Способ получения оптических изомеров мета-хлорбензгидриламина - RU2692684C1
Код документа: RU2692684C1
Чертежи
Описание
Изобретение относится к области органической химии, в частности к способу получения оптических изомеров и может быть использовано в фармацевтической промышленности.
Известен способ получения оптических изомеров через образование диастереомеров, которые, в отличие от энантиомеров, обладают индивидуальными физическими свойствами и могут быть отделены друг от друга, например, по различию в растворимости [Илиел Э. Основы органической стереохимии; Пер. с англ. / Илиел Э., Вайлен С., Дойл М. - М.: Бином, 2007. - С. 220].
Растворитель, используемый при кристаллизации, оказывает значительное влияние на полноту расщепления диастереомеров, поэтому выбор растворителя является самым трудоемким и ключевым этапом для проведения успешного разделения оптических изомеров при использовании данного метода.
Известен способ получения диастереомеров мета-хлорбензгидриламина (тартратов) [п. 4, 6 ф-лы RU 2537361 С1, МПК(2006.01) С07С 275/18, С07С 273/0, A61K 31/17, А61Р 25/08, опубл. 10.01.2015], в котором из рацемической смеси мета-хлорбензгидриламина в присутствии (R,R)-(+)- или (S,S)-(-)-винных кислот в органическом растворителе получают индивидуальные диастереомерные соли мета-хлорбензгидриламина (тартраты) с их последующей дискриминацией по различию в растворимости.
Недостатками этого способа являются трудности, связанные с выбором растворителя для отделения одного диастереомера от другого; использование многократных перекристаллизаций для диастереомерного обогащения тартратов, что снижает выход целевых энантиомеров мета-хлорбензгидриламина; использование больших объемов растворителей; ведение процесса при нагревании до 60-65°С.
Известен способ получения оптических изомеров мета-хлорбензгидриламина, выбранный в качестве прототипа, в котором рацемический амин обрабатывают оптически активной (R,R)-(+)-винной кислотой (L-(+)-винной кислотой) в условиях отсутствия растворителя [Куксенок В.Ю., Синтез производных мочевин, содержащих бензгидрильные и уреидные фармакофоры. Первый пример получения энантиомерно обогащенных форм препарата галодиф, диссертация на соискание ученой степени кандидата химических наук, Томск, 2016, 178 с.]. Мольное соотношение (R,R-(+)-винной кислоты к амину составляет 1:2. Реакцию проводят при помощи активного перетирания образующейся пасты при комнатной температуре в течение 30 мин. После исчезновения винной кислоты из реакционной массы полученную диастереомерную соль тщательно промывают гексаном от непрореагировавшего амина. Гексановый маточник отгоняют под вакуумом, получая энантиомерно обогащенный (S)-(+)-мета-хлорбензгидриламин. Полученный диастереомерно обогащенный (R)-(-)-мета-хлорбензгидриламина (+)-тартрат для дальнейшего обогащения перекристаллизовывают из метанола. Энантиомерно обогащенный амин повторно обрабатывают (R,R)-(+)-винной кислотой в тех же условиях для удаления примеси минорного (-)-энантиомера.
Однако в способе имеются трудности, связанные с использованием гексана для промывки (R)-(-)-мета-хлорбензгидриламина (+)-тартрата, что сужает возможность применения других растворителей; с использованием перекристаллизации (R)-(-)-мета-хлорбензгидриламина (+)-тартрата из метанола (лимитированный продукт) для его дальнейшего обогащения, что является дополнительными затратами в способе получения оптических изомеров мета-хлорбензгидриламина; с отсутствием способа получения (R)-(-)-мета-хлорбензгидриламина.
Технический результат предложенного изобретения заключается в получении индивидуальных оптических изомеров (энантиомеров) мета-хлорбензгидриламина в условиях отсутствия растворителя при комнатной температуре.
Способ получения оптических изомеров (R)-(-)- и (S)-(+)-мета-хлорбензгидриламина, также как в прототипе, заключается в перетирании рацемического мета-хлорбензгидриламина с оптически активной винной кислотой в условиях отсутствия растворителя в течение не менее 30 мин. при комнатной температуре.
Согласно изобретению перетирают рацемический мета-хлорбензгидриламин с 50 мол. % (S,S)-(-)-винной кислоты, затем к реакционной массе добавляют неполярный растворитель, перемешивают ее до растворения непрореагировавшего (R)-(-)-мета-хлорбензгидриламина, отфильтровывают кристаллический осадок тартрата и промывают его на фильтре неполярным растворителем от непрореагировавшего амина.
Полученный (S)-(+)-мета-хлорбензгидриламина (S,S-(-)-тартрат нейтрализуют неорганическим основанием до щелочной реакции, экстрагируют неполярным растворителем, отделяют органический слой, сушат полученный экстракт, отгоняют растворитель из экстракта и получают (S)-(+)-мета-хлорбензгидриламин. Из фильтрата отгоняют неполярный растворитель и получают (R)-(-)-мета-хлорбензгидриламин, который повторно перетирают с меньшим, чем 50 мол. %, количеством (S,S)-(-)-винной кислоты до исчезновения винной кислоты, получая (S)-(+)-мета-хлорбензгидриламина (S,S)-(-)-тартрат. Затем к реакционной массе добавляют неполярный растворитель, перемешивают ее до растворения непрореагировавшего (R)-(-)-мета-хлорбензгидриламина, отфильтровывают кристаллический осадок тартрата и промывают его на фильтре неполярным растворителем от непрореагировавшего амина. Из фильтрата отгоняют неполярный растворитель и получают энантиомерно обогащенный (R)-(-)-мета-хлорбензгидриламин. Процедуру обогащения амина повторяют до установления постоянного значения величины его удельного вращения.
Использование предложенного способа получения энантиомеров мета-хлорбензгидриламина позволяет: расширить ряд используемых расщепляющих агентов; расширить ряд используемых растворителей для промывки полученного тартрата; исключить стадию перекристаллизации тартрата из метанола для его дальнейшего обогащения; получить оба оптических изомера мета-хлорбензгидриламина.
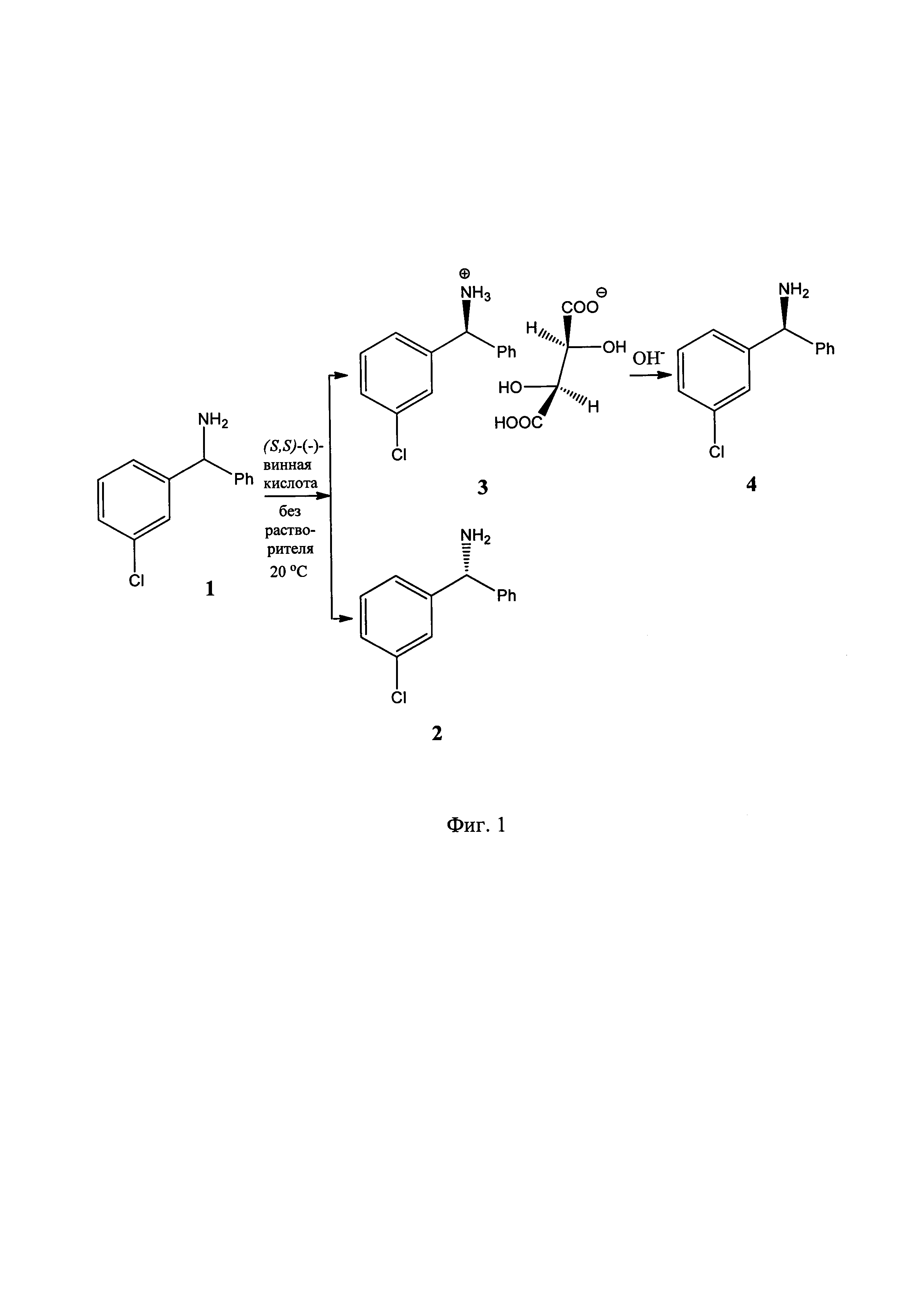
На фиг. 1 показана схема осуществления способа получения оптических изомеров мета-хлорбензгидриламина.
Далее предлагается пример, иллюстрирующий способ разделения рацемического мета-хлорбензгидриламина на оптические антиподы в отсутствие растворителя с использованием (S,S)-(-)-винкой кислоты.
Пример.
1. Расщепление рацемического мета-хлорбензгидриламина (S,S)-(-)-винной кислотой в отсутствие растворителя.
В агатовую ступку загрузили 3 г (13,8 ммоль) рацемического мета-хлорбензгидриламина (1) и 1.035 г (6,9 ммоль) (S,S)-(-)-винной кислоты и растирали полученную смесь в течение 30 минут при комнатной температуре. Конец реакции контролировали методом ТСХ (элюент н-С4Н9ОН:АсОН:H2O=(4:2:1) по исчезновению (S,S)-(-)-винной кислоты. Затем к реакционной массе добавили 15 мл гексана и перемешивали до растворения непрореагировавшего (R)-(-)-мета-хлорбензгидриламина (2), после чего отфильтровали кристаллический осадок тартрата (3), тщательно промыли его на фильтре гексаном (3 раза по 10 мл). Получили диастереомерно обогащенный тартрат (3) с выходом 48%.
Гексан из фильтрата отогнали на роторном испарителе и получили энантиомерно обогащенный (R)-(-)-мета-хлорбензгидриламин (2) с выходом 46%.
(S)-(+)-мета-хлорбензгидриламина (S,S)-(-)-тартрат (3). Выход 48%). Т. пл. 162-165°С. ЯМР1Н (D2O, 300 MHz), δм.д.: 4.44 (с, 2Н, СН-СН), 5.63 (с, 3Н), 7.29 (д, J=3.6 Hz, 1Н), 7.44 (м, 7Н).
(R)-(-)-мета-хлорбензгидриламин (2). Выход 46%. Масло. [α]D[20] (5%, EtOH)=- 6.12; е.е. 30%. ЯМР1Н (DMSO, 300 MHz), δм.д.: 5.11 (с, 1H, СН-NH2), 7.16-7.43 (м, 9Н, Ar), 7.58 (с, 2Н, NH2).
2. Нейтрализация (S)-(+)-мета-хлорбензгидриламина (S,S)-(-)-тартрата (3).
1 г (2,7 ммоль) тартрата (3) растворяли в 100 мл воды и нейтрализовали водным раствором NaOH до значения рН=8-9. Образовавшийся маслообразный амин экстрагировали гексаном (3 раза по 20 мл), отделяли органический слой от водного и сушили органический экстракт над безводным Na2SO4, после чего отогнали гексан и получили энантиомерно обогащенный (S)-(+)-мета-хлорбензгидриламин (4) с выходом 80%).
(S)-(+)-мета-хлорбензгидриламин (4). Выход 80%). Масло, е.е. 25%. ЯМР1Н (DMSO, 300 MHz), δм.д. : 5.11 (с, 1H, CH-NH2), 7.16-7.43 (м, 9Н, Ar), 7.58 (с, 2H,NH2).
3. Энантиомерное обогащение (R)-(-)-мета-хлорбензгидриламина (2) с использованием (S,S)-(-)-винную кислоты в отсутствие растворителя.
В агатовую ступку загрузили энантиомерно обогащенный (R)-(-)-мета-хлорбензгидриламин (2) с предыдущей стадии обогащения (см. пункт 1,
примера) и (S,S)-(-)-винную кислоту в количестве 25 мол. % в случае первого обогащения, либо 15 мол. % в случае второго и последующих обогащений.
Смесь растирали 30 мин. при комнатной температуре. Конец реакции контролировали методом ТСХ (элюент н-С4Н9ОН:АсОН:H2O (4:2:1) по исчезновению винной кислоты. Затем к реакционной массе добавили 15 мл гексана и перемешивали до растворения непрореагировавшего (R)-(-)-мета-хлорбензгидриламина (2), после чего отфильтровали кристаллический осадок тартрата (3), тщательно промыли его на фильтре гексаном (3 раза по 10 мл). Гексан из фильтрата отогнали на роторном испарителе и получили энантиомерно обогащенный (R)-(-)-мета-хлорбензгидриламин (2) с выходом 20%. Энантиомерное обогащение (R)-(-)-мета-хлорбензгидриламина (2) повторяли до постоянного значения его величины удельного вращения.
(R)-(-)-мета-хлорбензгидриламина (2). Выход 20%. Масло. [a]D[20] (5%, EtOH)=- 8.36; е.е. 73%. ЯМР1Н (DMSO, 300 MHz), δм.д. : 5.11 (с, 1Н, СН-NH2), 7.16-7.43 (м, 9Н, Ar), 7.58 (с, 2Н, NH2).
Реферат
Изобретение относится к области органической химии, а именно к способу получения оптических изомеров (R)-(-)- и (S)-(+)-мета-хлорбензгидриламина. Способ заключается в перетирании рацемического мета-хлорбензгидриламина с оптически активной винной кислотой в условиях отсутствия растворителя в течение не менее 30 мин при комнатной температуре. Способ характеризуется тем, что перетирают рацемический мета-хлорбензгидриламин с 50 мол.% (S,S)-(-)-винной кислоты, затем к реакционной массе добавляют неполярный растворитель, перемешивают ее до растворения непрореагировавшего (R)-(-)-мета-хлорбензгидриламина, отфильтровывают кристаллический осадок тартрата и промывают его на фильтре неполярным растворителем от непрореагировавшего амина. Полученный (S)-(+)-мета-хлорбензгидриламина (S,S)-(-)-тартрат нейтрализуют неорганическим основанием до щелочной реакции, экстрагируют неполярным растворителем, отделяют органический слой, сушат полученный экстракт, отгоняют растворитель из экстракта и получают (S)-(+)-мета-хлорбензгидриламин. Из фильтрата отгоняют неполярный растворитель и получают (R)-(-)-мета-хлорбензгидриламин, который повторно перетирают с меньшим чем 50 мол.% количеством (S,S)-(-)-винной кислоты до исчезновения винной кислоты и получают (S)-(+)-мета-хлорбензгидриламина (S,S)-(-)-тартрат. Затем к реакционной массе добавляют неполярный растворитель, перемешивают ее до растворения непрореагировавшего (R)-(-)-мета-хлорбензгидриламина, отфильтровывают кристаллический осадок тартрата и промывают его на фильтре неполярным растворителем от непрореагировавшего амина. Из фильтрата отгоняют неполярный растворитель и получают энантиомерно обогащенный (R)-(-)-мета-хлорбензгидриламин. Процедуру обогащения амина повторяют до установления постоянного значения величины его удельного вращения. Предлагаемый способ позволяет проводить процесс в отсутствие растворителя при комнатной температуре, снизить его продолжительность и увеличить энантиомерный избыток получаемых аминов. 1 ил., 1 пр.
Формула
Документы, цитированные в отчёте о поиске
Оптические изомеры (+) и (-)-бензгидрилмочевин и (+) и (-)-1-[(3-хлорфенил)-фенил-метил]мочевины, фармацевтическая композиция на их основе и способ их получения
Комментарии