Способ получения винилацетата - RU2197472C2
Код документа: RU2197472C2
Чертежи


Описание
Настоящее изобретение относится к способу получения винилацетата контактированием этилена, уксусной кислоты и кислородсодержащего газа с палладиевым катализатором на носителе.
Приготовление палладиевых катализаторов на носителе для получения винилацетата обычно включает пропитку соответствующего носителя палладиевым соединением с последующей конверсией палладиевого соединения до практически металлического палладия.
Способы приготовления катализаторов с пропитанной оболочкой описаны, например, в патентах США 3822308, 4048096, 5185308 и 5332710, Канады 2128162, США 4087622, Канады 2128154 и 2128161 и США 5422329.
Способы приготовления катализаторов необолочкового типа описаны, например, в патентах США 3743607, Великобритании 1333449, США 3939199 и 4668819, заявках ЕР 330853, ЕР 403950 и ЕР 431478 и патенте Канады 2071698.
В патенте США 5336803 описан способ предварительной обработки палладиевозолотых катализаторов, в котором катализатор выдерживают в присутствии окислителя, такого как воздух, при температуре, по меньшей мере достаточной для частичного окисления палладия; окислитель удаляют и вводят инертный газ, такой как азот; затем катализатор вновь выдерживают при температуре до 500oС в присутствии восстановителя, такого как водород или этилен. Описанный в этом патенте способ проиллюстрирован на примере "обычного катализатора, номинально содержащего 1% палладия и 0,5% золота".
Известно, что активность палладиевых катализаторов на носителях в процессе получения винилацетата по мере эксплуатации снижается. Если каталитическая активность, а следовательно, и производительность процесса снижается до технически неприемлемого уровня, необходимы регенерация и/или замена катализатора. Дезактивация винилацетатных катализаторов описана Abel и др. в Chem. Eng. Technol. 17 (1994), 112-118.
Простое увеличение количества палладия в катализаторе для увеличения срока службы этого катализатора сопряжено с определенной проблемой, состоящей в том, что начальная активность катализатора может оказаться слишком высокой для проведения безопасного и/или регулируемого процесса в промышленном масштабе, например, из-за ограниченной возможности теплоотвода установки.
Поэтому сохраняется необходимость в разработке позволяющего разрешить эту проблему способа приготовления палладиевого катализатора на носителе, предназначенного для получения винилацетата.
Таким образом, в соответствии с настоящим изобретением предлагается способ получения винилацетата, при осуществлении которого предусмотрено контактирование этилена, уксусной кислоты и кислородсодержащего газа с палладиевым катализатором на носителе, приготовленным по способу, включающему следующие стадии: (а) пропитку каталитического носителя палладиевым соединением, (б) конверсию палладиевого соединения до практически металлического палладия и (в) спекание палладия на носителе при температуре выше 500o С.
По настоящему изобретению описанная выше техническая задача решается за счет того, что палладий на носителе спекают при температуре выше 500oС.
Не основываясь на какой-либо теории, полагают, что эта стадия спекания вызывает рост частиц металлического палладия, который снижает первоначальную активность катализатора. Таким образом, предлагаемый способ обеспечивает приготовление катализаторов, характеризующихся высокой концентрацией палладия и технически приемлемой начальной активностью, и такие катализаторы обладают технически более длительным сроком службы по сравнению с известными катализаторами. Такая стадия спекания увеличивает также средний размер пор кремнийдиоксидных носителей. Было установлено также, что катализаторы по настоящему изобретению менее чувствительны к нежелательным эффектам избыточной концентрации промотора, такого как ацетат калия.
Стадию спекания (в) предпочтительно осуществляют с использованием газообразного восстановителя, но ее можно осуществлять в присутствии газообразного окислителя или в инертном газе. Приемлемыми газообразными восстановителями являются водород и монооксид углерода. Приемлемым газообразным окислителем является кислород. Его можно разбавлять инертным газом. Приемлемыми инертными газами для использования индивидуально или в сочетании с газообразными окислителями или восстановителями являются азот, диоксид углерода и гелий. Приемлемая температура для стадии спекания составляет от более 500 до 1000oС, предпочтительно от 650 до 1000oС. Предпочтительная продолжительность стадии спекания составляет 1-24 ч. В случае использования газообразного окислителя катализатор нуждается в последующем восстановлении. Катализатор можно продувать инертным газом перед спеканием и в период нагрева (для безопасности), а также во время охлаждения (до менее 100oС, более предпочтительно до менее 60o С) с целью предотвратить любое повторное диспергирование палладия. Нагрев и охлаждение можно проводить с любой приемлемой или реальной скоростью. Стадию спекания (в) в промышленном масштабе можно проводить в башне или сосуде, который соответствует вышеуказанным условиям осуществления способа. В ходе проведения процесса катализатор можно перемешивать с помощью газового потока. Можно применять печь с вращающимся шнеком. В лабораторном масштабе можно применять трубу, горизонтально или вертикально установленную в электропечи, при условии эффективного контактирования между газом и твердым материалом (обычно следует принимать во внимание соотношение длина/диаметр). Может потребоваться предварительный нагрев газового потока. Время и температура спекания взаимосвязаны: чем выше температура, тем короче требуемое время. Для специалистов в данной области техники очевидно, каким образом привести эти параметры в соответствие с масштабом проводимого процесса. Как правило, стадия спекания (в) вызывает рост частиц металлического палладия с 3-4 до 8-15 нм в диаметре.
Конверсия палладиевого соединения до практически металлического палладия на стадии (б) может осуществляться восстановлением, которое может непосредственно предшествовать стадии спекания (в), и осуществлением обеих технологический стадий в том же самом оборудовании.
Способ приготовления катализатора по настоящему изобретению можно применять для приготовления однородно пропитанных катализаторов или катализаторов с пропитанной оболочкой, предназначенных для проведения процессов получения винилацетата в псевдоожиженном слое или неподвижном слое.
Предлагаемый по настоящему изобретению способ приготовления катализаторов можно применять для приготовления катализаторов, обладающих высокой концентрацией палладия, например, превышающей 0,5 вес.%, предпочтительно более 1 вес.% в пересчете на общий вес катализатора. Концентрация палладия может достигать 5 вес.% в случае псевдоожиженного слоя или достигать 10 вес.% при применении в неподвижном слое. Можно было бы ожидать очень высокой начальной активности палладиевого катализатора на носителе, характеризующегося высокой концентрацией палладия, если его готовят по известному способу, и она могла бы быть даже настолько высокой, что стала бы небезопасной и/или нерегулируемой в случае применения в промышленном масштабе. Однако при приготовлении в соответствии со способом по настоящему изобретению начальная активность катализатора оказывается пониженной в сравнении с активностью обычно приготовленного катализатора, в то время как благодаря высокой концентрации палладия достигается технически приемлемая активность в течение более длительного срока службы катализатора.
Приемлемые для приготовления как катализаторов с пропитанной оболочкой, так и равномерно пропитанных катализаторов каталитические носители могут включать пористый диоксид кремния, оксид алюминия, диоксид кремния/оксид алюминия, диоксид титана, диоксид циркония или уголь, предпочтительно диоксид кремния. Приемлемый удельный объем пор носителя может составлять 0,2-3,5 мл/г носителя, удельная площадь их поверхности может составлять 5-800 м2/г носителя, а кажущаяся или объемная плотность может составлять 0,3-1,5 г/мл. В случае катализаторов, применяемых в процессах в неподвижном слое, размеры частиц носителя, как правило, равны 3-9 мм. В случае катализаторов, применяемых при проведении процессов в неподвижном слое, частицы носителя обычно могут иметь форму сфер, таблеток, экструдата, гранул или любую другую приемлемую форму. В случае применения катализаторов в процессах, проводимых в псевдоожиженном слое, носитель, как правило, может характеризоваться таким распределением частиц по размерам, что по меньшей мере 60% частиц катализатора имеют диаметр менее 200 мкм, предпочтительно по крайней мере 50% частиц имеют диаметр менее 105 мкм и не более 40% каталитических частиц имеют диаметр менее 40 мкм.
На стадии (а) носитель предпочтительно пропитывают палладиевым соединением в приемлемом растворителе. В качестве таких растворителей могут применяться вода, карбоновые кислоты, такие как уксусная кислота, бензол, толуол, спирты, такие как метанол или этанол, нитрилы, такие как ацетонитрил или бензонитрил, тетрагидрофуран или хлорированные растворители, такие как дихлорметан. В предпочтительном варианте в качестве растворителя применяют воду и/или уксусную кислоту. Носитель рекомендуется пропитывать ацетатом, сульфатом, нитратом, хлоридом палладия или другими галоидсодержащими солями палладия, такими как H2PdCl4, Na2PdCl4 или K2PdCl4. Предпочтительным водорастворимым соединением является Na2PdCl4. Предпочтительным растворимым в уксусной кислоте палладиевым соединением является ацетат палладия.
Пропитку носителя можно осуществлять контактированием носителя с раствором палладиевого соединения путем окунания, погружения или опрыскивания. Эту пропитку можно осуществлять в одну или несколько стадий или проведением непрерывного процесса. Носитель можно подвергать контактированию с пропиточным палладиевым раствором обработкой в барабане, в режиме вращения и вихревого движения или аналогичным путем с достижением равномерной пропитки. Пропитку, как правило, проводят при комнатной температуре. В случае ацетата палладия в уксусной кислоте можно создавать повышенную температуру, например, достигающую 120oС, предпочтительно 100oС, более предпочтительно 60oС. Такую пропитку проводят осторожно, избегая разрушения или абразивного износа носителя. Носитель можно заполнять пропиточным раствором на 5-100% объема его пор.
Помимо палладиевых соединений носитель можно также пропитывать на стадии (а) соединениями золота, меди и/или никеля, предпочтительно золота, которые конвертируют до металла совместно с палладием на стадии (б) и которые содержатся в частицах металлического палладия в виде смесей и/или сплавов с палладием. Приемлемые соединения золота включают золотохлористоводородную кислоту (HAuCl4), NaAuCl4, KAuCl4, диметилзолотоацетат, барийацетоаурат или ацетат золота, предпочтительно НАuСl4. Эти промоторы могут быть использованы в количестве 0,1-10 вес.% каждого металлического промотора, содержащегося в готовом катализаторе.
В дополнение к палладию и необязательно золоту, меди и/или никелю на любой соответствующей стадии процесса приготовления носитель может быть также пропитан одной или несколькими солями металлов группы I, группы II, лантанидов или переходных металлов, предпочтительно кадмия, бария, калия, натрия, железа, марганца, никеля, сурьмы и/или лантана, которые содержатся в готовом катализаторе в виде солей, как правило ацетатов. Обычно содержится калий. Приемлемыми солями этих соединений являются ацетаты и хлориды, но можно применять любую растворимую соль. Эти промоторы могут быть использованы в количестве 0,1-15 вес.%, предпочтительно 3-9 вес.% каждой промоторной соли, входящей в состав готового катализатора.
При необходимости ввести палладий или промотор в количестве, превышающем то количество, которое обеспечивается растворимостью соли в растворителе, пропитанный носитель необязательно можно сушить, а стадию пропитки повторять два или большее число раз. Стадию сушки можно осуществлять при температуре, достигающей 120oС, предпочтительно 100oС и наиболее предпочтительно при 60oС. Стадию сушки можно осуществлять при комнатной температуре и под пониженным давлением. На стадии сушки можно использовать воздух, азот, гелий, диоксид углерода или любой приемлемый инертный газ. С целью упростить сушку катализатор можно обрабатывать в барабане, приводить систему во вращательное движение или перемешивать с помощью газового потока.
Для приготовления катализаторов с пропитанной оболочкой осуществляют контактирование мокрого или сухого пропитанного носителя с основным раствором при обработке в барабане с приданием системе вихревого, вращательного движения, с перемешиванием или т.п. Этот основный раствор можно также наносить опрыскиванием пропитанного носителя во время обработки в барабане с приданием вращения, с перемешиванием или т.п. Основаниями могут служить гидроксиды, карбонаты или силикаты металлов групп I или II. Типичными примерами являются гидроксид натрия, метасиликат натрия, гидроксид калия, метасиликат калия и гидроксид бария. Основный раствор можно наносить в одну или несколько стадий с соответствующими интервалами выдержки между этими стадиями обработки. Стадию осаждения обычно проводят при комнатной температуре, но ее можно повысить до 100oС. Можно применять любой растворитель, в котором растворим основной материал, но предпочтительна вода. Контактирование основания с пропитанным носителем необходимо осуществлять в течение такого интервала времени, чтобы обеспечивалась возможность осаждения металлических солей в оболочке. Такой процесс обычно длится более одного часа, предпочтительно от 8 до 24 ч. Для осаждения обычно требуется оптимальное количество основания и, как правило, необходим его избыток, причем обычно оно равно 1,8-кратному номинальному количеству, необходимому для образования из металлических солей гидроксидов.
Пропитанный носитель можно промывать для удаления анионных примесей, например нитратных, сульфатных и обычно галогенидных. Для удаления хлоридов промывку деионизированной водой следует проводить до тех пор, пока испытание нитратом серебра не покажет отсутствие каких-либо хлоридов. Содержание анионных примесей должно быть минимальным. Содержание катионных примесей должно быть сведено к минимуму; так, например, количество натрия в сухом катализаторе должно составлять менее 0,5 вес.%, предпочтительно менее 0,2 вес. %. В столь низкой концентрации вполне допустимо наличие таких примесей, а их концентрация, равная абсолютному нулю, никакого существенного значения не имеет. В промышленном масштабе можно проводить периодический процесс промывки. Для ускорения процесса можно применять теплую воду. Кроме того, для замещения хлоридных и натриевых ионов можно применять ионообменные растворы (такие, как калийацетатные). Более того, реагенты для приготовления следует выбирать таким образом, чтобы избежать применения хлоридных и натриевых ионов, используя, например, метасиликат калия вместо натриевой соли.
В зависимости от используемых реагентов на стадии (б) палладиевое соединение можно превращать в металл до или после описанной необязательной стадии промывки. Можно применять жидкие восстановители, такие как водный гидразин, формальдегид, формиат натрия, метанол или спирты, предпочтительно водный гидразин. Восстановление можно также проводить с помощью газов, таких как монооксид углерода, водород и этилен. Их можно разбавлять инертным газом, таким как азот, диоксид углерода или гелий. Как правило, восстановление газами проводят при повышенных температурах порядка 100-500o С до восстановления материала. Восстановление обычно проводят в жидких восстановителях и при комнатной температуре, но можно проводить при температуре до 100oС.
После конверсии палладиевого соединения до металла его спекают по методике, представленной в настоящем описании. Эту стадию спекания (в) можно осуществлять вслед за стадией (б) последующим нагреванием катализатора в газообразном восстановителе до температуры выше 500oС. Далее материал можно пропитывать промоторными солями, как это описано выше.
Контактирование этилена, уксусной кислоты и кислородсодержащего газа с палладиевым катализатором на носителе, приготовленным в соответствии со способом приготовления катализатора по настоящему изобретению, можно осуществлять по методам, известным в данной области техники. Так, например, контактирование реагентов с катализатором можно проводить в неподвижном или в псевдоожиженном слое при температуре в интервале 145-195oС и под давлением в интервале 1-20 атм. Получаемый винилацетат можно выделять обычными методами, известными в данной области техники.
Ниже сущность изобретения проиллюстрирована со ссылками на фиг. 1-3, на примерах и экспериментах. На фиг. 1 схематически представлены некоторые из возможных способов приготовления катализаторов в соответствии с настоящим изобретением. На фиг. 2 представлен график, позволяющий сопоставить производительность катализатора, приготовленного в соответствии с изобретением, с производительностью катализатора, приготовленного не в соответствии с изобретением, в зависимости от времени эксплуатации. На фиг. 3 представлен график, позволяющий сопоставлять влияние количества промоторного ацетата калия на активность катализатора, приготовленного в соответствии с изобретением, с его влиянием на активность катализаторов, приготовленных не в соответствии с изобретением.
Как показано на фиг. 1, катализаторы однородного типа (необолочковые) могут быть приготовлены осуществлением стадий пропитки носителя палладиевыми солями и необязательными промоторами с последующими сушкой и восстановлением металлов. Далее такой материал можно подвергать необязательным промывке и сушке перед спеканием в соответствии с настоящим изобретением и заключительной пропитке необязательными промоторами, такими как ацетаты калия, натрия, кадмия или бария.
Для приготовления катализаторов оболочкового типа носитель, пропитанный палладием и необязательными промоторами, такими как золото, можно подвергать необязательной сушке. Затем осаждают металлы. Далее такой материал можно направлять либо (I) на восстановление до металлов, промывку и сушку, либо (II) на промывку и сушку с последующим восстановлением до металлов. После этого материал подвергают спеканию в соответствии с настоящим изобретением с последующей пропиткой промоторами, такими как ацетата калия, натрия, кадмия или бария.
Пример 1
Катализатор А готовили в
соответствии с настоящим изобретением таким образом, чтобы он характеризовался следующим номинальным составом (т.е. без каких-либо потерь в процессе приготовления): 1,8 вес.% палладия, 0,8 вес.%
золота и 7 вес.% ацетата калия.
1. Пропитка носителя
15 г сферических частиц диоксида кремния КА 160 в качестве носителя (4-6 мм, фирма SudChemie) вводили в раствор 1,0264 г
тригидрата тетрахлорпалладата натрия (фирма Johnson Matthey) и 0,2655 г тригидрата золотохлористоводородной кислоты (фирма Aldrich) в 9,1 г деионизированной воды. Материал добавляли в виде одной
порции и смесь перемешивали в вихревом режиме до тех пор, пока равномерно не абсорбировался весь раствор. Далее пропитанный носитель оставляли стоять в закрытом состоянии в течение двух часов при
комнатной температуре.
2. Осаждение соединений палладия и золота на носителе.
Раствор 1,7 г пентагидрата метасиликата натрия (фирма Fisons) в 18 г воды добавляли к пропитанному носителю со стадии 1. Смесь кратковременно несколько раз перемешивали в вихревом режиме в общей сложности в течение 15 мин, предотвращая образование "зон", а затем оставляли стоять в течение ночи.
3. Восстановление палладия и золота до практически металлического состояния
Водную фазу вышеуказанного материала со стадии 2 обрабатывали 5 г 55%-ного
гидразингидрата (фирма Aldrich).
4. Промывка соединений на носителе
Водную фазу декантировали и материал со стадии 3 четырежды промывали приблизительно 50 мл воды, декантируя
после каждой промывки. Полученный материал переносили в стеклянную колонну, снабженную запорным краном, а затем промывали деионизированной водой с расходом приблизительно 1 л за 12 ч до тех пор, пока
испытание нитратом серебра не давало отрицательного результата. Материал сушили при 60o С в течение ночи в сушильном шкафу с принудительной конвекцией и охлаждали.
5.
Спекание палладия (и золота)
Палладиевый материал на носителе со стадии 4 переносили в горизонтальную печь и заполняли им центральную часть кварцевой трубчатой вставки с заполнением
свободного пространства кварцевой ватой и носителем КА 160 (после предварительной тщательной сушки). Эту кварцевую трубчатую вставку помещали внутрь стальной трубки и подключали к источникам газов.
Температуру в печи повышали до 150oС со скоростью 10oС/мин и эту температуру поддерживали в течение 2 ч при постоянном токе азота. Вводили поток водорода при среднечасовой
скорости подачи 60/ч, а подачу азота прекращали. Температуру в печи повышали до 800oС со скоростью 30oС/мин и эту температуру поддерживали в течение 11 ч. По прошествии этого
периода полученному материалу давали остыть до комнатной температуры в токе водорода. Перед выгрузкой материала подачу потока азота возобновляли, а подачу потока водорода прекращали.
6. Пропитка ацетатом металла
Сухой материал со стадии 5 пропитывали 1,16 г безводного ацетата калия (фирма Aldrich), растворенного в 8,8 г воды. Смесь осторожно перемешивали в вихревом режиме
до абсорбции жидкости. Полученный материал вновь сушили в течение ночи при 60oС.
Пример 2 (сравнительный)
Катализатор Б готовили в соответствии со способом,
описанным в примере 1, за исключением того, что стадию спекания 5 не осуществляли.
Пример 3 (сравнительный)
Катализатор В готовили в соответствии со способом, описанным в
примере 1, за исключением того, что стадию спекания 5 не осуществляли и содержание металла уменьшали таким образом, чтобы достичь такой же начальной активности, что и у катализатора, приготовленного
согласно примеру 1.
Испытание катализатора в микрореакторе
Катализаторы, приготовленные по описанным выше методам, испытывали в микрореакторе с применением следующей общей
методики. Испытания проводили под абсолютным давлением 7,8 бар и при 150oС с применением гранулированного катализатора (приготовленного по описанному выше способу и в количестве, указанном
в таблице 1), разбавленного 60 мл стеклянных шариков диаметром 1 мм и загруженного в трубку из нержавеющей стали с внутренним диаметром 10-11 мм. Катализатор подготавливали к работе выдержкой при
160oС под абсолютным давлением 7,8 бар в течение 3 ч в токе азота, а затем при 150oС в токе этилена. Далее пары уксусной кислоты смешивали с этиленом и пропускали над
катализатором в течение по меньшей мере 50 мин. В исходный газ постепенно добавляли смесь 21% кислорода в гелие, одновременно поддерживая максимальную температуру каталитического слоя 150o
С. Температуру горячего участка катализатора поддерживали на уровне 150oС. Конечный состав смеси реагентов выражался объемным соотношением этилен:уксусная кислота:кислород:гелий, равным 53,
1: 10,4: 7,7: 28,6, а общая среднечасовая скорость подачи газа составляла 3850 ч-1. Поток продукта анализировали в паровой фазе через часовые интервалы с помощью смонтированного на
технологической линии газового хроматографа.
Активность катализатора рассчитывали в граммах полученного винилацетата на литр катализатора в час (объемная производительность, ОПр), а селективность катализатора рассчитывали по процентной доле превращенного этилена, содержавшегося в продукте. Данные приводили в пересчете на средние арифметические активности и селективности, которые определяли в промежутке от 17 до 22 часов после достижения полного расхода потока кислорода.
Результаты сопоставления активности катализаторов А, Б и В представлены в таблице 1.
Сопоставление активности катализаторов А и Б по данным таблицы 1 показывает, что стадия спекания (стадия 5) вызывала снижение активности катализатора А. Это соответствует росту размеров частиц палладия и снижению удельной площади поверхности частиц металлического палладия. Катализатор В готовили с более низким содержанием металла, чем у катализаторов А и Б, причем содержание металла подбирали с таким расчетом, чтобы начальная активность была такой же, как и у катализатора А. Таким образом, от катализаторов А и В можно было бы ожидать одинакового начального технологического поведения. Тем не менее ожидалось также, что катализатор А сохранит свою производительность в течение более длительного периода по сравнению с катализатором В, если рост размеров палладиевых частиц и уменьшение удельной площади поверхности частиц металлического палладия вызовут снижение начальной активности, что проиллюстрировано в примерах 7 и 8.
Испытание катализаторов в более крупногабаритных реакторах.
Катализаторы А и В испытывали в более крупногабаритных трубчатых реакторах следующим образом. 77,5 г катализатора А (пример 7) и 77,5 г катализатора В (пример 8, сравнительный) загружали в отдельные для каждого из них 6-футовые реакторные трубки. Эти две трубки помещали в одну и ту же баню с псевдоожиженным слоем песка. Температуру такой бани можно было регулировать, каждую трубку снабжали собственной системой подачи газообразного/жидкого сырья и осуществления манипуляций с продуктом. Начинали подавать поток азота с расходом 1106 мл/мин (при стандартных температуре и давлении) и поток этилена с расходом 2590 мл/мин (при стандартных температуре и давлении). Песчаную баню и трубки нагревали до 150oС и давление в реакторах повышали до 115 фунтов/кв.дюйм. В испаритель начинали подавать поток уксусной кислоты (содержавшей 2 вес. % воды) с расходом 155 г/ч и пары смешивали с азотом и этиленом. Небольшой поток уксусной кислоты (2 вес.% воды, 0,0285 вес.% ацетата калия) с расходом 13 г/ч вводили в зону предварительного нагрева с целью испарения вместе с основным газовым потоком. По истечении нескольких часов начинали подавать кислород с расходом 153 мл/мин (при стандартных температуре и давлении). Поток продукта анализировали с помощью смонтированного на технологической линии газового хроматографа, а затем конденсировали с получением сырого жидкого продукта, состоявшего из винилацетата, уксусной кислоты и воды, а оставшиеся газы сбрасывали в атмосферу, отбирая пробы с помощью смонтированного на линии газового хроматографа. Осуществляли контроль за ходом получения винилацетата с применением обоих катализаторов. Поскольку катализаторы дезактивировались, постоянную производительность первоначально поддерживали постепенным увеличением расхода кислородного сырья до максимального уровня 425 мл/мин (при стандартных температуре и давлении). При полном расходе потока кислорода состав газового сырья выражался объемным соотношением этилен:уксусная кислота:вода:кислород:азот, равным 49,7:19,6:1,3:8,2: 21,2, а общая среднечасовая скорость подачи газа составляла 2261 ч-1 (при стандартных температуре и давлении). После достижения полного расхода потока кислородного сырья постоянную производительность катализатора А в дальнейшем поддерживали постепенным повышением температуры песчаной бани от приблизительно 150 до 160oС. Поскольку обе трубки находились в одной и той же песчаной бане, производительность катализатора В опускалась до уровня, ниже чем у катализатора А, т.к. он дезактивировался быстрее. На фиг. 2 представлена нормализованная ежедневная производительность для катализаторов А и В в зависимости от числа дней эксплуатации. На фиг. 2 видно, что хотя начальная производительность обоих катализаторов была одинаковой, по прошествии пяти дней эксплуатации производительность сравнительного катализатора, т.е. катализатора В, оказывалась ниже производительности катализатора по изобретению, т. е. катализатора А. Изучение угловых коэффициентов кривых для производительности обоих катализаторов показывает, что катализатор А сохранял производительность приблизительно 1, в то время как производительность катализатора В с течением времени медленно снижалась, достигая в конце уровня 0,7. К концу эксперимента проводили испытание катализатора А на производительность в сравнении с катализатором В путем регулирования расхода потока кислородного сырья и/или температуры песчаной бани. Видно, что производительность поочередно повышалась и понижалась в соответствии с фиг. 2, причем следует отметить, что по производительности катализатор А всегда превосходил катализатор В. Катализатор А демонстрировал более низкую скорость дезактивации по сравнению с катализатором В даже несмотря на то, что по своей первоначальной активности они были очень похожими.
Пример 9. Дополнительные испытания катализаторов с применением микрореактора
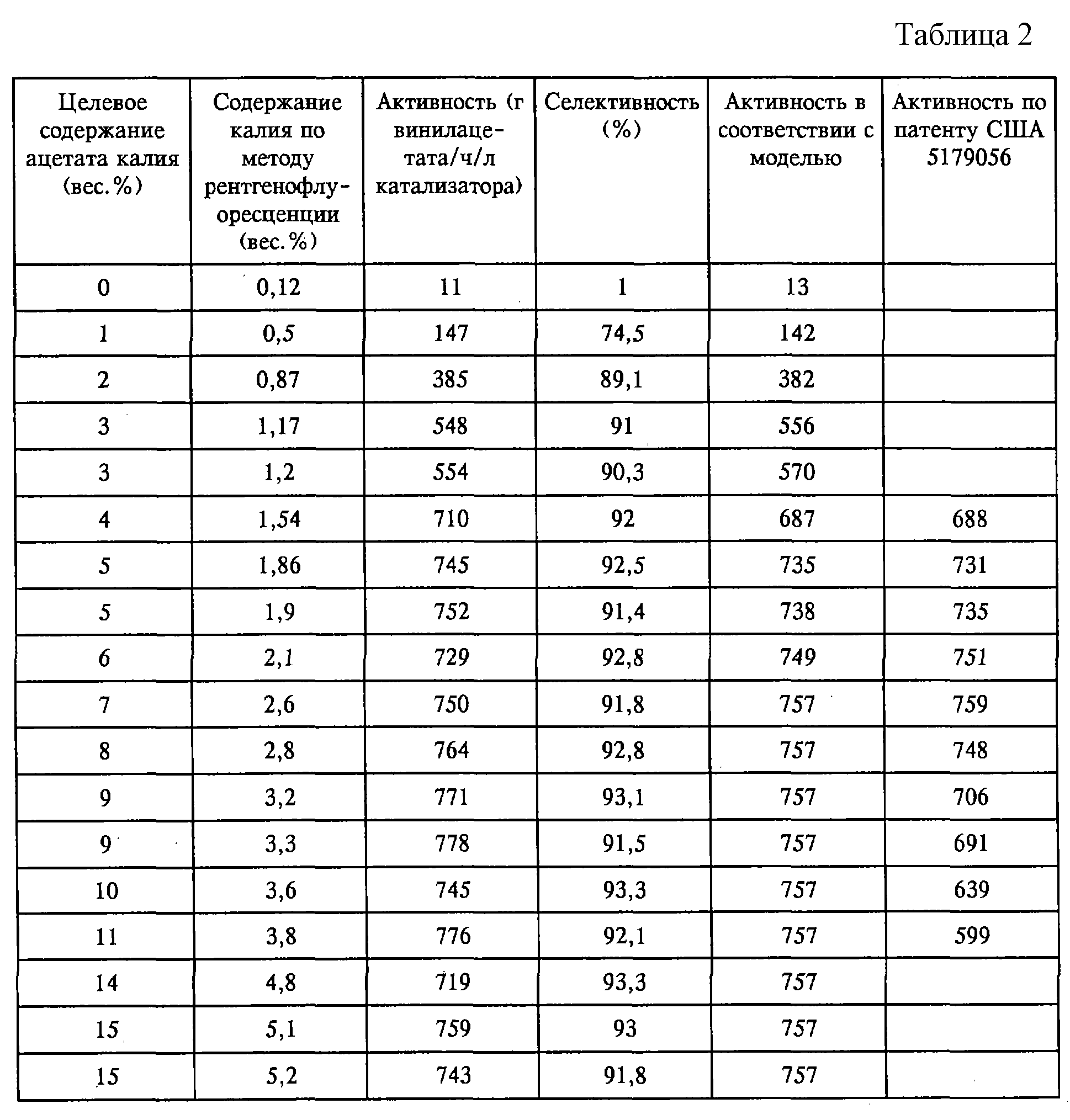
Две дополнительные порции катализатора готовили в соответствии со способом, который описан в примере 1, за исключением того, что количества используемых реагентов увеличивали в 9 раз. По
завершении стадии промывки и сушки каждую порцию катализатора точно делили на 9 равных частей и пропитывали целевым количеством (в вес.%) ацетата калия (см. таблицу 2). Эти образцы катализатора
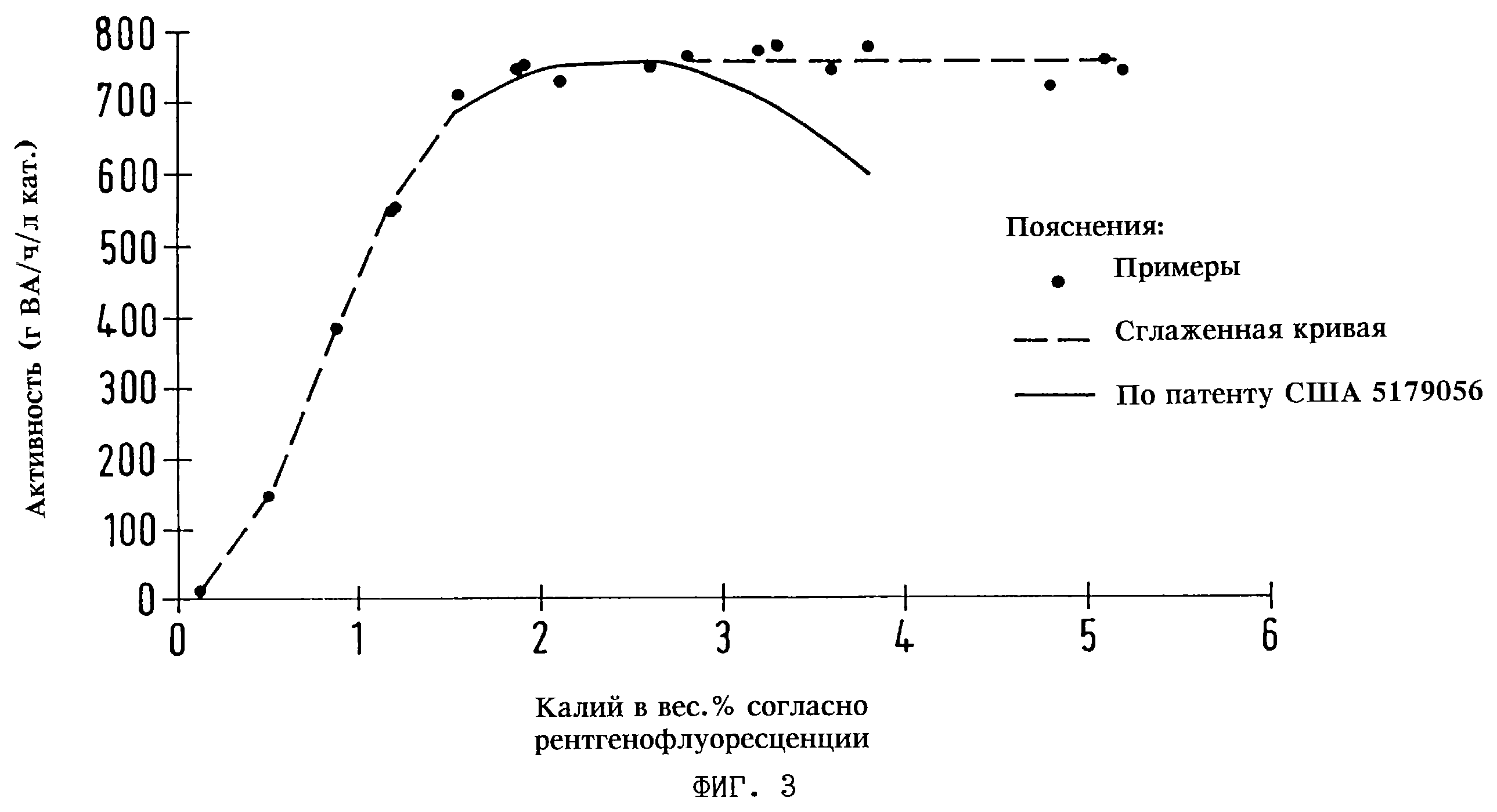
испытывали в соответствии с методикой испытаний для примеров 1-3. На фиг. 3 показана активность, которая достигалась с использованием таких каталитических образцов, в сравнении с активностью
соответствующих катализаторов, приведенной в патенте США 5179056 (эту активность определяли экстраполяцией в соответствии с моделью, описанной в патенте США 5179056). На фиг. 3 показано, что для
эффективной работы катализатору в соответствии с настоящим изобретением требуется минимальное количество калия, приблизительно 1,5 вес. %, тогда как катализатору согласно патенту США 5179056 для
достижения максимальной активности необходима добавка калия приблизительно 2,5 вес. %. В случае катализатора в соответствии с настоящим изобретением эффект калия остается примерно постоянным при
содержании этого промотора от приблизительно 1,5 до 5 вес.%. В случае катализатора в соответствии с патентом США 5179056 с повышением содержания промотора активность начинает падать.
Из вышеприведенных данных следует, что катализатор, приготовленный в соответствии с настоящим изобретением, менее чувствителен к избыточному содержанию в нем калийацетатного промотора.
Реферат
Изобретение относится к получению винилацетата контактированием этилена, уксусной кислоты и кислородсодержащего газа с палладиевым катализатором на носителе. Приготовление катализатора включает следующие стадии: (а) пропитку каталитического носителя палладиевым соединением, (б) конверсию этого палладиевого соединения до практически металлического палладия и (в) спекание палладия на носителе при температуре 650 - 1000oС в присутствии восстанавливающего газа, выбранного из группы, включающей азот, диоксид углерода, гелий, или в присутствии инертного газа, или смеси газов. Реакция протекает при температуре 145 - 195oС, давлении 1 - 20 атм. Предпочтительно в качестве палладиевого соединения используют ацетат, сульфат, нитрат, хлорид палладия или галоидсодержащую палладиевую соль. Носитель предпочтительно выбирают из группы: диоксид кремния, оксид алюминия, кремний - диоксид/алюминий оксид, диоксид титана, диоксид циркония или уголь. Технический результат: повышение активности и срока эксплуатации палладиевого катализатора. 12 з.п.ф-лы, 2 табл., 3 ил.

Формула
Документы, цитированные в отчёте о поиске
Способ получения винилацетата
Комментарии