Способ получения оптически активных эфиров эритро-3-амино-2-оксимасляных кислот и соответствующих исходных кислот - RU2169138C2
Код документа: RU2169138C2
Описание
Изобретение относится к способу получения оптически активных бутиратов, т. е. эфиров масляных кислот, преимущественно эритро-3-амино-2-оксимасляных кислот, а также соответствующих исходных кислот, имеющих, например, (2S, 3S)-конфигурацию, в качестве важных интермедиатных продуктов фармацевтических реагентов, в частности для ингибитора протеазы ВИЧ.
Уровень техники
Относительно
способов
получения оптически активных эфиров эритро-З-амино-2-оксимасляных кислот было опубликовано огромное количество публикаций, включающих способы, основанные на цианогидратации альдегида (см.,
например,
Bulletin of the Chemical Societe of Japan, 65, 360 (1992), Tetrahedron Letters, 33 (45), 6763 (1992)), способы, использующие в качестве исходного материала диэтилмалат (см. Tetrahedron
Letters, 33
(45), 6803 (1992)), а также способы, использующие в качестве исходного материала винную кислоту (см. Chemical & Pharmaceutical Bulletin, 39 (10), 2550 (1991)), однако большинство
способов
направлено на получение трео-соединений. Имели место и публикации относительно способов получения эритросоединений, включающие, например, способ на основе реакции асимметричного гидрирования
с
использованием асимметричных катализаторов (патент Японии N 1000/1993), способ на основе образования комплекса 2-окси-З-нитропропановых производных с альдегидом (патент Японии N 165678/1995),
способ,
использующий цианогидратацию, который обладает эритроселективностью, индуцируемой фталоиловой защитой аминогруппы (патент Японии N 309840/1995), и способ на основе нитроалдольной реакции с
использованием асимметричных катализаторов (Tetrahedron Letters, 35 (33), 6123 (1994).
Согласно способу, описанному в числе способов получения эритросоединений в патенте Японии N 1000/1993, асимметрия осуществляется путем реакции гидрирования с использованием асимметричных катализаторов, при этом требуется высокое давление водорода (100 атм) и образуется четыре изомера, т.к. исходный материал не является оптически активным веществом. С учетом того факта, что для введения аминогруппы получают в качестве интермедиатных продуктов химически нестабильные азидные соединения, при промышленном приложении способа возникает огромное число проблем. Кроме того, способ, описанный в японском патенте N 165678/1995, требует исследования асимметричного расположения гидроксильной группы в 2-положении 2-окси-3-нитропропановой кислоты, применяемой в качестве исходного материала, а также исследования процесса очистки конечного продукта, т. к. стереоселективность синтеза составляет приблизительно 8: 2, что не является достаточно высокой величиной. Способ, описанный в японском патенте N 309840/1995, не является благоприятным с точки зрения промышленного производства, т. к. стереоселективность цианогидратации составляет приблизительно 7:3, что требует оборудования для очистки получаемого вещества, сопровождаемой большими потерями изомеров в процессе очищения. Способ, описанный в Tetrahedron Letters, 35 (33), 6123 (1994), имеет высокие значения как выхода, так и стереоселективности, но не может быть признан пригодным для производства в промышленном масштабе, т.к. он требует большого периода времени для нитроалдольной реакции (3 дня) и гидролиза нитрогруппы (2 дня), а также использования комплексных соединений относительно дорогих редкоземельных элементов с 1,1'-би-2 нафтолом.
Для решения этих проблем были проведены интенсивные исследования. В результате согласно настоящему изобретению были получены хорошие результаты, описанные ниже.
Сущность изобретения
Настоящее изобретение относится к следующим способам (1) - (3):
(1) Способ получения оптически активного эфира
эритро-3-амино-2- оксимасляной кислоты,
представленного формулой III

где R1 - фенильная или циклогексильная группа, R2 - защитная группа, a R3 - замещенный или незамещенный низший алкил с числом углеродных атомов от 1 до 6; стерическая конфигурация*2 представляет собой S-конфигурацию в случае S- конфигурации*3 и R-конфигурацию в случае R-конфигурации*3), включающий окисление эфира трео- или трео-эритро-З-амино-2-оксимасляной кислоты, оптически активного в положении 3, как представлено в формуле I:
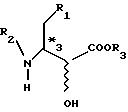
(где R1, R2 и R3 те же, что и выше, а стерическая конфигурация*3 представляет собой S- или R-конфигурацию),
с получением оптически активного эфира 3-амино-2-оксомасляной кислоты, представленного формулой II

где составы R1, R2, R3 и*3 независимо друг от друга те же, что и выше, и эритроселективное восстановление карбонильной группы в положении 2 путем применения алкоксида алюминия.
(2) Способ получения оптически активного эфира (2, 3)-эритро-3-амино-2-оксимасляной кислоты с формулой III, включающий эритро-селективное восстановление карбонильной группы в положении 2 оптически активного эфира 3-амино-2-оксомасляной кислоты путем применения алкоксида алюминия.
(3) Способ получения оптически активной (2, 3)-эритро-3-амино- или защищенной (R2-)амино-2-оксимасляной кислоты, включающий гидролиз оптически активного эфира (2, 3)-эритро-3-амино-2- оксимасляной кислоты с формулой (III), полученного согласно вышеуказанным пп. (1) или (2).
Наилучшая
модификация осуществления изобретения:
Здесь и далее будет приведено детальное описание настоящего
изобретения.
Согласно настоящему изобретению в качестве защитной группы (R2), выполняющей эту функцию относительно аминогруппы, может быть применена любая известная защитная группа такого рода; предпочтительные защитные группы включают низшие замещенные или незамещенные алкилкарбонилы с числом углеродных атомов от 1 до 6, такие как формил, ацетил, трифторацетил и пивалоил, замещенные и незамещенные бензоилы, представляющие собой защитные группы ацильного типа, замещенные или незамещенные бензилкарбонилы, алкоксикарбонилы с числом углеродных атомов от 1 до 6 и циклоалканоксикарбонилы, представляющие собой защитные группы уретанового типа, а также защитные группы, включающие (а) замещенные или незамещенные арилсульфонилы, (б) такие сульфонилы как замещенные или незамещенные бензолсульфонилы, например о- нитробензолсульфонил, и (в) замещенные или незамещенные фенилзамещенные низшие алкилы, такие как тритил (трифенилметил). Заместители в них включают атомы галогенов, нитрогруппу, гидроксильную группу, цианогруппу и т.п.
Эфирообразующая группа R3 включает замещенные или незамещенные низшие алкилы с числом углеродных атомов от 1 до 6, например метил, этил, пропил, изопропил, бутил, изобутил, третичный бутил, пентил и гексил.
Заместители в них включают атомы галогенов, нитрогруппу, гидроксильную группу и цианогруппу.
Говоря более конкретно, представленные формулой (1)
соединения включают описанные ниже
изопропил (3S)-3-(N-Вос)амино-4-фенил-2-оксибутират,
изопропил (3S)-3-(N-Z)амино-4-фенил-2-оксибутират,
изопропил
(3S)-3-N-ацетиламино-4-циклогексил-2-оксибутират,
где N-Z и N-Boc означают соответственно N-бензилоксикарбонил и
N-трет-бутоксикарбонил.
Согласно настоящему изобретению способ окисления гидроксильной группы в соединении формулы (I) с превращением в соединение формулы (II) не имеет специфических ограничений в рамках возможности окисления вторичного спирта в карбонильную группу согласно способу, кроме того, способ включает варианты способа окисления с применением хромовых кислот, диоксидов марганца, диметилсульфоксида (здесь и далее сокращенного обозначенного как ДМСО), нитроксильных соединений и т.п. Предпочтительными способами являются (а) для способа окисления с применением хромовых кислот - способ с применением комплексов хромовых кислот с пиридином, таких как хлорхромат пиридиния и дихромат пиридиния; (б) для способа окисления диметилсульфоксидом (ДМСО) - способ окисления, использующий ДМСО в форме активной сульфониевой соли путем применения соединений следующего вида: электрофильные реагенты/ДМСО или донор водорода - электрофильные реагенты/ДМСО, такие как уксусный ангидрид/ДМСО, триэтиламин - комплекс пиридина с серным ангидридом/ДМСО, дициклогексилкарбодиимид-трифторацетат пиридиния/ДМСО, водорастворимая гидрохлоридная соль карбодиимида-трифторацетат пиридиния и (в) для способа окисления нитроксильными соединениями - способ, использующий 2,2,6,6-тетраметилпиперидин-1-оксил (здесь и далее сокращенно обозначенный как ТЕМПО) или ТЕМПО, получаемый в реакционной системе из 2,2,6,6-тетраметилпиперидина (здесь и далее сокращенно обозначенного как TEMP) и окислителей, например перекиси водорода, органических пероксокислот (мета- хлорпероксобензойная кислота, пероксоуксусная кислота, пероксофталевая кислота и т.д.) или металлических окислителей (хлорид меди, нитрат меди, ферроцианатные соли и т. д. ). Среди этих способов предпочтительно применение способа окисления с низкотоксичным реагентом ДМСО или способа окисления с нитроксильными соединениями, но более предпочтительно осуществление способа, применяющего уксусный ангидрид/ДМСО, или способа с ТЕМПО, которые могут обеспечить высокий выход и проведение которых относительно несложно.
Для работы с уксусным ангидридом/ДМСО ДМСО применяют в качестве растворителя, а уксусный ангидрид добавляют в количестве от 2 до 10 эквивалентов, предпочтительно от 3 до 5 эквивалентов, относительно реакционного субстрата. Температура реакции обычно составляет от - 15o C до температуры дефлегмации растворителя, при этом предпочтительна комнатная температура, не требующая регулировки.
После окончания реакции проводили добавление воды с последующей экстракцией, промыванием, высушиванием и, далее, если необходимо, с концентрацией и очисткой путем хроматографии на силикагеле с получением соединения формулы (II).
При окислении с помощью ТЕМПО разновидности ТЕМПО, применяемые в качестве катализатора, альтернативно включают его замещенные, например 4-метокси-ТЕМПО, бензоат 4-гидрокси-ТЕМПО и 4-ацетамид-ТЕМПО, при этом используемые количества их составляют от 0,01 до 100 мол. % относительно субстрата реакции в виде соединения спирта и предпочтительно от 0,05 до 5 мол. %, более предпочтительно приблизительно от 0,1 до 1 мол. % относительно субстрата реакции при применении вместе с соокислителями. Применяемые соокислители включают гипогалогенит или галогенит, такие как гипохлорит натрия или гипохлорит кальция, бромид натрия, галогены, такие как хлор, и органические пероксокислоты, такие как мета-хлорпероксобензойная кислота, при этом используемые их количества составляют от 0,5 до 10 эквивалентов, предпочтительно от 1 до 5 эквивалентов. При применении гипогалогенитных солей, таких как гипохлорит натрия, в качестве способствующего реакции реагента добавляют ионы галогенов, такие как бромид-ион в форме, например, бромида натрия и бромида калия в количестве от 5 до 150 мол. % с последующим добавлением гидрокарбоната натрия для установления слабощелочного рН, например от 7 до 10, предпочтительно от 7 до 8, облегчающего протекание реакции.
Реакция эффективна только в органических растворителях, когда соокислители растворяются в органических растворителях или в двухслойной системе вода - органический растворитель и представляют собой водорастворимую неорганическую соль. Любой органический растворитель, способный отделяться хотя бы в какой-то степени от воды и растворять соединения с формулой (I), удовлетворителен без каких-либо специфических ограничений и включает углеводородные галоиды, такие как метиленхлорид и хлороформ, ароматические углеводороды, такие как бензол, толуол и ксилол, простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир и тетрагидрофуран, алифатические углеводороды, такие как пентан, гексан и гептан, сложные эфиры, такие как этилацетат, изопропилацетат и бутилацетат, причем все эти соединения применяются или сами по себе или в виде растворяющей смеси.
Температура реакции составляет от -15oC до температуры, соответствующей дефлегмированию растворителя, предпочтителен интервал от 0oC до комнатной температуры, не требующий температурной регулировки. При проведении реакции обеспечивают растворение соединения формулы (I) в растворителе с последующим добавлением ТЕМПО и аддитивным и постепенным добавлением соокислителя при интенсивном перемешивании. После окончания реакции соокислитель разлагают с помощью иодид-иона, например иодида натрия и иодида калия, с последующей нейтрализацией и далее проводят такие операции, как разделение слоев, экстракцию, промывание и высушивание. Соединение формулы (II) может быть получено концентрацией реакционного раствора после такой обработки с последующей очисткой, если это необходимо, с помощью хроматографии на силикагельной колонке. Эритроселективное восстановление полученной карбонильной группы в формуле (II) индуцируется реакцией переноса протона с применением алкоксида алюминия. Применяемый алкоксид алюминия не имеет специфических ограничений и включает, например, изопропоксид алюминия, этоксид алюминия и трет-бутоксид алюминия. Применяемые количества их составляют от 0,1 до 10 эквивалентов, предпочтительно от 0,5 до 5 эквивалентов. В качестве реакционного растворителя обычно применяют спирт, поскольку он участвует в реакции. Например, пригодны низшие спирты с числом атомов углерода от 1 до 6, такие как метанол, этанол, n-пропанол, изопропанол, n-бутанол и трет-бутанол. Температура реакции лежит обычно в интервале от комнатной температуры до температуры дефлегмации растворителя; для содействия реакции предпочтительно проводить ее с нагревом до температуры в интервале от 40oC до температуры перегонки растворителя.
Кроме того, алкоксид алюминия может быть получен в реакционной системе и затем использован по назначению. Способ получения алкоксида алюминия в реакционной системе содержит добавление алюминия к спирту, соответствующему алкоксиду, и далее добавление активирующего реагента, содействующего реакции, и растворение этого реагента при нагревании. Температура реакции лежит в интервале от комнатной температуры до температуры дефлегмации растворителя; обычно для содействия реакции ее температуру выбирают равной температуре дефлегмации растворителя. Активирующий реагент включает, например, хлорид ртути (II), иод и четыреххлористый углерод, причем любое из этих веществ может быть успешно применено. Их количество при применении составляет примерно от 0,1 до 10 мол. % по отношению к применяемому спирту. Остальные условия те же, что и в реакции c применением алкоксида алюминия. После окончания реакции гидроксид алюминия растворяют в растворе путем окисления полученной жидкости водным раствором соляной или серной кислот с последующей экстракцией, промыванием, высушиванием и концентрацией. Очисткой концентрата путем перекристаллизации и т.п. может быть получено соединение формулы (III) высокой чистоты.
Т. к. эфирный обмен в эфирной группе R3 в формуле (II) происходит благодаря присутствию триалкоксиалюминия во время восстановления, применяемый в реакции триалкоксиалюминий предпочтительно имеет форму (R3О)3Al, а в качестве реакционного растворителя предпочтительно применяют спирт, представленный формой R3OH.
Далее соединение формулы (i) в качестве исходного материала известно и может быть легко получено способом, использующим цианогидратацию оптически активного фенилаланина в качестве исходного материала; например, соединение, у которого R1 является фенилом, может быть получено путем защиты аминогруппы и карбоксильной группы 3-амино-2-окси-4-фенилмасляной кислоты, как это описано в Journal of Medicinal Chemistry, 20, 510 (1977); соединение, у которого R1 является циклогексилом, может быть получено путем защиты аминогруппы и карбоксильной группы 3-амино- 4-циклогексил-2-оксимасляной кислоты, как это описано в Journal Medicinal Chemistry, 33(10), 2707 (1990).
Согласно настоящему изобретению применяемый в качестве исходного материала эфир трео- или трео-эритро-3-амино-2-оксимасляной кислоты, оптически активный в положении 3, как показано в формуле (i), и легко синтезируемый из недефицитной оптически активной аминокислоты, может быть простым способом превращен в оптически активный эфир эритро-3-амино-2- оксимасляной кислоты при относительно мягких условиях реакции с высоким выходом и высокой оптической чистотой. Согласно настоящему изобретению, например, эфиры масляных кислот могут быть получены с оптической чистотой 90% и выше, предпочтительно 95% и выше.
Поскольку достигается более высокая степень превращения в эритросоединение, несмотря на то, что в соединении, представленном формулой (1) в качестве исходного материала, соотношение треосоединения выше, эффективность настоящего изобретения повышается.
Способ введения полученного эфирного соединения формулы (III) в соответствующую карбоновую кислоту включает гидролитические реакции под воздействием кислот и гидролитические реакции под воздействием оснований, но поскольку даже группа, защищающая аминогруппу, обычно гидролизуется путем гидролиза под воздействием кислот, в случае наличия такой группы обычно применяют гидролиз под воздействием оснований.
В случае гидролиза под воздействием кислот применяемые кислоты включают неорганические кислоты, такие как соляная кислота, серная кислота и бромистоводородная кислота, и органические кислоты, такие как уксусная кислота, трифторуксусная кислота, p-толуолсульфокислота и камфорная сульфокислота; обычно применяют относительно дешевые и недефицитные неорганические кислоты, такие как соляная и серная. Применяемое их количество изменяется в зависимости от используемых кислот и не имеет специфических ограничений, но обычно составляет величину приблизительно от 0,1 до 50-кратных эквивалентов, предпочтительно приблизительно от 1 до 20-кратных эквивалентов относительно соединения формулы (III). Что касается реакционного растворителя, может быть применен любой негидролизуемый растворитель без специфических ограничений, но часто применяют низшие спирты, имеющие от 1 до 4 атомов углерода, такие как метанол и этанол, тетрагидрофуран и диоксан, которые хорошо смешиваются с водой и содействуют реакции гидролиза. Для облегчения реакции добавляют воду в количестве одного или нескольких эквивалентов относительно соединения формулы (III). Температуру реакции изменяют в зависимости от применяемых кислот без специфических ограничений, но обычно температура составляет величину от 0oC до температуры дефлегмации растворителя, причем с точки зрения высокой скорости реакции предпочтительна последняя. После окончания реакции конечный продукт обрабатывают обычным способом для получения карбоновой кислоты.
В случае гидролиза под воздействием оснований в порядке альтернативы применяемые основания включают гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия и гидроксид лития, и органические амины, такие как триметиламин, триэтиламин и пиридин, при этом гидроксиды щелочных металлов, обладающие сильной основностью, предпочтительны, поскольку они относительно дешевы, недефицитны и обеспечивают быструю скорость реакции. Применяемое их количество изменяется в зависимости от используемых оснований без специфических ограничений, но обычно оно составляет величину приблизительно от 0,1 до 50-кратных эквивалентов, предпочтительно от 1 до 20-кратных эквивалентов, относительно соединения формулы (III). В качестве реакционного растворителя удовлетворителен любой негидролизуемый растворитель без специфических ограничений, но часто используют низшие спирты, такие как метанол и этанол, тетрагидрофуран и диоксан, которые смешиваются водой в степени, достаточной для содействия гидролизу. Для облегчения реакции добавляют воды в количестве одного или нескольких эквивалентов относительно соединения формулы (III). Температуру реакции изменяют в зависимости от применяемых оснований без специфических ограничений, но обычно реакция облегчается в температурном интервале от -20oC до температуры дефлегмации растворителя и протекает при относительно мягком режиме от -20 до 40oC в случае сохранения группы, защищающей аминогруппу.
После окончания реакции конечный продукт обрабатывают обычным способом для получения карбоновой кислоты.
Сведения,
подтверждающие возможность осуществления изобретения
Далее настоящее изобретение будем описано более детально со ссылками на соответствующие
примеры, однако его не следует рассматривать как
ограниченное этими примерами.
Пример 1
(А) Синтез изопропил (3S)-3-(N-трет- бутоксикарбонил)амино-4-фенил-2-оксобутирата.
В 50 мл толуола растворили 5,0 г
изопропил (2R, 3S)-3-(N-Вос)амино-4-фенил- оксибутирата (здесь и далее N-Вос означает N-трет-бутоксикарбонил) с последующим добавлением 50 мл воды к
полученной смеси, затем добавили 1,52 г бромида
натрия и 4,0 г гидрокарбоната натрия и полученную смесь охладили ниже 10oC. К смеси добавили 0,012 г ТЕМПО с последующим постепенным
добавлением по каплям 10,11 г водного 12% раствора
гипохлорита натрия при энергичном перемешивании. После окончания добавления по каплям полученную смесь перемешивали в течение приблизительно одного
часа и после подтверждения окончания реакции
добавили 0,3 г иодида калия с последующим добавлением 10% гидросульфата калия, чтобы довести pH полученной смеси до 7, и далее с разделением слоев, при
этом полученный водный слой экстрагировали в
толуоле. Экстрагированный раствор промыли водным 0,1 н. раствором тиосульфата натрия и далее водой и высушили над сульфатом магния с последующей
фильтрацией и концентрацией при пониженном давлении до
количественного получения 5,09 г изопропил (3S)-3-(N-Вос)амино-4-фенил-2-оксобутирата. Ниже приведены результаты его ЯМР-анализа:
1H-ЯМР(CDCl3)
δ (ppm) 1,34 (d,6H,
J= 6,3 Hz)
1,39(S,9H)
1,69 ~ 1,82(br, 1H)
2,92 ~ 3,28 (m, 2H)
4,98 ~ 5,10 (br, 1H)
5,15 (td,
1H, J=6,3: 12,6 Hz)
7,09 ~ 7,36 (m, 5H)
(Б)
Синтез изопропил (2S, 3S)-3-(N-Вос)амино-4-фенил-2-оксибутирата
В 10 мл изопропанола растворили 1,0 г изопропил
(3S)-3-(N-Вос)амино- 4- фенил-2-оксобутирата с последующим добавлением 0,7 г
изопропоксида алюминия и нагревали в течение 4 ч при температуре ниже температуры дефлегмации. После подтверждения
окончания реакции добавили к полученной смеси водный 1 н. раствор соляной кислоты,
чтобы довести pH смеси до 3,0, и полученную смесь сконцентрировали при пониженном давлении. Концентрат разбавили
этилацетатом и водой с последующим разделением слоев и полученный водный слой
экстрагировали в этилацетате. Экстрагированный раствор промыли в воде и в насыщенном растворе хлорида натрия и высушили
над безводным сульфатом магния с последующей фильтрацией и концентрацией,
получив 0,95 г изопропил (2S 3S)-3-(N-Вос)амино-4-фенил-2-оксобутирата с выходом 94,5%. Ниже приведены результаты его
ВЭЖХ-анализа.
Условия ВЭЖХ-анализа:
Колонка - Inertsil
ODS-2 (GL Science) 4,6 ⌀ х 250 мм
Температура колонки - 35oC
Элюент - ацетонитрил
водный 0,02 М раствор дигидрофосфатаата аммония (pH 2,5) = 6:5
Скорость
потока - 1,0 мл/мин
Время удерживания - 10,1 мин для (2S, 3S)-соединения - 11,9 мин для (2R,
3S)-соединения
Отношение выходов - (2S, 3S): (2R, 3S) = 93,8: 6,2
Полученный
изопропил (2S, 3S)-3-(N-Boc)aмино-4-фенил-2- оксибурат весом 0,95 г был подвергнут перекристаллизации с
применением n-гексана с получением смеси изомеров в отношении (2S, 3S): (2R, 3S) = 99,3: 0,7 с
выходом 83,2%, результаты ЯМР-анализа которой приведен ниже.
1H-ЯМР(CDCl3)
δ (ppm) 1,26 (d d, 6H, J = 6,3; 8,2 Hz)
1,35(S,9H)
2,67 ~ 2,78
(m,2H)
3,31 (br, 1H)
4,22 ~ 4,38 (m,2H)
4,82 ~ 4,93 (br, 1H)
5,00 (td,
1H, J= 6,2; 12,5 Hz)
7,14 ~ 7,32 (т, 5H)
(В) Синтез (2, S
3S)-3-(N-Вос)амино-4-фенил-2- оксимасляной кислоты
В 10 мл метанола растворили 0,5 г изопропил (2S,
3S)-3-(N-Вос)амино-4-фенил-2-оксибутирата с последующим добавлением 1,38 г водного 3 н.
раствора гидроксида натрия, полученную смесь промывали при комнатной температуре в течение 2 ч для содействия
реакции гидролиза. После подтверждения окончания реакции к полученной смеси добавили
водный 1 н. раствор соляной кислоты, чтобы довести pH смеси до 3,0, с последующей концентрацией при пониженном
давлении. Концентрат разбавили этилацетатом и водой с последующим разделением слоев и
полученный этилацетатный слой промыли насыщенным раствором хлорида натрия. Далее этилацетатный слой высушили над
безводным сульфатом магния, отфильтровали и сконцентрировали с получением 0,38 г (2S,
3S)-3-(N-Вос)амино-4-фенил-2-оксимасляной кислоты с выходом 86,9%. Ниже приведены результаты ЯМР-анализа:
1H-ЯМР(ДМСО-d6)
δ (ppm)1,26(S,9H)
2,62 ~ 2,78 (m,2H)
3,39 ~ 3,55 (m, 1H)
3,85 ~ 4,06 (m,2H)
6,67(br d, 1H)
7,10 ~ 7,30(m,5H)
Пример 2
(А) Синтез изопропил
(3S)-3-N-ацетиламино-4-циклогексил-2- оксобутирата
В 9 мл диметилсульфоксида растворили 2,0 г изопропил (2R,
3S)-3-N-ацетиламино-4-циклогексил-2-оксибутирата с последующим добавлением 3 мл
уксусного ангидрида, полученную смесь перемешивали в течение ночи при комнатной температуре. После подтверждения
окончания реакции к полученной смеси добавили воду с последующей экстракцией с
этилацетатом. Экстрагированный раствор промыли водным насыщенным раствором гидрокарбоната натрия и насыщенным раствором
хлорида натрия и высушили над безводным сульфатом магния. После высушивания были
легко проведены фильтрация и концентрация при пониженном давлении с количественным получением 2,06 г изопропил
(3S)-3-N-ацетиламино-4-циклогексил-2-оксобутирата. Ниже приведены результаты
ЯМР-анализа:
1H-ЯМР(CDCl3)
δ (ppm) 0,78 ~ 1,98 (m, 13H)
1,36 (dd, 6H, J=1,2;6,
3Hz)
2,04 (s, 3H)
5,11 ~ 5,30 (m, 2H)
6,22 ~ 6,
34(br,1H)
(Б) Синтез изопропил (38)-3-N-ацетиламино-4-циклогексил-2- оксибутирата
В 10 мл изопропанола
растворили 1,0 г изопропил (3S)- 3-N-ацетиламино-4- циклогексил-2-оксобутирата
с последующим добавлением 0,76 г изопропоксида алюминия, полученную смесь дефлегмировали при повышенной температуре в
течение 2 ч. После подтверждения окончания реакции к полученной смеси добавили
водный 1 н. раствор соляной кислоты, чтобы довести pH смеси до 3,0, с последующей концентрацией при пониженном давлении.
Концентрат разбавили этилацетатом и водой с последующим разделением слоев и
полученный водный слой экстрагировали с этилацетатом. Экстрагированный раствор промыли водой и насыщенным раствором хлорида
натрия, высушили над безводным сульфатом магния и далее отфильтровали и
сконцентрировали с получением 0,97 г изопропил (2S, 3S)-3-N- ацетиламино-4-циклогексил-2-оксибутирата с выходом 98,0%. Ниже
приведены результаты его
ВЭЖХ- и ЯМР-анализа:
Условия
ВЭЖХ-анализа
Колонка - Inertsil ODS-2 (GL Science) 4,6 ⌀ х 250 мм
Температура колонки - 35oC
Элюент - ацетонитрил: водный 0,02 М раствор дигидрофосфата
аммония (pH 2,5) = 4: 6
Скорость потока - 1,0 мл/мин
Время удерживания - 11,1 мин для (2S, 3S)- соединения - 11,3
мин для (2R, 3S)-соединения
Отношение выходов - (2S,3S):
(2R, 3S) = 96: 4
1H-ЯМР(CDCl3)
δ (ppm) 0,62 ~ 1,98 (m, 13H)
1,30 (d, 6H, J = 6,2 Hz)
2,02 (s, 3H)
3,45 (d, 1H,J = 5,3 Hz)
4,29
(dd, 1H, J=2,9; 5,3 Hz)
4,38 ~ 4,53 (m, 1H)
5,13 (td, 1H, J= 6,3; 12,5 Hz)
5,95 (br, 1H)
(В)
Синтез (2S, 3S)-3-N-ацетиламино-4-циклогексил-2- оксимасляной
кислоты
В 150 мл метанола растворили 190,1 г изопропил (2S, 3S)-3-N-ацетиламино-4-циклогексил-2-оксибутирата с последующим
добавлением 100 мл водного 1 н. раствора гидроксида натрия,
полученную смесь для содействия реакции гидролиза перемешивали при комнатной температуре в течение 2 часов. После подтверждения окончания
реакции к полученной смеси добавили водный 1 н. раствор
соляной кислоты, чтобы довести pH смеси до 3,0, с последующей концентрацией при пониженном давлении. Концентрат разбавили этилацетатом и водой
с последующим разделением слоев и полученный
этилацетатный слой промыли насыщенным раствором хлорида натрия. Далее этилацетатный слой высушили над безводным сульфатом магния, отфильтровали и
сконцентрировали с получением 154,0 г (2S,
3S)-3-N-ацетиламино-4-циклогексил-2-оксимасляной кислоты с выходом 95,0%. Ниже приведены результаты ЯМР-анализа:
1H-ЯМР(ДМСО-d6)
δ
(ppm) 0,68 ~ 1,78 (m, 13H)
1,77(s,
3H)
3,40 ~ 3,51 (m, 1H)
3,86 (d, 1H, J = 2,8 Hz)
4,10 ~ 4,26 (m, 1H)
7,43 (brd, 1H)
Промышленная
применимость
Согласно настоящему изобретению
эфиры эритро-3-амино-2- оксимасляной кислоты или эритро-3-амино-2-оксимасляная кислота с чистотой 90% или более, предпочтительно 95% или более,
могут быть легко получены с хорошими выходами
восстановлением эритроселективного оптически активного эфира трео- или трео-эритро-3-амино-2-оксомасляной кислоты, при этом указанный эфир может быть
легко синтезирован из недефицитной оптически
активной аминокислоты. Эти эритросоединения представляют собой важные интермедиатные продукты фармацевтических реагентов, например ингибиторов протеазы
ВИЧ, поэтому настоящее изобретение может иметь
промышленное применение как способ получения интермедиатных продуктов.
Реферат
Изобретение относится к разработке способа получения оптически активного эфира
эритро- 3-амино-2-оксимасляной кислоты, представляющего собой важный
интермедиатный продукт фармацевтических реагентов, в частности ингибитора протеазы ВИЧ. Описывается способ получения оптически
активных эфиров эритро-3-амино-2-оксимасляных кислот общей формулы III
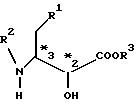
где R1 - фенильная или циклогексильная группа, R2 - защитная группа, a R3 - замещенный или незамещенный низший алкил с числом углеродных атомов от 1 до 6, стерическая конфигурация *3 представляет собой S- или R-конфигурацию, включающий окисление гидроксильной группы в положении 2-эфира 3-амино-2-оксимасляной кислоты, оптически активного в положении 3, как представлено в формуле I:
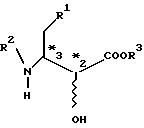
где R1, R2 и R3 те же, что и выше, а стерическая конфигурация *3 представляет собой S- или R-конфигурацию, с получением оптически активного эфира 3-амино-2-оксомасляной кислоты, представленного формулой II:

где R1, R2 , R3 и *3 независимо друг от друга те же, что и выше, и эритроселективное восстановление карбонильной группы в положении 2 путем применения алкоксида алюминия. Технический результат - повышение выхода и чистоты целевого продукта. 3 с.п.ф-лы.
Формула

где R1 - фенильная или циклогексильная группа,
R2 - защитная группа;
R3 - замещенный или незамещенный низший алкил с числом углеродных атомов от 1 до 6;
стерическая конфигурация *2 представляет собой S-конфигурацию в случае S-конфигурации *3 и R-конфигурацию в случае R-конфигурации *3,
включающий окисление эфира трео- или трео эритро-3-амино-2-оксимасляной кислоты, оптически активный в положении 3, как представлено в формуле I

где R1, R2 и R3 те же, что и выше;
стерическая конфигурации *3 представляет собой S- или R-конфигурацию,
с получением оптически активного эфира 3-амино-2-оксомасляной кислоты, представленного формулой II

где R1, R2, R3 и *3 независимо друг от друга те же, что и выше,
и эритроселективное восстановление карбонильной группы в положении 2 путем применения алкоксида алюминия.
Комментарии