Взрывчатый состав - RU2691721C1
Код документа: RU2691721C1
Чертежи




Описание
Для этого документа испрашивается приоритет по заявке AU 2015903557, все содержание которой включено в этот документ посредством ссылки.
Область техники, к которой относится изобретение
Настоящее изобретение в целом относится к нитратсодержащим взрывчатым веществам. В частности, настоящее изобретение относится к стабилизации нитратсодержащих взрывчатых веществ, предотвращению случайного разложения и повышению безопасности и стабильности нитратсодержащих взрывчатых веществ в шахтах с повышенной температурой и реакционно-способными горными породами.
Уровень техники
Взрывчатые составы, содержащие нитрат аммония (AN) или другие соли азотной кислоты, например, нитрат калия или нитрат натрия, широко используются в горнодобывающей промышленности.
«Взрывчатый состав» представляет собой взрывчатое вещество, относящееся к «третичным взрывчатым веществам». Иногда взрывчатым составам - или третичным взрывчатым веществам (иногда их называют просто взрывчатыми веществами) - отдают предпочтение из соображений безопасности благодаря отсутствию детонации при ударе или другом обычном инициировании взрыва. Взрывчатые составы обычно нуждаются в запальном заряде для инициирования взрыва. Для них требуется гораздо более мощный запальный заряд, чем для первичных взрывчатых веществ (например, фульмината серебра, этилазида или нитрида ртути), которые настолько чувствительны к удару, что могут детонировать при ударе молотком. Даже подрыв вторичных взрывчатых веществ (например, тринитротолуола (TNT) или циклонита (RDX)) можно инициировать с помощью капсюля-детонатора, обычно имеющего меньшую мощность, чем запальный заряд для взрывчатых составов.
Коммерчески используемые нитратсодержащие взрывчатые вещества представляют собой взрывчатые составы, поэтому они сравнительно малочувствительны к случайному инициированию взрыва. Эта нечувствительность к инициированию взрыва делает взрывчатые составы наиболее подходящими для использования в горнодобывающей промышленности. Однако безопасность и эффективность таких взрывчатых составов могут снижаться при их использовании в реакционно-способной горной породе, а тем более в горной породе с повышенной температурой (например, выше 55°С). Реакционноспособной горной породой считается горная порода, содержащая химические вещества, способные вступать в реакцию с нитратным компонентом взрывчатого вещества, включая породу, содержащую значительные количества сульфидов металлов, например, пирит (хотя присутствие пирита в самом шпуре не является обязательным, поскольку его реакционноспособные компоненты - Fe(II) и кислота - могут образоваться в другом месте и выщелачиваться в шпур). Когда нитратсодержащие взрывчатые вещества загружены в шпуры в реакционноспособной горной породе, нитратный компонент реагирует с сульфидом металла и кислотой, выделяя тепло. Если выделяется достаточно большое количество тепла, взрывчатый состав может преждевременно сдетонировать. Преждевременная детонация может привести к выбросу взрывчатого состава на поверхность и к детонации в других шпурах, а также к случайному травмированию и гибели людей на месте взрыва. Кроме того, при наличии реакционноспособной горной породы в шпурах с повышенной температурой скорость процесса разложения может дополнительно увеличиваться.
Описание терминов «реакционноспособная порода» и «горячая порода или порода с повышенной температурой» приведено в «Правилах Австралийской группы по безопасности взрывчатых веществ» (Australian Explosives Industry And Safety Group Inc (AEISG) Code of Practice, Edition 3, June 2012), содержание которых включено в настоящий документ посредством ссылки. Под «реакционноспособной породой» следует понимать материал с индукционной стадией, длительность которой меньше требуемого периода времени, при этом индукционная стадия представляет собой промежуток времени, достаточный для того, чтобы химическая система, состоящая из компонентов реакционноспособной породы и взрывчатого вещества, прореагировала так, чтобы вызвать термическое разложение нитрата. В целом, материал считается реакционноспособной породой, если его индукционная стадия длится менее одной недели или менее чем в четыре раза превышает требуемое время ожидания (sleep time) взрывчатого вещества.
Согласно определению в Правилах AEISG «горячая порода» может означать горную породу с температурой от 55°С до 100°С, а «высокотемпературная порода» представляет собой породу с температурой выше 100°С. Термин «порода с повышенной температурой» относится как к горячей породе, так и к высокотемпературной породе.
Повышенная температура и реакционноспособная порода были признаны источником проблем еще в 1963 г., когда игданит (ANFO) был заложен в реакционноспособную породу в Маунт-Айза, шт. Квинсленд, Австралия, что привело к его преждевременной детонации. Подобный инцидент произошел и в шахте Маунт-Уолбек, шт. Западная Австралия, Австралия, в 1983 году, когда в одном шпуре, заполненном игданитом, произошла преждевременная детонация. Спустя четыре года на шахте Маунт-Уолбек защитный рукав в шпуре прорвался, загруженный игданит вступил в прямой контакт с горной породой и произошла преждевременная детонация.
Соприкосновение нитратсодержащих взрывчатых веществ с горной породой при повышенной температуре или с реакционноспособной породой остается нерешенной проблемой. В 2010 г. на шахте Дрейтон, шт. Новый Южный Уэльс, Австралия, произошла авария, когда три человека были травмированы из-за преждевременной детонации взрывчатого состава, содержащей нитрат аммония, в реакционноспособной породе, имеющей повышенную температуру.
Поэтому было выполнено множество разработок, направленных на обеспечение безопасности при использовании нитратсодержащих взрывчатых смесей в условиях повышенной температуры или в реакционноспособной породе. Ряд способов известен и используется для того, чтобы предотвратить инициирование термического разложения нитратного взрывчатого вещества. Сначала для предотвращения контакта взрывчатого вещества и реакционноспособной породы использовали физические барьеры. Они могут представлять собой рукавные вкладыши, вводимые в шпур перед загрузкой взрывчатого вещества. Эти вкладыши хорошо работают в идеальных условиях, но не всегда эффективны на практике. Вкладыши могут быть повреждены при их введении в шпур или могут обеспечивать барьер неадекватного размера. Кроме того, буровые шламы из шпура на поверхности легко окисляются с образованием веществ, способных вступать в реакцию с нитратом аммония. Часть взрывчатого вещества, загружаемого во вкладыш, может оказаться на грунте возле шпура и взаимодействовать с буровым шламом. Поэтому при использовании подобных физических барьеров по-прежнему существуют риски.
Другим способом обеспечения безопасности при использовании нитратсодержащих взрывчатых смесей в реакционноспособных породах является введение во взрывчатое вещество добавки, ингибирующей опасные реакции, причем одной из наиболее известных добавок является мочевина. Одним из наиболее эффективных способов использования мочевины в качестве ингибитора является введение мочевины в окислительную фазу эмульсии или водного геля взрывчатого вещества. Вместо образования физического барьера мочевина вступает в химические реакции, позволяющие замедлять термическое разложение. Тем не менее, мочевина ограничена в применении, поскольку она подвергается гидролизу при повышенных температурах или просто с течением времени. Это приводит к снижению ее защитной способности и сопровождается образованием аммиака и двуокиси углерода, неблагоприятно воздействующих на здоровье в замкнутых пространствах, таких, как шахтные штреки.
Таким образом, существует потребность в способах и/или во взрывчатых составах, способных обеспечить повышение безопасности взрывчатых веществ, в том числе третичных взрывчатых веществ, таких как взрывчатые составы, в породе с повышенной температурой или в реакционноспособной породе.
Раскрытие изобретения
Согласно первому аспекту изобретения реализован способ стабилизации нитратсодержащего взрывчатого вещества, используемого при повышенной температуре или в реакционноспособной породе, включающий в себя стадию удаления оксидов NOx, образующихся во взрывчатом веществе при повышенной температуре или при взаимодействии с реакционноспособной породой, для предотвращения участия оксидов NOx в качестве катализатора или реагента в последующей химической реакции.
Настоящее изобретение направлено на такой лишь недавно выявленный фактор в химии взрывчатых веществ, связанный с горячей или реакционноспособной породой, как присутствие оксидов азота (NOx). Роль газообразных NOx в инициировании термического разложения нитратсодержащих взрывчатых веществ до сих пор не вполне ясна, но известно, что присутствие NOx ускоряет начало термического разложения взрывчатого вещества.
Таким образом, настоящее изобретение полезно тем, что обеспечивает средство для практически полного удаления или, по меньшей мере, уменьшения содержания NOx в химической системе взрывчатого вещества. В одном варианте осуществления изобретения предлагаемым способом удаляют, по меньшей мере, приблизительно 80%, 85%, 90%, 95% или 100% оксидов NOx. Кроме того, оно выгодно отличается тем, что это средство для удаления NOx обладает стойкостью по отношению к нитратам, используемым во взрывчатых веществах, термической стабильностью и обычно проявляет инертность по отношению к сульфидам металлов или к реакционноспособной породе в целом.
Эти и другие преимущества могут быть достигнуты в соответствии с настоящим изобретением, которое в широком смысле обеспечивает способ стабилизации нитратсодержащего взрывчатого вещества при его использовании в реакционноспособной породе путем добавления поглотителя NOx, который может представлять собой вещество или смесь веществ, способных удалять оксиды NOx, контактирующие с взрывчатым составом, или существенно снижать содержание оксидов NOx. Поглотитель NOx представляет собой химическое вещество, добавляемое для удаления или инактивации нежелательных оксидов NOx.
Изобретение основано на новой концепции, заключающейся в том, что удаление оксидов NOx, например, при взаимодействии пирита и нитрата аммония в шахтных шпурах, позволяет ингибировать реакции между нитратом аммония и реакционноспособной породой, увеличивая время до начала термического разложения нитрата аммония в шпуре. Таким образом, предлагаемые взрывчатые вещества способны обеспечить большую безопасность при использовании в реакционноспособной породе даже при повышенной температуре, чем существующие взрывчатые составы, содержащие нитрат аммония.
Настоящее изобретение направлено на удаление оксидов NOx, способных приводить к образованию азотистой кислоты HNO2, которая затем действует как катализатор, ускоряющий экзотермическую реакцию между пиритом и нитратом. Поглотитель NOx можно добавлять в виде отдельной фазы в масле к эмульсиям, которые уже могут содержать оптимальное количество мочевины в окислительной фазе. Поглощение оксидов NOx, растворенных в масле, позволяет замедлить их накопление во взрывчатом веществе и тем самым увеличить время до начала термического разложения нитрата (в одном из вариантов осуществления - нитрата аммония) во взрывчатом веществе. Таким образом, удаляя оксиды азота, можно разорвать цикл образования азотистой кислоты HNO2 путем устранения основной причины ее повторного образования.
Для возникновения проблемы, связанной с протеканием реакции между Fe(II) и нитратом, вовсе не обязательно наличие такой реакционноспособной породы, как пирит. В некоторых случаях быстрое разложение взрывчатого вещества происходит в горячей породе (температура более 55°С), где ускоряющим фактором является температура. Использование поглотителя NOx во взрывчатом веществе приносит пользу, позволяя предотвращать или существенно снижать накопление NOx во взрывчатом веществе. Оксиды NOx способны катализировать образование HNO2 в горячей породе. Вызванное ими снижение пороговой температуры термического разложения может быть опасным в горячей породе, поэтому в дополнение к поглотителю NOx в окислительную фазу эмульсии можно добавлять мочевину для взаимодействия с нитратом на молекулярном уровне. Как известно, мочевина увеличивает температуру начала термического разложения нитратов.
В одном из вариантов осуществления предлагаемого способа поглотитель NOxпредставляет собой пористое вещество, абсорбирующее и/или адсорбирующее NOx. Пористость поглотителя позволяет увеличить площадь поверхности поглотителя NOx, доступную для адсорбции оксидов NOx. В одном из вариантов осуществления пористый поглотитель NOx представляет собой цеолит. Цеолит может быть цеолитом А, цеолитом 4А или цеолитом 5А. Пористый поглотитель NOx может представлять собой молекулярную решетку. Молекулярная решетка может быть металлорганической решеткой Basolite С 300. Пористый поглотитель может представлять собой модифицированный глинистый минерал. Глинистый минерал может представлять собой слоистый двойной гидроксид. В одном из вариантов осуществления пористый двойной гидроксид для поглощения NOx является гидроталькитом. В способе согласно изобретению можно использовать и иные вещества, подобные гидроталькиту. Этот способ предусматривает также использование смесей пористых материалов.
В другом варианте осуществления предлагаемого способа поглотитель NOx представляет собой оксид переходного металла, способный вступать в реакцию с оксидами NOx или катализировать реакцию с этими оксидами. В результате протекания такой реакции может образоваться вещество, являющееся инертным (нереакционноспособным) в отношении нитратсодержащего взрывчатого вещества.
В одном из вариантов осуществления поглотитель NOx представляет собой кристаллический или аморфный диоксид марганца. Диоксид марганца можно использовать совместно с мочевиной.
В другом варианте осуществления предлагаемого способа стабилизированное нитратсодержащее взрывчатое вещество содержит масляную фазу и этот способ дополнительно включает в себя стадию введения поглотителя NOx в масляную фазу взрывчатого вещества перед его использованием. Это позволяет облегчить контакт оксидов NOx с поглотителем NOx, поскольку известно, что оксиды NOx лучше растворяются в гидрофобных фазах.
В одном из вариантов осуществления предлагаемого способа стабилизированное нитратсодержащее взрывчатое вещество представляет собой эмульсию типа «вода в масле», а поглотитель NOx диспергирован в масляной фазе эмульсии.
В альтернативном варианте осуществления предлагаемого способа стабилизированное нитратсодержащее взрывчатое вещество содержит нитратные гранулы, масляная фаза содержит нефтяное топливо, а способ дополнительно включает в себя стадию диспергирования частиц поглотителя NOx в нефтяном топливе для улучшения контакта поглотителя NOx с оксидами NOx.
Способ может включать в себя стадию гидрофобизации частиц поглотителя NOx для облегчения диспергирования этих частиц в масляной фазе. Гидрофобизацию можно проводить путем нанесения эмульгатора на поверхность частиц. Стадия гидрофобизации частиц поглотителя NOx может включать в себя стадию приготовления пасты поглотителя NOx. Пасту можно использовать для образования эмульсии взрывчатого вещества. Эмульгатор может представлять собой эмульгатор на основе полиизобутилен янтарного ангидрида (PIBSA).
В одном из вариантов осуществления предлагаемого способа он дополнительно включает в себя стадию добавления к взрывчатому веществу одного или более из следующего: мочевины, поглотителей кислоты, газовых пузырьков, стеклянных микрошариков и полимерных микрошариков для улучшения различных характеристик взрывчатого вещества, таких как взрывчатые свойства или стабильность, в зависимости от характера взрывных работ.
Согласно второму аспекту настоящего изобретения предлагается взрывчатый состав, предназначенный для использования в реакционноспособной породе и/или в породе с повышенной температурой, содержащий нитратсодержащее взрывчатое вещество и поглотитель NOx в количестве от приблизительно 0,5% до приблизительно 10% по массе.
Описание первого аспекта настоящего изобретения применимо и к другим аспектам настоящего изобретения, если из контекста явно не следует иное.
В одном из вариантов осуществления второго аспекта настоящего изобретения поглотитель NOx представляет собой неорганический поглотитель NOx, выбранный из следующего: цеолиты, молекулярные решетки, слоистые двойные гидроксиды и их смеси. Считается, что они способны адсорбировать и/или абсорбировать оксиды NOx из химической системы и тем самым потенциально ингибировать термическое разложение нитратсодержащего взрывчатого вещества во взрывчатом составе.
В одном из вариантов осуществления второго аспекта настоящего изобретения неорганический поглотитель NOx представляет собой слоистый двойной гидроксид. В одном из вариантов осуществления неорганический поглотитель NOx представляет собой гидроталькит.
В одном из вариантов осуществления второго аспекта настоящего изобретения неорганический поглотитель NOx находится в зернистом состоянии. В другом варианте осуществления частицы поглотителя имеют поперечный размер от приблизительно 0,5 мкм до приблизительно 50 мкм. В одном из вариантов осуществления средний размер частиц составляет, по меньшей мере, приблизительно 0,5 мкм; 5 мкм; 10 мкм; 20 мкм; 30 мкм; 40 мкм или 50 мкм. Размер этих частиц можно измерять как эквивалентный диаметр при рассеянии света.
В одном из вариантов осуществления поглотитель NOx может представлять собой вещество, вступающее в химическую реакцию с оксидами NOx, обеспечивая их инертность по отношению к нитратам. Под обеспечением инертности понимается придание оксидам NOx неспособности взаимодействовать в качестве катализатора или реагента с другими химическими веществами в системе. В одном из вариантов осуществления настоящего изобретения химически действующий поглотитель NOx представляет собой оксид переходного металла. Такой оксид металла может использоваться совместно с мочевиной. Оксид переходного металла может действовать в качестве катализатора. Оксид переходного металла может способствовать разложению оксидов NOx. Оксид переходного металла может представлять собой диоксид марганца. Оксид переходного металла может находиться в кристаллическом или аморфном состоянии. Оксид переходного металла может использоваться совместно с пористым поглотителем NOx. Когда пористый поглотитель насыщается оксидами NOx, диоксид марганца способен обеспечить их дополнительное поглощение.
Согласно третьему аспекту настоящего изобретения предлагается способ взрывания, включающий в себя стадию определения наличия реакционноспособной породы и/или породы с повышенной температурой в материале, подвергаемом взрыванию, и стадию загрузки в шпур, пробуренный в этом материале, взрывчатого состава, содержащего нитрат и поглотитель NOx.
В одном из вариантов осуществления третьего аспекта настоящего изобретения взрывание проводят с использованием взрывчатого состава, реализующего один или оба предыдущих аспекта настоящего изобретения.
В некоторых вариантах осуществления, по меньшей мере, часть шпура имеет температуру выше 55°С и шпур считается пробуренным, по меньшей мере, в «горячей породе». В некоторых вариантах осуществления, по меньшей мере, часть шпура имеет температуру выше 130°С и шпур считается пробуренным в «высокотемпературной породе». В некоторых вариантах осуществления настоящего изобретения шпур представляет собой обводненный шпур.
Краткое описание чертежей
Далее приведено описание вариантов осуществления изобретения со ссылкой на следующие чертежи.
На фиг. 1 приведен типичный график зависимости температуры от времени для реакции между пиритным черным сланцем и нитратом аммония. Участок А-В представляет собой начальную стадию (индукционную стадию или стадию I), участок В-С представляет собой промежуточную стадию или стадию II. Стадия воспламенения начинается в точке С (стадия III).
На фиг. 2 приведен рентгеноструктурный (XRD) спектр чистого пирита и реакционноспособной породы.
На фиг. 3 показано изменение концентрации мочевины и рН с течением времени в смесях реакционноспособной породы RG1, нитрата аммония и породоразрушающего раствора (WS), содержащих мочевину в количестве, составляющем 5% по массе, при нагревании до 55°С. Конец индукционного периода не был достигнут.
На фиг. 4 приведены ИК-спектры нитрата аммония и смеси нитрата аммония с пиритом.
На фиг. 5 приведен график изменения содержания NO и NO2 в пробах воздуха, отобранных над реакционной смесью, содержащей нитрат аммония, реакционноспособную породу RG1 и породоразрушающий раствор при 55°С. Начало стадии II зафиксировано приблизительно через 260 минут.
На фиг. 6 приведена зависимость продолжительности индукционного периода от содержания зернистого ингибитора в реакционной смеси, содержащей реакционноспособную породу RG1, нитрат аммония и породоразрушающий раствор после нагревания до 55°С в течение определенного времени.
Осуществление изобретения
Хорошо известно, что в реакционноспособной породе, содержащей пирит, естественным образом образуются серная кислота и ионы двухвалентного железа Fe(II). В реакционноспособной породе, содержащей аналогичные сульфиды металлов (такие как сульфид кадмия или сульфид меди), ионы железа отсутствуют, но серная кислота образуется иным образом. Учитывая способность как серной кислоты, так и ионов железа к переносу подземными водами и другими путями, эти компоненты не обязательно должны образовываться в месте их проявления. Когда шпур в реакционноспособной породе заполняют нитратсодержащим взрывчатым составом, Fe(II) и серная кислота медленно реагируют в шпуре с нитратами, образуя HNO2 и Fe(III). Значительного повышения температуры реакционной смеси в этот период, называемый «индукционной стадией», не наблюдается.
Нитратсодержащие взрывчатые вещества, в том числе и взрывчатые составы (содержащие, по меньшей мере, один нитрат в качестве основного компонента взрывчатого вещества), обычно подвергаются термическому разложению при температуре приблизительно начиная со 160°С, но в шпурах, где они контактируют с пиритом и серной кислотой, температура начала термического разложения может оказаться значительно ниже. Было установлено, что на индукционной стадии происходит накопление азотистой кислоты HNO2, которая действует как катализатор, увеличивая скорость реакции между реакционноспособной породой и нитратами на промежуточной стадии, поскольку присутствие азотистой кислоты способно снижать температуру начала реакции термического разложения.
По мере того, как концентрация азотистой кислоты и температура системы повышаются, реакция термического разложения (которая при обычной температуре окружающей среды протекает довольно медленно) начинает ускоряться, что приводит к «тепловому разгону», при котором температура химической системы быстро нарастает. В дальнейшем, значительное повышение температуры может привести к преждевременной детонации взрывчатого вещества, что в наилучшем сценарии развития событий приводит к нежелательному исходу, а в наихудшем сценарии представляет собой значительную угрозу безопасности.
Поэтому для безопасного проведения взрывных работ необходимо по возможности увеличивать продолжительность индукционной стадии. Как известно, азотистая кислота, образующаяся при разложения нитратов во взрывчатом составе, приближает начало термического разложения. При этом было обнаружено, что газообразные оксиды NOx, способные растворяться в одной или нескольких фазах, присутствующих в химической системе шпура и нитратсодержащего взрывчатого состава, оказывают такое же действие.
Взрывчатое вещество и взрывчатый состав
Нитратсодержащее взрывчатое вещество поставляется вместе с ингибирующей его разложение добавкой. Эта композиция может опционально содержать дополнительные компоненты, если эти дополнительные компоненты существенно не ухудшают свойства взрывчатого состава (например, его стабильность при хранении, технологичность и взрывчатые свойства).
Нитратсодержащее взрывчатое вещество, по меньшей мере, частично состоит из нитрата и может дополнительно содержать углеродистый материал, служащий источником топлива. Существует множество нитратов, обладающих взрывчатыми свойствами. Наиболее известным нитратом, пригодным для взрывных работ, является нитрат аммония, но используются и другие нитраты, в том числе нитрат натрия и нитрат калия.
Ингибирующая разложение добавка представляет собой поглотитель NOx. Этот поглотитель может быть пористым веществом, способным адсорбировать или абсорбировать оксиды NOx, и/или веществом, способным восстанавливать NOx. Восстановление NOx может означать, что это вещество предпочтительно обеспечивает избирательное восстановление NOx, а продукты, образовавшиеся при такой реакции восстановления, могут быть по существу инертными по отношению к нитратсодержащим взрывчатым смесям, реакционноспособной породе и/или породе с повышенной температурой.
Взрывчатое вещество может представлять собой взрывчатый состав. Взрывчатое вещество или взрывчатый состав может быть реализовано в любой подходящей форме. Например, взрывчатое вещество или взрывчатый состав может представлять собой эмульсию типа «вода в масле», смесь нитрата аммония и нефтяного топлива (ANFO) или смесь, содержащую два подобных взрывчатых состава.
Поглотители NOx
Поглотитель NOx способен более эффективно замедлять реакцию между сульфидами металлов и нитратами, чем используемые в настоящее время нейтрализаторы кислот (такие как оксид цинка, оксид магния и карбонат кальция). Нейтрализация кислоты способна обеспечить только один уровень защиты за счет удаления кислоты в шпурах. Однако было обнаружено, что удаление NOx еще в большей степени замедляет движение взрывчатой химической системы в направлении начала термического разложения.
В настоящем изобретении можно использовать один или несколько поглотителей оксидов NOx (т.е. оксидов NO и NO2) во взрывчатом веществе для предотвращения (или, по меньшей мере, замедления) накопления реакционноспособных оксидов NO и NO2 во взрывчатом веществе, когда оно находится в шпуре, пробуренном в реакционноспособной породе или в породе с повышенной температурой. Удаление оксидов NOx позволяет снизить доступность реагентов, участвующих в реакции термического разложения.
В некоторых вариантах осуществления поглотитель NOx можно покрывать гидрофобным поверхностно-активным веществом и непосредственно диспергировать в масле, используемом при изготовлении нитратсодержащих взрывчатых веществ для умеренно реакционноспособных пород.
Количество нитратов и поглотителя NOx, а также их соотношение во взрывчатом составе зависят от состояния среды, в которой будет использован взрывчатый состав. Специалисты в данной области техники могут определить эти соотношения на основе указаний, изложенных в данном документе, и путем проведения испытаний в полевых условиях. Обычно взрывчатый состав содержит нитратсодержащее взрывчатое вещество в количестве, составляющем от приблизительно 65% до приблизительно 94% по массе (т.е. от общей массы взрывчатого состава), и поглотитель NOx в количестве от приблизительно 1% до приблизительно 15% по массе (т.е. от общей массы взрывчатого состава). В некоторых вариантах осуществления взрывчатый состав содержит нитратсодержащее взрывчатое вещество в количестве от приблизительно 70% до приблизительно 90% от массы взрывчатого состава, от приблизительно 75% до приблизительно 85% от массы взрывчатого состава или от приблизительно 80% до приблизительно 85% от массы взрывчатого состава. В некоторых вариантах осуществления взрывчатый состав содержит поглотитель NOx в количестве от приблизительно 3% до приблизительно 12%, от приблизительно 5% до приблизительно 10%, от приблизительно 1% до приблизительно 10% или от приблизительно 7% до приблизительно 9% по массе. В одном из вариантов осуществления количество поглотителя NOx составляет, по меньшей мере, приблизительно 3%, 5%, 7%, 9%, 11% от общей массы взрывчатого состава. Количество поглотителя в композиции должно быть достаточно большим, чтобы не допустить участия оксидов NOx в качестве катализатора или реагента в последующей химической реакции. Во взрывчатом составе может оставаться некоторое количество оксидов NOx, но оно должно быть настолько малым, чтобы не оказывать существенного влияния на химические реакции.
Поглотители NOx адсорбционного или абсорбционного типа
Поглотитель NOx может быть любым, способным поглощать оксиды NOx (при условии, что он устойчив по отношению к нитратсодержащим взрывчатым веществам), например, путем адсорбции или абсорбции оксидов NOx (например, путем обеспечения их взаимодействия с поверхностью и/или связывания на поверхности подходящего поглотителя NOx). После поглощения оксиды NOx лишаются возможности участвовать в последующих реакциях.
В некоторых вариантах осуществления поглотитель NOx может представлять собой неорганический поглотитель NOx. Неорганические поглотители NOx удобны тем, что они обычно не дестабилизируют нитратсодержащую эмульсию. Поглотитель может быть пористым веществом. Подходящими неорганическими поглотителями NOx являются следующие вещества: цеолиты (например, цеолит А, 4А и 5А), молекулярные решетки (например, Basolite С300), слоистые двойные гидроксиды (например, гидроталькит и другие вещества, подобные гидроталькиту) и их смеси, но не только перечисленные вещества. В некоторых вариантах осуществления слоистые двойные гидроксиды могут подвергаться прокаливанию. Гидроталькит (НТ) был использован в качестве модельного поглотителя NOx в масляной фазе эмульсий нитрата аммония, тем не менее, специалисту в данной области на основании этого описания должно быть ясно, что принципы действия гидроталькита аналогичны принципам действия других пористых поглотителей NOx, упомянутых в этом документе.
В некоторых вариантах осуществления поглотитель NOx может содержать частицы, способные адсорбировать или абсорбировать оксиды азота. Эти частицы можно диспергировать в любой фазе, присутствующей во взрывчатом составе, не оказывая влияния на стабильность эмульсии. Частицы могут иметь любой размер, если они не настолько велики, чтобы ухудшать взрывчатые свойства взрывчатого состава, и не настолько малы, чтобы усложнять работу с ними. Размер частиц считается оптимальным, если он находится в диапазоне от приблизительно 0,5 мкм до приблизительно 50 мкм.
В целом предпочтительно, чтобы большая часть поглотителя NOx присутствовала в топливной фазе взрывчатого вещества, поскольку оксиды NOx в большей степени растворимы в гидрофобной фазе, чем в воде. Присутствие поглотителя NOx главным образом в топливной фазе повышает его способность предотвращать накопление NOx и тем самым снижает скорость индуцированной реакции.
В некоторых вариантах осуществления частицы поглотителя могут быть покрыты поверхностно-активным веществом или эмульгатором для повышения сродства частиц к маслу или топливной фазе взрывчатого вещества. Подходящим классом эмульгаторов являются эмульгаторы на основе полиизобутилен-янтарного ангидрида (PIBSA), которые обычно используются для производства эмульсионных взрывчатых веществ. Другими подходящими эмульгаторами или поверхностно-активными веществами являются жирные кислоты и амиды жирных кислот.
Было обнаружено, что когда поглотитель NOx смешивают с раствором эмульгатора, такого как PIBSA, молекулы эмульгатора обволакивают поглотитель NOx, обеспечивая его поверхности гидрофобные свойства. Модифицированный подобным образом или гидрофобизированный поглотитель NOx легче диспергируется в масляной фазе эмульсии, а также в масляной фазе игданита.
Следовательно, можно вводить поглотитель NOx, такой как гидроталькит, смешанный с поверхностно-активным веществом (предпочтительно тем же поверхностно-активным веществом, которое используется для получения эмульсионного взрывчатого вещества типа «вода в масле»), например, в виде пасты, в предварительно приготовленную эмульсию, перемешав ее до однородного состояния. Использование пасты, содержащей поглотитель и эмульгатор, позволяет устранить проблемы, связанные с обработкой тонкодисперсных порошков в промышленных масштабах. При введении пасты в эмульсию следует обеспечить надлежащее содержание в ней масла, не допуская, чтобы масло, добавленное вместе с поглотителем, привело к слишком большому общему количеству масла в эмульсии после смешивания. Другим преимуществом использования пасты является то, что ее можно легко нагнетать с помощью дозирующего насоса, обеспечивая непрерывность процесса.
Введение гидрофобизированного поглотителя NOx в гранулированный взрывчатый материал можно осуществлять, обеспечивая контакт гранул с нефтяным топливом, содержащим диспергированный поглотитель. Это может привести к изменению состава игданита. Поглотитель, такой как гидроталькит, сначала смешивают с маслом, содержащим, например, поверхностно-активное вещество PIBSA, а затем полученную дисперсию смешивают с гранулами.
Другим вариантом гидрофобизации является покрытие поглотителя NOx гидрофобным поверхностно-активным веществом, а затем его использование в виде сухого порошка для покрытия гранул. Это может быть выполнено во время изготовления, например, нитрата аммония. Возможно, что бентониты и другие порошки, используемые в настоящее время в качестве антикомкователей, возможно заменить гидрофобизированным поглотителем.
Гидрофобизация поглотителя NOx позволяет избежать кристаллизации, например, нитрата аммония во взрывчатом составе эмульсионного типа либо игданитового типа. Поэтому смесь поглотителя NOx с эмульгатором (как правило, того же эмульгатора, что используется для получения эмульсии, хотя можно использовать и другие эмульгаторы) можно вводить на месте в предварительно подготовленные эмульсионные взрывчатые вещества и перемешивать их до однородного состояния. Таким образом, поглотитель NOx согласно настоящему изобретению можно использовать с любым предварительно изготовленным взрывчатым веществом для получения взрывчатого состава согласно настоящему изобретению.
Поглотители NOx химически активного типа
Альтернативой пористым поглотителям NOx, удаляющим NOx путем адсорбции или абсорбции, являются поглотители NOx, удаляющие NOx путем химического превращения молекул NOx в соединение, являющееся инертным по отношению к нитратсодержащим взрывчатым веществам и/или к компонентам реакционноспособной породы или породы с повышенной температурой.
Как было упомянуто выше, способность мочевины функционировать в качестве восстановителя азотистой кислоты в действительности ограничена из-за ее склонности к разложению при повышенных температурах и с течением времени. Тем не менее, было установлено, что добавление оксида переходного металла, такого как диоксид марганца (MnO2), позволяет мочевине вступать в реакцию с оксидами NOx и восстанавливать их. Оксид переходного металла может способствовать каталитическому восстановлению NOx мочевиной, хотя может оказаться, что при протекании этой реакции расходуется, по меньшей мере, часть MnO2 или весь MnO2. Таким образом, уникальная и инновационная комбинация мочевины с катализатором или промотором на базе MnO2 способна обеспечивать восстановление мочевиной как азотистой кислоты, так и оксидов NOx. Это позволяет повысить скорость потребления мочевины, тем самым ограничивая разложение мочевины с образованием аммиака. Кроме того, образовавшийся аммиак (в сочетании с MnO2) также способен к каталитическому восстановлению газообразных оксидов NOx, обеспечивая дополнительное ингибирование термического разложения нитратов во взрывчатом составе.
Оксид MnO2 можно использовать в качестве катализатора или промотора в кристаллическом или аморфном состоянии. Оптимальное значение среднего размера частиц оксида металла может составлять от приблизительно 10 мкм до приблизительно 20 мкм. В вариантах осуществления с использованием диоксида марганца кристаллизация в эмульсионном взрывчатом веществе обнаружена не была.
Другие добавки
В некоторых вариантах осуществления взрывчатое вещество или взрывчатый состав может дополнительно содержать другие компоненты, такие как мочевина, газовые пузырьки, стеклянные или полимерные микрошарики или их смеси. Эти дополнительные компоненты могут придавать и другие полезные свойства, которые могут потребоваться в конкретных случаях (например, когда порода является более реакционноспособной или более горячей, чем обычно).
Мочевина увеличивает температуру начала термического разложения нитратов в контакте с сульфидными рудами, а также вступает в реакцию с азотистой кислотой при контакте с буровой водой, имеющий низкий показатель рН. Таким образом, в зависимости от содержания кислоты, Fe(II) и влаги в горной породе и от реакционной способности пород на месте взрывных работ, введение определенного количества мочевины во взрывчатый состав позволяет дополнительно удлинить индукционную стадию. Оптимальное содержание мочевины во взрывчатом составе увеличивает температуру начала термического разложения нитрата при контакте с сульфидами металла и удаляет образующуюся азотистую кислоту HNO2 на реакционноспособных участках с низким значением показателя рН.
В некоторых вариантах осуществления предлагаемый взрывчатый состав представляет собой эмульсию типа «вода в масле» и/или смесь нитрата аммония и нефтяного топлива (игданит). Эмульсия типа «вода в масле» может содержать не смешивающееся с водой углеводородное топливо в виде дисперсионной фазы и диспергированную воднокапельную фазу, содержащую пересыщенный раствор нитрата аммония (эта диспергированная фаза называется «окислительной фазой»). Диспергированные капли в дисперсионной фазе можно стабилизировать с помощью подходящего эмульгатора (например, эмульгатора PIBSA или сорбитанмоноолеата (SMO)).
Помимо капель нитрата, в масляной фазе могут быть диспергированы мелкие частицы ингибирующей разложение добавки. Эта диспергированная фаза может содержаться в количестве от приблизительно 1% до приблизительно 10% от массы взрывчатого состава.
В зависимости от реакционной способности и температуры породы, в окислительную фазу можно также вводить мочевину в количестве, составляющем до приблизительно 5%, 8%, 10% по массе для увеличения температуры начала термического разложения нитратсодержащего взрывчатого состава в присутствии сульфидов металлов и для замедления реакции нитратов с сульфидами. Ингибирующая разложение добавка в дисперсионной масляной фазе способна усиливать ингибирующее действие мочевины и позволяет значительно увеличивать задержку термического разложения нитрата аммония по сравнению с соответствующим взрывчатым составом, содержащим только мочевину.
В тех случаях, когда мочевина неблагоприятно влияет на энергию фрагментации взрывчатого вещества, можно поддерживать содержание мочевины на достаточно низком уровне и достигнуть требуемого ингибиторного действия за счет увеличения количества поглотителя оксидов NOx, например, гидроталькита, в масляной фазе. Таким образом, ингибиторы реакции можно вводить и в дисперсионную масляную среду, и в дисперсную окислительную фазу взрывчатого состава, причем эти ингибиторы дополняют друг друга и дают два типа или уровня защиты от реакции нитрата аммония с пиритом и продуктами его разрушения. Взрывчатый состав можно сенсибилизировать пузырьками газа в эмульсии, образованными химическим путем, либо добавлением стеклянных или полимерных микрошариков. Кроме того, в обводненных шпурах гранулированную мочевину в игданите можно заменить на гидроталькит, который нерастворим в воде.
Способ согласно настоящему изобретению
Настоящее изобретение относится также к способу удлинения индукционной стадии реакций, протекающих, когда нитратсодержащий взрывчатый состав подвергается воздействию со стороны реакционноспособной породы. Этот способ включает в себя введение во взрывчатый состав ингибирующей разложение добавки, представляющей собой поглотитель оксидов NOx.
Взрывчатый состав, используемый в предлагаемом способе, может быть таким же, что и подробно описанный выше взрывчатый состав. Взрывчатый состав можно изготавливать известными в данной области способами, зависящими от таких факторов, как тип взрывчатого состава (например, нитратная эмульсия, игданит и т.п.) и его предполагаемое назначение.
Как было указано выше, оксиды NOx лучше растворимы в масле, чем в воде. Таким образом, в тех вариантах осуществления, где взрывчатый состав представляет собой эмульсию типа «вода в масле», ингибирующую разложение добавку обычно вводят в масляную фазу эмульсии. Ингибирующую разложение добавку можно вводить в масляную фазу в любое подходящее время (перед образованием, во время образования или после образования эмульсии). Аналогичным образом, в тех вариантах осуществления, где взрывчатый состав представляет собой смесь нитрата аммония и нефтяного топлива, ингибирующую разложение добавку обычно вводят в нефтяное топливо. Ингибирующую разложение добавку можно вводить в нефтяное топливо в любое подходящее время (перед образованием, во время образования или после образования игданита).
В тех вариантах осуществления, где ингибирующая разложение добавка находится, по меньшей мере, частично в порошкообразном состоянии, порошок ингибирующей разложение добавки перед введением во взрывчатый состав можно покрыть связующим для увеличения сродства между его частицами и гранулированным нитратом или для повышения стабильности эмульсии.
В некоторых вариантах осуществления ингибирующую разложение добавку вводят во взрывчатый состав на месте взрывных работ. Например, передвижную установку для приготовления взрывчатого состава можно приспособить для смешивания ингибирующей разложение добавки с эмульсионной матрицей и/или игданитовой смесью. Настоящее изобретение также относится к способам взрывания. Эти способы включают в себя определение того, является ли подвергаемый взрыванию материал реакционноспособной породой, и загрузку в пробуренный в этом материале шпур взрывчатого состава, содержащего нитрат аммония и ингибирующую разложение добавку. Эти способы можно применять для обводненных и/или горячих шпуров (например, имеющих температуру свыше 55°С, включая шпуры, имеющие температуру, превышающую температуру разложения мочевины, приблизительно 130°С).
Примеры
Далее приведено химическое обоснование и экспериментальные данные, подтверждающие гипотезу о том, что удаление оксидов NOx является полезным. В примерах описаны взрывчатые составы, прошедшие испытание различными методами. Эти примеры предназначены для конкретизации вариантов осуществления изобретения, но изобретение не ограничивается использованными в них реагентами, их количествами и соотношениями.
Материалы и способы
Использовали пирит из нескольких источников. Образец RG1 был поставлен компанией Dyno Nobel и представлял собой образец реакционноспособной породы, содержащей адсорбированную воду в количестве приблизительно 2,5% и пирит в количестве менее 30% по массе. Остальной материал образца представлял собой смесь глины, кварца и органических веществ. Средний размер частиц составлял менее 50 мкм.
Чистый пирит (PY) был получен от компании Spectrum Chemicals и представлял собой 100% окисленный пирит с размером зерна 200-400 мкм. Пирит использовали в том виде, в котором он был получен, если не указано иное. В некоторых случаях его промывали водой для удаления остаточных солей и затем сушили при температуре 100°С.
Нитрат аммония (чистотой не менее 99%, от компании Acros Organics) использовали в том виде, в котором он был получен, но перед использованием его измельчали в ступке, чтобы избавиться от крупных комков. Додекан (чистотой не менее 99%, от компании Sigma), семиводный сульфат двухвалентного железа (чистотой не менее 99,5%, от компании BDH), пятиводный сульфат трехвалентного железа (от компании Fluka), мочевину (чистотой 99,5%, от компании Ajax chemicals), сульфат гидразиния (чистотой не менее 99,5%, от компании Ajax chemicals), каолин (от компании Kaolin Australia Pty Ltd, Eckafine BDF), гидроталькит (от компании Sigma) и Basolite C300 (от компании BASF) использовали в том виде, в котором они были получены. Нитрит натрия (от компании Mallinckrodt), диацетилмоноксим (DCM) (от компании Fluka), тиосемикарбазид (TSC) (от компании BDH), фосфорную кислоту (чистотой 85%, от компании Ajax Finechem Pty Ltd), серную кислоту (чистотой 96%, от компании Ajax Finechem Pty Ltd), шестиводный хлорид трехвалентного железа (от компании Merck) и эмульгатор PIBSA-DEEA (от компании Clariant) также использовали в том виде, в котором они были получены.
Определение содержания мочевины
Содержание мочевины определяли с использованием спектроскопии в видимой и ультрафиолетовой области при длине волны 525 нм с использованием диацетилмоноксима DCM и тиосемикарбазида TSC.31. Приготовили кислый раствор трехвалентного железа, содержащий фосфорную кислоту (100 мл), серную кислоту (300 мл), воду (600 мл) и хлорид железа (0,10 г). Смешали DCM и TSC (0,50: 0,01 г) и довели объем раствора до 100 мл. После подготовки к использованию их смешали с хромогенным реагентом, содержащим кислый раствор (2 части) и раствор DCM/TSC (1 часть). Приготовили маточные растворы мочевины, содержащие приблизительно 20 млн-1 мочевины.
Приготовили стандартные растворы мочевины путем разбавления маточных растворов мочевины водой. Раствор мочевины (0,32 мл) смешали с хромогенным раствором, довели объем до 10 мл, накрыли алюминиевой фольгой и нагревали на водяной бане в течение 10 минут. Образец быстро охладили в ледяной бане и измерили поглощение в видимой и ультрафиолетовой области спектра в диапазоне 400-600 нм.
Приготовили шесть образцов, содержащих мочевину в количестве 5% (от массы нитрата аммония), нитрат аммония (0,9 г), реакционноспособную породу RG1 (0,9 г) и породоразрушающий раствор, содержащий растворенную мочевину (0,245 г), поместили их в два термостата и нагревали до 55°С на песчаной бане. Образцы удаляли через определенные промежутки времени, первые через 5 минут, а последние через 20 дней. Образцы охлаждали водой (8,4 г) и измеряли показатель рН. Добавляли еще воды (общей массой 30 г), а затем суспензию фильтровали через фильтр с размером ячеек 0,2 мкм в мерную колбу объемом 50 мл, содержащую каплю концентрированной серной кислоты. Раствор дополнительно разбавляли (1,0 мл на 50 мл) и переносили по 0,32 мл пипеткой в мерные колбы объемом 10 мл, в которые добавляли хромогенный раствор до заполнения объема. Образец нагревали, как было описано выше, и после охлаждения измеряли поглощение в видимой и ультрафиолетовой области спектра в диапазоне 400-600 нм.
Породоразрушающий раствор
Был приготовлен свежий синтетический породоразрушающий раствор, содержащий семиводный сульфат двухвалентного железа (0,245 г), пятиводный сульфат трехвалентного железа (0,50 г) и воду (3,3 г). Смесь осторожно диспергировали ультразвуком до полного растворения. Обычно в экспериментах использовали 0,2 г этого раствора.
Измерения показателя рН
Смеси реакционноспособной породы (RG1), нитрата аммония и породоразрушающего раствора (0,9: 0,9: 0,2 г) помещали в небольшие стеклянные пробирки и нагревали на водяной бане при 55°С в течение 5 минут. По истечении этого времени образец охлаждали водой в количестве приблизительно 6,5 г и измеряли показатель рН. В некоторых случаях нагревание проводили до температуры 80°С.
Адсорбция NO
Реакционноспособную породу смешивали с нитратом аммония и породоразрушающим раствором (в соотношении 0,9: 0,9: 0,2 г) и помещали на дно небольшой пробирки объемом 5 мл. Потенциальные ингибиторы (поглотители) физически отделяли от реакционной смеси, чтобы они контактировали только через газовую фазу. Твердые ингибиторы диспергировали в додекане (в количестве приблизительно 40% по массе) и использовали приблизительно 0,7 г смеси. Реакционную смесь нагревали и перемешивали до однородного состояния, а затем помещали на дно реакционной пробирки.
Затем в середину пробирки устанавливали вырезанную по размеру пробку из пенополиэтилена и на нее помещали вырезанный по размеру стекловолоконный фильтр (с размером пор 250 мкм). Ингибиторы помещали поверх этого фильтра, чтобы предотвратить их непосредственный контакт с реакционной смесью. Фильтр не позволял мелким частицам попасть в реакционную смесь и ингибировать реакции при непосредственном контакте. Испытанию подвергали каолин, цеолит А и гидроталькит. Контрольный образец изготовили, поместив на стеклянный фильтр аналогичное количество додекана.
Реакционные пробирки закрывали пластиковой крышкой, имеющей булавочное отверстие, и погружали в водяную баню при температуре 55°С. Реакция начиналась, когда наблюдались первые признаки появления оксида NO2 коричневого цвета.
Анализ NOx
Накопление NOx в ходе стадии I и при переходе к стадии II определяли с помощью анализатора дымовых газов от компании Kane, Quintox. Были приготовлены четыре дублирующих образца, к которым добавили реакционноспособную породу, нитрат аммония и воду (в соотношении 0,9: 0,9: 0,2 г) в стеклянных пробирках внутренним диаметром 16 мм (длиной 15 см). Пробирки закрывали резиновой пробкой и нагревали на водяной бане до 55°С. Через определенные промежутки времени определяли содержание NO и NO2 в воздухе над каждым образцом, а затем пробирки продолжали нагревать. У некоторых образцов до завершения стадии I отбирали пробы на анализ до 10 раз, в то время как у других образцов анализ проводили только один раз или три раза.
ИК-спектроскопия, спектроскопия в видимой и ультрафиолетовой области и рентгеноструктурный анализ
ИК-спектры регистрировали спектрофотометром Bruker Tensor 27 с использованием методики диффузного отражения в диапазоне 400-4000 см-1 с использованием KBr в качестве фона. Изготовили также смеси нитрата аммония и пирита и определяли их ИК-спектры с использованием нитрата аммония в качестве фона.
Спектры поглощения в ультрафиолетовой и видимой области определяли с помощью спектрофотометра (компании Cary 1Е) в ультрафиолетовой и видимой областях спектра в диапазоне от 200 нм до 700 нм.
Данные рентгеноструктурного анализа были собраны с использованием излучения CuKa на дифрактометре X'Pert Pro (от компании Pan analytical). Источник меди был использован при 45 КэВ и 45 мА и измерения проводили под углом от 5° до 90°.
Общая процедура получения эмульсии
Эмульсии нитратсодержащих взрывчатых веществ, описанные в приведенных далее примерах, были изготовлены с использованием следующего общего метода. Ингредиенты окислительной фазы нагревали до 75°С для образования водного раствора. Компоненты топливной фазы смешивали отдельно при нагревании до 65°С. Затем горячую окислительную фазу медленно вливали в топливную фазу при перемешивании, обеспечиваемом смесителем Lightnin' Labmaster™ с перемешивающей лопастью Jiffy™ размером 65 мм, вращающейся с начальной скоростью 600 об/мин, в течение 30 секунд. Полученную грубую эмульсию гомогенизировали путем перемешивания со скоростью 1000 об/мин в течение 30 секунд, затем со скоростью 1500 об/мин в течение 30 секунд, а затем со скоростью 1700 об/мин до обеспечения заданной вязкости. Количество продукта, полученного в каждом образце, составляло 2,00 кг.
Процедура изотермического испытания
Процедура изотермического испытания, использованная в приведенных далее примерах, была разработана Австралийской группой по безопасности взрывчатых веществ (AEISG) и принята австралийскими поставщиками взрывчатых веществ для определения реакционноспособной породы (AEISG Code of Practice, Elevated Temperature and Reactive Ground, Edition 3, June 2012).
Образцы горной породы измельчали и просеивали до размера 250 мкм.
Измельченный и просеянный материал массой 18 г помещали в чистую сухую пробирку вместе с 18 г продукта и 4 г породоразрушающего раствора. Породоразрушающий раствор состоял из 2 г раствора сульфата двухвалентного железа с концентрацией 13,6% по массе и 2 г раствора сульфата трехвалентного железа с концентрацией 38,5% по массе. Все компоненты перемешивали и закрывали открытый конец пробирки алюминиевой фольгой.
Стеклянные пробирки помещали в алюминиевый блок, нагретый до необходимой температуры. Сквозь алюминиевую фольгу вводили в смесь термопару. Пробирку держали в алюминиевом блоке до тех пор, пока в образце не начинала протекать реакция, или в течение 28 дней, в зависимости от того, какое событие происходило раньше.
Считалось, что реакция происходит, когда наблюдался скачок температуры величиной не менее 2°С, а индукционным периодом считали время от начала испытания до максимума пика.
Процедура адиабатического испытания
Далее описана процедура адиабатического испытания, использованная в приведенных ниже примерах. Рассеяние тепла из области протекания реакции в шпуре зависит от теплопроводности окружающих пород, которая может быть весьма малой для некоторых типов пород. Поэтому самый худший сценарий явления самонагревания должен реализовываться в полуадиабатических условиях, а не в изотермических условиях. Учитывая этот практический аспект, изобретатели разработали полуадиабатический калориметр для оценки эффективности ингибированных взрывчатых смесей. Повышение температуры за счет протекания реакции между пиритом и нитратом аммония отслеживали по нагреванию реагентов в этом полуадиабатическом калориметре.
Калориметр был изготовлен с использованием вакуумного термоса из нержавеющей стали емкостью 350 мл (от компании Wellsense). Из керамической изоляционной бумаги, приобретенной в компании Mathews Industrial Products Pty Ltd (бумага FT толщиной 2 мм с теплопроводностью приблизительно 0,08 Вт/мК) изготовили полый цилиндр с толщиной стенки приблизительно 1,2 см. Наружный диаметр цилиндра составлял приблизительно 6 см, а высота - приблизительно 11 см. Перед сворачиванием керамическую бумагу обернули тонкой изоляционной лентой из тефлона для создания на цилиндре гладкой легко очищаемой поверхности. Этот цилиндр вставили в термос. На дно термоса поместили керамический диск толщиной приблизительно 0,8 см, который также обернули тефлоновой лентой. Образцы помещали в термос в тонкостенной пирексной пробирке (диаметр = 1,1 см).
Керамическую изоляцию использовали с целью предотвращения передачи тепла от нагревательной трубки к металлической стенке термоса циркулирующими конвекционными потоками во время быстрого саморазогрева образца. Крышку изготовили с использованием той же керамической бумаги. Эта керамическая крышка имела отверстие диаметром приблизительно 2 мм и свободно держалась в горловине колбы для обеспечения свободного выхода оксидов NOx без повышения давления в термосе. Горло реакционной пробирки (из пирекса) было неплотно закрыто куском керамической бумаги, чтобы он мог быть выброшен при быстром выделении оксидов NOx.
Термопару типа К в кожухе из тонкой нержавеющей стали (толщина стенки приблизительно 0,05 мм) вводили в середину образца или прикрепляли к нагревательной пробирке тефлоновой лентой. Термопара была подключена к регистратору данных ОСТТЕМР 2000 (от компании Omega), соединенному с компьютером для онлайн-записи. Калориметр нагревали до требуемой начальной температуры (обычно до 55°С), помещая его в водно-глицериновую баню с контролируемой температурой. В некоторых экспериментах пирексная пробирка, содержащая реакционную смесь, непосредственно соединялась со шприцем (объемом 60 мл) с использованием тефлоновой трубки для предотвращения потери оксидов NOx и влаги, а также для предотвращения роста давления в пробирке во время проведения реакции. Этот полуадиабатический калориметр позволил изобретателям оценивать ингибированные взрывчатые составы с использованием небольших образцов массой 5 г. В случае необходимости размеры калориметра могут быть увеличены для проведения испытаний более крупных образцов реакционноспособной породы.
Стабильность взрывчатых веществ в присутствии реакционноспособной породы можно оценить путем нагревания смеси пирита, продуктов его разрушения и взрывчатого состава. Нагревание можно проводить в изотермических или в адиабатических условиях. Изотермические методы легче в реализации, поэтому их обычно и используют в промышленности. Однако считается, что адиабатические методы обеспечивают наилучшее приближение к полевым условиям.
Химическое обоснование
Нитрат аммония разлагается с выделением тепла, образуя три моля газообразных продуктов на каждый моль твердого реагента:
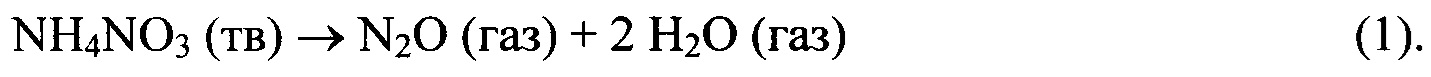
Количество выделяемого тепла и газообразных продуктов можно увеличить путем добавления некоторого количества окисляемого топлива:

Поэтому обычный аммониево-нитратный взрывчатый состав (игданит) называют ANFO, используя начальные буквы его английского названия «Ammonium Nitrate Fuel Oil». Температура разложения чистого нитрата аммония составляет 170°С, тем не менее, в последнее время было обнаружено, что однородная смесь нитрата аммония и пирита может разлагаться в шпурах диаметром более 0,2 м даже при такой низкой температуре, как 50°С. Это согласуется со многими полевыми наблюдениями детонации при низких температурах окружающей среды. Те же начальные реакции происходят в кислых шахтных водах, которые хорошо изучены. Можно провести параллель между этими двумя процессами и обнаружить их сходство. В обоих случаях требуется вода, следовательно, взаимодействуют растворимые вещества.
Первой стадией процесса является окисление пирита воздухом. Продукты окисления серы могут представлять собой различные вещества, например, SO2, SO3 или тиосульфат. Для наглядности выбран SO2, поскольку было обнаружено его образование в реакционноспособных горных породах; однако этот выбор не влияет на сделанные выводы. Например, кислород из воздуха окисляет дисульфидный анион до SO2:
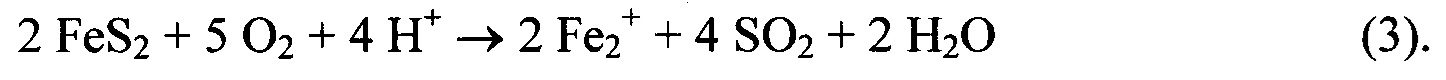
Затем Fe(II) окисляется до Fe(III), которое осаждается в виде нерастворимого гидроксида в растворах, показатель рН которых близок к нейтральному значению:

При протекании этих двух реакций протоны не расходуются и не высвобождаются:
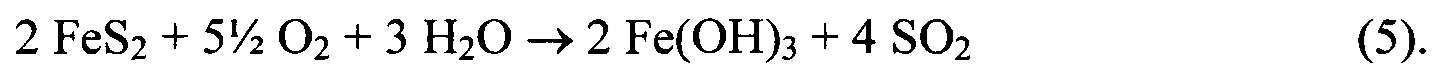
Тем не менее, SO2 легко растворяется в воде, образуя сернистую кислоту с pKa1=2:

Обе указанные реакции окисления протекают относительно медленно, но постепенно кислотность повышается и часть Fe(OH)3 начинает растворяться. Оказывается, что окисление пирита под действием Fe(III) происходит намного быстрее, чем под действием кислорода:
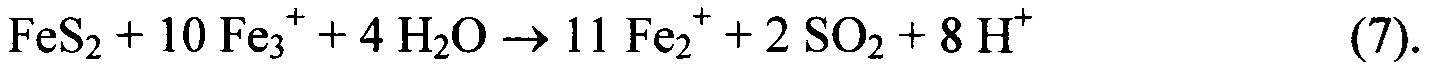
Далее процесс становится автокаталитическим, поскольку образуется все больше кислоты и растворяется все больше Fe(III). Стадией, ограничивающей скорость реакции в этом неорганическом цикле, становится окисление Fe(II) до Fe(III) кислородом, но в полевых условиях это окисление быстро осуществляется бактериями. В тех забоях, где присутствуют бактерии, значение показателя рН может составлять от 0,7 до 3,08, а концентрация Fe(III) - от 1 г/л до 20 г/л.
Термический профиль процесса разложения содержит три стадии: индукционный период, промежуточную стадию и конечную стадию выраженного экзотермического разложения (фиг. 1). Реакции, описанные выше, позволяют объяснить наличие индукционного периода при термическом разложении нитратсодержащих взрывчатых веществ под действием реакционноспособной породы. Некоторые предварительные исследования показали обратную зависимость между начальной кислотностью и продолжительностью индукционного периода. По мнению некоторых авторов, кислота увеличивает скорость на начальной стадии и практически не оказывает влияния на промежуточной стадии. Начальная стадия процесса интерпретируется как медленное снижение показателя рН до тех пор, пока быстрое экзотермическое окисление Fe(III) не ускорит его.
Предпочтительным способом регулирования кислотности как в кислых шахтных водах, так и в реакционноспособной породе считается поддержание высокого значения показателя рН за счет использования щелочных веществ. Однако использование твердых оснований, таких как известняк, оказалось малоэффективным, поскольку Fe(III) осаждается на поверхности и пассивирует остальную породу, образуя на ней плотный слой.
Соответственно, предпочтительным является использование мочевины, которая равномерно подвергается гидролизу, образуя слабые основания аммиак и карбонат и, следовательно, расходует протоны (уравнение 8):

Промышленность располагает убедительными эмпирическими доказательствами того, что мочевина является эффективным ингибитором термического разложения нитрата аммония в реакционноспособной породе. Механизм этого ингибирования остается неясым. Известно, что гидролиз мочевины является медленной реакцией, протекающей со скоростью, не зависящей от значения показателя рН. Длительность индукционного периода может быть ограничена общим расходом мочевины. Если же скорость образования кислоты больше, чем скорость гидролиза мочевины, то показатель рН системы может медленно снижаться, несмотря на частичную нейтрализацию за счет гидролиза мочевины, до тех пор, пока не накопится столько кислоты, что начнется автокаталитический разгон разложения. Наконец, мочевина может действовать как ингибитор иным путем, который не связан с ее щелочными свойствами.
Проверка гипотезы о нейтрализации кислоты
Реакционноспособную породу и чистый пирит исследовали с использованием ретгеноструктурного анализа (фиг. 2). Образец реакционноспособной породы содержал смеси минералов, состоящие преимущественно из кварца (Q) с небольшим количеством клинохлора (С) и пиритового минерала. Спектр пирита соответствовал 100% пириту. Приготовили шесть реакционных смесей, содержащих нитрат аммония, реакционноспособную породу (RG1), породоразрушающий раствор и мочевину в количестве 5% по массе, и отбирали пробы через каждые несколько дней.
После охлаждения образцов водой измеряли показатель рН и определяли общее содержание мочевины с использованием спектроскопии в видимой и ультрафиолетовой области. Полученные результаты показаны на фиг. 3.
На протяжении 17 дней на ингибирование реакции расходовалась лишь часть мочевины; количество мочевины уменьшалось от начальной массы 0,064 г до приблизительно 0,02 г. В то же время показатель рН суспензии снизился с 1,5 до 1,3. Если бы мочевина подвергалась гидролизу с образованием основания, то по истечении этого времени значение рН должно было составлять больше 1,3.
Аналогичные результаты были получены при более низких концентрациях мочевины. При содержании мочевины в количестве 0,2% по массе мочевина расходовалась на одну треть до начала процесса разложения в конце индукционного периода. На протяжении 25 дней при комнатной температуре в почти нейтральных растворах 17% мочевины в воде или 60% нитрата аммония в воде не наблюдалось значительного изменения показателя рН. Отсюда сделан вывод о том, что гидролиз мочевины протекает слишком медленно, чтобы в значительной степени нейтрализовать кислоту и что избыток мочевины не увеличивает скорость гидролиза. Это согласуется с публикациями о том, что реакция гидролиза протекает медленно, с константой скорости 8,4×10-10 сек-1 при 25°С.
Идентификация продукта реакции оксида NO
Предварительные исследования проводили в стеклянной реакционной ячейке, которую помещали на горячий предметный стол микроскопа при 55°С. Была создана граница раздела между реакционноспособной породой (RG1) и эмульсией нитрата аммония. Начальные стадии реакции наблюдали непосредственно с помощью видеомикроскопа. После индукционного периода длительностью приблизительно 20 минут в образце быстро образовывались пузырьки газа, если эмульсия не содержала ингибитора. Бесцветный газ в пузырьках сразу же становился коричневым при контакте с О2, свидетельствуя о том, что этот газ являлся оксидом азота.
На фиг. 4 приведены ИК-спектры нитрата аммония, пирита и смесей нитрата аммония и пирита. В области 1750-1800 см-1 видны диапазоны поглощения поверхностно-связанного оксида NO. Наличие колебательной моды при 1776 см-1 обусловлено валентным колебанием связи N=О адсорбированного оксида NO. Чтобы подтвердить начальное образование оксида NO в качестве предшественника газообразного NO2, реакцию между реакционноспособной породой (RG1) и нитратом аммония проводили в присутствии приблизительно 2% воды. Образец перемешивали, а затем герметизировали резиновой перегородкой и выдерживали на водяной бане при 55°С в течение 1 часа. Получали кислород, смешивая ионы перманганата с пероксидом и набирали газ в шприц. Затем кислород подавали сквозь резиновую перегородку. В пробирке сразу образовался коричневый оксид NO2. Образование газообразного NO наблюдали даже при комнатной температуре при смешивании равных количеств реакционноспособной породы и нитрата аммония в отсутствие дополнительной воды, закрыв пробирку с образцом пробкой. Когда удалили пробку приблизительно через 1 час, то наблюдали выделение прозрачного газа, который становился коричневым под воздействием воздуха.
Дальнейшие эксперименты проводили в реакционных пробирках, помещенных в водяную баню при 55°С. Продолжительность индукционного периода отсчитывали до того момента, когда над суспензией появлялись первые следы коричневого газа. Эта точка (конец стадии I) точно совпадала с расширением объема образца приблизительно в 2 раза (начало стадии II). После этого начального расширения объем продолжал увеличиваться приблизительно в 4 раза с выделением более темно-коричневого газа (стадия II). Стадия III начиналась, когда дальнейшее увеличение объема сопровождалось интенсивным газообразованием с последующим энергичным выделением темно-коричневого газа и иногда густого белого дыма. Присутствие ингибиторов обычно делало протекание стадии II (и III) более спокойным и увеличивало ее продолжительность. В подобных случаях продолжительность индукционного периода по-прежнему отсчитывали до того момента, когда начинал выделяться коричневый газ NO2, несмотря на задержку разгона реакции.
Вместо наблюдения за окислением оксида NO до коричневого оксида NO2, провели прямое измерение его образования с использованием устройства для контроля газообразных продуктов сгорания. Реакцию проводили при 55°С на водяной бане с отбором проб газовой среды для анализа в каждой точке сбора данных. Этот отбор проб газовой среды также тормозил реакцию, которая достигала стадии II только через четыре часа при отборе 10 проб за индукционный период, но если пробы не отбирали, то продолжительность индукционного периода составляла приблизительно 100 минут. Полученные результаты показывают, что накопление оксида NO остается низким, по меньшей мере, в газовой фазе, до конца индукционного периода (фиг. 5), когда он образуется в таком количестве, что превращается в оксид NO2.
Ингибиторы (поглотители) и оксид NO
Результаты, описанные выше, свидетельствуют о том, что разложение нитрата аммония происходит в присутствии оксида NO, но при этом оксиды NO и NO2 накапливаются в значительном количестве лишь в конце индукционной стадии, а их удаление задерживает реакцию.
Для проверки этого вывода было проведено стандартное испытание реакционноспособной породы с использованием нитрата аммония, RG1 и породоразрушающего раствора, но потенциальные ингибиторы были физически отделены от реакционной смеси, чтобы они могли контактировать с ней только через газовую фазу. Твердые ингибиторы диспергировали в масле, содержащем поверхностно-активное вещество, чтобы имитировать их фактическое состояние внутри эмульсии типа «вода в масле». Это испытание было проведено для определения того, оказывает ли образовавшийся при протекании реакции газ каталитическое действие на эту реакцию и если он оказывает такое действие, то какие материалы способны избирательно адсорбировать этот газ для увеличения продолжительности индукционного периода.
Реакционную смесь, состоящую из RG1, нитрата аммония и породоразрушающего раствора, перемешивали при нагревании до однородного состояния, а затем помещали на дно реакционной пробирки. Затем в середину пробирки устанавливали вырезанную по размеру пробку из пенополиэтилена и на нее помещали вырезанный по размеру стекловолоконный фильтр. Ингибиторы помещали поверх этого фильтра, чтобы предотвратить их непосредственный контакт с реакционной смесью. Фильтр не позволял мелким частицам попасть в реакционную смесь и ингибировать реакции при контакте. Поскольку диспергированные ингибиторы должны присутствовать в масляной фазе эмульсии, эти ингибиторы диспергировали в додекане до густого пастообразного состояния и полученную пасту помещали на фильтр. Использовали каолин, цеолит А и гидроталькит. Проводили испытание контрольного образца, помещая на стеклянный фильтр аналогичное количество додекана.
Реакционные пробирки закрывали пластиковыми крышками, имеющими булавочное отверстие, и погружали в водяную баню при 55°С. После нагревания в течение 71 минуты в образце цеолита А реакция уже прошла, а в образце каолина реакция начала проходить, как и в контрольном образце, о чем свидетельствует выделение коричневого газа NO2. Наконец, через 130 минут началась реакция и в образце гидроталькита. Через определенные промежутки времени выполнялось фотографирование и отмечалась стадия протекания реакции. Небольшие различия во времени между контрольными образцами и образцами ингибиторов объяснялись небольшими расхождениями в количествах ингибитора и масла в начальный момент, поскольку сложно было обеспечить добавление одинаковых количеств каждого из них.
Единственным механизмом ингибирования в этих системах являлась адсорбция газа, поскольку ингибиторы не контактировали с реакционной смесью, а были разделены друг с другом некоторым расстоянием. Поскольку в ингибиторе не было воды, оксид азота оставался в растворенном состоянии и не образовывал азотистой кислоты в сколько-нибудь значительной степени. В отсутствие ингибитора реакция разложения ожидается приблизительно через 20 минут. Увеличение продолжительности индукционного периода в присутствии додекана до 71-90 минут указывает на то, что адсорбция газа в масляной фазе связана с его значительной растворимостью. Хорошо известно, что оксид азота имеет гораздо большую растворимость в масле, чем в воде. Растворенный в масле оксид NO, видимо, не участвует в дальнейших реакциях разложения нитрата аммония.
Затем выбранные ингибиторы, включенные в металлорганическую решетку (MOF, Basolite С300), и мочевину нагревали с реакционноспособной породой, нитратом аммония и породоразрушающим раствором до 55°С (фиг. 6), чтобы отличить ингибирование путем удаления оксидов NOx от нейтрализации кислоты.
Предполагаемые механизмы
Теперь можно предположить механизмы многостороннего действия ингибиторов разложения нитрата аммония.
Пирит и/или Fe2+ взаимодействует с нитрат-ионом, образуя оксид NO. В присутствии ионов NO3- и кислоты часть растворенного оксида NO образует HNO2 за счет протекания обратимой реакции (уравнение 9) или окисляется молекулярным кислородом (уравнение 10):


Оксид NO является мощным автокатализатором, ускоряющим реакцию между пиритом и нитратами. Автокаталитические свойства оксида NO и его способность повышать скорость реакции используются для извлечения ценных металлов, вкрапленных в сульфидные минералы в виде включений, путем разрушения сульфидной решетки при быстром окислении.
Когда оксид NO растворяется в воде и превращается в HNO2, его можно восстановить мочевиной с образованием N2 и СО2 при низких температурах (5-60°С):

Поскольку при низких уровнях кислотности HNO2 разлагается с образованием газообразного оксида NO, процесс окисления мочевины проводят при значении показателя рН, приблизительно равном 1, чтобы предотвратить ее разложение. При значении показателя рН более 2 эффективность процесса резко снижается. Поэтому при использовании эмульсий мочевина в каплях эмульсии (при рН приблизительно равном 5) не удаляет оксид NO, диффундирующий в них через масляную фазу эмульсии.
Выводы
Активирует разложение, по-видимому, HNO2 с показателем pKa, приблизительно равным 2,818, а не нитрит-ион NO2-. Азотистая кислота образуется из оксида NO, поэтому связывание этого вещества обеспечивает еще одно средство ингибирования. Видимо, по этому механизму работает гидроталькит. Эффективными могут оказаться и другие модифицированные глинистые минералы. Изоляцию оксида NO обеспечивает лишь объем для его накопления, который в конечном итоге может оказаться заполненным. Надежным решением является разложение оксидов азота с образованием инертных N2 и Н2О, которое можно обеспечить мочевиной. При умеренно низкой температуре (мене приблизительно 60°С) мочевина действует как ингибитор, улавливая азотистую кислоту, а не подвергаясь медленному гидролизу с получением основания, как изначально предполагалось. Вероятно, кинетика именно этой реакции определяет время ожидания ингибированного продукта, и она станет предметом другого исследования в будущем.
Следующие примеры посвящены различным поглотителям оксидов NOx для иллюстрации вариантов осуществления изобретения.
Пример 1
Изготовили эмульсию, содержащую нитрат аммония в количестве 74,3%, мочевину в количестве, 4,9%, воду в количестве 14,4% и масляную фазу в количестве 6,3% по массе. Используемая масляная фаза представляла собой смесь эмульгатора PIBSA в количестве 15% и дизельного топлива в количестве 85% по массе. Эту эмульсию использовали в данном примере в качестве стандартной эмульсии.
Гидроталькит от компании Sigma прокаливали при 550°С в течение 4 часов. Прокаленный гидроталькит смачивали углеводородной смесью, содержащей эмульгатор PIBSA в количестве 15% по массе. Эта смесь гидроталькита с маслом содержала масляную фазу (включая эмульгатор) в количестве 33,3%. Затем этот покрытый маслом гидроталькит смешали со стандартной эмульсией и получили ингибированную эмульсию, содержащую гидроталькит в количестве 4,65% по массе.
Затем стандартную эмульсию и эмульсию с добавкой гидроталькита подвергли испытанию в соответствии со стандартной процедурой изотермического испытания при 130°С с использованием образцов горной породы из г. Ньюман, Западная Австралия. Промежуток времени от загрузки образца в нагревательный блок до максимального повышения температуры считали индукционным периодом.
Добавление гидроталькита увеличило продолжительность индукционного периода с 3,5 часов у стандартной эмульсии до 42 часов у эмульсии с добавлением гидроталькита.
Пример 2
Изготовили эмульсию, содержащую нитрат аммония в количестве 72,93%, мочевину в количестве 1,54%, воду в количестве 19,6% и масляную фазу в количестве 5,92% по массе. Используемая масляная фаза содержала додекан в количестве 65%, эмульгатор PIBSA DEEA в количестве 14% и дизельное топливо в количестве 21% по массе. Эту эмульсию использовали в данном примере в качестве стандартной эмульсии.
Затем непрокаленный гидроталькит смешивали с той же масляной фазой (содержащей эмульгатор PIBSA DEEA в количестве 14% по массе) для получения смеси, содержащей гидроталькит в количестве 71,3% по массе. Затем покрытый маслом гидроталькит тщательно смешивали с частью стандартной эмульсии для получения эмульсии, содержащей гидроталькит в количестве 1,2% по массе.
Для стандартной эмульсии и для эмульсии с добавлением гидроталькита определяли индукционный период, проводя испытание при 55°С в адиабатическом калориметре замкнутой системы. Вкратце, образцы для испытания (в двух экземплярах массой приблизительно 4,7 г) получали путем смешивания образцов стандартной эмульсии и эмульсии с добавлением гидроталькита с чистым пиритом от компании Spectrum. Пирит смачивали раствором, содержащим ионы Fe(II) и Fe(III), соответственно, согласно Правилам AEISG. Этот раствор, воспроизводящий продукты разрушения пирита, был получен путем растворения сульфатов Fe(II) и Fe(III), как описано в процедуре изотермического испытания. Один грамм раствора смешивали с 4,5 г пирита. Затем образцы порознь выдерживали при 55°С в адиабатическом калориметре, непрерывно регистрируя температуру образца, до возникновения экзотермической реакции. Продолжительность разогрева до экзотермической реакции считали индукционным периодом. Добавление гидроталькита увеличивало продолжительность индукционного периода с приблизительно 6,8 дней для стандартной эмульсии до 17 дней для эмульсии с добавлением гидроталькита.
Пример 3
Изготовили эмульсию, содержащую нитрат аммония в количестве 70,7% по массе, воду в количестве 19,9% по массе и масляную фазу в количестве 9,9% по массе. Используемой масляной фазой был додекан, содержащий эмульгатор PIBSA DEEA1100 в количестве 10,6% и дизельное топливо в количестве 16%. Эту эмульсию использовали в данном примере в качестве стандартной эмульсии.
Образец гидрофобного гидроталькита (от компании Sigma) массой 0,05 г тщательно смешали с частью эмульсии (10 г) для получения эмульсии с добавлением гидроталькита, в которой содержание гидроталькита, в конечном итоге, составило 0,50%. Этот гидрофобный гидроталькит не был смочен PIBSA перед добавлением в эмульсию.
Для стандартной эмульсии и для эмульсии с добавлением гидроталькита определяли продолжительность индукционного периода при 55°С. Образцы для испытания готовили путем смешивания эмульсий с реакционноспособной породой, полученной от компании Dyno Nobel, в соответствии с процедурой изотермических испытаний. Затем образцы (чистая эмульсия с реакционноспособной породой и эмульсия с добавлением гидроталькита и с реакционноспособной породой) выдерживались при 55°С в адиабатическом калориметре до возникновения реакции.
Оказалось, что добавление гидроталькита к чистой эмульсии в количестве 0,50%, увеличило продолжительность индукционного периода с 17 минут до 135 минут.
Пример 4
Изготовили смесь, содержащую кристаллы нитрата аммония (в количестве 89,9% по массе), масло (в количестве 7,5%) и прокаленный гидроталькит (в количестве 2,45%), сначала смешивая требуемое количество прокаленного гидроталькита с додеканом, содержащем эмульгатор PIBSA DEEA в количестве 14% по массе, а затем добавляя к этой смеси гидроталькита с маслом кристаллы нитрата аммония. Эту смесь использовали для приготовления смеси нитрат аммония-масло-гидроталькит-эмульсия, содержащей эмульсию в количестве 30% по массе. Используемая эмульсия содержала мочевину в количестве 2% по массе, нитрат аммония в количестве 69,56% по массе, смесь масла с PIBSA в количестве 11,6% по массе и воду в количестве 17,3% по массе. Стандартную смесь также получали смешиванием нитрата аммония с маслом и эмульсии в том же соотношении, что и раньше, но без гидроталькита.
Затем ингибированную смесь нитрат аммония-масло-гидроталькит-эмульсия и стандартную смесь подвергали взаимодействию с пиритом, содержащим породоразрушающий раствор, который получали методом, описанным в Правилах AEISG. Реакционные смеси (5 г) поместили в отдельные адиабатические калориметры, которые выдерживали при 55°С. Термический разгон стандартной смеси начался через 2,4 часа, а термический разгон образца, содержащего гидроталькит, начался через 57 часов.
Примеры с использованием смесей порошка нитрата аммония и ингибитора
Прокаленный и непрокаленный порошок гидроталькита смешивали с порошком нитрата аммония и определяли продолжительность их индукционного периода. Пирит, используемый в примерах 5-12, был получен от компании Spectrum Chemicals. Нитрат аммония (чистотой не менее 99%, от компании Acros Organics), семиводный сульфат железа (II) (чистотой 99,5%, от компании BDH) и пятиводный сульфат железа (III) (от компании Fluka) использовали в том виде, в котором они были получены.
Пример 5
Смешивали небольшие навески (общей массой 2 г) смесей нитрата аммония и пирита или нитрата аммония и измельченной породы, содержащих 0,9 г пирита или измельченной породы, 0,9 г нитрата аммония и 0,2 г породоразрушающего раствора. Добавляли гидроталькит с учетом содержания нитрата аммония и перемешивали суспензию при осторожном нагревании до 55°С, а затем герметизировали в полуадиабатической стеклянной ячейке с двойными стенками, содержащей шприц объемом 25 мл с иглой для выделяющегося газа. Образцы нагревали на глицериновых банях до 55°С, 80°С или 95°С (как описано в следующих примерах) до начала термического разгона.
Контрольная смесь нитрата аммония и пирита или нитрата аммония и измельченной породы без гидроталькита достигала разгона менее чем за 10 минут при 55°С, при 80°С реакция протекала приблизительно за 2 минуты, а при 95°С - за 1 минуту.
Пример 6. Гидроталькит при 55°С
Непрокаленный гидроталькит (HT-LD) смешали с чистым пиритом, нитратом аммония и породоразрушающим раствором с концентрацией 3,0% и 4,16% по массе, а затем нагрели до 55°С в закрытой пробирке. Образец с концентрацией 3% по массе вступил в реакцию через 15 часов, а образец с концентрацией 4,16% по массе вступил в реакцию через 6,75 дней.
Пример 7. Гидроталькит при 80°С
Когда пример 6 повторили при более высокой температуре 80°С, для ингибирования реакции потребовались более высокие концентрации гидроталькита. В отсутствие ингибитора разгон реакции начинался через 2 минуты. При содержании гидроталькита (HT-LD) 5,5% по массе продолжительность индукционного периода увеличилась до 5 дней, а при содержании гидроталькита 6,86% по массе продолжительность индукционного периода составила 7,5 дней.
Пример 8. Использование прокаленного гидроталькита при 80°С
Когда образец гидроталькита из примеров 6 и 7 заменили на прокаленный гидроталькит (прокаленный HT-LD) и подвергли взаимодействию при 80°С с 5,0% ингибитора по массе, то продолжительность индукционного периода увеличилась до 13,6 дней.
Пример 9. Использование 3% по массе мочевины и 1,9% по массе гидроталькита при 80°С
Чистый пирит, нитрат аммония и породоразрушающий раствор, содержащий мочевину в концентрации 3,0% по массе, смешали с непрокаленным гидроталькитом в концентрации 1,90% по массе и нагрели до 80°С, как описано в примере 5. Было установлено, что продолжительность индукционного периода возросла с приблизительно 10,4 дней при содержании только мочевины в количестве 3% по массе до 65,3 дней при добавлении гидроталькита.
Пример 10. Молекулярное сито 5А при 55°С
Молекулярное сито 5А измельчали в ступке пестиком и добавляли к смеси нитрата аммония с пиритом с концентрацией 5,11% по массе. Суспензию нагревали при перемешивании до 55°С в закрытой ячейке. Было установлено, что продолжительность индукционного периода составила приблизительно 12 часов.
Пример 11. Молекулярное сито 4А при 80°С
Молекулярное сито 4А измельчали в ступке пестиком и добавляли к смеси нитрата аммония с пиритом с концентрацией 6,22% по массе. Суспензию нагревали при перемешивании до 80°С в закрытой ячейке. Было установлено, что продолжительность индукционного периода составила приблизительно 5 часов.
Пример 12. Исследования с использованием молекулярных решеток
Молекулярную решетку Basolite С300 добавляли к смеси нитрата аммония с измельченной горной породой при содержании нитрата аммония в смеси от 1,74% до 2,4% по массе. Горная порода была получена из г. Ньюман. Эту смесь выдерживали при 55°С на водяной бане с контролируемой температурой до начала термического разгона реакции. При массовом содержании Basolite 2,4% продолжительность индукционного периода увеличилась с 15 минут до 247 минут. При массовом содержании Basolite 1,74% продолжительность индукционного периода увеличилась с 15 минут до 210 минут.
Примеры использования диоксида марганца в качестве поглотителя
Небольшие навески (общей массой приблизительно 5 г) проб игданита, состоящих из измельченного нитрата аммония (2,25 г), масла с эмульгатором PIBSA (приблизительно 0,17 г), измельченной мочевины (в количестве 0-5% от массы нитрата аммония), измельченного диоксида марганца (в количестве 0-5% от массы нитрата аммония) и пирита (2,25 г) смешивали с породоразрушающим раствором (0,5 г). Сначала частицы диспергировали в масляной фазе с помощью ультразвука. Затем при перемешивании добавляли мочевину и нитрат аммония. После этого добавляли смесь пирита с породоразрушающим раствором и суспензию тщательно перемешивали. Затем образец помещали в полуадиабатическую стеклянную пробирку с двойными стенками и прикрепляли термопары к наружной стенке внутренней пробирки. Пробирку закрывали пробкой и вводили шприц объемом 60 мл, а затем осторожно нагревали до 55°С. Нагрев образцов выполняли на глицериновой бане при 55°С до начала термического разгона.
Пример 13. Контрольное испытание игданита с мочевиной в количестве 1,86% по массе
Для сравнения приготовили чистую пробу игданита, содержащую только мочевину, и нагревали ее до 55°С. Перемешали контрольную смесь игданита (общей массой 5 г), содержащую мочевину в количестве 1,86% и масло с эмульгатором PIBSA (6,5% от масла). К ней добавили суспензию, содержащую пирит (2,25 г) в породоразрушающем растворе (0,5 г), тщательно перемешали и нагревали в полуадиабатических условиях до 55°С на глицериновой бане при скорости повышения температуры не более приблизительно 2,5°С/мин. Экзотермический пик был обнаружен после индукционного периода продолжительностью 9 часов.
Пример 14. Контрольное испытание игданита с мочевиной в количестве 2,18% по массе
Приготовили при перемешивании контрольную смесь игданита (общей массой 5 г), содержащую мочевину в количестве 2,18% и масло с эмульгатором PIBSA (6,5% от масла). К ней при перемешивании добавили суспензию, содержащую пирит (2,25 г) в породоразрушающем растворе (0,5 г), тщательно перемешали и нагревали в полуадиабатических условиях до 55°С на глицериновой бане при скорости повышения температуры не более приблизительно 2,5°С/мин. Экзотермический пик был обнаружен после индукционного периода продолжительностью 2 дня и 16 часов.
Пример 15. Испытание игданита с использованием мочевины с MnO2 в виде пиролюзита
Вышеуказанные эксперименты повторили в присутствии MnO2 в виде пиролюзита в количестве 2,5% по массе, диспергированного в масляной фазе. При использовании смеси 1,84% мочевины с MnO2 продолжительность индукционного периода увеличилось с 9 часов до 6 дней и 14 часов. При использовании смеси, содержащей 2,18% мочевины, продолжительность индукционного периода увеличилась с 2 дней и 16 часов до 8 дней и 13 часов.
Пример 16. Испытание игданита с использованием мочевины с аморфным MnO2 при 55°С
При добавлении 2,2% аморфного MnO2 к мочевине в количестве 1,70% продолжительность индукционного периода увеличилась с приблизительно 7 часов (только с мочевиной) до 3 дней и 7 часов (совместно с MnO2). При использовании мочевины в количестве 2,0% продолжительность индукционного периода составила 1 день и 21 час, а в присутствии аморфного MnO2 в количестве 2,2% она увеличилась до 9 дней и 9 часов.
Пример 17. Испытание игданита с использованием мочевины с MnO2 с размером частиц 10 мкм при 55°С
При добавлении MnO2 в количестве 2,48% к мочевине в количестве 1,92% продолжительность индукционного периода увеличилась с приблизительно 9 часов (только с мочевиной) до 6 дней и 2 часов (совместно с MnO2).
Пример 18. Контрольное испытание игданита с использованием мочевины
Реакции проводили при 100°С, нагревая образцы в изотермических условиях в алюминиевых блоках, вырезанных по размерам стеклянным пробирок. Внутрь образцов помещали термопары. Перемешали контрольную смесь игданита (общей массой 5 г), содержащую мочевину и масло с эмульгатором PIBSA (6,5% от масла). К ней добавили суспензию, содержащую пирит (2,25 г) в породоразрушающем растворе (0,5 г), тщательно перемешали и нагревали в изотермических условиях до 100°С в алюминиевом блоке при скорости повышения температуры не более приблизительно 2,5°С/мин. В контрольном эксперименте с добавлением 3,5% мочевины реакция в смеси возникла по истечении 142 минут. При добавлении 4,0% мочевины реакция возникла по истечении 4 дней и 22,5 часов.
Пример 19. Испытание игданита с использованием мочевины с MnO2 в виде пиролюзита при 100°С
Пример 5 повторили с добавлением 2,3% пиролюзита. Продолжительность индукционного периода при использовании 3,5% мочевины с MnO2 увеличилась с 142 минут до 5 дней и 15 часов, а при использовании 4,0% мочевины она увеличилась с 4 дней и 22,5 часов до 10 дней и 14,5 часов, соответственно.
Пример 20. Испытание игданита с использованием мочевины с аморфным MnO2 при 100°С
При добавлении 2,8% аморфного MnO2 к 3,6% мочевины при 100°С продолжительность индукционного периода увеличилась в присутствии MnO2 с 300 минут (только с мочевиной) до 3 дней и 5,5 часов.
Пример 21. Испытание игданита с использованием мочевины (нитрат аммония-масло-PIBSA-пирит-породоразрушающий раствор) при 120°С
Приготовили при перемешивании контрольную смесь игданита (общей массой 5 г), содержащую 5,36% мочевины и масло с эмульгатором PIBSA (6,5% от масла). К ней при тщательном перемешивании добавили суспензию, содержащую пирит (2,25 г) в породоразрушающем растворе (0,5 г) и нагревали в изотермических условиях до 120°С в алюминиевом блоке при скорости повышения температуры не более приблизительно 2,5°С/мин. Экзотермический пик был обнаружен после индукционного периода продолжительностью 3 дня и 20,5 часов.
Пример 22. Испытание игданита с использованием мочевины и MnO2 (нитрат аммония-масло-PIBSA-пирит-породоразрушающий раствор) при 120°С
При повторении предыдущего испытания (с использованием 5,30% мочевины) продолжительность индукционного периода увеличилась в присутствии 3,24% MnO2 с 3 дней и 20,5 часов до 7 дней и 8,5 часов.
Несмотря на то, что в данном документе описаны конкретные варианты осуществления настоящего изобретения, эти варианты осуществления должны рассматриваться во всех отношениях лишь как иллюстративные и следует понимать, что в них могут быть внесены изменения без отклонения от сущности и объема изобретения.
В приведенной далее формуле изобретения и в предшествующем описании изобретения, за исключением тех случаев, когда контекст требует иного из-за особенностей языка или логического построения, глагол «содержать» или такие варианты его использования, как «содержит» или «содержащий», имеют инклюзивный (охватывающий) смысл, то есть указывают на наличие определенных признаков, но не исключают наличие или добавление других признаков в различных вариантах осуществления изобретения.
Следует понимать, что если в данном документе упоминается какая-либо публикация из уровня техники, то такая ссылка не является признанием того, что эта публикация является частью общеизвестных знаний в данной области в Австралии или за ее пределами.
Реферат
Изобретение относится к способу стабилизации нитратсодержащего взрывчатого вещества, путем использования гидрофобизированного поглотителя NO, предотвращающего случайное разложение и повышающего безопасность и стабильность взрывчатого вещества при использовании его в шахтах с повышенной температурой и реакционноспособными горными породами. Также раскрыт взрывчатый состав, содержащий нитратсодержащее взрывчатое вещество и гидрофобизированный поглотитель NOв количестве от приблизительно 0,5 мас.% до приблизительно 10 мас.%, и способ его взрывания. 4 н. и 14 з.п. ф-лы, 6 ил., 22 пр.
Комментарии