Способ получения монатина - RU2351587C2
Код документа: RU2351587C2
Описание
Область техники
Настоящее изобретение относится к способу получения монатина, применимого в качестве подсластителя. В частности, данное изобретение относится к способу эффективного получения оптически активного монатина.
Уровень техники
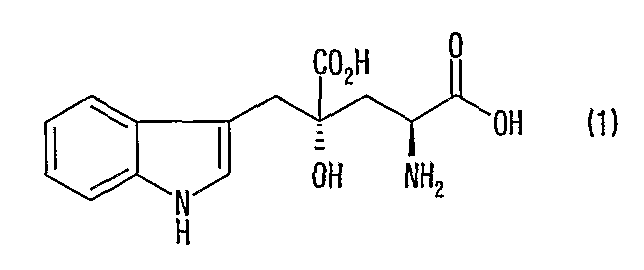
4-Гидрокси-4-(3-индолилметил)-2-аминоглутаровая кислота (иногда в дальнейшем упоминаемая как "монатин") в форме (2S, 4S), представленная формулой (5), содержится в кожице корней растения Schlerochitom ilicifolius, встречающегося в природе в Северном районе Южной Африки, а именно Северном Трансваале. Известно, что соединение в данной форме имеет уровень сладости в несколько сотен раз больший, чем уровень сладости сахарозы, и представляет собой производное аминокислоты, применимое в качестве подсластителя (см. официальный бюллетень -JP-A-64-25757).

Монатин имеет два асимметрических углеродных атома в положениях 2 и 4 и, поэтому включает наличие четырех типов оптических изомеров.
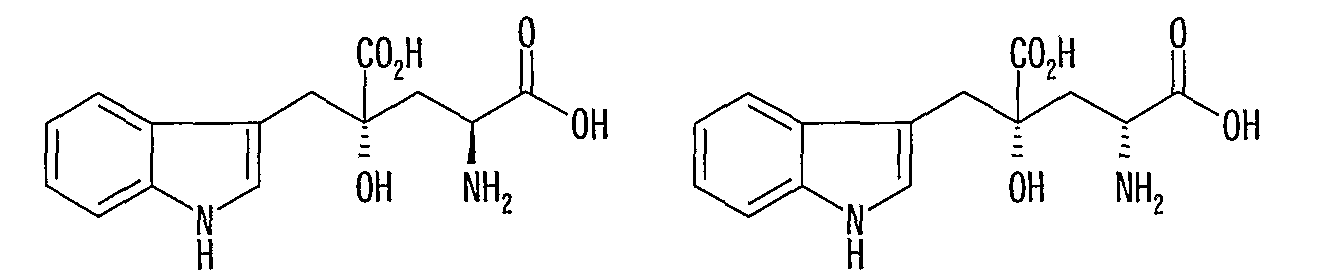
(2S, 4R)-монатин (2R, 4R)-монатин
(2R, 4S)-монатин (2S, 4S)-монатин
В различных публикациях сообщается о способах получения монатина (см., например, Tetrahedron Letters, 2001, Vol.42, No. 39, p. 6793-6796; Organic Letters, 2000, Vol.2, No. 19, p. 2967-2970; Описание патента США 5994559; Synthetic Communication, 1994, Vol.24, No.22, p. 3197-3211). Некоторые примеры сообщают об исследовании способов получения оптически активного монатина. Однако нельзя не отметить, что эти способы требуют такого большого числа стадий для получения, что делает их неподходящими для промышленного производства.
Альтернативно заявитель недавно обнаружил и сообщил о способе получения конкретного оптически активного монатина, включающем стадию синтеза предшественника монатина из индол-3-пировиноградной кислоты с образованием диастереомерной соли с конкретным оптически активным амином и в заключение стадию получения конкретного оптически активного монатина путем оптического разделения (см. описание международной заявки WO 03/059865). Например, упоминается (2R, 4R)-монатин. Данный способ представлен нижеследующей схемой.
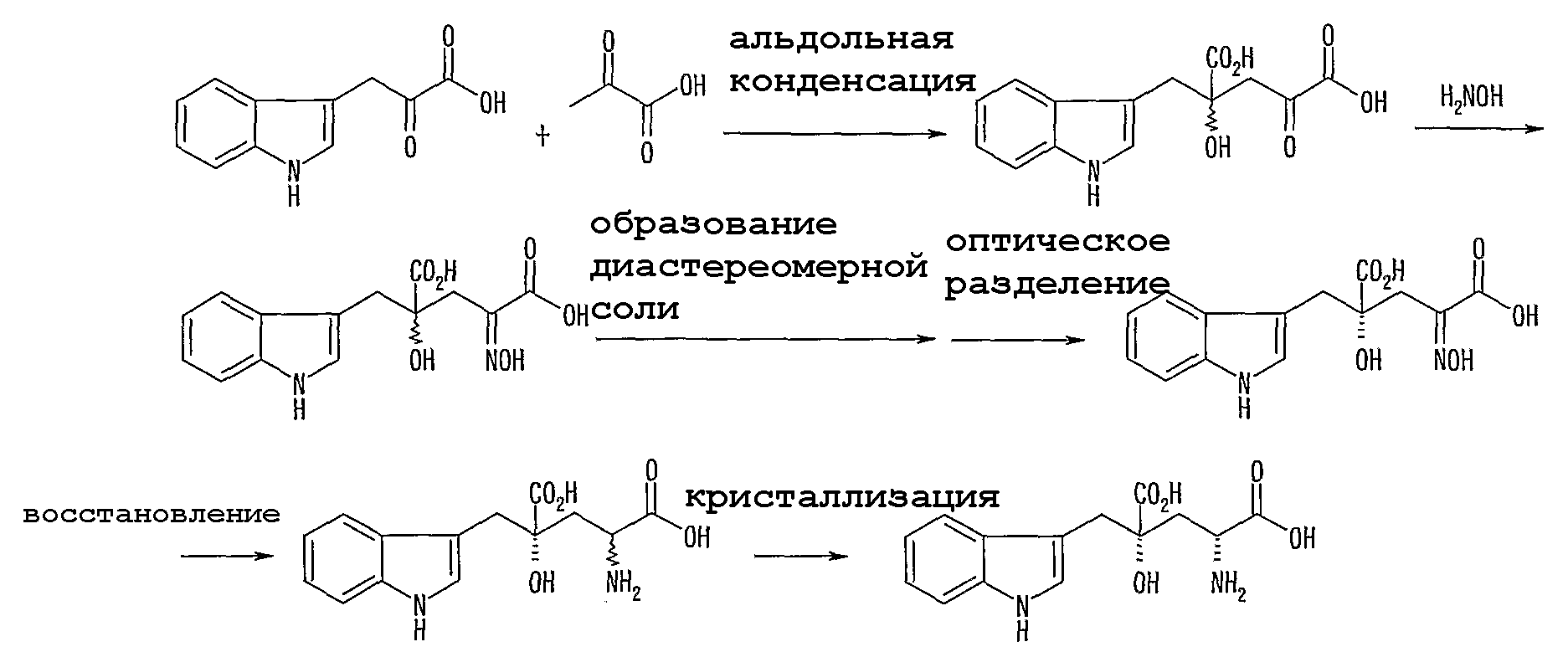
Данный способ получения требует меньшего числа стадий по сравнению с традиционными способами, и с его помощью может быть эффективно получен оптически активный монатин, поэтому данный способ является подходящим с точки зрения промышленного производства. Однако во время стадии кристаллизации в маточный раствор выделяется его нежелаемый оптический изомер в положении 2. Если бы данный оптический изомер мог быть изомеризован и превращен в предполагаемый оптически активный монатин, эффективность способа была бы дополнительно увеличена.
В качестве способов рацемизации оптически активных аминокислот были известны, например, способы рацемизации обработкой сильными кислотами или сильными щелочами или в условиях более высокой температуры или способы рацемизации при относительно умеренных условиях в присутствии альдегидов. Согласно этим способам максимальные выходы составляют около 50%. Поэтому об этих способах нельзя говорить как об эффективных способах рацемизации. Был известен способ рацемизации и осаждения для получения предполагаемого оптического изомера с более высоким выходом в комбинации с конкретным оптическим изомером (см. Tetrahedron, 1997, Vol. 53, No. 28, p. 9417-9476). Огромное число проб и ошибок было совершено при обнаружении такой комбинации.
Раскрытие изобретения
Целью изобретения является разработка способа эффективного получения монатина с одинаковыми конфигурациями в положениях 2 и 4 или его соли.
В результате проведения тщательных исследований и их анализа авторами было обнаружено, что путем осуществления реакции изомеризации в положении 2 монатина с различными конфигурациями в положениях 2 и 4 в присутствии альдегида в присутствии конкретного растворителя и при конкретном рН и при одновременном селективном осаждении только требуемого монатина с одинаковыми конфигурациями в положениях 2 и 4 равновесие реакции изомеризации значительно смещается в сторону требуемого оптически активного монатина, так что монатин может быть получен эффективным способом. Таким образом было осуществлено изобретение.
В частности, настоящее изобретение включает следующие аспекты.
[1] Способ получения монатина, имеющего одинаковые конфигурации в положениях 2 и 4, или его соли, включающий стадию реакции изомеризации в положении 2 монатина, имеющего различные конфигурации в положениях 2 и 4, в смешанном растворителе из воды и органического растворителя в присутствии альдегида при значении рН от 4 до 11 и одновременную кристаллизацию монатина, имеющего одинаковые конфигурации в положениях 2 и 4, или его соли.
[2] Способ получения монатина в конфигурации (2R, 4R) или его соли, включающий стадию реакции изомеризации в положении 2 монатина в конфигурации (2S, 4R) в смешанном растворителе из воды и органического растворителя в присутствии альдегида при значении рН от 4 до 11 и одновременную кристаллизацию монатина в конфигурации (2R, 4R) или его соли.
[3] Способ получения монатина в конфигурации (2S, 4S) или его соли, включающий стадию реакции изомеризации в положении 2 монатина в конфигурации (2R, 4S) в смешанном растворителе из воды и органического растворителя в присутствии альдегида при значении рН от 4 до 11 и одновременную кристаллизацию монатина в конфигурации (2S, 4S) или его соли.
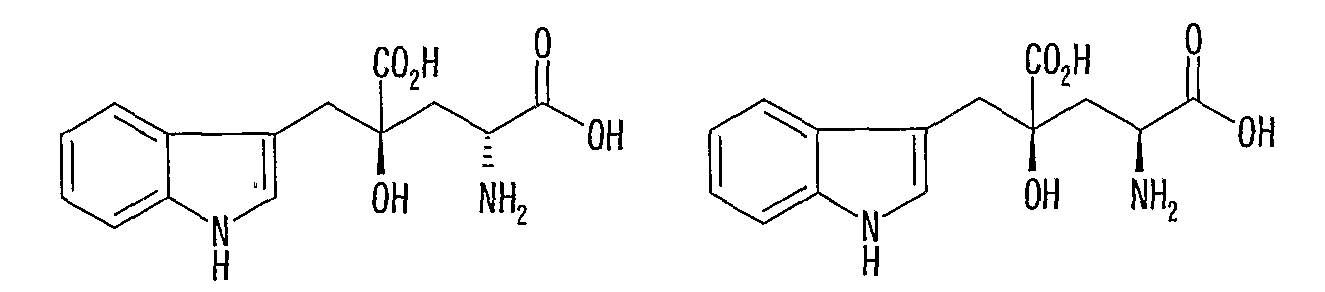
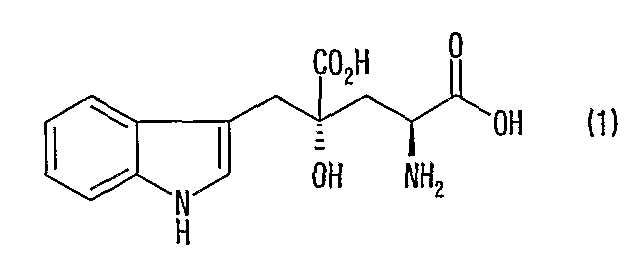
[4] Способ получения (2R, 4R)-монатина, представленного формулой (2), или его соли, включающий стадию реакции изомеризации в положении 2 (2S, 4R)-монатина, представленного формулой (1), в смешанном растворителе из воды и органического растворителя в присутствии альдегида при значении рН от 4 до 11 и одновременную кристаллизацию (2R, 4R)-монатина или его соли:

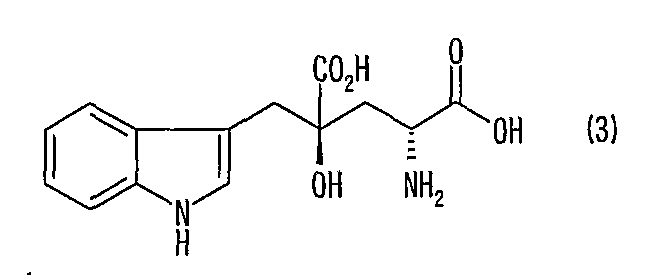
[5] Способ получения (2S, 4S)-монатина, представленного формулой (4), или его соли, включающий стадию реакции изомеризации в положении 2 (2R, 4S)-монатина, представленного формулой (3), в смешанном растворителе из воды и органического растворителя в присутствии альдегида при значении рН от 4 до 11 и одновременную кристаллизацию (2S, 4S)-монатина или его соли:

[6] Способ получения (2R, 4R)-монатина, представленного формулой (2), или его соли, включающий стадию реакции изомеризации в положении 2 (2S, 4R)-монатина, представленного формулой (1), в смеси (2S, 4R)-монатина и (2R, 4R)-монатина в смешанном растворителе из воды и органического растворителя в присутствии альдегида при значении рН от 4 до 11 и одновременную кристаллизацию (2R, 4R)-монатина или его соли:

[7] Способ получения (2S, 4S)-монатина, представленного формулой (4), или его соли, включающий стадию реакции изомеризации в положении 2 (2R, 4S)-монатина, представленного формулой (3), в смеси (2R, 4S)-монатина и (2S, 4S)-монатина в смешанном растворителе из воды и органического растворителя в присутствии альдегида при значении рН от 4 до 11 и одновременную кристаллизацию (2S, 4S)-монатина или его соли:
[8] Способ, описанный выше в п.п. 1-7, где органическим растворителем является спирт.
[9] Способ, описанный выше в п.п. 1-8, где реакцию изомеризации и кристаллизацию проводят при значении рН от 4,5 до 10.
[10] Способ, описанный выше в п.п. 1-8, где реакцию изомеризации и кристаллизацию проводят при значении рН от 5 до 9.
Согласно изобретению в дальнейшем фраза “одинаковые конфигурации в положениях 1 и 2” означает, что конфигурации в положениях 2 и 4 представляют собой обе конфигурации R или обе конфигурации S, если конфигурации выражают в виде R/S. Фраза "различные конфигурации в положениях 2 и 4" означает, что любая одна из конфигураций в положениях 2 и 4 является конфигурацией S, в то время как другая оставшаяся конфигурация представляет собой R, когда конфигурации выражают в виде R/S.
Наилучший способ осуществления данного изобретения
Способ получения (2R, 4R)-монатина или (2S, 4S)-монатина по данному изобретению включает стадию осуществления одновременно реакции изомеризации в положении 2 (2S, 4R)-монатина или (2R, 4S)-монатина и кристаллизации (2R, 4R)-монатина или (2S, 4S)-монатина в присутствии альдегида в смешанном растворителе из воды и органического растворителя, причем рН находится в диапазоне от 4 до 11.
Монатин, подлежащий использованию в способе по данному изобретению, включает не только (2S, 4R)-монатин или (2R, 4S)-монатин, взятые отдельно, но также смеси (2S, 4R)-монатина и (2R, 4R)-монатина при соответствующих соотношениях и смеси (2R, 4S)-монатина или (2S, 4S)-монатина при соответствующих соотношениях.
Способ по изобретению, в частности, предпочтительно используют при необходимости получения селективного выхода (2R, 4R)-монатина из монатина, оптически активного в положении 4, где (2S, 4R)-монатин и (2R, 4R)-монатин находятся в соответствующем соотношении или при необходимости получения селективного выхода (2S, 4S)-монатина из монатина, оптически активного в положении 4, где (2R, 4S)-монатин и (2S, 4S)-монатин находятся при соответствующем соотношении.
Монатин, подлежащий использованию в качестве исходного вещества для способа по изобретению, может быть в любой форме различных солей, такой как соль натрия, соль калия и соль аммония. Равно как и (2R, 4R)-монатин и (2S, 4S)-монатин, полученные способом по изобретению, могут быть также в любой форме различных солей.
Ниже иллюстрируется способ получения оптически активного монатина, описанный в описании международной публикации WO 03/059865. Например, (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровую кислоту, представленную формулой (6), гидрируют в присутствии катализатора, такого как родий на углероде, получая реакционную смесь, содержащую (2R, 4S)-монатин и (2S, 4S)-монатин:

Путем отфильтровывания катализатора от реакционной смеси и затем кристаллизации продукта в смешанном растворителе из воды и спирта можно селективно получить (2S, 4S)-монатин.
В этом случае (2R, 4S)-монатин, содержащийся в реакционной смеси, выделяется в маточный раствор. Однако, осуществляя одновременно кристаллизацию и реакцию изомеризации в конкретных условиях в соответствии с предлагаемым способом, (2R, 4S)-монатин превращают в (2S, 4S)-монатин, тем самым получая (2S, 4S)-монатин более эффективным способом.
Маточный раствор, оставшийся после кристаллизации, после выделения (2S, 4S)-монатина из реакционной смеси (2R, 4S)-монатина и (2S, 4S)-монатина, содержит (2R, 4S)-монатин в виде оптического изомера в положении 2 предполагаемого (2S, 4S)-монатина при более высоком отношении, чем предполагаемый монатин. Способ по данному изобретению может быть также применен к маточному раствору, чтобы изомеризовать (2R, 4S)-монатин в (2S, 4S)-монатин, который затем выделяют из маточного раствора. Учитывая эффективность реакции изомеризации, данное изобретение предпочтительно применяют не к реакционной смеси до кристаллизации, а к маточному раствору, содержащему предполагаемый изомер при более высоком отношении после кристаллизации. Как описано выше, кристалл (2S, 4S)-монатина, полученный из маточного раствора после кристаллизации способом по изобретению, рециркулируют на любой одной из стадий ряда обработок монатина, чтобы повысить продуктивность. Например, кристалл монатина добавляют к реакционной смеси на стадии кристаллизации (2S, 4S)-монатина из реакционной смеси (2R, 4S)-монатина и (2S, 4S)-монатина, чтобы более эффективно получить (2S, 4S)-монатин. Когда чистота (2S, 4S)-монатина высока, как получено из маточного раствора после кристаллизации, кристалл может быть смешан фактически с кристаллом (2S, 4S)-монатина, полученного кристаллизацией из реакционной смеси.
Случай получения (2S, 4S)-монатина иллюстрирован и описан выше. Это же справедливо и в случае получения (2R, 4R)-монатина.
Согласно способу по изобретению для реакции изомеризации используют альдегид. В качестве альдегида подходят алифатический или ароматический альдегид.
В качестве алифатического альдегида могут быть использованы насыщенные или ненасыщенные альдегиды с 1-7 углеродных атомов, например, формальдегид, ацетоальдегид, пропиональдегид, н-бутилальдегид, 1-бутилальдегид, н-валеральдегид, капрональдегид, н-гептилальдегид, акролеин и метакролеин.
В качестве ароматического альдегида могут быть использованы, например, бензальдегид, салицилальдегид м-гидроксибензальдегид, п-гидроксибензальдегид, о-нитробензальдегид, п-нитробензальдегид, 5-нитросалицилальдегид, анисовый альдегид, о-ванилин, ванилин, фурфураль и пиридоксаль.
В качестве альдегида особенно предпочтительны салицилальдегид, пиридоксаль и о-ванилин.
Такой альдегид может быть использован в диапазоне от 0,01 до 1 молярного эквивалента, предпочтительно от 0,05 до 0,5 молярного эквивалента по отношению к монатину, имеющемуся в системе.
Реакцию изомеризации и кристаллизацию согласно способу по изобретению осуществляют одновременно в присутствии альдегида, в то время как в качестве растворителей (растворитель для реакции и растворители для кристаллизации) используют смешанный растворитель из воды и органического растворителя. В качестве органического растворителя используют органический растворитель, смешивающийся с водой. В частности, предпочтительными являются спирты, такие как метанол, этанол, пропанол и изопропанол. В качестве органического растворителя можно смешать вместе и использовать два различных типа органических растворителей. Отношение органического растворителя и воды предпочтительно устанавливают в пределах от 1:0,01 до 1:1, более предпочтительно от 1:0,1 до 1:0,5, в виде объемного отношения органический растворитель:вода. Когда доля воды выше, кристаллизация, вероятно, происходит с большими трудностями. Если доля органического растворителя выше, это также неблагоприятно, поскольку исходный монатин хуже растворяется в таком растворителе.
Температуру реакции изомеризации и кристаллизации устанавливают в диапазоне предпочтительно от 0 до 100°C, более предпочтительно от 40 до 80°C. Продолжительность реакции изомеризации и кристаллизации находится в диапазоне предпочтительно от 10 часов до 2 недель, более предпочтительно от 15 часов до 10 дней.
pH устанавливают в диапазоне от 4 до 13, предпочтительно от 4,5 до 10, более предпочтительно от 5 до 9. pH можно регулировать, используя кислоту или щелочь. Кислота включает, но не ограничиваясь ими, органические кислоты, такую как уксусная кислота, или неорганические кислоты, такие как хлористоводородная кислота и серная кислота. Щелочь включает, например, но не ограничиваясь ими, гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия, или органические основания, такие как аммиак и амин. Когда значение pH слишком низкое, повышается вероятность кристаллизации (2R, 4S)-монатина или (2S, 4R)-монатина. Если значение pH слишком высокое, это невыгодно, поскольку кристаллы почти не осаждаются.
Далее изобретение подробно иллюстрируется нижеследующими примерами. Однако примеры никоим образом не ограничивают данное изобретение. В Примерах оптическую чистоту определяют методом ВЭЖХ при нижеследующих условиях.
<Колонка для выделения оптического изомера>
SUMICHIRAL OA-7100, производимая SUMIKA ANALYSIS CENTER
<Элюэнт>
20 мМ фосфатный буфер (pH 2,8) : ацетонитрил - 7:3
<Температура колонки>
10°C
<Скорость потока>
0,6 мл/мин
<Ссылочный пример 1>
Получение 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровой кислоты
Индолпировиноградную кислоту (12,30 г; 58,7 моль, с чистотой 97,0 %, по массе (в дальнейшем аббревиатура - мас.%)) добавляют к и растворяют в 2,45 г гидроксида натрия, растворенного в 209 ммоль воды. К полученному раствору в атмосфере азота при 35°C на протяжении 2 часов добавляют 47,61 г 25 мас.% водного раствора гидроксида натрия и раствор смеси 25,85 г (293,5 ммоль) пировиноградной кислоты и 25,85 г воды, в то время как реакционную систему поддерживают при рН 11,0. Затем полученную смесь перемешивают в течение 14 часов. Таким образом получают реакционный раствор, содержащий 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровую кислоту (выход по отношению к индолпировиноградной кислоте составлял 44,1%). К реакционному раствору для нейтрализации (рН 6,91) добавляют 3,60 г 1N хлористоводородной кислоты, получая 275 мл нейтрализованного реакционного раствора.
168 мл полученного таким образом нейтрализованного реакционного раствора загружают в колонку (диаметром 4,8 см) с полимером, содержащую 840 мл синтетического адсорбирующего агента (DIAION-SP207, изготовленного Mitsubishi Chemical), и элюируют чистой водой при скорости потока 23,5 мл/мин, выделяя от 1,7 до 2,9 L/L-R и получая 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровую кислоту с выходом 66,3%.
(ЯМР-спектр)
1Н-ЯМР (400 МГц, D2O) δ: 3,03 (д, 1Н, J=14,6 Гц), 3,11 (д, 1Н, J=14,6 Гц), 3,21 (д, 1Н, J=18,1 Гц), 3,40 (д, 1Н, J=18,1 Гц), 7,06-7,15 (м, 3Н), 7,39 (д, 1Н, J=7,8 Гц), 7,66 (д, 1Н, J=7,8 Гц).
13С-ЯМР (400 МГц, D2O) δ: 35,43, 47,91, 77,28, 109,49, 112,05, 119,44, 119,67, 121,91, 125,42, 128,41, 136,21, 169,78, 181,43, 203,58.
(Масс-спектр)
ЭРИ-МС (масс-спектрометрия с электрораспылительной ионизацией; ESI-MS;) теоретическое значение: C14H13N06 = 291,07
ЭРИ-МС экспериментальное значение: 290,02 [M-H]-
<Ссылочный пример 2>
Получение 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
1,0 г (4,92 ммоль) Индол-3-пировиноградной кислоты добавляют к и растворяют в 10 мл водного насыщенного раствора карбоната натрия. Затем полученный раствор доводят до рН 12,55, используя 25% водный раствор гидроксида натрия. После добавления к раствору 1,3 г (14,8 ммоль) пировиноградной кислоты полученный раствор доводят до pH 12,6, используя 25% водный раствор гидроксида натрия и подвергают взаимодействию при температуре окружающей среды в течение 2 часов, получая реакционный раствор, содержащий 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровую кислоту. В то время как раствор поддерживают около нейтрального рН, используя 25% водный раствор гидроксида натрия, к раствору добавляют 1,37 г (19,7 ммоль) гидрохлорида гидроксиламина и перемешивают при температуре окружающей среды в течение 4 часов. Используя концентрированную хлористоводородную кислоту, реакционный раствор доводят до кислого рН и экстрагируют органические вещества этилацетатом. Органический слой промывают насыщенным водным хлоридом натрия и сушат над безводным сульфатом магния. Затем полученный слой концентрируют, получая остаток. Остаток перекристаллизовывают из 28% водного аммиака и этанола, получая кристаллическую соль аммония (0,52 г; 1,5 ммоль) 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (при выходе 31% против индол-3-пировиноградной кислоты).
(ЯМР-спектр)
1Н-ЯМР (ДМСО-d6) δ: 2,66 (с, 2Н), 2,89 (д, 1Н, J=14,4 Гц), 3,04 (д, 1Н, J=14,4 Гц), 6,89-6,94 (м, 1Н), 6,97-7,03 (м, 1Н), 7,11 (д, 1Н, J=2,8 Гц), 7,27 (д, 1Н, J=7,8 Гц), 7,53 (д, 1Н, J=7,8 Гц), 10,71 (шир.с, 1Н).
(Масс-спектр)
ЭРИ-МС теретическое значение: C14H14N2О6=306,28
ЭРИ-МС экспериметальное значение: 305,17 [M-H]-
<Ссылочный пример 3>
Получение 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
10,0 г (49,2 ммоль) Индол-3-пировиноградной кислоты добавляют к и растворяют в 98 мл водного насыщенного раствора карбоната натрия. Затем полученный раствор доводят до pH 12,4, используя 25% водный раствор гидроксида натрия. После добавления к раствору 16,3 г (147,6 ммоль) пирувата натрия полученную смесь подвергают взаимодействию при температуре окружающей среды в течение 2 часов, получая реакционный раствор, содержащий 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровую кислоту. В то время как раствор поддерживают около нейтрального pH, используя 25% водный раствор гидроксида натрия, к раствору добавляют 13,7 г (197 ммоль) гидрохлорида гидроксиламина и перемешивают при температуре окружающей среды в течение 4 часов. Используя концентрированную хлористоводородную кислоту, реакционный раствор доводят до кислого pH и экстрагируют органические вещества в этилацетате. Органический слой промывают насыщенным водным хлоридом натрия и сушат над безводным сульфатом магния. Затем полученный слой концентрируют, получая остаток. Остаток перекристаллизовывают из 28% водного аммиака и этанола, получая кристаллическую соль диаммония (5,51 г; 16,2 ммоль) 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (при выходе 32% против индол-3-пировиноградной кислоты).
<Ссылочный пример 4>
Получение 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
73,8 г (352 ммоль) Индол-3-пировиноградной кислоты добавляют к и растворяют в 917 мл 1,6 мас.% водного раствора гидроксида натрия. Затем полученный реакционный раствор доводят до 35°C. В то время как раствор поддерживают при pH 11,1, используя 30% водный раствор гидроксида натрия, к смеси по каплям на протяжении 2 часов добавляют 310,2 г (1761 ммоль) 50% водного раствора пировиноградной кислоты. После протекания реакции в течение еще 4,5 часов получают реакционный раствор, содержащий 4-гидрокси-4-(3-индолилметил)-2-кетоглутаровую кислоту. В то время как раствор поддерживают при pH 7, используя 30% водный раствор гидроксида натрия, к раствору добавляют 367,2 г (2114 ммоль) 40% раствора гидрохлорида гидроксиламина и перемешивают при 5°C в течение 17,5 часов. Используя концентрированную хлористоводородную кислоту, реакционный раствор доводят до pH 2 и экстрагируют органические вещества этилацетатом. Органический слой промывают насыщенным водным хлоридом натрия и концентрируют, получая остаток. Остаток перекристаллизовывают из 60 мл 28% водного аммиака и 1350 мл 2-пропанола, получая кристаллическую соль диаммония (43,4 г; 142 ммоль) 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (при выходе 40% против индол-3-пировиноградной кислоты).
<Ссылочный пример 5>
Получение соли (R)-(+)-1-фенилэтиламина (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
44,7 г (0,131 моль) аммонийной соли 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты растворяют в 500 мл воды при 25°C. Полученный водный раствор доводят до pH 2, используя 25,5 г 36% хлористоводородной кислоты. Кислый раствор экстрагируют в 1300 мл этилацетата. Этилацетатный раствор промывают 200 мл водного насыщенного хлорида натрия. К полученному этилацетатному раствору добавляют 500 мл водного раствора карбоната натрия (13,9 г (0,131 моль) карбоната натрия) и перемешивают, чтобы разделить водный щелочной раствор от этилацетата. К полученному водному щелочному раствору добавляют 23,1 г 36% хлористоводородной кислоты, чтобы довести рН раствора до 2. К полученному кислому раствору по каплям добавляют 6,99 г (57,6 ммоль) (R)-(+)-1-фенилэтиламина, перемешивают при 25°C в течение одного часа. Образовавшиеся кристаллы отфильтровывают и сушат при пониженном давлении, получая соль (R)-(+)-1-фенилэтиламина и (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксиминоглутаровой кислоты (21,8 г; 47,8 ммоль) (с выходом 72,7% и оптической чистотой 87,4%).
1Н-ЯМР (400 МГц, ДМСО-d6) δ: 1,48 (д, 3Н, J=6,8 Гц), 2,63 (д, 1Н, J=14,0 Гц), 2,70 (д, 1Н, J=14,0 Гц), 2,90 (д, 1Н, J=14,1 Гц), 3,06 (д, 1Н, J=14,1 Гц), 4,40 (кв, 1Н, J=6,8 Гц), 6,91-7,54 (м, 10Н).
<Ссылочный пример 6>
Получение соли (S)-(-)-1-фенилэтиламина (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
После отделения кристаллов, полученных в Примере 5, к фильтрату далее по каплям добавляют 7,12 г (58,7 моль) (S)-(-)-1-фенилэтиламина и перемешивают при 25°C в течение одного часа. Образовавшиеся кристаллы отфильтровывают и сушат при пониженном давлении, получая (23,8 г; 53,3 ммоль) соль (S)-(-)-1-фенилэтиламина и (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (с выходом 81,1% и оптической чистотой 92,1%).
<Ссылочный пример 7а>
Получение аммонийной соли (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
200 мл воды и 18,5 г 28% водного аммиака добавляют к 21,8 г (51,0 моль) соли (R)-(+)-1-фенилэтиламина (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты для растворения кислоты и к этому раствору добавляют 200 мл толуола и перемешивают. Водный слой после экстракции нагревают до 60°C. К полученному водному раствору по каплям добавляют 900 мл 2-пропанола на протяжении 2 часов. Полученный водный раствор 2-пропанола охлаждают до 10°С на протяжении 5 часов. Затем перемешивание проводят при 10°C в течение 10 часов. Образовавшиеся кристаллы отфильтровывают и сушат при пониженном давлении, получая 14,75 г аммонийной соли (4S)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (с выходом 85,1% и оптической чистотой 99,0%).
Температура плавления: 205°C (разложение)
Удельное вращение; [α]20D+13,4 (c=1,00, H2О)
<Ссылочный пример 7b>
Получение аммонийной соли (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты
Как описано выше в Ссылочном примере, 16,2 г аммонийной соли (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты получают из 23,8 г (53,3 ммоль) соли (R)-(+)-1-фенилэтиламина (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты (с выходом 89,3% и оптической чистотой 99,9%).
Удельное вращение: [α]20D-13,6 (c=1,00, H2О)
<Ссылочный пример 8>
Получение (2R, 4R)-монатина
13,2 г (38,7 ммоль) аммонийной соли (4R)-4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты, полученной в Ссылочном примере 7, добавляют к и растворяют в 135 мл 28% водного аммиака и к этому раствору добавляют 6,93 г 5% родия на углероде (50%-влажный продукт) и подвергают взаимодействию при давлении водорода 1 МПа и 25°C. Через 24 часа катализатор отфильтровывают (при помощи 0,2-микронного фильтра). В полученном фильтрате растворяют 2,54 г (18,4 ммоль) карбоната калия. Раствор концентрируют. К 32,7 г полученного концентрата добавляют 20 мл воды и 45 мл этанола, перемешивают при 25°C, и к этому по каплям добавляют 60 мл этанола на протяжении 3 часов. Затем перемешивание проводят при 25°C в течение 20 часов для кристаллизации. 9,78 г образовавшихся влажных кристаллов растворяют в 12 мл воды и к этому раствору добавляют 24 мл этанола и дополнительно по каплям добавляют 51 мл этанола на протяжении 3 часов. Этанольный раствор охлаждают до 15°C на протяжении 4 часов и перемешивают при 15°C в течение 10 часов. 7,08 г полученного влажного кристаллического вещества сушат при пониженном давлении, получая 5,7 г калиевой соли предполагаемого (2R, 4R)-монатина.
1Н-ЯМР (400 МГц, D2О) δ: 2,06 (дд, 1Н, J=11,8, 15,3 Гц), 2,67 (дд, 1Н, J=2,0, 15,2 Гц), 3,08 (д, 1Н, J=14,4 Гц), 3,28 (д, 1Н, J=14,4 Гц), 3,63 (дд, 1Н, J=2,2, 12,2 Гц), 7,12-7,16 (м, 1Н), 7,20-7,24 (м, 2Н), 7,48-7,49 (м, 1Н), 7,71-7,73 (м, 1Н).
ЭРИ-МС теоретическое значение: C14H16N2О5=292,29
ЭРИ-МС экспериментальное значение: 291,28 [M-H]-
<Пример 1>
170 г маточного раствора для кристаллизации, полученного в Ссылочном примере 8, концентрируют до 25 г (содержащий 6,65 г (19,1 ммоль) монатина; (2S, 4R):(2R, 4R)=74:26), который затем доводят до рН 6,6, используя 0,69 г 35% хлористоводородной кислоты. К полученному раствору добавляют 0,467 г (3,82 ммоль) салицилальдегида и к этому добавляют 25 мл этанола и перемешивают при продувке азотом при 65°C в течение 189 часов, подвергая осуществлению одновременно реакции изомеризации и кристаллизации. Во время протекания реакции реакционный раствор периодически доводят до рН 6,6, используя 35% хлористоводородную кислоту. Соотношение изомеров монатина в реакционном растворе, в суспензии, составляло (2S, 4R):(2R, 4R)=11:89. Образовавшиеся кристаллы отделяют фильтрацией (5,14 г влажных кристаллов) и сушат при пониженном давлении, получая калиевую соль (3,98 г; 11,0 ммоль) требуемого (2R, 4R)-монатина (чистота 99,3% согласно ВЭЖХ).
<Пример 2>
Используя 0,788 г (3,82 ммоль) гидрохлорида пиридоксаля вместо салицилальдегида, используемого в Примере 1, перемешивание проводят при 65°C в течение 18 часов, для одновременного протекания реакции изомеризации и кристаллизации. Соотношение изомеров монатина в реакционном растворе, в суспензии, составляло (2S, 4R):(2R, 4R)=34:66. Образовавшиеся кристаллы отделяют фильтрацией, получая 19 г влажного кристаллического вещества. Соотношение изомеров монатина во влажном кристаллическом веществе составляло (2S, 4R):(2R, 4R)=27:73.
<Ссылочный пример 9>
Получение смеси (2R, 4R)-монатина и (2S, 4S)-монатина
Соль аммония (7,0 г; 20,6 ммоль) 4-гидрокси-4-(3-индолилметил)-2-гидроксииминоглутаровой кислоты, полученную в Ссылочном примере 4, растворяют в 60 мл 28% водного аммиака и к этому раствору добавляют 3,69 г 5% родия на углероде (50%-влажный продукт) и подвергают взаимодействию при давлении водорода 1 МПа и 25°C. Через 24 часа катализатор отфильтровывают (при помощи 0,2-микронного фильтра). Полученный раствор концентрируют. К 28,2 г полученного концентрата добавляют 28,8 мл воды и 1,18 г уксусной кислоты и перемешивают при 25°C в течение 1,5 часов для кристаллизации. Образовавшиеся кристаллы отфильтровывают и отделяют, получая 7,77 г влажных кристаллов. Влажные кристаллы сушат при пониженном давлении, получая свободную форму (5,45 г) предполагаемой смеси (2R, 4R)-монатина и (2S, 4S)-монатина (чистота 87,3%).
1Н-ЯМР (400 МГц, D2О) δ: 2,06 (дд, 1Н, J=11,8, 15,3 Гц), 2,67 (дд, 1Н, J=2,0, 15,2 Гц), 3,08 (д, 1Н, J=14,4 Гц), 3,28 (д, 1Н, J=14,4 Гц), 3,63 (дд, 1Н, J=2,2, 12,2 Гц), 7,12-7,16 (м, 1Н), 7,20-7,24 (м, 2Н), 7,48-7,49 (м, 1Н), 7,71-7,73 (м, 1Н).
ЭРИ-МС теоретическое значение: C14H16N2О5=292,29
ЭРИ-МС экспериментальное значение: 291,28 [M-H]-
<Пример 3>
55,2 г Маточного раствора для кристаллизации, полученного в Ссылочном примере 9, концентрируют до 7,7 г (содержащий 2,00 г (5,77 ммоль) монатина; (2S, 4R):(2R, 4S):(2R, 4R):(2S, 4S)=84,6: 84,6:15,4:15,4), который затем доводят до pH 5, используя 1,17 г уксусной кислоты. Добавляют 0,141 г (1,15 ммоль) салицилальдегида и затем добавляют 17,5 мл метанола и полученную смесь перемешивают в потоке азота при 65°C в течение 143 часов для одновременного осуществления реакции изомеризации и кристаллизации. Во время протекания реакции, реакционный раствор периодически доводят до pH 5, используя уксусную кислоту. Соотношение изомеров монатина в реакционном растворе, в суспензии, составляло (2S, 4R):(2R, 4S):(2R, 4R):(2S, 4S)=18,6:18,6:81,4:81,4. Образовавшиеся кристаллы отделяют фильтрацией (1,95 г влажных кристаллов) и сушат при пониженном давлении, получая кристаллы (1,31 г) свободной формы смеси монатина (с выходом 69,8%). Соотношение изомеров монатина составляло (2S, 4R):(2R, 4S):(2R, 4R):(2S, 4S)=13,0:13,0:87,0: 87,0.
Затем 1,31 г полученного выше кристаллического вещества смешивают с 5,45 г кристаллического вещества, полученного в Ссылочном примере 9. Затем смесь диспергируют в 15 мл воды и к этому добавляют 2,42 мл 8N водного раствора гидроксида натрия для растворения. К полученному раствору при 35°C добавляют 15 мл метанола и к этому дополнительно по каплям добавляют 70 мл метанола на протяжении 2 часов. После завершения капельного добавления метанольный раствор охлаждают до 10°C на протяжении 2,5 часов и перемешивают при 10°C в течение 30 минут для кристаллизации. Образовавшиеся кристаллы отфильтровывают и отделяют (5,47 г влажных кристаллов), сушат при пониженном давлении, получая натриевую соль (4,24 г) предполагаемой смеси (2R, 4R)-монатина и (2S, 4S)-монатина (с чистотой 99,1%).
1Н-ЯМР (400 МГц, D2О) δ: 2,06 (дд, 1Н, J=11,8, 15,3 Гц), 2,67 (дд, 1Н, J=2,0, 15,2 Гц), 3,08 (д, 1Н, J=14,4 Гц), 3,28 (д, 1Н, J=14,4 Гц), 3,63 (дд, 1Н, J=2,2, 12,2 Гц), 7,12-7,12 (м, 1Н), 7,20-7,24 (м, 2Н), 7,48-7,49 (м, 1Н), 7,71-7,73 (м, 1Н).
ЭРИ-МС теоретическое значение: C14H16N2О5 = 292,29
ЭРИ-МС экспериментальное значение; 291,28 [M-H]-
Сравнительный пример 1
170 г Маточного раствора для кристаллизации, полученного в Ссылочном примере 8, концентрируют до 25 г (содержащий 6,65 г (19,1 ммоль) монатина; (2S, 4R):(2R, 4R)=74:26), который затем доводят до pH 14, используя 1,75 г 50% гидроксида калия. К полученному раствору добавляют 0,467 г (3,82 ммоль) салицилальдегида и к этому добавляют 25 мл этанола и перемешивают при 65°C в течение 64 часов. Однако никаких кристаллов не осаждается. Соотношение изомеров монатина в реакционном растворе составляло (2S, 4R):(2R, 4R)=45:55.
Промышленная применимость
Согласно данному изобретению монатин с различными конфигурациями в положениях 2 и 4 может быть превращен эффективным способом в требуемый монатин с одинаковыми конфигурациями в положениях 2 и 4 посредством реакции изомеризации. Таким образом, (2R, 4R)-монатин и (2S, 4S)-монатин могут быть получены эффективным способом, что является полезным в промышленном отношении.
Настоящая заявка основана на японской патентной заявке 2004-053717, поданной в Японии, содержание которой в полном объеме включено в данное описание.
Реферат
Настоящее изобретение относится к способу получения оптически активного монатина с одинаковыми конфигурациями в положении 2 и 4, который может быть применен в качестве подсластителя. Способ изомеризации состоит в том, что монатин, имеющий разные конфигурации в положении 2 и 4, подвергают изомеризации во 2-м положении монатина в присутствии альдегида в смешанном растворителе из воды и органического растворителя при рН от 4 до 11 с одновременной кристаллизацией монатина, конфигурации в 2-положении и 4-положении которого идентичны друг другу. Технический результат - разработка эффективного способа получения оптически активных изомеров монатина. 7 н. и 5 з.п. ф-лы.
Формула




Комментарии