Тримеризация и олигомеризация олефинов с использованием катализатора, включающего источник хрома, молибдена или вольфрама и лиганд, содержащий по меньшей мере один атом фосфора, мышьяка или сурьмы, связанный с по меньшей мере одной (гетеро)углеводородной - RU2299096C2
Код документа: RU2299096C2
Описание
Настоящее изобретение относится к тримеризации олефинов, в частности к получению 1-гексена тримеризацией этилена.
В патенте US 5198563 и в указанных в описании патентах, выданных фирме Phillips, описаны хромсодержащие катализаторы, включающие монодентатные амидные лиганды, которые могут быть использованы для тримеризации олефинов.
В патенте US 5968866 описан способ олигомеризации/тримеризации этилена, при осуществлении которого для получения альфа-олефинов, которые обогащены 1-гексеном, применяют катализатор, включающий хромовый комплекс, который содержит координационный асимметричный тридентатный фосфановый, арсановый или стибановый лиганд (называемый в описании как фосфин, арсин или стибин и представленный в виде атома фосфора, мышьяка или сурьмы, связанного с тремя гидрокарбильными группами), и алюмоксан. В описании отсутствует указание на возможность замены любой из фосфановой, арсановой или стибановой групп: в самом деле, невозможно предсказать, какой эффект вызвала бы такая замена.
При создании настоящего изобретения были установлены другие лиганды, которые, когда их используют в сочетании с источником переходного металла групп с 3 по 10 в качестве катализаторов тримеризации, оказываются значительно более активными, чем те катализаторы, которые в настоящее время известны, и проявляют также другие ценные свойства. Объем изобретения включает, кроме того, новые катализаторы, содержащие такие лиганды в сочетании с источником хрома, молибдена или вольфрама.
В соответствии с первым объектом настоящего изобретения предлагается катализатор для тримеризации олефинов, включающий
(а) источник хрома, молибдена или вольфрама;
(б) лиганд, содержащий по меньшей мере один атом фосфора, мышьяка или сурьмы, связанный с по меньшей мере одной гидрокарбильной или гетерогидрокарбильной группой, у которой имеется полярный заместитель, за исключением случая, когда всеми такими полярными заместителями являются фосфановые, арсановые или стибановые группы; и необязательно
(в) активатор.
В настоящем описании понятие "тримеризация" означает катализируемую реакцию одного олефинового мономера или смеси олефиновых мономеров с получением продуктов, обогащенных теми компонентами, которые дериватизированы в результате взаимодействия (взаимодействий) трех олефиновых мономеров (в отличие от полимеризации или олигомеризации), которое как правило приводит к распределениям олефиновых продуктов, либо обусловленным уравнением геометрического ряда, либо соответствующим диаграмме распределения Пуассона. "Тримеризация" включает случаи, когда все мономерные звенья в продукте тримеризации оказываются идентичными, когда продукт тримеризации образуется из двух разных олефинов (т.е. два эквивалента одного мономера взаимодействуют с одним эквивалентом второго мономера), а также когда с образованием продукта взаимодействуют три разных мономерных звена. Реакцию, в которой принимают участие больше одного мономера, часто называют сотримеризацией.
Следует принять во внимание, что вышеуказанный катализатор можно либо готовить перед применением в реакции тримеризации, либо готовить in situ добавлением в реакционную смесь его отдельных компонентов.
По другому объекту изобретения предлагается способ тримеризации олефинов, включающий введение мономерного олефина или смеси олефинов в условиях тримеризации в контакт с катализатором, который включает
(а) источник переходного металла групп с 3 по 10;
(б) лиганд, содержащий по меньшей мере один атом фосфора, мышьяка или сурьмы, связанный с по меньшей мере одной гидрокарбильной или гетерогидрокарбильной группой, у которой имеется полярный заместитель, за исключением случая, когда всеми такими полярными заместителями являются фосфановые, арсановые или стибановые группы; и необязательно
(в) активатор.
При создании настоящего изобретения было также установлено, что катализаторы, используемые при осуществлении вышеописанного способа, обладают некоторыми новыми особенностями. Так, например, когда такие катализаторы наносят на носители, они утрачивают меньше своей активности относительно активности эквивалентного не нанесенного на носитель катализатора, чем известные катализаторы. Таким образом, еще одним объектом изобретения является катализатор на носителе, обладающий производительностью на моль катализатора, которая составляет по меньшей мере 50%, предпочтительно по меньшей мере 70%, его производительности, когда он не нанесен на носитель, причем в предпочтительном варианте этот катализатор включает
(а) источник переходного металла групп с 3 по 10;
(б) лиганд, содержащий по меньшей мере один атом фосфора, мышьяка или сурьмы, связанный с по меньшей мере одной гидрокарбильной или гетерогидрокарбильной группой, у которой имеется полярный заместитель, за исключением случая, когда всеми такими полярными заместителями являются фосфановые, арсановые или стибановые группы; и необязательно
(в) активатор.
Кроме того, при создании настоящего изобретения было установлено, что такие катализаторы обладают необычайно высокой производительностью и особенно хорошо сохраняют эту производительность. Соответственно еще один объект изобретения включает катализатор тримеризации олефинов, производительность которого составляет по меньшей мере 30000 г продукта на миллимоль катализатора в час при температуре 110°С или меньше и парциальном давлении этилена 21 бар или меньше. Другим объектом изобретения является катализатор тримеризации олефинов, производительность которого понижается со скоростью меньше 10%/ч.
В одном варианте осуществления способа по изобретению катализатор, используемый при выполнении настоящего изобретения, дополнительно включает другой катализатор (г), приемлемый для полимеризации, олигомеризации или других химических превращений олефинов. В процессах, проводимых в присутствии такого дополнительного катализатора, продукты тримеризации внедряют в полимер с более высокой молекулярной массы или другой химический продукт.
Катализаторы, используемые при осуществлении способа тримеризации по изобретению, проявляют исключительно высокую производительность и селективность в отношении 1-гексена внутри фракции продуктов, содержащих по 6 углеродных атомов. Высокая производительность катализаторов обуславливает более высокую эффективность процесса и/или более низкое характеристическое остаточное содержание катализатора в продуктах. Высокая селективность катализаторов дает возможность упростить очистку продукта (следствием чего является либо менее дорогостоящая очистка продуктов, либо более чистые продукты). Можно было бы рассчитывать на использование этих преимуществ как в процессах, в которых катализаторы в соответствии с изобретением включают по одному каталитическому компоненту, так и также в объединенных процессах, например при получении разветвленных полиолефинов, в которых применяют больше одного катализатора на основе переходного металла.
Что касается источников переходного металла групп с 3 по 10 (а), то они могут включать простые неорганические и органические соли, например галогениды, ацетилацетонаты, карбоксилаты, оксиды, нитраты, сульфаты и т.п., равно как и координационные и металлорганические комплексы, например хромтрихлоридтетрагидрофурановый комплекс, (бензол)трикарбонилхром, хромгексакарбонил, молибденгексакарбонил и т.п. Предпочтительным компонентом (а) является источник хрома, молибдена или вольфрама, а особенно предпочтителен хром.
Предпочтительный лиганд в качестве компонента (б) отвечает формуле
(R1)(R2)X-Y-X(R3)(R4), в которой
Х обозначает атом фосфора, мышьяка или сурьмы;
Y обозначает соединительную группу;
а каждый из R1, R2, R3 и R4 независимо обозначает гидрокарбильную, замещенную гидрокарбильную, гетерогидрокарбильную или замещенную гетерогидрокарбильную группу, по меньшей мере одна из которых содержит полярный заместитель, который не является фосфановой, арсановой или стибановой группой.
Другой предпочтительной структурой лиганда как компонента (б) является X(R1)(R2)(R3), где X и R1, R2 и R3 имеют указанные выше значения, причем по меньшей мере один из R1, R2 и R3 содержит полярный заместитель, который не является фосфановой, арсановой или стибановой группой.
Предпочтительным значением Х является атом фосфора. Что касается R1, R2, R3 и R4, то примерами подходящих гидрокарбильных групп служат метил, этил, этиленил, пропил, бутил, циклогексил, бензил, фенил, толил, ксилил, мезитил, дифенил, нафтил, антраценил и т.п. Примерами приемлемых гетерогидрокарбильных групп являются метокси, этокси, фенокси (т.е. -ОС6Н5), толилокси [т.е. -ОС6Н4(СН3)], ксилилокси, мезитилокси, диметиламино-, диэтиламино-, метилэтиламиногруппы, тиометил, тиофенил, триметилсилил, диметилгидразил и т.п.
Предпочтительны те группы с R1 по R4, содержащие полярные заместители, которые представляют собой замещенные арильные группы с по меньшей мере одним полярным заместителем каждая. Приемлемые замещенные арильные группы включают замещенную фенильную, замещенную нафтильную и замещенную антраценильную группы. Предпочтителен замещенный фенил. Полярные заместители включают метокси, этокси, изопропокси, С3-С20алкокси, фенокси, пентафторфенокси. триметилсилилокси, диметиламиногруппу, метилсульфанил, тозил, метоксиметил, метилтиометил, 1,3-оксазолил, метоксиметокси, гидроксил, амино-, сульфатную, нитрогруппы и т.п. Другие подходящие полярные заместители включают фосфаны, арсаны и стибаны, как изложено в US 5968866 (но с соблюдением вышеупомянутого условия, что по меньшей мере одна из групп с R1 по R4 содержит полярный заместитель, которым не является ни один из них). Наиболее предпочтительны ортозамещенные фенильные группы; предпочтительным ортозаместителем является алкокси, более предпочтителен метокси или метоксиметокси. Фенильные группы могут быть дополнительно замещены в мета- и пара- или других ортоположениях такими группами, как гидрокарбильная, гетерогидрокарбильная, замещенная гидрокарбильная, галогенидная и т.п., но предпочтительны те, которые в этих других положениях не замещены.
В предпочтительном варианте любые из групп с R1 по R4, у которых нет полярных заместителей, представляют собой независимо необязательно замещенные фенильные группы; заместителями могут служить гидрокарбил, гетерогидрокарбил, замещенный гидрокарбил, замещенный гетерогидрокарбил, галогенид и т.п. Однако в наиболее предпочтительном варианте все группы с R1по R4 содержат полярные заместители, которые представлены выше, которыми не являются фосфановые, арсановые или стибановые группы. Кроме того, в наиболее предпочтительном варианте группы с R1 по R4 являются одинаковыми.
Y может обозначать любую из мостиковых групп, например из гидрокарбильных, замещенных гидрокарбильных, гетерогидрокарбильных или замещенных гетерогидрокарбильных мостиковых групп, включая одноатомные звенья, такие как -О-. Группа Y может располагать необязательным дополнительным потенциальным донорным участком. Примеры значений Y включают метилен, 1,2-этан, 1,2-фенилен, 1,3-пропан, 1,2-катехин, 1,2-диметилгидразин, -N(R5)-, где R5 обозначает водородный атом, гидрокарбил или замещенный гидрокарбил и т.п. Предпочтительным значением Y является -N(R5)-; предпочтительным значением R5 является водородный атом, C1-С6алкил или фенил, более предпочтителен метил.
Любые из групп с R1 по R4 могут быть независимо связаны с любой одной или несколькими из других групп или с мостиковой группой Y с образованием вместе с Х или с Х и Y циклической структуры.
Лиганды могут быть получены с применением методов, которые специалисту в данной области техники известны и описаны в опубликованной литературе. Примерами предпочтительных соединений являются:
(2-метоксифенил)(фенил)PN(Ме)Р(фенил)2
(2-метоксифенил)2PN(Ме)Р(фенил)2
(2-метоксифенил)(фенил)PN(Ме)Р(2-метоксифенил)(фенил)
(2-метоксифенил)2PN(Ме)Р(2-метоксифенил)2
(2-этоксифенил)2PN(Ме)Р(2-этоксифенил)2
(2-изопропоксифенил)2PN(Ме)Р(2-изопропоксифенил)2
(2-гидроксифенил)2 PN(Ме)Р(2-гидроксифенил)2
(2-нитрофенил)2PN(Ме)Р(2-нитрофенил)2
(2,3-диметоксифенил)2PN(Ме)Р(2,3-диметоксифенил)2
(2,4-диметоксифенил)2PN(Ме)Р(2,4-диметоксифенил)2
(2,6-диметоксифенил)2PN(Ме)Р(2,6-диметоксифенил)2
(2,4, 6-триметоксифенил)2PN(Ме)Р(2,4,6-триметоксифенил)2
(2-диметоксифенил)(2-метоксифенил)PN(Ме)Р(2-метоксифенил)2
[2-(диметиламино)фенил]2PN(Ме)Р[2-(диметиламино)фенил]2
(2-метоксиметоксифенил)2PN(Ме)Р(2-метоксиметоксифенил)2
(2-метоксифенил)2 PN(этил)Р(2-метоксифенил)2
(2-метоксифенил)2PN(фенил)P(2-метоксифенил)2
(2-метоксифенил)2PN(Me)N(Me)P(2-метоксифенил)2
(2-метоксифенил)2РСН2Р(2-метоксифенил)2
(2-метоксифенил)2РСН2СН2Р(2-метоксифенил)2
три(2-метоксиметоксифенил)фосфан, т.е.
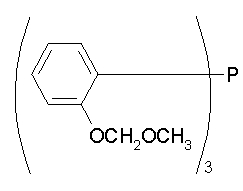
три(2-метоксифенил)фосфан.
Компоненты (а) и (б) могут содержаться в любом соотношении, предпочтительно в пределах 10000:1 и 1:10000, более предпочтительно соотношение в пределах 100:1 и 1:100, а особенно предпочтительно соотношение от 10:1 до 1:10, в частности от 3:1 до 1:3. Обычно количества компонентов (а) и (б) приблизительно равны, т.е. их соотношение находится в пределах 1,5:1 и 1:1,5.
В качестве активаторного соединения (в) в принципе можно использовать любое соединение, которое вместе с компонентами (а) и (б) образует активный катализатор. Могут быть также использованы смеси активаторов. Приемлемые соединения включают алюминийорганические соединения, борорганические соединения и минеральные кислоты и соли, такие как эфират тетрафторборной кислоты, тетрафторборат серебра, гексафторантимонат натрия и т.п. Подходящие алюминийорганические соединения включают соединения формуль AlR3, где каждый из R независимо обозначает С1-С12 алкил, атом кислорода или галогенидную группу, такие соединения как LiAlH4 и т.п. Примеры включают триметилалюминий (ТМА), триэтилалюминий (ТЭА), триизобутилалюминий (ТИБА), три-н-октилалюминий, метилалюминийдихлорид, этилалюминийдихлорид, диметилалюминийхлорид, диэтилалюминийхлорид, этилалюминийсесквихлорид, метилалюминийсесквихлорид и алюмоксаны. В данной области техники алюмоксаны хорошо известны в качестве как правило олигомерных соединений, которые могут быть получены регулируемым добавлением воды к алюминийалкильному соединению, например к триметилалюминию. Такие соединения могут быть линейными, циклическими, соединениями включения или их смесями. Обычно полагают, что технически доступные алюмоксаны представляют собой смеси линейных и циклических соединений. Циклические алюмоксаны могут быть представлены формулой [R6AlO]s, а линейные алюмоксаны - формулой R7(R8AlO)s, в которых s обозначает число от примерно 2 до 50 и в которых R6, R7 и R8 обозначают гидрокарбильные группы, предпочтительно C1-С6алкильные группы, например метильные, этильные или бутильные группы. Предпочтительны такие алкилалюмоксаны, как метилалюмоксан (МАО).
Особенно предпочтительны смеси алкилалюмоксанов с алюминийтриалкильными соединениями, такие как МАО с ТМА или ТИБА. В связи с этим необходимо отметить, что используемое в настоящем описании понятие "алкилалюмоксан" охватывает технически доступные алкилалюмоксаны, которые могут включать некоторую долю, как правило примерно 10 мас.%, но необязательно до 50 мас.%, соответствующего триалкилалюминия. Так, например, технический МАО обычно содержит приблизительно 10 мас.% триметилалюминия (ТМА), в то время как технический ММАО включает как ТМА, так и ТИБА. Указываемые в настоящем описании количества алкилалюмоксана включают такие алюминийтриалкильные примеси, вследствие чего указываемые в настоящем описании количества алюминийтриалкильных соединений рассматриваются как включающие количества соединений формулы AlR3 в дополнение к количествам любого соединения AlR3, включенного в алкилалюмоксан в случае его наличия.
Примерами подходящих борорганических соединений являются бороксины, NaBH4, триметилбор, триэтилбор, диметилфениламмонийтетра(фенил)борат, тритилтетра(фенил)борат, трифенилбор, диметилфениламмонийтетра(пентафторфенил)борат, тетракис[(бис-3,5-трифторметил)фенил]борат натрия, Н+ (OEt2)2[(бис-3,5-трифторметил)фенил]борат, тритилтетра(пентафторфенил)борат и трис(пентафторфенил)бор.
Активаторное соединение (в) может также представлять собой или содержать соединение, которое выполняет функции восстановителя или окислителя, такое как металлический натрий или цинк и т.п. и кислород и т.п.
При приготовлении катализаторов, используемых при выполнении настоящего изобретения, количество активирующего соединения, которое необходимо использовать, легко определить простым испытанием, например получением небольших испытательных образцов, которые могут быть использованы для тримеризации небольших количеств мономера (мономеров), и таким путем определить активность приготовленного катализатора. Как было установлено, используемого количества обычно достаточно для достижения соотношения от 0,1 до 20000 атомов, предпочтительно от 1 до 2000 атомов алюминия или бора на атом хрома. В некоторых случаях для конкретных сочетаний компонентов (а) и (б) активаторное соединение (в) может не понадобиться.
Компоненты с (а) по (в) каталитической системы, используемой при выполнении настоящего изобретения, можно добавлять совместно, одновременно или последовательно в любом порядке и в присутствии или в отсутствии мономера в любой подходящий растворитель, в результате чего получают активный катализатор. Так, например, компоненты (а), (б) и (в) и мономер можно вводить в контакт между собой одновременно или компоненты (а), (б) и (в) можно добавлять совместно, одновременно или последовательно в любом порядке, а затем вводить в контакт с мономером, или компоненты (а) и (б) можно добавлять совместно с получением пригодного для выделения комплекса металл-лиганд, а затем добавлять компонент (в) и вводить в контакт с мономером, или компоненты (а), (б) и (в) можно добавлять совместно с получением пригодного для выделения комплекса металл-лиганд, а затем вводить в контакт с мономером. Приемлемые для введения во взаимный контакт компоненты катализатора или каталитической системы включают, хотя ими их список не ограничен, углеводородные растворители, такие, как гептан, толуол, 1-гексен и т.п., и полярные растворители, такие, как диэтиловый эфир, тетрагидрофуран, ацетонитрил, дихлорметан, хлороформ, хлорбензол, метанол, ацетон и т.п.
Каталитические компоненты (а), (б) и (в), используемые при выполнении настоящего изобретения, можно не наносить или наносить на такой материал носителя, как, например, диоксид кремния, оксид алюминия, MgCl2 или диоксид циркония, или на полимер, например на полиэтилен, полипропилен, полистирол или полиаминостирол. Преимущество настоящего изобретения заключается в том, что когда катализатор нанесен на носитель, потеря его производительности (масса продукта на моль катализатора в час) очень мала. При необходимости катализаторы можно готовить in situ в присутствии материала носителя или материал носителя можно предварительно пропитывать или предварительно смешивать одновременно или поочередно с одним или несколькими каталитическими компонентами. Количество используемого материала носителя можно варьировать широко, например от 100000 до 1 г/г металла, содержащегося в соединении переходного металла. В некоторых случаях материал носителя может также действовать как активаторное соединение (в) или как его компонент. Примеры включают носители, содержащие алюмоксановые остатки и/или гидрокарбилбориловые остатки (см., например, Hlatky. G.G., Chem.Rev. 2000, 100, 1347).
Один вариант выполнения настоящего изобретения охватывает применение компонентов (а), (б) и необязательно (в) в сочетании с катализатором или каталитической системой (г) одного или нескольких типов для полимеризации олефинов с целью тримеризации олефинов и последующего внедрения части продукта (продуктов) тримеризации в более высокомолекулярный полимер.
Компонентом (г) может служить один или несколько приемлемых полимеризационных катализаторов или каталитических систем, примеры которых включают, хотя ими их список не ограничен, обычные катализаторы Циглера-Натта, металлоценовые катализаторы, моноциклопентадиенильные катализаторы или катализаторы "с затрудненной геометрией", активируемые нагреванием хромоксидные катализаторы на носителях (например, типа катализаторов фирмы "Филлипс"), недавно созданные катализаторы полимеризации с переходными металлами (например, дииминовые, дифосфиновые и салицилаль дииминовые никель/палладиевые катализаторы, железо- и кобальтпиридилдииминовые катализаторы и т.п.) и другие, так называемые "катализаторы с единственным участком" (КЕУ).
В общем катализаторы Циглера-Натта состоят из двух основных компонентов. Один компонент представляет собой алкил или гидрид металла групп с I по III, чаще всего Al(Et)3, Al(изо-Bu)3 или Al(Et)2Cl, но охватывает также реактивы Гриньяра, н-бутиллитий и цинкдиалкильные соединения. Второй компонент представляет собой соль переходного металла групп с IV по VIII, чаще всего галогениды титана или ванадия, такие как TiCl4, TiCl3, VCl4 или VOCl3. Каталитические компоненты, когда их смешивают, обычно в углеводородном растворителе, способны образовывать гомогенный или гетерогенный продукт. При необходимости с помощью средств, которые специалистам в данной области техники известны, такими катализаторами можно пропитывать носитель и, таким образом, использовать при осуществлении любого из основных методов получения полиолефинов с помощью координационных катализаторов, такого, как полимеризация в растворе, суспензии и газовой фазе. Для дальнейшей модификации полимеризационных характеристик или активности катализатора в дополнение к двум вышеописанным основным компонентам можно добавлять некоторые количества других соединений (как правило доноров электронов).
В общем металлоценовые катализаторы состоят из комплексов переходных металлов, чаще всего на основе металлов группы IV, лигированных группами циклопентадиенильного (Ср) типа. Известны катализаторы широкого диапазона структур этого типа, включая те, у которых имеются замещенные, связанные и/или гетероатомсодержащие Ср группы, Ср группы, сконденсированные с другими кольцевыми системами, и т.п. Часто используют дополнительные активаторы, такие как бораны и алюмоксаны, а при необходимости катализаторы могут быть нанесены на носители.
Моноциклопентадиенильные катализаторы или катализаторы "с затрудненной геометрией" обычно состоят из комплексов переходных металлов, чаще всего на основе металлов группы IV, лигированных одной группой циклопентадиенильного (Ср) типа, часто связанной с дополнительной донорной группой. Известны катализаторы широкого диапазона структур этого типа, включая те, у которых имеются замещенные, связанные и/или гетероатомсодержащие Ср группы, Ср группы, сконденсированные с другими кольцевыми системами, и ряд связанных и несвязанных дополнительных донорных групп, таких, как амидные, аминовые и алкоксидные. Часто используют дополнительные активаторы, такие как бораны и алюмоксаны, а при необходимости катализаторы могут быть нанесены на носители.
В составе типичного активируемого теплом катализатора хромоксидного типа (типа катализатора фирмы "Филлипс") применяют сочетание материала носителя, в который вначале добавляют хромсодержащий материал, где по меньшей мере часть хрома содержится в шестивалентном состоянии, путем нагревания в присутствии молекулярного кислорода. Носитель в общем включает от примерно 80 до 100 мас.% диоксида кремния, а остальную часть, если она имеется, выбирают из группы, включающей огнеупорные оксиды металлов, такие, как оксид алюминия, оксид бора, оксид магния, диоксид тория, диоксид циркония, диоксид титана и смеси двух или большего числа этих огнеупорных оксидов металлов. Материалы носителей могут также включать оксид алюминия, фосфат алюминия, фосфат бора и их смеси между собой или с диоксидом кремния. Соединение хрома как правило добавляют в материал носителя в виде соединения хрома(III), такого, как ацетат и ацетилацетонат, с целью избежать токсичности хрома(IV). Далее сырой катализатор кальцинируют на воздухе при температуре в пределах 250 и 1000°С в течение от нескольких секунд до нескольких часов. В результате этого по меньшей мере часть хрома переходит в шестивалентное состояние. Восстановление Cr(IV) до его активной формы обычно происходит во время реакции полимеризации, но его можно добиться в конце цикла кальцинирования с СО при примерно 350°С. В сырой катализатор фирмы "Филлипс" для его модификации можно добавлять дополнительные вещества, такие, как фтор, алюминий и/или титан.
Недавно созданные катализаторы на основе переходных металлов и катализаторы с единственным участком охватывают широкий диапазон каталитических структур на основе металлов переходного ряда (см., например, Britovsek, G.J.P. и др. Angew.Chem.Int.Ed.Engl. 1999, 38, 429 и Ittel, S.D. и др. Chem.Rev.2000, 100, 1169).
Компонент (г) может также включать один или несколько катализаторов или каталитических систем полимеризации совместно с одним или несколькими дополнительными катализаторами или каталитическими системами олигомеризации. Приемлемые катализаторы олигомеризации включают, хотя ими их список не ограничен, те, которые димеризуют (например, никельфосфиновые катализаторы димеризации) или тримеризуют олефины или олигомеризуют олефины каким-либо иным образом, приводя например, к распределению 1-олефинов, обусловленному уравнением геометрического ряда (например, железо- и кобальтпиридилдииминовые катализаторы олигомеризации).
Компонент (г) можно независимо наносить или не наносить на носитель. Когда компоненты (а) и (б) и необязательно (в) наносят на носитель, компонент (г) можно совместно наносить последовательно в любом порядке или одновременно на тот же носитель или можно наносить на отдельный носитель. Для некоторых сочетаний компоненты от (а) до (в) могут составлять часть или весь компонент (г). Так, например, если компонент (г) представляет собой активируемый теплом хромоксидный катализатор, тогда им может служить компонент (а), источник хрома, а если компонент (г) включает алюмоксановый активатор, тогда им может также служить необязательный активатор (в).
Компоненты (а), (б), (в) и (г) могут находиться в любом молярном соотношении. Что касается объединенного процесса, то соотношение между компонентами (а) и (г) имеет очевидное важное значение. Предпочтительное соотношение между компонентами (а) и (г) составляет от 10000:1 до 1:10000, а более предпочтительно от 100:1 до 1:100. Требующееся точное значение зависит от относительной реакционной способности компонентов, а также от целевых свойств продукта или каталитических систем.
Приемлемыми для применения в способе тримеризации по настоящему изобретению олефиновыми мономерами или их сочетаниями являются углеводородные олефины, например этилен, С2-С20-α-олефины, олефины с внутренними ненасыщенными связями, винилиденовые олефины, циклические олефины и диены, пропилен, 1-бутен, 1-пентен, 1-гексен, 4-метилпентен-1, 1-гептен, 1-октен, 1-нонен, 1-децен, 1-ундецен, 1-додецен, 1-тридецен,1-тетрадецен, 1-пентадецен, 1-гексадецен, 1-гептадецен, 1-октадецен, 1-нонадецен, 1-эйкозен, стирол, 2-бутен, 2-этил-1-гексен, циклогексен, норборнен, бутадиен и 1,5-гексадиен. Можно также использовать олефины с полярными функциональными группами, такие как метил(мет)акрилат, винилацетат, α,ω-ундеценол и т.п. Предпочтительным мономером является этилен. Кроме того, могут быть использованы смеси этих мономеров. Так, например, можно сотримеризовать 1-бутеновое звено с двумя этиленовыми звеньями с получением С8олефинов, сотримеризовать 1-гексен и этилен с получением С10олефинов или сотримеризовать 1-додецен и этилен с получением С16олефинов. Можно одновременно осуществлять сочетания этих реакций сотримеризации, в особенности когда in situ получают один или несколько мономеров (для получения смесей, включающих преимущественно гексены, октены и децены, может быть использована, например, смесь этилена с бутеном). Технология варьирования распределения продуктов этих реакций включает регулирование условий проведения процесса (например, концентрации, реакционной температуры, давления, продолжительности пребывания) и соответствующий выбор схемы процесса; она хорошо известна специалистам в данной области техники. Эти мономеры или их сочетания могут быть также использованы в присутствии компонента (г).
Олефиновые мономеры или смеси олефиновых мономеров можно применять для тримеризации в практически чистом виде или они могут содержать олефиновые примеси. Один вариант осуществления способа по изобретению включает тримеризацию содержащих олефины отходов из других химических процессов или других стадий того же процесса.
Когда процесс проводят в условиях растворной или суспензионной фазы, можно применять любой разбавитель или растворитель, которым служит олефин, смесь олефинов или материал, который в условиях тримеризации практически инертен. Можно было бы использовать также смеси инертных разбавителей с одним или несколькими олефинами или без них. Предпочтительными разбавителями или растворителями являются алифатические и ароматические углеводороды и галоидированные углеводороды, такие как, например, изобутан, пентан, толуол, ксилол, этилбензол, кумол, мезитилен, гептан, циклогексан, метилциклогексан, 1-гексен, 1-октен, хлорбензол, дихлорбензол и т.п., а также смеси, такие как изопар.
Условия тримеризации могут включать, например, растворную фазу, суспензионную фазу, газовую фазу или объемную фазу с температурой, находящейся в интервале от -100 до +300°С, предпочтительно от 0 до +300°С, а более предпочтительно от 35 до 200°С, и давления, равные атмосферному или повышенные, предпочтительно от атмосферного до манометрического в 800 бар, а более предпочтительно манометрическое от 1 до 100 бар. При необходимости процесс можно проводить при температурах выше 120°С и необязательно также под манометрическими давлениями ниже 30 бар. Высокая начальная производительность и низкая скорость дезактивации предлагаемой каталитической системы позволяют создавать более низкие давления, чем те, которые были бы экономически целесообразными при применении известных в данной области техники каталитических систем.
Независимо от применяемой технологии тримеризации процесс тримеризации как правило проводят в условиях, которые по существу исключают наличие кислорода, воды и других материалов, которые действуют как каталитические яды. Кроме того, тримеризацию можно проводить в присутствии добавок для регулирования селективности, повышения активности и уменьшения количества полимера, образующегося в процессах тримеризации. Приемлемые добавки включают, хотя ими их список не ограничен, водород или источник галогенида, такой как GeCl4. Примеры галогенидов включают, хотя ими их список не ограничен, фториды, хлориды, бромиды и/или иодиды.
Существует ряд вариантов тримеризационного реактора, включая варианты для проведения периодического, полупериодического и непрерывного процессов. Реакции тримеризации и сотримеризации по настоящему изобретению можно проводить в ряде технологических условий, которые вполне очевидны для специалистов в данной области техники, в частности в условиях гомогенной жидкофазной реакции в присутствии или отсутствии инертного углеводородного разбавителя, такого как толуол и гептаны; в условиях реакции в двухфазной системе жидкость/жидкость; в условиях суспензионного процесса, где катализатор находится в форме, которая проявляет слабую или отсутствие растворимости; в условиях процесса в объеме, в ходе проведения которого преобладающей средой служит практически чистый реагент и/или образующиеся олефины; в условиях газофазного процесса, в ходе проведения которого по меньшей мере часть реагента или получаемого олефина (олефинов) через газообразное состояние направляют к катализатору в нанесенной на носитель форме или от него. С целью обеспечить отвод тепла реакции можно прибегнуть к испарительному охлаждению с помощью одного или нескольких мономеров или инертных летучих жидкостей, что является не единственным методом. Реакции тримеризации можно проводить в газофазных реакторах известных типов, таких как реакторы с циркулирующим слоем, вертикальные или горизонтальные с перемешиваемым слоем, с неподвижным слоем или с псевдоожиженным слоем, жидкофазные реакторы, такие как с поршневым потоком, резервуарного типа непрерывного действия с мешалкой, реакторы с циркуляцией или их сочетания. Специалистам в данной области техники известно большое число приемлемых для применения методов выделения и/или очистки продукта, реагента и катализатора: дистилляция, фильтрование, разделение системы жидкость/жидкость, отстаивание суспензии, экстракция и т.д. Один или несколько этих методов можно осуществить отдельно от реакции тримеризации или может оказаться целесообразным объединение по меньшей мере некоторых из них с реакцией тримеризации. Неограничивающим примером этого послужил бы процесс с применением каталитической (или реакционной) дистилляции. Также эффективным может быть процесс, при проведении которого используют больше одного реактора, систему разрушения катализатора между реакторами или после последнего реактора или объединенные реактор/разделительная установка/очищающее устройство. Хотя при выполнении настоящего изобретения все каталитические компоненты, реагенты, инертные материалы и продукты можно было бы применять на однократной основе, часто один или несколько таких материалов экономически целесообразно возвращать в процесс; в случае каталитической системы это может потребовать восстановления одного или нескольких каталитических компонентов для приготовления активной каталитической системы. Объемом настоящего изобретения охватывается также возможность использования продукта тримеризации в качестве реагента. Так, например, 1-гексен, полученный тримеризацией этилена, проведением последующей реакции сотримеризации с этиленом может быть превращен в деценовые продукты.
Когда катализаторы по настоящему изобретению используют при проведении объединенного процесса, который включает последующее химическое превращение, т.е. в присутствии компонента (г), можно предусмотреть ряд технологических вариантов. Эти варианты включают "последовательные" процессы, в ходе проведения которых тримеризацию и последующую реакцию осуществляют в раздельных, связанных между собой реакторах с необязательным возвратом в процесс продуктов/реагентов между реакторами, и процессы "in situ", в ходе проведения которых обе реакционные стадии осуществляют в том же реакторе. Химические превращения, в которых участвуют олефины, специалистам в данной области техники хорошо известны; неограничивающие примеры химических реакций, которые можно было бы провести с использованием компонента (г), включают полимеризацию и сополимеризацию, олигомеризацию, галоидирование, гидроформилирование, окисление, гидратацию, сульфирование, эпоксидирование, изомеризацию, аминирование, циклизацию и алкилирование. Типичная продолжительность пребывания в полимеризационном реакторе составляет меньше 4 ч, предпочтительно меньше 3 ч.
В случае "последовательных" процессов между стадиями тримеризации и последующих реакций можно было бы включить самые разнообразные стадии очистки, анализа и регулирования для олигомерного продукта. Между размещенными последовательно реакторами также возможна рециркуляция. Примером такого процесса является, по-видимому, тримеризация этилена в единственном реакторе с использованием катализатора, включающего компоненты (а), (б) и необязательно (в), с последующей полимеризацией продукта тримеризации с этиленом в отдельном, связанным с первым реакторе с получением разветвленного полиэтилена. Другим примером послужила бы сотримеризация этилена с 1-бутеном и последующая полимеризация продукта тримеризации с получением полиоктена. Еще одним примером является, по-видимому, тримеризация этиленсодержащего отхода из процесса получения полиэтилена с последующим введением получаемого 1-гексена в процесс получения полиэтилена в качестве сомономера для получения разветвленного полиэтилена.
Примером процесса "in situ" служит получение разветвленного полиэтилена, катализируемое компонентами (а), (б), (г) и необязательно (в), добавляемыми в любом порядке, вследствие чего активные каталитические материалы, дериватизированные из компонентов (а), (б) и необязательно (в), находятся в реакторе в определенной точке совместно с компонентом (г).
Как "последовательные", так и "in situ" варианты могут послужить адаптациями современной технологии полимеризации для технологических стадий с участием компонента (г). Все основные существующие методы полимеризации олефинов, включая методы с несколькими реакторами, рассматривают как приемлемые для адаптации к данному техническому решению. Одной адаптацией является введение слоя катализатора тримеризации в возвратный контур процесса газофазной полимеризации, его можно было бы рассматривать как боковой или рецикловый контур внутри главного псевдоожижающего рециклового контура и/или внутри дегазирующей рекуперационной и рецикловой системы.
Когда присутствует компонент (г), полимеризационными условиями могут служить, например, те, которые создают в растворной фазе, суспензионной фазе, газовой фазе или объемной фазе, с температурами, находящимися в интервале от -100 до +300°С, и давлениями, равными атмосферному или повышенными, в частности от 1,40 до 41 бара. Реакционные условия как правило оказывают значительное влияние на свойства получаемого полимера (например, на плотность, индекс расплава, текучесть), вследствие чего предъявляемые к полимеру требования в значительной мере определяются реакционными переменными. Реакционную температуру, в частности в процессах, в ходе проведения которых важно, чтобы она была ниже температуры спекания полимера, как правило, а предпочтительно прежде всего, выбирают таким образом, чтобы оптимизировать условия реакции полимеризации. Высокая производительность и характеристики кинетического профиля этого нового катализатора тримеризации обуславливают повышенную техническую привлекательность получения "in situ" сомономера, предпочтительно гексена-1, во время получения полимера, предпочтительно полиэтилена, в сравнении с процессами, в которых используют известные в данной области техники каталитические системы. Сказанное справедливо даже при типичных реакционных температурах и давлениях при получении полиэтиленов с высоким содержанием сомономерных звеньев, таких как ЛПЭНП, ПЭОНП и ПЭСНП (предпочтительно в пределах 50 и 100°С в зависимости от плотности полимера), и даже при применении в суспензионных и газофазных процессах полимеризации (предпочтительные общие давления в газовой фазе находятся в пределах 15 и 30 бар, а давления этилена находятся в пределах 10 и 70% от давления в газовой фазе). При необходимости такой катализатор может быть использован для полимеризации этилена в процессе, проводимом в условиях высокого давления/высокой температуры, где полимерный материал образуется в сверхкритическом этилене в виде расплава. В предпочтительном варианте полимеризацию проводят в газовой фазе в условиях псевдоожиженного слоя или перемешиваемого слоя. Кроме того, полимеризацию или сополимеризацию можно проводить в присутствии добавок для регулирования молекулярных масс полимеров или сополимеров. Применение газообразного водорода в качестве средства регулирования средней молекулярной массы полимера или сополимера в общем приемлемо для способа полимеризации по настоящему изобретению.
Условия полимеризации в суспензионной фазе или условия газофазной полимеризации особенно эффективны при получении полиэтилена сортов высокой или низкой плотности и полипропилена. При осуществлении таких методов полимеризацию можно проводить в условиях периодического, непрерывного или полунепрерывного процесса. Более того, можно применять один или несколько реакторов, например от двух до пяти размещенных последовательно реакторов. В разных реакторах можно создавать разные реакционные условия, в частности разные температуры или концентрации водорода. В каскадном процессе катализатор тримеризации можно добавлять в любой или все соответствующие полимеризационные реакторы. Если катализатор тримеризации добавляют в первый реактор, откуда он переходит в последующие реакторы, и его можно дополнять или не дополнять в последующих реакторах свежим катализатором тримеризации или полимеризации, то в последующих реакторах этот катализатор можно дезактивировать добавлением обратимых или необратимых ядов, которые частично или полностью разрушают катализатор тримеризации, или добавлением дополнительных катализаторов полимеризации или модификаторов, которые дезактивируют катализатор тримеризации.
В ходе проведения процесса в суспензионной фазе или газофазного процесса катализатор обычно наносят на носитель, дозируют и направляют в полимеризационную зону в форме частиц твердого вещества либо как сухой порошок (например, с инертным газом, этиленом или другим олефином), либо как суспензию. Кроме того, в зону полимеризации можно подавать необязательный активатор, например в виде раствора, отдельно от твердого катализатора или совместно с ним. Компоненты от (а) до (г) можно добавлять в любую часть полимеризационного реактора либо на тех же частицах носителя, либо в виде физической смеси разных частиц носителя или можно добавлять раздельно в ту же или разные части реактора последовательно в любом порядке или одновременно. В другом варианте компоненты от (а) до (г) можно не наносить на носитель и независимо добавлять в любую часть полимеризационного реактора одновременно или последовательно, совместно или раздельно. Соотношение между звеньями основного мономера и других (со)мономеров оказывает заметное влияние на свойства получаемого полимера (например, на плотность), поэтому обычно его необходимо жестко контролировать. Это соотношение можно регулировать прежде всего изменением концентрации или парциального давления либо основного мономера, либо/и сомономера (сомономеров). Концентрацию звеньев основного мономера как правило регулируют независимо от соотношения с сомономерами (по другим причинам, таким как активность), а соотношение (соотношения) между звеньями основного мономера и сомономеров можно регулировать варьированием скорости введения катализатора тримеризации или изменением условий реакции, которые влияют преимущественно на реакцию тримеризации, а не на реакцию полимеризации, или которые влияют на распределение фактически образующихся сомономеров (например, с использованием обратимых каталитических ядов/активаторов). Для регулирования такого соотношения в полимеризационный реактор можно дополнительно вводить свежие исходные сомономеры. Может оказаться необходимым преимущественное вытеснение определенного (со)мономера или (со)мономеров, которые образуются во время реакции тримеризации, путем, например, нагревания или охлаждения пропускаемого потока паров (или жидкости) или рециклового потока внутри систем реакции полимеризации (или дегазации). Это может быть оптимизировано, например, путем регулирования knock-out компрессора или межстадийных условий в рецикловых или дегазирующих вентиляционных рекуперационных компрессорах или с применением специально предусмотренных конденсационных теплообменников или дистилляционных установок.
Скорость добавления каждого компонента можно регулировать независимо, что дает возможность варьировать соотношение между компонентами в получаемом полимере и его плотность. Для регулирования активности каждого компонента, а также с целью иметь возможность влиять на получаемый полимер можно варьировать также давление, температуру, добавление водорода, добавление галоидированных углеводородов, добавление доноров электронов, добавление активаторов/замедлителей и другие приемлемые параметры.
После выгрузки полимерного продукта из реактора практически все ассоциированные с ним и абсорбированные углеводороды удаляют или осуществляют дегазацию полимера, например понижением давления или вытеснением газа с помощью свежего материала или рециклового потока, азота или легких углеводородов (таких, как этилен). Рекуперированные газообразные или жидкие углеводороды можно возвращать в систему очистки или зону полимеризации.
В процессе полимеризации в суспензионной фазе используют полимеризационный разбавитель, который совместим с полимером (полимерами) и катализаторами и которым может служить алкан, такой, как гексан, гептан, изобутан или смесь углеводородов или парафинов. Зоной полимеризации может служить, например, автоклав или аналогичный реакционный сосуд, или заполненный жидкостью реактор непрерывного действия с циркуляцией, в частности такого типа, который хорошо известен в технологии получения полиэтилена по методу фирмы "Филлипс". Когда способ полимеризации по настоящему изобретению осуществляют в суспензионных условиях, в предпочтительном варианте полимеризацию проводят при температуре выше 0°С, наиболее предпочтительно выше 15°С. В предпочтительном варианте в суспензионных условиях полимеризационную температуру поддерживают ниже того уровня, при котором в присутствии полимеризационного разбавителя полимер начинает размягчаться или спекаться. Если температуре позволить повышаться до уровня, превышающего эту последнюю температуру, может произойти засорение реактора. Регулирование процесса полимеризации в этих указанных температурных интервалах может послужить эффективным средством регулирования средней молекулярной массы получаемого полимера. Другим эффективным средством регулирования молекулярной массы является проведение полимеризации в присутствии газообразного водорода, который выполняет функции регулятора степени полимеризации. Обычно чем выше концентрация используемого водорода, тем ниже средняя молекулярная масса получаемого полимера.
В процессах полимеризации в объеме в качестве полимеризационной среды используют жидкий мономер, такой как пропилен.
Методы проведения процессов газофазной полимеризации в данной области техники хорошо известны. Такие методы обычно включают перемешивание (например, с помощью механических средств, вибрации или псевдоожижения) слоя катализатора или слоя целевого полимера (т.е. полимера, обладающего такими же или теми же физическими свойствами, что и продукт, который необходимо получить в процессе полимеризации), содержащего катализатор, и подачу в него потока мономера (в таких условиях, в которых в контакте со слоем катализатора полимеризуется по меньшей мере часть мономера). Этот слой обычно охлаждают добавлением охлаждающего газа (например, рециклового газообразного мономера) и/или летучей жидкости (например, летучего инертного углеводорода или газообразного мономера, который предварительно конденсируют с получением жидкости). Что касается получаемого и удаляемого полимера, то в газофазных процессах в полимеризационной зоне образуется непосредственно твердое вещество, которое выгружают из нее свободным или практически свободным от жидкости. Как хорошо известно специалистам в данной области техники, если в процессе газофазной полимеризации допускают введение в полимеризационную зону какой-либо жидкости, то количество жидкости в полимеризационной зоне в сравнении с количеством содержащегося полимера мало. Это контрастирует с тем, что происходит при проведении процессов в "растворной фазе", когда образующийся полимер растворен в растворителе, и при проведении процессов в "суспензионной фазе", когда полимер образует в жидком разбавителе суспензию.
Газофазный процесс можно проводить как периодический, полупериодический или так называемый "непрерывный" процесс. В предпочтительном варианте его проводят в таких условиях, в которых при перемешивании в полимеризационную зону, содержащую катализатор полимеризации, непрерывно подают мономер, причем свежий мономер вводят взамен полимеризованного мономера, и полученный полимер из полимеризационной зоны непрерывно или периодически отводят со скоростью, сопоставимой со скоростью образования полимера, а взамен катализатора, отводимого из полимеризационной зоны вместе с полученным полимером, в эту полимеризационную зону добавляют свежий катализатор.
Методы проведения газофазных процессов в псевдоожиженном слое при получении полиэтилена, этиленовых сополимеров и полипропилена в данной области техники хорошо известны. Такой процесс можно проводить, например, в вертикальном цилиндрическом реакторе, оборудованном перфорированной распределительной плитой, которая служит опорой для слоя и распределения вводимого потока псевдоожижающего газа по всему слою. Псевдоожижающий газ, циркулирующий через слой, обеспечивает отвод тепла полимеризации из слоя и подачу мономера для полимеризации в этом слое. Таким образом, псевдоожижающий газ обычно включает мономер (мономеры), как правило вместе с каким-либо инертным газом (например, с азотом или инертными углеводородами, такими, как метан, этан, пропан, бутан, пентан и гексан) и необязательным водородом в качестве модификатора молекулярной массы. Горячий псевдоожижающий газ, отходящий из верхней части слоя, направляют необязательно через зону снижения скорости (она может составлять цилиндрическую часть реактора, имеющую больший диаметр) и, если необходимо, циклон и/или фильтры для выделения из газового потока уносимых им тонкодисперсных твердых частиц. Далее горячий газ направляют в теплообменник для отвода по меньшей мере части тепла полимеризации. В предпочтительном варианте катализаторы вводят в слой непрерывно или через равные интервалы. В начале процесса слой включает псевдоожижаемый полимер, который в предпочтительном варианте идентичен целевому полимеру. Вследствие полимеризации мономера (мономеров) внутри слоя непрерывно образуется полимер. В предпочтительном варианте с целью поддержания необходимой высоты псевдоожиженного слоя предусмотрены средства удаления полимера из этого слоя непрерывно или через равные промежутки времени. Процесс обычно проводят под относительно низким давлением, например от 10 до 50 бар, и при температуре, находящейся, например, в пределах от 50 до 135°С. Для того, чтобы избежать проблем агломерации, температуру слоя поддерживают на уровне ниже температуры спекания псевдоожиженного полимера.
В процессе газофазной полимеризации олефинов в псевдоожиженном слое тепло, выделяющееся в результате экзотермической реакции полимеризации, из полимеризационной зоны (т.е. из псевдоожиженного слоя) обычно отводят с помощью псевдоожижающего газового потока, как об этом сказано выше. Горячий реакторный газ, отходящей из верхней части слоя, пропускают через один или несколько теплообменников, в которых газ охлаждается. Далее охлажденный реакторный газ вместе со всем свежим газом возвращают в нижнюю часть слоя. При осуществлении способа газофазной полимеризации в псевдоожиженном слое по настоящему изобретению необходимо обеспечить дополнительное охлаждение слоя (и тем самым увеличить объемную производительность процесса) подачей в этот слой летучей жидкости в таких условиях, в которых жидкость испаряется в слое, поглощая таким образом у слоя дополнительное количество тепла полимеризации за счет эффекта "скрытой теплоты испарения". Когда горячий рецикловый газ, отходящий из слоя, попадает в теплообменник, летучая жидкость получает возможность конденсироваться из него. В одном варианте выполнения настоящего изобретения летучую жидкость из рециклового газа выделяют и вновь самостоятельно вводят в слой. Так, например, летучую жидкость можно выделять и распылять внутри слоя. В другом варианте выполнения настоящего изобретения летучую жидкость возвращают в слой совместно с рецикловым газом. Таким образом, летучую жидкость можно конденсировать из псевдоожижающего газового потока, отходящего из реактора, и можно возвращать в слой вместе с рецикловым газом или можно выделять из рециклового газа и затем возвращать в слой.
Метод конденсации жидкости в рецикловом газовом потоке и возврата в слой смеси газа и захватываемой жидкости описан в ЕР-А 0089691 и ЕР-А 0241947. В предпочтительном варианте конденсированную жидкость повторно вводят в слой отдельно от рециклового газа в соответствии со способом, описанным в US 5541270.
Когда катализаторы по настоящему изобретению используют при проведении объединенного процесса получения более высокомолекулярных полимеров, т.е. в случае присутствия компонента (г), можно предусмотреть ряд технологических вариантов. Эти варианты включают "последовательные" процессы, в ходе проведения которых тримеризацию и последующую полимеризацию осуществляют в раздельных, но связанных между собой реакторах, и процессы "in situ", в ходе проведения которых обе реакционные стадии осуществляют в одном и том же реакторе.
В случае газофазного процесса полимеризации "in situ" компонент (г) можно, например, вводить в полимеризационную реакционную зону в жидкой форме, в частности в виде раствора в практически инертном жидком разбавителе. Компоненты (а), (б), (в) и (г) можно независимо добавлять в любую часть полимеризационного реактора одновременно или последовательно, совместно или раздельно. В этих обстоятельствах в предпочтительном варианте жидкость, включающую такой компонент (компоненты), в виде тонкодисперсных капелек распыляют внутри полимеризационной зоны. Предпочтительный диаметр капелек находится в интервале от 1 до 1000 мкм. В ЕР-А 0593083 описан способ введения полимеризационного катализатора в зону газофазной полимеризации. При необходимости методы, которые представлены в ЕР-А 0593083, можно эффективно применять при осуществлении способа полимеризации по настоящему изобретению.
Хотя обычно это не требуется, при завершении полимеризации или сополимеризации или когда это необходимо для завершения полимеризации или сополимеризации, или с целью по меньшей мере временной дезактивации катализатора или каталитического компонента по настоящему изобретению катализатор можно вводить в контакт с водой, спиртами, ацетоном или другими подходящими каталитическими дезактиваторами таким путем, который специалистам в данной области техники хорошо известен.
В предпочтительном варианте (но необязательно) катализатор тримеризации добавляют перед введением катализатора полимеризации, благодаря чему целевое соотношение между основным мономером и сомономером (сомономерами) создают до введения катализатора полимеризации. Однако целевого состава для сомономеров в начале можно добиться введением свежих исходных сомономеров или путем приемлемого инициирования реакции тримеризации до или во время введения катализатора полимеризации.
При осуществлении способа полимеризации по настоящему изобретению в присутствии компонента (г) полимеры и сополимеры, преимущественно этиленовые полимеры, образуются с достижением высокой производительности (в пересчете на количество полимера или сополимера, получаемого на единицу массы комплекса, используемого в составе каталитической системы). Это означает, что в промышленных процессах, проводимых с применением способа по настоящему изобретению, расходуются относительно небольшие количества комплексов на основе переходных металлов. Это означает также, что когда процесс полимеризации по настоящему изобретению можно проводить в условиях удаления полимера, в которых не осуществляют стадии выделения катализатора, оставляя, таким образом, катализатор или его остатки в полимере (например, как это происходит в большинстве промышленных процессов полимеризации в суспензии и газовой фазе), количество комплекса на основе переходного металла в получаемом полимере может быть очень малым.
При варьировании соотношения компонентов (а), (б), необязательно (в) и (г) и/или добавлении дополнительных сомономеров для катализаторов по настоящему изобретению создается возможность обеспечить получение широкого разнообразия разветвленных полимеров, различающихся плотностью и другими важными физическими свойствами.
При этом доступным считают ряд полиэтиленовых полимеров, включая полиэтилен высокой плотности, полиэтилен средней плотности, полиэтилен низкой плотности, полиэтилен сверхнизкой плотности и эластомерные материалы. Особенно важное значение имеют полимеры, плотность которых находится в интервале от 0,91 до 0,93, в данной области техники обычно относимые к линейному полиэтилену низкой плотности. Такие полимеры и сополимеры широко применяют при изготовлении гибкой пленки экструзией с раздувкой или литьем.
Доступным считают также поли(1-гексен), поли(1-октен) и т.п., равно как и сополимеры, например 1-гексена и пропилена, 1-гексена и 1-октена, а также тройные сополимеры, например этилена, 1-гексена и винилацетата.
Для возможности сшивания с получением, например, эластомера и при изготовлении проводов и кабелей в такие полимерные продукты можно было бы также вводить диеновые звенья.
В зависимости от назначения полимерного продукта в состав полимера как правило вводят небольшие количества добавок, таких как очищающие агенты, антиоксиданты, стабилизаторы и т.п. Обычно эти добавки вводят в количествах от примерно 25 до 2000 част./млн, как правило от примерно 50 до примерно 1000 част./млн, а еще чаще от 400 до 1000 част./млн, в пересчете на полимер.
При применении полимеров или сополимеров, полученных в соответствии с изобретением, в форме порошка из них обычно изготавливают гранулы. Примеры применений полимерных композиций, приготовленных в соответствии с изобретением, включают их переработку с изготовлением волокон, экструдированных пленок, лент, холстов из спряденных волокон, отформованных или формованных с нагреванием изделий и т.п. Полимеры можно перерабатывать с изготовлением пленок экструзией с раздувкой или литьем или можно использовать для изготовления разнообразных формованных или экструдированных изделий, таких, как трубы, и контейнеров, таких, как бутылки и цилиндры. Конкретные пакеты добавок для каждой цели применения можно выбирать так, как это известно в данной области техники. Примеры дополнительных добавок включают добавки, понижающие трение, вещества, препятствующие слипанию, антистатики, смазки для форм, первичные и вторичные антиоксиданты, осветлители, зародышеобразователи, стабилизаторы УФ-излучения и т.п. Классы добавок в данной области техники хорошо известны, они включают фосфитные антиоксиданты, гидроксиламиновые (такие как N, N-диалкилгидроксиламин) и аминоксидные (такие как оксид диалкилметиламина) антиоксиданты, затрудненные аминовые светостабилизаторы и стабилизаторы УФ-излучения, фенольные стабилизаторы, бензофураноновые стабилизаторы и т.п. Различные добавки для олефиновых полимеров описаны в патентах US 4318845, 4325863, 4590231, 4668721, 4876300, 5175312, 5276076, 5326802, 5344860, 5596033 и 5625090.
В полимерные композиции можно, как это известно в данной области техники, добавлять наполнители, такие как диоксид кремния, стекловолокно, тальк и т.п., зародыши кристаллизации и красители.
Настоящее изобретение проиллюстрировано следующими примерами.
Примеры
Все операции проводили в анаэробных условиях. Растворители и газы сушили и растворители дегазировали по стандартным методам. Химикаты во всех случаях, если не указано иное, приобретали у фирмы Aldrich Chemical Company. Метилалюмоксан (МАО) и модифицированный метилалюмоксан (ММАО) приобретали у фирмы Witco в виде растворов соответственно в толуоле или гептанах концентрацией 10 мас.%. (2-метоксифенил)2PN(Ме)Р(2-метоксифенил)2 синтезировали по описанным в литературе методам (см. пример 12 в WO 97/37765). Cr(п-толил)Cl2(ТГФ)3 синтезировали по описанному в литературе методу (Daly, J.J.; Seeden, R.P.A.; J.Chem.Soc.A, 1967, 736). Реакционные продукты анализировали ГХМС с применением колонок с размерами 50 м × 0,3 мм (внутренний диаметр), СР sil. CBS-MS, df=0,4 мкм, в условиях начальной температуры -30°С, выдержки в течение 1 мин, скорости изменения температуры 7°С/мин, конечной температуры 280°С и конечной выдержки в течение 5 мин. Молярные количества катализатора указаны в пересчете на молярное количество источника хрома, использованного для его приготовления.
Пример 1
В трубку Шленка загружали 8 мг CrCl3 (ТГФ)3 (0,02 ммоля) и 10 мг (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2 (0,02 ммоля), добавляли 10 мл ТГФ и раствор перемешивали в течение 2 ч. По прошествии этого времени под пониженным давлением удаляли растворитель и полученное твердое вещество суспендировали в 50 мл толуола. Добавляли 4,2 мл МАО (6,0 ммоля, 300 экв.) и сразу же наблюдали образование зеленого раствора. Этот раствор помещали в атмосферу этилена (1 бар). Сразу же отмечали экзотермическую реакцию. Реакция протекала в течение 60 мин, причем в этот период времени сосуд оставляли открытым для подачи этилена под давлением 1 бар. Далее добавлением 50 мл разбавленного водного HCl катализатор разрушали, органический слой отделяли и сушили над MgSO4. Масса продукта, которую определяли взвешиванием по приросту массы реакционного сосуда Шленка, составляла 10,3 г.
В результате ГХМС анализа реакционных продуктов получали следующее распределение продуктов:
Пример 2
Эксперимент примера 1 повторяли, за исключением того, что вместо МАО использовали 300 экв. ММАО (4,2 мл, 6,0 ммоля). Масса продукта составляла 8,8 г.
Пример 3
Эксперимент примера 1 повторяли, за исключением того, что вместо МАО использовали 100 экв. (изо-Bu2AlO)2 (1,0 мл 2,0 М раствора в толуоле, 2,0 ммоля). Масса продукта составляла 1,3 г.
Пример 4
Эксперимент примера 1 повторяли, за исключением того, что вместо CrCl3(ТГФ)3 использовали 3 мг CrCl2 (0,02 ммоля). Масса продукта составляла 5,6 г.
Пример 5
В сосуд Шленка загружали 9 мг Cr(п-толил)Cl2(ТГФ)3 (0,02 ммоля) и 10 мг (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2 (0,02 ммоля), добавляли 50 мл толуола и раствор перемешивали в течение 5 мин. Добавляли 4,2 мл ММАО (6,0 ммоля, 300 экв) и раствор помещали в атмосферу этилена (1 бар). Реакция протекала в течение 60 мин, причем в этот период времени сосуд оставляли открытым для подачи этилена под давлением 1 бар. Реакционную смесь обрабатывали аналогично примеру 1. Масса продукта составляла 11,0 г.
Пример 6
Эксперимент примера 1 повторяли, за исключением того, что вместо 0,02 ммоля использовали 0,04 ммоля (20 мг) (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2. Масса продукта составляла 9,5 г.
Пример 7
Эксперимент примера 2 повторяли, за исключением того, что вместо 0,02 ммоля использовали 0,01 ммоля (5 мг) (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2. Масса продукта составляла 3,3 г.
Пример 8
В трубку Шленка загружали 415 мг (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2 (0,8 ммоля) и 300 мг CrCl3 (ТГФ)3 (0,8 ммоля) и добавляли 30 мл дихлорметана. Почти сразу же образовывался ярко-голубой раствор, который перемешивали в течение 2 ч. По прошествии этого времени под пониженным давлением удаляли растворитель с получением голубого твердого вещества, это последнее промывали диэтиловым эфиром и сушили под вакуумом. В другую трубку Шленка загружали миллиграмм этого соединения и добавляли 50 мл толуола. Добавляли 16,8 мл ММАО (24 ммоля, 300 экв) и раствор помещали в атмосферу этилена (1 бар). Реакция протекала в течение 60 мин, причем в этот период времени сосуд оставляли открытым для подачи этилена под давлением 1 бар. Реакционную смесь обрабатывали аналогично примеру 1. Масса продукта составляла 2,5 г.
Пример 9
Приготовление МАО на диоксиде кремния
200 мл толуола загружали в сосуд, содержавший в инертной атмосфере диоксид кремния (полученный в соответствии со способами, описанными в примере 37.1 WO 99/12981, диоксид кремния сорта ES70X, который поставляла фирма Crosfield, кальцинировали при 200°С в течение ночи; 20,5 г после кальцинирования). Суспензию механически перемешивали и с помощью шприца добавляли 41,4 мл МАО (1,5 М, 62,1 ммоля). Смесь перемешивали в течение 1 ч при 80°С, после чего под вакуумом удаляли избыток толуола и сушили с получением с количественным выходом продукта 15 мас.% МАО на диоксиде кремния.
Тримеризация с использованием каталитической композиции на носителе
В сосуд Шленка загружали 8 мг CrCl3(ТГФ)3 (0,02 ммоля) и 10 мг (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2 (0,02 ммоля), добавляли 10 мл ТГФ и раствор перемешивали в течение 2 ч. По прошествии этого времени под пониженным давлением удаляли растворитель и полученное твердое вещество суспендировали в 20 мл толуола. Добавляли 1,4 мл МАО (2 ммоля, 100 экв.) и сразу же наблюдали образование зеленого раствора. Далее с помощью полой иглы этот раствор переносили в трубку Шленка, содержавшую суспензию 15 мас.% МАО на диоксиде кремния (приготовлено по изложенному выше) в толуоле (1 г МАО/диоксида кремния в 30 мл толуола). Происходил быстрый перенос зеленой окраски раствора на диоксид кремния/МАО, а верхний слой оставался бесцветным. Эту суспензию перемешивали и помещали в атмосферу этилена (1 бар). Реакция протекала в течение 60 мин, причем в этот период времени сосуд оставляли открытым для подачи этилена под давлением 1 бар. Реакционную смесь обрабатывали аналогично примеру 1. Масса продукта составляла 8,9 г.
Пример 10
В сосуд Шленка загружали 8 мг CrCl3(ТГФ)3 (0,02 ммоля) и 10 мг (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2 (0,02 ммоля), добавляли 10 мл ТГФ и раствор перемешивали в течение 2 ч. По прошествии этого времени под пониженным давлением удаляли растворитель, полученное твердое вещество суспендировали в 10 мл толуола и добавляли 4,2 мл МАО (6,0 ммоля, 300 экв.). Затем этот раствор впрыскивали в автоклав при давлении этилена 8 бар и 50°С. Разбавителем служил изобутан. Реакция протекала в течение 1 ч под давлением этилена 8 бар и при 50°С, а по прошествии этого периода времени газообразные этилен и изобутан сбрасывали в атмосферу. Далее реакционные продукты обрабатывали аналогично примеру 1. Масса выделенного продукта составляла 40,0 г, а производительность в течение одного часа была равна 2000 г/ммоль·ч. В результате ГХМС анализа получали следующее распределение продуктов:
Пример 11
Эксперимент примера 10 повторяли со следующими исключениями: вместо изобутена использовали 500 мл толуольного разбавителя и использовали 0,01 ммоля катализатора. В реакторе в течение эксперимента продолжительностью 60 мин поддерживали следующие условия: 50°С и давление этилена 8 бар. За время эксперимента отмечали стабильность профиля поглощения газа. Масса выделенного продукта составляла 72,7 г, а производительность в течение одного часа была равна 7270 г/ммоль·ч (134700 г/г Cr·ч).
Пример 12
Эксперимент примера 11 повторяли, за исключением того, что в реакторе в течение эксперимента продолжительностью 60 мин поддерживали следующие условия: 80°С и давление этилена 20 бар. Использовали 0,0025 ммоля катализатора. Масса выделенного продукта составляла 141 г, а производительность в течение одного часа была равна 56400 г/ммоль·ч (1033200 г/г Cr·ч).
Пример 13
Эксперимент примера 11 повторяли, за исключением того, что в реакторе в течение эксперимента продолжительностью 60 мин поддерживали следующие условия: 108°С и давление этилена 8 бар. Использовали 0,01 ммоля катализатора. Масса выделенного продукта составляла 51,6 г, а производительность в течение одного часа была равна 5160 г/ммоль·ч (95900 г/г Cr·ч).
Пример 14
Эксперимент примера 11 повторяли за исключением того, что перед экспериментом в реакторе создавали давление водорода 1 бар. Использовали 0,01 ммоля катализатора. Масса выделенного продукта составляла 94,7 г, а производительность в течение одного часа была равна 9470 г/ммоль·ч (175300 г/г Cr·ч).
Пример 15
Эксперимент примера 11 повторяли, за исключением того, что использовали 0,01 ммоля катализатора на носителе, приготовленного аналогично примеру 8. Масса выделенного продукта составляла 49,8 г, а производительность в течение одного часа была равна 4980 г/ммоль·ч (90406 г/г Cr·ч).
Пример 16
Эксперимент примера 11 повторяли, за исключением того, что перед этим экспериментом в реактор добавляли 100 мл 1-бутена и использовали 400 мл толуольного разбавителя. В реакторе поддерживали следующие условия: 80° С и давление этилена 4 бара. Использовали 0,02 ммоля катализатора. Масса выделенного продукта составляла 49,4 г, а производительность в течение одного часа была равна 2470 г/ммоль·ч (46125 г/г Cr·ч).
Пример 17
Эксперимент примера 1 повторяли, за исключением того, что продолжительность эксперимента в данном случае составляла 90 мин, а массу продукта определяли по приросту массы реакционного сосуда Шленка по прошествии разных периодов времени по ходу эксперимента.
В результате ГХМС анализа продукта по прошествии 90 мин получали следующее распределение продуктов:
Пример 18
Эксперимент примера 2 повторяли, за исключением того, что использовали 20 мл толуола и перед началом эксперимента добавляли 20 мл 1-додецена. Масса продукта составляла 2,1 г.
Пример 19.
Эксперимент примера 2 повторяли, за исключением того, что использовали 20 мл толуола и перед началом эксперимента добавляли 20 мл 1-тетрадецена. Масса продукта составляла 3,2 г.
Пример 20.
Эксперимент примера 9 повторяли, за исключением того, что использовали 20 мл толуола и перед началом эксперимента добавляли 20 мл 1-додецена; в данном случае продолжительность эксперимента была равной 4,5 ч. Масса продукта составляла 7,5 г.
Пример А (сравнительный)
Эксперимент примера 1 повторяли, за исключением того, что вместо (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2 использовали 8 мг 1, 2-бис(дифенилфосфино)этана (0,02 ммоля). Никакого продукта не получали.
Пример 21
Эксперимент примера 1 повторяли, за исключением того, что вместо (2-метоксифенил)2PN(Ме)Р(2-метоксифенила)2 использовали 18 мг трис(2-метоксиметоксифенил)фосфана (0,04 ммоля). Масса продукта составляла 1,2 г.
В результате ГХМС анализа реакционных продуктов получали следующее распределение продуктов:
Пример Б (сравнительный)
Эксперимент примера 20 повторяли, за исключением того, что вместо трис(2-метоксиметоксифенил)фосфана использовали 11 мг трифенилфосфана (0,04 ммоля). Никакого продукта не получали.
Пример 22
(Со)полимеризация этилена
В автоклав загружали 500 мл изобутана и 1,5 мл 2,0 М раствора триэтилалюминия в толуоле (3 ммоля). Под давлением этилена давление в автоклаве доводили до 8 бар и нагревали до 50°С.
Далее в виде суспензии в 10 мл толуола инжектировали катализатор (0,02 ммоля), приготовленный аналогично примеру 8. Почти сразу же в виде суспензии в 10 мл толуола инжектировали 0,05 г катализатора Циглера, приготовленного в соответствии с изложенным в примере А описания к US 5470812. Реакция протекала в течение 1 ч под давлением этилена 8 бар и при 50°С, а по прошествии этого периода времени газообразные этилен и изобутан сбрасывали в атмосферу. Полученный полимер промывали разбавленным водным HCl, а затем метанолом и сушили под вакуумом. Масса выделенного полимера составляла 36,0 г. ЯМР-спектроскопия этого полимера показывала наличие бутильных ответвлений, что указывало на образование этилен/1-гексенового сополимера.
Пример 23
(Со)полимеризация этилена
Катализатор на носителе (0,01 ммоля) готовили аналогично примеру 9 в 40 мл толуола. В отдельной трубке Шленка миллиграмм рац-(связанный этиленовым мостиком бисинденил)цирконийдихлорида (0,01 ммоля) растворяли в 10 мл толуола и добавляли 7 мл ММАО (10,0 ммоля, 1000 экв.). С помощью полой иглы этот второй раствор добавляли в суспензию катализатора на носителе и образовавшуюся суспензию перемешивали в атмосфере этилена под давлением 1 бар. Реакция протекала в течение 60 мин, причем в этот период времени сосуд оставляли открытым для подачи этилена под давлением 1 бар. Далее осторожным добавлением 50 мл разбавленного водного HCl катализаторы разрушали. Затем как органическую, так и водную фракции добавляли в 500 мл ацетона, вызывая осаждение полученного полимера. Полимер промывали дополнительными порциями ацетона и сушили под вакуумом. Масса выделенного полимера составляла 3,4 г. ЯМР-спектроскопия этого полимера показывала наличие бутильных ответвлений, что указывало на образование этилен/1-гексенового сополимера.
Реферат
Изобретение относится к катализаторам для тримеризации, олигомеризации и полимеризации олефинов. Катализатор на носителе для тримеризации и олигомеризации олефинов обладает производительностью на моль катализатора по меньшей мере 50% его производительности, когда он не нанесен на носитель. Катализатор включает (а) источник переходного металла группы 6; (б) лиганд, отвечающий формуле (R1)(R2)X-Y-X(R3)(R4) или X(R1)(R2)(R3), в которых Х обозначает атом фосфора, мышьяка или сурьмы; Y обозначает соединительную группу; а каждый из R1, R2, R3, R4 независимо друг от друга обозначает углеводородную, замещенную углеводородную, гетероуглеводородную или замещенную гетероуглеводородную группу, по меньшей мере у одной из которых в каждой из формул имеется полярный заместитель, который не является фосфановой, арсановой или стибановой группой; и необязательно (в) активатор. Способ полимеризации олефинов или их смесей представляет собой объединенный процесс в одном или в разных реакторах, включающий введение мономерного олефина или смеси олефинов в условиях тримеризации в контакт с катализатором, охарактеризованным выше, и дополнительное контактирование с катализатором, приемлемым для полимеризации олефинов, в результате чего продукты тримеризации внедряют в полимер более высокой молекулярной массы, и проведение процесса возможно в реакционном контуре. Также заявлен катализатор на носителе для тримеризации и полимеризации 1-олефинов, который включает указанные компоненты (а), (б), необязательно (в) и дополнительно один или несколько катализаторов (г), приемлемых для полимеризации олефинов. Заявленные катализаторы не теряют свою активность при нанесении их на носитель. 4 н. и 32 з.п. ф-лы.
Формула
Комментарии