Способ получения изображения кровоснабжения миокарда - RU2648358C2
Код документа: RU2648358C2
Описание
Данное изобретение относится к новым соединениям, содержащим фрагменты, обеспечивающие изображение, и к их применению для диагностирования некоторых нарушений у пациента.
Митохондрии представляют собой окруженные мембранами органеллы, распределенные в цитозоле большинства эукариотных клеток. Митохондрии в основном сосредоточены в ткани миокарда.
Комплекс 1 ("МС-1") - это связанный с мембраной белковый комплекс 46 непохожих подъединиц. Этот комплекс является одним из трех комплексов, трансдуцирующих энергию, которые составляют респираторную цепь в митохондриях млекопитающего. Эта NADH - убихинон - оксоредуктаза представляет собой место входа большинства электронов, которые пересекают респираторную цепь, возможно приводя к восстановлению кислорода до воды (Q.Rev. Biophys. 1992, 25, 253-324).
Известные ингибиторы МС-1 включают дегелин, пиерицидин А, убицидин-3, роллиниастатин-1, роллиниастатин-2 (буллатацин), капсаицин, пиридабен, фенпироксимат, амитал, МРР+, хинолины и хинолоны (ВВА 1998, 1364, 222-235).
Данное изобретение частично основано на признании того, что прерывание нормальной функции митохондрий может благоприятно концентрировать некоторые соединения в митохондриях и, следовательно, в обогащенной митохондриями ткани миокарда.
Если в эти соединения введены фрагменты, обеспечивающие изображение, такой участок можно обнаружить, тем самым обеспечиваются ценные диагностические маркеры для получения изображения кровоснабжения миокарда. Для целей данной заявки такое соединение, содержащее фрагмент, обеспечивающий изображение, прикрепленный к этому соединению, называется "меченым".
Согласно одному из вариантов данное изобретение предусматривает способ получения изображения кровоснабжения миокарда, включающий введение пациенту контрастного агента, который содержит фрагмент, обеспечивающий изображение, и соединения, выбранного из дегелина, пиридабена, пиримидифена, тебуфенпирада, феназаквина, аналога дегелина, аналога пиридабена, аналога пиримидифена, аналога тебуфенпирада и аналога феназаквина, и сканирование пациента с использованием диагностического изображения. Согласно другому варианту фрагмент, обеспечивающий изображение, является радиоизотопом для ядерно-магнитной томографии, парамагнитными группами для применения в MRI, эхогенным фрагментом для применения в ультразвуковом исследовании, флуоресцентным фрагментом для применения в флуоресцентной томографии или светоактивным фрагментом для применения в оптической томографии.
Согласно еще одному варианту данное изобретение предусматривает контрастный агент, содержащий фрагмент, обеспечивающий изображение, и соединение, выбранное из дегелина, пиридабена, пиримидифена, тебуфенпирада, феназаквина, аналога дегелина, аналога пиридабена, аналога пиримидифена, аналога тебуфенпирада и аналога феназаквина.
Согласно еще одному варианту фрагмент, обеспечивающий изображение, является радиоизотопом для ядерно-магнитной томографии, парамагнитными группами для применения в MRI, эхогенным фрагментом для применения в ультразвуковом исследовании, флуоресцентным фрагментом для применения в флуоресцентной томографии или светоактивным фрагментом для применения в оптической томографии.
Согласно еще одному варианту парамагнитные группы для применения в MRI представляют собой Gd3+, Fe3+, In3+ или Mn3+.
Согласно еще одному варианту эхогенным фрагментом для применения в ультразвуковом исследовании является микросфера поверхностно-активного вещества, инкапсулированная во фторуглеводороде.
Согласно еще одному варианту радиоизотопов для ядерно-магнитной томографии представляет собой11С,13N,18F,123I,125I,99mTc,95Тс,111In,62Cu,64Cu,67Ga или68Ga. Согласно еще одному варианту фрагментом, обеспечивающим изображение является18F. Согласно другому варианту таким фрагментом служит99mTc.
Согласно другому варианту данное изобретение предусматривает контрастный агент, содержащий фрагмент, обеспечивающий изображение, и соединение, выбранное из дегелина, пиридабена, пиримидифена, тебуфенпирада, феназаквина, аналога дегелина, аналога пиридабена, аналога пиримидифена, аналога тебуфенпирада и аналога феназаквина, причем контрастный агент представляет собой соединение формулы (I).

где
каждый А независимо выбран из О, CHR1, S и NR1;
В выбран из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение;
С выбран из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение и связи с В;
D выбран из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение;
Е выбран из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение; или
Е и D вместе с атомом углерода, к которому они присоединены, образуют двойную связь, или
Е и D вместе с атомом углерода к которому они присоединены, образуют циклопропил;

а
R1, R2, R3, R4, R9, R10, R13 и R14 каждый независимо выбран из из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, галоида, гидроксильной группы и фрагмента, обеспечивающего изображение;
R5 и R6 каждый независимо выбран из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, галоида, гидроксильной группы и фрагмента, обеспечивающего изображение;
R7 и R8, если они содержатся, независимо выбраны из из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, галоида, гидроксильной группы и фрагмента, обеспечивающего изображение; или
R5 и R7 вместе образуют оксогруппу; или
R6 и R8 вместе образуют оксогруппу; или
R7 обозначает О или R8 обозначает связь с R7;
при условии, что когда
R11 обозначает водород или гидроксильную группу;
R12 выбран из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, или фрагмента, обеспечивающего изображение; или
R11 и R12 вместе образуют оксогруппу или =CHR1;
при условии, что в соединении формулы (I) имеется по меньшей мере один фрагмент, обеспечивающий изображение.
Согласно другому варианту А обозначает О;
В и С каждый независимо обозначает СН3 или CH218F;
D и Е каждый независимо обозначает СН3 или CH218F;
R5, R6, R9 и R10 каждый независимо обозначает водород или18F; и
R11 и R12 вместе образуют оксогруппу.
Согласно еще одному варианту контрастный агент выбран из







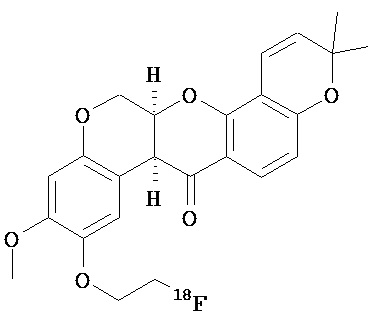
Согласно еще одному варианту данное изобретение предусматривает контрастный агент, содержащий фрагмент, обеспечивающий изображение, и соединение, выбранное из дегелина, пиридабема, пиримидифена, тебуфенпирада, феназаквина, аналога дегелина, аналога пиридабена, аналога пиримидифена, аналога тебуфенпирада, аналога феназаквина, причем контрастный агент является соединением формулы (II),

где G обозначает
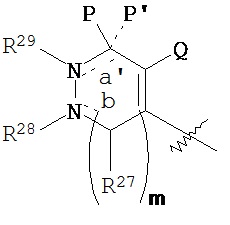

где m равен О или 1;


R27, R30, R31, R32, R33 и R34 каждый независимо выбран из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, или фрагмента, обеспечивающего изображение;
R28, если содержится, выбран из водорода и C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, при условии, что, когда является
R29, если содержится, обозначает C1-С6 алкил, возможно замещенный фрагментом, обеспечивающим изображение, при условии, что, когда

Р обозначает

независимо выбраны из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение;
Р’, если содержится, обозначает водород; или
Р и Р’ вместе образуют оксогруппу; при условии, что, когда

Q обозначает галоид или галоидалкил;
J выбран из N(R27), S, О, С(=О), С(=O)O, NHCH2CH2O, связи и С(=О) N(R27), причем каждая группа присоединена своим левым концом к G и правым концом присоединена к атому углерода, замещенному R21 и R22;
К, если содержится, выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение;
L, если содержится, выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение;
М выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение; или
L и М вместе с атомом, к которому они присоединены, образуют трех- или четырехчленное карбоциклическое кольцо; n равен 0, 1, 2 или 3;
R21, R22, R23, R24, R25 и R26 независимо выбраны из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение; и
Y выбран из связи, углерода и кислорода; при условии, что, когда Y обозначает связь, К и L отсутствуют и М выбран из арила и гетероарила; и при условии, что когда Y обозначает кислород, К и L отсутствуют и М выбран из из водорода, алкоксиалкила, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила;
при условии, что в соединении формулы (II) содержится по меньшей мере один фрагмент, обеспечивающий изображение.
Согласно другому варианту R29 обозначает C1-С6 алкил, где C1-С6 алкил является трет.бутилом.
Согласно еще одному варианту R28 обозначает C1-С6 алкил, где C1-С6 алкил является метилом.
Согласно еще одному варианту данное изобретение предусматривает контрастный агент, содержащий фрагмент, обеспечивающий изображение, и соединение, выбранное из дегелина, пиридабена, пиримидифена, тебуфенпирада, феназаквина, аналога дегелина, аналога пиридабена, аналога пиримидифена, аналога тебуфенпирада и аналога феназаквина, причем контрастный агент имеет формулу (III).

где
J выбран из N(R27), S, О, С(=О), С(=O)O, NHCH2CH2O, связи и С(=О) N(R27), причем каждая группа присоединена своим левым концом к G и правым концом присоединена к атому углерода, замещенному R21 и R22;
К, если содержится, выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение;
L, если содержится, выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение;
М выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение; или
L и М вместе с атомом, к которому они присоединены, образуют трех- или четырехчленное карбоциклическое кольцо;
Q обозначает галоид или галоидалкил;
правей 0, 1, 2 или 3;
R21, R22, R23, R24, R25, R26 и R27 независимо выбраны из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение;
R29 обозначает C1-С6 алкил, возможно замещенный фрагментом, обеспечивающим изображение и
Y выбран из связи, углерода и кислорода; при условии, что, когда Y обозначает связь, К и L отсутствуют и М выбран из арила и гетероарила; и при условии, что когда Y обозначает кислород, К и L отсутствуют и М выбран из из водорода, алкоксиалкила, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила;
при условии, что в соединении формулы (III) содержится по меньшей мере один фрагмент, обеспечивающий изображение.
Согласно другому варианту J обозначает О и R29 обозначает C1-С6 алкил, причем C1-С6 алкил является трет.бутилом.
Согласно еще одному варианту контрастный агент выбран из



Согласно еще одному варианту данное изобретение предусматривает контрастный агент, содержащий фрагмент, обеспечивающий изображение, и соединение, выбранное из дегелина, пиридабена, пиримидифена, тебуфенпирада, феназаквина, аналога дегелина, аналога пиридабена, аналога пиримидифена, аналога тебуфенпирада и аналога феназаквина, при этом контрастный агент имеет формулу (IV):

где
J выбран из N(R27), S, O, C(=O), C(=O)O, NHCH2CH2O, связи и С(=O) N(R27), причем каждая группа присоединена своим левым концом к G и правым концом присоединена к атому углерода, замещенному R21 и R22;
К, если содержится, выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение;
L, если содержится, выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение;
М выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение; или
L и М вместе с атомом, к которому они присоединены, образуют трех- или четырехчленное карбоциклическое кольцо;
Q обозначает галоид или галоидалкил;
правей 0, 1, 2 или 3;
R21, R22, R23, R24, R25, R26, R27, R28, R35, R36, R37, R38 и R39 независимо выбраны из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение; и
Y выбран из связи, углерода и кислорода; при условии, что, когда Y обозначает связь, К и L отсутствуют и М выбран из арила и гетероарила; и при условии, что когда Y обозначает кислород, К и L отсутствуют и М выбран из из водорода, алкоксиалкила, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила;
при условии, что в соединении формулы (IV) содержится по меньшей мере один фрагмент, обеспечивающий изображение.
Согласно еще одному варианту J обозначает С(=О) NH и R28 обозначает C1-С6 алкил, где C1-С6 алкил является метилом.
Согласно еще одному варианту контрастный агент выбран из








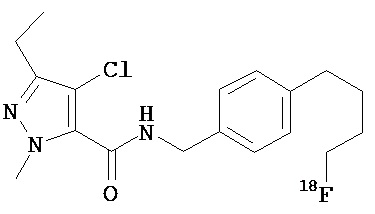
Согласно еще одному варианту данное изобретение предусматривает контрастный агент, содержащий фрагмент, обеспечивающий изображение, и соединение, выбранное из дегелина, пиридабена, пиримидифена, тебуфенпирада, феназаквина, аналога дегелина, аналога пиридабена, аналога пиримидифена, аналога тебуфенпирада и аналога феназаквина, при этом контрастный агент имеет формулу (V):
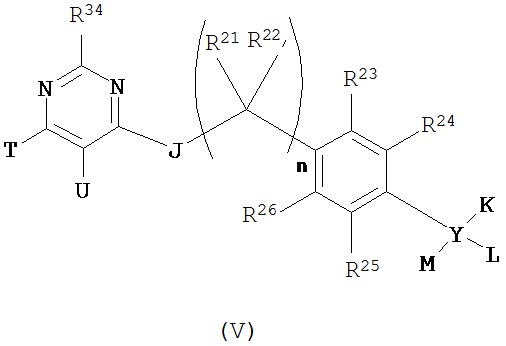
где
J выбран из N(R27), S, О, С (=О), С(=O)O, NHCH2CH2O, связи и С(=O)N(R27);
К выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение;
L, если содержится, выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение;
М, если содержится, выбран из водорода, алкоксиалкила, алкилокси, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, гетероарила и фрагмента, обеспечивающего изображение; или
L и М вместе с атомом, к которому они присоединены, образуют трех- или четырехчленное карбоциклическое кольцо;
Т и U независимо выбраны из водорода, алкокси, алкоксиалкила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, галоида и фрагмента, обеспечивающего изображение; или
Т и U вместе с атомами углерода, к которым они присоединены, образуют пяти- или шестичленное ароматическое или неароматическое кольцо, содержащее 0-2 гетероатома, выбранные из кислорода, азота и серы, причем это кольцо может быть замещено одним, двумя или тремя заместителями, независимо выбранными из C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение;
n равен 0, 1, 2 или 3; и
R21, R22, R23, R24, R25, R26, R27 и R34 независимо выбраны из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение;
Y выбран из связи, углерода и кислорода, при условии, что, когда Y обозначает связь, К и L отсутствуют и М выбран из арила и гетероарила; и при условии, что, когда Y обозначает кислород, К и L отсутствуют и М выбран из водорода, алкоксиалкила, арила, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и гетероарила; при условии, что соединение формулы (V) содержит по меньшей мере один фрагмент, обеспечивающий изображение.
Согласно другому варианту J обозначает О.
Согласно еще одному варианту данное изобретение предусматривает контрастный агент, содержащий фрагмент, обеспечивающий изображение, и соединение, выбранное из дегелина, пиридабена, пиримидифена, тебуфенпирада, феназаквина, аналога дегелина, аналога пиридабена, аналога пиримидифена, аналога тебуфенпирада и аналога феназаквина, при этом контрастный агент имеет формулу (VI):
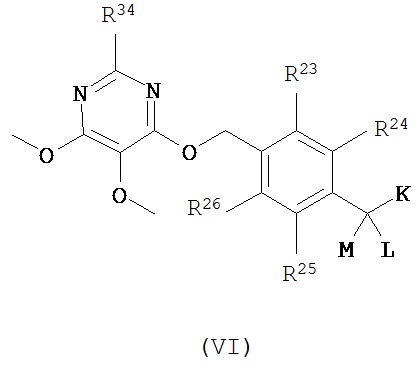
где
R23, R24, R25, R26 и R34 независимо выбраны из водорода, C1-С6 алкила, возможно замещенного фрагментом, обеспечивающим изображение, и фрагмента, обеспечивающего изображение;
при условии, что соединение формулы (VI) содержит по меньшей мере один фрагмент, обеспечивающий изображение.
Согласно другому варианту контрастный агент выбран из







Фрагменты, обеспечивающие изображение.
Контрастные агенты для ядерной томографии по данному изобретению включают11C,13N,18F,123I,125I,99mTc,95Tc,111In,62Cu,64Cu,67Ga и68Ga. Для проведения окисления жирных кислот применяли "С-пальмитат, а для достижения окислительного метаболизма в миокарде использовали11С-ацетат (Circulation 1987, 76, 687-696). Для получения изображения кровоснабжения миокарда широко применяется13N-аммиак (Circulation, 1989, 80, 1328-37). Для изображения гипоксии и рака использовали агенты на основе18F (Drugs of the Future 2002, 27, 655-667). Для получения изображения метаболизма миокарда применяли два иодированных агента - 15-(п-(123I) иодфенил)-пентадекановая кислота и 15-(п-(123I)-иодфенил)-3(R,S)-метилпентадекановая кислота. Согласно одному из вариантов фрагмент, обеспечивающий изображение в контрастных агентах согласно данному изобретению, является18F. Другие фрагменты, дающие изображение согласно изобретению, могут представлять собой один или несколько абсорбирующих Х-лучи или "тяжелых" атомов с атомным числом 20 или более, дополнительно содержащие возможный линкер L между фрагментом родительской молекулы и атомами, абсорбирующими Х-лучи. Часто в контрастных агентах в качестве тяжелого атома применяют иод. Недавно были описаны контрастные агенты, абсорбирующие Х-лучи, состоящие из хелатов металлов (патент США №5,417,959) и полихелатов, содержащих множество ионов металла (патент США №5,679,810). Совсем недавно в качестве контрастных агентов Х-лучей были описаны многоядерные кластерные комплексы (патент США №5,804,161, WO 91/14460 и WO 92/17125). Согласно некоторым вариантам данного изобретения металлы, используемые в контрастных агентах для Х-лучей включают Re, Sm, Ho, Lu, Pm, Y, Bi, Pd, Gd, La, Au, Yb, Dy, Cu, Rh, Ag и Ir.
Контрастные агенты для ядерно-магнитной томографии по данному изобретению могут состоять из одного или более аналогичных фрагментов, присоединенных к одному или нескольким ионам парамагнитных металлов, и дополнительно могут содержать линкер L между аналогичными фрагментами и ионами парамагнитных металлов. Ионы парамагнитных металлов могут быть в виде хелатов металлов или комплексов, или частиц окисей металлов. В патентах США №№5,412,148 и 5,760,191 описаны примеры хелатирующих агентов для ионов парамагнитных металлов, используемых в контрастных агентах для MRI.
В патентах США №№5,801,228, 5,567,411 и 5,281,704 описаны примеры полихелантов, используемых для получения комплексов с более чем одним ионом парамагнитного металла для использования в контрастных агентах для MRI.
В патенте США №5,520,904 раскрыты композиции, содержащие ионы парамагнитных металлов, для применения в контрастных агентах для MRI. Примеры конкретных металлов включают Gd3+, Fe3+, In3+ и Mn2+. Ультразвуковые контрастные агенты по данному изобретению могут включать множество аналогичных фрагментов, присоединенных к микропузырьку биосовместимого газа, жидкого носителя и микросферы поверхностно-активного вещества, или включенных в состав этого микропузырька, и могут также содержать линкер L между аналогичными фрагментами и микропузырьком. В данном контексте термин "жидкий носитель" означает водный раствор, а термин "поверхностно-активное вещество" означает любое амфифильное вещество, которое может приводить к снижению напряжения на границе раздела фаз в растворе. Перечень подходящих поверхностно-активных веществ для получения микросфер приведен, например, в заявке ЕР 0737225 А2. Термин "микросфера поверхностно-активного вещества относится к микросферам, наносферам, липосомам, пузырькам и т.п. Биосовместимый газ может быть любым физиологически приемлемым газом, включая, например, воздух, фторуглеводород, такой как С3-С5 перфторалкан, который обеспечивает разницу в эхогенности и тем самым контраст в ультразвуковом изображении.
Газ может быть инкапсулирован или содержаться в микросфере или каким-либо образом захватываться этой микросферой, к которой присоединен фрагмент, возможно через линкер. Присоединение может быть ковалентным, ионным или при попомщи Ван-дер-Ваальсовых сил. Конкретные примеры таких контрастных агентов включают, например, жидкие инкапсулированные углеводороды с множеством пептидов, связывающих неоваскулярные опухолевые рецепторы, полипептидов или пептидомиметиков. Примеры фрагментов, обеспечивающих изображение, наполненных газом, включают описанные в заявке США на патент №09/931,317, поданной 16 августа 2001 г. и патентах США №№5,088,499, 5,547,656, 5,228,446, 5,585,112 и 5,846,517.
Хелатирующие агенты
Известны многие подходы к мечению соединений при помощи99mTc, включая прямую метку соединения и включение хелатирующего фрагмента ("хелатора"). Согласно одному из вариантов хелатор является DADT, MAG3, МАМА, РАМА или DOTA.
Соединения по изобретению могут содержать хелатор ("С"). По некоторым вариантам изобретения хелатор представляет собой поверхностно-активное вещество, способное к образованию эхогенной наполненной веществом липидной сферы или микропузырька. Согласно другим вариантам хелатор является связующей единицей формулы, выбранной из







где
каждый А1 независимо выбран из -NR46R47, -NHR53, - SH, - S(Pg), - ОН, - PR46R47, -P(O)R48R49 и связи с соединением, которое связывает МС-1;
каждый А2 независимо выбран из N(R53), N(R46), S, О, P(R46) и OP(O)(R48)O-;
А3 обозначает N;
А4 выбран из ОН и ОС(=O)C1-С20 алкила;
А5 обозначает ОС(=O)C1-C20 алкил;
каждый Е независимо выбран из C1-C16 алкилена, замещенного 0-3 R50, С6-С10 арилена, замещенного 0-3 R50, С3-С10 циклоалкилена, замещенного 0-3 R50, гетероциклил - C1-С10 алкилена, замещенного 0-3 R50, С6-С10 арил - C1-С10 алкилена, замещенного 0-3 R50, и гетероциклилена, замещенного 0-3 R50;
Е1 выбран из связи и Е;
Каждый Е2 независимо выбран из C1-C16 алкила, замещенного 0-3 R50, С6-С10 арила, замещенного 0-3 R50, С3-С10 циклоалкила, замещенного 0-3 R50, гетероциклил-C1-С10-алкила, замещенного 0-3 R50, С6-С10арил - C1-С10 алкила, замещенного 0-3 R50, C1-С10 алкил - С6-С10 арила, замещенного 0-3 R50, и гетероциклила, замещенного 0-3 R50;
Е3 обозначает C1-С10 алкилен, замещенный 1-3 R59;
Pg - тиольная защитная группа;
R46 и R47 каждый независимо выбраны из связи с соединением, которое связывает МС-1, водорода, C1-С10 алкила, замещенного 0-3 R50, арила, замещенного 0-3 R50, С3-С10 циклоалкила, замещенного 0-3 R50, гетероциклил - C1-С10 алкила, замещенного 0-3 R50, С6-С10 арил - C1-С10 алкила, замещенного 0-3 R50, и гетероциклила, замещенного 0-3 R50;
R48 и R49 каждый независимо выбраны из связи с соединением, которое связывает МС-1, ОН, С1-С10 алкила, замещенного 0-3 R50, арила, замещенного 0-3 R50, С3-С10 циклоалкила, замещенного 0-3 R50, гетероциклил - C1-С10 алкила, замещенного 0-3 R50, С6-С10 арил - С1-С10 алкила, замещенного 0-3 R50, и гетероциклила, замещенного 0-3 R50;
каждый R5 независимо выбран из связи с соединением, которое связывает МС-1, =O, галоида, трифторметила, -CO2R51, -C(=O)R51, -С(=О)N(R51)2, -СНО, -CH2OR51; -ОС(=O)R51, -OC(=O)OR51, -OR51, -OC(=O)N(R51)2, -NR51C(=O)R51, -NR51C(=O)OR51, -NR51C(=O)N(R51)2, -NR51SO2N(R51), -NR51SO2R51, -SO3H, -SO2R51, -SR51, -S(=O)R51, SO2N(R51)2, -N(R51)2, -NHC(=S)NHR51, =NOR51, NO2, -C(=O)NHOR51, -C(=O)NHN(R51)2, -OCH2CO2H, 2-(1-морфолин)этокси, C1-C5 алкила, С2-С4 алкенила, С3-С6 циклоалкила, С3-С6 циклоалкилметила, С2-С6 алкоксиалкила, арила, замещенного 0-2 R51, и гетероциклила;
каждый R51 независимо выбран из связи с соединением, которое связывает МС-1, водорода, C1-С6 алкила, фенила, бензила и C1-С6 алкокси;
R53 обозначает координационную связь с металлом;
каждый R59 выбран из R61, =O, -CO2R60, -С(=O)R60, -C(=O)N(R60)2, CH2OR60, OR60, N(R60)2 и С2-С4 алкенила;
каждый R60 независимо выбран из R61, водорода, C1-С6 алкила, фенила, бензила, трифторметила и
R61 обозначает связь с соединением, которое связывает МС-1;
причем по меньшей мере один из A1, R46, R47, R48, R49, R50, R51 и R61 обозначает связь с соединением, которое связывает МС-1.
Способы получения
Обычно меченые +F соединения синтезируют Sn2 замещением соответствующей удаляемой группы. Эти удаляемые группы предпочтительно являются эфирными группами сульфоновой кислоты, такими как толуолсульфонатная (тозилатная, TsO), метансульфонатная (мезилатная, MsO) или трифторметансульфонатная (трифлатная, TfO).
Удаляемая группа может быть галоидом, фосфиноксидной (по реакции Мицунобу) или внутренней удаляемой группой (такой как эпоксидная или группа циклического сульфата). Указанные соединения получают из высокоактивированного сухого K18F, который делают "горячее" путем добавления криптандов, таких как критофикс [2.2.2]. Очистку проводят обычно путем удаления соли методом хроматографии с обращенной фазой (Sep-Pak).
Примеры получения контрастных агентов описаны в следующих примерах. Химические превращения можно проводить с применением методик, хорошо известных специалистам, знакомым с данной заявкой. Примеры растворителей включают, например, ДМФ, N-МП, ДМСО, ТГФ, этилацетат, дихлорметан и хлороформ. Реакционный раствор может быть нейтральным или щелочным, что достигается путем добавления амина, такого как триэтиламин или ДИЭА. Реакции могут проводиться при комнатной температуре и защищаться от кислорода и воды за счет использования атмосферы азота.
Для предотвращения реакции других групп, таких как аминные, спиртовые, фенольные и карбоксильные, можно применять временные защитные группы. Примеры защитных групп для аминов включают, например, трет.бутоксикарбонил и тритил (удаляются в умеренно кислых условиях), Fmoc (удаляется при применении вторичных аминов, таких как пиперидин) и бензилоксикарбонил (удаляемый сильной кислотой или каталитическим гидрогенолизом). Для защиты тиолов, фенолов и спиртов можно применять тритильную группу. Согласно некоторым вариантам для защиты карбоксильных групп используют защитные группы, включающие, например, трет.бутилэфирную (удаляется умеренной кислотой), бензилэфирную (обычно удаляется путем каталитического гидрогенолиза) и группы алкиловых эфиров, таких как метиловый или этиловый (обычно удаляются умеренным основанием). Все защитные группы можно удалить в конце синтеза, используя условия, описанные выше для отдельных защитных групп, а конечный продукт может быть очищен методами, которые очевидны для специалистов, знакомых с данным описанием.
Применение
Контрастные агенты согласно изобретению могут быть использованы в способе получения изображения (визуализации), включая получение изображения для пациента, включающем введение контрастного агента пациенту путем инъекции, инфузии или другим известным методом и визуализации области в организме пациента, где происходит исследуемое явление.
Применяемая доза вводимого агента и конкретный метод введения зависят от таких факторов, как возраст, вес, конкретная исследуемая область, а также вид конкретного контрастного агента, диагностическое применение и форма состава, например, суспензия, эмульсия, микросфера, липосома или т.п., что очевидно для специалистов.
Обычно используют минимальные дозы и увеличивают их, пока не будет достигнут желаемый диагностический эффект. По одному из вариантов вышеописанные контрастные агенты можно вводить путем внутривенной инъекции, обычно в физиологическом растворе, с дозой от примерно 0,1 до примерно 100 мКюри на 70 кг веса (все комбинации доз и конкретные дозы) или предпочтительно с дозой от примерно 0,5 до примерно 50 мКюри. Визуализация осуществляется методами, хорошо известными специалистам.
При применении в качестве контрастных агентов для магнитно-ядерной томографии композиций по изобретению, дозы, вводимые путем внутривенной инъекции, обычно находятся в пределах от примерно 0,5 мкмол/кг до примерно 1,5 ммол/кг (все комбинации доз и конкретные дозы), предпочтительно от примерно 0,8 мкмол/кг до примерно 1,2 ммол/кг.
В случае контрастных агентов для MRI композиции по изобретению могут быть использованы так же, как и другие агенты для MRI, описанные в патентах США №5,155,215, №5,087,440; Magn. Reson. Med. 1986, 3, 808; Radiology 1988, 166, 835; и Radiology 1988, 166, 693. Обычно пациенту можно вводить стерильные водные растворы контрастных агенту внутривенно в дозах от примерно 0,01 до примерно 1,0 ммол на кг вес (все комбинации доз и конкретные дозы).
Контрастные агенты для ультразвукового исследования можно вводить путем внутривенной инъекции в количестве от примерно 10 до примерно 30 мкл (все комбинации доз и конкретные дозы) эхогенного газа на кг веса или путем инфузии со скоростью около 3 мкл/кг/мин.
Другой аспект данного изобретения относится к диагностическим наборам для получения диагностических агентов для выявления, визуализации и/или мониторинга кровоснабжения миокарда.
Диагностические наборы по изобретению включают один или несколько сосудов, содержащих стерильный непирогенный состав, включающий заданное количество реагента по данному изобретению и, возможно, другие компоненты, такие как один или два вспомогательных лиганда, таких как трицин и 3-[бис(3-сульфофенил)фосфин) бензосульфокислота (TPPTS), восстановители, передающие лиганды, буферы, вспомогательные лиофилизирующие добавки, стабилизаторы, солюбилизаторы и бактериостатики. Наборы могут также содержать восстановитель, такой как, например, олово (II).
Буферы, применяемые при получении контрастных агентов и наборов, включают, например, фосфатные, цитратные, сульфосалицилатные и ацетатные буферы. Более полный перечень можно найти в United States Pharmacopoeia.
Лиофилизирующие средства, используемые при получении контрастных агентов и наборов, включают, например, манит, лактозу, сорбит, декстран, FICOLL® полимер и поливинилпирролидин (PVP).
Стабилизаторы, применяемые при получении контрастных агентов и наборов, включают, например, аскорбиновую кислоту, цистеин, монотиоглицерин, бисульфит натрия, метабисульфит натрия, гентисовую кислоту и инозитол.
Солюбилизирующие агенты, используемые для получения контрастных агентов и наборов, включают, например, этанол, глицерин, полиэтиленгликоль, пропиленгликоль, полиоксиэтиленсорбитана моноолеат, моноолеат сорбитана, полисорбаты, блок - сополимеры, поли(оксиэтилен) - поли(оксипропилен) - поли(оксиэтилен) ("Pluronics") и лецитин. Согласно некоторым вариантам солюбилизаторами являются полиэтиленгликоль и Pluronics.
Бактериостатики, применяемые для получения контрастных агентов и наборов, включают, например, бензиловый спирт, бензалкония хлорид, хлорбутанол и метил-, пропил- или бутилпарабен.
Компонент диагностического набора может выполнять более одной функции. Например, восстановитель для радионуклида может также служить стабилизатором или буфер может также служить лигандом, или лиофилизирующая добавка может также служить передающим, вспомогательным лигандом или солигандом.
Вышеописанные соединения могут иметь асимметричные центры. Если иное не указано, данное изобретение охватывает все хиральные, диастереомерные и рацемические формы. В соединениях по изобретению могут также содержаться многие геометрические изомеры олефинов, С=N двойных связей и т.п., все такие стабильные изомеры охвачены данным изобретением. Следует отметить, что соединения по изобретению могут содержать асимметрично замещенные атомы углерода и могут быть выделены в оптически активных или рацемических формах. Приготовление оптически активных форм, например, путем разрешения рацемических форм или путем синтеза их оптически активных исходных веществ хорошо известно из уровня механики. Известно, что существуют два различных изомера (цис и транс) пептидной связи; оба они также могут содержаться в соединениях по изобретению, и все такие стабильные изомеры охвачены данным изобретением. D- и L-изомеры конкретной аминокислоты в данном описании обозначаются при помощи принятого 3-х буквенного сокращения аминокислоты, как указано в следующих примерах: D-Leu или L-Leu.
Для упрощения места соединения ("-") не показаны. Когда указано, что атом или соединение означают переменную величину, это означает, что она заменяет переменную таким образом, чтобы насытить валентность атома или соединения. Например, если переменная "А" означает C(R80)=C(R80), оба атома углерода образуют часть цепи, чтобы насытить их соответствующие валентности.
Когда любая переменная содержится более одного раза в любом заместителе или в любой формуле, ее определение в каждом случае не зависит от ее определения в каждом другом случае.
Так, например, если группа или ряд групп замещены 0-2 R80, тогда указанная(-ые) группа(-ы) могут содержать до двух R80 и R80 в каждом случае в каждой группе выбирается независимо из указанного перечня возможных R80. Так же, например, в случае группы -N(R81)2 каждый из двух заместителей R81 у атома N независимо выбирается из указанного перечня возможных R81. Комбинации заместителей и/или переменных допускается только в том случае, если эта комбинация приводит к получению стабильных соединений. Когда показано, что связь, присоединяющая заместитель, пересекает связь, соединяющую два атома в кольце, тогда такой заместитель может присоединяться к любому атому в кольце.
Определения
Число атомов углерода в любой конкретной группе указывается перед названием группы. Например, термин "С6-С10 арил" означает арильную группу, содержащую от 6 до 10 атомов углерода, а термин "С6-С10 арил - C1-C10 алкил" относится к арильной группе, содержащей от 6 до 10 атомов углерода, присоединенных к части родительской молекулы через алкильную группу, содержащую от 1 до 10 атомов углерода.
Термин "алкенил" относится к линейному или разветвленному углеводороду, содержащему по меньшей мере одну углерод - углеродную двойную связь.
Термин "алкокси" относится к C1-С6 алкилу, присоединенному к части родительской молекулы через атом кислорода.
Термин алкоксиалкил" относится к C1-С6 алкилу, замещенному одной, двумя или тремя алкоксигруппами.
Термин "алкил" относится к группе на основе линейного или разветвленного насыщенного углеводорода.
Термин "алкиларил" относится к алкильной группе, присоединенной к части родительской молекулы через арильную группу.
Термин "алкилен" относится к двухвалентной группе на основе линейного или разветвленного насыщенного углеводорода.
Термин "алкилокси" относится к C1-С6 алкилу, присоединенному к части родительской молекулы через атом кислорода.
Термин "аналогичный фрагмент" означает соединения по изобретению, не содержащие части или частей, обеспечивающих изображение.
Термин "арил" относится к фенильной группе или бициклической конденсированной кольцевой системе, в которой одно или несколько колец являются фенильными. Бициклические конденсированные кольцевые системы содержат фенильную группу конденсированную с моноциклической циклоалкенильной группой, моноциклической циклоалкильной группой или другой фенильной группой. Арильные группы по данному изобретению могут быть присоединены к части родительской молекулы через любой замещаемый атом углерода в группе. Примеры арильных групп включают, без ограничения, антраценил, азуленил, флуоренил, инданил, инденил, нафтил, фенил и тетрагидронафтил.
Термин "арилалкил" относится к алкильной группе, замещенной одной, двумя или тремя арильными группами.
Термин "арилалкилен" относится к двухвалентной арилалкильной группе, в которой одно место присоединения к части родительской молекулы находится в арильной группе, а другое - в алкильной группе.
Термин "арилен" относится к двухвалентной арильной группе.
Используемые термины "вспомогательный" или "солиганды" относятся к лигандам, которые служат для завершения координационной сферы радионуклида вместе с хелатором или единицей реагента, связывающего радионуклид. В случае радиофармацевтиков, содержащих бинарную лигандную систему, координационная сфера радионуклида содержит один или несколько хелаторов или связующих единиц одного или нескольких реагентов и один или несколько вспомогательных лигандов или солигандов, при условии, что содержатся всего два типа лигандов, хелаторов или связующих единиц. Например, радиофармацевтик, состоящий из одного хелатора или связующей единицы одного реагента и двух одинаковых вспомогательных лигандов или солигандов, и радиофармацевтик, содержащий два хелатора или две связующие единицы одного или двух реагентов и один вспомогательный лиганд или солиганд, оба рассматриваются как бинарные лигандные системы. В случае радиофармацевтиков, содержащих тройную лигандную систему, координационная сфера радионуклида состоит из одного или нескольких хелаторов или связующих единиц одного или нескольких реагентов и одного или нескольких двух разных типов вспомогательных лигандов или солигандов, при условии, что содержится всего три типа лигандов, хелаторов или связующих единиц. Например, радиофармацевтик, содержащий один хелатор или одну связующую единицу одного реагента и два разных вспомогательных лиганда или солиганда, считается содержащим тройную лигандную систему.
Вспомогательные лиганды или солиганды, используемые для получения радиофармацевтиков и в диагностических наборах для получения указанных радиофармацевтиков, содержат один или несколько донорных атомов кислорода, азота, углерода, серы, фосфора, мышьяка, селена и теллура.
Лиганд может быть лигандом - переносчиком в синтезе радиофармацевтика, а также может служить вспомогательным лигандом или солигандом в другом радиофармацевтике. Определение лиганда как переносчика или вспомогательного лиганда или солиганда зависит от того, остается ли лиганд в координационной сфере радионуклида в радиофармацевтике, что определяется координационной химией радионуклида и хелатора или связующей единицы реагента или реагентов.
"Бактериостатик" представляет собой компонент, который ингибирует рост бактерий в препарате или во время хранения перед использованием или после того, как диагностический набор был использован для синтеза радиофармацевтика.
Термин "пузырьки" или "микропузырьки" относится к пузырькам, которые обычно характеризуются наличием одной или нескольких мембран или стенок, окружающих внутреннее пространство, которое заполнено газом или его предшественником.
Примеры пузырьков или микропузырьков включают, например, липосомы, мицеллы и т.п.
Термины "хелатор" или "связующая единица" относятся к фрагменту или группе реагента, который связывается с ионом металла через один или несколько донорных атомов.
Термин "контрастный агент" относится к агенту, используемому для освещения специфических областей, когда органы, кровеносные сосуды и/или ткани становятся более видимыми. Путем увеличения видимости исследуемых поверхностей можно определить наличие и степень развития болезни и/или повреждения.
Термин "циклоалкенил" относится к неароматической частично ненасыщенной моноциклической, бициклической или трициклической кольцевой системе, содержащей от 3 до 14 атомов углерода и не содержащей гетероатомов. Примеры циклоалкенильных групп включают, без ограничения. Циалогексенил, октагидронафталенил и норборниленил.
Термин "циклоалкил" обозначает насыщенную моноциклическую, бициклическую или перициклическую углеводородную кольцевую систему, содержащую от 3 до 14 атомов углерода и не содержащую гетероатомов. Примеры циклоалкильных групп включают, без ограничения, циклопропил, циклопентил, бицикло - [3.1.1] гептил и адамантил.
Термин "С3-С10 циклоалкилен" относится к двухвалентной циклоалкильной группе, содержащей от 3 до 10 атомов углерода.
Термин "диагностическое получение изображения" обозначает процедуру, используемую для обнаружения контрастного агента.
"Диагностический набор" или "набор" содержит ряд компонентов в одном или нескольких сосудах, которые используются практикующим специалистом в клинике или фармацевтике для синтеза диагностических радиофармацевтиков. Набор предпочтительно предусматривает наличие всех компонентов, требующихся для синтеза и применения фармацевтика, за исключением тех, которые общедоступны для практикующего специалиста, таких как вода или физиологический раствор для инъекции, раствор радионуклида, оборудование для нагревания набора в процессе синтеза радиофармацевтика, если это требуется, оборудование, необходимое для введения радиофармацевтика пациенту, такого как шприцы, экраны, оборудование для визуализации и т.п. Контрастные агенты предоставляются пользователю в их конечном виде в составе, обычно содержащемся в одном сосуде, в виде или лиофилизированного твердого вещества, или водного раствора. Пользователь восстанавливает лиофилизированный продукт водой или физиологическим раствором и извлекает дозу для пациента или извлекает дозу из имеющегося водного состава.
Термин "донорный атом" относится к атому, который непосредственно присоединен химической связью к металлу.
Термины "гало" и "галоген" относятся к F, Cl, Br или I.
Термин "галоалкил" означает C1-С6 алкил, замещенный одним, двумя, тремя или четырьмя атомами галоида.
Термин "гетероарил" относится к ароматическому пяти- или шестичленному кольцу, в котором по меньшей мере один атом выбран из N, О, S, Q остальные атомы являются атомами углерода. Термин "гетероарил" включает также бициклические системы, в которых гетероарильное кольцо конденсирован с четырех - шестичленным ароматическим или неароматическим кольцом, содержащим 0, 1 или 2 дополнительных гетероатома, выбранных из N, О и S. Гетероарильные группы присоединены к родительской молекуле через любой способный к замещению атом углерода или азота в группе. Примеры гетероарильных групп включают, без ограничения, бензоксадиазолил, бензоксазолил, бензофуранил, бензотиенил, фуранил, имидазолил, индазолил, индолил, изоксазолил, изохинолил, изотиазолил, нафтиридинил, оксадиазолил, оксазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, пиразолил, пирролил, хинолинил, тиазолил, тиенопиридинил, тиенил, триазолил, тиадиазолил и триазинил.
Термин "гетероциклил" охватывает пяти-, шести- или семичленные кольца, содержащие один, два или три гетероатома, независимо выбранные из группы, состоящей из азота, кислорода и серы. Пятичленное кольцо содержит 0-2 двойные связи, а шести- и семичленные кольца содержат 0-3 двойные связи. Термин "гетероциклил" включает также бициклические группы, в которых гетероциклил конденсирован с фенильной группой, моноциклической циклоалкенильной группой, моноциклической циклоалкильной группой или другой моноциклической гетероциклильной группой. Гетероциклильные группы по изобретению могут быть присоединены к родительской молекуле через атом углерода или атом азота в группе. Примеры гетероциклильных групп включают, без ограничения, бензотиенил, фурил, имидазолил, индолинил, индолил, изотиазолил, изоксазолил, морфолинил, оксазолил, пиперазинил, пиперидинил, пиразолил, пиридинил, пирролидинил, пирролопиридинил, пирролил, тиазолил, тиенил и тиоморфолинил.
Термин "гетероциклилалкил" относится к алкильной группе, замещенной одним, двумя или тремя гетероцклилами.
Термин "гетероциклилалкилен" относится к двухвалентной гетероциклилалкильной группе, в которой одно место присоединения к родительской молекуле находится в гетероциклильной части, а другое - в алкильной части.
Термин "гетероциклилен" означает двухвалентную гетероциклильную группу.
Термин "гидрокси" означает - ОН.
Термин "фрагмент, обеспечивающий изображение" относится к части или частям молекулы, которые обеспечивают обнаружение, изображение и/или мониторинг наличия и/или развития состояния(-ий), патологического нарушения(-и) и/или болезни(-ей).
Термин "связующая группа" относится к части молекулы, которая служит в качестве спейсера между двумя другими частями молекулы. Связующие группы могут также выполнять другие функции, описанные в данной заявке. Примеры связующих групп включают линейные, разветвленные или циклические алкильные, арильные, эфирные, полигидроксильные, полиэфирные, полиаминные, гетероциклические, ароматические, гидразидные, пептидные, пептоидные или другие физиологически совместимые ковалентные группы или их сочетания.
Используемый термин "липид" относится к синтетическому или природному амфипатическому соединению, которое содержит гидрофильный компонент и гидрофобный компонент. Липиды включают, например, жирные кислоты, нейтральные жиры, фосфатиды, гликолипиды, алифатические спирты и воски, терпены и стероиды. Примеры композиций, которые содержат липидное соединение, включают суспензии, эмульсии и пузырчатые композиции.
"Липосома" означает обычно сферический кластер или агрегат амфипатических соединений, включающих липидные соединения, обычно в виде одного или нескольких концентрических слоев, например, двухслойных. Они могут также называться в данном описании липидными пузырьками.
Термин "лиофилизирующее средство" обозначает компонент, который имеет благоприятные физические свойства для лиофилизации, такие как температура стеклования, и который обычно добавляется к составу для улучшения физических свойств сочетания всех компонентов в составе для лиофилизации.
Термин "оксо" относится k=О.
Используемое выражение "фармацевтически приемлемый" относится к таким соединениям, материалам, композициям и/или дозированным формам, которые, с точки зрения здравого смысла в медицине, подходят для применения при контакте с тканями людей и животных без излишней токсичности, раздражения, аллергических реакций или других проблем или осложнений с разумным соотношением выгода/риск.
Термин "фармацевтически приемлемая соль" означает соли или цвиттерионные формы соединений по изобретению, которые растворяются в воде или в масле или диспергируются, которые с точки зрения здравого смысла в медицине подходят для применения при контакте с тканями людей и животных без излишней токсичности, раздражения, аллергических реакций или других проблем или осложнений с разумным соотношением выгода/риск и эффективны для намеченного применения. Соли могут быть получены в процессе окончательного выделения и очистки соединений или путем взаимодействия подходящего атома азота с подходящей кислотой. Примеры солей присоединения включают ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензосульфонат, бисульфат, бутират, камфорат, камфосульфонат, диглюконат, глицерофосфат, гемисульфат, гептаноат, гексаноат, формиат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, мезитиленсульфонат, метансульфонат, нафтиленсульфонат, никотинат, 2-нафталинсульфат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, трихлорацетат, трифторацетат, фосфат, глутамат, бикарбонат, п-толуолсульфонат и ундеканоат. Примеры кислот, которые можно применять для получения фармацевтически приемлемых солей присоединения, включают неорганические кислоты, такие как соляная, бромистоводородная, серная и фосфорная, и органические кислоты, такие как щавелевая, малеиновая, янтарная и лимонная.
Под "реагентом" имеют в виду соединение по изобретению, способное к непосредственному превращению в металлофармацевтик. Реагенты могут применяться для получения металлофармацевтиков по изобретению или могут быть компонентами набора по изобретению.
"Восстановитель" является соединением, которое реагирует с радионуклидом и которое обычно получается как сравнительно нереакционноспособное соединение в высокой стадии окисления и при реакции переходит в состояние с меньшей степенью окисления при передаче электрона(-ов) радионуклиду, становясь при этом более реакционноспособным. Восстановители, пригодные для получения радиофармацевтиков и для диагностических наборов, подходящих для получения указанных радиофармацевтиков, включают, например, хлорид олова, фторид олова, формамидинсульфиновую кислоту, аскорбиновую кислоту, цистеин, фосфины и соли меди или железа. Другие восстановители описаны, например, в Brodack et al., заявка РСТ 94/22496.
"Стабилизирующее средство" означает компонент, который обычно добавляется к металлофармацевтику или в диагностический набор для стабилизации металлофармацевтика или для увеличения жизнеспособности набора перед его применением.
Стабилизирующие средства могут быть антиоксидантами, восстановителями или поглотителями радикалов и могут обеспечивать повышенную стабильность за счет предпочтительного взаимодействия с частицами, которые вызывают разложение других компонентов или металлофармацевтиков.
Под "стабильным соединением" или "стабильной структурой" подразумевают соединение, которое является достаточно объемным для выделения с достаточной степенью чистоты из реакционной смеси и для введения в состав эффективного фармацевтического агента.
"Средство для солюбилизации" означает компонент, который повышает растворимость одного или нескольких других компонентов в среде, треюущейся для получения состава.
Термин "группа, защищающая тиольную группу" относится к группе, предназначенной для защиты тиольной группы от нежелательных реакций в процессе синтеза. Может быть использована любая группа, защищающая тиольную группу, известная из уровня техники. Примеры таких защитных групп включают, без ограничения, ацетамидометил, бензамидометил, 1-этоксиэтил, бензоил и трифенилметил.
Термин "лиганд - переносчик" означает лиганд, который образует промежуточный комплекс с ионом металла, который является достаточно стабильным для предотвращения нежелательных побочных реакций, и достаточно лабильным для превращения в контрастный агент. Образование промежуточного комплекса является благоприятным кинетически, в то время как образование металлофармацевтика благоприятно термодинамически. Лиганды - переносчики, пригодные для получения контрастных агентов и для диагностических наборов, используемых для получения диагностических радиофармацевтиков, включают, например, глюконат, глюкогептаноат, маннит, глюкарат, N,N,N’,N’-этилендиаминтетрауксусную кислоту, пирофосфат и метилендифосфонат. В общем лиганды - переносчики содержат донорные атомы кислорода или азота.
Используемый термин "пузырек" относится к сферической частице, которая характеризуется наличием внутреннего пространства. Согласно одному из вариантов пузырьки образуются из липидов, включающих различные липиды, описанные в данной заявке. В любом данном пузырьке липиды могут быть в виде монослоя или двух слоев. В случае, когда имеется более одного или двух слоев, монослои или двойные слои обычно являются концентрическими. Пузырьки в жидкостях, описанные в данном описании, включают такие частицы, называемые обычно липосомами, мицеллами, пузырьками, микропузырьками, микросферами и т.п.
Так, для получения униламеллярной везикулы (пузырька), (состоящего из одного монослоя или двух слоев), олиголамеллярной везикулы (содержащей примерно два или примерно три монослоя или два слоя), или мультиламеллярной везикулы (содержащей более примерно трех монослоев или двух слоев) можно применять липиды. Внутренняя полость пузырьков может быть заполнена жидкостью, включая, например, водную жидкость, газом, предшественником газа и/или твердым или растворенным веществом, включая, например, биоактивный агент, если это желательно.
Используемый термин "везикулярная композиция" относится к композиции, которая получена из липидов и содержит везикулы (пузырьки).
Данное изобретение будет далее описано в примерах, которые не ограничивают его объем. Напротив, данная заявка охватывает все альтернативы, модификации и эквиваленты, которые могут быть описаны нижеследующей формулой изобретения. Таким образом, примеры иллюстрируют практическое воплощение изобретения, но следует понимать, что они приведены для описания только некоторых вариантов и представляют собой то, что считается наиболее полезным и легко понятным описанием методик и концептуальных аспектов изобретения.
Синтез аналога феназаквина
Пример 1 А
Синтез 4-[4-(2-гидроксиэтил)фенил]-4-оксомасляной кислоты метилового эфира

В сухую колбу объемом 250 мл в атмосфере азота добавляли фенетиловый спирт (2,50 г, 0,02 мол), безводный дихлорметан (150 мл) и метил-4-хлор-4-оксобутират (6,02 г, 0,04 мол). Содержимое колбы охлаждали до 0ºС на ледяной бане. К раствору порциями добавляли хлорид алюминия (25 г, 0,2 мол), избегая быстрого выделения тепла. Полученную смесь желтоватого цвета перемешивали в течение 3 ч. В этот момент реакцию обрывали ледяной водой. Смесь разбавляли дихлорметаном и перемещали в разделительную воронку. Органический слой промывали насыщенным раствором бикарбоната натрия, рассолом и затем сушили над сульфатом магния. После фильтрования и концентрации фильтрата при пониженном давлении получали сырое масло желтого цвета. Масло суспендировали в безводном метаноле (100 мл) и к смеси добавляли металлический натрий до достижения рН равного 9. Смесь перемешивали в течение 3 ч. Объем уменьшался, затем смесь разбавляли этилацетатом. Раствор перемещали в разделительную воронку и промывали водным 0,05 N раствором соляной кислоты, рассолом и высушивали над сульфатом магния. Раствор концентрировали при пониженном давлении с получением сырого масла желтого цвета в количестве 5,88 г. Хроматография на колонке [силикагель; элюент гексаны - этилацетат (3:2)] позволила получить желаемый продукт (2.69 г, 57%).1Н (CDCl3) δ (ppm): 2.65 (t, 2H); 2.81 (t, 2H); 3.19 (t, 2H); 3.6 (s, 3Н); 3.75 (t, 2H); 7.22 (d, 2H); 7.81 (d, 2H).13С (CDCl3) 8 (ppm): 27.76, 33.03, 38.66, 51.52, 62.68, 127.97, 128.99, 134.47, 144.78, 173.21, 197.64.
Пример 1 В
Синтез 4-[4-(2-гидроксиэтил)фенил) масляной кислоты метилового эфира

Смесь соединения, полученного в примере 1 А (250 г, 11 ммол), 10% Pd/C (0,25 г, 0,23 ммол металлического Pd) в безводном метаноле (25 мл) дегазировали для удаления воздуха (два цикла вакуум/Н2), после чего герметизировали и использовали баллон, наполненный Н2, в течение 12 ч. Затем реакционную смесь отфильтровывали через диатомовую землю (Celite®) и фильтрат концентрировали при пониженном давлении с получением 2,32 г сырого продукта. После хроматографии на колонке [силикагель; элюент гексаны - этилацетат (2:1)] получали желательный продукт (0.92 г, 39%).1Н (CDCl3) δ (ppm): 1.91-1.96 (m, 2H); 2.32 (t, 2H); 2.62 (t, 2H); 2.83 (t, 2H); 3.66 (s, 3Н); 3.85 (t, 2H); 7.11-7.15 (m, 4H).
Пример 1 С
Синтез 4-{4-[2-(хиназолин-4-илокси)этил] фенил масляной кислоты метилового эфира

Сухую колбу объемом 50 мл снабжали воронкой для добавления реагентов. В колбу добавляли 4-хлорхиназолин (592 мг, 3,6 ммол), безводный тетрагидрофуран (10 мл) и 60 вес.% гидрид натрия (187 мг, 4,7 ммол). При помощи воронки добавляли по каплям раствор соединения, полученного в примере 1 В (800 мг, 3,6 ммол), в безводном тетрагидрофуране (10 мл). Смесь перемешивали в течение 3,5 ч. Полученную реакционную смесь разбавляли этилацетатом и обрывали реакцию путем добавления водного 0,1 N раствора соляной кислоты. Смесь переносили в разделительную воронку и промывали рассолом. Органический слой высушивали над сульфатом магния, отфильтровывали и концентрировали. После хроматографии на колонке [силикагель; элюент гексаны - этилацетат (4:1)] получали нужный продукт (538 мг, 43%).1Н (CDCl3) δ (ppm): 1.92-1.98 (m, 2H); 2.33 (t, 2H); 2.64 (t, 2H); 3.19 (t, 2H); 3.66 (s, 3H); 4.79 (t, 2H); 7.15 (d, 2H); 7.27 (d, 2H); 7.57 (t, 1H); 7.83 (t, 1H); 7.94 (d, 1H); 8.15 (d, 1H); 8,80 (s, 1H). 26.68, 33.59, 34.93, 35.03; 51.67, 67.89, 116.48, 123.72, 127.23, 127.82, 128.87, 129.24, 133.74, 135.76, 139.90, 151.08, 154.56, 166.89, 174.10.
Пример 1 D
Синтез 4-{4-[2-(хиназолин-4-илокси) этил]фенил}бутан-1-ола

В сухую колбу объемом 15 мл добавляли литийалюминийгидрид (233 мг, 6,0 ммол) и безводный диэтиловый эфир (3 мл). Смесь охлаждали на ледяной бане. При энергичном перемешивании медленно добавляли раствор соединения, полученного в примере 1 С (538 мг, 1,54 ммол) в безводном диэтиловом эфире (3 мл). Баню удаляли и суспензию перемешивали в течение 15 мин. реакции обрывали водой (0,233 мл), водным 15% раствором гидроокиси натрия (0,233 мл) и водой (0,699 мл). Продукт белого цвета отфильтровывали, фильтрат высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением прозрачного масла. Это масло затем растворяли в безводном дихлорметане (10 мл) и к раствору добавляли окись марганца (IV) (500 мг, 5,8 ммол). Смесь перемешивали в течение 12 ч. После фильтрации через диатомовую землю (Celite®) с последующей концентрацией фильтрата при пониженном давлении получали 395 мг сырого продукта. После хроматографии на колонке [силикагель; элюент пентан - этилацетат (2:3)] получали нужный продукт (225 мг, 49%).1Н (CDCl3) δ (ppm): 1.55-1.61 (m, 2H); 1.65-1.68 (m, 2H); 2.61 (t, 2H); 3.17 (t, 2H); 3.64 (t, 2H); 4.79 (t, 2H); 7.12 (d, 2H); 7.23 (d, 2H); 7.56 (t, 1H); 7.82 (t, 1H); 7.93 (d, 1H); 8.14 (d, 1H); 8.77 (s, 1H).13C (CDCl3) δ (ppm): 27.52, 32.31, 34.89, 35.21; 62.81, 67.74, 116.67, 123.54, 127.08, 127.49, 128.63, 128.98, 133.61, 135.23, 140.64, 150.68, 154.29, 166.79.
Пример 1 Е
Синтез 4-{4-[2-(хиназолин-4-илокси)этил]фенил}бутилового эфира толуол-4-сульфокислоты

В сухую колбу объемом 10 мл добавляли п-толуолсульфонилхлорид (32,5 мг, 0,17 ммол), 4-(диметиламино)пиридин (20,7 мг, 0,17 ммол), соединение, полученное в примере ID (50,0 мг, 0,16 ммол), безводный дихлорметан (1 мл) и триэтиламин (17,2 мг, 0,17 ммол). полученный раствор перемешивали в течение 2 ч, концентрировали при пониженном давлении и очищали методом хроматографии на колонке [силикагель; элюент пентан - этилацетат (1,86:1)] с получением нужного продукта (52 мг, 70%).1Н (CDCl3) δ (ppm): 1.64-1.68 (m, 4H); 2.44 (s, 3Н); 2.56 (t, 2H); 3.19 (t, 2H); 4.04 (t, 2H); 4.78 (t, 2H); 7.08 (d, 2H); 7.26 (d, 2H); 7.57 (t, 1H); 7.78 (d, 2H); 7.84 (t, 1H); 8.14 (d, 1H); 8.80 (s, 1H).
Пример 1 F
Синтез 4-{2-[4-(4-фторбутил)фенил]этокси}хиназолина
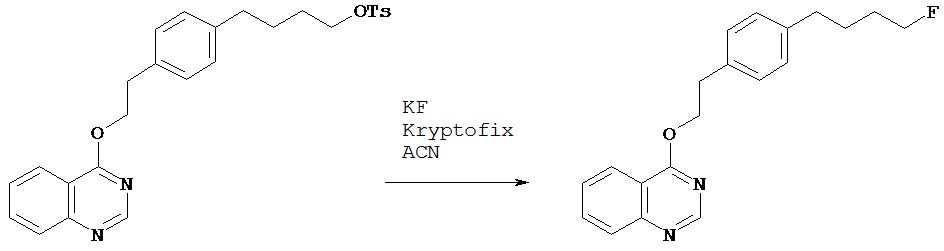
Сухую колбу объемом 5 мл снабжали обратным конденсатором. В колбу добавляли фторид калия (6,1 мг, 0,1 ммол), криптофикс (kryptofix) (40 мг, 0,1 ммол) и безводный ацетонитрил (0,5 мл). К полученному раствору добавляли раствор соединения, полученного в примере 1 Е (25 мг, 0,05 ммол) в безводном ацетонитриле (1 мл). Колбу помещали в масляную баню с температурой 90ºС. Раствор перемешивали в течение 1 ч. После охлаждения реакционную смесь разбавляли диэтиловым эфиром, помещали в разделительную воронку и промывали водным 0,1 N раствором соляной кислоты, насыщенным водным раствором бикарбоната натрия и затем рассолом. Органический слой высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении. После хроматографии на колонке [силикагель; элюент гексаны - этилацетат (3:1)] получали нужный продукт (10,7 мг, 63%).1Н (CDCl3) δ (ppm): 1.65-1.73 (m, 4H); 2.63 (t, 2H); 3.17 (t, 2H); 4.40 (t, 1H); 4.48 (t, 1H); 4.77 (t, 2H); 7.13 (d, 2H); 7.24 (d, 2H); 7.55 (1H); 7.82 (t, 1H); 7.92 (d, 1H); 8.13 (d, 1H); 8.78 (s, 1H).13С (CDCl3) δ (ppm): 27.19 (d,4JCF=4.5), 30.20 (d,3JCF=19.5), 35.15 (d,2JCF=27.0), 67.94, 84.17 (d,1JCF=163.3), 116.93, 123.75, 127.26, 127.84, 128.82, 129.23, 129.42, 133.77, 135.62, 138.21, 140.54, 151.08, 154.59.
19F (CDCl3, CFCl3 внутр. стандарт) δ (ppm): -218.59 (t of t, J=-27.6, -50.4).
Синтез аналогов пиридабена
Пример 2 А
Синтез 4-фенилбутилового эфира масляной кислоты

К 4-фенил-1-бутанолу (7,0 г, 0,047 мол) добавляли безводный дихлорметан (20 мл). По каплям добавляли раствор бутирилхлорида (4,79 г, 0,045 мол) в безводном дихлорметане. полученный раствор перемешивали в течение 36 ч. Затем смесь концентрировали при пониженном давлении с получением сырого масла. После хроматографии [силикагель; элюент гексаны - этилацетат (3:1)] получали нужный продукт (9,8 г, 94%) в виде прозрачной вязкой жидкости.1Н (CDCl3) δ (ppm): 0.94 (t, 3H); 1.61-1.71 (m, 6H); 2.27 (t, 2H); 2.64 (t, 2H); 4.08 (t, 2H); 7.16-7.19 (m, 3H); 7.25-7.29 (m, 2H).
Пример 2 В
Синтез 4-(4-гидроксибутил) бензойной кислоты метилового эфира

К хлориду алюминия (6,7 г, 0,05 мол) в сухой круглодонной колбе объемом 250 мл добавляли безводный дихлорметан (100 мл). Колбу охлаждали на ледяной бане с температурой 0ºС. По каплям добавляли оксалилхлорид (6,4 г, 0,05 мол). Смесь перемешивали в течение 5 мин. Затем по каплям добавляли раствор соединения, полученного в примере 2 А (9,8 г, 0,044 мол) в безводном дихлорметане (50 мл). Полученную смесь перемешивали в течение 4 ч при 0ºС. Реакционную смесь выливали в разделительную воронку, содержащую лед и рассол. Органический слой промывали рассолом и высушивали над сульфатом магния. После фильтрации и концентрирования при пониженном давлении получали 9,1 г желтого масла. 9,0 г этого масла суспендировали в метаноле, доводили рН до 2 и перемешивали в течение 48 ч. Полученную смесь концентрировали при пониженном давлении. После хроматографии [силикагель; элюент гексаны - этилацетат (2,57:1)] получали нужный продукт (2,80 г, 31%) в виде прозрачной вязкой жидкости.1Н (CDCl3) δ (ppm): 1.56-1.61 (m, 2H); 1.63-1.73 (m, 2H); 2.67 (t, 2H); 3.64 (t, 2H); 3.88 (s, 3H); 7.23 (d, 2H); 7.93 (d, 3Н).
Пример 2 С
Синтез 4-[4-(трет.бутилдиметилсиланилокси)бутил]бензойной кислоты метилового эфира
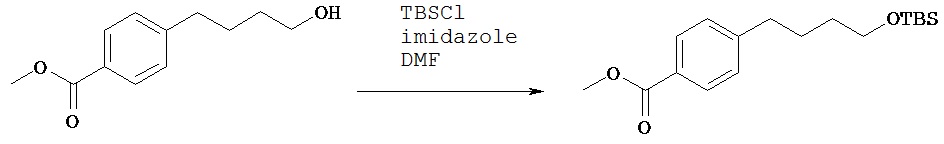
К соединению, полученному в примере 2 В (1,0 г, 4,8 ммол) добавляли безводный диметилформамид (10 мл), имидазол (0,5 г, 7,2 ммол) и трет.бутилдиметилсилилхлорид (1,08 г, 7,3 ммол). Раствор перемешивали на водяной бане в течение 2 ч. Полученную реакционную смесь разбавляли этилацетатом, выливали в разделительную воронку, промывали водой (20 мл, 5х), затем промывали насыщенным раствором бикарбоната натрия (20 мл, 2х). Органический слой высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением нужного продукта (1,17 г, 75%), который использовали на следующей стадии без очистки.
Пример 2 D
Синтез {4-[4-(трет.бутилдиметилсиланилокси)бутил]фенил}метанола

К соединению, полученному в примере 2 С (1,17 г, 3,6 ммол) добавляли безводный диэтиловый эфир (14 мл). Раствор охлаждали до 0ºС на ледяной бане. К этому раствору частями добавляли литийалюминийгидрид (0,28 г, 7,2 ммол). Эту смесь перемешивали в течение 1 ч. Затем добавляли дистиллированную воду (0,28 мл) и перемешивали смесь в течение 5 мин. Затем добавляли водный 15% раствор гидроокиси натрия и перемешивали смесь в течение 5 мин. Добавляли свежую дистиллированную воду (0,84 мл) и перемешивали смесь в течение 5 мин. Фильтрованием отделяли твердый продукт белого цвета. Фильтрат высушивали при помощи сульфата магния, фильтровали и концентрировали с получением 1,23 г сырого продукта. После хроматографии [силикагель; элюент гексаны - этилацетат (4:1)] получали нужный продукт (1,02 г, 96%) в виде прозрачной вязкой жидкости.
Пример 2 Е
Синтез 2-трет.бутил-5-{4-[4-(трет.бутилдиметилсиланилокси)бутил]-бензилокси}-4-хлор-2Н-пиридазин-3-она

В сухую круглодонную колбу объемом 25 мл, снабженную обратным конденсатором, добавляли соединение, полученное в примере 2 D (0,41 г, 1,4 ммол), 2-трет.бутил-4,5-дихлор-2Н-пиридазин-3-он (0,93 г, 4,2 ммол), карбонат цезия (1,37 г, 4,2 ммол) и безводный диметилформамид (11 мл). Колбу помещали на масляную баню с температурой 68ºС и перемешивали реакционную смесь в течение 12 ч. Колбу удаляли из масляной бани и давали охладиться. Смесь разбавляли этилацетатом, помещали в разделительную воронку и промывали водой (25 мл, 5х). Органический слой высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением 1,3 г сырого продукта. После хроматографии на колонке [силикагель; элюент гексаны - этилацетат (9:1)] получали нужный продукт (594 мг, 89%).1Н (CDCl3) δ (ppm): 0.05 (s, 6H); 0.90 (s, 9H); 1.64 (s, 9H); 2.65 (t, 2H); 3.64 (t, 2H); 5.23 (s, 2H); 7.23 (d, 2H); 7.33 (d, 2H); 7.74 (s, 1H).13С (CDCl3) δ (ppm): 18.57, 26.19, 27.75, 28.09; 32.58, 35.61, 63.14, 66.57, 72.14, 118.46, 125.41, 127.44, 129.23, 132.38, 143.72, 154.02, 159.30
Пример 2 F
Синтез 2-трет.бутил-4-хлор-5-[4-(4-гидроксибутил)бензилокси]-2H-пиридазин-3-она
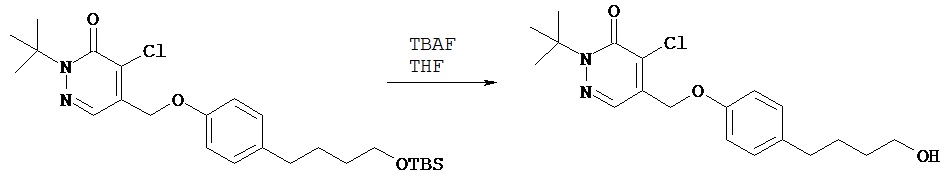
К соединению, полученному в примере 2 Е (594 мг, 1,45 ммол), добавляли безводный тетрагидрофуран (3 мл) и 1,0 М раствор трет.бутиламмонийфторида в тетрагидрофуране (2,9 мл, 2,9 ммол). Раствор перемешивали в течение 1 ч, затем концентрировали при пониженном давлении. После хроматографии на колонке [силикагель; элюент пентан - этилацетат (1,8:1)] получали нужный продукт (410 мг, 77%).1Н (CDCl3) δ (ppm): 1.61-1.64 (m, 11H); 1.67-1.74 (m, 2H); 2.68 (t, 2H); 3.68 (t, 2H); 5.23 (s, 2H); 7.23 (d, 2H); 7.33 (d, 2H); 7.74 (s, 1H).13С (CDCl3) δ (ppm): 27.43, 27.86, 32.56, 35.35, 62.74, 66.36, 71.88, 118.27, 125.18, 127.27, 128.99, 132.28, 143.17, 153.78, 159.07.
Пример 2 G
Синтез бутилового эфира 4-[4-(1-трет.бутил-5-хлор-6-оксо-1,6-дигидро-пиридазин-4-илоксиметил)фенил]толуол-4-сульфокислоты

В круглодонную колбу объемом 5 мл добавляли соединение, полученное в примере 2 F (200 мг, 0,55 ммол), п-толуол-сульфонилхлорид (125 мг, 0,66 ммол), 4-(диметиламино)пиридин (80 мг, 0,66 ммол), диизопропилэтиламин (85 мг, 0,66 ммол) и безводный дихлорметан (2 мл). Полученный раствор перемешивали в течение 2 ч. Реакционную смесь разбавляли этилацетатом, помещали в разделительную воронку и промывали водным 0,1 N раствором соляной кислоты и затем промывали рассолом. Органический слой сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением 299 мг сырого продукта. После хроматографии [силикагель; элюент пентанэтилацетат (3:1)] получали нужный продукт (197 мг, 69%).1Н (CDCl3) δ (ppm): 1.62-1.70 (m, 13H); 2.43 (s, 3H); 2.58 (t, 2H); 4.03 (t, 2H); 7.15 (d, 2H); 7.29-7.33 (m, 4H); 7.72 (s, 1H); 7.77 (d, 2H).13С (CDCl3) δ (ppm): 21.63, 26.98, 27.86, 28.34, 34.80, 66.37, 70.23, 71.81, 118.25, 125.12, 127.32, 127.87, 128.93, 129.82, 132.48, 133.15, 142.40, 144.72, 153.75, 159.05.
Пример 2 Н
Синтез 2-трет.бутил- 4-хлор-5-(4-(4-фторбутил) бензил) окси-3(2Н)-пиридазинона
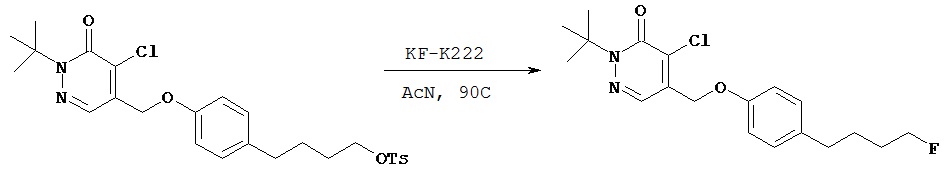
Соединение, полученное в примере 2 G (57 мг, 0,10 ммол) растворяли в 1 мл ацетонитрила и добавляли смесь KF-К222 (1:1, 0,167 ммол), растворенную в 1 мл ацетонитрила. Полученную смесь погружали затем в масляную баню с температурой 90ºС и нагревали с обратным холодильником в течение 15 мин, затем методом ТСХ определяли завершение реакции. Летучие компоненты удаляли в вакууме, сырое масло очищали методом флэш-хроматографии на силикагеле (гексаны - этилацетат (4:1)) с получением 28 мг желательного продукта в виде масла, которое затвердевало при стоянии.1Н (CDCl3) δ (ppm): 1.6 (s, 9H); 1.7 (m, 4H); 2.6 (t, 2Н); 4.44 (d of t, 2Н, J=47.4 & 6 Hz); 5.2 (s, 2Н); 7.2 (d, 2Н, J=8.4 Hz); 7.3 (d, 2Н, J=8.4 Hz); 7.71 (s, 1H).13C (CDCl3) δ (ppm): 26.8 (3JCF=4.65 Hz), 27.8, 29.8 (2JCF=19.8 Hz), 35.1, 66.3, 71.8, 83.8 (1JCF=163.8 Hz), 118.2, 125.1, 127.2, 128.9, 132.3, 142.8, 153, 159.19F (CDCl3, CFCl3 в качестве внутреннего стандарта 5 (ppm): - 218.6 (t of t, J=-27.6, -50.4).
Пример 3 А
Синтез (±)-1-трет.бутилдиметилсилил-окси-2-гидроксибутана
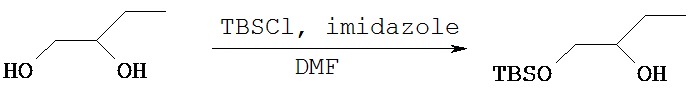
В круглодонную колбу объемом 50 мл загружали (±)-1,2-бутандиол (1 г, 11,09 ммол) и добавляли диметилформамид (8 мл) с последующим добавлением трет.бутилдиметилсилилхлорида (2,5 г, 16,64 ммол) и имидазола (1,88 г, 27,7 ммол). Реакционную смесь перемешивали в течение 10 ч, затем разбавляли ее дихлорметаном и выливали в разделительную воронку и промывали водой (80 мл) и рассолом и высушивали над сульфатом магния. После фильтрования и концентрирования сырое масло очищали методом флэш-хроматографии на силикагеле (гексаны: этилацетат) и получали 1 г чистого желательного продукта с выходом равным 45%.1Н (CDCl3) δ (ppm): 3.6 (m, 1H); 3.5 (m, 1H), 3.4 (m, 1H), 2.4 (s, 1H), 1.44 (m, 2H), 0.99 (t, 3H), 0.9 (s, 9H), 0.06 (s, 6H).
Пример 3 В
Синтез (±)-4-(1-трет.бутилдиметилсилилоксибут-2-окси)метилбензоата
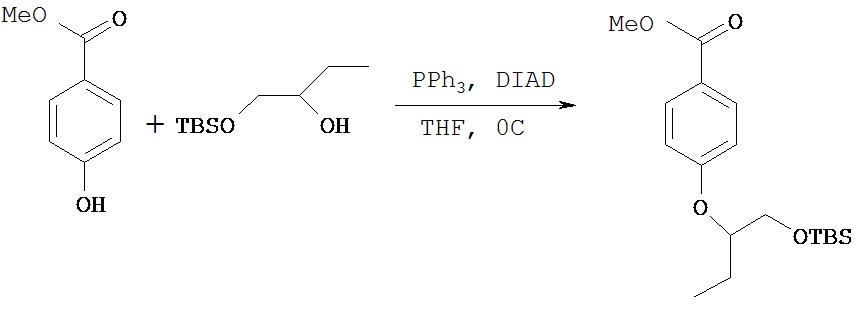
4-Гидроксиметилбензоат (1,1 г, 7,34 ммол), соединение, полученное в примере 3 А (0,75 г, 3,67 ммол), и трифенилфосфин (1,972 г, 7,34 ммол) добавляли в круглодонную колбу и добавляли 8 мл тетрагидрофурана. Колбу охлаждали на ледяной бане при 0ºС, затем при помощи шприца добавляли диизопропилазодикарбоксилат (1,485 г, 7,34 ммол). Реакционную смесь перемешивали в течение 2 ч, после чего реакция была завершена, что показала ТСХ. Все количество растворителя удаляли при пониженном давлении и сырое масло сразу же подвергали очистке методом флэш-хроматографии на силикагеле (гексаны: диэтиловый эфир) с получением 1,0 г (83%) желательного соединения в виде густого масла.1Н (CDCl3) δ (ppm): 7.9 (d, 2H); 6.9 (d, 2H); 4.3 (p, 1H, J=5.4 Hz); 3.9 (s, 3H); 3.7 (2H), 1.78 (m, 1H), 1.7 (m, 1H), 0.9 (t, 3H, J=7.8 Hz), 0.89 (s, 9H), 0.05 (s, 3H), 0.01 (s, 3H).13С (CDCl3) δ (ppm): 166.8, 162.8, 131.5, 122.3, 115.2, 80, 64.5, 51.7, 25.8, 24.1, 18.2, 9.5,-5.3.
Пример 3 С
Синтез (±)-4-(1-трет.бутилдиметилсилилоксибут-2-окси)бензиловый спирт

К раствору соединения. Полученного в примере 3 В (1 г, 2,95 ммол), в простом эфире (15 мл) добавляли литийалюминийгидрид (0,336 г, 8,8 ммол) и перемешивали смесь в атмосфере азота в течение 1,5 ч. В этот момент реакция, как показала ТСХ, была завершена, ее обрывали последовательным добавлением 0,336 мл воды, 0,336 мл 15% раствора NaOH и 1,00 мл воды. Полученную смесь перемешивали еще 20 мин, после чего образовавшийся осадок белого цвета отфильтровывали и промывали простым эфиром. Фильтрат высушивали над сульфатом магния. После фильтрации и удаления растворителя получали 0,50 г (54%) желательного продукта в виде твердого вещества белого цвета.1Н (CDCl3) δ (ppm): 7.2 (d, 2H); 6.9 (d, 2H); 4.3 (р, 1Н); 3.77 (d of d, 1H), 3.66 (d of d, 1H), 1.77-1.72 (m, 1H), 1.68-1.61 (m, 1H), 1.5 (t, 1H, J=5.4 Hz), 0.9 (t, 3H, J=7.8 Hz), 0.89 (s, 9H), 0.04 (s, 3H), 0.01 (s, 3H).13C (CDCl3) δ (ppm): 158.5, 133, 128.4, 116.1, 80.1, 65, 64.5, 25.8,24.1, 18.2, 9.5, -5.3.
Пример 3 D
Синтез (±)-2-трет.бутил-4-хлор-5-(4-(1-трет.бутилдиметилсилилокси-бут-2-окси)бензил)-окси-3(2Н)-пиридазинона
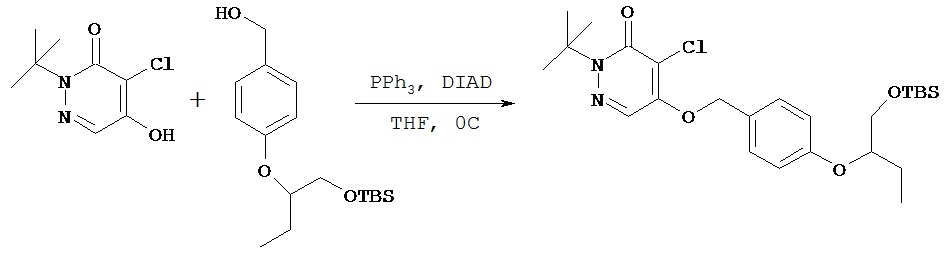
(±)-2-трет.бутил-4-хлор-5-гидрокси-3(2H)-пиридазинон (0,48 г, 2,417 ммол) загружали в круглодонную колбу объемом 100 мл и добавляли тетрагидрофуран (40 мл). Полсе того, как раствор стал прозрачным, в колбу добавляли соединение, полученное в примере 3 С (0,5 г, 1,611 ммол), и трифенилфосфин (0,633 г, 2,417 ммол) и охлаждали колбу до 0ºС. При помощи шприца вводили диизопропилазодикарбоксилат (0,488 г, 2,417 ммол, 0,468 мл) и перемешивали смесь в течение 2 ч, после чего реакция завершалась, о чем свидетельствовала ТСХ. Содержимое колбы концентрировали в вакууме и полученное сырое масло очищали методом флэш-хроматографии на силикагеле (гексаны: этилацетат) с получением 0,33 г желательного соединения в виде масла.1Н (CDCl3) δ (ppm): 7.72 (s, 1H); 7.2 (d, 2H), 6.9 (d, 2H); 5.2 (s, 2H), 4.2 (р, 1H); 3.75 (d of d, 1H), 3.68 (d of d, 1H), 1.75 (m, 2H), 1.65 (m, 1H), 1.6 (s, 9H), 0.99 (t, 3H), 0.85 (s, 9H), 0.04 (s, 3H), 0.02 (s, 3H).13С (CDCl3) δ (ppm): 159.6, 159.3, 154, 129, 126.9, 125, 118.5, 116.5, 80.3, 72.1, 66.5, 64.8, 28.1, 26, 24.4, 18.4, 9.6, -5.3.
Пример 3 Е
Синтез (±)-2-трет.бутил-4-хлор-5-(4-(1-гидрокси-бут-2-окси)бензил)окси-3(2Н)пиридазинона

К соединению, полученному в примере 3 D (0,3 г, 0,6 ммол), в круглодонной колбе объемом 10 мл добавляли тетрагидрофуран (2 мл). После растворения добавляли тетрабутиламмонийфторид (1,8 ммол, 1,8 мл, 1 М раствор в ТГФ) и перемешивали реакционную смесь в течение 90 мин. Содержимое колбы затем концентрировали при пониженном давлении и очищали сырую смесь методом флэш-хроматографии на силикагеле (гексаны: этилацетат) с получением 185 мг (80%) чистого желательного продукта.
1Н (CDCl3) δ (ppm): 7.74 (s, 1H); 7.3 (d, 2Н), 6.9 (d, 2Н); 5.2 (s, 2Н), 4.3 (m, 1Н); 3.81-3.77 (two br s, 2Н), 1.84 (br t, 1H), 1.77-1.69 (m, 2Н), 1.64 (s, 9H), 0.98 (t, 3H).13С (CDCl3) δ (ppm): 159.2, 158.9, 153.9, 129.2, 127.5, 125.4, 116.6, 80.4, 71.9, 66.5, 64.2, 28, 23.5, 9.7.
Пример 3 F
Синтез (±)-2-трет.бутил-4-хлор-5-(4-(1-тозилокси-бут-2-окси)-бензил)окси-3(2Н)-пиридазинона

В круглодонную колбу объемом 10 мл добавляли соединение, полученное в примере 3 Е (0,05 г, 0,13 ммол) и затем дихлорметан (2 мл). Затем к реакционной смеси последовательно добавляли толуолсульфонилхлорид (0,075 г, 0,39 ммол), 4-N,N-диметиламинопиридин (0,048 г, 0,39 ммол) и диизопропилэтиламин (0,05 г, 0,39 ммол, 68,7 мкл) и перемешивали в течение 35 мин. Затем к смеси добавляли воду, выливали раствор в разделительную воронку и разделяли слои. Органический слой промывали водой и рассолом и сушили над сульфатом магния. Сырое масло, полученное после фильтрации и концентрирования, очищали методом флэш-хроматографии на силикагеле (гексаны: этилацетат) с получением 54 мг (77%) желательного соединения в виде густого бесцветного масла.1H (CDCl3) δ (ppm): 7.74 (3Н, two singlets); 7.3 (m, 4H), 6.8 (d, 2H); 5.2 (s, 2H), 4.38 (p, 1H); 4.15 (m, 2H), 2.44 (s, 3Н), 1.72 (m, 2H), 1.6 (s, 9H), 0.95 (t, 3Н).13С (CDCl3) δ (ppm): 159.2, 158.5, 153.9, 145.1, 133, 130, 129, 128.1, 127.2, 125.4, 118.5, 116.5, 71.9, 70.2, 66.6, 28.1, 24.2, 21.8, 9.4.
Пример 3 G
Синтез (±)-2-трет.бутил-4-хлор-5-(4-(1-фтор-бут-2-окси)бензил)-окси-3(2Н)-пиридазинона

Продукт, полученный в примере 3 F (28 мг, 52,4 мкмол), растворяли в 0,5 мл ацетонитрила в колбе объемом 5 мл и к этому раствору добавляли раствор фторида калия (4,5 мг, 78,6 мкмол) и Kryptofix 222 (29,6 мг, 78,6 мкмол) в 0,5 мл ацетонитрила. Затем этот раствор погружали в масляную баню, предварительно нагретую до 90ºС. Реакционная смесь перемешивалась в течение 90 мин, затем все летучие удаляли при пониженном давлении и сырую смесь очищали методом препаративной тонкослойной хроматографии с получением 13 мг (65%) чистого желательного соединения.1Н (CDCl3) δ (ppm): 7.72 (s, 1H); 7.3 (d, 2H), 6.9 (d, 2H); 5.23 (s, 2H), 4.57-4.59 (m, 2H); 4.4 (m, 4H), 1.74 (m, 2H), 1.6 (s, 9H), 1.0 (t, 3Н).13С (CDCl3) δ (ppm): 159, 158.7, 153.7, 129, 127.5, 125.2, 118.3, 116.4, 83.85 (d,1JCF=172.2), 78, 71.1, 66.3, 27.8, 23.2, 9.48.19F (CDCl3, CFCl3 в качестве внутреннего стандарта) δ (ppm): - 228 (d of t, J=-19, -60 Hz).
Пример 4 А
Синтез 4-(3-гидроксипропокси)бензойной кислоты метилового эфира

В колбу объемом 250 мл добавляли 3-бром-1-пропанол (4,17 г, 0,03 мол), безводный диметилформамид (40 мл), метил-4-гидроксибензоат (3,0 г, 0,02 мол) и карбонат калия (4,15 г, 0,03 мол). Колбу помещали в масляную баню с температурой 50ºС и перемешивали ее содержимое в течение 12 ч. После охлаждения смесь разбавляли этилацетатом, перемещали в разделительную воронку, промывали 0,1 N водным раствором соляной кислоты, водой и затем рассолом. Органический слой высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением 5,14 г сырого масла. После хроматографии [силикагель; элюент гексаны - этилацетат (1,68:1)] получали желательный продукт (1,25 г, 30%) в виде белого порошка.1H (CDCl3) δ (ppm): 2.04-2.08 (m, 2H); 3.86-3.88 (m, 5H); 4.17 (t, 2H); 6.91 (d, 2H); 7.98 (d, 2H);13С (CDCl3) δ (ppm): 31.89, 51.81, 59.88, 65.50, 114.06, 122.67, 131.57, 162.60, 166.84.
Пример 4 В
Синтез 4-[3-(трет.бутилдиметилсиланилокси)пропокси]бензойной кислоты метилового эфира

В колбу объемом 50 мл добавляли соединение, полученное в примере 4 А (300 мг, 1,4 ммол), безводный диметилформамид (4 мл), трет.бутилдиметилсилилхлорид (317 мг, 2,1 ммол) и имидазол (146 мг, 2,1 ммол). Полученный раствор перемешивали в течение 2 ч. Затем смесь разбавляли этилацетатом и помещали в разделительную воронку. Органическую фазу промывали водным 0,1 N раствором соляной кислоты (2х), водой (2х), затем рассолом. Органический слой высушивали над сульфатом магния, фильтровали и концентрировали. После хроматографии [силикагель; элюент гексаны - этилацетат (9,5:1)] получали желательный продукт (413 мг, 91%).1Н (CDCl3) δ (ppm): 0.03 (s, 6H); 0.87 (s, 9H); 1.97-2.01 (m, 2H); 3.79 (t, 2H); 3.87 (s, 3H); 4.11 (t, 2H); 6.90 (d, 2H); 7.97 (d, 2H);13С (CDCl3) δ (ppm): 18.30, 25.89, 32.3, 51.78, 59.27, 64.67, 114.08, 122.43, 131.56, 162.90, 166.90.
Пример 4 С
Синтез {4-[3-(трет.бутилдиметилсиланилокси)пропокси]фенил}метанол

Соединение, полученное в примере 4 В (396 мг, 1,22 ммол), добавляли в сухую колбу объемом 50 мл вместе с безводным диэтиловым эфиром (10 мл). Колбу погружали в ледяную баню. Затем в колбу порциями добавляли литийалюминийгидрид (93 мг, 2,44 ммол). Смесь перемешивали в бане в течение 2 ч. Реакцию обрывали водой (0,093 мл), водным 15% раствором гидроокиси натрия (0,093 мл), затем водой (0,279 мл). Твердый продукт белого цвета отфильтровывали и фильтрат сушили над сульфатом магния, отфильтровывали и концентрировали с получением желательного продукта (291 мг, 80%).1Н (CDCl3) δ (ppm): 0.04 (s, 6H); 0.88 (s, 9H); 1.95-1.99 (m, 2H); 3.79 (t, 2H); 4.05 (t, 2H); 4.60 (s, 2H); 6.88-6.89 (m, 2H); 7.25-7.27 (m, 2H); (CDCl3) δ (ppm): 18.30, 25.91, 32.41, 59.50, 64.57, 65.10, 114.59, 128.60, 132.97, 158.75.
Пример 4 D
Синтез 2-трет.бутил-4-хлор-5{4-[3-(трет.бутилдиметилсиланилокси)-пропокси]бензилокси}-2H-пиридазин-3-она
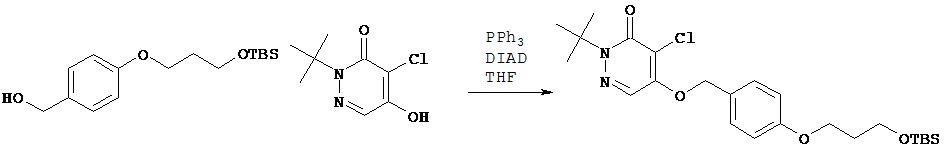
В сухую колбу объемом 25 мл добавляли соединение, полученное в примере 4 С (211 мг, 0,71 ммол), и безводный тетрагидрофуран (3 мл). Колбу охлаждали в ледяной бане. Затем в колбу добавляли трифенилфосфин (187 мг, 0,71 ммол). Последним добавляли диизопропилазадикарбоксилат (144 мг, 0,71 ммол). Реакционную смесь перемешивали на ледяной бане в течение 1 ч. Затем смесь разбавляли диэтиловым эфиром и перемещали в разделительную воронку. Органический раствор промывали водой и затем рассолом, высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении. После хроматографии на колонке [силикагель; элюент гексаны - этилацетат (9:1)] получали целевой продукт (106 мг, 31%).1Н (CDCl3) δ (ppm): 0.03 (s, 6H); 0.87 (s, 9H); 1.62 (s, 9H); 1.95-1.99 (m, 2H); 3.79 (t, 2H); 4.06 (t, 2H); 5.23 (s, 2H); 6.91-6.92 (m, 2H); 7.30-7.31 (m, 2H); 7.72 (s, 1H);13С (CDCl3) δ (ppm): 18.29, 25.90, 27.87, 32.34, 59.41, 64.63, 66.30, 71.89, 114.90, 118.34, 125.34,126.68, 128.92, 153.79, 159.07, 159.55.
Пример 4 Е
Синтез 2-трет.бутил-4-хлор-5-[4-(3-гидроксипропокси)бензилокси]-2Н-пиридазин-3-она

В сухую колбу объемом 10 мл добавляли соединение, полученное в примере 4 D (100 мг, 0,21 ммол) вместе с безводным тетрагидрофураном (2 мл). К содержимому колбы добавляли 1,0 М раствор тетрабутиламмонийфторида в тетрагидрофуране (0,42 мл, 0,42 ммол). Раствор перемешивали в течение 2 ч. Затем реакционную смесь концентрировали при пониженном давлении. После очистки методом тонкослойной хроматографии (силикагель; элюент гексаны - этилацетат (1:1)] получали желательный продукт (57,8 мг, 76%).1Н (CDCl3) 6 (ppm): 1,62 (s, 9H); 2.02-2.06 (m, 2Н); 3.86 (t, 2Н); 4.13 (t, 2Н); 5.30 (s, 2Н); 6.92-6.93 (m, 2Н); 7.31-7.32 (m, 2Н); 7.71 (s, 1H);13C (CDCl3) δ (ppm): 27.87, 31.97, 60.24, 65.67, 66.34, 71.81, 114.91, 118.37, 125.31, 127.06, 128.98, 153.76, 159.07, 159.27.
Пример 4 F
Синтез 3-[4-(1-трет.бутил-5-хлор-6-оксо-1,6-дигидропиридазин-4-илоксиметил)фенокси]пропилового эфира толуол-4 сульфокислоты

В сухую колбу объемом 5 мл добавляли соединение, полученное в примере 4Е (40 мг, 0,11 ммол), 4-метилбензосульфонилхлорид (31 мг, 0,16 ммол), 4-(диметиламино)-пиридин (20 мг, 0,16 ммол), диизопропилэтиламин (16,6 мг, 0,16 ммол) и безводный дихлорметан (0,6 мл). Полученный раствор перемешивали в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении. После очистки методом ТСХ [силикагель; элюент пентан - этилацетат (3:2)] получали желательный продукт (18,6 мг, 33%).1Н (CDCl3) δ (ppm): 1,62 (s, 9H); 2.09-2.13 (m, 2Н); 2.37 (s, 3H); 3.95 (t, 2H); 4.23 (t, 2Н); 5.22 (s, 2Н); 6.78 (d, 2Н); 7.23 (d, 2Н); 7.29 (d, 2Н); 7.73 - 7.75 (m, 3H);13C (CDCl3) δ (ppm): 21.60, 27.85, 28.81, 63.15, 66.35, 66.87, 71.75, 114.76, 118.27, 125.18, 127.11, 127.83, 128.94, 129.80, 132.79, 144.80, 163.72, 158.90, 159.03.
Пример 4 G
Синтез 2-трет.бутил-4-хлор-5-[4-(3-фтор-пропокси)бензилокси]-2Н-пиридазин-3-она
В сцинтилляционную колбу, содержащую суспензию соединения, полученного в примере 4 F (4,5 мг, 8,64×10-3 ммол) в безводном ацетонитриле (0,25 мл) добавляли раствор фторида калия (1,6 мг, 4,07×10-2 ммол) и Kryptofix (15,0 мг, 4,07×10-2 ммол) в безводном ацетонитриле (0,25 мл). Колбу закрывали и погружали в масляную баню с температурой 90ºС. Реакционную смесь перемешивали в течение 40 мин. Затем смесь охлаждали и концентрировали при пониженном давлении. После препаративной тонкослойной хроматографии [силикагель; элюент пентан - этилацетат (3:2)] получали желательный продукт (0,8 мг, 25%).1Н (CDCl3) δ (ppm): 1,62 (s, 9H); 2.14-2.20 (m, 2H); 4.09-4.11 (m, 2H); 4.60 (t, 1H); 4.68 (t, 1H); 5.24 (s, 2H); 6.92 (d, 2H); 7.32 (d, 2H); 7.72 (s, 1H);19F (CDCl3, CFCl3 в качестве внутреннего стандарта) δ (ppm): -222.66 (t of t, J=28.2, -50.4).
Пример 5 А
Синтез 4-(2-гидроксиэтоксиметил) бензойной кислоты метилового эфира

В двухгорлую круглодонную колбу, снабженную конденсатором Дьюара, помещали раствор 4-гидроксиметилбензойной кислоты метиловый эфир (2,50 г, 0,015 мол) в безводном дихлорметане (30 мл) и охлаждали до -10ºС в бане, содержащей соль со льдом. К охлажденному перемешиваемому раствору по каплям добавляли окись этилена (1,10 мл) с последующим введением эфирата трехфтористого бора (0,51 мл). Реакционную смесь перемешивали в течение 45 мин и затем нагревали до комнатной температуры в течение 30 мин для удаления избытка окиси этилена из реакционной смеси. Затем реакционную смесь разбавляли рассолом. Водный слой экстрагировали дихлорметаном (3 раза). Все органические слои соединяли, сушили над Na2SO4, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом хроматографии на силикагеле (4:1 пентан:этилацетат) и получали желательный продукт (537 мг, 2,56 ммол), выход 17%.1Н (CDCl3 8.36, 600 MHz): δ (2H, d, J=8.4 Hz), 7.41 (2H, d, J=8.5 Hz), 4,62 (3H, s), 3.92 (2H, s), 3.78 (m, 2H), 3.63 (2H, m);13C (CDCl3 167.1, 143.5, 130.0, 129.8, 127.5, 72.9, 72.0, 150 MHz): δ 62.1, 52.3.
Пример 5 В
Синтез 4-[2-(трет.бутилдиметилсиланилокси)этоксиметил] бензойной кислоты метилового эфира

К раствору продукта, полученного в примере 5 А (544,5 мг, 2,59 ммол), в безводном ДМФ (26 мл) добавляли имидазол (264 мг, 3,89 ммол) и TBDMS-Cl (586 мг, 3,89 ммол). Реакционную смесь перемешивали при комнатной температуре в течение ночи и добавляли воду. Водный слой экстрагировали этилацетатом (3x). Все соединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали при помощи хроматографии на силикагеле (4:1 пентан:этилацетат) с получением желательного продукта (677,5 мг, 2,19 ммол), выход 84%.1Н (CDCl3 8.01, 600 MHz): δ (2H, d, J=8.3 Hz), 7.42 (2H, d, J=8.4 Hz), 4,63 (2H, s), 3.91 (2H, s), 3.82 (2H, t, J=5.0), 3.58 (2H, t, J=5.1 Hz), 0.91 (9H, s), 0.07 (6H, s);13C (CDCl3 166.5, 143.5, 129.2, 128.8, 126.5, 72.1, 71.6, 150 MHz): δ 62.3, 51.5, 25.4, 17.9, -5.8.
Пример 5 С
Синтез {4-[2-(трет.бутилдиметилсиланилокси)этоксиметил]фенил}-метанола
К раствору продукта, полученного в примере 5 В (670 мг, 2,18 ммол), растворенного в безводном ТГФ (22 мл) по каплям добавляли раствор LiAlH (1,0 N раствор в ТГФ, 2,18 мл, 2,18 ммол). После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли водой. Водный слой обрабатывали этилацетатом (3x). Все соединенные органические слои сушили над Na2S04, фильтровали и концентрировали с получением масла (587 мг, 1,98 ммол), которое использовали на следующей стадии без очистки (выход 91%).1Н (CDCl3 7.34 4Н, s), 4.68 (2Н, s), 4.57 (2Н, s), 3.80, 600 MHz): δ (2H, t. J=5.2 Hz), 3.56 (2H, t, J=5.3 Hz), 1.69 (1H, br s), 0.90 (9H, s), 0.07 (6H, s);13C (CDCl3 140.4, 138.3, 128.0, 127.2, 73.2, 71.9, 65.4, 150 MHz): δ 63.0, 26.2, 18.6, -5.0.
Пример 5 D
Синтез 2-трет.бутил-5-{4-[2-(трет.бутилдиметилсиланилокси)-этоксиметил]бензилокси}-4-хлор-2H-пиридазин-3 она
К раствору соединения, полученного в примере 5 С (437 мг, 1,48 ммол), и 2-трет.бутил-4-хлор-5-гидрокси-2H-пиридазин-3-она (250 мг, 1,23 ммол) в безводном ТГФ (12 мл) добавляли твердый PPh3 (485 мг, 1,85 ммол) и диизопропилазодикарбоксилат (DIAD, 0,358 мл, 1,85 ммол). После окончания добавления реакционную смесь продолжали перемешивать при комнатной температуре. Через 20 ч реакционную смесь разбавляли водой. Водный слой отделяли и экстрагировали этилацетатом (3x). Все соединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом хроматографии на силикагеле (4:1 пентан:этилацетат) с получением желательного продукта (528 мг, 1,10 ммол), выход 89%.1Н (CDCl3 7.70 (1Н, s), 7.38 (4H, m), 5.30 (2H, s) 4.58, 600 MHz): δ (2H, s), 3.80 (2H, t, J - 5.4 Hz), 3.57 (2H, t, J=5.4 Hz), 1.63 (9H, br s), 0.90 (9H, s), 0.07 (6H, s);13C (CDCl3 159.0, 153.7, 138.8, 134.4, 128.3, 127.3, 150 MHz): δ 125.1, 118.5, 72.8, 71.7, 71.6, 66.4, 61.9, 29.7, 27.9, 25.6, -5.1.; HRMS вычислено для C24H37ClN2O4Si: 481.228389, найдено 481.2282.
Пример 5Е
Синтез 2-трет.бутил-4-хлор-5-[4-(2-гидроксиэтоксиметил)-бензилокси]-2H-пиридазин-3-она

К раствору соединения, полученному в примере 5D (528 мг, 1,09 ммол) в безводном ТГФ (11 мл) по каплям добавляли раствор TBAF (1,0 М раствор в ТГФ, 1,65 мл, 1,65 ммол). После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем реакцию обрывали водой. Водный слой отделяли и экстрагировали этилацетатом (3x). Все соединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом хроматографии на силикагеле (4:1 гексаны:этилацетат) с получением желательного продукта (311 мг, 0,850 ммол), выход 78%.1Н (CDCl3, 600 MHz): δ 7.70 (1Н, s), 7.38 (4H, m), 5.30 (2H, s), 4.56 (2H, s), 3.76 (2H, t, J=4.9 Hz), 3.60 (2H, t, J=4.8 Hz), 2.00 (1Н, br s), 1.61 (9H, br s);13C (CDCl3 159.0, 153.6, 150 MHz): δ 138.8, 134.4, 128.2, 127.2, 125.1, 118.3, 72.8, 71.6, 71.6, 66.4, 61.9, 27.8; HRMS вычислено для C18H23ClN2O4: 367.141911, найдено 367.1419.
Пример 5 F
Синтез 2-[4-(1-трет.бутил-5-хлор-6-оксо-1,6-дигидропиридазин-4-илоксиметил)-бензилокси этилового эфира толуол-4-сульфокислоты

К раствору соединения, полученного в примере 5Е (200 мг, 0,546 ммол) в безводном дихлорметане (5,50 мл) добавляли TsCl (125 мг, 0,656 ммол), DMAP (100 мг, 0,819 ммол) и триэтиламин (0,091 мл, 0,656 ммол). Реакционную смесь перемешивали при комнатной температуре. Через 22 ч реакционную смесь разбавляли водой. Водный слой отделяли и экстрагировали этилацетатом (3x). Все соединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом хроматографии на силикагеле (3:2 пентан:этилацетат) с получением желательного продукта (232 мг, 0,447 ммол), выход 82%.1Н (CDCl3, 7.79, 600 MHz): δ (2Н, d, J=8.3 Hz), 7.71 (1H, s), 7.38 (2H, d, J=8.2 Hz), 7.32 (4H, m), 5.30 (2H, s), 4.50 (2H, s), 4.21 (2H, m), 3.69 (2H, m), 2.43 (3H, s), 1.63 (9H, br s);13C (CDCl3 159.0, 153.7, 144.8, 138.8, 150 MHz): δ 134.4, 133.1, 129.8, 128.1, 128.0, 127.2, 125.1, 118.4, 72.8, 71.7, 69.2, 68.8, 66.4, 27.9, 21.6; HRMS вычислено для C25H29ClN2O6: 521.150762, найдено 521.1503.
Пример 5 G
Синтез 2-трет.бутил-4-хлор-5-[4-(2-фтор-этоксиметил)бензилоил]-2H-пиридазин-3-она

К раствору соединения, полученного в примере 5F (50 мг, 0,096 ммол) в безводном ацетонитриле (1,0 мл) добавляли KF (11,2 мг, 0,192 ммол) и Kryptofix (72,4 мг, 0,192 ммол). После окончания добавления реакционную смесь нагревали до 90ºС. Через 10 мин реакционную смесь охлаждали до комнатной температуры и разбавляли водой. Водный слой отделяли и экстрагировали этилацетатом (3x). Все соединенные органические слои высушивали над Na2S04, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом хроматографии на силикагеле (4:1 пентан:этилацетат) с получением желательного продукта (28 мг, 0,076 ммол), выход 79%.1Н (DMSO-d6, 600 MHz): δ 8.22 (1H, s), 7.45 (2H, d, J=8.20 Hz), 7.39 (2H, d, J=8.24 Hz), 5.42 (2H, s), 4.60 (1H, m), 4.54 (2H, s), 4.52 (1H, m), 3.71 (1H, m), 3.66 (1H, m), 1.57 (9H, s); 157.8, 153.8, 138.6;13С (DMSO-d6, 150 MHz): δ 134.6, 127.8, 127.7, 126.2, 115.6, 83.5, (82.4), 71.6, 71.2, 69.1 (69.0), 65.3, 27.4;19F (DMSO-d6 - 221.74 (1F, m), 564 MHz): δ HRMS вычислено для C18H22ClAN2O3: 369.137575, найдено 369.1377.
Пример 6 А
Синтез 1-(4-гидроксиметилфенокси)пропан-2-она

К перемешиваемому раствору 4-гидроксибензилового спирта (1,0 г, 8,06 ммол) в ацетоне (80 мл) добавляли карбонат калия (1,34 г, 9,68 ммол) и хлорацетон (0,771 мл, 9,68 ммол). После окончания добавления реакционную смесь нагревали с обратным холодильником. Через 20 ч реакционную смесь охлаждали до комнатной температуры и удаляли растворитель. К сырому продукту добавляли воду и этилацетат. Водный слой отделяли и экстрагировали этилацетатом (3x). Все соединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом хроматографии на силикагеле (градиент от 4:1 до 1:1 пентан:этилацетат) с получением желательного продукта (0,981 г, 5,45 ммол), выход 98%.1Н (CDCl3, 600 MHz): δ 7.30 (2Н, d, J=8.7 Hz), 6.87 (2H, d, J=8.7 Hz), 4.63 (2H, d, J=5.7 Hz), 4.54 (2H, s), 2.27 (3H, s), 1.66 (1H, t, J=5.8 Hz);13С (CDCl3, 150 MHz): 8 205.7, 157.3, 134.3, 128.8, 114.6, 73.1, 64.8, 25.6.
Пример 6 В
Синтез 1-(4-гидроксиметилфенокси)пропан-2-ола

К раствору 1-(4-гидроксиметилфенокси)-пропан-2-она (1,26 г, 6,99 ммол) в метаноле (60 мл) добавляли твердый NaBH4 (0,32 г, 8,39 ммол). После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой, водный слой экстрагировали этилацетатом (3x). Все соединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением масла (1,24 г, 6,81 ммол), которое использовали на следующей стадии без очистки (выход 98%).1Н (CDCl3, 7.29, 600 MHz): δ (2H, d, J=8.4 Hz), 6.90 (2H, d, J=8.5 Hz), 4.62 (2H, s), 4.21 (1H, m), 3.94 (1H, dd, J=9.2, 3.1 Hz), 3.82 (1Н, m), 1.29 (3Н, d, J=6.4 Hz).
Пример 6 С
Синтез 2-трет.бутил-4-хлор-5-[4-(2-гидроксипропокси)бензилокси]-2H-пиридазин-3-она
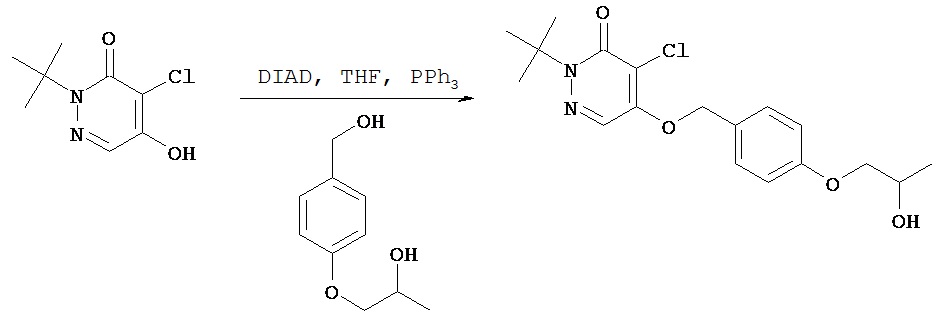
К раствору соединения, полученного в примере 6 В (269 мг, 1,48 ммол) и 2-трет.бутил-4-хлор-5-гидрокси-2H-пиридазин-3-она (250 мг, 1,23 ммол) в безводном ТГФ (18,5 мл) добавляли твердый PPh3 (485 мг, 1,85 ммол) и DIAD (0,358 мл, 1,85 ммол). После окончания добавления реакционную смесь перемешивали при комнатной температуре. Через 20 ч реакционную смесь разбавляли водой. Водный слой отделяли и экстрагировали этилацетатом (3x). Все соединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом хроматографии на силикагеле (1:1 пентан:этилацетат) с получением желательного продукта (234 мг, 0,634 ммол), выход 51%.
1Н (CDCl3, 7.71 (1H, s), 7.33 (2H, d, 600 MHz): δ (J=8.7 Hz), 6.94 (2H, d, J=8.7 Hz), 5.24 (2H, s), 4.19 (1H, m), 3.95 (1H, dd, J=9.2, 3.1 Hz), 3.81 (1H, dd, J=9.2, 7.7 Hz), 1.62 (9H, s), 1.29 (3H, d, J=6.4 Hz).
Пример 6 D
Синтез 2-[4-(1-трет.бутил-5-хлор-6-оксо-1,6-дигидропиридазин-4-илоксиметил)фенокси]-1-метилэтилового эфира толуол-4-сульфокислоты
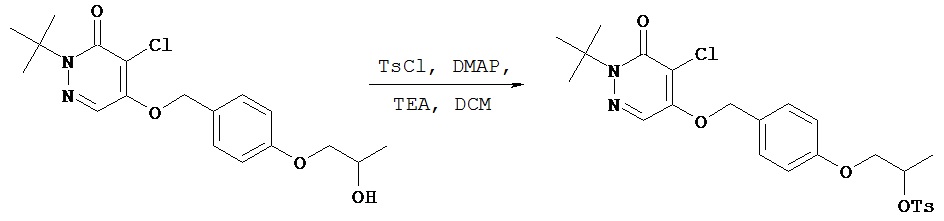
К раствору соединения, полученного в примере 6С (200 мг, 0,546 ммол) в безводном дихлорметане (6,0 мл) добавляли TsCl (125 мг, 0,656 ммол), DMAP (100 мг, 0,819 ммол) и триэтиламин (0,0914 мл, 0,656 ммол). Реакционную смесь перемешивали при комнатной температуре. Через 22 ч реакционную смесь разбавляли водой. Водный слой отделяли и экстрагировали этилацетатом (3x). Все соединенные органические слои сушили над Na2S04, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом хроматографии на силикагеле (70: 30 пентан: этилацетат) с получением желательного продукта (166 мг, 0,319 ммол), выход 58%.1Н (CDCl3, 7.80 (2Н, d, 600 MHz): δ (J=8.3 Hz), 7.72 (1H, s), 7.32 (2H, d, J=7.9Hz). 7.29 (2H, d, J=8.7 Hz), 6.74 (2H, d, J=8.7 Hz), 5.22 (2H, s), 4.19 (1H, m), 4.02 (1H, dd, J=10.4, 6.0 Hz), 3.93 (1H, dd, J=10.4, 4.5 Hz), 2.44 (3H, s), 1.63 (9H, s), 1.42 (3H, d, J=6.5 Hz);13С (CDCl3 158.9, 150 MHz): δ 158.3, 153.6, 144.6, 133.8, 129.6, 128.8, 127.8, 127.4, 125.1, 118.0, 114.7, 76.8, 71.5, 69.7, 66.2, 27.7, 21.5, 17.6; HRMS вычислено для C25H29ClN2O6S: 521.150762, найдено 521.1505.
Пример 6 Е
Синтез 2-трет.бутил-4-хлор-5-[4-(2-фторпропокси)бензилоил]-2H-пиридазин-3-она
К раствору соединения, полученного в примере 6 D (50 мг, 0,096 ммол) в безводном ацетонитриле (1,0 мл) добавляли KF (11,2 мг, 0,192 ммол) и Kryptofix (72,4 мг, 0,192 ммол). После окончания добавления реакционную смесь нагревали до 90ºС. Через 40 мин смесь охлаждали до комнатной температуры и разбавляли водой. Водный слой отделяли и экстрагировали этилацетатом (3x). Все соединенные органические слои сушили над Na2S04, фильтровали и концентрировали с получением масла. Сырой продукт очищали методом тонкослойной хроматографии на силикагеле (4:1 пентан:этилацетат) с выделением жлеательного продукта (12,5 мг, 0,034 ммол), выход 41% (в расчете на выделенное исходное соединение). Кроме непрореагировавшего исходного соединения (5,8 мг, 0,011 ммол).1Н (CDCl3, 600 MHz): δ 7.73 (1H, s), 7.34 (2H, d, J=8.6Hz), 6.95 (2H, d, J=8.6 Hz), 5.25 (2H, s), 5.06-4.96 (1H, m), 4.06 (2H, m), 1.63 (9H, s), 1.47 (3H, dd, J=6.4, 23.6 Hz);13С (DMSO-d6, 158.4, 157.8, 153.9, 129.8, 127.6, 126.2, 115.5, 114.6, 89.01, 150 MHz): δ (88.0), 71.2, 70.4, (70.3), 65.3, 27.4, 16.9, (16.8);19F (DMSO-d6 - 178.20 (1F, m); 564 MHz): δ HRMS вычислено для C18H22ClFN2O3: 369.137575, найдено 369.1370.
Пример 7 А
Синтез 4-(3-оксобутил) бензойной кислоты метилового эфира

К раствору метил-4-бромбензоата (1,0 г, 4,05 ммол) в триэтиламине (13 мл) добавляли 3-бутен-2-ол (1 мл, 11,63 ммол), ацетат палладия (II) (0,104 г, 0,465 ммол) и затем трифенилфосфин (0,244 г, 0,93 ммол). Реакционную смесь перемешивали на масляной бане с температурой 75ºС в течение ночи. Мониторинг методом ТСХ (3:1 гексан:этилацетат) показал наличие целевого продукта и арилбромида. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали. Затем добавляли воду с последующей экстракцией этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2S04, фильтровали и концентрировали. Сырой продукт очищали методом флэш-хроматографии на колонке (от 5:1 до 3:1 гексан:этилацетат) и получали нужный продукт (250 мг, выход 26%).1Н NMR (600 MHz, CDCl3): δ 7.95 (d, 2H, J=8.4 Hz), 7.25 (d, 2H, J=8.4 Hz), 3.90 (s, 3H), 2.95 (t, 2H, J=7.45 Hz), 2.77 (t, 2H, J=7.68 Hz), 2.14 (s, 3H).
Пример 7 В
Синтез 2-трет.бутил-4-хлор-5-[4-(3-гидроксибутил)бензилокси]-2H-пиридазин-3-она
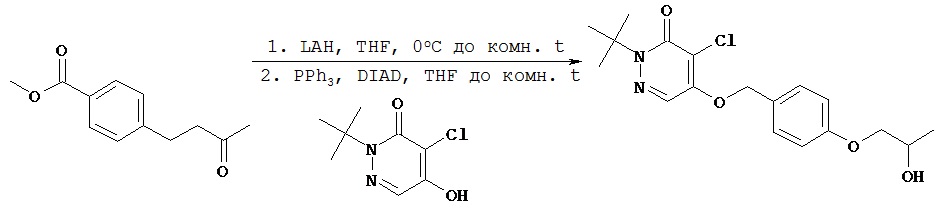
К раствору соединения, полученного в примере 7А (505 мг, 2,447 ммол) в ТГФ (19 мл) при 0ºС по каплям добавляли 1 М раствор (в ТГФ) литийалюминийгидрида (12,2 мл, 12,237 ммол). После окончания добавления удаляли ледяную баню и перемешивали реакционную смесь при комнатной температуре в течение 1 ч в атмосфере азота. Затем последовательно добавляли воду (183 мкл), 15% раствор NaOH (183 мкл) и воду (548 мкл). Реакционную смесь перемешивали в течение 15 мин перед фильтрованием и промывкой ТГФ. Затем фильтрат концентрировали при пониженном давлении с получением 4-(4-гидроксиметилфенил)-бутан-2-ола в виде масла коричневого цвета (314 мг, выход 71%). Затем к раствору 2-трет.бутил-4-хлор-5-гидрокси-2Н-пиридазин-3-она (234 мг, 1,155 ммол) в ТГФ (45 мл) добавляли 4-(4-гидроксиметилфенил)бутан- 2-ол (312 мг, 1,732 ммол), трифенилфосфин (454 мг, 1,732 ммол) и затем диизопропилазодикарбоксилат (DIAD, 335 мкл, 1,732 ммол). Реакционную смесь перемешивали при комнатной температуре в течение ночи в атмосфере азота. Тонкослойная хроматография (100% этилацетата) показала поглощение исходного пиридазинона, затем реакционную смесь концентрировали. Сырой продукт очищали методом флэш-хроматографии (4:1 гексан:этилацетат до 100% этилацетата) с получением прозрачного масла (200 мг, выход 48%).1Н NMR (600 MHz, CDCl3): δ 7.73(s, 1Н), 7.32 (d, 2Н, J=8.0), 7.24 (d, 2Н, J=8.0), 5.30 (s, 1H), 5.27 (s, 2Н), 3.83 (m, 1H), 2.80 -2.76 (m, 1H), 2.71-2.66 (m, 1H), 1.63 (s, 9H), 1.23 (d, 3H, J=6.2);13С (CDCl3 159.3, 153.9, 143.2, 132.5, 129.2, 127.6, 125.4, 118.5, 73.4, 67.6, 66.6, 40.9, 32.0, 28.1, 23.9, 150 MHz): δ HRMS вычислено для C19H25ClN2O3: 365.162647, найдено 365.1624.
Пример 7 С
Синтез 3-[4-(1-трет.бутил-5-хлор-6-оксо-1,6-дигидропиридазин-4-илоксиметил)фенил]-1-метилпропилового эфира толуол-4-сульфокислоты

К раствору соединения, полученного в примере 7 В (200 мг, 0548 ммол), в пиридине (10 мл) добавляли п-толуолсульфонилхлорид (209 мг, 1,096 ммол). Реакционную смесь перемешивали при комнатной температуре в течение ночи в атмосфере азота. Мониторинг методом LC - MS показал смесь 1:1 исходного вещества и желательного продукта. Реакционную смесь разбавляли этилацетатом и промывали 5% CuSO4 до получения водного раствора бледно-синего цвета. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали методом флэш-хроматографии (3:1 гексан:этилацетат до 100% этилацетата, выделяя исходное соединение (90 мг) и желательный продукт в виде прозрачного масла (74 мг, выход 47% в расчете на выделенное исходное соединение).1Н NMR (600 MHz, CDCl3): 7.80 (d, 2H, J=8.3 Hz), 7.72 (s, 1H), 7.33 (d, 2H, J=8.0 Hz), 7.30 (d, 2H, J=8.1 Hz), 7.13 (d, 2H, J=8.1 Hz), 5.27 (s, 2H), 4.66 (m, 1H), 2.65 (m, 1H), 2.54 (m, 1H), 2.45 (s, 3H), 1.94 (m, 1H), 1.81 (m, 1H), 1.63 (s, 9H), 1.26 (s, 3H).
Пример 7 D
Синтез 2-трет.бутил-4-хлор-5-[4-(3-фторбутил)бензилокси]-2H-пиридазин-3-она
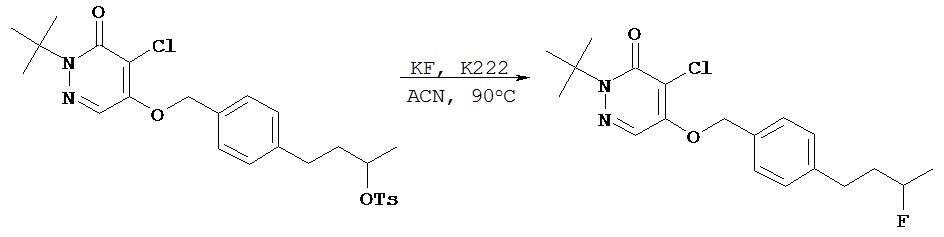
К раствору соединения, полученного в примере 7С (18,2 мг, 0,035 ммол), в ацетонитриле (400 мкл) добавляли фторид калия (4,1 мг, 0,070 ммол) и К222 (26,4 мг, 0,070 ммол). Реакционную смесь перемешивали при 90ºС в течение 20 мин в атмосфере азота, проводя мониторинг методом LC - MS. Затем реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Сырой продукт очищали методом препаративной тонкослойной хроматографии (4:1 гексан:этилацетат в качестве элюента) с получением желательного продукта в виде масла (5 г, выход 39%).1Н NMR (600 MHz, CDCl3): 5 7.70 (s, 1H), 7.34 (d, 2H, J=7.9 Hz), 7.24 (d, 2H, J=8.0 Hz), 5.28 (s, 2H), 4.71-4.60 (m, 2H), 2.84-2.80 (m, 1H), 2.73-2.69 (m, 1H), 2.02-1.93 (m, 1H), 1.87-1.77 (m, 1H), 1.63 (s, 9H), 1.35 (dd, 3H, J=6.2 и 23.9 Hz);13C (CDCl3 159.1, 153.8, 150 MHz): 5 142.4, 132.5, 129.0, 127.4, 125.2, 118.3, 90.4, (89.3), 71.9, 66.3, 38.5 (38.4), 31.1 (31.0), 27.9, 21.1 (21.0);19F (CDCl3 - 174.7, 564 MHz): δ (1F, m); HRMS вычислено для C19H23ClFN2O2: 367.158310, найдено 367.1582.
Пример 8 А
Синтез тетрадейтерата 4-[2-гидроксиэтоксиметил]бензойной кислоты метилового эфира
В высушенную пламенем двухгорлую колбу добавляли раствор метил-4-(гидроксиметил)бензоата (2,5 г, 15 ммол) в дихлорметане (30 мл). Реакционную смесь промывали азотом и охлаждали до -5ºС. На колбу устанавливали конденсатор Дьюара (также высушенный пламенем), содержащий баню со смесью сухой лед / ацетон (-78ºС) и добавляли тетрадейтерат окиси этилена (~55 капель). Затем по каплям добавляли BF3⋅Et2O (510 мкл, 0,0041 ммол) и перемешивали реакционную смесь при -5ºС в течение 35 мин в атмосфере азота. Мониторинг методом ТСХ (100% этилацетат) показала полное израсходование исходного материала. Реакционную смесь нагревали до комнатной температуры и сдували избыток газообразной окиси этилена. Затем реакционную смесь разбавляли рассолом и экстрагировали дихлорметаном (2 раза). Соединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая сырое масло. После очистки методом флэш-хроматографии (4:1 пентан:этилацетат) получали желательный продукт в виде прозрачного масла (520 мг, выход 16%).1Н NMR (600 MHz, CDCl3): δ 8.02 (d, 2H, J=8.2 Hz), 7.41 (d, 2H, J=8.1 Hz), 4.62 (s, 2H), 3.92 (s, 3Н);13С NMR (150 MHz, CDCl3 167.1, 143.5, 130.8) δ 129.9, 127.5, 72.8, 52.4.
Пример 8 В
Синтез тетрадейтерата 4-[2-(трет.бутилдиметилсилилокси)-этоксиметил]-бензойной кислоты метилового эфира

К раствору соединения, полученного в примере 8А (500 мг, 2,334 ммол), в ДМФ (23 мл) добавляли трет.бутилдиметилсилилхлорид (528 мг, 3,501 ммол) и имидазол (238 мг, 3,501 ммол). Смесь перемешивали при комнатной температуре в течение 5 ч в атмосфере азота, осуществляя мониторинг методом ТСХ (3:1 пентан:этилацетат). Добавляли другую 0,5 экв. часть трет.бутилдиметилсилилхлорида (176 мг) и имидазол (79 мг) и полученную смесь перемешивали при комнатной температуре в течение ночи. Через 16 ч большая часть исходного соединения была израсходована, о чем свидетельствовала ТСХ. Реакционную смесь разбавляли водой и экстрагировали этилацетатом (2 раза). Соединенные органические слои сушили над Na2SO4, фильтровали при пониженном давлении с получением сырого масла, которое очищали пропусканием через толстую прокладку из силикагеля (3:1 пентан:этилацетат) с получением желательного продукта в виде прозрачного масла (602 мг).1Н NMR (600 MHz, CDCl3): 8.00 (d, 2H, J=8.3 Hz), 7.40 (d, 2H, J=8.5 Hz), 4.62 (s, 2H), 3.90 (s, 3H), 0.90 (s, 3H), 0.06 (s, 6H).
Пример 8 С
Синтез {4-[2-(трет.бутилдиметилсилилокси)этоксиметил]фенил}-метанола гексадейтерата

К раствору продукта, полученного в примере 8 В (610 мг, 1,857 ммол), в ТГФ (19 мл) при 0ºС по каплям добавляли 1 М раствор (в ТГФ) литийалюминий дейтерида (1,9 мл, 1,857 ммол). После окончания добавления ледяную баню убирали и перемешивали реакционную смесь при комнатной температуре в течение 3,5 ч в атмосфере азота, осуществляя мониторинг методом ТСХ (3:1 пентан:этилацетат). Реакционную смесь затем разбавляли водой и экстрагировали этилацетатом (2 раза). Соединенные органические слои сушили над Na2S04, фильтровали и концентрировали при пониженном давлении с получением прозрачного масла (482 мг, выход 86%). Продукт применяли на следующей стадии без очистки.1Н NMR (600 MHz, CDCl3): 7.33 (s, 4H), 4.56 (s, 2H), 0.89 (s, 9H), 0.66 (s, 6H).
Пример 8 D
Синтез гексадейтерата 2-трет.бутил-4-хлор-5-{4-[2-(трет.бутилдиметилсиланилокси)этоксиметил]бензилокси}-2Н-пиридазин-3-она

К раствору 2-трет.бутил-4-хлор-5-гидрокси-2Н-пиридазин-3-она (212 мг, 1,047 ммол) в ТГФ (15 мл) добавляли продукт, полученный в примере 8 С (475 мг, 1,570 ммол), трифенилфосфин (412 мг, 1,570 ммол) и затем диизопропилазодикарбоксилат (DIAD, 304 мкл, 1,570 ммол). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч в атмосфере азота. ТСХ (1:1 гексан:этилацетат) показала израсходование исходного пиридазинона, реакционную смесь концентрировали в вакууме. Сырой продукт очищали методом флэш-хроматографии (90:10 пентан:этилацетат) с получением прозрачного масла (336 мг, выход 66%).1Н NMR (600 MHz, CDCl3): 7.70 (s, 1H), 7.39 (m, 4H), 4.58 (s, 2Н), 1.63 (s, 9H), 0.90 (s, 9H), 0.07 (s, 6H); HRMS вычислено для C24H31D6ClN2O4Si: 509.24738, найдено 509.2480.
Пример 8 Е
Синтез гексадейтерата 2-трет.бутил-4-хлор-5-[4-(2-гидроксиэтоксиметил)бензилокси]-2Н-пиридазин-3-она

К раствору соединения, полученного в примере 8 D (330 мг, 0,677 ммол), в ТГФ (7 мл) по каплям добавляли 1 М раствор (в ТГФ) тетрабутиламмонийфторида (1 мл, 1,016 ммол). Смесь перемешивали при комнатной температуре в течение 2 ч в атмосфере азота, проводя мониторинг методом ТСХ (1:1 гексан:этилацетат). Затем реакционную смесь концентрировали при пониженном давлении и пропускали через толстую прокладку из окиси кремния (100% этилацетат), получая продукт в виде масла, содержащего небольшое количество соответствующего силанола. Продукт применяли на следующей стадии без очистки.1Н NMR (600 MHz, CDCl3): 7.72 (s, 1H), 7.41 (s, 4H), 4.59 (s, 2Н), 1.64 (s,9H);13C NMR (150 MHz, rt, CDCl3): 159.2, 153.9, 139.5, 134.5, 128.5, 127.5, 125.3, 118.6, 73.0, 66.6,28.1; HRMS вычислено для C25H23D6ClN2O6S: 549.169754, найдено 549.1705.
Пример 8 F
Синтез гексадейтерата 2-[4-(1-трет.бутил-5-хлор-6-оксо-1,6-дигидро-пиридазин-4-илоксиметил)бензилокси] этилового эфира толуол-4-сульфокислоты

К раствору продукта, полученного в примере 8 Е (250 мг, 0,670 ммол), в дихлорметане (7 мл) добавляли п-толуолсульфонилхлорид (153 мг, 0,805 ммол), N,N-диметиламинопиридин (DMAP, 98 мг, 0,805 ммол) и триэтиламин (140 мкл, 1,005 ммол). Смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. ТСХ (1:1 гексан:этилацетат) показала израсходование исходного пиридазинона, реакционную смесь концентрировали в вакууме. Сырой продукт очищали методом флэш-хроматографии (2:1 гексан:этилацетат до 100% этилацетата) с выделением исходного соединения (9 мг) и желательного продукта (261 мг, выход 77% в расчете на выделенное исходное соединение).1H NMR (600 MHz, CDCl3): 7.76 (d, 2H, J=8.3 Hz), 7.73 (s, 1H), 7.36 (d, 2H, J=8.1 Hz), 7.29 (m, 4H), 4.47 (s, 2H), 2.40 (s, 3H), 1.61 (s, 9H);13C NMR (150 MHz, rt, CDCl3): 159.0, 153.8, 145.0, 138.5, 134.4, 133.1, 129.9, 128.1, 128.0, 127.3, 125.2, 118.1, 72.7, 71.0, 37.0. 63.4, 28.0, 21.7.
Пример 8 G
К раствору продукта, полученного в примере 8 F (14 мг, 0,027 ммол), в ацетонитриле (300 мкл) добавляли фторид калия (3,1 мг, 0,053 ммол) и K222 (20 мг, 0,053 ммол). Реакционную смесь перемешивали при 90ºС в течение 10 мин в атмосфере азота, осуществляя мониторинг методом ТСХ (1:1 гексан:этилацетат). Затем реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Сырой продукт очищали методом препаративной ТСХ (2:1 гексан:этилацетат) с получением продукта в виде масла (6,2 мг, выход 62%).1Н NMR (600 MHz, CDCl3): 7.70 (s, 1H), 7.40 (s, 4H), 4.61 (s, 2H), 1.63 (s, 9H);13С NMR (150 MHz, rt, CDCl3): 158.5, 153.1, 138.2, 133.8, 127.7, 126.8, 124.6, 117.8, 72.4, 65.9, 27.3;19F NMR (564 MHz, CDCl3): -225.2 (m, 1F).
Методы радиосинтеза и очистки при получении комплексов феназаквина и пиридабена, радиомеченых радионуклидом фтором-18
Фтор-18 (18F), используемый в процессе исследования, получали путем бомбардировки протонами обогащенного кислорода-18 (18О) в виде Н218О с применением примерно 10 MeV протонов и PETnet (Wobum, МА). Для этой ядерной реакции: О18 (р, γ)18F
Для всех реакций радиосинтеза применяли одну и ту же процедуру. Всю стеклянную посуду силанизировали с целью исключения адгезии материала к стенкам сосуда и оптимизации его передачи. Специфическая установка ЖХВР была использована для очистки всех соединений. Для радиоаналитических анализов конечного продукта применяли особую установку ЖХВР.
Обычно18F получали от поставщика в виде осажденного на колонке (колонка18F), заключенной в свинцовый кожух. В колонке18F содержалась натриевая соль, координированная или с алюминием, или с четвертичной аммониевой солью в стеклянной колонке. Концы колонок были соединены с трубкой Tygon™ с помощью входящих и охватывающих зажимов Luer™.18F удаляли из колонки, используя следующий метод.
1. Соединяли раствор 15 мг карбоната калия (К2СО3) в 1 мл дистиллированной/деионизированной воды (H2O) и раствор 90 мг 4, 7, 13, 16, 21, 24 - гексаокса - 1, 10 - диазабицикло [8. 8. 8] гексакозана (Kryptofix™; K222) в 4 мл безводного ацетонитрила (CH3CN) и аккуратно перемешивали смесь, следя за тем, чтобы слои не разделялись, получали элюирующий раствор для колонки (CES).
2. Из сосуда, описанного на стадии 3, извлекали один мл аликвоты CES с помощью шприца 3 мл, шприц был прикреплен к входящему Luer™ трубки Tygon™, связанной с колонкой18F.
3. К охватывающему зажиму Luer™ другой трубки Tygon™, соединенной с колонкой18F, была присоединена узкая игла, эта игла была вставлена через каучуковую перегородку, которой снабжена грушевидная стеклянная колба 24/40 Pyrex™ объемом 15 мл.
4. Грушевидная колба объемом 15 мл освобождалась от воздуха при помощи иглы и колбу промывали сухим азотом. Иглу соединяли с линией вакуума и поток регулировали таким образом, что CES медленно проходила через колонку18F в грушевидную колбу объемом 15 мл.
5. Вакуум и поток N2 регулировали таким образом, что содержимое колбы извлекалось досуха. При помощи шприца в колбу добавляли безводный CH3CN (1 мл), используя для этого вакуум. Затем регулировали вакуум, и поток N2 уравновешивали для удаления ацетонитрила. Эту процедуру повторяли дважды, после чего снимали вакуум.
6. Содержимое колбы удаляли при помощи шприца и количественно оценивали радиоактивность. Раствор18F непосредственно использовали в процессе радиомечения.
Следующие стадии описывают радиомечение аналогов феназаквина и пиридабена18F. Как указано выше, эти стадии были одинаковыми для каждого из соединений. Нижеследующая схема описывает этот метод для всех аналогов18F - феназаквина и пиридабена:

7. Предшественник (толуолсульфонатный эфир) желательного аналога феназаквина или пиридабена (2,5 мг) растворяли в CH3CN (0,5 мл) в коническом силанизированном стеклянном сосуде Wheaton™ объемом 5 мл, снабженном магнитной мешалкой. Сосуд погружали в масляную баню, нагретую до 90ºС. Раствор18F, описанный выше, добавляли в реакционный сосуд и полученную смесь нагревали 30 мин при 90ºС.
8. Содержимое перемещали в силанизированную круглодонную колбу объемом 50 мл, содержащую дистиллированную/деионизированную воду (25 мл) и содержимое колбы удаляли при помощи шприца и осаждали в колонке Waters™ Oasis HLB (гидрофильно-липофильный баланс), давая непрореагировавшему фториду и непрореагировавшим солям проходить вместе с элюатом.
9. Органические компоненты элюировали из колонки в коническую колбу объемом 5 мл, используя дихлорметан (3 мл, CH2Cl2). Элюент очищали методом препаративной ЖХВР (колонка Phenomex LUNA С 18, 250×10 мм, частицы 5 мк, поры 100 А, градиент элюирования 90/10 H2O/CH3CN-CH3CN). Соответствующие фракции концентрировали и анализировали для определения радиохимического выхода и радиохимической чистоты (аналитическая ЖХВР). Раствор концентрировали под вакуумом досуха и растворяли в подходящем объеме 10% этанольного физиологического раствора для инъекции и/или биологических исследований.
Кроме того, нижеследующие соединения могут быть получены следующими методами:
Пример 1 - Аналоги дегелина
Синтез 4’-бром-рот-2’-еноновой кислоты
Ротенон (5,0 г, 12,7 ммол), растворенный в дихлорметане (30 мл), добавляли быстро к охлажденному (-10ºС) раствору трехбромистого бора (3,15 г, 12,7 ммол) в дихлорметане (32,7 мл). Реакционную смесь перемешивали точно в течение 2 мин и затем выпаривали досуха. Полученный сырой продукт коричневого цвета растворяли в минимальном количестве метанола и охлаждали до 0ºС для начала кристаллизации. Коричневые кристаллы собирали и высушивали с получением 4’-бром-рот-2’-еноновой кислоты (3,24 г).

Синтез 4’-гидрокси-рот-2’-еноновой кислоты
Окись серебра (1,0 г, 4,24 ммол) добавляли к раствору 4’-бром-рот-2’-еноновой кислоты (2,0 г, 4,24 ммол) в ацетоне (80 мл). После окончания добавления реакционную смесь продолжали перемешивать в темноте. Через 24 ч смесь фильтровали через целит, фильтрат концентрировали с получением желтого масла. Сырой продукт растворяли в минимальном количестве дихлорметана и охлаждали до 0ºС для начала кристаллизации. 4’-гидрокси-рот-2’-еноновую кислоту (1 г) собирали в виде желтых кристаллов.

Синтез (6aS, 12aS)-7’-гидроксидегелина
Твердый PhSe-Cl (370,87 мг, 1,94 ммол) добавляли к охлажденному (-30ºС) раствору 4-гидрокси-рот-2’-еноновой кислоты (725,5 мг, 1,71 ммол) в дихлорметане (20 мл). После окончания добавления реакционной смеси давали нагреться до комнатной температуры в течение 2 ч и продолжали перемешивать еще 1 ч при комнатной температуре. Через три часа, составивших все время протекания реакции, реакционную смесь концентрировали с получением желтого масла. Сырой продукт растворяли в ТГФ (20 мл) и охлаждали до 0ºС. Добавляли перекись водорода (30% в воде, 0,354 мл). После окончания добавления реакционную смесь перемешивали при 0ºС в течение часа и затем в течение ночи при комнатной температуре. На следующий день реакционную смесь разбавляли диэтиловым эфиром. Органический слой отделяли и промывали 5% NaHCO3 (2х), сушили над Na2SO4 и концентрировали с получением (6aS, 12aS)-7’-гидроксидегелина в виде твердого аморфного вещества желтого цвета.

Синтез (6aS, 12aS)-7’-толуолсульфонилдегелина
К перемешиваемому раствору (6aS, 12aS)- 7’-гидроксидегелина (30 мг, 0,073 ммол) в дихлорметане (1,5 мл) добавляли TsCl (15,3 мг, 0,080 ммол) и пиридин (6,47 мкл, 0,080 ммол). После окончания добавления реакционную смесь продолжали перемешивать при комнатной температуре. Через 48 ч реакция была завершена примерно на -50% (данные LCMS), ее концентрировали. Сырой продукт очищали методом хроматографии на силикагеле (градиент от 100% дихлорметана до 25% ацетона в дихлорметане) с получением (6aS, 12aS)-7’-толуолсульфонилдегелина в виде масла желтого цвета.

Синтез (6aS, 12aS)-7’-метансульфонилдегелина
К перемешиваемому раствору (6aS, 12aS)-7’-гидроксидегелина (50 мг, 0,122 ммол) в дихлорметане (0,5 мл) добавляли MsCl (9,48 мкл, 0,12 ммол) и триэтиламин (17,0 мкл, 0,122 ммол). После завершения добавления реакционную смесь продолжали перемешивать при комнатной температуре. Через 3 ч, поскольку реакция была завершена только на -80%, добавляли дополнительные эквиваленты MsCl и триэтиламина. Через 24 ч реакция была завершена, реакционную смесь разбавляли водой. Водный слой экстрагировали дихлорметаном. Все соединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением желтого масла. После хроматографии на силикагеле (градиент от 100% дихлорметана до 5% ацетона в дихлорметане) получали (6aS, 12aS)-7’-метансульфонилдегелин (48 мг) в виде желтого масла.

Синтез (6aS, 12aS)-7’-[18F]фтордегелина
В тонкостенный силанизированный контейнер с вакуумом объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% об/вес раствор в воде) и раствор18F- в воде (10 микроКюри, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушили путем повторяющегося добавления и выпаривания CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали смесь под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), сосуд открывали и одной порцией добавляли (6aS, 12aS)-7’-метансульфонилдегелин (2 мг). Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения содержимое разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light C-18 Sep-Pak). Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и остаток очищали методом ЖХВР с получением чистого (6aS, 12aS)-7’-[18F] фтордегелина.

Синтез (6aS, 12aS)-7’-[18F] фтордегелина
В тонкостенный силанизированный контейнер с вакуумом объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% об/вес раствор в воде) и раствор18F- в воде (10 микрокюри, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушили путем повторяющегося добавления и выпаривания CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали смесь под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), сосуд открывали и одной порцией добавляли (6aS, 12aS)-7’-толуолсульфонилдегелин (2 мг). Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения содержимое разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light C-18 Sep-Pak). Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и остаток очищали методом ЖХВР с получением чистого (6aS, 12aS)-7’-[18F] фтордегелина.
Синтез (-)-рот-2’-еноновой кислоты

Твердый цианборгидрид натрия (264 мг, 4,20 ммол) добавляли к раствору 4’-бром-рот-2’-еноновой кислоты (500 мг, 1,05 ммол), растворенной в НМРА. После завершения добавления реакционную смесь нагревали до 70ºС. Через 2,5 ч

реакционную смесь охлаждали до комнатной температуры и разбавляли водой. Водный слой экстрагировали смесью диэтиловый эфир / гексан (3/1). Органический слой сушили над Na2SO4, фильтровали и концентрировали с получением прозрачного масла. После хроматографии на силикагеле (градиент от 20% гексана в дихлорметане до 5% ацетона в дихлорметане) получали (-)-рот-2’-еноновой кислоты (162,2 мг) в виде прозрачного масла.
Синтез (6aS, 12aS)-дегелина
Твердый PhSe-Cl (185 мг, 0,972 ммол) добавляли к охлаждаемому (-30ºС) раствору (-)-рот-2’-еноновой кислоты (350 мг, 0,884 ммол) в дихлорметане (10,5 мл). После окончания добавления реакционной смеси давали нагреться до комнатной температуры в течении 2 ч и продолжали ее перемешивать при комнатной температуре еще 1 ч. После завершения реакции (общее время реакции составило 3 ч) реакционную смесь концентрировали с получением желтого масла. Сырой продукт растворяли в ТГФ (10,5 мл) и охлаждали до 0ºС. Добавляли перекись водорода (30% в воде, 0,177 мл). После окончания добавления реакционную смесь продолжали перемешивать при 0ºС в течение 1 ч и затем продолжали перемешивать при комнатной температуре в течение ночи. На следующий день смесь разбавляли диэтиловым эфиром. Органический слой отделяли и промывали 5% NaHCO3 (2x), сушили над Na2S04 и концентрировали с получением (6aS, 12aS)-дегелина в виде твердого аморфного вещества желтого цвета.
Синтез (6aS)-дегелиненольного эфира


К раствору дегелина (245 мг, 0,622 ммол) в метаноле (20 мл) добавляли п-TsOH моногидрат (118,3 мг, 0,622 ммол) и триметилортоформиат (68,14 мкл, 0,622 ммол). После окончания добавления реакционную смесь нагревали с обратным холодильником в течение 8 ч и затем продолжали перемешивать при комнатной температуре в течение ночи. На следующий день реакционную смесь разбавляли водой. Водный слой экстрагировали этилацетатом. Соединенные органические слои промывали насыщенным NaHCO3, сушили над Na2SO4 и концентрировали с получением (6aS)-дегелиненольного эфира в виде твердого аморфного вещества желтого цвета.

Синтез (6aS)-4’,5’-дигидро-4’,5’-эпоксидегелиненольного эфира
К охлажденному (0ºС) раствору (6aS)-дегелиненольного эфира (50 мг, 0,123 ммол) в дихлорметане (0,5 мл) добавляли м-СРВА (45 мг, 0,184 ммол). После завершения добавления реакционную смесь продолжали перемешивать при комнатной температуре. Через 6,5 ч реакционную смесь разбавляли водой. Водный слой экстрагировали дихлорметаном. Все соединенные органические слои сушили над Na2SO4, концентрировали и очищали методом хроматографии на силикагеле (градиент: 100% дихлорметана до 30% в дихлорметане) с получением (6aS)-4’,5’-дигидро-4’,5’-эпоксидегелиненольного эфира.

Синтез (6aS, 12aS)-4’,5’-дигидро-4’[18F]-фтор-5’-гидроксидегелина
В тонкостенный силанизированный контейнер с вакуумом объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% об/вес раствор в воде) и раствор18F- в воде (10 микроКюри, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушили путем повторяющегося добавления и выпаривания CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали смесь под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), сосуд открывали и одной порцией добавляли (6aS)-4’,5’-дигидро-4’,5’-экоксидегелиненольного эфира (2 мг). Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения до комнатной температуры медленно добавляли раствор трифторуксусной кислоты (500 мл) и воду (300 мл). Реакционный сосуд закрывали и оставляли на 2 мин при 60ºС. После охлаждения до комнатной температуры содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light C-18 Sep-Pak). Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и остаток очищали методом ЖХВР с получением чистого (6aS, 12aS)-4’,5’-дигидро-4’[18F]-5’-гидроксидегелина.
Синтез (6aS, 12aS)-2-0-дезметилдегелина

(6aS, 12aS)-дегелин (251 мг, 0,638 ммол) и метантиолят натрия (12 мг, 1,78 ммол) растворяли в 4 мл N,N-диметилацетамида и нагревали 2 ч при 80ºС. Реакционную смесь разбавляли до 50 мл водой и экстрагировали дихлорметаном. Водный слой подкисляли 5% HCl и снова экстрагировали дихлорметаном. Все органические слои сушили над Na2SO4, концентрировали и очищали методом хроматографии на силикагеле (100% дихлорметана - 30% ацетона в дихлорметане) с получением (6aS, 12aS)-2-0-дезметилдегелина.
Синтез (6aS, 12aS)-2-[18F] фторметоксидегелина
[18F]F получали облучением [18О] воды (>94 ат.%; 400 мкл) в серебряных камерах с 17 meV протонами из AVF циклотрона 103 см. Типичное облучение проводили в течение 45 мин пучком тока 10 мА с выходом примерно 18 ГБк [18F] фторида. После облучения воду передают через силиконовую трубку в аппарат для синтеза. Этот аппарат состоит из сосуда, выполненного из боросиликата (5 мл), который содержит карбонат калия (5 мг, 36 мкмол) и К2.2.2 (18 мг, 48 мкмол) в ацетонитриле (1 мл).

Воду выпаривали при пониженном давлении и в токе Не. При 110ºС добавляли три части ацетонитрила. Камере давали охладиться до комнатной температуры и к сухой смеси18F/К2.2.2 добавляли дибромметан (50 мкл) в ацетонитриле (1 мл). Реакционную смесь снова нагревали при 110ºС и летучие передавали на колонку препаративной Г-Х при помощи Не в качестве носителя. Колонку нагревали до 100ºС и [18F] CH2BrF отделяли от растворителей и других реагентов.
Полученный [18F] CH2BrF добавляли в сосуд, содержащий (6aS, 12aS)-2-O-дезметилдегелин (2 мг) в ацетонитриле (150 мкл). Сосуд закрывали и нагревали 30 мин при 65ºС. После охлаждения содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light С-18 Sep-Pak). Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и очищали остаток методом ЖХВР с получением чистого (6aS, 12aS)-2-[18F] фторметоксидегелина на носителе.
Синтез (6aS, 12aS)-2-[18F]фторэтоксидегелина

Толуолсульфонилхлорид (38,3 г, 0,201 мол) и пиридин (15,9 г, 0,201 мол) добавляли к раствору этан-1,2- диола (5 г, 0,081 мол) в дихлорметане (100 мл) при 0ºС. После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение ночи. Утром смесь разбавляли водой. Водный слой экстрагировали дихлорметаном, сушили над Na2SO4 и концентрировали. Сырой продукт очищали методом хроматографии на силикагеле (4:1 гексаны:этилацетат - 100% этилацетат) с получением дитозилэтана с хорошим выходом.
В тонкостенный силанизированный контейнер с вакуумом объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (8,5 мкл, 40% вес/об раствор в воде) и раствор18F- в воде (10 мкК, 340 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушили путем повторяющегося добавления и выпаривания CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали под вакуумом без нагревания. Перед полным удалением растворителя добавляли ТГФ (150 мкл), сосуд открывали и одной порцией добавляли 1,2 - дитолуолсульфонатэтан (3,4 мг). Сосуд снова закрывали и нагревали при 85ºС в течение 30 мин. После охлаждения до комнатной температуры растворитель удаляли при пониженном давлении с получением предшественника [18F] фторэтилтозилата (2,0 мг, 0,010 ммол). (6aS, 12aS)-2-O-дезметилдегелин (3,8 мг, 0,010 ммол) и тетрабутиламмония гидроокись (2,6 мг, 0,010 ммол) добавляли в ДМФ (0,25 мл) и реакционную смесь нагревали снова до 60ºС. Через 15 мин смесь охлаждали до комнатной температуры, разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light С-18 Sep-Pak) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и после ЖХВР получали чистый (6aS, 12aS)-2-[18F]-фторэтоксидегелин на носителе.
Синтез (6aS)-4’,5’-дигидро-5’- гидроксидегелиненольного эфира

(6aS) - 4’,5’-дигидро-4’,5’-эпоксидегелиненольный эфир (1,0 г, 2,35 ммол) растворяют в ТГФ (20 мл) и охлаждали до 0ºС. К перемешиваемому раствору по каплям добавляли литийалюминий гидрид (2,35 мл 1 М раствора в ТГФ). После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение ночи. Утром реакцию обрывали водой. Водный слой экстрагировали этилацетатом. Все органические слои сушили над Na2SO4, концентрировали и очищали методом хроматографии на силикагеле (100% дихлорметан - 30% ацетона в дихлорметане) с получение (6aS)-4’,5’-дигидро-5’-гидроксидегелиненольного эфира.

Синтез (6aS)-4’,5’-дигидро-5’-толуолсульфонилдегелиненольного эфира
К перемешиваемому раствору (6aS)-4’,5’-дигидро-5’-гидроксидегелиненольного эфира (31 мг, 0,073 ммол) в дихлорметане (1,5 мл) добавляли TsCl (15,3 мг, 0,080 ммол) и пиридин (6,47 мкл, 0,080 ммол). После окончания добавления смесь перемешивали при комнатной температуре. Через 28 ч реакция завершалась (данные LCMS) и смесь концентрировали. Сырой продукт очищали методом хроматографии на силикагеле (градиент от 100% дихлорметана до 25% ацетона в дихлорметане) с получением (6aS)-4’,5’-дигидро-5’- толуолсульфонилдегелиненольного эфира.
Синтез (6aS, 12aS)-4’,5’- дигидро-5’-[18F]-фтордегелина

В тонкостенный силанизированный контейнер с вакуумом объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% об/вес раствор в воде) и раствор18F- в воде (10 микрокюри, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушили путем повторяющегося добавления и выпаривания CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали смесь под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), сосуд открывали и одной порцией добавляли (6aS)-4’,5’-дигидро-5’-толуолсульфонилдегелиненольного эфира (2 м). Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения до комнатной температуры медленно добавляли раствор трифторуксусной кислоты (500 мкл) и воду (300 мкл). Реакционный сосуд закрывали и оставляли стоять 2 мин при 60ºС. После охлаждения до комнатной температуры содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загружен Waters Light С-18 Sep-Pak) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и остаток очищали методом ЖХВР с получением чистого (6aS, 12aS)-4’,5’-дигидро-5’-[F]-фтордегелина на носителе.
Синтез (6aS)-4’,5’-дигидро-5’-карбонилдегелиненольного эфира

(6aS)-4’,5’-дигидро-5’-гидроксидегелиненольный эфир (1,0 г, 2,3 ммол) растворяли в дихлорметане (20 мл) и добавляли к раствору РСС (0,51 г, 2,3 ммол) в дихлорметане (20 мл). После перемешивания при комнатной температуре в течение 2 ч реакционную смесь фильтровали через целит и концентрировали. Сырой продукт очищали методом хроматографии на силикагеле (100% дихлорметана - 90% ацетона в дихлорметане) с получением (6aS)-4’,5’-дигидро-5’- карбонилдегелиненольного эфира.
Синтез (6aS)-5’-триметилстаннилдегелиненольного эфира

К раствору 2, 4, 6 - триизопропилбензосульфонилгидразида (33,0 г, 0,10 мол) в ACN (100 мл) добавляли (6aS)-4’,5’-дигидро-5’-карбонилдегелиненольный эфир (42,4 г, 0,10 мол) и 10 мл концентрированной соляной кислоты. Раствор перемешивали при комнатной температуре и затем охлаждали до 0ºС в течение 4 ч. Тризилгидразоновое производное собирали в виде твердого продукта.
Раствор тризилгидразонового производного (38,3 ммол, 22,67 г) в 200 мл TMEDA - гексанов (1:1) обрабатывали точно 2,0 экв. втор. бутиллития/циклогексана (76,6 ммол втор - BuLi, -80ºС) и давали нагреться до -10ºС до прекращения выделения N2 (40 мин). Одной порцией добавляли раствор свежесублимированного хлорида триметилолова (50 ммол, 9,97 г, 1,3 экв) в 30 мл гексана. После обработки водой и последующей дистилляции через аппарат с коротким путем пробега при пониженном давлении получали (6aS)-5’- триметилстаннилдегелиненольный эфир.
Синтез (6aS, 12aS)-5’-[18F]-фтордегелина

В тонкостенный силанизированный контейнер с вакуумом объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% об/вес раствор в воде) и раствор18F- в воде (10 микрокюри, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушили путем повторяющегося добавления и выпаривания CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали смесь под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), сосуд открывали и одной порцией добавляли (6aS)-5’-триметилстаннилдегелиненольный эфир (2 мг). Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения до комнатной температуры медленно добавляли раствор трифторуксусной кислоты (500 мкл) и воду (300 мкл). Реакционный сосуд закрывали и оставляли стоять 2 мин при 60ºС. После охлаждения до комнатной температуры содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загружен Waters Light С-18 Sep-Pak) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и остаток очищали методом ЖХВР с получением чистого (6aS, 12aS)-5’-[18F]-фтордегелина.
Синтез (6aS)-4’,5’-дигидро-4’-гидроксидегелиненольного эфира
(6aS)-дегелиненольный эфир (155,0 мг, 0,38 ммол) и катехинборан (0,40 мл 1,0 М раствора в ТГФ, 0,40 ммол) добавляли к раствору катализатора А (0,003 г, 1 мол. %) в ТГФ (0,5 мл). Катализатор А получали способом, описанным в WO 95/13284. Смесь перемешивали в атмосфере азота в течение 2 ч, затем реакцию обрывали EtOH (0,5 мл), NaOH (2,0 М в воде, 0,5 мл) и перекисью водорода (30% в воде, 0,5 мл) при перемешивании еще в течение 2 ч. Реакционную смесь экстрагировали диэтиловым эфиром. Органический слой промывали 1,0 М NaOH, сушили над Na2SO4 и очищали методом хроматографии на силикагеле (100% дихлорметана - 30% ацетона в дихлорметане) с получением (6aS)-4’,5’-дигидро-4’-гидроксидегелиненольного эфира.
Синтез (6aS)-4’,5’-дигидро-4’-карбонилдегелиненольного эфира
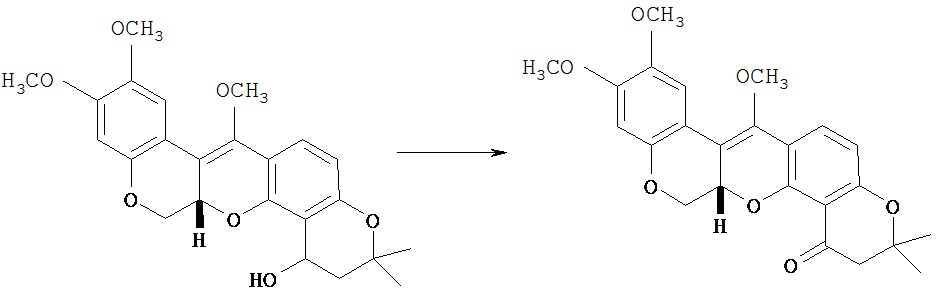
(6aS)-4’,5’-дигидро-5’-гидроксидегелиненольный эфир (1,0 г, 2,3 ммол) растворяли в дихлорметане (20 мл) добавляли к раствору РСС (0,51 г, 2,3 ммол) в дихлорметане (20 мл). После перемешивания при комнатной температуре в течение 2 ч реакционную смесь отфильтровывали через целит и концентрировали. Сырой продукт очищали методом хроматографии на силикагеле (100% дихлорметане - 30% ацетона в дихлорметане) с получением (6aS)-4’,5’-дигидро-4’-карбонилдегелиненольного эфира. Синтез (6aS)-4’-триметилстаннилдегелиненольного эфира

К раствору 2, 4, 6 - триизопропилбензосульфонилгидразида (33,0 г, 0,10 мол) в ацетонитриле (100 мл) добавляли (6aS)-4’,5’-дигидро- 4’-карбонилдегелиненольный эфир (42,4 г, 0,10 мол) и 10 мл концентрированной соляной кислоты. Раствор перемешивали при комнатной температуре и затем охлаждали до 0ºС в течение 4 ч. Тризилгидразоновое производное собирали в виде твердого продукта.
Раствор тризилгидразонового производного (38,3 ммол, 22,67 г) в 200 мл TMEDA - гексанов (1:1) обрабатывали точно 2,0 экв. втор. бутиллития/циклогексана (76,6 ммол втор - BuLi, -80ºС) и давали нагреться до -10ºС до прекращения выделения N2 (40 мин). Одной порцией добавляли раствор свежесублимированного хлорида триметилолова (50 ммол, 9,97 г, 1,3 экв) в 30 мл гексана. После обработки водой и последующей дистилляции через аппарат с коротким путем пробега при пониженном давлении получали (6aS)-4’-триметилстаннилдегелиненольный эфир. Синтез (6aS, 12aS)-4’-[18F]-фтордегелина

В тонкостенный силанизированный контейнер с вакуумом объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% об/вес раствор в воде) и раствор18F- в воде (10 микрокюри, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушили путем повторяющегося добавления и выпаривания CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали смесь под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), сосуд открывали и одной порцией добавляли (6aS)-5’-триметилстаннилдегелиненольный эфир (2 мл). Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения до комнатной температуры медленно добавляли раствор трифторуксусной кислоты (500 мкл) и воду (300 мкл). Реакционный сосуд закрывали и оставляли стоять 2 мин при 60ºС. После охлаждения до комнатной температуры содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загружен Waters Light С-18 Sep-Pak) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и остаток очищали методом ЖХВР с получением чистого (6aS, 12aS)-4’-[18F]-фтордегелина на носителе.
Синтез 2,4-дигидрокси-6-нитробензальдегида

2,4-диметокси-6-нитробензальдегид (135 мг, 0,638 ммол) и метантиолат натрия (125 мг, 1,78 ммол) растворяли в 4 мл N,N-диметилацетамида и нагревали при 80ºС в течение 26 ч. Реакционную смесь разбавляли до 50 мл водой и экстрагировали дихлорметаном. Затем водный слой подкисляли 5% HCl и снова экстрагировали дихлорметаном. Все органические слои сушили над Na2SO4, концентрировали и очищали методом хроматографии на силикагеле (100% дихлорметана - 30% ацетона в дихлорметане) с получением 2,4-дигидрокси-6-нитро-бензальдегида.
Синтез 2,4-дигидрокси-5-нитро-бензальдегида

2,4-диметокси-5-нитро-бензальдегид (135 мг, 0,638 ммол) и метантиолат натрия (125 мг, 1,78 ммол) растворяли в 4 мл N, N - диметилацетамида и нагревали при 80ºС в течение 26 ч. Реакционную смесь разбавляли до 50 мл водой и экстрагировали дихлорметаном. Затем водный слой подкисляли 5% HCl и снова экстрагировали дихлорметаном. Все органические слои сушили над Na2SO4, концентрировали и очищали методом хроматографии на силикагеле (100% дихлорметана - 30% ацетона в дихлорметане) с получением 2,4-дигидрокси-5-нитро-бензальдегида.

Синтез 5-гидрокси-2,2-диметил-8-нитро-2Н-хромей-6-карбальдегида
Раствор 2,4-дигидрокси-5-нитробензальдегида (10,61 г, 58 ммол) в Me2CO (6 мл) добавляли в течение 5,5 ч к перемешиваемому раствору 3-метил-бут-2-еналя (4,00 г, 29 ммол) в пиридине (2,29 г, 2,34 мл, 29 ммол) при 120ºС. После окончания добавления нагревание продолжали в течение 18 ч. Ме2СО выпаривали и удаляли пиридин азеотропной отгонкой с толуолом, получая сырой продукт. Сырой продукт очищали методом хроматографии на силикагеле с применением 1% этилацетата в гексанах с получением 5-гидрокси-2,2-диметил-8-нитро-2Н-хромен-6-карбальдегида.
Синтез 5-гидрокси-2,2-диметил-7нитро-2Н-хромен-6-карбальдегида
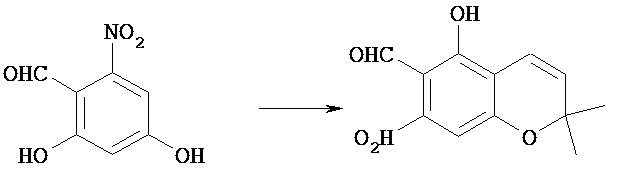
Раствор 2,4-дигидрокси-6-нитро-бензальдегида (10,61 г, 58 ммол) в Ме2СО (6 мл) добавляли в течение 5,5 ч к перемешиваемому раствору 3-метил-бут-2-еналя (4,00 г, 29 ммол) в пиридине (2,29 г, 2,34 мл, 29 ммол) при 120ºС. После окончания добавления нагревание продолжали в течение 18 ч. Ме2СО выпаривали и удаляли пиридин азеотропной отгонкой с толуолом, получая сырой продукт. Сырой продукт очищали методом хроматографии на силикагеле с применением 1% этилацетата в гексанах с получением 5-гидрокси-2,2-диметил-7-нитро-2Н-хромен-6-карбальдегида.
Синтез 5-метокси-2,2-диметил-7-нитро-2Н-хромен-6-карбальдегида

Смесь 5-гидрокси-2,2-диметил-7-нитро-2Н-хромен-6-карбальдегида (2,34 г, 10 ммол), К2СО3 (4,12 г, 29,8 ммол) и MeI (2,13 г, 0,94 мл, 15 ммол) в Ме2СО (40 мл) нагревали с обратным холодильником в течение 4 ч и перемешивали при комнатной температуре в течение ночи. Смесь концентрировали, обрабатывали водой (15 мл) и экстрагировали дихлорметаном. Соединенные органические слои промывали водой, сушили над Na2SO4 и удаляли растворитель в вакууме, получая масло, которое очищали методом хроматографии (3% Ме2СО3 в гексане) с получением 5-метокси-2,2-диметил-7-нитро-2Н-хромен-6-карбальдегида.
Синтез 5-метокси-2,2-диметил-8-нитро-2Н-хромей-6-карбальдегида

Смесь 5-гидрокси-2,2-диметил-8-нитро-2Н-хромен-6-карбальдегида (2,34 г, 10 ммол), К2СО3 (4,12 г, 29,8 ммол) и MeI (2,13 г, 0,94 мл, 15 ммол) в Ме2СО (40 мл) нагревали с обратным холодильником в течение 4 ч и перемешивали при комнатной температуре в течение ночи. Смесь концентрировали, обрабатывали водой (15 мл) и экстрагировали дихлорметаном. Соединенные органические слои промывали водой, сушили над Na2S04 и удаляли растворитель в вакууме, получая масло, которое очищали методом хроматографии (3% Ме2СО3 в гексане) с получением 5-метокси-2,2-диметил-8-нитро-2Н-хромен-6-карбальдегида.
Синтез 4-бут-2-инилокси-1,2-диметоксибензола

К 3,4-диметоксифенолу (15,4 г, 0,1 мол) в ДМФ (100 мл) добавляли пропаргилбромид (14,15 г, 0,12 мол) и карбонат калия (11,88 г, 0,12 мол). Смесь перемешивали при комнатной температуре в течение 12 ч и добавляли насыщенный NH2Cl и диэтиловый эфир. Органические слои промывали водой, рассолом и сушили над Na2SO4. Сырой продукт фильтруют через прокладку из двуокиси кремния (1:1 гексаны:дихлорметан) с получением 4-бут-2-инилокси-1,2-диметоксибензола в виде масла желтого цвета.
Синтез 4-(3,4-диметоксифенокси)-1-(5-метокси-2,2-диметил-8-нитро-2Н-хромен-6-ил)-бут-2-ин-1-она

К раствору 4-бут-2-инилокси-1,2-диметоксибензола (1,66 г, 8,66 ммол) в ТГФ (75 мл) добавляли н-бутиллитий (5,54 мл 1,6 М раствора в ТГФ, 8,86 ммол) при -78ºС. Через 30 мин добавляли 5-метокси-2,2-диметил-8-нитро-2Н-хромен-6-карбальдегид (2,17 г, 8,25 ммол) в ТГФ (50 мл). Смесь перемешивали в течение 1 ч и затем добавляли насыщенный NH4Cl и этилацетат. Органические слои промывали рассолом и сушили над Na2SO4. Полученный сырок продукт растворяли в дихлорметане (20 мл) и добавляли MnO2 (5,3 г, 61 ммол). После перемешивания смеси при комнатной температуре в течение ночи добавляли простой эфир и фильтровали суспензию через целит и силикагель с получением 4-(3,4-диметоксифенокси)-1-(5-метокси-2,2-диметил-8-нитро-2Н-хромен-6-ил)-бут-2-ин-1-она.
Синтез 4-(3,4-диметоксифенокси)-1-(5-метокси-2,2-диметил-7-нитро-2Н-хромен-6-ил)-бут-2-ин-1-она

К раствору 4-бут-2-инилокси-1,2-диметоксибензола (1,66 г, 8,66 ммол) в ТГФ (75 мл) добавляли н-бутиллитий (5,54 мл 1,6 М раствора в ТГФ, 8,86 ммол) при -78ºС. Через 30 мин добавляли 5-метокси-2,2-диметил-7-нитро-2Н-хромен-6-карбальдегид (2,17 г, 8,25 ммол) в ТГФ (50 мл). Смесь перемешивали в течение 1 ч и затем добавляли насыщенный NH4Cl и этилацетат. Органические слои промывали рассолом и сушили над Na2SO4. Полученный сырок продукт растворяли в дихлорметане (20 мл) и добавляли MnO2 (5,3 г, 61 ммол). После перемешивания смеси при комнатной температуре в течение ночи добавляли простой эфир и фильтровали суспензию через целит и силикагель с получением 4-(3,4-диметоксифенокси)-1-(5-метокси-2,2-диметил-7-нитро-2Н-хромен-6-ил)-бут-2-ин-1-она.
Синтез (6,7-диметокси-2Н-хромай-3-ил)-(5-метокси-2,2-диметил-7-нитро

В круглодонную колбу объемом 10 мл, высушенную пламенем, добавляли 4-(3,4-диметоксифенилокси)-1-(5 метокси-2,2-диметил-7-нитро-2Н-хромен-6 ил)-бут-2-ин-1-он (61,6 мг, 0,135 ммол) и PtCl2 (1,8 мг, 5 мол. %). Колбу вакуумировали и промывали три раза аргоном с последующим добавлением толуола (1,8 мл, 0,1 м). Реакционную смесь перемешивали при 55ºС в течение 10 ч и затем концентрировали. Сырой продукт очищали методом хроматографии на силикагеле (7:3 гексаны:этилацетат) с получением (6,7-диметокси-2Н-хроман-3-ил)-(5-метокси-2,2-диметил-7-нитро-2Н-хромен-6-ил)-метанона.

Синтез (6,7-диметокси-2Н-хроман-3-ил)-(5-метокси-2,2-диметил-8- нитро-2Н-хромен-6-ил)-метанона
В круглодонную колбу объемом 10 мл, высушенную пламенем, добавляли 4-(3,4-диметоксифенилокси)-1-(5 метокси-2,2-диметил-8- нитро-2Н-хромен-6 ил)-бут-2-ин-1-он (61,6 мг, 0,135 ммол) и PtCl2 (1,8 мг, 5 мол. %). Колбу вакуумировали и промывали три раза аргоном с последующим добавлением толуола (1,8 мл, 0,1 м). Реакционную смесь перемешивали при 55ºС в течение 10 ч и затем концентрировали. Сырой продукт очищали методом хроматографии на силикагеле (7:3 гексаны:этилацетат) с получением (6,7-диметокси-2Н-хроман-3-ил)-(5-метокси-2,2-диметил-8-нитро-2Н-хромен-6-ил)-метанона.
Синтез (+/-)-10-нитродегелина
В круглодонную колбу объемом 10 мл, высушенную пламенем, добавляли (6,7-диметокси-2Н-хроман-3-ил)-(5-метокси-2,2-диметил-8-нитро-2Н-хромен-6-ил)-метанон (50,2 мг, 0,111 ммол) и дихлорметан (2,0 мл). Раствор охлаждали до -78ºС и добавляли трихлорид бора (0,133 мл, 1 М раствор в дихлорметане, 0,133 ммол). После перемешивания в течение 1 ч реакцию обрывали насыщенным NH4Cl, экстрагировали

этилацетатом, сушили над Na2SO4 и концентрировали. Сырой продукт растворяли в EtOH, насыщали ацетатом калия и нагревали с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры к реакционной смеси добавляли этилацетат и воду. Водный слой экстрагировали этилацетатом. Соединенные органические слои промывали

рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт фильтровали через двуокись кремния (3:1 гексаны:этилацетат) с получением (+/-)-10-нитродегелина.
Синтез (+/-)-11-нитродегелина
В круглодонную колбу объемом 10 мл, высушенную пламенем, добавляли (6,7-диметокси-2Н-хроман-3-ил)-(5-метокси-2,2-диметил -7-нитро-2Н-хромен-6-ил)-метанон (50,2 мг, 0,111 ммол) и дихлорметан (2,0 мл). Раствор охлаждали до -78ºС и добавляли трихлорид бора (0,133 мл, 1 М раствор в дихлорметане, 0,133 ммол). После перемешивания в течение 1 ч реакцию обрывали насыщенным NH4Cl, экстрагировали этилацетатом, сушили над Na2S04 и концентрировали. Сырой продукт растворяли в EtOH, насыщали ацетатом калия и нагревали с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры к реакционной смеси добавляли этилацетат и воду. Водный слой экстрагировали этилацетатом. Соединенные органические слои промывали рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт фильтровали через двуокись кремния (3:1 гексаны:этилацетат) с получением (+/-)-11-нитродегелина.
Синтез (+/-)-11-[18F]-фтордегелина
В тонкостенный силанизированный вакуумный контейнер объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% вес/об раствор в воде) и раствор18F- в воде (10 мкК, 200 мкл).

Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушат путем повторяющегося добавления и испарения CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), открывали сосуд и добавляли одной порцией (+/-)-11-нитродегелин (2 мг). Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения до комнатной температуры содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light С-18 Sep-Рак) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и очищали остаток методом ЖХВР с получением чистого не содержащего носителя (+/-)-11-[18F]-фтордегелина.
Синтез (+/-)-10-[18F]-фтордегелина

В тонкостенный силанизированный вакуумный контейнер объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% вес/об раствор в воде) и раствор18F- в воде (10 мкК, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушат путем повторяющегося добавления и испарения CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), открывали сосуд и добавляли одной порцией (+/-)-10-нитродегелин (2 мг). Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения до комнатной температуры содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light С-18 Sep-Pak) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и очищали остаток методом ЖХВР с получением чистого не содержащего носителя (+/-)-10-[18F]-фтордегелина.
Пример 2 - Аналоги тебуфенпирада
Синтез 5-N-(4-трет.бутилбензил)-карбоксамидо-3-(метоксикарбонил)-1-метилпиразола

Смесь 3-(метоксикарбонил)-1-метил-5-карбоновой кислоты (20 ммол) и тионилхлорида (30 ммол) нагревали с обратным холодильником в течение 30 мин. Избыток тионилхлорида удаляли под вакуумом и остаток сушили отгонкой в виде азеотропа с сухим бензолом. Полученный сырой ацилхлорид растворяли в ТГФ (10 мл) и перемешивали, охлаждая при 0ºС, добавляя по каплям раствор 4-трет.бутилбензиламина (22 ммол) и диизопропилэтиламина (25 ммол) в ТГФ (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и нагревали кратковременно до кипения с обратным холодильником для завершения реакции. Смесь охлаждали и выливали в ледяную воду (100 мл) и экстрагировали простым эфиром (3×100 мл). Соединенные органические слои высушивали (насыщенный водный раствор NaCl, Na2SO4), отфильтровывали и концентрировали. После очистки остатка методом флэш-хроматографии (силикагель, градиент элюирования 0-20% этилацетата/гексаны) с получением 5-N-(4-трет.бутилбензил)-карбоксамидо-3-(метоксикарбонил)-1-метилпиразола.
Синтез метил-5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолилкарбоксилата

Раствор 5-N-(4-трет.бутилбензил)карбоксамидо-3-(метоксикарбонил)-1-метилпиразола (0,1 мол) и тионилхлорида (0,13 мол) в 1,2-дихлорэтане (15 мл) нагревали с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали и концентрировали в вакууме. Остаток распределяли между дихлорметаном (100 мл) и насыщенным водным раствором NaHCO3 (100 мл) при рН водной фазы >7. Водный слой отделяли и экстрагировали дихлорметаном (2×100 мл) и соединенные органические слои высушивали (насыщенный водный раствор NaCl, Na2SO4), фильтровали и концентрировали. Перекристаллизацией остатка (EtOH - вода) получали чистый метил-5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолилкарбоксилат.
Синтез 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолил-карбоновой кислоты

Раствор 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолил-карбоксилата (50 ммол) в диоксане (33 мл) и воде (75 мл) обрабатывали раствором Нз804 (конц., 1 мл) в воде (1,5 мл). Полученную смесь нагревали с обратным холодильником до исчерпания исходного материала. Полученную смесь концентрировали в вакууме до точки насыщения (удаление диоксана) и охлаждали при 0ºС в течение ночи. Полученный осадок собирали фильтрованием и высушивали. Фильтрат экстрагировали дихлорметаном (3×100 мл) и соединенные органические слои высушивали (насыщенный водный раствор NaCl, Na2SO4), отфильтровывали и концентрировали. Перекристаллизацией остатка (этилацетат-метанол) получали чистую 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолил-карбоновую кислоту.
Синтез 1-(5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолил)-1-этанона

Раствор 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолил-карбоновой кислоты (20 ммол) в тионилхлориде (30 ммол) нагревали с обратным холодильником в течение 15 мин. Смесь охлаждали и концентрировали в вакууме. Добавляли бензол (10 мл) и удаляли вначале при атмосферном давлении и затем под вакуумом. Полученный ацилхлорид использовали непосредственно на следующей стадии.
В колбу загружали твердую безводную бромистую медь (I) (25 ммол) и промывали аргоном. Добавляли тетрагидрофуран (125 мл). Полученную суспензию охлаждали при -78ºС, добавляя по каплям бромид метилмагния (17,8 мл, 2,9 М в диэтиловом эфире). Смесь перемешивали, охлаждая при -78ºС в течение 20 мин. Полученный хлорангидрид кислоты растворяли в ТГФ (10 мл) и охлаждали до -78ºС. Хлорангидрид медленно при помощи канюли добавляли к купрату при протекании добавляемого раствора по боковой стенке реакционной колбы для охлаждения. Колбу с хлорангидридом промывали ТГФ (5 мл), который снова охлаждали и добавляли при помощи канюли. Баню удаляли и смесь перемешивали при комнатной температуре в течение 30 мин. Для обрыва реакции добавляли метанол и выливали смесь в насыщенный водный раствор NH4Cl (200 мл). Смесь перемешивали в течение 1 ч для растворения солей меди и отделяли органический слой. Водную фазу промывали дихлорметаном (2×200 мл) и соединенные органические слои сушили (насыщенный водный раствор NaCl, Na2SO4), фильтровали и концентрировали. Остаток очищали методом хроматографии (силикагель, градиент элюирования 10-30% этилацетата-гексаны), получая чистый 1-(5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолил)-1-этанон.
Синтез 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-3-(1-гидроксиэтил)-1-метилпиразолина
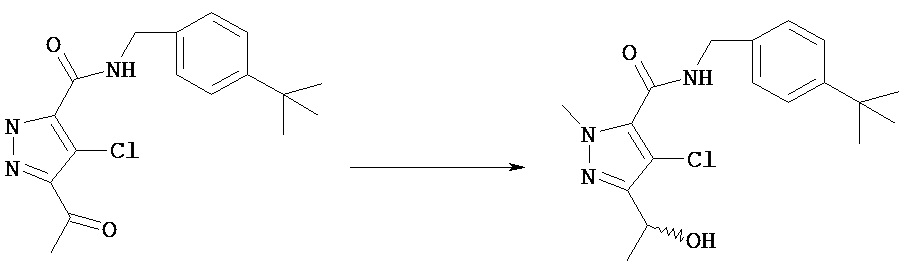
Одной порцией добавляли твердый боргидрид бора (20 ммол) в перемешиваемый раствор 1-(5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-пиразолил)-1-этанона (10 ммол) в этаноле (15 мл) при комнатной температуре. Смесь перемешивали до исчерпания исходного кетона. Если необходимо, добавляют снова боргидрид натрия. Добавляли воду (2 мл), смесь концентрировали, и смесь распределяли между водой (100 мл) и дихлорметаном (2×100 мл). Соединенные органические слои сушили (насыщенный водный раствор NaCl, Na2SO4), фильтровали и концентрировали. Остаток очищали методом хроматографии (силикагель, элюент 10-30% этилацетата-гексаны) с получением 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-3-(1-гидроксиэтил)-1-метилпиразолина.
Синтез 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-(1-п-толуолсульфонатэтил)пиразолина
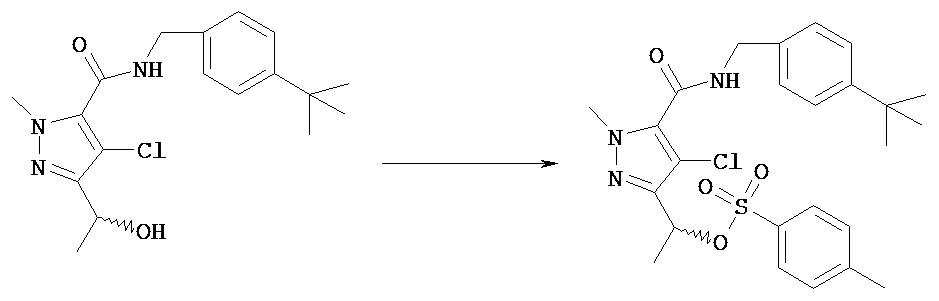
Раствор 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-3-(1-гидроксиэтил)-1-метилпиразолина (5 ммол) и п-толуолсульфонилхлорида (5,5 ммол) в пиридине (12 мл) перемешивали при комнатной температуре в течение 4 ч. Раствор концентрировали и распределяли между водой (100 мл) и дихлорметаном (2×100 мл). Соединенные органические слои сушили (насыщенный водный раствор NaCl, Na2SO4), фильтровали и концентрировали. Остаток очищали методом хроматографии (силикагель, элюент 2-20% этилацетата-гексаны) с получением чистого 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-(1-п-толуолсульфонатэтил)пиразолина.
Синтез 5-N-(4-трет.бутил)бензил-карбоксамидо-4-хлор-1-метил-3- (1-[18F]фторэтил)пиразолина (через тозилат)

В тонкостенный силанизированный вакуумный контейнер объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% вес/об раствор в воде) и раствор18F- в воде (10 мкК, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушат путем повторяющегося добавления и испарения CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), открывали сосуд и одной порцией в твердом виде добавляли чистый 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-(1-п-толуолсульфонатэтил)пиразолин. Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения до комнатной температуры содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light С-18 Sep-Рак) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и очищали остаток методом ЖХВР с получением чистого не содержащего носителя 5-N-(4-трет.бутил)бензил-карбоксамидо-4-хлор-1-метил-3-(1-[18F]фторэтил)пиразолина.
Синтез 5-N-(4-трет.бутил)бензил-карбоксамидо-4-хлор-1-метил-3-(1-метансульфонатэтил)пиразолина.

Раствор 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-3-(1- гидроксиэтил)-1-метилпиразолина (5 ммол) и метансульфонилхлорид (5,5 ммол) в пиридине (12 мл) перемешивали при комнатной температуре в течение 4 ч. Раствор концентрировали и распределяли между водой (100 мл) и дихлорметаном (2×100 мл). Соединенные органические слои сушили (насыщенный водный раствор NaCl, Na2SO4), фильтровали и концентрировали. Остаток очищали методом хроматографии (силикагель, элюент 2-20% этилацетата-гексаны) с получением чистого 5-N-(4-трет.бутил)бензил-карбоксамидо-4-хлор-1-метил-3-(1-метансульфонатэтил)пиразолина.
Синтез 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-(1-[18F]-фторэтил)пиразолина (через мезилат)

В тонкостенный силанизированный вакуумный контейнер объемом 10 мл с силанизированной пробкой загружали гидроокись тетрабутиламмония (5 мкл, 40% вес/об раствор в воде) и раствор18F- в воде (10 мкК, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем сушат путем повторяющегося добавления и испарения CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали под вакуумом без нагревания. До полного удаления растворителя добавляли ТГФ (150 мкл), и добавляли одной порцией твердый 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-(1-метансульфонатэтил) пиразолин. Сосуд снова закрывали и нагревали при 65ºС в течение 30 мин. После охлаждения до комнатной температуры содержимое сосуда разбавляли водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light С-18 Sep-Pak) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и очищали остаток методом ЖХВР с получением чистого не содержащего носителя 5-N-(4-трет.бутил)бензилкарбоксамидо-4-хлор-1-метил-3-(1-[18F]-фторэтил)пиразолина.

Синтез 4-трет.бутил-3-нитробензамида
Смесь 4-трет.бутил-3-нитробензойной кислоты (0,1 М), гидроксибензотриазола (HOBt, 0,12 мол) и дициклогексилкарбодиимида (DCC, 0,11 мол) в дихлорметане (100 мл) перемешивали при комнатной температуре, добавляя быстро раствор аммиака в 2-пропаноле (2,0 М, 75 мл, 0,12 мол). Смесь перемешивали в течение 2 ч при комнатной температуре и выливали в водный раствор NaHCO3 (5%, 200 мл). Слои разделяли и экстрагировали водную фазу дихлорметаном (2×200 мл). Соединенные органические слои промывали (2×200 мл, 5% NaHCO3), сушили (насыщенный водный раствор NaCl, Na2SO4), фильтровали и концентрировали. Продукт перекристаллизовывали из смеси EtOH - вода с получением чистого 4-трет.бутил-3-нитробензамида. Синтез 4-трет.бутил-3-[18F]фторбензиламина

В тонкостенный силанизированный вакуумный контейнер объемом 10 мл, снабженный силанизированной пробкой, загружали гидроокись тетрабутиламмония (5 мкл, 40% вес/об раствор в воде) и раствор18F- в воде (10 мКюри, 200 мкл). Полученную смесь выпаривали досуха в токе азота при 100ºС. Остаток затем высушивали путем повторяющегося добавления и выпаривания CH3CN (3×200 мкл). Добавляли дополнительную аликвоту CH3CN и концентрировали под вакуумом без нагревания. Перед полным удалением растворителя добавляли диоксан (150 мкл), открывали сосуд и одной порцией добавляли твердый 4-трет.бутил-3-нитробензамид (1 мг, около 4,5 мкмол). Сосуд закрывали и нагревали при 100ºС в течение 25 мин. После охлаждения добавляли раствор бис-(тетрагидрофураната)литийалюминийгидрида в толуоле (1,0 М, 50 мкл, 50 мкмол) и смесь нагревали при 50ºС в течение 5 мин. Сосуд охлаждали и разбавляли содержимое водой (4 мл) и пропускали через картридж с силикагелем (предварительно загруженный Waters Light С-18 Sep-Pak) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и остаток очищали методом ЖХВР с получением чистого не содержащего носителя 4-трет.бутил-3-[18F]фторбензиламина. Растворитель выпаривали, и продукт использовали при осуществлении следующего способа.
Синтез 5-N-(4-трет.бутил-3-[18F]фтор)-бензилкарбоксамидо-4-хлор-3-этил-1-метилпиразолина

К перемешиваемой смеси 3-этил-1-метилпиразол-5-карбоновой кислоты (50 мкмол), дициклогексилкарбодиимида (DCC, 50 мкмол, используется как аликвота из исходного раствора в дихлорметане), гидроксибензотриазола (HOBt, 60 мкмол) в метиленхлориде (200 мкл) добавляли раствор 4-трет.бутил-3-[18F]-фторбензиламина (полученного как описано выше) в дихлорметане (100 мкл). Смесь перемешивали при комнатной температуре в течение 10 мин, концентрировали и растворяли в смеси ацетонитрил - вода (1: 4,3 мл). Смесь пропускали через картридж с силикагелем (предварительно загруженный Waters Light С-18 Sep-Pak) для загрузки образца. Картридж промывали водой и элюировали CH3CN (2 мл). Ацетонитрил выпаривали и остаток очищали методом ЖХВР с получением чистого не содержащего носителя 5-N-(4-трет.бутил-3-[18F]фтор)-бензилкарбоксамидо-4-хлор-3-этил-1-метилпиразолина.
Пример 3 - Аналоги пиридабена
Синтез 2-трет.бутил-4,5-дихлор-3(2Н)-пиридазинона

К мукохлорной кислоте (4,0 г, 23,6 ммол) в воде (35 мл) при 0ºС добавляли безводный Na2CO3 (1,21 г, 11,5 ммол). Смесь перемешивали до получения прозрачного раствора и добавляли к нему гидрохлорид трет.бутилгидразина (2,94 г, 23,6 ммол). Осадок начинал образовываться через несколько минут. Реакционную смесь перемешивали еще 2,5 ч, после чего отфильтровывали. Желтый осадок промывали холодной водой и сушили с получением 4,81 г сырого гидразона.
К 4,32 г сырого гидразона добавляли 40 мл уксусной кислоты и раствор нагревали с обратным холодильником в течение 25 мин. Затем раствор охлаждали и концентрировали. Полученный продукт обрабатывали дихлорметаном и промывали 1 М раствором карбоната натрия и водой. Органический слой сушили и концентрировали с получением твердого продукта желтого цвета, который очищали методом хроматографии на колонке, используя в качестве элюента смесь гексаны:хлороформ (от 1:1 до 0:100). Получали 2,4 г желательного соединения.
Синтез 2-трет.бутил-4 хлор-5-тио-3(2Н)-пиридазинона

К 0,5 г 2-трет.бутил-4,5-дихлор-3(2Н)-пиридазинона добавляли 7 мл воды и сульфид натрия (0,53 г, 6,81 ммол) и нагревали смесь до 80ºС, пока не растворился твердый продукт. Раствор охлаждали до комнатной температуры и аккуратно добавляли концентрированную HCl с получением осадка желтого цвета, который отфильтровывали и промывали холодной водой. После кристаллизации из гексанов получали продукт в виде твердого вещества белого цвета (270 мг).

Синтез 2-трет.бутил-4-хлор-5-(4-трет.бутилбензил)-тио-3(2Н)-пиридазинона
К 220 мг 2-трет.бутил-4-хлор-5-тио-3(2Н)-пиридазинона в 4 мл ДМФ добавляли 4-трет.бутилбензилбромид (226 мг, 1 ммол) и Na2CO3. Реакционную смесь перемешивали в течение 16 ч при комнатной температуре и затем экстрагировали этилацетатом, промывали водой и очищали методом хроматографии (силикагель этилацетат/гексаны). Получали желательное соединение.
Синтез 2-трет.бутил-4-фтор-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинона
В круглодонную колбу помещали 2-трет.бутил-4-хлор-5(4-трет.бутилбензил)-тио-3(2Н)-пиридазинон (100 мг, 0,27 ммол) и добавляли фторид калия (23,4 мг, 0,40 ммол) и 2 мл диметилсульфоксида. Смесь нагревали до 120ºС в течение 6 ч. Затем реакционную смесь выливали в воду и проводили экстрагирование этилацетатом. Продукт промывали водой и сушили. После очистки методом флэш-хроматографии (силикагель; этилацетат/гексаны) получали желательное соединение.

Синтез 2-трет.бутил-4-(18F]фтор-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинона
В реакционную колбу объемом 5 мл, содержащую 500 мКюри18F в 350 мг18О воды, добавляли 1 мл раствора, содержащего 10 мг Kryptofix, 1 мг карбоната калия, 0,005 мл воды и 0,95 мл ацетонитрила. Колбу нагревали для удаления всех растворителей и добавляли сухой ацетонитрил (1 мл). Этот растворитель также удаляли выпариванием. Затем добавляли 2-трет.бутил-4-хлор-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинон (5 мг) в ацетонитриле. Сосуд герметизировали и нагревали при 100ºС в течение 30 мин. Смесь разбавляли дихлорметаном и пропускали через Sep-Pak и элюировали тетрагидрофураном. Растворитель выпаривали, получая желательное соединение.
Синтез 4-(4-метилфенил)бутанола

К литийалюминийгидриду (427 мг, 11,2 ммол), суспендированному в сухом простом эфире (5 мл) при 0ºС добавляли 1 г 4-(4-метилфенил) бутановую кислоту (5,614 ммол), растворенную в сухом эфире (10 мл) в течение 30 мин. Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 ч. Затем последовательно добавляли воду (0,43 мл), NaOH (15% раствор, 0,43 г) и воду (1,29 мл) и полученный раствор перемешивали в течение 30 мин. Осадок отфильтровывали и промывали эфиром и сушили. Затем продукт концентрировали и очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны).
Синтез 4-(4-метилфенил)бутил-трет.бутилдиметилсилилового эфира

4-(4-метилфенил)бутанол (0,5 г, 3,04 ммол) растворяли в 5 мл ДМФ и добавляли имидазол (310 мг, 4,56 ммол) и трет.бутилдиметилсилилхлорид (685 мг, 4,56 ммол). Смесь перемешивали в течение 4 ч, затем экстрагировали этилацетатом и промывали водой для удаления всего ДМФ. Затем органический слой высушивали и концентрировали. Сырой продукт затем очищали методом флэш-хроматографии, используя в качестве элюента смесь этилацетата с гексанами, и получали желательный продукт.
Синтез 4-(4-бромметилфенил)бутил-трет.бутилдиметилсилилового эфира
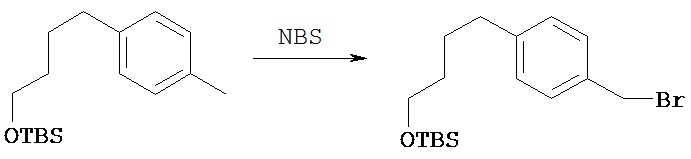
В кругло донную колбу объемом 50 мл добавляли 4-(4-метилфенил)бутил-трет.бутилдиметилсилиловый эфир (0,25 г, 0,89 ммол), N-бромсукцинимид (0,158 г, 0,89 ммол), перекись бензоила (2,17 мг, 0,0089 ммол) и 10 мл четыреххлористого углерода. Эту смесь нагревали с обратным холодильником в течение ночи, затем охлаждали и фильтровали. Фильтрат концентрировали и полученный сырой остаток очищали методом флэш-хроматографии при помощи смеси этилацетат - гексаны, получая нужный продукт.
Синтез 2-трет.бутил-4-хлор-5-(4-(4-трет.бутилдиметилсилилоксибутил)бензил)-тио-3-(2Н)-пиридазинона

В колбу, содержащую 2-трет.бутил-4-хлор-5-тио-3(2Н)-пиридазинон (0,2 г, 0,917 ммол), добавляли 5 мл ДМФ и затем карбонат цезия (0,358 мг, 1,1 ммол) и 4-(4-бромметилфенил)-бутил-трет.бутилдиметилсилиловый эфир (0,391 г, 1,1 ммол). Смесь нагревали до 60ºС в течение 2 ч и затем охлаждали, экстрагировали этилацетатом, промывали, сушили и концентрировали. Сырой продукт затем очищали методом хроматографии с применением силикагеля и смеси этилацетат - гексаны в качестве элюента. Получали нужный продукт.
Синтез 2-трет.бутил-4-хлор-5-(4-(4-гидроксибутил)бензил)-тио-3(2Н)-пиридазинона

К 0,2 г 2-трет.бутил-4-хлор-5-(4-(4-трет.бутилдиметилсилилоксибутил)-бензил)-тио-3(2Н)-пиридазинона (0,404 ммол) добавляли 5 мл 1% концентрированной HCl в этаноле.

Реакционную смесь перемешивали в течение 30 мин, затем экстрагировали этилацетатом, промывали водой и высушивали. Очистка (силикагель; EtOAc/гексаны) сырого продукта, полученного после концентрации, привела к получению нужного продукта.
Синтез 2-трет.бутил-4-хлор-5(4-(4-толуолсульфонилоксибутил)-бензил)тио-3(2Н)-пиридазинона
В круглодонную колбу объемом 15 мл, содержащую 2-трет.бутил-4-хлор-5-(4-(4-гидроксибутил)бензил)-тио-3(2Н)-пиридазинон (0,15 г, 0,39 ммол) добавляли пиридин. Затем туда же добавляли толуолсульфонилхлорид (88,9 мг, 0,42 ммол) и перемешивали смесь в течение 2 ч. Смесь разбавляли этилацетатом, промывали 5% раствором сульфата меди и затем водой и высушивали. После удаления растворителя в роторном испарителе сырой продукт очищали методом флэш-хроматографии, используя в качестве элюента смесь этилацетат - гексаны.
Синтез 2-трет.бутил-4-хлор-5-(4-(4-фторбутил)бензил)тио-3(2Н)-пиридазинона

В круглодонную колбу загружали 2-трет.бутил-4-хлор-5-(4-(4-толуолсульфонилоксибутил)бензил)тио-3(2Н)-пиридазинон (0,05 г, 0,093 ммол) и добавляли фторид тетрабутиламмония (1,0 М раствор в ТГФ, 0,93 мкл, 0,93 ммол) и затем 0,2 мл ТГФ. Смесь нагревали до 60ºС и перемешивали при этой температуре в течение 30 мин. Затем смесь охлаждали и концентрировали и сырой продукт очищали методом флэш-хроматографии для получения нужного соединения.
Синтез 2-трет.бутил-4-хлор-5-(4-(4-[18F]-фторбутил)бензил)тио-3(2Н)-пиридазинона

Водный18F (16 мКюри, 0,1 мл) добавляли в вакуумный сосуд, содержащий 5 мкл гидроокиси тетрабутиламмония (40% вес. раств. в воде). Смесь концентрировали в атмосфере азота в масляной бане и добавляли 250 мкл ацетонитрила и эту смесь тоже концентрировали в атмосфере азота. Затем добавляли 100 мкл ТГФ и 5 мг 2-трет.бутил-4-хлор-5-(4-(4-толуолсульфонилоксибутил)бензил)тио-3(2Н)-пиридазинона. Эту смесь затем нагревали на масляной бане при 70ºС в течение 30 мин. Полученный продукт затем разбавляли водой, использовали С18 Sep-Pak и ацетонитрил в качестве элюента и получали нужное соединение.
Синтез (4-трет.бутилфенил)этан-1,2-диола
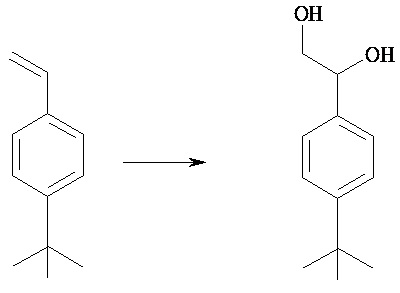
В круглодонную колбу объемом 100 мл добавляли 20 мл трет.бутанола, 20 мл воды и 5,6 г AD - mix - β. Раствор перемешивали и охлаждали до 0ºС. К смеси добавляли трет.бутилстирол (0,64 г, 4 ммол) и полученный раствор перемешивали в течение ночи при 0ºС. Добавляли твердый сульфит натрия (6 г) и перемешивали смесь еще 30 мин. Затем раствор экстрагировали этилацетатом, промывали водой и сушили. Сырой продукт очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны) и получали нужное соединение.
Синтез 1-трет.бутилдиметилсилилокси-2-гидрокси-2-(4-трет.бутилфенил)этана

(4-трет.бутилфенил)этан-1,2-диол (0,5 г, 2,57 ммол) растворяли в ДМФ в круглодонной колбе объемом 25 мл и добавляли имидазол (0,210 г, 3,09 ммол) и трет.бутилдиметилсилилхлорид (0,46 г, 3,09 ммол). Смесь перемешивали в течение 6 ч, затем экстрагировали дихлорметаном и промывали органический слой водой и сушили. После очистки методом флэш-хроматографии (силикагель; этилацетат/гексаны) получали нужный продукт.
Синтез 2-трет.бутил-4-хлор-5-(2-трет.бутилдиметилсилилокси-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинона

К раствору 2-трет.бутил-4,5-дихлор-3(2Н)-пиридазинона (0,5 г, 2,27 ммол) в ДМФ (10 мл) добавляли безводный карбонат цезия (0,74 г, 2,27 ммол) и 1-трет.бутил-диметилсилилокси-2-гидрокси-2-(4-трет.бутилфенил) этан (0,7 г, 2,27 ммол). Смесь перемешивали 2 ч при 70ºС и затем охлаждали до комнатной температуры и добавляли этилацетат. Затем раствор промывали водой, сушили и концентрировали, остаток подвергали очистке методом флэш-хроматографии (силикагель; этилацетат/гексаны) и получали нужный продукт.
Синтез 2-трет.бутил-4-хлор-5-(2-гидрокси-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинона

В круглодонную колбу объемом 25 мл загружали 2-трет.бутил-4-хлор-5-(2-трет.бутилдиметилсилилокси-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинон (0,5 г, 1,01 ммол) и добавляли 5 мл 1% конц. HCl в этаноле. Раствор перемешивали в течение 1 ч, затем выливали в воду и экстрагировали этилацетатом. Этилацетат удаляли в роторном испарителе, остаток подвергали флэш-хроматографии, используя силикагель и смесь этилацетат/гексаны в качестве элюента.
Синтез 2-трет.бутил-4-хлор-5-(2-п-толуолсульфонилокси-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинона

В круглодонную колбу объемом 15 мл, содержащую 2-трет.бутил-4-хлор-5-(2-гидрокси-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинон (0,25 г, 0,66 ммол), добавляли пиридин. Затем добавляли толуолсульфонилхлорид (0,15 г, 0,79 ммол) и перемешивали 4 ч. Реакционную смесь разбавляли этилацетатом, промывали 5% раствором сульфата меди, затем водой и сушили. Этилацетат удаляли в роторном испарителе, остаток подвергали флэш-хроматографии, используя силикагель и смесь этилацетат/гексаны в качестве элюента.
Синтез 2-трет.бутил-4-хлор-5-(2-фтор-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинона
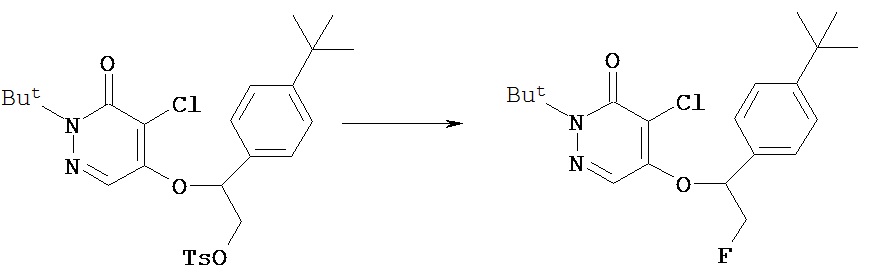
В круглодонную колбу объемом 15 мл, содержащую 2-трет.бутил-4-хлор-5-(2-п-толуолсульфонилокси-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинон (0,2 г, 0,375 ммол) добавляли 3,75 мл раствора фторида тетрабутиламмония (1 М в ТГФ, 3,75 ммол). Смесь вначале перемешивали 15 мин при комнатной температуре, затем нагревали 15 мин при 100ºС. Затем охлаждали раствор до комнатной температуры и добавляли к нему дихлорметан и затем воду. Слои разделяли, органический слой промывали водой и затем сушили. Органический слой затем концентрировали и подвергали очистке с применением флэш-хроматографии на силикагеле (этилацетат/гексаны) с получением нужного соединения.
Синтез 2-трет.бутил-4-хлор-5-(2-[18F]-фтор-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинона

Водный18F (16 мКюри, 0,1 мл) добавляли в вакуумный сосуд, содержащий 5 мкл гидроокиси тетрабутиламмония (40% вес. раствор в воде). Смесь концентрировали в атмосфере азота на масляной бане и добавляли 250 мкл ацетонитрила и концентрировали эту смесь в атмосфере азота. Затем добавляли 100 мкл ТГФ и затем 5 мг 2-трет.бутил-4-хлор-5-(2-п-толуолсульфонилокси-1-(4-трет.бутилфенил)-1-этил)-окси-3(2Н)-пиридазинона. Затем смесь нагревали на масляной бане при 70ºС в течение 30 мин. Затем смесь разбавляли водой и применяли С18 Sep-Pak и ацетонитрил в качестве элюента, получали нужное соединение.
Синтез 2-трет.бутил-4-метил-5-хлор-3(2Н)-пиридазинона

2-трет.бутил-4,5-дихлор-3(2Н)-пиридазинон (5 г, 22,72 ммол). Растворенный в 12 мл простого эфира, по каплям добавляли к 15 мл эфирного раствора бромида метилмагния (3 М в эфире) при 5ºС. После окончания добавления раствор перемешивали при 5ºС в течение 2 ч. Затем медленно добавляли 10 мл 6 N раствора HCl и раствор перемешивали в течение 10 мин. Затем смесь экстрагировали диэтиловым эфиром. Эфирный слой затем промывали водой и сушили. Сырой продукт после концентрирования эфира подвергали флэш-хроматографии (силикагель; этилацетат/гексаны 9:1) с получением нужного продукта.
Синтез 2-трет.бутил-4-бромметил-5-хлор-3(2Н)-пиридазинона

2-трет.бутил-4-метил-5-хлор-3(2Н)-пиридазинон (3 г, 15 ммол) растворяли в 25 мл четыреххлористого углерода и добавляли N - бромсукцинимид (2,6 г, 15 ммол) и перекись бензоила (14 мг). Смесь затем нагревали с обратным холодильником в течение 6 ч, затем охлаждали и фильтровали. Фильтрат промывали водой и сушили. После удаления органического растворителя сырой остаток очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны 9:1) с получением нужного продукта.
Синтез 2-трет.бутил-4-гидроксиметил-5-хлор-3(2Н)-пиридазинона

2-трет.бутил-4-бромметил-5-хлор-3(2Н)-пиридазинон (2 г, 7,19 ммол) и карбонат кальция (3,5 г) добавляли к смеси 1:1 диоксан - вода (40 мл). Смесь нагревали с обратным холодильником в течение 6 ч, затем добавляли 30 мл 3 N раствора HCl. Раствор перемешивали в течение 10 мин, затем диоксан удаляли при пониженном давлении. Полученный раствор затем экстрагировали дихлорметаном, слой дихлорметана промывали и сушили. Сырой продукт, полученный после концентрации, очищали методом флэш-хроматографии (этилацетат/гексаны 1:2).
Синтез 2-трет.бутил-4-трет.бутилдиметилсилилоксиметил-5-хлор-3(2Н)-пиридазинона
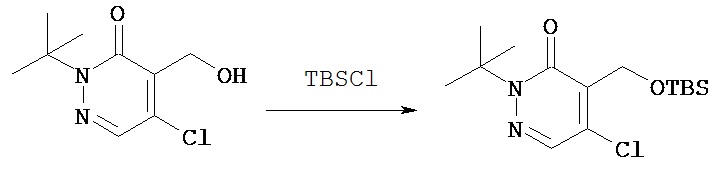
2-трет.бутил-4-гидроксиметил-5-хлор-3(2Н)-пиридазинон (1 г, 4,62 ммол) растворяли в ДМФ в круглодонной колбе объемом 25 мл и добавляли имидазол (0,377 г, 5,0 ммол) и трет.бутилдиметилсилилхлорид (0,762 г, 3,09 ммол). Смесь перемешивали в течение 10 ч, затем экстрагировали дихлорметаном, органический слой промывали водой и сушили. После очистки методом флэш-хроматографии (силикагель; этилацетат/гексаны) получали нужный продукт.
Синтез 2-трет.бутил-4-трет.бутилдиметилсилилоксиметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинона

К раствору 2-трет.бутил-4-трет.бутилдиметилсилилоксиметил-5-хлор-3(2Н)-пиридазинона (1,5 г, 4,54 ммол) в ДМФ (10 мл) добавляли безводный карбонат цезия (2,9 г, 9,09 ммол) и 4-трет.бутилбензилмеркаптан (1,02 г, 4,54 ммол). Смесь перемешивали 2 ч при 70ºС и затем охлаждали до комнатной температуры и добавляли этилацетат. Затем раствор промывали водой, сушили и концентрировали, остаток очищали флэш-хроматографией (силикагель; этилацетат/гексаны) с получением нужного соединения.
Синтез 2-трет.бутил-4-гидроксиметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинона
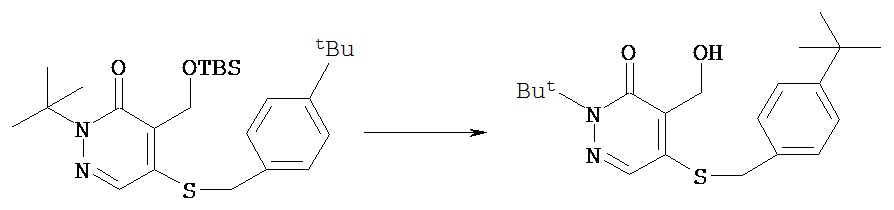
В круглодонную колбу объемом 15 мл загружали 2-трет.бутил-4-трет.бутилдиметилсилилоксиметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинон (2 г, 4,2 ммол) и добавляли раствор фторида тетрабутиламмония (1 М в ТГФ, 21 мл, 21 ммол). Смесь сначала перемешивали при комнатной температуре в течение 5 ч и добавляли дихлорметан и затем воду. Слои разделяли и органический слой промывали водой и сушили. Органический слой затем концентрировали и очищали флэш-хроматографией на силикагеле (этилацетат/гексаны) с получением нужного соединения.
Синтез 2-трет.бутил-4-п-толуолсульфонилоксиметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинона

В круглодонную колбу объемом 15 мл загружали 2-трет.бутил-4-гидроксиметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинон (1,0 г, 2,77 ммол) и добавляли пиридин. Затем добавляли п-толуолсульфонилхлорид (0,79 г, 4,15 ммол) и смесь перемешивали в течение 4 ч. Реакционную смесь разбавляли этилацетатом, промывали 5% раствором сульфата меди и затем водой и сушили. После удаления растворителя в роторном испарителе сырой продукт очищали флэш-хроматографией с применением силикагеля и смеси этилацетат/гексаны в качестве элюента и получали нужное соединение.
Синтез 2-трет.бутил-4-фторметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинона

В круглодонную колбу объемом 15 мл, содержащую 2-трет.бутил-4-п-толуолсульфонилоксиметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинон (0,5 г, 0,972 ммол) добавляли 4,86 мл раствора фторида тетрабутиламмония (1 М в ТГФ, 4,86 ммол). Смесь сначала перемешивали при комнатной температуре в течении 15 мин, затем нагревали в течение 15 мин при 100ºС. Затем раствор охлаждали до комнатной температуры, добавляли дихлорметан и затем воду. Слои разделяли и органический слой промывали водой и затем сушили. Затем органический слой концентрировали и подвергали очистке, применяя флэш-хроматографию на силикагеле (этилацетат/гексаны), с получением нужного соединения.
Синтез 2-трет.бутил-4-[18F]фторметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинона

Водный18F (50 мкКюри, 0,1 мл) добавляли в вакуумный сосуд, содержащий 5 мкл гидроокиси тетрабутиламмония (40% вес.раствор в воде). Смесь концентрировали в атмосфере азота в масляной бане, добавляли 250 мкл ацетонитрила и эту смесь тоже концентрировали в атмосфере азота. Затем добавляли 100 мкл ТГФ и затем 5 мг 2-трет.бутил-4-п-толуолсульфонилоксиметил-5-(4-трет.бутилбензил)тио-3(2Н)-пиридазинон. Затем смесь нагревали на масляной бане при 70ºС в течение 30 мин. Смесь затем разбавляли водой, использовали С18 Sep-Pak и ацетонитрил в качестве элюента и получали нужное соединение.
Пример 4 - Аналоги феназаквина
Синтез 4-хлор-хиназолина

4-хиназолон (5 г, 34,2 ммол), пентахлорид фосфора (10,26 г, 47,9 ммол) и оксихлорид фосфора (40 мл) нагревали с обратным холодильником 2 ч при 115-118ºС. Оксихлорид фосфора удаляли в вакууме и остаток экстрагировали простым эфиром. Всю смесь выливали в сосуд, содержащий колотый лед, и экстрагировали простым эфиром. Эфирный слой промывали бикарбонатом натрия и сушили. Эфир удаляли при пониженном давлении и сырой продукт перекристаллизовывали из гексанов, получая нужный продукт.
Синтез 4-(4-метилфенил)бутанола
К литийалюминийгидриду (427 мг, 11,2 ммол), суспендированному в сухом эфире (5 мл) при 0ºС добавляли 1 г 4-(4-метилфенил) бутановую кислоту (5,614 ммол), растворенную в сухом эфире (10 мл) в течение 30 мин. Затем реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 4 ч. Затем последовательно добавляли воду (0,43 мл), NaOH (15% раствор, 0,43 г) и воду и полученный раствор перемешивали в течение 30 мин. Полученный осадок фильтровали и промывали эфиром и сушили. Фильтрат концентрировали и очищали флэш-роматографией, используя в качестве элюента смесь этилацетат - гексаны.
Синтез 4-(4-метилфенил) бутил-трет.бутил-диметилсилилового эфира

4-(4-метилфенил)бутанол (0,5 г, 3,04 ммол) растворяли в 5 мл ДМФ и добавляли имидазол (310 мг, 4,56 ммол) и трет.бутилдиметилсилилхлорид (685 мг, 4,56 ммол). Реакционную смесь перемешивали в течение 4 ч, затем экстрагировали этилацетатом и промывали водой для удаления всего ДМФ. Органический слой сушили и концентрировали. Сырой продукт очищали затем флэш-хроматографией, используя смесь этилацетат - гексаны в качестве элюента, получали нужный продукт.
Синтез 4-(4-бромметилфенил)бутил-трет.бутилдиметилсилилового эфира
В круглодонную колбу объемом 50 мл загружали 4-(4-метилфенил)бутил-трет.бутилдиметилсилиловый эфир (0,25 г, 0,89 ммол), N-бромсукцинимид (0,158 г, 0,89 ммол), перекись бензоила (2,17 мг, 0,0089 ммол) и 10 мл четыреххлористого углерода. Эту смесь нагревали с обратным холодильником в течение ночи, затем охлаждали и фильтровали. Фильтрат концентрировали и полученный сырой остаток очищали флэш-хроматографией с применением смеси этилацетат - гексаны, получая нужный продукт.
Синтез 4-(4-трет.бутилдиметилсилилоксибутил)фенилуксусной кислоты

4-(4-бромметилфенил)бутил-трет.бутилдиметилсилиловый эфир (0,2 г, 0,561 ммол) в сухом эфире по каплям добавляли к стружке Mg (13,77 мг, 0,561 ммол). Затем для начала реакции добавляли несколько кристаллов йода и смесь нагревали с обратным холодильником в атмосфере азота в течение ночи. Раствор охлаждали и пропускали через него газообразный СО2 в течение 10 мин. Перемешивание продолжали еще 2 ч и затем к реакционной смеси добавляли воду. Затем смесь экстрагировали этилацетатом, промывали и сушили. После удаления органического растворителя при пониженном давлении сырой продукт очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны) и получали нужный продукт.
Синтез 2-гидроксиэтил-4-(4-трет.бутилдиметилсилилоксибутил)бензола

4-(4-трет.бутилдиметилсилилоксибутил)-фенилуксусную кислоту (0,25 г, 0,775 ммол) растворяли в сухом эфире (44,2 мг, 1,16 ммол). Реакционную смесь перемешивали в течение 5 ч, после чего последовательно добавляли воду (45 мкл), NaOH (15% раствор, 45 мкл) и воду, затем смесь перемешивали еще 30 мин. Полученный осадок фильтровали и промывали эфиром. Эфирный фильтрат промывали водой и сушили. После концентрирования эфира полученный продукт очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны).
Синтез 4-(2-(4-(4-трет.бутилдиметилсилилоксибутил)фенил)этокси)-хиназолина

2-гидроксиэтил-4(4-трет.бутилдиметилсилилоксибутил)бензол (0,3 г, 0,97 ммол) растворяли в сухом тетрагидрофуране и добавляли гидрид натрия (24 мг, 1 ммол). Полученный раствор перемешивали при комнатной температуре в течение 30 мин, затем к этому раствору добавляли 4-хлорхиназолин (0,164 г, 1 ммол). Раствор далее перемешивали в течение 6 ч, затем к смеси добавляли воду. Раствор экстрагировали дихлорметаном. Органический слой промывали, сушили и концентрировали с получением сырого продукта, который очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны) с получением нужного продукта.
Синтез 4-(2-(4-(4-гидроксибутил)фенил)этокси)хиназолина

К 4-(2-(4-(4-трет.бутилдиметилсилилоксибутил)фенил)этокси)хиназолину (0,4 г, 0,916 ммол) добавляли раствор фторида тетрабутиламмония (1 М TBAF в ТГФ, 4,58 мл, 4,58 ммол). Раствор перемешивали в течение 2 ч, затем добавляли воду и экстрагировали продукт этилацетатом. Органический слой затем промывали водой, сушили и концентрировали. Полученный остаток очищали флэш-хроматографией (силикагель; этилацетат/гексаны).
Синтез 4-(2-(4-(4-п-толуолсульфонилоксибутил)фенил)этокси)-хиназолина

В круглодонную колбу объемом 15 мл, содержащую 4-(2-(4-(-гидроксибутил)фенил)-этокси)хиназолин (0,25 г, 0,77 ммол), растворенный в пиридине (5 мл). Затем добавляли п-толуолсульфонилхлорид (0,15 г, 0,79 ммол) и перемешивали смесь в течение 4 ч. Реакционную смесь разбавляли этилацетатом, промывали 5% раствором сульфата меди, затем водой и высушивали. После удаления растворителя в роторном испарителе сырой продукт очищали флэш-хроматографией с применением силикагеля (этилацетат/гексаны) и получали нужный продукт.
Синтез 4-(2-(4-(4-фторбутил)фенил)этокси)хиназолина
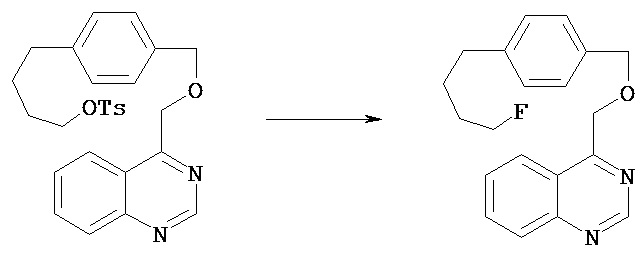
4-(2-(4-(4-п-толуолсульфонилоксибутил)фенил)этокси)хиназолин (0,3 г, 0,63 ммол) добавляли к раствору фторида калия / Kryptofix 222 в 5 мл ТГФ (отношение 1:1, по 3,15 ммол каждого). После перемешивания при комнатной температуре в течение 15 мин раствор нагревали с обратным холодильником в течение 20 мин. Затем смесь охлаждали и добавляли воду. Затем осуществляли экстракцию дихлорметаном и промывали водой и сушили полученный продукт. Этот продукт очищали методом флэш-хроматографии на силикагеле (этилацетат/гексаны) и получали нужный продукт.
Синтез 4-(2-(4-(4-[18F]-фторбутил)фенил)этокси)хиназолина

В реакционный сосуд объемом 5 мл, содержащий 100 мкКюри18F в 300 мг18О воды добавляли 1 мл раствора, содержащего 10 мг Kryptofix, 1 мг карбоната калия, 0,005 мл воды и 0,95 мл ацетонитрила. Сосуд нагревали для удаления всех растворителей и добавляли в сосуд сухой ацетонитрил (1 мл). Последний также удаляли выпариванием и затем добавляли 4-(2-(4-(4-п-толуолсульфонилоксибутил)фенил)этокси)хиназолин (5 мг) в ацетонитриле. Сосуд герметизировали и нагревали в течение 30 мин при 100ºС. Смесь разбавляли дихлорметаном и пропускали через Sep-Pak и элюировали тетрагидрофураном. Растворитель выпаривали, получая нужное соединение.
Синтез 4-хлор-2-хиназолона

2-цианфенилизоцианат (5 г, 34,7 ммол) суспендировали в ди-н-бутиловом эфире. Затем при 80ºС в течение 7 ч пропускали через суспензию газообразный HCl. Полученный осадок фильтровали, сушили и перекристаллизовывали из хлорбензола, получали нужный продукт.
Синтез 4-(2-(4-трет.бутилфенил)этокси)-2-хиназолона

2-(4-трет.бутилфенил) этанол (0,3 г, 1,68 ммол) растворяли в сухом тетрагидрофуране (7 мл) и добавляли гидрид натрия (48,5 мг, 2,02 ммол). Полученный раствор перемешивали при комнатной температуре в течение 30 мин, затем к этому раствору добавляли 4-хлор-2-хиназолон (0,302 г, 1,68 ммол). Затем раствор перемешивали в течение 6 ч, затем добавляли воду. После этого проводили экстракцию дихлорметаном. Органический слой промывали, сушили и концентрировали с получением сырого продукта, который очищали флэш-хроматографией (силикагель; этилацетат/гексаны) с получением нужного продукта.
Синтез 4-(2-(4-трет.бутилфенил)этокси)-2(трифторметансульфонилокси)хиназолина
4-(2-(4-трет.бутилфенил)этокси)-2-хиназолон (0,25 г, 0,775 мол) растворяли в дихлорметане (5 мл) и добавляли трифторметансульфоновый ангидрид (0,328 г, 1,16 ммол) и диизопропилэтиламин (0,3 г, 2,32 ммол). Смесь перемешивали в течение ночи, затем разбавляли ее дихлорметаном и промывали водой. Органический слой сушили и концентрировали. Сырой продукт выделяли методом флэш-хроматографии (силикагель; этилацетат/гексаны).
Синтез 4-(2-(4-трет.бутилфенил)-этокси)-2-фтор-хиназолина

В круглодонную колбу объемом 15 мл загружали 4-(2-(4-трет.бутилфенил)этокси)-2-(трифторметансульфонилокси)хиназолин (0,3 г, 0,66 ммол). Затем добавляли раствор фторида тетрабутиламмония (1 М в ТГФ, 3,3 мл, 3,3 ммол) и раствор нагревали с обратным холодильником в течение 60 мин. Смесь затем охлаждали и добавляли воду. Затем проводили экстракцию дихлорметаном, промывали водой и сушили. Полученный после концентрирования сырой продукт очищали путем флэш-хроматографии на силикагеле (этилацетат/гексаны) с получением нужного продукта.
Синтез 4-(2-(4-трет.бутилфенил)-этокси)-2-[18F]-фтор-хиназолина

Водный 18F (16 мкКюри, 0,1 мл) добавляли в вакуумный сосуд, содержащий 5 мкл гидоокиси тетрабутиламмония (40% вес. раствор в воде). Смесь концентрировали в атмосфере азота на масляной бане при 100ºС и добавляли 250 мкл ацетонитрила и затем снова концентрировали смесь в атмосфере азота. Эту процедуру повторяли дважды, затем добавляли 100 мкл ацетонитрила и вакуумировали содержимое сосуда. Затем добавляли сухой ТГФ и затем 5 мг 4-(2-(4-трет.бутилфенил)-этокси)-2-(трифторметансульфонилокси)-хиназолина. Затем смесь нагревали на масляной бане при 70ºС в течение 30 мин. Затем смесь разбавляли водой, пропускали через С18 Sep-Pak, промывали водой и элюировали ацетонитрилом, получая нужное соединение.
Синтез 6-нитро-4(3Н)-хиназолона

Смесь 5-нитроантраниловой кислоты (2 г, 14,6 ммол) и формамида (2,9 мл, 72 ммол) облучали при 150ºС в микроволновой печи (мощность 60 В) до завершения реакции по данным ТСХ (20 мин). После охлаждения реакционную смесь промывали этилацетатом и выпаривали при пониженном давлении. Сырой продукт очищали флэш-хроматографией (силикагель; этилацетат/гексаны) с получением нужного продукта.
Синтез 6-нитро-4-хлорхинолина

6-нитро-4(3Н)-хиназолон (1 г, 5,23 ммол) и РОС1з (7,1 мл) смешивали и облучали при 100ºС (мощность 70 В) в течение 10 мин. POCl3 выпаривали в вакууме и растворяли остаток в этилацетате и промывали насыщенным NaHCO3, сушили и концентрировали. Полученный продукт очищали флэш-хроматографией (силикагель; этилацетат/гексаны) с получением нужного соединения.
Синтез 6-нитро-4-(2-(4-трет.бутилфенил)-этокси)-хиназолина

2-(4-трет.бутилфенил)этанол (1,0 г, 5,59 ммол) растворяли в сухом тетрагидрофуране (7 мл) и добавляли гидрид натрия (48,5 мг, 2,02 ммол). Полученный раствор перемешивали при комнатной температуре в течение 30 мин, затем к этому раствору добавляли 6-нитро-4-хлорхиназолин (1,17 г, 5,6 ммол). Затем раствор перемешивали в течение 6 ч, затем к смеси добавляли воду. После этого раствор обрабатывали дихлорметаном. Органический слой промывали, сушили и концентрировали с получением сырого продукта, который очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны), получали нужное соединение.
Синтез 6-фтор-4-(2-(4-трет.бутилфенил)этокси)хиназолина

В круглодонную колбу объемом 25 мл добавляли фторид калия (82,6 мг, 1,42 ммол) и Kryptofix 222 (0,53 г, 1,42 ммол). Смесь перемешивали в среде ТГФ в течение 20 мин, затем добавляли 6-нитро-4-(2-(4-трет.бутилфенил)этокси)хиназолин (0,1 г, 0,284 ммол). Раствор нагревали с обратным холодильником в течение 30 мин, затем его охлаждали и добавляли воду. Затем проводили экстракцию дихлорметаном, промывали водой и сушили. После очистки методом флэш-хроматографии (силикагель; этилацетат/гексаны) получали нужное соединение.
Синтез 6-[18F]-фтор-4-(2-(4-трет.бутилфенил)этокси)хиназолина

В сосуд объемом 5 мл, содержащий 50 микроКюри18F в 300 мг18О воды добавляли 1 мл раствора, содержащего 10 мг Kryptofix, 1 мг карбоната калия, 0,005 мл воды и 0,95 мл ацетонитрила. Сосуд нагревали для удаления растворителей и добавляли сухой ацетонитрил (1 мл). Последний также удаляли выпариванием. Затем добавляли 6-нитро-4-(2-(4-трет.бутилфенил)этокси)хиназолин (5 мг) в ацетонитриле. Сосуд герметизировали и нагревали 30 мин при 100ºС. Смесь разбавляли дихлорметаном и пропускали через Sep-Pak и элюировали тетрагидрофураном. Растворитель выпаривали и получали нужное соединение.
Синтез (4-трет.бутилфенил)этан-1,2-диола
В круглодонную колбу объемом 100 мл загружали 20 мл трет.бутанола, 20 мл воды и 5,6 г AD - mix - β. Раствор перемешивали и охлаждали до 0ºС. К этой смеси добавляли трет.бутилстирол (0,64 г, 4 ммол) и полученный раствор перемешивали при 0ºС в течение ночи. Добавляли твердый сульфит натрия (6 г) и смесь перемешивали еще в течение 30 мин. Раствор обрабатывали этилацетатом, промывали водой и сушили. Сырой продукт очищали флэш-хроматографией (силикагель; этилацетат/гексаны) и получали нужное соединение.
Синтез 1-трет.бутилдиметилсилилокси-2-гидрокси-2-(4-трет.бутилфенил)этана
(4-трет.бутилфенил)этан-1,2-диол (0,5 г, 2,57 ммол) растворяли в ДМФ в кругло донной колбе объемом 25 мл и добавляли имидазол (0,210 г, 3,09 ммол) и трет.бутилдиметилсилилхлорид (4,46 г, 3,09 ммол). Смесь перемешивали в течение 6 ч, затем экстрагировали дихлорметаном и промывали органический слой водой и высушивали. После очистки методом флэш-хроматографии (силикагель; этилацетат/гексаны) получали нужный продукт.
Синтез 1-трет.бутилдиметилсилилокси-2-тетрагидропиранилокси-2-(4-трет.бутилфенил)этана

1-трет.бутилдиметилсилилокси-2-гидрокси-2-(4-трет.бутилфенил)этан (0,5 г, 1,622 ммол) растворяли в дихлорметане и добавляли дигидропиран (0,163 г, 1,94 ммол) и толуолсульфокислоту (33 мг, 0,194 ммол). Смесь перемешивали в течение 2 ч, затем промывали водой и сушили. Сырой остаток, полученный после концентрирования, очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны) с получением нужного соединения.
Синтез 1-гидрокси-2-тетрагидропиранилокси-2-(4-трет.бутилфенил)этана

К 1-трет.бутилдиметилсилилокси-2-тетрагидропиранилокси-2-(4-трет.бутилфенил)этану (0,4 г, 1,01 ммол) добавляли раствор фторида тетрабутиламмония (1 М TBAF в ТГФ, 5 мл, 5,0 ммол). Раствор перемешивали в течение 2 ч, затем добавляли воду и экстрагировали продукт этилацетатом. Органический слой промывали водой, высушивали и концентрировали. Полученный остаток очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны).
Синтез 4-(2-тетрагидропиранилокси)-2-(4-трет.бутилфенил)этокси-)хиназолина

1-гидрокси-2-тетрагидропиранилокси-2-(4-трет.бутилфенил)этан (0,3 г, 1,07 ммол) растворяли в сухом тетрагидрофуране (7 мл) и добавляли гидрид натрия (30,96 мг, 1,29 ммол). Полученный раствор перемешивали при комнатной температуре в течение 30 мин, затем к этому добавляли 4-хлорхиназолин (0,175 г, 1,07 ммол). Затем этот раствор перемешивали в течение 6 ч, затем к смеси добавляли воду. Раствор экстрагировали дихлорметаном. Органический слой промывали, сушили и затем концентрировали, получая продукт, который очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны), получали нужное соединение.
Синтез 4-(2-гидрокси-2-(4-трет.бутил-фенил)этокси)хиназолина

4-(2-тетрагидропиранилокси)-2-(4-трет.бутилфенил)этокси)хиназолин (0,25 г, 0,615 ммол) растворяли в 5 мл этанола и добавляли п-толуолсульфонат пиридиния (15,4 мг, 0,061 ммол). Раствор нагревали до 55ºС и перемешивали при этой температуре в течение 4 ч. Этанол удаляли и сырой продукт очищали методом флэш-хроматографии (силикагель; этилацетат/гексаны).
Синтез 4-(2-п-толуолсульфонилокси-2-(4-трет.бутилфенил)этокси)-хиназолина

В круглодонную колбу объемом 15 мл загружали 4-(2-гидрокси-2-(4-трет.бутилфенил)этокси)хиназолин (0,25 г, 0,77 ммол), растворенный в пиридине (5 мл). Затем добавляли п-толуолсульфонилхлорид (0,15 г, 0,79 ммол) и смесь перемешивали в течение 4 ч. реакционную смесь разбавляли этилацетатом, затем промывали 5% раствором сульфата меди, затем водой и сушили. После удаления растворителя в роторном испарителе сырой продукт очищали методом флэш-хроматографии, используя силикагель (этилацетат/гексаны).
Синтез 4-(2-фтор-2-(4-трет.бутилфенил)этокси)хиназолина

В круглодонную колбу объемом 15 мл загружали 4-(2-п-толуолсульфонилокси-2-(4-трет.бутилфенил)этокси)хиназолин (0,3 г, 0,84 ммол). Затем добавляли раствор фторида тетрабутиламмония (1 М в ТГФ, 4,2 мл, 4,2 ммол) и нагревали с обратным холодильником в течение 60 мин. Затем смесь охлаждали и добавляли к ней воду. Затем проводили экстракцию дихлорметаном, промывали водой и сушили. Сырой продукт после концентрирования очищали методом флэш-хроматографии на силикагеле (этилацетат/гексаны) с получением нужного соединения.
Синтез 4-(2-[18F]-фтор-2-(4-трет.бутилфенил)этокси)хиназолина

Водный18F (16 мкКюри, 0,1 мл) добавляли в вакуумный сосуд, содержащий 5 мкл гидроокиси тетрабутиламмония (40% вес. раствор в воде). Смесь концентрировали в атмосфере азота на масляной бане при 100ºС и добавляли 250 мкл ацетонитрила и концентрировали снова эту смесь в атмосфере азота. Процедуру повторяли дважды, добавляли 100 мкл ацетонитрила и содержимое вакуумировали. Затем добавляли сухой ТГФ и затем 5 мг 4-(2-п-толуолсульфонилокси-2-(4-трет.бутилфенил)этокси)хиназолина. Затем смесь нагревали на масляной бане при 70ºС в течение 30 мин. Затем смесь разбавляли водой, применяли Sep-Pak, промывали водой и элюировали ацетонитрилом, получали нужное соединение.
Специалистам очевидно, что данное изобретение не ограничено приведенными иллюстративными примерами, оно может быть воплощено в других конкретных формах. Поэтому желательно, чтобы эти примеры рассматривались во всех аспектах как иллюстративные, но не ограничивающие и внимание должно уделяться формуле изобретения в большей степени, чем этим примерам, и чтобы все изменения с учетом смысла и эквивалентов признаков, приведенных в формуле изобретения, рассматривались как охваченные данным изобретением.
Реферат
Изобретение относится к области медицины, а именно к контрастному агенту структурной формулы:где J представляет собой О; Y представляет собой углерод; K и L независимо выбраны из водорода и C-Салкила; М выбран из водорода, C-Салкилокси или C-Салкила, незамещенных или замещенныхF; Т и U вместе с атомами углерода, к которым они присоединены, образуют шестичленное ароматическое кольцо, которое незамещено или замещеноF; R, Rи Rнезависимо выбраны из водорода и фрагментаF; R, R, Rи Rявляются водородом; n=2; причем по меньшей мере одинF присутствует в структуре указанного контрастного агента и является фрагментом, обеспечивающим изображение. Изобретение также относится к применению указанного контрастного агента, композиции или диагностического набора на его основе для выявления, визуализации и/или мониторинга перфузии миокарда. Группа изобретений обеспечивает расширение арсенала диагностических маркеров для получения изображения кровоснабжения миокарда. 5 н. и 4 з.п. ф-лы, 55 пр.
Формула

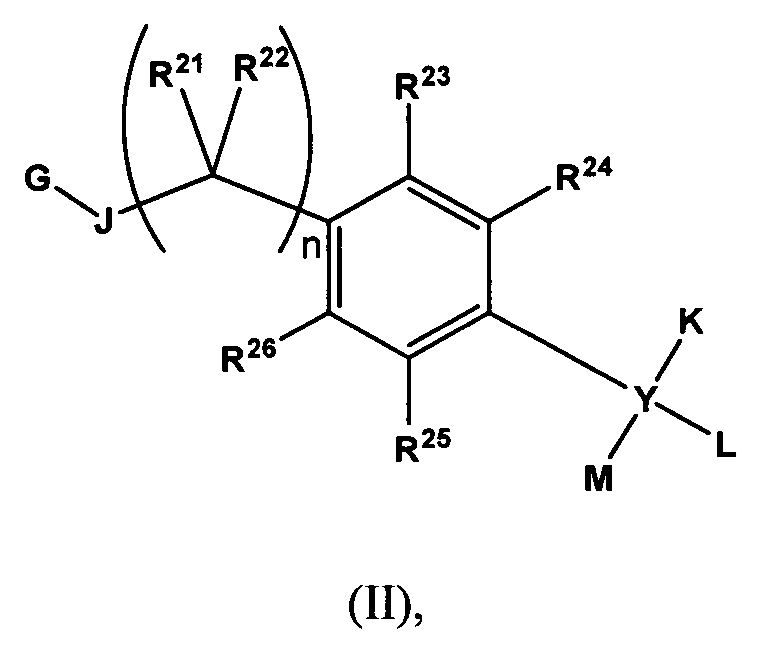






Комментарии