Разделение энантиомеров 3-этилбицикло [3.2.0] гепт-3-ен-6-она - RU2734475C2
Код документа: RU2734475C2
Описание
Область технического применения
Настоящее изобретение относится к способу получения оптически активного промежуточного соединения, полезного для получения оптически активного бициклического производного γ-аминотетразола или его фармакологически приемлемой соли, особенно соединения, имеющего активность в качестве лиганда α2δ, а также для получения производного тетразола.
Предпосылки
Каналы кальциевого типа с формированием напряжения формируются комбинациями порообразующей α1 субъединицы и вспомогательных белков α2δ, β и γ (Lateral (2000) Annu. Rev. Cell Dev. Biol. 16: 521-555). Известно, что белок α2δ регулирует плотность как кальциевых каналов, так и зависящую от напряжения кинетику этих каналов (Felix et al., 1997) J. Neuroscience 17: 6884-6891, Klugbauer et al (1999) J. Neuroscience 19: 684-691, Hobom et al (2000) Eur. J. Neuroscience 12: 1217-1226; и Qin et al (2002) Mol. Pharmacol. 62: 485-496).
Габапентин (GBP) является противоэпилептическим, противогипергенным и анксиолитическим препаратом, который связывается с высоким сродством с двумя подтипам субъединиц α2δ кальциевого канала α2δ1 и α2δ2. GBP был первоначально разработан для лечения эпилепсии и нашел применение для лечения боли и беспокойства (Taylor et al (1998) Epilepsy Res. 29: 223-249). Механизм, лежащий в основе действия GBP, до сих пор плохо изучен. GBP был первоначально разработан как липофильный аналог γ-аминомасляной кислоты (ГАМК), но впоследствии было показано, что он не взаимодействует c каким-либо из ферментов на метаболическом пути ГАМК и не взаимодействует непосредственно с GABAA или GABAB рецепторами. Тем не менее, он способен эффективно пересекать гематоэнцефалический барьер через L-системный аминокислотный транспортер. Прегабалин (PGB) является более мощным преемником GBP второго поколения для лечения тех же состояний, которые перечислены выше. GBP (структура GBP ниже) и PGB (структура PGB ниже) связываются с субблоком α2δ-1 с IC50 значениями 140 и 80 нМ, соответственно (Dolphin (2013) Bioch Biophys Acta 1828: 1541-1549).
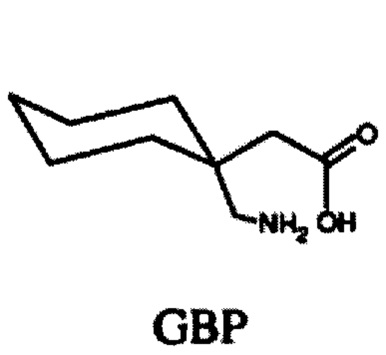
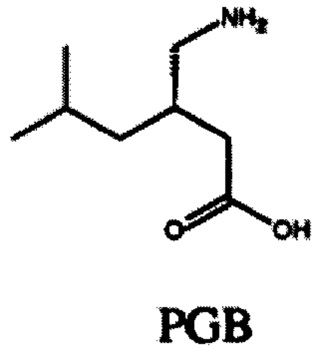
GBP имеет несколько, если таковые имеются, токсичных побочных эффектов в клинически значимых дозах. Однако он обладает относительно коротким периодом полураспада, выводясь из организма без изменений, возможно, из-за очень высокой растворимости в воде и очевидного отсутствия связывания белка in vivo. Слабые седации, головокружение и атаксия являются основными дозозависимыми побочными эффектами, и они, как полагают, являются централизованно опосредованными.
GBP и PGB, в отличие от многих других центрально действующих препаратов, являются гидрофильными и двузарядными при нейтральном рН, что делает их нерастворимыми в липидах, таких как клеточные мембраны. Однако оба соединения, по-видимому, пересекают мембранные барьеры кишечника, гематоэнцефалический барьер и клеточные мембраны через специализированную транспортную систему (система L), которая также переносит эндогенные аминокислоты, такие как L-лейцин, L-изолейцин и L-валин (Su et al (2005) J. Pharm. Exp. Ther. 313,1-10).
У млекопитающих существуют четыре связанных подтипа белка α2δ, каждый из которых кодируется другим геном. Каждый подтип белка имеет молекулярную массу приблизительно 150 кДа (кД) и состоит из 997-1150 аминокислотных остатков. Только α2δ подтипов 1 и 2 связывают PGB с высоким сродством; подтипы 3 и 4 лишены значительного связывания лекарственных средств (Fink et al., 2002) Neuropharmacology, 42, 229-236). Аффинность связывания PGB аналогична для рекомбинантных белков α2δ типа 1 и типа 2, демонстрируя, что PGB не является селективным типом (Piechan et al., 2004) Soc. Neuroscience Abstr., 111 (программа №115)).
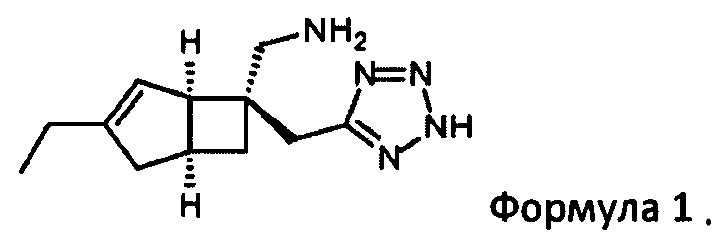
В WO 2015/091463, среди прочего, раскрывается польза бициклических производных γ-аминотетразола формулы 1
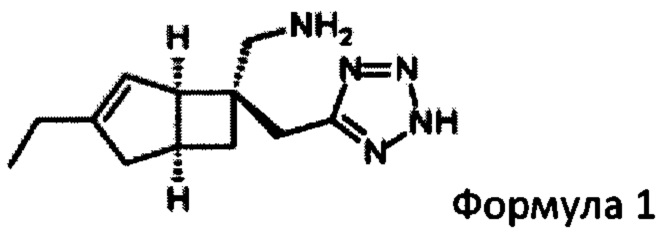
для лечения боли и способы их производства.
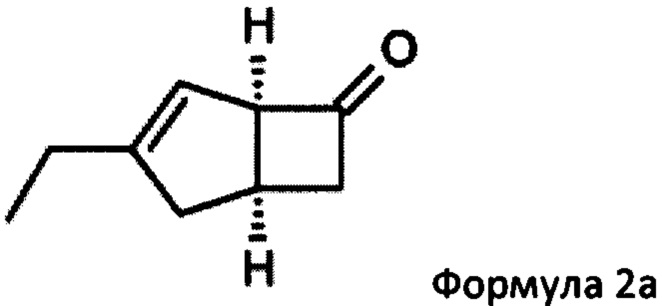
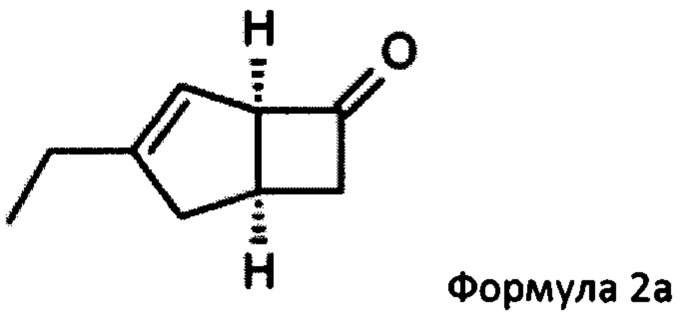
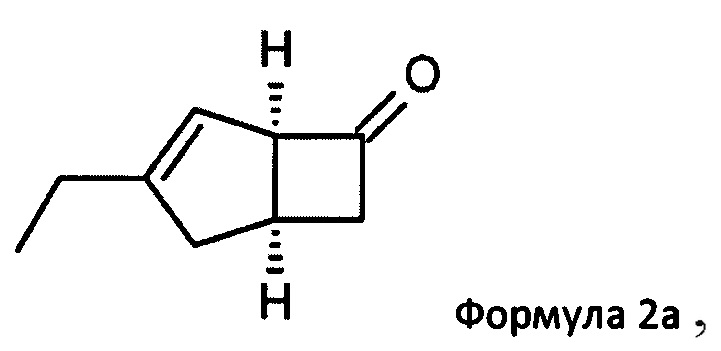
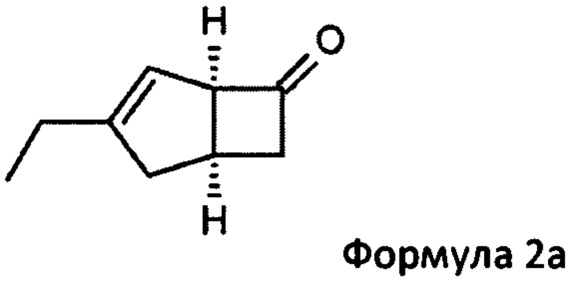
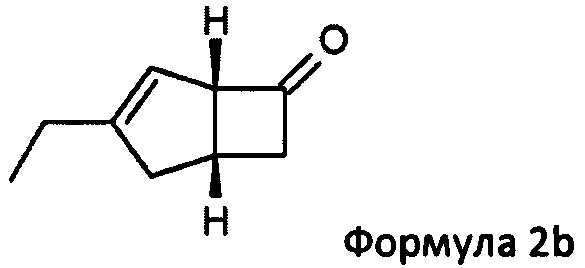
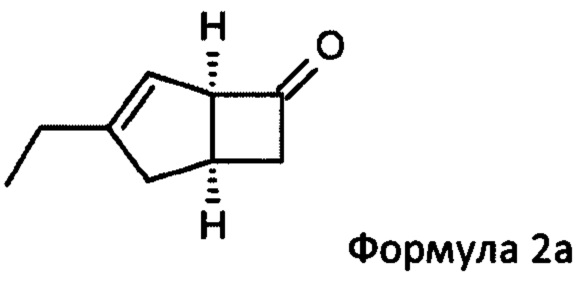
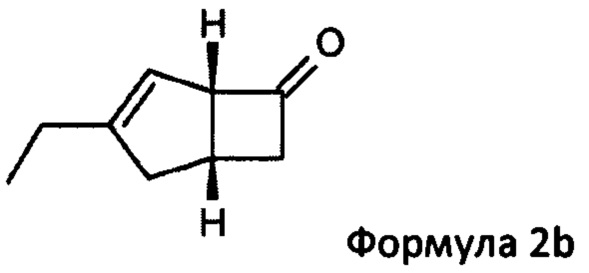
US 2012/0071685 относится к производству бициклических производных γ-аминокислот, обладающих активностью в качестве α2δ-лиганда и его промежуточных продуктов, включая синтез диастереомерной смеси соединений формул 2а, 2b.
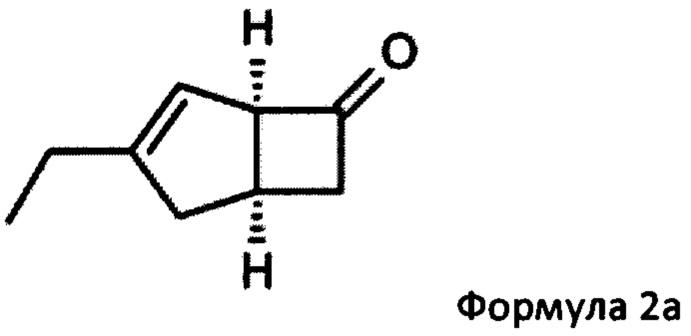
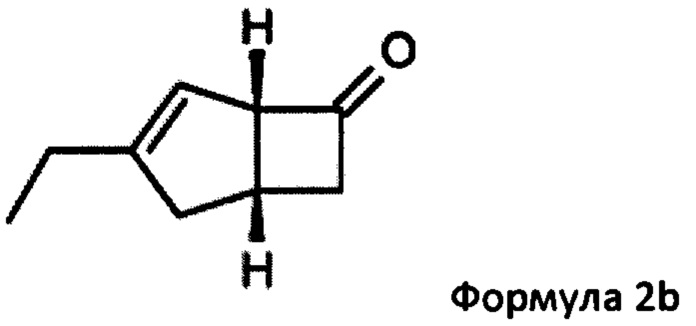
Однако эта диастереоизомерная смесь 2а, 2b не разлагается на отдельные изомеры.
В US 2014/0094623 описан трехстадийный способ получения соединения формулы А и соединения формулы В из их смеси
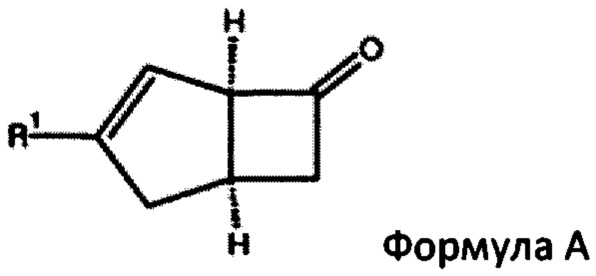
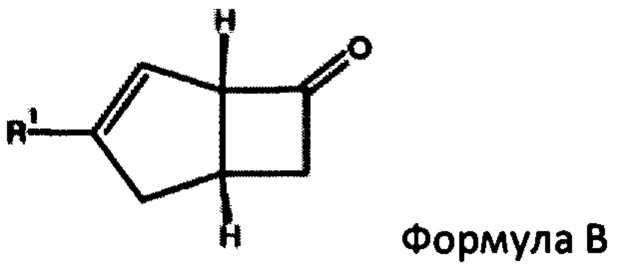
Путем (а) взаимодействия бис-аллилового ацеталя с кислотным или кислотным ангидридом и кислотой с получением альдегида путем перегруппировки Клайзена, (б) нагревания продукта стадии (а) с помощью малоновой кислоты с получением альфа-бета-ненасыщенной кислоты и (с) нагревания продукта стадии (b) ангидридом кислоты и третичным амином для получения 4-5 бициклической кольцевой системы с помощью реакции [2+2] циклоприсоединения. Выделение вышеуказанных соединений формулы А или формулы В из их диастереоизомерной смеси также описано в US 2015/0038738, в котором раскрыт ферментативный способ отделения соединений, и в US 2014/0296569, который использует реакцию диастереоизомерной смеси с кислотным бензальдегидным реагентом, оптически активным амином и растворителем.
Краткое описание изобретения
(I) Техническая проблема
Целью настоящего изобретения является создание способа получения бициклического производного
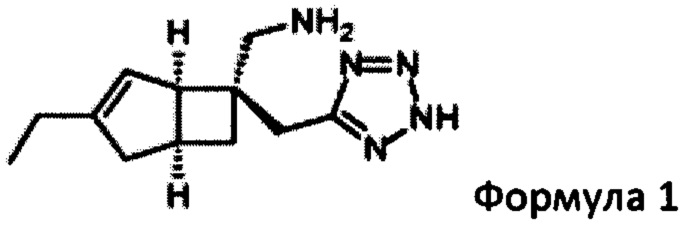
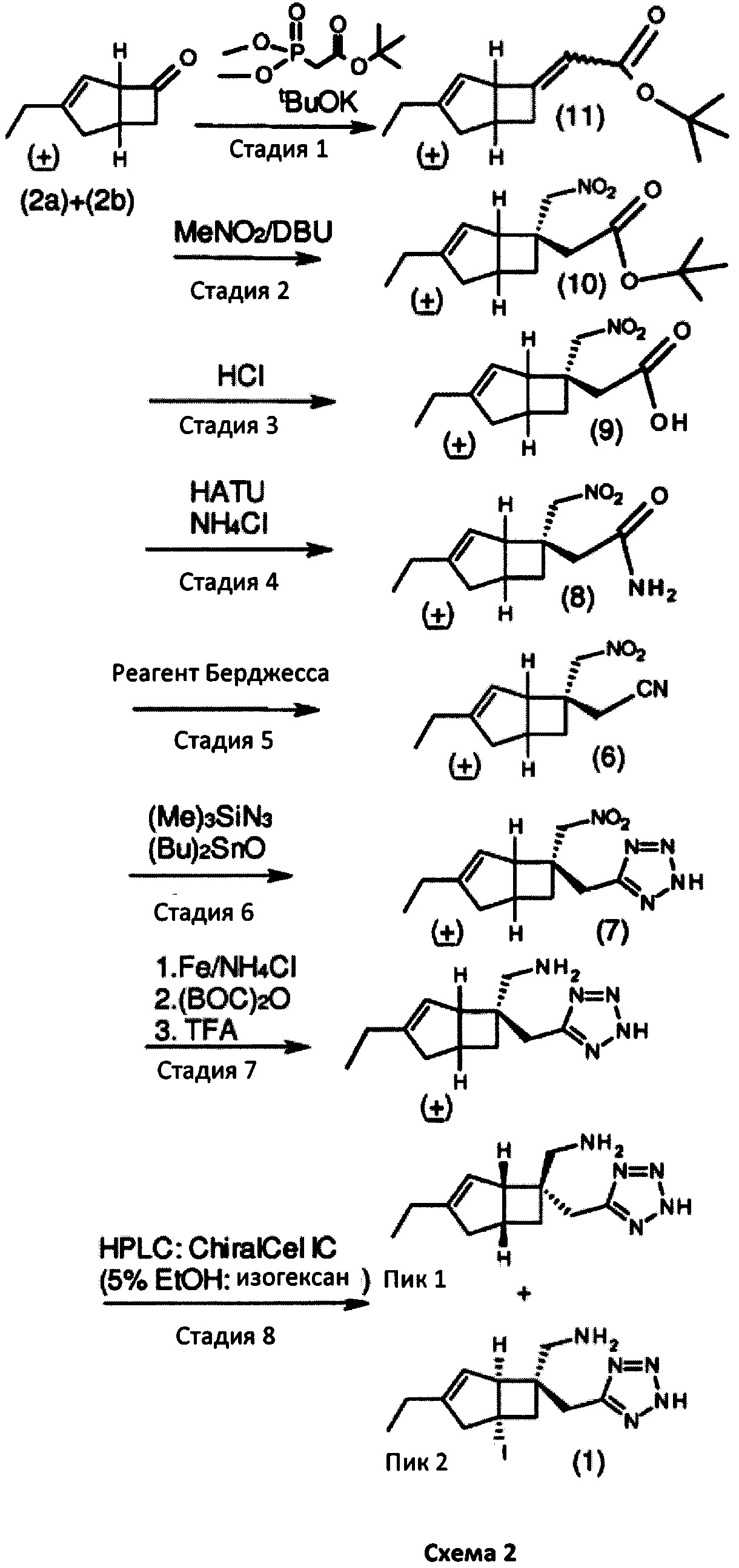
имеющего превосходную активность в качестве α2δ-лиганда и промежуточного соединения для получения того же соединения и его фармакологически приемлемых солей. В предыдущем способе получения соединение формулы 1 было получено с помощью семи стадий синтеза из рацемического кетона формул 2а и 2b с дополнительным оптическим разрешением, выполненным в качестве конечной стадии, с использованием хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ), как показано на схеме 1 (см. WO 2015/091463 и US 2012/0071685).
(II) Решение проблемы
Технической задачей, которая должна быть решена в соответствии с настоящим изобретением, является разработка способа производства, который включает получение промежуточного соединения при получении соединений формулы 1 в качестве оптически активного соединения на более ранней стадии с последующим меньшим синтетическим превращением при получении соединения формулы 1.
Однако авторы настоящего изобретения предположили, что более эффективный способ производства будет устанавливаться путем осуществления оптического разрешения на более ранней стадии и использования последующих синтетических превращений, которые более эффективно производят соединение формулы 1. Таким способом можно получить соединение формулы 1 с помощью четырех общих стадий синтеза.
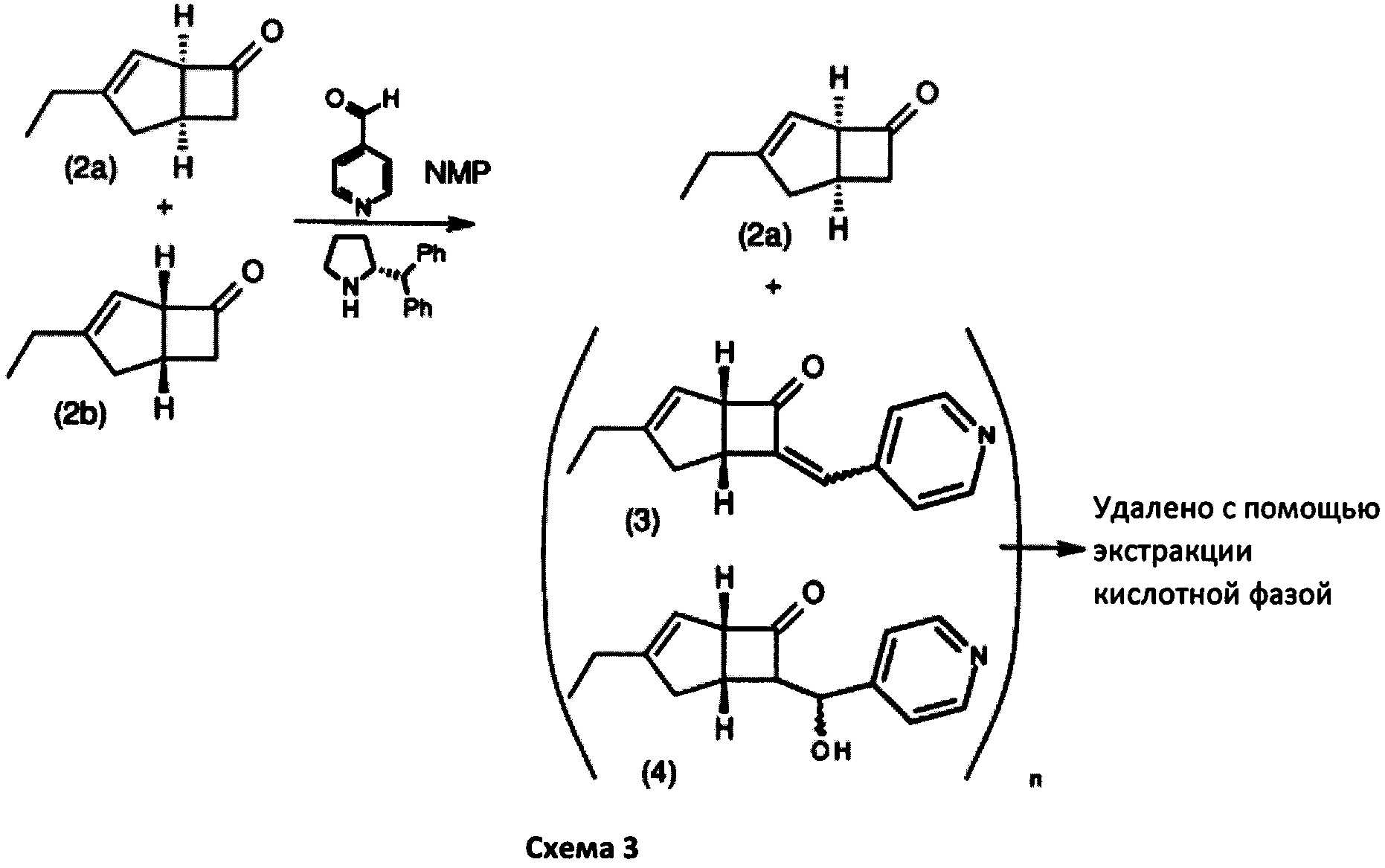
Изобретение будет описано ниже. Способ дает соединение формулы 1 или его соль с помощью оптического кинетического разрешения, выполняемого на ранней стадии синтетической последовательности. Сосредоточив внимание на методе стереоконтроля для асимметричного углерода в способе получения соединения формулы 2а, авторы настоящего изобретения продолжали прилежные исследования для разработки собственного эффективного способа. Авторы настоящего изобретения обнаружили, что такой способ получения соединения формулы 2а имеет важное значение для получения соединения формулы 1, как показано на схеме 2.
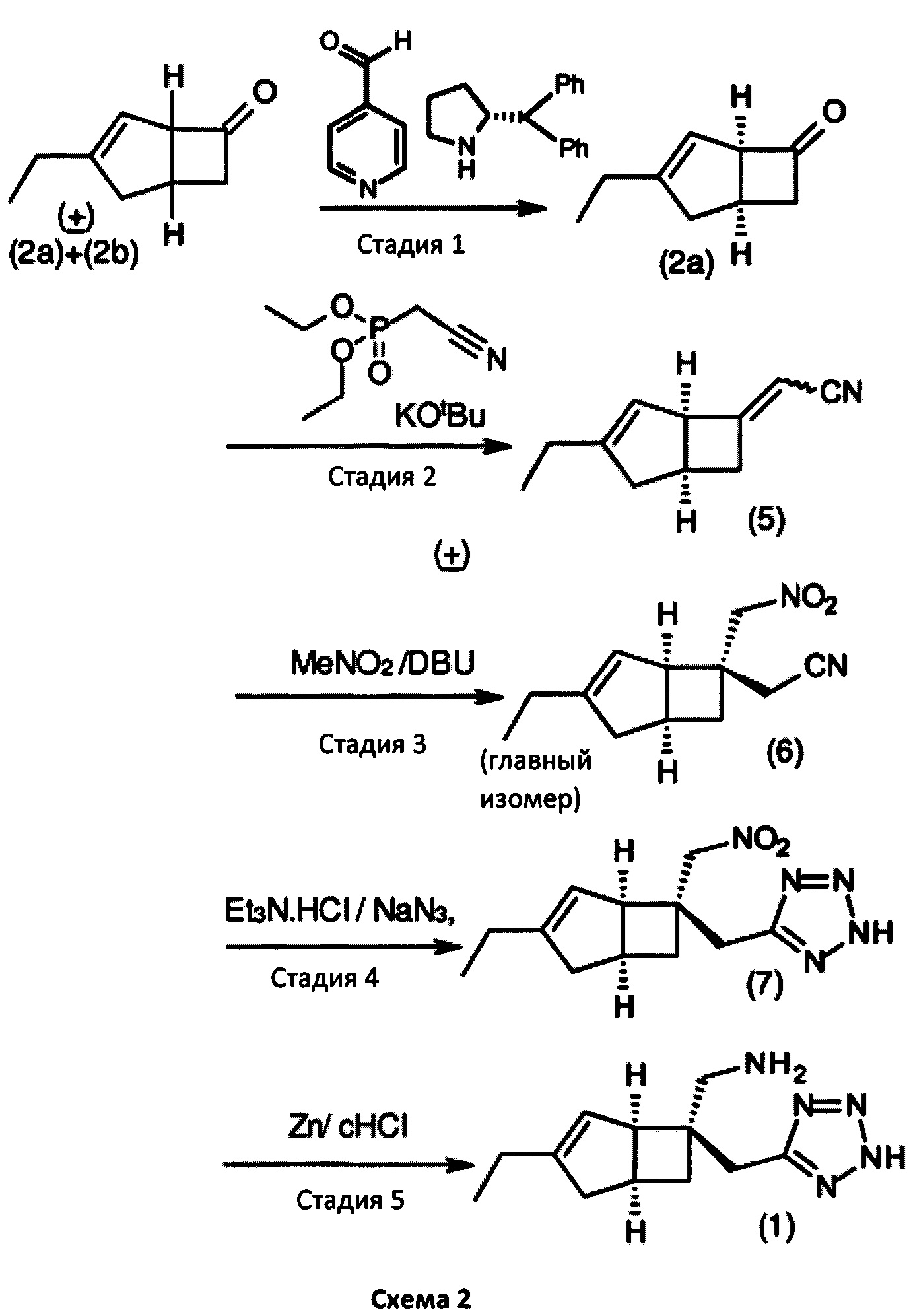
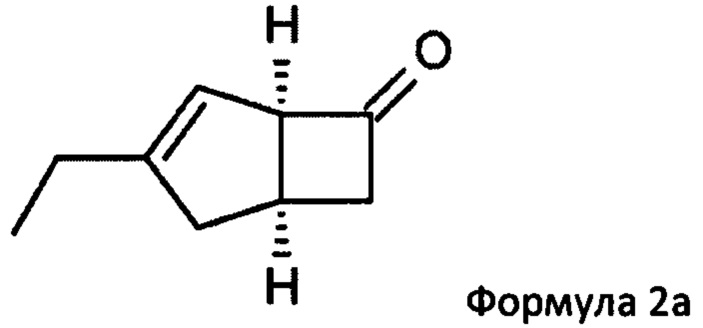
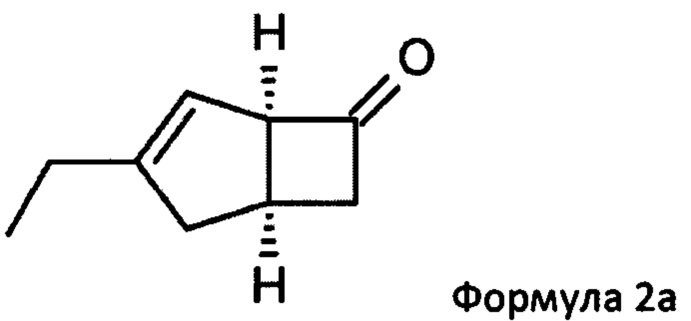
Соответственно, настоящее изобретение относится к способу выделения соединения формулы 2а
или его соли или сольвата, включая а) взаимодействие смеси диастереоизомеров формул 2а, 2b
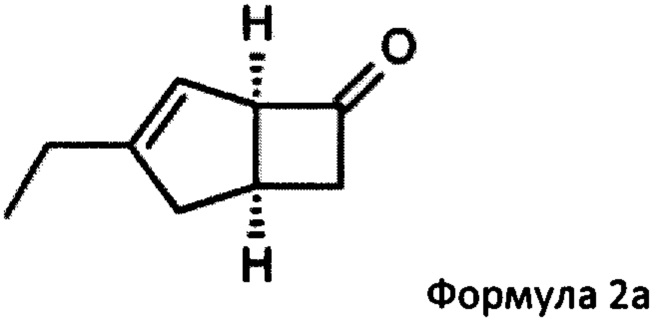
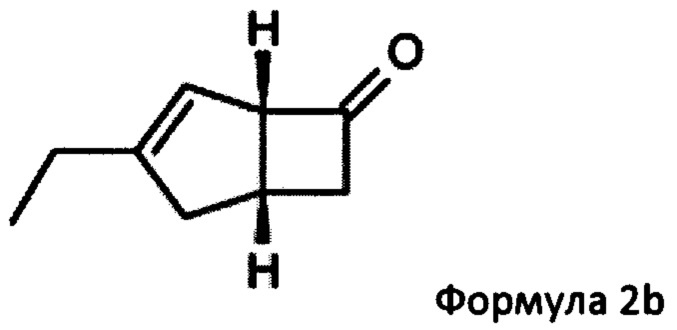
с основным гетероциклическим альдегидным соединением в присутствии оптически активного амина и основания; а также б) отделение соединения формулы 2а от продукта стадии а) путем экстракции кислотой.
Способ может приводить к получению соединения формулы 2а с энантиомерным избытком, превышающим 90%, предпочтительно более 95% или даже 98%.
Предпочтительные аспекты настоящего изобретения будут описаны ниже. Способ включает взаимодействие смеси диастереоизомеров формул 2а и 2b,
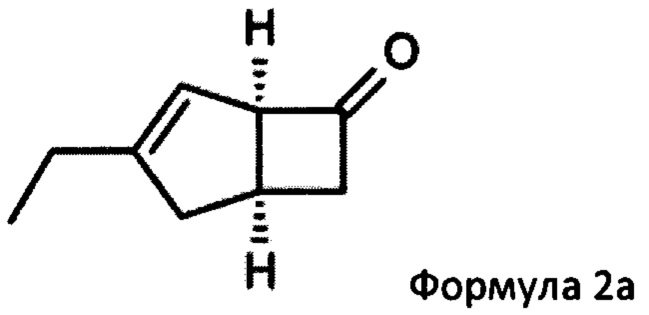
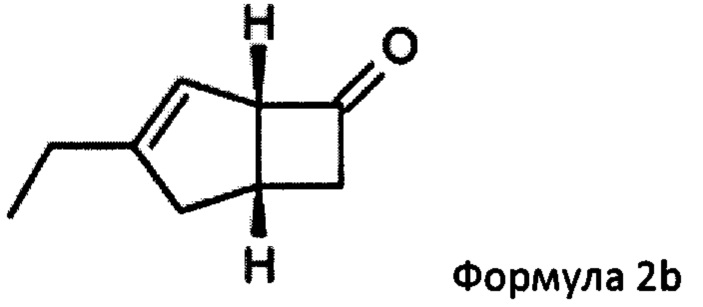
особенно рацемической смеси соединений формул 2а и 2b с основным гетероциклическим альдегидным соединением в присутствии оптически активного амина и основания. В способе согласно изобретению считается, что соединение формулы 2b реагирует с основным гетероциклическим альдегидным соединением в присутствии оптически активного амина с образованием гетероциклических арильных производных, тогда как соединение формулы 2а остается непрореагировавшим. Соединение формулы 2а легко может быть разделено обычными методами кислотной экстракции.
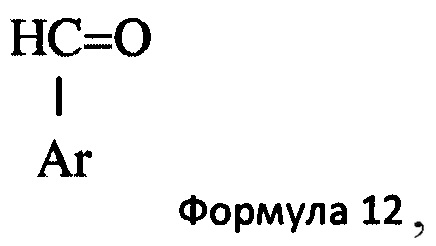
Основное гетероциклическое альдегидное соединение предпочтительно представляет собой соединение формулы 12,
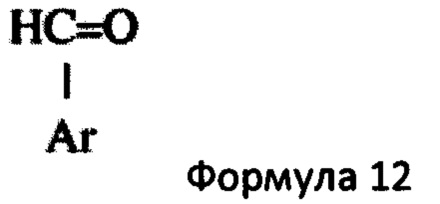
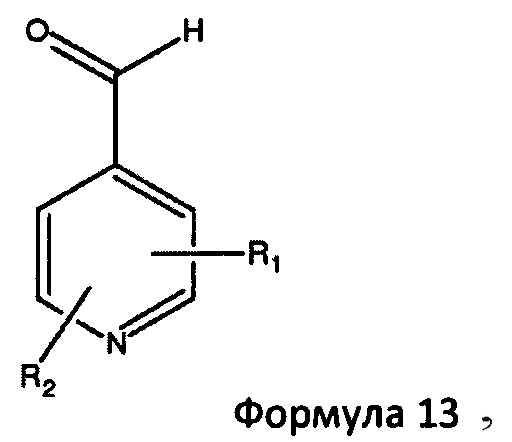
в которой Ar представляет собой гетероциклическую 5- или 6-членную гетероарильную кольцевую структуру, необязательно замещенную одним или двумя заместителями, выбранными из С1-6алкила и С1-6алкокси. Предпочтительно Ar представляет собой имидазолильную или пиридильную кольцевую структуру. Типичные примеры включают, но не ограничиваются, такие соединения, как 1-метил-5-имидазолкарбоксальдегид, 1-метил-4-имидазолкарбоксальдегид, 3-пиридинкарбоксальдегид и 2-пиридинкарбоксальдегид. Более предпочтительно, основное гетероциклическое альдегидное соединение представляет собой соединение формулы 13,
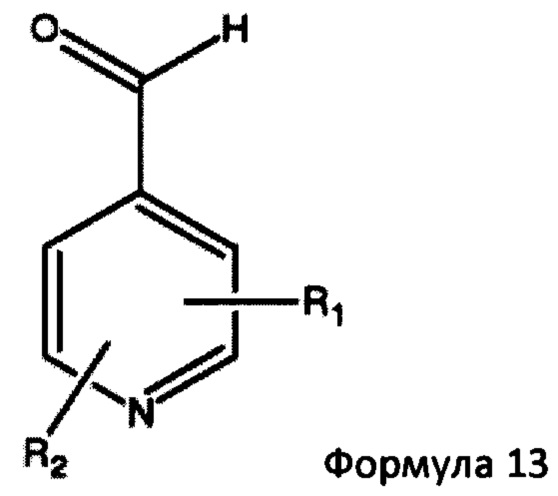
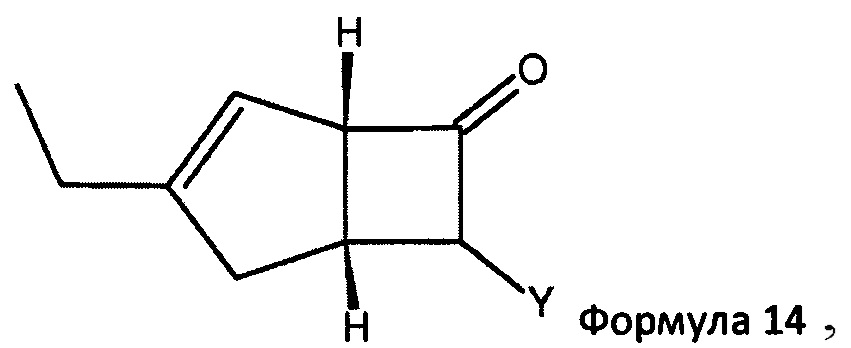
в которой R1 представляет собой водород или С1-6 алкил и R2 представляет собой водород или С1-6 алкокси, такой как 4-пиридинкарбоксиальдегид. Когда реакцию проводят с соединением формулы 13, соединение формулы 2b превращают в аддукты, обозначенные смесью соединений формулы 14 (изображенных формулами 3 и 4 на схеме 3 ниже).
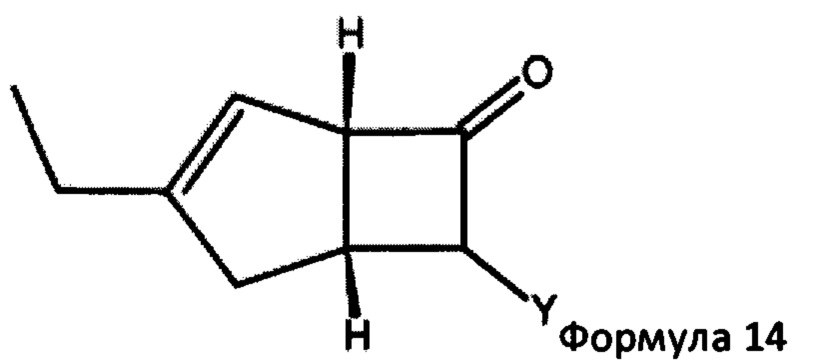
в которой Y представляет собой = СН-(4-пиридил) или -СН(ОН)-(4-пиридил).
После этого соединения формулы 14 могут быть эффективно отделены от непрореагировавшего соединения формулы 2а методами кислотного разделения фаз, как показано на схеме 3. Процесс разделения удобно проводить с соединениями формулы 13, поскольку основная пиридиновая функциональность облегчает разделение, особенно в случае, когда оптически активный амин содержит (R)-2-(дифенилметил) пирролидин, а основание включает 4-метилморфолин.
Предпочтительно, в способе согласно настоящему изобретению стехиометрическое соотношение основного гетероциклического альдегидного соединения к смеси диастереоизомеров формул 2а, 2b находится в диапазоне от 0,5:1 до 2:1, более предпочтительно в соотношении 1:1. Как правило, реакцию между смесью диастереоизомеров формул 2а, 2b, основным гетероциклическим альдегидным соединением и оптически активным амином проводят в течение периода времени до 24 часов, обычно в интервале от 15 до 24 ч, при температуре в диапазоне от окружающей среды до 80°С, более конкретно от 30 до 60°С. В предпочтительном варианте осуществления реакцию проводят при 35-45°С в течение периода от 15 до 20 часов.
Соответственно, оптически активный амин представляет собой вторичный амин. Типичные примеры оптически активного амина включают, но не ограничиваются, (R, R)-2,5-бис (метоксиметил) пирролидин, (R)-(2-пирролидинил)-1Н-тетразол, (R)-2-(метоксиметил) пирролидин, (R)-2-(этоксиметил) пирролидин, (R)-2-(изопропоксиметил) пирролидин, (R)-2-(трет-бутоксиметил) пирролидин, (R)-2-(феноксиметил) пирролидин, (R)-2-(дифенилметил) пирролидин, N-[(2R)-2-пирролидинилметил]-рифторметансульфонамид, (R)-2-[бис (4-метилфенил) метил] пирролидин, (R)-2-[бис (3, 5-диметилфенил) метил] пирролидин, (R)-2-[бис (4-фторфенил) метил] пирролидин и (S)-4,5-дигидро-3Н-динафто [2,1-с: 1',2'-е] азепин-2,6-диилбис (дифенилметанол). Предпочтительные оптически активные амины включают (R)-2-(дифенилметил) пирролидин, (R)-2-[бис (4-метилфенил) метил] пирролидин, (R)-2-(бис (3,5-диметилфенил) метил] пирролидина и (R)-2-[бис (4-фторфенил) метил] пирролидина. Особенно предпочтительным оптически активным амином является (R)-2-(дифенилметил) пирролидин.
Предпочтительно стехиометрическое соотношение оптически активного амина к смеси диастереоизомеров формул 2а, 2b находится в интервале от 0,01:1 до 1:1, более предпочтительно в диапазоне 0,01:0,3.
Типичные примеры основания включают, но не ограничиваются, 4-метилморфолин, N, N-диизопропилэтиламин, триметиламин, трибутиламин, N-метилпиррол, N-метилпиролидин, N-метилпиперадин, пиридин, 4-пиколин, 2,6-лутидин, N-метилимидазол, N, N-диэтиланилин, фосфат калия, 1,8-диазобицикло [5,4,0] ундец-7-ен и 1,4-диазобицикло [2,2,2] октан. Предпочтительным основанием является 4-метилморфолин.
Предпочтительно стехиометрическое отношение основания к смеси диастереоизомеров формул 2а, 2b находится в диапазоне от 0,5:1 до 1:1,5, более предпочтительно в диапазоне от 0,8:1 до 1:1,3.
Реакцию можно преимущественно проводить в присутствии растворителя, в частности полярного растворителя. Типичные примеры включают, но не ограничиваются, 1-метил-2-пирролидинон, N, N-диметилформамид, N, N-диметилацетамид, ацетонитрил, 2-пропанол, тетрагидрофуран, 1,2-иметоксиэтан и диметилсульфоксид. Особенно предпочтительным растворителем является 1-метил-2-пирролидинон.
В еще одном предпочтительном аспекте настоящего изобретения основное гетероциклическое альдегидное соединение, основание и оптически активный амин отделяют от непрореагировавшего соединения формулы 2а путем экстракции кислотной фазой. В особенно предпочтительном аспекте настоящего изобретения 4-метилморфолин (основание), любой непрореагировавший 4-пиридинкарбоксиальдегид (основной гетероциклический альдегид) и оставшийся (R)-2-(дифенилметил) пирролидин (оптически активный амин) отделяют от непрореагировавшего соединения формулы 2а путем экстракции кислотной фазой. Как правило, экстракцию кислотой проводят с помощью водного раствора кислоты, например, соляной кислоты, серной кислоты, фосфорной кислоты, лимонной кислоты, щавелевой кислоты или винной кислоты при температуре окружающей среды. Соответственно, экстракцию кислотой проводят соляной кислотой или серной кислотой в присутствии растворителя и воды. Примеры растворителя включают диэтиловый эфир, трет-бутилметиловый эфир, этилацетат, 2-метилтетрагидрофуран, изогексан, дихлорметан. После перемешивания и для разделения фаз собирают фазу, содержащую соединение формулы 2а. В предпочтительном варианте осуществления, реакционная смесь промывается соляной кислотой (обычно 1М), водой, соленой водой и диэтиловым эфиром. Водный слой можно дополнительно промыть изогексаном и другим соединением.
Предпочтительный вариант осуществления изобретения может быть изображен на схеме 3 ниже.
В следующем аспекте изобретения предлагается способ получения соединения формулы 1
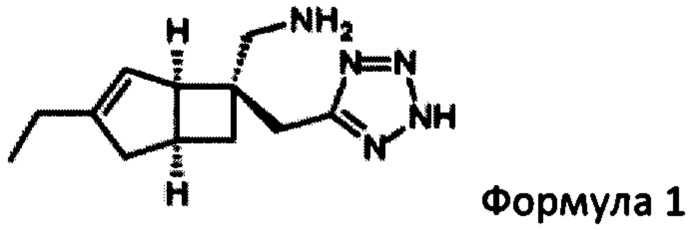
или его пролекарства, включая стадию разделения смеси диастереоизомеров формул 2а, 2b
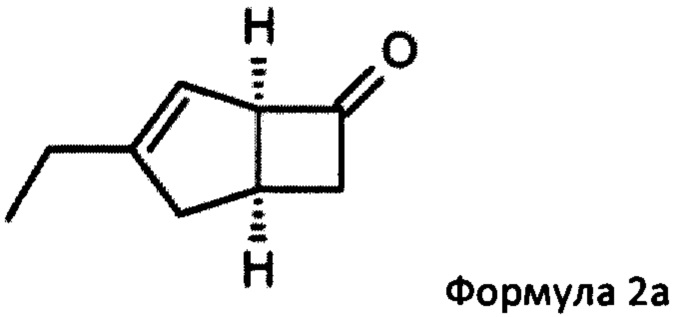
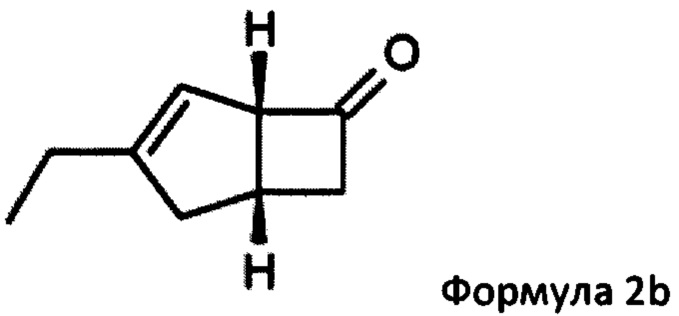
в состав формулы 2а
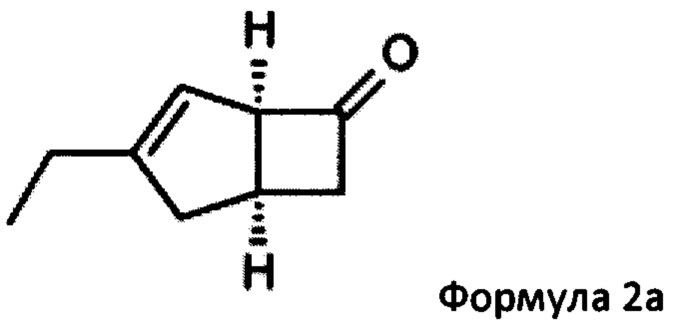
путем взаимодействия с основным гетероциклическим альдегидным соединением в присутствии оптически активного амина и основания, и отделения соединения формулы 2а с последующим превращением соединения формулы 2а в соединение формулы 1.
В предпочтительном способе осуществления указанное превращение включает взаимодействие соединения формулы 2а
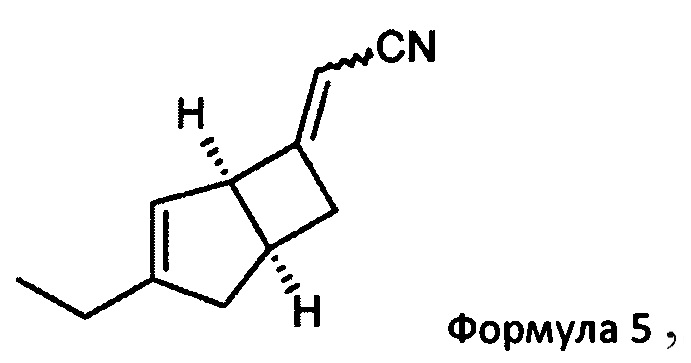
с двойным связующим реагентом в присутствии основания для получения соединения формулы 5
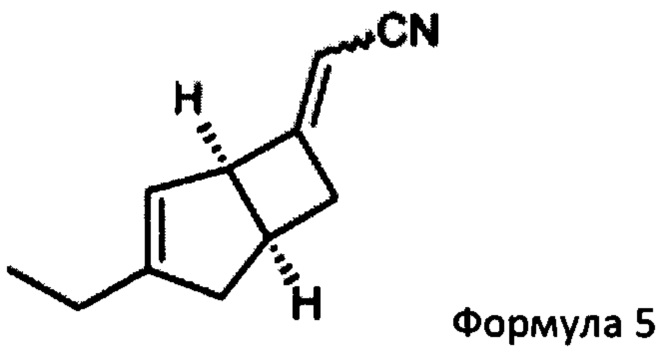
Предпочтительно, подходящий двойной связывающий агент включает, например, диэтилцианометилфосфат, и подходящее основание включает, например, трет-бутоксид калия. Реакцию удобно проводить в подходящем растворителе, например тетрагидрофуране, при температуре в диапазоне от 0°С до температуры окружающей среды.
Указанное превращение может дополнительно включать взаимодействие соединения формулы 5
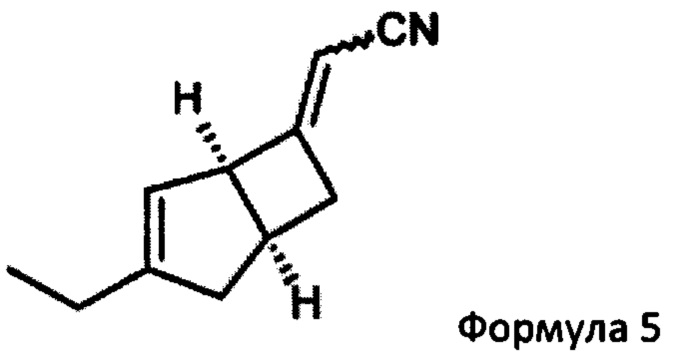
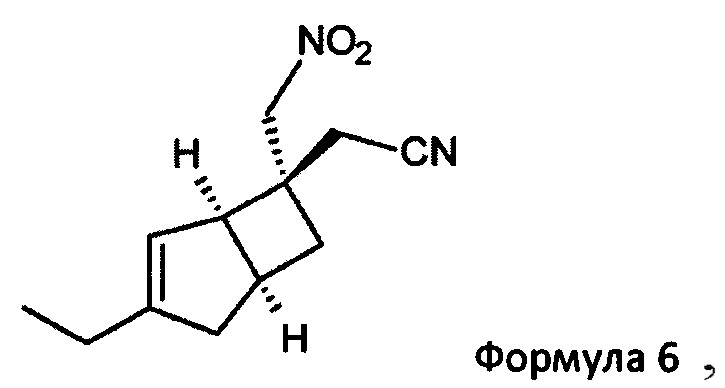
с нитрометаном в присутствии основания для получения соединения формулы 6
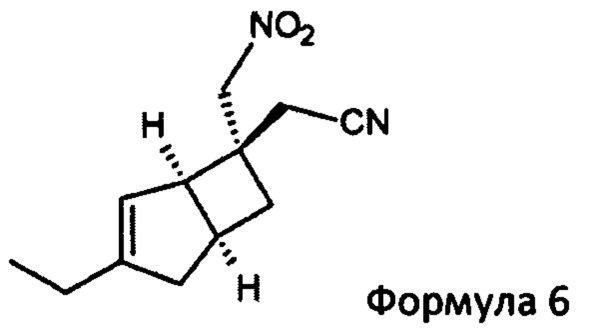
Указанное превращение может дополнительно включать взаимодействие соединения формулы 6
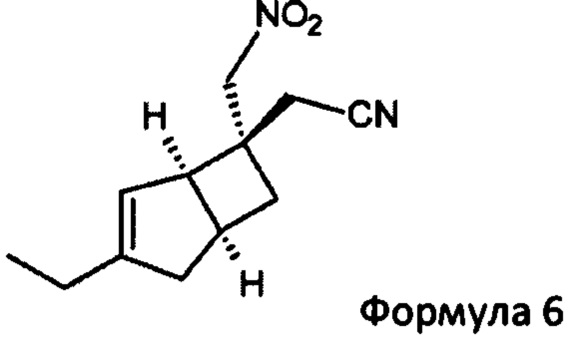
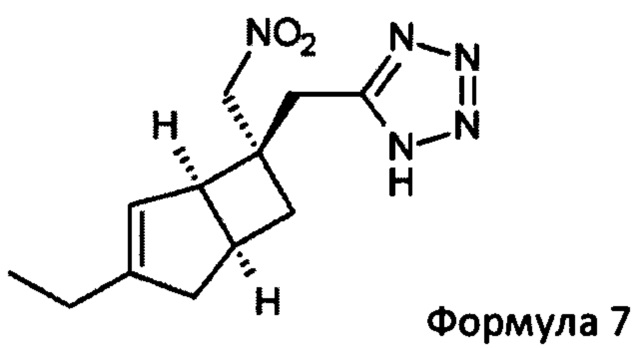
с азидным соединением, например, реакция образования кольца, необязательно в присутствии катализатора и при повышенной температуре, с получением соединения формулы 7
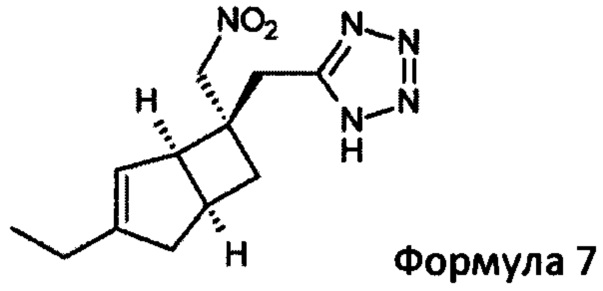
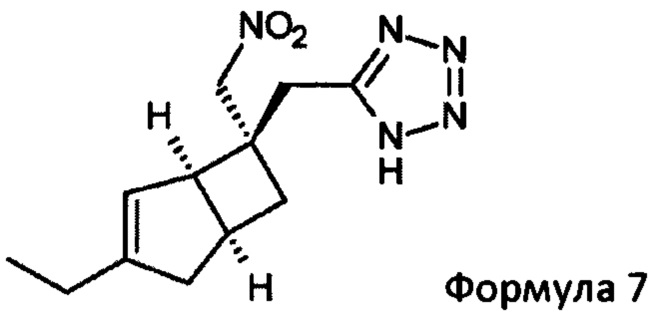
Подходящим азидом может быть, например, натрий-, калий- или триметилсилилазид вместе с общеизвестными подходящими катализаторами, такими как дибутилоловооксид, гидрохлорид пиридина, гидрохлорид триэтиламина или хлорид аммония в растворителе, таком как диметилформамид (ДМФ), N-метил-2-пирролидинон (NMP) или толуол. Реакцию можно проводить при повышенных температурах в интервале от 60 до 120°С
Указанное превращение может дополнительно включать восстановление соединения формулы 7
для получения соединения формулы 1
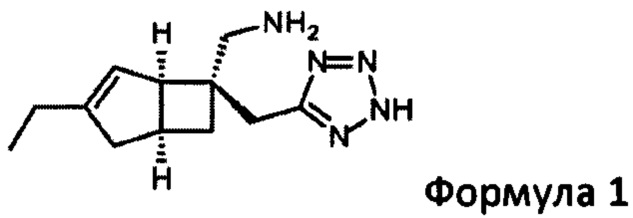
Подходящие восстанавливающие агенты включают использование смеси подходящих металлов, таких как цинк, железо или олово, в присутствии подходящей кислоты и растворителя, таких как хлористоводородная кислота, серная кислота, уксусная кислота или хлорид аммония в воде или этаноле при 0°С до температуры окружающей среды. Альтернативно, подходящие восстановители включают гидразингидрат в присутствии металла, такого как никель Ренея, в подходящем растворителе, таком как смеси этанола и воды. Предпочтительный вариант осуществления включает использование цинка и концентрированной соляной кислоты при температурах между 0°С и 35°С.
Преимущественно, пролекарство может быть гидролизуемым карбаматом аминогруппы соединений формулы 1.
В следующем аспекте настоящее изобретение относится к способу получения соединений формулы 14
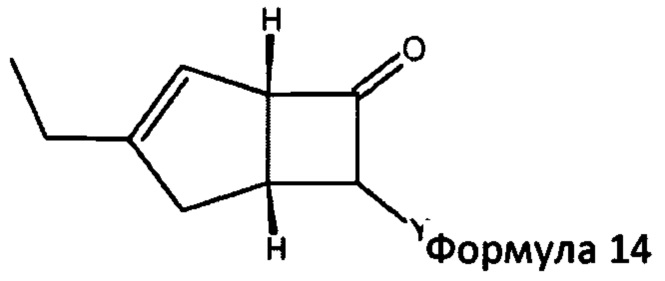
в которой Y = СН-(4-пиридил) или -СН(ОН)-(4-пиридил), включающий обработку смеси диастереоизомеров формул 2а, 2b
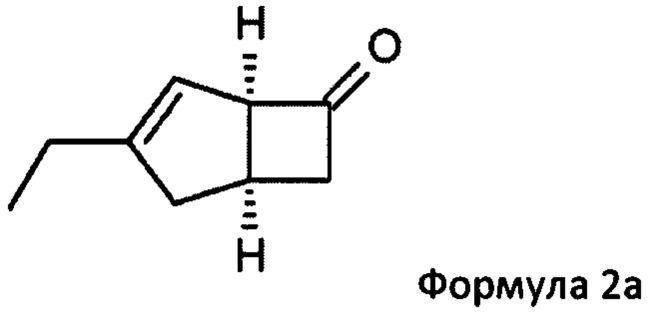
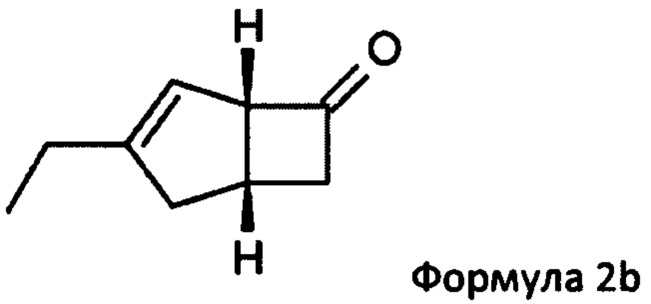
с основным гетероциклическим альдегидным соединением формулы 13
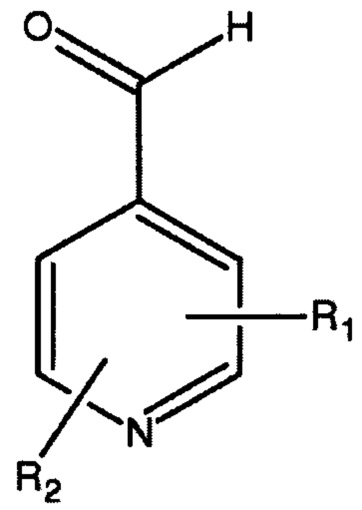
как описано выше
в присутствии оптически активного амина и основания.
Выгодные эффекты изобретения
Способ получения в соответствии с настоящим изобретением может обеспечить бициклическое производное γ-аминотетразола формулы 1, имеющее превосходную активность в качестве α2δ-лиганда, промежуточного соединения для получения того же или его соли. Кроме того, основные гетероциклические альдегиды, такие как 4-пиридинкарбоксальдегид, реагируют с нежелательным изомером формулы 2b с получением основных производных, например формулы 14, в которой Y = СН-(4-пиридил) или -СН(ОН)-(4 пиридил), которые эффективно удаляются с помощью методов разделения кислых фаз с получением желаемого соединения формулы 2а с энантиомерным избытком (э. и.) ≥98, как показано на схеме 3. Последующая модификация с помощью четырех стадий синтеза обеспечивает соединение формулы 1, представленное на схеме 2.
Описание
Используемый здесь термин «соединение формулы 1» включает ее фармацевтически приемлемые соли и сольваты. Ссылки на промежуточные соединения также включают соли и сольваты. Фармацевтически приемлемые соли соединений по изобретению могут включать основные аддитивные соли соединения. Такие соли могут быть образованы с неорганическим основанием, которое дает фармацевтически приемлемый катион, например соль щелочного металла, такую как натриевая или калиевая соль, или соль щелочноземельного металла, такую как соль кальция или магния. Фармацевтически приемлемые соли по изобретению могут также включать кислотно-аддитивные соли. Такие соли могут быть образованы с неорганической или органической кислотой, которая дает фармацевтически приемлемый анион, например гидрогалоидную соль, такую как соль хлорида или бромида, сульфатную или фосфатную соль или соль органической кислоты, например соль с ацетатом, фумарат, малеат, тартрат, лактат, цитрат, пируват, сукцинат, оксалат, метансульфонат или β-толуолсульфонат.
Термин «сольват» относится к соединению по изобретению в твердом состоянии, где молекулы подходящего растворителя включены в кристаллическую решетку. Подходящий растворитель для терапевтического введения физиологически приемлем в веденной дозе. Примерами подходящих растворителей для терапевтического введения являются этанол и вода. Когда вода является растворителем, сольват упоминается как гидрат. Как правило, сольваты образуются путем растворения соединения в соответствующем растворителе и выделения сольвата путем охлаждения или использования антирастворителя. Сольват обычно сушат или подвергают азеотропной перегонке в условиях окружающей среды.
Типичные сольваты включают гидраты, такие как моногидрат, дигидрат или тригидрат.
Кроме того, настоящее изобретение относится к способу получения пролекарств соединения формулы 1, например, гидролизуемых карбаматов in vivo по аминофункции соединения формулы 1. Гидролизуемый карбамат in vivo соединения формулы 1, который содержит карбоксильные, эфирные или гидроксильные группы, представляет собой, например, фармацевтически приемлемый карбамат, который расщепляется в организме человека или животного с образованием родительского амина. Такие карбаматы могут быть идентифицированы, например, путем внутривенно введения испытуемому животному тестируемого соединения и последующего анализа жидкости организма тестируемого животного.
Типичные фармацевтические композиции включают терапевтически эффективное количество соединения формулы 1 вместе с фармацевтически приемлемым носителем.
Соединение формулы 1 используется в количестве, эффективном для лечения, уменьшения или улучшения невропатической боли у субъекта, особенно человека, страдающего болезненным состоянием. Такое лечение боли может или не может быть связано с расстройством центральной нервной системы (ЦНС) или периферической нервной системы (ПНС). Соединение формулы 1 также эффективно для лечения, уменьшения или улучшения любых других расстройств ЦНС, не связанных с болью.
Композиции включают терапевтически эффективное количество соединения формулы 1, которое обычно находится в диапазоне 0,1-95 мас. % соединения формулы 1, но зависит от точной природы активного вещества и способа введения. Как правило, доза активного вещества находится в диапазоне от 0,1 до 500 мг в виде разовой или разделенной дозы, в зависимости от точной природы активного вещества и способа введения.
В терапевтическом применении соединение формулы 1 можно вводить перорально, ректально, парентерально или локально. Фармацевтические композиции могут принимать форму любого перорального, ректального, парентерального или местного состава, известного специалистам в данной области, с использованием носителей, хорошо известных в области фармации. Такие композиции обычно готовят в виде стандартной лекарственной формы. Композиции для перорального введения могут включать твердые лекарственные формы, такие как таблетки, капсулы или жидкие лекарственные формы, такие как сиропы и водные или масляные суспензии. Твердые лекарственные формы, такие как таблетки и капсулы, могут быть получены путем смешивания соединения формулы 1 с инертным разбавителем в присутствии дезинтегрирующих агентов и других вспомогательных средств для составов, таких как смазки. Капсулы могут быть в виде твердых капсул, например твердых желатиновых капсул или мягких капсул, которые получают обычными способами, в которых активный ингредиент включен в носитель и инкапсулирован. Необязательно, такие дозы могут включать энтеросолюбильное покрытие, полученное в соответствии с обычными процедурами, которые могут быть использованы для изменения скорости высвобождения, или эксципиент, который задерживает высвобождение, чтобы обеспечить отсроченное высвобождение или композицию с замедленным высвобождением.
Жидкие лекарственные формы могут быть получены путем растворения активного вещества в подходящем жидком носителе, таком как вода или масляный эксципиент, необязательно в присутствии одного или нескольких растворителей, поверхностно-активных веществ и/или суспендирующих средств. Композиции для ректального введения представляют собой известные фармацевтические формы для такого введения, например, суппозитории с восковым или полиэтиленгликолевым основанием. Композиции для парентерального введения также являются известными фармацевтическими формами для такого введения, например, для стерильных растворов или суспензий в подходящей системе растворителей.
Композиции для местного введения могут включать кремы, лосьоны, мази, гели или другие формы, которые можно вводить путем нанесения композиции непосредственно на пораженный участок или путем включения композиции в носитель, такой как трансдермальный пластырь или в виде композиции, содержащейся в проницаемой мембране для применения в болезненной области. Обычные водные и неводные носители, такие как минеральные масла и воски, могут использоваться отдельно или в комбинации для приготовления кремов, лосьонов или мазей. Гели могут быть получены путем смешивания соединения формулы 1 с местным носителем, содержащим гелеобразующий агент, например, карбомер в присутствии воды. Дополнительно могут быть также добавлены дополнительные средства для составов, такие как трансдермальные ускорители, загустители. В другом варианте осуществления соединение по изобретению может использоваться в комбинации с подходящим фармацевтическим эксципиентом для местного лечения болей в спине. Комбинация соединения и фармацевтического эксципиента может быть в форме геля, гелеобразной формы и приспособлена для размещения на коже пациента с болью.
В другом варианте осуществления, комбинация соединения и фармацевтического эксципиента может быть включена в ткань пластыря и приспособлена для размещения и/или адгезии к коже субъекта при боли. В более предпочтительном варианте осуществления соединение высвобождается с низкой скоростью из фармацевтического эксципиента в ткани пластыря.
Соединение формулы 1 может быть включено в фармацевтические композиции, которые полезны в условиях, указанных ниже.
Настоящее открытие предполагает, что соединение формулы 1 может использоваться в клинических условиях для лечения невропатической боли. В другом варианте осуществления соединение может использоваться для лечения боли в центральной нервной системе (ЦНС). В другом варианте осуществления соединение формулы 1 может быть использовано для лечения боли, которая не связана с ЦНС. В следующем варианте осуществления соединение формулы 1 может использоваться для лечения боли, которая связана с периферической нервной системой (ПНС). В еще одном варианте осуществления соединение формулы 1 может быть использовано для лечения расстройства ЦНС. В одном варианте расстройство ЦНС выбрано из группы, состоящей из эпилепсии, ишемического цереброваскулярного заболевания, инсульта, церебральных новообразований, болезни Альцгеймера, болезни Пика, болезни Хантингтона, деменции, болезни Паркинсона и других экстрапирамидных расстройств, амиотрофического бокового склероза и других заболеваний двигательных нейронов, прогрессирующей нейронной мышечной атрофии, пигментной дегенерации сетчатки, наследственных атаксий, рассеянного склероза и других демиелинизирующих заболеваний, бактериального и вирусного менингита, абсцесса головного мозга, субдуральной эмпиемы, эпидурального абсцесса, гнойного внутричерепного тромбофлебита, миелита и радикулита, вирусного заболевания центральной нервной системы, прионных заболеваний, включая куру, болезнь Крейтцфельдта-Якоба и синдром Герстмана-Штрауслера-Шейнкера, фатальной семейной бессонницы, пищевых и метаболических заболевание нервной системы, нейрофиброматоза, клубневого склероза, мозжечкового гемангиобластоматоза, энцефалотригеминного синдрома, умственной отсталости и других нарушений центральной нервной системы, включая синдром Дауна, церебральный паралич, нейроскелетные расстройства, расстройства вегетативной нервной системы, нарушения черепных нервов, заболевания спинного мозга, мышечную дистрофию и другие нервно-мышечные расстройства, нарушений периферической нервной системы, дерматомиозита и полимиозита, унаследованных, метаболических, эндокринных, токсических миопатий, миастении, периодического паралича, психических расстройств, включая изменение настроения, тревогу и шизофренические расстройства, сезонного аффективного расстройство (САД), акатезии, амнезии, кататонии, диабетической невропатии, поздней дискинезии, дистонии, параноидальных психозов, постгерпетической невралгии, синдрома Туретта, прогрессирующего супрануклеарного паралича, кортикобазальной дегенерации и семейной лобно-височной деменции. В другом варианте осуществления соединение формулы 1 может использоваться для лечения болей в ЦНС, таких как, но не ограничиваясь, головная боль и мигрень.
В другом варианте осуществления соединение формулы 1 можно использовать в сочетании с подходящим лосьоном в фармацевтической композиции для местного лечения боли в спине. В другом варианте осуществления соединение по изобретению может использоваться для местного лечения боли в суставах.
Примеры
Оптическое разрешение соединений формул 2а и 2b
Шаг 1:
(1R, 5S)-3-этилбицикло [3.2.0] гепт-3-ен-6-он (2а)
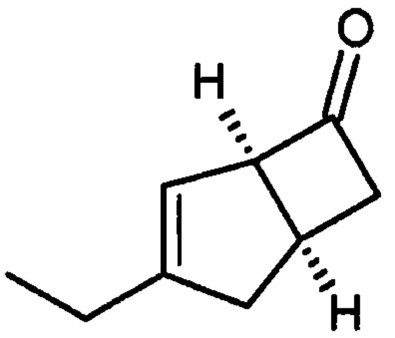
К перемешиваемому раствору 4-пиридинкарбоксальдегида (59,29 г, 554 ммоль) и 4-метилморфолина (55,9 г, 553 ммоль) в 1-метил-2-пирролидиноне (188 мл) при комнатной температуре добавляли рацемическую смесь 3-этилбицикло [3,2,0] гепт-3-ен-6-он (WO 210116757) (75,34 г, 553 ммоль) с последующим добавлением (R)-2-(дифенилметил) пирролидина (13,11 г, 55,3 ммоль) в 1-метил-2-пирролидинон (37,7 мл). Смесь перемешивали при 40°С в течение 18 часов. Реакционной смеси давали остыть до комнатной температуры и затем добавляли диэтиловый эфир (960 мл). Смесь затем промывали 1М HCl (2×820 мл), водой (600 мл) и рассолом (600 мл). Органический слой отделяли и сушили над сульфатом магния. Полученный раствор фильтровали и выпаривали при пониженном давлении (200 мбар, температура бани 28°С), получая 26,4 г масла. Водную фазу дополнительно экстрагировали изогексаном (300 мл), который затем промывали водой (100 мл) и рассолом (100 мл). Полученный раствор сушат и выпаривают, как ранее, и остаток объединяют с первой порцией продукта с получением энантио-обогащенного (1R, 5S)-3-этилбицикло [3.2.0] гепт-3-ен-6-он (28,8 г, 39%) в виде бесцветного масла.
1Н ЯМР (300 МГц, CDCl3): δ 5,21 (1Н, m), 4,23-4,14 (1Н, m), 3,30-3,12 (1Н, m), 2,85-2,70 (3Н, m), 2,38-2,25 (1Н, m), 2,13 (2Н, q, J=7,4), 1,06 (3Н, t, J=7,4).
Определение энантиомерной чистоты вышеуказанного продукта проводили путем получения соответствующего 1,3-диоксолана, полученного из реакции между (1R, 5S)-3-этилбицикло [3.2.0] гепт-3-ен-6-он и (2R, 3R)-(-)-2,3-бутандиол. Интеграция1Н-ЯМР-сигналов показывает, что энантиомерный избыток (э. и.) составил ≥98% и подтвержден с помощью анализа ГХ-МС, как описано ниже:
Определение энантиомерной чистоты (1R, 5S)-3-этилбицикло [3.2.0] гепт-3-ен-6-он.
Два диастереоиомерных 1,3-диоксолана, полученных в результате реакции между рацемическим (1RS, 5RS)-3-этилбицикло [3.2.0] гепт-3-ен-6-он и (2R, 3R)-(-)-2,3-бутандиол также синтезировали.
Сравнивая и интегрируя сигналы1H-ЯМР, было показано, что энантиомерный избыток (э.и.) составил ≥98%.
(1'R, 4R, 5R, 5'S)-3'-этил-4,5-диметил-спиро [1,3-диоксолан-2,6'-бицикло [3.2.0] гепт-3-ен] (одиночный энантиомер)
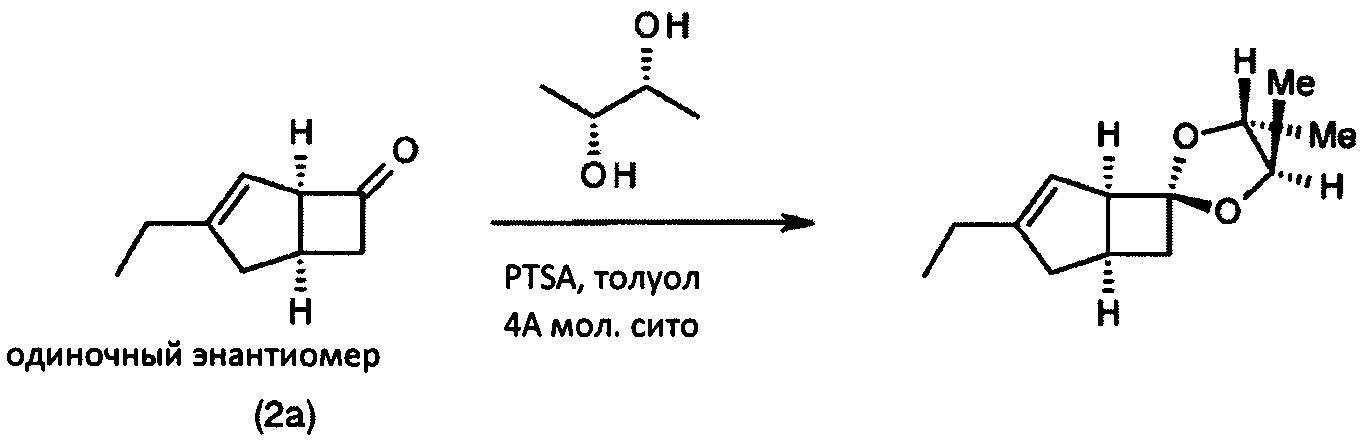
Перемешиваемую смесь (1R, 5S)-3-этилбицикло [3.2.0] гепт-3-ен-6-он (100 мг, 0,73 ммоль), (2R, 3R)-(-)-2,3-бутандиола (131 мг, 1,46 ммоль) и моногидрата пара-толуолсульфоновой кислоты (14 мг, 0,073 ммоль) в толуоле (2 мл), содержащем молекулярные сита 4А, нагревали при 120°С в течение 1 часа. После охлаждения до комнатной температуры большую часть растворителя выпаривали. Остаток очищали хроматографией на двуокиси кремния (5% диэтилэтилизогексана) с получением (1'R, 4R, 5R, 5'S)-3'-этил-4,5-диметил-спиро [1,3-диоксолан-2,6'-бицикло [3.2.0] гепт-3-ен].
1H ЯМР (300 МГц, CDCl3): δ 5,33 (1Н, m), 3,58-3,72 (2Н, m), 3,37-3,43 (1Н, m), 2,48-2,59 (3Н, m), 2,05-2,20 (4Н, m), 1,31 (3Н, d, J=6,0), 1,25 (3Н, d, J=6,0), 1,09 (3Н, t, J=7,4).
Интеграция1Н-ЯМР-сигналов показывает, что энантиомерный избыток (э. и.) составил ≥98%.
ГХ-МС (Hewlett-Packard 5972, HP-5MS 25М × 0,25 мм × 0,25 мкм, газообразный носитель гелия (температура в печи ГХ 60°С в течение 1 мин, затем градиент 60-300°С в течение 24 мин, затем 300°С в течение 20 минут). m/z (EI) 208 [М]+ при 10,36 мин.
(1'S, 4R, 5R, 5'R)-3'-этил-4,5-диметил-спиро [1,3-диоксолан-2,6'-бицикло [3,2,0] гепт-3-ен] и (1'R, 4R, 5R, 5'S)-3'-этил-4,5-диметил-спиро [1,3-диоксолан-2,6'-бицикло [3,2,0] гепт-3-ен] (смесь 1:1 двух диастереоизомеров)
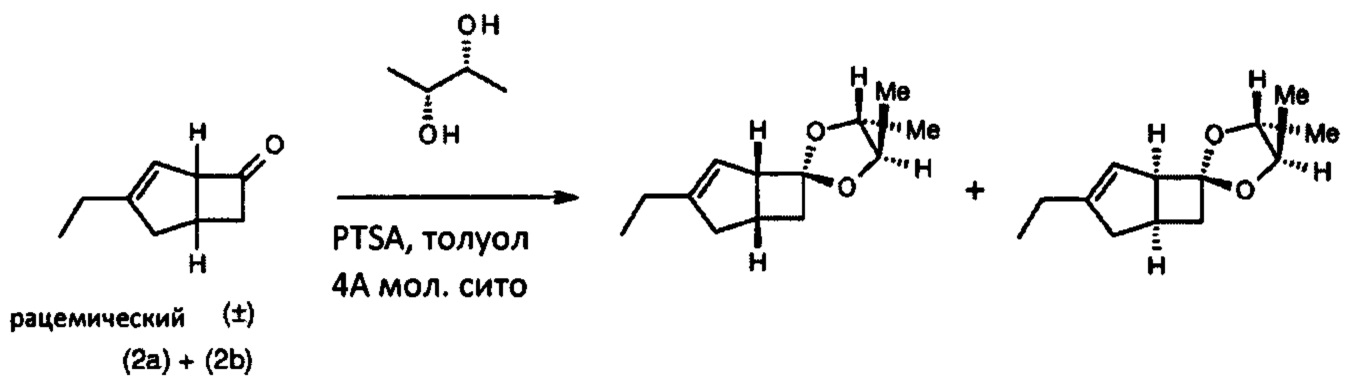
Перемешиваемую смесь рацемического (1RS, 5RS)-3-этилбицикло [3.2.0] гепт-3-ен-6-он (100 мг, 0,73 ммоль), (2R, 3R)-(-)-2,3-бутандиол (131 мг, 1,46 ммоль) и моногидрата пара-толуолсульфоновой кислоты (14 мг, 0,073 ммоль) в толуоле (2 мл), содержащем молекулярные сита 4А, нагревали при 120°С в течение 1 часа. После охлаждения до комнатной температуры большую часть растворителя выпаривали. Остаток очищали хроматографией на двуокиси кремния (5% диэтилэтилизогексана) с получением смеси 1:1 (1'S, 4R,5R, 5'R)-3'-этил-4,5-диметил-спиро [1,3] диоксолан-2,6'-бицикло [3.2.0] гепт-3-ен] и (1'R, 4R, 5R, 5'S)-3'-этил-4,5-диметил-спиро [1,3-диоксолан-2,6'-бицикло [3.2.0] гепт-3-ен].
1H ЯМР (300 МГц, CDCl3): δ; 5,37 (0,5Н, m), 5,33 (0,5Н, m), 3,58-3,72 (2Н, m), 3,49-3,54 (0,5Н, m) 3,37-3,43, m), 2,42-2,60 (3Н, m), 2,02-2,21 (4Н, m), 1,30-1,32 (3Н, m), 1,23-1,26 (3Н, m), 1,09 (3Н, t, J=7,4).
Интеграция1H-ЯМР-сигналов показывает соотношение диастереоизомеров 1:1.
ГХ-МС (Hewlett-Packard 5972, HP-5MS 25М × 0,25 мм × 0,25 мкм, гелий-носитель-газ (температура в печи ГХ 60°С в течение 1 мин, затем градиент 60-300°С в течение 24 мин, затем 300°С в течение 20 минут), m/z (EI) 208 [М]+ при 10,28 и 10,36 мин (соотношение диастереоизомеров 1:1).
Получение соединения формулы 1
Шаг 2:
(2E/Z)-2-((1R, 55)-3-этил-6-бицикло [3,2,0] гепт-3-енилиден) ацетонитрила (5)
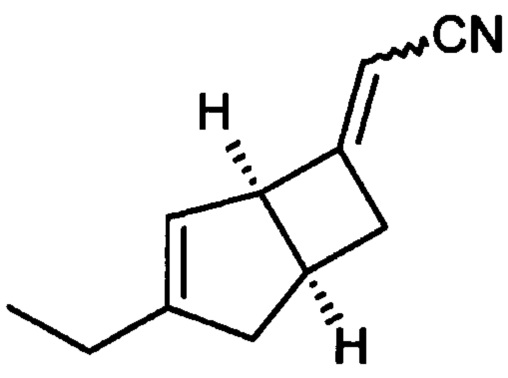
К раствору 1М трет-бутоксида калия в тетрагидрофуране (179 мл, 179 ммоль) при 0°С добавляли диэтилцианометилфосфонат (33,19 г, 187 ммоль). Реакционную смесь перемешивали при 0°С в течение 10 минут, оставляли нагреваться до комнатной температуры и перемешивали еще 30 минут. Смесь переносили в капельную воронку, уравнивающую давление, и по каплям добавляли к раствору (1R, 5S)-3-этилбицикло [3.2.0] гепт-3-ен-6-он (продукт стадии 1) (23,23 г, 170,6 ммоль) в тетрагидрофуране (219,5 мл) при 0°С. Смеси давали нагреться до комнатной температуры и перемешивали в течение 18 часов.
Смесь разбавляли насыщенным водным раствором хлорида аммония (200 мл) и этилацетатом (400 мл), и слои разделяли. Водный слой экстрагировали этилацетатом (3×100 мл) и объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (75 мл), рассолом (75 мл) и сушили над сульфатом магния. Остаток после фильтрации и выпаривания проверяли с помощью ЯМР, и было показано, что они содержат этилфосфатные побочные продукты. Неочищенный продукт распределяли между изогексаном (200 мл) и водой (350 мл). Слои разделяли и водный реактор экстрагировали изогексаном (4×100 мл). Объединенные органические слои сушили над сульфатом магния и выпаривали с получением (2E/Z)-2-((1R, 55)-3-этил-6-бицикло [3,2,0] гепт-3-енилиден) ацетонитрила в виде а~60:40 смеси изомеров E/Z (31,3 г в сочетании с продуктом, полученным из предыдущей партии из 5,5 г исходного материала, 93%).
ЖХМС (Agilent, Waters SunFire С18, 4,6×30 мм, кислота (0,05% муравьиная кислота, 6 минутный метод, 3-97% ацетонитрил/вода): m/z 160,2 (М+Н)+ (ES+) 2,88 мин.
1Н ЯМР (300 МГц, CDCl3):~60:40 смесь изомеров алкена δ 5,43 (0,4Н, m), 5,23 (0,6Н, m), 5,09 (0,6Н, m), 4,98 (0,4Н, m) 4,12 (0,4Н, br s), 3,93 (0,6Н, br s), 3,19-2,90 (2Н, m), 2,74-2,46 (2Н, m), 2,29-2,07 (3Н, m), 1,14-1,06 (3Н, m).
Шаг 3:
2-((1R, 55,65)-3-Этил-6-(нитрометил) бицикло [3,2,0] гепт-3-ен-6-ил) ацетонитрил (6)
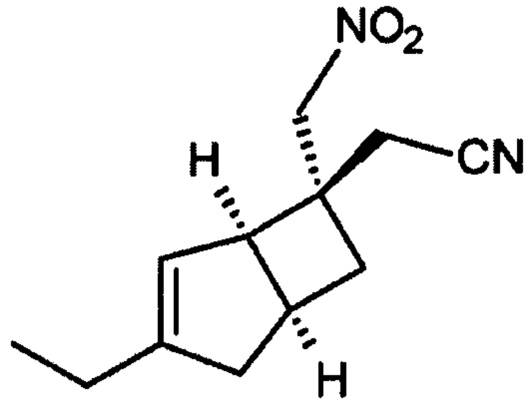
К раствору (2E/Z)-2-((1R, 5S)-3-этил-6-бицикло [3,2,0] гепт-3-енилиден) ацетонитрила (продукт стадии 2) (31,2 г, 196 ммоль) в нитрометан (273 мл, 307 г, 5,04 моль) в атмосфере азота добавляли 1,8-диазабицикло [5.4.0] ундец-7-ен (32 мл, 32,5 г, 213,4 ммоль) и смесь перемешивали в течение 18 часов при комнатной температуре.
Реакционную смесь выливают в 5%-ный водный раствор дигидро-ортофосфата калия (1270 мл) и добавляют этилацетат (950 мл). Слои разделяли и водный слой дополнительно экстрагировали этилацетатом (2×400 мл). Объединенные органические слои сушили над сульфатом магния и выпаривали с получением неочищенного продукта. Остаток очищали хроматографией на подушке из диоксида кремния (35% этилацетатсогексана) с получением 2-((1R, 5S, 6S)-3-этил-6-(нитрометил) бицикло [3,2,0] гепт-3-ен-6-ил) ацетонитрила (38,38 г, выход 89% + менее чистой фракции 3,3 г) в виде смеси диастереомеров~70:30. Данные для основного диастереомера: 2-((1R, 5S, 6S)-3-этил-6-(нитрометил) бицикло [3,2,0] гепт-3-ен-6-ил) ацетонитрила.
ЖХМС (Agilent, Waters SunFire С18, 4,6×30 мм, кислота (0,05% муравьиная кислота, 6 минутный метод, 3-97% ацетонитрил/вода): m/z 221 (М+Н)+ (ES+) 2,81 мин.
1Н ЯМР (300 МГц, DMSO-d6): δ 5,33 (1Н, m), 4,86 (2Н, s), 3,16 (1Н, br. s), 3,02-2,82 (1Н, m), 2,65 (2Н, s), 2,48-2,40 (1Н, m), 2,23 (1Н, ddd, J=12,4, 8,8, 2,5), 2,16-2,02 (3Н, m), 1,56 (1H, dd, J=12,5, 7,2), 1,06, t, J=7,5) ч. н. м.
Шаг 4:
5-(((1R, 5S, 6S)-3-Этил-6-(нитрометил)-6-бицикло [3.2.0] гепт-3-енил) метил)-1Н-тетразол (7)
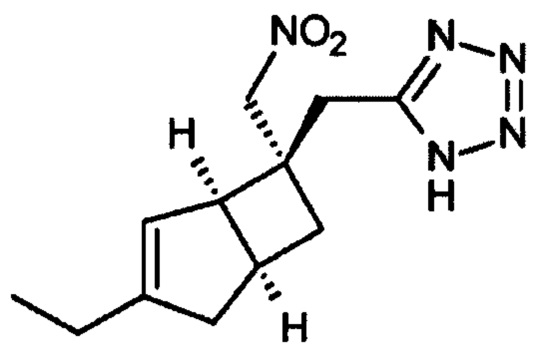
К раствору 2-((1R, 5S, 6S)-3-этил-6-(нитрометил) бицикло [3,2,0] гепт-3-ен-6-ил) ацетонитрила (продукт стадии 3) (11 г, 50 ммоль) в 1-метил-2-пирролидиноне (158 мл) добавляли гидрохлорид триэтиламина (26,55 г, 192 ммоль) и азид натрия (12,54 г, 192 ммоль). Колбу нагревали в атмосфере азота при 110°С в течение 18 часов и затем оставляли охлаждаться до комнатной температуры. Смесь разбавляли водой (200 мл) и осторожно доводили до рН 11-12 с использованием водного 2М раствора гидроксида натрия. Полученный раствор экстрагировали этилацетатом (2×350 мл) и органический слой обратно экстрагировали водным 1М раствором гидроксида натрия (2×40 мл). К объединенным основным водным фазам добавляли 20% водный раствор нитрита натрия (100 мл) и смесь охлаждали на ледяной бане.
20%-ную водную серную кислоту добавляли по каплям (выделение газа) до тех пор, пока смесь не подкислялась, а выделение газа не прекращалось (~рН 1-2). Затем смесь перемешивали еще 1 час. Полученный водный раствор экстрагировали этилацетатом (3×300 мл).
Объединенные органические слои промывали водой (3×250 мл) и рассолом (2×100 мл) и сушили над сульфатом магния. Фильтрация и выпаривание дают сырой продукт, который очищают хроматографией на двуокиси кремния (этилацетат : изогексан : уксусная кислота 250:750:1) с получением (одиночного диастереоизомера) 5-((1R, 5S, 6S)-3-этил-6-(нитрометил)-6-бицикло [3.2.0] гепт-3-енил) метил)-1Н-тетразол (4,7 г, 17,8 ммоль, 35%) (и еще 0,5 г смеси~95:5 диастереомеры для повторной очистки).
ЖХМС (Agilent, Waters SunFire С18, 4,6×30 мм, кислота (0,05% муравьиная кислота, 6 минутный метод, 3-97% ацетонитрил/вода): m/z 264 (М+H)+ (ES+), 262 (М-Н)- (ES-), в 2,38 мин.
1Н ЯМР (300 МГц, DMSO-d6): δ 16,10 (1Н, br. s), 5,37 (1Н, d, J=1,5), 4,79 (2H, s), 3,22 (1H, 2H, s), 2,94-2,81 (1H, m), 2,48-2,40 (1H, m), 2,19-2,02 (4H, m), 1,64 (1H, dd, J=12,4, 7,5), 1,05 (3H, t, J=7,5) ч. н. м.
Шаг 5:
[(1R, 5S,6S)-3-Этил-6-(1Н-тетразол-5-илметил)-6-бицикло [3,2,0] гепт-3-енил] метанамин (1)
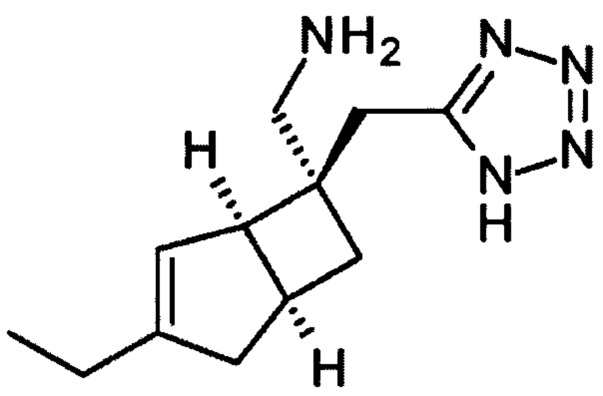
К раствору 5-((1R, 5S,6S)-3-этил-6-(нитрометил)-6-бицикло [3.2.0] гепт-3-енил) метил)-1Н-тетразола (продукт стадии 4) (1,377 г, 5,23 ммоль) в этаноле (27,3 мл) в атмосфере азота добавляли концентрированную соляную кислоту (7,85 мл). Цинковая пыль (6,08 г, 93,5 ммоль) добавлялась по частям в течение 10 минут (при внешнем охлаждении на водяной/ледяной бане для обеспечения того, чтобы внутренняя температура реакции не превышала 35°С). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов.
Реакционную смесь выливают в 50 г SCX-картриджа (предварительно промывают метанолом 200 мл), который затем элюируют метанолом (160 мл), а затем водным метанолом (1:1,120 мл) и метанолом (120 мл). Затем смолу элюировали 0,7М аммиаком в метанольном растворе (360 мл) и собирали фракции. Фракции, содержащие продукт, объединяли и выпаривали с получением [(1R, 5S, 6S)-3-этил-6-(1Н-тетразол-5-илметил)-6-бицикло [3,2,0] гепт-3-енил] метанамин (1,159 г, 95%) в виде белого порошка.
ЖХМС (Agilent, Waters SunFire С18, 4,6×30 мм, кислота (0,05% муравьиная кислота, 6 минутный метод, 3-97% ацетонитрил/вода): m/z 234 (М+Н)+ (ES+), 232 (МН)- (ES-), на 0,86 мин.
1Н ЯМР (400 МГц, CD3OD): δ 5,42 (1Н, br. m), 3,08-3,15 (3Н, m), 3,07 (1Н, d, J=13), 3,03 (1H, d, J=16), 2,82 (1H, m), 2,54 (1H, br. dd, J=16, 8), 2,18 (2H, q, J=7), 2,10-2,16 (1H, br. d, J=16), 1,93 (1H, ddd, J=12, 9, 3), 1,63 (1H, dd, J=12,7), 1,12 (3H, t, J=8) ч. н. м.
ЯМР-присвоение: (CD3OD)
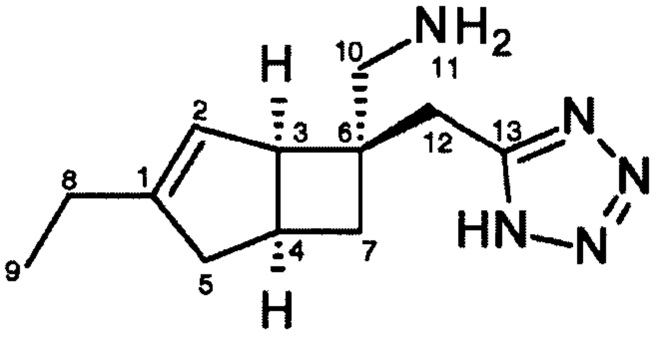
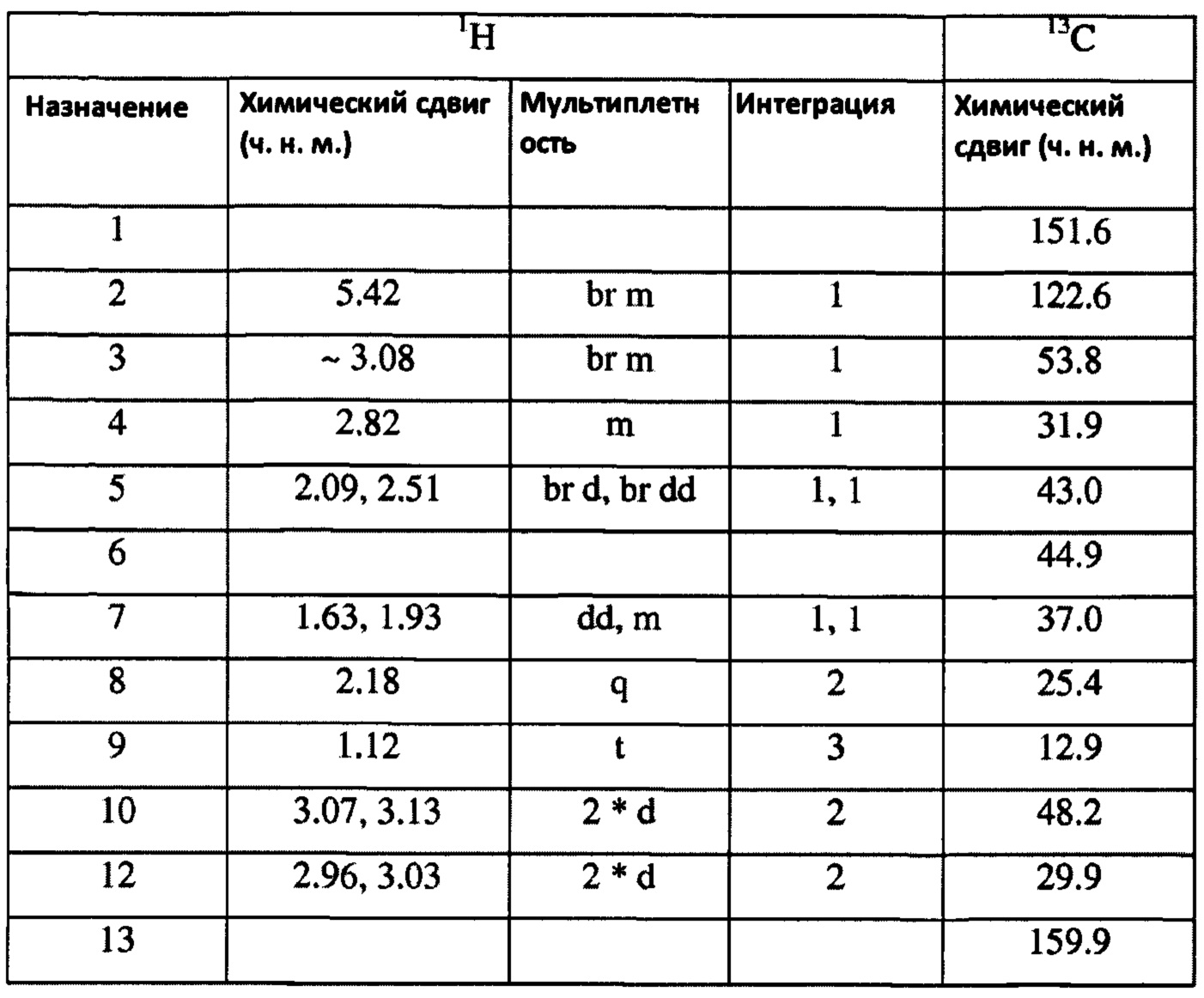
Ключ к сокращениям множественности: s = синглет, d = дублет, t = триплет, q = квартет, m = мультиплет, (может быть объединен, например, dd дублет дублетов, либо префикс br - широкий, например, широкий синглет)
Протонные химические сдвиги, относящиеся к остаточной воде при 4,90 ч. н. м.
Углеродные химические сдвиги, на которые делается ссылка, с использованием внутреннего спектрометра, ссылающегося на
Чистота ВЭЖХ: 99,3% (% AuC при 210 нм).
Колонка: Waters XBridge С18, 150×4,6 мм, 3,5 мкм
Растворитель А: вода + 0,1% TFA
Растворитель В: ацетонитрил + 0,1% TFA
Расход: 1.0 мл/мин
Температура: 40°С
Объем инжекции: 5 мкл раствора 1 мг/мл в ацетонитриле/воде (1:1)
Длина УФ-волны: 210 нм
Градиент растворителя:
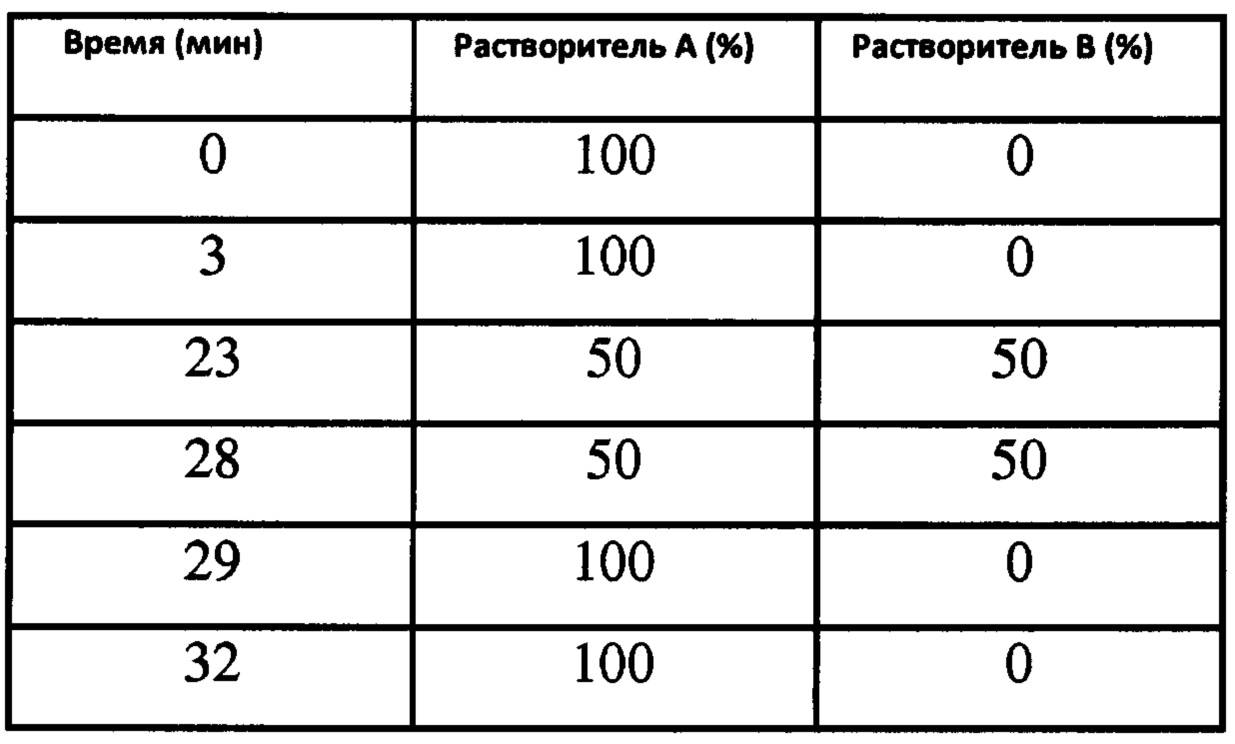
Время отстаивания: ок. 15.7 мин.
Чистая чистота ВЭЖХ: > 99,9.
Колонка: Daicel Chiralpak IC, 250×4,6 мм, 5 мкм
Мобильная фаза: изо-гексан : этанол (70:30)
Условия: изократический анализ, 30-минутное время выполнения.
Скорость потока: 1,5 мл/мин
Температура: окружающая среда
Объем инъекции: 10 мкг раствора 0,5 мг/мл в этаноле.
УФ: 215 нм
Длина волны:
Время отстаивания: Желаемый изомер элюируется прибл. 20 минут
Нежелательный изомер элюируется прибл. 10 минут
Оптическое вращение: [α]D23-101,5 (с = 27,4 мг в EtOH (2 мл)).
Температура плавления: 203-206°С.
Альтернативная методика восстановления нитрогруппы (стадия 5) в рацемической смеси.
Рацемический [(1R, 5S, 6S)-3-этил-6-(1Н-тетразол-5-илметил)-6-бицикло [3,2,0] гепт-3-енил] метанамин
К раствору рацемического 5-((1R, 5S, 6S)-3-этил-6-(нитрометил)-6-бицикло [3.2.0] гепт-3-енил) метил)-1Н-тетразола (275 мг, 1,04 ммоль) в этаноле (2,2 мл) в атмосфере азота добавляли гидразингидрат (201 мкл, 207 мг, 4,13 ммоль). Добавляли суспензию никеля Ренея в воде (67 мкл) и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли дополнительную аликвоту суспензии никеля Ренея (100 мкл) и гидразина гидразина (200 мкл) и смесь перемешивали еще 18 часов. Добавляли еще одну аликвоту суспензии никеля Ренея (200 мкл) и гидразингидрата (200 мкл) и смесь перемешивали еще в течение 2 часов, после чего реакционную смесь фильтровали через целит и промывали этанолом. Полученный раствор выпаривали и очищали на картридже SCX, элюируя метанолом. Затем смолу элюировали 0,7М аммиаком в метанольном растворе и собирали фракции. Фракции, содержащие продукт, объединяли и выпаривали с получением рацемического [(1R,5S,6S)-3-этил-6-(1Н-тетразол-5-илметил)-6-бицикло [3,2,0] гепт-3-енил] метанамин (216 мг, 89%).
ЖХМС и1Н-ЯМР, как сообщалось на шаге 5 выше.
Реферат
Настоящее изобретение относится к способу выделения соединения формулы 2авключающему а) взаимодействие смеси диастереоизомеров формул 2а, 2bс основным гетероциклическим альдегидным соединением и оптически активным амином в присутствии основания; и б) отделение соединения формулы 2а от продукта стадии а) путем экстракции кислотой. Также изобретение относится к способу соединения формулы 1, имеющего превосходную активность в качестве α2δ-лиганда, одной из стадий которого является способ выделения соединения формулы 2а. Предлагаемое изобретение позволяет получить соединение формулы 1 с использованием упрощенной технологии. 2 н. и 16 з.п. ф-лы, 1 пр.
Формула


Комментарии