Способ и промежуточные соединения для получения прегабалина - RU2628298C2
Код документа: RU2628298C2
Чертежи


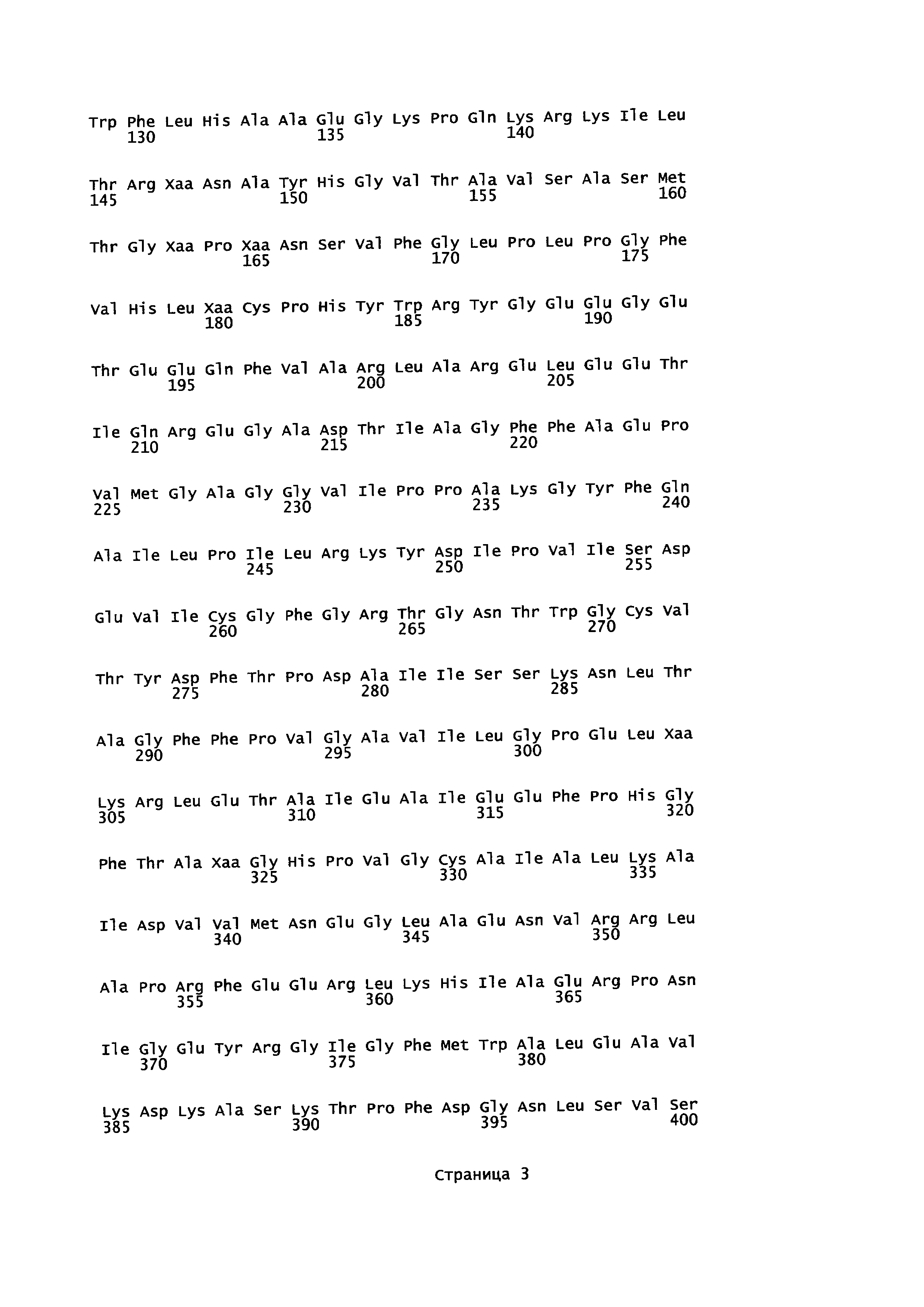



















Описание
Область изобретения
Настоящее изобретение относится к получению 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5H-2-фуранона и его производных для использования в получении (S)-(+)-3-аминометил-5-метилгексановой кислоты (прегабалина). Изобретение дополнительно относится к улучшенному способу превращения 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона в прегабалин. Прегабалин представляет собой γ-аминокислоту, проявляющую связывающее сродство к субъединице α2δ кальциевого канала человека.
Предшествующий уровень техники
Прегабалин или (S)-(+)-3-аминометил-5-метил-гексановая кислота ((S)-II),
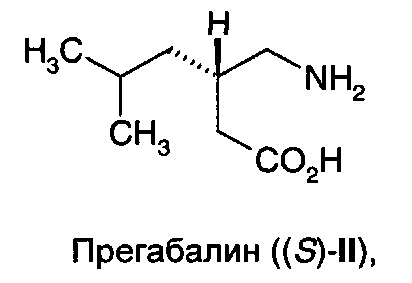
является активным агентом в препарате Лирика®, одобренном для лечения эпилепсии, нейропатической боли, фибромиалгии и генерализованного тревожного расстройства. Он проявляет противосудорожную активность, как обсуждается в патенте США №5,563,175, и антиноцицептивную активность, как обсуждается в патенте США №6,001,876. Выдвинута гипотеза, что фармакологическая активность прегабалина (II) является результатом связывания с альфа-2-дельта (α2δ) субъединицей кальциевого канала. Также описано, что прегабалин (II) применим при других состояниях, таких как физиологические состояния, обусловленные психомоторными стимуляторами, воспалением, желудочно-кишечным повреждением, алкоголизмом, бессонницей и различными психиатрическими расстройствами, включая тревогу, депрессию, манию и биполярное расстройство.
Раскрыт ряд способов получения прегабалина. 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранон (IА) идентифицировали как пригодный предшественник.

Понятно, что соединение (IА) вместе с рядом других соединений, обсуждаемых в данном документе, можно рассматривать как циклизованный изомер производного 4-оксобутановой кислоты. Производные 4-оксобутановой кислоты могут существовать либо в виде формы с открытой цепью, либо в виде циклической формы.

Эти две изомерные формы могут сосуществовать в равновесии, и относительный вклад этих двух форм зависит от точной химической природы соединений.
В Международной заявке на патент PCT/US 2008/004699 (опубликованной как WO 2008/127646 A2) предложено превращение 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона (IА) или сложного эфира изомерной формы с раскрытым циклом (XIII, где RA представляет собой алкил) в соединение (II) с использованием химического или ферментативно опосредованного восстановительного аминирования. Предполагают, что использование фермента трансаминазы селективно обеспечит предпочтительный ((S)-II) энантиомер.

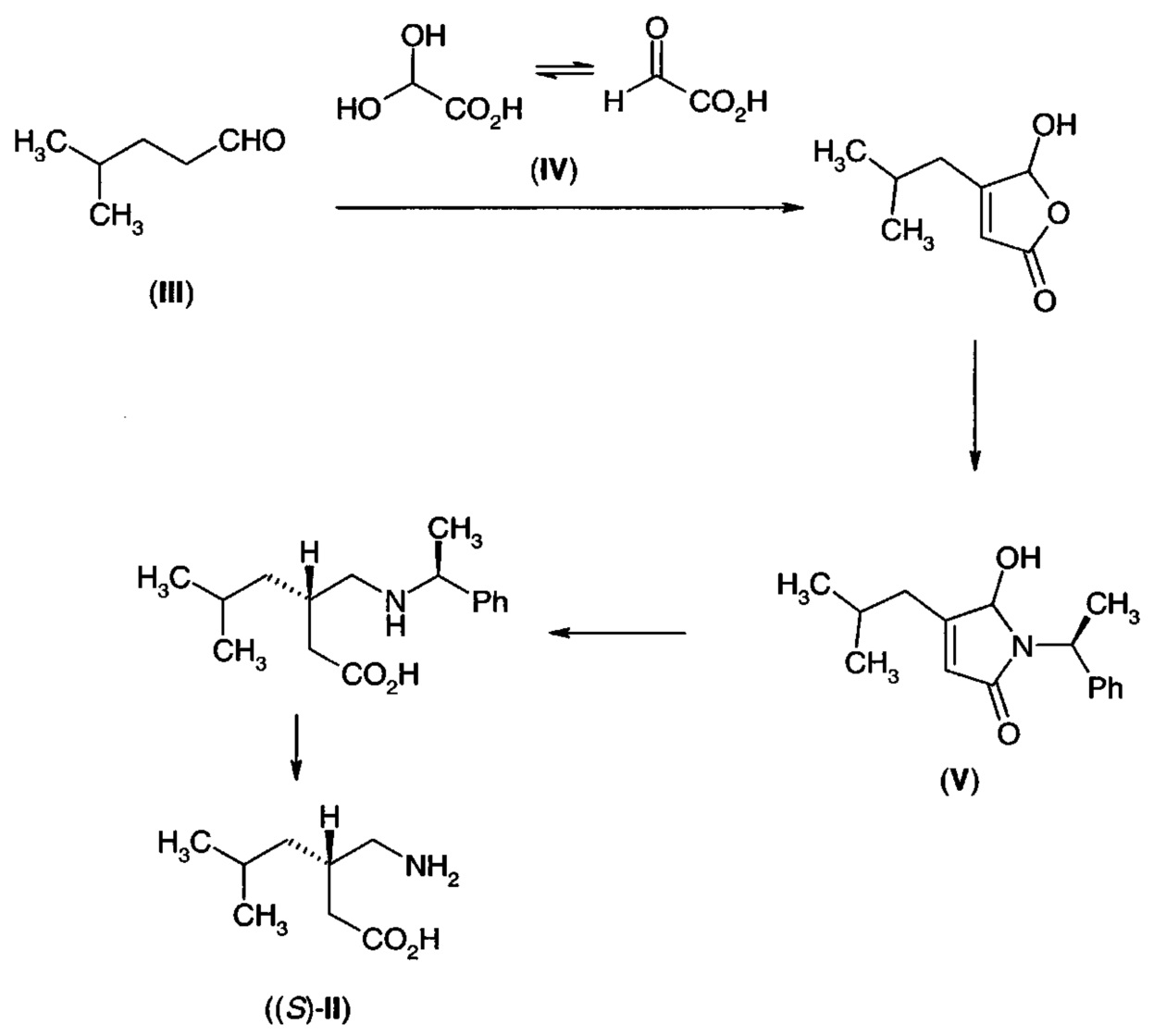
Сложный эфир (XIII) получают из 4-метилпентаналя (III) путем алкилирования соответствующего галогенацетата в присутствии диизобутиламина. Предшественник (IА) получают либо путем гидролиза сложноэфирной группы сложного эфира (XIII), либо путем конденсации 4-метилпентаналя (III) с глиоксалевой кислотой (IV) и последующего восстановления двойной связи. Применение ферментативно опосредованного восстановления также предложено в качестве пути внедрения желаемой стереохимии.

В Международной заявке на патент PCT/IN 2010/000140 (опубликованной как WO 2011/086565) раскрыт подобный способ. Продукт конденсации 4-метилпентаналя (III) и глиоксилевой кислоты (IV) подвергают взаимодействию с хиральным амином, таким как α-метилбензиламин, с получением пирролона (V). В результате гидрогенизации получают некоторую степень стереоселективности, и в результате удаления защиты получают прегабалин (II) в хиральной форме.
Существует потребность в еще более улучшенных способах синтеза прегабалина (II). В частности, желательно разработать способ, который является экономически эффективным и безопасным. В частности, важно разработать синтез прегабалина (II), который можно выполнять в коммерческом масштабе, при котором используют легкодоступные, недорогостоящие и безопасные исходные вещества и реагенты, и при котором нет необходимости в трудных разделениях.
Краткое изложение сущности изобретения
В настоящем изобретении предложены улучшенные способы получения 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона (IА) и промежуточные соединения, полезные в этих улучшенных способах.
В первом аспекте А1 в изобретении предложено соединение формулы (VI)
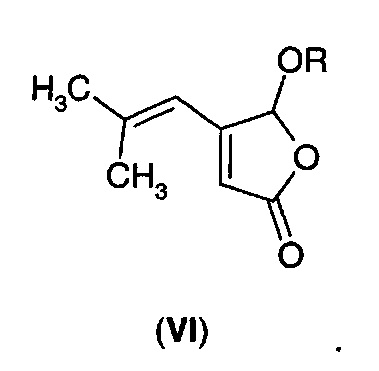
В первом воплощении А1Е1 в изобретении предложено соединение формулы (VI), где R выбран из:
водорода,
(С1-С6)алкила,
(С1-С6)галогеналкила,
(С1-С3)алкокси(С2-С6)алкила,
(С2-С6)алкенила,
(С3-С10)циклоалкила, который может быть возможно замещен 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
(С3-С10)циклоалкил(С1-С6)алкила, где циклоалкильная группа может быть возможно замещена 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
арила, который может быть возможно замещен 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
арил(С1-С6)алкила, где арильная группа может быть возможно замещена 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
R1 представляет собой -С(О)- и
R2 представляет собой -SO2-;
R1 выбран из:
водорода,
(С1-С6)алкила,
(С1-С6)галогеналкила,
(С1-С3)алкокси(С2-С6)алкила,
(С2-С6)алкенила,
(С3-С10)циклоалкила, который может быть возможно замещен 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
(С3-С10)циклоалкил(С1-С6)алкил, где циклоалкильная группа может быть возможно замещена 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
арила, который может быть возможно замещен 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси, и
арил(С1-С6)алкила, где арильная группа может быть возможно замещена 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
и R2 выбран из:
(С1-С6)алкила,
(С1-С6)галогеналкила,
(С1-С3)алкокси(С2-С6)алкила,
(С2-С6)алкенила,
(С3-С10)циклоалкила, который может быть возможно замещен 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
(С3-С10)циклоалкил(С1-С6)алкила, где циклоалкильная группа может быть возможно замещена 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси,
арила, который может быть возможно замещен 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси, и
арил(С1-С6)алкила, где арильная группа может быть возможно замещена 1, 2 или 3 группами, независимо выбранными из галогена, (С1-С3)алкила и (С1-С3)алкилокси.
В еще одном воплощении А1Е2 в изобретении предложено соединение согласно воплощению А1Е1, где R представляет собой водород, так что соединение формулы (VI) представляет собой 5-гидрокси-4-(2-метил-1-пропенил)-5Н-2-фуранон формулы (VIA).

В еще одном воплощении А1Е3 в изобретении предложено соединение согласно воплощению А1Е1 формулы (VIе)
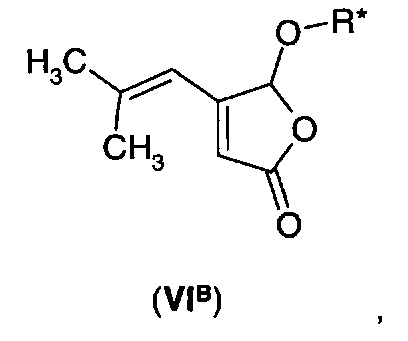
где R* представляет собой хиральную (С5-С15) углеводородную группу.
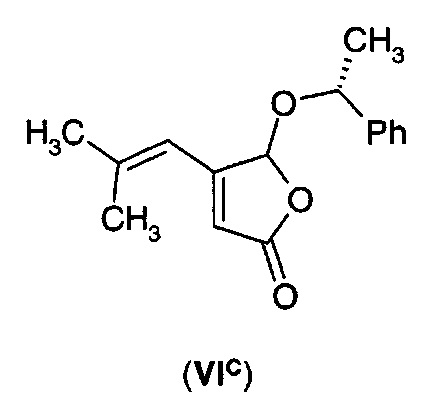
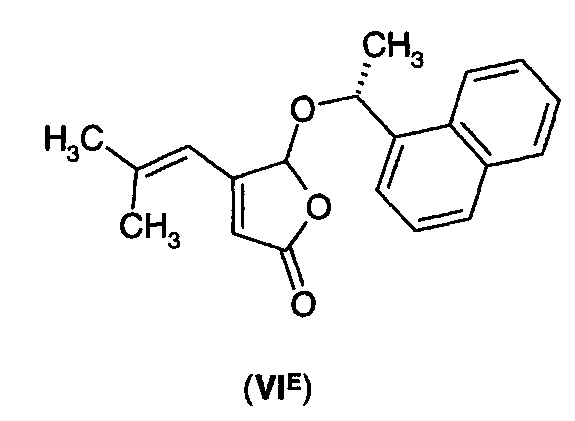
В еще одном воплощении А1Е4 в изобретении предложено соединение согласно воплощению А1Е3, где R* выбран из (R)- или (S)-α-метилбензила, (R)- или (S)-1-(1-нафтил)этила, (R)- или (S)-1-(2-нафтил)этила, ментила и борнила, так что соединение формулы (VI) представляет собой соединение формулы (VIC)-(VIK).






В еще одном воплощении А1Е5 в изобретении предложено соединение согласно воплощению А1Е1, где R представляет собой R1-C(O)- или R2-SO2-, и R1 и R2 представляют собой хиральные (С5-С15) углеводородные группы.
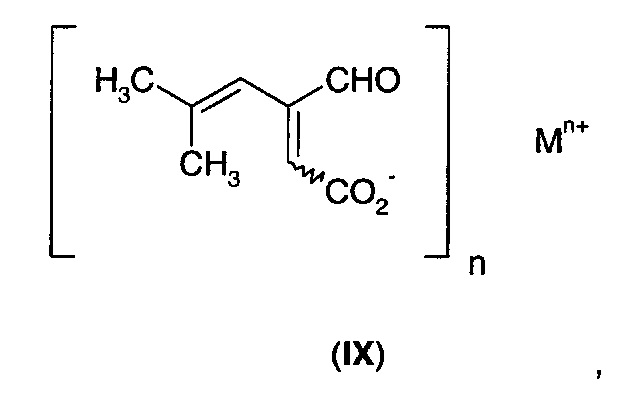
В другом аспекте А2 в изобретении предложено соединение формулы (IX)
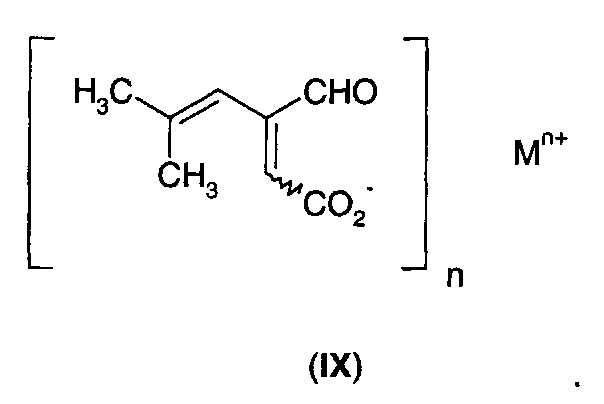
Соединение формулы (IX) может существовать в виде либо (Е)-, либо (Z)-геометрического изомера, либо в виде смеси геометрических изомеров.
В первом воплощении А2Е1 в изобретении предложено соединение формулы (IX), где:
n равно 1, и М+ выбран из Li+, Na+, К+, Rb+, NH4+, ((С1-С3)алкил)NН3+, ((С1-С3)алкил)2NН2+, ((С1-С3)алкил)3NН+ и ((С1-С3)алкил)4N+; или
n равно 2, и М2+ выбран из Мg2+, Са2+ и Zn2+.
В еще одном воплощении А2Е2 в изобретении предложено соединение согласно воплощению А2Е1, где n равно 1, и М+ выбран из NH4+ и ((С1-С3)алкил)NН3+.
В еще одном воплощении А2Е3 в изобретении предложено соединение согласно воплощению А2Е1, где n равно 1, и М+ выбран из Li+, Na+ и К+.
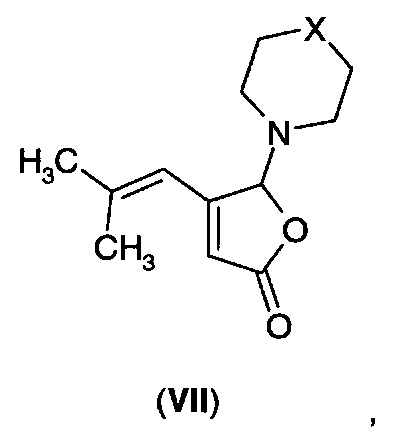
В другом аспекте A3 в изобретении предложено соединение формулы (VII).

В первом воплощении А3Е1 в изобретении предложено соединение формулы (VII), где -Х- представляет собой одиночную связь, -СН2-, -О-; -NН-,-N((С1-С3)алкил)-, -N(бензил)- или
В следующем воплощении А3Е2 в изобретении предложено соединение согласно воплощению А3Е1, выбранное из:
4-(2-метилпропенил)-5-пирролидин-1-ил-5Н-фуран-2-она;
4-(2-метилпропенил)-5-пиперидин-1-ил-5H-фуран-2-она;
4-(2-метилпропенил)-5-морфолин-4-ил-5Н-фуран-2-она; и
1,4-бис-(4-(2-метилпропенил)-5Н-фуран-2-он-5-ил)пиперазина.
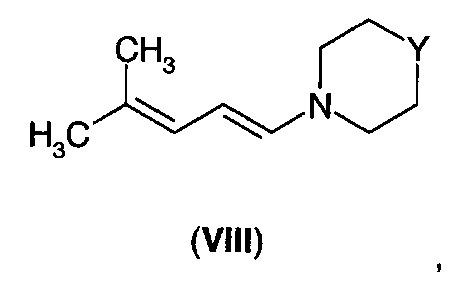
В другом аспекте А4 в изобретении предложено соединение формулы (VIII).

В первом воплощении А4Е1 в изобретении предложено соединение формулы (VIII), где -Y- представляет собой одиночную связь, -СН2-, -О-; -NH-, -N((С1-С3)алкил)-, -N(бензил)- или

В следующем воплощении А4Е2 в изобретении предложено соединение согласно воплощению А4Е1, выбранное из:
4-(4-метил-1,3-пентадиен-1-ил)морфолина;
1-(4-метил-1,3-пентадиен-1-ил)-пиперазина,
1-(4-метил-1,3-пентадиен-1-ил)-4-метилпиперазина,
4-этил-1-(4-метил-1,3-пентадиен-1-ил)-пиперазина,
4-бензил-1-(4-метил-1,3-пентадиен-1-ил)-пиперазина и
1,4-бис-(4-метил-1,3-пентадиен-1-ил)пиперазина.
В другом аспекте А5 в изобретении предложен способ получения соединения формулы (VIA), включающий стадию обработки соединения формулы (VII) водой в присутствии кислотного катализатора.
В первом воплощении А5Е1 в изобретении предложен способ получения соединения формулы (VIA), включающий стадии:
а) получение соединения формулы (VII)
где -Х- представляет собой одиночную связь, -СН2-, -О-, -NH-, -N((С1-С3)алкил)-, -N(бензил)- или
и
б) обработку соединения формулы (VII) водой в присутствии кислотного катализатора.
В еще одном воплощении А5Е2 в изобретении предложен способ согласно воплощению А5Е1, где соединение формулы (VII) со стадии (а) выделяют перед стадией гидролиза (б).
В следующем воплощении А5Е3 в изобретении предложен способ согласно воплощению А5Е1, где стадию гидролиза (б) выполняют непосредственно после стадии (а), так что соединение формулы (VII) или (VIIB) не выделяют перед стадией гидролиза (б).
В следующем воплощении А5Е4 в изобретении предложен способ согласно воплощениям А5Е1, А5Е2 или А5Е3, где соединение формулы (VII) получают путем обработки соединения формулы (VIII)
где -Y- представляет собой одиночную связь, -СН2-, -О-; -NH-, -N((С1-С3)алкил)-, -N(бензил)- или
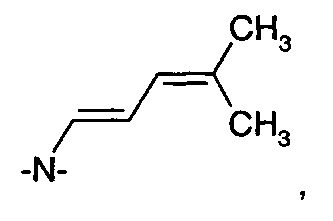
глиоксалевой кислотой или ее гидратом.
В другом аспекте А6 в изобретении предложен способ получения соединения формулы (VI), где R не является водородом, R1-C(O)- и R2-SO2-, включающий стадию обработки соединения формулы (VIA) спиртом R-OH в присутствии кислотного катализатора.
В воплощении А6Е1 в изобретении предложен способ получения соединения формулы (VI), где R не является водородом, R1-C(O)- и R2-SO2-, включающий стадии:
а) получение соединения формулы (VIA) с помошью способа по любому из воплощений А5Е1, А5Е2, А5Е3 и А5Е4, как определено выше; и
б) обработку соединения формулы (VIA) спиртом R-OH в присутствии кислотного катализатора.
В другом аспекте А7 в изобретении предложен способ получения соединения формулы (VI), где R не является водородом, R1-C(O)- и R2-SO2-, включающий стадию обработки соединения формулы (VII) спиртом R-OH в присутствии стехиометрического количества кислоты.
В воплощении А7Е1 в изобретении предложен способ получения соединения формулы (VI), где R не является водородом, R1-C(O)- и R2-SO2-, включающий стадии:
а) получение соединения формулы (VII); и
б) обработку соединения формулы (VII) спиртом R-OH в присутствии стехиометрического количества кислоты.
В другом аспекте А8 в изобретении предложен способ получения соединения формулы (VI), где R представляет собой R1-C(O)-, включающий стадию обработки соединения формулы (VIA) хлорангидридом R1-C(O)-Cl или ангидридом кислоты (R1-C(O))2O.
В воплощении А8Е1 в изобретении предложен способ получения соединения формулы (VI), где R представляет собой R1-C(O)-, включающий стадии:
а) получение соединения формулы (VIA) с использованием способа согласно любому из воплощений А5Е1, А5Е2, А5Е3 и А5Е4, как определено выше; и
б) обработку соединения формулы (VIA) хлорангидридом R1-C(O)-Cl или ангидридом кислоты (R1-C(O))2O.
В другом аспекте А9 в изобретении предложен способ получения соединения формулы (VI), где R представляет собой R2-SO2-, включающий стадию обработки соединения формулы (VIA) сульфонилхлоридом R2-SO2-Cl.
В воплощении А9Е1 в изобретении предложен способ получения соединения формулы (VI), где R представляет собой R2-SO2-, включающий стадии:
а) получение соединения формулы (VIA) с помощью способа согласно любому из воплощений А5Е1, А5Е2, А5Е3 и А5Е4, как определено выше; и
б) обработку соединения формулы (VIA) сульфонилхлоридом R2-SO2-Cl.
В другом аспекте А10 в изобретении предложен способ получения енаминного производного 4-метил-2-пентеналя.
В первом воплощении А10Е1 в изобретении предложен способ получения енаминного производного 4-метил-2-пентеналя, включающий взаимодействие ацетальдегида с изобутиральдегидом в присутствии подходящего амина.
В еще одном воплощении А10Е2 в изобретении предложен способ согласно воплощению А10Е1, где подходящий амин представляет собой вторичный амин.
В еще одном воплощении А10Е3 в изобретении предложен способ согласно воплощению А10Е2, где вторичный амин выбран из: ((С1-С4)алкил)2NН, пирролидина, пиперидина, морфолина, пиперазина, N-метилпиперазина, N-этилпиперазина и N-бензилпиперазина.
В еще одном воплощении А10Е4 в изобретении предложен способ согласно воплощению А10Е3, где вторичный амин выбран из пирролидина, пиперидина, морфолина и пиперазина.
В еще одном воплощении А10Е5 в изобретении предложен способ согласно любому из воплощений А10Е1, А10Е2, А10Е3 и А10Е4, где взаимодействие выполняют в присутствии кислотного катализатора.
В еще одном воплощении А10Е6 в изобретении предложен способ согласно любому из воплощений А10Е1, А10Е2, А10Е3, А10Е4 и А10Е5, где изобутиральдегид объединяют с подходящим амином перед добавлением ацетальдегида.
В следующем воплощении А10Е6 в изобретении предложен способ согласно любому из воплощений А10Е1, А10Е2, А10Е3, А10Е4 и А10Е5, где изобутиральдегид и ацетальдегид добавляют в реакционный сосуд одновременно.
В другом аспекте А11 в изобретении предложен способ получения 3-аминометил-5-метилгексановой кислоты (II).
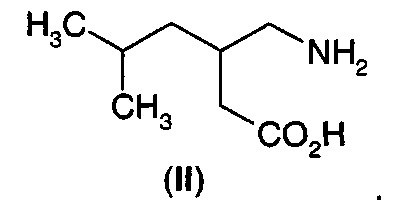
В первом воплощении А11Е1 в изобретении предложен способ получения 3-аминометил-5-метилгексановой кислоты (II) или ее фармацевтически приемлемой соли, включающий стадии:
а) получения 5-гидрокси-4-(2-метил-1-пропенил)-5Н-2-фуранона (VIA)
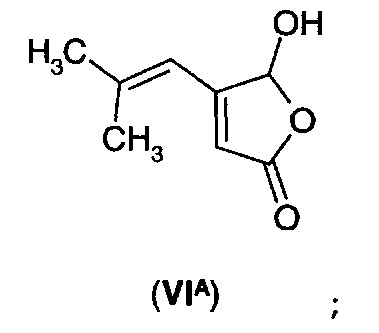
б) преобразования указанного 5-гидрокси-4-(2-метил-1-пропенил)-5Н-2-фуранона (VIA) в 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранон (IА)

и
в) преобразования указанного 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона (IА) в 3-аминометил-5-метилгексановую кислоту (II).
В еще одном воплощении А11Е2 в изобретении предложен способ согласно воплощению А11Е1, где 3-аминометил-5-метилгексановая кислота (II) представляет собой (S)-3-аминометил-5-метилгексановую кислоту ((S)-II)
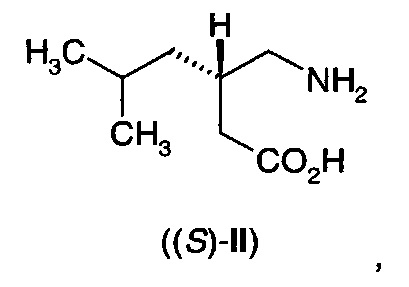
где указанный (S)-3-аминометил-5-метилгексановая кислота имеет энантиомерный избыток по меньшей мере 80%.
В еще одном воплощении А11Е3 в изобретении предложен способ согласно воплощению А11Е1 или А11Е2, где стадия (а) включает способ согласно воплощениям А5Е1, А5Е2, А5Е3 или А5Е4, как описано выше.
В еще одном воплощении А11Е4 в изобретении предложен способ согласно воплощению А11Е1, А11Е2 или А11Е3, где стадия (б) включает стадии:
(б1) обработки 5-гидрокси-4-(2-метил-1-пропенил)-5H-2-фуранона (VIA) оксидом, гидроксидом, карбонатом или бикарбонатом металла, аммиаком, моно-, ди- или три-(С1-С3)алкиламина или гидроксидом тетра-(С1-С3)алкиламмония с образованием соли формулы (IX)
где:
n равно 1, и М+ выбран из Li+, Na+, К+, Rb+, NH4+, ((С1-С3)алкил)NН3+, ((С1-С3)алкил)2NН2+, ((С1-С3)алкил)3NН+ и ((С1-С3)алкил)4N+; или
n равно 2, и М2+ выбран из Мg2+, Са2+ и Zn2+;
(б2) гидрорования соли формулы (IX) с получением соли формулы (X)

(б3) обработку соли формулы (X) кислотой.
В еще одном воплощении А11Е5 в изобретении предложен способ согласно воплощению А11Е1, А11Е2 или А11Е3, где стадия (б) включает стадии:
(б1) преобразования 5-гидрокси-4-(2-метил-1-пропенил)-5Н-2-фуранона (VIA) в соединение формулы (VI), как определено в воплощении А1Е3, где R представляет собой хиральную (С5-С15)углеводородную группу;
(б2) гидрирования соединения формулы (VI) с получением соединения формулы (XI)

где R* представляет собой хиральную (С5-С15)углеводородную группу; и
(б3) обработку соединения формулы (XI) кислотой с получением ((S)-IA).
В другом аспекте А12 в изобретении предложен еще один способ получения 3-аминометил-5-метилгексановой кислоты (II).
В первом воплощении А12Е1 в изобретении предложен способ получения 3-аминометил-5-метилгексановой кислоты (II) или ее фармацевтически приемлемой соли, включающий стадии:
а) получения 5-гидрокси-4-(2-метил-1-пропенил)-5Н-2-фуранона (VIA)
б) обработки 5-гидрокси-4-(2-метил-1-пропенил)-5Н-2-фуранона (VIA) аммиаком или моно-(С1-С3)алкиламином с образованием соли формулы (IХА)
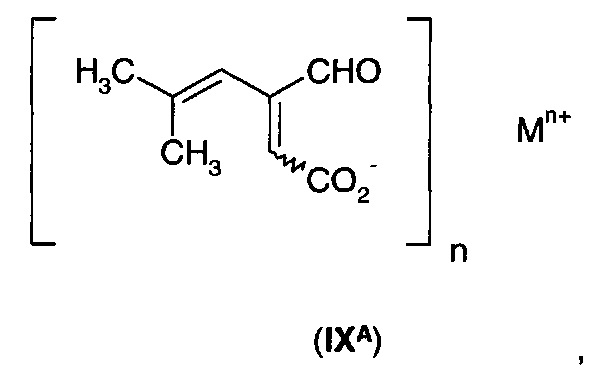
где n равно 1, и М+ выбран из NH4+ и ((С1-С3)алкил)NН3+;
в) гидрирования соли формулы (IХА) с получением соли формулы (ХА)
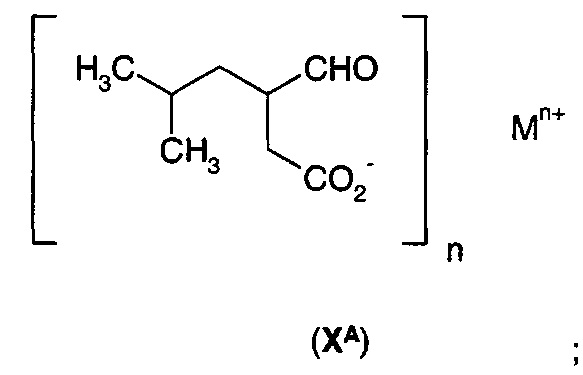
г) обработки соли формулы (ХА) ферментом трансаминазой или аминоксидазой/иминоредуктазой с получением 3-аминометил-5-метилгексановой кислоты (II).
В еще одном воплощении А12Е2 в изобретении предложен способ согласно воплощению А12Е1, где 3-аминометил-5-метилгексановая кислота (II) представляет собой (S)-3-аминометил-5-метилгексановую кислоту ((S)-II)
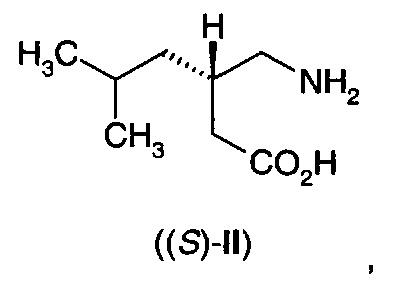
где указанный (S)-3-аминометил-5-метилгексановая кислота имеет энантиомерный избыток по меньшей мере 80%.
В еще одном воплощении А12Е3 в изобретении предложен способ согласно воплощению А12Е1 или А12Е2, где стадия (а) включает способ согласно воплощениям А5Е1, А5Е2, А5Е3 или А5Е4, как описано выше.
В другом аспекте А13 в изобретении предложен еще один способ получения 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона (IA).
В первом воплощении А13Е1 в изобретении предложен дополнительный способ получения 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона (IA), включающий стадии:
а) получения 3-изобутилиден-2-оксопентандикарбоновой кислоты (ХIIА) или ее циклизованного изомера (XIIB)

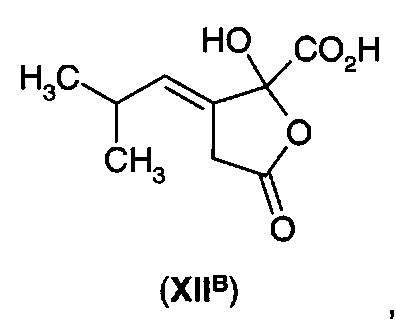
б) последовательного или одновременного восстановления углерод-углеродной двойной связи и декарбоксилирование α-кетокислотной функциональной группы.
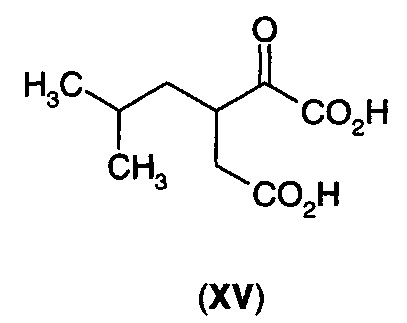
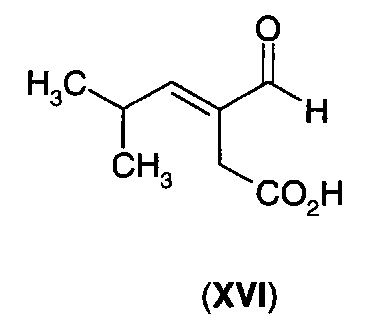
В следующем воплощении А13Е2 в изобретении предложен способ согласно воплощению А13Е1, где углерод-углеродную двойную связь восстанавливают с получением 3-изобутил-2-оксопентандикарбоновой кислоты (XV) или ее циклизованного изомера (XVA)

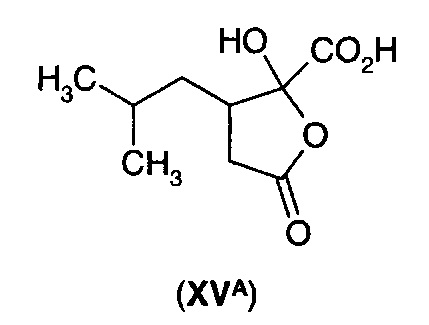
перед декарбоксилированием α-кетокислотной функциональной группы.
В еще одном воплощении А13Е3 в изобретении предложен способ согласно воплощению А13Е1, где α-кетокислотную функциональную группу декарбоксилируют с получением 3-формил-5-метил-3-пентеновой кислоты (XVI) или ее циклизованного изомера (XVIA)
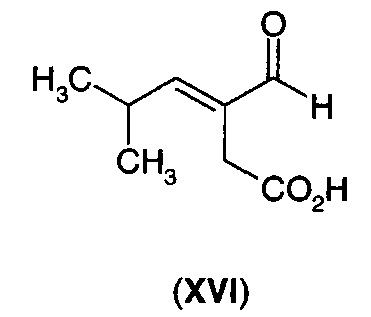

перед восстановлением углерод-углеродной двойной связи.
В еще одном воплощении А13Е4 в изобретении предложен способ согласно воплощению А13Е1, где α-кетокислотную функциональную группу декарбоксилируют, и углерод-углеродную двойную связь восстаналивают одновременно.
В еще одном воплощении А13Е5 в изобретении предложен способ согласно воплощениям А13Е1, А13Е2, А13Е3 или А13Е4, где декарбоксилирование выполняют в присутствии фермента декарбоксилазы.
В еще одном воплощении А13Е6 в изобретении предложен способ согласно воплощениям А13Е1, А13Е2, А13Е3, А13Е4 или А13Е5, где восстановление углерод-углеродной двойной связи выполняют в присутствии фермента еноатредуктазы.
В другом аспекте А14 в изобретении предложено соединение, выбранное из:
3-изобутилиден-2-оксопентандикарбоновой кислоты;
3-изобутил-2-оксопентандикарбоновой кислоты; и
3-формил-5-метил-3-пентеновой кислоты,
или их соли, (С1-С6)алкилового эфира или циклизованного изомера.
В другом аспекте А15 в изобретении предложен способ получения (S)-3-аминометил-5-метилгексановой кислоты ((S)-II)

или ее фармацевтически приемлемой соли, включающий стадии:
а) получения 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5H-2-фуранона (IA) с использованием способа согласно любому из воплощений А13Е1, А13Е2, А13Е3, А13Е4, А13Е5 или А13Е6, как определено выше, и
б) преобразования указанного 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона в (S)-3-аминометил-5-метилгексановую кислоту.
В другом аспекте А16 в изобретении предложен способ преобразования (R)-3-аминометил-5-метилгексановой кислоты в (S)-3-аминометил-5-метилгексановую кислоту, включающий обработку (R)-3-аминометил-5-метилгексановой кислоты ферментом трансаминазой или ферментом аминоксидазой/имидоредуктазой.
В другом аспекте А17 в изобретении предложен способ увеличения доли (S)-3-аминометил-5-метилгексановой кислоты в смеси (R)- и (S)-3-аминометил-5-метилгексановой кислоты, включающий обработку смеси ферментом трансаминазой или ферментом аминоксидазой/имидоредуктазой.
В другом аспекте А18 в изобретении предложен фермент трансаминаза, который пригоден для преобразования 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона (IA) в прегабалин.
В первом воплощении А18Е1 в изобретении предложен фермент трансаминаза, имеющий аминокислотную последовательность, которая обладает по меньшей мере 95% гомологией с аминокислотной последовательностью


где
X27 выбран из глутамина (Q) и глутаминовой кислоты (Е);
X41 выбран из изолейцина (I) и валина (V);
X45 выбран из аспарагина (N) и гистидина (Н);
X147 выбран из аспарагина (N) и глутамина (Q);
X163 выбран из лейцина (L) и метионина (М);
X165 выбран из тирозина (Y) и гистидина (Н);
X180 выбран из треонина (Т); глицина (G) и серина (S);
X304 выбран из аланина (А) и серина (S);
X324 выбран из глицина (G) и серина (S);
X401 выбран из лизина (К) и глутаминовой кислоты (Е);
X408 выбран из треонина (Т) и глутамина (Q);
X415 выбран из серина (S) и аланина (А);
X416 выбран из пролина (Р) и аланина (А);
X417 выбран из лейцина (L) и метионина (М); и
X424 выбран из цистеина (С) и серина (S).
В еще одном воплощении А18Е2 в изобретении предложен фермент трансаминаза согласно воплощению А18Е1, имеющий аминокислотную последовательность SEQ ID NO. 1.
В еще одном воплощении А18ЕЗ в изобретении предложен фермент трансаминаза согласно воплощению А18Е1 или А18Е2, где:
X27 представляет собой глутаминовую кислоту (Е);
X147 представляет собой глутамин (Q);
X165 представляет собой гистидин (Н);
X304 представляет собой серии (S);
X324 представляет собой глицин (G);
X401 представляет собой лизин (К);
X408 представляет собой глутамин (Q);
X416 представляет собой аланин (А);
X417 представляет собой метионин (М); и
X424 представляет собой серии (S).
В еще одном воплощении А18Е4 в изобретении предложен фермент трансаминаза согласно воплощению А18Е2, имеющий аминокислотную последовательность, выбранную из:







В другом аспекте А19 в изобретении предложен способ получения (S)-3-аминометил-5-метилгексановой кислоты ((S)-II)

или ее фармацевтически приемлемой соли, включающий стадию обработки 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона (IA) и амина ферментом трансаминазой согласно любому из воплощений А18Е1, А18Е2, А18Е3 и А18Е4.
Подробное описание предпочтительных воплощений изобретения
Термин "алкил" означает прямоцепочечный или разветвленный насыщенный алифатический углеводородный радикал, содержащий конкретное число атомов углерода. Примеры алкильных радикалов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, неопентил и н-гексил.
Термин "алкокси" означает группу, состоящую из алкильной группы, как определено выше, соединенной с атомом кислорода. Примеры алкоксигрупп включают метокси, этокси и изопропокси.
Термин "алкокси-алкил" означает прямоцепочечный или разветвленный насыщенный алифатический углеводородный радикал, в котором алкоксигруппа заменяет атом водорода алкила. Примером алкокси-алкильной группы является 2-метоксиэтил.
Термин "галогеналкил" означает алкильную группу, как определено выше, где один или более чем один атом водорода заменены на атом фтора, хлора, брома или йода. Когда заменено более одного атома водорода, заменяющие атомы галогена могут быть одинаковыми или разными. Примеры галогеналкильных групп включают фторметил, дифторметил, трифторметил, хлордифторметил, 2,2,2-трифторэтил и 3-бромпропил.
Термин "арил" означает фенильную или нафтильную группу.
Термин "арил-алкил" означает прямоцепочечный или разветвленный насыщенный алифатический углеводородный радикал, в котором арильная группа замегяет атом водорода алкила. Примером арил-алкильной группы является бензил.
Термин "циклоалкил" означает насыщенное моноциклическое или полициклическое карбоциклическое кольцо, содержащее конкретное число атомов углерода. Примеры моноциклических циклоалкильных групп включают циклопропил, циклобутил, циклопентил и циклогексил. Примеры полициклических циклоалкильных групп включают бицикло[2,2,1]гептил и бицикло[3,2,1]октил.
Термин "возможно замещенный" в отношении алкильной или арильной группы означает, что атом водорода алкильной или арильной группы может быть заменен одной из перечисленных групп. Замещение можно осуществлять в любом положении в пределах алкильной или арильной группы. Когда возможное замещение осуществляется "одной или более группой", любое число атомов водорода алкильной или арильной группы, вплоть до максимального числа, равного числу атомов водорода, присутствующих в алкильной или арильной группе, может быть заменено, и каждое замещение является независимым от других.
Термин "энантиомерный избыток", иногда обозначаемый с помощью сокращения как "э.и.", является для данного образца мерой избытка одного энантиомера по отношению к его антиподу и выражается в виде процентов. Энантиомерный избыток определяют следующим образом:
100×(еr-1)/(еr+1),
где "еr" представляет собой отношение более часто встречающегося энантиомера к менее часто встречающемуся энантиомеру.
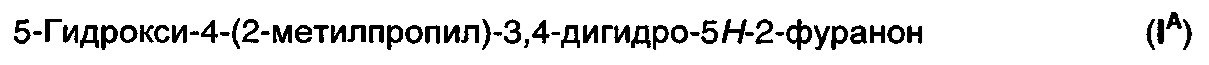
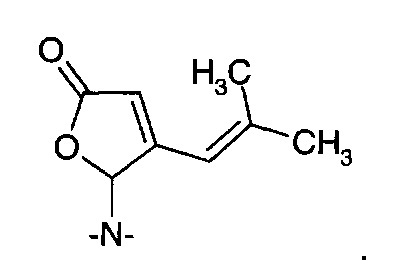
представляет собой традиционное промежуточное соединение при получении 3-аминометил-5-метилгексановой кислоты (II). Обработка рацемического соединения (IА) аммиаком в присутствии химического восстанавливающего агента обеспечивает получение рацемической 3-аминометил-5-метилгексановой кислоты ((R/S)-II) (см. WO 2008/127646A2). Предполагают, что в реакцию вовлечен изомер с раскрытым циклом (IB).

Восстановительное аминирование донором амина в присутствии фермента трансаминазы может обеспечить получение энантиомерно обогащенной 3-аминометил-5-метилгексановой кислоты. Подходящими донорами амина являются первичные амины, такие как моно-алкиламины, в частности, изопропиламин, и α-аминокислоты.
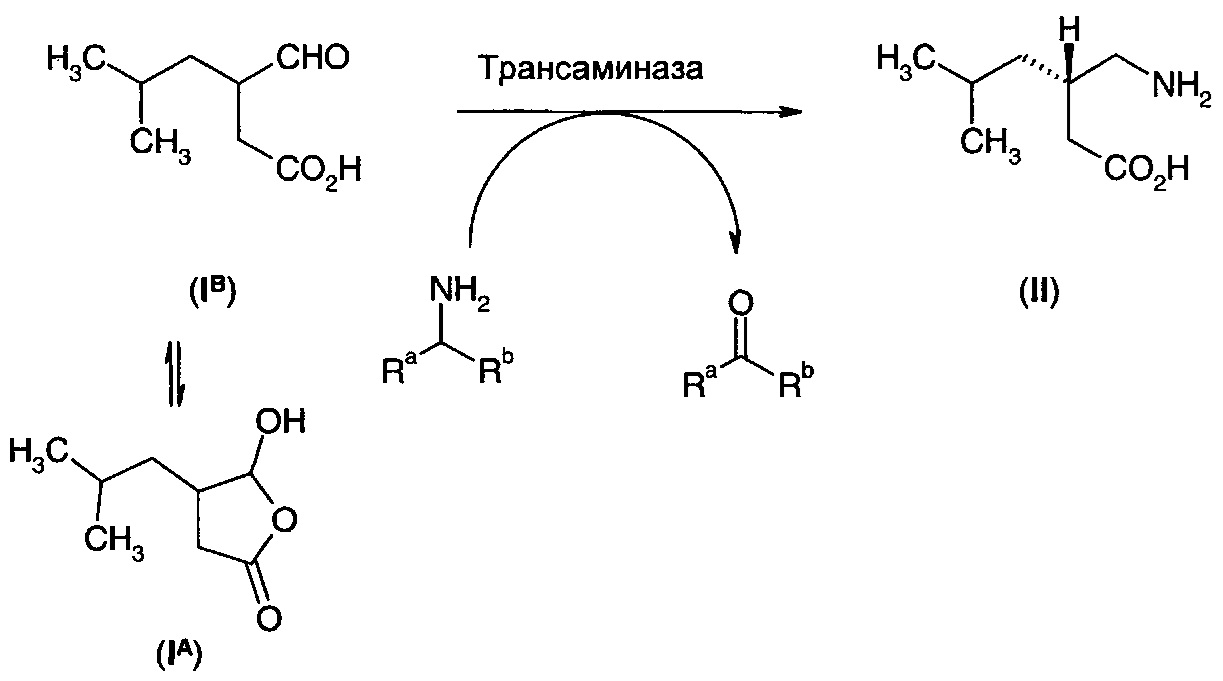
Взаимодействие соединения (IА) с подходящей аминодегидрогеназой/иминоредуктазой в присутствии аммиака может также представлять собой подходящий путь получения прегабалина. Кофактор, такой как NADH или NADPH, может быть необходим в стехиометрическом количестве, или вторая оксидоредуктаза, такая как формиатдегидрогеназа, может быть включена для замыкания цикла кофактора.

В случаях, когда дигидрофуранон (IА) имеет определенную стереохимию в С-4 положении, данная стереохимия может быть сохранена в процессе реакции восстановительного аминирования. Поскольку прегабалин имеет (S)-стереохимию, может быть предпочтительно, чтобы эта стереохимия уже присутствовала в дигидрофураноне.
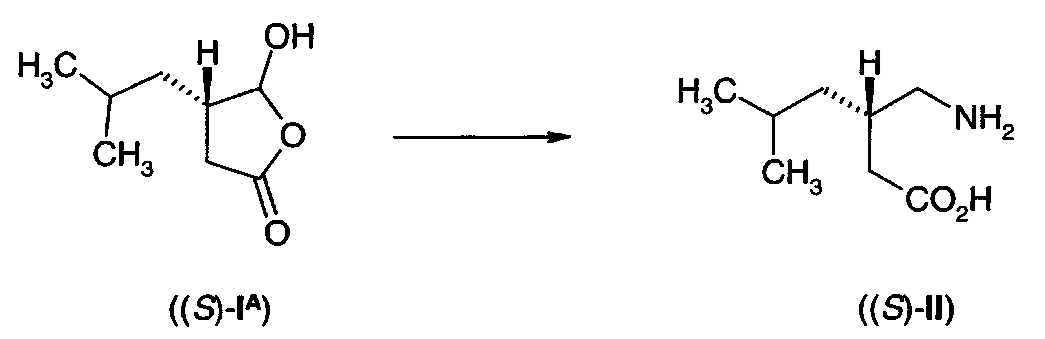
Альтернативно, дигидрофуранон (IА) можно использовать в виде рацемической формы. Затем желаемый стероизомер продукта может быть получен либо путем реакции восстановительного аминирования в условиях, дающих возможность стереоселективного образования одного стереоизомера, например, путем выполнения превращения в присутствии фермента трансаминазы, или путем подвергания продукта отдельной стадии разделения, такой как кристаллизация с хиральной кислотой или основанием.
Дигидрофуранон (IА) может быть легко получен в рацемической или энантиомерно обогащенной форме с использованием способом, описанных ниже.
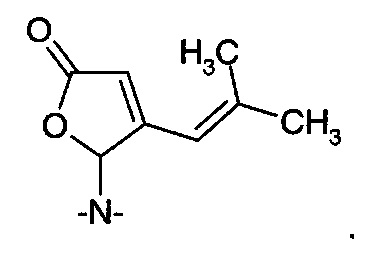
В первом способе дигидрофуранон (IА) получают путем восстановления 5-гидрокси-4-(2-метил-1-пропенил)-5Н-2-фуранона (VIA).

Восстановление можно удобно осуществлять путем гидрирования в присутствии подходящего катализатора. Подходящие катализаторы включают гомогенные и гетерогенные катализаторы. Катализатор типично содержит переходный металл, такой как палладий, платина, родий, рутений или никель, или их соль или оксид. Гетерогенные катализаторы включают тонкоизмельченные металлы, а также металлы и оксиды металлов, нанесенные на подложку, где подложка может представлять собой углерод, диоксид кремния, оксид алюминия или любое подходящее инертное вещество. Гомогенные катализаторы включают комплексы переходных металлов с фосфиновым лигандом. Когда фосфиновый лиганд является хиральным, катализатор является хиральным. Когда используют ахиральный катализатор, тогда продукт представляет собой рацемический дигидрофуранон (IА). Использование хирального катализатора может обеспечить энантиоселективное получение дигидрофуранона (IА).
Селективность реакции гидрирования и общий выход можно улучшить при осуществлении реакции в щелочной среде. Без связи с теорией полагают, что в присутствии основания фуранон преимущественно существует в виде соли с разомкнутым циклом (IX).
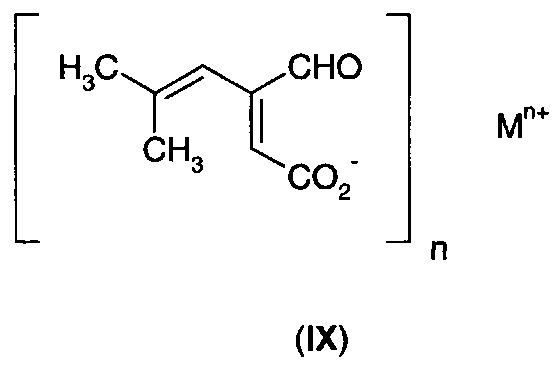
Можно использовать любое подходящее основание при условии, что оно не препятствует процессу гидрирования, например, за счет загрязнения катализатора. Примеры подходящих оснований включают оксиды, гидроксиды, карбонаты и бикарбонаты щелочного (такого как Li, Na, К и Rb) и щелочноземельного (такого как Са и Мg) металла. Соли других металлов, такие как соли цинка, также могут быть использованы. Соли щелочных металлов могут быть предпочтительны, благодаря их хорошей растворимости и/или низкой токсичности. Аминные основания, такие как аммиак и первичные, вторичные и третичные амины могут быть использованы для получения солей аммония. Гидроксид тетра-алкиламмония также может быть использован, что приведет к образованию солей тетра-алкиламмония.
Гидрирование соли формулы (IX) обеспечивает получение соли формулы (X).

Дигидрофуранон (IА) выделяют после реакции гидрирования путем обработки этой соли подходящей кислотой. Альтернативно, эта соль может быть непосредственно превращена в 3-аминометил-5-метилгексановую кислоту (II) путем обработки ферментом трансаминазой или аминоксидазой/иминоредуктазой. В данном случае может быть предпочтительным использование соли аммония (М+=NH4+) или соли первичного алкиламмония (М+=алкил-NН3+), поскольку ион аммония или алкиламмония является субстратным кофактором для фермента. Использование соли изопропиламмония в комбинации с ферментом трансаминазой представляет собой предпочтительный пример.

Фураноны формулы (VI), где R не является водородом, можно также восстанавливать путем гидрирования. В случае, когда R представляет собой алкильную, галогеналкильную, алкоксиалкильную, алкенильную, циклоалкильную, циклоалкил-алкильную, арильную или арил-алкильную группу, эти фураноны могут быть получены из соединения формулы (VIA) путем взаимодействия со спиртом R-OH в присутствии кислотного катализатора. В случае, когда R представляет собой R1-C(O)-, фураноны могут быть получены из соединения формулы (VIA) путем взаимодействия с ангидридом кислоты (R1-C(O))2O или хлорангидридом R1-C(O)-Cl, возможно в присутствии основания, например, третичного амина. В случае, когда R представляет собой R2-SO2-, фураноны могут быть получены из соединения формулы (VIA) путем взаимодействия с сульфонилхлоридом R2-SO2-Cl, возможно в присутствии основания, например, третичного амина.
После стадии гидрирования дигидрофуранон формулы (IА) может быть образован из продукта восстановления путем обработки кислотой (в случае, когда R представляет собой алкильную, галогеналкильную, алкоксиалкильную, алкенильную, циклоалкильную, циклоалкил-алкильную, арильную или арил-алкильную группу) или основанием (в случае, когда R представляет собой R1-С(О)- или R2-SO2-)-
Использование хиральной группы R может обеспечить получение хирального продукта гидрирования без необходимости в хиральном катализаторе.
Например, фуранон (VIA) подвергают взаимодействию с хиральным спиртом R*-OH с получением хирального эфирного производного (VIB).
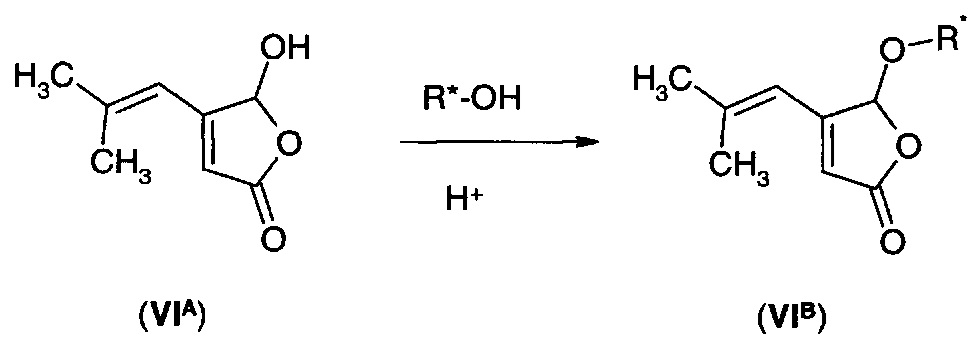
Подходящие хиральные спирты могут включать α-арильные спирты, такие как 1-фенилэтанол и 1-нафтилэтанол, а также терпеновые спирты, такие как ментол и борнеол. Гидрирование производного (VIB) может протекать энантиоселективным путем, и затем в результате обработки полученного продукта подходящей кислотой и в присутствии воды получают дигидрофуранон (IА) в хиральной форме.
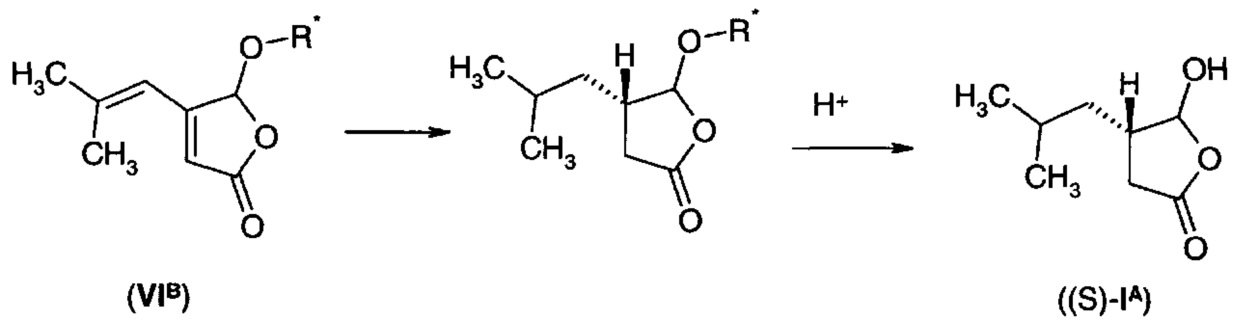
Фуранон (VIA) может быть получен из соединения формулы (VII)

где -Х- представляет собой одиночную связь, -СН2-, -О-, -NH-, -N((С1-С3)алкил)-, -N(бензил)-, или
путем обработки соединения формулы (VII) водой в присутствии кислотного катализатора. Подходящие кислоты включают минеральные кислоты, такие как серная кислота. Альтернативно, соединение формулы (VII) можно обработать спиртом R-OH с получением непосредственно соединения формулы (VI), где R представляет собой алкильную, галогеналкильную, алкоксиалкильную, алкенильную, циклоалкильную, циклоалкил-алкильную, арильную или арил-алкильную группу.
Соединения формулы (VII) могут быть получены путем взаимодействия диенамина формулы (VIII).
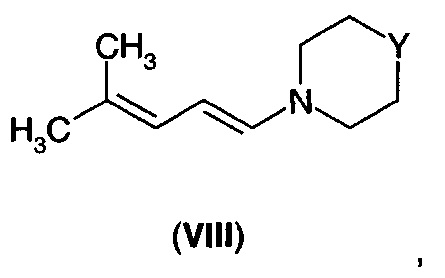
где -Y- представляет собой одиночную связь, -СН2-, -О-; -NH-, -N((С1-С3)алкил)-, -N(бензил)- или

с глиоксалевой кислотой или ее гидратом.

Должно быть понятно, что -Х- в формуле (VII) соответствует -Y- в исходном веществе формулы (VIII) за исключением того случая, когда -Y-представляет собой

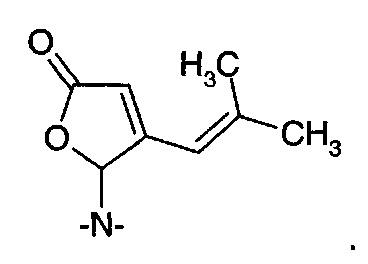
Соединения формулы (VIII), где -Y- представляет собой одиночную связь или -СН2-, получали путем взаимодействия 4-метил-2-пентеналя с пирролидином или пиперидином (Kienzle, F. et al., Helv. Chim. Acta 1985, 68(5), 1133-39). Другие соединения формулы (VIII) могут быть получены аналогично.
Альтернативно, конденсация изобутиральдегида и ацетальдегида в присутствии подходящего амина, такого как пирролидин, пиперидин или морфолин, с каталитической кислотой в растворителе, таком как ацетонитрил, обеспечивает получение производных диенамина (VIII).

Если пиперазин используют в качестве амина, то получают бис-диенамин.
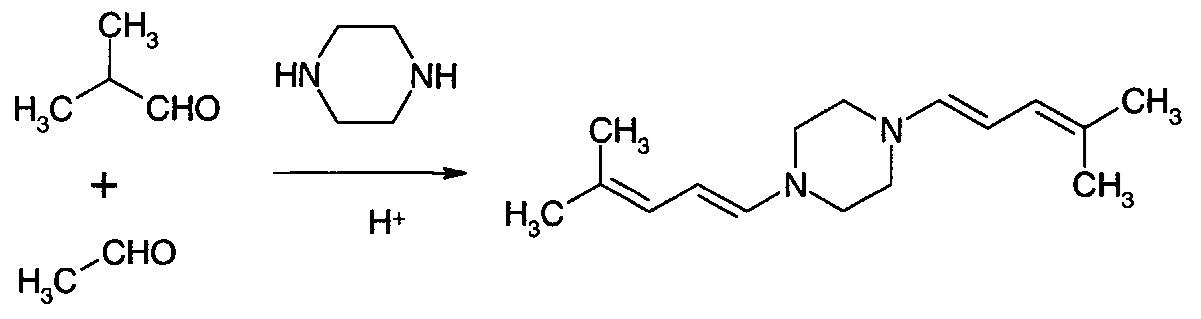
Соединение формулы (VIII), где Y представляет собой NH, может быть получено путем использования моно-защищенного пиперазина в качестве амина с последующей стадией удаления защиты.
Ациклические вторичные амины, такие как диэтиламин и диизопропиламин, также могут быть использованы, но циклические вторичные амины являются предпочтительными.
Данный способ взаимодействия изобутиральдегида и ацетальдегида отличается от прямого взаимодействия, катализируемого основанием (например, при использовании карбоната калия в качестве основания: патент Великобритании GB 834100), при котором только образуется аддукт 2,2-диметил-3-гидроксибутаналь, где изобутиральдегид действует как нуклеофил.

Не желая быть связанными какой-либо конкретной теорией, утверждают, что и ацетальдегид, и изобутиральдегид первоначально преобразуются в свои енаминные производные. В присутствии кислотного катализатора более основный енамин изобутиральдегида преобразуется в его иминиевый ион. Эти электрофильные соединения преимущественно взаимодействуют с менее стерически затрудненным нуклеофилом, который представляет собой енамин ацетальдегида, что гарантирует, что взаимодействие протекает желаемым путем.
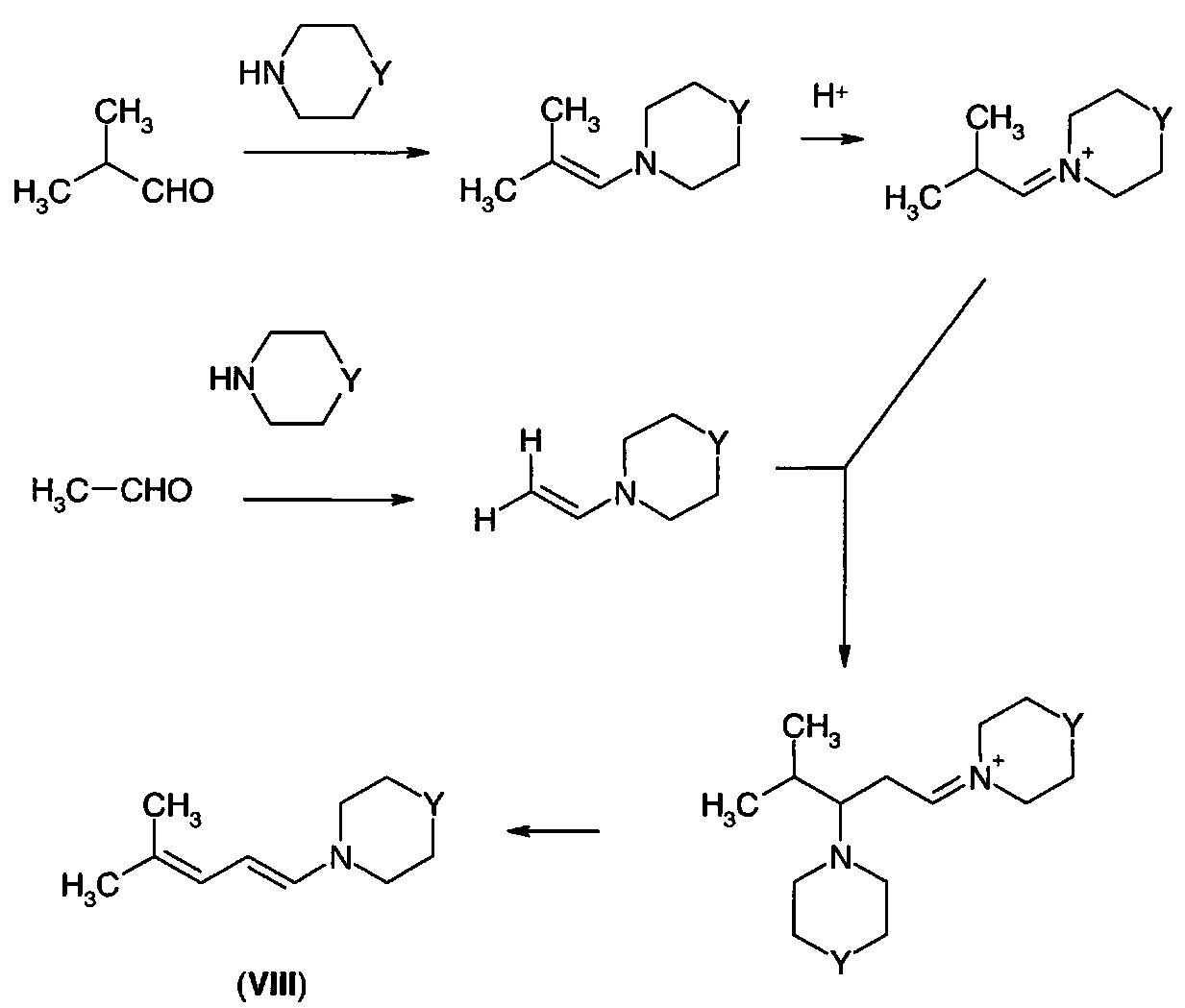
Способ по настоящему изобретению является более экономичным и более пригодным для масштабирования, чем описанные в литературе способы осуществления данного «обращения поляризации» (umpolung) реакционной способности нормального ацетальдегида, при которых ацетальдегид преобразуют в О-силилированное енольное производное и подвергают сочетанию с изобутиральдегидом в условиях альдольной реакции Мукаямы. В настоящем изобретении оба партнера сочетания активируются одновременно, поскольку электронные и стерические эффекты направляют наблюдаемый паттерн реакционной способности. Другое преимущество состоит в том, что продукт диенамин представляет собой желаемую «активированную» форму 4-метил-2-пентеналя для взаимодействия с глиоксалевой кислотой с образованием желаемых 5-аминофуранонов (VII).
Эти диенаминные производные формулы (VIII) могут быть выделены и очищены, или, альтернативно, они могут быть непосредственно обработаны глиоксалевой кислотой (или ее гидратом), что обеспечивает получение производного фуранона (VII) непосредственно.
Производные фуранона (VII) можно быть выделены и очищены. Затем обработка водной кислотой обеспечивает получение фуранона (VIA).
В целом, описанные выше превращения обеспечивают короткий путь для получения прегабалина с использованием недорогостоящих и безопасных исходных веществ.
В альтернативном воплощении дигидрофуранон (I) получают из 3-изобутилиден-2-оксопентандикарбоновой кислоты (XII), которую легко получают путем конденсации изобутиральдегида с 2-оксопентандикарбоновой кислотой (α-кетоглутаровой кислотой) (XIV).
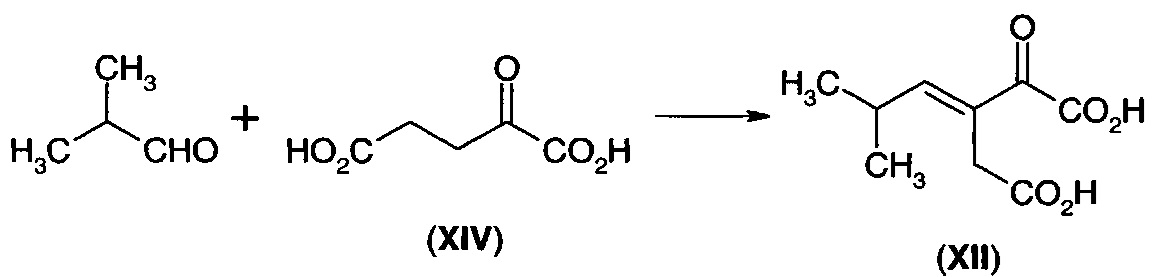
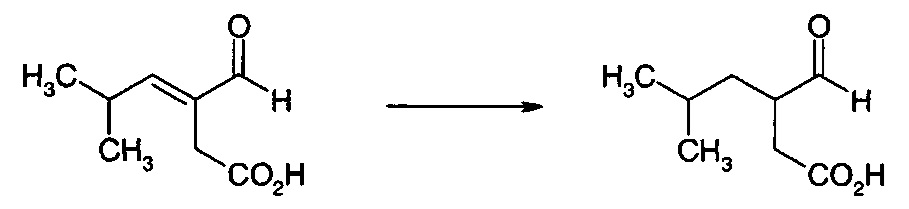
Для преобразования дикарбоновой кислоты (XII) в дигидрофуранон (I) требуется стадия декарбоксилирования и стадия восстановления. Эти две стадии способа можно осуществлять по отдельности, и в этом случае либо стадия декарбоксилирования, либо стадия восстановления может быть первой стадией, либо эти два процесса можно осуществлять одновременно. Когда стадию восстановления осуществляют первой, то получают промежуточное соединение (XV). Когда стадию декарбоксилирования осуществляют первой, то получают промежуточное соединение (XVI).
Стадию восстановления можно выполнять химическим путем, например, путем гидрирования, но оно предпочтительно достигается с использованием опосредованного ферментом восстановления, например, путем обработки ферментом еноил-редуктазой. Стадию декарбоксилирования предпочтительно выполняют путем обработки соединения ферментом декарбоксилазой.
В случае, когда рассматриваются опосредованные ферментами превращения, тогда фермент может представлять собой изолированный фермент, включая фермент, иммобилизованный на носителе, он может представлять собой препарат частично изолированного фермента, такой как клеточный гомогенат, или он может представлять собой неизолированный фермент, и в этом случае используют цельноклеточный препарат. Клетки могут включать клетки, экспрессирующие желаемый фермент естественным путем, и клетки, которые были подвергнуты таким манипуляциям, чтобы они экспрессировали желаемый фермент.
Опосредованное ферментом восстановительное аминирование соединения формулы (IА) является обратимым, и поэтому обработка 3-аминометил-5-метилгексановой кислоты (II) ферментом трансаминазой или ферментом аминоксидазой/иминоредуктазой может приводить к образованию дигидрофуранона (IА). Изомер этого соединения с разомкнутым циклом представляет собой соединение (IB), которое является эпимеризуемым. С учетом этого, возможно преобразовать ((R)-II) в ((S)-II) или увеличить оптическую чистоту смеси ((R)-II) и ((S)-II) с использованием такого фермента.

ПРИМЕРЫ
Изобретение проиллюстрировано приведенными ниже неограничивающими примерами, в которых используют следующие сокращения и определения:


Имеющиеся в продаже химические вещества использовали в том виде, в котором они были получены, если не указано иное. Тонкослойную хроматографию проводили на предварительно покрытых полимерных пластинах (Merck, силикагель 60F254) и визуализировали с помощью УФ света и добавления КМnO4. Спектры протонного (1Н) и углеродного (13С) ЯМР снимали на спектрофотометре Varian INOVA 300 МГц. Химические сдвиги оценивали относительно тетраметилсилана и по необходимости соотносили с пиками остаточного растворителя. Если не указано иное, анализ с помощью хиральной HPLC проводили с использованием системы Agilent 1200 HPLC, и данные обрабатывали с помощью программного обеспечения Chemstation, или с использованием полупрепаративной/аналитической HPLC Varian с помощью программного обеспечения Galaxie.
Пример 1
Получение 4-(2-метил-1-пропенил)-5-морфолино-5Н-2-фуранона из 4-метил-2-пентеналя
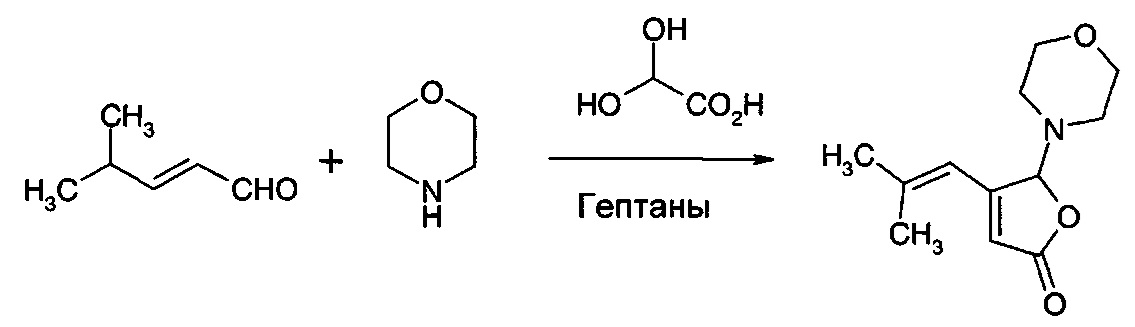
50% раствор глиоксалевой кислоты в воде (29,6 г, 0,2 моль) добавляли к двухфазной перемешанной смеси морфолина (17,8 г, 0,2 моль) и гептанов (75 мл), предварительно охлажденной до 0-10°С. Температуру поддерживали ниже 10°С. Смесь подогревали до 20°С и добавляли 4-метил-2-пентеналь (19,6 г, 0,2 моль). Смесь перемешивали при 45°С в течение 20 ч. Образовывалось большое количество твердого вещества. Добавляли воду (100 мл) при температуре окружающей среды, и смесь перемешивали в течение 2 ч. Смесь экстрагировали циклопентилметиловым эфиром (100 мл), и органический раствор дважды промывали водой (100 мл) и концентрировали с получением 30,4 г неочищенного продукта. Это твердое вещество очищали перекристаллизацией из метанола (100 мл) с получением 17,7 г (40%) чистого 4-(2-метил-1-пропенил)-5-морфолино-5H-2-фуранона.
GS/MS: m/z=223
1Н ЯМР: δ 6.0 (s, 1Н); 5.9 (s, 1Н); 5.5 (s, 1Н), 3.7 (d, 4Н), 2.7 (d, 4Н), 2.00 (s, 3Н), 1.95 (s, 3Н).
Пример 2
Получение 4-(4-метил-1,3-пентадиен-1-ил)морфолина

Изобутиральдегид (46,90 г, 0,65 моль, 1,43 экв.) перемешивали в ацетонитриле (300 мл). Морфолин (56,63 г, 0,65 моль, 1,43 экв.), а затем pTsOH (8,63 г, 0,1 экв.) медленно добавляли при комнатной температуре к раствору изобутиральдегида. Раствор ацетальдегида (20 г, 0,454 моль) в ацетонитриле (100 мл) добавляли по каплям в течение 1 ч при мониторинге внутренней температуры при 50°С. После завершения добавления смесь перемешивали в течение 30 мин при 50°С, а затем охлаждали до комнатной температуры, после чего выпаривали растворитель в вакууме с получением оранжевого масла (123,3 г). Анализ неочищенного продукта с помощью количественного ЯМР с использованием бензилбензоата в качестве внутреннего стандарта показал 40%, что дает 65% выход желаемого диенамина.
GS/MS: m/z=167
1Н ЯМР: δ 6.01 (d, 1Н); 5.69 (d, 1Н); 5.31 (dd, 1Н), 3.70 (m, 4Н), 2.89 (m, 4Н), 1.71 (s, 3Н), 1.63 (s, 3Н).
Пример 3
Получение 4-(2-метил-1-пропенил)-5-морфолино-5H-2-фуранона из неочищенного 4-(4-метил-1,3-пентадиен-1-ил)морфолина
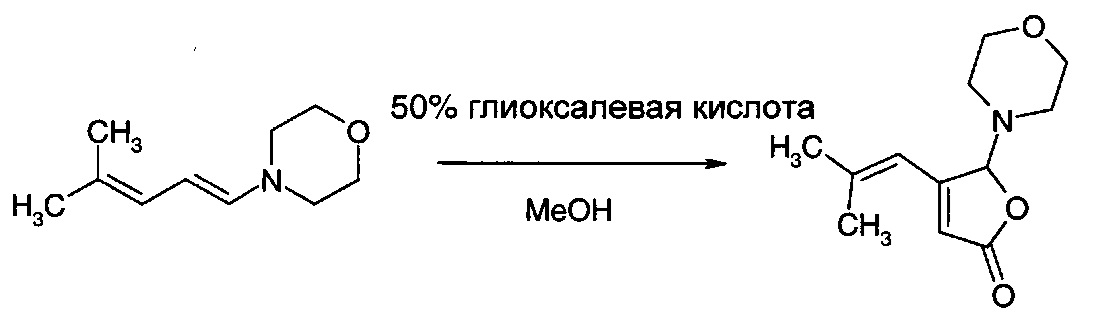
Неочищенный 4-(4-метил-1,3-пентадиен-1-ил)морфолиндиенамин из примера 2 (123,3 г, анализ 40%) растворяли в метаноле (300 мл) при комнатной температуре. После полного растворения добавляли глиоксалевую кислоту (50 масс. %, 60 г, 1,1 экв.), и полученную в результате двухфазную смесь перемешивали при 50°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли роторным выпариванием. Остаток распределяли между этилацетатом (200 мл) и насыщенным раствором карбоната натрия (200 мл). Водную фазу экстрагировали этилацетатом (100 мл), и объединенные органические фазы промывали рассолом и упаривали с получением густого масла, которое отвердевало при стоянии, (89,0 г, анализ 67% по qЯМР, 60% выход).
GS/MS: m/z=223
ЯМР: как в примере 1.
Пример 4
Получение 4-(2-метил-1-пропенил)-5-морфолино-5Н-2-фуранона в одном сосуде из изобутиральдегида и ацетальдегида

Изобутиральдегид (102,90 г, 1,43 моль) перемешивали в ацетонитриле (600 мл). Морфолин (124,3 г, 1,43 моль), а затем pTsOH (19,0 г, 0,1 экв.), медленно добавляли при комнатной температуре к раствору изобутиральдегида. Раствор ацетальдегида (44,05 г, 1,0 моль) в ацетонитриле (150 мл) добавляли по каплям в течение 1 ч при мониторинге внутренней температуры при 50°С. После завершения добавления смесь перемешивали в течение 30 мин при 50°С, а затем охлаждали до температуры менее 10°С. Добавляли глиоксалевую кислоту (50% по масс, 211,3 г, 1,43 моль), и полученную в результате двухфазную смесь перемешивали при 50°С в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры, и растворитель удаляли роторным выпариванием. Остаток распределяли между этилацетатом (1 л) и насыщенным раствором карбоната натрия (1 л). Водную фазу экстрагировали этилацетатом, и объединенные органические фазы промывали рассолом и упаривали с получением густого масла, которое отвердевало при стоянии, (166 г). Неочищенный продукт растирали с МТВЕ при комнатной температуре. Продукт собирали фильтрованием и промывали МТВЕ с получением чистого 4-(2-метил-1-пропенил)-5-морфолино-5H-2-фуранона (84 г, выход 37%), идентичного веществу, полученному в Примере 1. Анализ неочищенного твердого вещества (166 г) показал выход приблизительно 50%.
Пример 5
Получение 1,4-бис-(4-метил-1,3-пентадиен-1-ил)пиперазина
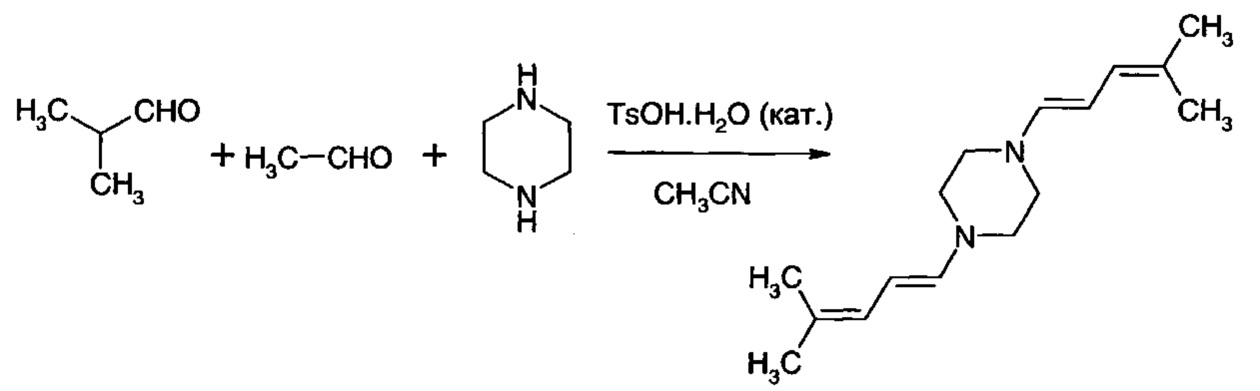
Готовили раствор пиперазина (43,0 г, 0,5 моль) и моногидрата 4-толуолсульфоновой кислоты (4,4 г, 0,023 моль) в ацетонитриле (600 мл). Этот раствор нагревали до 50°С и добавляли изобутиральдегид (100 мл, 1,10 моль) в течение 10 минут. Цвет раствора переходил в красно-оранжевый, и появлялся неустойчивый белый осадок. Затем добавляли раствор ацетальдегида (30,8 г, 0,70 моль) в ацетонитриле (30 мл) через шприцевой насос в течение 3 ч при 50°С. Образовывалась суспензия, которую перемешивали при 50°С в течение 0,5 ч. Растворитель удаляли, и остаточное твердое вещество выделяли из метанола (500 мл) при -5°С. Вещество фильтровали и промывали охлажденным метанолом и сушили с получением 52,9 г (60%) 1,4-бис-(4-метил-1,3-пентадиен-1-ил)пиперазина.
GS/MS: m/z=246.
1Н ЯМР: δ 6.00 (d, 2Н); 5.70 (d, 2Н); 5.28 (dd, 2Н); 2.92 (s, 8Н); 1.73 (s,6H); 1.68 (s, 6Н).13С ЯМР (CDCl3): 140.6 (С), 126.3 (СН), 123.4 (СН), 100.2 (СН), 48.2 (СН2), 25.8 (СН3), 18.1 (СН3).
Пример 6
Получение 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она) из 1,4-бис-(4-метил-1,3-пентадиен-1-ил)пиперазина

1,4-бис-(4-Метил-1,3-пентадиен-1-ил)пиперазин (37,3 г, 0,151 моль; см. пример 5) загружали в метанол (300 мл). Температуру доводили до 34°С и быстро добавляли 50% раствор глиоксалевой кислоты в воде (44,9 г, 0,302 моль) (в течение 5 минут). Полученную в результате суспензию перемешивали при 45°С в течение 15 ч; охлаждали до 0-10°С в течение 2 ч и фильтровали. Твердое вещество промывали метанолом (100 мл) и сушили с получением чистого 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она) (32,3 г, 60%). Дополнительные 5% можно было получить из маточного раствора путем концентрирования.
1Н ЯМР: δ 5.96 (d, 2Н), 5.87 (d, 2Н), 5.56 (m, 2Н), 2.71 (s, 8Н), 2.07-1.90 (m, 12Н).13С ЯМР (CDCl3): 172.3 (С), 159.2 (С), 150.9 (С), 116.6 (СН), 115.4 (СН), 99.2 (СН), 46.7 (СН2), 28.2 (СН3), 21.4 (СН3).
Эту реакцию также можно проводить в изопропаноле, смеси ацетонитрил-вода, смеси толуол-вода или гептан-вода с аналогичными выходами.
Пример 7
Получение 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-он) в одном сосуде из изобутиральдегида и ацетальдегида

Толуолсульфоновую кислоту (6,5 г, 0,03 моль) загружали в раствор пиперазина (59 г, 0,69 моль) в ацетонитриле (240 мл). Реакционную смесь нагревали до 50°С и перемешивали, пока не наблюдалось растворение твердых веществ, после чего добавляли изобутиральдегид (138 мл, 1,51 моль). Реакционную смесь выдерживали при 50°С, после чего в сосуд загружали раствор ацетальдегида (60 мл; 1,07 моль) в ацетонитриле (30 мл) в течение 3 ч через шприцевой насос. После завершения добавления реакционную смесь перемешивали в течение следующих 30 минут, после чего к реакционной смеси добавляли 50% по масс, раствор глиоксалевой кислоты в воде (148 г, 1,0 моль) в течение 5 мин с последующим добавлением воды (50 мл). Затем реакционную смесь нагревали до 70°С в течение 2 ч, затем до 50°С в течение ночи. Затем реакционную смесь охлаждали до 5°С и выдерживали в течение 30 минут. 5,5'-(Пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-он) осаждали, выделяли фильтрованием и промывали ацетонитрилом (2×100 мл) с получением желаемого продукта (126 г, анализ 93,5%, 66% выход из ацетальдегида).
Пример 8
Получение 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-она) без растворителя
Изобутиральдегид (79 г, 100 мл, 1,1 моль) загружали в 3-горлую круглодонную колбу, и через систему продували аргон. Пиперазин (43 г, 0,5 моль) и TsOH-H2О (4,4 г; 0,023 моль) делили на 4 равные порции, содержащие пиперазин (10,75 г) и TsOH-H2O (1,1 г). Добавление первой порции пиперазина (10,75 г) в результате приводило к экзотермическому эффекту от 24°С до 35°С. После этого добавляли первую порцию TsOH (1,1 г). Реакционную смесь перемешивали до растворения всего пиперазина (t=35°C), после чего добавляли вторую порцию пиперазина (10,75 г), а затем TsOH (1,1 г) (t=41°C). Перемешивание продолжали до растворения всего пиперазина (t=41°С), затем добавляли третью порцию пиперазина (10,75 г), а затем TsOH (1,1 г). После 3ей загрузки образовался прозрачный раствор, а затем добавляли четвертую порцию пиперазина (10,75 г) и TsOH-H2O, а затем TsOH (1,1 г) (t=52°C). После завершения добавления реакционную смесь перемешивали при 50°С в течение 30 мин.
В другую колбу (50 мл) загружали ацетальдегид (30,8 г, 39 мл, 0,7 моль) и помещали в баню лед-вода (0-2°С). Колбу с ацетальдегидом соединяли с 3-горлой колбой с помощью канюли через мембрану. Затем через колбу с ацетальдегидом пропускали поток аргона со скоростью, гарантирующей, что добавление ацетальдегида завершено в пределах 3 ч. После добавления всего ацетальдегида реакционную смесь перемешивали в течение 30 мин при 50°С, затем охлаждали до 40°С и добавляли по каплям 50% глиоксалевую кислоту (104 г, 78 мл) в течение 45 мин с такой скоростью, чтобы поддерживать температуру ниже 50°С. Затем добавляли воду (100 мл), и реакционную смесь нагревали при 70°С в течение 5 ч. Реакционную смесь охлаждали до 4°С в бане лед-вода. Выпавший в осадок продукт фильтровали, и фильтрационный осадок промывали холодной водой (100 мл). После сушки в вакууме при 50°С получили 88,9 г (71%) желаемого продукта. Осадок содержит 78,5% указанного в заголовке соединения (анализ HPLC).
Пример 9
Альтернативное получение 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она) с одновременным добавлением обоих альдегидов
Пиперазин (236,6 г) загружали в чистый сухой сосуд на 3 л, оборудованный термометром, капельной воронкой (100 мл) и обратным холодильником. В сосуд загружали pTsOH (25,3 г), а затем ацетонитрил (550 мл). Включали мешалку, и в сосуде создавали инертную атмосферу азотом. Изобутиральдегид (150 мл, 27% суммарной загрузки) загружали в перемешиваемую суспензию пиперазин/pTsOH, и наблюдали подъем температуры до приблизительно 43°С. Затем белую суспензию нагревали до 50°С (+/- 5°С). Предварительно перемешанный охлажденный раствор изобутиральдегида (400 мл, 73% суммарной загрузки) и охлажденный ацетальдегид (260 мл) загружали в чистый сухой сосуд на 1 л, и эту смесь выдерживали в ледяной бане. Смесь изобутиральдегид/ацетальдегид загружали в содержимое 3 л сосуда аликвотами по 100 мл в течение 5-6 ч при 50°С. Содержимое реакционной колбы изменялось с белой суспензии на раствор винно-красного цвета и, наконец, на оранжевую суспензию в процессе добавления ацетальдегида. После завершения добавления суспензию перемешивали при 50°С в течение приблизительно от 0,5 до 1,0 ч.
В суспензию загружали водный раствор глиоксалевой кислоты (50% масс/масс.) в течение 10-15 мин, и температура поднималась до приблизительно 75°С. Наблюдали кристаллизацию из раствора 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она). Суспензию перемешивали при 70°С (+/- 5°С) в течение 6 ч, затем охлаждали при перемешивании до температуры окружающей среды. Затем партию охлаждали до -5-0°С и выдерживали в течение 3-4 ч, после чего суспензию фильтровали, и фильтрационный осадок промывали охлажденным метанолом (1×500 мл), затем 2×500 мл метанола при температуре окружающей среды. Промытый продукт сушили в вакуумной печи при 40-50°С до постоянной массы с получением 474 г 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она) 98% чистоты (70%).
Пример 10
Получение 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она из 4-(2-метил-1-пропенил)-5-морфолино-2(5H)-фуранона или 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-она)

10% по масс, водный раствор серной кислоты (110 г) загружали в 4-(2-метил-1-пропенил)-5-морфолино-2(5H)-фуранон (20,0 г, 0,896 моль), и смесь перемешивали при кипячении с обратным холодильником в течение 4 ч или до тех пор, пока TLC (100:3 СРМЕ:НОАс) не показала завершение реакции. Смесь все время оставалась в виде суспензии. Затем ее охлаждали до 5°С, выдерживали в течение 2 ч и фильтровали. Белое твердое вещество промывали водой и сушили с получением 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она (12,9 г, 93%).
Альтернативно, 10% по масс, водный раствор серной кислоты (412 г) загружали в сосуд, содержащий 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-он) (60 г, 0,167 моль). Содержимое перемешивали, а затем нагревали до температуры дефлегмации. Реакционную смесь выдерживали при кипячении с обратным холодильником до израсходования исходного вещества, что определяли на основании растворения. После завершения реакции партию охлаждали до 35°С, и в это время целевое соединение начало кристаллизоваться. Суспензию дополнительно охлаждали до 0-5°С, а затем переносили на фильтр. Продукт фильтровали, промывали водой (2×100 мл), а затем сушили в вакууме при температуре менее 50°С с получением 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она (42,0 г, 81%).
GS/MS: m/z=154
1Н ЯМР (CDCl3): δ 6.10 (s, 1Н); 5.92 (s, 1Н); 5.88 (s, 1Н); 5.35 (bs, 1Н, О-Н); 1.98 (s, 3Н); 1.93 (s, 3Н).13C ЯМР (CDCl3): 172.8 (С), 161.4 (С), 152.3 (С), 115.3 (СН), 114.9 (СН), 99.5 (СН), 28.2 (СН3), 21.4 (СН3).
Пример 11
Получение 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она в одном сосуде из изобутиральдегида и ацетальдегида
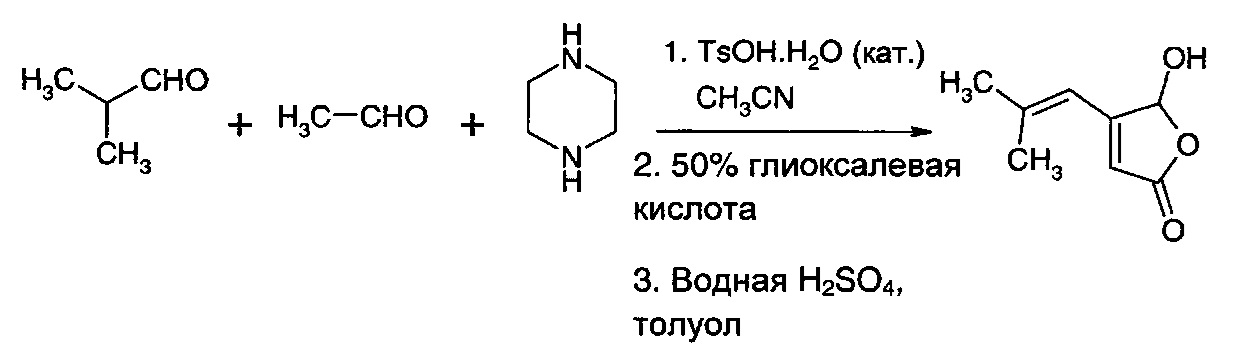
Смесь пиперазина (43,0 г, 0,5 моль) и изобутиральдегида (200 мл) кипятили с обратным холодильником с ловушкой Дина-Старка до сбора теоретического количества воды (18 мл). Избыток изобутиральдегида отгоняли с получением в остатке кристаллической массы 1,4-бис(2-метилпропен-1-ил)пиперазина. Его растворяли в ацетонитриле (1,2 л) и добавляли дополнительное количество изобутиральдегида (100 мл, 1,0 моль). Добавляли дополнительное количество пиперазина (4,4 г), а затем пара-толуолсульфоновую кислоту (4,4 г, 23,2 ммоль). Температуру доводили до 40°С и добавляли раствор ацетальдегида (44 г, 1,0 моль) в ацетонитриле (40 мл) в течение 4 ч. Реакционную смесь перемешивали при 40°С в течение 1 ч и при температуре окружающей среды в течение 4 ч. Ацетонитрил отгоняли, и остаток суспендировали в толуоле (400 мл). Добавляли смесь 50% глиоксалевой кислоты (148 г) и воды (150 мл) в течение 0,5 ч. Затем смесь перемешивали при 45°С в течение 15 ч. Твердое вещество выпадало в осадок. Добавляли разбавленную серную кислоту (10%, 1 л), и смесь кипятили с обратным холодильником в течение 3 ч. В результате получили двухфазную смесь. Фазу толуола отделяли. Она содержала приблизительно 50% по выходу (на основе ацетальдегида) 5-гидрокси-4-(2-метилпропен-1-ил)-2-фуранона на основании анализа.
Пример 12
Получение 5-метокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-она

4-(2-Метил-1-пропенил)-5-морфолино-2(5H)-фуранон (7,70 г, 34,5 ммоль) перемешивали в МеОН (50 мл) с H2SO4 (4 мл, 2,2 экв.) при кипячении с обратным холодильником в течение 3 ч до исчезновения исходного вещества по GC. Реакционную смесь охлаждали до комнатной температуры. Затем растворитель выпаривали в вакууме, и остаток разбавляли ЕtOАс (50 мл). Органическую фазу промывали Н2O (3×50 мл). Затем растворитель выпаривали в вакууме с получением 5-метокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она в виде оранжевого масла (5,50 г, 95%), которое использовали без очистки.
Альтернативно, 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-он) (130 г, 363 ммоль) и МеОН (690 мл) загружали в круглодонную колбу на 2 л при механическом перемешивании с образованием суспензии. Добавляли по каплям концентрированную H2SO4 (43 мл, 762 ммоль) при перемешивании при 20-25°С, наблюдали небольшую экзотермию, и природа присутствующих твердых веществ изменялась с диспергируемой и легко перемешиваемой на более густую взвесь. Реакционную смесь кипятили с обратным холодильником в течение 5 ч. Через 0,5 ч наблюдали полное растворение (оранжевую жидкость). Через 5 ч LCMS показала порядка 99% желаемого продукта. Смесь охлаждали до температуры окружающей среды и оставляли для кристаллизации сульфата пиперазина. Осадки фильтровали, и фильтрационный осадок промывали охлажденным на льду МеОН (400 мл). Фильтрат концентрировали в вакууме (температура бани 40°С), и остаток распределяли между водой (50 мл) и МТВЕ (300 мл). Водный слой отделяли и дополнительно экстрагировали (2×300 мл МТВЕ). Объединенные органические экстракты промывали насыщенным водным раствором NaHCO3 (200 мл). Слой МТВЕ сушили над Na2SO4 при перемешивании в течение 1,5 ч, фильтровали и упаривали с получением 5-метокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она в виде оранжевого масла (120 г, 98%).
Перегоняли в вакууме при 0,2-0,3 мбар (20-30 Па) с Ткип 100-105°С. GS/MS: m/z=168;1Н ЯМР (400 МГц, CDCl3) δ 5.59 (1Н, s) 5.88-5.86 (1Н, m) 5.75 (1Н, d, J=0.6) 3.53 (3Н, s) 2.01-2.00 (3Н, m) 1.97-1.96 (3Н, m).13С ЯМР (CDCl3): 171.4 (С), 159.2 (С), 151.9 (С), 115.8 (СН), 115.2 (СН), 104.4 (СН), 56.1 (СН3), 28.2 (СН3), 21.4 (СН3).
Следующие соединения были получены из 4-(2-метил-1-пропенил)-5-морфолино-2(5H)-фуранона в соответствии с первым из описанных выше способов путем замены метанола 3-метилбутанолом (изоамиловым спиртом), н-пентанолом (амиловым спиртом) или н-бутанолом:
Пример 12А
5-(3-Метилбутокси)-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-он
Перегоняли в вакууме при 0,4 мбар (40 Па) с Ткип 139-140°С; GS/MS: m/z=224;1Н-ЯМР (400 МГц, CDCl3) δ 5.94 (1Н, s) 5.87-5.84 (1Н, m) 5.80 (1Н, s) 3.86-3.80 (1Н, m) 3.70-3.64 (1Н, m) 2.00 (3Н, s) 1.96 (3Н, s) 1.77-1.66 (1Н, m) 1.57-1.50 (2Н, m) 0.93-0.91 (3Н, m) 0.91-0.89 (3Н, m).
Пример 12В
4-(2-Метилпроп-1-ен-1-ил)-5-пентилоксифуран-2(5Н)-он
Перегоняли в вакууме при 0,08 мбар (8 Па) с Ткип 123-134°С; GS/MS: m/z=224;1Н-ЯМР (400 МГц, CDCl3) δ 5.94 (s, 1Н); 5.86 (s, 1Н); 5.80 (s, 1Н); 3.84-3.75 (m, 1Н); 3.68-3.59 (m, 1Н); 2.01 (s, 3Н); 1.95 (s, 3Н); 1.69-1.59 (m, 2Н); 1.38-1.29 (m, 4Н); 0.95-0.85 (m, 3Н).
Пример 12С
5-Бутокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-он
Перегоняли в вакууме при 0,05 мбар (5 Па) с Ткип 136-146°С; GS/MS: m/z=210;1Н-ЯМР (400 МГц, CDCl3) δ 5.94 (s, 1Н); 5.86 (s, 1Н); 5.80 (s, 1Н); 3.85-3.76 (m, 1Н); 3.69-3.60 (m, 1Н); 2.00 (s, 3Н); 1.95 (s, 3Н); 1.68-1.58 (m, 2Н); 1.45-1.32 (m, 2Н); 0.93 (t, 3Н, J=7.4 Гц).
Пример 13
Получение 5-метокси-4-(2-метилпропил)-дигидрофуран-2(3Н)-она
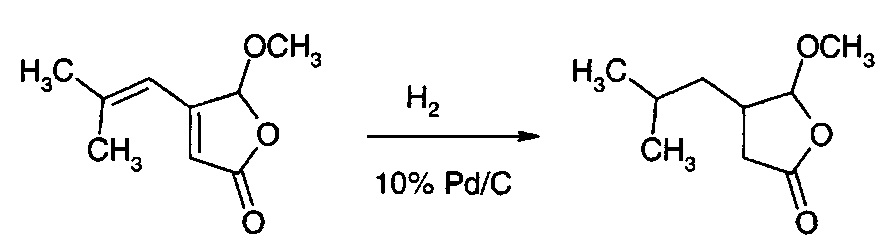
Раствор 5-метокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она (100 г, см. пример 12) в МТВЕ (700 мл), 5 г (5 масс. %) 10% Pd/C загружали в сосуд для гидрирования. Вводили 1 атм. газа водорода, и давление поддерживали постоянным в процессе реакции. Через 7 ч при 20°С реакционную смесь фильтровали через слой целита (∅60 мм, Н=30 мм) и промывали МТВЕ (3×50 мл). Фильтрат промывали 1 М раствором NаНСО3 (200 мл), водой, рассолом и сушили над Na2SO4. После удаления растворителя получали 5-метокси-4-(2-метилпропил)-дигидрофуран-2(3Н)-он в виде бесцветной жидкости (98 г, 96%).
GS/MS: m/z=172
1Н ЯМР (400 МГц, CDCl3) δ 5.25 (d, J=5.0 Гц, 0.53 Н, основной изомер), 5.04 (d, J=2.4 Гц, 0.26Н, минорный изомер), 3.48 (s, 0.92Н, минорный изомер), 3.46 (s, 1.66Н, основной изомер), 2.78 (dd, J=17.7, 8.6 Гц, 0.33Н), 2.59-2.42 (m, 1.27Н, основной изомер), 2.42-2.34 (m, 0.33Н, минорный изомер), 2.29 (dd, J=16.7, 11.8 Гц, 0.63Н, основной изомер), 2.15 (dd, J=17.7, 4.6 Гц, 0.33Н, минорный изомер), 1.67-1.31 (m, 2.63Н оба изомера), 1.28-1.19 (m, 0.33Н, минорный изомер), 0.95-0.86 (m, 6Н оба изомера).13С ЯМР (CDCl3): 177.7 (С, основной изомер), 176.04 (С, минорный изомер), 110.1 (СН, минорный изомер), 106.1 (СН, основной изомер), 57.0 (СН3, минорный изомер), 56.6 (СН3, основной изомер), 41.2 (СН2, минорный изомер), 39.1 (СН2, минорный изомер), 38.3 (СН2, основной изомер), 37.1 (СН2, основной изомер), 33.9 (СН, минорный изомер), 32.8 (СН, основной изомер), 26.0 (СН, основной изомер), 25.8 (СН, минорный изомер), 23.0 (СН3, основной изомер), 22.9 (минорный изомер), 22.7 (основной изомер), 22.6 (минорный изомер).
С использованием того же общего способа и соединений примеров 12А, 12В и 12С, соответственно, в качестве исходных веществ были также получены следующие соединения:
Пример 13А
5-(3-Метилбутокси)-4-(2-метилпропил)-дигидрофуран-2(3Н)-он
GS/MS: m/z=228;1Н-ЯМР (400 МГц, CDCl3) δ 5.35 (0.86Н, d, J=5.0 Гц) 5.13 (0.14Н, d, J=2.7 Гц) 3.87-3.79 (m, 1Н) 3.57-3.45 (m, 1Н) 2.58-2.37 (2Н, m) 2.35-2.27 (0.79Н, m) 2.20-2.11 (0.21 H, m) 1.74-1.62 (1H, m) 1.61-1.44 (4H, m) 1.42-1.31 (1H, m) 0.95-0.86 (12H, m).
Пример 13B
4-(2-Метилпропил)-5-пентилоксидигидрофуран-2(3Н)-он
GS/MS: m/z=228;1Н-ЯМР (400 МГц, CDCl3) δ 5.35 (0.77H, d, J=5.0 Гц) 5.13 (0.23H, d, J=2.6 Гц,) 3.83-3.75 (1H, m) 3.55-3.40 (1H, m) 2.59-2.37 (2H, m) 2.32 (0.73H, dd, J=16.6, 11.8 Гц) 2.15 (0.27H, dd, J=17.7, 5.0 Гц) 1.67-1.45 (4H, m) 1.41-1.25 (5H, m) 0.96-0.86 (9H, m).
Пример 13C
5-Бутокси-4-(2-метилпропил)-дигидрофуран-2(3Н)-он
GS/MS: m/z=214;1Н-ЯМР (400 МГц, CDCl3) δ 5.35 (0.8H, d, J=5.0 Гц) 5.13 (0.2H, d, J=2.6 Гц,) 3.85-3.75 (1H, m) 3.56-3.41 (1H, m) 2.59-2.38 (2H, m) 2.31 (0.76H, dd, J=16.5, 11.7 Гц) 2.15 (0.24H, dd, J=17.6, 5.0 Гц) 1.66-1.45 (4H, m) 1.43-1.30 (3H, m) 0.95-0.87 (9H, m).
Пример 14
Получение и гидрирование 5-(L-ментилокси)-4-(2-метил-1-пропенил)-2(5Н)-фуранона

Смесь 5-гидрокси-4-(2-метил-1-пропенил)-2-фуранона (59,5 г, 0,386 моль), L-ментола (89,5 г, 1,5 экв.) и метансульфоновой кислоты (1,5 г) перемешивали при 70-80°С в вакууме (для удаления воды) в течение 100 ч. Жидкую массу вливали (горячей) в ацетонитрил (450 мл), и указанное в заголовке соединение выделяли путем охлаждения, фильтрования и промывания. Выход составлял 76,2 г (67%). Подходящие кристаллы для XRD выращивали путем медленного упаривания раствора в ацетоне. Конфигурация атома углерода ацеталя являлась (R) конфигурацией.
В результате гидрирования 5-(L-ментилокси)-4-(2-метил-1-пропенил)-2-фуранона в этилацетате при использовании условий примера 13, получили насыщенное производное при количественном выходе. На основании1Н ЯМР соединение представляло собой смесь диастереомеров 1:1.
Пример 15
Получение 5-ацетокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-она

Суспензию 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-она (19 г, 0,123 моль) в этилацетате (100 мл) обрабатывали твердым карбонатом натрия (13,1 г, 0,123 моль) и тетрабутиламмония гидросульфатом (0,5 г). Добавляли уксусный ангидрид (18,8 г, 1,5 экв.) одной порцией. Протекала умеренно экзотермическая реакция. Смесь перемешивали в течение ночи, добавляли воду (100 мл), и органическую фазу отделяли (рН водной фазы составлял 6,0). Фазу этилацетата промывали водой и концентрировали с получением в остатке твердого вещества (24,6 г, 100%). Это вещество было чистым по TLC (100:3 СРМЕ : уксусная кислота). При проведении данной реакции в изопропилацетате чистое соединение можно выделить путем охлаждения его раствора в изопропилацетате после промывания водой при выходе приблизительно 70%.
m/z: 196
Пример 16
Гидрирование 5-ацетокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она
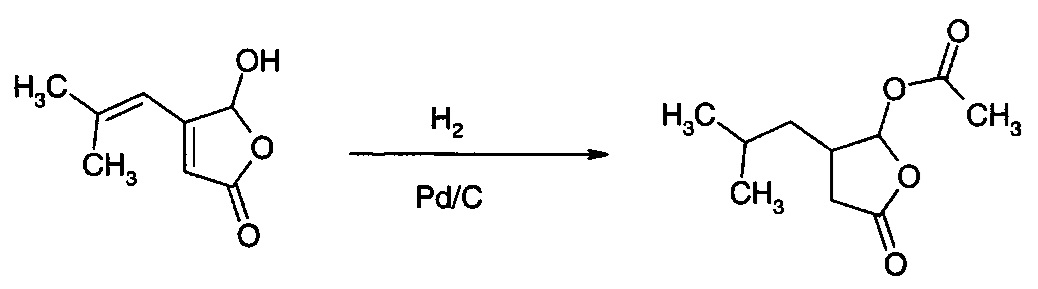
Вещество примера 34 гидрировали в условиях, подобных условиям, описанным в примере 13, с получением выхода более 90% 5-ацетокси-4-(2-метилпропил)-3,4-дигидро-2(5H)-фуранона в виде смеси диастереомеров 1:1. Продукту сопутствует приблизительно 5% 4-(2-метилпропил)-3,4-дигидрофуран-2(5Н)-она. Продукт можно подвергать гидролизу, как в примере 30, с получением 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5Н-2-фуранона/3-формил-5-метилгексановой кислоты.
Пример 17
Получение 5-гидрокси-4-(2-метилпропил)дигидрофуран-2(3Н)-она (IA) путем гидрирования 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-она в щелочном растворе

5-Гидрокси-4-(2-метилпропен-1-ил)-2-фуранон (3,35 г, 21,7 ммоль) и воду (20 мл) загружали в гидрогенизатор. Затем в сосуд загружали гидроксид калия (1,21 г, 1 экв.), и содержимое нагревали до 40°С, пока не произошло растворение. В сосуд загружали 10% Pd/углерод (0,67 г), а затем содержимое реактора гидрогенизировали при 40°С и 5 бар (500 кПа) давления водорода. После завершения реакции реакционную смесь охлаждали, и катализатор удаляли фильтрованием. Значение рН доводили до рН 2 добавлением 36% соляной кислоты, и водный слой промывали толуолом для экстракции желаемого продукта. Объединенные толуольные экстракты концентрировали с получением указанного в заголовке соединения (3,17 г, 92%).
Пример 18
Гидрирование 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она в нейтральном растворе
5-Гидрокси-4-(2-метилпропен-1-ил)-2-фуранон гидрировали в 6 мл воды на грамм исходного вещества 10% по масс, катализатора Pd/C (22 ч, 40°С, 10 бар (1000 кПа)). После фильтрования масляную фазу отделяли от воды. Эта фаза представляла собой смесь 1:1 5-гидрокси-4-(2-метилпропил)дигидрофуран-2(3Н)-она (IА) и 4-(2-метилпропил)-дигидрофуран-2-она. Соединение (IА) можно легко выделить в чистом виде путем кислотно-основной экстракции. Эту реакцию повторяли в органических растворителях с 2-пропанолом с получением относительно чистого соединения (IА).
Пример 19
Получение 5-гидрокси-4-(2-метилпропил)дигидрофуран-2(3Н)-она (IA) из 5,5'-(пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5Н)-она) в одном сосуде
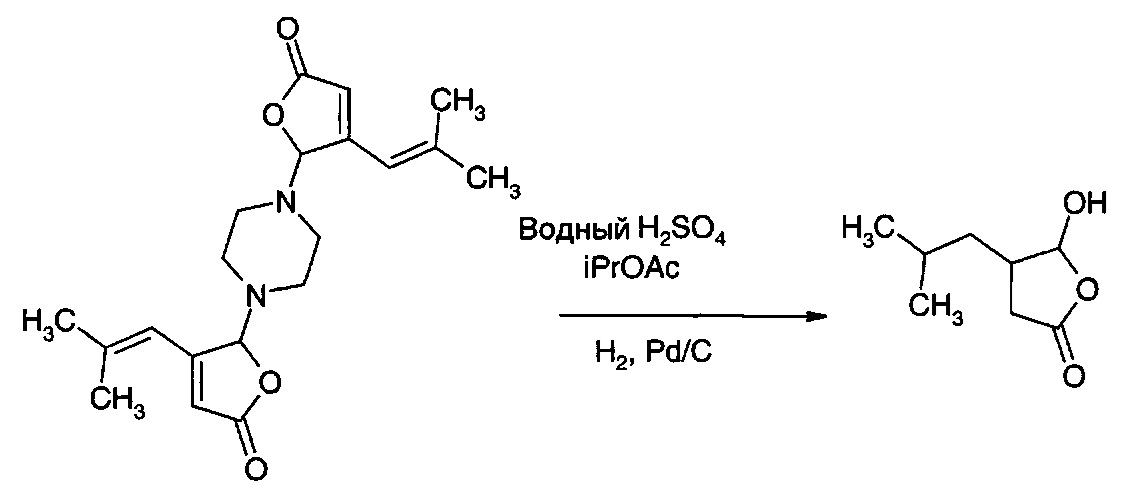
5,5'-(Пиперазин-1,4-диил)бис(4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-он) (50,0 г, 0,14 моль) загружали в воду (300 мл), содержащую серную кислоту (16,0 г, 0,163 моль). Добавляли изопропилацетат (200 мл). Добавляли 5% палладий на углероде, увлажненный 50% воды (3,0 г), и смесь гидрировали (5 бар (500 кПа) водорода) при температуре окружающей среды в течение 15 ч. Реакционную смесь отфильтровывали от катализатора, промывали изопропилацетатом (300 мл), который также использовали для промывания сосуда. Органическую фазу отделяли, промывали водой (100 мл) и концентрировали на роторном испарителе с получением 38,8 г прозрачного масла. Это масло представляет собой смесь желаемого соединения (IА) и сверхвосстановленного лактона (4-(2-метилпропил)-дигидрофуран-2-она). Его легко очищали путем экстракции водным раствором карбоната калия и промывания толуолом. В результате подкисления экстракта карбоната калия муравьиной кислотой получили желаемый продукт (IА) (25,6 г, 58%).
1Н ЯМР (D2O с добавлением К2СO3): δ 9.5 (1Н), 2.8 (m, 2Н), 2.40 (m, 2Н), 1.55 (m, 2Н), 1.30 (m, 2Н), 0.95 (m, 6Н).
Пример 20
Получение монокалиевой соли 3-изобутилиден-2-оксопентандикарбоновой кислоты
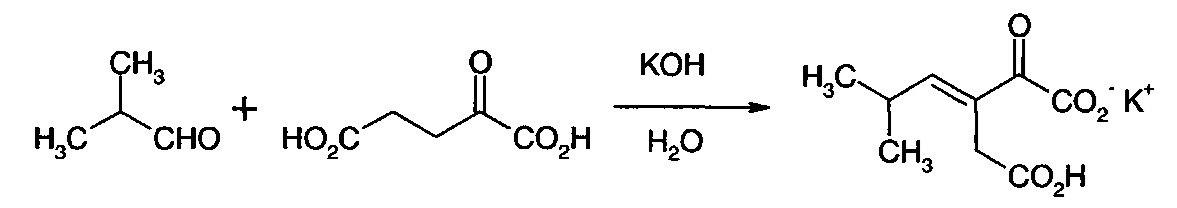
В круглодонную колбу на 2 л загружали 300 г α-кетоглутаровой кислоты и 450 мл ледяной воды. При перемешивании и охлаждении загружали 314 г гидроксида калия в 450 мл воды, поддерживая температуру в сосуде ниже 25°С. Реактор помещали в атмосферу азота и добавляли изобутиральдегид при 0,2 мл/мин в течение 50 ч. Полученный в результате двухфазный желтый раствор разделяли и промывали 100 мл МТВЕ. Значение рН нижней водной фазы доводили до 3,2-3,4 и концентрировали до 1/3 объема при 65°С в вакууме. После охлаждения до температуры менее 5°С полученные в результате твердые вещества собирали и промывали несколькими небольшими объемами воды с получением после высушивания 230 г белого твердого вещества, содержащего 60% монокалиевой соли 3-изобутилиден-2-оксопентандикарбоновой кислоты и остаточный KCl.
1Н ЯМР (D2O при рН 8-10, 400 МГц) δ 6.4 (d, 1Н), 3.3 (s, 2Н), 2.7 (m, 1Н), 0.96 (d, 6Н).13С ЯМР (D2O при рН 8-10, 400 МГц) δ 23 (2СН3), 31 (СН), 35 (СН2), 133 (С), 164 (СН), 170 (С), 176 (С), 182 (С).
Пример 21
Получение монокалиевой соли 3-(2-метилпропил)-2-оксопентандикарбоновой кислоты
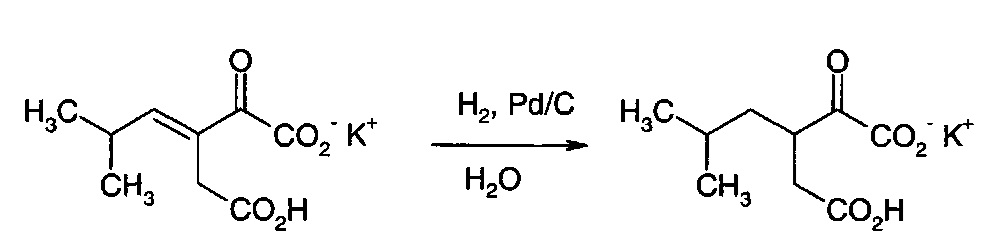
Водный раствор монокалиевой соли 3-изобутилиден-2-оксопентандикарбоновой кислоты, полученный в примере 20, доводили до рН 6-8 и добавляли 5% Pd/C, и раствор гидрировали при 10 бар Н2 при 25°С в течение 10 часов. Катализатор удаляли фильтрованием и рН раствора доводили до рН 3,8. Смесь охлаждали до температуры менее 5°С, фильтровали и промывали небольшим количеством холодной воды. Полученные в результате белые кристаллы высушивали при 40°С в вакууме с получением указанного в заголовке соединения, 286 г, 58% выход за 2 стадии.
1Н ЯМР (D2O при рН 8-10, 400 МГц) δ 3.5 (m, 1Н), 2.5 (dd, 1Н), 2.3 (dd, 1Н), 1.6-1.5 (m, 2Н) 1.3-1.2 (m, 1Н) 0.9 (d, 6Н).13С ЯМР (D2O при рН 8-10, 400 МГц) δ 21.7 (СН3), 22.2 (СН3), 25.5 (СН), 38.4 (СН2), 39.3 (СН2), 43.4 (СН), 167.8 (С), 170.6 (С), 180.7 (С).
Пример 22
Экспрессия Ферментов декарбоксилаз в Е. coli
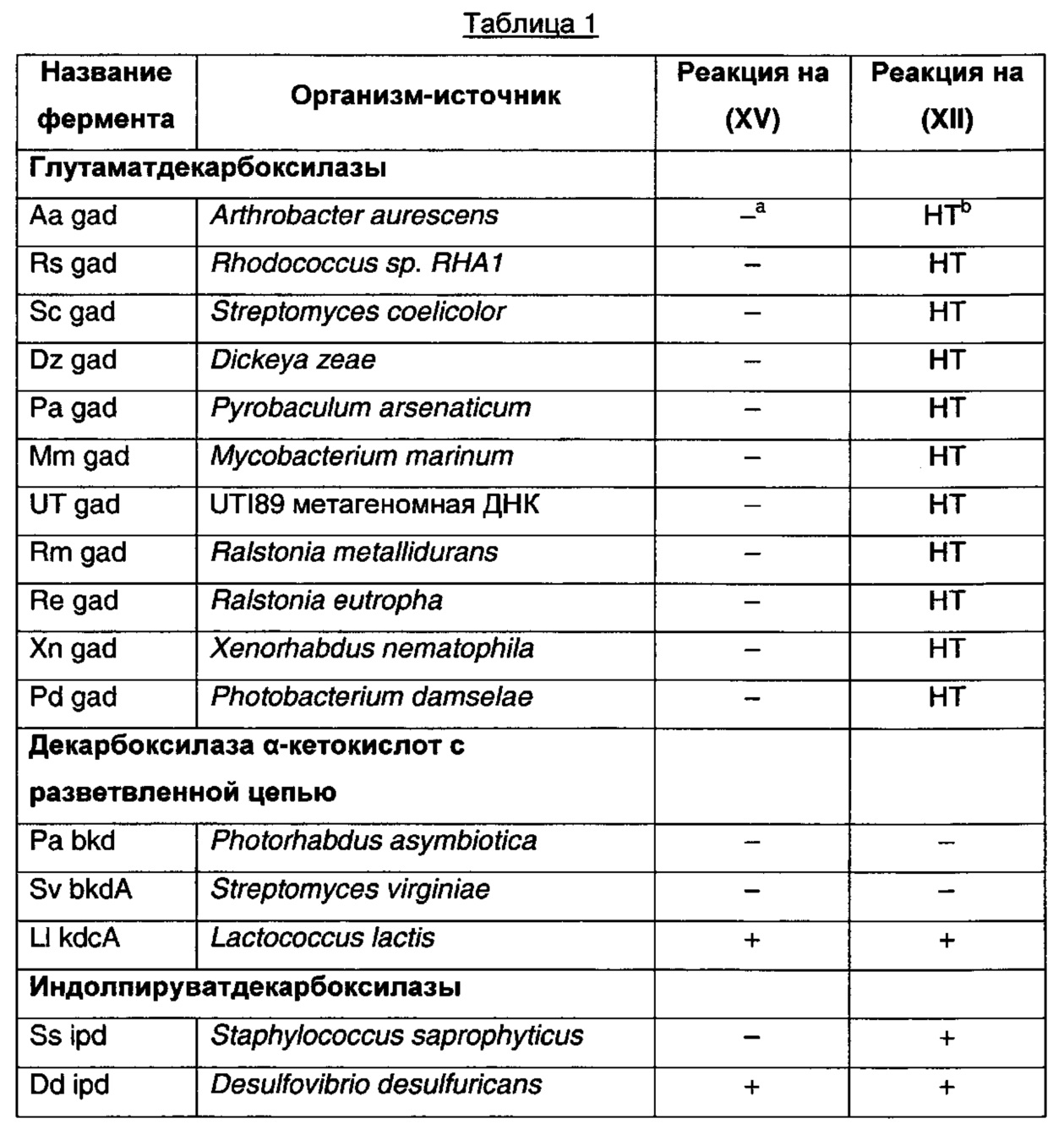
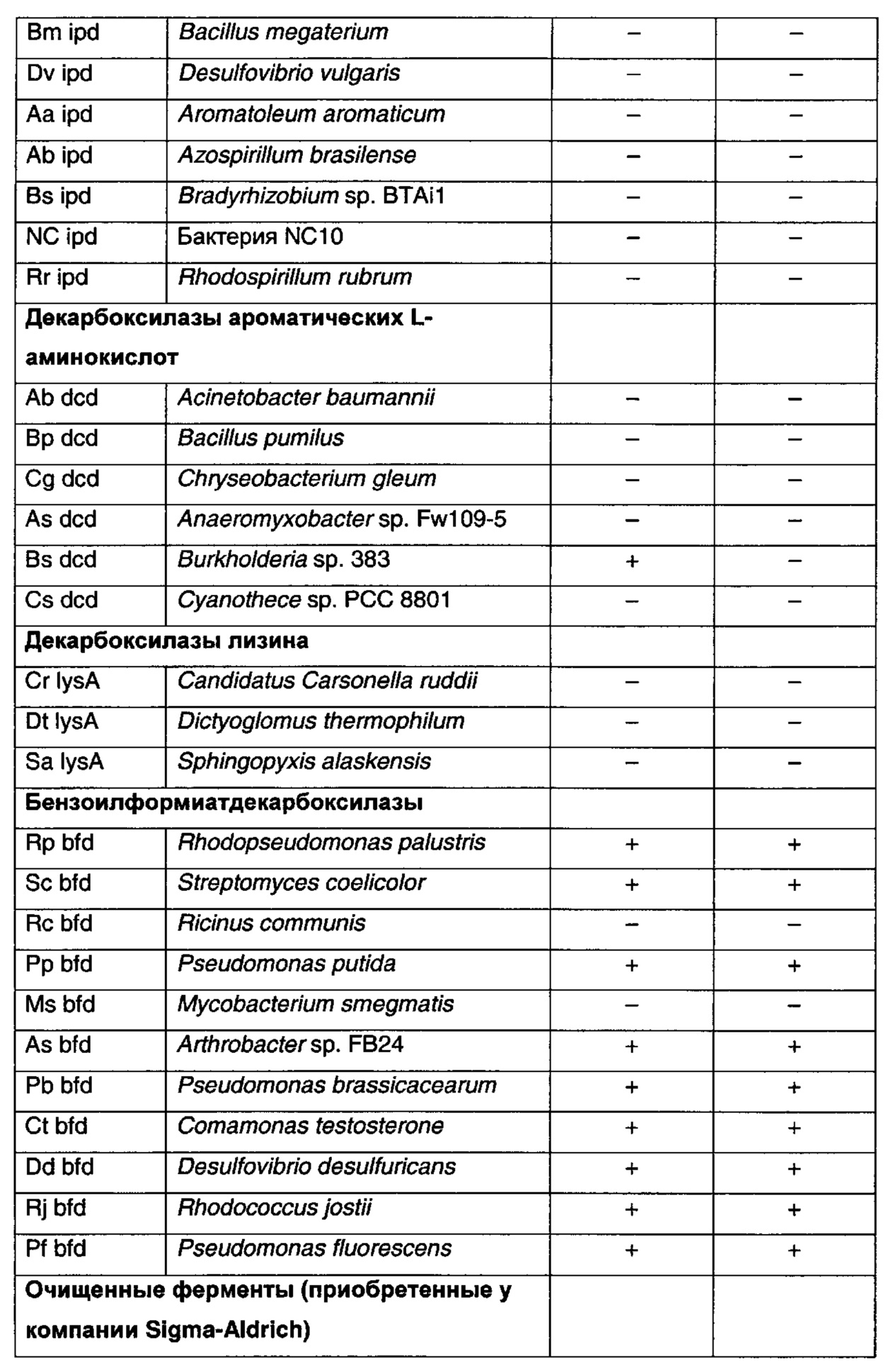
PubMed использовали для поиска литературы по ферментам декарбоксилазам с широким спектром активности. Для поиска декарбоксилаз микроорганизмов с активностью на соединениях, в некоторой стерени подобных по структуре 3-(2-метилпропил)-2-оксопентандикарбоновой кислоте или 3-изобутилиден-2-оксопентандикарбоновой кислоте, использовали программу KEGG (Kyoto Encyclopedia of Genes and Genomes). Для исследования было выбрано семь классов декарбоксилаз на основании сообщений об активности на соединениях с определенным структурным подобием. Классы декарбоксилаз представляли собой глутаматдекарбоксилазу, диаминопимелатдекарбоксилазу, индолпируватдекарбоксилазу, декарбоксилазу α-кетокислот с разветвленной цепью, декарбоксилазу ароматических L-аминокислот, декарбоксилазы лизина и бензоилформиатдекарбоксилазу. Была выбрана сорок одна последовательность генов доказанных или предполагаемых ферментов декарбоксилаз. Эти гены были оптимизированы по кодонам для экспрессии в Е. coli, синтезированы компанией GeneArt (Германия), DNA2.0 (Menlo Park, СА USA), или Blue Heron Biotechnology (Ботелл, Вашингтон, США), клонированы в экспрессионном векторе pSTRC52 (Pfizer Inc., США) и помещены в экспрессирующий штамм Е. coli BDG62 (Pfizer Inc., США) (таблица 1 ниже). Два гена амплифицировали с геномной ДНК из соответствующих организмов с помощью полимеразной цепной реакции (ПЦР) и клонировали в том же экспрессионном векторе и штамме Е. coli. Четыре фермента декарбоксилазы были приобретены у компании Sigma и протестированы на активность на 3-(2-метилпропил)-2-оксопентандикарбоновой кислоте.
Каждый штамм Е. coli, содержащий клонированный ген декарбоксилазы, выращивали в течение ночи в жидкой среде Лурия-Бертани (LB) с соответствующим антибиотиком. Небольшое количество (50 мкл - 100 мкл) затравочной культуры, культивированной в течение ночи, использовали для инокуляции 4,0 мл среды Terrific Broth с соответствующими антибиотиками в культуральных пробирках 20 мм. Культуры выращивали во встряхивателе-инкубаторе при 32°С и 300 об/мин. После 5 ч роста добавляли изопропилтиогалактозид (IPTG) до конечной концентрации 0,4 мМ для индуцирования экспрессии фермента. Культуры возвращали в инкубатор и выращивали дополнительно в течение 19 ч.
Пример 23
Декарбоксилирование калиевой соли 3-(2-метилпропил)-2-оксопентандикарбоновой кислоты и калиевой соли 3-изобутилиден-2-оксопентандикарбоновой кислоты Ферментами декарбоксилазами

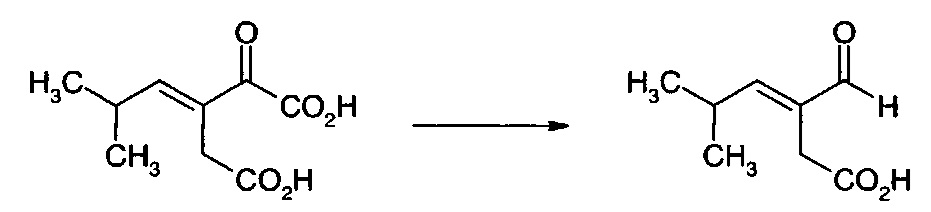
Рекомбинантные ферменты декарбоксилазы тестировали на декарбоксилирование 3-(2-метилпропил)-2-оксопентандикарбоновой кислоты (XV) и 3-изобутилиден-2-оксопентандикарбоновой кислоты (XII), используя клетки Е. coli, полученные, как описано в примере 22. Реакции (1 мл) проводили при 37°С в калийфосфатном буфере (100 мМ, рН 6,4), содержащем декарбоксилазу (60 мг влажных клеток), 50 мМ 3-(2-метилпропил)-2-оксопентандикарбоновую кислоту или 3-изобутилиден-2-оксопентандикарбоновую кислоту, ThDP (0,1 мМ) и MgSO4 (2,5 мМ). Декарбоксилирование 3-(2-метилпропил)-2-оксопентандикарбоновой кислоты и 3-изобутилиден-2-оксопентандикарбоновой кислоты определяли с помощью анализа UPLC. Аликвоту (0,1 мл) реакционной смеси обрабатывали 0,5 мл калийфосфатного буфера (100 мМ, доведенного до рН 2,2 ортофосфорной кислотой) и 0,3 мл раствора 2,4-динитрофенилгидразина (20 мМ в смеси 1 М НСl:ацетонитрил, 3:1, об/об) в течение 30 мин при 50°С. Дериватизированные образцы разводили 0,5 мл ацетонитрила, фильтровали и анализировали с помощью UPLC на колонке Agilent Eclipse Plus С18 (100 мм × 3,0 мм, 1,8 мкм), элюировали 0,1% трифторуксусной кислотой в смеси вода:ацетонитрил (55:45, об/об) при 1,1 мл/мин. Колонку поддерживали при 40°С, и мониторинг элюата проводили при 360 нм с помощью масс-спектроскопии ES+. На положительные результаты указывало присутствие пика 3-формил-5-метилгексановой кислоты от 3-(2-метилпропил)-2-оксопентандикарбоновой кислоты или 3-формил-5-метилгекс-3-еновой кислоты от 3-изобутилиден-2-оксопентандикарбоновой кислоты. Результаты анализов представлены в таблице 1.

Пример 24
Получение 3-Формил-5-метилгекс-3-еновой кислоты

В круглодонную колбу на 250 мл загружали 80 г концентрата клеток, содержащего Е. coli, которые экспрессируют бензоилформиатдекарбоксилазу Pseudomonas putida (см. пример 23). К этому концентрату добавляли доведенный до pH 6,2 раствор монокалиевой соли 3-изобутилиден-2-оксопентандикарбоновой кислоты (10 г, см. пример 20) в 50 мл воды с 0,5 г сульфата магния и 0,5 г тиаминпирофосфата. Полученную в результате взвесь с pH 6,2 нагревали до 50°C и перемешивали в течение 145 ч, поддерживая pH от 6,2 до 7,2 концентрированной HCl. Реакционную смесь охлаждали до кт и центрифугировали. Водные декантаты промывали 50 мл MTBE при минимальном перемешивании. Значение pH водной фазы доводили до 4 и фильтровали через целит. Фильтрат экстрагировали при минимальном перемешивании три раза 50 мл MTBE. MTBE высушивали над безводным сульфатом натрия и концентрировали до густого красного масла. Это масло экстрагировали несколькими порциями горячих гексанов; объединенные гексаны охлаждали до температуры ниже 0°, и полученные в результате кристаллы выделяли и высушивали на воздухе с получением 3-формил-5-метилгекс-3-еновой кислоты в виде белого твердого вещества (0,6 г).
1H ЯМР (D2O при pH 8-10, 400 МГц) δ 9.2 (s, 1H), 6.6 (d, 1H), 3.1 (s, 2H), 2.7 (m, 1H), 1 (d, 6H)
Пример 25
Экспрессия гомологов еноатредуктазы в Е. coli
Последовательность ДНК, соответствующую гену 12-оксофитодиеноатредуктазы 1 (OPR1) Lycopersicon esculentum (томата) получали из базы данных Genbank (номер доступа AC Q9XG54) и синтезировали в компании GeneArt (Германия). Осуществляли оптимизацию последовательности по кодонам для экспрессии в Е. coli и ее субклонировали в экспрессионной плазмиде Е. coli pSTRC18 (Pfizer Inc., США). Последовательность белка приведена ниже. Экспрессионную конструкцию OPR1 трансформировали в BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США) в соответствии с инструкциями, и культуры инкубировали в течение ночи в среде LB со стрептомицином. Культуру LB использовали для инокуляции экспрессионных культур (LB, M9Y или TB), которые инкубировали при 37°C (210 об/мин). Когда культура достигала подходящей концентрации биомассы (OD (оптическая плотность) 1 при A600), добавляли IPTG (1 мМ), и культуры инкубировали еще в течение 20 ч (30°C, 210 об/мин). Клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C.
Для поиска последовательностей генов, родственных гену 12-оксофитодиеноатредуктазы (OPR1) из Lycopersicon esculentum в неизбыточной базе данных белковых последовательностей Национального центра биотехнологической информации (NCBI) использовали программу BLASTP. Тридцать восемь последовательностей родственных генов были отобрано, оптимизированы по кодонам для экспрессии в E. coli и субклонированы в экспрессионной плазмиде pET28b(+) Е. coli (Novagen, EMD Chemicals, Гиббстон, Нью-Джерси, США). Экспрессионные конструкции, родственные OPR1, трансформировали в BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США), в соответствии с инструкциями, и культуры инкубировали в течение ночи в среде LB с канамицином. Культуры LB использовали для инокуляции экспрессионных культур, выращиваемых в среде TB для быстрой экспрессии в течение ночи (Novagen, EMD Chemicals, Гиббстон, Нью-Джерси, США). Культуры инкубировали в течение 20 ч при 30°C. Клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C.
Последовательность белка 12-оксофитодиеноатредуктазы 1 Lycopersicon esculentum (томата):

Последовательность 12-оксофитодиеноатредуктазы 1 Lycopersicon esculentum (томата), оптимизированная по кодонам:


Пример 26
Восстановление (E)-3-формил-5-метилгекс-3-еновой кислоты рекомбинантными редуктазами
Рекомбинантные еноатредуктазы были протестированы на восстановление (E)-3-формил-5-метилгекс-3-еновой кислоты с использованием клеток Е. coli, полученных, как описано в примере 25. Реакции (0,5 мл) проводили при 30°C в калийфосфатном буфере (100 мМ, pH 7,0), содержащем клетки Е. coli (100 мг влажных клеток/мл), NADPH (10 мМ), NADH (10 мМ) и (E)-3-формил-5-метилгекс-2-еновую кислоту (10 мМ). Через 16 ч в каждую реакционную смесь добавляли ацетонитрил (0,5 мл), и полученные в результате смеси центрифугировали (2000 об/мин × 5 мин). Аликвоты (0,1 мл) полученных в результате супернатантов обрабатывали 0,1 мл калийфосфатного буфера (100 мМ, доведен до pH 2,2 ортофосфорной кислотой) и 0,225 мл раствора 2,4-динитрофенилгидразина (20 мМ в смеси 1 М HCl : ацетонитрил, 3:1, об/об) в течение 30 мин при 50°C. Полученные образцы разводили 0,225 мл ацетонитрила и анализировали с помощью HPLC на колонке Amylose-2 Phenomenex Lux 5 мкм (250 мм × 4,6 мм внутренний диаметр), элюируя 0,1% трифторуксусной кислотой в смеси вода : ацетонитрил (65:35, об/об) при 2 мл/мин. Колонку поддерживали при 50°C, и мониторинг элюата проводили при 360 нм. Результаты анализов HPLC представлены в таблице 2.




Пример 27
Восстановление (E)-3-формил-5-метилгекс-3-еновой кислоты рекомбинантными редуктазами и формиатдегидрогеназой

Рекомбинантные еноатредуктазы оценивали на восстановление (E)-3-формил-5-метилгекс-3-еновой кислоты с NAD+ и формиатдегидрогеназой. Еноатредуктазы экспрессировали в клетках Е. coli, как описано в примере 25. Формиатдегидрогеназу экспрессировали в клетках Е. coli, как описано ниже. Экспрессионную конструкцию формиатдегидрогеназы pET26b трансформировали в BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США) согласно инструкции, и культуры выращивали в течение ночи в среде LB с канамицином. Культуры LB использовали для инокуляции экспрессионных культур, выращиваемых в среде ТМ для быстрой экспрессии в течение ночи с канамицином (Novagen, EMD Chemicals, Гиббстон, Нью-Джерси, США). Культуры инкубировали в течение 20 ч при 30°C, и клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C. Вариант (D74M) оксофитодиеноатредуктазы 3 (номер доступа Q9FEW9) экспрессировали в клетках Е. coli, как описано ниже: Для создания варианта оксофитодиеноатредуктазы 3 D74M использовали набор для сайтнаправленного мутагенеза QuikChange от компании Stratagene (Ла-Хойя, Калифорния, США) согласно инструкции. Праймеры заказывали у компании Integrated DNA Technologies (Коралвилл, Айова, США). Экспрессионной конструкцией pSTRC18 оксофосфодиеноатредуктазы 3 (OPR3) трансформировали BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США) согласно инструкции, и ночные культуры выращивали в среде для размножения со стрептомицином (Zymo Research, Ирвин, Калифорния, США). Культуры LB использовали для инокуляции экспрессионных культур, выращиваемых в среде для гиперэкспрессии со стрептомицином (Zymo Research, Ирвин, Калифорния, США). Культуры инкубировали в течение 20 ч при 23°C, и клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C.
Реакции (0,5 мл) проводили при 30°C в калийфосфатном буфере (100 мМ, pH 7,0), содержащем еноатредуктазы (40 мг влажных клеток/мл), формиатдегидрогеназу (80 мг влажных клеток/мл), NAD+ (0,02 мМ), формиат аммония (30 мМ) и (E)-3-формил-5-метилгекс-3-еновую кислоту (20 мМ). Через 24 ч реакционные смеси подкисляли 0,025 мл 4 н HCl и экстрагировали 1 мл этилацетата. Аликвоты (0,5 мл) этилацетатных экстрактов (0,5 мл) сушили над безводным сульфатом натрия и обрабатывали метанолом (0,02 мл) и (триметилсилил)диазометаном (0,01 мл 2 М раствора в диэтиловом эфире), для превращения карбоновокислотных группировок в их соответствующие метиловые эфиры. Полученные образцы анализировали с помощью GC на колонке Chiraldex™ G-TA (30М × 0,25 мм, температура колонки: 135°C изотермическая, температура инжектора: 200°C, газ-носитель: гелий, скорость тока приблизительно 1 мл/мин) с получением результатов, представленных в таблице 3.
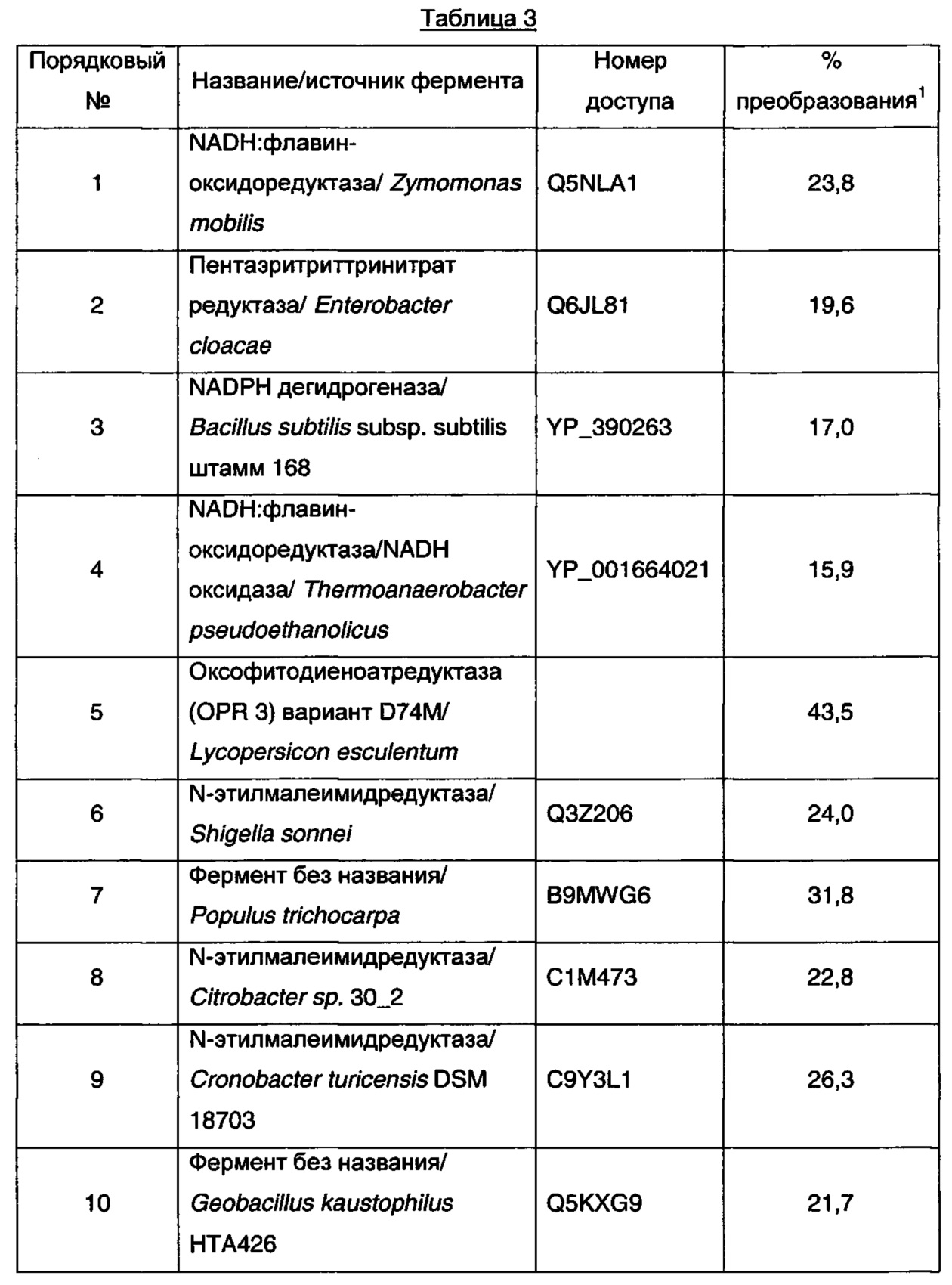
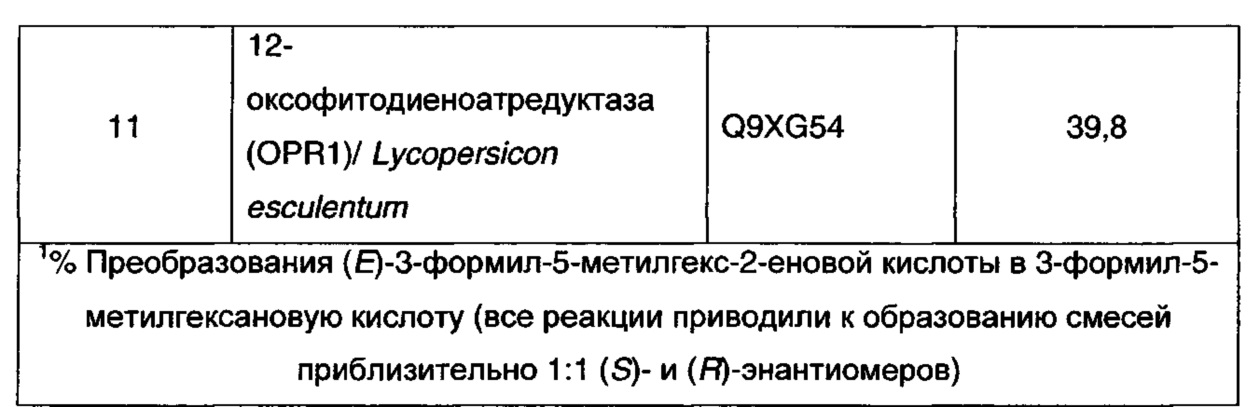
Пример 28
Восстановление (E)-3-Формил-5-метилгекс-3-еновой кислоты рекомбинантными редуктазами

Рекомбинантные еноатредуктазы оценивали на восстановление (E)-3-формил-5-метилгекс-3-еновой кислоты с NADP+ и алкогольдегидрогеназой Lactobacillus brevis (X-zyme). Еноатредуктазы экспрессировали в клетках Е. coli, как описано в примере 25. Вариант (D74M) оксофитодиеноатредуктазы 3 (OPR3) был получен, как описано в примере 27. Реакции (0,5 мл) проводили при 30°C в калийфосфатном буфере (100 мМ, pH 7,0), содержащим еноатредуктазы (40 мг влажных клеток/мл), алкогольдегидрогеназу Lactobacillus brevis (32 ед./мл), NADP+ (0,02 мМ), 2-пропанол (3 об. %) и (E)-3-формил-5-метилгекс-2-еновую кислоту (20 мМ). Через 24 ч реакционные смеси подкисляли 0,025 мл 4 н HCl и экстрагировали 1 мл этилацетата. Аликвоты (0,5 мл) этилацетатных экстрактов (0,5 мл) сушили над безводным сульфатом натрия и обрабатывали метанолом (0,02 мл) и (триметилсилил)диазометаном (0,01 мл 2 М раствора в диэтиловом эфире) для превращения карбоновокислотных группировок в их соответствующие метиловые эфиры. Полученные образцы анализировали с помощью GC на колонке Chiraldex™ G-TA (30М × 0,25 мм, температура колонки: 135°C изотермическая, температура инжектора: 200°C, газ-носитель: гелий, скорость тока приблизительно 1 мл/мин) с получением результатов, представленных в таблице 4.

Пример 29
Получение 3-формил-5-метилгексановой кислоты
В реакционный сосуд загружали 7,5 мл калийфосфатного буфера (0,1 М, pH 7,0), 8,4 мг NADP+, 0,2 мл алкогольдегидрогеназы Lactobacillus brevis (32 ед./мл, X-zyme), 0,3 мл 2-пропанола, пентаэритриттринитратредуктазу (2 мл суспензии 200 мг/мл клеток E. coli в калийфосфатном буфере) и 156 мг (E)-3-формил-5-метилгекс-3-еновой кислоты и перемешивали при 40°C. Через 6,75 ч реакционную смесь центрифугировали, и супернатант доводили до pH 2 4 н HCl и экстрагировали этилацетатом (2×10 мл). Этилацетатный экстракт сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением 141 мг бесцветного масла (89% выход).
1H ЯМР (D2O при pH 8-10) δ 2.53-2.43 (m, 2H), 2.34-2.28 (m, 1H), 1.53-1.42 (m, 1H), 1.41-1.34 (m, 1H), 1.20-1.12 (m, 1H), 0.76 (d, 3H), 0.74 (d, 3H).
Пример 30
Биотрансформация 3-формил-5-метилгексановой кислоты в прегабалин рекомбинантными ω-трансаминазами
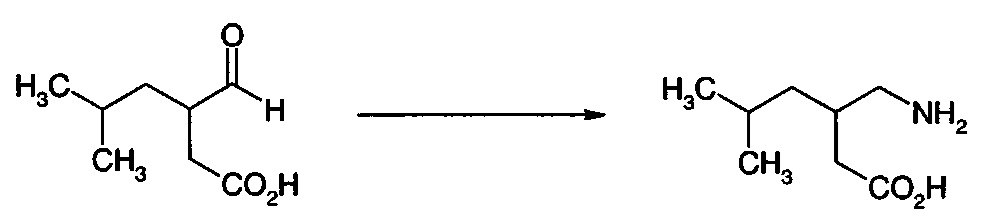
Трансаминирование 3-формил-5-метилгексановой кислоты с образованием прегабалина оценивали с различными рекомбинантными трансаминазами. Рекомбинантные ω-трансаминазы из Vibrio fluvialis, Rhodobacter sphaeroides и Paracoccus denitrificans экспрессировали в E. coli, как описано ниже. Экспрессионные конструкции ω-трансаминаз в pET28b трансформировали в BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США) согласно инструкции, и культуры выращивали в течение ночи в среде LB с канамицином. Культуры LB использовали для инокуляции экспрессионных культур, выращиваемых в среде TB для быстрой экспрессии в течение ночи с канамицином (Novagen, EMD Chemicals, Гиббстон, Нью-Джерси, США). Культуры инкубировали в течение 20 ч при 30°C, и клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C.
Реакции (0,5 мл) проводили при 30°C в калийфосфатном буфере (100 мМ, pH 7,0), содержащем пиридоксальфосфат (2 мМ), изопропиламин (150 мМ), 3-формил-5-метилгексановую кислоту (50 мМ) и ω-трансаминазу (40 мг влажных клеток/мл) из Vibrio fluvialis, Rhodobacter sphaeroides или Paracoccus denitrificans. Через 24 ч образцы реакционных смесей (0,1 мл) разводили 0,4 мл смеси ацетонитрил : вода (1:1, об/об). Аликвоты (0,05 мл) разведенных образцов реакционных смесей обрабатывали насыщенным водным раствором бикарбоната натрия (0,01 мл) и реагентом Марфи (N-α-(2,4-динитро-5-фторфенил)аланинамид, 0,2 мл раствора 5 г/л в ацетонитриле) при 40°C. Через 1 ч полученные реакционные смеси гасили 0,01 мл 1 н водным раствором соляной кислоты и разводили 0,23 мл ацетонитрила. Образцы полученных реакционных смесей анализировали с помощью колоночной UPLC (колонка: BEH C18, 50 мм × 2,1 мм внутренний диаметр, градиентное элюирование: от 70% A:30% B до 55% A:45% B за 5 мин (A = 1% триэтиламин (pH 3, ортофосфорная кислота); B = ацетонитрил), скорость тока: 0,8 мл/мин, температура колонки: 30°C, детектирование: 210-400 нм) с получением результатов, представленных в таблице 5.


Пример 31
Биотрансформация 3-формил-5-метилгексановой кислоты в прегабалин рекомбинантными ω-трансаминазами
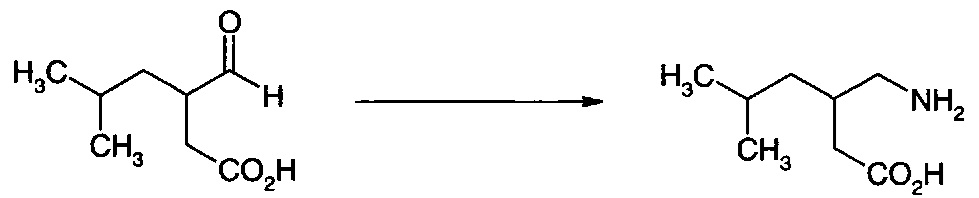
Рекомбинантные варианты Vibrio fluvialis тестировали на восстановительное аминирование 3-формил-5-метилгексановой кислоты с образованием прегабалина. Варианты ω-трансаминазы V. fluvialis (номер доступа AEA39183) экспрессировали в Е. coli, как описано ниже. Для создания вариантов аминотрансферазы V. fluvialis использовали наборы для сайтнаправленного мутагенеза QuikChange от Stratagene (Ла-Хойя, Калифорния, США) согласно инструкции. Праймеры заказывали в Integrated DNA Technologies (Коралвилл, Айова, США). Экспрессионные конструкции ω-трансаминазы в pET28b трансформировали в BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США) согласно инструкции, и культуры выращивали в течение ночи в среде LB с канамицином. Культуры LB использовали для инокуляции экспрессионных культур, выращиваемых в среде ТВ для быстрой экспрессии в течение ночи с канамицином (Novagen, EMD Chemicals, Гиббстон, Нью-Джерси, США). Культуры инкубировали в течение 20 ч при 30°C, и клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C.
Реакции (0,5 мл) проводили при 30°C в калийфосфатном буфере (100 мМ, pH 7,0), содержащем пиридоксальфосфат (2 мМ), изопропиламин (300 мМ), 3-формил-5-метилгексановую кислоту (100 мМ) и ω-трансаминазу V. fluvialis дикого типа или ее варианты (40 мг влажных клеток/мл). Через 28 ч образцы реакционных смесей (0,1 мл) разводили 0,4 мл смеси ацетонитрил : вода (1:1, об/об). Аликвоты (0,05 мл) разведенных образцов реакционных смесей обрабатывали насыщенным водным раствором бикарбоната натрия (0,01 мл) и реагентом Марфи (N-α-(2,4-динитро-5-фторфенил)аланинамид, 0,2 мл раствора 5 г/л в ацетонитриле) при 40°C. Через 1 ч полученные реакционные смеси гасили 0,01 мл 1 н водного раствора соляной кислоты и разводили 0,23 мл ацетонитрила. Образцы полученных реакционных смесей анализировали с помощью UPLC (колонка: BEH C18, 50 мм × 2,1 мм внутренний диаметр, градиент элюирования: от 70% A:30% B до 55% A:45% B за 5 мин (A = 1% триэтиламин (pH 3, ортофосфорная кислота); B = ацетонитрил), скорость тока: 0,8 мл/мин, температура колонки: 30°C, детектирование: 210-400 нм) с получением результатов, представленных в таблице 6.

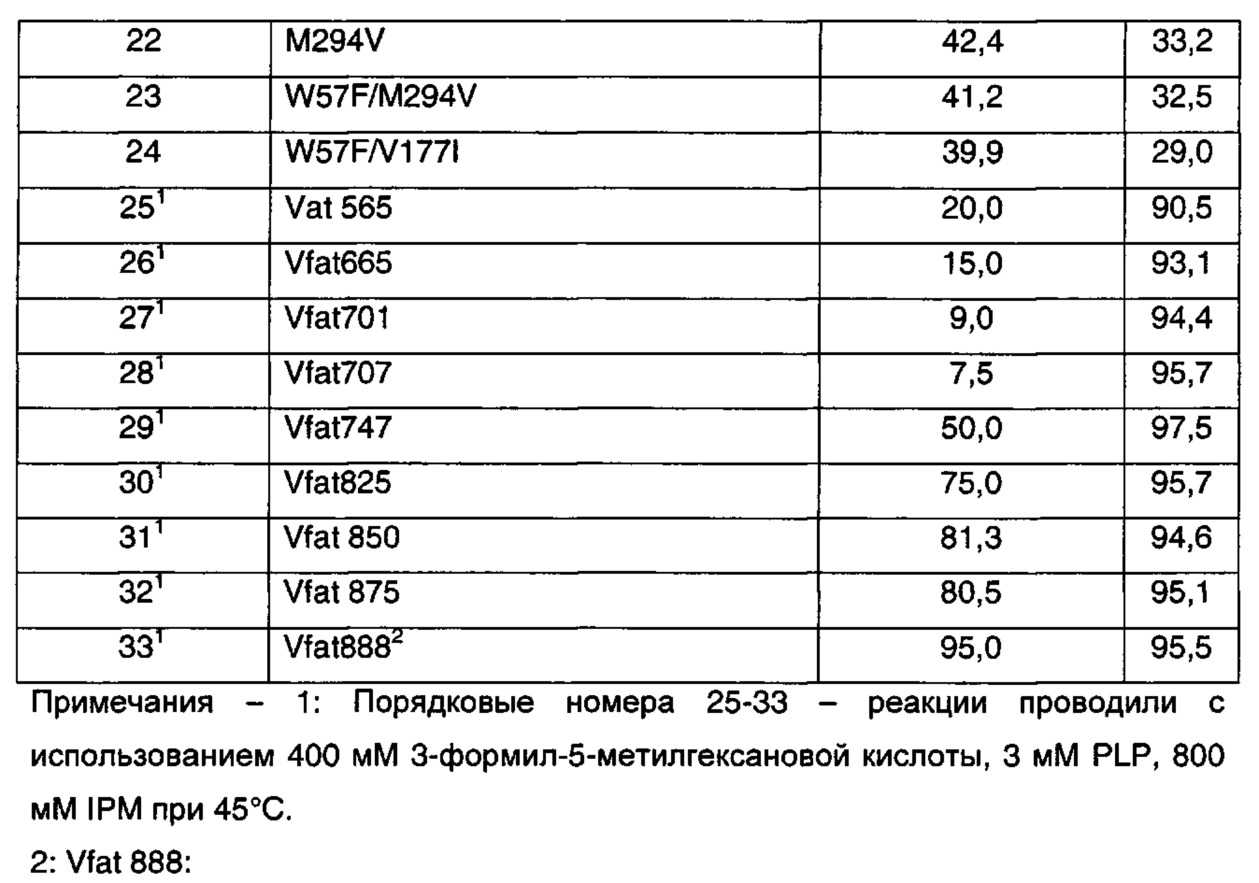
ПОСЛЕДОВАТЕЛЬНОСТЬ ДНК


АМИНОКИСЛОТНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ

Пример 32
Ферментативное восстановление (E)-3-формил-5-метилгекс-2-еновой кислоты до 3-формил-5-метилгексановой кислоты и преобразование in situ в прегабалин ω-трансаминазой
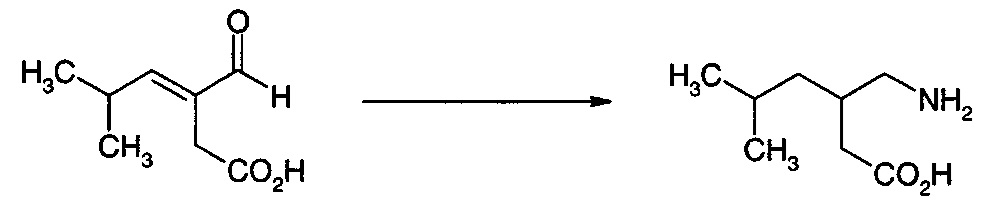
Восстановление (E)-3-формил-5-метилгекс-3-еновой кислоты до 3-формил-5-метилгексановой кислоты с помощью пентаэритриттринитратредуктазы и преобразование in situ в прегабалин оценивали с различными ω-трансаминазами. Рекомбинантные ω-трансаминазы из Vibrio fluvialis, Rhodobacter sphaeroides и Paracoccus denitrificans экспрессировали в E. coli, как описано ниже. Экспрессионные конструкции ω-трансаминазы в pET28b трансформировали в BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США) согласно инструкции, и культуры выращивали в течение ночи в среде LB с канамицином. Культуры LB использовали для инокуляции экспрессионных культур, выращиваемых в среде TB для быстрой экспрессии в течение ночи с канамицином (Novagen, EMD Chemicals, Гиббстон, Нью-Джерси, США). Культуры инкубировали в течение 20 ч при 30°C, и клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C.
Реакции (0,5 мл) проводили при 30°C в калийфосфатном буфере (100 мМ, pH 7.0), содержащем пентаэритриттринитратредуктазу (40 мг влажных клеток/мл), алкогольдегидрогеназу Lactobacillus brevis (32 ед./мл), NADP+ (0,02 мМ), 2-пропанол (3% по об.), пиридоксальфосфат (2 мМ), изопропиламин (100 мМ), (E)-3-формил-5-метилгекс-2-еновую кислоту (20 мМ) и ω-трансаминазу (40 мг влажных клеток/мл) из Vibrio fluvialis, Rhodobacter sphaeroides или Paracoccus denitrificans. Через 43 ч образцы реакционных смесей (0,1 мл) разводили 0,1 мл смеси ацетонитрил : вода (1:1, об/об). Аликвоты (0,1 мл) разведенных образцов реакционной смеси обрабатывали насыщенным водным раствором бикарбоната натрия (0,01 мл) и реагентом Марфи (N-α-(2,4-динитро-5-фторфенил)аланинамид, 0,4 мл раствора 5 г/л в ацетонитриле) при 40°C. Через 1 ч полученные реакционные смеси гасили 0,1 мл 1 н водного раствора соляной кислоты. Образцы полученных реакционных смесей анализировали с помощью UPLC (колонка: BEH C18, 50 мм × 2,1 мм внутренний диаметр, градиентное элюирование: от 70% A:30% B до 55% A:45% B за 5 мин (A = 1% триэтиламин (pH 3, ортофосфорная кислота); B = ацетонитрил), скорость тока: 0,8 мл/мин, температура колонки: 30°C, детектирование: 210-400 нм) с получением результатов, представленных в таблице 7.


Пример 33
Ферментативное восстановление (E)-3-формил-5-метилгекс-2-еновой кислоты до 3-формил-5-метилгексановой кислоты и преобразование in situ в прегабалин ω-трансаминазой V. Fluvialis
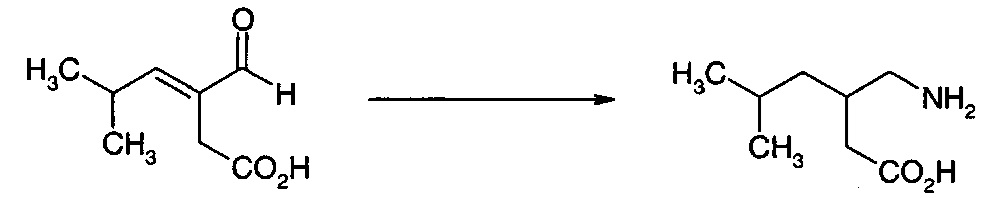
Восстановление (E)-3-формил-5-метилгекс-3-еновой кислоты до 3-формил-5-метилгексановой кислоты с помощью пентаэритриттринитратредуктазы и преобразование in situ в прегабалин оценивали с вариантами аминотрансферазы V. fluvialis. Варианты ω-трансаминазы V. fluvialis (номер доступа AEA39183) экспрессировали в Е. coli, как описано ниже. Для создания вариантов аминотрансферазы V. fluvialis использовали наборы для сайтнаправленного мутагенеза QuikChange от Stratagene (Ла-Хойя, Калифорния, США) согласно инструкции. Праймеры заказывали в Integrated DNA Technologies (Коралвилл, Айова, США). Экспрессионные конструкции ω-трансаминазы в pET28b трансформировали в BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США) согласно инструкции, и культуры выращивали в течение ночи в среде LB с канамицином. Культуры LB использовали для инокуляции экспрессионных культур, выращиваемых в среде TB для быстрой экспрессии в течение ночи с канамицином (Novagen, EMD Chemicals, Гиббстон, Нью-Джерси, США). Культуры инкубировали в течение 20 ч при 30°C, и клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C.
Реакции (0,5 мл) проводили при 30°C в калийфосфатном буфере (100 мМ, pH 7,0), содержащем пентаэритриттринитратредуктазу (40 мг влажных клеток/мл), алкогольдегидрогеназу Lactobacillus brevis (32 ед./мл), NADP+ (0,1 мМ), 2-пропанол (3% по об.), пиридоксальфосфат (2 мМ), изопропиламин (300 мМ), (E)-3-формил-5-метилгекс-2-еновую кислоту (100 мМ) и ω-трансаминазу V. fluvialis дикого типа или варианты (40 мг влажных клеток/мл). Через 48 ч образцы реакционных смесей (0,02 мл) разводили 0,18 мл смеси ацетонитрил : вода (1:1, об/об). Аликвоты (0,1 мл) разведенных образцов реакционной смеси обрабатывали насыщенным водным раствором бикарбоната натрия (0,01 мл) и реагентом Марфи (N-α-(2,4-динитро-5-фторфенил)аланинамид, 0,4 мл раствора 5 г/л в ацетонитриле) при 40°C. Через 1 ч полученные реакционные смеси гасили 0,1 мл 1 н водного раствора соляной кислоты. Образцы полученных реакционных смесей анализировали с помощью UPLC (колонка: BEH C18, 50 мм × 2,1 мм внутренний диаметр, градиентное элюирование: от 70% A:30% B до 55% A:45% B за 5 мин (A = 1% триэтиламин (pH 3, ортофосфорная кислота); B = ацетонитрил), скорость тока: 0,8 мл/мин, температура колонки: 30°C, детектирование: 210-400 нм) с получением результатов, представленных в таблице 8.

Пример 34
Альтернативные варианты ω-трансаминазы Vibrio fluvalis
Описанные ниже дополнительные рекомбинантные варианты ω-трансаминазы Vibrio fluvialis экспрессировали в Е. coli, как описано ниже. Экспрессионные конструкции ω-трансаминазы в pET28b трансформировали в BL21(DE3) Е. coli (Stratagene, Agilent Technologies, Санта Клара, Калифорния, США) согласно инструкции, и культуры выращивали в течение ночи в среде LB с канамицином. Культуры LB использовали для инокуляции экспрессионных культур, выращиваемых в среде TB для быстрой экспрессии в течение ночи с канамицином (Novagen, EMD Chemicals, Гиббстон, Нью-Джерси, США). Культуры инкубировали в течение 20 ч при 30°C, и клетки собирали центрифугированием (4000 x g, 30 мин, 4°C) и хранили при -20°C.
Пример 34a: Vfat906
Последовательность ДНК:

Аминокислотная последовательность:

Пример 34b: Vfat999
Последовательность ДНК:


Аминокислотная последовательность:

Пример 34c: Vfat1010
Последовательность ДНК:


Аминокислотная последовательность:

Пример 34d: Vfat1020
Последовательность ДНК:


Аминокислотная последовательность:

Пример 34e: Vfat1030
Последовательность ДНК:


Аминокислотная последовательность:

Пример 35
Биотрансформация 3-изобутилиден-2-оксопентандикарбоновой кислоты в прегабалин

Гены бензоилформиатдекарбоксилазы Pseudomonas putida, пентаэритриттринитратредуктазы Enterobacter cloacae, алкогольдегидрогеназы Lactobacillus brevis и ω-трансаминазы Vibrio fluvialis клонировали в экспрессионном векторе pDSTRC52 (Pfizer Inc., США) и помещали в экспрессирующий штамм Е. coli BDG62 (Pfizer Inc., США). Культуру выращивали и индуцировали продукцию фермента, как описано в примере 22. Экспрессию фермента определяли с помощью электрофореза в полиакриламидном геле с использованием гелей и штаммов Novex (Invitrogen Corporation, Карлсбад, Калифорния).
Реакции (1,0 мл) проводили при 40°C в фосфатном буфере (100 мМ, pH 6,4) с декарбоксилазой P. putida, трансаминазой V. fluvialis, алкогольдегидрогеназой L. brevis, пентаэритриттринитратредуктазой Е. cloacae и NADP (0,1 мМ) в одной ячейке. 200 мкл 1 М 3-изобутилиден-2-оксопентандикарбоновой кислоты, 100 мкл 1 мМ ThDp + 25 мМ MgSO4, 40 мкл 50 мМ PLP, 30 мкл изопропанола, 250 мкл 2 М изопропиламина. Доводили pH до 6,4 в течение 24 часов, затем доводили pH до 6,8 в течение дополнительных 24 часов. Аликвоту (0,5 мл) реакционной смеси обрабатывали 1 М водным раствором бикарбоната натрия (0,05 мл) и реагентом Марфи (N-α-(2,4-динитро-5-фторфенил)аланинамид, 0,5 мл раствора 5 г/л в ацетонитриле) при 40°C. Через 1 ч полученные реакционные смеси гасили 0,05 мл 1 н водным раствором соляной кислоты. Образцы полученных реакционных смесей фильтровали и анализировали с помощью UPLC (колонка: Agilent Eclipse Plus С18 (100 мм × 3,0 мм, 1,8 мкм), элюируя 0,1% трифторуксусной кислотой в смеси вода : ацетонитрил (60:40, об/об) при 1,3 мл/мин. Колонку поддерживали при 30°C, и мониторинг элюата проводили при 340 нм с помощью ES+ масс-спектроскопии. В результате реакции получили небольшое количество прегабалина, 82% которого составлял желаемый S-изомер.
Пример 36
Биотрансформация 3-(2-метилпропил)-2-оксопентандикарбоновой кислоты в прегабалин

Гены бензоилформиатдекарбоксилазы Pseudomonas putida и ω-трансаминазы Vibrio fluvialis клонировали в экспрессионном векторе pDSTRC52 (Pfizer Inc., США) и помещали в экспрессирующий штамм Е. coli BDG62 (Pfizer Inc., США). Культуру выращивали и индуцировали продукцию фермента, как описано в примере 22. Экспрессию фермента определяли с помощью электрофореза в полиакриламидном геле с использованием гелей и штаммов Novex (Invitrogen Corporation, Карлсбад, Калифорния).
Реакции (2,0 мл) проводили при 40°C в фосфатном буфере (100 мМ, pH 6,4), содержащем 500 мкл культуры (42,5 мг сухой массы клеток) из 24-часовых ферментеров, декарбоксилазу P. putida и ω-трансаминазу V. fluvialis, клонированные в одной плазмиде, 400 мкл 0,5 М 3-(2-метилпропил)-2-оксопентандикарбоновой кислоты, 200 мкл 10x ThDP (конечная концентрация 0,1 мМ) и MgSO4 (конечная концентрация 2,5 мМ), 80 мкл 50 мМ PLP (конечная концентрация 2 мМ), 300 мкл 2 М изопропиламин (конечная концентрация 0,3 М). Доводили pH до 6,4 и инкубировали при 45°C в течение 24 часов, затем доводили pH до 6,8 в течение дополнительных 24 часов. Аликвоту (0,5 мл) реакционной смеси обрабатывали 1 М водным раствором бикарбоната натрия (0,05 мл) и реагентом Марфи (N-α-(2,4-динитро-5-фторфенил)аланинамид, 0,5 мл раствора 5 г/л в ацетонитриле) при 40°C. Через 1 ч полученные реакционные смеси гасили 0,05 мл 1 н водным раствором соляной кислоты. Образцы полученных реакционных смесей фильтровали и анализировали с помощью UPLC (колонка: Agilent Eclipse Plus C18 (100 мм × 3,0 мм, 1,8 мкм), элюируя 0,1% трифторуксусной кислотой в смеси вода : ацетонитрил (60:40, об/об) при 1,3 мл/мин. Колонку поддерживали при 30°C, и мониторинг элюата проводили при 340 нм с помощью ES+ масс-спектроскопии. В результате реакции получили 100 мкг/мл прегабалина, 65% которого составлял желаемый S-изомер.
Пример 37
Получение (S)-прегабалина посредством гидролиза 5-метокси-4-(2-метилпропил)-дигидрофуран-2(3H)-она и ферментативного трансаминирования
5-Метокси-4-(2-метилпропил)-дигидрофуран-2(3H)-он (2,58 г, 15 ммоль, см. пример 13) суспендировали в DIW (5,2 г) и охлаждали в бане лед/вода. Водный раствор KOH (50 масс. %, 1,77 г, 1,05 экв.) добавляли по каплям через шприц в течение 5 мин. Реакционную смесь удаляли из бани лед/вода и перемешивали при кт в течение 90 мин. Доводили pH до 7,0, используя муравьиную кислоту. Затем реакционную смесь использовали в качестве сырья в последующей трансаминазной реакции.
Раствор фермента трансаминазы (12,5 г), PLP (35 мг), DIW (15 мл) и 4,0 М изопропиламина в водном растворе HCl (7,5 мл, 30 ммоль) загружали в колбу на 100 мл и подогревали до 45°C. Гидролизную реакционную смесь добавляли одной порцией с последующим добавлением DIW (6 мл, использовали для промывания сосуда). Значение pH реакционной смеси доводили до 7,25 с использованием чистого изопропиламина, и реакционную смесь перемешивали при 45°C до завершения реакции. Затем реакционную смесь нагревали до внутренней температуры 55°C, и pH доводили до 4,0, используя 95% муравьиную кислоту. Добавляли углерод Darco (125 мг), и смеси давали возможность охладиться до комнатной температуры, после чего охлаждали на бане лед/вода в течение 20 минут. Затем смесь фильтровали через бумагу Whatman no. 3. Фильтрат концентрировали до 1/3 его массы, а затем нагревали до 55°C. Затем pH раствора доводили до pH 7,5 с использованием 50% KOH, после чего раствор охлаждали до температуры окружающей среды, а затем до 0-5°C в бане лед/вода. При охлаждении наблюдали осаждение продукта. Суспензию фильтровали и промывали смесью DIW/EtOH (10 мл, 1:1, 0°C). Белый осадок сушили в течение 12 часов в вакуумной печи (45°C) с получением (S)-прегабалина при выходе 61%, чистоте 98,6% по массе и э.и. 99,8% (предпочтительный S-изомер).
Пример 38
Получение (S)-прегабалина посредством гидролиза 5-бутокси-4-(2-метилпропил)-дигидрофуран-2(3H)-она и ферментативного трансаминирования

5-Бутокси-4-(2-метилпропил)-дигидрофуран-2(3H)-он (3,21 г, 15 ммоль, см. пример 13C) суспендировали в DIW (8,5 г) и охлаждали в бане лед/вода. Водный раствор KOH (50 масс. %, 2,02 г, 1,2 экв.) добавляли по каплям через шприц в течение 5 мин. Реакционную смесь удаляли из бани лед/вода и перемешивали при кт в течение 90 мин. Доводили pH до 7,0, используя муравьиную кислоту. Затем реакционную смесь использовали в качестве сырья в последующей трансаминазной реакции.
Раствор фермента трансаминазы (12,5 г), PLP (35 мг), DIW (15 мл) и 4,0 М изопропиламина в водном растворе HCl (7,5 мл, 30 ммоль) загружали в колбу на 100 мл и подогревали до 45°C. Гидролизную реакционную смесь добавляли одной порцией с последующим добавлением DIW (6 мл, использовали для промывания сосуда). Значение pH реакционной смеси доводили до 7,25 с использованием чистого изопропиламина, и реакционную смесь перемешивали при 45°C.
Реакционную смесь нагревали до внутренней температуры 55°C, и pH доводили до 4,0, используя 95% муравьиную кислоту. Добавляли углерод Darco (125 мг), и смеси давали возможность охладиться до комнатной температуры, после чего охлаждали на бане лед/вода в течение 20 минут. Затем смесь фильтровали через бумагу Whatman no. 3. Фильтрат концентрировали до 1/3 его массы, а затем нагревали до 55°C. Затем pH раствора доводили до pH 7,5 с использованием 50% KOH, после чего раствор охлаждали до температуры окружающей среды, а затем до 0-5°C в бане лед/вода. При охлаждении наблюдали осаждение продукта. Суспензию фильтровали и промывали смесью DIW/EtOH (10 мл, 1:1, 0°C). Белый осадок сушили в течение 12 часов в вакуумной печи (45°C) с получением (S)-прегабалина при выходе 51%, чистоте 98,4% по массе и э.и. 99,9% предпочтительного S-изомера.
Пример 39
Получение (S)-прегабалина из 5-гидрокси-4-(2-метилпропил)-дигидрофуран-2(3H)-она посредством ферментативного трансаминирования
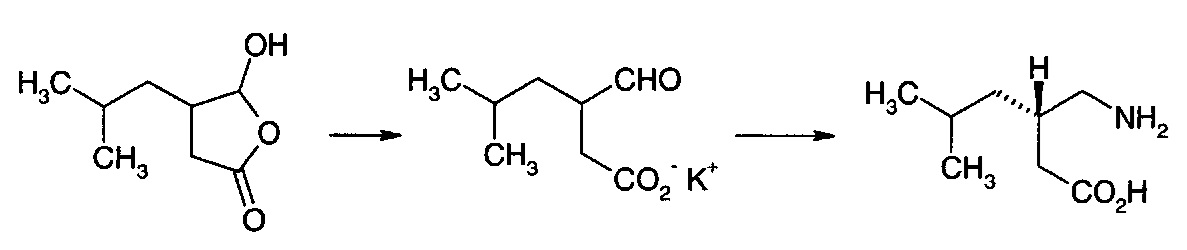
5-Гидрокси-4-(2-метилпропил)-дигидрофуран-2(3H)-он (2,0 г, 12,5 ммоль) суспендировали в DIW (8,5 г) и охлаждали в бане лед/вода. Добавляли порциями карбонат калия (0,863 г, 6,3 ммоль) в течение 5 мин. Реакционную смесь удаляли из бани лед/вода и перемешивали при кт в течение 90 мин. Доводили pH до 7,0 с использованием муравьиной кислоты, а затем реакционную смесь использовали в качестве сырья в последующей трансаминазной реакции.
Раствор фермента трансаминазы (10,4 г), PLP (30 мг), DIW (12,5 мл) и 4,0 М изопропиламина в водном растворе HCl (6,3 мл, 30 ммоль) загружали в колбу на 100 мл и подогревали до 45°C. Гидролизную реакционную смесь добавляли одной порцией с последующим добавлением DIW (5 мл, использовали для промывания сосуда). Значение pH реакционной смеси доводили до 7,25 с использованием чистого изопропиламина, и реакционную смесь перемешивали при 45°C. Реакционную смесь нагревали до внутренней температуры 55°C, и pH доводили до 4,0, используя 95% муравьиную кислоту. Добавляли углерод Darco (125 мг), и смеси давали возможность охладиться до комнатной температуры, после чего охлаждали на бане лед/вода в течение 20 минут. Затем смесь фильтровали через бумагу Whatman no. 3. Фильтрат концентрировали до 1/3 его массы, а затем нагревали до 55°C. Затем pH раствора доводили до pH 7,5 с использованием 50% KOH, после чего раствор охлаждали до температуры окружающей среды, а затем до 0-5°C в бане лед/вода. При охлаждении наблюдали осаждение продукта. Суспензию фильтровали и промывали смесью DIW/EtOH (10 мл, 1:1, 0°C). Белый осадок сушили в течение 12 часов в вакуумной печи (45°C) с получением (S)-прегабалина при выходе 61%, чистоте 98,3% по массе и э.и. 99,9% предпочтительного S-изомера.
Пример 40
Получение (R/S)-3-аминометил-5-метилгексановой кислоты посредством восстановительного аминирования 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-она

0,01 М раствор 5-гидрокси-4-(2-метилпроп-1-ен-1-ил)фуран-2(5H)-она в 30% водном растворе аммиака гидрировали (10 бар (1000 кПа), температура окружающей среды) в течение 48 ч в присутствии 10% по мол. катализатора никеля Ренея. Катализатор отфильтровывали, и раствор концентрировали с получением в остатке твердого вещества. Продукт выделяли путем добавления соляной кислоты к метанольной суспензии, и обнаружили, что он представляет собой чистый гидрохлорид 3-аминометил-5-метилгексановой кислоты.
В результате использования палладия в качестве катализатора получили смеси 3-аминометил-5-метилгексановой кислоты и соответствующего вторичного амина, 3-[(2-карбоксиметил-4-метил-пентиламино)-метил]-5-метил-гексановой кислоты.
Пример 41
Преобразование R-3-аминометил-5-метилгексановой кислоты в 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5H-2-фуранон с использованием трансаминазы

Раствор R-3-аминометил-6-метилгексановой кислоты в деионизированной воде при pH 7,5/45°C перемешивают с лизатом фермента трансаминазы или с цельноклеточным препаратом, содержащим пиридоксальфосфат (PLP) и ацетон. Полученный изопропиламин удаляют посредством вентиляции азотом. Анализ раствора через 24 ч показывает 3-аминометил-5-метилгексановую кислоту с низким э.и. и присутствие соединения 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5H-2-фуранона. Снижение хиральной чистоты 5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5H-2-фуранона является следствием селективного преобразования (4S)-5-гидрокси-4-(2-метилпропил)-3,4-дигидро-5H-2-фуранона в S-прегабалин.
Пример 42
Дерацемизация рацемического прегабалина с использованием трансаминазы

Раствор рацемического прегабалина в DIW при pH 7,5 обрабатывают лизатом подходящей трансаминазы. Добавляют PLP, и реакция протекает в течение 12 ч при 45°C с вентиляцией азотом. Затем добавляют изопропиламин, и реакцию продолжают еще в течение 12 ч. Продукт выделяют, как в примере 38, с получением энантиомерно обогащенного прегабалина.
Возможно также осуществлять превращения, проиллюстрированные в примерах 41 и 42, с использованием подходящей системы ферментов аминоксидазы/иминоредуктазы с кофактором, таким как NADP.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
SEQ ID NO. 1
Аминокислотная последовательность типичной Vfat

X27 выбран из глутамина (Q) и глутаминовой кислоты (E); X41 выбран из изолейцина (I) и валина (V); X45 выбран из аспарагина (N) и гистидина (H); X147 выбран из аспарагина (N) и глутамина (Q); X163 выбран из лейцина (L) и метионина (M); X165 выбран из тирозина (Y) и гистидина (H); X180 выбран из треонина (T); глицина (G) и серина (S); X304 выбран из аланина (A) и серина (S); X324 выбран из глицина (G) и серина (S); X401 выбран из лизина (K) и глутаминовой кислоты (E); X408 выбран из треонина (T) и глутамина (Q); X415 выбран из серина (S) и аланина (A); X416 выбран из пролина (P) и аланина (A); X417 выбран из лейцина (L) и метионина (M); и X424 выбран из цистеина (C) и серина (S).
SEQ ID NO. 2
Аминокислотная последовательность Vfat888

SEQ ID NO. 3
Аминокислотная последовательность Vfat906

SEQ ID NO. 4
Аминокислотная последовательность Vfat999

SEQ ID NO. 5
Аминокислотная последовательность Vfat1010

SEQ ID NO. 6
Аминокислотная последовательность Vfat1020

SEQ ID NO. 7
Аминокислотная последовательность Vfat1030

SEQ ID NO. 8
Последовательность белка 12-оксофитодиеноатредуктазы 1 Lycopersicon esculentum (томата):

SEQ ID NO. 9
Последовательность 12-оксоФитодиеноатредуктазы 1 Lycopersicon esculentum (томата), оптимизированная по кодонам:

SEQ ID NO. 10
Последовательность ДНК Vfat 888

SEQ ID NO. 11
Последовательность ДНК Vfat 906


SEQ ID NO. 12
Последовательность ДНК Vfat 999


SEQ ID NO. 13
Последовательность ДНК Vfat 1010


SEQ ID NO. 14
Последовательность ДНК Vfat 1020

SEQ ID NO. 15
Последовательность ДНК Vfat 1030

Реферат
Изобретение относится к способу получения соединения формулы (I) и промежуточным соединениям, используемым для осуществления способа, а также к способу получения прегабалина. Способ может быть осуществлен в коммерческом масштабе с использованием легкодоступных, безопасных исходных веществ и реагентов и при отсутствии необходимости в трудных разделениях. 8 н. и 12 з.п. ф-лы, 8 табл., 41 пр.
Формула
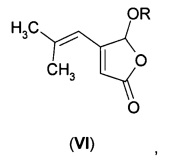
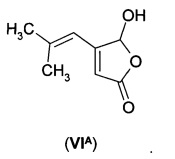



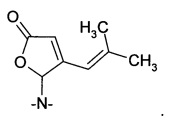


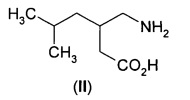

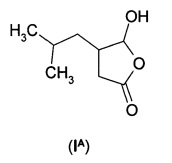


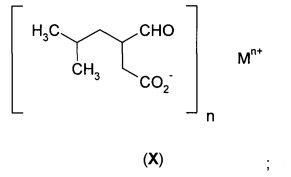
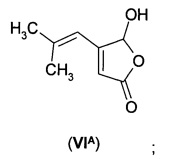

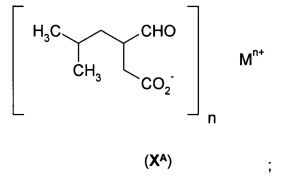

Комментарии