Способ получения радиоактивного фтор-меченного органического соединения - RU2434846C2
Код документа: RU2434846C2
Чертежи



Описание
Область техники
Настоящее изобретение относится к способу получения радиоактивного фтор-меченного органического соединения. Более конкретно, изобретение относится к способу получения радиоактивного фторорганического соединения, пригодного при обнаружении опухолей позитронно-эмиссионной томографией.
Предшествующий уровень техники
Исследование ядерной медицины, представленное позитронно-эмиссионной томографией (далее в настоящем документе обозначается как РЕТ) и однофотонной эмиссионной компьютерной томографией (далее в настоящем документе обозначается как SPECT), является эффективным при диагностировании разнообразия заболеваний, включая заболевание сердца и рак. Эти способы включают введение агента, помеченного определенным радиоизотопом (далее в настоящем документе обозначается как радиофармацевтическое средство), пациенту с последующим обнаружением γ-лучей, излучаемых непосредственно или косвенно из агента. Исследование ядерной медицины характеризуется тем, что оно имеет не только высокую специфичность и чувствительность к заболеваниям, но также преимущество предоставления информации о функциональных свойствах повреждений, сравнимую с другими способами исследования.
Например, [18F] 2-фтор-дезокси-D-глюкоза (далее в настоящем документе обозначается как «[18F]-FDG») одно из радиофармацевтических средств, используемых для РЕТ исследования, может концентрироваться в областях, где метаболизм глюкозы является усиленным, делая возможным, таким образом, конкретно определить опухоли, в которых метаболизм глюкозы является усиленным.
Исследование ядерной медицины выполняют измерением радиоактивным способом распределения введенного радиофармацевтического средства, и данные, полученные в результате исследования, изменяются в зависимости от природы радиофармацевтического средства. Таким образом, были разработаны различные радиофармацевтические средства для различных заболеваний и некоторые из них положены в клиническое применение. Например, были разработаны разнообразные диагностические агенты для опухолей, диагностические агенты кровотока и рецепторные картирующие агенты.
В последние годы серии радиоактивных галоген-меченных аминокислотных соединений, включая [18F] 1-амино-3-фторциклобутанкарбоновую кислоту (далее в настоящем документе обозначается как [18F]FACBC), были задуманы в качестве новых радиофармацевтических средств и их клиническое применение находится на изучении (патентный документ 1 и непатентные документы 1 и 2). [18F]FACBC считается эффективной в качестве диагностического агента для высокопролиферативных опухолей, поскольку она имеет свойство быть специфически поглощенной аминокислотным переносчиком.
В качестве способов получения [18F]FACBC раскрываются способы, которые включают: обеспечение сложного эфира 1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1 карбоновой кислоты в качестве меченого предшественника, замещение трифлатной группы в положении 3 предшественника радиоактивным фтором и выполнение реакций элиминирования сложноэфирной группы и Boc группы путем подвергания полученного соединения в форме раствора кислым условиям (патентный документ 1 и непатентные документы 1 и 2).
Для получения [18F]-FDG раскрыт способ синтеза, где стадию снятия защитной группы выполняют в твердой фазе, который дает возможность сократить время синтеза, уменьшенное число реагентов и уменьшенное число компонентов в аппарате получения (патентный документ 2).
Патентный документ 1: опубликованная японская патентная заявка №2000-500442.
Патентный документ 2: опубликованная японская патентная заявка №11-508923.
Непатентный документ 1: Jonathan McConathy и др., “Improved synthesis of anti-[18F]FACBC: improved preparation of labeling precursor and automated radiosynthesis.”, Applied Radiation and Isotopes, (Netherlands), 2003, 58, p. 657-666.
Непатентный документ 2: Timothy M. Shoup и др., “Synthesis and Evaluation of [18F]1-Amino-3-fluorocyclobutane-1-carboxylic Acid to Image Brain Tumors.”, The Journal of Nuclear Medicine, 1999, 40, p. 331-338.
Раскрытие изобретения
Проблемы, решаемые изобретением
Таким образом, давно раскрытые способы получения [18F]FACBC достигали выходов продукта от 12 до 24% (J. McConathy и др., Applied Radiation and Isotopes, 2003, 58, p. 657-666), которые не могут считаться как являющимися достаточно высокими с точки зрения промышленного получения. То есть, с целью промышленного получения [18F]FACBC является желательным использовать способ получения или условия, которые могут стабильно обеспечить более высокий выход.
Получение [18F]FACBC, главным образом, включает стадию радиофторирования, в которой добавляют радиоактивный фтор для меченого предшественника; и стадию расщепления сложного эфира и снятия защиты, в которой промежуточное соединение, полученное на стадии радиофторирования, подвергают расщеплению сложного эфира и снимают защиту. Авторы настоящего изобретения провели изучение на стадии радиофторирования с целью улучшения выхода продукта и широко известного способа, в силу чего выход стадии фторирования может быть улучшен до 73,79%, который был 24,16% согласно традиционным способам. В результате, авторы изобретения сделали возможным улучшить выход продукта [18F]FACBC до 54,8 ± 4,8% (N = 15). Однако, подробное изучение, проведенное авторами изобретения, выявило, что полученный водный [18F]FACBC раствор содержал большое количество нерадиоактивных примесей (см. сравнительные примеры, описанные ниже). Количество примесей в фармацевтических средствах должно быть сдержано до определенного уровня или ниже. Таким образом, если после завершения реакции примеси присутствуют на определенном уровне или выше, то примеси должны быть удалены на последующей стадии. Однако, добавление последующей стадии очистки для уменьшения примесей вызывает удлинение времени, требуемого для стадий получения вслед за мечением радиоактивным фтором. Из-за того, что период полураспада радиоактивного фтора является коротким - приблизительно, 110 минут, не является предпочтительным удлинять время, требуемое для стадий после мечения радиоактивным фтором, с точки зрения промышленного получения меченых радиоактивным фтором соединений.
Настоящее изобретение было выполнено ввиду вышеописанных обстоятельств и имеет целью обеспечение способа получения [18F]FACBC, который может уменьшить получаемое количество нерадиоактивных примесей.
Способы решения проблем
В качестве результата исследования авторами изобретения было найдено, что количество примесей в целевом продукте может быть легко и эффективно уменьшено выполнением стадии расщепления сложноэфирной группы сложного эфира, которая является карбоксильно защищенной группой в обращенной фазе колонки для твердофазной экстракции и, таким образом, выполнили настоящее изобретение. Твердофазный способ снятия защитной группы традиционно применялся, главным образом, для целей снижения времени получения (см., например, опубликованный японский патент №11-508923). Авторами настоящего изобретения было найдено, что использованием твердофазного способа снятия защитной группы можно достичь уменьшения количества примесей, представленных в целевом продукте, и применили это открытие.
В соответствии с настоящим изобретением обеспечивают способ получения радиоактивного фтор-меченного органического соединения, содержащего стадию расщепления сложного эфира, удерживаемого в колонке с обращенной фазой, соединения, представленного следующей формулой (1):
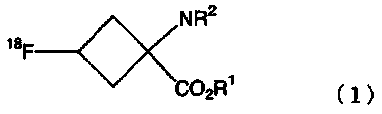
где R1 является С1-С10 линейной или разветвленной алкильной цепью или ароматическим заместителем; и R2является защитной группой;
загрузку в колонку раствора щелочи для расщепления сложного эфира вышеупомянутого соединения и последующую выгрузку щелочного раствора из колонки для получения соединения, представленного последующей формулой (2):

где Х является натрием или калием; и R2 является защитной группой; и
стадию снятия защитной группы аминозащитной группы соединения, полученного на стадии расщепления сложного эфира для получения соединения, представленного последующей формулой (3):

В формулах, показанных выше, R1 является С1-С10 линейной или разветвленной алкильной цепью или ароматическим заместителем и, предпочтительно, является заместителем, выбираемым из метила, этила, т-бутила и фенильных групп.
В формулах, показанных выше, R2 является защитной группой и не является особенно ограниченной, при условии, что она может предотвратить реакцию между радиоактивным фтором и аминогруппой. В частности, могут быть использованы защитные группы, выбираемые из группы, состоящей из различных карбаматных заместителей, различных амидных заместителей, различных имидных заместителей и различных аминных заместителей. Предпочтительно, могут быть использованы защитные группы, выбираемые из группы, состоящей из линейных или разветвленных С2-С7 алкилоксикарбонил заместителей; линейных или разветвленных С3-С7 алкенилоксикарбонил заместителей; С7-С12 бензилоксикарбонил заместителей, которые могут иметь модифицированную группу; С2-С7 алкилдитиооксикарбонил заместителей; линейных или разветвленных С1-С6 алкиламидных заместителей; линейных или разветвленных С2-С6 алкениламидных заместителей; С6-С11 бензамидных заместителей, которые могут иметь модифицированную группу; С4-С10 циклических имидных заместителей; С6-С11 ароматических иминных заместителей, которые могут иметь заместитель; линейных или разветвленных С1-С6 алкиламинных заместителей; линейных или разветвленных С2-С6алкениламинных заместителей; и С6-С11 бензиламинных заместителей, которые могут иметь модифицированную группу. Более предпочтительно, может быть использована защитная группа, выбираемая из т-бутоксикарбонильной группы, аллилоксикарбонильной группы, фталимидной группы и N-бензилиденаминным заместителем; и, наиболее предпочтительно, может быть использована т-бутоксикарбонильная группа или фталимидная группа.
В формулах, показанных выше, Х является катионом, содержащимся в щелочи, использованной на стадии расщепления сложного эфира, и является выбранным согласно типу щелочи. Например, с гидроксидом натрия Х является натрием, с гидроксидом калия Х является калием.
На стадии расщепления сложного эфира в качестве колонки с обращенной фазой могут быть использованы различные колонки с наполнителем, чьи функциональные группы являются гидрофобными группами, такими как фенил, циклогексил, и алкильными группами. Предпочтительно, используют колонны с обращенной фазой с наполнителем, имеющим структуру, в которой С2-С8 алкильные цепи прикреплены к подложке посредством кремния. Частный пример колонки с обращенной фазой включает колонку, имеющую в качестве функциональных групп октадецилсилиловые группы.
Удерживание соединения вышеописанной формулы (1) в колонке с обращенной фазой может быть выполнено различными способами. В частности, может быть использован способ, в котором раствор соединения вышеописанной формулы (1), полученный стадией радиофторирования, разводят водой и пропускают через колонку с обращенной фазой. Вода для разведения может быть использована в количестве, подходящем для связывания соединения вышеобозначенной формулы (1) в колонке с обращенной фазой.
В качестве щелочного раствора могут быть использованы различные растворы, но использование раствора гидроксида натрия является предпочтительным. Количество щелочного раствора для использования, предпочтительно, составляет равное или превышающее заполняющую способность колонки для твердофазной экстракции. Концентрация щелочного раствора не является ограниченной, при условии, что щелочь может быть введена в колонну в количестве, достаточном для выполнения расщепления сложного эфира; однако необходимо обратить внимание, поскольку, если его количество является слишком большим, будет необходимо использовать большее количество кислоты в последующей стадии снятия защитной группы. На стадии расщепления сложного эфира колонку с обращенной фазой выдерживают в течение определенного периода времени удерживания соединения вышеобозначенной формулы (1), в то время как заливают щелочной раствор. Время, в которое колонку с обращенной фазой выдерживают, заливая щелочной раствор, не является особенно ограниченным, при условии, что оно является достаточным для выполнения реакции расщепления сложного эфира.
Когда щелочной раствор сливают из колонны, соединение, представленное обозначенной выше формулой (2), сливают вместе со щелочным раствором. В это время через колонку может быть дополнительно пропущена вода после слива щелочного раствора так, чтобы смыть любое оставшееся соединение (2). Эта операция смыва может дополнительно улучшить выход соединения (2).
Стадия снятия защитной группы может быть выполнена использованием известных способов, например способом, описанным в литературе “J. McConathy и др., Applied Radiation and Isotopes, 2003, 58, p. 657-666”; и, в частности, способом, в котором кислые условия придают реакционному раствору после завершения стадии расщепления сложного эфира.
Стадия радиофторирования может быть выполнена применением известного способа или комбинации известного способа с условиями, которые установили авторы настоящего изобретения. В частности, соединение, представленное последующей формулой (4):

и инертный органический растворитель добавляют в смесь, содержащую катализатор межфазного переноса с18F ионами и ионами калия, так, чтобы получить реакционный раствор, и к реакционному раствору в процессе перемешивания применяют реакционные условия, такие как нагрев.
В формуле (4) R1 и R2 являются такими, как определены выше; R3 является элементом, выбираемым из группы, состоящей из линейных или разветвленных С1-С10 галогеналкил сульфоновая кислота заместителей; линейных или разветвленных С1-С10 алкил сульфоновая кислота заместителей; фторсульфоновая кислота заместителей и ароматическая сульфоновая кислота заместителей. Предпочтительно, используемым может быть заместитель, выбираемый из метансульфоновой кислоты, толуолсульфоновой кислоты, нитробензолсульфоновой кислоты, бензолсульфоновой кислоты, трифторметансульфоновой кислоты, фторсульфоновой кислоты и перфторалкилсульфоновой кислоты.
На стадии радиофторирования могут быть использованы различные инертные органические растворители, но должен быть использован амфифильный органический растворитель. В частности, может быть использован растворитель, выбираемый из группы, состоящей из тетрагидрофурана, 1,4-диоксана, ацетона, диметилформамида, диметилсульфоксида и ацетонитрила, ацетонитрил является наиболее предпочтительным. Количество инертного органического растворителя для использования, предпочтительно, регулируют так, чтобы концентрация меченого предшественника в реакционном растворе под условиями реакции радиофторирования составляла 40 ммоль/л или более, с целью значительного улучшения выхода в реакции радиофторирования.
В качестве реакционных условий для стадии радиофторирования могут быть использованы различные условия, например, может быть использовано условие, в котором реакционный раствор нагревают в процессе перемешивания. В этом случае температура нагрева не должна быть выше, чем температура кипения инертного органического растворителя, добавленного в реакционный раствор, например, когда в качестве инертного органического растворителя используют ацетонитрил, температура нагрева может быть от 70 до 90°С.
Эффекты изобретения
Способ получения настоящего изобретения способен уменьшить количество нерадиоактивных примесей, полученных при получении радиоактивных фтор-меченных аминокислотных соединений, таких как [18F]FACBC и также является пригодным в качестве способа очистки таких радиоактивных фтор-меченных аминокислотных соединений.
Лучший вариант осуществления изобретения
Далее в данном описании описывают в подробностях способ получения радиоактивной фтор-меченной аминокислоты согласно изобретению.
В наиболее предпочтительном осуществлении способ получения настоящего изобретения включает стадии: (1) реакции меченого предшественника со смесью, содержащей катализатор межфазного перехода,18F ионы и ионы калия для мечения меченого предшественника радиоактивным фтором, таким образом получая сложный эфир радиоактивным фтором-меченного предшественника (стадия радиофторирования); (2) стадию расщепления сложного эфира радиоактивного фтор-меченного предшественника в колонке для твердофазной экстракции (стадия расщепления сложного эфира); и (3) снятие защитной группы аминозащищенной группы соединения, полученного на стадии расщепления сложного эфира (стадия снятия защитной группы).
Радиоактивный фтор может быть получен известным способом, например способом, в котором Н218О обогащенную воду используют в качестве мишени и подвергают протонному облучению. В этом примере радиоактивный фтор существует в Н218О, обогащенной воде, используемой в качестве мишени. Н218О, обогащенной воде, содержащей радиоактивный фтор, позволяют пройти через анионообменную колонку так, чтобы радиоактивный фтор адсорбировался и собирался на колонке, таким образом отделяясь от Н218О, обогащенной воды. Соответственно, раствору карбонат калия позволяют пройти через колонку для элюирования радиоактивного фтора и элюат дополняют катализатором межфазного переноса и испаряют досуха для получения смеси, содержащей катализатор межфазного переноса так же, как18F ионы и ионы калия.
Количество карбоната калия для использования здесь в качестве иона калия может быть эквивалентным или больше, чем количество меченого предшественника, используемого на последующей стадии радиофторирования; однако избыточное количество карбоната калия не является предпочтительным, потому что продукт реакции может разлагаться под воздействием карбонатных ионов. В наиболее предпочтительном осуществлении концентрацию и количество раствора карбоната калия регулируют так, чтобы сделать количество иона калия приблизительно эквивалентным количеству меченого предшественника.
В качестве катализатора межфазного переноса могут быть использованы различные соединения, имеющие свойство образовывать клатрат с18F ионом. В частности, могут быть использованы различные соединения, используемые для получения радиоактивных фтор-меченных органических соединений, могут быть использованы 18-краун-эфир и другие различные аминополиэфиры. В наиболее предпочтительном осуществлении может быть использован 4, 7, 13, 16, 21, 24-гексаокса-1, 10-диазобицикло [8.8.8] гексакозан.
Большее количество катализатора межфазного переноса - станет больше выход, но избыточное количество не является предпочтительным, потому что удаление избыточно добавленного катализатора межфазного переноса часто будет являться недостаточным. В предпочтительном осуществлении общее количество катализатора межфазного перехода может быть 0,2 моль или меньше, например, когда количество меченого предшественника для использования равно 80 µмоль, мольное отношение катализатора межфазного перехода к меченому предшественнику составляет 2,5 или меньше.
После того как была получена смесь, содержащая катализатор межфазного перехода, а также18F ионы и ионы калия, выполняют радиофторирование реакцией меченого предшественника и18F ионов. Для стадии радиофторирования могут быть использованы различные способы, например, может быть использован способ, в котором этиловый эфир 1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты и инертный органический растворитель добавляют в вышеупомянутую смесь для получения реакционного раствора и затем обеспечивают для реакционного раствора такие реакционные условия, как нагревание при перемешивании для получения этилового эфира [18F]1-(N-(т-бутоксикарбонил)амино)-3-фторциклобутан-1-карбоновой кислоты (далее в данном описании упоминается как «[18F]Boc-FACBC»). В наиболее предпочтительном осуществлении меченый предшественник, этиловый эфир 1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты может быть растворен в инертном органическом растворителе прежде, чем его добавляют в смесь.
В качестве инертного органического растворителя, используемого на стадии радиофторирования, являются используемыми различные растворители, которые не имеют способности вступать в химическую реакцию с [18F] ионом фтора, катализатором межфазного перехода, ионом калия и веществом, меченным предшественником; и, предпочтительно, может быть использован растворитель, выбранный из группы, состоящей из тетрагидрофурана, 1,4-диоксана, ацетона, диметилформамида, диметилсульфоксида и ацетонитрила, с ацетонитрилом, являющимся наиболее предпочтительным. Количество инертного органического растворителя для использования, предпочтительно, регулируют так, чтобы концентрация меченого предшественника в реакционном растворе при реакции радиофторирования составляла 40 ммоль/л или более с целью значительного улучшения выхода в реакции радиофторирования.
В качестве реакционных условий для стадии радиофторирования могут быть использованы различные условия, например, может быть использовано условие, в котором реакционный раствор нагревают в процессе перемешивания. Температура нагрева в этом случае, предпочтительно, не выше, чем температура кипения инертного органического растворителя, добавленного в реакционный раствор, например, когда в качестве инертного органического растворителя используют ацетонитрил, температура нагрева может быть от 70 до 90°С. Реакционное время зависит от реакционной температуры, когда, например, реакционная температура равна 83°С, достаточное время реакции равно 3 минутам или более. Большее время реакции предполагает, дополнительно, протекание реакции радиоактивного фтор-мечения, но необходимо принять во внимание то, что в то же самое время протекает расщепление радиоактивного фтора.
После того как стадия радиофторирования была завершена, выполняют стадию расщепления сложного эфира для получения [18F]1-(N-(т-бутоксикарбонил)амино)-3-фторциклобутан-1-карбоновой кислоты (далее в данном описании упоминается как «[18F]DE-Boc-FACBC»). Настоящее изобретение характеризуется тем, что реакцию расцепления сложного эфира на этой стадии выполняют в колонке для твердофазной экстракции. В наиболее предпочтительном осуществлении пример для расщепления сложного эфира, а именно [18F]DE-Boc-FACBC улавливают на колонке для твердофазной экстракции разведением реакционного раствора, содержащего [18F]Boc-FACBC, полученного на стадии радиофторирования, водой и пропускание полученного раствора в качестве образца через колонку для твердофазной экстракции. Разведение реакционного раствора выполняют для предотвращения [18F]Boc-FACBC от извлечения из адсорбента, не будучи уловленным на колонке, когда образец пропускают через колонку для твердофазной экстракции. По этой причине вода, используемая для разведения, может быть использована в количестве, достаточном для улавливания [18F]Boc-FACBC на наполнителе колонки для твердофазной экстракции, когда растворителем реакционного раствора является ацетонитрил, достаточное количество воды равно пятикратному количеству растворителя.
Колонка для твердофазной экстракции, используемая на стадии расщепления сложного эфира, должна быть колонкой для твердофазной экстракции, наполненной наполнителем с обращенной фазной. Предпочтительно, наполнитель колонки является наполнителем, имеющим гидрофобную функциональную группу, такую как фенил, циклогексил и алкильные группы, и, более предпочтительно, наполнителем, имеющим структуру с подложкой, в которой С2-С18 алкильные группы прикреплены посредством кремния. В наиболее предпочтительном осуществлении может быть использована колонка, наполненная наполнителем, имеющим в качестве функциональных групп октадецилсилильные группы. Кроме того, предпочтительным является использование наполнителя колонки, имеющего структуру, в которой является затруднительным отделить функциональные группы от подложки при водных условиях реакции и в процессе длительной реакции расщепления сложного эфира.
После того как образец был уловлен на колонке для твердофазной экстракции, в колонку загружают щелочной раствор. В наиболее предпочтительном осуществлении щелочной раствор загружают непосредственным введением щелочного раствора в колонку, останавливая подачу щелочного раствора после подтверждения, что щелочной раствор начал вытекать через выходное отверстие колонки. Примеры щелочей, используемых здесь, включают гидроксид натрия и гидроксид калия, с гидроксидом натрия, являющимся, предпочтительно, рассматриваемым с целью получения изобретения, используемым в качестве вводимой пробы.
В наиболее предпочтительном осуществлении объем щелочного раствора приблизительно равен объему колонки. В этом примере должно быть принято во внимание что, если объем щелочного раствора для использования является избыточным, то образец ранее расщепленного сложного эфира может быть выгружен вместе с сбросным раствором, таким образом вызывая увеличение выхода.
Концентрация используемого щелочного раствора не является ограниченной при условии, что щелочь может быть введена в колонку в количестве, достаточном для выполнения расщепления сложного эфира. Концентрацию щелочного раствора определяют в рассмотрении пригодного объема щелочного раствора и необходимого количества щелочи. В этом примере необходимо принять во внимание, что, если используют избыточное количество щелочи, будет необходимо использовать большее количество кислоты для нейтрализации на последующей стадии снятия защитных групп.
После того как в колонку для твердофазной экстракции был загружен щелочной раствор, колонку выдерживают бездействующей в течение определенного периода времени с намерением расщепить сложный эфир в колонке. В этом примере температура колонки необязательно должна быть специфически контролируемой, но действия могут быть выполнены при комнатной температуре. Продолжительность, в которой колонку выдерживают бездействующей, может быть промежутком времени, достаточным для выполнения расщепления сложного эфира. Большая продолжительность - дополнительно реакция расщепления сложного эфира будет продолжаться, но необходимо обратить внимание на то, что в то же самое время протекает расщепление радиоактивного фтора. Например, когда [18F]Boc-FACBC удерживают в ODS колонке содержащей 400 мг смолы и 0,8 мл 4 моль/л раствора гидроксида натрия вводят в колонку для выполнения расщепления сложного эфира, достаточным является промежуток времени от 1 до 5 минут.
После завершения расщепления сложного эфира открывают выпускное отверстие колонки, таким образом вызывая выгрузку [18F]DE-Boc-FACBC, полученного расщеплением сложного эфира, вместе с щелочным раствором. После того как щелочной раствор был выгружен, щелочной раствор может быть дополнительно добавлен в колонку, с последующим повторением тех же операций, как описано выше, так, чтобы [18F]Boc-FACBC остающийся в колонке с обращенной фазой, мог быть более основательно подвергнут расщеплению сложного эфира, таким образом улучшая выход. Является предпочтительным, что после выгрузки колонку далее промывают водой так, чтобы выгрузить остаток [18F]DE-Boc-FACBC из колонки, таким образом дополнительно улучшая выход.
После завершения стадии расщепления сложного эфира выполняют стадию снятия защитных групп для снятия амино-защитной группы, таким образом производя [18F]FACBC, которое является целевым продуктом настоящего изобретения. Стадия снятия защитной группы может быть выполнена согласно известным способам, например, способом, описанным в литературе «J. McConathy и др., Applied Radiation and Isotopes, 2003, 58, p. 657-666». В предпочтительном осуществлении стадия снятия защитной группы может быть выполнена обеспечением кислых условий в реакционном растворе, содержащем [18F]DE-Boc-FACBC. Кислые условия могут быть обеспечены различными способами, например способом, в котором кислоту добавляют к раствору, содержащему [18F]DE-Boc-FACBC. Кислота для использования здесь не является, в частности, ограниченной, но, предпочтительно, включает кислоту, выбираемую из неорганических кислот, таких как соляная кислота, серная кислота и азотная кислота, и органических кислот, таких как перфторалкил карбоновая кислота (например, трифторуксусная кислота). Количество кислоты, которое добавляют, должно быть достаточным для приведения рН раствора, содержащего [18F]DE-Boc-FACBC, к 1 или меньше. В частности, количество кислоты должно быть таким, чтобы нейтрализовать щелочь в [18F]DE-Boc-FACBC растворе, полученную на стадии расщепления сложного эфира, и привести достаточные кислотные условия в образце раствора. Например, когда [18F]Boc-FACBC подвергают расщеплению сложного эфира, повторенному дважды, используя 0,8 мл 4 моль/л раствора гидроксида натрия, может быть добавлено 2,2 мл 6 моль/л соляной кислоты к извлеченному из адсорбента реакционному раствору. На стадии снятия защитной группы реакционный раствор, предпочтительно, нагревают, чтобы позволить реакции протекать более быстро. Реакционное время зависит от реакционной температуры или других условий, но когда реакцию снятия защитной группы при описанных выше условиях выполняют при 60°С, достаточное реакционное время равно 5 минут. [18F]FACBC раствор, полученный на стадии снятия защитной группы, может быть необязательно очищен, использованием ионообменной колонки, колонки окиси алюминия или колонки с обращенной фазой.
Примеры
Далее в описании настоящее изобретение будет описано более детально посредством примеров и сравнительных примеров; однако изобретение не ограничено этими примерами.
Справочный пример 1
Синтез этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3--[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты
Гидролиз син-гидантоина (Фиг. 1, стадия 1)
250 мл насыщенного водного раствора гидроксида бария добавляли к 6,15 г (соответственно 25 ммоль) син-5-(3-бензилоксициклобутан)гидантоина и смесь нагревали в колбе с обратным холодильником на масляной бане при 114°С в течение 24 часов или более. Затем выполняли анализ ТСХ, используя в качестве подвижных растворителей два вида систем, а именно хлороформ:метанол = 5:1 (Rf значение син-гидантоина равно, приблизительно, 0,3), и подтверждали завершение реакции (основанное на окрашивании с UV и фосфомолибденовой кислотой).
После подтверждения того, что реакция была завершена, полученный реакционный раствор охлаждали до комнатной температуры и добавляли, приблизительно, 24 мл 1 моль/мл серной кислоты для нейтрализации реакционного раствора. После нейтрализации реакционный раствор дополнительно перемешивали при комнатной температуре в течение 5 минут и полученный осадок отфильтровывали. Затем фильтрат собирали с получением 5,67 г син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты в виде бесцветных кристаллов.
Образование сложного этилового эфира (Фиг. 1, Стадия 2)
5,6 г син-1-амино-бензилоксициклобутан-1-карбоновой кислоты, которую полностью высушили для удаления воды, растворяли в 200 мл этанола. К этому раствору добавляли 9,5 мл (соответственно, 75 ммоль) триэтиламина и смесь охлаждали до -78°С в течение 20 мин, с последующим добавлением туда 4,6 мл (соответственно, 62,5 ммоль) тионилхлорида. Реакционный раствор перемешивали при 0°С в течение 1 часа и при комнатной температуре в течение 1 часа, с последующим нагреванием в колбе с обратным холодильником на масляной бане при 95°С в течение ночи. Затем завершение реакции подтверждали анализом ТСХ, который выполняли, используя в качестве подвижных растворителей два вида систем, а именно хлороформ:метанол = 95:1 (Rf значение целевого продукта равно, приблизительно, 0,6) (в котором подтверждение основывалось на окрашивании с UV и фосфомолибденовой кислотой). После подтверждения, что реакция завершена, полученный реакционный раствор собирали при пониженном давлении с получением 7,64 г этилового эфира син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты в виде бесцветных кристаллов.
Добавление Boc (Фиг. 1, Стадия 3)
7,64 г этилового эфира син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты растворяли в 250 мл смешанного раствора этанол:триэтиламин = 9:1. Полученный раствор охлаждали на ледяной бане в течение 15 минут, затем туда добавляли 8,6 мл (соответственно, 37,5 ммоль) ди-трет-бутил дикарбоната и смесь оставляли на ночь при перемешивании. Затем завершение реакции подтверждали анализом ТСХ, который выполняли, используя в качестве подвижного растворителя гексан:этилацетат = 1:1 (Rf значение целевого продукта равно, приблизительно, 0,6) (в котором подтверждение основывалось на окрашивании с UV и фосфомолибденовой кислотой). После подтверждения, что реакция завершена, полученный реакционный раствор собирали при пониженном давлении с получением остатка в виде бесцветных кристаллов. К остатку добавляли 150 мл холодного этилацетата и 150 мл 0,5 моль/л холодной соляной кислоты, смесь перемешивали при комнатной температуре в течение 5 минут и выстаивали для разделения. Органический слой экстрагировали и дважды промывали 150 мл воды, 150 мл насыщенного водного раствора гидрокарбоната натрия, 150 мл воды дважды и 150 мл насыщенного соляного раствора дважды в этом порядке, экстракт высушивали безводным сульфатом натрия и затем собирали при пониженном давлении, чтобы дать желтое масляное вещество. Отдельно водный слой экстрагировали и дважды промывали 150 мл этилацетата, дважды 150 мл воды и 150 мл насыщенного соляного раствора в этом порядке и экстракт высушивали безводным сульфатом натрия и затем собирали при пониженном давлении, таким образом, собрали небольшое количество желтого масляного вещества. Серия операций дала 8,82 г светло-желтого масляного вещества. Остаток отделяли и очищали хроматографией на колонке силикагеля (гексан:этилацетат = 1:1) с получением 8,04 г (соответственно, 23 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты в виде бесцветных кристаллов.
Дебензилирование (Фиг. 2, Стадия 4)
150 мл этанола добавляли к 8,04 г (соответственно, 23 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты и затем туда добавляли 960 мг палладия на активированном угле (10% палладия), продутом водородом, и смесь оставляли перемешиваемой при комнатной температуре на ночь. После реакции палладий на активированном угле отфильтровывали, используя Celite, и полученный фильтрат собирали при пониженном давлении, чтобы дать 5,74 г остаток в виде бесцветных кристаллов. Реакцию отслеживали анализом ТСХ, используя в качестве подвижного растворителя гексан:этилацетат = 1:1 (Rf значение целевого реакционного продукта равно, приблизительно, 0,2)(подтверждение основывалось на окрашивании с UV и нингидрином) для подтверждения завершения реакции. Затем осадок отделяли и очищали хроматографией на колонке силикагеля (гексан:этилацетат = 1:1, гексан:этилацетат = 4:1) для получения 5,36 г (соответственно, 20,7 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты в виде бесцветных кристаллов.
Введение трифлата (Фиг. 3, Стадия 5)
2,07 г (8 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты растворяли в 26 мл пиридина и раствор перемешивали на ледяной бане в течение 20 минут. Туда добавляли 2,0 мл (соответственно, 12 ммоль) безводной трифторметансульфокислоты и смесь перемешивали как есть в течение 30 минут. Реакцию отслеживали анализом ТСХ, используя в качестве подвижного растворителя гексан:диэтиловый эфир = 1:1 (Rf значение целевого реакционного продукта равно, приблизительно, 0,6) (подтверждение основывалось на окрашивании с нингидрином) для подтверждения завершения реакции. Для подтверждения того, что реакция завершена, в реакционный раствор добавляли 100 мл воды и 100 мл эфира, полученную смесь экстрагировали и промывали 100 мл 1 моль/л соляной кислоты дважды, 100 мл воды дважды и 100 мл насыщенного соляного раствора дважды в этом порядке. Полученный экстракт высушивали безводным сульфатом натрия и затем собирали при пониженном давлении, чтобы дать 2,78 г светло-желтых кристаллов. Реакционную смесь разделяли и очищали хроматографией на колонке силикагеля (гексан:диэтиловый эфир = 3:1) для получения белых кристаллов и полученные белые кристаллы снова перекристаллизировали, используя пентан:диэтиловый эфир, чтобы получить 1,84 г (соответственно, 4,7 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты.
Сравнительный пример
Н218О, содержащую [18F] ионы фтора (13 до 182 ГБк), пропускали через анионообменную колонку так, чтобы [18F] ионы фтора были адсорбированы и уловлены на колонке. Затем через колонку пропускали раствор карбоната калия для извлечения из адсорбента [18F] ионов фтора, затем колонку промывали водой и водный раствор смешивали с элюатом. В полученный раствор добавляли раствор в ацетонитриле 4,7,13,16,21,24-гексаокса-1,10-диазобицикло[8.8.8]гексакозана (торговое название: Kryptofix 222, производимое Merck) и смесь нагревали и выпаривали до сухости.
К осушенной смеси добавляли раствор, полученный растворением 32 мг этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты в 1 мл ацетонитрила, и полученную смесь перемешивали в течение 3 минут при 83°С так, чтобы позволить реакции радиофторирования продолжаться. Затем смесь охлаждали при комнатной температуре в течение 5 минут и в нее добавляли 4 мл диэтилового эфира. Полученную смесь пропускали через Silica Sep-Pak (зарегистрированный товарный знак Waters Investments Limited или торговое название, доступное от Nihon Waters K. K.), чтобы получить ацетонитрил/диэтиловый эфир раствор [18F]Вос-FACBC.
В полученный раствор ацетонитрил/диэтиловый эфир [18F]Вос-FACBC добавляли 1,5 мл 4 моль/л соляной кислоты и смесь нагревали при 120°С в течение 15 минут для снятия защитных групп. Полученный раствор далее очищали пропусканием через ионообменную колонку (торговое название: AG11A8, производимое Bio-Rad Laboratories Japan, Inc.), колонку оксида алюминия (торговое название: Sep-Pak (зарегистрированный товарный знак, производимый Waters Investments Limited) light ALUMN, производимый Nihon Waters K. K.) и колонку с обращенной фазой (торговое название: Oasis HLB Plus EXTRACTION Cartridge Column, производимое Nihon Waters K. K.) в этом порядке для получения раствора [18F]FACBC. Выход [18F]FACBC раствора составил от 9,4 до 13,4 мл. Полученное [18F]FACBC подвергали анализу ТСХ при следующих условиях и радиохимическую чистоту подтверждали согласно последующему уравнению (1).
Условия анализа ТСХ:
Подвижная фаза: ацетонитрил/метанол/вода/уксусная кислота = 20/5/5/1.
TLC пластина: Silica Gel 60F254 (торговое название, толщина пленки: 0,25 мм, произведенная Merck)
Проявляющая длина: 10 см
Анализатор ТСХ: Rita Star (произведенный Raytesr)

В дополнение количества нерадиоактивных примесей в целевом продукте были сравнены, используя значения полученные введением поправок в соответствии с последующим уравнением (2), значение пиковой области каждой примеси, подтвержденное HPLC анализом при последующих условиях (далее в данном описании обозначенные как «скорректированные значения области»). Образец раствора, подвергнутого HPLC анализу, достаточно разводили, используя физиологический солевой раствор (коэффициент разведения равен от 2,1 до 9,9).

(2)

HPLC условия измерения:
Колонка: CAPCELLPAK C18 MG (торговая название, произведенное Shiseido Co., Ltd, размер: 5 µм, 4,6 мм I.D. × 250 мм)
Температура колонки: комнатная температура (приблизительно, 25°С)
Подвижная фаза: использовали 5 ммоль/л натрий октансульфонат, содержащий фосфатный буфер (рН 2,1), как раствор А и ацетонитрил как раствор В, контроль градиента концентрации выполняли варьированием соотношения компонентов смеси растворов А и В как показано в Таблице 1.
Объемный расход подвижной фазы: 1,0 мл/мин
Количество введенного образца: 10 µл
Условия послеколоночного получения производных:
Реакционный раствор: 0,3 моль/л буфера борной кислоты (рН 10,4), 6 ммоль/л о-фталевого альдегида и 6 ммоль/л N-ацетил-L-цистеина.
Объемный расход реакционного расхода: 1,0 мл/мин
Реакционная температура: 50°С
Детектор: флуоресцентный детектор (тип: Waters 2475 модель, произведенная Nihon Waters K. K.); возбуждающая длина волны: 330 нм; флуоресцентная длина волны: 430 нм)
Опыт Сравнительного Примера 1 повторяли 19 раз.
Радиохимическая чистота полученного [18F]FACBC составляла 98,8 ± 0,4%. Пики примесей, подтвержденные HPLC хроматограммами, были определены, как показано в Таблице 2. Скорректированное значение области пика каждой примеси показано в Таблице 3.
Примеры 1 и 2
Н218О, содержащую [18F] ионы фтора (7-36 ГБк), пропускали через анионообменную колонку так, что [18F] ионы фтора адсорбировались и улавливались на колонке. Затем через колонку пропускали, для извлечения из адсорбента [18F] ионов фтора, раствор карбоната калия и колонку промывали водой и промывной раствор смешивали с элюатом. В полученный раствор добавляли раствор в ацетонитриле 4,7,13,16,21,24-гексаокса-1,10-диазобицикло[8.8.8]гексакозана (торговое название: Kryptofix 222, произведенное Merck) и смесь нагревали и выпаривали до сухости.
В осушенную смесь добавляли раствор, полученный растворением 32 мг этилового эфира 1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты в 1 мл ацетонитрила, и полученную смесь нагревали при 83°С при перемешивании в течение 3 минут.
Полученный реакционный раствор охлаждали при комнатной температуре в течение 5 минут и затем разводили 14 мл воды и полученный раствор пропускали через каждый Sep-Pak (зарегистрированный товарный знак, Waters Investments Limited) картридж (произведенный Nihon Waters K. K.), показанный в Таблице 4, и затем колонку дополнительно промывали 10 мл воды.
Колонку для твердофазной экстракции осушали пропусканием через нее воздуха и затем в колонку загружали 0,8 мл/л 4 моль/л раствора гидроксида натрия с последующим закупориванием выходного отверстия колонки. После истечения 3 минут выходное отверстие колонки открывали для извлечения из адсорбента щелочного раствора из колонки для проведения твердофазной экстракции и элюат собирали в виалу. В колонку дополнительно загружали 0,8 мл 4 моль/л раствора гидроксида натрия и повторяли ту же процедуру, как описана выше. Колонку для твердофазной экстракции затем промывали 3 мл воды и промывной раствор смешивали с ранее собранным щелочным раствором.
В упомянутый выше собранный раствор добавляли 2,2 мл 6 моль/л соляной кислоты, и выполнялась реакция снятия защитной группы при 60°С в течение 5 минут. Полученный продукт далее очищали пропусканием через ионообменную колонку (торговое название: AG11A8, произведенную Bio-Rad Laboratories Japan, Inc.), колонку оксида алюминия (торговое название: Sep-Pak (зарегистрированный товарный знак, Waters Investments Limited) light ALUMN, произведенную Nihon Waters K. K.) и колонку с обращенной фазой (торговое название: Oasis HLB Plus EXTRACTION Cartridge Column, произведенную Nihon Waters K. K.) в этом порядке для получения [18F]FACBC раствора. Выход [18F]FACBC растворов составил от 11,9 до 17,0 мл.
Полученный [18F]FACBC раствор оценивали на радиохимическую чистоту [18F]FACBC и скорректированное значение области каждой примеси при тех же условиях как в Сравнительном Примере. Образцы растворов, подвергнутые HPLC анализу, соответствующе разводили, используя физиологический солевой раствор (коэффициент разбавления равен 3,0-4,7).
Радиохимические чистоты [18F]FACBC, полученного в Примерах 1 и 2, составляли 99,4 и 99,3% соответственно. Таблица 5 показывает скорректированное значение области пиков каждой примеси. Как показано в Таблице 5, в каждом Примере 1 и 2 количества всех нерадиоактивных примесей, за исключением примеси D, сокращены по сравнению с образцом, полученным согласно традиционному способу (Сравнительный Пример 1) и общие скорректированные значения области сокращены до менее чем половины. Эти результаты подтвердили, что количество нерадиоактивных примесей может быть уменьшено способом получения [18F]FACBC согласно настоящему изобретению.
Промышленная применимость
Способ получения радиоактивного фтор-меченного органического соединения согласно изобретению может быть использован в области производства радиофармацевтических средств.
Краткое описание чертежей
Фиг.1 является схемой синтеза этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты.
Фиг.2 является схемой синтеза этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты.
Фиг.3 является схемой синтеза этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты.
Реферат
Изобретение относится к способу получения радиоактивного фтор-меченного органического соединения формулы . Способ включает стадию расщепления сложного эфира, представленного формулой , где R1 является линейным или разветвленным C1-С10 алкилом и R2 является защитной группой, выбранной из линейных или разветвленных C2-C7 алкилоксикарбонильных групп, пропускаемого и удерживаемого в колонке с обращенной фазой, содержащей наполнитель, имеющий структуру, в которой C2-C18 алкильные группы прикреплены к подложке посредством кремния. Для расщепления вышеуказанного сложного эфира в колонку загружают раствор щелочи, после чего щелочной раствор выгружают из колонки с получением соединения, представленного формулой , где является натрием или калием и R2 является защитной группой, выбранной из линейных или разветвленных С2-С7 алкилоксикарбонильных групп. На следующей стадии осуществляют снятие защитной группы R2 соединения формулы (2), полученного на стадии расщепления сложного эфира, с получением соединения формулы (3). Способ позволяет снизить количество нерадиоактивных примесей и получить соединение формулы (3) с хорошим выходом. 1 з.п. ф-лы, 3 ил., 5 табл.
Формула

который включает:
стадию расщепления сложного эфира, представленного следующей формулой (1):

где R1 является линейным или разветвленным C1-С10 алкилом, и R2 является защитной группой, выбранной из линейных или разветвленных C2-C7 алкилоксикарбонильных групп, пропускаемого и удерживаемого в колонке с обращенной фазой, содержащей наполнитель, имеющий структуру, в которой C2-C18 алкильные группы прикреплены к подложке посредством кремния, посредством загрузки в колонку раствора щелочи для расщепления сложного эфира вышеуказанного соединения и последующей выгрузки щелочного раствора из колонки с получением соединения, представленного следующей формулой (2):

где X является натрием или калием; и R2 является защитной группой, выбранной из линейных или разветвленных C2-C7 алкилоксикарбонильных групп; и
стадию снятия защитной группы R2 соединения формулы (2), полученного на стадии расщепления сложного эфира, с получением соединения, представленного формулой (3).
Комментарии