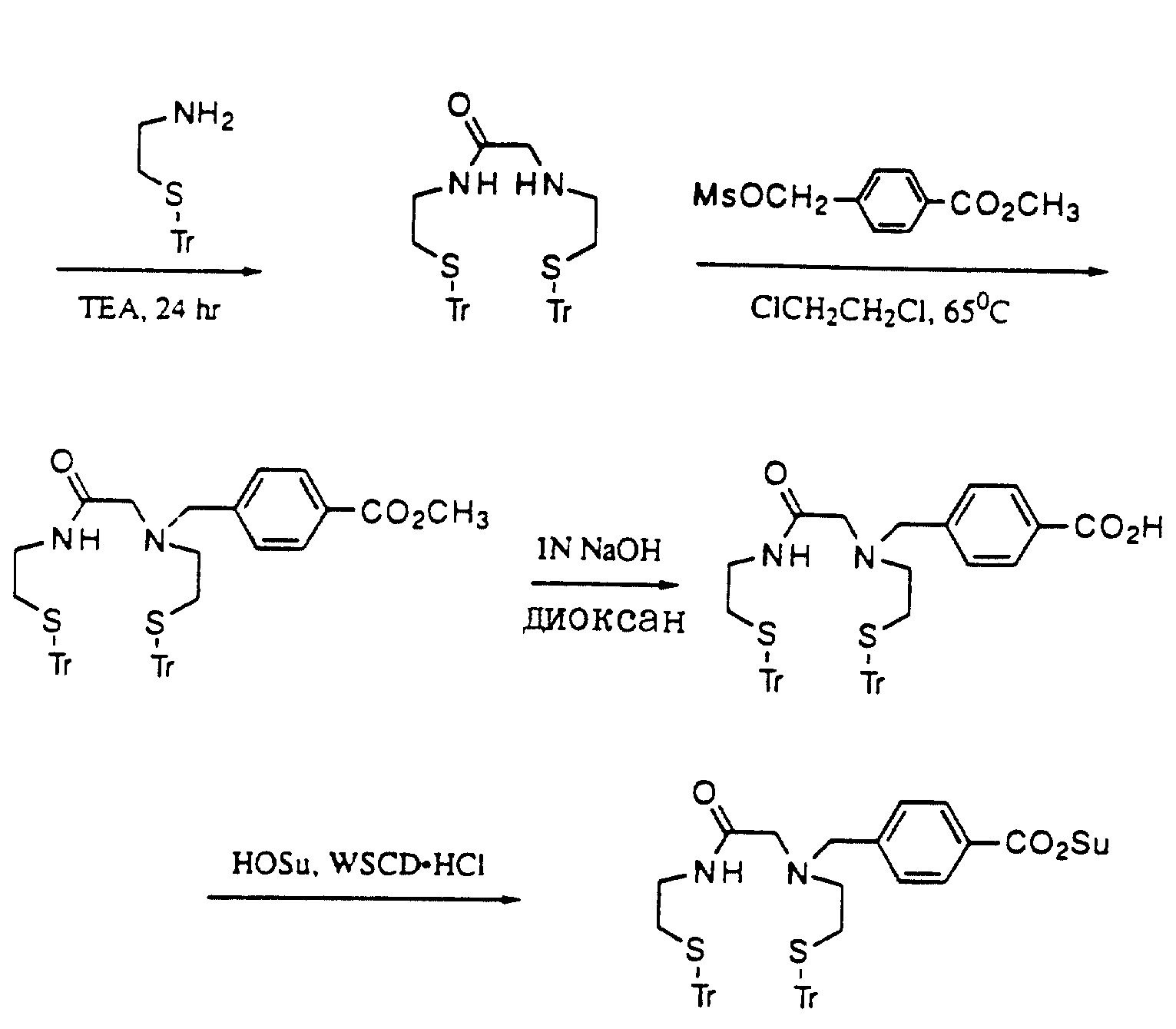
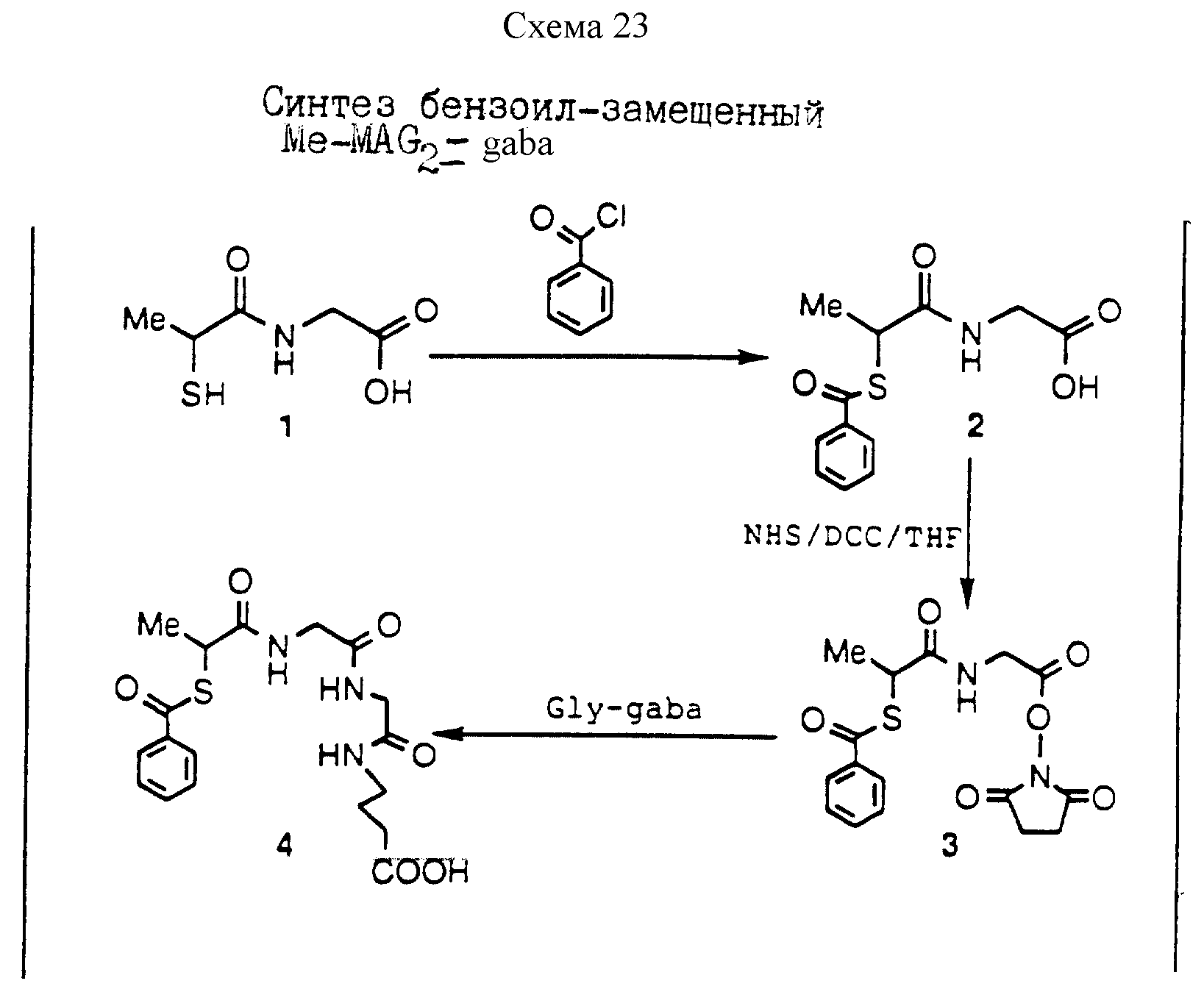
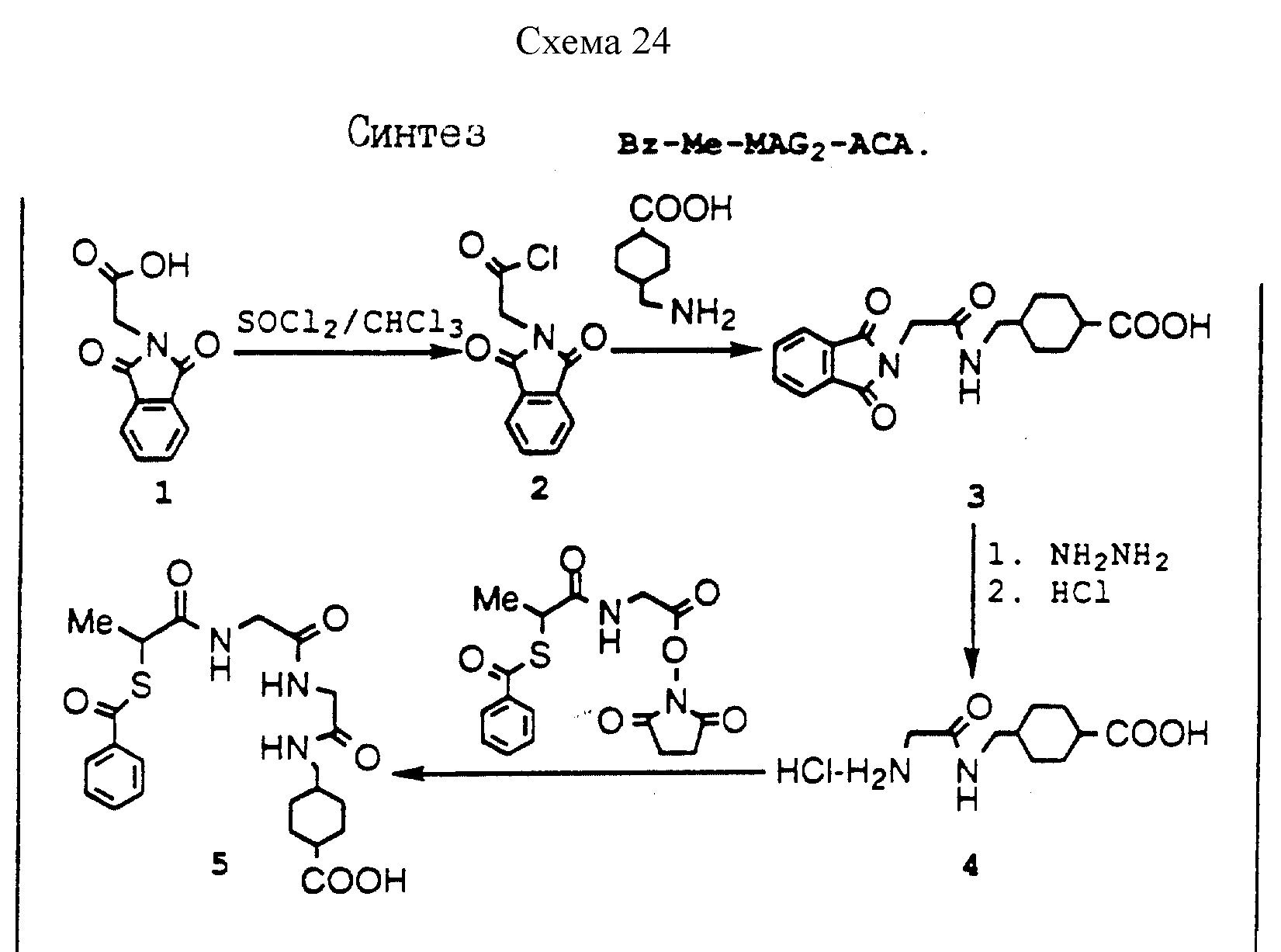


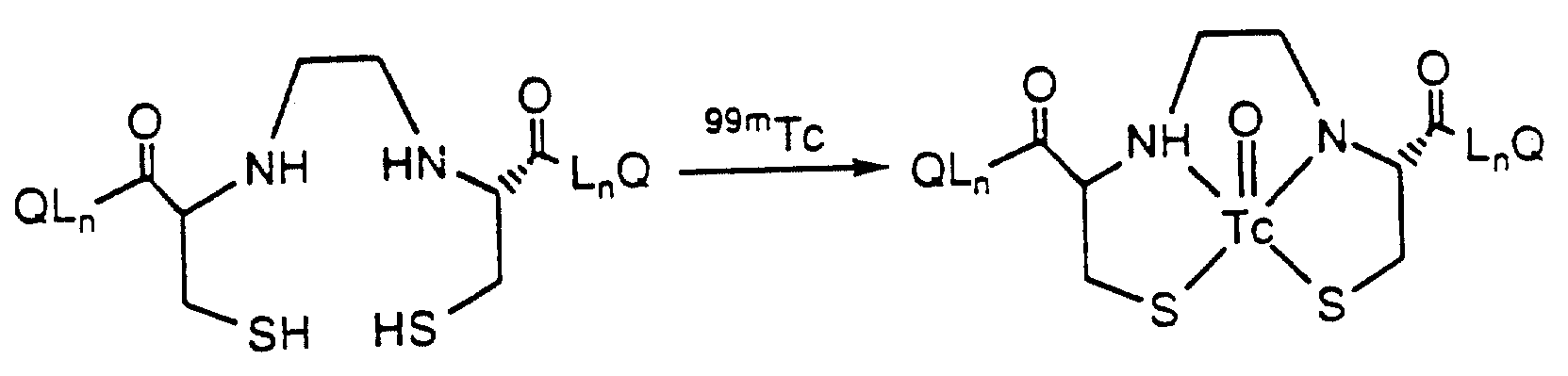
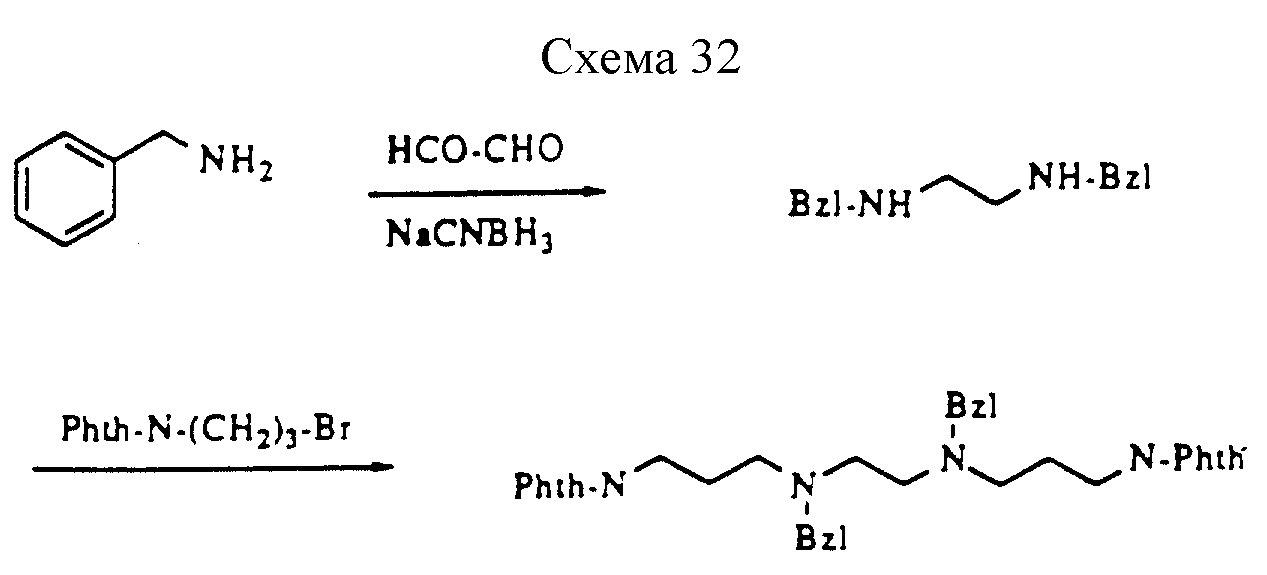
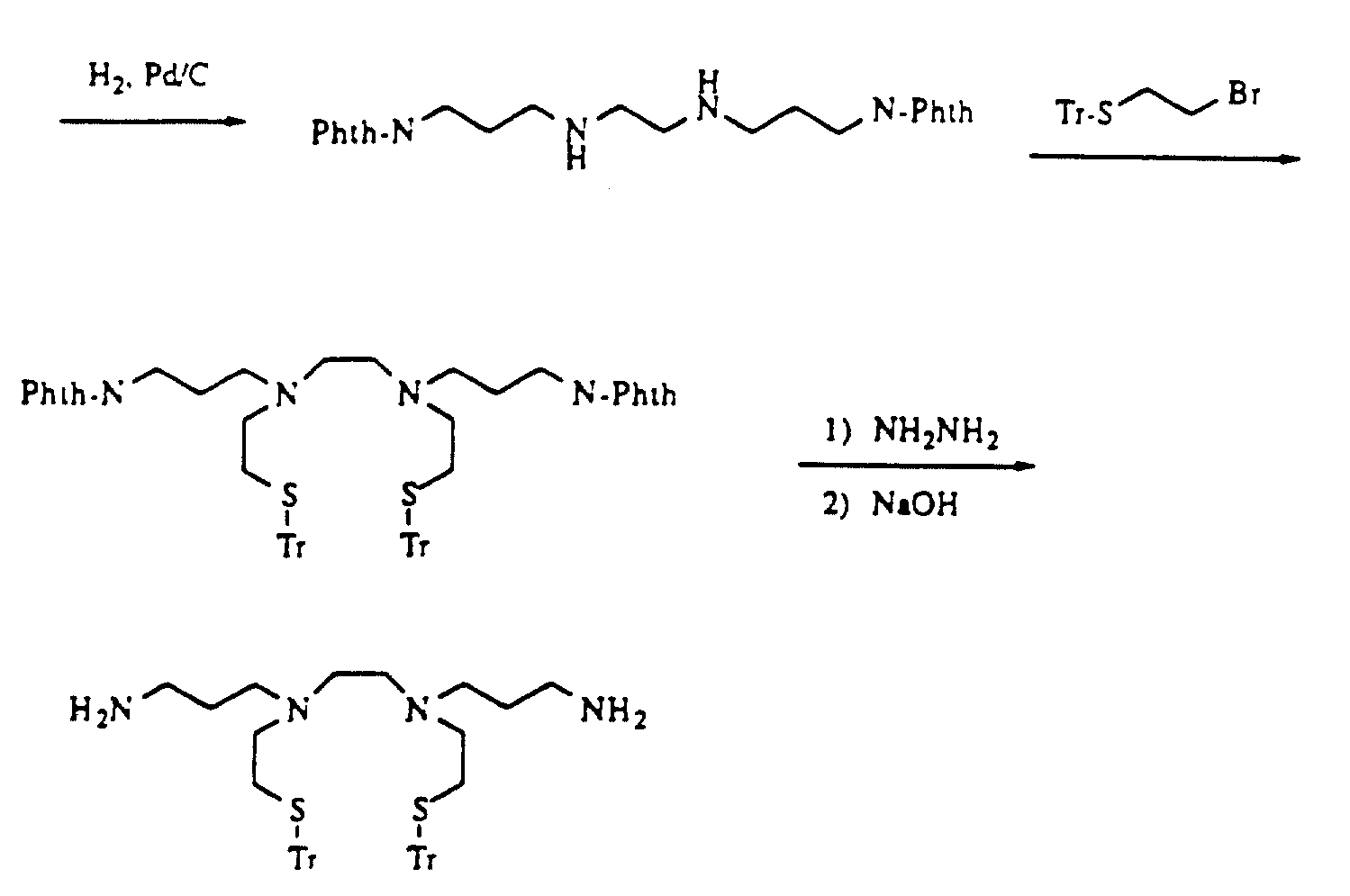

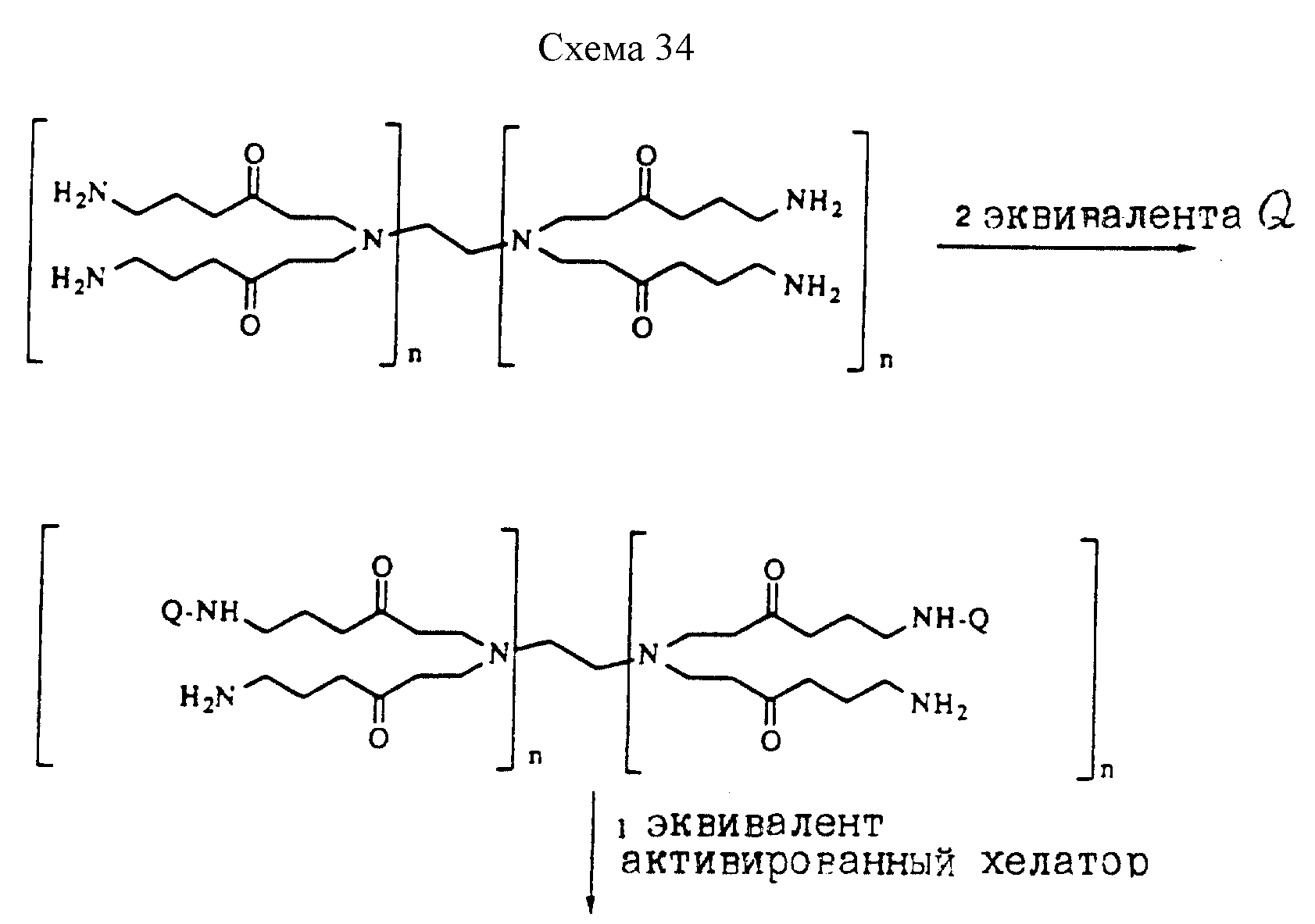
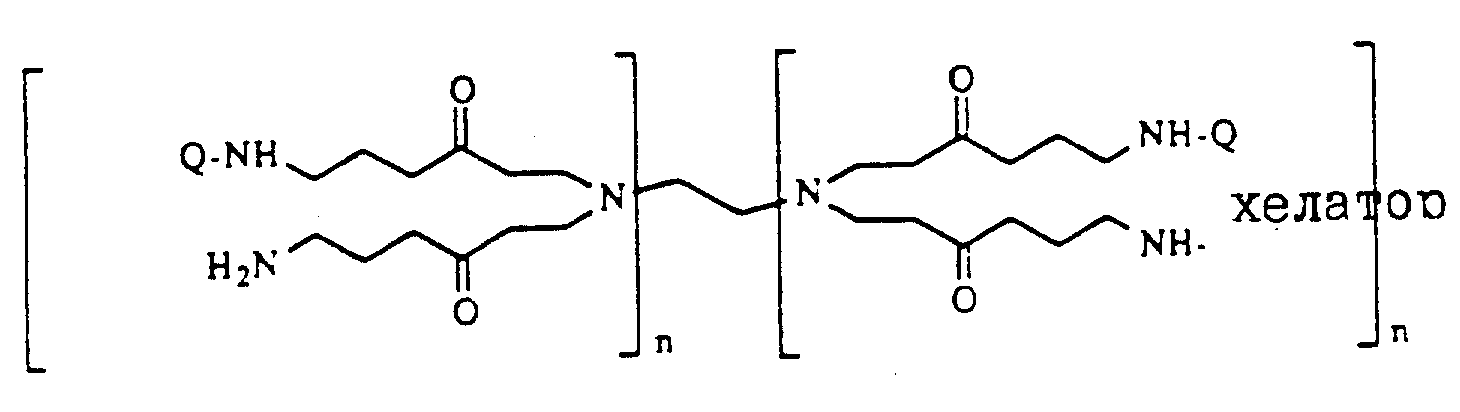
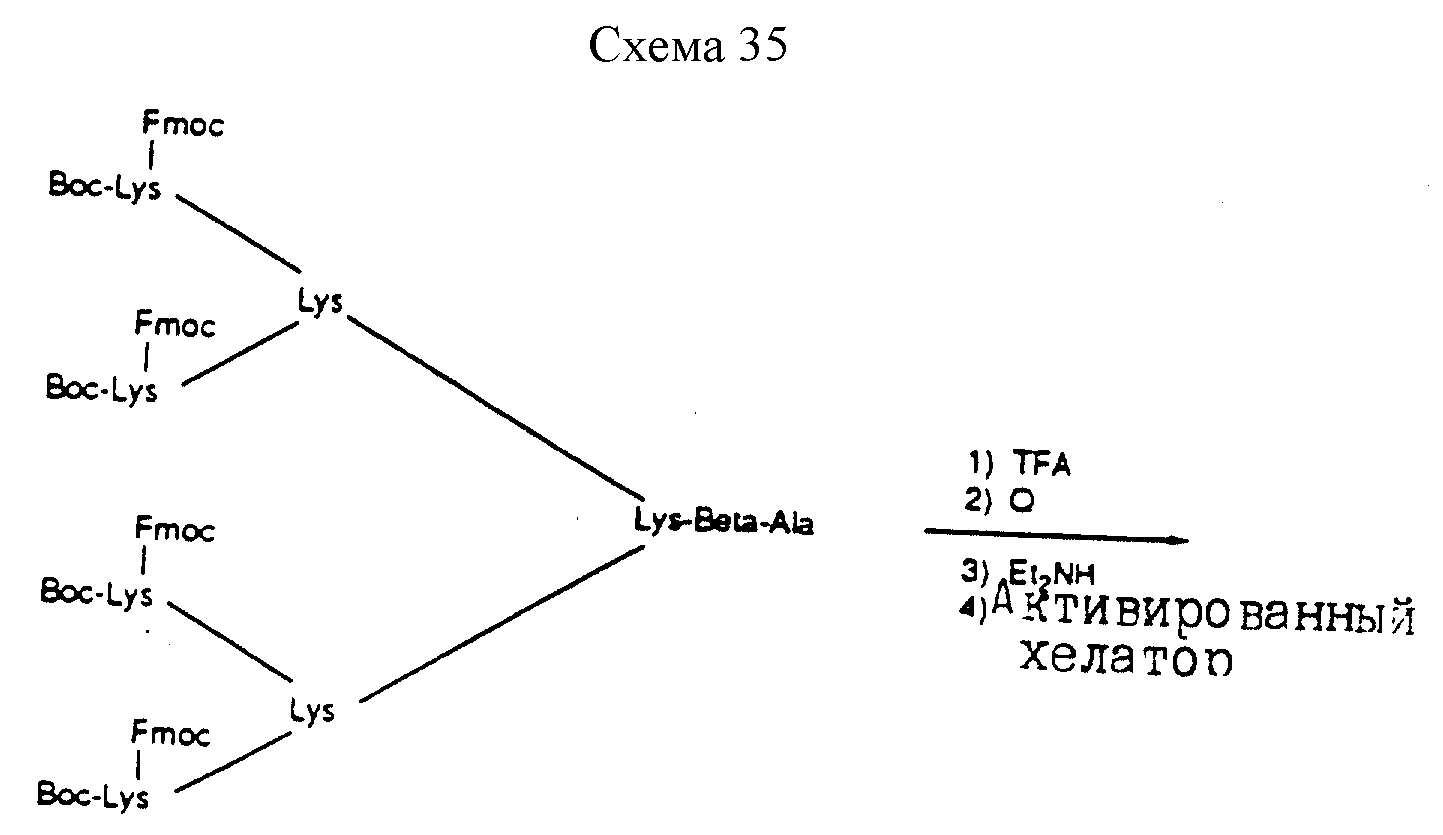
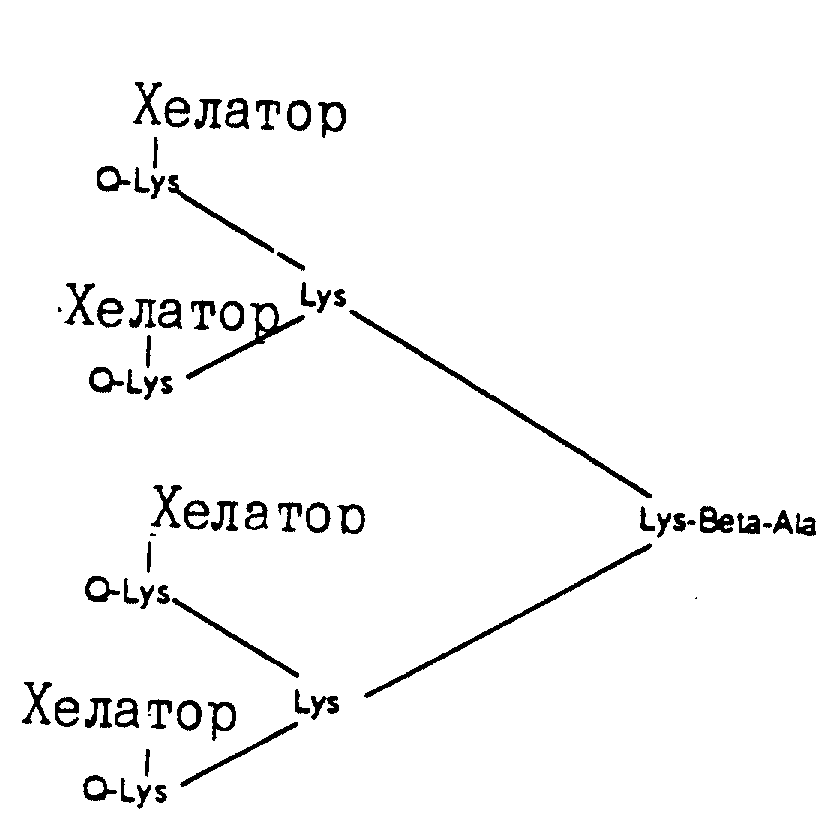
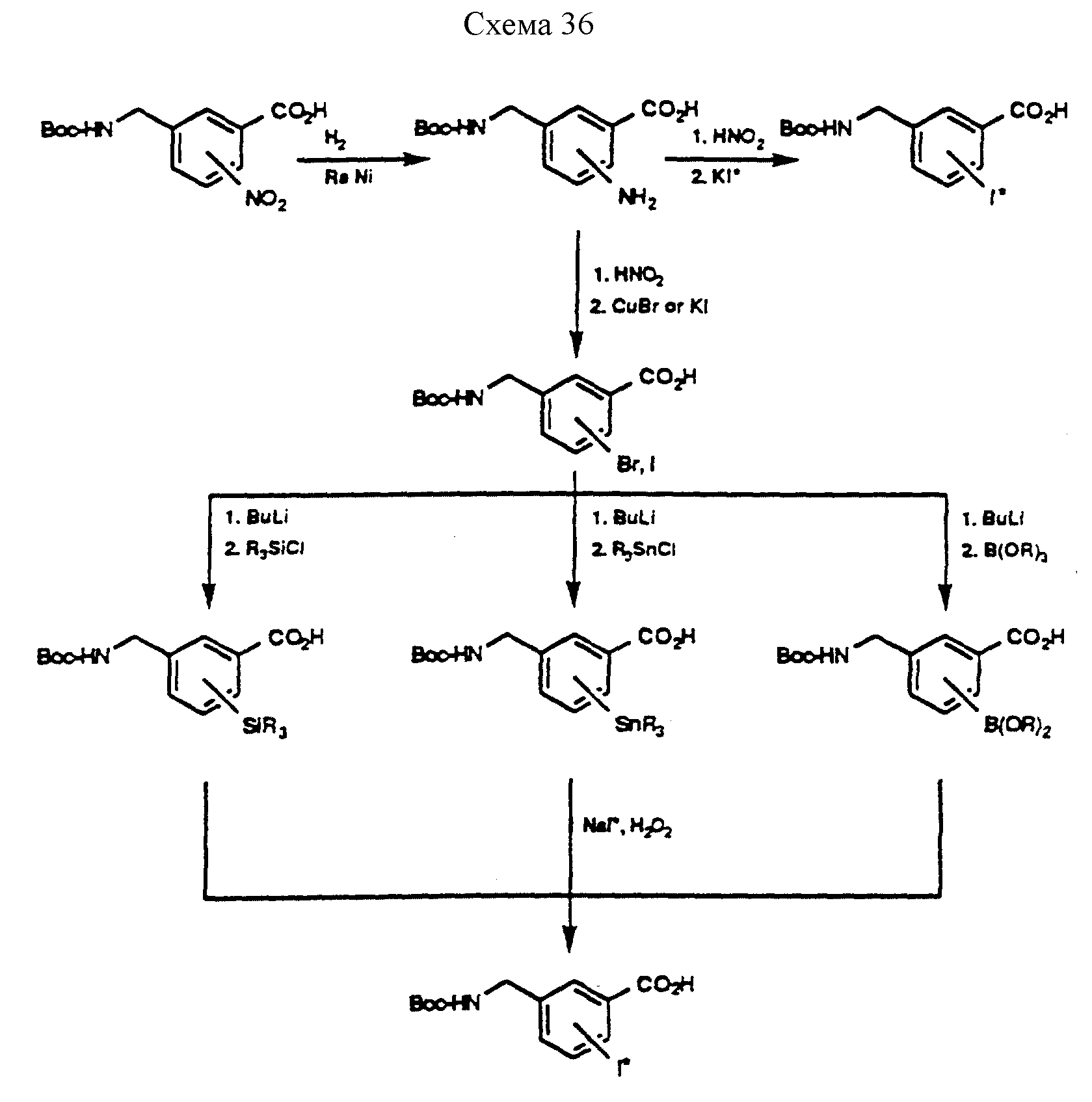
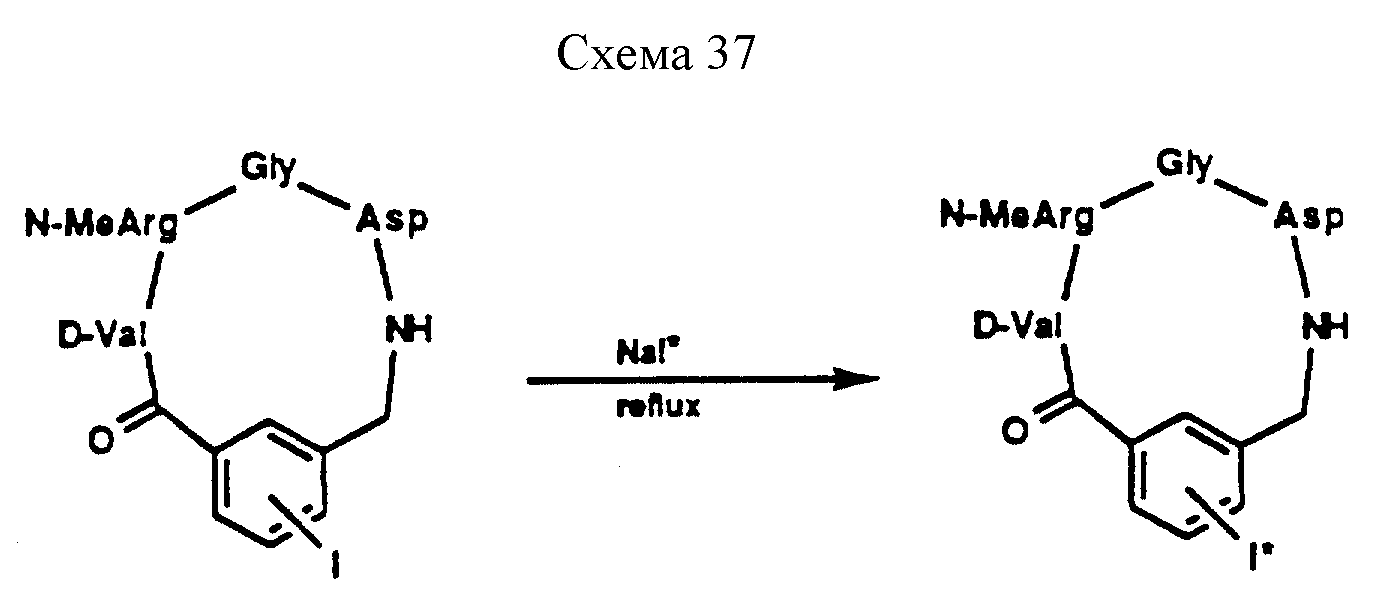


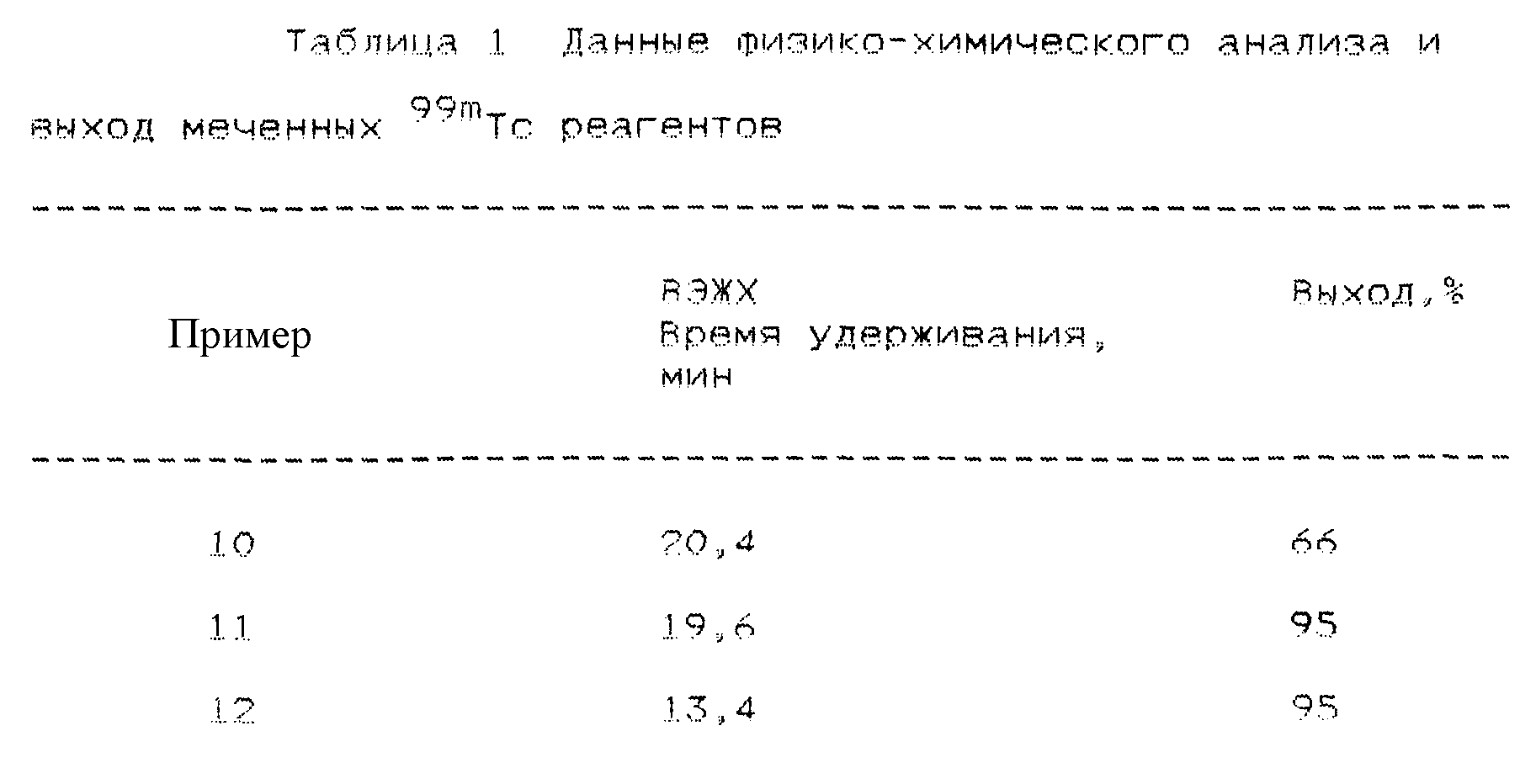

Описание
Изобретение относится к новым радиофармацевтическим препаратам, представляющим собой радиоактивномеченые циклические или гетероциклические кольцевые системы. Кроме того, изобретение относится к способам применения указанных радиофармацевтических препаратов в качестве создающих изображение соединений для диагностики артериального и венозного тромбоза, а также к новым реагентам для получения данных радиофармацевтических препаратов и к диагностическим наборам, содержащим указанные реагенты.
Клиническая диагностика венозных и артериальных тромбоэмболических заболеваний затруднена с точки зрения как чувствительности, так и специфичности. При разрешении критических для жизни ситуаций необходимо иметь в распоряжении врачей быстрый и неинвазивный метод выявления тромбоэмболических заболеваний. Показано, что активация и последующая агрегация тромбоцитов связана с различными патофизиологическими состояниями, включая тромбоэмболические кардиоваскулярные и цереброваскулярные нарушения, в том числе нестабильную стенокардию, инфаркт миокарда, преходящее нарушение мозгового кровообращения, мозговой удар, атеросклероз и диабет. Роль тромбоцитов в развитии указанных заболеваний обусловлена их способностью формировать агрегаты или тромбы, особенно на артериальных стенках после их повреждения. В особенности см. Fuster et al., JACC, vol. 5, N 6, pp. 175B-183B (1985); Rubenstein et al. , Am. Heart. J., vol. 102, pp. 363-367 (1981); Hamm et al., J. Am. Coll. Cardiol. , vol. 10, pp. 998-1006 (1987); и Dsvies et al., Circulation, vol. 73, pp. 418-427 (1986). Недавно был идентифицирован гликопротеиновый комплекс тромбоцитов IIb/IIIa (GPIIb/IIIa), представляющий собой мембранный белок, который опосредует агрегацию тромбоцитов обычным путем для известных агонистов рецепторов тромбоцитов. См. Philips et al., Cell, vol.65, pp. 359-362 (1991).
Считается, что активация и агрегация тромбоцитов имеют также существенное значение в развитии венозных тромбоэмболических заболеваний, таких как венозный тромбофлебит и последующая легочная эмболия. Кроме того, известно, что у больных, чья кровь контактирует с искусственными поверхностями, например простетическими сердечными клапанами из синтетических материалов, имеют повышенный риск развития тромбозов, эмболии и образования тромбоцитарных бляшек. В особенности см. Fuster et al., JACC, vol.5, N. 6, pp. 175B-183B (1985); Rubenstein et al., Am. Heart. J., vol. 102, pp. 363-367 (1981); Hamm et al. , J. Am. Coll. Cardiol., vol. 10, pp. 998-1006 (1987); и Dsivies et al., Circulation, vol. 73, pp. 418-427 (1986).
Подходящие средства для неинвазивной диагностики из мониторинга больных с такого рода возможными тромбоэмболическими заболеваниями могли бы оказаться чрезвычайно полезными, и были предприняты многочисленные попытки получения радиоактивномеченых соединений, выявляющих тромбоциты путем создания изображения при неинвазивной радионуклидной диагностике. Например, проводились экспериментальные исследования по использованию 99mIc-моноклональных антител к фибрину для диагностики предполагаемого артериального тромбоза. См. Cerqueira et al., Cerculation, vol. 85, pp. 298-304 (1992). Авторы сообщили о возможном использовании указанных соединений для создания изображения недавно образовавшихся артериальных тромбов. Кроме того, сообщалось о возможном применении моноклональных антител, меченных131J и специфичных по отношению к активированным тромбоцитам человека, для диагностики артериального и венозного тромбоза. Однако, достоверное отношение тромб/кровь (мишень/фон) достигалось только через 4 часа после введения радиоактивномеченого антитела. См. Wu et al. , Clin. Med. J., vol. 105, pp. 533-539 (1992). Кроме того, недавно обсуждалось использование радиоактивномеченых125J131J99mIc и111In моноклональных антител 7EЗ к тромбоцитам для создания изображения предполагаемых тромбов. См. Coller et al., Заявка PCT N 89/11538 (1989). Однако, применение радиоактивномеченого 7EЗ-антитела неудобно из-за большой ол. массы. Другие исследователи использовали энзиматически инактивированный t-PA, меченный радиоактивными изотопами иода123J,125J и131J, для определения локализации тромбов. См. Ordm et al., Cerculation, vol.85, pp. 288-297 (1992). Кроме того, известны другие исследования, касающиеся радиологического определения тромбоэмболических заболеваний. См., например, Koblik et al., Semin. Nucl. Med., vol. 19, pp. 221-237 (1989).
Выявление локализации артериальных и венозных тромбов имеет существенное значение для достоверной диагностики тромбоэмболических заболеваний и разработки методов соответствующей терапии. Существует необходимость получения новых и улучшенных радиоактивномеченых соединений для неинвазивного создания изображения тромбов при радионуклидной диагностике.
Настоящее изобретение направлено на решение этой важной проблемы.
Настоящее изобретение относится к новым радиофармацевтическим препаратам, которые представляют собой радиоактивномеченые циклические соединения, содержащие карбоциклические или гетероциклические кольцевые системы, причем указанные соединения функционируют как антагонисты гликопротеинового комплекса IIb/IIIa тромбоцитов. Кроме того, изобретение относится к способам использования данных радиофармацевтических препаратов в качестве создающих изображение соединений при диагностике артериального и венозного тромбоза. Изобретение также относится к новым реагентам для приготовления указанных радиофармацевтических препаратов, а также к диагностическим наборам, содержащим эти соединения.
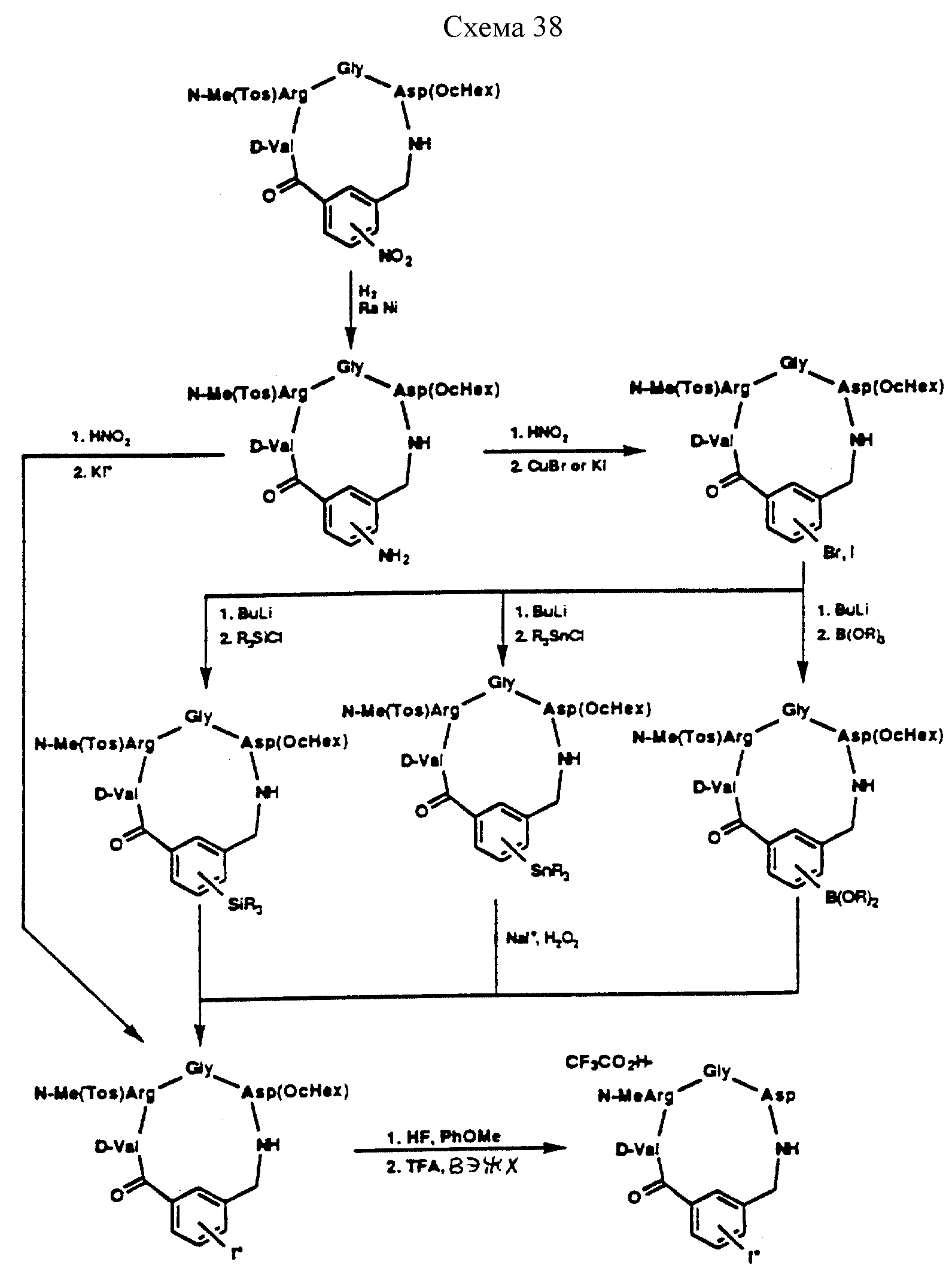
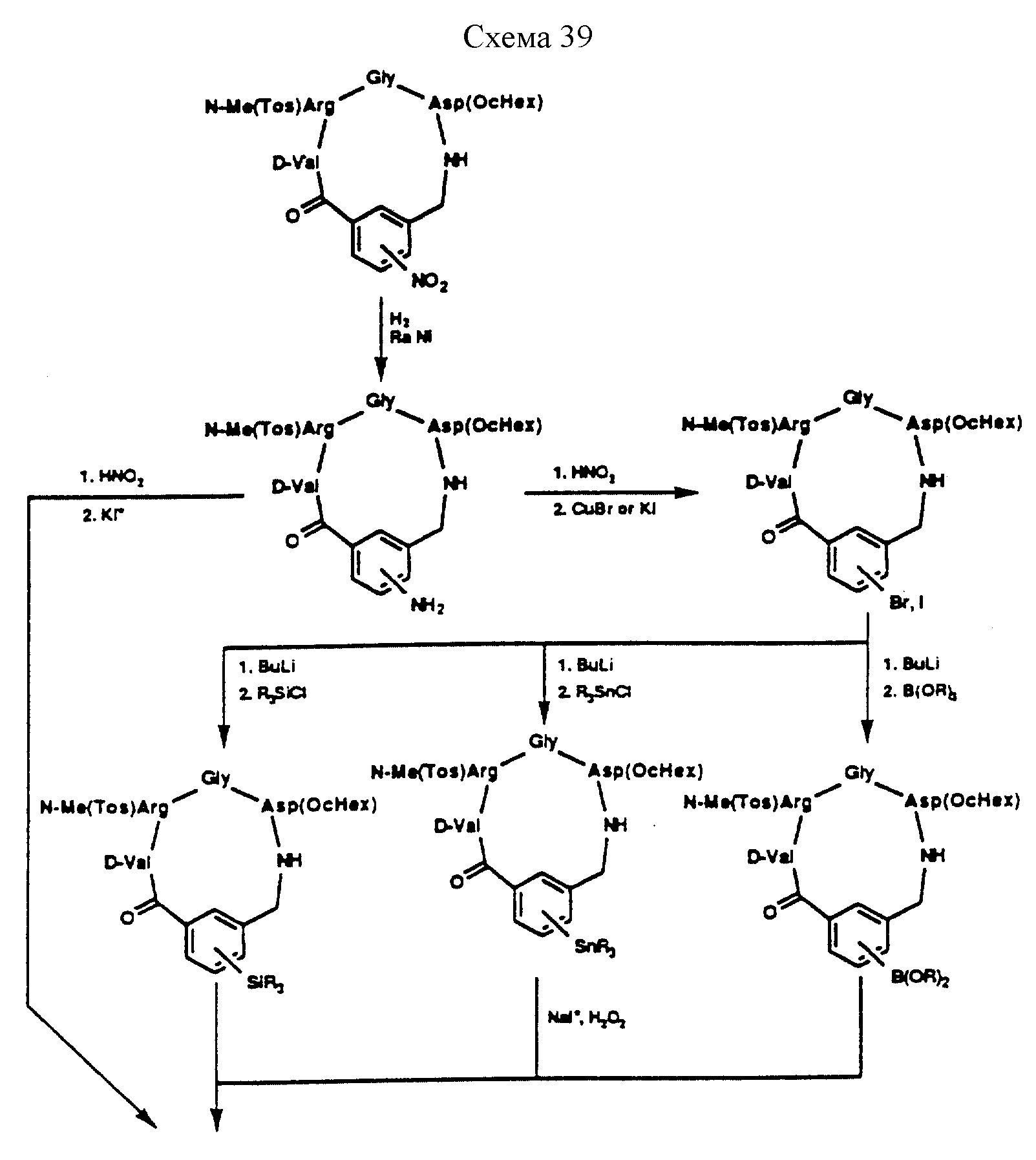
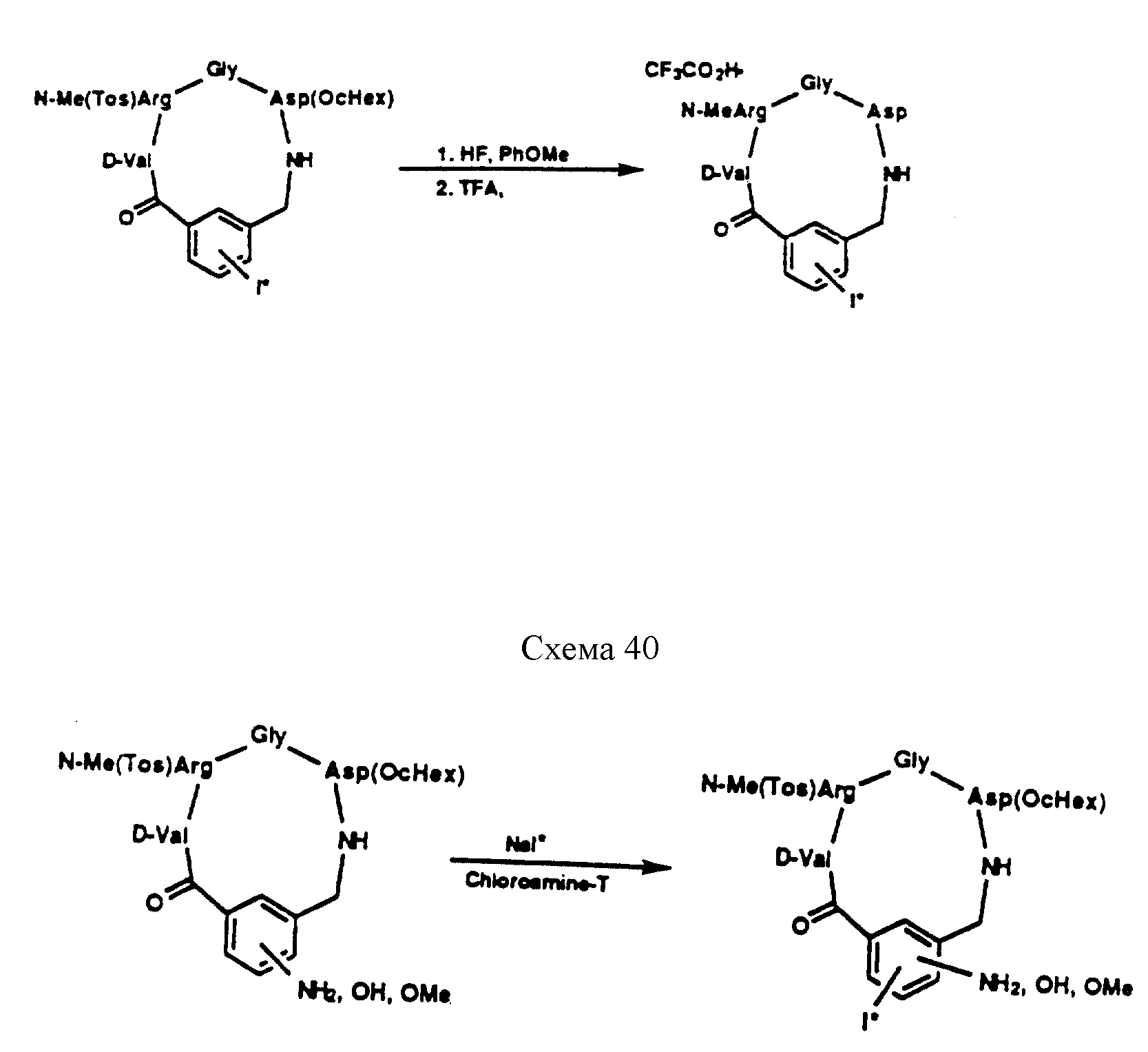
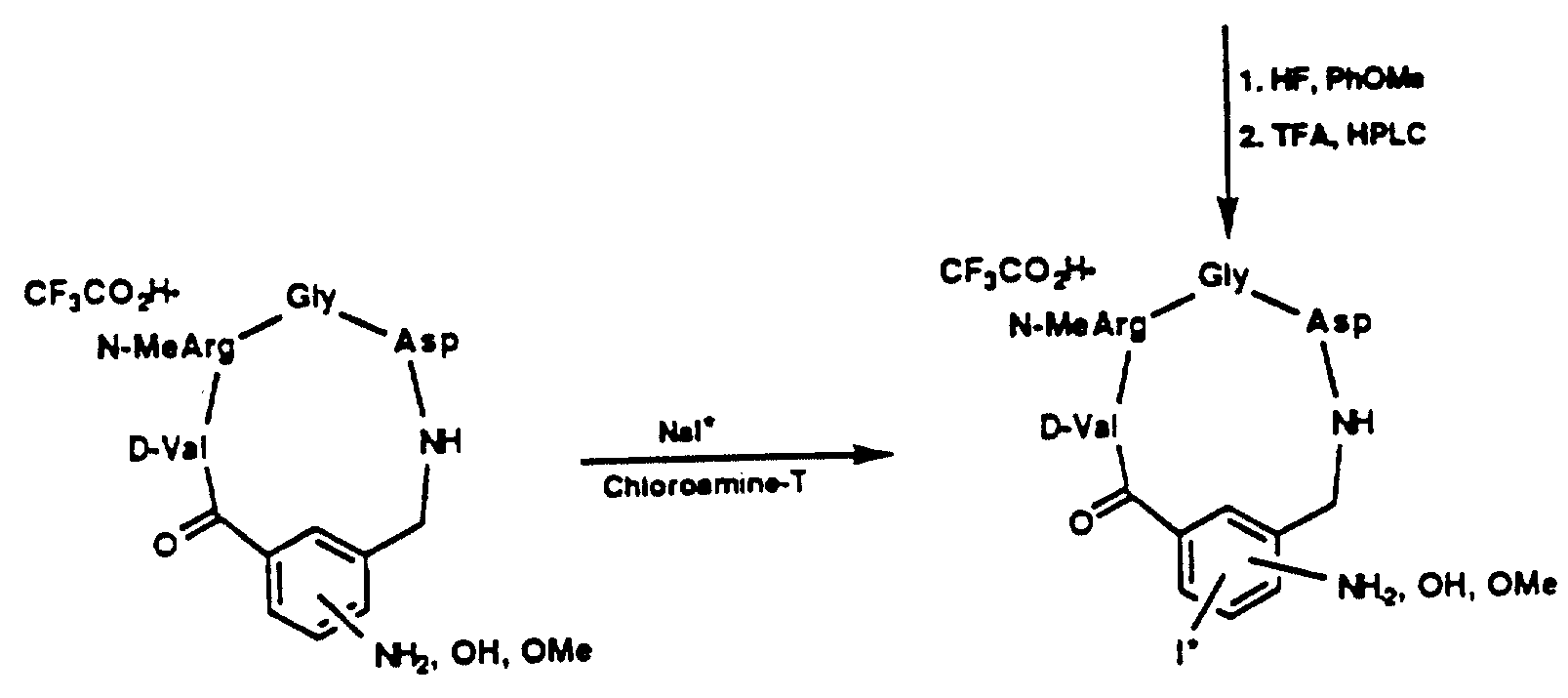
Фиг. 1а. Показано обычное изображение,
полученное при использовании радиофармацевтического соединения (пример 12), введенного внутривенно (1 мкKu/кг веса) собаке при диагностике глубокого венозного тромбоза на экспериментальной модели.
Данная модель предусматривает формирование тромбов в яремных венах во время стаза, а затем ток крови возобновляют. Соединения вводят с возобновленным током крови. Приведены изображения, полученные при
включении диагностических соединений в быстро растущие венозные тромбы через 15, 60 и 120 мин после введения соединений
Фиг. 1b. Показано обычное изображение, полученное при использовании
радиофармацевтического соединения (пример 19), введенного внутривенно (1 мкKu/кг веса) собаке при диагностике глубокого венозного тромбоза на экспериментальной модели. Данная модель предусматривает
формирование тромбов в яремных венах во время стаза, а затем ток крови возобновляют. Соединения вводят с возобновленным током крови. Приведены изображения, полученные при включении диагностических
соединений в быстро растущие венозные тромбы через 15, 60 и 120 мин после введения соединений.
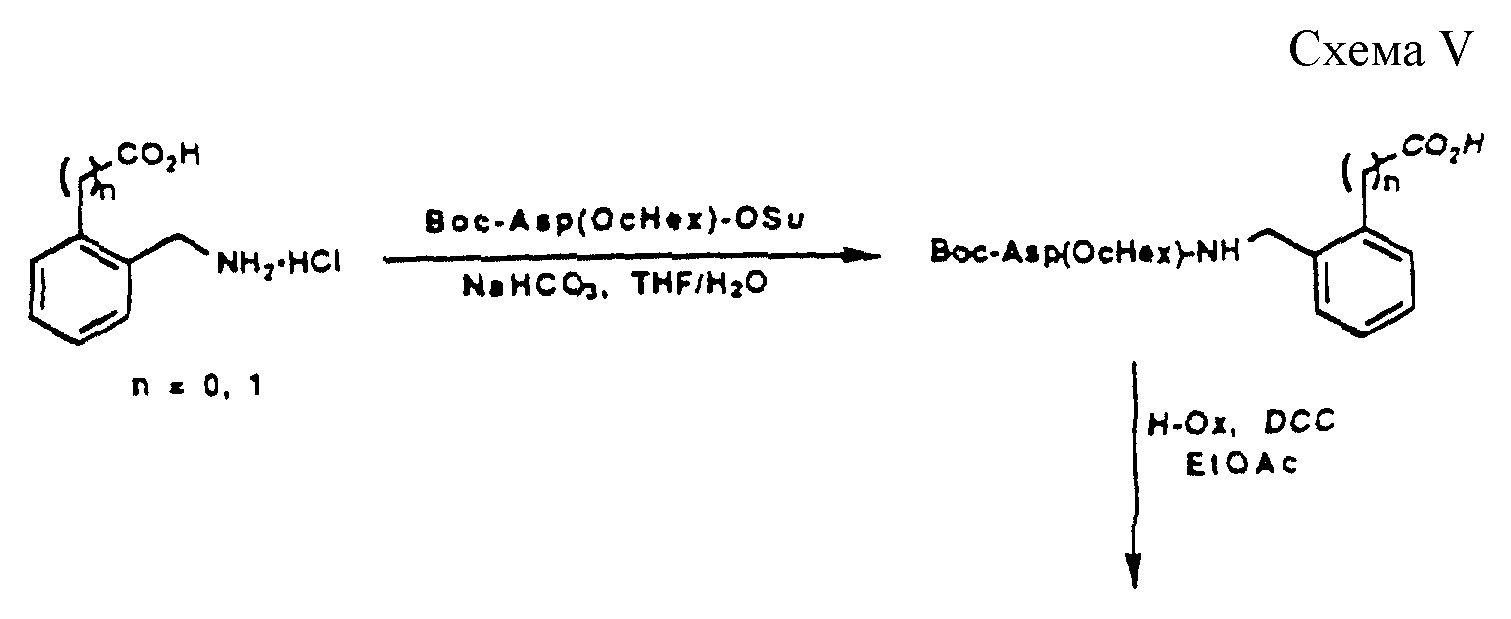
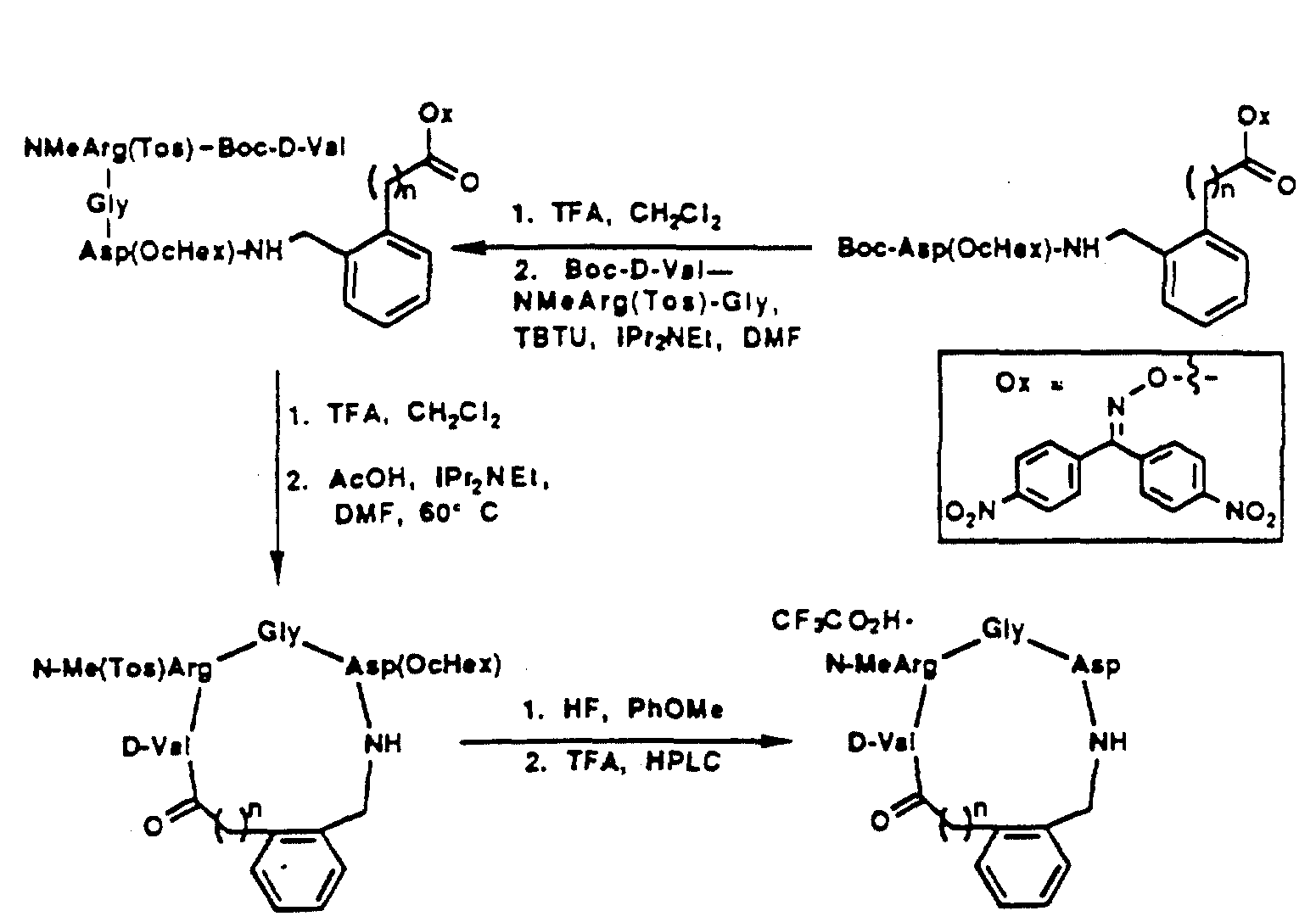
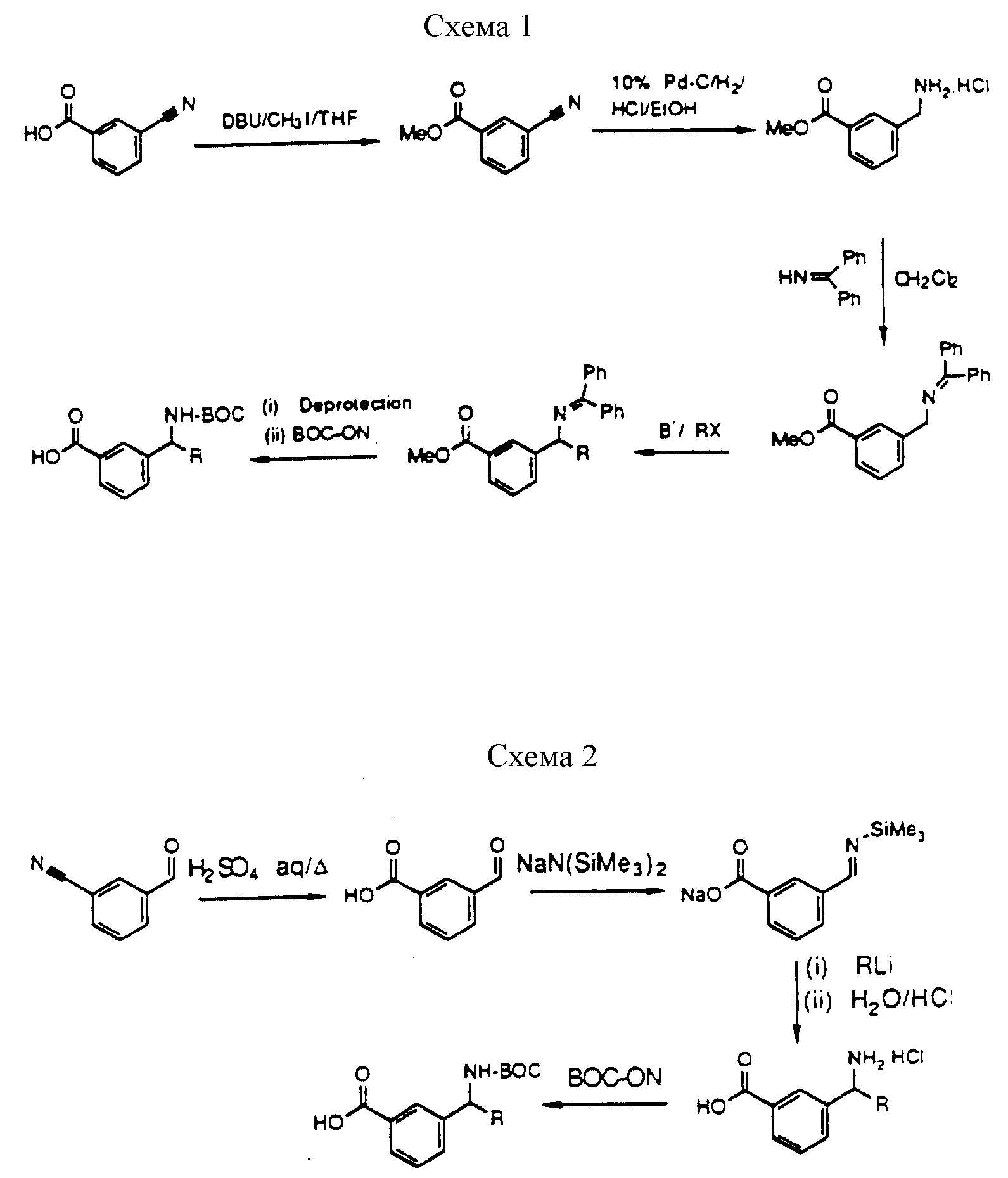
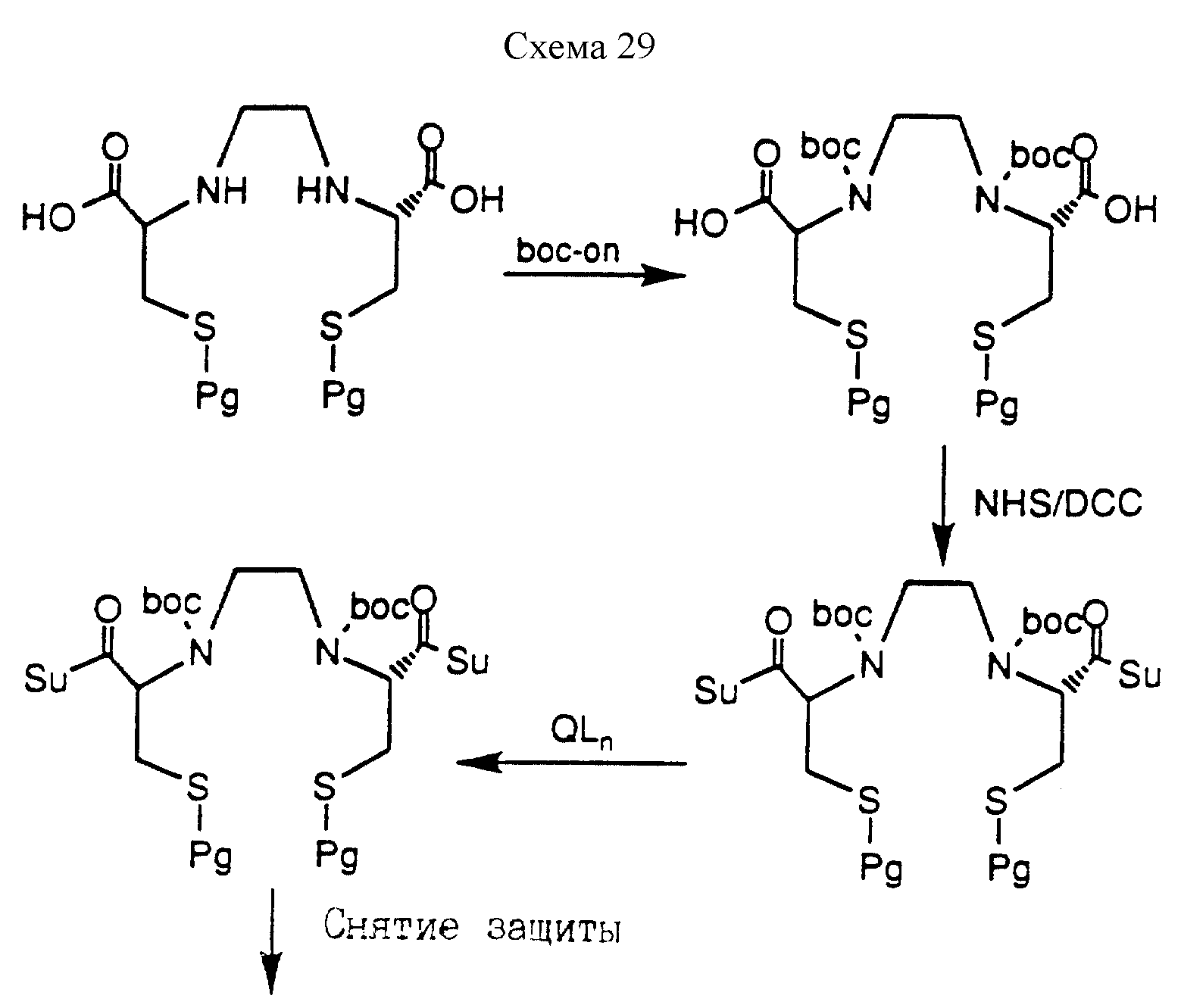
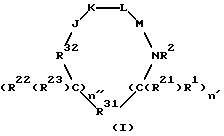
[1] Настоящее изобретение относится к новым реагентам для получения радиофармацевтических
препаратов формулы: (QLn)dCh; (Q)d'Ln-Ch, причем d = 1-3, d' = 2 - 20, Ln - связывающая группа; Ch - хелатор металла и Q - соединение
формулы (I)
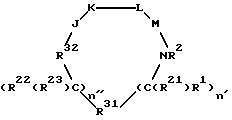
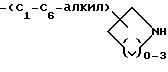
или фармацевтически пригодная соль или ее неактивная форма, причем R31 представляет собой C6-C14 насыщенную, частично насыщенную или ароматическую карбоциклическую кольцевую систему, замещенную 0-4 R10 или R10a и факультативно имеющую связь с Ln; гетероциклическую кольцевую систему, факультативно замещенную 0-4 R10 или R10a и факультативно имеющую связь с Ln;
R32 выбирают из соединений -C(=O)-; -C(=S)-; -S(=O)2-; -S(=O)-; -P(= Z)(ZR13)-; Z - S или O; n'' и n' = 0-2; R1 и R22 независимо выбирают из следующих групп: водород, C1-C8-алкил, замещенный 0-2 R11; C2-C8-алкенил, замещенный 0-2 R11; C2-C8-алкинил, замещенный 0-2 R11; C3-C10-циклоалкил, замещенный 0-2 R11; связь с Ln; арил, замещенный 0-2 R12; 5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатомов, независимо выбранных из N, S и O, причем указанная кольцевая система замещена 0-2 R12; =O, F, Cl, Br, I, -CF3, -CN, -CO2R13, -C(=O)R13, -C(=O)N(R13)2, -CHO, -CH2OR13, -OC(= O)R13, -OC(= O)OR13a, -OR13, -OC(=O)N(R13)2 , -NR13C(=O)R13, -NR14C(= O)OR13a, -NR13C(=O)N(R13)2, -NR14SO2N(R13)2, -NR14SO2R13a, -SO3H, -SO2R13a,
-SR13, -S(=O)R13a, -SO2N(R13)2, -N(R13)2, -NHC(=NH)NHR13, -C(=NH)NHR13, = NOR13, NO2, -C(= O)NHOR13, -C(= O)NHNR13R13a, -OCH2CO2H, 2-(1-морфолино)этокси; R1 и R21 могут быть альтернативно соединены с образованием 3-7-членного карбоциклического кольца, замещенного 0-2 R12; когда n' = 2, R1 или R21 могут быть альтернативно взяты вместе с R1 или R21 на соседнем атоме углерода с образованием прямой связи, таким образом формируя двойную или тройную связь между соседними атомами углерода; R21 и R23 независимо выбирают из водорода; C1-C4-алкила, факультативно замещенного 1-6 галогеном; бензила; R22 и R23 могут быть альтернативно соединены с образованием 3-7-членного карбоциклического кольца, замещенного 0-2 R12; когда n'' = 2, R22 или R23 могут быть взяты вместе с R22 или R23 на соседнем атоме углерода с образованием прямой связи, таким образом формируя двойную или тройную связь между соседними атомами углерода; R1 и R2, где R21 - H, могут быть альтернативно соединены с образованием 5-8-членного карбоциклического кольца, замещенного 0-2 R12; R11 выбирают из одного или более следующих соединений: =O, F, Cl, Br, I, -CF3, -CN, -CO2R13, -C(=O)R13, -C(=O)N(R13)2, -CHO, -CH2OR13, -OC(= O)R13, -OC(=O)OR13a, -OR13, -OC(=O)N(R13)2, -NR13C(=O)R13, -NR14C(=O)OR13a, -NR13C(= O)N(R13)2, -NR14SO2N(R13)2, -NR14SO2R13a, -SO3H, -SO2R13a , -SR13, -S(= O)R13a, -SO2N(R13)2, -N(R13)2, -NHC(=NH)NHR13, -C(=NH)NHR13, =NOR13, NO2, -C(= O)NHOR13, -C(= O)NHNR13R13a, -OCH2CO2H, 2-(1-морфолино)этокси, C1-C5-алкил, C2-C4 -алкенил, C3-C6-циклоалкил, C3-C6-циклоалкилметил,
C2-C6-алкоксиалкил, C3-C6-циклоалкокси, C1 -C4алкил/алкил, замещенный 1-5 группами, независимо выбранными из -NR13R14, -CF3, NO2, -SO2R13a или -S(= O)R13a), арил, замещенный 0-2 R12, 5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатома, независимо выбранных из N, S и O, причем указанное гетероциклическое кольцо замещено 0-2 R12; R12 выбирают из одного или более следующих соединений: фенил, бензил, фенетил, фенокси, бензилокси, галоген, гидрокси, нитро, циано, C1-C5-алкил, C3-C6-циклоалкил; C3-C6-циклоалкилметил, C7-C10-арилалкил, C1-C5-алкокси, -CO2R13, -C(= O)NHOR13a, -C(=O)NHN(R13)2, =NOR13, -B(R34)(R35), C3-C6-циклоалкокси, -OC(= O)R13, -C(= O)R13, OC(=O)OR13a, -OR13, -(C1-C4-алкил)-OR13, -N(R13)2, -OC(= O)N(R13)2, -NR13 C(=O)R13, -NR13C(=O)OR13a, -NR13C(=O)N(R13)2, -NR13SO2N(R13)2, -NR13SO2R13a, -SO3H, -SO2R13a, -S(=O)13a, -SR13, -SO2(R13)2,
C2-C6 -алкоксиалкил, метилендиокси, этилендиокси, C1-C4-галоалкил, C1-C4-галоалкокси, C1-C4-алкилкарбонилокси, C1-C4 -алкилкарбонил, C1-C4-алкилкарбониламино, -OCH2CO2H, 2-(1-морфолино)этокси, C1-C4-алкил/алкил, замещенный -N(R13)2, -CF3, NO2 или -S(=O)R13a); R13 независимо выбирают из H, C1-C10-алкила, C3-C10-циклоалкила, C4-C12-алкилциклоалкила, арила, -(C1-C10-алкил)-арила или C3-C10-алкоксиалкила;
R13a - C1-C10 -алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
когда две R13 группы связаны с одиночным N, указанные R13-группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2 )O(CH2)-;
DM-6591-A
R14 - OH, H, C1-C4-алкил или бензил;
R2 - H или C1-C8-алкил;
R10 и R10a независимо выбирают из одного или более соединений: фенил, бензил, фенэтил, фенокси, бензилокси, галоген, гидрокси, нитро, циано, C1-C5-алкил, C3-C6-циклоалкил, C3-C6циклоалкилметил; C7-C10-арилалкил,
C1-C5-алкокси, -CO2R13, C(=O)N(R13)2, -C(=O)NHOR13a, -C(=O)NHN(R13)2, = NOR13, -B(R34)(R35), C3-C6-циклоалкокси, -OC(= O)R13, -C(= O)R13, -OC(= O)OR13a, -OR13, -(C1-C4-алкил)-OR13, -N(R13)2, -OC(=O)N(R13 )2, -NR13C(= O)R13, -NR13C(= O)OR13a, -NR13C(=O)N(R13)2, -NR13SO2N(R13)2, -NR13SO2R13a, -SO3H, -SO2R13a, -S(=O)R13a, -SR13, -SO2N(R13)2, C2-C6-алкоксиалкил, метилендиокси, этилендиокси, C1-C4-галоалкил (включая -CvFw, где v = 1-3 и w = 1-(2v+1)), C1-C4 -галоалкокси, C1-C4-алкилкарбонилокси, C1-C4-алкилкарбонил,
C1-C4-алкилкарбониламино, -OCH2CO2H, 2-(1-морфолино)этокси, C1-C4-алкил, замещенный -N(R13)2, CF3, NO2, или -S(=O)R13a);
J - β- Ala или L-изомер или D-изомер аминокислоты, имеющей структуру -N(R3)C(R4)(R5)C(=O)-, причем R3 - H или C1-C8-алкил; R4 - H или C1 -C3-алкил;
R5 выбирают из соединений: водород; C1-C8-алкил, замещенный 0-2 R11; C2-C8-алкенил, замещенный 0-2 R11; C2-C8-алкенил, замещенный 0-2 R11; C3-C10-циклоалкил, замещенный 0-2 R11; связь с Ln; арил, замещенный 0-2 R12; 5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатома, независимо выбранных из N, S или O, причем указанное гетероциклическое кольцо замещено 0-2 R12; =O, F, Cl, Br, I, -CF3, -CN, -CO2R13, -C(=O)R13, -C(= O)N(R13)2, -CHO, -CH2OR13, -OC(= O)R13, -OC(=O)OR13a, -OR13, -OC(= O)N(R13)2, -NR13C(=O)R13, -NR14C(=O)OR13a, -NR13C(=O)N(R13)2, -NR14SO2N(R13)2, -NR14SO2R13a, -SO3H, -SO2R13a, -SR13, -S(= O)R13a, -SO2(R13)2, -N(R13)2, -NHC(= NH)NHR13, -C(= NH)NHR13, =NOR13, NO2, -C(= O)NHOR13 , -C(= O)NHNR13R13a, =NOR13, -B(R34)(R35), -OCH2CO2H, 2-(1-морфолино)этокси, -SC(= NH)NHR13, N3, -Si(CH3)3, (C1-C5-алкил)NHR16; -(C0-C6 алкил)X;
где q = 0, 1;
-(CH2)mS(O)p'(CH2)2X, где m = 1, 2 и p' = 0-2;
причем X определен ниже; и
R3 и R4 также могут быть взяты вместе с образованием
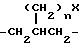
n = 0, 1 и X представляет собой

R3 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)t- или -CH2S(O)p'C(CH3)2,
где t = 2-4 и p' = 0-2; или
R4 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)u-, где u = 2-5;
R16 выбирают из соединений: аминозащитная группа; 1-2 аминокислоты; 1-2 аминокислоты, замещенные аминопротективной группой;
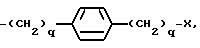
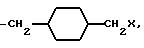
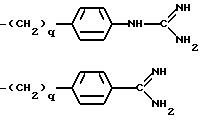
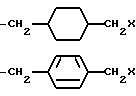
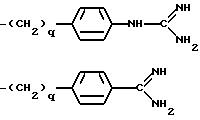
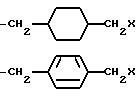
K представляет собой D-изомер или L-изомер аминокислоты формулы -N(R6)CH(R7)C(= O)-, причем R6 представляет собой H или C1-C8-алкил; R7 выбирают из соединений: -(С1 -С7 алкил)X;

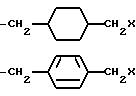
причем каждое q = 0-2 и замещение на фенил происходит в третьей или четвертой позиции;
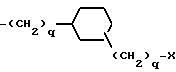
причем каждое q = 0-2 и замещение на циклогексил происходит в третьей или четвертой позиции;
-(CH2)mO-(C1-C4-алкил)-X, где m = 1 или 2;
-(CH2)mS(O)p'-(C1-C4-алкил)-X, где m = 1 или 2 и p' = 0-2; и X выбирают из соединений
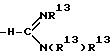
-N(R13)R13; -C(= NH)(NH2); -SC(= NH)-NH2; -NH-C(= NH)(NHCN); -NH-C(= NCN)(NH2); -NH-C(= N-OR13)(NH2); R6 и R7 могут быть альтернативно взяты вместе с образованием
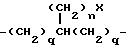
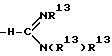
причем каждое q = 1 или 2 и причем n = 0 или 1 и X представляет собой -NH2 или
L - -Y(CH2)vC(=O)-, причем Y - NH; N(C1-C3-алкил), O или S; и v = 1 или 2;
M - D-изомер или L-изомер аминокислоты формулы
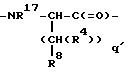
причем q' = 0-2;
R17 - H; C1 -C3-алкил;
R8 выбирают из соединений -CO2R13, -SO3R13, -SO2NHR14, -B(R34)(R35), -NHSO2CF3, -CONHNHSO2CF3, -PO(OR13)2, -PO(OR13)R13, -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбранных из N, S или O), -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбранных из N, S или O), -SO2NHCOR13, -CONHSO2R13a, -CH2CONHSO2R13a, -NHSO2NHCOR13a, -NHCONHSO2R13a, -SO2NHCONHR13; R34 и R35 независимо выбирают из соединений: -OH, -F, -N(R13)2 или C1-C8-алкокси; R34 и R35 могут быть альтернативно взяты вместе с образованием циклического эфира бора, где указанная цепь или кольцо содержат от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбранных из N, S или O; бивалентного циклического амида бора, где указанная цепь или кольцо содержат от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбранных из N, S или O; циклического амидоэфира бора, где указанная цепь или кольцо содержат от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбранных из N, S или O.
[2] В настоящее изобретение включены указанные в [1] реагенты, причем R31 связан с (C(R23)R22)n'' и (C(R21)R1)n' на двух различных атомах указанного карбоциклического кольца.
[3] В настоящее изобретение включены указанные в [1] реагенты, причем:
n'' = 0 и n' = 0;
n'' = 0 и n' =
1;
n'' = 0 и n' = 2;
n'' = 1 и n' = 0;
n'' = 1 и n' = 1;
n'' = 1 и n' = 2;
n'' = 2 и n' = 0;
n'' = 2 и n' = 1 или
n'' = 2 и n' = 2.
[4] В настоящее изобретение включены указанные в [1] реагенты, причем R6 представляет собой метил, этил или пропил.
[5] В настоящее изобретение включены указанные
в [1] реагенты, причем R32 выбирают из соединений: -C(=O)-; -C(=S)-; -S(=O)2-; R1 и R22 независимо выбирают из следующих групп: водород; C1-C8-алкил, замещенный 0-2 R11, C2-C8-алкенил, замещенный 0-2 R11, C2-C8-алкинил, замещенный 0-2 R11, C3
-C8-циклоалкил, замещенный 0-2 R11, C6-C10-бициклоалкил, замещенный 0-2 R11, связь с Ln; арил, замещенный 0-2 R12;
5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатома, независимо выбранных из N, S или O, указанное гетероциклическое кольцо замещено 0-2 R12; =O, F, Cl, Br, I,
-CF3, -CN, -CO2R13, -C(=O)R13, -C(=O)N(R13)2, -CHO, -CH2OR13, -OC(=O)R13, -OC(= O)OR13a,
-OR13, -OC(= O)N(R13)2, -NR13C(=O)R13, -NR14C(=O)OR13a, -NR13C(= O)N(R13)2, -NR14SO2N(R13)2, -NR14SO2R13a, -SO3H, -SO2R13a, -SR13, -S(= O)R13a, -SO2N(R13)2, -CH2N(R13)2, -N(R13)2, -NHC(= NH)NHR13, -C(= NH)NHR13, NO2; R1
и R2 могут альтернативно соединяться с образованием 5-7-членного карбоциклического кольца, замещенного 0-2 R12; когда n' = 2, R1 или R11 могут альтернативно
быть взяты вместе с R1 или R21 на соседнем атоме углерода с образованием прямой связи, таким образом формируя двойную или тройную связь между указанными углеродными атомами;
R22 и R23 могут альтернативно соединяться с образованием 3-7-членного карбоциклического кольца, замещенного 0-2 R12; когда n'' = 2, R22 или R23
могут альтернативно быть взяты вместе с R22 или R23 на соседнем атоме углерода с образованием прямой связи, таким образом формируя двойную или тройную связь между соседними
атомами углерода; R1 и R2, R21 представляет собой H, могут альтернативно соединяться с образованием 5-8-членного карбоциклического кольца, замещенного 0-2 R12; R11 выбирают из одного или более следующих соединений: =O, F, Cl, Br, I, -CF3, -CN, -CO2R13, -C(=O)R13, -C(=O)N(R13)2, -CHO, -CH2OR13, -OC(= O)R13, -OC(=O)OR13a, -OR13, -OC(=O)N(R13)2, -NR13C(=O)R13, -NR14C(=O)OR13a, -NR13C(= O)N(R13)2, -NR14SO2N(R13)2, -NR14SO2R13a, -SO3H, -SO2R13a, -SR13, -S(= O)R13a, -SO2N(R13)2, -CH2N(R13)2, -N(R13)2, -NHC(= NH)NHR13, -C(= NH)NHR13, = NOR13, NO2; C1-C5-алкил, C2-C4-алкенил, C3-C6
-циклоалкил, C3-C6-циклоалкилметил, C2-C6-алкоксиалкил, C1-C4-алкил (замещенный -NR13R14, -CF3,
NO2, -SO2R13 или -S(=O)R13а), арил, замещенный 0-2 R12; 5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатома, независимо
выбранных из N, S или O, указанное гетероциклическое кольцо замещено 0-2 R12;
R3 - H или CH3;
R5 - H, C1-C8-алкил,
C3-C6-циклоалкил, C3-C6-циклоалкилметил, C1-C6-циклоалкилэтил, фенил, фенилметил, CH2OH, CH2SH, CH2
OCH3, CH2SCH3, CH2CH2SCH3, (CH2)sNH2, (CH2)sNHC(= NH)(NH2), (CH2)sNHR16, где s = 3-5; связь с Ln;
R3 и R5 могут альтернативно быть взяты вместе с образованием -(CH2)t- (t
= 2-4) или -CH2SC(CH3)2-; или
R7 выбирают из: -(C1-C7-алкил)X;

причем каждое q = 0-2 и замещение на фенил происходит в третьей или четвертой позиции;
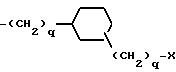
причем каждое q = 0-2 и замещение на циклогексил происходит в третьей или четвертой позиции;
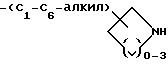
-(CH2)mO-(C1-C4)-X, где m = 1 или 2; -(CH2)mS-(C1-C4 )-алкил, где m = 1 или 2; и X выбирают из -NH-C(=NH)(NH2), -NHR13, -C(=NH)(NH2), -SC(NH)-NH2;
R6 и R7 могут быть альтернативно взяты вместе с образованием

где n = 0 или 1 и X представляет собой -NH2 или -NH-C(=NH)(NH2);
L - Y(CH2)vC(=O)-, причем Y - NH, N(C1-C3 алкил), O или S; и v = 1 или 2;
M - D-изомер или L-изомер аминокислоты формулы
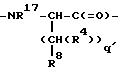
причем q' = 0-2;
R17 представляет собой H, C1-C3-алкил;
R8 выбирают из соединений -CO2R13, -SO3R13, -SO2NHR14, -B(R34)(R35), -NHSO2CF3, -CONHNHSO2CF3, -PO(OR13)2, -PO(OR13)R13, -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбранных из N, S или O), -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбранных из N, S или O), -SO2NHCOR13, -CONHSO2R13a, -CH2CONHSO2R13a, -NHSO2NHCOR13a, -NHCONHSO2R13a, -SO2NHCONHR13;
R34 и R35 независимо выбирают из -OH, -F, -NR13R14 или C1 -C8-алкокси;
R34 и R35 могут быть альтернативно взяты вместе с образованием: циклического эфира бора, где указанная цепь или кольцо содержит от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбранных из N, S или O; бивалентного циклического амида бора, где указанная цепь или кольцо содержит от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбранных из N, S или O; циклического амидоэфира бора, где указанная цепь или кольцо содержит от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатомов, независимо выбранных из N, S или O.
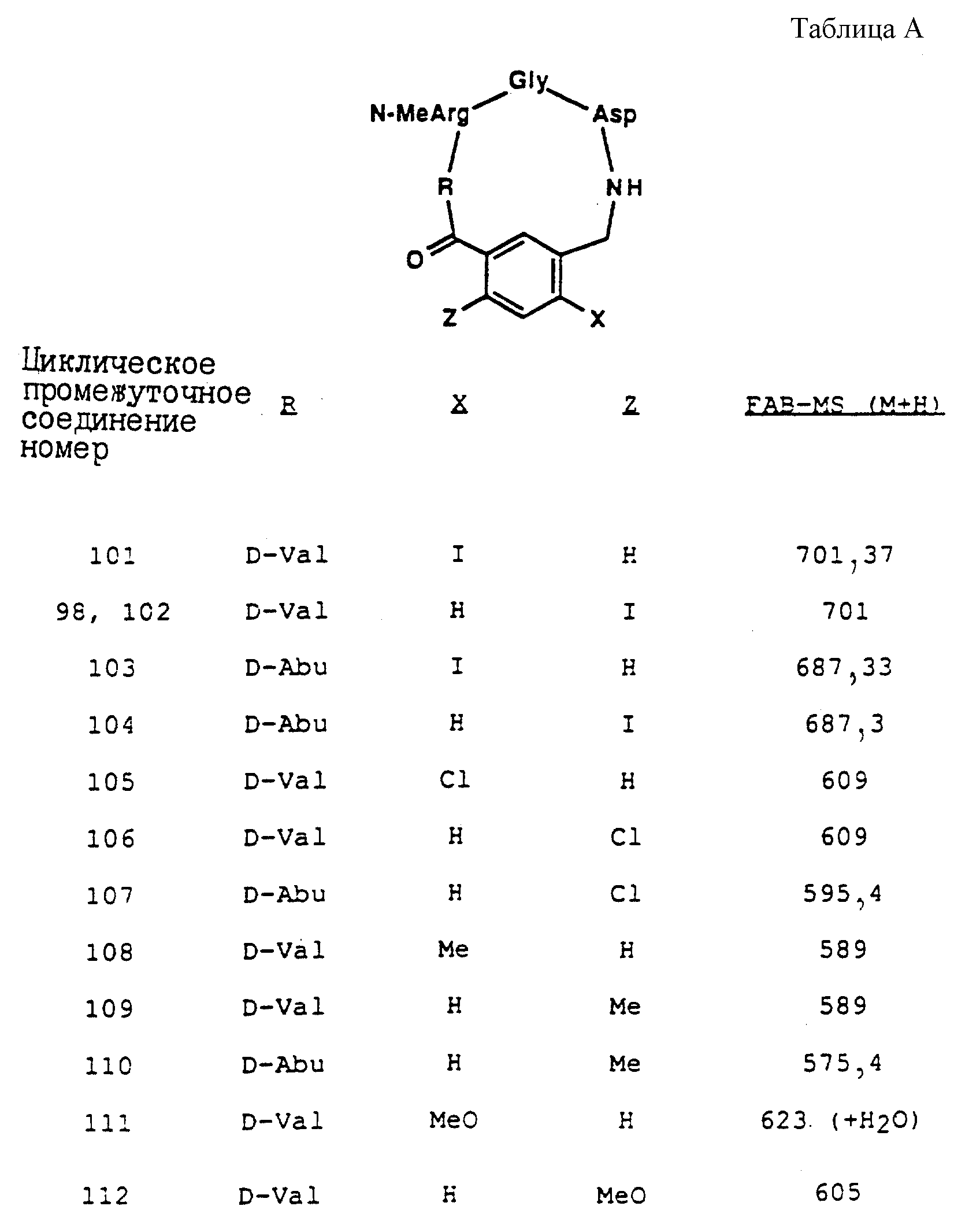
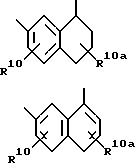
[6] В настоящее изобретение включены указанные в [1] реагенты, причем R31 выбирают из группы, состоящей из
(а) 6-членного насыщенного,
частично насыщенного или ароматического карбоциклического кольца, замещенного 0-3 R10 или R10a и факультативно имеющего связь с Ln;
(б) 8-11-членного
насыщенного, частично насыщенного или ароматически слитого бициклического карбоциклического кольца, замещенного 0-3 R10 или R10a и факультативно имеющего связь с Ln;
или
(в) 14-членного насыщенного, частично насыщенного или ароматически слитого трициклического карбоциклического кольца, замещенного 0-3 R10 или R10a и факультативно
имеющего связь с Ln.
[7] В настоящее изобретение включены указанные в [1] реагенты, причем R31 выбирают из группы, состоящей из (а) 6-членного насыщенного,
частично насыщенного или ароматического карбоциклического кольца формулы
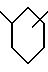
причем какая-либо связь, образующая карбоциклическое кольцо, может быть одинарной или двойной и причем указанное карбоциклическое кольцо замещено 0-3 R10 и факультативно имеет связь с Ln;
(б) 10-членного насыщенного, частично насыщенного или ароматического бициклического карбоциклического кольца формулы
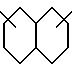
причем какая-либо связь, образующая карбоциклическое кольцо, может быть одинарной или двойной, а указанное карбоциклическое кольцо независимо замещено 0-4 R10 и факультативно имеет связь с Ln;
(в) 9-членного насыщенного, частично насыщенного или ароматического бициклического карбоциклического кольца формулы
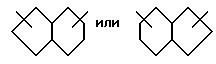
причем какая-либо связь, образующая карбоциклическое соединение, может быть одинарной или двойной, а указанное карбоциклическое кольцо независимо замещено 0-4 R10 и факультативно имеет связь с Ln.
[8] В настоящее изобретение включены указанные выше в [1] реагенты,
причем R31 выбирают из (пунктирная линия может означать как одинарную, так и двойную связь):
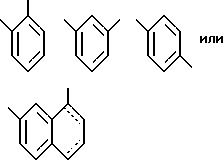
причем R31 может быть независимо замещен 0-3 R10 или R10a и факультативно иметь связь с Ln;
n'' = 0 или 1 и n' = 0-2.
[9] В настоящее изобретение включены указанные выше в [1] реагенты, причем R1 и R22 независимо выбирают из соединений фенил, бензил, фенетил, фенокси, бензилокси,
галоген, гидрокси, нитро, циано, C1-C5-алкил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C7-C10-арилалкил, C1-C5-алкокси, -CO2R13, -C(=O)NHOR13a, -C(=O)NHN(R13)2, =NOR13, -B(R34)(R35), C3
-C6-циклоалкокси, -OC(=O)R13, -С(=O)R13, -OC(=O)OR13a, -OR13, -(C1-C4-алкил)-OR13, -N(R13)2
, -OC(= O)N(R13)2, -NR13C(= O)R13, -NR13C(= O)OR13a, -NR13C(=O)N(R13)2, -NR13SO2
(R13)2, -NR13SO2R13a, -SO3H, -SO2R13a, -S(= O)R13a, -SR13, -SO2N(R13
)2, -C2-C6-алкоксиалкил, метилендиокси, этилендиокси, -C1-C4-галоалкил, C1-C4-галоалкокси, C1-C4
-алкилкарбонилокси,
C1-C4-алкилкарбонил, C1-C4-алкилкарбониламино, -OCH2CO2H, 2-(1-морфолино)этокси, C1-C4-алкил (алкил замещен -N(R13)2, -CF3, NO2 или -S(=O)R13a).
[10] В настоящее изобретение включены указанные выше в [1]
реагенты, причем R31 выбирают из
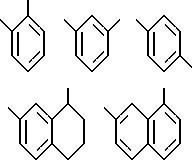
причем R31 может быть независимо замещен 0-3 R10 или R10a и может факультативно иметь связь с Ln;
R32 представляет собой -C(=O)-;
n'' = 0 или 1;
n' = 0-2;
R1 и R22 независимо выбирают из соединений H, C1-C4-алкил, фенил, бензил, фенил-(C2-C4)-алкил, C1-C4-алкокси, связь с Ln;
R21 и R23 независимо - H или C1-C4-алкил;
R2 представляет собой H или C1-C8-алкил;
R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил, или C3-C10-алкоксиалкил;
R13a представляет собой C1-C10-алкил, C3-C10 -циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил; когда две R13-группы связаны с единичным N, указанные R13-группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2)O(CH2)-;
R14 представляет собой OH, H, C1-C4-алкил или бензил;
R10 и R10a независимо выбирают из соединений, H, C1-C8-алкил, фенил, галоген или С1-С4-алкокси;
J - β- Ala или L-изомер или D-изомер аминокислоты формулы -N(R3)C(R4)(R5)C(=O)-, причем R3 - H или CH3;
R4 - H или C1-C3-алкил;
R5 - H, C1-C8-алкил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C1-C6-циклоалкилэтил, фенил, фенилметил, CH2OH, CH2SH, CH2OCH3, CH2SCH3, CH2CH2SCH3, (CH2)sNH2, -(CH2)sNHC(=NH)(NH2), -(CH2)sNHR16, где s = 3-5; связь c Ln;
или R3 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)t- (t = 2-4) или -CH2SC(CH3 )2-; или
R4 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)u-, где u = 2-5;
R16 выбирают из соединений аминозащитная группа;
1-2 аминокислоты или 1-2 аминокислоты, замещенные аминопротективной группой;
K - L-изомер аминокислоты формулы -N(R6)CH(R7 )C(=O)-, причем R6 - H или C1-C8-алкил; R7 представляет собой
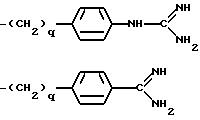
где q = 0 или 1;
-(CH2)rX, где r = 3-6;

-(CH2)mS(CH2)2X, где m = 1 или 2;
-(C3-C7-алкил)-NH-(C1-C6-алкил);
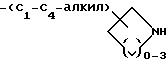
-(CH2)m-O-(C1-C4-алкил)- NH-(C1-C6-алкил), где m = 1 или 2;
-(CH2)m-S-(C1-C4-алкил)- NH-(C1-C6-алкил), где m = 1 или 2; и X представляет собой -NH2 или -NHC(=NH)(NH2); или
R6 и R7 могут быть альтернативно взяты вместе с образованием
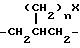
где n = 0 или 1 и X - -NH2 или -NHC(=NH)(NH2);
L - -Y(CH2)vC(=O)-, причем Y - NH, O или S и v = 1 или 2;
M - D-изомер или L-изомер аминокислоты формулы
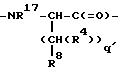
причем q' = 0-2;
R17 - H; C1 -C3-алкил;
R8 выбирают из соединений -CO2R13, -SO3R13, -SO2NHR14, -B(R34)(R35), -NHSO2CF3, -CONHNHSO2CF3, -PO(OR13)2, -PO(OR13)R13, -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбранных из N, S или O), -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбранных из N, S или O), -SO2NHCOR13, -CONHSO2R13a, -CH2CONHSO2R13a, -NHSO2NHCOR13a, -NHCONHSO2R13a, -SO2NHCONHR13.
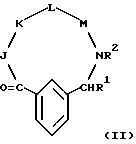
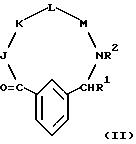
[11] В настоящее изобретение включены реагенты, указанные выше в [1], причем Q представляет собой 1,3-замещенное соединение фенила
формулы (II)
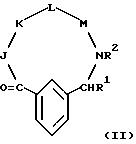
причем приведенное в формуле (II) фениловое кольцо может быть замещено 0-3 R10 и может факультативно иметь связь с Ln;
R10 независимо выбирают из соединений: H, C1-C8-алкил, фенил, галоген или C1-C4-алкокси;
R1 - H, C1-C4-алкил, фенил, бензил, фенил-(C1-C4)-алкил или связь с Ln;
R2 - H или метил;
R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
R13a - C1-C10-алкил, C3-C10-циклоалкил, C4 -C12-алкилцилкоалкил, арил, -(C1-C10алкил)арил или C3-C10алкоксиалкил; когда две R13-группы связаны с единичным N, указанные R13-группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2)O(CH2)-;
R14 - OH, H, C1-C4-алкил или бензил;
J - β- Ala или L-изомер, или D-изомер аминокислоты формулы -N(R3)C(R4)(R5)C(=O)-, причем R3 - H или CH3;
R4 - H или C1-C3-алкил;
R5 - H, C1-C8-алкил, C3-C6-циклоалкил, C3-C6 -циклоалкилметил, C1-C6-циклоалкилэтил, фенил, фенилметил, CH2OH, CH2SH, CH2OCH3, CH2SCH3, CH2CH2SCH3, (CH2)sNH2, (CH2)sNHC(= NH)(NH2), -(CH2)sNHR16, где s = 3-5, или связь с Ln;
R3 и R5 могут быть альтернативно взяты вместе с образованием -CH2CH2CH2- или R4 и R5 могут быть альтернативно взяты с образованием -(CH2)u, где u = 2-5;
R16 выбирают из соединений аминозащитная группа; 1-2 аминокислоты или 1-2 аминокислоты, замещенные аминопротективной группой;
K - L-изомер аминокислоты формулы -N(R6)CH(R7)C(=O)-, причем R6 - H или C1-C8-алкил; R7 представляет собой
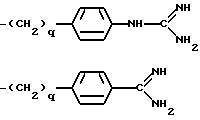
где q = 0 или 1;
-(CH2)rX, где r = 3-6;
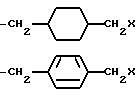
-(CH2)mS(CH2)2X, где m = 1 или 2;
-(C3-C7-алкил)-NH-(C1-C6-алкил)
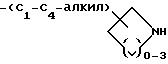
-(CH2)m-O-(C1-C4-алкил)-NH- (C1-C6-алкил), где m = 1 или 2;
-(CH2)m-S-(C1-C4-алкил)-NH- (C1 -C6-алкил), где m = 1 или 2; и X - -NH2 или -NHC(=NH)(NH2), причем X не является -NH2, когда r = 4; или
R6 и R7 могут быть альтернативно взяты вместе с образованием

где n = 0, 1;
X - -NH2 или -NHC(=NH)(NH2);
L - -Y(CH2)vC(=O)-, причем Y - NH, O или v и v = 1, 2;
M - D-изомер или L-изомер аминокислоты формулы
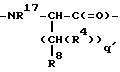
причем q' = 0-2; R17 - H, C1-C3-алкил; R8 выбирают из соединений -CO2R13, -SO3R13, -SO2NHR14, -B(R34)(R35), -NHSO2CF3-, -CONHNSO2 CF3, -PO(OR13)2, -PO(OR13)R13, -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбранных из N, S или O), -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбранных из N, S или O), -SO2NHCOR13, -CONHSO2R13a, -CH2CONHSO2R13, -NHSO2NHCOR13a, NHCONHSO2R13a, -SO2NHCONHR13.
[12] В настоящее изобретение включены реагенты, указанные выше в [1], причем Q представляет собой 1,3-замещенное соединение фенила формулы (II)
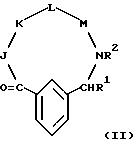
причем фенильное кольцо формулы (II) может быть замещено 0-3 R10 и R10a;
R10 и R10a независимо выбирают из соединений H, C1-C8-алкил, фенил, галоген или C1-C4-алкокси;
R1 - H, C1-C4-алкил, фенил, бензил или фенил-(C1-C4)-алкил;
R2 - H или метил;
R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
когда две R13-группы связаны с одиночным N, указанные R13 группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2)O(CH2 )-;
R13a - C1-10-алкил, C3-C10-циклоалкил, C4-12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-10-алкоксиалкил;
R14 - OH, H, C1-C4-алкил или бензил;
J - β- Ala или L-изомер, или D-изомер аминокислоты формулы -N(R3)C(R4)(R5)C(= O)-, причем R3 - H или CH3; R4 - H; R5 - H, C1-C8-алкил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C1-C6-циклоалкилэтил, фенил, фенилметил, CH2OH, CH2SH, CH2OCH3, CH2SCH3, CH2CH2SCH3, (CH2)sNH2, (CH2)sNHC(= NH)(NH2), (CH2)sNHR16, где s = 3-5; или связь с Ln;
R3 и R5 могут быть альтернативно взяты вместе с образованием -CH2CH2CH2 -;
R16 выбирают из соединений аминопротективная группа; 1-2 аминокислоты; 1-2 аминокислоты, замещенные аминопротективной группой;
K - L-изомер аминокислоты формулы -N(R6)CH(R7)C(=O)-, причем R6 - H или C3-C8-алкил; R7 представляет собой
где q = 0 или 1;
-(CH2)rX, где r = 3-6;
-(CH2)mS(CH2)2X, где m = 1 или 2;
-(C4-C7-алкил)-NH-(C1-C6 -алкил)
-(CH2)m-O-(C1-C4-алкил)-NH- (C1 -C6-алкил), где m = 1 или 2;
-(CH2)m-S-(C1-C4-алкил)-NH- (C1-C6-алкил), где m = 1 или 2; и
X представляет собой -NH2 или -NHC(=NH)(NH2), причем X не может быть -NH2, когда r = 4;
или L представляет собой -YCH2C(=O)-, причем Y - NH или O; M - D-изомер или L-изомер аминокислоты формулы
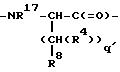
причем q' = 1;
R17 - H, C1 -C3-алкил;
R8 выбирают из соединений -CO2H или -SO3R13.
[13] В настоящее изобретение включены указанные выше в [1]
реагенты, причем: фениловое кольцо формулы (II) имеет связь с Ln и может быть в дальнейшем замещено 0-2 R10 или R10a;
R10 и R10a
независимо выбираются из соединений H, C1-C8-алкил, фенил, галоген или C1-C4-алкокси;
R1 - H;
R2 - H;
R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
R13a - C1-C10-алкил, C3-C10-циклоалкил, C4-C12
-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
когда две R13 группы связаны с одинаковым N, указанные R13 группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2)O(CH2)-;
R14 - OH, H, C1-C4-алкил или бензил;
J - β- Ala, или L-изомер, или D-изомер аминокислоты формулы -N(R3)CH(R5)C(=O)-, причем
R3 - H и R5 - H,
CH3, CH2CH3, CH(CH3)2, CH(CH3)CH2CH3, CH2CH2CH3, CH2CH2
CH2CH3, CH2CH2SCH3, CH2CH(CH3)2, (CH2)4NH2, (C3-C5
-алкил)NHR16; или R3 - CH3 и R5 - H; или R3 и R5 могут быть альтернативно взяты вместе с образованием -CH2CH2
CH2-;
R16 выбирают из соединений аминопротективная группа; 1-2 аминокислоты; 1-2 аминокислоты, замещенные аминопротективной группой;
K - L-изомер аминокислоты
формулы N(CH3)CH(R7)C(=O)-, причем R7 - -(CH2)3NCH=(NH)(NH2);
L - -NHCH2C(=O)-;
M - D-изомер или L-изомер
аминокислоты формулы
причем q' = 1;
R4 - H или CH3;
R17 - H;
R8 - -CO2H7, -SO3H.
[14] В настоящее изобретение включены указанные выше в [1] реагенты, причем фениловое
кольцо формулы (II) имеет связь с Ln;
R1 и R2 независимо выбирают из H и метила;
J выбирают из D-Val, D-2-аминомасляной кислоты, D-Leu, D-Ala, Gly,
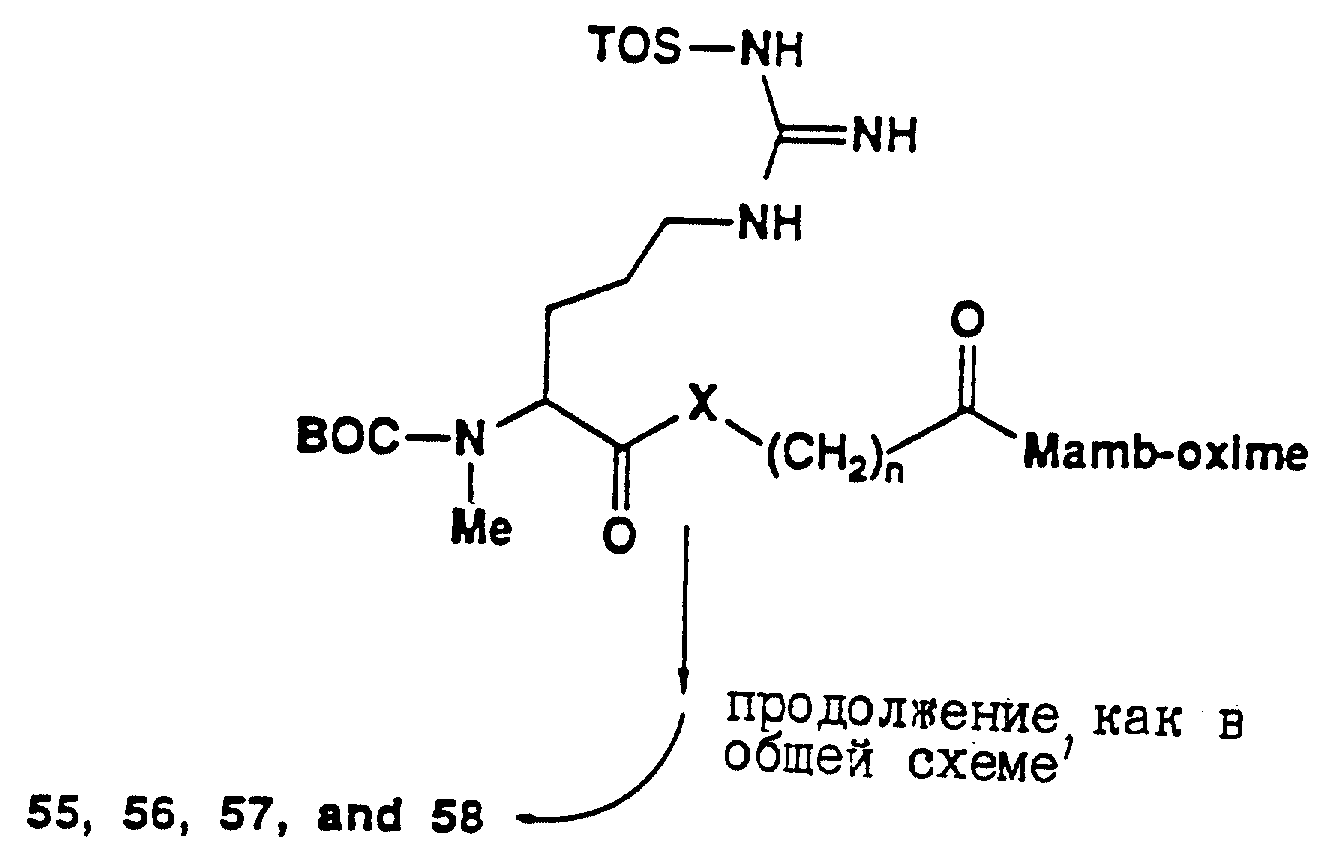
D-Pro, D-Ser, D-Lys, β- Ala, Pro, Phe, NMeGly, D-Nle, D-Phg, D-Ile, D-Phe, D-Tyr, Ala, Nδ- -p-азидобензоил-D-Lys, Nε- p-бензоилбензоил-D-Lys, Nε- триптофанил-D-Lys, Nε- o-бензоилбензоил-D-Lys, Nε- p-ацетилбензоил-D-Lys, Nε- дансил-D-Lys, Nε- глицил-D-Lys, Nε- глицил-p-бензоилбензоил-d-Lys, Nε- p-фенилбензоил-D-Lys, Nε- m-бензоилбензоил-D-Lys, Nε- o-бензоилбензоил-D-Lys;
K
выбирают из NMeArg, Arg;
L выбирают из Gly, β- Ala, Ala;
M выбирают из Asp; α MeAsp; β MeAsp; NMeAsp; D-Asp.
[15] В настоящее изобретение
включены указанные выше в [1] реагенты, причем R31 - фенильное кольцо и имеет связь с Ln;
R1 и R2 независимо выбирают из H, метила;
J
выбирают из D-Val, D-2-аминомасляной кислоты, D-Leu, D-Ala, Gly, D-Pro, D-Ser, D-Lys, β- Ala, Pro, Phe, NMeGly, D-Nle, D-Phg, D-Ile, D-Phe, D-Tyr, Ala;
K выбирают из NMeArg;
L
выбирают из Gly;
M выбирают из Asp; α MeAsp; β MeAsp; NmeAsp; D-Asp.
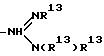
[16] В настоящее изобретение включены указанные выше в [1]-[15] реагенты, причем Ch выбирают из группы соединений
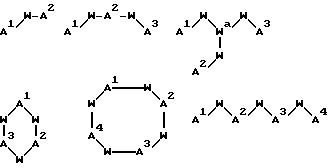
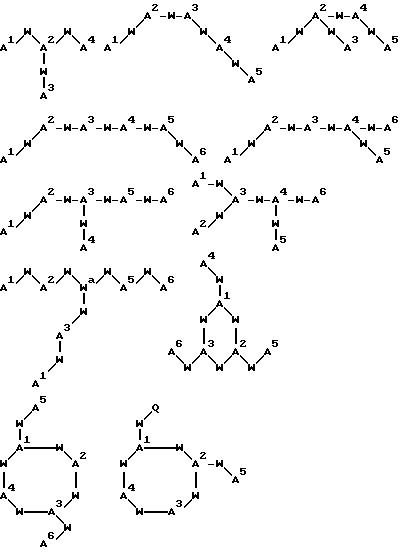
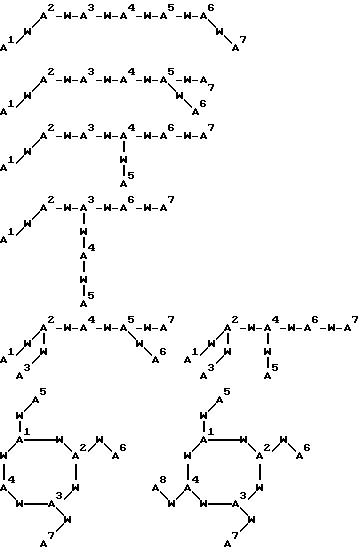
причем A1 - A7 независимо выбирают в каждом случае из группы соединений NR40R41, S, SH, S(Pg), O, OH, PR42R43, P(O)R42R43, P(S)R42R43, P(NR44)R42R43;
W - связь, CH или спейсерная группа, выбираемая из соединений C1-C10-алкил, замещенный 0-3 R52; арил, замещенный 0-3 R52; циклоалкил, замещенный 0-3 R52; гетероциклоалкил, замещенный 0-3 R52; аралкил, замещенный 0-3 R52, и алкарил, замещенный 0-3 R52;
Wa представляет собой C1C10-алкильную группу или C3-C14-карбоцикл;
R40 - R44 каждый независимо выбирают из группы соединений: связь с Ln; водород; C1-C10-алкил, замещенный 0-3 R52; арил, замещенный 0-3 R52; циклоалкил, замещенный 0-3 R52; гетероциклоалкил, замещенный 0-3 R52; аралкил, замещенный 0-3 R52; алкарил, замещенный 0-3 R52 и электроном, причем, когда один из радикалов R40 или R41 представляет собой электрон, другой также является электроном, и причем, когда один из радикалов R42 или R43 представляет собой электрон, другой также является электроном;
дополнительно R40 и R41 могут соединяться с образованием = C(C1-C3-алкил)(C1-C3-алкил);
R52 независимо выбирают в каждом случае из группы соединений; связь с Ln, = O, F, Cl, Br, I, -CF3, -CN, -CO2R53, -C(=O)R53, -C(=O)N(R53 )2, -CHO, -CH2OR53, -OC(= O)R53, -OC(=O)OR53a, -OR53, -OC(=O)N(R53)2, -NR53C(=O)R53, NR54C(= O)OR53a, -NR53C(= O)N(R53)2, -NR54SO2N(R53)2, -NR54SO2R53a, -SO3H, -SO2R53a, -SR53, -S(= O)R53a, -SO2N(R53)2, -N(R53)2, -NHC(=NH)NHR53, -C(= NH)NHR53, = NOR53, NO2, -C(=O)NHOR53, -C(=O)NHNR53R53a, -OCH2CO2H, 2-(1-морфолино)этокси, C1 -C5-алкил, C2-C4-алкенил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C2-C6-алкоксиалкил, арил, замещенный 0-2 R53, 5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатома, независимо выбираемых из N, S и O;
R53, R53a и R54 независимо выбирают в каждом случае из группы соединений: связь с Ln, C1-C6-алкил, фенил, бензил, C1-C6-алкокси, галид, нитро, циано и трифторметил; и
Pg представляет собой триоловую защитную группу.
[17] В настоящее изобретение включены реагенты, указанные выше в [1]-[15], причем Ch выбирают из
соединений
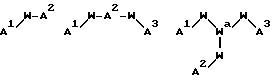
причем A1 - A7 независимо выбирают в каждом случае из группы соединений: NR40R41, S, SH, S(Pg), OH;
W представляет собой связь, CH или спейсерную группу, выбираемую из соединений C1-C3-алкил, замещенный 0-3 R52;
Wa представляет собой метиленовую группу или C3-C6-карбоцикл;
R40 - R44 каждый независимо выбирают из группы соединений; связь с Ln; водород; C1-C10-алкил, замещенный 0-3 R52 и электрон, причем, когда один из радикалов R40 или R41 представляет собой электрон, другой также является электроном, и причем, когда один из радикалов R42 или R43 представляет собой электрон, другой также является электроном;
дополнительно R40 и R41 могут соединяться вместе с образованием = C(C1-C3-алкил)(C1-C3-алкила);
R52 независимо выбирают в каждом случае из группы соединений: связь с Ln, = O, F, Cl, Br, I, -CF3, -CN, -CO2R53, -C(=O)R53, -C(=O)N(R53)2, -CHO, -CH2OR53, -OC(= O)R53, -OC(=O)OR53a, -OR53, -OC(=O)N(R53)2, -NR53C(=O)R53, -NR54C(= O)OR53a, -NR53C(= O)N(R53)2, -NR54SO2 N(R53)2, -NR54SO2R53a, -SO3H, -SO2R53a, -SR53, -S(= O)R53a, -SO2N(R53 )2, -N(R53)2, -NHC(=NH)NHR53, -C(= NH)NHR53, = NOR53, NO2, -C(=O)NHOR53, -C(=O)NHNR53R53a, -OCH2CO2H, 2-(1-морфолино)этокси,
R53, R53a и R54 независимо выбирают в каждом случае из группы соединений: связь с Ln, C1-C6-алкил.
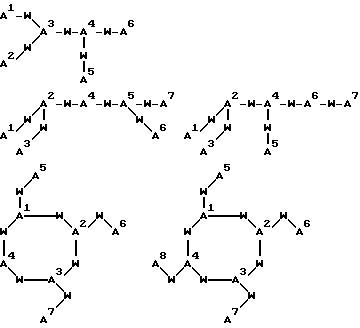
[18] В настоящее изобретение включены указанные выше в [1]-[15] реагенты формулы (QLn)dCh, причем d = 1 и Ch
выбирают из
причем A1 и A4 - SH или SPg;
A2 и A3 - NR41;
W независимо выбирают из группы соединений CHR52, CH2CHR52, CH2CH2CHR52 и CHR52C=O; и
R41 и R52 независимо выбирают из водорода и связи c Ln, и
причем A1 - NH2 или
N=C(C1-C3-алкил)(C1-C3-алкил);
W - связь;
A2 - NHR40, причем R40 - гетероцикл, замещенный R52, причем гетероцикл выбирают из соединений группы пиридин, пиразин, пролин, фуран, тиофуран, тиазол и диазин и R52 представляет собой связь с Ln.
[19] В настоящее изобретение включены указанные выше в [1]-[15] реагенты формулы (QLn)dCh, причем d = 1 и Ch
представляет собой
причем A1 - NH2 или N=C(C1-C3 -алкил)(C1-C3-алкил);
W - связь;
A2 - NHR40, причем R40 - гетероцикл, замещенный R52, причем гетероцикл выбирают из пиридина, тиазола, а R52 представляет собой связь с Ln.
[20] В настоящее изобретение включены указанные выше в [1]-[15] реагенты, причем Ln
представляет собой связь между Q и Ch или соединение формулы
M1-[Y1(CR55R56)h (Z1)h''Y2]h'-M2,
причем M1 - [(CH2)gZ1]g' - (CR55R56)g''-;
M2 - (CR55R56)g''- [Z1(CH2)g]g'-;
g = 0-10;
g' = 0-1;
g'' = 0-10;
h = 0-10;
h' = 0-10;
n'' = 0-1;
Y1 и Y2 в каждом случае независимо выбирают из соединений: связь, O, NR56 , C= O, C(=O)O, OC(=O)O, C(=O)NH-, C=NR56, S, SO, SO2, SO3, NHC(=O), (NH)2C(=O), (NH)2C=S;
Z1 независимо выбирают в каждом случае из C6-C14 насыщенной, частично насыщенной
или ароматической карбоциклической кольцевой системы, замещенной 0-4 R57; гетероциклической кольцевой системы, факультативно замещенной 0-4 R57;
R55 и R56 независимо выбирают в каждом случае из соединений: водород; C1-C10-алкил, замещенный 0-5 R57; (C1-C10-алкил)арил, причем арил замещен
0-5 R57;
R57 независимо выбирают в каждом случае из группы соединений: водород, OH, NHR58, C(= O)R58, OC(= O)R58, OC(=O)OR58,
C(=O)OR58, C(=O)NR58-, C≡N, SR58, SOR58, SO2R58, NHC(=O)R58, NHC(=O)NHR58, NHC(=S)NHR58; или
альтернативную, будучи прикрепленным к дополнительной молекуле Q, R57 независимо выбирают в каждом случае из группы соединений O, NR58, C=O, C(=O)O, OC(=O)O, C(=O)N-, C=NR58, S, SO, SO2, SO3, NHC(=O), (NH)2C(=O), (NH)2C=S; и R58 независимо выбирают в каждом случае из группы соединений: водород; C1
-C6-алкил; бензил и фенил.
[21] В настоящее изобретение включены указанные выше в [1]-[15] реагенты, причем Ln представляет собой соединение формулы
M1-[Y1(CR55R56)h (Z1)h''Y2]h'-M2,
причем M1 - [(CH2)gZ1]g'- (CR55R56)g''-;
M2 - (CR55R56)g''- [Z1(CH2)g
]g'-;
g = 0-10;
g' = 0-1;
g'' = 0-10;
h = 0-10;
h'= 0-10;
h'' = 0-1;
Y1 и Y2 в каждом случае независимо
выбирают из соединений: связь, O, NR56 , C= O, C(=O)O, OC(=O)O, C(=O)NH-, C=NR56, S, SO, SO2, SO3, NHC(=O), (NH)2C(=O), (NH)2C=S;
Z1 независимо выбирают в каждом случае из C6-C14 насыщенной, частично насыщенной или ароматической карбоциклической кольцевой системы, замещенной 0-4 R57;
гетероциклической кольцевой системы, факультативно замещенной 0-4 R57;
R55 и R56 независимо выбирают в каждом случае из соединений: водород; C1
-C10-алкил, замещенный 0-5 R57; (C1-C10-алкил)арил, причем арил замещен 0-5 R57;
R57 независимо выбирают в каждом случае из
группы соединений: водород, OH, NHR58, C(=O)R58, OC(=O)R58, OC(=O)OR58, C(=O)OR58, C(=O)NR58, C≡N, SR58, SOR58, SO2R58, NHC(= O)R58, NHC(=O)NHR58, NHC(=S)NHR58; или альтернативно, будучи прикрепленным к дополнительной молекуле Q, R57
независимо выбирают в каждом случае из группы соединений O, NR58, C=O, C(=O)O, OC(=O)O, C(=O)N-, C= NR58, S, SO, SO2, SO3, NHC(=O), (NH)2C(=O),
(NH)2C=S, и R57 прикреплен к дополнительной молекуле Q; и
R58 независимо выбирают в каждом случае из группы соединений: водород; C1-C6
-алкил; бензил и фенил.
[22] В настоящее изобретение включены указанные выше в [1]-[15] реагенты, причем Ln представляет собой
-(CR55R56)g''- [Y1(CR55R56)hY2]h'- (CR55R56)g''-,
причем g'' = 1-10; h = 0-10; h' = 1-10; Y1 и Y2 в каждом случае независимо выбирают из соединений: связь, a bond, O, NR56, C=O, C(=O)O, OC(=O)O, C(= O)NH-, C=NR56, S, SO, SO2, SO3,
NHC(=O), (NH)2C(=O), (NH)2C=S; R55 и R56 независимо выбирают в каждом случае из соединений: водород; C1-C10-алкил, замещенный 0-5
R57; (C1-C10-алкил)арил, причем арил замещен 0-5 R57; R57 независимо выбирают в каждом случае из группы соединений: водород, OH, NHR58,
C(= O)R58, OC(= O)R58, OC(=O)OR58, C(=O)OR58, C(=O)NR58-, C≡N, SR58, SOR58, SO2R58, NHC(=
O)R58, NHC(=O)NHR58, NHC(=S)NHR58; или альтернативно, будучи прикрепленным к дополнительной молекуле Q, R57 независимо выбирают в каждом случае из группы
соединений O, NR58, C=O, C(=O)O, OC(=O)O, C(=O)N-, C=NR58, S, SO, SO2, SO3, NHC(=O), (NH)2C(=O), (NH)2C=S, и R57 прикреплен
к дополнительной молекуле Q; и R58 независимо выбирают в каждом случае из группы соединений: водород; C1-C6-алкил; бензил и фенил.
[23] В настоящее
изобретение включены указанные выше в [1]-[15] реагенты, причем Ln представляет собой
-(CR55R56)g''- [Y1(CR55R56
)hY2]h'- (CR55R56)g''-,
причем g'' = 1-5; h = 0-5; h' = 1-5; Y1 и Y2 в каждом случае независимо
выбирают из соединений: O, NR56, C=O, C(=O)O, OC(=O)O, C(=O)NH-, C=NR56, S, SO, SO2, SO3, NHC(=O), (NH)2C(=O), (NH)2C=S; R55
и R56 независимо выбирают в каждом случае из соединений: водород; C1-C10-алкил, (C1-C10-алкил)арил.
[24] В настоящее изобретение
включены указанные выше в [1]-[15] реагенты, причем Ln представляет собой
-(CR55R56)g''- [Y1(CR55R56)h
Y2]h'- (CR55R56)g''-,
причем g'' = 1-5; h = 0-5; h' = 1-5; Y1 и Y2 в каждом случае независимо выбирают из
соединений: O, NR56, C=O, C(=O)O, OC(=O)O, C(=O)NH-, C=NR56, S, NHC(=O), (NH)2C(=O), (NH)2C=S; R55 и R56 независимо выбирают в каждом
случае из водорода.
[25] В настоящее изобретение включены указанные выше в [1] реагенты, которые представляют собой
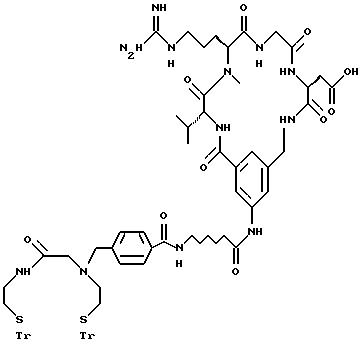
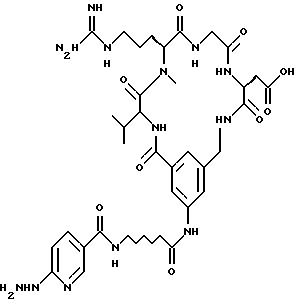
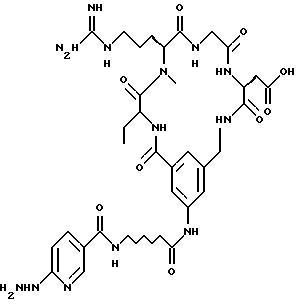
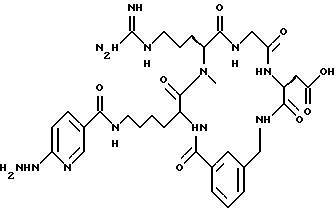
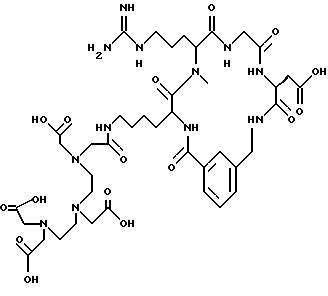
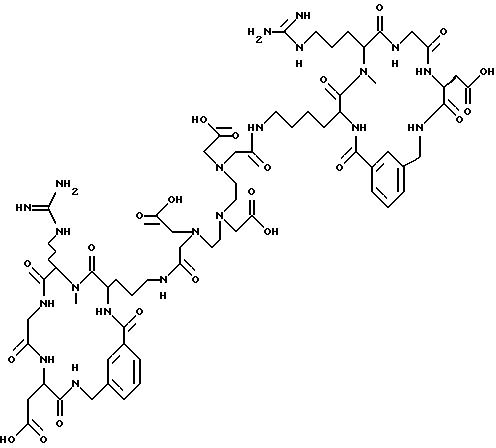
[26] Кроме того, в настоящее изобретение включен набор для приготовления радиофармацевтического препарата, состоящего из определенного количества стерильного, фармацевтически пригодного реагента, охарактеризованного в [23] .
[27] Кроме того, в настоящее изобретение включен набор для приготовления радиофармацевтического препарата, состоящего из определенного количества стерильного, фармацевтически пригодного реагента, охарактеризованного в [24] .
[28] Кроме того, в настоящее изобретение включен набор для приготовления радиофармацевтического препарата, состоящего из определенного количества стерильного, фармацевтически пригодного реагента, охарактеризованного в [25] .
[29] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [1]-[15], и радионуклида, выбранного из группы соединений99mTc,94mTc,95Tc,111In,62Cu,43Sc,45Ti,67Ga,68Ga,97Ru,72As,82Rb и201Tl.
[30] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [16], и радионуклида, выбранного из группы соединени99mTc,94mTc,95Tc,111In,62Cu,43Sc,45Ti,67Ga,68Ga,97Ru,72As,82Rb и201Tl.
[31] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [17], и радионуклида, выбранного из группы соединений99mTc,94mTc,95Tc,111In,62Cu,43Sc,45Ti,67Ga,68Ga,97Ru,72As,82Rb и201Tl.
[32] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [18], и радионуклида, выбранного из группы соединений99mTc,94mTc,95Tc,111In,62Cu,43Sc,45Ti,67Ga,68Ga,97Ru,72As,82Rb и201 Tl.
[33] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [19], и радионуклида, выбранного из группы соединений99mTc,94mTc,95Tc,111In,62Cu,43Sc,45Ti,67Ga,68Ga,97Ru,72As,82Rb и201Tl.
[34] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [20], и радионуклида, выбранного из группы соединений99mTc,94mTc,95Tc,111In,62Cu,43Sc,45Ti,67Ga,68Ga,97Ru,72As,82Rb и201Tl.
[35] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [21], и радионуклида, выбранного из группы соединений:99mTc,111In и62Cu.
[36] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [22], и радионуклида, выбранного из группы соединений99mTc,111In и62Cu.
[37] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [23], и радионуклида, выбранного из группы соединений99mTc,111In и62Cu.
[38] Кроме того, в настоящее изобретение включен радиофармацевтический препарат, содержащий комплекс реагента, охарактеризованного в [24], и радионуклида, выбранного из группы соединений:99mTc и111In.
[39] Кроме того, в настоящее изобретение включены радиофармацевтические препараты,
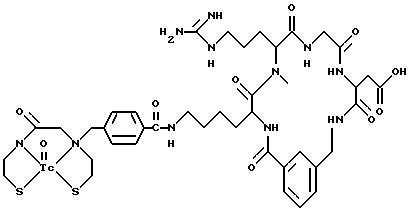
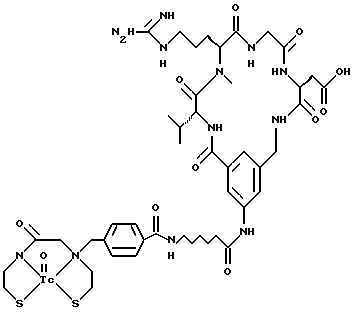
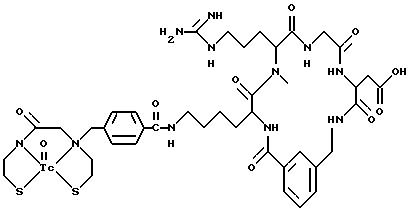
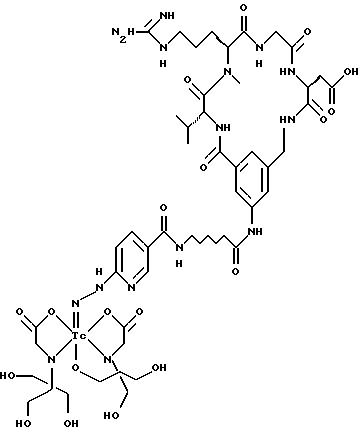
охарактеризованные в [29], которые представляют собой
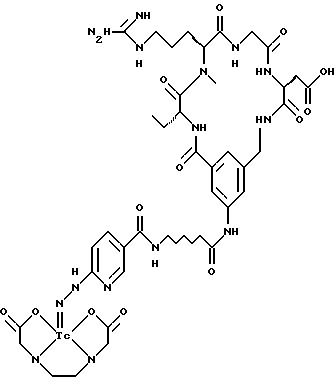

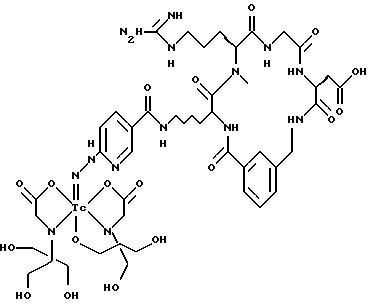
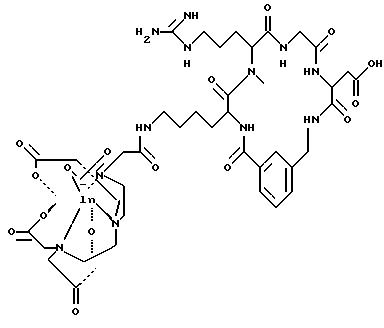
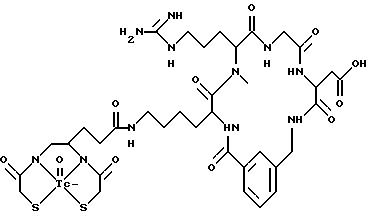
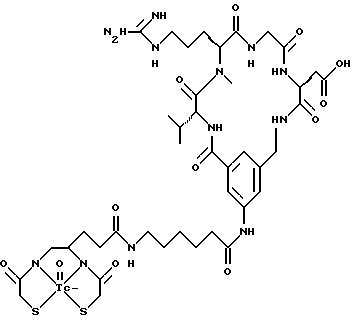

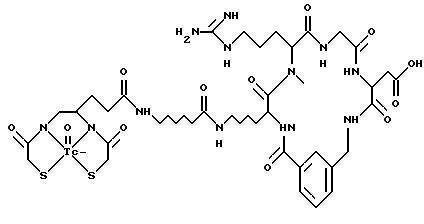
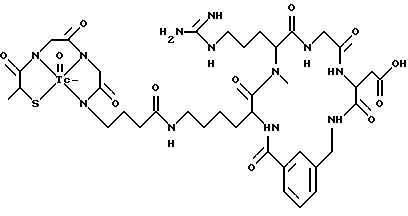

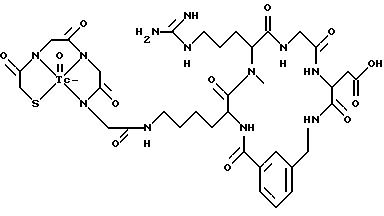
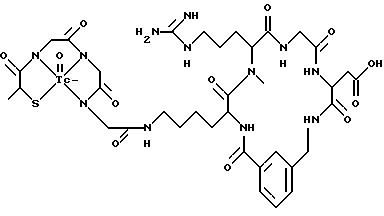
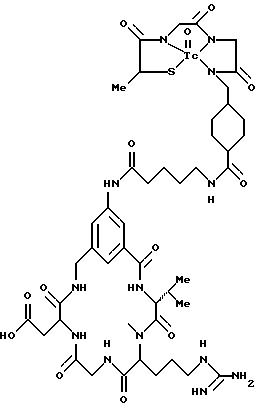
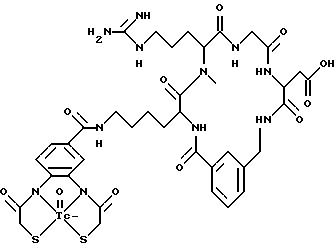
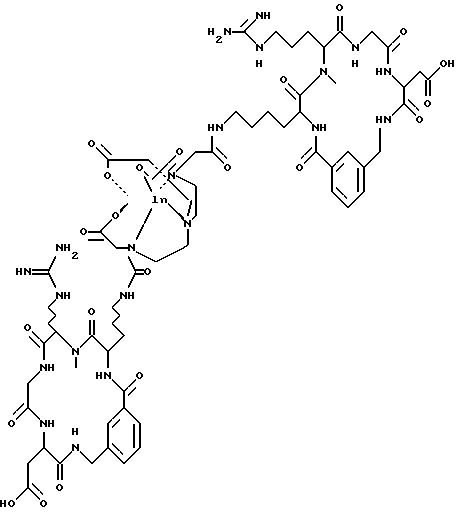
[40] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [29], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[41] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [30], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[42] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [31], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[43] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [32], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[44] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [33], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[45] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [34], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[46] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [35], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[47] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [36], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[48] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [37], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[49] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [38], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[50] Кроме того, в настоящее изобретение включен способ визуального определения участков отложения тромбоцитов у млекопитающих путем создания изображения с помощью радиофармацевтических препаратов, включающий (i) введение указанному млекопитающему эффективного количества радиофармацевтического препарата, охарактеризованного в [39], и (ii) сканирование млекопитающего с помощью устройства, выявляющего изображение, полученное при использовании радиофармацевтического препарата.
[51] Кроме того, настоящее изобретение
относится к меченным соединениям формулы (I)
или фармацевтически подходящей соли или ее предшественнику, причем R31 представляет собой C6-C14 насыщенную, частично насыщенную или ароматическую карбоциклическую кольцевую систему, замещенную 0-4 R10 или R10a;
R32 выбирают из соединений -C(=O)-; -C(=S)-; -S(=O)2-; -S(=O)-; -P(= Z)(ZR13)-; Z - S или O; n'' и n' = 0-2;
R1 и R22 независимо выбирают из следующих групп соединений:
водород: C1-C8-алкил, замещенный 0-2 R11; C2-C8-алкенил, замещенный 0-2 R11; C2-C8-алкинил, замещенный 0-2 R11; C3-C8-циклоалкил, замещенный 0-2 R11; арил, замещенный 0-2 R12; 5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатома, независимо выбираемых из N, S и O, указанное гетероциклическое кольцо, замещенное 0-2 R12; =O, F, Cl, Br, I, -CF3, -CN, -CO2R13, -C(=O)R13, -C(=O)N(R13)2, -CHO, -CH2OR13, =OC(=O)R13, -OC(= O)OR13a, -OR13, -OC(= O)N(R13)2, -NR13C(=O)R13, -NR14C(=O)OR13a, -NR13C(= O)N(R13)2, -NR14SO2N(R13)2, -NR14SO2R13a, -SO3H, -SO2R13a, -SR13, -S(= O)R13a, -SO2N(R13)2, -N(R13)2, -NHC(=NH)NHR13, -C(=NH)NHR13, =NOR13, NO2, -C(=O)NHOR13, -C(=O)NHNR13R13a, -OCH2CO2H, 2-(1-морфолино)этокси;
R1 и R21 могут быть альтернативно соединены с образованием 3-7-членного карбоциклического кольца, замещенного 0-2 R12; когда n' составляет 2, R1 и R21 могут быть альтернативно взяты вместе с R1 и R21 на соседнем атоме углерода с образованием прямой связи, таким образом формируя двойную или тройную связь между указанными атомами углерода;
R22 и R23 могут альтернативно соединяться с образованием 3-7-членного карбоциклического кольца, замещенного 0-2 R12; когда n'' = 2, R22 и R23 могут быть альтернативно взяты вместе с R22 и R23 на соседнем атоме углерода с образованием прямой связи, таким образом формируя двойную или тройную связь между соседними атомами углерода;
R1 и R2, когда R21 представляет собой H, могут альтернативно соединяться с образованием 5-8-членного карбоциклического кольца, замещенного 0-2 R12;
R11 выбирают из одного или более следующих соединений =O, F, Cl, Br, I, -CF3, -CN, -CO2R13, -C(=O)R13, -C(=O)N(R13)2, -CHO, -CH2OR13, -OC(=O)R13, -OC(= O)OR13a, -OR13, -OC(= O)N(R13)2, -NR13C(=O)R13, -NR14C(=O)OR13a, -NR13 C(= O)N(R13)2, -NR14SO2N(R13)2, -NR14SO2R13a, -SO3H, -SO2R13a, -SR13, -S(= O)R13a, -SO2N(R13)2, -N(R13)2, -NHC(=NH)NHR13, -C(=NH)NHR13, =NOR13, NO2, -C(= O)NHOR13, -C(= O)NHNR13R13a, -OCH2CO2H, 2-(1-морфолино)этокси; C1-C5-алкил, C1-C4 -алкенил, C3-C6-циклоалкил, C3-C6-циклоалкилметил,
C2-C6-алкоксиалкил, C3-C6-циклоалкокси, C1 -C4-алкил (алкил, замещенный 1-5 группами, которые независимо выбирают из -NR13R14, -CF3, NO2, -SO2R13a или -S(= O)R13a, арил, замещенный 0-2 R12, 5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатома, независимо выбираемых из N, S и O, указанное гетероциклическое кольцо, замещенное 0-2 R12, R12 выбирают из одного или более следующих соединений: фенил, бензил, фенэтил, фенокси, бензилокси, галоген, гидрокси, нитро, циано, C1-C5-алкил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C7-C10-арилалкил, C1-C5-алкокси, -CO2R13, -C(= O)NHOR13a, -C(=O)NHN(R13)2, =NOR13, -B(R34)(R35), C3-C6-циклоалкокси, -OC(= O)R13, -C(=O)R13, -OC(=O)OR13a, -OR13, -(C1-C4-алкил)-OR13, -N(R13)2, -OC(= O)N(R13)2, -NR13C(=O)R13, -NR13C(=O)OR13a, -NR13C(=O)N(R13)2, -NR13SO2N(R13)2, -NR13 SO2R13a, -SO3H, -SO2R13a, -S(= O)R13a, -SR13, -SO2N(R13)2, C2-C6 -алкоксиалкил, метилендиокси, этилендиокси, C1-C4-галоалкил, C1-C4-галоалкокси, C1-C4-алкилкарбонилокси, C1-C4 -алкилкарбонил, C1-C4-алкилкарбониламино, -OCH2CO2H, 2-(1-морфолино)этокси, C1-C4-алкил (алкил, замещенный -N(R13)2, -CF3, NO2 или -S(= O)R13a);
R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10 -циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
R13a - C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
когда две R13-группы связаны с единичным N, указанные R13-группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2 )O(CH2)-;
R14 - OH, H, C1-C4-алкил или бензил;
R21 и R23 независимо выбирают из соединений: водород; C1 -C4-алкил, факультативно замещенный 1-6 галогеном; бензил; R2 - H или C1-C8-алкил; R10 и R10a независимо выбирают из одного или более следующих соединений: фенил, бензил, фенэтил, фенокси, бензилокси, галоген, гидрокси, нитро, циано, C1-C5-алкил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C7-C10-арилалкил,
C1-C5-алкокси, -CO2R13, -C(=O)N(R13)2, -C(=O)NHOR13a, -C(=O)NHN(R13)2, = NOR13, -B(R34)(R35), C3-C6-циклоалкокси, -OC(= O)R13, -C(= O)R13, -OC(= O)OR13a, -OR13, -(C1-C4-алкил)-OR13, -N(R13)2, -OC(=O)N(R13)2, -NR13C(= O)R13, -NR13C(= O)OR13a, -NR13C(=O)N(R13)2, -NR13SO2N(R13)2, -NR13SO2R13a, -SO3H, -SO2R13a, -S(=O)R13a, -SR13, -SO2N(R13)2, C2-C6-алкоксиалкил, метилендиокси, этилендиокси, C1-C4-галоалкил (включая -CvFw, где v = 1-3 и w = от 1 до (2v+1), C1-C4-галоалкокси, C1-C4 -алкилкарбонилокси, C1-C4-алкилкарбонил, C1-C4-алкилкарбониламино, -OCH2CO2H, 2-(1-морфолино)этокси, C1-C4-алкил (алкил, замещенный -N(R13)2, -CF3, NO2 или -S(= O)R13a);
J - β- Ala, или L-изомер, или D-изомер аминокислоты формулы -N(R3)C(R4)(R5)C(=O)-, причем R3 - H или C1-C8-алкил; R4 - H или C1-C3-алкил;
R5 выбирают из соединений: водород; C1-C8-алкил, замещенный 0-2 R11; C2-C8-алкенил, замещенный 0-2 R11; C2-C8-алкинил, замещенный 0-2 R11; C3-C8-циклоалкил, замещенный 0-2 R11; арил, замещенный 0-2 R12; 5-10-членная гетероциклическая кольцевая система, содержащая 1-4 гетероатома, независимо выбираемых из N, S и O, причем указанное гетероциклическое кольцо, замещенное 0-2 R12; = O, F, Cl, Br, I, -CF3, -CN, -CO2R13, -C(=O)R13, -C(= O)N(R13)2, -CHO, -CH2OR13, -OC(=O)R13, -OC(=O)OR13a, -OR13, -OC(=O)N(R13)2, -NR13C(= O)R13, -NR14C(= O)OR13a, -NR13C(= O)N(R13)2, -NR14SO2N(R13)2, -NR14SO2R13a, -SO3H, -SO2R13a, -SR13, -S(=O)R13a, -SO2N(R13)2, -N(R13)2, -NHC(= NH)NHR13, -C(=NH)NHR13, =NOR13, NO2, -C(=O)NHOR13, -C(=O)NHNR13R13a, = NOR13, -B(R34 )(R35), -OCH2CO2H, 2-(1-морфолино)этокси; -SC(=NH)NHR13, N3, Si(CH3)3, (C1-C5-алкил)NHR16; -(C0-C6-алкил)X;
где q = 0,1;
-(CH2)mS(O)p'(CH2)2X,
где m = 1, 2 и p' = 0-2;
причем X определен ниже, и
R3 и R5 также могут быть взяты вместе с образованием

где n = 0, 1 и X представляет собой
R3 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)t- или -CH2S(O)p'C(CH3)2-, где t = 2-4 и p' = 0-2; или
R4 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)u-, где u = 2-5;
R16 выбирают из соединений: аминозащитная группа; 1-2 аминокислоты; 1-2 аминокислоты, замещенные аминозащитной группой;
K - D-изомер или L-изомер аминокислоты формулы -N(R6)CH(R7)C(=O)-, причем R6 - H или C1-C8-алкил;
R7 выбирают из соединений:
-(C1-C7-алкил)X;
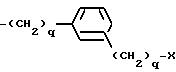
причем каждое q = 0-2, и замещение на фенил происходит в третьем или четвертом положении;
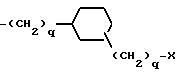
причем каждое q = 0-2, и замещение на циклогексил происходит в третьем или четвертом положении;
-(CH2)m-O-(C1-C4-алкил)-X, где m = 1 или 2;
-(CH2)mS(O)p'-(C1 -C4-алкил)-X, где m = 1 или 2 и p' = 0-2; и X выбирают из соединений
-N(R13)R13; -C(= NH)(NH2); -SC(= NH)-NH2; -NH-C(= NH)(NHCN); -NH-C(= NCN)(NH2); -NH-C(= N-OR13)(NH2); R6 и R7 могут быть альтернативно взяты вместе с образованием
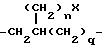
причем каждое q = 1 или 2, причем n = 0 или 1 и X представляет собой -NH2 или
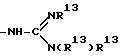
L представляет собой -Y(CH2)vC(=O)-, причем Y - NH, N(C1-C3-алкил), O или S и v = 1 или 2;
M - D-изомер или L-изомер аминокислоты формулы
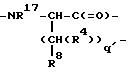
причем q = 0-2; R17 - H, C1-C3-алкил;
R8 выбирают из соединений
-CO2 R13; -SO3R13; -SO2NHR14, -B(R34)(R35), -NHSO2CF3, -CONHNHSO2CF3, -PO(OR13)2, -PO(OR13)R13, -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбираемых из N, S или O), -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбираемых из N, S или O); -SO2NHCOR13, -CONHSO2R13a, -CH2CONHSO2R13a, -NHSO2NHCOR13a, -NHCONHSO2R13a, -SO2NHCONHR13;
R34 и R35 независимо выбирают из соединений -OH, -F, -N(R13)2 или C1-C8-алкокси;
R34 и R35 могут быть альтернативно взяты вместе с образованием циклического эфира бора, где указанная цепь или кольцо содержит от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбираемых из N, S или O; бивалентного циклического амида бора, где указанная цепь или кольцо содержит от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбираемых из N, S или O; циклического амидоэфира бора, где указанная цепь или кольцо содержит от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатомов, независимо выбираемых из N, S или O; и причем радиоактивномеченое соединение выбирают из123J,125J,131J,18F,11C,13N,15O,75Br.
[52] В настоящее изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, причем R31 связан с (C(R23)R22)n'' и (C(R21R1)n' посредством двух различных атомов указанного карбоциклического кольца.
[53] В настоящее изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые
соединения, причем
n'' = 0 и n' = 0;
n'' = 0 и n' = 1;
n'' = 0 и n' = 2;
n'' = 1 и n' = 0;
n'' = 1 и n' = 1;
n'' = 1 и n' = 2;
n'' = 2 и
n' = 0;
n'' = 2 и n' = 1 или
n'' = 2 и n' = 2,
[54] В настоящее изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, причем
R6 - метил, этил или пропил.
[55] В настоящее изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, причем R31
выбирают из группы соединений, состоящей из
(а) 6-членного насыщенного, частично насыщенного или ароматического карбоциклического кольца, замещенного 0-3 R10 или R10a;
(б) 8-11-членного насыщенного, частично насыщенного или ароматически слитого бициклического карбоциклического кольца, замещенного 0-4 R10 или R10a; или
(в)
14-членного насыщенного, частично насыщенного или ароматически слитого трициклического карбоциклического кольца, замещенного 0-4 R10 или R10a.
[56] В настоящее
изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, причем R31 выбирают из группы соединений, состоящей из:
(а) 6-членного
насыщенного, частично насыщенного или ароматического карбоциклического кольца формулы
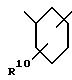
причем любая связь, формирующая карбоциклическое кольцо, может быть одинарной или двойной, а указанное карбоциклическое кольцо независимо замещено 0-4 R10;
(б) 10-членного насыщенного, частично насыщенного или ароматического бициклического карбоциклического кольца формулы
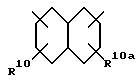
причем любая связь, формирующая карбоциклическое кольцо, может быть одинарной или двойной,
а указанное карбоциклическое кольцо независимо замещено 0-4 R10 или R10a;
(в) 9-членного насыщенного, частично насыщенного или ароматического бициклического карбоциклического кольца формулы
причем любая связь, формирующая карбоциклическое кольцо, может быть одинарной или двойной,
а указанное карбоциклическое кольцо независимо замещено 0-4 R10 или R10a.
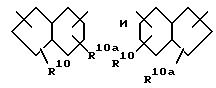
[57] В настоящее изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, причем R31 выбирают
из соединений (прерывистая линия может обозначать как одинарную, так и двойную связь):
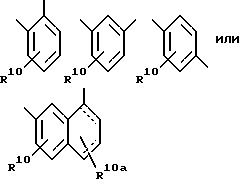
n'' = 0 или 1 и n' = 0-2
[58] В настоящее изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, причем R1 и R22 независимо выбирают из соединений: фенил, бензил, фенэтил, фенокси, бензилокси, галоген, гидрокси, нитро, циано, C1-C5-алкил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C7-C10-арилалкил, C1-C5-алкокси, -CO2R13, -C(= O)NHOR13a, -C(=O)NHN(R13)2, =NOR13, -B(R34)(R35), C3-C6-циклоалкокси, -OC(= O)R13, -C(= O)R13, -OC(=O)OR13a, -OR13, -(C1-C4алкил)-OR13, -N(R13)2, -OC(= O)N(R13)2, -NR13C(=O)R13, -NR13C(=O)OR13a, -NR13C(=O)N(R13)2, -NR13SO2N(R13)2, -NR13SO2R13a, -SO3H, -SO2R13a, -S(= O)R13a, -SR13, -SO2N(R13)2, C2-C6-алкоксиалкил, метилендиокси, этилендиокси, C1-C4-галоалкил, C1-C4-галоалкоси, C1-C4-алкилкарбонилокси, C1-C4-алкилкарбонил, C1-C4-алкилкарбониламино, -OCH2CO2H, 2-(1-морфолино)этокси, C1-C4-алкил (алкил, замещенный -N(R13)2, -CF3, NO2 или -S(= O)R13a);
[59] В настоящее изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, причем R31 выбирают из соединений
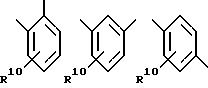
причем R31 может быть независимо замещен 0-3 R10 или R10a;
R32 - -C(=O)-,
n'' = 0 или 1,
n' = 0-2;
R1 и R22 независимо выбирают из соединений H, C1-C4-алкил, фенил, бензил, фенил-(C2-C4)алкил, C1-C4-алкокси;
R21 и R23 независимо выбирают из соединений H или C1-C4-алкил;
R2 - H или C1-C8-алкил;
R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10 -алкоксиалкил;
R13a - C1-C10-алкил, C3-C10-циклоалкил, Ct4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
когда две R13-группы соединяются с единичным N, указанные R13-группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2)O(CH2)-;
R14 - OH, H, C1-C4-алкил или бензил;
R10 и R10a независимо выбирают из соединений H, C1-C8-алкил, фенил, галоген или C1-C4-алкокси;
J - β- Ala, или L-изомер, или D-изомер аминокислоты формулы -N(R3)C(R4)(R5)C(=O)-, причем R3 - H или CH3;
R4 - H или C1-C3-алкил;
R5 - H, C1-C8-алкил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C1-C6-циклоалкилэтил, фенил, фенилметил, CH2OH, CH2SH, CH2OCH3, CH2SCH3, CH2CH2SCH3, (CH2)sNH2, -(CH2)sNHC(=NH)(NH2), -(CH2)sNHR16, где s = 3-5; или
R16 выбирают из соединений: аминозащитная группа; 1-2 аминокислоты или 1-2 аминокислоты, замещенные аминозащитной группой;
R3 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)t- (t = 2-4) или -CH2SC(CH3)2-; или
R4 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)u-, где u = 2-5;
K - L-изомер аминокислоты формулы -N(R6)CH(R7)C(=O)-, причем R6 - H или C1-C8-алкил;
R7 представляет собой
где q = 0 или 1;
-(CH2)rX, где r = 3-6;
-(CH2)mS(CH2)X, где m = 1 или 2, -(C3-C7-алкил)-NH-(C1-C6-алкил)
-(CH2)m-O-(C1-C4-алкил)-NH- (C1-C6-алкил), где m = 1 или 2;
-(CH2)m-S-(C1-C4-алкил)-NH- (C1-C6)-алкил), где m = 1 или 2; и
X представляет собой -NH2 или -NHC(=NH)(NH2);
или
R6 и R7 могут быть альтернативно взяты вместе с образованием
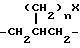
где n = 0 или 1; X представляет собой -NH2 или -NHC(=NH)(NH2);
L - -Y(CH2)vC(=O)-, причем Y - NH, O или S; и v = 1 или 2; M - D-изомер или L-изомер аминокислоты формулы
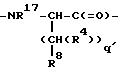
причем q = 0-2; R17 - H, C1-C3-алкил; R8 выбирают из соединений -CO2R13, -SO3R13, -SO2NHR14, -B(R34)(R35), -NHSO2CF3, -CONHNHSO2CF3, -PO(OR13)2,
-PO(OR13)R13, -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбираемых из N, S или O), -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбираемых из N, S или O); -SO2NHCOR13, -CONHSO2R13a, -CH2 CONHSO2R13a, -NHSO2NHCOR13a, -NHCONHSO2R13a, -SO2NHCONHR13.
[60] В настоящее изобретение
включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, представляющие собой радиоактивномеченые 1,3-двузамещенные соединения фенила формулы (II)
причем показанное фенильное кольцо формулы (II) может быть далее замещено 0-3 R10;
R10 независимо выбирают из соединений H, C1-C8-алкил, фенил, галоген или C1-C4-алкокси; R1 - H, C1-C4-алкил, фенил, бензил или фенил-(C1-C4)алкил; R2 - H или метил; R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил; R13a - C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил; когда две R13-группы связываются с единичным N, указанные R13-группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или (CH2 )O(CH2)-;
R14 - OH, H, C1-C4-алкил или бензил;
J - β- Ala, или L-изомер, или D-изомер аминокислоты формулы -N(R3)C(R4)(R5)C(=O)-, причем R3 - H или CH3; R4 - H или C1-C3-алкил; R5 - H, C1-C8-алкил, C3 -C6-циклоалкил, C3-C6-циклоалкилметил, C1-C6-циклоалкилэтил,
фенил, фенилметил, CH2OH, CH2SH, CH2 OCH3, CH2SCH3, CH2CH2SCH3, (CH2)sNH2, -(CH2)sNHC(=NH)(NH2), -(CH2)sNHR16, где s = 3-5; или
R16 выбирают из соединений: аминозащитная группа; 1-2 аминокислоты или 1-2 аминокислоты, замещенные аминозащитной группой;
R3 и R5 могут быть альтернативно взяты вместе с образованием -CH2CH2CH2-; или R4 и R5 могут быть альтернативно взяты вместе с образованием -(CH2)u, где u = 2-5; K - L-изомер аминокислоты формулы -N(R6)CH(R7)C(=O)-, причем R6 - H или C1-C8 -алкил; R7 представляет собой
где q = 0 или 1;
-(CH2)rX, где r = 3-6;
-(CH2)mS(CH2)X<где m = 1 или 2, -(C3-C7-алкил)-NH-(C1-C6-алкил),
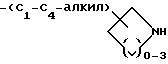
-(CH2 )m-O-(C1-C4-алкил)- NH-(C1-C6-алкил), где m = 1 или 2;
-(CH2)m-S-(C1-C4-алкил)- NH-(C1-C6-алкил), где m = 1 или 2; и
X - -NH2 или -NHC(=NH)(NH2);
причем X не представляет собой NH2, когда r = 4; или R6 и R7 альтернативно взяты вместе с образованием
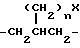
где n = 0, 1 и X - -NH2 или -NHC(=NH)(NH2); L - -Y(CH2)vC(=O), причем Y - NH, O или S; и v = 1, 2; M - D-изомер или L-имзомер аминокислоты формулы

причем q = 0-2; R17 - H, C1-C3-алкил; R8 выбирают из соединений -CO2R13, -SO3R13, -SO2NHR14, -B(R34)(R35), -NHSO2CF3, -CONHNHSO2CF3, -PO(OR13 )2,
-PO(OR13)R13, -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбираемых из N, S или O), -SO2NH-гетероарил (указанный гетероарил является 5-10-членным и имеет 1-4 гетероатома, независимо выбираемых из N, S или O); -SO2NHCOR13, -CONHSO2R13a, -CH2CONHSO2R13a, -NHSO2NHCOR13a, -NHCONHSO2R13a, -SO2NHCONHR13.
[61] В
настоящее изобретение включены описанные выше в [51] непосредственным образом радиоактивномеченые соединения, представляющие собой радиоактивномеченые 1,3-двузамещенные соединения фенила формулы
(II)
причем фенильное кольцо формулы (II) может быть далее замещено 0-3 R10 или R10a;
R10 или R10a независимо выбирают из соединений H, C1-C8-алкил, фенил, галоген или C1-C4-алкокси;
R1 представляет собой H, C1-C4-алкил, фенил, бензил или фенил-(C1-C4)алкил;
R2 - H или метил;
R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10 -алкил)арил или C3-C10-алкоксиалкил;
когда две R13-группы связаны с единичным N, указанные R13-группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2)O(CH2)-;
R13a - C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
R14 - OH, H, C1-C4 -алкил или бензил;
J - β- Ala, или L-изомер, или D-изомер аминокислоты формулы N(R3)C(R4)(R5)C(O)-, причем R3 - H или CH3; R4 - H; R5 - H, C1-C8-алкил, C3-C6-циклоалкил, C3-C6-циклоалкилметил, C1-C6-циклоалкилэтил, фенил, фенилметил, CH2OH, CH2SH, CH2OCH3, CH2SCH3, CH2CH2SCH3, (CH2)sNH2, (CH2)sNHC(= NH)(NH2), (CH2)sR16, где s = 3-5; R3 и R5 могут быть альтернативно взяты вместе с образованием -CH2CH2CH2-;
R16 выбирают из соединений: аминозащитная группа; 1-2 аминокислоты или 1-2 аминокислоты, замещенные аминозащитной группой
K - L-изомер аминокислоты формулы -N(R6)CH(R7)C(=O)-, причем R6 - H или C3-C8-алкил; R7 представляет собой
где q = 0 или 1;
-(CH2)rX, где r = 3-6;
-(CH2)mS(CH2)X, где m = 1 или 2,
-(C4-C7 -алкил)-NH-(C1-C6-алкил)
-(CH2)m-O-(C1 -C4-алкил)- NH(C1-C6-алкил), где m = 1 или 2;
-(CH2)m-S-(C1-C4-алкил)- NH-(C1-C6-алкил), где m = 1 или 2; и
X представляет собой -NH2 или -NHC(=NH)(NH2);
причем X не представляет собой NH2, когда r = 4; или
L - -YCH2 C(=O)-, причем Y - NH или O; M - D-изомер или L-изомер аминокислоты формулы
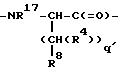
причем q = 1; R17 - H, C1-C3-алкил; R8 выбирают из соединений -CO2H или -SO3R13.
[62] В настоящее изобретение включены
непосредственным образом радиоактивномеченые соединения формулы (II), приведенной выше, причем фенильное кольцо формулы (II) может быть далее замещено 0-2 R10 или R10a;
R10 или R10a независимо выбирают из соединений H, C1-C8-алкил или C1-C4-алкокси;
R1 - H,
R2
- H;
R13 независимо выбирают из соединений H, C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил,
-(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
R13a - C1-C10-алкил, C3-C10-циклоалкил, C4-C12-алкилциклоалкил, арил, -(C1-C10-алкил)арил или C3-C10-алкоксиалкил;
когда две R13-группы связаны с единичным N,
указанные R13-группы могут быть альтернативно взяты вместе с образованием -(CH2)2-5- или -(CH2)O(CH2)-;
R14 - OH, H, C1-C4-алкил или бензил;
J - β- Ala, или L-изомер, или D-изомер аминокислоты формулы -N(R3)C(R4)(R5)C(= O)-, причем R3 - H и
R5 - H, CH3, CH2CH3, CH(CH3)2, CH(CH3)CH2CH3, CH2CH2CH3, CH2CH2CH2CH3, CH2CH2SCH3, CH2CH(CH3)2, (CH2)4NH2, (C3
-C5-алкил)NHR16; или R3 - CH3 и R5 - H; или R3 и R5 могут быть альтернативно взяты вместе с образованием -CH2
CH2CH2-;
R16 выбирают из соединений: аминозащитная группа; 1-2 аминокислоты или 1-2 аминокислоты, замещенные аминозащитной группой;
K - L-изомер
аминокислоты формулы -N(R6)CH(R7)C(=O)-, причем R7 представляет собой -(CH2)3NHC(=NH)(NH2);
L представляет собой -NHCH2C(= O)- и M - D-изомер или L-изомер аминокислоты формулы
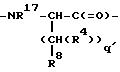
причем q = 1; R4 - H или CH3;
R17 - H;
R8 - -CO2H; -SO3H.
[63] В настоящее изобретение включены непосредственным образом
радиоактивномеченые соединения формулы (II), приведенной выше, причем R1 и R2 независимо выбирают из соединений H, метил;
J выбирают из D-Val, D-2-аминомасляной кислоты,
D-Leu, D-Ala, Gly, D-Pro, D-Ser, D-Lys, β- Ala, Pro, Phe, NMeGly, D-Nle, D-Phg, D-Ile, D-Phe, D-Tyr, Ala, Nε- p-азидобензоил-D-Lys, Nε
- p-бензоилбензоил-D-Lys, Nε- триптофанил-D-Lys, Nε- o-бензоилбензоил-D-Lys, Nε- p-ацетилбензоил-D-Lys, Nε- данзил-D-Lys, Nε- глицил-D-Lys, Nε- глицил-p-бензоилбензоил-d-Lys, Nε- p-фенилбензоил-D-Lys, Nε- m-бензоилбензоил-D-Lys, Nε
- o-бензоилбензоил-D-Lys;
K выбирают из NMeArg, Arg;
L выбирают из Gly, β- Ala, Ala;
M выбирают из Asp; α MeAsp; β MeAsp; NMeAsp; D-Asp.
[64] В настоящее изобретение включены непосредственным образом радиоактивномеченые соединения формулы (II), приведенной выше, причем R1 и R2 независимо выбирают из соединений H,
метил;
J выбирают из D-Val, D-2-аминомасляной кислоты, D-Leu, D-Ala, Gly, D-Pro, D-Ser, D-Lys, β- Ala, Pro, Phe, NMeGly, D-Nle, D-Phg, D-Ile, D-Phe, D-Tyr, Ala,
K выбирают из
NMeArg;
L выбирают из Gly;
M выбирают из Asp; α MeAsp; β MeAsp; NMeAasp; D-Asp.
[65] В настоящее изобретение включены описанные выше в [51]
непосредственным образом радиоактивномеченые соединения, представляющие собой:
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Val; K - NMeArg; L
- Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-2-аминомасляная кислота; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Leu; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Ala; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Gly; K - NMeArg; L - Gly и M
- Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Pro; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II),
причем R1 и R2 - H; J - D-Lys; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - β- Ala; K
- NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - NMeGly; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое
соединение формулы (II), причем R1 - метил (изомер 1); R2 - H; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1
- метил (изомер 2); R2 - H; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 - фенил (изомер 1); R2 - H; J
- D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем J - D-Met, K - NMeAry, L - Gly, M - Asp, R1 - H, R2 - H;
радиоактивномеченое соединение формулы (II), причем J - D-Abu, K - диNMe-гуанидинил-Орн, L - Gly, M - Asp, R1 - H, R2 - H;
радиоактивномеченое соединение формулы (II),
причем J - D-Abu, K - диNMe-Lys, L - Gly, M - Asp, R1 - H, R2 - H;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Nε- p-азидобензоил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Nε
- p-бензоилбензоил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Nε- триптофанил-D-Лизин;
K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Nε- o-бензилбензоил-D-Лизин; K - NMeArg; L - Gly и
M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Nε- p-ацетилбензоил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Nε- данзил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение
формулы (II), причем R1 и R2 - H; J - Nε- глицил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Nε- глицил-p-бензоилбензоил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и
R2 - H; J - Nε- p-фенилбензоил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J
- Nε- m-бензоилбензоил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - Nε
- o-бензоилбензоил-D-Лизин; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (III)
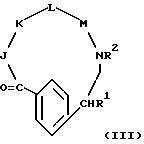
причем R1 и R2 - H; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Val; K - D-NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Nle; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Phg; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Phe; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (V)
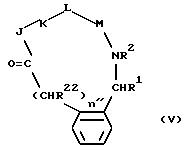
причем R1 и R2 - H; J - D-Ile; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (V), причем n''=1; R1, R2 и R22 - H; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (V), причем n''=0; R1 и R2 - H; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VI)

причем R1 и R22 - H; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII)

причем R1 и R2 и R10 - H; R10a - Cl; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10 - H; R10a - I; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10 - H; R10a - I; J - D-Abu; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10 - H; R10a - Me; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10a - H; R10 - Cl; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10a - H; R10 - MeO; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10a - H; R10 - Me; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10 - H; R10a - Cl; J - D-Abu; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10 - H; R10a - I; J - D-Abu; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VII), причем R1, R2 и R10 - H; R10a - Me; J - D-Abu; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Tyr; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Val; K - NMeAmf; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (II), причем R1 и R2 - H; J - D-Val; K - NMeArg; L - Gly и M - β MeAsp;
радиоактивномеченое соединение формулы (II), причем R1 - H; R2 - CH3; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (III), причем R1 и R2 - H; J - D-Val; K - NMeArg; L - Gly и M - Asp;
радиоактивномеченое соединение формулы (VIII), причем J - D-Val; K - NMeArg; L - Gly и M - Asp;
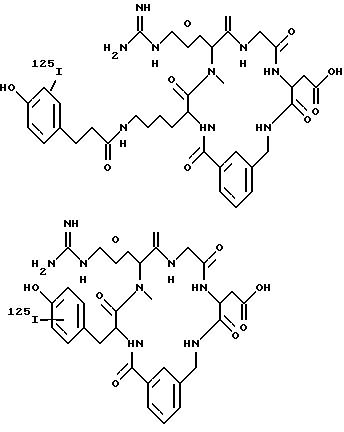
[66] В настоящее изобретение включены радиоактивномеченые соединения, описанные в любой из рубрик [51]-[65], причем радиоактивную метку выбирают из группы соединений:18F,11C,123J и125J.
[67] В настоящее изобретение включены радиоактивномеченые соединения, описанные в [66], причем радиоактивная метка представляет собой123J.
[68] В настоящее изобретение включена радиофармацевтическая композиция, содержащая радиофармацевтически приемлемый носитель и радиоактивномеченое соединение, описанное в любой из рубрик [51]-[67].
[69] В настоящее изобретение включен способ выявления отложений тромбоцитов у млекопитающего, заключающийся в том, что указанному млекопитающему вводят радиофармацевтическую композицию, содержащую соединение, описанное в любой из рубрик [51]-[67], и выявляют полученное изображение у данного млекопитающего.
[70] В настоящее изобретение включен способ диагностики заболевания, связанного с отложением тромбоцитов у млекопитающего, заключающийся в том, что указанному млекопитающему вводят радиофармацевтическую композицию, содержащую соединение, описанное в любой из рубрик [51]-[67], и выявляют полученное изображение у данного млекопитающего.
Как отмечается выше, циклические соединения, заявленные согласно данному изобретению, являются радиоактивномечеными.
Понятие "радиоактивномеченый" означает, что циклические гликопротеиновые соединения IIb/IIIa тромбоцитов содержат радиоизотоп, который пригоден для введения млекопитающему. Такие изотопы известны специалистам в данной области исследований и включают, например, изотопы галогенов (хлора, фтора, брома и йода) и металлов, в том числе технеция и индия. Предпочтительно используют следующие радиоизотопы:11C,18F,123J,125J,131J,99mTc,94mTc,95Tc,111In,62Cu,43Sc,45Ti,67Ga,68Ga,97Ru,72As,82Rb и201Tl. Наиболее предпочтительными являются изотопы123J,111In и99mTc. Радиоактивномеченые соединения согласно данному изобретению могут быть получены с помощью обычных процедур введения радиоактивных меток, хорошо известных специалистам в данной области исследований.
Подходящие методики синтеза детально описаны ниже. Как отмечалось выше, циклические соединения гликопротеина IIb/IIIa тромбоцитов согласно настоящему изобретению могут быть помечены радиоактивной меткой как непосредственным образом (то есть радиоактивная метка включается в само соединение), так и опосредованно (то есть радиоактивная метка включается в соединение через хелатирующий агент, который вводится в соединение).
Кроме того, радиоактивная метка может быть изотопной или неизотопной. При изотопном радиоактивном мечении, одна группа в составе циклического соединения, описанного выше, замещается (обменивается) на радиоизотоп. При неизотопном радиоактивном мечении, радиоизотоп добавляется к циклическому соединению без замещения (обмена) на какую-либо существующую группу. Таким образом, используемый согласно настоящему изобретению термин "радиоактивномеченые соединения" означает соединения, меченные как непосредственным образом, так и опосредованно, причем метка может быть как изотопной, так и неизотопной. Такие радиоактивные метки должны быть стабильными как с химической, так и с метаболической точки зрения в соответствии с принятыми стандартами. Более того, несмотря на то, что соединения в соответствии с настоящим изобретением могут быть мечены различным образом с помощью различных радиоизотопов, как это известно специалистам в данной области исследований, радиоактивное мечение должно осуществляться таким образом, чтобы высокая связывающая способность и специфичность немеченных циклических соединений гликопротеина GP IIb/IIIa тромбоцитов согласно настоящему изобретению, по отношению к GP IIb/IIIa-рецептору значительным образом не изменялась. Это означает, что связывающая способность и специфичность не изменяются более, чем примерно на 3 логарифмических единицы, предпочтительно более чем примерно на 2 логарифмических единицы, предпочтительнее более чем на 1 логарифмическую единицу, еще предпочтительнее более чем примерно на 500% и даже еще предпочтительнее более чем примерно на 250% и совсем предпочтительно, чтобы связывающая способность и специфичность не изменялись совсем.
Для радиоактивномеченых соединений метка может локализоваться по любому положению Q. Согласно настоящему изобретению предпочтение отдается радиоактивномеченым соединениям, в которых радиоактивная метка локализуется на карбоциклической кольцевой системе R31, R5-заместителе J и на R1 или R22. Еще более предпочтительными соединениями являются соединения формулы (II), причем радиоактивная метка локализуется на карбоциклической кольцевой системе R31 или R5 - заместителе J. При этом для предпочтительных и наиболее предпочтительных соединений, меченных непосредственным образом, предпочтительной меткой является галогеновая метка, особенно радиоактивный йод. Для радиоактивномеченых соединений, полученных опосредованно, предпочтительными радиоактивными металлами являются99mTc и111In. Предпочтительные связывающие группы, Ln и хелаторы металлов, Ch, описаны ниже.
Обнаружено, что радиоактивномеченые соединения согласно настоящему изобретению могут быть использованы в качестве радиофармацевтических препаратов для неинвазивной создающей изображение диагностики существующих или потенциально возможных тромбоэмболических заболеваний, таких как артериальный или венозный тромбоз, включая, например, нестабильную стенокардию, инфаркт миокарда, преходящее нарушение мозгового кровообращения, мозговой удар, атеросклероз, диабет, тромбофлебит, легочную эмболию или образование тромбоцитарных бляшек, а также тромбы и эмболии, вызванные сердечными протезами, например искусственными клапанами сердца. Радиоактивномеченные соединения в соответствии с настоящим изобретением могут быть использованы для выявления как вновь образованных, так и более старых тромбов. Кроме того, с их помощью можно диагностировать другие имеющиеся или потенциально возможные заболевания, связанные с чрезмерной экспрессией рецепторов GPIIb/IIIa, например при образовании метастатических раковых клеток.
Заявленные соединения могут быть эффективно применены в низких дозах, чтобы минимизировать любой возможный риск проявления токсичности.
Кроме того, данные соединения имеют значительно меньший размер по сравнению, например, с радиоактивномечеными антителами 7E3, известными специалистам, что позволяет уменьшить отношение "мишень/фон" (М/Ф) для выявляемых тромбов. Использование заявленных радиоактивномеченых соединений показано ниже в разделе, посвященном промышленной применимости.
Кроме того, при осуществлении настоящего изобретения обнаружено, что указанные выше радиоактивномеченые соединения пригодны для использования в качестве ингибиторов гликопротеина IIb/IIIa (GPIIb/IIIa) и таким образом могут быть применены в терапевтических целях, помимо диагностического использования, как описано выше. Как уже отмечалось, GPIIb/IIIa опосредует процессы активации и агрегации тромбоцитов. Заявленные согласно настоящему изобретению радиоактивномеченые соединения ингибируют активацию и агрегацию тромбоцитов, вызываемую всеми известными эндогенными агонистами тромбоцитов.
Описанные соединения могут иметь асимметрические центры. Если не указано иное, в настоящее изобретение включены все хиральные, диастереометрические и рацемические формы. Кроме того, в указанных соединениях могут присутствовать многочисленные геометрические изомеры олефинов, C=N двойные связи и подобные им структуры, а также в объем настоящего изобретения включены все стабильные изомеры. Существенно, что заявленные соединения содержат асимметрически замещенные атомы углерода и могут быть выделены в оптически активной или же рацемической форме. Хорошо известно, каким образом можно получить оптически активные формы, например путем разложения рацемических форм или путем синтеза, используя в качестве исходного материала оптически активные соединения. Описаны два различных изомера (цис и транс) пептидной связи, оба могут присутствовать в описанных здесь соединениях и все такого рода стабильные изомеры включены в настоящее изобретение. Если специально не указано иное, L-изомер аминокислоты находится в положениях J, K, L и M заявленных согласно данному изобретению соединений. За исключением того, что указано в предыдущем предложении, все хиральные, диастереометрические, рацемические формы и все геометрические изомерные формы включены в объем изобретения, если специально не отмечаются специфические стереохимические или изомерные формы заявленных соединений. D- и L-мзомеры определенной аминокислоты определены здесь в соответствии с трехбуквенным кодом, как показано в следующих примерах: D-Leu, D-Leu, L-Leu или L-Leu.
Когда какой-либо символ (например, R1-R8, m, n, p, X, Y и так далее) встречается в одной формуле или ее составляющей более одного раза, его определение в каждом случае не зависит от определения в других. Так, например, если отмечается, что группа замещена 0-2 R11, то указанная группа может быть факультативно замещена максимально двумя R11 и R11 в каждом случае независимо выбирают из всех возможных значений R11. Таким же образом, как показано на примере группы -N(R13)2, каждый из двух R13-заместителей N независимо выбирают из всех возможных значений R13.
Когда связь с заместителем показана пересекающей связью между двумя атомами в кольце, то такой заместитель может быть связан с любым атомом кольца.
Комбинации заместителей и/или переменных допускаются только в тех случаях, если такие комбинации приводят к образованию стабильных соединений.
Под стабильным соединением или "стабильной структурой" понимается соединение, которое в достаточной степени устойчиво к выделению из реакционной смеси и имеет необходимую чистоту и может быть включено в состав эффективного терапевтического средства.
Понятие "замещенный", используемое здесь, означает, что один или более атомов водорода, связанных с определенным атомом, заменены на соединение (структуру), выбранные из указанной группы, так что обычная валентность данного атома не увеличивается, и само замещение приводит к образованию стабильного соединения. Когда заместителем является кетон (=O), то замещаются 2 атома водорода.
Используемое здесь понятие "алкил" означает наличие как разветвленной, так и прямолинейной цепи насыщенных алифатических углеводородных групп, имеющих определенное количество атомов углерода; "галоалкил" предполагает наличие как разветвленной, так и прямолинейной цепи насыщенных алифатических углеводородных групп, имеющих определенное количество атомов углерода, замещенных одним или более галогеном (например, -CvFw, где v=1-3 и w=1-(2v+1)); "алкокси" обозначает наличие алкильной группы у определенного числа атомов углерода, прикрепленной посредством кислородного мостика; "циклоалкил" означает наличие насыщенных кольцевых групп, включая моно-, би- или полициклические кольцевые системы, такие как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и адамантил; и "бициклоалкил" предполагает наличие насыщенных бициклических кольцевых групп, таких как [3.3.0] бициклооктан, [4.3.0] бициклононан, [4.4.0] бициклодекан (декалин), [2.2.2] бициклооктан и так далее. "Алкенил" предполагает наличие углеводородных цепей как прямолинейной, так и разветвленной конфигурации, и одной или более углерод-углеродных связей, которые могут находиться в какой-либо стабильной точке, цепи, например к данной группе соединений относятся этенил, пропенил и подобные им вещества; и "алкинил" предполагает наличие углеводородных цепей как прямолинейной, так и разветвленной конфигурации, и одной или более тройных углерод-углеродных связей, которые могут находиться в какой-либо стабильной точке цепи, например к данной группе соединений относятся этинил, пропинил и подобные им вещества.
Понятие "борная кислота", используемое здесь, обозначает группу формулы -B(R34)(R35), причем R34 и R35 независимо выбирают из соединений -OH; -F; -NR13 R14; или C1-C8-алкокси; или R34 и R35 могут быть альтернативно взяты вместе с образованием: циклического эфира бора, где указанная цепь или кольцо содержат от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбираемых из N, S или O; бивалентного циклического амида бора, где указанная цепь или кольцо содержат от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбираемых из N, S или O; циклического амидо-эфира бора, где указанная цепь или кольцо содержат от 2 до 20 атомов углерода и, факультативно, 1-4 гетероатома, независимо выбираемых из N, S или O. Такие циклические эфиры бора, амиды бора или амидо-эфиры бора могут также быть факультативно замещены 1-5 группами, независимо выбираемых из R11.
Эфиры бора включают защитные группы борной кислоты, в том числе структуры, происходящие от диолов, например пинандиол и пинакол с образованием пинандиолового эфира борной кислоты и пинаколовой борной кислоты соответственно. Другие примеры диолов, пригодных для образования эфиров борной кислоты, включают перфторпинакол, этиленгликоль, диэтиленгликоль, 1-2-этандиол, 1,3-пропандиол, 1,2-пропандиол, 1,2-бутандиол, 1,4-бутандиол, 2,3-бутандиол, 2,3-гександиол, 1,2-гександиол, катехол, 1,2-диизопропилэтандиол, 5,6-дикандиол, 1, 2-дициклогексилэтандиол.
Понятие "гало" или "галоген" относится к фтору, хлору, брому и йоду; а термин "противоион" означает небольшую, отрицательную заряженную частицу, такую как хлорид, бромид, гидроксид, ацетат, такую как хлорид, бромид, гидроксид, ацетат, сульфат и тому подобное.
Используемое здесь понятие "арил" или "ароматический остаток" означает фенил или нафтил. Термин "карбоцикл" или "карбоциклический остаток" означает какое-либо стабильное 3- или 7-членное моноциклическое или бициклическое или 7- или 14-членное бициклическое или трициклическое или полициклическое (максимально 26-членное) углеродное кольцо, каждое из которых может быть насыщенным, частично ненасыщенным или ароматическим. Примерами таких карбоциклов могут служить (но не ограничиваются указанными) циклопропил, циклопентил, циклогексил, фенил, бифенил, нафтил, инданил, адамантил или тетрагидронафтил (тетралин).
Используемое здесь понятие "гетероцикл" или "гетероциклическая кольцевая система" означает стабильное 5-7-членное моноциклическое или бициклическое или 7-10-членное бициклическое гетероциклическое кольцо, которое может быть насыщенным, частично ненасыщенным или ароматическим и которое содержит атомы углерода и от одного до четырех гетероатомов, независимо выбираемых из N, O и S, и причем гетероатомы азота и серы могут быть факультативно окислены, и атом азота может быть факультативно четвертичным, причем указанное понятие включает любую бициклическую группу, в которой любое из описанных выше гетероциклических колец слито с бензольным кольцом. Гетероциклическое кольцо может быть соединено со своей парной группой посредством любого гетероатома или атома углерода с образованием стабильной структуры. Указанные здесь гетероциклические кольца могут быть замещены по атому углерода или азота, если образующиеся в результате этого соединения являются стабильными. Примерами таких гетероциклов служат бензопиранил, тиадиазин, тетразолил, бензофуранил, бензотиофенил, индолен, хинолин, изохинолинил или бензимидазолил, пиперидинил, 4-пиперидол, 2-пирролидон, тетрагидрофуран, тетрагидрохинолин, тетрагидроизохинолин, декагидрохинолин, октагидроизохинолин, азоцин, триазин (включая 1,2,3-, 1,2,4- и 1,3,5-триазин), 6H-1,2,5-тиадиазин, 2H, 6H-1,5,2-дитиазин, тиофен, тетрагидротиофен, тиантрен, фуран, пиран, изобензофуран, хромен, ксантен, феноксантин, 2H-пиррол, имидазол, пиразол, тиазол, изотиазол, оксазол (включая 1,2,4- и 1,3,4-оксазол), изоксазол, триазол, пиридин, пиразин, пиримидин, пиридазин, индолизин, изоиндол, 3H-индол, индол, 1H-индазол, пурин, 4H-хинолизин, изохинолин, хинолин, фталазин, нафтиридин, хиноксалин, хиназолин, циннолин, птеридин, 4aH-карбазол, карбазол, β- карболин, фенантридин, акридин, пиримидин, фенантролин, феназин, фенарсазин, фенотиазин, фуразан, феноксазин, изохроман, хроман, пирролидин, пирролин, имидазолидин, имидазолин, пиразолидин, пиразолин, пиперазин, индолин, изоиндолин, хинуклидин или морфолин (приведенные соединения не ограничивают объем изобретения). Кроме того, в объем притязаний включены слитые кольцевые и спиросоединения, содержащие, например, указанные выше гетероциклы.
Используемое здесь понятие "любая группа, которая при введении млекопитающему расщепляется с образованием свободного гидроксила, амино или сульфгидрила", означает какую-либо группу, связанную с атомом O, N или S соответственно, которая отщепляется от атома O, N или S, когда соединение вводится млекопитающему, с образованием вещества, имеющего оставшийся свободный гидроксил, амино или сульфгидрильную группу соответственно. Примерами групп, которые при введении млекопитающему расщепляются с образованием свободного гидроксила, амино или сульфгидрил, служат (но не ограничиваются указанными) C1-C6-алкил, замещенный 0-3 R11; C3-C6-алкоксиалкил, замещенный 0-3 R11; C1-C6-алкилкарбонил, замещенный 0-3 R11; C1-C6-алкоксикарбонил, замещенный 0-3 R11; C1-C6-алкиламинокарбонил, замещенный 0-3 R11; бензиол, замещенный 0-3 R12; феноксикарбонил, замещенный 0-3 R12; фениламинокарбонил, замещенный 0-3 R12. Примерами групп, которые при введении млекопитающему, расщепляются с образованием свободного гидроксила, амино или сульфгидрила, включают гидрокси, амино или сульфгидрильные защитные группы соответственно.
Используемый здесь термин "аминозащитная группа" означает какую-либо группу, известную специалистам в области органического синтеза для защиты аминогрупп. Такие аминозащитные группы, например,
приведены в книге Grune "Защитные группы в органическом синтезе" издательства John Wiley & Sons, New York (1981) и в книге "Пептиды: анализ, синтез, биология, vol. 3, Academic Press, New York
(1981), которые приведены здесь в качестве ссылки. Могут быть использованы и любые другие защитные группы, известные из уровня техники. Примерами аминозащитных групп служат (но не ограничиваются
указанными) следующие соединения:
1) ацилы, такие, как формил, трифторацетил, фталил и p-толуэнсульфонил;
2) ароматические карбаматы, такие, как бензилоксикарбонил (Cbz или Z) и
замещенные бензилоксикарбонилы, 1-(p-бифенил)-1-метилэтоксикарбонил, и 9-фторенилметилоксикарбонил (Fmoc);
3) алифатические карбаматы, такие, как третбутилоксикарбонил (Boc), этоксикарбонил,
диизопропилметоксикарбонил и аллилоксикарбонил;
4) циклические алкилкарбаматы, такие, как циклопентилоксикарбонил и адамантилоксикарбонил;
5) алкилы, такие, как трифенилметил и
бензил;
6) триалкилсилан, такой, как триметилсилан; и
7) тиолосодержащие соединения, такие, как фенилтиокарбонил и дитиосукциноил. Кроме того, под аминозащитными группами понимаются
ациловые группы, такие как азидобензоил, p-бензоилбензоил, o-бензилбензоил, p-ацетилбензоил, дансил, глицил-p-бензоилбензоил, фенилбензоил, m-бензоилбензоил, бензоил-бензоил.
Используемый здесь термин "фармацевтически приемлемые соли" относится к производным описываемых соединений, причем исходное соединение формулы (I) модифицировано путем образования кислых или основных солей соединения формулы (I). Примерами фармацевтически приемлемых солей служат (но не ограничиваются указанными) минеральные или органические кислые соли основных остатков, например аминов; щелочные или органические соли кислотных остатков, например карбоксиловых кислот; и тому подобные соединения.
Фармацевтически приемлемые соли заявленных соединений могут быть получены при взаимодействии свободных кислых или основных форм указанных соединений со стехиометрическим количеством соответствующего основания или кислоты в воде или органическом растворителе, или в их смеси; в основном, безводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил являются предпочтительными. Список подходящих солей приведен в книге Remington's Pharmacentical Sciences, 17 th ed., Mack Publishing Company, Easton, PA, 1985, p. 1418, которая указывается здесь в качестве ссылки.
Термин "аминокислота", используемый в данном описании, обозначает органическое соединение, содержащее как основную аминогруппу, так и кислую карбоксильную группу. Кроме того, данное понятие относится к модифицированным и необычным аминокислотам, таким, например, которые приведены в книге Roberts and Vellaccio (1983) The Peptides, 5: 342-429, приведенной здесь в качестве ссылки. Модифицированные или необычные аминокислоты, которые могут быть использованы при осуществлении изобретения, включают (однако, перечень не ограничивается указанными аминокислотами) D-аминокислоты, гидроксилизин, 4-гидроксипролин, орнитин, 2,4-диаминомасляная кислота, гомоаргинин, норлейцин, N-метиламиномасляная кислота, нафтилаланин, фенилглицин, β- фенилпролин, третичный лейцин, 4-аминоциклогексилаланин, N-метил-норлейцин, 3,4-дегидропролин, 4-аминопиперидин-4-карбоксиловая кислота, 6-аминокапроновая кислота, транс-4-(аминометил)-циклогексанкарбоксиловая кислота, 2-, 3- и 4-(аминометил)-бензойная кислота, 1-аминоциклопентанкарбоксиловая кислота, 1-аминоциклопропанкарбоксиловая кислота и 2-бензил-5-аминопентаноевая кислота.
Термин "аминокислотный остаток", используемый в данном описании, обозначает участок аминокислоты (определенной выше), который входит в состав пептида.
Термин "пептид", используемый в данном описании, означает соединение линейной структуры, которое состоит из двух или более аминокислот (определенных выше), соединенных пептидной связью. Кроме того, данный термин относится к соединениям, содержащим как пептидные, так и непептидные компоненты, например, псевдопептид или остатки, имитирующие пептиды или другие неаминокислотные компоненты. Такие соединения, содержащие как пептидные, так и непептидные компоненты, также могут быть обозначены термином, "пептидный аналог".
"Псевдопептид" или "имитирующий пептид" означает соединение, которое имитирует структуру аминокислотного остатка или пептида, например, при использовании связывающих групп, иных, чем амидные связи между имитирующим пептидом и аминокислотным остатком (псевдопептидные связи) и/или при использовании заместителей неаминокислотной природы и/или модифицированного аминокислотного остатка.
Понятие "псевдопептидный остаток" означает участок псевдопептида или имитирующего пептида (определенного выше), который входит в состав пептида.
Понятие "пептидная связь" означает ковалентную амидную связь, которая образуется при высвобождении молекулы воды между контактирующими карбоксильной группой одной аминокислоты и аминогруппой другой аминокислоты.
Понятие "псевдопептидная связь" означает изостерическую пептидную связь, которая может быть использована вместо или в качестве заместителя для нормальной амидной связи. Такие заместители или амидные "эквивалентные" связи образованы комбинациями атомов, в норме не встречающихся в пептидах или белках, причем эти заместители или связи отвечают пространственным требованиям амидной связи и обеспечивают стабилизацию молекулы по отношению к энзиматическому расщеплению.
Понятия "Ln", "связывающая (линкерная) группа" и "линкер", используемые здесь как взаимозаменяемые, обозначают группу атомов, разделяющих Q от хелатора металла, Ch.
Понятия "активированная Ln группа", "активированная Ln", "активированная связывающая группа" или "активированный линкер", используемые здесь как взаимозаменяемые, относятся к связывающей группе, которая имеет одну или более реактивных групп, способных реагировать или образовывать связь с хелатором или Q.
Понятие "Ch", "хелатор металла" и "хелатор", используемые здесь как взаимозаменяемые, обозначают химическое вещество, способное связываться или образовывать комплексы с нуклидом металла.
Понятие "циклизующее вещество" относится к промежуточному соединению, которое служит в качестве предшественника R31-группы Q.
Термин "кольцо, замещенное циклизующим фрагментом" обозначает циклический фрагмент, несущий замещающую группу одного или более его карбоциклического или гетероциклического колец.
Термин "циклизующий фрагмент, модифицированный линкером" соответствует циклизирующему фрагменту, который имеет в своем составе Ln-группу.
Термин "циклическое промежуточное соединение" означает промежуточное соединение, которое служит предшественником Q группы в упомянутых соединениях.
Термин "циклическое промежуточное соединение, модифицированное линкером" означает циклическое промежуточное соединение, которое несет активированную Ln-группу.
Соединения настоящего изобретения могут быть получены путями, хорошо известными квалифицированному специалисту в области органического синтеза. Предпочтительные методы описаны, но они не ограничивают данное изобретение.
Используются следующие аббревиатуры и сокращения:
Acm - ацетамидометил;
D-Abu - Д-2-аминомасляная кислота;
5-Aca
- 5-аминокапроамид(5-аминогексанамид);
b-Ala, b-Ala или b-Ala - 3-аминопропионовая кислота;
Boc - трет-бутилоксикарбонил;
Boc-iodo-Mamb
- трет-бутилоксикарбонил-3-аминометил-4-иодобензойная кислота;
Boc-Mamb acid - трет-бутилоксикарбонил-3-аминометилбензойная кислота;
Boc-ON
- [2-(трет-бутилоксикарбонилоксилимино)-2-фенилацетонитрил;
Cl2Bzl - дихлорбензил;
CBz, Cbz или Z - карбобензилокси;
DCC - дициклогексилкарбодиимид;
DIEA
- диизопропилэтиламин;
Di-NMeOrn - N-aMe-N-gMe-орнитин;
DMAP - 4-диметиламинопиридин;
HBTU - 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурониум гексафторфосфат;
NMeArg или MeArg - α- N-метиларгинин;
MeArg - N-метиламинометилфенилаланин;
NMeAsp - α- N-метиласпарагиновая кислота;
NMeGly или MeGly - N-метилиглицин;
NMe-Mamb - N-метил-3-аминометилбензойная кислота;
MMM - N-метилморфолин;
OcHex - O-циклогексил;
OBzl - O-бензил;
oSu - O-сукцинимидил;
pNP
- n-нитрофенил;
TBTU - 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурониумтетрафторборат;
Teoc - 2-(триметилсилил)этилоксикарбонил;
Tos - тозил;
Tr - тритил.
Используются следующие трехбуквенные обозначения аминокислот: Ala - аланин; Arg - аргинин; Asn - аспарагин; Asp - аспарагиновая кислота; Cys - цистеин; Gln - глютамин; Glu - глутаминовая кислота; Gly - глицин; His - гистидин; Ile - изолейцин; Leu - лейцин; Lys - лизин; Met - метионин; Nle - норлейцин; Phe - фенилаланин; Phg - фенилглицин; Pro - пролин; Ser - серин; Thr - треонин; Trp - триптофан; Tys - тирозин; Val - валин.
Соединения, относящиеся к данному изобретению, могут быть синтезированы стандартными методами, известными среднему специалисту в данной области. Предпочтительные методы включают нижеследующие, но не ограничиваются ими.
В основном, пептиды наращивают разрушением - аминогруппы C-конца и связыванием со следующей соответствующей защищенной аминогруппой с помощью пептидной связи, используя описанные методы. Такая процедура снятия защиты и связывания повторяется до тех пор, пока не будет достигнута желаемая последовательность. Это связывание может выполняться как последовательное попарное соединение аминокислот как конденсация (соединение) фрагментов (двух или нескольких аминокислот) или как комбинацию обоих этих процессов либо путем твердофазного пептидного синтеза по методу Merrifield, J.Am.Chem.Soc., 85, 2149-2154 (1963), сущность которого здесь кратко приведена.
Соединения настоящего изобретения могут быть также синтезированы с помощью специального автоматизированного пептидсинтезирующего оборудования. В соответствии с вышесказанным процедуры синтеза пептидов описаны в: Stewart и Young, "Твердофазный синтез пептидов", 2-е изд, Pirce Chemical Co., Rockford, IL (1984); Gross, Meienhofer, Udenfriend, Eds, "Пептиды: анализ, синтез, биология", тт. 1, 2, 3, 5 и 9, Academic Press, New York, 1980-1987; Bodanzky, "Пептидная химия: практический учебник", Springer Verlag, New York (1988) и Bodanzky et al. "Практика синтеза пептидов", Springer-Verlag, New York (1984), суть которых здесь кратко изложена.
Связывание между двумя аминокислотными производными, аминокислотой и пептидом, двумя пептидными фрагментами или циклизация пептида может быть осуществлена с помощью стандартных методов связывания, таких как азидный метод, смешанный кислотно-ангидридный метод (на основе изобутилового эфира хлормуравьиной кислоты), карбодиимидный метод (на основе дициклогексилкарбодиимида, диизопропилкарбодиимида или водорастворимых карбодиимидов), метод активных эфиров (n-нитрофенильный эфир, N-гидроксисукцинимидный эфир), метод Вудворда с реагентом K, карбонилдиимидазольный метод, метод на основе фосфорсодержащих реагентов, таких как BOP-Cl, метод окисления-восстановления. Некоторые их этих методов (особенно, карбодиимидный) могут быть усилены при добавлении 1-гидроксибензотриазола. Эти реакции связывания могут проводиться в любом растворе (жидкая фаза) или твердой фазе.
Функциональные группы связываемых аминокислот могут быть
защищены при реакциях связывания с целью избежания образования нежелательных связей. Защищающие агенты, которые могут быть использованы, указаны в Greene, "Защищающие группы в органическом синтезе",
John Wiley and Sons, New York (1981) и "Пептиды: Анализ, синтез, биология, т. 3, Academic Press, New York (1981), суть которых здесь кратко изложена. -карбоксильную группу C-концевого остатка обычно
защищают эфирной, которая затем может быть преобразована в карбоксильную. Эти защищающие группы включают: 1) алкильные эфиры, такие как метиловый и трет-бутиловый,
2) арильные эфиры, такие
как бензиловый и замещенный бензиловый,
3) эфиры, расщепляемые при слабощелочной обработке или обработке слабым окислителем, таким как трихлорэтиловый и фенациловый эфиры. При твердофазном
синтезе аминокислота C-конца присоединяется к нерастворимому носителю (обычно - полистирол). Эти нерастворимые носители содержат группу, способную реагировать с карбоксильной с образованием связи,
устойчивой в условиях наращивания пептидной цепочки и легко расщепляемой после этого. Примерами таковых являются: оксим-смола (DeGrado and Kaiser (1980) J.Org.Chem., 45, 1295-1300), хлор- или
бромметил-смола, гидроксиметил-смола и аминометил-смола. Многие из этих смол с уже включенной коммерчески требуемой C-концевой аминокислотой вполне доступны.
α- аминогруппа
каждой аминокислоты должна быть защищена. Для этого могут использоваться любые известные защищающие группы. Примерами таковых являются:
1) ацилы - например, формил, трифторацетил, фталил,
p-толуолсульфонил,
2) ароматические карбаматы, такие как бензилоксикарбонил (Cbz) и замещенные бензилоксикарбонилы, 1-(n-бифенил)-1-метилэтоксикарбонил и 9-фторенилметилоксикарбонил (Fmoc);
3) алифатические карбаматы, такие как трет-бутоксикарбонил (Boc), этоксикарбонил, диизопропилметоксикарбонил и аллилоксикарбонил;
4) циклические алкильные карбаматы, например,
циклопентилоксикарбонил и адамантилоксикарбонил;
5) алкильные, такие как трифенилметил и бензил,
6) триалкилсиланы, например, триметилсилан, и
7) тиолсодержащие, например,
фенилтиокарбонил и дитиосукциноил.
Предпочтительными α- аминозащищающими группировками являются "Boc" и "Fmoc". Многие производные аминокислот, соответствующе защищенные для синтеза пептидов, являются доступными.
α- Аминозащищающая группа удаляется перед взаимодействием со следующей аминокислотой. При использовании Boc-группы предпочтительными являются: трифторуксусная кислота, чистая или в дихлорметане, либо HCl в диоксане. Образовавшаяся соль аммония затем нейтрализуется (или перед связыванием или в процессе его) щелочными растворами, например водными буферными растворами или растворами третичных аминов в дихлорметане или диметилформамиде. При использовании Fmoc предпочтительными реагентами являются пиперидин или замещенные пиперидины в диметилформамиде, однако могут быть использованы любой вторичный амин или водные щелочные растворы. Удаление защищающей группы проводится при температуре от 0oC до комнатной.
Любая аминокислота, несущая функциональные группы в боковой цепи, должна быть защищена в процессе синтеза пептида с помощью упомянутых выше защищающих групп. Специалист учтет тот факт, что выбор и использование соответствующих защищающих групп для вышеупомянутых функциональных групп боковых цепей будет зависеть от самой аминокислоты и наличия (присутствия) других защищающих групп в пептиде. Избрание такой защищающей группировки является важным по той причине, что она не должна быть удалена при снятии защиты и связывании α- аминогруппы.
Например, если выбрана Boc для защиты α- аминогруппы, то приемлемыми защищающими группами являются следующие: n-толуолсульфонил (тозил) кольца и нитрогруппа для аргинина; бензилоксикарбонил, замещенные бензилоксикарбонилы, тозил или трифторацетил для лизина; бензил или алкильные эфиры, такие, как циклопентил для глутаминовой и аспарагиновой кислот; бензильные эфиры для серина и треонина; бензиловые эфиры, замещенные бензиловые эфиры или 2-бромбензилоксикарбонил для тирозина; n-метилбензил, n-метоксибензил; ацетамидометил, бензил или трет.бутилсульфонил для цистеина; а индольное кольцо триптофана может быть оставлено незамещенным или защищенным формальной группой.
Если для α- аминозащиты выбран Fmoc, обычно применяют защищающие группы на основе третбутильного радикала. Например, Boc может быть использован для лизина, третбутиловый эфир для серина, треонина и тирозина и трет-бутиловый эфир для глутаминовой и аспарагиновой кислот. Когда наращивание и циклизация пептида закончены, все защищающие группировки удаляются. При жидкофазном синтезе защищающие группировки удаляются любым путем, который зависит от природы самих удаляемых групп. Эти способы хорошо знакомы специалистам.
При использовании твердофазного синтеза пептид должен быть удален с подложки без одновременного снятия защиты с функциональных групп, чтобы не помешать процессу циклизации пептида. Таким образом, если пептид должен быть подвергнут циклизации в растворе, условия отщепления защитных групп нужно выбрать таким образом, чтобы свободная - карбоксилатная группа и свободная α- аминогруппа возникли без одновременного удаления защищающих групп.
Например, пептид может быть удален со смолы с помощью так называемого "гидразинолиза" и затем присоединен азидным методом.
Другой подходящий метод представляет собой синтез пептидов на оксимовой смоле, протекающий за счет внутримолекулярных нуклеофильных перемещений со смолы, в результате чего синтезируется циклический пептид (Osapay, Profit and Taylor (1990) Tetrahedron Letters, 43, 6121-6124). При использовании оксимовой смолы, схема Boc защиты особенно предпочтительна. Далее, предпочтительный метод снятия защитных групп боковых цепей, в основном, включает обработку ангидридом HF, содержащим добавки диметилсульфида, анизола, тиоанизола или n-крезола при 0oC. Расщепление защитных групп пептида может быть также завершено другими кислотными реагентами, такими как смесь трифторметансульфокислоты и трифторуксусной кислоты.
Редкие аминокислоты, используемые в настоящем изобретении, могут быть синтезированы стандартными методами, описанными в работах данной области ("Пептиды: анализ, синтез, биология, т. 5, с. 342-449, Academic Press, New York, 1981). N-алкилсодержащие аминокислоты могут быть получены с помощью методов, описанных ранее (Cheung et al., 1977, Can. J., Chem. 55, 906; Freidinger et al., 1982, J., Org. Chem. 48, 77, 1982).
Соединения настоящего изобретения могут быть получены нижеописанными методами.
Исходные материалы и методики для получения соединений будут описаны ниже дополнительно.
Процедура твердофазного пептидного синтеза выполнялась в 25-миллилитровых полипропиленовых фильтрационных трубках от BioRad Inc. или в 60-миллилитровых стеклянных сосудах от Peptides International. Оксимовая смола (уровень замещения = 0,96 ммоль/г) была получена в соответствии с методикой, описанной: (Degrado and Kaiser (1980) J.Org.Chem., 45, 1295) или закуплена у Novabiochem (уровень замещения = 0,62 ммоль/г). Все реагенты и растворители использовались без предварительной очистки. Трет-бутилоксикарбонил (Boc) аминокислоты и другие исходные аминокислоты могут быть получены на коммерческой основе у фирм Bachem Inc., Bachem Biosciences Inc. (Филадельфия, PA), Advanced ChemTech (Луневилль, KY), Peminsula Laboratories (Belmont, Ca) или Sigma (St. Louis, MO). 2-(1H-Бензотриазол-1-ил)-1,1,3,3-тетраметилурониум гексафторфосфат (НВТИ) и ТВТИ были закуплены у Advanced ChemTech. N-Метилморфолин (МММ) м-крезол, D-2-аминомасляная кислота (Abu), триметилацетилхлорид, диизопропилэтиламин (DIEA), 3-цианобензойная кислота и [2-(трет-бутилоксикарбонилоксилимино)-фенилацетонитрил] (Boc-ON) были приобретены у Aldrich Chemical Company. Диметилформамид (DMF), этилацетат, хлороформ (CHCl3), метанол (MeOH), пиридин и соляная кислота (HCl) были получены от Baker. Ацетонитрил, дихлорметан (DCM), уксусная кислота (HOAc), трифторуксусная кислота (TFA), сложный этиловый эфир, триэтиламин, ацетон и сульфат магния были получены от EM Scince. Палладий на угле - катализатор (10% Pd) был приобретен у Fluka Chemical Company. Абсолютный этанол был получен от Quantum Chemical Corporation. Тонкослойная хроматография выполнялась на силикагельных 60 F254 TCL пластинках (толщина слоя 0,2 мм), которые были закуплены у EM Separations. TCL визуализация была выполнена с использованием ультрасвета, впрыскивания йода, нингидрина и/или с помощью впрыскивателя Sakaguchi. Точки замерзания определялись с помощью аппаратуры Thomas Hoover или Electrothermal 9200 melting point и не проверялись. HPLC-анализы были проделаны с помощью Hewlett Packard 1090, Waters Delta Prep 3000, Rainin или Du Pont 8800 систем. Спектры ЯМР были получены на 300 MHZ General Electric QE-300, Varian 300 или Varian 400 - спектрометрах. Масс-спектрометрия ("бомбардировка быстрым атомом") (FAB-MS) была выполнена на VG Zab-E-масс-спектрометра с двойной фокусировкой с использованием Xenon FAB-пушки как источника ионов или Finnigan MAT 8230.
BOC-D-2-аминомасляная кислота (BOC-D-Abu) была получена модификацией метода, ранее
описанного в литературе (Itoh, Hagiwara, and Kamiya (1975) Tett. Lett., 4393), как показано на схеме
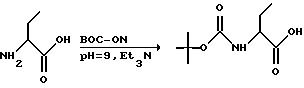
D-2-аминомасляная кислота.
D-2-Аминомасляную кислоту (1,0 г, 9,70 ммоль) растворяли в 20 мл H2O и добавляли раствор Boc ON (2,62 г, 10,6 ммоль) в 20 мл ацетона. Получившийся белый осадок растворяли прибавлением триэтиламина (3,37 мл, 24,2 ммоль) до образования бледно-желтого раствора (pH 9, на влажной pH-бумаге). При комнатной температуре раствор перемешивали в течение ночи, при этом под вакуумом был отогнан ацетон. Оставшуюся водную фазу трижды экстрагировали эфиром, подкисляли до pH 2 концентрированной HCl и затем экстрагировали трижды этилацетатом.
Собранный органический слой высушивали над безводным сульфатом магния и подвергали выпариванию при пониженном давлении, чтобы получить т-бутилоксикарбонил-D-2-аминомасляную кислоту, напоминающую масло (2,05 г, более чем количественный выход, содержит растворитель), которая была использована далее без дополнительной очистки.1HNMR (CDCl3) 0,98 (t, 3H), 1,45 (s, 9H), 1,73 (m, 1H), 1,90 (m, 1H), 4,29 (m, 1H), 5,05 (m, 1H).
Синтез R31 циклизующих разъясняет синтез определенных циклизующих, которые являются промежуточными от группы R31 до Q. Другие разделы поясняют синтез других циклизующих.
Синтез производных BOC-аминометилбензойной кислоты, BOC-аминофенилуксусной кислоты и BOC-аминометилфенилуксусной кислоты.
Производные BOC-аминометилбензойной кислоты, используемые как циклизующие в синтезе соединений данного изобретения, получают известными способами,
например, описанными в Tell.Lett., 4393 (1975); Modern Synthetic Reactions, H. O. House (1972); или Harting et al. J. Am. Chem. Soc., 50: 3370 (1928) и как показано на схеме
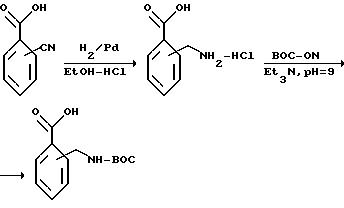
Гидрохлорид 3-аминометилбензойной кислоты
3-цианобензойную кислоту (10,0 г, 68 ммоль) растворяли в 200 мл этанола при нагревании на водяной бане при 35-50oC. Добавляли концентрированную HCl (6,12 мл, 73 ммоль) и полученный раствор перенесли в 500-миллилитровую обработанную азотом круглодонную колбу, содержащую катализатор - палладий на угле (1,05 г, 10% Pd/C). Суспензию перемешивали в атмосфере водорода в течение 38 часов, фильтровали через стеклянную воронку и тщательно промывали водой. Этанол удалялся при пониженном давлении и оставшийся водный слой, который содержал белый осадок, растворяли в добавленных сюда же 250 мл воды. Диэтиловый эфир (250 мл) добавлялся и суспензию переносили на фильтр. При энергичном встряхивании все твердые частицы растворялись и водный слой был затем дважды промыт эфиром, упарен при пониженном давлении до 150 мл и лиофилизирован до получения вышеуказанного соединения (хлоргидрат 3-аминометилбензойной кислоты) (8,10 г, 64%) в виде осадка бежевого цвета.1HNMR (D2O), 4,27 (s, 2H), 7,60 (t, 1H), 7,72 (d, 1H), 8,06 (d, 2H).
Т-бутилоксикарбонил-3-аминометилбензойная кислота (Boc-Mamb)
Указанное соединение получают в соответствии с модифицированными стандартными процедурами,
описанными в литературе (Itoh, Hagiwara and Kamiya (1975) Tett. , Lett., 4393). 3-Аминометилбензойную кислоту (в форме гидрохлоридной соли) (3,0 г, 16,0 ммоль) растворяли в 60 мл H2O. Сюда
же добавляли раствор Boc ON (4,33 г, 17,6 ммоль) в 60 мл ацетона, после чего триэтиламин (5,56 мл, 39,9 ммоль). Раствор становился желтым и pH достигал 9 (на влажный pH-бумаге) при добавлении 1,0 мл
(7,2 ммоль) триэтиламина. Раствор перемешивался в течение ночи при комнатной температуре, в процессе чего ацетон отгонялся при пониженном давлении, а оставшийся водный слой промывали трижды эфиром.
Водный слой затем подкисляли до pH 2 2H HCl и затем трижды экстрагировали этилацетатом. Объединенные органические слои трижды промывали водой, высушивали над безводным сульфатом магния и выпаривали
под вакуумом до сухого состояния. Продукт был перекристаллизован из смеси этилацетат/гексан и получали указанное вещество дважды (2,58 г, 64%) в виде белого порошка tпл 123-125oC;1HNMR (CDCl3) 1,47 (s, 9H), 4,38 (br s, 2H), 4,95 (br s 1H), 7,45 (t, 1H), 7,55 (d, 1H), 8,06 (d, 2H).
Синтез m-бутилоксикарбонил-3-аминофенилуксусной кислоты T-бутилоксикарбонил-3-аминофенилуксусные кислоты, используемые как промежуточные соединения в синтезе соединений настоящего изобретения, получают известными способами, например, описанными Collman and Groh (1982), J. Am.Chem. Soc., 104; 1391, и как показано на схеме.
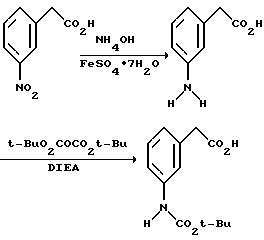
T-бутилоксикарбонил-3-аминофенилуксусная кислота.
Раствор 3-аминофенилуксусной кислоты (Aldrich, 10 г, 66 ммоль), ди-трет-бутилдикарбонат (15,8 г, 72 ммоль) и DIEA (8,6 г, 66 ммоль) в 50 мл дихлорметана перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют, при разделении между дихлорметаном и водой, водный слой отделяют, подкисляют 1H HCl до pH 3 и экстрагируют дихлорметаном. Экстракты промывают водой, раствором соли, высушивают над безводным сульфатом натрия и окончательно высушивают при пониженном давлении. Полученный материал был очищен перекристаллизацией из гептана, в результате было получено вышеуказанное соединение (3,7 г, 22%) в виде порошка белого цвета, точка плавления 105oC;1H NMR (CDCl3) 7, 35 (s, 1H), 7,25 (m, 3H), 6,95 (m, 1H), 6,60 (br s, 1H), 3,65 (s, 2H), 1,50 (s, 9H).
Синтез гидрохлорида 2-аминометилбензойной кислоты и гидрохлорида 2-аминометилфенилуксусной
кислоты
Гидрохлорид 2-аминометилбензойной кислоты и гидрохлорид 2-аминометилфенилуксусной кислоты, используемые как промежуточные соединения в синтезе заявленных соединений, получают
известными методами, например, как описано Naito et al. J.Antibiotics, 30: 698 (1977); или Young and Sweet J. Am.Chem. Soc., 80: 800 (1958), и как показано ниже
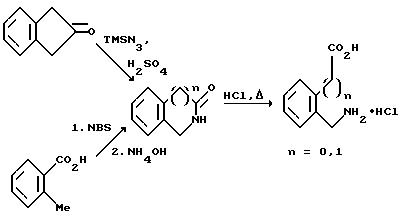
d-Лактам 2-аминометилфенилуксусной кислоты
Данное соединение получают при модификации метода, описанного ранее в литературе (Naito et al. 1977) J.Antibiotics, 30: 698). К охлажденной льдом суспензии 2-инданона (10,8 г, 82 ммоля) и азидотриметилсилана (9,4 г, 82 ммоля) в 115 мл хлороформа приливали 25 мл концентрированной серной кислоты таким образом, чтобы температура удерживалась в интервале 30-40oC. После выдерживания в течение 3 ч реакционная смесь выливалась на лед и водный слой подщелачивали концентрированным гидроксидом аммония, хлороформный слой отделяли, промывали водой, солевым раствором, высушивали над безводным сульфатом магния и досушивали в вакууме. Продукт очищали сублимацией (145oC, < 1 мм), после чего подвергали перекристаллизации из бензола до получения вышеуказанного соединения (5,4 г, 45%) в виде бледно-желтых кристаллов. Tпл 149-150oC;1 NMR (CDCl3) 7,20 (m, 5H), 4,50 (s, 2H), 3,60 (s, 2H).
Гидрохлорид 2-аминометилфенилуксусной кислоты
Указанное соединение
получают при модификации метода, ранее описанного в литературе ((Naito et al. 1977) J.Antibiotics, 30: 698). Смесь d-лактама 2-аминометилфенилуксусной кислоты (6,4 г, 44 ммоля) и 21 ил 6H HCl
нагревали с обратным холодильником 4 ч. Реакционную смесь обрабатывали активированным углем (Norit A), фильтровали, высушивали, а кубовые остатки растирали с ацетоном. В результате фильтрации получали
вышеназванное вещество (5,5 г, 62%) в виде бесцветных кристаллов. Tпл 168oC (dec);1H NMR (D6-DMSO) 12,65 (br s, 1H), 8,35 (br s, 3H), 7,50 (m, 1H), 7,35 (m,
3H), 4,05 (ABq, 2H), 3,80 (s, 2H).
d-Лактам 2-аминометилбензойной кислоты
Указанное соединение получают при модификации метода, ранее описанного в литературе (Danishefsky et
al. (1975) J.Org. Chem. 40: 796).
Смесь o-метилового эфира толуиловой кислоты (45 г, 33 моля), N-бромсукцинимида (57 г, 21 моля) и дибензоилпероксид (0,64 г) в 175 мл четыреххлористого углерода нагревали с обратным холодильником 4 часа. Охлажденную реакционную смесь фильтровали, высушивали при пониженном давлении, вновь растворяли в 250 мл метанола, концентрировали добавлением гидроксида аммония (74 мл, 1,11 моль). Реакционную смесь затем кипятили с обратным холодильником в течение 5 часов, концентрировали, фильтровали, осадок промывали водой, затем эфиром. Полученный продукт очищали перекристаллизацией из воды и получали вышеназванное вещество (11,0 г, 26%) в виде белого осадка Tпл 150oC;1H NMR (CDCl3) 7,90 (d, 1H), 7,60 (t, 1H), 7,50 (t, 2H), 7,00 (br s, 1H), 4,50 (s, 2H).
Гидрохлорид 2-аминометилбензойной кислоты
Данное соединение получают по способу, описанному для
гидрохлорида 2-аминометилфенилуксусной кислоты. Лактам (3,5 г, 26 ммоль) был превращен в указанное соединение (2,4 г, 50%) в виде бесцветных кристаллов.
Tпл 233o C (dec);1H NMR (D6 - DMSO), 13,40 (br s, 1H), 8,35 (br s, 3H), 8,05 (d, 1H), 7,60 (m, 3H), 4,35 (br s, 2H).
Синтез промежуточных циклических соединений
Этот раздел раскрывает синтез определенных промежуточных циклических соединений, они выполняют роль предшественника Q группы в заявленных соединениях (QLn)dCn; (Q)d, Ln-Ch. Эти соединения могут быть непосредственно мечены радиоизотопами или модифицированы присоединением линкерной группы (групп) или хелатора (хелаторов).
T-Бутилоксикарбонил-3-аминометилбензойная кислота (Boc-Mamb) связывается с оксимовой смолой с помощью модифицированного метода, описанного DeGrado and Kaiser (1980). J.Org. Chem. 45, 1295, при использовании 1 эквивалента 3-аминометилбензойной кислоты (с учетом степени замещения смолы, 1 эквивалента HBTU и 3 эквивалентов NMM). Boc-Mamb (1 эквивалент) может быть присоединен к оксимовой смоле при использовании 1 эквивалента DCC и DMAP (каждого из них) в метиленхлориде. Время связывания 15 - 96 ч. Степень связывания затем определяется или тестом пикриновой кислоты (Sarin, Rent, Tam and Merrifield (1981), Anal. Biochem, 117, 145-157), или количественным нингидриновым анализом (Gisin (1972) Anal.Chim. Acta, 58, 248-249). Непрореагировавшие оксим-группы блокируют 0,5 триметилацетилхлоридом/0,5 М диизопропилэтиламином в DMF в течение 2 ч.
Снятие Boc защищающей группы осуществляют 25% TFA в DCM в течение 30 мин. Оставшиеся аминокислоты или производные аминокислот связываются при 2-10-кратном избытке (связанном с "нагруженностью" первой аминокислоты и аминокислотного производного) соответствующей аминокислоты или аминокислотных производных и HBTU в приблизительно 8 мл DMF. Смолу затем нейтрализуют 3 эквивалентами NMM (в соответствии с количеством используемой аминокислоты) и продолжительность связывания составляет от 1 ч до нескольких дней. Полнота связывания контролируется качественным нингидриновым анализом или тестом пикриновой кислоты в случаях, когда аминокислота взаимодействовала с вторичным амином. Отсутствие связывания аминокислот, если необходимо, определяли по этим же результатам.
После того, как линейный пептид был собран, N-концевая группа Boc удалялась обработкой 25% TFA в DCM в течение 30 мин.
Смолу затем нейтрализовали обработкой 10% DIEA в DCM. Циклизацию с сопутствующим расщеплением пептида выполняли с помощью метода Osapay и Taylor ((1990) J.Am. Chem. Soc, 112, 6046) суспендированием смолы в приблизительно 10 мл/г DMF, добавляя 1 эквивалент HOAc (в соответствии с загруженностью первой аминокислоты) и перемешивая при 50-60oC в течение 60-72 ч. Затем фильтровали через стеклянную воронку, фильтрат DMF выпаривали, перерастворяли в HOAc или смеси ацетонитрил : H20 (1:1) и лиофилизовали до получения защищенного циклического продукта. Он затем обрабатывается с помощью стандартных процедур безводной HF (Stewart and Joung (1984) "Твердофазный пептидный синтез" 2 изд-е, Pierce Chemical Co., 85), содержащей 1 мл/г м-крезола или анизола как поглотителя при 0oC в течение 20-60 мин для удаления защитных групп боковых цепей. Неочищенный продукт можно очистить обратно-фазовой HPLC хроматографией, используя 2,5 см препаратов Vydac C18 колонку с линейным градиентом ацетонитрила, содержащим 0,1% TFA до получения чистого циклического продукта. Следующие N-a-Boc-защищенные аминокислоты могут использоваться для синтеза: Boc-Arg(Tos), Boc-N-a-MeArg(Tos), Boc-Gly, Boc-Asp (OcHex), BOC-3-аминометил-H-иодо-бензойная кислота, Boc-D-Ile, Boc-NmeAsp (OcHex), Boc-NMe-Mamb, Boc-D-Phg, Boc-D-Asp(OBzl), Boc-L-Asp(OcHex), Boc-aMe-Asp(OcHex), Boc-bMe-Asp(OcHex), Boc-L-Ala, Boc-L-Pro, Boc-D-Nle, Boc-d-Leu, Boc-D-Val, Boc-D-2-аминомасляная кислота (Boc-D-Abu), Boc-Phe, Boc-D-Ser(Bzl), Boc-D-Ala, Boc-3-аминометилбензойная кислота (Boc-Mamb), Boc-D-Lys (2-Clz), Boc-b-Ala, Boc-D-Pro, Boc-D-Phe, Boc-D-Tyr(Cl2Bzl), Boc-NMe-Amf(CBZ), Boc- аминотетралинкарбоксильная кислота, Boc-аминометилнафтойная кислота, Boc-4-аминометилбензойная кислота или Boc-NMeGly.
Предпочтительные N-a-Boc-защищенные аминокислоты, используемые в этом синтезе: Boc-Arg(Tos), Boc-N-a-MeArg(Tos), Boc-Gly, Boc-Asp (OcHex), Boc-D-Deu, Boc-D-Val-Boc-D-2-аминомасляная кислота (Boc-D-Abu), Boc-Phe, Boc-D-Ser(Bzl), Boc-D-Ala, Bос-3-аминометилбензойная кислота (Boc-Mamb), Boc-D-Lys (2-Clz) Boc-Ala, Boc-D-Pro или Boc-NMeGly.
Синтез заявленных соединений ниже проиллюстрирован. Нижеследующие таблицы дают представление о соединениях, защищаемых в настоящем изобретении.
Циклическое соединение - фрагмент 1 (промежуточное N1)
Цикло-(Gly-NMeArg-Gly-Asp-Mamb); соединение формулы II где J - Gly, K - NMeArg, L - Gly, M - Asp, R1=R2 - H
Указанное соединение получают, используя основной метод, описанный ниже для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид получали исходя из расчета 0,336 ммоля, для
того, чтобы получить защищенный циклопептид в количестве 218 мг, 84%. Пептид (200 мг) в 200 мл м-крезола были обработаны безводной HF при 0oC в течение 1 ч. Неочищенный материал осаждали
эфиром, перерастворяли в водной HOAc, лиофилизировали и получали указанное соединение в виде бледно-желтого осадка (158 мг, больше, чем количественный выход, вычислено через ацетатную соль). Очистка
завершалась обращеннофазовым методом ВЭЖХ с использованием препаративной Vydac С18 колонки (2,5 см) и 0,23%/мин градиента 2-11% ацетонитрила, содержащего 0,1% TFA, и лиофилизацией до получения TFA
соли указанного соединения в виде пушистого белого осадка (21% - выделено, окончательный выход 16,3%).
Масс-спектр: M+H=533,26.
Циклический фрагмент 2
Цикло-(D-Ala-NMeArg-Gly-Asp-Mamb); соединение формулы II, где J - D-Ala, K - NMeArg, L - Gly, M - Asp, R1=R2 - H
Указанное соединение получают, используя основной метод,
описанный ниже для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Установлено, что отсоединение остатка Boc-N-MeArg(Tos) необходимо. Пептид получали исходя из расчета 0,244 ммоля для того, чтобы получить
защищенный циклопептид (в количестве 117 мг; 61%). Пептид (110 мг) и 100 мл м-крезола обрабатывали безводной HF при 0oC в течение 1 ч. Неочищенный материал осаждали эфиром, перерастворяли в
водной HOAc, лиофилизировали и получали указанное соединение в виде бледно-желтого осадка. Очистку завершали обращеннофазовым методом ВЭЖХ в обратной фазе на препаративной Vydac C18 колонке (2,5 см) с
использованием 0,25%/мин градиента 2-11% ацетонитрила, содержащего 0,1% TFA соли указанного соединения в виде пушистого белого осадка.
Масс-спектр: M+H=547,33.
Циклическое промежуточное соединение 3
Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb); соединение формулы II, где J - D-Abu, K - NMeArg, L - Gly, M - Asp, R1=R2 - H.
Указанное соединение получают, используя основной метод, описанный ниже для циклического соединения N 4. Пептид получают исходя из расчета 0,101 ммоля для того, чтобы получить защищенный циклопептид (51 мг, 63%). Пептид (43 мг) и 50 мкл м-крезола обрабатывались безводной HF при 0oC в течение 30 ч. Неочищенный продукт осаждали эфиром, перерастворяли в водной HOAc, лиофилизировали, чтобы получить указанное соединение в виде бледно-желтого осадка (23 мг, 68,7% вычислено как ацетатная соль). Очистку заканчивали обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке с использованием 0,23%/мин градиента 7-14% ацетонитрила, содержащего 0,1% трифторуксусной кислоты и затем лиофилизировали до получения TFA соли указанного соединения в виде пушистого белого осадка (31%-выделено); конечный выход 12,4%). Масс-спектр: M+H=561,46.
Циклическое промежуточное соединение 3a.
Цикло-(Abu-NMeArg-Gly-Asp-Mamb); соединение формулы II где J - Abu, K - NMeArg, L - Gly, M - Asp, R1 - H; R2 - H.
Указанное соединение получают, используя основной метод, описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (Циклическое промежуточное соединение 4). DCC/DMAP использовался для присоединения Boc-Mamb к оксимовой смоле. TBTU используется как связывающий агент. Пептид получают исходя из расчета 0,596 ммоль, чтобы получить защищенный циклопептид (182 мг, 38,4%). Пептид (176 мг) и 0,176 мл анизола обрабатывали безводной HF при 0oC в течение 20 мин. Неочищенный материал осаждали эфиром, растворяли в водном ацетонитриле, лиофилизировали и получали указанное соединение (116 мг, 90,4% вычислено на основе фтористой соли). Очистку заканчивали ВЭЖХ хроматографией (обращеннофазовый метод) на препаративной Vydac C18 колонке (2,5 см) с использованием 0,45%/мин градиента 9-27% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизировали до получения TFA соли указанного соединения в виде пушистого белого осадка (выделено 1,92%, конечный выход 0,574%); FAB-MS. Масс-спектр: M+H=561,39.
Циклическое промежуточное соединение 4.
Цикло-(D-Val-NMeArg-Gly-Asp-Mamb); соединение формулы II, где J - D-Val, K - NMeArg, L - Gly, M - Asp, R1=R2 - H.
В 25-миллилитровую полипропиленовую пробирку, снабженную стеклянной фриттой, добавляли Boc-Mamb (0,126 г, 0,5 ммоля) и 6 мл DMF. Сюда же добавляли HBTU (0,194 г, 0,5 ммоля), оксимовую смолу (0,52 г, степень замещения = 0,96 ммоль/г) и N-метилморфолин (0,165 мл, 1,50 ммоль). Суспензию перемешивали при комнатной температуре в течение 24 часов. Затем смолу тщательно промывают (10-12 объемами): DMF (3X), MeOH (1X), DCM (3X), MeOH (2X) и DCM (3X). Количественным нингидриновым тестом был определен уровень замещения: 0,389 ммоль/г. Непрореагировавшие оксимгруппы были блокированы в результате обработки 0,5 M триметилацетилхлорид/0,5 М DIEA в DMF в течение 2 ч.
Дальнейшее выполнялось следующим образом: (Стадия 1). Смолу промывали DMF (3X), MeOH (1X), DCM (3X), MeOH (2X) и DCM (3X) (Стадия 2). С третичных Boc-групп была снята защита 25% TFA в DCM в течение 30 мин. (Стадия 3). Смолу промывали DCM (3X), MeOH (1X), DCM (2X), MeOH (3X) и DMF (3X) (Стадия 4) Boc-Asp(OcHex) (0,613 г, 1,94 ммоль), HBTU (0,753 г, 1,99 ммоль), 8 мл DMF и N-метилморфолин (0,642 мл, 5,84 ммоль) были добавлены к смоле и реакция продолжалась в течение 2,5 ч (Стадия 5). Завершение реакции связывания контролировалось качественным нингидриновым тестом. Стадии 1-5 повторяли до тех пор, пока не получали желаемую последовательность. Связывание Boc-D-Val с NMeArg контролировалось тестом пикриновой кислоты.
После того как линейный пептид был собран, N-терминальная T-Boc группа удалялась обработкой 25% TFA в DMM (30 мин). Смолу тщательно промывали DCM (3X), MeOH (2X) и DCM (3X) и затем нейтрализовали 10% DIEA в DCM (2x1 мин). Смолу тщательно промывали DCM (3X) и MeOH (3X) и высушивали. Половину смолы (0,101 ммоль) подвергали "сшивке" (циклизовали) в результате обработки 6 мл DMF, содержащим HOAc (5,8 мл, 0, 101 ммоль) и нагревания при 50oC в течение 72 ч. Смолу затем отфильтровывали через стеклянный фильтр и тщательно промывали DMF. DMF-фильтрат выпаривали в масле, перерастворяли в смеси ацетонитрил: H2O (1:1) лиофилизировали до получения желаемого циклопептида (49 мг, 60%).
Пептид (42 мг) обрабатывали безводной HF при 0oC в присутствии 50 мл м-крезола (в качестве поглотителя) в течение 30 мин для удаления защищающих групп боковых цепей. Неочищенный материал осаждали эфиром, перерастворяли в водной HOAc и лиофилизировали до получения указанного соединения в виде слабо-желтого осадка (23 мг, 70%, в расчете на ацетатную соль). Очистка производилась обращеннофазовым методом ВЭЖХ в обратной фазе на препаративной Vydac C18 колонке (2,5 см) и при 0,23%/мин градиенте 7-18% ацетонитрила, содержащего 0,1% трифторуксусной кислоты, и получали трифторацетат вышеназванного соединения в виде пушистого белого осадка (выделено 24%, окончательный выход 9,4%). FAB-MS: [M+H]=575,45.
Жидкофазный синтез циклического промежуточного соединения 4
Следующие аббревиатуры используются далее для TLC систем
растворителей: хлороформ (метанол 95:5=СМ;
хлороформ/уксусная кислота 95:5=СА;
хлороформ/метанол/уксусная кислота 95:5=СМА.
BocNMeArg(Tos)-Gly-OBzl - 25 ммоль BocNMeArg(Tos) (11,07 г, BacHem), 30 ммоль Gly-OBzl тозилата (10,10 г, Bachem), 25 ммоль HBTU (O-бензотриазол-N, N,N',N'- тетраметил-урониумгексафторфосфат; 9,48 г, Advanced Chemtech), и 75 ммоль DIEA (диизопропилэтиламин: Aldrich) были растворены в 25 мл CH2Cl2. Реакция протекала 1 ч, растворитель отгонялся под вакуумом в виде сиропа, который затем растворяли в 400 мл этилацетата. Полученный раствор подвергали экстракции (порциями по 150 мл) 2 • 5% лимонной кислотой, 1 • водой, 2 • насыщенным раствором NaHCO3, 1 • насыщенным раствором NaCl. Органический слой высушивали над MgSO4, а растворитель отгонялся под вакуумом. Получившийся масляный остаток растирали с петролейным эфиром и высушивали в глубоком вакууме минимум в течение 1 ч, выход 14,7 г (99,5%) TLC Rf(см) = 0,18, Rf(СА) = 0,10; NMR соответствует структуре; FABMS M+H+ = 590,43 (Теоретически ожидаемое 590,26).
NMeArg(Tos)-Gly-OBzl - 14 ммоль (BocNMeArg(Tos)-Gly-OBzl (24,5 ммоль) растворяли в 30 мл TFA, продолжительность взаимодействия 5 мин, затем растворитель отгоняли при комнатной температуре и остаточном давлении 1 мм.
Получившийся сироп растворяли в 400 мл ледяного этилацетата и экстрагировали 100 мл ледяного насыщенного раствора NaHCO3, водную фазу экстрагировали дважды 200 мл этилацетата, а смесь органических фаз экстрагировали 25 мл насыщенного раствора NaCl. Растворитель отгонялся при пониженном давлении до получения вязкого кубового остатка, который растирали с 300 мл эфира. Получившийся осадок растирали эфиром, получая гигроскопичное вещество, которое высушивали в вакууме: выход 10,33 г (86,2%) TLC Rf(см) = 0,03, Rf(СМА) = 0,20; NMR соответствует структуре: FABMS M+H+ = 490,21 (Теоретически 490,20).
Boc-D-Val-NMeArg(Tos)-Gly-OBzl - 9,80 ммоль, NMeArg(Tos)-Gly-OBzl (4,80 г), 9, 82 ммоль Boc-D-Val (2,13 г, Bachem) и 10,0 ммоль HBTU (3,79 г) растворяли в 10 мл метиленхлорида. Колбочку помещали на ледяную ванночку и добавляли 20 ммоль DIEA (3,48 мл). Реакция шла 15 мин при 0oC и 2 дня при комнатной температуре. К реакционной смеси приливали 400 мл этилацетата, экстрагировали (по 200 мл) дважды 5% лимонной кислотой, 1 раз насыщенным раствором NaCl, высушивали над MgSO4, выпаривали при пониженном давлении. Получившийся масляный остаток растирали с 50, затем с 30 мл эфира в течение 30 мин со следующими показателями: выход 4,58 г (69%) TLC Rf(см) = 0,27 (содержит также пятно рядом с исследуемым, которое соответствует ароматическому загрязнению, удаляемому при растирании продукта на следующей стадии); NMR соответствует структуре; FABMS M+H+ = 689,59 (Теоретически 689,43).
Boc-D-Val-NMeArg(Tos)-Gly - 4,50 г, Boc-D-Val-NMeArg(Tos)-Gly-OBzl (4,44 г), растворенного в 80 мл метанола, продували азотом (N2) в течение 10 мин. Затем добавляли 1,30 г Pd/C катализатора (10% Fluka lof# 273890) и пропускали H2 непосредственно над поверхностью реакции. TLC показала, что реакция заканчивается приблизительно через 0,5 ч. Спустя 1 ч катализатор удаляли фильтрацией через целитовый фильтр, а растворитель отгоняли при 40oC при пониженном давлении. Получаемый осадок хорошо растирали с 50 мл перегнанного эфира, фильтровали и промывали петролейным эфиром: выход 3,05 г (78%); TLC Rf(см) = 0,03; Rf(СМА) = 0,37; NMR соответствует структуре; FABMS M+H+ = 599,45 (Теоретически 599,29).
L-нитробензофенон оксим (Ox) - 50 г 4-нитробензофенона (220 мл, Aldrich) и 30,6 г гидрохлорида гидроксиламина (Aldrich, 440 ммоль) нагревали с обратным холодильником в 0,5 л смеси метанол/пиридин (9:1) в течение 1 ч. Реакционная смесь выпаривалась при пониженном давлении, растворялась в 500 мл эфира и экстрагировалась (200 мл порциями) 5% лимонной кислоты (дважды) и насыщенным раствором NaCl (1 раз), высушивалась над MgSO4, выпаривалась при пониженном давлении и растиралась с эфиром до получения 44,35 г (83%) оксима в виде смеси цис- и трансизомеров: TLC Rf(см) = 0,50; Rf(СМА) = 0,82; NMR соответствует структуре; FABMS M+H+ = 242,07 (Теоретически 242,07).
BocMamb-Ox - 22 ммоля BocMamb (5,522 г), 20 ммоль нитробензофенон оксима (4,84 г) и 20 ммоль DMAP (4-диметиламинопиридин; Aldrich) растворяли в 40 мл CH2Cl2. Колбу помещали в ванночку со льдом и добавляли 21 ммоль DCC (дициклогексилкарбодиимид) 4,33 г). Реакция шла на льду 30 мин при комнатной температуре в течение ночи. Образовавшуюся дициклогексилмочевину отфильтровывали и промывали 40 мл метиленхлорида. Фильтрат выпаривали при пониженном давлении при комнатной температуре до получения сиропообразного остатка, который растворяли в 400 мл этилацетата. Этот раствор экстрагировали (по 150 мл каждый раз): дважды 5% лимонной кислотой, 1 раз - водой, 2 раза насыщенным раствором NaHCO3, 1 раз насыщенным раствором NaCl. Органический слой высушивают над MgSO4, растворитель отгоняют при пониженном давлении. Получаемый остаток растирают с петролейным эфиром и высушивают в глубоком вакууме как минимум 1 ч.
Выход 7,51 г (79%); TLC Rf(см) = 0,41; Rf(СМА) = 0,66; NMR соответствует структуре; FABMS M+H+ = 476,30 (Теоретически 476,18).
TFA-Mamb-Ox-BocMamb-Ox, 7,4 г (15,5 ммоль) растворяли в 30 мл метиленхлорида + 10 мл TFA (25% TFA). Реакция шла при комнатной температуре 1 ч, растворитель выпаривали при пониженном давлении 20 мин при комнатной температуре, затем 15 мин при 40oC. Получившийся сироп растирали с эфиром (200 мл) при 5oC. Спустя 1 ч образовавшиеся кристаллы отфильтровывали и хорошо промывали эфиром: выход 7,22 г (95%); Rf(СМА) = 0,25; NMR соответствует структуре; FABMS M+H+ = 376,22 (Теоретически 376,12).
Boc-Asp(OcHex)-Mamb-Ox - 20 ммоль, Boc-Asp(OcHex) (6,308 г Bachem) и 44 ммоль DIEA (7,66 ммоль) растворяли в 20 мл DMF. Добавляли 20 ммоль HBTU (7,58 г Aldrich Chtm Tech) и реакция шла 2 мин при энергичном встряхивании. Добавляли TFA-Mamb-Ox (7,13 г, 15 ммоль) и продолжали реакцию в течение ночи при комнатной температуре. Растворитель выпаривали при пониженном давлении до маслообразного остатка, который затем растворяли в 500 мл этилацетата и этот раствор экстрагировали порциями по 150 мл: дважды 5% лимонной кислотой, 1 раз водой, дважды насыщенным раствором NaHCO3, раз насыщенным раствором NaCl. Органический слой высушивали над MgSO4, растворитель отгоняли при пониженном давлении. Образовавшийся остаток растирали с петролейным эфиром и высушивали в глубоком вакууме: выход 9,76 г (97%); TLC Rf(см) = 0,55; NMR соответствует структуре; FABMS M+H+ = 673,45 (Теоретически 673,23).
TFA Asp(OcHex)-Mamb-Ox - 15 ммоль Boc-Asp(OcHex)-Mamb-Ox было растворено в 50 мл 35% в CH2 Cl2 и оставлено для реакции в течение 90 мин. Растворитель отгоняли при пониженном давлении при комнатной температуре в течение 10 мин. Затем при 40oC в течение 15 мин. Для удаления следов TFA было добавлено 25 мл DMF и растворитель отгоняли при 50oC. Оставшийся сироп растирали с эфиром (200 мл), затем высушивали в глубоком вакууме: выход 9,61 г (93%); Rf(СМА) = 0,45; NMR соответствует структуре; FABMS M+H+ = 573,56 (Теоретически 573,23).
Boc-D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-Mamb-Ox 10 ммоль TFA Asp(OcHex)-Mamb-Ox + 10 ммоль Boc-D-Val-NMeArg(Tos)-Gly + 10 ммоль HBTU + 30 ммоль DIEA растворяли в 20 мл DMF. Спустя 4 ч растворитель отгоняли при пониженном давлении, а остаток переносили в 600 мл этилацетата, после чего экстрагировали 300 мл порциями: 5% лимонной кислоты, воды и насыщенного раствора NaCl. Органический слой высушивали над MgSO4, выпаривали при пониженном давлении, растирали с эфиром и высушивали в вакууме: выход 9,90 г (86%); Rf(см) = 0,10; NMR соответствует структуре; FABMS M+H+ = 1153,22 (Теоретически 1153,47).
TFA
D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-Mamb-Ox
Это соединение было получено из Boc-D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-Mamb-Ox (9,8 г, 8,5 ммоль) обработкой его TFA/CH2Cl2 (1:1)
в течение 45 мин. Растворитель отгоняли и продукт растирали с эфиром: выход 9,73 г (98%); Rf(см) = 0,10; NMR соответствует структуре; FABMS M+H+ = 1053,22 (Теоретически 1053,
4).
Цикло (D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-Mamb)
TFA D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-Mamb-Ox (1,80 г, 1,54 ммоль) + 2 ммоля DIEA + 2 ммоля уксусной кислоты были растворены
в 200 мл DMF. Смесь нагревалась в течение 2 дней (50oC) и затем подвергалась выпариванию при пониженном давлении. Остаток перерастворялся в 400 мл смеси этилацетат/н-бутанол (1: 1) и
экстрагировался порциями по 200 мл: 5% лимонной кислоты (3х); насыщенным раствором NaCl. Органический слой высушивали над MgSO4 и дважды растирали с 200 мл эфира: выход 1,07 г (86%); Rf(см) = 0,10; NMR соответствует структуре; FABMS M+H+ = 811,25 (Теоретически 811,38).
Цикло (D-Val-NMeArg-Gly-Asp-Mamb) 0,50 г цикло (D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-Mamb) обрабатывали 5 мл HF при 0oC в присутствии 0,5 мл анизола в течение 30 мин, HF удаляли при пониженном давлении и полученный пептид растирали с эфиром, этилацетатом и вновь эфиром. Полученный осадок растворяли в 10% уксусной кислоте и лиофилизировали: выход 0,321 г (82% из расчета на ацетатную соль). Продукт выделили со степенью очистки 40% с использованием метода, описанного для продукта, полученного твердофазным синтезом.
Кристаллизация циклического промежуточного соединения 4.
Получение солей на основе циклического промежуточного соединения 4.
Обнаружено, что соединения настоящего изобретения могут быть выделены кристаллизацией продукта из органических и водных растворителей. Цвиттер-ион циклического промежуточного соединения 4 превращалось в мезиловую (метансульфонатную) соль циклического промежуточного соединения 4 путем кипячения с обратным холодильником и при встряхивании в изопропаноле из расчета 25 мг/мл и медленном добавлении раствора 1,0 молярного эквивалента метансульфоновой кислоты (соответствующего содержанию воды), растворенной в изопропаноле. Нагревание отключали и раствор охлаждался до 5oC в ледяной бане. После 1-часового перемешивания раствор фильтровали и осадок промывали 3 раза холодным изопропанолом и высушивали под вакуумом до постоянного веса.
Следующие соли соединения (циклическое промежуточное соединение 4) были получены этим способом при добавлении 1,0 эквивалента соответствующей кислоты:
Циклическое промежуточное соединение 4 (бифенилсульфонат): цвиттер-ион + 1,0 эквивалент бифенилсульфоновой кислоты.
Циклическое промежуточное соединение 4 (a-нафталинсульфонат): цвиттер-ион + 1,0 эквив. a-нафталинсульфокислоты.
Циклическое промежуточное соединение 4 (b-нафталинсульфонат): цвиттер-ион + 1,0 эквив. b-нафталинсульфокислоты.
Циклическое промежуточное соединение 4 (соль бензолсульфокислоты): цвиттер-ион + 1,0 эквив. бензолсульфокислоты.
Циклическое промежуточное соединение 4 (n-толуолсульфонат): цвиттер-ион + 1,1 эквив. n-толуолсульфокислоты.
Следующие соли соединения (циклическое промежуточное соединение 4) были получены кристаллизацией из водных систем.
Циклическое промежуточное соединение 4 (в виде сульфата):
10 мг аморфного циклического промежуточного соединения 4 (полученного при лиофилизации амфотерного продукта из раствора 2 мольных
эквивалентов уксусной кислоты в воде), растворенные на 1 мл 1H H2SO4, pH до 2,5.
При выдерживании при комнатной температуре образуется осадок. Его отфильтровывают на стеклянном фильтре и высушивают под вакуумом до постоянного веса.
Циклическое промежуточное соединение 4 (в виде метансульфоната (мезила)):
100 мг
аморфного DMP 728, растворенные на 1 мл воды + 1,2 мольных эквивалента метансульфокислоты (полученной в виде 4M водного раствора). При выдерживании при комнатной температуре образуется большой плоский
кристалл.
Циклическое промежуточное соединение 4 (в виде соли бензолсульфокислоты):
100 мг цвиттер-иона, растворенные на мл воды + 1,2 эквивалента бензолсульфокислоты. При
выдерживании при комнатной температуре образуется осадок. Он отфильтровывается через стеклянный фильтр, промывается небольшим объемом изопропанола и высушивается под вакуумом до постоянного веса.
Циклическое промежуточное соединение 4 (в виде n-толуолсульфоната):
100 мг цвиттер-иона, растворенные на мл воды + 1,2 мольных эквивалента толуолсульфокислоты. При выдерживании
при комнатной температуре образуется осадок. Он отфильтровывается через стеклянный фильтр, высушивается под вакуумом до постоянного веса.
Циклическое промежуточное соединение 4b
Цикло-(D-Val-D-NMeArg-Gly-Asp-Mamb); J - D-Val; K - D-NMeArg, L - Gly; M - Asp, R1 - H, R2 - H.
Указанное соединение получают с помощью основного способа, описанного для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (то есть циклическое промежуточное соединение 4). DCC/DMAP метод был использован для присоединения Boc-Mamb к оксимовой смоле. Получают пептид (0,596 ммоль) из расчета для получения защищенного циклопептида в количестве 186 мг, 38,6%. Пептид (183 мг) и 0,183 мл анизола обрабатывали безводной HF от 0oC в течение 30 мин. Неочищенный продукт осаждали эфиром, растворяли в водном ацетонитриле. Лиофилизировали и получали указанное соединение (145 мг, больше чем количественный выход из расчета на соль HF). Очистка выполнялась HPL-хроматографией на препаративной Vydac C18 колонке (2,5 см) с использованием 0,23%/мин градиента 9 - 22,5% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизировали продукт, до получения TFA-соли указанного соединения в виде пушистого белого осадка (14,8% выделено; конечный выход 5,3%); FABMS: [M+H] = 575,31.
Циклическое промежуточное соединение 5.
Цикло-(D-Leu-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Leu; K - NMeArg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя основной способ, описанный выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид получали в пределах 0,115 ммоль, из расчета получить циклопептид в количестве 92,4 мг, 98%. Пептид (92,4 мг) и 93 мл м-крезола обрабатывали безводной HF при 0oC в течение 20 мин. Неочищенный продукт осаждали эфиром, растворяли в водной HOAc, лиофилизировали и получали указанное соединение в виде слабо-желтого осадка (45,7 мг, 69% из расчета на ацетатную соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке (2,5 см) с использованием 0,23%/мин градиента 7 - 21% ацетонитрила, содержащего 0,1% TFA и затем лиофилизировали продукт до получения TFA-соли указанного соединения в виде пушистого белого осадка (29% выделено, конечный выход 16,5%); FAB-MS: [M+H] = 589,48.
Циклическое промежуточное соединение 7.
Цикло-(D-Nle-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Nle; K - NMeArg; L - Gly; M - Asp, R1 - H, R2 - H.
Указанное соединение получают, используя способ, описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклосоединение 4), DCC/DMAP метод использовался для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен из расчета 0,586 ммоль с тем, чтобы получить защищенный циклопептид (305 мг, 63,3%). Пептид (295 мг) и 0,295 мл анизола обрабатывали безводной HF при 0oC в течение 30 мин. Полученный продукт осаждали эфиром, перерастворяли в водном ацетонитриле и лиофилизировали, чтобы получить указанное соединение (207 мг, 95,4% из расчета на фтористую соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке (2,5 см) с использованием 0,23%/мин градиента 5,4 - 18% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизировали продукт до получения TFA-соли указанного соединения в виде пушистого белого осадка (выделено 44%, конечный выход 22,9%); FAB-MS: [M+H] = 589,26.
Циклическое промежуточное соединение 11.
Цикло-(D-Phg-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Phg; K - NMeArg, L - Gly; M - Asp, R1 - H, R2 - H.
Указанное соединение получали на основе способа, описанного для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). DCC/DMAP метод использовался для присоединения Boc-Mamb к оксимовой смоле. Пептид получали из расчета 0,611 ммоль, чтобы получить защищенный циклопептид (296 мг, 57,4%). Пептид (286 мг) и 286 мл анизола обрабатывали безводной HF при 0oC в течение 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали, чтобы получить указанное соединение (210 мг, 98,9% в расчете на фтористую соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке (2,5 см) с использованием 0,23%/мин градиента 5,4 - 18% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизировали продукт до получения TFA-соли указанного соединения в виде пушистого белого осадка (выделено 24,2%, конечный выход 11,9%); FAB-MS: [M+H] = 609,27.
Циклическое промежуточное соединение 12.
Цикло-(D-Phe-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Phe; K - NMeArg, L - Gly; M - Asp, R1 - H, R2 - H.
Указанное соединение получают на основе способа, описанного для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое соединение 4). DCC/DMAP метод использовался для присоединения Boc-Mamb к оксимовой смоле. Пептид получали из расчета 0,611 ммоль с тем, чтобы получить защищенный циклопептид в количестве 140 мг (26,7%). Пептид (135 мг) и 0,135 мл анизола обрабатывали безводной HF при 0oC в течение 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали до получения указанного соединения (108 мг, больше, чем количественный выход в расчете на фтористую соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке (2,5 см) с использованием 0,23%/мин градиента 7,2 - 22,5% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизировали продукт до получения TFA-соли указанного соединения в виде пушистого белого осадка (выделено 35%, конечный выход 8,7%); FAB-MS: [M+H] = 623,28.
Твердофазный
синтез циклического промежуточного соединения 13 f
Цикло-(D-Lys-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Lys; K - NMeArg, L - Gly; M - Asp, R1=R2 - H.
Данное соединение получали, используя основной метод, описанный выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид получали из расчета 0,586 ммоль, чтобы получить защищенный циклопептид (349 мг, 58,9%). Пептид (334 мг) и 334 мл анизола обрабатывали безводной HF при 0oC в течение 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали, чтобы получить указанное соединение в виде бледно-желтого осадка (168 мг, 79,1% из расчета на фтористую соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке (2,5 см) с использованием 0,23%/мин градиента 5,4 - 14,4% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизировали продукт и получали TFA-соли указанного соединения в виде пушистого белого осадка (выделено 33,6%, конечный выход 12,1%); FAB-MS: [M+H] = 604,32.
Жидкофазный синтез
циклического промежуточного соединения 13 f
Схема, иллюстрирующая синтез, описанный ниже, приводится сразу после описания
Цикло-(D-Lys-NMeArg-Gly-Asp-Mamb); соединение формулы (VV),
где
Часть A-Boc-Asp(OBzl)
К раствору Boc-Asp(OBzl) (45,80 г, 140 ммоль) и HOSu(N-гидроксисукцинимид; 16,10 г, 140 ммоль) в 300 мл n-диоксана при 5-10oC добавляли DCC (30,
20 г; 140 ммоль). Раствор перемешивали 30 мин при 5-10oC, затем отфильтровывали осадок и промывали его диоксаном (3 х 50 мм). Собранную органическую фазу концентрировали при пониженном
давлении до получения чистого маслянистого остатка, который кристаллизовали до получения бесцветного осадка (42,98 г, 73%) при растирании с диэтиловым эфиром (3 х 100 мл), NMR соответствует структуре;
Tпл = 98 - 99oC; DCI-MS: [M+NH4] = 438.
Часть B-Boc-Asp-(OBzl)-Mamb
3-Аминометилбензойная кислота • HCl (Mamb; 13,08 г, 70,0 ммоль)
была растворена в 120 мл DMF и добавлен DIEA (24,32 мл, 140 ммоль) при изменении pH от 4 до 7,5. Белая суспензия перемешивалась в течение 30 мин при комнатной температуре перед добавлением раствора
Boc-Asp(OBzl)-OSu (49,40 г, 70,0 ммоль) в DMF (50 мл). Смесь перемешивалась 24 часа, в течение которых она превратилась в раствор золотистого цвета. Раствор добавили к 5% лимонной кислоте (2000 мл) и
охлаждали при 5oC 3 ч. Полученный осадок отфильтровали, промыли ледяной водой (200 мл) и ледяным диэтиловым эфиром (100 мл), высушили при пониженном давлении до получения указанного
соединения в виде бесцветного осадка (29,62 г, 92%); Tпл = 149 - 151oC; DCI-MS: [M+NH4] = 474.
Часть C - HCl • H • Asp(OBzl)-Mamb
Boc-Asp(OBzl)-Mamb (7,92 г, 17,4 ммоль) растворили в 4N HCl в диоксане (50 мл), перемешивали 2 ч, раствор концентрировали при пониженном давлении до получения указанного соединения в виде
бесцветного осадка (6,80 г; 99%). DCI-MS: [M+NH4] = 374.
Часть D-Boc-Lys-(Tfa)-NMeArg(Tos)-Gly-OBzl.
NMeArg(Tos)-Gly-OBzl (14,40 г, 29,4 ммоль), Boc-Lys-(Tfa) (10,0 г, 29,4 ммоль) и HBTU (11,37 г, 62,0 ммоль) растворили в метиленхлориде (40 мл). После охлаждения до 0oC добавили DIEA (10,44 г, 62,0 ммоль) и реакция шла в течение 20 мин при 0oC и 2 дня при комнатной температуре. Реакционная смесь перерастворялась в этилацетате (800 мл), экстрагировалась 200 мл порциями 0,2 N HCl (1x), насыщенным раствором NaHCO3 (1x), насыщенным раствором NaCl (2x), сушилась (MgSO4) и выпаривалась при пониженном давлении до осадка желтого цвета. Очистка производилась однократной хроматографией (силикагель; 5: 1 EtOAc : ацетонитрил), давала указанное соединение в виде бесцветного осадка (20,34 г, 85%). Tпл 78 - 85oC; DCI-MS: [M+NH4].
Часть E
- Boc-D-Lys(Tfa)-NMeArg(Tos)-Gly
Раствор Boc-D-Lys(Tfa)-NMeArg(Tos)-Gly-OBzl (11,0 г, 13,5 ммоль) в метаноле (200 мл) помещали в баллон аппарата для встряхивания, пропускали N2 в
течение 10 мин и обрабатывали катализатором 10% "палладий на угле" (10% Pd/C, 3,6 г). В процессе встряхивания баллон 7 раз подвергали процедуре нагнетания и спуска давления, избыточное давление
снимали и встряхивали еще в течение 90 мин, во время которых вычисленное количество водорода было израсходовано.
Катализатор отделяли фильтрацией через фильтр из целита, а фильтрат концентрировали при пониженном давлении, получая осадок. Растирание с дефлегмированным диэтиловым эфиром (75 мл) дает чистый продукт (9,18 г; 94%) в виде бесцветного осадка. DCI-MS: [M+H] = 724.
Часть F - Boc-D-Lys(Tta)-NMeArg(Tos)-Gly-OSu
Boc-D-Lys(Tta)-NMeArg(Tos)-Gly (8,00 г, 11,0 ммоль), HOSu (1,25 г, 10,8 ммоль) и DCC (2,22 г, 10,8 ммоль) растворяли в DMF (75 мл) и
перемешивали при комнатной температуре 2 дня. Осадок удаляли фильтрацией и промывали DMF (2 x 15 мл). Фильтрат концентрировали при пониженном давлении и полученный концентрат высушивали при пониженном
давлении при 40oC до получения осадка рыжевато-коричневого цвета (6,50 г, 72%). Tпл = 66 - 69oC; FAB-MS: [M+H] = 821.
Часть G - Boc-D-Lys(Tfa)-N-MeArg(Tos)-Gly-Asp(OBzl)-Mamb.
Суспензия Boc-D-Lys(Tfa)-N-MeArg(Tos)-Gly-OSu (8,85 г, 10,8 ммоль) и HCl • Asp(OBzl)-Mamb (4,24 г, 10,8 ммоль) в 100 мл смеси диоксан: DMF (4:1) обрабатывалась DIEA (1,39 г, 10,8 ммоль) в течение 10 мин. Полученная смесь перемешивалась в течение 2-х дней при комнатной температуре и концентрировалась при пониженном давлении до сиропообразного состояния. Этот остаток растворяли в этилацетате (300 мл) и промывали 75 мл порциями 0,2 N HCl (3x), насыщенным раствором NaHCO3 (2x), водой (1x) и насыщенным раствором NaCl (1x). Органический слой высушивали (MgSO4) и концентрировали при пониженном давлении при 40oC до получения янтарного осадка (9,13 г, 78%). Tпл. = 90-93o C; FAB-MS: [M+H]=1062.
Часть H - HCl • D-Lys(Tfa)-N-MeArg(Tos)-Gly-Asp(OBzl)-Mamb.
Boc-D-Lys(Tfa)-N-MeArg(Tos)-Gly-Asp(OBzl)-Mamb (8,30 г, 7,8 ммоль) был частично растворен в 4N HCl в диоксане (50 мл), перемешивался при комнатной температуре 30 мин, затем выпаривался при пониженном давлении до получения желтого осадка. Растирание с теплым EtOAc (60 мл) позволяло получить продукт (7,65 г, 98%) в виде осадка желтого цвета. FAB-MS: [M+H]=962.
Часть I - цикло-(D-Lys(Tfa)-N-MeArg(Tos)-Gly-Asp(OBzl)-Mamb.
HCl • D-Lys(Tfa)-N-MeArg(Tos)-Gly-Asp(OBzl)-Mamb (3,00 г, 3,00 ммоля), DIEA (0,77 г, 6,0 ммоля) и TBTU (0,98 г, 3,0 ммоля) растворяли в DMF (100 мл). Смесь перемешивалась при комнатной температуре в течение 22 часов и pH поднимался до 7-8 добавлением DIEA при необходимости. Реакционную смесь выпаривали при пониженном давлении, и получившийся остаток перерастворили в 110 мл смеси этилацетата и 1-бутанола (3,75:1). Органическую фазу промывали 50 мл порциями 0,2 N HCl (2x), насыщенным раствором NaHCO3 (1x), водой (1x), насыщенным раствором NaCl (1x), высушивали (MgSO4), концентрировали до коричневого масляничного остатка. Растирание с диэтиловым эфиром (100 мл) давало коричневый остаток, который очищали хроматографией (силикагель, 5:1 EtOAc:EtOH) до получения названного соединения (1,62 г, 57%) в виде бесцветного осадка, Tпл = 128 - 130oC; FAB-MS: [M+H]=944.
Часть J -цикло-(D-Lys(Tfa)-N-MeArg(Tos)-Gly-Asp-Mamb.
Цикло-(D-Lys(Tfa)-N-MeArg(Tos)-Gly-Asp(OBzl)-Mamb (0,85 г, 0,9 ммоль) растворяли в TFA (10 мл) и охлаждали до минус 10oC. Трифликовую кислоту (трифторметансульфокислоту 10 мл) медленно добавляли к перемешиваемой реакционной смеси до тех пор, пока температура не начинала подниматься до -5oC. Добавляли анизол (2 мл) и перемешивание продолжали 3 ч при -5o C. Если температура реакции опускалась до -78oC, добавляли диэтиловый эфир (200 мл) и реакционная смесь перемешивалась в течение 1 ч. Осадок белого цвета отделяли фильтрацией и промывали ледяным эфиром (50 мл). Осадок перерастворяли в смеси ацетона и воды (1:1) (10 мл) и лиофилизировали до получения продукта (0,63 г; 100%) в виде бесцветного осадка. FAB-MS: [M+H]=700.
Часть K -цикло-(D-Lys-N-MeArg-Gly-Asp-Mamb).
Цикло-(D-Lys(Tfa)-N-MeArg-Gly-Asp-Mamb (0,63 г, 0,9 ммоль) растворяли в 1,0 М водном пиперидине (10 мл) при 0oC и реакционной смеси позволяли медленно нагреваться до комнатной температуры в течение 3 ч. Раствор лиофилизировали и получили осадок желтого цвета. Очистка выполнялась обращеннофазовым методом ВЭЖХ на Vydac C18 колонке (21 см) с использованием 0,36%/мин градиента 9-18% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизацией получали указанное соединение (0,20 г, 90%) в виде бесцветных кристаллов. Tпл = 138 - 142oC; FAB-MS: [M+H]=604.
Жидкофазный синтез соединения 13f см. в конце описания.
Циклическое промежуточное соединение 13 r
Цикло-(D-Ile-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Ile; K - NMeArg, L - Gly; M - Asp, R1 - H, R2 - H.
Данное соединение получают, используя способ, описанный для цикло-(D-Val-NMeArd-Gly-Asp-Mamb) (циклическое соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид получали из расчета 0,611 ммоль для того, чтобы получить защищенный циклопептид (349 мг, 69,2%). Пептид (342 мг) и 342 мл анизола обрабатывали безводной HF при 0oC в течение 30 мин. Продукт далее осаждали эфиром, перерастворяли в водном ацетонитриле и лиофилизировали, чтобы получить указанное соединение (227 мг, 90% вычислено в расчете на фтористую соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ в обратной фазе на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 10,8-19,8% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизировали продукт до получения TFA-соли указанного соединения в виде белого осадка (выделено 22,5% конечный выход 12,1%); FAB-MS: [M+H]= 589,34.
Циклическое промежуточное соединение 17
Цикло-(D-Met-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Met; K - NMeArg, L - Gly, M - Asp, R1 - H, R2 - H.
Указанное соединение получают, используя способ, описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к смоле. Пептид получали из расчета 0,179 ммоль, чтобы получить защищенный циклопептид (105 мг, 69,7%). Пептид (105 мг) и 0,105 мл анизола обрабатывали безводной HF при 0oC в течение 20 мин. Полученный продукт осаждали эфиром, перерастворяли в водном ацетонитриле и лиофилизировали до получения указанного соединения (72 мг, 92,3% из расчета на фтористую соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке (2,5 см) с использованием 0,23%/мин градиента 14,4-23,4% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли указанного соединения в виде белого осадка (выделено 13,2%, конечный выход 7,4%); FAB-MS: [M+H]=607,3.
Циклическое промежуточное соединение 18
Цикло-(NMeGly-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J
- NMeGly; K - NMeArg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя основной метод, описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид получали из расчета 0,43 ммоль, с тем, чтобы получить защищенный циклопептид (205 мг, 60%). Пептид (200 мг) и 200 мл м-крезола обрабатывали безводной HF при 0oC в течение 30 мин. Полученный продукт осаждали эфиром, перерастворяли в водной HOAc и лиофилизировали, чтобы получить соединение 18 в виде бледно-желтого осадка (148 мг, 97% в расчете на ацетатную соль). Очистку заканчивали обращеннофазовым методом ВЭЖХ в обратной фазе на препаративной колонке Vydac C18 (2,5 см) с использованием 0, 23%/мин градиента 7-22% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизируют до получения трифторуксусной соли соединения (18) в виде белого осадка (выделено 14,7%, конечный выход 7,9%); FAB-MS: [M+H]= 547,34.
Циклическое промежуточное соединение 24
Цикло-(Pro-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - Pro; K - NMeArg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя основной метод, описанный выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). DCC/DMAP метод использовался для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве исходя из расчета (0,43 ммоль) для получения защищенного циклопептида (170 мг, 48,8%). Пептид (164 мг) и 164 мл м-крезола обрабатывали безводной HF при 0oC в течение 30 мин. Неочищенный продукт осаждали эфиром, растворяли в водной HOAc и лиофилизировали для получения соединения (24) в виде желтого осадка (101 мг, 79% в расчете на ацетатную соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 7-22% ацетонитрила, содержащего 0,1% TFA. Затем проводили лиофилизацию для получения TFA-соли соединения (24) в виде белого осадка (выделено 5,8%, конечный выход 2,1%); FAB-MS: [M+H]=573,46.
Циклическое
промежуточное соединение 25
Цикло-(D-Pro-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Pro; K - NMeArg, L - Gly; M - Asp, R1=R2 - H.
Метод, описанный выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). DCC/DMAP метод использовался для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,43 ммоля из расчета, чтобы получить защищенный циклопептид (211 мг, 60,8%). Пептид (200 мг) и 200 мл м-крезола обрабатывали безводной HF при 0oC в течение 30 мин. Полученный продукт осаждали эфиром, перерастворяли в HOAc, лиофилизировали для получения соединения (25) в виде желтых кристаллов (145 мг, 93% в расчете на ацетатную соль). Очистка выполнялась обращеннофазовым методом ВЭЖХ в обратной фазе на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 7-22% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли указанного соединения (25) в виде белого осадка (выделено 6,4% конечный выход 3,3%); FAB-MS: [M+H]=573,35.
Циклическое промежуточное соединение 28c
Цикло-(b-Ala-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J
- b-Ala, K - NMeArg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb).
DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,586 ммоль из расчета, чтобы получить защищенный циклопептид (264 мг, 57,5%). Пептид (258 мг)
и 258 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения в виде
желтого осадка (231 мг, больше чем количественный выход из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,
23%/мин градиента 5,4-14,4% ацетонитрила,
содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 53,2%, конечный выход
32,5%); FAB-MS: [M+H]=547,28.
Циклическое промежуточное соединение 28f
Цикло-(D-Tyr-NMeArg-Cly-Asp-Mamb); соединение формулы (II), где J - D-Tyr; K - NMeArg, L - Gly; M - Asp,
R1 - H, R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,313 ммоль из расчета, чтобы получить защищенный циклопептид (342 мг, больше чем количественный выход). Пептид (331 мг) и 0,330 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (218 мг, больше, чем количественный выход; из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ в обратной фазе на препаративной колонке Vydac C18 (2, 5 см) с использованием 0,23%/мин градиента 9-18% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали, до получения TFA-соли данного соединения в виде белого осадка (выделено 11,3%, конечный выход 10,8%); FAB-MS: [M+H]=639,54.
Циклическое промежуточное соединение 29
Цикло-(Gly-Arg-Cly-Asp-Mamb); соединение формулы (II), где J - Gly; K - Arg, L - Gly; M
- Asp, R1=R2 - H.
Указанное соединение получали на основе способа, описанного ранее для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,283 ммоль и половина его была циклизована с тем, чтобы получить защищенный циклопептид (62 мг, 58%). Пептид 60 мг и 60 мл м-крезола обрабатывали безводной HF при 0oC 60 мин. Полученный продукт осаждали эфиром, растворяли в водном HOAc и лиофилизировали для указанного соединения в виде желтого осадка (48 мг, больше чем количественный выход из расчета на ацетатную соль). Очистку производили обращеннофазовым методом ВЭЖХ в обратной фазе на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 0-9% ацетонитрила, содержащего 0,1% TFA, и затем лиофилизировали для получения TFA-соли соединения в виде белого осадка (выделено 36%, конечный выход 19,9%); FAB-MS: [M+H]=519,26.
Циклическое промежуточное соединение 30
Цикло-(D=
Ala-Arg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Ala; K - Arg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,189 ммоль из расчета, чтобы получить защищенный циклопептид (211 мг, больше чем количественный выход). Пептид (195 мг) и 195 мл м-крезола обрабатывали безводной HF при 0oC 60 мин. Полученный продукт осаждали эфиром, растворяли в водном HOAc и лиофилизировали для получения указанного соединения в виде желтого осадка (125 мг, 83% из расчета на ацетатную соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 2-11% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 12,5%, конечный выход 13,8%); FAB-MS: [M+H]=533,26.
Циклическое промежуточное соединение 31
Цикло-(Ala-Arg-Gly-Asp-Mamb); соединение формулы (II), где J - Ala; K - Arg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,324 ммоль из расчета, чтобы получить защищенный циклопептид (191 мг, 76,4%). Пептид (100 мг) и 100 мл м-крезола обрабатывали безводной HF при 0oC 1 ч. Полученный продукт осаждали эфиром, растворяли в водном HOAc и лиофилизировали для получения указанного соединения в виде желтого осадка (75 мг, 97,4% из расчета на ацетатную соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 2-11% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 15,5%, конечный выход 10,5%); FAB-MS: [M+H]=533,25.
Циклическое промежуточное соединение 32
Цикло-(D-Val-Arg-Gly-Asp-Mamb); соединение формулы (II), где J -D-Val; K - Arg, L - Gly; M - Asp,
R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,193 ммоль из расчета, чтобы получить защищенный циклопептид (199 мг, больше количественного выхода). Пептид (193 мг) и 193 мл м-крезола обрабатывали безводной HF при 0oC 1 ч. Полученный продукт осаждали эфиром, растворяли в водном HOAc и лиофилизировали для получения указанного соединения в виде желтого осадка (130 мг, 86% из расчета на ацетатную соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 2-13% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 57%, конечный выход 58,1%); FAB-MS: [M+H]= 561,22.
Циклическое промежуточное соединение 33
Цикло-(D-Leu-Arg-Gly-Asp-Mamb); соединение формулы
(II), где J - D-Leu; K - Arg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,202 ммоль из расчета, чтобы получить защищенный циклопептид (152 мг, 93%). Пептид (150 мг) и 150 мл м-крезола обрабатывали безводной HF при 0oC 1 час. Полученный продукт осаждали эфиром, растворяли в водной HOAc и лиофилизировали для получения указанного соединения в виде желтого осадка (78 мг, 66% из расчета на ацетатную соль). Очистку производили ВЭЖХ-хроматографией методом обращенных фаз на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 5-18% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 26%, конечный выход 14,8%); FAB-MS: [M+H]=575,45.
Циклическое
промежуточное соединение 34
Цикло-(D-Abu-Arg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Abu; K - Arg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получали, используя способ, описанный выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,193 ммоль с тем, чтобы получить защищенный циклопептид (210 мг, больше количественного выхода). Пептид (206 мг) и 206 мл м-крезола обрабатывали безводной HF при 0oC 1 ч. Полученный продукт осаждали эфиром, перерастворяли в водном HOAc и лиофилизировали для получения указанного соединения в виде желтого осадка (153 мг, 99% в расчете на ацетатную соль). Очистку производили методом обращеннофазовой высокоэффективной жидкостной хроматографии на препаративной Vydac C18 колонке (2,5 см) с использованием 0,23%/мин градиента 2-11% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 57%, конечный выход 72,2%); FAB-MS: [M+H]=547,21.
Циклическое промежуточное соединение 35
Цикло-(D-Ser-Arg-Gly-Asp-Mamb); соединение формулы (II), где
J - D-Ser; K - Arg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,193 ммоль из расчета, чтобы получить защищенный циклопептид (224 мг, больше количественного выхода). Пептид (210 мг) и 210 мг м-крезола обрабатывали безводной HF при 0oC 1 ч. Полученный продукт осаждали эфиром, растворяли в водной HOAc и лиофилизировали для получения указанного соединения в виде желтого осадка (145 мг, 89% из расчета на ацетатную соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 2-13% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 22% конечный выход 27%); FAB-MS: [M+H]=549,31.
Циклическое промежуточное соединение 36
Цикло-(D-Phe-Arg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Phe; K - Arg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,266 ммоль из расчета, чтобы получить защищенный циклопептид (202 мг, 90%). Пептид (157 мг) и 157 мл м-крезола обрабатывали безводной HF при 0oC 1 час. Полученный продукт осаждали эфиром, растворяли в водной HOAc и лиофилизировали для получения указанного соединения в виде желтого осадка (125 мг, > количественного выхода из расчета на ацетатную соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 7-23% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 35%, конечный выход 29,3%) FAB-MS: [M+H]= 609,25.
Циклическое промежуточное соединение 37
Цикло-(Phe-Arg-Gly-Asp-Mamb); соединение формулы (II), где J - Phe; K - Arg, L - Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Пептид был получен в количестве 0,335 ммоль из расчета, чтобы получить защищенный циклопептид (306 мг, > количественного выхода). Пептид (275 мг) и 275 мл м-крезола обрабатывали безводной HF при 0oC 1 ч. Полученный продукт осаждали эфиром, растворяли в водной HOAc и лиофилизировали для получения указанного соединения в виде желтого осадка (214 мг, 98% из расчета на ацетатную соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 9-23% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 32%, конечный выход 31,5%); FAB-MS: [M+H]=609,26.
Циклическое промежуточное соединение 40
Цикло-(D-Val-NMeAmf-Gly-Asp-Mamb); соединение формулы (II), где J - D-Val; K - NMeAmf, L
- Gly; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,586 ммоль из расчета, чтобы получить защищенный циклопептид (189 мг, 39,9%). Пептид (189 мг) и 189 мл анизола обрабатывали безводной HF при 0oC 30 час. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (212 мг, > количественного выхода из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0, 23%/мин градиента 10,8-22,5% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 8,1%, конечный выход 4,1%); FAB-MS: [M+H]=595,23.
Циклическое промежуточное соединение 48a.
Указанное соединение может быть синтезировано с помощью методов, описанных Mosher et al., Tett. Lett. 29: 3183-3186, и в соответствии со схемой I (см. в конце описания). Этот же метод часто используется для превращения первичной аминогруппы в функциональную группу гуанидина.
Циклические промежуточные соединения 42-45
Синтез циклических промежуточных соединений 42-45 показан на схеме II.
Циклические промежуточные соединения 46 и 47
Циклические промежуточные соединения 46 и 47 получают известными методами, например, описанными Garigipati, Tett. Lett. (1990) 31: 1969-197 и патенте Канады N 2008311, приведены на схеме III. Группа
аспарагиновой кислоты может быть защищена (например, фенацильной защищающей группой), чтобы избежать побочной реакции.
Циклическое промежуточное соединение 54
Цикло-(D-Val-NMeArg-b-Ala-Asp-Mamb); соединение формулы (II), где J - D-Val; K - NMeArg, L - A-Ala; M - Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,586 ммоль из расчета, чтобы получить защищенный циклопептид (227 мг, 46,9%). Пептид (219 мг) и 219 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (54) в виде желтого осадка (150 мг, 93,2 из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 7,2-16,2% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения (54) в виде белого осадка (выделено 43,6%, конечный выход 16,5%): FAB-MS: [M+H]=589,32.
Циклические промежуточные соединения 55-58
Синтез циклических промежуточных соединений
55-58 показан на схеме IV.
Циклическое промежуточное соединение 58c
Цикло-(D-Val-NMeArg-L-Ala-Asp-Mamb); соединение формулы (II), где J - D-Val; K - NMeArg, L - L-Ala; M
- Asp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,611 ммоль из расчета, чтобы получить защищенный циклопептид (375 мг, 74,6%). Пептид 360 мг и 0,360 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (220 мг, 83% из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 9-18% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 19,9%, конечный выход 10,6%); FAB-MS: [M+H] = 589,31.
Циклическое промежуточное соединение 63 и 63a
Цикло-(D-Val-NMeArg-Gly-a-MeAsp-Mamb); соединение формулы (II), где J - D-Val; K - NMeArg, L - Gly; M - a-MeAsp, R1=R2
- H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Asp-Mamb). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,794 ммоль из расчета, чтобы получить защищенный циклопептид (237 мг, 36,1%). Пептид (237 мг) и 0,237 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (165 мг, 94,3 из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 9-18% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения (54) в виде белого осадка, изомер #1 (выделено 8,36%, конечный выход 2,5%); FAB=MS: [M+H] = 589,29; изомер # 2 (выделено 9,16%, конечный выход 2,7%); FAB=MS: [M+H] = 589, 27.
Циклическое промежуточное соединение 64 и 64a
Цикло-(D-Val-NMeArg-Gly-B-MeAsp-Mamb); соединение формулы (II), где J - D-Val; K - NMeArg, L - Gly; M - B-MeAsp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,611 ммоль из расчета, чтобы получить защищенный циклопептид (201 мг, 40,0%). Пептид (200 мг) и 0,200 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (162 мг, > количественного выхода из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 9-18% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения (54) в виде белого осадка, изомер #1 (выделено 12,7%, конечный выход 4,8%); FAB=MS: [M+H] = 589,43; изомер # 2 (выделено 13,9%, конечный выход 5,3%); FAB=MS: [M+H] = 589,45.
Циклическое промежуточное соединение 64b
Цикло-(D-Val-NMeArg-Gly-MeAsp-Mamb); соединение
формулы (II), где J - D-Val; K - NMeArg, L - Gly; M - NMeAsp, R1=R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,611 ммоль из расчета, чтобы получить защищенный циклопептид (232 мг, 46,1%). Пептид (225 мг) и 0,225 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (160 мг, 96,4% из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 9-18% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 28, 2%, конечный выход 10,9%); FAB-MS: [M+H] = 589,42.
Циклическое промежуточное соединение 64c
Цикло-(D-Val-NMeArg-Gly-D-Asp-Mamb); соединение формулы (II), где J - D-Val; K
- NMeArg, L - Gly; M - D-Asp, R1 - H, R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,611 ммоль из расчета, чтобы получить защищенный циклопептид (257 мг, 51,9%). Пептид (250 мг) и 0,250 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (192 мг, > количественного выхода из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 9-18% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 44,4%, конечный выход 20,7%); FAB=MS: [M+H] = 575,42.
Циклическое промежуточное соединение 89e
Цикло-(D-Abu-di-NMeOrn-Gly-Asp-Mamb); соединение формулы (II), где J - D-Val; K - di-NMeOrn, L - Gly; M - Asp, R1=R2
- H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,498 ммоль из расчета, чтобы получить защищенный циклопептид (150 мг, 39,3%). Пептид (150 мг) и 0,150 мл анизола обрабатывали безводной HF при 0oC 30 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (93 мг, 86% из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 3,6-18% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 49,3% конечный выход 14,2%); FAB=MS: [M+H] = 533,34.
Циклическое промежуточное соединение
89f
Цикло-(D-Abu-NMeArg-Gly-D-Asp-Mamb); соединение формулы (II), где J - D-Abu; K - NMeArg, L - Gly; M - D-Asp, R1 - H, R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. Пептид был получен в количестве 0,596 ммоль из расчета, чтобы получить защищенный циклопептид (273 мг, 57,6%). Пептид (263 мг) и 0,263 мл анизола обрабатывали безводной HF при 0oC 20 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали для получения указанного соединения (218 мг, > количественного выхода из расчета на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 10,8-19,8% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 40,4% конечный выход 21,9%); FAB=MS: [M+H] = 561,37.
Циклическое промежуточное соединение
89g
Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb); соединение формулы (II), где J - D-Abu; K - NMeArg, L - Gly; M - Asp, R1 - H, R2 - H.
Указанное соединение получают, используя способ, ранее описанный для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение (4). DCC/DMAP метод использовали для присоединения Boc-Mamb к оксимовой смоле. TBTU использовался как связывающий агент. Пептид получали в пределах 0,596 ммоль, с тем чтобы получить защищенный циклопептид (241 мг, 50,8%). Пептид (235 мг) и 0,235 мл анизола обработаны безводной HF при 0oC в течение 20 мин. Полученный продукт осаждали эфиром, растворяли в водном ацетонитриле и лиофилизировали, чтобы получить указанное соединение (168 мг, 98,3% в расчете на фтористую соль). Очистку производили обращеннофазовым методом ВЭЖХ на препаративной колонке Vydac C18 (2,5 см) с использованием 0,23%/мин градиента 12,6-21,6% ацетонитрила, содержащего 0,1% TFA, и затем продукт лиофилизировали до получения TFA-соли данного соединения в виде белого осадка (выделено 2,3%, конечный выход 0,99%); FAB=MS: [M+H] = 561,36.
Циклическое промежуточное
соединение 89h
Цикло-(D-Ala-p-гуанидинил-Phe-Gly-Asp-Mamb); соединение формулы (II), где J - D-Ala; K - p-гуанидинил-Phe, L - Gly; M - B-MeAsp, R1 - H, R2 - H.

Растворили 25 мг (38,3 ммоля) цикло-(D-Ala-n-амино-Phe-Gly-Asp-Mamb) в виде TFA-соли, 14,3 мг (114,9 ммоль) формамидина сульфокислоты и 18,7 мг (153,2 ммоль) 4-диметиламинопиридина в 5 мл этанола в 10-миллилитровой круглодонной колбе. Смесь кипятили с обратным холодильником в течение 3 ч, затем дополнительно добавили 14,3 мг формамидина сульфокислоты и 18,7 мг 4-диметил-аминопиридина. После отгонки в течение еще 3 ч было установлено (с помощью обращеннофазового метода ВЭЖХ), что реакция завершалась на ≈ 75%. Этанол выпаривали при пониженном давлении, а остаток очищался на препаративной колонке Vydac C18 (2,5 см) с использованием 0,45%/мин градиента 0-18% ацетонитрила с добавкой 0,1% TFA, лиофилизация позволяла выделить TFA-соль указанного соединения как белый порошок (28% выделено; 26,4% - конечный выход); FAB=MS: [M+H] = 581,30.
Циклическое
промежуточное соединение 89i
Цикло-(D-Abu-(DiNMe, гуанидинил-Orn)-Gly-Asp-Mamb); соединение формулы (II), где J - D-Abu; K - diNMe, гуанидинил-Orn, L - Gly; M - B-MeAsp, R1 - H,
R2 - H.
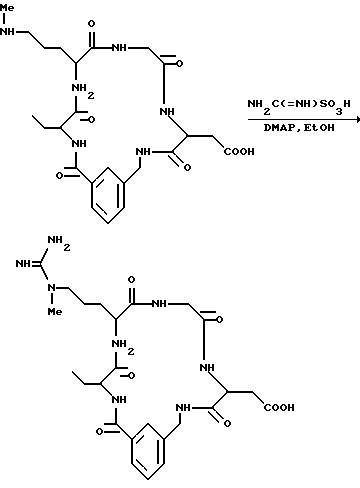
Растворили 10,53 мг (16,3 ммоля) цикло-(D-Abu-diNMeOrn-Gly-Asp-Mamb) (в виде TFA-соли), 6,08 мг (48,99 ммоль) формамидина сульфокислоты и 8,00 мг (65,57 ммоль) 4-диметиламинопиридина в 2,5 мл этанола в 10-миллилитровой круглодонной колбе. Смесь кипятили с обратным холодильником 2 ч и затем в течение ночи перемешивали при комнатной температуре. Кипятили еще 1 ч и дополнительно добавили 6,08 мг формамидина сульфокислоты и 8,00 мг 4-диметил-аминопиридина, еще кипятили 2 ч. Этанол выпаривали при пониженном давлении и очищали остаток на препаративной Vydac C18 колонке (2,5 см) с использованием 0,45%/мин градиента 3,6-18% ацетонитрила с добавкой 0,1% TFA. Лиофилизация позволяла выделить TFA-соль указанного соединения как осадок белого цвета (выделено 57,2%; конечный выход - 53,5%); FAB=MS: [M+H] = 575,34.
Циклическое промежуточное соединение 89j
Цикло-(D-Abu-(Di-NMeLys-Gly-Asp-Mamb) соединение формулы (II), где J - D-Abu; K - di-NMeLys, L - Gly; M - Asp, R1 - H, R2 - H.
цикло-(D-Abu-NMeLys-Gly-Asp-Mamb) соединение формулы (II), где J - D-Abu; K - NMeLys, L - Gly; M - Asp, R1 - H, R2 - H.
Di-N-метильное производное аминокислоты может быть получено на основе методов, описанных ранее: (Olsen, J. Org. Chem. (1970 35:1912) или, с другой стороны, в результате использования NaH/CH3 I. Моно-NMe-лизин аминокислота была получена как побочный продукт в процессе синтеза соответствующего di-NMe-производного лизина. Указанные вещества получали с помощью установившихся правил и методик жидкофазной пептидной химии, описанных ранее.
Цикло-(D-Abu-diNMeLys-Gly-Asp-Mamb) был получен с 0,31% выходом, FAB=MS: [M+H] = 547,3.
Цикло-(D-Abu-NMeLys-Gly-Asp-Mamb) был получен при 0,25% конечным выходом, FAB=MS: [M+H] = 533,3.
Циклическое промежуточное соединение 90
Цикло-(D-Val-NMeArg-Gly-Asp-2-аминометилфенилацетоуксусная кислота).
Указанное соединение получали модифицированным обычным жидкофазным химическим способом. Этот подход предусматривает связывание сукцинимидного эфира аминокислоты с ароматическим циклическим фрагментом и динитробензофенон оксимом, как показано на схеме V (n = 1).
Boc-Asp(OcHex)-2-аминометилфенилуксусная кислота
К суспензии 2-аминометилфенилуксусной кислоты • HCl (4,0 г, 20 ммоль) в воде (20 мл) добавляли NaHCO3 (5,0 г, 60 ммоль),
затем раствор Boc-Asp(OcHex)-OS4 (7,5 г, 18 ммоль) в THF (20 мл). Реакционную смесь перемешивали в течение 3 часов, фильтровали, разбавляли водой, подкисляли 1 н HCl, экстрагировали
этилацетатом. Экстракты промывали водой, раствором соли, высушивали над MgSO4 и выпаривали при пониженном давлении. Этот продукт растирали с эфиром и получали вышеназванное соединение (7,0
г, 83%) в виде белого порошка.
1H NMR (D6-DMSO) 12,40 (br s, 1H), 8,30 (br t, 1H), 7,20 (m, 5H), 4,65 (m, 1H), 4,35 (q, 1H), 4,25 (m, 2H), 3,65 (s, 2H), 2,70 (dd, 1H), 2,55 (dd, 1H), 1,70 (m, 4H), 1,40 (s, 9H), 1,35 (m, 6H).
4,4'-Динитробензофеноноксим
Указанное соединение получают модификацией метода, ранее описанного в литературе:
Chapman and Fidler (1936) J. Chem. Soc, 448; Kulin and Leffek (1973) Can. J. Chem. 51: 687). Раствор хромового ангидрида (20 мг, 200 ммоль) в 125 мл воды добавляли по каплям в течение 4 ч к суспензии
бис-(4-нитрофенил)метана (25 г, 97 ммоль) в 300 мл уксусной кислоты, кипятящейся с отгонкой. Реакционную смесь кипятили с обратным холодильником 1 ч, охлаждали до комнатной температуры и выливали в
воду. Твердый осадок отделяли фильтрацией, промывали водой, 5% раствором бикарбоната соды, водой, высушивали на воздухе и получали смесь (1:1) бис(4-нитрофенил)метана и 4,4'-динитробензофенона с1H NMR. Этот продукт окисляли второй порцией хромового ангидрида (20 г, 200 ммоль), затем проводили те же процедуры до получения неочищенного продукта. Растирание с 200 мл бензола, который
кипятили 16 час с обратным холодильником, приводило к получению 4,4'-динитробензофенона (20,8 г, 79%) в виде желтого порошка. Раствор гидрохлорида гидроксиламина (10,2 г, 147 ммоль) добавляли к
суспензии 4,4'-динитробензофенона (19 г, 70 ммоль) в 100 мл этанола. Реакционную смесь нагревали до кипячения с обратным холодильником в течение 2 часов, охлаждали при комнатной температуре и
образовавшийся осадок отфильтровывали. Перекристаллизацией из этанола получали указанное соединение (14,0 г, 70%) в виде желтых кристаллов. Tпл. = 194oC;1H NMR (D6-DMSO) 12,25 (s, 1H), 8,35 (d, 2H), 8,20 (d, 2H), 7,60 (d, 4H).
4,4'-Динитробензофенон оксим Boc-Asp(OcHex)-2- аминометилфенилацетат
К охлажденному льдом раствору
Boc-Asp(OcHex)-2- аминометилфенилуксусной кислоты (3,5 г, 7,6 ммоль) и 4,4'-динитробензофенон оксим (2,2 г; 7,5 ммоль) в 50 мл этилацетата и 5 мл DMF добавляли DCC (1,6 г, 7,8 ммоль). Реакционную
смесь перемешивали при комнатной температуре в течение 8 ч, фильтровали, разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия, водой, солевым раствором, высушивали над безводным
MgSO4, выпаривали до абсолютно сухого состояния при пониженном давлении. Полученный продукт очищали колоночной хроматографией на силикагеле (EM Science, 230-400 меш), используя 10: 1
дихлорметан/этилацетат, до получения указанного соединения (4,3 г, 78%) в виде желтых кристаллов.
1H NMR (D6-DMSO) 8,30 (dd, 5H), 7,80 (d, 2H), 7,65 (d, 2H), 7,15 (m, 5H), 4,65 (m, 1H), 4,35 (q, 1H), 4,15 (m, 2H), 3,90 (s, 2H), 2,70 (dd, 1H), 2,50 (dd, 1H), 1,70 (m, 4H), 1,40 (s, 9H), 1,35 (m, 6H).
4,4'-Динитробензофенон оксим
Boc-D-Val-NMeArg(Tos)-Gly- Asp(OcHex)-2-аминометилфенилацетат
К раствору 4,4-динитробензофенон оксим Boc-Asp(OcHex)-2-аминометилфенилацетат (1,5 г, 2 ммоль) в 4 мл дихлорметана было добавлено
2 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, разбавили дихлорметаном, выпаривали до сухого состояния при пониженном давлении. Маслянистый
осадок концентрировали в сильном вакууме для удаления следов избытка трифторуксусной кислоты.
К раствору неочищенной TFA-соли и Boc-D-Val-NMeArg(Tos)-Gly (1,2 г, 1 ммоля) в 5 мл DMF добавили TBTU (640 мг, 2 ммоля) и DIEA (780 мг, 6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, концентрировали в сильном вакууме, разбавили этилацетатом, промыли 5% раствором лимонной кислоты, H2O солевым раствором, высушили над безводным сульфатом магния и выпаривали до сухого состояния при пониженном давлении. Полученный продукт растирали с эфиром и получили указанное соединение (2,3 г, 95%) в виде желтого порошка. Полученный продукт использовался без дальнейшей очистки.
Цикло-(D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-2-аминометилфенилуксусная кислота)
К раствору 4,4'-динитробензофенон оксим Boc-D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-2-аминометилфенилацетата (1,2 г, 1
ммоль) в 4 мл дихлорметана добавили 2 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, разбавили дихлорметаном и высушивали выпариванием при
пониженном давлении. Маслянистый осадок концентрировали в глубоком вакууме для удаления следов избытка трифторуксусной кислоты.
К раствору неочищенной TFA-соли в 100 мл DMF добавляли уксусную кислоту (0,50 мл, 8,7 ммоль) и DIEA (1,52 мл, 8,7 ммоль). Реакционную смесь перемешивали при 60oC в течение 3 дней, концентрировали в глубоком вакууме, разбавили этилацетатом и раствор подвергли кристаллизации в течение ночи. В результате фильтрации получили целевое соединение (563 мг, 68%) в виде желтого порошка.
1H NMR (D6-DMSO) 8,70 (d, 1H), 8,40 (br s, 1H), 8,30 (br s, 1H), 8,05 (t, 1H), 7,65 (d, 2H), 7,25 (d, 2H), 7,20 (m, 4H), 7,10 (br d, 1H), 6,80 (br s, 1H), 6,60 (br s, 1H), 5,10 (dd, 1H), 4,65 (m, 1H), 4,55 (m, 1H), 4,40 (m, 2H), 3,85 (m, 2H), 3,65 (d, 1H), 3,45 (m, 2H), 3,05 (m, 2H), 2,80 (s, 3H), 2,80 (m, 1H), 2,60 (dd, 1H), 2,30 (s, 3H), 1,70 (m, 6H), 1,30 (m, 9H), 0,95 (d, 3H), 0,80 (d, 3H); DCl-(NH3)-MS: [M+H]=825.
Цикло-(D-Val-NMeArg-Gly-Asp-2-аминометилфенилуксусная кислота)
Смесь 352 мг (0,43 ммоль) цикло-(D-Val-NMeArg(Tos)-Gly-Asp(OcHex)-2-аминометилфенилуксусной кислоты
и 352 мкл анизола обрабатывали при 0oC 5 мл HF в течение 20 мин. Избыток HF удаляли при пониженном давлении, остаток растирали с эфиром, растворяли в (50%) ацетонитрил/вода и
лиофилизировали до получения неочищенной HF-соли циклопептида в виде белого порошка.
Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке (2,5 см) с
использованием 0,8% град/мин 10-38% ацетонитрила, содержащего 0,1% трифторуксусной кислоты до получения TFA-соли указанного соединения (225 мг, 75%) в виде белого осадка;
1H NMR
(D6-DMSO) 8,70 (d, 1H), 8,35 (d, 1H), 8,20 (t, 1H), 8,00 (t, 1H), 7,45 (t, 1H), 7,20 (m, 3H), 7,10 (m, 1H), 7,00 (br s, 1H), 5,10 (dd, 1H), 4,50 (dt, 1H), 4,40 (m, 2H), 3,85 (dt, 2H), 3,65
(d, 1H), 3,50 (dd, 1H), 3,45 (d, 1H), 3,10 (m, 2H), 2,90 (s, 3H), 2,75 (dd, 1H), 2,55 (dd, 1H), 2,00 (m, 1H), 1,85 (m, 1H), 1,65 (m, 1H), 1,30 (m, 2H), 0,95 (d, 3H), 0,95 (d, 3H), 0,8 (d, 3H); FAB-MS:
[M+H]=589.
Циклическое промежуточное соединение 91
Цикло-(D-Val-NMeArg-Gly-Asp-2-аминометилбензойная кислота)
Указанное соединение получали основным
жидкофазным
способом, описанным выше для цикло-(D-Val-NMeArg-Gly-Asp-2- аминометилфенилуксусной кислоты), и как было показано на ранее приведенной схеме для циклического промежуточного соединения 90 (n=0).
Циклический пептид (192 мг, 0,24 ммоля) был подвергнут снятию "защиты" избытком HF в присутствии анизола. Очистка выполнялась обращеннофазовым методом ВЭЖХ на препаративной Vydas C18 колонке (2,5 см)
с использованием 0,8% град/мин. 10-38% ацетонитрила, содержащего 0,1% трифторуксусной кислоты до получения TFA-соли указанного соединения (20 мг, 12%) в виде белого осадка;
1H NMR
(D6-DMSO) 8,75 (d, 1H), 8,50 (d, 1H), 7,65 (t, 1H), 7,60 (t, 1H), 7,50 (m, 2H), 7,40 (m, 3H), 7,00 (br s, 4H), 5,05 (dd, 1H), 4,50 (t, 1H), 4,30 (m, 2H), 4,10 (dd, 1H), 3,70 (m, 2H), 3,15
(q, 1H), 1,95 (m, 1H), 1,60 (m, 1H), 1,40 (m, 2H), 1,05 (d, 3H), 0,95 (d, 3H), 0,95 (d, 3H); FAB-MS: [M+H]=575.
Циклическое промежуточное соединение 92
Цикло-(D-Val-NMeArg-Gly-Asp-3-аминометилбензойная кислота)
Указанное соединение получали основным жидкофазным методом, описанным выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), и как схематически
показано на схеме ниже. Циклический пептид (360 мг, 0,44 ммоль) подвергался снятию "защиты" избытком HF в присутствии анизола. Очистку проводили обращеннофазовым методом ВЭЖХ на препаративной
LiChrosher RP-18 колонке (5 см) с использованием градиента 2,3%/мин 22-90% ацетонитрила, содержащего 0,1% трифторуксусной кислоты до получения TFA-соли указанного соединения (150 мг, 50%) в виде
белого осадка;
1H NMR (D6-DMSO) 12,40 (br s, 1H), 8,95 (s, 1H), 8,55 (m, 1H), 8,45 (t, 1H), 7,90 (d, 1H), 7,50 (m, 1H), 7,20 (t, 1H), 7,00 (br, s, 4H), 6,90 (m, 2H), 5,15
(dd, 1H), 4,65 (q, 1H), 4,55 (t, 1H), 3,65 (m, 2H), 3,60 (dd, 1H), 3,10 (m, 2H), 2,85 (s, 3H), 2,85 (d, 1H), 2,70 (dd, 2H), 2,00 (m, 2H), 1,75 (m, 1H), 1,35 (m, 2H), 0,90 (d, 3H), 0,85 (d, 3H);
FAB-MS: [M+H]=575.
Циклическое промежуточное соединение 87, 88
Цикло-(D-Val-NMeArg-Gly-Asp-4-аминометилбензойная кислота); соединение формулы (II), где J - D-Val; K - NMeArg,
L - Gly; M - Asp, R1 - H, R2 - H.
Указанное соединение получали, используя основной метод, описанный выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). DCC/DMAP метод
использовался для присоединения Boc-4-аминометилбензойной кислоты к оксимовой смоле. Пептид получали в пределах 0,43 ммоль с тем, чтобы получить защищенный циклопептид (212 мг, 60,8%). Пептид (200 мг)
и 0,200 мл м-крезола обрабатывали безводной HF при 0oC в течение 30 мин. Неочищенный продукт осаждали эфиром, растворяли в водной HOAc и лиофилизировали до образования пептида в виде
желтого осадка (152 мг, 97% из расчета на ацетатную соль). Очистку выполняли обращеннофазовым методом ВЭЖХ на препаративной Vydac C18 колонке (2,5 см) с использованием градиента 0,23%/мин 7-22%
ацетонитрила, содержащего 0,1% TFA. Двумя пиками были выделены два изомера #1 (87) (17,1% - выделено, конечный выход 9,3%) и # 2 (88) (13,4% - выделено, конечный выход 7,3%); FAB-MS: [M+H]= 575,41
(изомер #1, 87); 575,44 (изомер #2, 88)
R1 или R2 - замещенные фрагменты
Циклические промежуточные соединения, которые имеют заместители R1 или R2, синтезируются на основе соответствующих замещенных циклических фрагментов.
Нижеследующие схемы, пояснения и примеры раскрывают получение этого класса циклических фрагментов и соответствующих циклических промежуточных соединений.
t-Бутилоксикарбонил-N-метил-3-аминометилбензойная кислота (Boc-NMeMamb)
Указанное соединение получают в соответствии со
стандартным способом, например, как описано Olsen, J. Org. Chem (1970) 35:1912) и как показано схематически ниже:
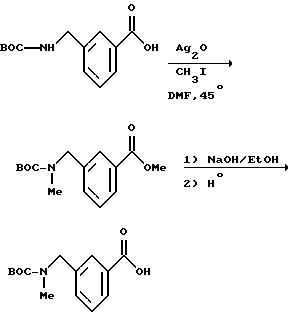
Синтез аналогов аминометилбензойной кислоты
Циклические фрагменты, соответствующие нижеприведенной формуле, могут быть получены обычными стандартными методами, например, как показано в нижеследующей схеме
При R-CH3, CH2 CH3, CH2CH2CH3, CH2CH2CH2CH3, CH(CH3)2, C(CH3)3, CH(CH3 )CH2CH3, бензил, циклопентил, циклогексил; см. схему 1.
при R = CH3, CH2CH2CH2CH3, фенил, см. схему 2,
при R = CH3, фенил, см. схемы 3 и 4.
3-[1'-(трет-бутилоксикарбонил)амино]этилбензойная кислота (Boc-MeMamb)
Данное соединение получают для целей настоящего
изобретения в соответствии с вышеприведенной схемой 4.
3-Ацетилбензойную кислоту (0,50 г, 3 ммоль), гидроксиламин гидрохлорид (0,70 г, 10 ммоль) и пиридин (0,70 мл, 9 ммоль) нагревают с обратным холодильником в течение 2 ч в 10 мл этанола. Реакционную смесь концентрируют, полученный продукт растирают с водой, фильтруют и высушивают. Оксим выделяют в виде белого твердого вещества (0, 51 г, выход 94,4%).
Данные1H ЯМР спектроскопии (CD3OD): 7,45-8,30 (m, 4H), 2,3 (s, 3H). Масс-спектр (CH4-Cl): [M+H-O]=164.
Раствор оксима (0,51 г, 3 ммоль) в этаноле, содержащий 10% Pd, насыщенного на углерод (1,5 г), концентрируют HCl (0,25 мл, 3 ммоль), гидрируют при давлении водорода 30 фунт/кв.дюйм в автоклаве Парра в течение 5 ч. Катализатор отфильтровывают, фильтрат концентрируют. Остаток растирают с эфиром. Амин гидрохлорид выделяют в виде белого твердого вещества (0,48 г, выход 85,7%). Спектр1H ЯМР (CD3OD): 7,68-8,15 (m, 4H), 4,55 (q, 1H), 1,70 (s, 3H). Масс-спектр: [M+H]=166.
Амин гидрохлорид (0,40 г, 2 ммоль) растворяют в 15 мл воды. Добавляют раствор BOC-ON (0,52 г, 2,1 ммоль) в 15 мл ацетона, а затем - триэтиламин (0,8 мл, 0,6 ммоль). Реакция протекает в течение 20 ч. Реакционную смесь концентрируют, разделяют между этилацетатом и водой. Водный слой подкисляют до pH 2, используя 10% раствором HCl. Продукт экстрагируют этилацетатом. После выделения по обычным методикам и перекристаллизации из смеси этилацетат/гексан получают BOC-MeMamb в виде белого твердого вещества (0,30 г, выход 57%). Температура плавления 116-118oC. Данные спектроскопии1H ЯМР (CDCl3): 7,35-8,2 (m, 4H), 4,6 (bs, 1,5H), 1,50 (d, 3H), 1,40 (s, 9H). Масс-спектр (NH3-Cl): [M+NH4]=283.
3-[1'-(трет. бутилоксикарбонил)амино] этилбензилбензойная кислота (BOC-PhMamb)
Данное соединение получают для целей
настоящего изобретения в соответствии с вышеприведенной схемой 4 по методике, аналогичной методике получения метильного производного.
Раствор 3-бензоилбензойной кислоты (2,00 г, 9 ммоль), гидроксиламина гидрохлорида (2,00 г, 29 ммоль) и пиридина (2,00 мл, 25 ммоль) в этаноле нагревают с обратным холодильником в течение 12 ч. После выделения обычными методами получают белое твердое вещество (2,41 г). Этот продукт содержит следы пиридина, но используется на последующих стадиях без дополнительной очистки.
Полученный продукт (без дополнительной очистки) (2, 00 г, ≈ 8 ммоль) растворяют в 200 мл этанола. Добавляют 10% Pd-C (2,00 г) и концентрируют HCl (1,3 мл, 16 ммоль). Реакционную смесь гидрируют при давлении 30 фунт/кв. дюйм в течение 1 час. Катализатор отфильтровывают, реакционную смесь концентрируют. После растирания с эфиром и сушки в вакууме получают амин гидрохлорид в виде белого твердого вещества (2,12 г, выход 97%). Данные спектроскопии ЯМР1H (CD3OD): 7,4-8,15 (m, 10H), 5,75 (s, 1H). Масс-спектр (CH4-Cl): [M+H-OH]=211.
Амин гидрохлорид (1,00 г, 4 ммоль) переводят в его BOC-производное по методике, аналогичной методике получения метильного производного. Получают 0,60 г (выход 48%) перекристаллизованного из смеси этанол/гексан вышеуказанного соединения BOC-PhMamb в виде белого твердого вещества. Tпл 190-192oC. Данные спектроскопии ЯМР1H (CD3OD): 7,2-8,0 (m, 10H), 5,90 (2s, 1H, 2 изомера), 1,40 (s, 9H). Масс-спектр (NH3-Cl): [M+NH4-C4H8]=289.
Циклические промежуточные соединения 68 и 68a
Цикло-(D-Val-NMeArg-Gly-Asp-MeMamb); соединение формулы
(II), где J - D-Val, K - NMeArg, L - Gly, M - Asp, R1 - CH3, R2 - H.
Содержащие MeMamb циклизующиеся фрагменты получают в соответствии со схемой 4 (приведена ранее). Указанные в заглавии соединения синтезируют методом присоединения MeMamb к трипептиду в растворе. После циклизации получают защищенный циклический пептид. Снятие защиты осуществляют обработкой пептида (390 мг) и анизола (0,390 мл) безводным HF при 0oC в течение 30 мин. Продукт осаждают эфиром, перерастворяют в 10% водном растворе уксусной кислоты и лиофилизуют. Получают смесь двух изомеров (330 мг, выход более чем количественный, вычислено в пересчете на соль уксусной кислоты). Очистку и разделение изомеров завершают обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 7-23% ацетонитрил, содержащий 0,1% TFA, градиент 0,48% мин. Фракции, собранные при Rf = 24,1 мин и Rf = 26,8 мин, лиофилизуют и получают соли трифторуксусной кислоты изомеров 1 и 2, соответственно. Масс-спектр (FAB), изомер 1: [M+H]=589,31; масс-спектр (FAB), изомер 2: [M+H]=589,31.
Циклические промежуточные соединения
76 и 76a
Цикло-(D-Val-NMeArg-Gly-Asp-PhMamb); соединение формулы (II), где J - D-Val, K - NMeArg, L - Gly, M - Asp, R1 - Ph, R2 - H.
Содержание PhMamb циклизующиеся фрагменты получают в соответствии со схемой 4 (описана ранее). Указанное в заглавии соединение синтезируют методом присоединения PhMamb к трипептиду в растворе. После циклизации получают защищенный циклический пептид. Снятие защиты осуществляют обработкой пептида (470 мг) и анизола (0,470 мл) безводным HF при 0oC в течение 30 мин. Продукт осаждают эфиром, перерастворяют в 10% водном растворе уксусной кислоты и лиофилизуют. Получают смесь двух изомеров (310 мг, суммарный выход 82,4%). Очистку и разделение изомеров завершают обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 18-36% ацетонитрил, содержащий 0,1% TFA, градиент 0,55%/мин. Фракции, собранные при Rf = 22 мин и Rf - 24,6 мин, лиофилизуют и получают соли трифторуксусной кислоты изомеров 1 и 2, соответственно. Масс-спектр (FAB), изомер 1: [M+H]=651,33; масс-спектр (FAB), изомер 2: [M+H]=651,33.
Циклическое промежуточное соединение
79
Цикло-(D-Val-NMeArg-Gly-Asp-NMeMamb); соединение формулы (II), где J - D-Val, K - NMeArg, L - Gly, M - Asp, R1 - H, R2 - H3.
Указанное в заглавии соединение получают по общей методике, описанной для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (промежуточное соединение 4). Для введения группировки Boc-NMeArg в смолоподобный оксим используют метод DCC/DMAP. Из пептида, взятого в количестве 0,456 ммоль, получают защищенный циклический пептид (406 мг, выход более количественного). Пептид (364 мг) и 0,364 мл анизола обрабатывают безводным фтористым водородом при 0oC в течение 30 мин. Продукт осаждают эфиром, перерастворяют в водном ацетонитриле, лиофилизуют и получают указанное в заглавии соединение (251 мг, 93,5%, вычислено в расчете на фторид). Очистку завершают обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 9-18% ацетонитрил, содержащий 0,1% TFA, градиент 0,23%/мин. После выделения лиофилизацией получают соль трифторуксусной кислоты указанного в заглавии соединения в виде пушистого белого твердого вещества (выход 34,2%, суммарный выход 29,9%). Масс-спектр (FAB), [M+H]=589,33.
Циклические фрагменты, содержащие замещенное радикалом R31 ароматическое ядро.
Циклические фрагменты, включающие ароматическое ядро, содержащее заместители, могут быть получены с использованием методов, которые иллюстрируют нижеследующие примеры и схемы.
Синтез 4,5 и 6-замещенной аминометилбензойной кислоты • HCl и 4,5 и
6-замещенных производных трет-бутилоксикарбонил-3-аминоэтилбензойной кислоты
4,5 и 6-Замещенную 3-аминоэтилбензойную кислоту • HCl и 4,5 и 6-замещенные производные
бутилоксикарбонил-3-аминоэтилбензойной кислоты, применяемые как промежуточные соединения для синтеза заявленных в настоящем изобретении соединений, получают по известным методикам, например, как
описано в Felder et. al, Helv. Chim. Acta, 48: 259 (1965); de Diesbach Helv. Chim. Acta, 23: 1232 (1949); Truitt and Creagn J. Org.Chem., 27: 1066 (1962); или Sekiya et al. Chem. Pharm. Bull., 11: 551
(1963) и как схематически показано ниже
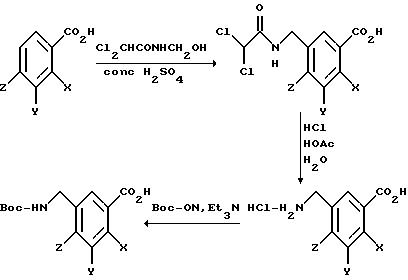
Синтез 4-хлор-3-аминометилбензойной кислоты • HCl
Указанное в заглавии соединение получают в соответствии с модификацией ранее описанной в литературе методики (Felder et al. Helv. Chim. Acta, 48: 259). К раствору 4-хлорбензойной кислоты (15,7 г, 100 ммоль) в 150 мл концентрированной серной кислоты постепенно добавляют N-гидроксиметилдихлорацетамид (23,7 г, 150 ммоль). Реакционную смесь перемешивают в течение 2 дней при комнатной температуре, охлаждают 375 г льда, перемешивают 1 ч, осадок отделяют фильтрацией и промывают водой. Влажный осадок растворяют в 5% растворе бикарбоната натрия, фильтруют и подкисляют до pH 1 концентрированной HCl. Осадок отфильтровывают, промывают водой и после сушки на воздухе получают 4-хлор-3-дихлорацетиламинометилбензойную кислоту (26,2 г, 89%) в виде белого порошка.
Суспензию 4-хлор-3-дихлорацетиламинометилбензойной кислоты (26,2 г, 88 ммоль) в 45 мл уксусной кислоты, 150 мл концентрированной HCl и 150 мл H2O нагревают с обратным холодильником в течение 3 ч, фильтруют в горячем состоянии и дают остыть до комнатной температуры. Осадок отделяют фильтрацией, промывают эфиром и затем смесью ацетона с эфиром. После сушки на воздухе в течение ночи получают указанное соединение (7,6 г, 39%) в виде беловатых кристаллов. Tпл = 278-279oC. Спектр ЯМР1H (D6 - DMSO): 13,40 (br s, 1H), 8,75 (br s, 3H), 8,20 (s, 1H), 7,95 (dd, 1H), 7,70 (d, 1H), 4,20 (br s, 2H).
Трет-бутилоксикарбонил-4-хлор-3-аминометилбензойная кислота
Суспензию 4-хлор-3-аминометилбензойной кислоты • HCl
(6,7 г, 30 ммоль) и триэтиламина (9,3 г, 92 ммоль) в 50 мл H2O добавляют к раствору Boc-ON (9,2 г, 38 ммоль) в 50 мл тетрагидрофурана, охлажденному до 0oC. Реакционную смесь
перемешивают при комнатной температуре в течение ночи, затем удаляют летучие вакуумным концентрированием. Остаток растворяют в воде, промывают эфиром, подкисляют до pH 3 1N HCl и экстрагируют
этилацетатом. Экстракт промывают водой, упаривают, сушат над безводным сульфатом магния в течение ночи и упаривают до сухого остатка при пониженном давлении. Полученный продукт растирают со смесью
эфира и гексана и получают указанное в заглавии соединение в виде белого твердого вещества (7,4 г, выход 87%). Tпл= 159oC (с разл.).
Спектр ЯМР1H (D6 - DMSO): 13,20 (br s, 1H), 7,90 (s, 1H), 7,80 (dd, 1H), 7,60 (br s, H), 7,55 (d, 1H), 4,20 (br d, 2H), 1,40 (s, 9H).
Синтез 3-аминометил-6-йодобензойная кислота •
HCl
Указанное соединение получают, используя модификацию ранее описанной в литературе методики (Felder et al. (1965) Helv, Chim. Acta, 48: 259). К раствору 6-йодобензойной кислоты (24,8 г,
100 ммоль) в 150 мл концентрированной серной кислоты постепенно добавляют N-гидроксиметилдихлорацетамид, реакционную смесь перемешивают при комнатной температуре в течение 7 дней, охлаждают 375 г льда,
перемешивают 1 ч. Осадок отделяют фильтрованием и промывают водой. Влажный осадок растворяют в 5% растворе бикарбоната натрия, фильтруют и подкисляют до pH 1 концентрированной HCl. Осадок отделяют
фильтрованием, промывают водой, сушат на воздухе в течение ночи. Получают 3-дихлорацетил-аминометил-6-йодобензойную кислоту (32,0 г, 82%) в виде белого порошка.
Суспензию 3-дихлорацетиламиноэтил-6-бензойной кислоты (32,0 г, 82 ммоль) в 51 мл уксусной кислоты, 170 мл концентрированной HCl и 125 мл H2O нагревают с обратным холодильником в течение 3 ч, фильтруют в горячем состоянии и дают остыть до комнатной температуры. Осадок отделяют фильтрованием, промывают эфиром, затем смесью ацетона с эфиром. После сушки на воздухе в течение ночи получают указанное в заглавии соединение (13,2 г, 51%) в виде бежевого порошка. Спектр ЯМР1H в D6 - DMSO: 13,50 (br s, 1H), 8,50 (br s, 3H), 8,05 (d, 1H), 7,85 (s, 1H), 7,40 (d, 1H), 4, 05 (br s, 3H).
Трет-бутилоксикарбонил-3-аминоэтил-6-йодобензойная кислота
Суспензию 3-аминоэтил-6-йодобензойной кислоты • HCl (8,0 г, 26 ммоль) и триэтиламина (8,7 г,
86 ммоль) в 32 мл H2O добавляют к раствору Boc-ON (8,0 г, 32 ммоль) в 23 мл тетрагидрофурана, охлажденному до 0oC. Реакционную смесь перемешивают при комнатной температуре в
течение ночи, удаляют летучие соединения вакуумным концентрированием. Остаток растворяют в воде, промывают эфиром и подкисляют до pH 3 1N HCl, экстрагируют этилацетатом. Экстракт промывают водой,
упаривают, сушат над безводным сульфатом магния и упаривают до сухого остатка при пониженном давлении. Полученный продукт растирают с эфиром, получают указанное соединение (5,7 г, 59%) в виде белого
порошка. Tпл= 182oC (с разл.). Спектр ЯМР1H в D6 - DMCO: 13,35 (br s, 1H), 7,95 (d, 1H), 7,60 (s, 1H), 7,50 (br, 1H), 7,10 (d, 1H), 4,10 (d, 2H), 1,40 (s,
9H).
Другие примеры циклических соединений, содержащих замещенное радикалом R31 ароматическое ядро, полученных по общей методике, описанной выше для трет-бутилоксикарбонил-3-аминометил-6-йодобензойной кислоты, сведены в таблицу I.
4-Бромо- и 6-бромопроизводные, применяемые как промежуточные соединения для получения заявленных в настоящем изобретении веществ, могут быть получены по методике, аналогичной приведенной выше методике получения трет-бутилоксикарбонил-3-аминоэтил-6-йодобензойной кислоты. 4-Гидрокси и 6-гидроксипроизводные, пригодные как промежуточные соединения для синтеза веществ настоящего изобретения, могут быть получены как описано в: Sekiya et al., Chem, Pharm, Bull., 11: 551 (1963). 5-Нитро и 5-аминопроизводные, пригодные как промежуточные соединения для синтеза веществ настоящего изобретения, могут быть получены, как описано в: Felder et al., Helv. Chim. Acta, 48: 259 (1965). Аминопроизводные могут быть переведены в 5-йодо, 5-бромо-, 5-хлоро- или 5-фторопроизводные через соль диазония, как описано в Org. Syn. Coll. Vol. 2: 130 (1943); 2: 299 (1943); 2: 351 (1943) и 3: 185 (1955) (см. табл. II).
Синтез циклических промежуточных соединений с использованием циклических фрагментов, содержащих замещенное радикалом R31 ароматическое ядро.
Циклические промежуточные соединения, в которых циклический фрагмент содержит ароматическое ядро, несущее заместители, могут быть получены как показано в следующих примерах.
Циклическое промежуточное соединение 93
Цикло-(D-Val-NMeArg-Gly-Asp-3-аминометил-4-хлорбензойная кислота)
Указанное в заглавии соединение получают по общей методике синтеза в
растворе, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Защиту циклического пептида (240 мг, 0,28 ммоль) снимают действием избытка HF в присутствии анизола в качестве отнимающего агента.
Очистку завершают обращеннофазной ВЭЖХ на препаративной колонке LiChrospher RP-18 (5 см), используя 22-90% ацетонитрил, содержащий 0,1% трифторуксусной кислоты (градиент 1,4%/мин), и получают соль
трифторуксусной кислоты указанного в заглавии соединения (80 мг, 39%) в виде пушистого белого вещества.
Спектр ЯМР1H в D6 - DMSO: 9,00 (d, 1H), 8,45 (t, 1H), 7, 60 (d, 2H), 7,45 (s, 1H), 7,45 (d, 2H), 7,00 (br s, 4H), 5,15 (dd, 1H), 4,45 (m, 2H), 4,20 (m, 2H), 4,10 (d, 1H), 3,55 (d, 1H), 3,10 (m, 2H), 2,90 (s, 3H), 2,65 (dd, 1H), 2,50 (m, 1H), 2,05 (m, 2H), 1, 50 (m, 1H), 1,30 (m, 2H), 1,05 (d, 3H), 0,85 (d, 3H).
Масс-спектр (FAB): [M+H]=609,00.
Циклическое промежуточное соединение 94
Цикло-(D-Val-NMeArg-Gly-Asp-йодо-Mamb): соединение формулы (VII), где J - D-Val, K - NMeArg, L - Gly, M - Asp, R1=R2 - H, R10 - H, R10a - I.
Указанное в заглавии соединение получают по общей методике, описанной для синтеза цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (промежуточное соединение 4). Для введения группы Boc-йодо-Mamb в смолоподобный оксим используют метод DCC/DMAP. Из пептида, взятого в количестве 1,05 ммоль, получают защищенный циклический пептид (460 мг, 46,8%). Пептид (438 мг) и 0,5 мл анизола обрабатывают безводным HF при 0oC в течение 30 мин. Продукт осаждают эфиром, перерастворяют в водной уксусной кислоте, лиофилизуют. Получают указанное в заглавии соединение (340 мг, 95,6%, вычислено в расчете на уксуснокислую соль). Очистку завершают обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 12-22,5% ацетонитрил, содержащий 0,1% TFA (градиент 0,23%/мин).
После выделения лиофилизацией получают соль трифторуксусной кислоты указанного в заглавии соединения в виде пушистого белого твердого вещества (выход 39,7%, суммарный выход 16,8%). Спектр ЯМР1H в D6 - DMSO: 9,05 (d, 1H), 8,55 (d, 1H), 8,55 (t, 1H), 7,90 (d, 1H), 7,65 (d, 1H), 7,55 (t, 1H), 7,20 (d, 1H), 7,15 (s, 2H), 7,00 (br, s, 4H), 5,15 (dd, 1H), 4,50 (g, 1H), 4,30 (m,
3H), 3,95 (dd, 1H), 3,60 (d, 1H), 3,10 (m, 2H), 3,00 (s, 3H), 2,75 (dd, 1H), 2,55 (dd, 1H), 2,10 (m, 2H), 1,60 (m, 1H), 1,35 (m, 2H), 1,10 (d, 3H), 0,90 (d, 3H);
Масс-спектр (FAB), [M+H]=701,
37.
Циклическое промежуточное соединение 95
Цикло-(D-Val-NMeArg-Gly-Asp-3-аминоэтил-4-метоксибензойная кислота)
Указанное в заглавии соединение получают по общей
методике, описанной для синтеза в растворе, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Защиту циклического пептида (600 мг, 0,71 ммоль) снимают избытком HF в присутствии анизола в качестве
отнимающего агента. Очистку проводят методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 7-18% ацетонитрил, содержащий трифторуксусную кислоту (градиент 0,33%/мин), и
получают соль трифторуксусной кислоты указанного в заглавии соединения (104 мг, 32%) в виде белого пушистого твердого вещества. Спектр ЯМР1H в D6 - DMSO: 12,40 (br s, 1H), 8,25
(d, 1H), 8,20 (br s, 1H), 8,00 (br s, 2H), 7,85 (s, 1H), 7,65 (br s, 1H), 7,05 (d, 1H), 7,05 (br s, 4H), 5,00 (dd, 1H), 4,60 (q, 1H), 4,30 (d, 1H), 4,25 (d, 2H), 3,85 (s, 3H), 3,85 (dd, 1H), 3,70 (dd,
1H), 3,10 (q, 2H), 3,00 (s, 3H), 2,70 (m, 1H), 2,50 (m, 1H), 2,10 (m, 1H), 1,90 (m, 1H), 1,65 (m, 1H), 1,35 (m, 2H), 1,00 (d, 3H), 0,90 (d, 3H);
Масс-спектр (FAB), [M+H2
O+H]=623.
Циклическое промежуточное соединение 96
Цикло-(D-Val-NMeArg-Gly-Asp-3-аминоэтил-4-метилбензойная кислота)
Указанное в заглавии соединение получают по общей
методике синтеза в растворе, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Защиту циклического пептида (210 мг, 0,25 ммоль) снимают избытком HF в присутствии анизола в качестве отнимающего
агента. Очистку проводят методом обращеннофазной ВЭЖХ на препаративной колонке LiChrosper RP-18 (5 см), используя 22-90% ацетонитрил, содержащий 0,1% трифторуксусной кислоты (градиент 2,3%/мин).
Получают соль трифторуксусной кислоты указанного в заглавии соединения (75 мг, 42%) в виде пушистого белого твердого вещества. Спектр ЯМР1H в D6 - DMSO: 12,30 (br s, 1H), 8,85
(d, 1H), 8,55 (d, 1H), 8,30 (t, 1H), 7,75 (d, 1H), 7,55 (m, 1H), 7,40 (s, 1H), 7,20 (s, 1H), 7,00 (br s, 4H), 5,20 (dd, 1H), 4,55 (q, 1H), 4,45 (dd, 1H), 4,30 (m, 2H), 4,05 (dd, 1H), 3,60 (d, 1H), 3,
10 (q, 2H), 3,00 (s, 3H), 2,70 (dd, 1H), 2,50 (m, 1H), 2,25 (s, 3H), 2,10 (m, 2H), 1,60 (m, 1H), 1,35 (m, 2H), 1,10 (d, 3H), 0,90 (d, 3H);
Масс-спектр (FAB), [M+H]=589.
Циклическое промежуточное соединение 97
Цикло-(D-Val-NMeArg-Gly-Asp-3-аминометил-6-хлорбензойная кислота)
Указанное в заглавии соединение получают по общей методике синтеза в
растворе, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), с тем исключением, что используют оксим 4,4'-динитробензофенона. Защиту циклического пептида (550 мг, 0,65 ммоль) снимают избытком HF в
присутствии анизола в качестве отнимающего агента. Очистку проводят методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 10-38% ацетонитрил, содержащий 0,1%
трифторуксусной кислоты (градиент 0,8%/мин). Получают соль трифторуксусной кислоты указанного в заглавии соединения (254 мг, 54%) в виде белого пушистого твердого вещества. Спектр ЯМР1H в
D6 - DMSO: 12,30 (br s, 1H), 9,05 (d, 1H), 8,45 (m, 2H), 7,50 (t, 1H), 7,35 (d, 1H), 7,30 (m, 2H), 7,10 (s, 1H), 7,05 (br s, 4H), 5,15 (dd, 1H), 4,45 (dd, 1H), 4,40 (q, 2H), 4,05 (dt, 2H),
3,55 (dd, 1H), 3,15 (q, 2H), 3,10 (s, 3H), 2,70 (dd, 1H), 2,50 (m, 1H), 2,05 (m, 2H), 1,65 (m, 1H), 1,35 (m, 2H), 1,10 (d, 3H), 0,90 (d, 3H);
Масс-спектр (FAB), [M+H]=609.
Циклическое промежуточное соединение 99
Цикло-(D-Val-NMeArg-Gly-Asp-3-аминометил-6-метоксибензойная кислота)
Указанное в заглавии соединение получают по общей методике синтеза в
растворе, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), с тем исключением, что используют оксим 4,4'-динитробензофенона. Защиту циклического пептида (256 мг, 0,30 ммоль) снимают избытком HF в
присутствии анизола в качестве отнимающего агента. Очистку проводят методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 10-38% ацетонитрил, содержащий 0,1%
трифторуксусной кислоты (градиент 0,8%/мин). Получают соль трифторуксусной кислоты указанного в заглавии соединения (137 мг, 63%) в виде белого пушистого твердого вещества. Спектр ЯМР1H в
D6 - DMSO: 8,45 (d, 1H), 8,40 (d, 1H), 8,30 (t, 1H), 7,65 (d, 1H), 7,50 (t, 1H), 7,40 (s, 1H), 7,35 (d, 1H), 7,05 (d, 1H), 7,00 (br s, 4H), 5,20 (dd, 1H), 4,55 (dd, 1H), 4,50 (q, 1H), 4,35
(dd, 1H), 4,25 (dd, 1H), 3,95 (dd, 1H), 3,90 (s, 3H), 3,55 (d, 1H), 3,10 (q, 2H), 3,00 (s, 3H), 2,70 (dd, 1H), 2,50 (m, 1H), 2,05 (m, 1H), 1,60 (m, 1H), 1,35 (m, 2H), 1,10 (d, 3H), 0,95 (d, 3H).
Масс-спектр (FAB), [M+H]=605.
Циклическое промежуточное соединение 100
Цикло-(D-Val-NMeArg-Gly-Asp-3-аминоэтил-6-метилбензойная кислота)
Указанное в
заглавии соединение получают по общей методике синтеза в растворе, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), с тем исключением, что используют оксим 4,4'-динитробензофенона. Защиту
циклического пептида (230 мг, 0,28 ммоль) снимают избытком HF в присутствии анизола в качестве отнимающего агента. Очистку проводят методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5
см), используя 10-38% ацетонитрил, содержащий 0,1% трифторуксусной кислоты (градиент 0,8%/мин). Получают соль трифторуксусной кислоты указанного в заглавии соединения (54 мг, 27%) в виде белого
пушистого твердого вещества. Спектр ЯМР1H в d6 - DMSO: 12,30 (br s, 1H), 8,80 (d, 1H), 8,40 (d, 1H), 8,30 (t, 1H), 7,45 (m, 2H), 7,15 (q, 2H), 7,00 (s, 1H), 7,00 (br s, 4H), 5,
15 (dd, 1H), 4,45 (m, 3H), 4,05 (m, 2H), 3,55 (dd, 1H), 3,10 (q, 2H), 3,05 (s, 2H), 2,70 (dd, 1H), 2,50 (m, 1H), 2,30 (s, 3H), 2,05 (m, 2H), 1,60 (m, 1H), 1,35 (m, 2H), 1,05 (d, 3H), 0,90 (d, 3H);
Масс-спектр (FAB), [M+H]=589.
Циклическое промежуточное соединение 100a
Цикло-(D-Abu-NMeArg-Gly-Asp-3-аминоэтил-6-хлорбензойная кислота)
Указанное в заглавии
соединение получают по общей методике синтеза в растворе, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), с тем исключением, что используют оксим 4,4'-динитробензофенона. Защиту циклического
пептида (330 мг, 0,40 ммоль) снимают избытком HF в присутствии анизола в качестве отнимающего агента. Очистку проводят методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см),
используя 10-38% ацетонитрил, содержащий 0,1% трифторуксусной кислоты (градиент 1,0%/мин). Получают соль трифторуксусной кислоты указанного в заглавии соединения (114 мг, 41%) в виде белого пушистого
твердого вещества. Спектр ЯМР1H в D6 - DMSO: 9,00 (d, 1H), 8,40 (m, 2H), 7,50 (m, 1H), 7,40 (d, 1H), 7,30 (m, 2H), 7,15 (s, 1H), 7,00 (br s, 4H), 5,15 (dd, 1H), 4,65 (q, 1H), 4,
50 (dd, 1H), 4,40 (q, 1H), 4,05 (dd, 1H), 3,95 (dd, 1H), 3,65 (dd, 1H), 3,10 (q, 2H), 3,05 (s, 3H), 2,75 (dd, 1H), 2,50 (m, 1H), 1,95 (m, 1H), 1,75 (m, 2H), 1,60 (m, 1H), 1,35 (m, 2H), 0,95 (t,
3H).
Масс-спектр (FAB), [M+H]=595,4.
Циклическое промежуточное соединение 89d
Цикло-(D-Abu-NMeArg-Gly-Asp-йода-Mamb); соединение формулы (VII), где J - D-Abu,
K - NMeArg, L - Gly, M - Asp, R1=R2 - H, R10- H, R10a - I.
Указанное в заглавии соединение получают по общей методике, описанной для синтеза цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). Для введения группы Boc-йодо-Mamb в смолоподобный оксим используют метод DCC/DMAP. Из пептида, взятого в количестве 3,53 ммоль, получают защищенный циклический пептид (4,07 г, выход выше количественного). Пептид (4,07 мг) и 4,5 мл анизола обрабатывают безводным HF при 0oC в течение 30 мин. Продукт осаждают эфиром, перерастворяют в водном растворе уксусной кислоты и выделяют лиофилизацией указанное в заглавии соединение (2,97 г, выход выше количественного, рассчитано на уксуснокислую соль). Очистку завершают обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 16,2-22,5% ацетонитрил, содержащий 0,1% TFA (градиент 0,16%/мин). После лиофилизации получают соль трифторуксусной кислоты указанного в заглавии соединения в виде пушистого белого твердого вещества (выход 28,7%, суммарный выход 30,2%). Масс-спектр (FAB), [M+H]=687,33.
Циклическое
промежуточное соединение 100b
Цикло-(D-Abu-NMeArg-Gly-Asp-3-аминометил-6-йодобензойная кислота)
Указанное в заглавии соединение получают по общей методике синтеза в растворе,
описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), с тем исключением, что используют оксим 4,4'-динитробензофенона. Защиту циклического пептида (350 г, 0,38 ммоль) снимают избытком HF в присутствии
анизола в качестве отнимающего агента. Очистку осуществляют методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 10-38% ацетонитрил, содержащий 0,1% трифторуксусной
кислоты (градиент 1,0%/мин). Получают соль трифторуксусной кислоты указанного в заглавии соединения (150 мг, 49%) в виде пушистого белого вещества. Спектр ЯМР1H в D6-DMSO: 8,90
(d, 1H), 8,40 (m, 2H), 7,70 (d, 1H), 7,50 (m, 1H), 7,30 (m, 1H), 7,05 (s, 1H), 7,00 (d, 1H), 7,00 (bs, s, 4H), 5,15 (dd, 1H), 4,65 (q, 1H), 4,45 (dd, 1H), 4,40 (q, 1H), 4,00 (q, 1H), 3,90 (q, 1H), 3,
65 (dd, 3H), 3,10 (q, 2H), 3,05 (s, 3H), 2,70 (dd, 1H), 2,50 (m, 1H), 1,95 (m, 1H), 1,75 (m, 2H), 1,60 (m, 1H), 1,40 (m, 2H), 0,95 (t, 3H);
Масс-спектр (FAB), [M+H] = 687,3.
Циклическое
промежуточное соединение 100c
Цикло-(D-Abu-NMeArg-Gly-Asp-3-аминометил-6-метилбензойная кислота); соединение формулы (VII), где J - D-Abu, K - NMeArg, L - Gly, M - Asp, R10 - Me.
Указанное в заглавии соединение получают по общей методике синтеза в растворе, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), с тем исключением, что используют
оксим 4,4'-динитробензофенона. Защиту циклического пептида (130 мг, 0,16 ммоль) снимают избытком HF в присутствии анизола в качестве отнимающего агента. Очистку проводят методом обращеннофазной ВЭЖХ
на препаративной колонке Vydac C18 (2,5 см), используя 10-38% ацетонитрил, содержащий 0,1% трифторуксусной кислоты (градиент 1,0%/мин). Получают соль трифторуксусной кислоты указанного в заглавии
соединения (31 мг, 28%) в виде белого пушистого твердого вещества. Спектр ЯМР1H в D6-DMSO: 8,70 (d, 1H), 8,40 (d, 1H), 8,30 (t, 1H), 7,50 (m, 1H), 7,45 (m, 1H), 7,15 (q, 2H), 7,
05 (s, 1H), 7,00 (br, s, 4H), 5,15 (dd, 1H), 4,65 (q, 1H), 4,45 (m, 2H), 4,00 (m, 2H), 3,65 (dd, 1H), 3,10 (q, 2H), 3,05 (s, 3H), 2,75 (dd, 1H), 2,50 (m, 1H), 2,30 (s, 3H), 2,00 (m, 1H), 1,75 (m, 2H),
1,60 (m, 1H), 1,35 (m, 2H), 0,95 (t, 3H);
Масс-спектр (FAB), [M+H] = 575,4.
Твердофазный синтез циклического промежуточного соединения 101
Цикло-(D-Val-NMeArg-Gly-Asp-аминометил-4-йодобензойная кислота)
Указанное в заглавии соединение получают по общей методике, ранее описанной для цикло-(D-Val-NMeArg-Gly-Asp-Mamb). Для введения
группы Boc-йодо-Mamb в смолоподобный оксим используют метод DCC/DMAP. Из пептида, взятого в количестве 1,05 ммоль, получают защищенный циклический пептид (460 мг, 46,8%). Пептид (438 мг) и анизол
обрабатывают безводным HF при 0oC в течение 30 мин. Продукт осаждают эфиром, перерастворяют в водной уксусной кислоте и выделяют лиофилизацией указанное в заглавии соединение (340 мг, 95,6%,
рассчитано на уксуснокислую соль). Очистку осуществляют методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 12,6-22,5% ацетонитрил, содержащий 0,1% трифторуксусной
кислоты (градиент 0,23%/мин), и затем лиофилизацией получают соль трифторуксусной кислоты указанного в заглавии соединения в виде белого пушистого твердого вещества (выход 39,7%, суммарный выход 16,
6%). Спектр ЯМР1H в D6 - DMSO: 9,05 (d, 1H), 8,55 (d, 1H), 8,55 (t, 1H), 7,90 (d, 1H), 7,65 (d, 1H), 7,55 (t, 1H), 7,20 (d, 1H), 7,15 (s, 2H), 7,00 (br, s, 4H), 5,15 (dd, 1H), 4,
50 (q, 1H), 4,30 (m, 3H), 3,95 (dd, 1H), 3,60 (d, 1H), 3,10 (m, 2H), 3,00 (s, 3H), 2,75 (dd, 1H), 2,55 (dd, 1H), 2,10 (m, 2H), 1,60 (m, 1H), 1,35 (m, 2H), 1,10 (d, 3H), 0,90 (d, 3H);
Масс-спектр (FAB), [M+H] = 701,37.
Синтез циклического промежуточного соединения 102 в растворе
Цикло-(D-Val-NMeArg-Gly-Asp-3-аминометил-6-йодобензойная кислота)
Указанное в заглавии соединение получают в соответствии с приведенным на схеме 6 методом.
1. Boc-Asp(OcHex)-3-аминометил-6-йодобензойная кислота
К суспензии
3-аминометил-6-иодобензойной кислоты • HCl (4,9 г, 16 ммоль) в H2O (16 мл) добавляют HaHCO3 (3,9 г, 47 ммоль), а затем раствор Boc-Asp(OcHex)-OSu (5,9 г, 14 ммоль) в ТГФ.
Реакционную смесь перемешивают при комнатной температуре в течение ночи, фильтруют, разбавляют H2O, подкисляют 1Н HCl и экстрагируют этилацетатом. Экстракт промывают водой, упаривают, сушат
над безводным сульфатом магния и окончательно высушивают в вакууме. Полученный продукт растирают с эфиром, получают указанное в заглавии соединение (6,7 г, 82%) в виде белого порошка. Спектр1H ЯМР в D6 - DMSO: 8,45 (br t, 1H), 7,90 (d, 1H), 7,60 (s, 1H), 7,15 (m, 2H), 4,65 (m, 1H), 4,35 (m, 1H), 4,25 (d, 2H), 2,70 (m, 1H), 2,55 (m, 1H), 1,70 (m, 4H), 1,40 (s, 9H), 1,35
(m, 1H).
2. 4,4'-динитробензофеноноксим
Указанное в заглавии соединение получают в соответствии с модификацией ранее описанной в литературе методики (Chapman and Fielder
(1936), J. Chem. Soc, 448, Kulin and Leffek (1973), Can. J. Chem, 51: 867). Раствор хромового ангидрида (20 г, 200 ммоль) в 125 мл H2O в течение 4 ч прикапывают к суспензии
бис-(4-нитрофенил)метана (25 г, 97 ммоль) в 300 мл уксусной кислоты, нагреваемой с обратным холодильником. После этого реакционную смесь нагревают с обратным холодильником 1 ч, охлаждают до комнатной
температуры и выливают в воду. Осадок отделяют фильтрацией, промывают водой, 5% раствором бикарбоната натрия, H2O и после сушки на воздухе получают смесь 1:1 бис-(4-нитрофенил)метана и 4,
4'-динитробензола (по данным ЯМР1H). Этот продукт окисляют второй порцией хромового ангидрида (20 г, 200 ммоль), после чего по аналогичной методике выделяют продукт реакции, растирают с
200 мл бензола. После нагревания в течение 16 ч с обратным холодильником получают 4,4'-динитробензофенон (20,8 г, 79%) в виде желтого порошка.
Раствор гидроксиламина гидрохлорида (10, 2 г, 147 ммоль) добавляют к суспензии 4,4'-динитробензофенона (19 г, 70 ммоль) в 100 мл этанола. Реакционную смесь нагревают с обратным холодильником в течение 2 ч, охлаждают до комнатной температуры, отделяют твердый продукт фильтрованием. После перекристаллизации из этанола получают указанное в заглавии соединение (14,0 г, 70%) в виде бледно-желтых кристаллов.
Tпл. = 194oC, Спектр ЯМР1H в D6 - DMSO:d 12,25 (s, 1H), 8,35 (d, 2H), 8,20 (d, 2H), 7,60 (d, 4H).
4,4'-Динитробензофеноноксим
Boc-Asp(OcHex)-3-аминометил-6- йодобензоат
К охлажденному льдом раствору Boc-Asp(OcHex)-3-аминометил-6- йодобензойной кислоты (3,3 г, 5,75 ммоль) и 4,4'-динитробензофеноксима (1,7 г, 5,9
ммоль) в 32 мл этилацетата добавляют DCC (1,2 г, 5,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 час, фильтруют, разбавляют этилацетатом, промывают насыщенным
раствором бикарбоната натрия, водой, упаривают, высушивают над безводным сульфатом магния и окончательно высушивают в вакууме. Полученный продукт очищают на колонке с силикагелем (EM Science, 230-400
меш.), используя смесь 10:1 дихлорметана и этилацетата. Получают указанное в заглавии соединение (1,8 г, 36%) в виде бледно-желтых кристаллов.
Спектр ЯМР1H в D6 - DMSO:d 8,40 (dd, 5H), 7,90 (m, 5H), 7,45 (s, 1H), 7,20 (m, 2H), 4,65 (m, 1H), 4,35 (m, 1H), 4,20 (m, 2H), 2,75 (dd, 1H), 2,50 (dd, 1H), 1,70 (m, 4H), 1,40 (s, 9H), 1,35 (m, 6H).
4. Boc-D-Val-NmeArg(Tos) (11,07 г, 25 ммоль) и Gly-OBzl тозилата (10,10 г, 30 ммоль) в 25 мл дихлорметана добавляют HBTU (9,48 г, 25 ммоль) и DIEA (9,69 г, 75 ммоль).
Реакционную смесь перемешивают при комнатной температуре в течение 1 ч, концентрируют в вакууме, разбавляют этилацетатом, промывают 5% раствором лимонной кислоты, насыщенным раствором бикарбоната натрия, упаривают, сушат над безводным сульфатом магния и окончательно высушивают при пониженном давлении. Полученный маслянистый продукт растирают с петролейным эфиром. Получают Boc-NmeArg(Tos)-Gly-OBzl (14,7 г, 100%); масс-спектр (FAB): [M+H] = 590,43. Этот продукт используют без дополнительной очистки.
Раствор Boc-NmeArg(Tos)-Gly-OBzl (14,5 г, 24,6 ммоль) в 30 мл трифторуксусной кислоты перемешивают при комнатной температуре в течение 5 мин, сушат при пониженном давлении. Полученный маслянистый продукт разбавляют холодным этилацетатом, промывают холодным насыщенным раствором бикарбоната натрия, водную фазу экстрагируют этилацетатом. Объединенный органический продукт промывают, упаривают и окончательно высушивают в вакууме, полученный маслянистый продукт промывают, упаривают и окончательно высушивают в вакууме, полученный маслянистый продукт растирают с эфиром. Твердую часть отфильтровывают, промывают эфиром и высушивают в вакуумном эксикаторе. Получают NmeArg(Tos)-Gly-OBzl (10,3 г, 86%); масс-спектр (FAB): [M+H] = 490,21. Этот продукт используют без дополнительной очистки.
К раствору NmeArg(Tos)-Gly-OBzl (4,80 г, 9,8 ммоль) и Boc-D-Val (2,13 г, 9,8 ммоль) в 10 мл дихлорметана, охлажденному на базе со льдом, добавляют HBTU (3,79 г, 10,0 ммоль) и DIEA (2,58 г, 20,0 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 48 ч, разбавляют этилацетатом, промывают 5% лимонной кислотой, упаривают, сушат над безводным сульфатом магния и окончательно высушивают при пониженном давлении. Полученный маслянистый продукт растирают с эфиром, после чего получают Boc-D-Val, NmeArg(Tos)-Gly-OBzl (4,58 г, 68%); масс-спектр (FAB): [M+H] = 689,59. Этот продукт используют без дополнительной очистки.
Раствор Boc-D-Val-NmeArg(Tos)-Gly-OBzl (4,50 г, 6,53 ммоль) продувают азотом, добавляют 1,30 г 10% Pd/c и пропускают в реактор водород. Через 1 ч катализатор отфильтровывают через слой целита, удаляют растворитель при пониженном давлении. Полученное твердое вещество растирают с эфиром, отфильтровывают и промывают петролейным эфиром. Получают Boc-D-Val-NmeArg(Tos)-Gly (3,05 г, 78%); Спектр ЯМР1H в D6 - DMSO:d 7,90 (br t, 1H), 7,65 (d, 2H), 7,30 (d, 2H), 7,00 (d, 1H), 6,85 (br d, 1H), 6,60 (br s, 1H), 5,00 (dd, 1H), 5,00 (dd, 1H), 4,15 (t, 1H), 3,70 (m, 2H), 3,05 (m, 2H), 2,90 (s, 3H), 2,35 (s, 3H), 1,90 (m, 2H), 1,55 (m, 2H), 1,35 (s, 9H), 1,25 (m, 2H), 1,80 (br t, 6H).
масс-спектр (FAB): [M+H] = 599,45.
5. 4,
4'-Динитробензофеноноксим Boc-D-Val-NmeArg(Tos)-Gly-(OcHex)-3-аминометил-6-йодобензоат
К раствору 4,4'-динитробензофеноноксим Boc-Asp(OcHex)-3-аминометил-6-йодобензоата (0,5 г, 0,59 ммоль) в
1 мл дихлорметана добавляют 0,5 мл трифторуксусной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 90 мин, разбавляют дихлорметанолом и окончательно высушивают при
пониженном давлении. Маслянистый остаток концентрируют в высоком вакууме для удаления следов избытка трифторуксусной кислоты.
К раствору полученной соли трифторуксусной кислоты и Boc-D-Val-NmeArg(Tos)-Gly (0,52 г, 0,57 ммоль) в 3,8 мл DMF добавляют TBTU (0,28 г, 0,87 ммоль) и DIEA (0,33 г, 2,58 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, концентрируют в высоком вакууме, разбавляют этилацетатом, промывают 5% лимонной кислотой, H2O, упаривают, сушат над безводным сульфатом магния и окончательно досушивают при пониженном давлении. Полученный продукт растирают с эфиром, после чего получают указанное в заглавии соединение (0,48 г, 61%) в виде порошка. Этот продукт используют без дополнительной очистки.
6. Цикло-(D-Val-NmeArg(Tos)-Gly-Asp(OcHex)-3-аминометил-6- йодобензойная кислота
К раствору 4,4'-динитробензофеноноксим Bos-(D-Val-NmeArg(Tos)-Gly-Asp(OcHex)-3-аминометил-6-йодобензойной
кислоты (0,48 г, 0,36 ммоль) в 1 мл дихлорметана добавляют 0,5 мл трифторуксусной кислоты. Реакционную смесь перемешивают при комнатной температуре в течение 45 мин, разбавляют дихлорметаном и
окончательно высушивают при пониженном давлении. Маслянистый остаток концентрируют в высоком вакууме для удаления следов избытка трифторуксусной кислоты.
К раствору неочищенной соли трифторуксусной кислоты в 38 мл DMF добавляют уксусную кислоту (0,09 г, 1,57 ммоль) и DIEA (0,26 г, 1,49 ммоль). Реакционную смесь перемешивают при 60oC в течение 3 дней, концентрируют в высоком вакууме, разбавляют этилацетатом, промывают 5% лимонной кислотой, упаривают, высушивают над безводным сульфатом магния и окончательно досушивают при пониженном давлении. Этот продукт очищают колоночной хроматографией на силикагеле (FM Seince, 230-400 меш. ), используя хлороформ и изопропанол, взятые в соотношении 10:1. Получают указанное в заглавии соединение (0,13 г, 38%) в виде порошка.
Спектр ЯМР1H (в D6 - DMSO):d 8,95 (d, 1H), 8,50 (t, 1H), 8,45 (d, 2H), 7,70 (d, 1H), 7,60 (d, 2H), 7,30 (d, 3H), 7,05 (d, 1H), 7,00 (s, 1H), 6,80 (br s, 1H), 6,60 (br s, 1H), 5,10 (dd, 1H), 4,65 (m, 1H), 4,45 (m, 1H), 4,35 (m, 1H), 4,00 (m, 1H), 3,55 (d, 1H), 3,05 (m, 2H), 3,00 (s, 3H), 2,70 (dd, 1H), 2,55 (dd, 1H), 2,35 (s, 3H), 2,05 (m, 1H), 1,90 (m, 1H), 1,75 (m, 1H), 1,65 (m, 1H), 1,35 (m, 13H), 1,15 (d, 3H), 0,85 (d, 3H); масс-спектр (FAB(GLYC):[M+H] = 937.
7. Цикло-(D-Val-NMeArg-Gly-Asp-3-аминометил-6-йодобензойная
кислота)
Снятие защиты циклического пептида (490 мг, 0,52 ммоль) осуществляют избытком HF в присутствии анизола в качестве акцептора. Очистку проводят методом обращеннофазовой ВЭЖХ на
препаративной колонке Vydac C18 (2,5 см), используя 10-38% ацетонитрил, содержащий 0,1% трифторуксусной кислоты (градиент 0,8% мин). Получают соль трифторуксусной кислоты указанного в заглавии
соединения (194 мг, 46%) в виде пушистого белого твердого вещества;
Спектр ЯМР1H (в D6 - DMSO):d 12,30 (br s, H), 9,00 (d, 1H), 8,40 (m, 2H), 7,70 (d, 1H), 7,50 (m, 1H),
7,30 (m, 1H), 7,05 (d, 1H), 7,00 (s, 1H), 7,00 (br s, 4H), 5,15 (dd, 1H), 4,40 (d, 1H), 4,40 (q, 2H), 4,00 (m, 2H), 3,55 (dd, 1H), 3,15 (q, 2H), 3,10 (s, 3H), 2,70 (dd, 1H), 2,50 (m, 1H), 2,05 (m,
2H), 1,65 (m, 1H), 1,35 (m, 2H), 1,15 (d, 3H), 0,90 (d, 3H); масс-спектр (FAB):[M+H] = 701.
В таблице A приведены данные масс-спектроскопии (FAB) для некоторых циклических промежуточных соединений.
Другие фрагменты, содержащие заместители в ароматическом ядре, могут быть синтезированы, как показано ниже на схемах и комментариях к схемам. Фрагмент вышеприведенной формулы, где Z=NH2, может быть получен, по меньшей мере, двумя различными способами. Например, используя в качестве исходного соединения 4-ацетамидбензойную кислоту (Aldrich Chemical Co.), алкилированием по Фриделю Крафтсу N-гидроксиметилдихлорацетамидом можно получить дихлорацетилпроизводное 3-аминоэтил-4-ацетамидбензойной кислоты (Felder, Pitre and Fumagall: (1964), Helv. Chim. Acta, 48, 259-274). В результате гидролиза двух амидных групп получают 3-аминометил-4-аминобензойную кислоту.
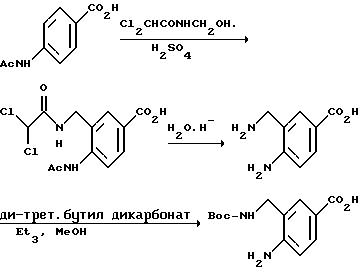
Иначе, используя в качестве исходного соединения 3-циано-4-нитротолуол, окислением трехокисью хрома с последующим восстановлением получают 3-аминометил-4-аминобензойную кислоту.
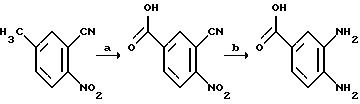
a] CrO3 b] H2 - катализ
Фрагмент вышеприведенной формулы, где Y=CH2NH2, может быть получен из 3,5-дицианотолуола, путем окисления метиловой группы с триоксидом хрома с последующим восстановлением.
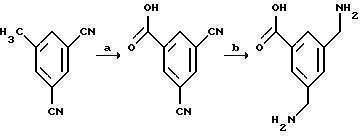
a] CrO3, b] H2 - катализ
Фрагмент вышеприведенной формулы, где Z=CH2NH2, может быть получен из 3-циано-4-метилбензойной кислоты (K и K Rare and Fine Chemicals). Бромированием N-бромсукцинимидом можно получить 4-бромометил-3-цианобензойную кислоту. Реакция нуклеофильного замещения бромометильной группы амидным анионом приводит к получению защищенного амина. В качестве амидного аниона в этой реакции можно использовать фталимид калия (Gabriel Synthesis) и анион трифторацетамида (Usui (1991), Nippon Kagaku Kaishi, 206-212), применяемый в данном примере. Восстановление нитрильной группы позволяет получить вторичную аминометильную группу, которая может быть защищена по реакции с ди-трет-бутил-дикарбонатом. Удаление трифторацетамидной защитной группы водным раствором пиперидина приводит к образованию вышеуказанного фрагмента.
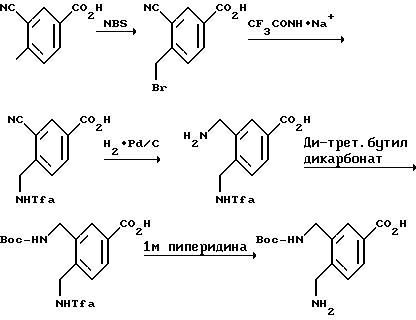
Этот фрагмент также может быть получен из 4-бромобензойной кислоты, как показано на схеме
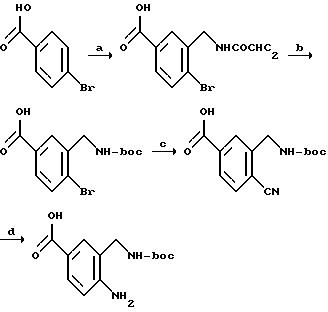
a] H2SO4, HOCH2NHCOCHCl2, b] H+, boc-ON,
c] CuCN, DMF d] H2-catalyst
Эти замещенные в ароматическое ядро циклические фрагменты могут быть использованы для синтеза циклических промежуточных соединений.
Циклическое промежуточное соединение 113
Цикло-(D-Val-NMeArg-Gly-Asp-Mamb(4-NH2)
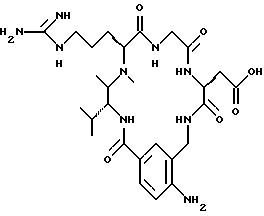
Это соединение может быть получено по методике, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), замещением в циклических фрагментах, содержащих заместители в ароматическом ядре, при Z=NH2.
Циклические промежуточные соединения 114, 115 и 116
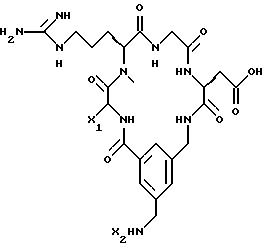
x1 - 2-пропил, этил или p-гидроксифенилметил;
x2 - H.
Соединения Цикло-(D-Val-NMeArg-Gly-Asp-Mamb(5-CH2NHX2), Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(5-CH2NHX2) и цикло-(D-Tyr-NMeArg-Gly-Asp-Mamb(5-CH2NHX2) могут быть получены по описанной выше методике с использованием замещенного в ароматическое ядро фрагмента с Y-CH2NH2.
Циклические промежуточные соединения 117,
118 и 119
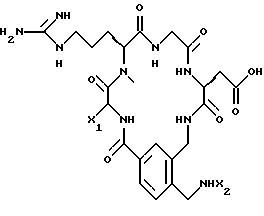
x1 - 2-пропил, этил или p-гидроксифенилметил;
x2 - H.
Соединения цикло-(D-Val-NMeArg-Gly-Asp-Mamb(4-CH2NHX2), цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(4-CH2NHX2) и цикло-(D-Tyr-NMeArg-Gly-Asp-Mamb(4-CH2NHX2) получены по описанной выше методике с использованием замещенного в ароматическое ядро фрагмента с Z=CH2NH2.
Другие циклические фрагменты, содержащие заместитель R31
Вместо Mamb в качестве образующего цикл фрагмента R31 в циклических пептидах настоящего
изобретения могут быть использованы остатки аминоалкилнафтойной кислоты или тетрагидронафтойной кислоты.
Типичные примеры производных аминоалкилнафтойной кислоты и аминоалкилтетрагидронафтойной кислоты, используемых в синтезе циклических пептидов настоящего изобретения, приведены ниже. Синтез этих производных иллюстрирует приведенная схема 7.
8-Амино-5,6,7,8-тетрагидро-2-нафтойная кислота гидрохлорид (8).
Для получения указанного в названии соединения используют модификацию обычной методики, ранее описанной в литературе (Earnest, I, Kalvoda, J.Rihs, G., Mutter, M., Tett. Lett., vol. 31, N 28, pp. 4011-4014, 1990).
Как показано на схеме 7, 4-фенилмасляную кислоту (1) переводят в этиловый эфир (2), который затем ацилируют ацетилхлоридом в присутствии хлористого алюминия, и получают этиловый эфир 4-ацетилфенилмасляной кислоты (3). Этот эфир омыляют, и получают 4-ацетилфенилмасляную кислоту (4). Далее ацетильную группу окисляют с образованием 4-карбоксифенилмасляной кислоты (5), которую затем превращают в 1-тетралин-7-карбоновую кислоту (6) циклизацией по Фриделю-Крафтсу с использованием хлористого алюминия, с довольно высоким выходом.
На этом этапе тетралон разделяют на две порции, и одну из них превращают в оксим (7), используя ацетат натрия и гидроксиламин гидрохлорид. Оксим восстанавливают действием водорода, и получают рацемическую смесь 8-амино-5,6,7,8-тетрагидро-2-нафтойной кислоты в виде гидрохлорида (8), которую используют как промежуточное соединение для введения в циклический пептид.
Часть A. Раствор 4-фенилмасляной кислоты (50,0 г, 0,3 моль) в этаноле (140 мл) с добавлением концентрированной серной кислоты (0,53 мл) перемешивают с обратным холодильником в течение 5 ч. Охлажденный раствор выливают в ледяную воду и экстрагируют этилацетатом. Объединенные органические слои промывают, высаливают и высушивают над безводным сульфатом магния, окончательно досушивают при пониженном давлении. Получают этиловый эфир 4-фенилмасляной кислоты (56,07 г, 0,29 моль, 97%) в виде желтой жидкости.
Спектр ЯМР1H (в CDCl3):d 7,3-7,1 (m, 5H), 4,1 (q, 2H, J = 7,1 Гц), 2,7 (t, 2H, J = 7,7 Гц), 2,3 (t, 2H, J = 7,5 Гц), 1,95 (квинтет, 2H, J = 7,5 Гц), 1,25 (t, 3H, J = 7,1 Гц).
Часть B. К раствору, содержащему алюминий хлорид (153 г, 1,15 моль) и ацетилхлорид (38,5 мл, 42,5 г, 0,54 моль) в дихлорметане (1500 мл), прикапывают раствор этилового эфира 4-фенилмасляной кислоты (50,0 г, 0,26 моль) в дихлорметане (500 мл). Реакционную смесь перемешивают при комнатной температуре в течение 15 мин. Раствор выливают в холодную концентрированную соляную кислоту (2000 мл) и затем экстрагируют дихлорметаном. Объединенные органические слои подвергают обратной промывке, упаривают, сушат над безводным сульфатом магния и окончательно досушивают при пониженном давлении. Получают этиловый эфир 4-ацетилфенилмасляной кислоты (53,23 г, 0,23 моль, 88%) в виде темно-желтой жидкости.
Спектр ЯМР1H (в CDCl3):d 7,9 (d, 2H, J = 8,1 Гц), 7, 25 (d, 2H, J = 8,4 Гц), 4,1 (q, 2H, J = 7,1 Гц), 2,75 (t, 2H, J = 7,6 Гц), 2,6 (s, 3H), 2,35 (t, 2H, J = 7,6 Гц), 2,0 (квинтет, 2H, J = 7,5 Гц), 1,25 (t, 3H, J = 7,1 Гц).
Часть C. К раствору этилового эфира 4-ацетилфенилмасляной кислоты (50,0 г, 0,21 моль) в этаноле (1250 мл) прикапывают раствор гидроокиси натрия (50,0 г) в воде (1250 мл). Все перемешивают с обратным холодильником в течение 4 час. Раствор концентрируют до половины объема и затем подкисляют 1H HCl до значения pH, равного 1. Полученный осадок собирают и промывают водой. Получают 4-ацетилфенилмасляную кислоту (53,76 г, 0,26 моль, 99%) в виде белого твердого вещества. Tпл = 50 - 52oC.
Спектр ЯМР1H (в CDCl3):d 7,9 (d, 2H, J = 8,1 Гц), 7,25 (d, 2H, J = 9,1 Гц), 2,75 (t, 2H, J = 7,7 Гц), 2,26 (s, 3H), 2,4 (t, 2H, J = 7,3 Гц), 2,0 (квинтет, 2H, J = 7,4 Гц).
Часть D. К раствору гидрохлорита натрия (330 мл, 17,32 г, 0,234 моль) в растворе гидроокиси натрия (50%, 172 мл), нагретому до 55oC, постепенно добавляют 4-ацетилфенилмасляную кислоту (16,0 г, 0,078 моль) в виде твердого вещества, поддерживая температуру между 60 - 70oC. Все перемешивают при 55oC в течение 20 ч. Охлажденный раствор гасят прикапыванием раствора бисульфита натрия (25%, 330 мл). Затем смесь переносят в стакан и осторожно подкисляют добавлением концентрированной соляной кислоты. Полученное твердое вещество собирают, промывают водой и высушивают, затем растирают последовательно с хлоробутаном и гексаном. Получают 4-карбоксифенилмасляную кислоту (15,31 г, 0,074 моль, 95%) в виде белого твердого вещества, Tпл = 190-195oC.
Спектр ЯМР1H в DMCO:d 12,55 (bs, 1H), 8,1 (s, 1H), 7,85 (d, 2H, J = 8,1 Гц), 7,3 (d, 2H, J = 8,1 Гц), 2,7 (t, 2H, J = 7,5 Гц), 2,2 (t, 2H, J = 7,4 Гц), 1,8 (квинтет, 2H, J = 7,5 Гц).
Часть E. Смесь 4-карбоксифенилмасляной кислоты (10,40 г, 0,05 моль), хлористого алюминия (33,34 г, 0,25 моль) и хлористого натрия (2,90 г, 0,05 моль) нагревают при непрерывном перемешивании до 190oC в течение 30 мин. Как только смесь охладится до 60oC, осторожно добавляют холодную соляную кислоту (1H, 250 мл). Смесь экстрагируют дихлорметаном. Объединенные органические слои подвергают обратной промывке разбавленной соляной кислотой и водой, высушивают над безводным сульфатом магния и окончательно досушивают при пониженном давлении. Полученное твердое вещество растирают с хлорбутаном. Получают 1-тетралон-7-карбоновую кислоту (9,59 г, 0,05 моль, 100%) в виде коричневого твердого вещества.
Tпл = 210 - 215oC. Спектр ЯМР1H в DMCO:d 8,4 (s, 1H), 8,1 (d, 2H, J = 8,0 Гц), 7,5 (d, 2H, J = 7,9 Гц), 3,0 (t, 2H, J = 6,0 Гц), 2,65 (t, 2H, J = 6,6 Гц), 2,1 (квинтет, 2H, J = 6,3 Гц).
Часть F. Раствор, содержащий 1-тетралон-7-карбоновую кислоту (1,0 г, 0,0053 моль), ацетат натрия (1,93 г, 0,024 моль) и гидроксиламин гидрохлорид (1,11 г, 0,016 моль) в смеси метанола с водой (1:1,15 мл), перемешивают с обратным холодильником в течение 4 ч. Смесь охлаждают и затем добавляют дополнительно воду (50 мл). Твердое вещество собирают, промывают водой и высушивают, затем растирают с гексаном. Получают 1-тетралоноксим-7-карбоновую кислоту (0,78 г, 0,0035 моль, 72%) в виде белого твердого вещества.
Tпл 205 - 215oC. Спектр ЯМР1H в DMSO:d 11,3 (s, 2H), 8,4 (s, 1H), 7,8 (d, 1H, J = 7,7 Гц), 7,3 (d, 1H, J = 7,7 Гц), 2,8 (t, 2H, J = 5,9 Гц), 2,7 (d, 2H, J = 6,6 Гц), 1,9 - 1,7 (m, 2H).
Часть G. Смесь 1-тетралоноксима-7-карбоновой кислоты (0,75 г, 0,0037 моль) в метаноле (25 мл) с концентрированной соляной кислотой (0,54 мл, 0,20 г, 0,0056 моль) и катализатор-палладий, нанесенный на углерод (0,10 г, 5% Pd/C), встряхивают в течение 20 ч при температуре окружающей среды в атмосфере водорода (60 фунт/кв. дюйм). Реакционную смесь фильтруют через Celite@ и промывают метанолом. Фильтрат упаривают до сухости при пониженном давлении, и остаток очищают хроматографически, используя гексан и этилацетат в соотношении 1:1. Получают рацемическую смесь 8-амино-5,6,7,8-тетрагидро-2-нафтойной кислоты гидрохлорида (0, 255 г, 0,001 моль, 27%) в виде белого твердого вещества.
Tпл = 289 - 291oC. Спектр ЯМР1H в DMSO:d 8,55 (bs, 3H), 8,2 - 8,1 (m, 1H), 7,85 - 7,8 (m, 1H), 7,35 - 7,25 (m, 1H), 4,5 (m, 1H), 2,9 - 2,8 (m, 2H), 2,1 - 1,9 (m, 3H), 1,85 - 1,7 (m, 1H).
N-Boc-8-аминометил-5,6,7,8-тетрагидро-2-нафтойная кислота (12)
Как показано
на схеме 7, оставшийся тетралон затем переводят в метиловый эфир (9). Используя методику, описанную Gregory, G.B. and Johnson, A.L., JOC, 1990, 55, 1479, метиловый эфир тетралона (9) сначала
превращают в циангидрин обработкой триметилсилилцианидом и йодидом цинка, а затем in situ осуществляют дегидратацию хлорокисью фосфора в пиридине, и получают метил-8-циано-5,6-дигидро-2-нафтоат (11).
Этот нафтоат разделяют на две части. Одну часть восстанавливают действием водорода, вводят N-BOC-защиту, омыляют и получают N-(BOC)-8-аминометил-5,6,7,8-тетрагидро-2-нафтойную кислоту (12)
- промежуточное соединение, предназначенное для введения в циклические пептиды.
Часть A. Смесь 1-тетралон-7-карбоновой кислоты (7,0 г, 0,037 моль) в метаноле (13,6 мл, 10,8 г, 0,30 моль) с каталитическим количеством соляной кислоты (0,07 мл, 0,12 г, 0,0012 моль) перемешивают с обратным холодильником в течение 5 ч. Охлажденную реакционную смесь выливают в ледяную воду и экстрагируют этилацетатом. Объединенные органические слои подвергают обратной промывке и упаривают, сушат над безводным сульфатом магния и окончательно высушивают при пониженном давлении. Полученное твердое вещество растирают с гексаном. Получают метиловый эфир 1-тетралон-7-карбоновой кислоты (3,61 г, 0,018 моль, 49%) в виде желтого твердого вещества.
Tпл = 170 - 172oC. Спектр ЯМР1H (в CDCl3):d 8,7 (s, 1H), 8,15 (d, 1H, J = 8,1 Гц), 7,35 (d, 1H, J = 8,1 Гц), 3,95 (s, 3H), 3,05 (d, 2H, J = 6,1 Гц), 2,7 (t, 2H, J = 6,4 Гц), 2, 15 (квинтет, 2H, J = 6,2 Гц).
Часть B. Раствор метилового эфира 1-тетралон-7-карбоновой кислоты (3,50 г, 0,017 моль), триметилсилилцианида (1,98 г, 0,02 моль) и йодида цинка (0,10 г) в бензоле (20 мл) перемешивают при температуре окружающей среды в течение 15 ч. Затем добавляют последовательно, по каплям, пиридин (20 мл) и хлорокись фосфора (4,0 мл, 6,55 г, 0,0425 моль). Реакционную смесь перемешивают с обратным холодильником в течение 1 ч, затем полностью удаляют летучие при пониженном давлении. Остаток соединяют с хлороформом, промывают водой, высушивают над безводным сульфатом магния и окончательно высушивают при пониженном давлении. Получают метил-8-циано-5,6-дигидро-2-нафтоат (1,70 г, 0,008 моль, 47%) в виде желтого твердого вещества.
Tпл = 73 - 75oC. Спектр ЯМР1H (в CDCl3):d 8,0 - 7,9 (m, 1H), 7,3 - 7,2 (m, 1H), 6,95 (t, 1H, J = 4,8 Гц), 3,95 (s, 3H), 2,9 (t, 2H, J = 8,3 Гц), 2,6 - 2,4 (m, 3H).
Часть C. Смесь метил-8-циано-5,6-дигидро-2-нафтоата (0,80 г, 0,0038 моль) в метаноле (25 мл) с концентрированной соляной кислотой (0,56 мл) и катализатором-палладий, нанесенный на углерод (0,40 г, 5% Pd/C) - встряхивают в течение 20 ч при комнатной температуре в атмосфере азота (60 фунт/кв. дюйм). Реакционную смесь фильтруют через Celite и промывают метанолом. Фильтрат полностью высушивают при пониженном давлении, остаток растирают с гексаном. Получают рацемическую смесь метил-8-аминометил-5,6,7,8-тетрагидро-2-нафтоата (0,80 г, 0,0037 моль, 97%) в виде белого твердого вещества.
Tпл = 170 - 179oC. Спектр ЯМР1H в DMSO:d 8,2 (m, 4H), 7,9 - 7,7 (m, 6H), 7,5 - 7,2 (m, 4H), 3,9 - 3,8 (m, 7H), 3,3 - 3,2 (m, 10H), 2,0 - 1,6 (m, 8H).
Часть D. К раствору метил-8-аминометил-5,6,7,8-тетрагидро-2-нафтоата (0,78 г, 0,0036 моль) и триэтиламина (0,55 мл, 0,40 г, 0,004 моль) в водном тетрагидрофуране (50%, 75 мл) добавляют частями, в виде твердого вещества, 2-(трет-бутоксикарбонилоксиимино)-2-фенилацетонитрил. Все перемешивают при комнатной температуре в течение 3 ч. Раствор концентрируют до половины объема и экстрагируют диэтиловым эфиром. Затем водный слой подкисляют до pH 1,0 соляной кислотой (1H) и экстрагируют этилацетатом. Объединенные органические слои высушивают над безводным сульфатом магния и упаривают до сухого состояния при пониженном давлении. Остаток очищают хроматографически, используя гексан и этилацетат в соотношении 8:2. Получают метил N-(BOC)-8-аминометил-5,6,7,8-тетрагидро-2-нафтоат (0,54 г 0,0017 моль, 47%) в виде белого твердого вещества.
Tпл = 72 - 80oC. Спектр ЯМР1H (в DMSO):d 13,8 (s, 1H), 7,8 - 7,65 (m, 3H), 7,6 - 7,5 (m, 3H), 7,25 - 7,20 (m, 1H), 7,15 - 7,05 (m, 1H), 3,9 - 3,8 (m, 1H), 3,2 - 2,8 (m, 4H), 1,8 - 1,6 (m, 3H), 1,4 (s, 6H).
Часть E. К раствору метил N-(BOC)-8-аминометил-5,6,7,8-тетрагидро-2-нафтоата (0,50 г, 0,0016 моль) в этаноле (12,5 мл) прикапывают раствор гидроокиси натрия (0,50 г) в воде (12,5 мл). Все перемешивают с обратным холодильником в течение 4 ч. Реакционную смесь концентрируют до половины объема и затем подкисляют до pH 1 соляной кислотой (1H). Остаток очищают хроматографически, используя градиент гексан:этилацетат (1:1), этилацетат, этилацетат: метанол (9:1). Получают рацемическую смесь указанного в заглавии соединения, N-(BOC)-2-амино-этил-5,6,7,8-тетрагидро-2-нафтойной кислоты (0,19 г, 0, 00062 моль, 39%) в виде белого твердого вещества.
Tпл = 172 - 176oC. Спектр ЯМР1H (в DMSO):d 7,8 (s, 1H), 7,65 (d, 1H, J = 8,1 Гц), 7,15 (d, 1H, J = 8,1 Гц), 7,1 - 7,0 (m, 1H), 3,2 - 3,1 (m, 2H), 3,0 - 2,7 (m, 4H), 1,8 - 1,6 (m, 4H), 1,4 (s, 9H).
N-(BOC)-8-аминометил-2-нафтойная кислота (14)
Оставшийся нафтоат (11)
обрабатывают 2,3-дихлоро-5,6,-дициано-1,4-бензохиноном (DDQ) в диоксане для получения конденсированной бициклической системы - метил-8-циано-2-нафтоата (13). Затем метильную группу восстанавливают
действием водорода, и метиловый эфир омыляют до карбоновой кислоты. В эту кислоту вводят защитную N-BOC-группу, и получают N-(BOC)-8-аминометил-2-нафтойную кислоту (14) - промежуточное соединение для
синтеза циклического пептида.
Часть A. Раствор метил 8-циано-5,6-дигидро-2-нафтоата (1,0 г, 0,0047 моль) и 2,3-дихлоро-5,6-дициано-1,4-бензохинона (1,07 г, 0,0047 моль) в диоксане (50 мл) перемешивают при 120oC в течение 16 ч. Реакционную смесь выливают в ледяную воду и экстрагируют этилацетатом. Объединенные органические слои высушивают над безводным сульфатом магния и упаривают до сухого остатка при пониженном давлении. Сухой остаток очищают хроматографически, используя этилацетат.
Получают метил 8-циано-2-нафтоат (0,72 г, 0,0034 моль, 73%) в виде рыжевато-коричневого твердого вещества.
Tпл = 178 - 182oC. Спектр ЯМР1H (в CDCl3):d 8,95 (s, 1H), 8,3 - 8,2 (m, 1H), 8,15 - 8,10 (m, 1H), 8,0 - 7,95 (m, 2H), 7,7 - 7,6 (m, 1H), 4,05 (s, 1H).
Часть B. Смесь метил-8-циано-2-нафтоата (1,0 г, 0,0047 моль) в метаноле (35 мл) с концентрированной соляной кислотой (0,69 мл) и катализатором-палладий, нанесенный на углерод (0,20 г, 5% Pd/C), встряхивают в течение 6 ч при комнатной температуре в атмосфере водорода (50 фунт/дюйм). Реакционную смесь фильтруют через Celite@ и промывают метанолом. Фильтрат упаривают до сухого остатка при пониженном давлении, сухой остаток растирают с гексаном. Получают метил-8-аминометил-2-нафтоат (0,76 г, 0,0035 моль, 75%) в виде масла. Спектр ЯМР1H (в DMSO):d 8,75 (s, 1H), 8,5 (bs, 2H), 8,2 - 8,05 (m, 3H), 7,57 - 7,70 (m, 2H), 4,6 (s, 2H), 3,95 (m, 3H).
Часть C. К раствору метил-8-аминометил-2-нафтоата (0,75 г, 0,0035 моль) в сухом тетрагидрофуране, охлажденному до 0oC, добавляют раствор гидроокиси лития (0,5 М, 5,83 мл). Все перемешивают при комнатной температуре в течение 20 ч. Добавляют следующую аликвоту гидроокиси лития, и все перемешивают еще 20 ч. Отфильтровывают твердый продукт, фильтрат упаривают до сухости.
Твердые продукты растирают с диэтиловым эфиром.
Получают 8-аминометил-2-нафтойную кислоту (0,67 г, 0,0033 моль, 95%) в виде белого твердого вещества.
Tпл = 223 - 225oC. Спектр ЯМР1H (в DMSO):d 8,6 (s, 1H), 8,1 - 7,9 (m, 1H), 7,8 - 7,7 (m, 4H), 7,55 - 7,5 (m, 1H), 7,45 - 7,35 (m, 2H), 4,2 (s, 2H).
Часть D. К раствору 8-аминометил-2-нафтойной кислоты (0,50 г, 0,00025 моль) и триэтиламина (0,038 мл, 0,028 г, 0,000275 моль) в водном тетрагидрофуране (50%, 5 мл), добавляют частями, в виде твердого вещества, 2-(трет-бутоксикарбонилоксиимино)-2-фенилацетонитрил (0,068 г, 0,000275 моль). Все перемешивают при комнатной температуре в течение 5 ч. Раствор концентрируют до половины объема и экстрагируют диэтиловым эфиром. Затем водный слой подкисляют до pH 1 соляной кислотой (1H) и экстрагируют этилацетатом. Объединенные органические слои высушивают над безводным сульфатом магния и упаривают до сухого остатка при пониженном давлении. Получают указанное в заглавии соединение, N-(BOC)-8-аминометил-2-нафтойную кислоту (0,050 г, 0,00017 моль) в виде белого твердого вещества.
Tпл = 190 - 191oC. Спектр ЯМР1H (в DMSO):d 13,11 (s, 1H), 8,8 (s, 1H), 8,0 (q, 2H, J = 7,9 Гц), 7,9 (d, 1H, J = 8,1 Гц), 7,5 (t, 1H, J = 7,5 Гц), 7,65 - 7,55 (m, 2H), 4,6 (d, 2H, J = 5,5 Гц), 1,4 (s, 9H).
Циклическое промежуточное соединение 89a и 89b
Цикло-(D-Val-NMeArg-Gly-Asp-аминотетралинкарбоновая кислота), соединение формулы (VII),
где J - D-Val, K - NMeArg, L - Gly, M - Asp, R1=R2 - H.
Указанное в заглавии соединение получают по общей методике, описанной для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). Для присоединения Boc-аминотетралин-карбоновой кислоты к оксиму используют метод DCC/DMAP. Из пептида, используемого в количестве 0,164 ммоль, получают защищенный циклический пептид (69 мг, 49,3%). Пептид (69 мг) в присутствии 0,069 мл анизола обрабатывают безводным фтористым водородом при 0oC в течение 30 мин. Полученный продукт осаждают эфиром, перерастворяют в водном ацетонитриле и выделяют лиофилизацией указанное в заглавии соединение (59,7 мг, выход выше количественного, рассчитано на фтористую соль).
Очистку осуществляют методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 16,2 - 27% ацетонитрил, содержащий 0,1% TFA, градиент 0,23%/мин с последующей лиофилизацией. Получают соль трифторуксусной кислоты указанного в заглавии соединения в виде белого пушистого твердого вещества, 2 изомера, изомер #1 (выход 12,5%, суммарный выход 6,2%), масс-спектр (FAB): [M+H] = 615,34; изомер #2 (выход 18,6%, суммарный выход 9,3%, масс-спектр (FAB): [M+H] = 615,35.
Циклическое промежуточное соединение 89c
Цикло-(D-Val-NMeArg-Gly-Asp-аминометилнафтойная кислота), соединение формулы (IX), где J - D-Val, K - NMeArg, L - Gly, M - Asp, R1- H, R2 - H.
Указанное в заглавии соединение получают по общей методике, описанной для цикло-(D-Val-NMeArg-Gly-Asp-Mamb) (циклическое промежуточное соединение 4). Для присоединения Boc-аминометилнафтойной кислоты к оксиму используют метод DCC/DMAP. Из пептида, используемого в количестве 0,737 ммоль, получают защищенный циклический пептид (463 мг, 73,1%). Пептид (463 мг) в присутствии 0,463 мл анизола обрабатывают безводным фтористым водородом при 0oC в течение 20 мин. Полученный продукт осаждают эфиром, перерастворяют в водном ацетонитриле и выделяют лиофилизацией указанное в заглавии соединение (349 мг, выход выше количественного, рассчитано на фтористую соль).
Очистку осуществляют методом обращенно-фазной ВЭЖХ на препаративной колонке Vydac C18 (2,5 см), используя 4,5 - 22, 5% ацетонитрил, содержащий 0,1% TFA, градиент 0,45%/мин и затем лиофилизуют, в результате чего получают соль трифторуксусной кислоты указанного в заглавии соединения в виде белого пушистого твердого вещества (выход 12,1%, суммарный выход 7,8%), масс-спектр (FAB): [M+H] = 625,32.
Синтез циклических промежуточных соединений, модифицированных введением "связки".
Циклические промежуточные соединения, модифицированные ведением "связки", могут быть получены либо присоединением предварительно защищенной "связки" к фрагменту цикла с последующим включением этого фрагмента в циклическую систему, либо введением "связки" в циклическое промежуточное соединение.
Фрагменты цикла, модифицированные введением "связки"
Фрагменты цикла,
модифицированные введением "связок", могут быть синтезированы либо присоединением "связки" к фрагменту цикла, содержащему заместители в ароматическом ядре, как описано выше, либо присоединением
предварительно защищенной "связки" в процессе синтеза фрагмента.
Например, описанные выше циклические фрагменты, содержащие заместители в ароматическом ядре, где X=NH2, могут взаимодействовать с сукцинимидильной "связкой", RCOOSu (R= -(CH2)5-NH2 или CH2-C6H5-p-NH2), в результате чего "связка" присоединяется в положение X через амидную группу.
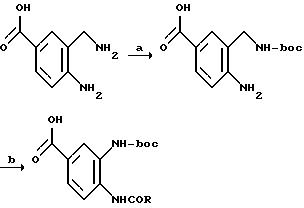
a] Boc-ON, b] RCOOSu
Замещенные в ароматическое ядро фрагменты циклических промежуточных соединений с X=OH могут взаимодействовать со "связкой", полученной из тетраэтиленгликоля. Эта "связка" состоит из четырех этиленов единиц, разделенных эфирными группами, и содержит Z-замещенную аминогруппу на одном конце и отщепляющуюся группу, такую как тозилат, - на другом.
Таким образом может быть получена "связка", присоединенная в положение X через эфирную группу.
Циклические фрагменты, содержащие заместители в ароматическом ядре, где Z-NH2, могут взаимодействовать с (Z-NH(CH2)5CO)2O с образованием "связки", присоединенной в положение Z через амидную группу.
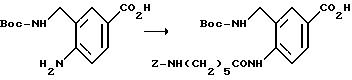
"Связки" могут быть присоединены к фрагменту цикла, содержащему заместители в ароматическом ядре, при Z=OH. Для присоединения "связки" к ароматическому кольцу необходимо использовать "связку", содержащую отщепляющуюся группу, способную взаимодействовать с фенолят-ионом.
Такие отщепляемые группы включают галиды, арилсульфонаты (то есть тозилаты), алкилтозилаты (то есть мезилаты). Например, используют алкильную цепь, содержащую тозильную группу на одном конце и защищенную аминогруппу - на другом. В литературе приводится несколько примеров алкилирования фенильной группы в присутствии карбоксильной группы (например, Brakmann, Kluge and Muxfeldt (1957), Ber. Deutsh. Chem. Ges., 90, 2302).

Фрагменты цикла, содержащие заместители в ароматическом ядре, при Z= CH2NH2 могут взаимодействовать с Z-NH(CH2)nCOOS
с образованием "связки", присоединенной в положении Z через амидометильную группу.
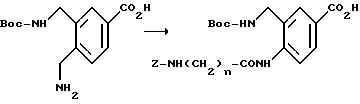
Предшествующие примеры иллюстрируют использование "связок", содержащих концевые защищенные аминогруппы. "Связки", содержащие концевые карбоксильные или эфирные группы, также могут представлять интерес. Некоторые такие "связки" могут быть присоединены к циклическим фрагментам, описанным выше. Например, по следующей схеме, 3-аминометил-4-гидроксибензойная кислота, защищенная трет-Boc-группой, взаимодействует с бензилхлороацетатом и основанием, в результате чего вводится короткая "связка", оканчивающаяся эфирной группой.
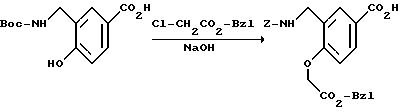
"Связка" может быть присоединена к фрагменту цикла, содержащему заместители в ароматическом ядре, при Y=NH2. Как показано на схеме 8, мягкий щелочной гидролиз метилового эфира 3-аминометил-5-аминобензоата, защищенного трет-Boc-группой с последующим взаимодействием с бензилакрилатом (Lancaster Synthesis, Inc) в присутствии катализатора - уксусной кислоты - приводит к получению продукта присоединения по Михаэлю. Даже если фрагмент цикла, модифицированный введением "связки", содержит незащищенную вторичную аминогруппу, он может быть непосредственно использован в твердофазном синтезе. Однако, если необходимо, может быть осуществлена защита аминогруппы действием бензилхлороформиата и мягкого основания.
Связка также может быть введена в процессе синтеза циклического фрагмента. Примером является синтез циклического фрагмента, модифицированного "связкой" 5-Aca-Mamb.
Синтез Boc-Mamb (Z-5-Aca).
Этот синтез показан на схеме 9.
Часть A. Метил-3-нитро-5-гидроксиметилбензоат
К раствору монометил-3-нитроизофталата добавляют 2,0 M RMS (борный комплекс метил-сульфида) в ТГФ (880 мл, 1,76 моль), по каплям в течение 1 ч. Полученный раствор нагревают с обратным
холодильником в течение 12 ч и затем медленно добавляют MeOH (750 мл), чтобы погасить реакцию. После концентрирования получают желтое твердое вещество, которое перекристаллизовывают из толуола (297,5
г, 80%).
Спектр ЯМР1H (в CDCl3): 8,71 - 8,70 (m, 1H), 8,41 - 8,40 (m, 1H), 8,31 - 8,30 (m, 1H), 4,86 (s, 1H), 3,96 (s, 3H), 2,47 (s, 1H). Тпл. 76,5 - 77,5oC. Масс-спектр (DCl): [M+H]=212.
Часть B. 3-карбометокси-5-нитробензилметансульфокислота
Метил 3-нитро-5-гидроксиметилбензоат (296,0 г, 1,40 моль) и
акцептор протонов (360,8 г, 1,68 моль) растворяют в этилендихлориде (150 мл). Ангидрид трифторметансульфокислота (292,3 г, 1,68 моль), растворенный в этилендихлориде (800 мл), по каплям добавляют к
суспензии в течение 90 мин и перемешивают смесь 18 ч в атмосфере азота. Реакцию гасят водой (2000 мл), разделяют два слоя, затем органический слой промывают порциями по 1000 мл, 1H HCl, H2O,
насыщенным раствором NaHCO3, H2O и насыщенным раствором NaCl.
Органический слой высушивают (MgSO4) и концентрируют при пониженном давлении. Полученное желтое твердое вещество перекристаллизовывают из толуола и получают указанное в заглавии соединение в виде рыжевато-коричневого твердого вещества (366,8 г, 91%).
Спектр ЯМР1H в CDCl3: 8,84 - 8,85 (m, 1H), 8,45 - 8,46 (m, 1H), 8,40 - 8,39 (m, 1H), 5,35 (s, 2H), 3,98 (s, 3H), 3,10 (s, 3H). Тпл. 96 - 97oC. Масс-спектр (DCl): [M+NH4]=307.
Часть C. Метил-3-азидометил-5-нитробензоат
3-Карбометокси-5-нитробензилметансульфонат (300,0 г, 1,04 моль) и азид натрия (81,0 г, 1,25 моль)
суспендируют в DMF (1700 мл) и перемешивают при комнатной температуре в течение 5 ч. Реакционную смесь разбавляют этилацетатом (2000 мл), промывают, порциями по 1000 мл, H2O (2 раза) и
насыщенным раствором NaCl (1 раз), высушивают (MgSO4) и затем концентрируют при пониженном давлении. Полученный янтарный сироп высушивают в вакууме при 40oC и получают указанное
в заглавии соединение в виде рыжевато-коричневого твердого вещества (226,5 г, 92%).
Спектр ЯМР1H (в CDCl3): 8,60 (s, 1H), 8,26 (s, 1H), 8,20 (s, 1H), 4,52 (s, 2H), 3,88 (s, 3H). Тпл. 44 - 46oC.
Часть D. Метил-3-амино-5-аминометилбензоат
Раствор метил-3-азидометил-5-нитробензоата (15,50 г, 65,7 ммоль) и
бензолсульфокислоты (22,14 г, 140 ммоль) в теплом метаноле помещают во встряхивающийся реактор Парра и продувают азотом в течение 15 мин. Добавляют катализатор-палладий, нанесенный на углерод (10%
Pd/C, 4,0 г), и проводят 7 раз продувку встряхивающегося реактора водородом по циклической схеме - подача давления - сброс давления, после этого сбрасывают давление и встряхивают 18 ч, в течение этого
времени поглощается требуемое количество водорода. Катализатор удаляют фильтрацией через слой Calite, после концентрирования фильтрата при пониженном давлении получают рыжевато-коричневое масло.
Растирают с перегнанным EtOAc (2х150 мл), затем охлаждают 12 ч при -5oC и получают рыжевато-коричневое твердое вещество, которое собирают фильтрованием, промывают EtOAc (2х150 мл) и
высушивают под вакуумом (25,82 г, 80%).
Спектр ЯМР1H (в CD3OD): 8,25 - 8,33 (m, 1H), 8,07 - 8,06 (m, 1H), 7,86 - 7,80 (m, 5H), 7,49 - 7,42 (m, 6H), 4,29 (s, 2H), 3,97 (s, 3H).
Часть E. Метил-3-амино-5-(трет-бутоксикарбониламино)-метилбензоат
Проводят взаимодействие в растворе MeOH (350 мл) метил-3-амино-5-аминометилбензоата (19,32 г,
39,0 ммоль), TFA (7,89 г, 78,0 ммоль) и ди-трет-бутилдикарбоната (8,51 г, 39,0 моль) в течение 24 ч при комнатной температуре. После концентрирования получают бесцветное твердое вещество.
Очистку проводят хромографически (силикагель, смесь гексана и EtOAc, 1: 1). Получают целевой продукт (9,21 г, 84%) в виде бесцветного твердого вещества.
Спектр ЯМР1H (в CD3OD): 7,26 - 7,25 (m, 2H), 6,86 - 6,85 (m, 1H), 4,16 (s, 2H), 3,88 (s, 3H), 1,48 (s, 9H). Тпл. 57 - 65oC. Масс-спектр (ESI): [M+H)=281.
Часть F. Boc-Mamb(Z-5-Aca)-OMa
N-CBZ-e-аминокапроновую кислоту (7,77 г, 29,3 ммоль) и TEA (2,97 г, 23,3 моль) растворяют в безводном ТГФ (250 мл) и охлаждают до -20oC. Добавляют по
каплям изобутилхлороформиат (4,00 г, 29,3 ммоль) и проводят реакцию в течение 5 мин при -20oC. Метил-3-амино-5-(трет-бутоксикарбониламино)метилбензоат (8,20 г, 29,3 ммоль), растворенный в
безводном ТГФ (50 мл), охлаждают до -20oC и добавляют к реакционной смеси. Реакционной смеси дают медленно нагреться до комнатной температуры и перемешивают еще 2 дня. Твердые продукты
удаляют фильтрацией, фильтрат концентрируют при пониженном давлении. Полученный остаток растворяют в EtOAc (125 мл) и промывают, порциями по 50 мл, 0,2 H HCl, насыщенным раствором NaHCO3 и
насыщенным раствором NaCl. Органический слой высушивают (MgSO4) и концентрируют при пониженном давлении. Полученный продукт очищают хроматографически (силикагель, смесь гексан: EtOac, 1:2)
и перекристаллизовывают из CCl4, получают указанное в заглавии соединение (10,09 г, 65%) в виде бесцветного твердого вещества.
Спектр ЯМР1H в CDCl3: 8,03 - 7,63 (m, 3H), 7,32 - 7,28 (m, 5H), 5,12 - 4,92 (m, 4H), 4,27 - 4,25 (m, 2H), 3,85 (s, 3H), 3,17 - 3,12 (m, 2H), 2,34 - 2,28 (m, 2Y), 1,72 - 1,66 (m, 2H), 1,48 - 1,53 (m, 2H), 1,43 (s, 9H), 1,36 - 1,34 (m, 2H). Тпл. 52 - 54oC.
Масс-спектр (ESI): [M+H]=528.
Часть G. Boc-Mamb(Z-5-Aca)
Boc-Mamb(Z-5-Aca)-OMe (22,58 г, 43,0 ммоль)
растворяют в смеси 1:1 1N NaOH и MeOH (500 мл) и перемешивают при комнатной температуре 18 ч. Реакционную смесь разделяют между EtOAc (300 мл) и H2O (200 мл) и отделяют слои. Значение pH
водного слоя понижают до 4,5 и полученный маслянистый осадок экстрагируют EtOAc (2х300 мл). Органический экстракт высушивают (MgSO4), после концентрирования получают желтое твердое
вещество. Твердое вещество растирают с перегнанным CCl4 (3х100 мл) и получают целевой продукт (14,47 г, 64%) в виде бесцветного твердого вещества. Спектр ЯМР1H (в CD3
OD): 8,04 (s, 2H), 7,71 - 7,66 (m, 2H), 7,30 - 7,23 (m, 5H), 5,02 (s, 2H), 4,24 (s, 2H), 3,32 (s, 3H), 3,11 (t, J = 6,8 Гц, 2H), 2,34 (t, J = 6,8 Гц, 2H), 1,74 - 1,35 (m, 15H). Тпл. 168
- 169oC. Масс-спектр (DCl):[M+NH4)=531.
На схеме 10 показано, каким образом может быть синтезирован фрагмент цикла, содержащий "связку", присоединенную через противоположную амидную функциональную группу. Восстановление нитрогруппы монометил-3-нитроизофталата (Fluka) с использованием палладия, нанесенного на углерод, позволяет получить монометил-3-аминоизофталат, который может быть превращен в соответствующий нитрил по методике Sandmeyer. Обработка этого эфира монозащищенным диамином позволяет получить соответствующий амид. Защитная группа диамина должна быть устойчивой при гидрировании. На схеме показано использование Теос(2-триметилсилилэтилоксикарбонильной)группы, но могут быть использованы и другие известные специалистам в этой области подобные группы. Восстановление нитрильной группы с использованием палладия, нанесенного на углерод, позволяет получить циклический фрагмент, модифицированный "связкой" (см. схему 10).
"Связки", присоединенные в положении Y ароматического кольца циклического фрагмента через эфирную группу, могут быть получены, используя в качестве исходного соединения 3-гидрокси-5-аминобензойную кислоту. Для перевода амина в 3-гидрокси-5-цианобензойную кислоту может быть использована реакция, предложенная Sandmeyer. "Связку" вводят алкилированием. Восстановление нитрильной группы с использованием катализатора-палладия, нанесенного на углерод, позволяет получить аминометильную группу. Защита аминогруппы трет-Boc-группой, используя ди-трет-бутилдикарбонат, позволяет получить циклические фрагменты, содержащие "связку", готовые для применения в твердофазном синтезе. Это показано на схеме 11.
"Связки", оканчивающиеся на группу карбоновой кислоты, могут быть получены с использованием циклических ангидридов. Схема 12 иллюстрирует такой синтез с использованием янтарного ангидрида. Реакция метил-3-аминометил-5-аминобензоата, защищенного трет-Boc-группой, с янтарным ангидридом, дает "связку" из карбоновой кислоты. Активация карбоновой кислоты и конденсация с бензилгидразином (Lancaster Synthesis, Inc) позволяет получить замещенный гидразид. Этот гидразид служит для защиты карбоновой кислоты во время синтеза. Гидролиз метилового эфира приводит к получению циклического фрагмента, модифицированного "связкой" в форме, готовой для твердофазного синтеза. После окончания синтеза отщепление Cbz-защитной группы от гидразида открывает путь для синтеза азида и присоединения азида к комплексообразователю (Hofmann, Magee and Lindenmann (1950) J.Amer. Chem. Soc., 72, 2814). Это показано на схеме 12.
"Связки" также могут введены в процессе синтеза других циклических фрагментов. Например, "связка", модифицирующая гетероциклический фрагмент циклической системы, может быть получена из 4-амино-6-карбэтокси-1-гидроксиметилпиримидина (Boger (1994), J.Amer, Chem. Soc, 116, 82 - 92). Спирт может быть превращен в амин в три стадии.
Сначала взаимодействие с толуолсульфонил хлоридом и основанием позволяет получить тозилат, который после обработки азидом натрия дает азид. Восстановление азида с использованием катализатора-палладия, нанесенного на углерод, приводит к диамину. Значительное различие в нуклеофильности двух аминогрупп позволяет осуществить селективную защиту аминометильной группы, используя ди-трет-бутилдикарбонат. Присоединение защищенной "связки", такой как Z-5-Aca, к оставшейся аминогруппе может быть проведено с использованием смешанного ангидрида или симметричного ангидрида. Наконец, гидролиз этилового эфира позволяет получить гетероциклический фрагмент циклической системы, модифицированный "связкой", который используется в твердофазном синтезе. Это показано на схеме 13.
"Связки"
Синтез "связки" на основе тетраэтиленгликоля, рассмотренный выше, показан на схеме 14. В качестве исходного соединения используют 1-амино-11-азидо-3,6,
9-триоксаундекан (Bertozzi and Bednarski (1990), J.Org.Chem, 56, 4326-4329). Восстановление азида с использованием катализатора-палладия, нанесенного на углерод, приводит к амину, который защищают
Cbz-группой (обозначено как Z на схеме 14, и далее). Спирт превращают в тозилат действием толуолсульфохлорида и основания.
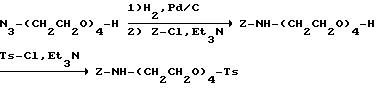
Другой тип "связок", включающих этиленгликольные остатки, показан на следующий схеме. Эта "связка" имеет на одном конце группу карбоновой кислоты, вследствие чего может быть присоединена к фрагменту цикло, содержащему функциональную аминогруппу. Синтез начинается с описанного выше введения Cbz-защиты в аминоспирт. Обработка спирта этилдиазоацетатом и (Rh)2(OAC)4 приводит к образованию эфира гликолевой кислоты, включающего тетраэтиленгликолевый "хвост". В результате гидролиза этилового эфира получают "связку", готовую для введения в циклический фрагмент. Это показано на схеме 15:
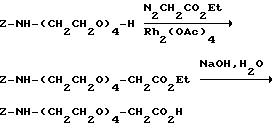
Приведенные ниже фрагменты циклической структуры, модифицированные введением "связки", могут быть использованы в синтезе промежуточных циклических соединений, модифицированных введением "связки".
Циклическое
соединение 1, модифицированное введением "связки" цикло-(D-Val-NMeArg-Gly-Asp-Mamb(5-Aca))
Синтез указанного в заглавии соединения показан на приведенной ниже схеме 16.
В 60 мл - колбу для реакции пептида добавляют смолу - оксим (1,61 г, степень замещения = 0,62 ммоль/г). После однократного смачивания DMF (30 мл) смола набухает. В реакционную колбу добавляют Boc-Mamb(Z-5-Aca) (513 мг, 1,0 ммоль), HBTU (379 мг, 1,0 ммоль) и DIEA (0,52 мл, 3 ммоль). Суспензию перемешивают при комнатной температуре 96 час. Смолу тщательно промывают, порциями по 30 мл, DMF (3 раза), MeOH (1 раза), DCM (3 раза), MeOH (2 раза) и DCM (3 раза). Уровень замещения, определенный по тесту с пикриновой кислотой, составляет 0,381 ммоль/г.
Затем проводят следующие стадии. Стадия 1 - смолу промывают, порциями по 30 мл, DMF (3 раза), MeOH (1 раз), DCM (3 раза), MeOH (2 раза) и DCM (3 раза).
Стадия 2 - смолу замачивают 30 мл 50% TFA в DCM и удаляют трет-Boc-защиту действием 30 мл 50% TFA (трифторуксусной кислоты) в течение 30 мин. Стадия 3 - смолу тщательно промывают DCM (3 раза), MeOH (1 раз), DCM (2 раза), MeOH (3 раза) и DMF (3 раза). Стадия 4 - к смоле добавляют Boc-Asp(OBz1) (0,982 г, 3,04 ммоль), HBTU (1,153 г, 3,04 ммоль), DIEA (1,59 мл, 9,14 ммоль) и DMF (14 мл) и проводят реакцию в течение 22 час. Стадия 5 - Завершенность реакции присоединения контролируют по тесту с пикриновой кислотой. Стадии 1-5 повторяют до тех пор, пока не будет достигнут требуемый результат.
После того, как собран линейный пептид, удаляют N-концевую трет-Boc-группу промывкой 50% TFA в DCM, а затем обработкой 30 мл 50% TFA в DCM в течение 30 мин. Смолу затем тщательно промывают DCM (3 раза), MeOH (2 раза), DCM (3 раза) и затем нейтрализуют порциями по 30 мл 10 DIEA в DCM (2 раза по 1 мин). Смолу промывают DCM (3 раза) и MeOH (3 раза) и после сушки в вакууме получают 1,965 г коричневой смолы. Смолу циклизуют суспендированием в DMF (20 мл), содержанием HOAc (35 мкл, 0,609 ммоль) и нагревают при 50oC в течение 72 ч. Смолу отфильтровывают через воронку со стеклянным фильтром и тщательно промывают 10 мл DMF (3 раза). DMF - фильтрат выпаривают, и полученное масло перерастворяют в смеси ацетонитрила и H2O (1:1, 20 мл), после выделения лиофилизацией получают защищенный циклический пептид (342 мг). Очистку проводят методом "обращеннофазной" ВЭЖХ на препаративной колонке Vydac C18 (2,1 см) с изократной подвижной фазой ацетонитрил : H2O (1:1), содержащей 0,1% TFA. Лиофилизация фракции целевого продукта позволяет получить очищенный защищенный пептид (127 мг).
Защиту пептида (120 мг, 0,11 ммоль) снимают действием TFA (1 мл) и ангидрида трифторметилсульфоновой кислоты (1 мл) в присутствии анизола (0,2 мл) в течение 3 час при - 10oC. Пептид осаждают добавлением эфира и охлаждением до - 35oC в течение 1,5 час. Пептид собирают фильтрацией, промывают эфиром и высушивают. Полученное твердое вещество растворяют в смеси 1:1 ацетона и H2O (12 мл) и pH доводят до 4-6 обработкой ионообменной смолой Rad AGI-8x ацетат. Смолу отфильтровывают и промывают водой. Фильтрат лиофилизуют и получают хроматографически чистый пептид (75 мг, суммарный выход 13,5%); масс-спектр (FAB): [M+H]=703,3951.
Циклическое
соединение 2, модифицированное введением "связки" Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(5-Aca))
Указанное в заглавии соединение получают по общей методике, описанной для
цикло-(D-Val-NMeArg-Gly-Asp-Mamb(5-Aca)). Из пептида, используемого в количестве 1,35 ммоль, получают неочищенный циклический защищенный пептид (1,05 г, 73%). Удаление защиты пептида (500 мг)
осуществляют действием TFA (4 мл) и ангидрида трифторметилсульфоновой кислоты (4 мл) в присутствии анизола (0,8 мл) в течение 3 час при -10oC. Пептид осаждают эфиром и охлаждают до - 35oC в течение 1,5 час. Пептид отделяют фильтрацией, промывают эфиром и высушивают. Полученное твердое вещество растворяют в смеси (50 мл) 1:1 ацетона и воды и лиофилизуют. Очистку проводят
методом обращеннофазной ВЭЖХ на препаративной колонке Vydac C18 (2,1 см), используя 9-18% ацетонитрил, содержащий 0,1% TFA (градиент 0,36%/мин), и затем лиофилизацией. Получают соль трифторуксусной
кислоты указанного в заглавии соединения в виде бесцветного пушистого твердого вещества (218 мг, выход 69%, суммарный выход 37%); масс-спектр (FAB):[M+H]=689,3735.
Модифицированные
введением "связки" циклические соединения 3-8
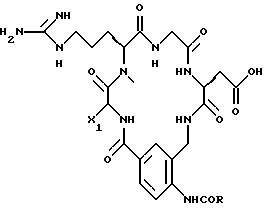
R - -(CH2)5-NH2 или CH2-C6H5-p-NH2
x1 - 2-пропил, этил или n-гидроксифенилметил.
Соединения цикло-(D-Val-NMeArg-Gly-Asp-Mamb(4-NHCOR), цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(4-NHCOR) и цикло-(D-Tyr-NMeArg-Gly-Asp-Mamb(4-NHCOR) могут быть получены по методике, описанной выше.
"Связки" могут быть введены в процессе синтеза циклических промежуточных соединений.
Модифицированные введением "связки" циклические соединения 9, 10 и 11
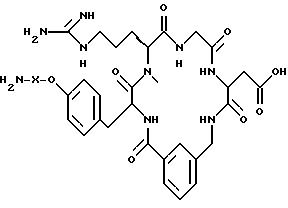
x - CH2CH2, CH2CH2CH2, CH2CH2CH2CH2,
Цикло(O-2-аминоэтил-D-Tyr)-NMeArg-Gly-Asp-Mamb),
Цикло(O-3-аминопропил-D-Tyr)-NMeArg-Gly-Asp-Mamb),
Цикло(O-4-аминобутил-D-Tyr)-NMeArg-Gly-Asp-Mamb):
Эти соединения могут быть получены по методике, описанной выше для цикло-(D-Val-NMeArg-Gly-Asp-Mamb), используя модифицированный введением "связки" D-Tyr. O-замещенный D-Tyr может быть получен алкилированием Boc-D-Tyr 2-бромэтиламином (или 3-бромопропиламином, 4-бромобутиламином) с защищенной аминогруппой в присутствии основания.
"Связки" также могут быть присоединены уже к готовому циклическому промежуточному соединению.
Модифицированное введением "связки" циклическое соединение 12 цикло-(D-Lys(5-Aca)-NMeArg-Gly-Asp-Mamb).
Синтез этого соединения показан на схеме 17.
Проводят взаимодействие в растворе цикло-(D-Lys-NMeArg-Gly-Asp-Mamb) (100 мг, 0,12 ммоль), Boc-5-аминокапроновая кислота гидроксилсукцинимид эфира (47 мг, 0,144 моль) и Et3N (50 мл, 0,36 ммоль) в DMF (1,50 мл) при комнатной температуре в течение 60 мин. Ход реакции контролируют "нормальнофазной" TLC (90:8:2, CHCl3:MeOH:HOAc), используя нингидрин и пробу Sakaguehi. DMF удаляют при пониженном давлении. Неочищенный продукт обрабатывают TFA (3 мл) при комнатной температуре в течение 45 мин для удаления трет-Boc-защитных групп. TFA удаляют при пониженном давлении и конъюгат очищают методом "обращеннофазной" ВЭЖХ на препаративной колонке Vydac C18 (2,1 см), используя 6% ацетонитрил, содержащий 0,1% TFA, в течение 20 мин, затем 6-36% ацетонитрила, содержащий 0,1% TFA с градиентом 3,0%/мин. После выделения лиофилизацией получают соль трифторуксусной кислоты указанного в заглавии соединения в виде бесцветного пушистого твердого вещества (80 мг, 70%).
Модифицированное введением "связки" циклическое соединение 13 цикло([3-(4-гидроксифенил)пропил-D-Lys]-NMeArg-Gly-Asp-Mamb)
Раствор
N-сукцинимидил-3-(4-гидроксифенил)-пропионата (Bolton-Hunter rcagent; 0,022 г, 0,08 ммоль) и DIEA (0,02 мл, 0,10 ммоль) в диоксане (5 мл) добавляют к раствору цикло-(D-Lys-NMeArg-Gly-Asp-Mamb) (0,026
г, 0,04 ммоль) в фосфатном буфере с pH 9 (5 мл) и проводят реакцию при перемешивании в течение 2 дней. Раствор лиофилизуют, и полученное белое твердое вещество очищают методом "обращеннофазной" ВЭЖХ
на препаративной колонке Vydac C18 (2,1 см), используя 9-18% ацетонитрил, содержащий 0,1% TFA (градиент 0,36%/мин), и получают целевой продукт (0,018 г, 60%) в виде бесцветного твердого вещества.
Тпл. = 146 = 155oC.
Масс-спектр (ESI):[M] = 751.
Модифицированное введением "связки" циклическое соединение 14
цикло((N-E-Tyr-D-Lys)-NMeArg-Gly-Asp-Mamb)
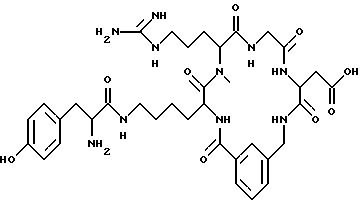
Данное соединение может быть получено по реакции цикло-(D-Lys-NMeArg-Gly-Asp-Mamb) с Boc-Tyr-OSu в растворителе, таком как DMF, в присутствии основания, такого как триэтиламин, с последующим снятием защиты.
Модифицированное
введением "связки" циклическое соединение 15 цикло-(N-E-(4-аминофенилацетил)D-Lys)-NMeArg-Gly-Asp-Mamb)
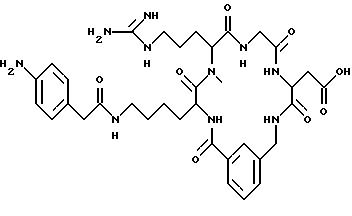
Указанное соединение может быть получено по реакции цикло-(D-Lys-NMeArg-Gly-Asp-Mamb) с сукцинимидил-fmoc-4-аминофенилацетатом в растворителе, таком как DMF, в присутствии основания, такого как триэтиламин с последующим снятием защиты.
Модифицированное введением "связки" циклическое соединение 16
цикло-(N-E-(4-амино-2-гидроксибензоил)-D-Lys)-NMeArg-Gly-Asp-Mamb)

Указанное соединение может быть получено по реакции цикло-(D-Lys-NMeArg-Gly-Asp-Mamb) с сукцинимидил-4-амино-2-гидроксибензоатом в растворителе, таком как DMF или TNF, в присутствии основания, такого как триэтиламин.
Ряд модифицированных введением "связки" циклических соединений может быть получен использованием бифункциональных сшивающих агентов, применяемых для модификации белков. Эти реагенты содержат две электрофильные группы, такие как активные эфирные или изоцианатные, разделенные пространственной группировкой. Такие реагенты могут быть гомобифункциональными, если две реагирующие группы идентичны, или гетеробифункциональными. Пространственная группировка может быть алифатической или ароматической и может содержать дополнительные функциональные группы для модификации липофильности конъюгатов или для разделения цепи. Следующие примеры проиллюстрируют использование некоторых выпускаемых сшивающих агентов с использованием в качестве исходного соединения циклического промежуточного соединения, полученного с использованием 4-аминометил-Mamb структурных единиц.
В соответствии с первым примером циклическое соединение обрабатывают избытком DSS (дисукцинимидил-пробковая кислота, Pierca Chemical Co.) в водном или органическом растворителе при pH между 7 и 8. Это типичные условия реакции для таких сшивающих реагентов. Избыток сшивающего реагента сводит к минимуму образование димеров. Величина pH 7-9 позволяет аминогруппе взаимодействовать с достаточной скоростью, но не позволяет пройти заметному гидролизу второй реакционноспособной группы и препятствует взаимодействию с гуанидиновой группой аргинина. Активная эфирная группа на конце "связки" достаточно стабильна, что делает возможным проведение очистки методом ВЭЖХ или испарительной хроматографии. После очистки модифицированное введением "связки" циклическое соединение может вступать во взаимодействие с комплексообразователем, содержащим нуклеофильные группы, такие как аминогруппа или тиольная группа. Это показано на схеме 18.
Гетеробифункциональные реагенты обычно используют для проведения весьма селективной активации пептидов и белков. В следующем примере SMPB (сукцинимид 4-(n-малеинимидофенил)бутират, Pierce Chemical Co.) используют для модификации аминофункционального циклического соединения и подготовки его к взаимодействию с тиолсодержащим комплексообразователем. Обработка циклического соединения SPMB в слабощелочной среде приводит к образованию модифицированного введением "связки" соединения, в котором "связка" имеет на конце малеинимидную группу. Селективность достигается за счет того, что малеинимидная группа обладает низкой реакционной способностью по отношению к аминогруппе, и димеризация минимальна. После очистки малеинимидная группа может соединяться с тиолсодержащим комплексообразователем. Это показано на схеме 19.
"Связки", содержащие внутренние функциональные группы, могут быть получены с использованием реагентов, показанных на схеме 20, EGS (этиленгликоль-бис-(сукцинимидилсукцинимидат), Sigma Chemical Co. является биссукцинимидил-эфиром, который преимущественно реагирует с амином. Диметил 3,3'-дитио-бис-пропионимидат (DTBP, также называемый реагентом Ванга и Ричардса; Pierce Chemical Co.) также преимущественно взаимодействует с аминами. Дисульфид расцепляется тиолами. Meares и cjmp. показали (Int. J.Cancer: Supplement 2, 1988, 99-102), что конъюгаты антитело-хелат, меченные111In, присоединенные дисульфидсодержащей "связкой", характеризуются более быстрым снижением радиоактивности (на мышах), чем конъюгаты; которые не содержат способной расщепляться "связки". Третий пример на схеме 20 демонстрирует использование BSOCOES (бис-[2-(сукцинимидоксикарбонилокси)этил]сульфон, Pierce Chemical Co), гомобифункционального сшивающего агента, который содержит внутреннюю сульфоновую группу. При присоединении этого реагента к амину образуется фрагмент карбаминовой кислоты.
Схема 21 иллюстрирует использование бисизоцианатов и бистиоцианатов в синтезе модифицированных введением "связки" циклических соединений. Эти реагенты взаимодействуют с аминами с образованием мочевинных и тиомочевинных групп, соответственно. Эти реагенты используются в избытке с целью уменьшения образования димеров.
Изоцианатные и тиоизоцианатные группы на конце "связки" достаточно стабильны для проведения очистки продукта.
Комплексообразователи
Настоящее изобретение также позволяет
получить новые реагенты, необходимые для синтеза радиоактивных фармацевтических препаратов. Эти реагенты состоят из комплексообразователя, Ch, присоединенного через связующую группу, Ln, к циклическому промежуточному соединению Q. Эти реагенты могут быть получены несколькими путями, либо присоединением комплексообразователя к модифицированному введением "связки" циклическому
промежуточному соединению, или присоединением комплексообразователя, несущего связующую группу, к циклическому промежуточному соединению. Преимущественно комплексообразователь присоединяют к
циклическому промежуточному соединению, модифицированному введением "связки".
Самые различные комплексообразователи могут быть использованы в данном изобретении, при условии, что они образуют стабильный комплекс с радиоактивными изотопами. Типичными радиоактивными изотопами являются металлы или переходные металлы, координационное соединение с комплексообразователем является комплексом металл-хелат. Примеры комплексов металл-хелатт можно найти в недавнем обзоре (S. Jurisson et al., Chem.Rev., 1993, 93, 1137-1156), приведенном здесь в качестве ссылки.
Комплексообразователи могут быть присоединены к "связкам" множеством способов, известных специалистам в данной области. В общем, реакционноспособная группа "связки" может взаимодействовать с комплексообразователем или, иначе, реакционноспособная группа комплексообразователя может взаимодействовать со "связкой". Подходящие реакционноспособные группы включают активированные эфиры, изотиоцианаты, алкил- и арилгалилы, амины, тиолы, гидразины, малеинимиды и т.п. Некоторые модифицированные введением "связки" циклические соединения, содержащие реакционноспособные группы, описаны в приведенных ниже примерах.
Типичные комплексообразователи: диэтилентриамин-пентауксусная кислота (DTPA) этилендиамин-тетрауксусная кислота (EDTA), 1,4,7,10-тетраазоциклододекан-N, N', N'', N'''-тетрауксусная кислота (DOTA), 1,4,7,10-тетраазациклододекан-N, N', N''-триуксусная кислота, гидроксибензилэтилен-диамин диуксусная кислота, N,N'-бис(пиридоксил-5-фосфат)этилендиамин, N, N'-диацетат, 3,6,9-триаза-12-окса-3,6,9-трикарбоксиметилен-10-карбокси-13-фенил -тридекановая кислота, 1,4,7-триазациклононан-N, N', N''-триуксусная кислота, 1,4,8,11-тетраазацикло-тетрадекан-N, N', N'', N'''-тетрауксусная кислота, 2,3-бис(S-бензоил) меркаптоацетамидопропановая кислота и описанные ниже комплексообразователи.
Другие комплексообразователи могут включать связывающие
металл фрагменты, полученные из белков, связывающих металл, таких например, как богатый сульфгидрильными группами белок цитоплазмы, содержащийся в позвоночных животных, беспозвоночных животных и
грибах
Синтез комплексообразователей
Синтез 4,5-бис((S-бензоил)меркаптоацетамидо) пентановой кислоты (mapt)
Комплексообразователь получают, как описано в: Fritzberg et al.,
Appl. Radiat. Isot. 1991, 42, 525-530.
Синтез (S-бензоил)меркаптоацетилглицидил-глицидилглицина (MAG3)
Комплексообразователь получают, как описано в: Brandau, W,
et al., Appl. Radiat. Isot. 1988, 39, 121-129.
Синтез сукцинимидил 6-Boc-гидразинопиридин-3-карбоксилат (SHNH)
Комплексообразователь получают, как описано в: Schwartz et al.,
1990, European Patent Application 90301949.5.
Синтез эфира N-[4-(карбокси)бензил]-N,N'- бис[(2-трифенилметилтио)этил] глицинамид N-гидроксисукцинимида
Синтез указанного в
заглавии соединения проиллюстрирован ниже на схеме 22
Часть A. S-трифенилметил-2-аминоэтантиол
Раствор цистеинамина гидрохлорида (79,5 г, 0,7 моль) в трифторуксусной кислоте (TFA)
(500 мл) обрабатывают трифенилметианолом (182 г, 0,7 моль) и перемешивают при комнатной температуре 1 час. Затем TFA удаляют при пониженном давлении при 45oC и полученное темно-оранжевое
масло растворяют в EtOAc (700 мл). Раствор в EtOAc промывают холодной 2N NaOH (3х350 мл), H2O (2х350 мл), насыщенным раствором бикарбоната натрия (350 мл) и насыщенным раствором NaCl (350
мл). Объединенные промывочные воды повторно экстрагируют EtOAc (350 мл). Объединенные органические слои высушивают (MgSO4) и после концентрирования получают желтое твердое вещество. После
растирания с эфиром (500 мл) получают целевой продукт (97,2 г, 43%) в виде бесцветного твердого вещества. Tпл. = 90-92oC (по D.Brenner et. al., J. Inorg. Chem. 1984, 23,
3793-3797, Tпл. = 93-94oC).
Концентрирование эфирной вытяжки до объема 100 мл с последующим охлаждением позволяет дополнительно получить 40,9 г продукта с Tпл. = 89-91oC, суммарный выход составляет 62%.
Часть B. N-2-бромацетил-S-трифенилметил-2-аминоэтантиол
Раствор S-трифенилметил-2-аминоэтантиола (48 г, 0,15
моль) и Et3N (20,9 мл, 0,15 моль) в DOM (180 мл) медленно добавляют при перемешивании к раствору бромацетилбромида (13,9 мл, 0,15 моль) в DCM (100 мл) при температуре -20oC.
Затем реакционной смеси дают нагреться до комнатной температуры в течение 1 час, после чего промывают, порциями по 500 мл, водой, 0,2 H HCl, насыщенным раствором NaHCO3 и насыщенным
раствором NaCl. Органический раствор высушивают (MgSO4). После концентрирования получают маслянистый продукт, который перекристаллизовывают из смеси ДСМ-гексан и получают бесцветное твердое
вещество Tпл. = 137-139,5oC (по J.A.Wolff, Ph.D.Thesis, Massachusetts Institute of Technology, February 1992, Tпл. = 130-135oC.
Часть C - N,
N'-бис[(2-трифенилметилтио)этил]глицинамид
Раствор N-2-бромацетил-S-трифенилметил-2-аминоэтантиола (35,2 г, 0,08 моль), S-трифенилметил-2-аминоэтантиола (25,5 г, 0,08 моль) и Et3N
(16,7 мл, 0,12 моль) в DCM (375 мл) выдерживают при комнатной температуре 24 час. Затем раствор промывают, порциями по 200 мл, водой (1 раз), насыщенным раствором NaHCO3 (2 раза), водой (1
раз), насыщенным раствором NaCl (1 раз), высушивают (MgSO4) и после концентрирования получают вязкое масло, которое растворяют в смеси DCM : EtOH (70:30, 150 мл) и охлаждают на бане со
льдом. Образовавшееся твердое вещество удаляют фильтрацией. Фильтрат концентрируют до состояния вязкого масла. Это масло очищают хроматографически на силикагеле (200-400 меш, 60 A), используя в
качестве подвижной фазы смесь DCM:EtOAc в соотношении 70:30, и получают целевой продукт (34,4 г, 63%) в виде бесцветного аморфного пенистого твердого вещества.
Спектр ЯМР1 H (в CDCl3): 7,42 - 7,18 (m, 30H), 3,12 - 3,01 (m, 4H), 2,48 - 2,27 (m, 6H).
Часть D - Метил-4-(метансульфонилметил)бензоат
Раствор метил 4-(гидроксиметил)бензоата
(10,8 г, 0,065 моль) и акцептора протонов (19,5 г, 0,091 моль) в ДСМ (200 мл) обрабатывают ангидридом метансульфоновой кислоты (13,94 г, 0,08 моль) и перемешивают при комнатной температуре в течение
20 час. Реакционную смесь промывают, порциями по 100 мл, водой (1 раз), 1H HCl (2 раза), H2O (1 раз), насыщенным раствором NaHCO3 (1 раз) и H2O (1 раз). Органическую
фазу высушивают, и после концентрирования получают 15,5 г бледно-желтого твердого вещества. Перекристаллизовывают и CCl4 (150 мл) с обесцвечиванием активированным углем. Получают целевой
продукт (14,2 г, 90%) в виде бесцветных игл. Tпл. = 91-94oC.
Часть F - N-[4-карбометокси)бензил]-N,N-бис-[(2-трифенилметилтио)этил] глицинамид
Раствор
метил N, N'-бис[(2-трифенилметилтио)этил]глицинамида (16,27 г, 0,024 моль) и метил-4-(метансульфонилметил) бензоата (4,88 г, 0,02 моль) в этилендихлориде (200 мл) нагревают с обратным холодильником в
течение 28 час. Реакционную смесь промывают, порциями по 200 мл, насыщенным раствором NaHCO3 и H2O, высушивают (MgSO4), после концентрирования получают
светло-коричневое масло (30 г). Это масло очищают хроматографией на силикагеле (200-400 меш, 60 A), используя в качестве подвижной фазы смесь DCM:EtOAc, получают целевой продукт (9,9 г, 60%) в виде
бесцветного, аморфного пенистого твердого вещества. Спектр ЯМР1H (в CDCl3): 7,90 (d, 2H, J=6,5 Гц), 7,49 - 7,18 (m, 32H), 3,91 (s, 3H), 3,47 (s, 2H), 3,01 (q, 2H, J = 6,2 Гц), 2,
88 (s, 2H), 2,43 (t, 2H, J = 6,2 Гц), 2,39 - 2,27 (m, 4H),
Часть F - N-[(карбокси)бензил]-N,N'-бис[(2-трифенилметилтио)этил]глицинамид
Смесь N-[4-(карбометокси)бензил] -N,
N'-бис-[(2-трифенилметилтио)этил] глицинамида (6,0 г, 7,26 ммоль) в диоксане (65 мл) и 1H NaOH (65 мл) перемешивают при комнатной температуре в течение 24 ч. Смесь подкисляют 2,5 M лимонной кислотой
(100 мл) и образующийся резиноподобный осадок экстрагируют EtOAc (400 мл). Раствор в EtOAc промывают H20 (3х200 мл) и насыщенным раствором NaCl (100 мл), высушивают (MgSO4) и
после концентрирования получают целевой продукт (5,90 г 100%) в виде бесцветного, аморфного пенистого твердого вещества.
Спектр ЯМР1H (в CDCl3): 7,96, (d, 2H, J = 8,1 Гц), 7,40 - 7,16 (m, 32H), 3,71 (s, 3H), 3,49 (s, 2H), 3,00 (q, 2H, J = 5,4 Гц), 2,91 (s, 2H), 2,44 (t, 2H, J = 5,4 Гц), 2,38 - 2,30 (m, 4H).
Часть G
- Эфир-N-[4-(карбокси)бензил] -N, N'-бис- [(2-трифенилметилтио)этил]глицинамид N-гидроксисукцинимида
Раствор N-[4-(карбокси)бензил]-N,N'-бис[(2-трифенилметилтио)этил]глицинамида (450 мг, 0,55
ммоль) и N-гидроксисукцинимида (76 мг, 6 ммоль) в DCM (7 мл) обрабатывают раствором WSCD•HCl (122 мг, 0,66 ммоль) в DCM (7 мл) и перемешивают при комнатной температуре в течение 22 ч.
Реакционную смесь концентрируют, и твердый остаток перерастворяют в EtOAc (60 мл). Раствор в EtOAc промывают водой (2х25 мл), 0,1 H NaOH (35 мл), H2O (2х25 мл) и насыщенным раствором NaCl
(35 мл), высушивают (Na2SO4), и после концентрирования получают целевой продукт (469 мг, 93%) в виде бесцветного твердого вещества.
Синтез
N[2-(бензоилтио)пропионил] глицилглицил-g-аминомасляной кислоты (Bz-Me-MAG2-gaba)
Указанное в заглавии соединение получают в соответствии со схемой 23 из
N-(2-меркаптопропионил)-глицина (1), который выпускается фирмой Aldrich. Защита тиольной группы в соединении 1 достигается взаимодействием с бензоилхлоридом в щелочных условиях с образованием
соединения 2. Карбоксильная группа может быть активирована образованием ее сукцинимидного эфира (3), который взаимодействует с глицил-g-аминомасляной кислотой в 90% метанольном растворе с образованием
бензоил-замещенного Me-MaG2-gaba (4). Спектральные данные (ИК, ЯМР и масс-спектроскопия (FAB) полностью согласуются с предложенной схемой 23.
Стадия 1: N-[2-(бензоилтио)пропионил]глицин (2).
Гидроокись натрия (4,5 г, 0,109 моль) и N-(2-меркаптопропионил)глицин (8,20 г, 0,05 моль) растворяют в смеси воды (40 мл) и толуола (30 мл). Температуру понижают до 5-15oC, используя баню со льдом. Добавляют по каплям бензоилхлорид (4,6 мл, 0,051 моль) в толуоле (10 мл) при энергичном перемешивании. После этого смесь перемешивают при 5-15oC еще 30 мин и затем при комнатной температуре - 2 час. Органический слой отделяют, промывают водой (2х20 мл) и выбрасывают. Водные фракции соединяют и подкисляют до pH 1,5, используя конц, HCl, при этом образуется белый осадок, отделяют фильтрованием, промывают водой и небольшим количеством этанола, высушивают в вакууме. Выход составляет 13,0 г (97%). Элементный анализ: вычислено (найдено) для C12H13NO4S: C, 53,90 (53,89); H, 4,90 (4,81); N, 5,24 (5,22). ИК-спектр (таблетка с KBr, см-1): 3375 (s, nN-н, 3200-2500 (br, nо-н); 1745 (vs, тиоэфир nc=o); 1663, 1625 (vs, амидный и карбоксильный nc=o). Спектр ЯМР1H в d6-DMCO (d, в м.д.): 1,47 (d, 3H, CH3, J = 7,0 Гц); 3,79 (d, 2H, CH2, J = 5,9 Гц); 4,40 (q, 1H, CH, J = 7,0 Гц); 7,53 (m, 2H, =CH); 7,69 (m, 1H, =CH); 7,90 (dd, 2H, =CH, J=7,0 Гц), 8,59 (t, 1H, NH, J = 5,8 Гц); 12,6 (bs, 1H, COOH). Масс-спектр (DCl): m/z=268 ([M+H]+).
Стадия 2: N-[2-бензоилтио)пропионил]глицин
Сукцинимид эфир (3). К суспензии N-гидроксисукцинимида (5,80 г,
0,05 моль) и N-[2-бензоилтио)пропионил]глицина (13,35 мг, 0,05 моль) в сухом ТГФ (400 мл) добавляют DCC (12,0 мг, 0,052 моль) в том же растворителе (1000 мл ТГФ) при 5-10oC. Смесь
перемешивают при 5-10oC в течение 2 ч и затем при комнатной температуре 2 дня. К реакционной смеси добавляют 2-3 мл уксусной кислоты и затем перемешивают еще 2 часа. Осадок отфильтровывают,
промывают ТГФ (2х150 мл). Органические фракции соединяют, и после удаления растворителя при пониженном давлении получают белое твердое вещество, которое собирают, промывают диэтиловым эфиром и
высушивают на воздухе. Выход составляет 14,5 г (80%). Элементный анализ, вычислено (найдено): для C16H16N2O6S: C, 52,72 (52,70); H, 4,43 (4,21); N, 7,69 (7,
69). ИК-спектр (таблетка с KBr, см-1); 3290 (s, nN-H), 1820 (m, сукцинимид, nc-o); 1785 (m, эфир nс=o); 1735 (vs, тиоэфир, nс=o), 1600 (vs,
амидный, nс=o). Спектр ЯМР1H (в CDCl3) (d в м.д.): 1,57 (d, 3H, CH3, J = 7,0 Гц); 2,79 (s, 4H, CH2); 4,33 (q, 1H, CH3, J = 7,0 Гц), 4,
39 (m, 2H, CH2); 7,00 (t, 1H, NH, J = 5,8 Гц); 7,44 (m, 2H, = CH), 7,59 (m, 1H, =CH); 7,93 (dd, 2H, =CH, J = 7,0 Гц). Масс-спектр (DCT): m/z=365 ([M+H]+).
Стадия 3: Синтез N[2-(бензоилтио)пропионил]-глицилглицил-g- аминомасляная кислота (Bz-Me-MAG2-gaba, 4). N[2-(бензоилтио)пропионил]глицин-сукцинимид эфир (1,82 г, 5 ммоль) и глицил-g-аминомасляную кислоту (0,80 г, 5 ммоль) суспендируют в смеси метанола (150 мл) и воды (30 мл). Смесь нагревают с обратным холодильником в течение 5 час, за это время мутная смесь превращается в прозрачный раствор. Затем раствор охлаждают до комнатной температуры и оставляют при перемешивании на ночь. Упаривание при пониженном давлении позволяет получить белое твердое вещество, которое очищают промывкой водой и сушкой в вакууме. Выход составляет 1,85 г (93%). Элементный анализ, вычислено (найдено) для C18H23N3O6S: C, 52,78 (52,69); H, 5,66 (5,70); N, 10,27 (10,17). ИК-спектр (таблетка с KBr, см-1); 3380, 3320 (s, nN-H), 3100-2500 (br, nO-H); 1725 (vs, тиоэфир, nс=o), 1680, 1640, 1624 (vs, амид. Nс=o). Спектр ЯМР1H (в d6-DMSO) (d в м.д.): 1,49 (d, 3H, CH3, J = 7,0 Гц); 1,62 (qin, 2H, CH2, J = 7,1 Гц); 2,21 (t, 2H, CH2COOH, J = 7,5 Гц), 3,05 (qart, 2H, NH-CH2, J = 7,0 Гц); 3,67 (d, 2H, NHCH2, J = 5,7 Гц); 3,75 (d, 2H, NH-CH2, J = 7,0 Гц), 4,42 (q, 1H, CH, J = 7,0 Гц); 7,57 (m, 2H, =CH); 7,70 (m, 1H, =CH); 7,80 (t, 1H, NH, J = 3,0 Гц); 7,90 (dd, 2H, =CH, J = 7,0 Гц); 8,14 (t, 1H, NH, J = 5,70 Гц); 8,57 (t, 1H, NH, J = 5,90 Гц), 12,0 (bs, 1H, COOH). Масс-спектр (DCl): m/z=410 ([M+H]+).
Синтез N[2-(бензоилтио)пропионил]глицилглицилглицина(Bz-Me-MAG3)
Указанное в заглавии соединение получают по методике, приведенной выше для Bz-Me-MAG2-gaba, заменяя
глицилглицин на глицил-g-аминомасляную кислоту. Выход составляет 83%. Элементный анализ, вычислено (найдено) для C16H19N3O6S: C, 50,39 (50,59); H, 5,02 (5,
78); N, 11,02 (10,70). ИК-спектр (таблетка с KBr, см-1): 3380, 3300 (s, nN-H), 3100-2500 (br, nO-H); 1738 (vs, тиоэфир, nс=o), 1680, 1660 (vs, амид, nс=o). Спектр ЯМР1H (в d6-DMCO) (d в м.д.): 1,48 (d, 3H, CH3, J = 7,0 Гц); 3,78 (m, 4H, CH2); 3,85 (d, 2H, CH2, J = 6,00 Гц); 4,41 (m, 1H,
CH); 7,52 (m, 2H, =CH); 7,70 (m, 1H, =CH); 7,90 (m, 2H, =CH), 8,15 (t, 1H, NH, J = 3,00 Гц), 8,51 (t, 1H, NH, J = 3,00 Гц), 8,80 (t, 1H, NH, J=3,00 Гц). Масс-спектр (ESI): m/z=381,9 ([M+H]+
).
Синтез N[2-(бензоилтио)пропионил] глицилглицил-4-аминометилциклогексан карбоновой кислоты (Bz-Me-MAG2-ACA)
Синтез Bz-Me-MAG2-ACA состоит из
нескольких стадий (схема 24). Соединение 1 может быть легко переведено в соответствующий хлорид 2, который взаимодействует с 4-трансаминометилциклогексанкарбоновой кислотой с образованием соединения
3. Защита соединения 3 с использованием гидразина в этаноле с последующим присоединением HCl приводит к образованию соединения 4. Взаимодействие соединения 4 с Bz-Me-MAG-Succ в метаноле в присутствии
Et3N дает Bz-Me-MAG2-ACA- соединение 5.
Стадия 1: фталоилглицилхлорид. Фталоилглицин (40 г) суспендируют в хлороформе (400 мл) и затем добавляют тионилхлорид (60 мл). Смесь нагревают с обратным холодильником 2 ч, в течение этого времени смесь превращается в гомогенный прозрачный раствор. Растворитель и избыток тионилхлорида удаляют при пониженном давлении, и получают беловатое твердое вещество, которое высушивают в вакууме и используют без дополнительной очистки. Спектр ЯМР1H соответствует предполагаемой структуре.
Стадия 2:
4-транс-[(фталоилглицил)аминометил] циклогексанкарбоновая кислота
В DMF (150 мл) суспендируют 4-транс-аминометилциклогексанкарбоновую кислоту (7,85 г, 509 ммоль) и K2CO3
(5 г, 50 ммоль). К суспензии добавляют фталоилглицидилхлорид (11,85 г, 50 ммоль) в ацетонитриле (150 мл). Реакционную смесь нагревают с обратным холодильником в течение 3 ч и затем фильтруют в горячем
состоянии. Растворитель удаляют при пониженном давлении и получают маслянистый продукт. После добавления диэтилового эфира (50 мл) формируется белый осадок. Твердое вещество отфильтровывают, промывают
диэтиловым эфиром и высушивают на воздухе. Выход составляет 10,32 г (60%). Спектр ЯМР1H (в d6-DMSO) (d в м.д. относительно тетраметилсилана): 0,87-2,00 (m, 9H, CH2 и
CH и циклогексанового кольца): 2,10 (m, 1H, CHCOOH): 2,92 (t, 2H, CH2, J = 4,6 Гц); 4,19 (s, 2H, CH2); 7,85 (m, 4H, -CH=); 8,21 (t, 1H, NH, J = 4,1 Гц).
Стадия 3. Глицил-4-транс-(аминометил)циклогексанкарбоновая кислота гидрохлорид (Gly-ACA•HCl).
К суспензии 4-транс-[(фталоилглицил)аминометил)циклогексанкарбоновой кислоты (10,32 г, 30 ммоль) в этаноле (300 мл) добавляют 85% гидразин гидрата (100 мл). Смесь нагревают с обратным холодильником 12 ч, в течение этого времени образуется белый осадок. После удаления растворителя к остатку добавляют 2N HCl (200 мл). Смесь нагревают до 60-70oC в течение 20 мин, осадок отфильтровывают и выбрасывают. Фильтрат концентрируют до 1/3 первоначального объема. Смесь охлаждают на бане со льдом в течение 2 ч. Осадок отделяют фильтрацией, промывают небольшим количеством воды и этанола и высушивают в вакууме. Выход - 3,45 г (45%).
Спектр ЯМР1H в D2O (d выражено в м.д., относительно тетраметилсилана): 1,04 (m, 2H, CH2); 1,45 (m, 2H, CH2); 1,57 (m, 1H, CH); 1,81-2,05 (m, 4H, CH2); 2,35 (m, 1H, - CHCOOH); 3, 15 (d, 2H, CH2, J = 4,9 Гц), 3,84 (s, 2H, CH2).
Стадия 4: N-[2-(бензоилтио)пропионил]глицилглицил-4-амино- метилциклогексан карбоновая кислота (Bz-Me-MAG2ACA). Gly-ACA•HCl (1,25 г, 5 ммоль). Et3N (1,0 г, 10 ммоль) и Bz-Me MAG-SuCC (1,82 г, 5 ммоль) суспендируют в смеси метанола (200 мл) и ацетонитрила (100 мл). Смесь нагревают с обратным холодильником с вечера до утра. Растворитель удаляют при пониженном давлении и получают белый твердый остаток, к которому добавляют 6H HCl (10 мл). Твердый продукт отделяют фильтрацией, промывают водой и небольшим количеством этанола, высушивают в вакууме. Выход - 1,35 г (58%). Элементный анализ, вычислено (найдено) для C22C29N3O6S: C 57,00 (58,41), H, 6,31 (6,70); N, 9,06 (9,72). ИК-спектр (таблетка с KBr, см-1); 3600-2000 (br, OH---N) 3270 (s, nN-H), 1720, 1656, 1625 и 1565 (vs, nc=o): 1738 (vs, тиоэфир, nс=o), 1680, 1660 (vs, амид, nс=o). Масс-спектр (FAB): m/z=464 (M+1). Спектр ЯМР1H (в d6-DMSO) (d выражено в м.д. относительно тетраметилсилана): 0,81-1, 90 (m, 9H, CH2 и CH циклогексанового кольца): 1,48 (d, 3H, CH3, J = 5,2 Гц); 2,10 (t, 1H, CHCOOH, J = 9,00 Гц); 2,91 (t, 2H, CH2, J = 4,6 Гц); 3,68 (d, 2H, CH2, 4,2 Гц); 3,75 (d, 2H, =CH2, J = 4,1 Гц); 4,42 (q, 1H, CH, J = 5,2 Гц), 7,50 (t, 2H, -CH, J = 5,8 Гц), 7,71 (t, 2H=, J = 5,4 Гц), 7,91 (d, 1H, -CH, J = 6,4 Гц), 8,14 (t, 1H, NH, J = 4,2 Гц); 8,60 (t, 1H, NH=CH=, J = 4,1 Гц); 12,00 (bs, 1H, COOH).
Синтез 3,4-бис[3-(бензоилтиоацетил)амидо]бензойная кислота (Bz-MABA)
К раствору S-бензоилтиоацетилхлорида (8,
69 г, 40 ммоль), свежеприготовленного по реакции S-бензоилтиоуксусной кислоты с избытком тионилхлорида в хлороформе, в сухом ТГФ (300 мл) добавляют 4,4-диаминобензойную кислоту (3,04 г, 20 ммоль), при
этом раствор становится коричневым. Раствор нагревают с обратным холодильником с вечера до утра, в течение этого времени формируется осадок. Смесь охлаждают, твердое вещество отделяют фильтрацией,
промывают ТГФ, этанолом и диэтиловым эфиром и высушивают в вакууме. Получают светло-серое твердое вещество. Выход - 5,8 г (54%). Элементный анализ, вычислено (найдено для C25H20
N2O6S2: C 59,04 (58,82); H 3,96 (4,04); N 5,51 (s, 46). ИК-спектр (таблетка с KBr, см-1): 3600-2000 (br, OH---N), 3340 (s, nN-H), 1690,
1670, 1655, 1610 и 1595 (s или m, nс=о. Масс-спектр (FAB):m/z= 509 (M+1). Спектр ЯМР1H (в COCl3) (d выражено в м.д. относительно тетраметилсилана): 4,12 и 4,14 (s,
4H, CH2); 7,50-8,30 (m, 13H, ароматические протоны); 9,85 и 9,89 (s, 2H, NH); 12,99 (bs, 1H, COOH).
S-трифенилметил-L-цистеин этиловый эфир (2). К раствору L-цистеин этилового эфира гидрохлорида (18,6 г, 0,1 ммоль) в 200 мл трифторуксусной кислоты добавляют трифенилметанол (52 г, 0,2 моль). Полученный темно-коричневый раствор перемешивают в течение 2 час при комнатной температуре в атмосфере азота. Растворитель удаляют в вакууме и к остатку добавляют этанол (100 мл). 1М раствор этилата натрия добавляют к этанольному раствору и перемешивают 90 мин, в течение этого времени раствор мутнеет. Смесь фильтруют, фильтрат концентрируют в вакууме и получают маслянистый остаток. После хроматографии на колонке с использованием смеси этилацетата и гексана (1:3) и этилацетата получают целевой продукт (содержащий некоторое количество этилацетата, которое трудно удалить), который хранят под вакуумом.
N-бромоацетил-S-трифенилметил-L-цистеин этиловый эфир (3): Раствор S-трифенилметил-L-цистеин этилового эфира (18 г, 46 ммоль) и триэтиламина (6,4 мл, 46 ммоль) в сухом ТГФ (250 мл) в атмосфере азота охлаждают до 0oC. Добавляют по каплям раствор бромоацетилбромида (9,28 г, 46 ммоль) в сухом (60 мл), в это время раствор мутнеет. Реакционную смесь перемешивают при 0oC в течение 1 ч и затем при комнатной температуре 1 ч. Реакционную смесь фильтруют, и фильтрат концентрируют в вакууме, в результате чего получают масло. Это масло фракционируют между метиленхлоридом и водой (по 60 мл), органический слой промывают 5% HCl, NaHCO3, высушивают (сульфат магния), фильтруют, и после удаления летучих получают целевой продукт (69%).
2-(S-трифенилметилмеркапто)этиламиноацетил-S-трифенилметил-L-цистеин этиловый эфир (4): К раствору N-бромацетил-S-трифенилметил-L-цистеин этилового эфира (1,0 г, 1,98 ммоль) и триэтиламина (0,4 мл, 2, 9 ммоль) в метиленхлориде (10 мл) добавляют S-трифенилметил-2-аминоэтантиол (0,64 г, 2,0 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение семи дней. Добавляют воду (10 мл). Органический слой промывают NaHCO3 (2x10 мл), водой (2х10 мл) и раствором соли (10 мл), высушивают (сульфат магния) и после концентрирования в вакууме получают пенистый продукт. После хроматографической очистки с использованием смеси этилацетат : гексан (3:1) получают целевой продукт с выходом 22%. Масс-спектр: (M+H)=751. Вычислено 751,3.
Синтез комплексообразователя, содержащего одну группу карбоновой кислоты, способную к присоединению к "связке", показан на схеме 26. Синтез начинают с N-алкилирования Cys (Acm)OMa диметилацетальной группой бромоацетальдегида. Вторичную аминогруппу алкилированного продукта защищают от участия в дальнейших превращениях группой Teoc. Также могут быть использованы другие защитные группы, устойчивые в слабощелочной и слабокислой среде и удаляемые в присутствии серы. Teoc-группа вводится действием 2-(триметилсилил)этил n-нитрофенил карбоната. Итак, ацеталь гидролизуют в слабокислой среде, альдегид восстановительно аминируют S-трифенилметил-2-амино-этантиолом. Единственная свободная аминогруппа комплексообразователя защищается Teoc-группой, метиловый эфир гидролизуют водным раствором щелочи, в результате чего получают карбоновую кислоту, готовую к взаимодействию с реакционноспособной группой циклического соединения, модифицированного введением "связки".
Комплексообразователь, содержащий одну дополнительную аминогруппу, способную к взаимодействию с циклическим соединением, модифицированным введением "связки", может быть получен по методике, показанной на схеме 27. Acm - защищенная тиогликолевая кислота может быть присоединена к N-трет-бутоксикарбонилэтилендиамину с использованием стандартных методов присоединения пептидного синтеза. Защитная Boc-группа может быть удалена действием трифторуксусной кислоты, и полученный амин может вступать во взаимодействие с Boc-Cys(Acm)-OH. Удаление защитной Boc-группы приводит к получению S-замещенного комплексообразователя в форме, пригодной для реакции с реакционноспособной группой циклического соединения, модифицированного введением "связки".
Предметом настоящего изобретения также являются реагенты формулы (QLn)dCh для введения радиоактивных меток, которые состоят более чем из одного циклического промежуточного соединения, модифицированного введением "связки", присоединенного к комплексообразователю, а также реагенты формулы (Q)d•Ln-Ch, содержащие два или более промежуточных циклических соединения, присоединенных к общей "связке", которая также присоединена к комплексообразователю.
Пример реагента, содержащего два циклических промежуточных соединения, модифицированные введением "связки" и присоединенные к комплексообразователю, приведен ниже (схемы 28 и 29). Другие характерные примеры показаны на следующих схемах. По этой схеме аминогруппы двух модифицированных введением "связки" промежуточных соединений взаимодействуют с двумя активированными эфирными группами, в результате чего образуется соединение данного изобретения формулы (QLn)2Ch.
Защита меркаптогруппы, Pg, показанная выше, также как и другие Pg-группы, заявленные здесь, может представлять собой любую защиту меркаптогруппы, способную отщепляться под действием радиоактивных изотопов металлов. Такие защитные группы хорошо известны специалистам. Примеры подходящих защитных групп приведены в патентах США NN 4897255, 4965392 и 4980147, которые указаны здесь как ссылки.
Катализаторы, которые могут быть использованы в синтезе этих реагентов, описаны в: Chervu et al., патент США N 4883862; Bergstein et al., патент США N 5279811. Синтез других подходящих комплексообразователей показан на следующих схемах.
Следующий пример показывает, каким образом могут быть получены три комплексообразователя такого типа. На схеме 30 приведен синтез N2S2-лиганда, содержащего две группы карбоновой кислоты, к которым может быть присоединено нужное соединение. Синтез начинается с реакции алкилирования двух аминогрупп DL-2,3-диаминоянтарной кислоты (Sigma Chemical Co.) s-трифенилметил-2-бромоэтантиолом. Здесь необходимо провести защиту вторичных аминогрупп для того, чтобы избежать автоконденсации при активации групп карбоновых кислот. Это осуществляют по стандартным методикам защиты аминогрупп; z - группу подбирают таким образом, чтобы она могла быть удалена в кислой среде (HBr/HOAC или трифторуксусная кислота/трифторметансульфоновая кислота) одновременно с тритильной защитой меркаптогруппы.
Синтез другого соединения N2 S2, содержащего две группы карбоновой кислоты, показан на схеме 31. Алкилирование этилендиамин-N,N'-дипропионовой кислоты (American Tokyo kasei) S-трифенилметил-2-бромоэтантиолом дает готовое к сопряжению соединение N2S2. Аминогруппы являются третичными и не требуют дополнительной защиты.
Схема 32 иллюстрирует синтез N2S2 -лиганда, содержащего две дополнительные аминогруппы для присоединения требуемого циклического соединения, включающего реакционноспособные электрофильные группы (например, активированные эфиры). Реакция восстановительного аминирования между бензиламином и глиоксалем приводит к образованию N,N'-дибензилэтилендиамина. Алкилирование двух аминогрупп N-(3-бромопропил)фталимидом приводит к получению полностью защищенного тетраамина. Бензильная защита двух вторичных аминогрупп может быть удалена каталитическим восстановлением, после чего три свободные аминогруппы могут быть проалкилированы S-трифенилметил-бромоэтантиолом с образованием полностью защищенного лиганда. Селективное снятие защиты первичных аминогрупп может быть проведено гидразином.
Реагенты, содержащие две заданные группы и один комплексообразователь, присоединенные к общей "связке", могут быть синтезированы в соответствии с методикой, показанной на схеме 33. Реакция бензиламина с N-(3-бромопропил)фталимидом приводит к образованию N,N-бис(3-фталимидопропил)бензиламина (Niitsu and Samejima (1986), Chem. Pharm. Bull, 34, 1032 - 1038). Обработка гидразином снимает фталимидные защитные группы, N, N-бис(3-аминопропил)бензиламин может затем реагировать с янтарным ангидридом с образованием дикислоты, которая затем может быть переведена в активированный бис(эфир)действием DCC и N-гидроксисукцинимида. Этот активированный бис(эфир) может затем взаимодействовать с модифицированным введением "связки" циклическим соединением. Гидрирование снимает бензильные защитные группы, и взаимодействие с активированным комплексообразователем приводит к образованию целевого продукта.
Более чем два соединения Q и более, чем один комплексообразователь, могут быть соединены вместе при использовании звездообразной или дендритной "связки". Дендриты конструируются добавлением разветвляющих сегментов к функциональному ядру, при этом образуется продукт, содержащий в два раза больше функциональных групп, чем исходное ядро. Такое добавление разветвляющих фрагментов может быть осуществлено несколько раз и приводит к образованию объемной полифункциональной молекулы. Одним из примеров является PAMAM (полиамидоамин) - дендрит (Aldrich Chemical Co.), в котором функциональным ядром служит этилендиамин. Схема 34 иллюстрирует общую методику получения радиоактивного фармацевтического препарата на основе PAMAM-дендрита, содержащего заданные циклические соединения и комплексообразователь в соотношении 2: 1. По этой схеме при n = 1 исходный дендрит будет содержать два заданных циклических соединения и один комплексообразователь, а следующее поколение дендритов, при n = 2, будет содержать четыре заданных циклических соединения и два комплексообразователя. Соотношение и общее число заданных циклических соединений и комплексообразователей будет контролироваться стехиометрией реакции.
Подобная система, называемая мультиплетной антигенной пептидной (MAP) системой, разработана: Posnett, MeGrath and Tam, J. Biol. Chem., 263 (1988), 1719) для ускорения образования антител. Эта система построена из разветвленной сети на твердом носителе, с использованием двух аминогрупп лизина. Так как две различные аминогруппы лизина могут быть ортогонально защищены, эта система делает возможным контролировать реакцию образования конъюгатов. На схеме 35 MAP-система, оканчивающаяся на четыре группы лизина, первоначально присоединяет 4 заданных циклических соединения по альфа-аминогруппам, а затем - 4 комплексообразователя по эпсилон-аминогруппам.
Синтез соединений, содержащих радиоактивную метку
Содержащие радиоактивную метку циклические соединения гликопротеина IIb/IIIa тромбоцитов, являющиеся
предметом настоящего изобретения, могут быть получены с использованием стандартных методов, известных специалистам, используя радиоактивные изотопы галогенов (таких как хлор, фтор, бром и йод),
технеция и индия, и другие. Преимущественные радиоактивные изотопы включают123I,125I,131I,99mTc иIIIIn.
В циклические соединения гликопротеина IIb/IIIa тромбоцитов радиоактивная метка может быть введена либо прямо (а именно, связыванием радиоактивной метки непосредственно соединением), либо косвенно (то есть введением радиоактивной метки в соединение с помощью комплексообразователя, уже включенного в соединение). При прямом введении радиоактивной метки, как известно специалистам, введение метки может быть изотопным или неизотопным. Если осуществляют изотопное введение метки, то одну из групп, уже имеющихся в циклическом соединении, замещают (обменивают) на радиоактивный изотоп. Если осуществляют неизотопное введение метки, радиоактивный изотоп добавляют к циклическим соединениям без замещения (обмена) уже имеющейся группы.
В общем меченые соединения получают по методикам, которые включают введение метки на заключительных стадиях синтеза. Это позволяет повысить радиохимический выход и сократить время работы с радиоактивным материалом. При работе с короткоживущими изотопами существенным фактором является время, необходимое для проведения операций синтеза и очистки. Методики синтеза радиоактивных фармацевтических препаратов описаны в: Tubis and Wolf, Eds., "Radiopharmacy", Wiley-Interscience, New York (1976); Wolf, Christman, Fowler, Lanbrecht, "Synthesis of Radiopharmaceuticals and Labeled Compounds, vol. 1, p. 345 - 381 (1973), которые приведены здесь в качестве ссылок.
Для получения содержащих радиоактивную метку соединений настоящего изобретения в случае, когда радиоактивной меткой является галоген, могут быть использованы разнообразные методики.
Наиболее общими синтетическими приемами для введения иода в качестве радиоактивной метки в ароматические соединения являются реакция замещения иодом радикалов диазония, боро-, станно-, сило-, талло-групп и обменные реакции галогенов. Наиболее принятыми для неизотопного введения радиоактивной метки в ароматические соединения такого типа, как описанные в настоящем изобретении, являются реакции иододепротонирования и электрофильного замещения ароматических соединений.
Эти методы и дополнительные методики, описанные в: Merkushev, Syntesis, 923 (1988) and Seevers et al., Chem. Rev., 82: 575 (1982), приведены здесь в качестве ссылок.
Например, меченые радиоактивными изотопами производные 4,5 и 6-гало-трет-бутилоксикарбонил-3-аминометилбензойной кислоты могут быть получены с использованием описанной выше общей методики синтеза не содержащих радиоактивную метку соединений. Для такого введения радиоактивной метки важно, чтобы время полураспада выбранного изотопа было много больше, чем время практического осуществления синтеза. Известные исходные материалы включают 2,3 и 4-иодо (123I,125I,131I) бензойные кислоты.
Меченые радиоактивным иодом производные Mamb могут быть также получены из аналина по реакции Сэндмейера, как описано в: Ellis et al. Aust J. Chem, 26: 907 (1973).
Кроме того, такие соединения могут быть получены введением радиоактивной изотопной метки в не содержащие такой метки бромо- и йодопроизводные различными двухстадийными реакциями, например, с использованием триалкилсилильных реакционноспособных групп, как описано в: Wilson et al. J.Org.Chem., 51: 483 (1986) and Wilbur et al. J.Label. Compound. Radiopharm., 19: 1171 (1982), применение триалкилсилильных реагентов описано в: Chumpradit et al. J.Med. Chem. , 34: 877 (1991) and Chumpradit et al. J.Med.Chem., 32: 1431 (1989). Использование производных борной кислоты описано в: Kabalka et al. J.Label. Compound. Radiopharm. , 19: 795 (1982) and Koch et al. Chem. Rer., 124:2091 (1991). Превращения в ходе синтеза иллюстрирует схема 36.
Хотя приведенные выше методы могут быть использованы для получения меченых радиоактивным изотопом соединений настоящего изобретения, для повышения радиохимического выхода, сокращения времени работы с радиоактивными материалами и получения короткоживущих меченых галогеном соединений предпочтительно осуществлять введение изотопной метки галогена на заключительных стадиях синтеза циклического соединения. Далее приводятся такие методики введения радиоактивной метки на заключительных стадиях синтеза.
Не содержащие метку иодо-соединения являются удобными исходными соединениями, которые могут быть переведены в меченые производные по любой из двухстадийных реакций, приведенных выше. Подходящими функциональными группами для введения в Mamb-содержащее циклическое соединение являются бромо-, нитро-, триалкилсилил-, триалкилстанно-группы и группы борной кислоты. Синтез и применение таких исходных соединений описаны выше.
Наименее сложным способом радиойодирования циклических соединений настоящего изобретения путем изотопного обмена на заключительной стадии синтеза является замещение радиоактивным иодом стабильного атома йода, уже имеющегося в молекуле, это часто может быть сделано путем нагревания соединения с радиоактивным иодом в подходящем растворителе, как описано в: Ellis et al., Aust. J. Chem. , 26: 907 (1973). Для ароматических йодидов очень маленькое количество и низкая концентрация используемого радиоактивного йодида приводят к получению весьма умеренной удельной активности. Последовательность операций показана на схеме 37.
Радиоактивная метка может быть также введена в циклические соединения на последней стадии их синтеза из анилинов по реакции Сэндмейера, как описано в Ellis et al. Aust J. Chem, 26: 907 (1973). Этот подход позволяет получить меченые циклические соединения с высокой удельной активностью. Для устранения затруднений при синтезе циклического соединения идеально использовать нитрогруппу, как исходную для получения замещенного анилина.
Иначе, в циклические соединения может быть введена радиоактивная метка по реакции не содержащих метку бромо- и йодопроизводных различными двухстадийными методами, как описано выше, например, используя триалкилсилильные реагенты, как описано в: Wilson et al, J. Org. Chem., 51: 4833 (1986) and Wilbur et al., J. Label. Compound. Radiopharm., 19: 1171 (1982), используя триалкилсилильные реагенты, как описано в: Chumpradit et al., J. Med. Chem, 34: 877 (1991) and Chumpradit et al., J. Med. Chem., 32: 1431 (1989), а также с использованием производных борной кислоты, как описано в: Kabalka et al. , J. Label. Compound. Radiopharm., 18: 795 (1982) and Koch et al., Chem. Ber., 124: 2091 (1991).
Сходный подход, когда осуществляют изотопное введение радиоактивной метки галогена на заключительных этапах синтеза, включает превращение замещенного Mamb-группой производного в циклическое соединение, которое уже включает триалкилсилильные, триалкилстанногруппы или группы борной кислоты. Синтез отдельных Mamb-производных был описан в предыдущих разделах. Синтетическая трансформация на циклических соединениях указана на схеме 38.
Меченые иодопроизводные могут быть легко получены неизотопно из амино-, гидрокси-, метокси-замещенных циклических соединений, как описано Arora et al. , J. med. Chem., 30: 918 (1987). Реакции электрофильного ароматического замещения облегчаются при наличии таких электроно-донорных заместителей. Последовательность операций синтеза показана на схемах 39 и 40.
Другой вариант введения радиоактивного галогена состоит в том, что метилзамещенное циклическое соединение может быть переведено в галоидзамещенное толуольное производное действием NBS или NCS в условиях свободно-радикального галогенирования. Бензилгалиды могут быть мягко замещены на радиоактивный йод по реакции нуклеофильного замещения. Последовательность операций показана на схеме 41.
Хотя примеры иллюстрируют получение главным образом меченых йодом соединений, приведенные выше методы могут быть использованы для введения радиоактивных изотопов любого галогена.
18F - Производные этих циклических соединений могут быть получены сопряжением с циклическими соединениями18F - замещенных фенильных промежуточных соединений.18F-замещенные циклические соединения могут быть получены, как показано на схеме 42 (R.H.March et al., J. Med. Chem., 1993, 36, 3707-3720). Реакция n-триметиламмонийбензальдегида с [18F] CsF в водном DMF при 120oC в течение 10 мин (водный [18F] KF/kryptofix/ACN также может быть использован для получения18F-фенильных соединений из соответствующего триметиламмоний- или нитропроизводного) с последующей обработкой смесью LAH/THF/пентан и 57% водным раствором HI приводит к образованию n-18F-бензилиодида.
Для получения содержащих радиоактивную метку соединений настоящего изобретения, в случае, когда радиоактивной меткой является металл, а также технеций или индий, могут быть использованы разнообразные методики. Эти методики используют для получения соединений настоящего изобретения формулы (QLn)dCh и (Q)d'Ln-Ch. Типовые методики введения технеция и индия в качестве радиоактивной метки приведены, например, в Cerqueira et al., Circulation, vol. 85, N. 1, pp. 298-304 (1992), Pak et al., J. Nucl. Med., Vol. 30, N. 5, p. 793, 36th Ann. Meet. Soc. Nucl. Med. (1989), Epps et al., J. Nucl. Med., Vol. 30, N. 5, p. 794, 36th Ann. Meet. Soc. Nucl. Med. (1989), Pak et al., J. Nucl. Med. , Vol. 30, N. 5, p. 794, 36 th Ann. Meet. Soc. Nucl. Med. (1989), and Dean et al., J. Nucl. Med., vol. 30, N. 5, p. 794, 36th Ann. Meet. Soc. Nucl. Med. (1989), которые приведены здесь только в качестве ссылок. В дополнение, специальные методики приведены ниже в примерах.
Другой подходящий метод введения радиоактивной метки в циклическое соединение настоящего изобретения включает синтез99mTc-содержащего (в качестве метки) комплексообразователя и его взаимодействие с либо циклическим промежуточным соединением, либо с модифицированным введением "связки" циклическим соединением. Этот метод предполагает введение метки до заключительного комплексообразования.
Как показано, например, на приведенной ниже схеме, 4,5-бис(S-бензоил)меркаптоацетамидопентановая кислота (1) взаимодействует с99mTcO4 в восстановительных условиях с образованием (2). Затем соединение (2) переводят в активированный эфир (3), содержащий тетрафторфенильную группу. Комплекс (3) затем может взаимодействовать с соответствующим промежуточным циклическим соединением, таким как (5) или (6) с образованием меченого радиоактивным изотопом соединения (4). Другой подходящий комплексообразователь с технецием - 2,3-бис(S-бензоил)меркаптоацетамидопропановая кислота (7). Очистка методом ВЭЖХ комплекса99mTc может проводиться на каждой стадии. Этот метод показан на схеме 43.
Примеры.
Раздел A. Реагенты для
введения радиоактивной метки
Пример 1.
Соединение цикло-(D-Val-NMeArg-Gly-Asp-Mamb (5-ACA))-N- [4-(карбокси)бензил]-N,N-бис[(2-трифенилметилтио)этил]-глицинамид
Раствор N-[4-(карбокси)бензил]-N,N'-бис[(2-трифенилметиолтио) этил]-глицинамид-N-гидроксисукцинимид эфира (0,017 ммоль), цикло-(D-Val-NMeArg-Gly-Asp-Mamb (5-ACA)) (13,9 мг, 0,015 моль) и Et3
N (6,25 мкл, 0,045 ммоль) в DMF (350 мкл) перемешивают при комнатной температуре 14 ч. Ход реакции контролируют "нормально-фазной" ТСХ (CHCl3:MeOH:HOAc, 90:8:2), используя нингидрин и тест
Sakaguchi. DMF удаляют при пониженном давлении. Соединение очищают методом "обращеннофазной" ВЭЖХ на препаративной колонке Vydac C18 (2,1 см), используя 18-36% ацетонитрил, содержащий 0,1% TFA
(градиент 1,0%/мин), после лиофилизации получают соль трифторуксусной кислоты указанного в заглавии соединения в виде бесцветного пушистого твердого вещества (11 мг, 53%).
Пример 2.
Соединение цикло-(D-Lys-NMeArg-Gly-Asp-Mamb)-N-[4-(карбокси) бензил]-N, N'-бис[(2-трифенилметилтио)этил]-глицинамид
Раствор N-[4-(карбокси)бензил]-N,
N'-бис[(2-трифенилметилтио) этил]-глицинамид-N-гидроксисукцинимид эфира (30 мг, 0,033 моль), цикло-(D-Lys-NMeArg-Gly-Asp-Mamb) (23,8 мг, 0,029 ммоль) и Et3N (12 мкл, 0,087 ммоль) в DMF (0,
60 мл) перемешивают при комнатной температуре в течение 63 час. Ход реакции контролируют "нормально-фазной" ТСХ (CHCl3: MeOH:HOAc, 90:8:2), используя нингидрин и тест Sakaguchi. DMF удаляют
при пониженном давлении. Соединение очищают методом "обращеннофазной" ВЭЖХ на препаративной колонке Vydac C18 (2,1 см), используя 18-36% ацетонитрил, содержащий 0,1% TFA (градиент 0,9%/мин), после
лиофилизации получают соль трифторуксусной кислоты указанного в заглавии соединения в виде пушистого бесцветного твердого вещества (24 мг, 60%). Масс-спектр (ESI):[M] = 1397,3.
Пример 3.
Цикло-(D-Val-NMeArg-Asp-Mamb(N-гидразино-никотинил-5-Aca)) TFA соль
Часть A. Синтез цикло-(D-Val-NMeArg-Gly-Asp-Mamb (N-boc- гидразино-никотинил-5-Aca)) TFA соли
К раствору цикло-(D-Val-NMeArg-Gly-Asp-Mamb(N-boc- гидразино-никотинил-5-Aca) (10 мг, 0,011 ммоль), сукцинимидил boc-гидразиноникотината (4,6 мг, 0,0132 ммоль), в DMF (0,3 мл) добавляют триэтиламин (0,
0061 мл, 0,044 ммоль) и перемешивают реакционную смесь в атмосфере азота при комнатной температуре в течение 24 ч. Растворитель удаляют в вакууме, и остаток растворяют в водном растворе ацетонитрила.
После выделения лиофилизацией получают беловатое твердое вещество. Очистку части продукта осуществляют "общеннофазной" ВЭЖХ на препаративной колонке Vydac C18 (2,1 см), используя 6,3-72% водный
ацетонитрил, содержащий 0,1% TFA (градиент 2,0%/мин). После лиофилизации соль трифторуксусной кислоты указанного в заглавии соединения в виде пушистого вещества. Масс-спектр: [M+H] = 938,4849,
вычислено 938,4848.
Часть B. Снятие защиты и синтез соединения цикло-(D-Val-NMeArg-Gly-Asp-Mamb(N-гидразиноникотинил-5-Aca)) TFA соль
Цикло-(D-Val-NMeArg-Gly-Asp-Mamb(N-boc-гидразиноникотинил-5-Aca)) TFA соль растворяют в смеси (98:2) TFA и анизола (2 мл) и перемешивают реакционную смесь в течение 15 мин. Растворитель удаляют в
вакууме и остаток растворяют в водном растворе ацетонитрила. После лиофилизации получают белое твердое вещество. Очистку завершают "общеннофазной" ВЭЖХ на препаративной колонке Vydac C18 (2,1 см),
используя 6,3-72% водный ацетонитрил, содержащий 0,1% TFA (градиент 2,0%/мин). После лиофилизации получают соль трифторуксусной кислоты - указанное в заглавии соединение в виде пушистого твердого
вещества. Масс-спектр: [M+H] = 838,4849, вычислено 838,4324.
Пример 4.
Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(N-гидразиноникотинил-5-Aca)) TFA соль
Часть A. Синтез
цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(N-boc-гидразино-никотинил-5-Aca)) TFA соли
К раствору цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(5-Aca) (10 мг, 0,0019 ммоль), сукцинимидил boc-гидразиноникотината (4,
55 мг, 0,0131 ммоль), в DMF (0,4 мл) добавляют триэтиламин (0,0061 мл, 0,044 ммоль) и реакционную смесь перемешивают в атмосфере азота при комнатной температуре 24 час. Растворитель удаляют в вакууме,
остаток растворяют в водном растворе ацетонитрила. После выделения лиофилизацией в течение ночи получают беловатое твердое вещество. Очистка части продукта завершается "обращеннофазной" ВЭЖХ на
препаративной колонке Vydac C18 (2,1 см), используя 6,3-72% водный ацетонитрил, содержащий 0,1% TFA (градиент 2,0%/мин). После лиофилизации получают соль трифторуксусной кислоты указанного в заглавии
соединения в виде пушистого твердого вещества. Масс-спектр: [M+H] = 924,4699, вычислено 924,4692.
Часть B. Снятие защиты и получение соединения
цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(N-гидразино-никотинил-5-Aca)) TFA соль
Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(N-гидразиноникотинил-5-Aca) TFA соль:
цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(N-boc-гидразиноникотинил-5-Aca) TFA соль растворяют в смеси (98:2) TFA и анизола (2 мл) и перемешивают реакционную смесь 15 мин. Растворитель удаляют в вакууме,
остаток растворяют в водном растворе ацетонитрила. После лиофилизации получают белое твердое вещество. Очистку завершают "обращеннофазной" ВЭЖХ на препаративной колонке Vydac C18 (2,1 см), используя 6,
3-85,5% водный ацетонитрил, содержащий 0,1% TFA (градиент 2,07%/мин), после лиофилизации получают соль трифторуксусной кислоты - указанное в заглавии соединение в виде пушистого твердого вещества.
Масс-спектр: [M+H] = XX, вычислено XX.
Пример 5.
Цикло-((N-E-гидразиноникотинил-D-Lys)-NMeArg-Gly-Asp-Mamb) TFA соль
Часть A. Синтез соединения
цикло-((N-E-boc-гидразиноникотинил-D-Lys)-NMeArg-Gly-Asp-Mamb) TFA соль
К раствору цикло-(D-Lys-NMeArg-Gly-Asp-Mamb • 2TFA (4,2 мг, 0,005 ммоль), сукцинимидил boc-гидразиноникотината
(2,1 мг, 0,006 ммоль), в DMF (0,15 мл) добавляют триэтиламин (0,003 мл, 0,02 ммоль), и реакционную смесь перемешивают при комнатной температуре в атмосфере азота 48 ч. Растворитель удаляют в вакууме,
остаток растворяют в водном растворе ацетонитрила. После лиофилизации в течение ночи получают беловатое твердое вещество. Очистку завершают "обращеннофазной" ВЭЖХ на препаративной колонке Vydac C18,
используя 6,3-85,5% водный ацетонитрил, содержащий 0,1% TFA (градиент 1,7%/мин). После лиофилизации получают соль трифторуксусной кислоты - указанное в заглавии соединение в виде пушистого твердого
вещества. Масс-спектр: [M+H] = 839,4157, вычислено 839,4164.
Часть B. Снятие защиты и получение соединения цикло-(N-E-гидразиноникотинил-D-Lys)-NMeArg-Gly-Asp-Mamb) TFA соль
Цикло-(N-E-гидразиноникотинил-D-Lys)-NMeArg-Gly-Asp-Mamb) TFA соль: цикло-((N-E-boc-гидразиноникотинил-D-Lys)-NMeArg-Gly-Asp-Mamb) TFA соль (3 мг) растворяют в смеси (98:2) TFA и анизола (2 мл) и
перемешивают реакционную смесь в течение 15 мин. Растворитель удаляют в вакууме, а остаток перерастворяют в водном растворе ацетонитрила, после выделения лиофилизацией получают белое твердое вещество.
Очистку завершают "обращеннофазной" ВЭЖХ на препаративной колонке Vydac C18, используя 6,3 - 72% водный ацетонитрил, содержащий 0,1% TFA (градиент 2,0%/мин). После лиофилизации получают соль
трифторуксусной кислоты - указанное в заглавии соединение в виде пушистого твердого вещества. Масс-спектр: [M+H] = 739,3629, вычислено 739,3640.
Пример 6
Соединение
цикло-([DTPA-D-Lys]-NMeArg-Gly-Asp-Mamb)
К раствору 250 мг (2 ммоль) цикло(D-Lys-NMeArg-Gly-Asp-Mamb) в 208 мл 0,1 М бората (pH 9,88) при комнатной температуре добавляют при непрерывном
перемешивании DTPA-ангидрид (743 мг, 10 ммоль). Реакционную смесь перемешивают 2 часа. Неочищенную смесь продуктов, полученных после удаления растворителя, очищают препаративной ВЭЖХ (колонка Vydac
C18, градиент 0 - 50% ACN, содержащий 0,1% TFA в течение 60 мин, скорость потока 20 мл/мин). Выделяют два основных компонента. Компонент A - цикло([DTPA-D-Lys]-NMeArg-Gly-Asp-Mamb).
Масс-спектр: 979,1 (M+H)+.
Пример 7.
Цикло-(D-Lys-NMeArg-Gly-Asp-Mamb)]2-DTPA
Компонент B, полученный описанным в примере 6 синтезом,
представляет собой [цикло-(D-Lys-NMeArg-Gly-Asp-Mamb)]2-DTPA. Масс-спектр: 1565,4 (M+).
Раздел B. Соединения меченые радиоактивным изотопом
Прямое
введение метки
Пример 8.
Цикло-((125I)D-Tyr-NMeArg-Gly-Asp-Mamb)
В пятимиллилитровый пузырек добавляют 22 мКи (45 мкл) водного раствора Na125I,
100 мкл 0,5 М фосфатного буфера (pH 7,5), 4,5 мкл 1H HCl, 75 мкг циклического промежуточного соединения цикло-(D-Tyr-NMeArg-Gly-Asp-Mamb), растворенного в 75 мкл 0,1% водного раствора TFA и 50 мкг
хлорамина T, растворенного в 50 мкл H2O. После протекания реакции в течение 1 мин добавляют 50 мкг метабисульфита натрия, растворенного в H2O. Полученный продукт очищают
препаративной ВЭЖХ (колонка Zorbax-Rx C18, скорость потока 1 мл/мин, градиент - от 100% A до 100% B за 30 мин; растворитель A = 0,1% TFA в H2O, растворитель B = 40% этанол в A.
Продукт характеризуется временем удерживания, равным 30 мин.
Пример 9.
[(125I)N-3-(4-гидроксифенил)пропионил] -цикло- (D-Lys-NMeArg-Gly-Asp-Mamb)
В пятимиллилитровый пузырек добавляют 11,4 мКи (25 мкл) водного раствора Na125I, 100 мкл 0,5 М фосфатного буфера (pH 7,5), 4,5 мкл 1H HCl, 50 мкг модифицированного введением "связки"
циклического соединения [N-3(4-гидроксифенил)пропионил]-цикло- (D-Tyr-NMeArg-Gly-Asp-Mamb), растворенного в 50 мкл H2O. Через 1 мин после начала реакции добавляют 50 мкг метабисульфита
натрия, растворенного в H2O. Продукт очищают препаративной ВЭЖХ (условия описаны в примере 10). Продукт характеризуется временем удерживания, равным 32 мин. Непрямое введение метки.
Пример 10.
99mTcO (MAMA)-цикло-(D-Val-NMeArg-Gly-Asp-Mamb-(5-Aca))
Часть A. Снятие защиты
Удаляют тритильные защитные группы реагента,
описанного в примере 1: в отдельный чистый пузырек объемом 10 см3 добавляют реагент и 0,1 мл трифторуксусной кислоты (TFA). При растворении твердого вещества образуется желтый раствор.
Часть В. Синтез99mTc-глюкогептоната
В пузырек GlucoscanR вводят 1,0 мл милли-Q-H2O.
Отбирают 0,2 мл этого раствора в чистый пузырек объемом 10 см3 и добавляют ~ 20 мКи99mTcO4. Реакция протекает при комнатной температуре 20 мин.
Часть C. Синтез99mTcO
(MAMA)-цикло- (D-Val-NMeArg-Gly-Asp-Mamb-(5-Aca))
К раствору реагента из части A, с удаленной защитной группой, добавляют 0,2 мл 5N NaOH и 0,4 мл 0,2 M фосфатного буфера (pH 6). Измеряют pH,
и если необходимо, доводят до 6.
Этот раствор сразу же добавляют в пузырек с99mTc-глюкогептонатом, закрывают и нагревают при 100oC 15 мин. После охлаждения ~ 2 мин 20 мкл этого раствора анализируют методом ВЭЖХ, используя методику 1 (см.таблицу 1).
Пример 11
99mTcO (MAMA)-цикло-(D-Lys-NMeArg-Gly-Asp-Mamb)
Часть A.
Снятие защиты
Удаляют тритильные защитные группы реагента, описанного в примере 2: в отдельный чистый пузырек объемом 10 см3 добавляют реагент и 0,1 мл трифторуксусной кислоты
(TFA). При растворении твердого вещества образуется желтый раствор.
Часть B. Синтез99mTcO (MAMA)-цикло-(D-Lys-NMeArg-Gly-Asp-Mamb)
К раствору реагента из части A,
с удаленной защитной группой, добавляют 0,2 мл 5N NaOH и 0,4 мл 0,2 М фосфатного буфера (pH 6). Измеряют pH, и если необходимо, доводят до 6. Этот раствор сразу же добавляют в пузырек с раствором99mTc-глюкогептоната, полученным, как описано в примере 11, часть B, закрывают и нагревают при 100oC 15 мин. После охлаждения ~ 2 мин 20 мкл этого раствора анализируют методом ВЭЖХ по
методике 1 (см.таблицу 1).
Пример 12
99mTc (трицин)2-цикло- (D-Val-NMeArg-Gly-Asp-Mamb(гидразино-никотинил-5-Аса))
К раствору 70 мг
трицина(N-[трис(гидроксиметил)метил]глицин) в 1,0 мл воды добавляют 0,05 мл 1,0 N NaOH, чтобы повысить pH до 7,0, затем 0,1-1,0 мл99mTcO4 в физиологическом растворе 10-100 мКи,
10 мкг реагента, получение которого описано в примере 3, растворенного в 100 мкл 0,1 Н HCl, и 100 мкг SnCl2 • 2H2O, растворенного в 0,1 H HCl. Реакция протекает при
комнатной температуре 45 мин. Полученный продукт анализируют методом ВЭЖХ по методике 1 и методом TIX по методике 2 (см.таблицу 1).
Пример 13.
99mTc
(EDDA)-цикло-(D-Val-NMeArg-Gly-Asp-Mamb (гидразино-никотинил-5-Aca))
К раствору 10 мг этилендиамин-N,N'-диуксусной кислоты (EDDA) в 1,0 мл воды добавляют 0,05 мл NaOH для создания pH до 7,0,
затем 0,1-1,0 мл99mTcO4 - в виде соли (10-100 мКи) и затем 50 мкг реагента, описанного в примере 3, растворенного в 100 мкл 0,1 Н HCl, и 100 мкг SnCl2 • 2H2O, растворенного в 0,1 Н HCl. Реакцию проводят при комнатной температуре в течение 45 мин. Полученный продукт анализируют методом ВЭЖХ, используя методику 1, и методом TIX, используя методику 2
(см.таблицу 1).
Пример 14.
99mTc (трицин)2-цикло-(D-Abu-NMeArg-Gly-Asp-Mamb (гидразино-никотинил-5-Aca))
К раствору 70 мг
трицина(N- [трис(гидроксиметил)метил]глицин) в 1,0 мл воды добавляют 0,05 мл 1,0 Н NaOH, чтобы повысить pH до 7,0, затем 0,1-1,0 мл99mTcO4 - в физиологическом растворе (10-100
мКи), затем 10 мкг реагента, методика получения которого описана в примере 4, растворенного в 100 мкл 0,1 Н HCl, и 100 мкг SnCl2 • 2H2O, растворенного в 0,1 Н HCl. Реакция
протекает при комнатной температуре 45 мин. Полученный продукт анализируют методом ВЭЖХ, используя методику 1, и методом TIX, используя методику 2 (см.таблицу 1).
Пример 15
99mTc (трицин)2-цикло-(D-Lys-NMeArg-Gly-Asp-Mamb (гидразино-никотинил-5-Aca))
К раствору 70 мг трицина(N- [трис(гидроксиметил)метил]глицин) в 1,0 мл воды добавляют 0,05
мл 1,0 Н NaOH, чтобы повысить pH до 7,0, затем 0,1-1,0 мл99mTcO4 - в физиологическом растворе (10-100 мКи) и затем 10 мкг реагента, получение которого описано в примере 5,
растворенного в 10 мкл 0,1 Н NaOH, и 100 мкг SnCl2 • 2H2O, растворенного в 0,1 Н HCl. Реакция протекает при комнатной температуре 45 мин. Полученный продукт анализируют
методом ВЭЖХ по методике 1 и методом TIX по методике 2 (см.таблицу 1).
Пример 16
Цикло-([IIIIn-DTPA-D-Lys]-NMeArg-Gly-Asp-Mamb)
50 МклIIIIn
Cl3 (~100 куб.мдюйм/мл 0,05 М HCl), полученного от Du Pont-NTN Products, Billerica, MA, смешивают с равным объемом свежеприготовленного 1,0 М раствора ацетата аммония. Примерно через 5 мин
добавляют 0,1-1 мг реагента, описанного в примере 6, растворенного в 0,25 мл воды. Реакция протекает при комнатной температуре 30 мин. Продукт анализируют методом ВЭЖХ по методике 3.
Пример 17.
IIIIn-DTPA-[цикло-(D-Lys-NMeArg-Gly-Asp-Mamb)]2
К 0,5 мл раствора реагента,
описанного в примере 7, в воде (0,9 мг/1 мл) добавляютIIIIn Cl3 (~3 mKt) в 0,5 мл 1Н раствора NH4OAC. Смесь выдерживают при комнатной температуре в течение 30 мин, после чего анализируют методом ВЭЖХ, используя методику 3 (см.
таблица 2).
Раздел C. Меченые99mTc реагенты, полученные введением метки через координационное соединение
Меченые99mTc реагенты, описанные в этих
примерах, синтезируют, используя метод введения метки до комплексообразования. Такой метод включает следующие стадии: (1) координирование99mTc с комплексообразователем; (2) активация
некоординированной карбоксильной группы полученного комплекса образованием его тетрафторфенильного (TFP) эфира; (3) взаимодействие TFP-эфирного комплекса с циклическим промежуточным соединением или
модифицированным введением "связки" циклическим соединением путем образования амидной связи.
Пример 18
Цикло-([(99mTcO(mapt)]-
DLys]-NMeArg-Gly-Asp-Mamb)
Часть A. Образование координационного соединения.
В чистый флакон емкостью 10 см3 помещают 0,35 мл Bz-mapt (3,0 мг/мл в 1H NaOH), 0,10 мл SnCl2 • 2H2O (10 мг/мл в 1H HCl) и 200 мКи99mTcO4 в физиологическом растворе. Флакон закрывают и помещают на водяную баню (100oC). После охлаждения в течение ~2 мин 10 мкл раствора анализируют методом ВЭЖХ по методике 1.
Часть В. Активация
К раствору, полученному в части A, добавляют 0,3 мл 0,5 М
раствора фосфата натрия с pH 6,0, 0,3 мл 2,3,5,6- тетрафторфенола (100 мг/мл в 90% ацетонитриле), 0,3 мл 1-(3- диметиламино-пропил)-3-этилкарбодиимида (100 мг/мл в 90% ацетонитрила) и ~0,1 мл 1H HCl.
Если необходимо, pH доводят до 6. Пузырек закрывают и нагревают при 40oC 25 мин. После охлаждения в течение ~2 мин 20 мкл раствора анализируют методом ВЭЖХ, используя методику 1.
Часть C. Сопряжение
1,0 - 2,5 мг промежуточного циклического соединения цикло-(D-Lys-NMeArg-Gly-Asp-Mamb) растворяют в 0,3 мл 0,5 М фосфатного буфера (pH 9) и добавляют раствор,
получение которого описано в части B. Величину pH раствора доводят до 9 действием 1H NaOH. Реакционную смесь нагревают при 40oC в течение 30 мин. После охлаждения в течение ~2 мин 25 мкл
раствора анализируют методом ВЭЖХ, используя методику 1 (см. таблицу 3).
Пример 19.
Цикло-(D-Val-NMeArg-Gly-Asp-Mamb([99mTcO(mapt)]-5Aca))
1,0 - 2,
5 мг модифицированного введением "связки" циклического соединения цикло-(D-Val-NMeArg-Gly-Asp-Mamb(5-Aca)) растворяют в 0,3 мл 0,5 M фосфатного буфера (pH 9) и добавляют раствор, получение которого
описано в примере 18 часть B. Используя 1H NaOH, доводят pH до 9. Реакцию проводят при 40oC в течение 30 мин. После охлаждения (~2 мин) 25 мкл раствора анализируют методом ВЭЖХ, используя
методику 1 (см. таблицу 3).
Пример 20
Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb([99mTcO(mapt)]-5Aca))
1,0 - 2,5 мг модифицированного введением "связки"
циклического соединения цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(5-Aca)) растворяют в 0,3 мл 0,5 M фосфатного буфера (pH 9) добавляют раствор, получение которого описано в примере 18 часть B. Используя 1H
NaOH, доводят pH до 9. Реакцию проводят при 40oC в течение 30 мин. После охлаждения (~2 мин) 25 мкл раствора анализируют методом ВЭЖХ, используя методику 1 (см. таблицу 3).
Пример 21
Цикло-([(99mTcO(mapt)]--5-Aca)D-Lys]-NMeArg- Gly-Asp-Mamb)
1,0 - 2,5 мг модифицированного введением "связки" циклического соединения
цикло-((5-Aca)D-Lys-NMeArg-Gly-Asp-Mamb) растворяют в 0,3 мл 0,5 M фосфатного буфера (pH 9) и добавляют раствор, получение которого описано в примере 18 часть B. Используя 1H NaOH, доводят pH до 9.
Реакцию проводят при 40oC в течение 30 мин. После охлаждения (~2 мин) 25 мкл этого раствора анализируют методом ВЭЖХ, используя методику 1 (см. таблицу 3).
Пример 22.
Цикло-([(99mTcO(MeMAG2gaba)]--D-Lys-NMeArg -Gly-Asp-Mamb)
Часть A. Образование координационного соединения
В десятимиллилитровый пузырек
добавляют 100 - 250 мКи99mTcO4- в 1,0 мл физиологического раствора, 1,0 мл раствора Bz-MeMAG2gaba (1 мг/мл в 0,5 M фосфатном буфере с pH 12), а
затем 0,15 - 0,20 мл раствора SnCl2•2H2O (15 мг/3мл в 1H HCl). Величину pH доводят до ~11, а затем смесь нагревают при 100oC в течение 30 мин. Раствор
анализируют методом ВЭЖХ, используя методику 1.
Часть B. Активация
К раствору, полученному по методике, описанной в части A, добавляют 0,2 мл 0,1 H HCl, 0,5 мл раствора
тетрафторфенола (100 мг/мл в 90% CH3CN), затем 0,5 мл раствора (1-[3-(диметиламино)пропил]-3-этилкарбодиимид хлорида (100 мг/мл в 90% CH3CN).
Значение pH доводят до 6,0, затем смесь нагревают при 50oC в течение 30 мин.
Часть C. Сопряжение
1,0 - 2,5 мг промежуточного циклического соединения цикло-(D-Lys-NMeArg-Asp-Mamb)
растворяют в 0,3 мл 0,5 M фосфатного буфера (pH 9) и добавляют к раствору, получение которого описано в части B. Используя 1H NaOH, pH доводят до 9. Реакционную смесь нагревают при 40oC в
течение 30 мин. После охлаждения (~ 2 мин) 25 мкл этого раствора анализируют методом ВЭЖХ, используя методику 1 (см. таблицу 3).
Пример 23
Цикло-(D-Val-NMeArg-Gly-Asp-Mamb([99mTcO(MeMAG2gaba)]--5-Aca))
1,0 - 2,5 мг модифицированного введением "связки" циклического соединения
цикло-(D-Val-NMeArg-Gly-Asp-Mamb(5-Aca)) растворяют в 0,3 мл 0,5 M фосфатного буфера (pH 9) и добавляют раствор, получение которого описано в примере 22 часть B. Используя 1H NaOH, pH доводят до 9.
Реакционную смесь нагревают при 40oC 30 мин. После охлаждения (~2 мин) 25 мкл раствора анализируют методом ВЭЖХ, используя методику 1 (см. таблицу 3).
Пример 24
Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb([99mTcO(MeMAG2gaba)]--5-Aca))
1,0 - 2,5 мг модифицированного введением "связки" циклического соединения
цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(5-Aca)) растворяют в 0,3 мл 0,5 M фосфатного буфера с pH 9 и добавляют раствор, получение которого описано в примере 22 часть B. Используя 1H NaOH, значение pH доводят
до 9. Реакционную смесь нагревают при 40oC в течение 30 мин. После охлаждения (~2 мин) 25 мкл раствора анализируют методом ВЭЖХ, используя методику 1 (см. таблицу 3).
Пример 25
Цикло-([[99mTcO(MAG2)]--D-Lys]-NMeArg-Gly -Asp-Mamb)
Это соединение получают по методике, описанной в примере 22, используя в качестве
комплексообразователя Bz-MAG3 (см. таблицу 3).
Пример 26
Цикло-([[99mTcO(MeMAG2)]--D-Lys]-NMeArg-Gly -Asp-Mamb)
Это
соединение получают по методике, описанной в примере 22, используя в качестве комплексообразователя Bz-Me-MAG3 (см. таблицу 3).
Пример 27
Цикло-(D-Val-NMeArg-Gly-Asp-Mamb([99mTcO(MeMAG2 ACA)]--5-Aca))
Указанное в заглавии соединение получают по методике, описанной в примере 22, используя в части
A в качестве комплексообразователя Bz-Me-MAG2-ACa, а в части C в качестве модифицированного введением "связки" циклического соединения цикло-(D-Val-NMeArg-Gly-Asp-Mamb(5-Aca)) (см. таблицу
3).
Пример 28.
Цикло-([99mTcO(MABA)]--D-Lys]-NMeArg-Gly-Asp-Mamb)
Часть A. Образование координационного соединения
В
десятимиллилитровый пузырек добавляют 50 - 300 мКи99mTcO4- в 0,5 мл физиологического раствора, затем 0,5 мл раствора Bz-MABA (1 мг/мл в 0,5 M фосфатном буфере
с pH 12), а затем 0,15 мл раствора Na2S2O4 (5 мг/мл в 0,5 M фосфатном буфере с pH 11,5). Используя 1H NaOH, pH доводят до 10 - 12, и нагревают смесь 30 мин при 100oC, затем анализируют методом ВЭЖХ по методике 1.
Часть B. Активация
К раствору, получение которого описано в части A, добавляют 0,2 мл 1H HCl, 0,5 мл раствора TFP (50
мг/0,5 мл в 90% CH3CN) и 0,5 мл раствора DCI (50 мг в 0,5 90% CH3CN). Если необходимо, pH доводят до 6, и смесь нагревают при 45 - 50oC в течение 30 мин, затем
анализируют методом ВЭЖХ по методике 1.
Часть C. Сопряжение
К раствору, получение которого описано в части B, добавляют 2 - 3 мг промежуточного циклического соединения
цикло-(D-Lys-NMeArg-Gly-Asp-Mamb), растворенного в 0,5 мл 0,5 M фосфатного буфера с pH 9, и доводят pH до 9,5 - 10. Раствор нагревают при 50oC в течение 30 мин, затем анализируют методом
ВЭЖХ по методике 1 (см. таблицу 3).
Пример 29.
Цикло-(D-Val-NMeArg-Gly-Asp-Mamb([99mTcO(MABA]- -5-Aca))
Указанное в заглавии соединение
получают по методике, описанной в примере 28, используя в части C модифицированное введением "связки" циклическое соединение цикло-(D-Val-NMeArg-Gly-Asp-Mamb(5-Aca)) вместо промежуточного циклического
соединения цикло-(D-Lys-NMeArg-Gly-Asp-Mamb).
Пример 30
Цикло-(D-Abu-NMeArg-Gly-Asp-Mamb([99mTcO(MABA]- -5-Aca))
Указанное в заглавии
соединение получают по методике, описанной в примере 28, используя в части C модифицированное введением "связки" циклическое соединение цикло-(D-Abu-NMeArg-Gly-Asp-Mamb(5-Aca)) вместо промежуточного
циклического соединения цикло-(D-Lys-NMeArg-Asp-Mamb).
Пример 31
Цикло-([[99mTcO(MA-MAMA)]--D-Lys]-NMeArg-Gly-Asp-Mamb)
Часть A. Снятие
защиты
Тритильные группы комплексообразователя MA•MAMA удаляют растворением 6 мг в 1 мл безводной трифторуксусной кислоты (TFA). Полученный желтый раствор выдерживают при комнатной
температуре 5 мин. К желтому раствору добавляют триэтилсилан (0,5 мл) и получают прозрачную двухслойную смесь. Летучие удаляют при пониженном давлении, и получают белый остаток.
Часть B. Гидролиз этилового эфира.
К белому осадку, полученному по описанной в части A методике, добавляют 0,5 мл 5H NaOH и 1 мл THF. Смесь нагревают на водяной бане (100oC) 5 мин, в течение этого времени большая часть THF испаряется. К реакционной смеси добавляют 3 мл 0,5 M фосфатного буфера с pH 11,5. Значение pH доводят до 10-12 и добавляют дитионит натрия (15-30 мг). Смесь фильтруют и доводят общий объем до 6 мл 0,5 М фосфатным буфером с pH 11,5.
Часть C. Образование координационного соединения
В десятимиллилитровый пузырек помещают
50-150 мКи99mTcO4- в 0,5 мл физиологического раствора, затем 0,5 мл раствора лиганда, полученного по методике, описанной в части B. Используя 1H NaOH,
значение pH доводят до 10-12, затем смесь нагревают при 100oC в течение 30 мин и анализируют методом ВЭЖХ по методике 1.
Часть D. Активирование
К раствору,
полученному по методике, описанной в части C, добавляют 0,2 мл 1H HCl, 0,5 мл раствора TFP (50 мг/0,5 мл 90% CH3CN). Если необходимо, значение pH доводят до 6, и нагревают смесь при
45-50oC в течение 30 мин, затем анализируют методом ВЭЖХ по методике 1.
Часть E. Сопряжение
К раствору, полученному по методике, описанной в части D, добавляют 2,5
мг промежуточного циклического соединения цикло-(D-Lys-NMeArg-Gly-Asp-Mamb), растворенного в 0,5 мл 0,5 М фосфатного буфера с pH 9, и доводят значение pH до 9,5 - 10. После нагревания при 50oC в течение 30 мин раствор анализируют методом ВЭЖХ по методике 1.
Пример 32.
Цикло-(D-Val-NMeArg-Gly-Asp-Mamb([99mTcO(MA•MABA]-
-5-Aca))
Указанное в заглавии соединение получают по методике, описанной в примере 31, используя в части Е модифицированное введением "связки" соединение цикло-(D-Lys-NMeArg-Gly-Asp-Mamb).
Методы анализа
ВЭЖХ, методика 1
Колонка: Vydac C18, 250 мм х 4,6 мм, размер 300 A
Растворитель A: 10 мМ фосфат натрия, pH 6
Растворитель B:
100% ацетонитрил
Градиент:
0%B 30%B 75%B
O' 15' 25'
Скорость потока: 1,0 мл/мин
Детектирование: проба с NaI.
TLC, методика 2
Полоска ITLC-SG, 1 см х 7,5 см, элюент смесь ацетона и воды, 1:1.
ВЭЖХ, методика 2
Колонка: Vydac C18, 250 мм х 4 мм, размер пар 300 A
Растворитель A: 10
мМ фосфат натрия, pH 6,0
Растворитель B: 75% ацетонитрил в растворителе A
Градиент:
5%B 5%B 100%B
O' 5' 40'
Скорость потока: 1,0 мл/мин
Детектирование: проба с NaS.
Полезность
Содержащие радиоактивную метку соединения настоящего изобретения могут быть полезны в качестве радиофармпрепаратов для получения
изображения тромбов, которые могут присутствовать у больных с неустойчивыми ангинами, инфарктом миокарда, транзиторным ишемическим приступом, внезапным приступом, атеросклерозом, диабетом,
тромбофлебитом, эмболией легочной артерии или с кардиологическими протезирующими устройствами, такими как сердечный клапан, а также для диагностики таких имеющихся или потенциальных нарушений.
Пациентом может быть любой тип млекопитающих, но предпочтительно человек. Содержащие радиоактивную метку соединения могут использоваться самостоятельно или в виде композиции с радиофармацевтически
приемлемыми носителями и/или в сочетании с другими диагностическими или терапевтическими средствами. Подходящие радиофармацевтические носители и количество, в котором их следует использовать, хорошо
известны специалистам и могут быть найдены, например, в: Remington's Pharmaceutical Sciences, Gennaro, A.R., ed, Mack Publishing Company, Easton, PA (1985), and The United States Pharmacopia - The
National Formulary, 22nd Revision, Mack Printing Company, Easton, PA (1990) - общепринятых руководствах по фармацевтике. Также могут быть использованы другие известные специалистам добавки для
стабилизации композиции включая антиокислительные агенты, такие как бисульфит натрия, сульфит натрия, аскорбиновая кислота, гентизиновая кислота или лимонная кислота (или их соли), а также
этилендиаминтетрауксусная кислота (натрий ЭДТА), общеизвестные добавки. Другие такие материалы, а также количества, в которых их используют, также описаны в Remington's Pharmaceutical Sciences and The
United States Pharmacopia - The National Formulary, цитированных выше.
Настоящее изобретение также содержит радиофармацевтические наборы, содержащие меченые соединения данного изобретения. Такие наборы могут содержать меченое соединение в стерильной лиофилизованной форме и могут содержать стерильный резервуар с радиофармацевтически приемлемой разбавляющей жидкостью. Подходящие разбавляющие жидкости описаны в Remington's Pharmaceutical Sciences and The United States Pharmacopia - The National Formulary, цитированных выше. Такие наборы, возможно, могут включать стерильный резервуар с композицией соединения настоящего изобретения, содержащего радиоактивную метку. Такие наборы, если необходимо, могут включать другие обычные компоненты таких наборов, такие, например, как один или более носителей, один или более дополнительных флаконов для смешения. В набор также могут быть включены инструкции (вкладыши или этикетки), характеризующие количество меченого соединения и носитель, условия смешения компонентов и методику введения. Стерилизация резервуаров и других материалов, входящих в набор, и лиофилизация (также называемая лиофильной сушкой) меченых соединений настоящего изобретения могут быть осуществлены по общепринятым методикам стерилизации и лиофилизации, известным специалистам.
Для осуществления способа настоящего изобретения меченые радиоактивными изотопами соединения могут быть, как правило, введены внутривенно или в виде пилюль, хотя они также могут быть введены любыми средствами, обеспечивающими контакт этих соединений с тромбоцитами. Подходящие количества для введения могут быть легко установлены специалистами, использующими изобретение. Вводимые дозы, конечно, варьируются в зависимости от таких факторов, как вид соединения, возраст, состояние здоровья и вес пациента, а также симптомы, имеющиеся у пациента, количество радиоактивной метки, скорость выведения меченого радиоактивным изотопом соединения из крови.
Подходящие дозы для введения меченых радиоактивными изотопами материалов приведены, например, в руководстве Physicians Desk Refevence (PDR) for Nuclear Medicine, опубликованном Medical Exonomics Company, хорошо известном специалистам. Обсуждение некоторых из указанных выше факторов приводится в Eckelman et al., J.Nucl. Med., Vol. 209. pp. 350-357 (1979). Как обычно рекомендуют, дозы меченых радиоактивным изотопом соединений настоящего изобретения могут изменяться от приблизительно 1 до приблизительно 40 мKu.
После того, как меченое радиоактивным изотопом соединение введено, присутствие тромба может быть визуализировано обычными радиосцинтиллографическими системами получения изображения, такими как гамма-камера или компьютерный томографический прибор, в результате чего будет выявлено тромбоэмболическое нарушение. Такие системы получения изображения хорошо известны и описаны, например, в Macovski, A., Medical Imaging Systems, Information and Systems Science Series, Kailath, T., ed., Prentice-Hall, Inc., Englewood Cliffs, NJ (1983). Предпочтительными являются однофотонная эмиссионная компьютерная томография (SPECT) и позитронная эмиссионная томография (PET). В частности, получение изображения может проводиться сканированием всего пациента или отдельного участка пациента, в котором подозревается образование тромба, используя радиосцинтиллографическую систему, и детектированием сигнала радиоактивного изотопа. Детектируемый сигнал затем преобразуется системой в изображение тромба. Полученное изображение должно быть прочитано опытным специалистом, таким, например, как врачом - специалистом в области ядерной медицины. Вышеописанный процесс называется здесь снимком пациента. В общем, получение снимка пациента осуществляется в течение от примерно 1 мин до примерно 48 час после введения меченного радиоактивным изотопом соединения настоящего изобретения. Точное время получения изображения будет зависеть от таких факторов, как период полураспада используемого радиоактивного изотопа, скорость вывода из организма введенного соединения, и может быть установлено специалистом в этой области. Преимущественно, получение изображения проводится в течение от примерно 1 мин до примерно 4 час после введения препарата.
Преимущества использования содержащих радиоактивную метку соединений настоящего изобретения, которые обладают свойством специфической локализации и высоким сродством к тромбам, что позволяет определить наличие тромба и/или диагносцировать тромбоэмболические нарушения у пациента, будут вполне очевидны специалисту в данной области, познакомившемуся с настоящим изобретением.
Модель - артериовенозный анастомоз. Взрослые беспородные собаки различного пола (9-13 кг) были наркотизированы пентобарбиталом натрия (35 мг/кг, внутривенно) и вентилированы окружающим воздухом через интубационную трубку (12 раз/мин, 25 мл/кг). Для определения артериального давления левую сонную артерию канюлируют полиэтиленовым катетером (РЕ-240), заполненным физиологическим раствором, и подсоединяют к датчику давления Statham (P23ID; Oxnard, CA). Среднее артериальное кровяное давление определяют по затуханию пульсирующего сигнала. Частоту сердечных сокращений контролируют, используя кардиотахометр (Biotach, Grass Quincy, MA), подсоединенный ко II отведению электрокардиограммы, от конечностей. Шейную вену канюлируют (РЕ 240) для введения лекарства. Как бедренные артерии, так и бедренные вены канюлируют обработанными силиконом (Sigmacote, Sigma Chemical Co., St. Louis, Mo), наполненными физиологическим раствором трубками (PE-200), и соединяют с 5 см - секцией обработанной силиконом трубки (РЕ-240) для образования экстракорпоральных артериально-венозных шунтов (A-V). Раскрытое состояние шунта контролируют, используя доплер-систему контроля потока (модель VF-1, Crystac Biotech Ine, Hopkinton, MA) и пробу количества проходящей жидкости (2-2,3 мм, Titronies Med. Inst., Iowa City, IA), расположенную в непосредственной близости к шунту. Все параметры непрерывно записывают многоканальным самописцем (модель 7D Grass), скорость подачи бумаги 10 мм/мин или 25 мм/сек.
После окончания 15-минутного пост-хирургического стабилизационного периода формируют обтурирующий тромб введением тромбогенной поверхности (шелковая нить в 4-0 сплетений, длина 5 см, hicon Inc., Somervillie, NJ) в один шунт, другой шунт служит для контроля. Исследование проводят в течение двух последовательных 1 ч периодов с введенным инфузией в течение 5 мин исследуемым агентом, начиная отсчет за 5 мин до введения тромбогенной поверхности. В конце каждого одночасового периода шунтирования шелк тщательно удаляют, взвешивают и определяют подсчетом % инкорпорирования. Вес тромба рассчитывают вычитанием веса шелка до введения из суммарного веса шелка после его удаления из шунта. Результаты приведены в таблице 4. Артериальную кровь отбирают до первого шунта каждые 30 мин для определения коэффициента очищения крови, полной коллаген-индуцированной аггрегации тромбоцитов, тромбин-индуцированной дегрануляции тромбоцитов (выделение тромбоцитами АТР), протромбинового времени и количества тромбоцитов. Время кровотечения также определяют каждые 30 мин по стандартной методике.
Модель глубокого тромбоза вены (на собаках). Эта модель включает триаду явлений (состояние гиперкоагуляции, период стаза, незначительное изменение среды), существенных для образования венозного обогащенного фибрином активно растущего тромба. Методика заключается в следующем. Взрослых беспородных собак обоего пола (9- 13 кг) наркотизируют пентобарбиталом натрия (35 мг/кг, внутривенно) и вентилируют окружающим воздухом через интубационную трубку (12 раз/мин, 25 мл/кг). Для определения артериального давления правую бедренную артерию канюлируют наполненным физиологическим раствором полиэтиленовым катетером и подсоединяют к датчику давления Statham (P23ID, Oxnard, Ca). Среднее артериальное давление крови определяют по затуханию пульсирующего сигнала. Частоту сердечных сокращений контролируют, используя кардиотахометр (Biotach, Grass Quin-Су, MA), подсоединенный по II отведению электрокардиограммы, от конечностей. Правую бедренную артерию канюлируют для введения лекарства. Сегмент (5 см) обеих шейных вен выделяют, освобождают от фасции и накладывают шелковый шов. Микротермистер помещают на сосуд, что служит для косвенного измерения венозного потока. Для того, чтобы вызвать 15-минутный период стаза, используют катетер-баллон для эмболектомии, в это время наблюдается состояние гиперкоагуляции, вызываемое использованием 5U тромбина (American Diagnosticia, Grttnwich CT), введенного в блокированный сегмент. Через 15 мин кровоток восстанавливают, убирая баллон. Агент вводят в течение первых 5 мин после восстановления потока и контролируют скорость инкорпорирования, используя гамма-сцинтиллограф. Результаты для образцов по примерам 12 и 19 показаны на фиг.1.
Пример 33 (см. табл. 4).
Анализ аггрегации тромбоцитов. Собачью кровь собирают в 10 мл цитратные пробирки Vacutainer. Кровь центрифугируют в течение 15 мин при 150хg при комнатной температуре. Отделяют обогащенную тромбоцитами плазму (PRP). Оставшуюся кровь центрифугируют еще 15 мин при 150хg при комнатной температуре и отделяют обедненную тромбоцитами плазму (PPP). Образцы анализируют на аггрегометре (PAP-4 Platelet Affregation Profiler), используя PPP в качестве стандарта (100% пропускания). В каждую микропробирку для анализа добавляют 200 мкл PRP и пропускание устанавливают на 0%. В каждую пробирку добавляют 20 мкл различных активаторов (ADP, коллаген, арахидонат, адреналин, тромбин) и строят кривую аггрегации (% пропускания в зависимости от времени). Результаты выражают как % ингибирования вызванной активаторами аггрегации тромбоцитов. Для определения IC50 исследуемые соединения добавляют в различных концентрациях до активации тромбоцитов.
Анализ связывания тромбоцитами фибриногена
Связывание125I-фибриногена и тромбоцитов
осуществляют, как описано в: Btnnett et al. (1983) Proc. Natl. Acad. Sci. USA 80: 2417-24122, с некоторыми модификациями, приведенными ниже. Для очистки фракций тромбоцитов плазму (h-PRP) вводят в
колонку с Сефарозой. Аликвоту тромбоцитов (5•108 клеток) вместе с 1 мМ хлорида кальция добавляют до активации очищенных гель-хроматографически тромбоцитов (h-GPP).
Активацию очищенных гель-хроматографически тромбоцитов проводят, используя ADP, коллаген, арахидонат, адреналин и/или тромбин в присутствии лиганда,125I-фибриногена.125 I-фибриноген, присоединенный к активированным тромбоцитам, отделяют от несвязанной формы центрифугированием и затем измеряют гамма-счетчиком. Для оценки IC50 исследуемые соединения добавляют в различных концентрациях до активации тромбоцитов.
Новые циклические соединения гликопротеина IIb/IIIa настоящего изобретения могут также обладать тромболитической активностью, то есть они способны к лизису (разрушению) уже сформировавшихся обогащенных тромбоцитами фибринсодержащих сгустков крови, и это может быть полезным при лечении тромбообразования, как видно из данных по активности этих соединений в описанных ниже тестах. Преимущественными циклическими соединениями настоящего изобретения для разрушения (лизиса) тромбов являются соединения, характеризующиеся величиной IC50 (это молярная концентрация циклического соединения, способного вызывать 50% разрушение сгустков) менее чем примерно 1 мМ, более предпочтительно значение IC50 менее чем примерно 0,1 мМ, даже более предпочтительно значение IC50 менее чем примерно 0,01 мМ, еще более предпочтительно значение IC50 менее чем примерно 0,001 мМ и самым предпочтительным является значение IC50 примерно 0,0005 мМ.
Оценка величины IC50 может быть проведена с использованием стандартных методов анализа тромболиза, как описано ниже. Другой класс предпочтительных тромболитических соединений настоящего изобретения может включать такие соединения, которые характеризуются Kd < 100 нМ, преимущественно < 10 нМ и наиболее предпочтительно 0,1-1,0 нМ.
Анализ тромболиза. Венозную кровь, полученную от людей-доноров, не принимавших лекарства и аспирин по меньшей мере две недели по сдаче крови, помещают в 10 мл цитратные пробирки Vacutainer. Кровь центрифугируют 15 мин (1500хg) при комнатной температуре, отделяют обогащенную тромбоцитами плазму (PRP).
К PRP затем добавляют 1•10-3 М активатора - ADP, адреналин, коллаген, арахидонат, серотонин или тромбин, или их смесь, и инкубируют PRP в течение 30 мин. Центрифугируют PRP 12 мин (2500хg) при комнатной температуре. Выливают надосадочную жидкость, и тромбоциты, оставшиеся в пробирке, суспендируют в объединенной тромбоцитами плазмер (PPP), который служит источником плазминогена. Суспензию затем анализируют, используя счетчик Coutler Counter (Coutler Electronics, Inc., Hialeah, FL) для определения количества тромбоцитов в начальный момент - нулевая точка. После определения начала отсчета добавляют исследуемые соединения в различных концентрациях. Образцы исследуют в различные моменты времени и подсчитывают число тромбоцитов, используя счетчик Coulter Counter. Для того, чтобы определить процент лизиса, количество тромбоцитов, определенное в момент после введения исследуемого соединения, вычитают из количества тромбоцитов в нулевой точке, и полученную величину делят на количество тромбоцитов в нулевой точке. Умножая результат на 100, получают процент лизиса сгустков, вызываемого добавлением исследуемого соединения.
Для оценки величины IC50 исследуемые соединения добавляют в различных концентрациях, и подсчитывается процент лизиса, вызываемого добавлением исследуемых соединений.
Патенты и публикации, цитируемые в этом документе, приводятся только в качестве ссылок.
Из предшествующего описания специалистам будут вполне понятны различные модификации изобретения, в дополнение к тому, что показано и описано здесь. Такие модификации предполагаются всем объемом формулы изобретения.
Реферат
Изобретение относится к новым радиофармацевтическим препаратам, которые представляют собой радиоактивномеченые циклические соединения, содержащие карбоциклические или гетероциклические кольцевые системы и действующие как антагонисты гликопротеинового комплекса IIb/IIIa. Кроме того, изобретение относится к способу применения указанных радиофармацевтических препаратов в качестве предполагаемых агентов для диагностики артериального или венозного тромбоза, а также к новым реагентам для получения данных радиофармацевтических препаратов и к диагностическим наборам, содержащим указанные реагенты. 3 с. и 26 з.п. ф-лы, 7 табл., 2 ил.
Формула
(QLn)dCh,
где d = 1 - 2; Ln - связывающая группа;
Ch - хелатор металла;
Q - соединение формулы (I)
или его фармацевтически приемлемая соль или неактивная форма,
где J - D - Val, D - Lys, содержащий связь с Ln, и D - Abu, когда J представляет собой иной, чем D - Lys, содержащий связь с Ln, показанное фенильное кольцо может иметь факультативную связь с Ln-Ch в положении *, Ln - NHC(O) - (CH2)h - Y2 - M2 или C(O) - (CH2)h - Y2 - M2; M2 отсутствует или выбран из фенилен-CH2, циклогексил-CH2-, (CH2)2 и (CH2)3; h = 0, 1, 2, 3 или 5, Y2 отсутствует или представляет собой NHC(O),
Ch выбран из

где A1 - NH2, N = C(C1 - C3алкил)(C1 - C3алкил), N(-Ln)(CH2CO2H), S, SH или S(Pg);
A2 - N, NH, NH-пиридил-Ln или N(CH2CO2H);
A3 - N, NH, N - Ln, N(-Ln)(CH2CO2H) или N(CH2CO2H)2;
A4 - N - Ln, S, SH или S(Pg);
W отсутствует или выбран из (CH2)2, CH2C(O), CH(CH3)C(O), C(O)CH2, CH2CH - Ln или
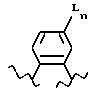
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
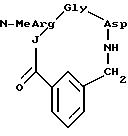
или его фармацевтически приемлемая соль, или неактивная форма,
где J - D - Lys, содержащий связь с Ln, Ln - C(O) - (CH2)n - Y2 - M2, M2 отсутствует или выбран из фенилен-CH2 или (CH2)2, h = 0, 1, 2, 3 или 5, Y2 отсутствует или представляет собой NHC(O), Ch выбран из
где A1 - NH2, N = C(C1 - C3алкил)(C1 - C3алкил), N(-Ln)(CH2CO2H), S, SH или S(Pg);
A2 - N, NH, NH-пиридил-Ln или N(CH2CO2H);
A3 - N, NH, NH-Ln, N(-Ln)(CH2CO2H) или N(CH2CO2H)2;
A4 - N-Ln, S, SH или S(Pg);
W отсутствует или выбран из (CH2 )2, CH2C(O), CH(CH3)C(O), C(O)CH2, CH2CH - Ln и
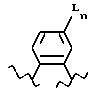
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
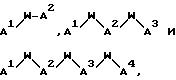
где A1 - NH2, N = C(C1 - C3алкил)(C1 - C3алкил), N(-Ln)(CH2CO2H), S, SH или S(Pg);
A2 - N, NH, NH-пиридил-Ln или N(CH2 CO2H);
A3 - N, NH, N-Ln или N(CH2CO2H)2;
A4 - N-Ln, S, SH или S(Pg);
W отсутствует или выбран из (CH2)2, CH2C(O), CH(CH3)C(O), C(O)CH2, CH2CH - Ln и
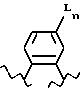
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
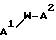
A1 - NH2 или N = C(C1 - C3алкил)(С1 - С3алкил), A2 - NH-пиридил-Ln и W отсутствует.
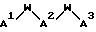
A1 - N(-Ln)(CH2CO2H);
A2 - N(CH2CO2H);
A3 - N(CH2CO2H)2;
W - (CH2)2.
A1 - S, SH или S(Pg);
A2 - N или NH;
A3 - N - Ln;
A4 - S, SH или S(Pg);
W - (CH2)2 или C(O)CH2;
Pg тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
где A1 - S, SH или S(Pg);
A2 - N или NH;
A3 - N или NH;
A4 - N - Ln;
W - (CH2)2, CH2C(O) и CH(CH3)C(O);
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
где A1 - S, SH или S(Pg);
A2 - N или NH;
A3 - N или NH;
A4 - S, SH или S(Pg);
W отсутствует или выбран из CH2C(O), C(O)CH2 и CH2CH - Ln,
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
где A1 - S, SH, S(Pg);
A2 - N или NH;
A3 - N или NH;
A4 - S, SH или S(Pg);
W отсутствует или выбран из CH2C(O), C(O)CH2 и
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
где A1 - N(-Ln)(CH2CO2H);
A2 - N(CH2CO2H);
A3 - N(-Ln)(CH2CO2H)2;
W - (CH2)2.
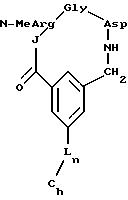
где J - D - Val или D - Abu;
Ln - NHC(O) - (CH2)5 - NHC(O) - M2;
M2 отсутствует или выбран из фенилен-CH2, циклогексил-CH2-, (CH2)2 и (CH2)3;
Ch -
A1 - NH2, N = C(C1 - C3алкил)(C1 - C3алкил), S, SH или S(Pg);
A2 - N, NH, NH-пиридил-Ln;
A3 - N, NH или N - Ln;
A4 - N, NH, N - Ln, S, SH или S(Pg);
W отсутствует или выбран из (CH2)2, CH2C(O), CH(CH3)C(O), C(O)CH2 и CH2CH - Ln;
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
где A1 - NH2 или N = C(C1 - C3алкил)(C1 - C3алкил);
A2 - NH-пиридил-Ln;
W отсутствует.
где A1 - S, SH или S(Pg);
A2 - N или NH;
A3 - N, NH или N-Ln;
A4 - N, NH, N-Ln, S, SH S(Pg);
W - (CH2)2, CH2C(O), CH(CH3)C(O), C(O)CH2 или CH2CH-Ln;
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
где A1 - S, SH или S(Pg);
A2 - N или NH;
A3 - N-Ln;
A4 - S, SH или S(Pg);
W - (CH2)2, C(O)CH2;
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
A1 - S, SH или S(Pg);
A2 - N или NH;
A3 - N, NH;
A4 - N-Ln;
W - CH2C(O) или CH(CH3)C(O);
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
где A1 - S, SH или S(Pg);
A2 - N или NH;
A3 - N или NH;
A4 - S, SH или S(Pg);
W - CH2C(O), C(O)CH2 или CH2CH-Ln;
Pg - тиоловая защитная группа, которая может быть заменена при взаимодействии с радионуклидом.
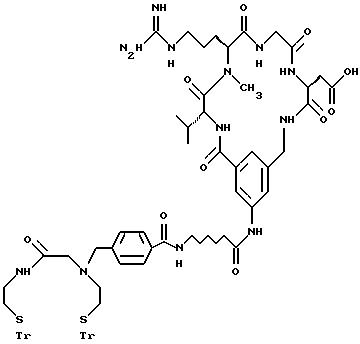
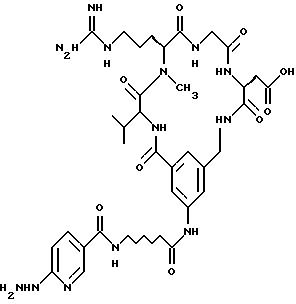
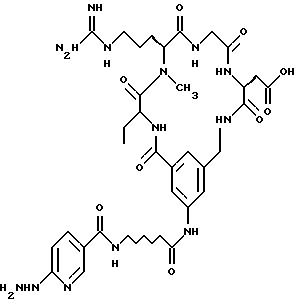
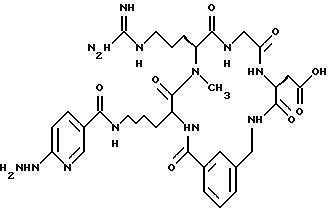
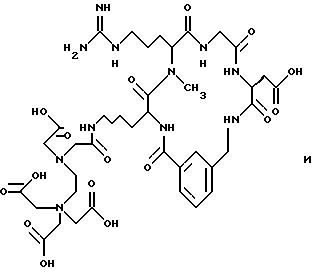
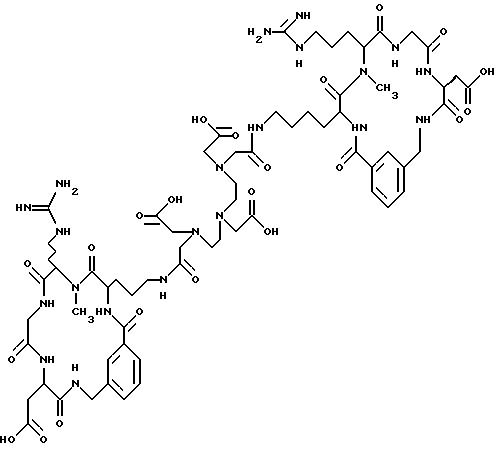
20. Радиофармацевтический препарат, представляющий собой комплекс соединения по одному из пп.1 - 19 и радионуклида, выбранного из группы99mTc,99mTcO и111In.
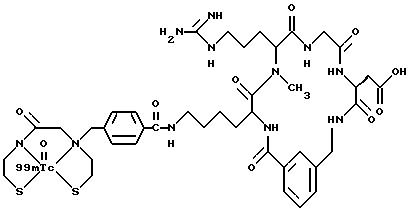
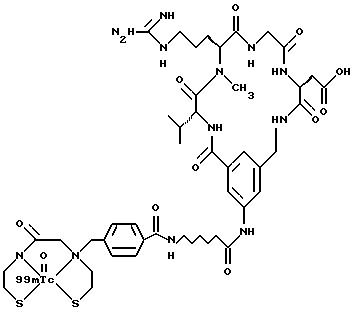
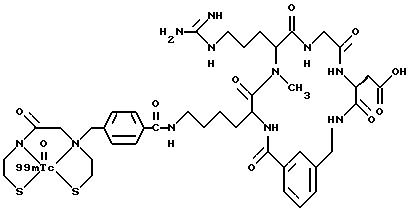
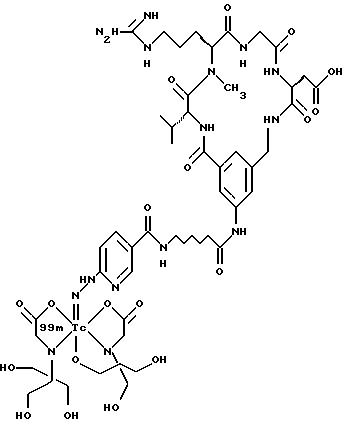
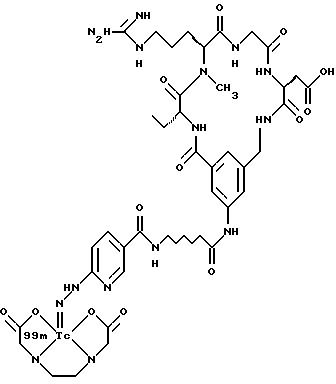

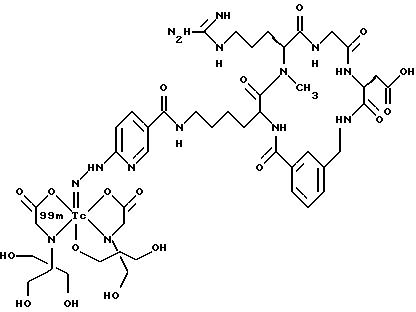
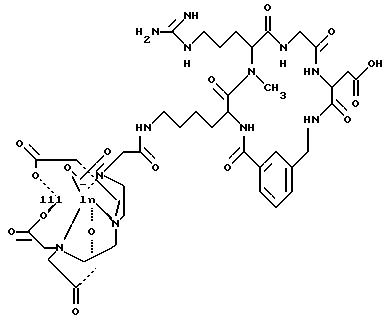
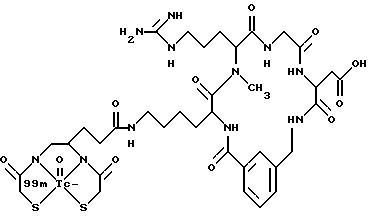
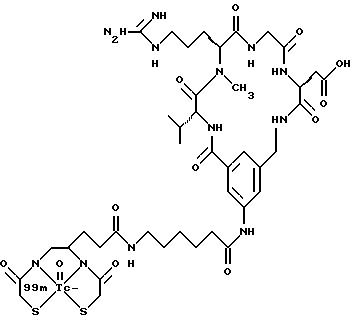
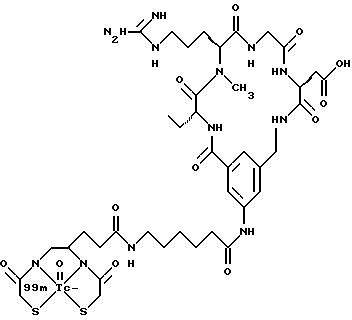
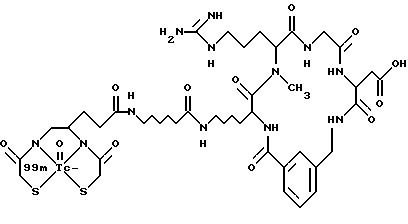
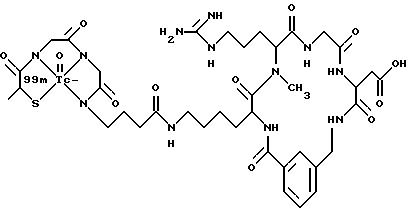
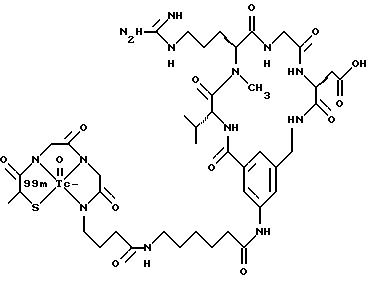

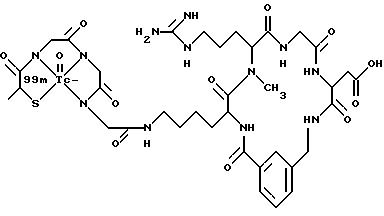
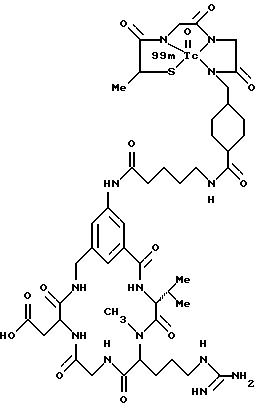
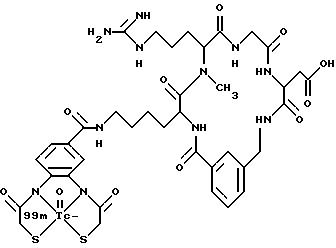
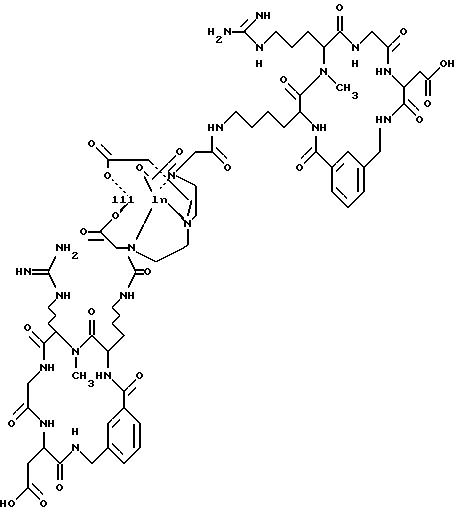
22. Циклический пептид, имеющий радиоактивную метку, введенную непосредственно, формулы (II)
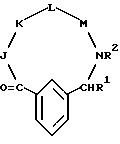
или его фармацевтически приемлемая соль или неактивная форма,
где R1 и R2 - водород;
J - D-изомер аминокислоты структуры -N(R3)C(R4) (R5)C(=O)-, где R3 - H, R4 - H, R5 - C1-C8алкил, с незамещенным или замещенным R11, R11 выбран из одного или нескольких -NHC(= O)R13 и арила, незамещенного или замещенного R12, R12 представляет собой -OH, R13 - (гидроксифенил)-C1-C10алкил,
К - L-изомер аминокислоты структуры -N(R6)CH(R7)C(= O)-, где R6 - C1-C4алкил, R7 - (C1-C7алкил)Х, Х -

где R13 - H;
L - -NHCH2C(=O)-;
M - L-изомер аминокислоты структуры

где q' = 1 - 2;
R4 - H;
R17 - H;
R8 - -CO2H;
где радиометка представляет собой125I или123I и она присоединена к арильной группе, представленной в формуле I.
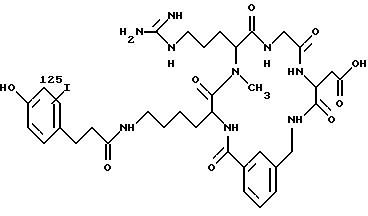
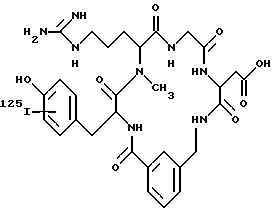
26. Циклический пептид, имеющий радиоактивную метку, по одному из пп.22 - 25, который используют для определения отложения тромбоцитов у млекопитающих.
30.03.93 по пп.22 - 27;
28.03.94 по пп.1 - 21, 28 и 29.
Комментарии