Получение частично гидрированных анса-металлоценовых комплексов - RU2333215C2
Код документа: RU2333215C2
Описание
Настоящее изобретение относится к способу получения гидрированных или частично гидрированных рацемических анса-металлоценовых комплексов, к самим комплексам и их производным и к их использованию в качестве катализаторов, или компонента катализаторов полимеризации олефино-ненасыщенных соединений, или в качестве реагентов или катализаторов стереоселективного синтеза.
Помимо стереоспецифической полимеризации олефинов, энантиоселективный синтез предлагает все более интересные возможные применения хиральных металлоценовых комплексов металлов III-VI переходных групп периодической системы элементов. Как правило, в упомянутых применениях требуется использование металлоценового комплекса в форме рацемата, то есть без мезо-соединений. В случае смеси диастереомеров (рацемической и мезо-формы), получаемой обычно при синтезе металлоценов предшествующей области техники, мезо-форму необходимо отделять. Поэтому в прошлом были предприняты попытки разработать рацемоселективный синтез анса-металлоценов и были описаны соответствующие общие синтетические подходы, например, в WO 99/15538 и в DE 10030638. Данный рацемоселективный синтез анса-металлоценов протекает через промежуточное соединение бисфеноксид анса-металлоцена, или дифеноксид анса-металлоцена.
Гидрированные металлоцены, такие как дихлорид этилен-бис(тетрагидроинденил)циркония и дихлорид диметилсиландиил бис(тетрагидроинденил)циркония описаны в J. Am. Chem. Soc. (1996), 118, 2105, J. Mol. Katal. A. Chem. (1995), 102, 59, EPA 0643079 и JPA 07292019. Они подходят для получения полиолефинов, таких как изотактический полипропилен, сополимеров и эластомеров. Кроме того, известен ряд других гидрированных металлоценов, см. ссылку EPA 0581754, JPA 07041521. Гидрированные и частично гидрированные металлоцены были также описаны в качестве предшественников катализаторов полимеризации олефинов, см., например, ссылку EPA 0344887, ЕРА 0185918 и ЕРА 0537686. Гидрированные и частично гидрированные металлоцены обладают различной трехмерной структурой и различными полимеризационными свойствами по сравнению со своими ненасыщенными аналогами, и подобные измененные структура и свойства приводят к выгодным эффектам и возможным использованиям в конкретных применениях.
В предшествующей области техники получение гидрированных или частично гидрированных анса-металлоценов, в частности металлоценов, содержащих частично гидрированные инденильные системы в качестве лигандов, обычно проводят через получение бисинденилцирконоценовых дихлоридов, их выделение и последующее гидрирование с последующим выделением гидрированного или частично гидрированного продукта кристаллизацией (см, например, ссылку ЕР 839822 и цитированные там ссылки). Данный синтетический подход имеет ряд недостатков.
Обычно синтез анса-металлоценовых дихлоридных комплексов проводят общим способом с использованием тетрахлорида циркония в качестве источника металла: при этом анса-лиганд растворяют в толуоле, депротонируют действием сильных оснований, а затем вводят во взаимодействие с тетрахлоридом циркония, получая соответствующий дихлорид анса-металлоцена (плюс два эквивалента хлорида лития). Дихлорид анса-металлоцена, который преимущественно выпадает в осадок при образовании, отделяют от хлорида лития известными способами, а комплекс, в случае необходимости, затем очищают кристаллизацией.
У данного «классического» реакционного пути есть две существенные проблемы. Помимо желательного рацемата, в большинстве случаев в фактически эквивалентных количествах образуется зеркально-симметричный мезо-диастереомер. Кроме того, достигаемые по данной стратегии выходы смеси рац/мезо сравнительно невысоки (порядка от около 30 до 40%). Однако в случае, когда дихлорид анса-металлоцена нужно применять в качестве катализаторов полимеризации замещенных олефинов, можно использовать только рацематы (как указано выше). В рамках синтеза дихлоридов анса-металлоценов для этого часто требуется трудоемкое и невыгодное разделение рац/мезо, или даже разрушение мезо-комплекса. В результате этого конечный выход чистого дихлорида рац-анса-металлоцена обычно не превышает 15-20%.
Прочие недостатки связаны с эффективностью синтеза. Как описано выше, дихлорид анса-металлоцена нужно отделить от одновременно образующегося хлорида лития. Данная стадия часто сравнительно трудна, поскольку для отделения малорастворимого (в частности, в толуоле) дихлорида анса-металлоцена от также фактически нерастворимого (в толуоле) хлорида лития требуются большие количества растворителя и, следовательно, ограничиваются производительность и эффективность данного синтетического подхода.
Более того, вследствие малой растворимости дихлорида анса-металлоцена в углеводородных растворителях, концентрации комплекса в реакции гидрирования данного комплекса в большинстве случаев относительно невысоки. Это также ограничивает производительность и эффективность данного общего синтетического подхода.
По этой причине частично гидрированные анса-металлоцены до сих пор получались только при допущении значительных потерь в выходе и условий неэкономичного синтеза.
Задача настоящего изобретения заключается во избежании недостатков предшествующей области и обеспечении простого и экономичного, но в то же время высокоэффективного способа получения гидрированных и частично гидрированных рацемических металлоценовых комплексов, которые фактически не содержат мезо-изомера (в пределах точности измерения ЯМР). Особая задача настоящего изобретения состоит в разработке рацемоселективного синтетического подхода к гидрированным и/или частично гидрированным рацемическим металлоценовым комплексам, приводящего простым и недорогим способом к конечным продуктам, которые можно выделить в чистом виде. Еще одной задачей является получение гидрированных или частично гидрированных рацемических металлоценовых комплексов, которые можно использовать либо непосредственно в качестве катализаторов, в первую очередь, для полимеризации олефинов, либо использовать после модификации, например после замещения «вспомогательного лиганда», в катализаторах, в первую очередь, для полимеризации олефинов, или которые можно использовать в качестве реагентов или катализаторов стереоселективного синтеза.
Авторами изобретения найдено, что данная задача решается при помощи способа, определенного в формуле изобретения, конечных гидрированных или частично гидрированных рацемических металлоценовых комплексов (VI) и их использования в качестве катализаторов полимеризации олефино-ненасыщенных соединений, или в качестве реагентов или катализаторов стереоселективного синтеза.
Термин «мезо-форма», «рацемат» и, следовательно, также «энантиомеры» в контексте металлоценовых комплексов известны и охарактеризованы, например, у Rheingold et al., Organometallics 11 (1992), p. 1869-1876.
В настоящей заявке термин «фактически мезо-форма» означает, что более 80%, предпочтительно, по меньшей мере, 90% соединения находится в форме рацемата, в частности, предпочтительно, по меньшей мере, 95%.
В настоящем контексте термин «гидрированный или частично гидрированный» означает, что, по меньшей мере, одна пара или большая часть пар ненасыщенных атомов углерода, то есть sp2-гибридизованных атомов углерода, присутствующих в лиганде до стадии гидрирования, гидрируются, то есть присутствуют после гидрирования в водородно-насыщенном виде, а именно в виде sp3-гибридизованных атомов углерода.
Способ настоящего изобретения получения гидрированных или частично гидрированных рацемических анса-металлоценовых комплексов включает в себя следующие стадии:+
взаимодействие мостиковых или не мостиковых ароматических комплексов переходных металлов формулы I
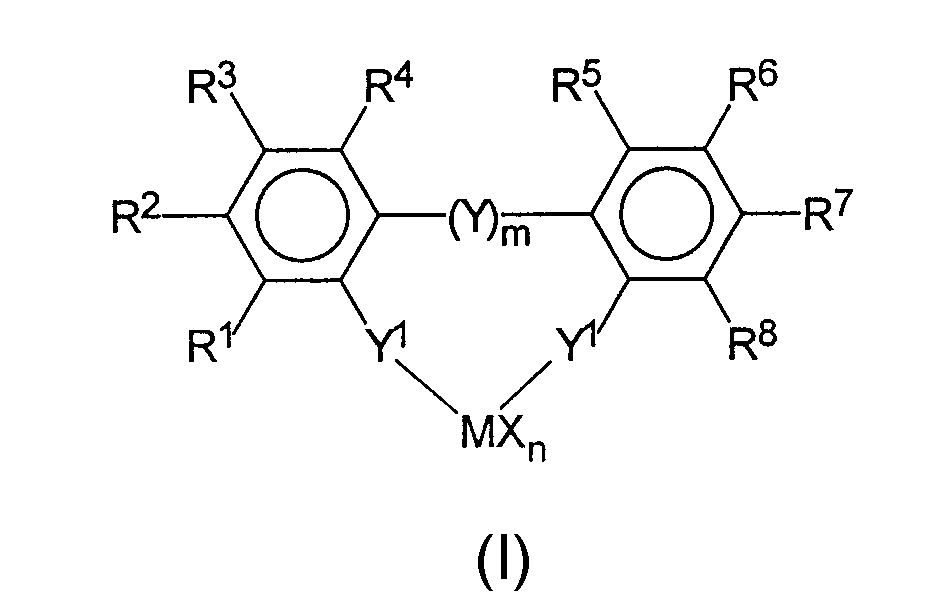
где заместители и индексы имеют следующие значения:
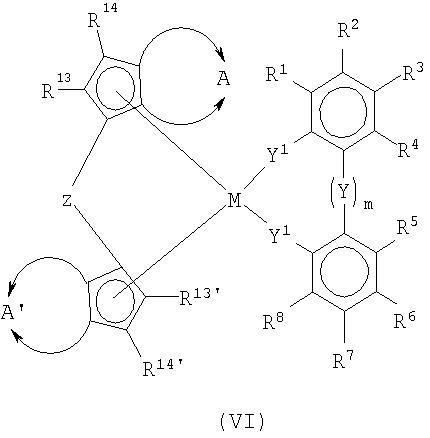
М представляет собой титан, цирконий, гафний, ванадий, ниобий, тантал, хром, молибден, вольфрам, или любой элемент III переходной группы периодической системы и лантаниды;
Х являются одинаковыми или различными и каждый из них представляет собой фтор, хлор, бром, йод, водород, С1-С10-алкил, С6-С15-арил, алкиларил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, -OR10 или -NR10R11,
n является целым числом от 1 до 4 и соответствует валентности М минус 2,
R1 по R8 являются одинаковыми или различными и каждый из них представляет собой водород, галоген, С1-С20-алкил, 3-8-членный циклоалкил, который, в свою очередь, может содержать С1-С10-алкильную группу в качестве заместителя, С6-С15-арил, алкиларил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, арилалкил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, где соседние радикалы от R2 по R7 могут также образовывать насыщенные, или частично ненасыщенные циклические группы, содержащие от 4 до 15 атомов углерода, Si(R9)3, -OR10, -SR10, -N(R10)2, Р(R10)2 и все вышеуказанные радикалы могут быть полностью или частично замещены гетероатомами,
R9 являются одинаковыми или различными и каждый представляет собой С1-С20-алкил, С3-С10-циклоалкил, С6-С15-арил, где упомянутые радикалы могут быть полностью или частично замещены гетероатомами,
R10 являются одинаковыми или различными и каждый представляет собой С1-С10-алкил, С6-С15-арил, С3-С10-циклоалкил, алкиларил или Si(R11)3,
R11 являются одинаковыми или различными и каждый представляет собой С1-С10-алкил, С6-С15-арил, С3-С10-циклоалкил, алкиларил,
Y, Y1 являются одинаковыми или различными и каждый представляет собой
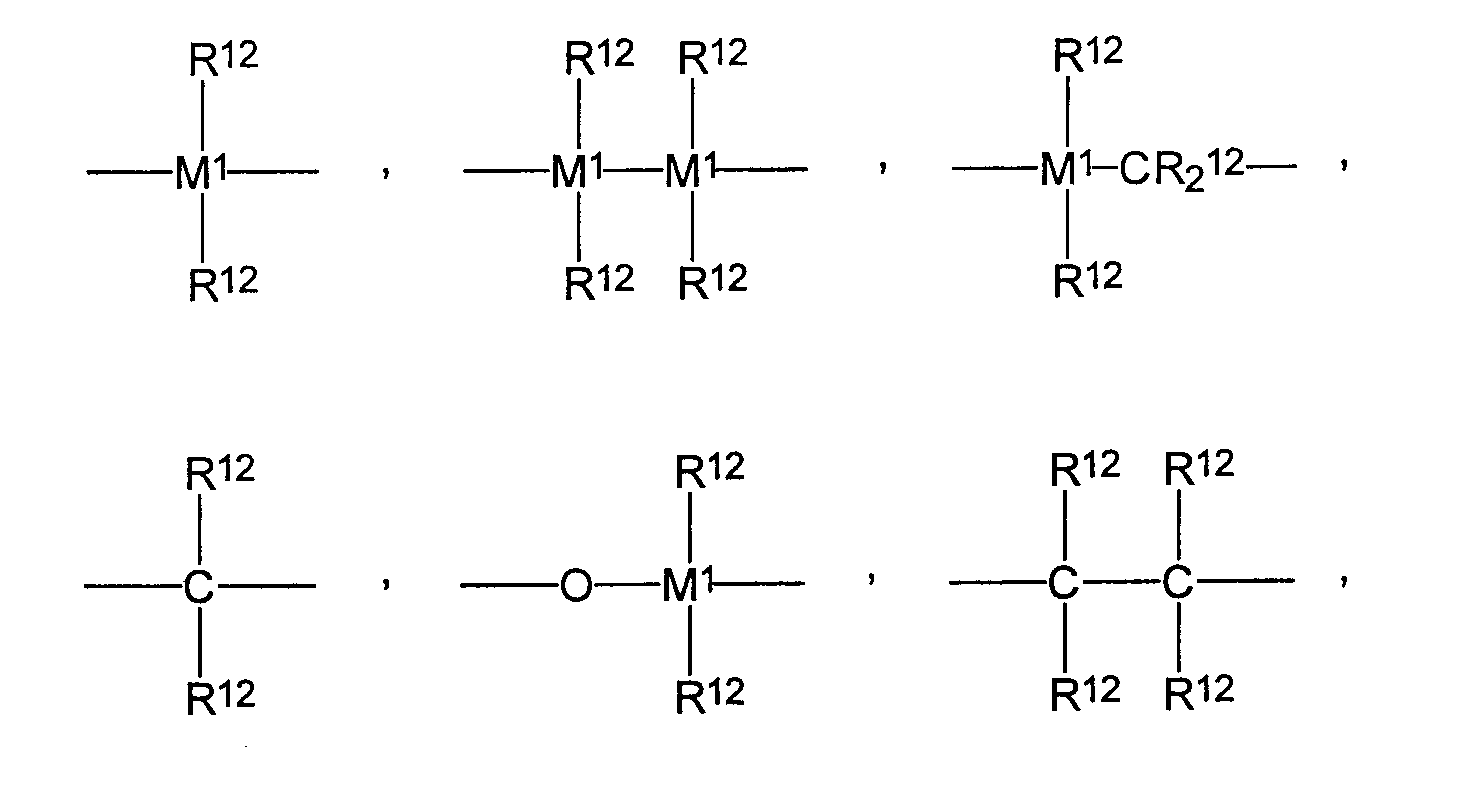
или =BR12, =AlR12, -Ge-, -Sn-, -O-, -S-, =SO, =SO2, =NR12, =CO, =PR12 или =P(O)R12, где
R12 являются одинаковыми или различными и каждый представляет собой водород, галоген, С1-С10-алкил, С1-С10-фторалкил, С6-С10-фторарил, С6-С10-арил, С1-С10-алкокси, С2-С10-алкенил, С7-С40-арилалкил, С8-С40-арилалкенил, С7-С40-алкиларил, или два радикала R12вместе со связывающими их атомами образуют цикл,
М1 представляет собой кремний, германий или олово, а
m равен 0, 1, 2 или 3, или
Y является несвязывающим и представляет собой два радикала R' и R'', где
R' и R'' аналогичны определенным для R1 по R8, а R' и R'' вместе с соседними радикалами R4, R5 могут также образовывать насыщенные, частично насыщенные или ненасыщенные циклические группы, содержащие от 4 до 15 атомов углерода,
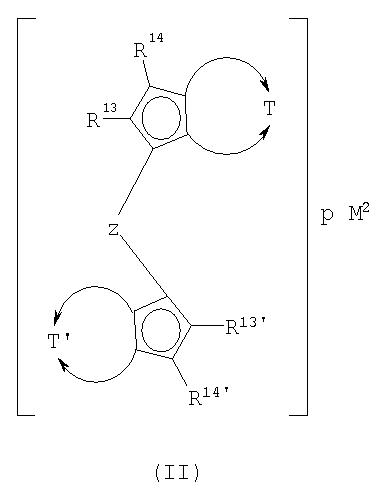
с циклопентадиенильными производными формулы II

где

представляет собой двухвалентную группу, такую как

а

представляет собой двухвалентную группу, такую как
а заместители и индексы имеют следующие значения:
R13, R13', R14, R14' являются одинаковыми или различными и каждый представляет собой водород, галоген, С1-С20-алкил, 3-8-членный циклоалкил, который, в свою очередь, может содержать С1-С10-алкильную группу в качестве заместителя, С6-С15-арил, алкиларил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, арилалкил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, -OR10, -SR10, -N(R10)2, -Р(R10)2 или Si(R9)3,
Z представляет собой -[Q(R15)(R16)]q-группу, в которой
Q могут быть одинаковыми или различными и каждый представляет собой кремний, германий, олово или углерод,
R15, R16, каждый, представляет собой водород, С1-С10-алкил, С3-С8-циклоалкил или С6-С15-арил, а
q равен 1, 2, 3 или 4,
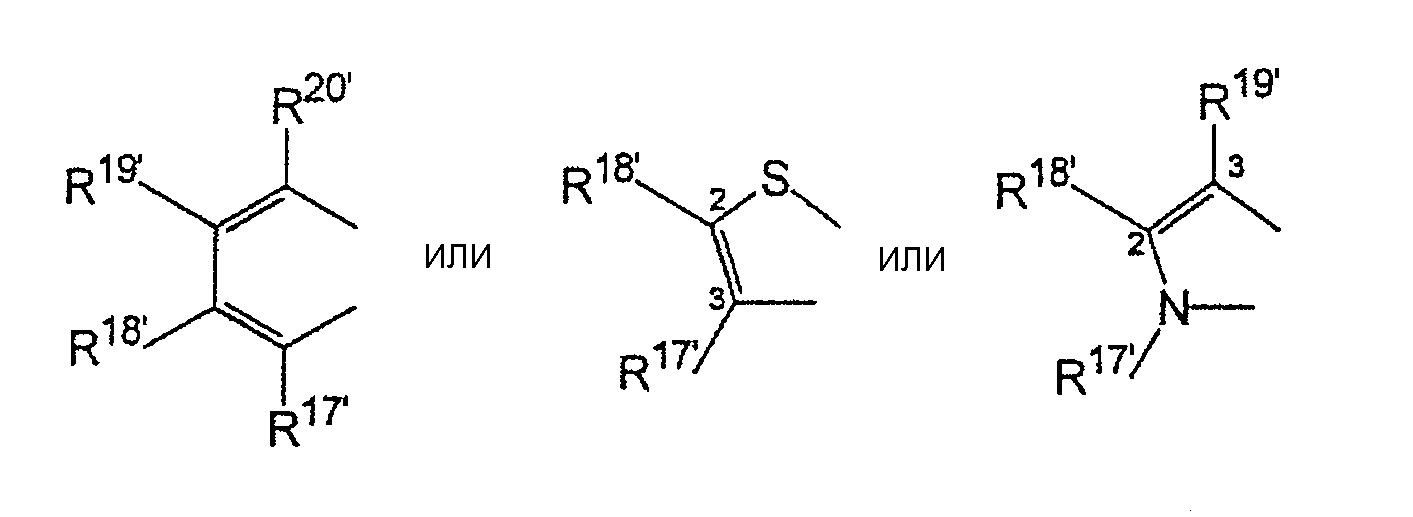
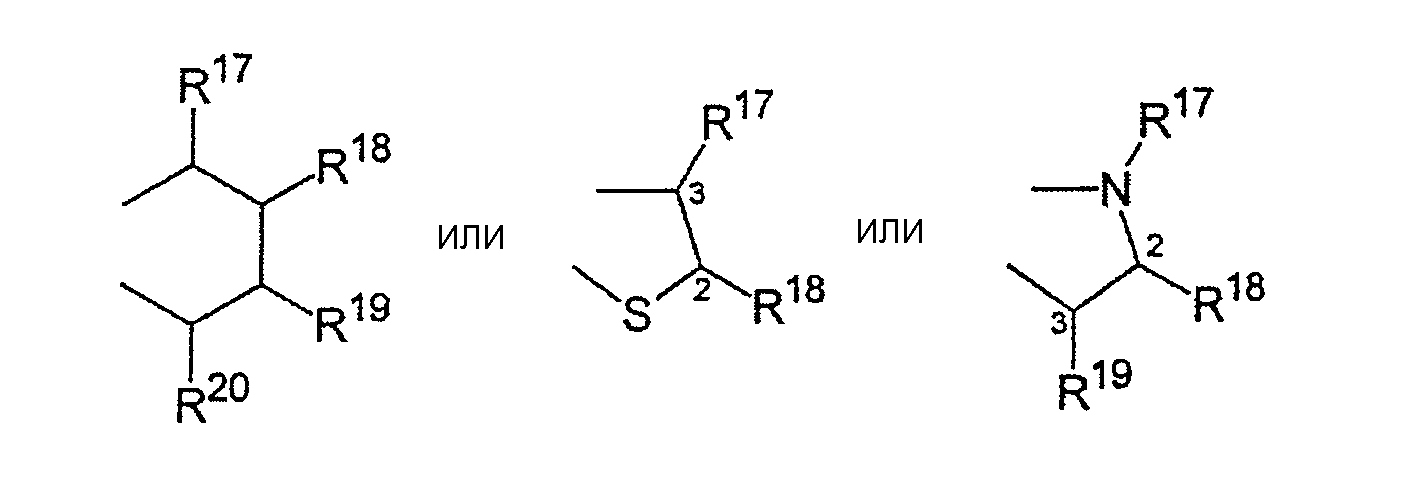
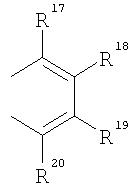
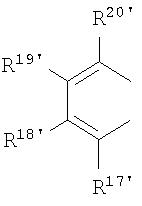
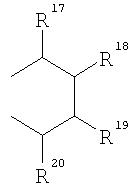
R17-R20, R17'-R20' являются одинаковыми или различными и каждый представляет собой водород, С1-С20-алкил, 3-8-членный циклоалкил, который, в свою очередь, может содержать С1-С10-алкильную группу в качестве заместителя, С6-С15-арил или арилалкил, где соседние радикалы могут совместно образовывать циклические группы, содержащие от 4 до 15 атомов углерода, или Si(R11)3, и
М2 представляет собой ион щелочного металла или ион щелочно-земельного металла, а
р равен 1, если М2 представляет собой ион щелочно-земельного металла, и 2, если М2 представляет собой ион щелочного металла;
и нагревание полученной реакционной смеси до температуры в интервале от минус 78°С до 250°С с добавлением или без добавления свободных радикалов или источников свободных радикалов с получением комплекса формулы III
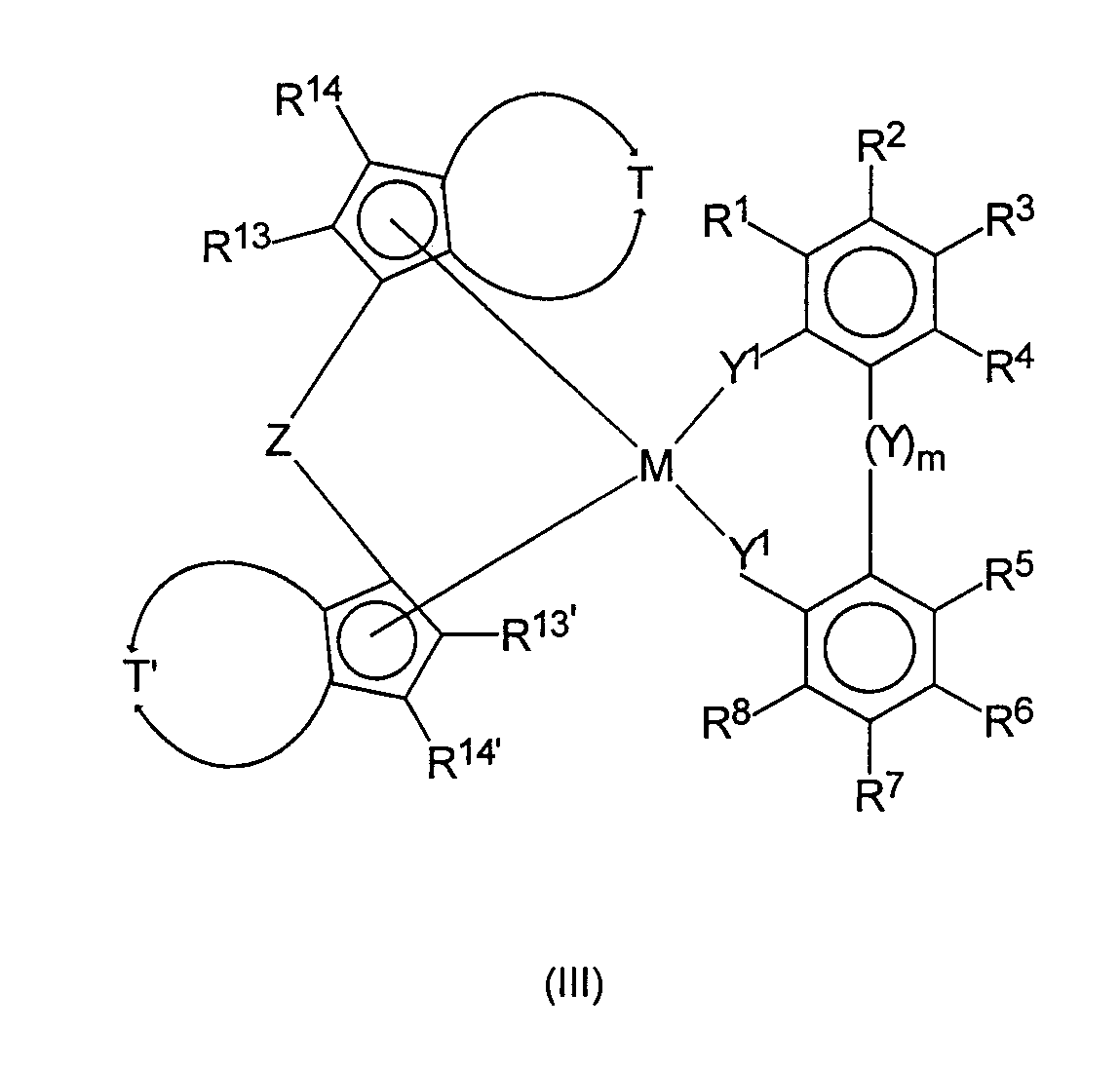
и, по меньшей мере, частичное гидрирования III при действии водорода в присутствии подходящего катализатора.
К удивлению, было найдено, что рацемоселективный синтез гидрированных или частично гидрированных металлоценовых комплексов можно успешно осуществить при получении комплексов переходных металлов формулы I, предпочтительно, без выделения промежуточных соединений, из соответствующих мостиковых лигандов дифеноксидного типа, или не мостиковых лигандов бисфеноксидного типа, реакцией с галогенидами переходных металлов способом, который известен как таковой, при этом данные комплексы вводят во взаимодействие с циклопентадиенильными производными формулы II, получая дифеноксид- или бисфеноксидзамещенные металлоцены, содержащие ароматические бисинденильные лиганды, или их гетероатомсодержащие аналоги, формулы III, которые затем каталитически гидрируют в реакционной смеси. Было найдено, что данный реакционный подход приводит рацемоселективно и с высокими выходами к соответствующим гидрированным или частично гидрированным, рацемическим дифеноксид- или бисфеноксидзамещенным анса-металлоценам. Таким образом избегают снижающего выход, сложного разделения диастереомеров.
Гидрированные или частично гидрированные рацемические дифеноксид- или бисфеноксидзамещенные анса-металлоцены можно использовать в качестве катализаторов либо непосредственно, либо после превращения в соответствующие частично гидрированные анса-металлоценовые дихлоридные комплексы за счет замещения феноксидных лигандов. Поскольку, в отличие от соответствующих гидрированных или частично гидрированных анса-металлоценовых дихлоридных комплексов, дифеноксид- или бисфеноксидзамещенные анса-металлоцены сравнительно легко растворимы в органических растворителях, их отделение от LiCl, или других солей, или катализатора гидрирования, значительно проще. Более того, синтез можно проводить в растворах высокой концентрации, что дополнительно повышает экономичность данного реакционного подхода.
В способе настоящего изобретения предпочтение отдают использованию мостиковых или не мостиковых ароматических комплексов с переходными металлами формулы I,
которые получают способом, известным как таковой, из соединений формулы IV
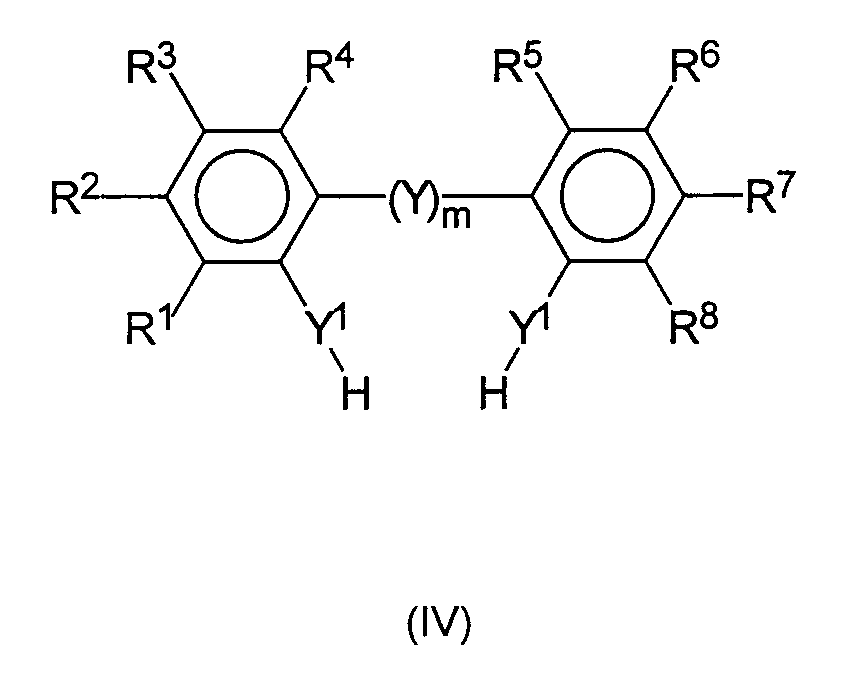
депротонированием при действии подходящих депротонирующих агентов и последующей реакцией депротонированных соединений с подходящим соединением переходного металла формулы V
MX4(основание Льюиса)k, (V)
где k равен 0, 1 или 2, причем заместители и индексы в формулах (I), (V) и (IV) имеют следующие значения:
М представляет собой титан, цирконий, гафний, в частности, цирконий; а
Х являются одинаковыми или различными и каждый представляет собой фтор, хлор, бром, йод, предпочтительно, хлор, или С1-С6-алкил, такой как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изо-бутил, предпочтительно, трет-бутил, или алкоксид -OR9, или амид -N(R9)2, где R9 являются одинаковыми или различными и каждый предпочтительно представляет собой С1-С10-алкил, С6-С15-арил, алкиларил, арилалкил, фторалкил, или фторарил, каждый из которых содержит от 1 до 10 атомов углерода в алкильном радикале и от 6 до 20 атомов углерода в арильном радикале, например метил, этил, изо-пропил, трет-бутил, фенил, нафтил, п-толил, бензил, трифторметил, пентафторфенил.
Заместители от R1 по R8 являются одинаковыми или различными и каждый из них предпочтительно представляет собой водород, фтор, хлор, бром, йод, С1-С20-алкил, -OR10, -SR10, -N(R10)2, -Р(R10)2 или Si(R9)3, где R9 и R10 являются одинаковыми или различными и каждый из них представляет собой С1-С10-алкил, С6-С15-арил, С3-С10-циклоалкил, алкиларил.
Кроме того, каждый из заместителей от R1 по R8 может представлять собой 3-8-членный циклоалкил, который, в свою очередь, может содержать С1-С10-алкильный радикал, такой как метил, этил или пропил, в качестве заместителя. Примерами подобных циклоалкильных радикалов являются циклопропил, циклопентил, предпочтительно, циклогексил, норборнил. В конкретных вариантах осуществления от R1 по R8 могут также представлять собой С6-С15-арил, такой как фенил или нафтил, или алкиларил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, например, п-толил; арилалкил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, например бензил, неофил; или триорганосилил, такой как Si(R9)3, где R9 являются одинаковыми или различными и каждый из них представляет собой С1-С20-алкил, С3-С10-циклоалкил, С6-С15-арил, например триметилсилил, трет-бутилдиметилсилил, трифенилсилил. Конечно, упомянутые радикалы могут быть частично или полностью замещены гетероатомами, например S-, N-, O-, или галогенсодержащими структурными элементами. Примерами подобных замещающих радикалов от R1 по R8 являются трифторметильная, пентафторэтильная, гептафторпропильная, гептафторизопропильная и пентафторфенильная группы.
Предпочтительными заместителями R1 и R8 для мостикового Y и R1, R', R'' и R8 для не мостикового Y являются, независимо друг от друга, С1-С10-алкильные группы, такие как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, предпочтительно, просто метил. Особое предпочтение отдают случаю, когда все заместители R1, R', R'' и R8 для не мостикового Y являются одинаковыми и каждый из них представляет собой метил и когда заместители R1 и R8 для мостикового Y являются одинаковыми и каждый из них представляет собой трет-бутильные группы.
В предпочтительных вариантах осуществления все R1, R', R'' и R8 для не мостикового Y являются метилами, при этом особенно предпочтительно, чтобы Y1 представлял собой -О-, то есть в формуле (IV) в качестве лигандов используются 2 молекулы 2,6-диметилфенола. Кроме того, использование 2,4,6-триметилфенола и 2,4-ди-трет-бутилфенола также предпочтительно согласно настоящему изобретению.
Заместители R3 и R6 могут меняться в широких пределах с целью изменения растворимости металлоценовых комплексов (III) и (VI), полученных по способу настоящего изобретения и согласно настоящему изобретению они являются одинаковыми или различными и каждый из них предпочтительно представляет собой водород, фтор, хлор, бром, йод, предпочтительно, хлор, алкоксид -OR10, тиолат -SR10, амин -N(R10)2, -Р(R10)2, или Si(R9)3, где R9 и R10являются одинаковыми или различными и каждый из них представляет собой С1-С10-алкил, С3-С10-циклоалкил, в частности 3-8-членный циклоалкил, который, в свою очередь, может содержать С1-С10-алкильный радикал, такой как метил, этил, или пропил в качестве заместителя. Примерами подобных циклоалкильных радикалов являются циклопропил, циклопентил, предпочтительно циклогексил, норборнил. Кроме того, R9 и R10 могут также представлять собой галогензамещенные алкильные или циклоалкильные радикалы, например трифторметил, пентафторэтил, гептафторпропил или гептафторизопропил.
Специалист в данной области техники выберет алкильные, циклоалкильные или ароматические заместители R3 и R6 для повышения растворимости металлоценовых комплексов в неполярных растворителях и выберет полярные заместители R3 и R6, например галоген, алкоксид, тиолат, амин и так далее, для уменьшения растворимости данных комплексов в неполярных растворителях.
В последнем случае R3 и R6 предпочтительно представляют собой атомы галогена, такие как хлор, или бром, алкоксиды -OR10, тиолаты -SR10, или амины -N(R10)2, где R10 представляет собой метил, этил, н-пропил, изопропил, трет-бутил, циклопропил, циклопентил, циклогексил, норборнил.
Особое предпочтение отдают случаю, когда R9 представляет собой метил. R3 и R6 в формуле (I) особенно предпочтительно представляют собой хлор, бром, метокси, этокси, изопропилокси, трет-бутилокси, циклопропилокси, или циклогексилокси.
Варьирование заместителей R3 и R6 в широком интервале не оказывает неблагоприятного эффекта на рацемоселективность синтеза, так что соответствующий выбор заместителей, принимая во внимание выбранные условия реакции, дает возможность повысить и улучшить выход синтеза заданным образом.
В целях настоящего изобретения все феноксиды, дифеноксиды или бисфеноксиды (или их аналогичные производные) представляют собой соединения формулы IV и их аналогичные производные, заявленные согласно настоящему изобретения, то есть как те конкретные соединения, так и лиганды, имеющие данную основную структуру, и другие элементы или группы, определенные в случае Y1 вместо фенольного атома кислорода в положении Y1.
Помимо упомянутых выше мостиков Y и Y1, предпочтительными мостиками в конкретных вариантах осуществления изобретения являются метилен -СН2-, -S-, -O-, -C(CH3)2-; особое предпочтение отдается мостикам Y1, которые являются одинаковыми и каждый из которых представляет собой кислород -О-. Кроме того, особое предпочтение отдается дифеноксидам, в которых m равен нулю, а оба Y1 являются кислородом.
В случае, когда Y является не мостиковым и представляет собой два радикала R', R'', эти радикалы предпочтительно определены, как в случае R1 по R8, включая радикалы, указанные там как предпочтительные. Кроме того, R', R'', наряду с соседними радикалами R4, R5 могут образовывать насыщенные, частично насыщенные или ненасыщенные циклические группы, содержащие от 4 до 15 атомов углерода.
Подходящие депротонирующие агенты для депротонирования (IV) представляют собой, например, н-бутиллитий, трет-бутиллитий, гидрид натрия, трет-бутилоксид калия, реагенты Гриньяра, полученные из магния, магниевые соединения, такие как ди-н-бутилмагний, (н,втор)-дибутилмагний, или другие подходящие алкильные соединения щелочно-земельных металлов или щелочных металлов.
Мостиковые или не мостиковые комплексы переходных металлов формулы (I) впоследствии вводят во взаимодействие с циклопентадиенильными производными формулы (II):
в которой заместители и индексы являются определенными выше, с использованием способов, которые обычно известны специалистам в данной области техники.
Что касается циклопентадиенильных производных формулы (II), предпочтение отдают производным, в которых
представляют собой

и в которых М2 представляет собой ион щелочного металла, или ион щелочно-земельного металла, в частности Li, Na, K, Rb, Cs, Be, Mg, Ca, Sr или Ba, где р=1 в случае Be, Mg, Ca, Sr, Ba, и р=2 в случае Li, Na, K, Rb, Cs, при этом радикалы от R17 по R20 и от R17' по R20' аналогичны определенным выше.
Было найдено, что хорошо подходящая методика является следующей, обычно осуществляемой в интервале температур от -78 до 110°С, предпочтительно первоначально примерно при 20°С, и в котором реакцию можно, при необходимости, завершить кипячением с обратным холодильником.
Сначала соединения формулы (IV) депротонируют в подходящем растворителе, или смеси растворителей, например, в толуоле, или тетрагидрофуране (ТГФ) при действии, например, гидрида натрия, или н-бутиллития, а затем добавляют соединение переходного металла, например галогенид, такой как тетрахлорид титана, тетрахлорид циркония или тетрахлорид гафния, преимущественно, в виде бис-ТГФ аддукта, или ДМЭ аддукта формулы (V). В качестве альтернативы, депротонированное соединение формулы (IV) можно добавлять к раствору соединения переходного металла. По завершении реакции продукт (I) можно, как правило, получить кристаллизацией после отделения солей. Однако предпочтение отдают использованию реакционной смеси, освобожденной от солей, для дальнейшей методики без выделения (I).
Обычно мостиковые или не мостиковые комплексы переходных металлов (I) все еще содержат от 1 до 4 эквивалентов основания Льюиса, которое, как правило, вводится синтетическим путем. Примерами таких оснований Льюиса являются простые эфиры, такие как диэтиловый эфир, или тетрагидрофуран (ТГФ), а также амины, такие как ТМЭДА. Однако возможно также получать комплексы переходных металлов (II), не содержащие оснований Льюиса, например, высушиванием при пониженном давлении или за счет выбора других растворителей для синтеза. Подобные приемы известны специалистам в данной области техники.
Комплексы переходных металлов формулы (I) вводят во взаимодействие, как описано выше для случая циклопентадиенильных производных формулы (II), при последующем нагревании полученной реакционной смеси, или без него, и в присутствии или в отсутствие свободных радикалов или источников свободных радикалов, как описано далее.
Предпочтение отдают использованию комплексов переходных металлов (I), в которых М представляет собой цирконий, а радикалы от R1 по R8 имеют описанные выше предпочтительные значения, и в которых каждый из Y1 является кислородом. Весьма подходящими комплексами являются бис(2,6-диметилфеноксид) дихлорциркония, бис(2,4,6-триметилфеноксид) дихлорциркония, бис(2,6-диметил-4-хлорфеноксид) дихлорциркония, бис(2,6-диметил-4-бромфеноксид) дихлорциркония, бис(2,6-диметил-4-метоксифеноксид) дихлорциркония, бис(2,6-диметил-4-этоксифеноксид) дихлорциркония, бис(2,6-диметил-4-трет-бутоксифеноксид) дихлорциркония, бис(2,4-ди-трет-бутилфеноксид) дихлорциркония, бис(3,5-ди-трет-бутилфеноксид) дихлорциркония, 3,3'-ди-трет-бутил-5,5'-диметокси-1,1'-ди-2-феноксид дихлорциркония, 3,3'-ди-трет-бутил-5,5'-диэтокси-1,1'-ди-2-феноксид дихлорциркония, 3,3'-ди-трет-бутил-5,5'-дипропокси-1,1'-ди-2-феноксид дихлорциркония, 3,3'-ди-трет-бутил-5,5'-диметилтио-1,1'-ди-2-феноксид дихлорциркония, 3,3'-ди-трет-бутил-5,5'-диэтилтио-1,1'-ди-2-феноксид дихлорциркония, 3,3'-ди-трет-бутил-5,5'-дипропилтио-1,1'-ди-2-феноксид дихлорциркония, их аддукты с растворителями и соединения бисфеноксида циркония и дифеноксида циркония, упомянутые в примерах.
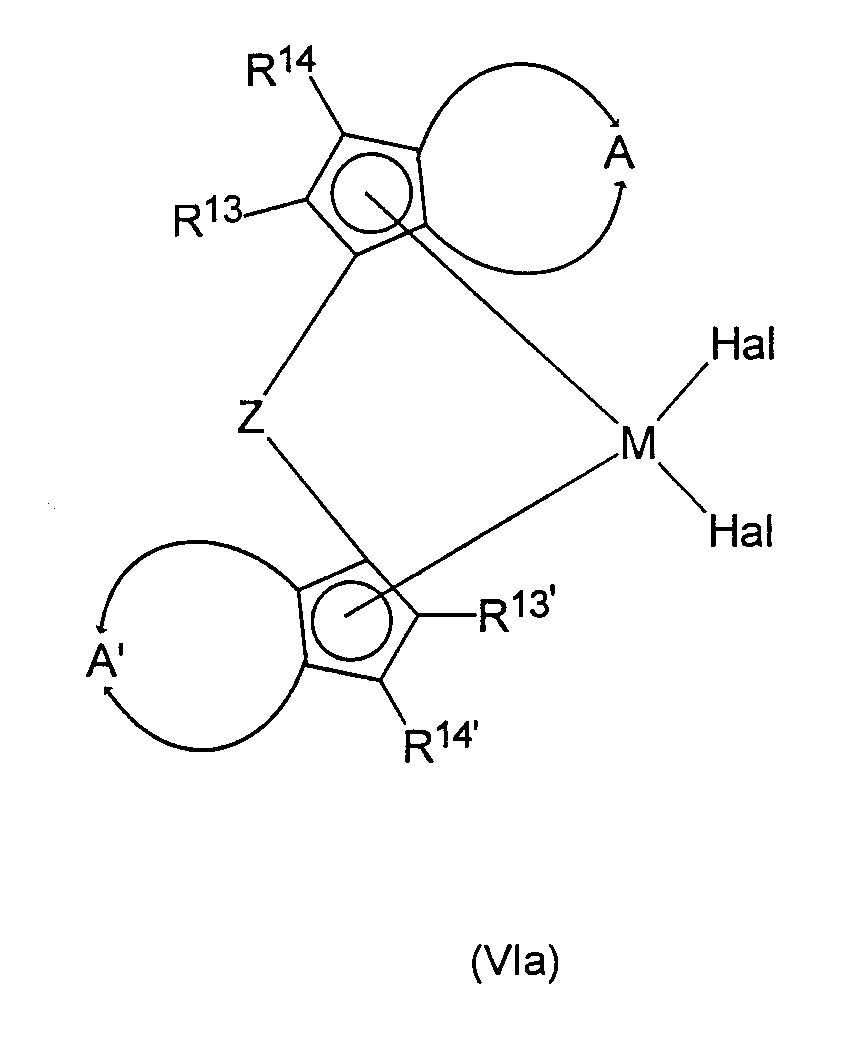
В соответствии с настоящим изобретением реакция соединений формулы (I) с циклопентадиенильными производными формулы (II) в способе настоящего изобретения первоначально приводит известными образом к комплексам переходных металлов формулы (III)
в которой все заместители аналогичны определенным выше, включая предпочтительные заместители.
На следующей стадии способа комплексы формулы (III), по меньшей мере, частично гидрируют в присутствии подходящего катализатора, получая гидрированные или частично гидрированные рацемические анса-металлоценовые комплексы формулы (VI):

в которой все заместители и индексы имеют определенные выше значения, в частности предпочтительные значения, а
представляет собой двухвалентную группу, такую как
а
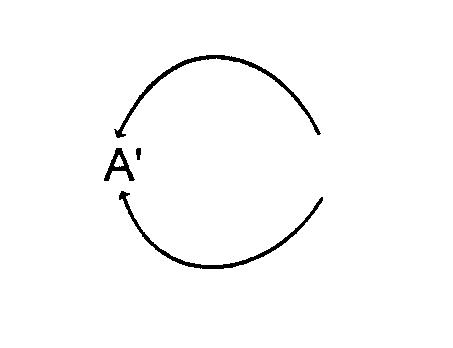
представляет собой двухвалентную группу, такую как

в которой
R13, R13', R14, R14' являются одинаковыми или различными и каждый представляет собой водород, галоген, С1-С20-алкил, 3-8-членный циклоалкил, который, в свою очередь, может содержать С1-С10-алкильную группу в качестве заместителя, С6-С15-арил, алкиларил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, арилалкил, содержащий от 1 до 10 атомов углерода в алкильной части и от 6 до 20 атомов углерода в арильной части, -OR10, -SR10, -N(R10)2, -Р(R10)2 или Si(R9)3, и
Z представляет собой -[Q(R15)(R16)]q- группу, в которой
Q может быть одинаковой или различной и представляет собой кремний, германий, олово или углерод,
каждый из R15, R16 представляет собой водород, С1-С10-алкил, С3-С10-циклоалкил или С6-С15-арил, а
q равен 1, 2, 3 или 4;
R17-R20, R17'-R20' являются одинаковыми или различными и каждый представляет собой водород, С1-С20-алкил, 3-8-членный циклоалкил, который, в свою очередь, может содержать С1-С10-алкильную группу в качестве заместителя, С6-С15-арил, или алкиларил, где соседние радикалы могут, кроме того, совместно образовывать циклические группы, содержащие от 4 до 15 атомов углерода, или Si(R11)3.
Особенное предпочтение отдается
представляющим собой
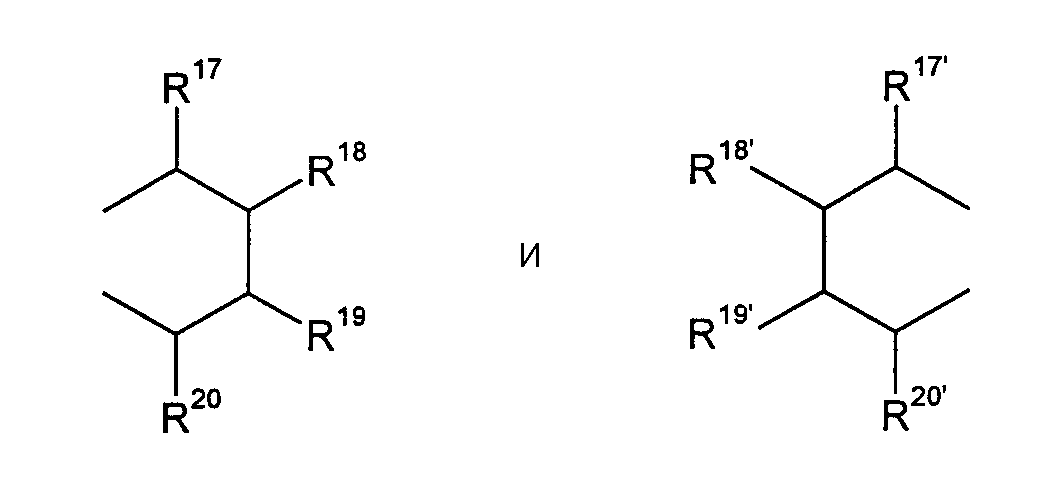
где R17-R20, R17'-R20' являются одинаковыми или различными и каждый предпочтительно представляет собой водород, С1-С10-алкил, где соседние радикалы могут, кроме того, совместно образовывать циклические группы, содержащие от 4 до 15 атомов углерода.
Гидрирование можно проводить в присутствии гомогенного или гетерогенного катализатора, предпочтительно гетерогенного катализатора. Подходящими катализаторами являются Pt, Pd, Rh, Ru, Os, а также никель, никель Ренея, их оксиды, соли или комплексы, их смеси, при желании, на подходящих носителях катализаторов. Особенное предпочтение отдается проведению гидрирования в присутствии гетерогенных палладиевых катализаторов, в частности палладия на угле или на активированном угле.
Дополнительные примеры катализаторов гидрирования, подходящих для целей настоящего изобретения, представляют собой палладий на сульфате бария, палладий на оксиде алюминия, палладиевую чернь, губчатый палладий, оксид платины, платиновую чернь, губчатую платину, диоксид платины и так далее.
В принципе, подходящими катализаторами гидрирования являются такие соединения или элементы, которые не гидрируют или лишь частично гидрируют растворитель в используемых условиях гидрирования.
Каталитическое гидрирование предпочтительно проводят в интервале температуры от 0°С до 150°С, в частности в интервале от 15°С до 100°С. Используемые в реакции растворители являются подходящими устойчивыми к гидрированию растворителями, в особенности предпочтительно, не содержащими галогена растворителями. Растворители, подходящие для данной цели, представляют собой ароматические растворители, такие как бензол, толуол, ксилол (в виде смеси изомеров), о-ксилол, м-ксилол, п-ксилол, мезитилен, тетралин, анизол, кумол, 1,2-диэтилбензол, 1,3-диэтилбензол, 1,4-диэтилбензол, 1-этил-2-метилбензол, 1-этил-3-метилбензол, 1-этил-4-метилбензол. Предпочтение отдается анизолу, толуолу, бензолу, ксилолам (в виде смеси чистых соединений) и тетралину.
Кроме того, подходящими растворителями являются ароматические или алифатические простые эфиры, такие как анизол, этилфениловый эфир, изопропилфениловый эфир, диэтиловый эфир, ди-н-бутиловый эфир, трет-бутилметиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан (ДМЭ). Можно также использовать сложные эфиры алифатических или ароматических карбоновых кислот, например этилацетат или пропилбутират.
Кроме того, можно также использовать галогенсодержащие растворители, такие как хлористый метилен. Однако предпочтение отдается использованию не содержащих галогена растворителям, поскольку использование галогенсодержащих растворителей в сравнительно больших количествах возможно лишь в случае следования строгим мерам безопасности и защиты окружающей среды. Кроме того, если нужно избежать реакций галогенирования, в хлорированных растворителях можно использовать только катализаторы гидрирования, обладающие низкой активностью, например платиновую чернь или диоксид платины. Реакции галогенирования приводят к разложению продукта и коррозионным проблемам в используемых аппаратах.
Гетерогенно катализируемое гидрирование в способе настоящего изобретения обычно проводят в соответствующих сосудах высокого давления, то есть автоклавах, которые заполняют газообразным водородом (Н2) под давлением. Подходящие значения давления водорода находятся в интервале до 100 бар, предпочтительно, до 30 бар, особенно предпочтительно до 20 бар.
Рацемические металлоценовые комплексы формулы (VI), которые гидрировали или частично гидрировали в соответствии с настоящим изобретением, можно использовать либо непосредственно в качестве катализаторов или в качестве компонентов катализаторов для полимеризации олефиноненасыщенных соединения, либо в качестве реагентов или катализаторов стереоселективного синтеза, или обычно можно дополнительно модифицировать.
В частности, один или два феноксидных лиганда, или единственный дифеноксидный лиганд в комплексе (VI), например, можно заменить путем замещения перед дальнейшим использованием. Подходящим способом замещения является реакция рацемических металлоценовых комплексов формулы (VI) с SOCl2, четыреххлористым кремнием, дихлоридом метилалюминия, хлоридом диметилалюминия, треххлористым алюминием, диалкилалюминийхлоридами, полуторными хлоридами алюминия, в особенности предпочтительно, дихлоридом этилалюминия, или кислотой Бренстеда, такой как галогеноводород, то есть HF, HBr, HI, предпочтительно HCl, который обычно используют как таковой или в виде раствора в воде, или любом органическом растворителе, таком как диэтиловый эфир, или ТГФ. Хорошо подходящими растворителями являются алифатические углеводороды, такие как пентан, гексан, гептан, ароматические углеводороды, такие как толуол, орто-, мета- или параксилол, или изопропилбензол (кумол), простые эфиры, такие как тетрагидрофуран (ТГФ), диэтиловый эфир, метил-трет-бутиловый эфир, или диметоксиэтан (ДМЭ), амины, такие как диизопропиламин, тетраметилэтандиамин (ТМЭДА), или пиридин.
Кроме того, подходят содержащие основание Льюиса смеси растворителей, включающие в себя углеводороды и простые эфиры, или амины, или и то, и другое, например смеси толуола и ТГФ, толуола и ДМЭ, или толуола и ТМЭДА, при этом основание Льюиса обычно присутствует в количестве 0,01-50 мол.%, предпочтительно 0,1-10 мол.%, из расчета на смесь растворителей. Особенно подходящими замещающими реагентами являются галогенангидриды карбоновых кислот, такие как ацетилхлорид, фенилацетилхлорид, 2-тиофенацилхлорид, трихлорацетилхлорид, триметилацетилхлорид, О-ацетилманделилхлорид, хлорангидрид 1,3,5-бензолтрикарбоновой кислоты, хлорангидрид 2,6-пиридинкарбоновой кислоты, трет-бутилацетилхлорид, хлорацетилхлорид, 4-хлорфенилацетилхлорид, дихлорацетилхлорид, 3-метоксифенилацетилхлорид, ацетилбромид, бромацетилбромид, ацетилфторид, бензоилфторид, причем их, как правило, используют в упомянутых выше растворителях или как таковые.
Это обычно приводит к аналогичному моногалогениду или дигалогениду формулы (VIa)
в которой Hal представляет собой фтор, хлор, бром или йод.
Предпочтительные металлоцены формулы VIa, которые можно получить способом настоящего изобретения, без ограничения представляют собой:
дихлорид рац-диметилсиландиилбис(4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис(2-метил-4,5,6,7-тетрагидроинденил)циркония,
дибромид рац-диметилсиландиилбис(2-метил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис(2-метил-4-фенил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис[(2-метил-4-(1-нафтил)-4,5,6,7-тетрагидроинденил]циркония,
дихлорид рац-диметилсиландиилбис[(2-метил-4-(1-нафтил)-4,5,6,7-тетрагидроинденил]гафния,
дихлорид рац-диметилсиландиилбис(2-метил-6,7-дигидро-4,5-бензоинденил)циркония,
дихлорид рац-диметилсиландиилбис(4,5,6,7-тетрагидро-4,5-бензоинденил)циркония,
дихлорид рац-диметилсиландиилбис(2-метил-4,6-диизопропил-4,5,6,7-тетрагидроинденил)циркония,
дифторид рац-диметилсиландиилбис(2-метил-4,6-диизопропил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис(2-этил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис(2-этил-4-фенил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис(2-этил-4-фенил-4,5,6,7-тетрагидроинденил)гафния,
дихлорид рац-диметилсиландиилбис[2-этил-4-(1-нафтил)-4,5,6,7-тетрагидроинденил]циркония,
дихлорид рац-диметилсиландиилбис(2-этил-6,7-дигидро-4,5-бензоинденил)циркония,
дихлорид рац-диметилсиландиилбис(4,5-бензо-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис(2-этил-4,6-диизопропил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис(2-этил-4,6-диметил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-диметилсиландиилбис(2,4,6-триметил-4,5,6,7-тетрагидроинденил)гафния,
дихлорид рац-изопропилиденбис(4,5,6,7-тетрагидроинденил)циркония,
дифторид рац-этандиилбис(4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-этандиилбис(4,5,6,7-тетрагидроинденил)циркония,
дибромид рац-этандиилбис(4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-этандиилбис(2-метил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-этандиилбис(2-этил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-этандиилбис(2-этил-4,6-диизопропил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-этандиилбис(2-метил-6,7-дигидро-4,5-бензоинденил)циркония,
дихлорид рац-этандиилбис(2-этил-6,7-дигидро-4,5-бензоинденил)циркония,
дихлорид рац-этандиилбис(2-этил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-этандиилбис(2-этил-4-фенил-4,5,6,7-тетрагидроинденил)циркония,
дихлорид рац-этандиилбис(2-этил-4-фенил-4,5,6,7-тетрагидроинденил)гафния,
дихлорид рац-этандиилбис[2-этил-4-(1-нафтил)-4,5,6,7-тетрагидроинденил]циркония,
а также соединения, упомянутые в примерах.
Следующим подходящим способом замещения является реакция гидрированных или частично гидрированных рацемических металлоценовых комплексов формулы (VI) с алюминийорганическими соединениями, такими как три-С1-С10-алкилалюминий, то есть триметилалюминием, триэтилалюминием, три-н-бутилалюминием, триизобутилалюминием. Исходя из настоящего уровня знаний, это обычно приводит к органическому соединению, аналогичному (VI) (органические радикалы вместо би(с)феноксида, например, С1-С10-алкил, такой как метил, этил, н-бутил, изобутил) и, например, алюминийорганическому динафтоксиду.
В реакциях замещения компоненты обычно используют в стехиометрическом соотношении, что определяется тем, нужно ли получить монозамещенный или дизамещенный продукт.
В особенно предпочтительных вариантах осуществления дихлорид рац-этандиилбис(4,5,6,7-тетрагидроинденил)циркония получают по способу настоящего изобретения путем взаимодействия бис(2,4,6-триметилфеноксид)дигалогенциркониевого соединения, или бис(2,6-диметилфеноксид)дигалогенциркониевого соединения, или бис(2,4-ди-трет-бутилфеноксид)дигалогенциркониевого соединения с этан-1,2-диилбисинденилдилитием, получая этандиилбис(инденил)цирконий бис(2,4,6-триметилфеноксид), или этандиилбис(инденил)цирконий бис(2,4-диметилфеноксид), или этандиилбис(инденил)цирконий бис(2,4-ди-трет-бутилфеноксид), которые впоследствии гидрируют до этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксид), или этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4-диметилфеноксид), или этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4-ди-трет-бутилфеноксид), а затем замещают феноксидные группы.
Способ настоящего изобретения дает возможность получать рацемическую форму металлоценовых комплексов (III), а также получаемые из них гидрированные или частично гидрированные комплексы (VI), а также получаемые из них соответствующие дигалогениды (VIa) и аналогичные соединения очень селективно.
Гидрированные или частично гидрированные рацемические металлоценовые комплексы (VI) и получаемые на их основе соединения можно получить селективно и с высокими выходами в соответствии с настоящим новым способом. Кроме того, способ настоящего изобретения дает возможность простым образом регулировать растворимость конечных продуктов, выбирая подходящие заместители в феноксидном или дифеноксидном лигандах, что упрощает выделение продуктов и повышает выход синтеза. Кроме того, хорошая растворимость подлежащих гидрированию комплексов (III) в неполярных растворителях дает возможность работать в концентрированных растворах, что существенно улучшает экономичность и эффективность описанного синтеза по сравнению со способами предшествующей области техники.
Еще одним значительным преимуществом является то, что способ настоящего изобретения можно осуществлять рацемоселективно однореакторным методом. В целях настоящего изобретения однореакторный метод означает, что после отдельных стадий способа промежуточные соединения не выделяют. Следующую реакцию можно проводить непосредственно с использованием реакционной смеси продуктов от предшествующей стадии.
Гидрированные или частично гидрированные рацемические металлоценовые комплексы, получаемые в соответствии с настоящим изобретением, в частности комплексы формулы (VI), или их описанные выше производные формулы (VIa), которые получают, например, замещением (ди)феноксидных лигандов, можно использовать в качестве катализаторов или в каталитических системах для полимеризации олефиноненасыщенных соединений, таких как этилен, пропилен, 1-бутен, 1-гексен, 1-октен, стирол. Они особенно выгодны для стереоселективной полимеризации прохиральных олефиноненасыщенных соединений, таких как пропилен, стирол. Подходящие катализаторы или каталитические системы, в которых рацемические металлоценовые комплексы, получаемые в соответствии с настоящим изобретением, могут функционировать в качестве «металлоценового компонента», обычно получают при помощи соединений, образующих металлоцениевые ионы, например, как описано в ЕР-А-0700935, со страницы 7, строка 34 по страницу 8, строка 21, и приведенных там формулах (IV) и (V). Еще одними соединениями, способными образовывать металлоцениевые ионы, являются алюмоксаны (RAlO)n, такие как метилалюмоксан, или также борные активаторы.
Рацемические металлоценовые комплексы, получаемые в соответствии с настоящим изобретением, в частности комплексы формулы (VI), или их описанные выше производные формулы (VIa), которые можно получить, например, замещением ди(феноксидных) лигандов, можно также использовать в качестве реагентов или в качестве катализаторов, или в каталитических системах в стереоселективном синтезе, в частности в стереоселективном органическим синтезе. Примерами, которые можно упомянуть, являются стереоселективное восстановление, или стереоселективное алкилирование С=С двойных связей, или С=О и С=N двойных связей.
Примеры
Общие методики: Получение и манипуляции с металлоорганическими соединениями проводили в отсутствие воздуха и влаги в атмосфере аргона (аппаратура Шленка, или сухой бокс). Все необходимые растворители продували аргоном и сушили над молекулярными ситами перед использованием.
Использованные в примерах растворители и реагенты получали из коммерческих источников, например:
Получение мостиковых бисинденильных лигандов проводили обычными методами, известными специалистам в данной области техники из предшествующей области техники; некоторые из использованных бисинденилов также представляют собой коммерчески доступные соединения. Концентрация использованного раствора BuLi составляла около 20 массовых % бутиллития в толуоле (примерно 2,6 молярная).
Пример 1А
Получение этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида)
А) Получение ZrCl4(ДМЭ)
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, суспендировали 10,6 г (45,47 ммоль) тетрахлорида цирконий в 50 г толуола. Суспензию охлаждали примерно до 4°С на бане со льдом, после чего медленно добавляли из капельной воронки 4,2 г ДМЭ в течение 15 минут. Суспензию оставляли нагреваться до комнатной температуры и перемешивали еще в течение часа.
b) Получение Li(2,4,6-Ме3-С6Н2О)
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 12,8 г (91 ммоль) 2,4,6-триметилфенола растворяли в 50 г толуола и 8,4 г ДМЭ. Раствор охлаждали примерно до 4°С на бане со льдом, после чего добавляли из капельной воронки 28,3 г раствора BuLi (20 массовых %) в течение 15 минут. По окончании добавления раствор оставляли нагреваться до комнатной температуры и перемешивали еще в течение часа при комнатной температуре.
с) Получение (ДМЭ)Cl2Zr(2,4,6-Ме3-С6Н2О)2
Раствор со стадии b) добавляли при помощи шприца в течение нескольких минут к суспензии со стадии а) при комнатной температуре в атмосфере азота. Остаточный феноксид лития, оставшийся в круглодонной колбе, промывали 10 мл толуола.
d) Получение этан-1,2-диилбисинденилдилития
В сухой трехгорлой круглодонной колбе на 1000 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 9,80 г (37,9 ммоль) 1,2-этандиилбисинденила растворяли в 50 г толуола и 8,4 г ДМЭ. При комнатной температуре медленно, по каплям, добавляли 25,6 г раствора BuLi (20 массовых %) в течение 20 минут. Полученную суспензию перемешивали еще в течение 2,5 часов при комнатной температуре.
е) Получение этандиилбис(инденил)цирконий бис(2,4,6-триметилфеноксида)
Суспензию со стадии с) добавляли при помощи шприца к суспензии со стадии d) в атмосфере азота. Остающиеся в круглодонной колбе остатки промывали 10 мл толуола. Объединенную суспензию перемешивали в течение ночи при комнатной температуре, а затем нагревали до 60°С и переносили при помощи шприца на стеклянный фильтр № 4, продутый инертным газом. Суспензию фильтровали в круглодонную колбу на 1000 мл с краном, а остаток на фильтре промывали 10 г толуола.
f) Синтез этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) гидрированием
Раствор, полученный на стадии е), переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствор добавляли 2,3 г палладия на угле и 2,8 г NHEt2. Включали перемешивание и продували реактор три раза азотом, а затем три раза водородом. Реактор нагревали до 60°С и нагнетали 20 бар Н2. Спустя два часа давление снижалось примерно до 17 бар и вновь доводилось до 20 бар. Через 4,5 часов давление водорода падало до 19 бар и вновь повышалось до 20 бар. Перемешивание и нагревание прекращали и оставляли реактор на ночь. Через 12 часов давление водорода падало до 17 бар и вновь доводилось до 20 бар. Спектр ЯМР указывал на неполное протекание реакции. Добавляли 2,5 мол.% Pd/C в виде суспензии в 30 г толуола. Реактор снова нагревали до 60°С в течение четырех часов, при этом водород не расходовался. Добавляли 10,9 мол.% Pd/C в виде суспензии в 50 г толуола. Через 19 часов в отсутствие перемешивания и нагревания давление водорода снижалось до 18 бар. Реактор снова нагревали до 60°С, а спустя 2,5 часа охлаждали до комнатной температуры. Полученный раствор фильтровали. Масса фильтрата составляла 353 г.1Н-ЯМР спектр указывал на полное гидрирование исходного соединения в целевой комплекс. После этого фильтрат разделяли на две части. Вторая часть весила 172,5 г и содержала приблизительно 20 ммоль гидрированного целевого комплекса этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида).
Пример 1В
Получение дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония
К раствору, полученному в примере 1А (172,5 г), медленно добавляли из капельной воронки 3,5 г ацетилхлорила в 10 г толуола при комнатной температуре. При добавлении раствора образовывался осадок. Затем смесь оставляли стоять в течение пяти часов при комнатной температуре. После этого из капельной воронки добавляли 0,8 г ацетилхлорила в 5 г толуола. В1Н-ЯМР спектре не наблюдалось резонансного сигнала, который можно было бы приписать исходному комплексу (бисфеноксиду). Суспензию оставляли стоять на ночь, а затем фильтровали. Остаток с фильтра сушили при пониженном давлении и масса его составляла 3 г. В1Н-ЯМР спектре наблюдали чистый рацемический дихлорид этандиилбис(4,5,6,7-тетрагидроинденил)циркония. Общий выход составляет около 35%. Масса фильтрата составляла 188,7 г.
Впоследствии фильтрат концентрировали до 21,7 г, при этом образовывалось небольшое количество осадка. Добавляли небольшие количества гептана, но больше осадка не образовывалось. Общий выход из примера 1А плюс пример 1В: 35%.
Пример 2:
Улучшенное получение этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) и дихлорида этандиил бис(4,5,6,7-тетрагидроинденил)циркония
а) Получение ZrCl4(ТГФ)2
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 10,6 г (45,47 ммоль) ZrCl4 суспендировали в 50 г толуола. Суспензию охлаждали приблизительно до 4°С на бане со льдом, после чего медленно добавляли из капельной воронки 6,9 г ТГФ в течение 15 минут. Суспензию оставляли нагреваться до комнатной температуры и перемешивали еще 40 минут.
b) Получение Li(2,4,6-Me3-C6H2О)
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 12,8 г 2,4,6-триметилфенола (91 ммоль) растворяли в 50 г толуола и 6,6 г ТГФ. Раствор охлаждали примерно до 4°С на бане со льдом, а затем добавляли по каплям из капельной воронки 28,3 г 20%-ного по массе раствора BuLi в течение 15 минут. По окончании добавления смесь оставляли нагреваться до комнатной температуры и перемешивали еще 40 минут.
с) Получение (ТГФ)2Cl2Zr(2,4,6-Me3-C6H2О)2
К суспензии со стадии а) добавляли раствор со стадии b) в атмосфере азота при помощи шприца при комнатной температуре в течение нескольких минут и перемешивали полученную суспензию еще 2,5 часа при комнатной температуре.
d) Получение этан-1,2-диилбисинденилдилития
В сухой трехгорлой круглодонной колбе на 1000 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 9,8 г (37,9 ммоль) 1,2-этандиилбисинденила растворяли в 50 г толуола и 3,9 г ТГФ. При комнатной температуре медленно добавляли по каплям 25,6 г 20%-ного по массе раствора BuLi в течение 20 минут. По окончании добавления суспензию перемешивали еще 2,5 часа при комнатной температуре.
е) Получение этандиилбис(инденил)цирконий бис(2,4,6-триметилфеноксида)
К суспензии со стадии d) добавляли суспензию со стадии с) в атмосфере азота при помощи шприца. Суспензию перемешивали 48 часов при комнатной температуре, а затем помещали в атмосфере азота при помощи шприца на стеклянный фильтр № 4, продутый инертным газом. Суспензию фильтровали в круглодонную колбу на 1000 мл с краном.
f) Синтез этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) гидрированием
Суспензию, полученную на стадии е), переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствору добавляли 2,3 г палладия на угле и 7,7 г триэтиламина. Начинали перемешивание и трижды продували реактор азотом, а затем трижды водородом. Реактор нагревали до 80°С, а затем нагнетали 20 бар водорода. Через 2,5 часа давление падало примерно до 17,5 бар. По данным1Н-ЯМР спектра, гидрирование исходного соединения прошло полностью. Перемешивание и нагревание прекращали и охлаждали реактор до комнатной температуры в течение четырех часов. Для отделения от катализатора гидрирования суспензию фильтровали на фильтре Зейца. Масса фильтрата составляла 290 г, и его концентрировали до 131,5 г при 50°С и 150-200 мбар.
g) Получение дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония
К раствору, полученному на стадии f), добавляли по каплям 7,5 г ацетилхлорида в 50 г толуола при комнатной температуре из капельной воронки. Во время добавления раствора образовывался осадок. После добавления всего раствора ацетилхлорида суспензию оставляли стоять при комнатной температуре в течение четырех часов. В течение этого времени образовывался плотный осадок. Реакционную смесь нагревали до 30°С, при этом осадок не растворялся. Реакционную смесь перемешивали при комнатной температуре еще в течение 48 часов. Затем к раствору добавляли 30 г гептана. Образовавшийся осадок отфильтровывали и трижды промывали 20 г гептана и один раз 10 г гептана. Осадок сушили при пониженном давлении несколько часов и его конечная масса составляла 8,3 г. По данным1Н-ЯМР спектра отфильтрованный осадок состоял из чистого дихлорида рац-этандиилбис(4,5,6,7-тетрагидроинденил)циркония. Общий выход: 50% (из расчета на количество лиганда).
Пример 3
Синтез этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) и дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония на большую загрузку
а) Получение ZrCl4(ТГФ)2
Синтез проводили, как на стадии а) примера 2, но использованные количества составляли 46,6 г ZrCl4 (199,97 ммоль), 219,8 г толуола и 30,3 г ТГФ.
b) Получение Li(2,4,6-Me3-C6H2O)
Синтез проводили, как на стадии b) примера 2, но использованные количества составляли 56,3 г 2,4,6-триметилфенола (413,3 ммоль), 220 г толуола, 29 г ТГФ и 124,3 г 20%-ного раствора BuLi.
с) Получение (ТГФ)2Cl2Zr(2,4,6-Me3-C6H2О)2
Синтез проводили, как на стадии с) примера 2.
d) Получение этан-1,2-диилбисинденилдилития
Синтез проводили, как на стадии d) примера 2, но использованные количества составляли 43,1 г 1,2-этандиилбисинденила (166,82 ммоль) в 220 г толуола и 17,1 г ТГФ. Добавляли по каплям 112,5 г 20%-ного раствора BuLi.
е) Получение этандиилбис(инденил)цирконий бис(2,4,6-триметилфеноксида)
Синтез проводили, как на стадии е) примера 2, смешивая суспензии со стадий с) и d). Оставшиеся остатки промывали 10 мл толуола. Суспензию перемешивали в течение двух часов при комнатной температуре и фильтровали в атмосфере азота на стеклянном фильтре № 4; остаток на фильтре промывали 50 г толуола. Общая масса фильтрата составляла 1097,4 г, а масса остатка на фильтре составляла 31 г. Фильтрат разделяли на три части.
f1) Синтез этандиилбис(инденил)цирконий бис(2,4,6-триметилфеноксида) гидрированием в присутствии триэтиламина
274,35 г (Теоретически 41,8 ммоль) раствора фильтрата, полученного на стадии е) переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом азота. К данному раствору добавляли 8,5 г триэтиламина, затем 2,5 г палладия на угле. Включали перемешивание и три раза продували реактор азотом, а затем три раза водородом. После этого реактор нагревали до 80°С и нагнетали 20 бар водорода. Спустя 40 минут давление водорода снижалось до 18 бар и вновь доводилось до 20 бар. Спустя еще два часа давление все еще составляло 20 бар. По данным ЯМР спектра произошло полное гидрирование до желательного соединения. Реактор оставляли остывать в течение 1,5 часов, а суспензию переносили в круглодонную колбу; реактор промывали 20 г толуола. Для отделения катализатора гидрирования суспензию фильтровали через фильтр Зейца и промывали фильтр 20 г толуола. Масса фильтрата составляла 390,8 г.
g1) Синтез дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония
К раствору, полученному на стадии f1), добавляли по каплям из капельной воронки 3,3 г ацетилхлорида в 10 мл толуола при комнатной температуре. Во время добавления образовывался осадок. По окончании добавления суспензию оставляли стоять при комнатной температуре в течение одного часа. Затем добавляли по каплям еще 3,3 г ацетилхлорида в 10 мл толуола. Реакционную смесь оставляли стоять при комнатной температуре в течение 12 часов. После этого реакционную смесь нагревали при 40°С в течение пяти часов, а потом оставляли стоять при комнатной температуре в течение 72 часов. Затем добавляли еще 1,8 г ацетилхлорида в 10 мл толуола и перемешивали реакционную смесь при комнатной температуре в целом 17 часов. После этого реакционную смесь нагревали при 45°С в течение четырех часов, а потом перемешивали реакционную смесь при комнатной температуре еще в течение 96 часов. Затем смесь концентрировали и 100 мл перегоняли. Смесь фильтровали на фильтре № 3 и промывали остаток на фильтре 15 г толуола. Остаток с фильтра сушили при пониженном давлении несколько часов и масса его составляла 11,4 г. В1Н-ЯМР спектре наблюдали чистый рацемический дихлорид этандиилбис(4,5,6,7-тетрагидроинденил)циркония.
Фильтрат концентрировали до 260 г и хранили при -20°С в течение 12 часов. Образовавшийся осадок отфильтровывали и промывали остаток на фильтре небольшими количествами толуола. Осадок сушили при пониженном давлении и масса составляла еще 1,7 г. В1Н-ЯМР спектре наблюдали чистый рацемический продукт. Масса фильтрата составляла 94 г и в его1Н-ЯМР спектре наблюдали лишь небольшие количества комплекса.
Общий выход: 44% из расчета на количество лиганда.
f2) Получение этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) гидрированием без добавления амина.
274,35 г (Теоретически 41,8 ммоль) раствора, полученного на стадии реакции е), переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствору добавляли 2,5 г палладия на угле. При перемешивании реактор трижды продували азотом, а затем три раза водородом. После этого реактор нагревали до 80°С и нагнетали 20 бар водорода. Через один час давление водорода снижалось примерно до 17,5 бар и вновь доводилось до 20 бар. Спустя 1,5 часа давление снижалось примерно до 19 бар и снова доводилось до 20 бар. Через четыре часа давление водорода снижалось примерно до 18 бар и вновь доводилось до 20 бар. По данным1Н-ЯМР спектра произошло полное гидрирование до этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида).
После этого реактор охлаждали до комнатной температуры при давлении водорода 10 бар в течение получаса. Суспензию переносили в колбу и фильтровали на фильтре Зейца, чтобы избавиться от катализатора гидрирования. Фильтрат сохраняли.
g2) Синтез дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония
К раствору, полученному на стадии f2), медленно добавляли из капельной воронки 6,6 г ацетилхлорида при комнатной температуре. По окончании добавления суспензию перемешивали в течение 17 часов. Затем суспензию фильтровали, а остаток на фильтре дважды промывали 10 г толуола, а потом 10 г гептана, а затем снова промывали 10 г толуола. Остаток с фильтра сушили при пониженном давлении несколько часов и масса его составляла 6,9 г. В1Н-ЯМР спектре наблюдали чистый рацемический дихлорид этандиилбис(4,5,6,7-тетрагидроинденил)циркония. Масса фильтрата составляла 331,5 г и его концентрировали до 97,2 г при 40°С и 100-150 мбар. К концентрированному фильтрату добавляли гептан. Сразу же образовывался белый осадок, который отфильтровывали и сушили при пониженном давлении несколько часов. Его масса составляла 0,5 г. В1Н-ЯМР спектре наблюдали чистый рацемический комплекс.
Общий выход: 42% из расчета на количество лиганда.
f3) Синтез этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) гидрированием.
548,7 г (Теоретически 83,6 ммоль) раствора, полученного на стадии е), переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствору добавляли 5 г палладия на угле. При перемешивании реактор трижды продували азотом, а затем три раза водородом. После этого реактор нагревали до 80°С и нагнетали 20 бар водорода. Через 1,5 часа давление водорода снижалось до 6 бар. Реакцию останавливали и1Н-ЯМР спектр свидетельствовал о полном гидрировании до KL050. Перемешивание и нагревание прекращали и охлаждали реактор до комнатной температуры. Суспензию переносили в круглодонную колбу и фильтровали на фильтре Зейца для отделения от катализатора гидрирования. Масса фильтрата составляла 585,6 г. Фильтрат разделяли на три части, масса каждой из которых составляла 146,4 г.
g3a) Синтез дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония при добавлении ацетилхлорида в две стадии
К 146,4 г раствора, полученного на стадии f3), при комнатной температуре медленно добавляли 1,95 г ацетилхлорида в 5 г толуола из капельной воронки. После этого реакционную смесь перемешивали при комнатной температуре в течение одного часа, после чего добавляли еще 1,95 г ацетилхлорида в 5 г толуола и перемешивали реакционную смесь при комнатной температуре еще в течение 5,5 часа. За это время примерно через четыре часа образовывался осадок. Реакционную смесь перемешивали еще 12 часов. После этого суспензию концентрировали до 30% первоначальной массы, а затем фильтровали на фильтре № 3. Осадок сушили при пониженном давлении несколько часов, а масса его составляла 3,1 г. В1Н-ЯМР спектре наблюдали чистый рацемический комплекс. Масса фильтрата составляла 46,9 г. Общий выход: 35% из расчета на использованный лиганд.
g3b) Синтез дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония при добавлении ацетилхлорида в одну стадию
Синтез проводили, как на стадии g3а), за исключением того, что все количество ацетилхлорида (3,7 г) добавляли из капельной воронки в одну стадию при комнатной температуре и реакционную смесь перемешивали в течение пяти часов при 45°С, а не при комнатной температуре. Осадок образовывался примерно через два часа. Суспензию концентрировали до 46% первоначальной массы, а затем фильтровали на фильтре № 3. Осадок промывали небольшими количествами толуола, а затем сушили при пониженном давлении несколько часов. В результате получали массу 2,3 г.1Н-ЯМР спектр свидетельствовал об образовании чистого рацемического комплекса. Масса фильтрата составляла 89,7 г. Общий выход: 26% из расчета количество лиганда.
g3с) Синтез дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония при добавлении неразбавленного ацетилхлорида
К 146,4 г реакционного раствора, полученного на стадии f3), добавляли из капельной воронки 3,7 г ацетилхлорида при 45°С. Реакционную смесь перемешивали при данной температуре в течение двух часов. Образовывался осадок. Суспензию концентрировали приблизительно до 60% первоначальной массы, а затем фильтровали на фильтре № 3. Осадок сушили при пониженном давлении несколько часов и его масса составляла 2,2 г. По данным1Н-ЯМР спектра образовывался чистый рацемический комплекс. Масса фильтрата составляла 114,2 г. Общий выход: 26% из расчета на количество использованного лиганда.
Пример 4
Получение этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) с выделением промежуточного соединения
а) Получение ZrCl4(ТГФ)2
Синтез проводили, как на стадии а) предшествующих примеров. Использованные количества составляли 46,6 г ZrCl4 (199,97 ммоль), 80 г толуола и 30,3 г ТГФ.
b) Получение Li(2,4,6-Me3-C6H2O)
Синтез проводили, как на стадии b) предшествующего примера. Использованные количества составляли 56,3 г (413,42 ммоль) 2,4,6-триметилфенола, 100 г толуола, 29 г ТГФ и 124,3 г раствора BuLi.
с) Получение (ТГФ)2Cl2Zr(2,4,6-Me3-C6H2O)2
Синтез осуществляли, как на стадии с) предшествующего примера.
d) Получение этан-1,2-диилбисинденилдилития
Синтез осуществляли, как на стадии d) предшествующего примера. Использованные количества составляли 46,5 г (179,98 ммоль) 1,2-этандиилбисинденила, 80 г толула, 17,1 г ТГФ, 112,5 г раствора BuLi.
е) Получение этандиилбис(инденил)цирконий бис(2,4,6-триметилфеноксида)
Синтез осуществляли, как на стадии е) предшествующего примера, но остаток на фильтре промывали 40 г толуола и еще 35 г толуола. Теоретическая концентрация: 14,6%, масса фильтрата: 750,7 г.
Выделение этандиилбис(инденил)цирконий бис(2,4,6-триметилфеноксида)
Примерно 200 г раствора, полученного на стадии е), концентрировали менее чем до половины его массы (выпаривали 123 г растворителя). Комплекс кристаллизовался из данного раствора после нескольких часов при комнатной температуре. Комплекс выделяли фильтрованием, промывали 5 мл толуола и сушили при пониженном давлении. В результате получали 13,35 г комплекса. Маточный раствор концентрировали дополнительно при пониженном давлении и получали в результате еще 2,61 г кристаллов после нескольких дней при комнатной температуре. Общий выход: 15,61 г (25,26 ммоль); таким образом, выход в общей реакции 52% (94,81 ммоль).
f1) Получение этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) гидрированием
557,6 г (Теоретически 134 ммоль) раствора, полученного на стадии е), переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствору добавляли 8 г палладия на угле. Включали перемешивание и трижды продували реактор азотом, а затем три раза водородом. После этого реактор нагревали до 80°С и нагнетали 20 бар водорода. Через 60 минут давление снижалось примерно до 0 бар и вновь доводилось до 20 бар. Через 1,5 часа давление снижалось до 17 бар. Данные1Н-ЯМР спектра свидетельствовали о полном гидрировании до этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида) лишь с небольшими следами примесей. Реактор охлаждали до 37°С в течение 1,5 часа. Суспензию переносили в круглодонную колбу и фильтровали на фильтре Зейца для отделения от катализатора гидрирования. Фильтр промывали 20 г толуола. Масса фильтрата составляла 567,9 г.
Выделение этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4,6-триметилфеноксида)
155,36 г раствора, полученного на стадии f1), концентрировали в значительной степени (примерно до 30 мл растворителя). Комплекс кристаллизовался медленно при комнатной температуре из данного раствора через несколько часов. Раствор концентрировали дополнительно примерно до 20 мл и получали вязкое масло. Добавляли 50 мл гептана при комнатной температуре. Комплекс выпадал в виде белых кристаллов. Колбу хранили при -20°С в течение трех дней, и затем отфильтровывали осадок. Оказалось возможным выделить только 7,34 г комплекса из-за его крайне высокой растворимости даже в углеводородах. Выход 7,34 г (32,5%).
Пример 5:
Получение этандиилбис(4,5,6,7-тетрагидроинденил)цирконий 3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-феноксида через этандиилбис(инденил)цирконий 3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-феноксид
а) Получение ZrCl4(ТГФ)2
Синтез проводили, как на стадии а) предшествующих примеров. Использованные количества составляли 8,93 г ZrCl4(38,32 ммоль), 130 мл толуола и 8,0 г ТГФ.
b) Получение Li2(3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-феноксида)
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 15,7 г (38,23 ммоль) 3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-фенола растворяли в 130 мл толуола и 8 г ТГФ. Раствор охлаждали примерно до 4°С на бане со льдом, а затем добавляли по каплям из капельной воронки 28,4 мл 20%-ного по массе раствора BuLi в течение одного часа. По завершении добавления реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали еще час.
с) Получение (ТГФ)2Cl2Zr(3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-феноксида)
Раствор со стадии b) вводили в атмосфере азота при помощи шприца в суспензию со стадии а) в течение нескольких минут при комнатной температуре. Суспензию перемешивали еще в течение четырех часов.
d) Получение этан-1,2-диилбисинденилдилития
В сухой трехгорлой круглодонной колбе на 1000 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 9,5 г (36,77 ммоль) инденила суспендировали в 120 мл толуола и 7,0 г ТГФ. Суспензию охлаждали на бане со льдом, а затем медленно добавляли по каплям 27,5 мл раствора BuLi. После этого суспензию дополнительно перемешивали при комнатной температуре еще 1,5 часа.
е) Получение этандиилбис(инденил)цирконий 3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-феноксида
Суспензию со стадии с) вводили в атмосфере азота при помощи шприца в суспензию со стадии d). Оставшиеся остатки промывали 10 мл толуола. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, после чего ее нагревали до 80°С. При данной температуре суспензию помещали в атмосфере азота на стеклянный фильтр № 4 и фильтровали в круглодонную колбу с вакуумным отводом. Фильтрат концентрировали при пониженном давлении, выпаривая 370 мл растворителя. Концентрированный фильтрат оставляли стоять при комнатной температуре на несколько дней, при этом комплекс не выкристаллизовывался. Растворитель из фильтрата удаляли полностью при пониженном давлении, получая пену, которую измельчали в порошок.
Выделяли 30,5 г сырого комплекса, загрязненного LiCl.
Пример 6:
6,1 г (Теоретически 9,2 ммоль) сырого этандиилбис(инденил)цирконий 3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-феноксида из предшествующего примера растворяли в 232 г толуола и переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствору добавляли 0,11 г палладия на угле. Включали мешалку и продували реактор трижды азотом, а затем три раза водородом. После этого реактор нагревали до 40°С и нагнетали 20 бар водорода. Через два часа давление падало примерно до 18 бар и снова доводилось до 20 бар. Через три часа давление падало до 18 бар и снова доводилось до 20 бар. Еще через шесть часов давление падало до 18 бар и снова повышалось до 20 бар. Добавляли еще 0,9 г Pd/C в толуоле. В реактор снова нагнетали 20 бар водорода в течение одного часа, после чего давление падало до 18 бар. Еще через два часа реакцию останавливали. Данные1Н-ЯМР спектра свидетельствовали о полном образовании гидрированного комплекса этандиилбис(4,5,6,7-тетрагидроинденил)цирконий 3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-феноксида и наблюдались резонансные сигналы, которые могли быть отнесены к 3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-фенолу.
b) Реакцию предшествующей стадии повторяли, используя 8,5 г (теоретически 11 ммоль) сырого исходного комплекса, которые растворяли в 300 г CH2Cl2, а затем переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствору добавляли 0,15 г палладия на угле, включали мешалку и продували реактор три раза азотом, а потом три раза водородом. Реактор нагревали до 50°С и нагнетали 20 бар водорода. Через 90 минут давление падало до 17 бар и снова доводилось до 20 бар. Через 11 часов давление снова повышали до 20 бар и увеличивали температуру до 60°С. Еще через шесть часов давление оставалось постоянным. Перемешивание и нагревание прекращали и охлаждали реактор до 25°С и выдерживали при этой температуре 48 часов. После этого реактор снова нагревали до 60°С и нагнетали 20 бар водорода. Данное давление поддерживалось в течение 4,5 часов. Затем реактор охлаждали до комнатной температуры и переносили суспензию в круглодонную колбу и фильтровали на фильтре Зейца для отделения от катализатора гидрирования. Растворитель из фильтрата выпаривали, получая бежевую пену. К этой пене добавляли 10 мл гептана. Через несколько минут образовывались белые кристаллы. Колбу охлаждали до 2°С и выдерживали при этой температуре 4 часа. После этого осадок отфильтровывали, промывали небольшими количествами гептана, а затем сушили при пониженном давлении. Выделяли 1,5 г продукта. По данным1Н-ЯМР спектра белый осадок представлял собой чистый рац-этандиилбис(4,5,6,7-тетрагидроинденил)цирконий 3,3',5,5'-тетра-трет-бутил-1,1'-ди-2-феноксид. Из маточного раствора выделяли еще 0,1 г. Выход: 18% (из расчета на количество сырого исходного вещества).
Пример 7:
Синтез дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония из этандиилбис(инденил)цирконий бис(2,4-ди-трет-бутилфеноксида)
а) Получение ZrCl4(ТГФ)2
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 18 г (77,4 ммоль) тетрахлорида циркония суспендировали в 40 г толуола. При комнатной температуре медленно добавляли из капельной воронки 11,9 г ТГФ в течение 15 минут. Суспензию перемешивали еще один час при комнатной температуре.
b) Получение Li(2,4-tBu2-C6H3O)
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 32,9 г (154,8 ммоль) 2,4-ди-трет-бутилфенола растворяли в 40 г толуола и 9 г ТГФ. При комнатной температуре медленно добавляли из капельной воронки 50,6 г раствора BuLi (20% по массе) в течение 15 минут. Полученную суспензию перемешивали еще один час при комнатной температуре.
с) Получение (ТГФ)2Cl2Zr(2,4-tBu2-C6H3O)2
Раствор со стадии b) добавляли через канюлю в течение нескольких минут к суспензии со стадии а) при комнатной температуре в атмосфере азота. Остаток феноксида лития, оставшийся в колбе, промывали 10 мл толуола.
d) Получение этан-1,2-диилбисинденилдилития
В сухой трехгорлой круглодонной колбе на 1000 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 20 г (77,4 ммоль) 1,2-этандиилбисинденила растворяли в 40 г толуола и 7 г ТГФ. При комнатной температуре медленно добавляли по каплям 50,8 г раствора BuLi (20% по массе) в течение 20 минут. Полученную суспензию перемешивали еще 2,5 часа при комнатной температуре.
е) Получение этандиилбис(инденил)цирконий бис(2,4-ди-трет-бутилфеноксида)
Суспензию со стадии с) добавляли через канюлю к суспензии со стадии d) в атмосфере азота. Остатки, оставшиеся в колбе, промывали 10 мл толуола. Полученную суспензию перемешивали в течение ночи при комнатной температуре, а затем помещали при помощи канюли на стеклянный фильтр № 4, продутый инертным газом. Суспензию фильтровали в круглодонную колбу на 1000 мл с вакуумным отводом с краном, а остаток на фильтре промывали 10 г толуола.
f) Синтез этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4-ди-трет-бутилфеноксида) гидрированием
Раствор, полученный на стадии е), переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствору добавляли 4,2 г палладия на угле. Включали мешалку и продували реактор три раза азотом, а потом три раза водородом. Реактор нагревали до 80°С и нагнетали 20 бар Н2. Через один час давление падало примерно до 8 бар и вновь доводилось до 20 бар. Через два часа давление падало примерно до 12 бар и вновь доводилось до 20 бар. Через 3 часа давление падало до 10 бар и вновь доводилось до 20 бар. Через 4 часа давление падало до 11 бар и перемешивание и нагревание прекращали. По данным1Н-ЯМР спектра произошло полное гидрирование исходного соединения в желательный комплекс. Полученный раствор фильтровали. Масса фильтрата составляла 306,4 г.
g) Получение дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония
К раствору, полученному на стадии f), медленно добавляли из капельной воронки при комнатной температуре 10,9 г ацетилхлорида. Во время добавления ацетилхлорила образовывался осадок. После этого смесь оставляли стоять на 56 часов при комнатной температуре. В1Н-ЯМР спектре не наблюдалось резонансного сигнала, который можно было бы отнести к исходному комплексу (бисфеноксиду). После этого суспензию фильтровали. Остаток с фильтра сушили при пониженном давлении, и масса его составляла 17,5 г. В1Н-ЯМР спектре наблюдался чистый рацемический дихлорид этандиилбис(4,5,6,7-тетрагидроинденил)циркония. Общий выход составляет примерно 55% (из расчета на количество исходного вещества).
Сравнительный пример А:
Синтез дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония из хлорида этандиилбис(инденил)цирконий (2,4-ди-трет-бутилфеноксида)
а) Получение ZrCl4(ТГФ)2
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 18 г (77,4 ммоль) тетрахлорида циркония суспендировали в 40 г толуола. При комнатной температуре медленно добавляли из капельной воронки 12 г ТГФ в течение 15 минут. Суспензию перемешивали еще один час при комнатной температуре.
b) Получение Li(2,4-tBu2-C6H3O)
В сухой трехгорлой круглодонной колбе на 500 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 16,5 г (77,6 ммоль) 2,4-ди-трет-бутилфенола растворяли в 20 мл толуола и 4,5 г ТГФ. При комнатной температуре медленно добавляли из капельной воронки 24,1 г раствора BuLi (20% по массе) в течение 15 минут. Полученную суспензию перемешивали еще один час при комнатной температуре.
с) Получение (ТГФ)2Cl3Zr(2,4-tBu2-C6H3O)
Раствор со стадии b) добавляли через канюлю в течение нескольких минут к суспензии со стадии а) при комнатной температуре в атмосфере азота. Остаток феноксида лития, оставшийся в круглодонной колбе, промывали 10 мл толуола.
d) Получение этан-1,2-диилбисинденилдилития
В сухой трехгорлой круглодонной колбе на 1000 мл, продутой инертным газом и снабженной магнитной мешалкой, капельной воронкой и вакуумным отводом с краном, 20 г (77,4 ммоль) 1,2-этандиилбисинденила растворяли в 40 г толуола и 7 г ТГФ. При комнатной температуре медленно добавляли по каплям 50,8 г раствора BuLi (20% по массе) в течение 20 минут. Полученную суспензию перемешивали еще 2,5 часа при комнатной температуре.
е) Получение хлорида этандиилбис(инденил)цирконий бис(2,4-ди-трет-бутилфеноксида)
Суспензию со стадии с) добавляли через канюлю в течение нескольких минут к суспензии со стадии d) в атмосфере азота. Остатки, оставшиеся в круглодонной колбе, промывали 10 мл толуола. Полученную суспензию перемешивали в течение ночи при комнатной температуре, а затем помещали при помощи канюли в атмосфере азота на стеклянный фильтр № 4, продутый инертным газом. Суспензию фильтровали в круглодонную колбу на 1000 мл с краном, а остаток на фильтре промывали 10 г толуола.
f) Синтез хлорида этандиилбис(4,5,6,7-тетрагидроинденил)цирконий бис(2,4-ди-трет-бутилфеноксида) гидрированием
Раствор, полученный на стадии е), переносили в атмосфере азота в автоклав на 1000 мл, снабженный вводом водорода. К данному раствору добавляли 4,2 г палладия на угле. Включали мешалку и продували реактор три раза азотом, а потом три раза водородом. Реактор нагревали до 80°С и нагнетали 20 бар Н2. Через один час давление падало примерно до 10 бар и вновь доводилось до 20 бар. Через два часа давление падало примерно до 12 бар и вновь доводилось до 20 бар. Через 3 часа давление падало до 16 бар, а затем перемешивание и нагревание прекращали. По данным1Н-ЯМР спектра происходило полное гидрирование исходного соединения в желательный комплекс. Полученный раствор фильтровали. Масса фильтрата составляла 248 г.
g) Получение дихлорида этандиилбис(4,5,6,7-тетрагидроинденил)циркония
К раствору, полученному на стадии f), медленно добавляли из капельной воронки при комнатной температуре 5,5 г ацетилхлорида. Во время добавления ацетилхлорила образовывался осадок. После этого смесь оставляли стоять на 18 часов при комнатной температуре. Затем из капельной воронки добавляли 0,6 г ацетилхлорида и после этого суспензию нагревали до 45°С в течение 2 часов. В1Н-ЯМР спектре не наблюдалось резонансного сигнала, который можно было бы отнести к исходному комплексу (монофенолксиду). После этого суспензию фильтровали. Остаток с фильтра сушили при пониженном давлении и масса его составляла 5 г. В1Н-ЯМР спектре наблюдался чистый рацемический дихлорид этандиилбис(4,5,6,7-тетрагидроинденил)циркония. Общий выход составляет примерно 15% (из расчета на количество исходного вещества).
Реферат
Данное изобретение относится к улучшенному способу получения гидрированных или частично гидрированных рацемических анса-металлоценовых комплексов путем введения во взаимодействие мостиковых или не мостиковых комплексов переходных металлов с соединениями щелочных металлов или соединениями щелочно-земельных металлов, нагревания полученной реакционной смеси до температуры в интервале от комнатной до 80°С и, по меньшей мере, частичного гидрирования продуктов реакции в присутствии подходящего катализатора в соответствующие гидрированные или частично гидрированные металлоцены, и к их использованию в качестве катализаторов, или в качестве компонента катализаторов полимеризации олефино-ненасыщенных соединений, или в качестве реагентов или катализаторов стереоселективного синтеза. 4 з.п. ф-лы.
Формула








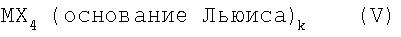
Комментарии