Высокоэффективный способ аммоксидирования и катализаторы на основе смешанных оксидов металлов - RU2575933C2
Код документа: RU2575933C2
Чертежи



Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение относится к способу и катализатору процесса аммоксидирования ненасыщенного углеводорода с получением соответствующего ненасыщенного нитрила. В частности, данное изобретение направлено на создание способа и катализатора для аммоксидирования пропилена с получением акрилонитрила, цианистого водорода и ацетонитрила.
УРОВЕНЬ ТЕХНИКИ
Катализаторы, содержащие оксиды железа, висмута и молибдена, промотированные подходящими элементами, уже давно используются для конверсии пропилена и/или изобутилена в присутствии аммиака и кислорода (обычно в виде воздуха) при повышенных температурах с целью получения акрилонитрила и/или метакрилонитрила. В частности, каждый из следующих патентов: патента Великобритании 1436475, патентов США 4766232, 4377534, 4040978, 4168246, 5223469 и 4863891 относится к катализаторам на основе висмута-молибдена-железа, которые могут быть промотированы элементами II группы и применяются для получения акрилонитрила. Кроме того, патенты США 5093299, 5212137, 5658842 и 5834394 относятся к промотированным катализаторам на основе висмута-молибдена, которые обеспечивают получение высоких выходов акрилонитрила.
В частности, настоящее изобретение относится к катализаторам на основе висмута-молибдена-железа, которые получают, как описано в данной заявке. Обычно эти катализаторы получают периодическим способом путем простого соединения и взаимодействия исходных соединений (исходных) для получения различных металлических компонентов. Однако обычно используют более сложные и многостадийные способы. Например, в патенте США 4040978 раскрыт способ получения катализатора, в котором молибдаты каждого металла получали в отдельности и затем соединяли для осуществления их взаимодействия; патент США 4148757 описывает способ получения катализатора, согласно которому висмут и молибден сначала реагируют с получением молибдата висмута и затем молибдат висмута соединяют со смесью соединений, являющихся источниками получения различных других металлических компонентов.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к усовершенствованному способу и катализатору для аммоксидирования пропилена с получением акрилонитрила, цианистого водорода и ацетонитрила. Эти способ и катализатор характеризуются большей степенью конверсии пропилена в акрилонитрил, цианистый водород и ацетонитрил, чем ранее известные способы и катализаторы. Исторически способы и катализаторы, которые обеспечивали получение акрилонитрила с большим выходом, сопровождались соответствующим уменьшением выхода цианистого водорода и/или ацетонитрила.
Согласно одному из вариантов данное изобретение относится к способу получения акрилонитрила, цианистого водорода и ацетонитрила, включающему приведение в контакт при повышенной температуре пропилена, аммиака и кислорода в паровой фазе в присутствии катализатора, при этом катализатор включает комплекс оксидов металлов, и отношения элементов в указанном катализаторе выражены следующей формулой:
Mo12 Bia Feb Ac Dd Ее Ff Gg Ceh Ox,
где:
А обозначает по меньшей мере один элемент, выбранный из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути;
а, b, с, d, e, f, g, h и n, соответственно, обозначают атомные отношения висмута (Bi), железа (Fe), А, D, Е, F церия (Се) и кислорода к 12 атомам молибдена (Мо), где:
а имеет значение от 0,05 до 7,
в имеет значение от 0,1 до 7,
с имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, которое требуется для насыщения валентности других имеющихся элементов; и где относительные величины выхода акрилонитрила, ацетонитрила и цианистого водорода при осуществлении указанного способа определяются следующим уравнением:
α=[(%AN+(3×%HCN)+(1.5×%ACN))÷%РС]×100
где:
%AN обозначает выход акрилонитрила и %AN≥81,
%HCN означает выход цианистого водорода,
%ACN означает выход ацетонитрила и %
PC означает конверсию пропилена и
где α больше 100.
Согласно другому варианту изобретение относится к каталитической композиции, содержащей комплекс оксидов металлов, при этом относительные количества элементов в указанной каталитической композиции выражены следующей формулой:
Mo12BiaFebAcDdEeFfGgCehOx
где А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; а, b, с, d, е, f, g, h и n, соответственно, обозначают атомные отношения висмута (Bi), железа (Fe), A, D, Е, F церия (Се) и кислорода к 12 атомам молибдена (Мо), где:
а имеет значение от 0,05 до 7,
в имеет значение от 0,1 до 7,
с имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, которое требуется для насыщения валентности других имеющихся элементов; при этом относительные величины выхода акрилонитрила, ацетонитрила и цианистого водорода при осуществлении указанного способа определяются следующим уравнением:
α=[(%AN+(3×%HCN)+(1.5×%ACN))÷%РС]×100
где % AN обозначает выход акрилонитрила и % AN≥81,
%HCN означает выход цианистого водорода,
%ACN означает выход ацетонитрила и
%PC означает конверсию пропилена и
где α больше 100.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фигуре 1 показан график, отражающий исторические тенденции разработки катализаторов для получения акрилонитрила. На оси X показан выход акрилонитрила, а на оси Y показан выход цианистого водорода. Этот график показывает, что со временем величина выхода акрилонитрила возрастала, соответствующий выход цианистого водорода снижался. Катализаторы согласно данному изобретению не соответствуют указанной тенденции. Катализаторы согласно данному изобретению позволяют повысить выход акрилонитрила без значительного снижения выхода цианистого водорода, показанного на графике.
На Фигуре 2 отражена картина рентгеновской (XRD) дифракции или рентгеновская (XRD) дифрактограмма катализатора в соответствии с данным изобретением. На этой дифрактограмме видны интенсивный дифракционный пик с величиной угла дифракции 2θ, равной 28±0,3 градуса (интенсивность обозначена как X) и интенсивный дифракционный пик с величиной угла дифракции 2θ, равной 26,5±0,3 градуса (интенсивность обозначена как Y). Отношение X/Y равно 0,97.
На Фигуре 3 показана кривая, характеризующая износ катализатора от трения в зависимости от отношения Ce/Fe в катализаторе. Было установлено, что катализаторы с составом, описанным в данной заявке, и имеющие отношение Ce/Fe, превышающее или равное 0,8 и меньшее или равное 5, являются более прочными и имеют меньшие потери при трении, определенные при действии затопленной струи воздуха.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к способу и новому катализатору для получения акрилонитрила, ацетонитрила и цианистого водорода, характеризующимся тем, что относительные величины выхода акрилонитрила, ацетонитрила и цианистого водорода при осуществлении этого способа и применении этого катализатора выражаются следующим уравнением:
α=[(%AN+(3×%HCN)+(1.5×%ACN))÷%PC]×100
где %AN обозначает выход акрилонитрила и %AN≥81,
%HCN означает выход цианистого водорода,
%ACN означает выход ацетонитрила и
% PC означает конверсию пропилена и
где α больше 100.
Согласно другим вариантам данного изобретения %AN больше или равен 82, % PC больше 90, % PC больше 95, % PC больше 98, α больше 101, α больше 101,3, α больше 101,5. Применяемый в данной заявке термин выход "акрилонитрила" означает молярный выход акрилонитрила в % (выражается числом без указания процентов), рассчитанный следующим образом: (число молей полученного акрилонитрила÷число молей пропилена, подаваемого в реактор)х 100. Термин "выход цианистого водорода" означает молярный выход цианистого водорода в % (выражается числом без указания процентов), рассчитанный следующим образом: (число молей полученного цианистого водорода÷число молей пропилена, подаваемого в реактор)х 100. Термин выход "ацетонитрила" означает молярный выход ацетонитрила в % (выражается числом без указания процентов), рассчитанный следующим образом: (число молей полученного ацетонитрила÷число молей пропилена, подаваемое в реактор) х 100.
Катализатор
Данное изобретение частично относится к многокомпонентной каталитической композиции для аммоксидирования на основе смешанных оксидов металлов, содержащей комплекс каталитических оксидов, при этом элементы и относительные количества элементов в указанной каталитической композиции определяются по следующей формуле:
Mo12 Bia Feb Ac Dd Ее Ff Gg Ceh Ox,
где А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; а, b, с, d, е, f, g, h и n соответственно обозначают атомные отношения висмута (Bi), железа (Fe), A, D, Е, F церия (Се) и кислорода к 12 атомам молибдена (Мо), где:
а равен от 0,05 до 7,
b равен от 0,1 до 7,
с равен от 0,01 до 5,
d равен от 0,1 до 12,
е равен от 0 до 5,
f равен от 0 до 5,
g равен от 0 до 0,2,
h равен от 0,01 до 5 и
х равен количеству атомов кислорода, которое требуется для насыщения валентности других имеющихся элементов; и где относительные выхода акрилонитрила, ацетонитрила и цианистого водорода при осуществлении указанного способа определяются следующим уравнением:
α=[(%AN+(3×%HCN)+(1.5×%ACN))÷%РС]×100
где %AN обозначает выход акрилонитрила и %AN≥81,
%HCN означает выход цианистого водорода,
%ACN означает выход ацетонитрила и
%PC означает конверсию пропилена и где α больше 100.
Согласно другим вариантам данного изобретения %AN больше или равен 82, %PC больше 90, %PC больше 95, %PC больше 98, α больше 101, α больше 101,3, α больше 101,5.
Согласно одному варианту описанной выше каталитической композиции 0,15≤(a+h)≤1. Согласно другому варианту описанной выше каталитической композиции . Еще в одном варианте описанной выше каталитической композиции картина дифракции рентгеновских лучей вышеуказанной каталитической композиции включает дифракционные пики при угле 2θ 28±0,3 градусов и угле 2θ 26,5±0,3 градусов, и, если отношение интенсивности наиболее интенсивного дифракционного пика при 2θ угле 28±0,3 градуса к интенсивности наиболее интенсивного дифракционного пика при угле 2θ 26.5±0,3 градуса определяется как X/Y, тогда отношение X/Y больше или равно 0,7. Согласно другим независимым вариантам описанной выше каталитической композиции ; ; ; ; X/Y больше или равно 0,8; X/Y больше или равно 0,9; .
Согласно варианту, когда , означает отношение церия к железу в катализаторе и для любого состава катализатора оно означает количество молей церия (обозначенное нижним индексом у церия в формуле), деленное на количество молей железа (обозначенное нижним индексом у железа в формуле). Было обнаружено, что катализаторы, описываемые указанной выше формулой, где являются более прочными, что сказывается на меньшей величине потерь при истирании (большей износостойкости), определенной в тесте на истирание при действии затопленной струи.
В соответствии с вариантом, охарактеризованным рентгеновской дифракционной картиной идентифицированной выше каталитической композиции, на которой имеются дифракционные пики при угле дифракции 2θ, равном 28±0,3, и при угле дифракции 2θ, равном 26,5±0,3, следует указать, что, если отношение интенсивности наиболее интенсивного пика на рентгеновской дифрактограмме при угле дифракции 2-тета, равном 28±0,3, к интенсивности наиболее интенсивного пика на рентгеновской дифрактограмме при угле дифракции 2θ, равном 26,5±0,3, обозначено как X/Y, и X/Y больше или равно 0,7, то было обнаружено, что такие катализаторы обеспечивают большую степень общей конверсии при аммоксидировании пропилена и/или бутилена с получением нитрилов (то есть соединений, содержащих функциональную группу -CN, таких как акрилонитрил, метакрилонитрил, ацетонитрил и цианистый водород.
Используемые в данной заявки термины "каталитическая композиция" и "катализатор" представляют собой синонимы и применяются как взаимозаменяемые. Применяемый в данной заявке термин "редкоземельный элемент" означает по меньшей мере один элемент из лантана, церия, празеодима, неодима, прометия, самария, европия, гадолиния, тербия, диспрозия, гольмия, эрбия, тулия, иттербия, скандия и иттрия. Применяемые обозначения и «29» являются синонимами.
Катализатор согласно данному изобретению может применяться в виде катализатора на носителе и в виде катализатора без носителя (то есть катализатор может содержать носитель). Подходящие носители представляют собой диоксид кремния, оксид алюминия, диоксид циркония, диоксид титана или их смеси. Носитель обычно служит как связующее для катализатора и обеспечивает получение более прочного (то есть более устойчивого к истиранию, износостойкого) катализатора. Однако для коммерческих целей соответствующая смесь как активной фазы (то есть комплекса каталитических оксидов, описанного выше), так и носителя является важной для получения у катализатора приемлемой активности и прочности (стойкости к износу). Обычно количество носителя составляет между 40 и 60 вес.% в расчете на катализатор с носителем. Согласно одному из вариантов изобретения количество носителя может быть небольшим, например, равным примерно 30 вес.% в расчете на катализатор на носителе. Согласно другому варианту данного изобретения количество носителя может составлять примерно 70% в расчете на катализатор на носителе.
Согласно одному из вариантов изобретения в качестве носителя для катализатора применяют золь диоксида кремния (силикатный золь). Обычно золи диоксида кремния содержат некоторое количество натрия. Согласно одному из вариантов золь диоксида кремния содержит менее 600 м. д. (частей на миллион) натрия. Согласно другому варианту золь диоксида кремния содержит менее 200 м. д. натрия. Обычно средний диаметр коллоидных частиц золя диоксида кремния находится в интервале между примерно 15 нм и примерно 50 нм. Согласно одному из вариантов изобретения средняя величина диаметра коллоидных частиц золя диоксида кремния составляет примерно 10 нм и может быть такой маленькой, как 4 нм. В соответствии с другим вариантом данного изобретения средняя величина диаметра коллоидных частиц золя диоксида кремния составляет примерно 100 нм. Согласно еще одному варианту данного изобретения средняя величина диаметра коллоидных частиц золя диоксида кремния составляет примерно 20 нм.
Согласно одному из вариантов катализатор имеет "степень износа", равную величине менее примерно 8,0, определенную при действии затопленной струи. Степень износа выражается числом, соответствующим потере массы катализатора при истирании в течение промежутка времени между 1 и 20 ч. Степень износа является мерой прочности частиц катализатора. Желательно достигать меньших величин степени износа. В случае промышленных катализаторов величина степени износа более примерно 8,0 не является предпочтительной из-за высокой скорости потерь катализатора. Устройство для проведения теста при действии затопленной струи описано в ASTM D5 75 7-00 Standard Test Method for Determination of Attrition and Abrasion of Powdered Catalysts by Air Jets.
Получение катализатора
Согласно одному из вариантов изобретения элементы каталитической композиции, описанной выше, соединяются вместе в водной суспензии предшественника катализатора, полученная водная суспензия сушится с получением предшественника катализатора и затем предшественник катализатора подвергается прокаливанию с образованием катализатора. Однако уникальными для данного процесса являются следующие признаки:
(i) соединение в водном растворе исходных соединений Bi и Се и возможно одного из Na, К, Rb, Cs, Са, редкоземельного элемента, Pb, V, Y с получением смеси;
(ii) добавление к смеси (то есть к первой смеси) исходного для Мо для взаимодействия со смесью и образования "суспензии осадка";
(iii) соединение суспензии осадка с исходными для остальных элементов и для остального молибдена в катализаторе для получения водной суспензии предшественника катализатора.
Применяемый в данной заявке термин "исходные соединения" (исходные, источники) означает соединения, которые содержат один или более металлов или обеспечивают наличие одного или более металлов для каталитической композиции на основе смешанных оксидов металлов. Применяемые в данной заявке термины "остальные соединения" или "остальные соединения в катализаторе" относятся к тем элементам и их количеству, которые обозначены как A, D, Е, F и G в приведенной выше формуле, которые не были включены в первую смесь. Согласно одному из вариантов некоторые элементы могут быть частями первой и второй смеси. Кроме того, используемые термины "остальной молибден" или "молибден, оставшийся в катализаторе" относятся к необходимому количеству молибдена, требующемуся в конечном катализаторе, которое не входит в состав (то есть оно не было введено при получении) в суспензии осадка. Наконец, суммарное количество молибдена, обеспеченного исходными соединениями для молибдена, добавленными на стадиях (ii) и (iii), равно всему количеству молибдена, содержащемуся в катализаторе.
В способе получения катализатора, описанном в данной заявке, исходные соединения (источники) для остальных элементов и остального (оставшегося) молибдена, которые соединяются в суспензии осадка, могут объединяться в любом порядке или в любой комбинации остальных элементов и остального молибдена. Согласно одному из вариантов смесь исходных соединений для остальных элементов и остального молибдена соединяется с суспензией осадка с образованием водной суспензии предшественника катализатора. Согласно другому варианту (i) смесь исходных соединений остальных элементов соединяется с суспензией осадка и (ii) исходные соединения для остального молибдена отдельно добавляются к суспензии осадка с образованием водной суспензии предшественника катализатора. Порядок добавления компонентов не является решающим. Согласно еще одному варианту исходные соединения для остальных элементов и для остального молибдена добавляются по отдельности (то есть один компонент в данное время) для осаждения суспензии. Согласно другому варианту совокупность (то есть более, чем одна) смесей исходных соединений для остальных элементов и для остального молибдена, при этом в каждой смеси содержится одно или более исходных соединений для остальных элементов и для остального молибдена, добавляется поочередно (то есть одна смесь в данное время или одновременно добавляется несколько смесей) к суспензии осадка с образованием водной суспензии предшественника катализатора. Согласно еще одному варианту смесь исходных соединений для остальных элементов соединяется с исходным соединением для остального молибдена, и полученная смесь затем добавляется к суспензии осадка с образованием водной суспензии предшественника катализатора. Согласно другому варианту носитель представляет собой диоксид кремния (SiO2), и диоксид кремния соединяется с исходным соединением для остального молибдена до объединения остального молибдена с суспензией осадка (то есть диоксид кремния и исходное соединение для остального молибдена соединяются с образованием смеси, и затем эта смесь добавляется к суспензии осадка, в отдельности или в комбинации с одним или более исходными соединениями для остальных элементов).
В способе получения катализатора, описанном в данной заявке, молибден добавляется и при получении суспензии осадка, и при получении водной суспензии предшественника катализатора. На атомном уровне минимальное количество молибдена, добавляемое для образования суспензии осадка, определяется по следующему уравнению:
Mo=1,5(Bi+Ce)+0,5(Rb+Na+K+Cs)+(Ca)+l,5(сумма числа атомов редкоземельных элементов)+(Pb)+3(W)+1,5(Y).
В приведенном выше уравнении "Мо" обозначает количество атомов молибдена, которое должно быть добавлено к первой смеси, и "Bi", "Се", "Rb", "Na", "К", "Cs", "Ca", "Pb","W" и "Y" обозначают количество атомов висмута, церия, рубидия, натрия, калия, цезия, кальция, свинца, вольфрама и иттрия соответственно, содержащееся в первой смеси.
Обычно количество молибдена, добавляемого в первую смесь для образования суспензии осадка, составляет примерно от 20% до 35% от всего количества молибдена в полученном катализаторе. Согласно одному из вариантов исходное соединение для остального молибдена, содержащегося в катализаторе, добавляется к исходным соединениям для остальных элементов (то есть ко второй смеси) до соединения смеси остальных элементов с суспензией остатка для получения суспензии предшественника катализатора. Согласно другим вариантам исходное для молибдена, содержащее остальной молибден, имеющийся в конечном катализаторе, добавляется к суспензии осадка до, после или одновременно со смесью исходных соединений для остальных элементов (то есть ко второй смеси) с целью получения суспензии предшественника катализатора.
В начале осуществления способа исходные соединения для Bi и Се и, необязательно, один или более элементов из Na, К, Rb, Cs, Ca, редкоземельного элемента, Pb, W и Y соединяются в водном растворе с получением их смеси. Согласно одному из вариантов нитрат висмута и, необязательно, другие нитраты металлов (то есть, нитраты Na, К, Rb, Cs, Ca, редкоземельного элемента, Pb, W и/или Y) растворяются в водном растворе нитрата церия-аммония.
К смеси, содержащей висмут и церий (и, необязательно, один или более элементов из Na, К, Rb, Cs, Ca, редкоземельного элемента, Pb, W и Y), добавляется исходное для молибдена. Согласно одному из вариантов это исходное соединение для молибдена представляет собой гептамолибдат аммония, растворенный в воде. После добавления соединения, являющегося источником молибдена, к смеси, содержащей висмут и церий, будет происходить реакция, которая приводит к получению осадка, и полученная смесь является суспензией осадка.
Суспензия осадка затем соединяется со смесью исходных соединений для остальных элементов и исходного соединения для молибдена с образованием водной суспензии предшественника катализатора. Смесь исходных соединений для остальных элементов и исходного соединения для молибдена может быть приготовлена путем соединения исходных соединений для остальных элементов в водном растворе (например, исходные соединения соединяются в воде) и затем добавления исходного соединения для молибдена. Согласно одному из вариантов это исходное соединение для молибдена представляет собой гептамолибдат аммония, растворенный в воде. При соединении суспензии осадка со смесью остальных элементов/молибдена порядок добавления не является решающим, то есть суспензия осадка может быть добавлена к смеси остальные элементы/молибден или смесь остальные элементы/молибден может быть добавлена к суспензии осадка. Водная суспензия предшественника катализатора поддерживается при повышенной температуре.
Количество водного растворителя в каждой из описанных выше водных смесей и суспензий может меняться в зависимости от растворимости исходных соединений, объединяемых с образованием смешанных оксидов металлов. Количество водного растворителя должно быть по меньшей мере достаточным для получения суспензии или смеси твердых веществ и жидкостей, которые можно перемешивать.
В любом случае исходные соединения предпочтительно соединяются и взаимодействуют по протоколу, который включает смешение исходных соединений во время их объединения и/или стадию взаимодействия. Конкретный механизм смешения не является критическим и может включать, например, смешение (то есть перемешивание или взбалтывание) компонентов во время реакции любым эффективным методом. Такие методы включают, например, взбалтывание содержимого сосуда, например, путем встряхивания, опрокидывания или покачивания сосуда, содержащего введенные компоненты. Эти методы включают также, например, перемешивание при помощи перемешивающего элемента, который расположен, по меньшей мере частично, внутри реакционного сосуда, и движущей силы, которая подводится к перемешивающему элементу или к реакционному сосуду для обеспечения движения мешалки и реакционного сосуда относительно друг друга. Перемешивающий элемент может быть с приводом от вала и/или без привода. Движущая сила может непосредственно подводиться к перемешивающему элементу или подводиться косвенно (например, при помощи магнитов). Смешение должно быть предпочтительно достаточным для смешения компонентов с целью осуществления эффективной реакции между компонентами реакционной смеси, например, с образованием более гомогенной реакционной среды (и получения более гомогенного предшественника смешанных оксидов металлов) по сравнению с реакцией, осуществляемой без перемешивания. Это приводит к более эффективному использованию исходных материалов и к получению более однородного продукта, представляющего собой смешанные оксиды металлов. Перемешивание суспензии осадка во время реакции также вызывает осаждение продукта в растворе, а не на стенках реакционного сосуда. Преимущественно получить осадок в растворе, так как это позволяет частицам расти по всей поверхности, а не на ограниченной поверхности, когда рост начинается от стенки реакционного сосуда.
Исходное соединение для молибдена может представлять собой оксид молибдена (VI) (МоО3), гептамолибдат аммония или молибденовую кислоту. Источником молибдена может быть любой оксид молибдена, такой как диоксид, триоксид, пентаоксид или гептаоксид молибдена. Однако предпочтительно, когда в качестве источника молибдена применяют гидролизуемую или разлагаемую соль молибдена.
Типичными исходными соединениями для висмута, церия и остальных элементов катализатора являются нитратные соли металлов. Такие нитраты являются легкодоступными и хорошо растворяются.
Исходное соединение для висмута может включать оксид или соль, которая при прокаливании образует оксид. Предпочтительными являются водорастворимые соли, которые легко диспергируются, но образуют стабильные оксиды при нагревании. Согласно одному из вариантов изобретения источником висмута является нитрат висмута, Вi(NО3)3×5Н2O.
Исходное соединение для церия может включать оксид или соль, которая при прокаливании образует оксид. Предпочтительными являются водорастворимые соли, которые легко диспергируются, но образуют стабильные оксиды при нагревании. Согласно одному из вариантов изобретения источником церия является нитрат церия-аммония, (NН4)2Се(NО3)6.
Исходное соединение для железа может быть получено из любого соединения железа, которое после прокаливании приведет к образованию оксида. Как и в случае других элементов предпочтительными являются водорастворимые соли из-за легкости, с которой они диспергируются в катализаторе. Наиболее предпочтительным является нитрат железа.
Исходные для остальных элементов могут быть получены из любого подходящего источника. Например, кобальт, никель и магний могут быть введены в катализатор при помощи нитратных солей. Кроме того, магний может быть введен в катализатор в виде нерастворимого карбоната или гидроокиси, которая при нагревании образует оксид. Фосфор может быть введен в катализатор с помощью соли щелочного металла или соли щелочноземельного металла или соли аммония, но предпочтительно вводится с помощью фосфорной кислоты.
Щелочные компоненты могут быть введены в катализатор в виде оксида или соли, которая при нагревании образует оксид.
Растворители, которые в дополнение к воде могут быть использованы для получения смешанных оксидов металлов согласно данному изобретению, включают, но без ограничения, спирты, такие как метиловый спирт, этиловый спирт, пропиловый спирт, диолы (например, этиленгликоль, пропиленгликоль и т.д.), органические кислоты, такие как уксусная кислота, а также другие полярные растворители, известные из уровня техники. Исходные соединения для введения металлов по меньшей мере частично растворяются в таком растворителе.
Как отмечалось ранее, катализатор согласно данному изобретению можно использовать как на носителе, так и без него (то есть катализатор может содержать носитель). Подходящие носители представляют собой диоксид кремния, оксид алюминия, диоксид циркония, диоксид титана или их смеси. Момент добавления носителя при получении катализатора не является критическим. Носитель может быть добавлен в любое время до сушки суспензии предшественника катализатора. Носитель может быть добавлен в любое время во время или после получения любой смеси элементов, суспензии осадка или суспензии предшественника катализатора. Далее, не является необходимым добавление носителя в одной точке или на одной стадии (то есть носитель может быть добавлен во многих точках в процессе получения катализатора). Согласно одному из вариантов носитель соединяется с другими ингредиентами во время приготовления водной суспензии предшественника катализатора. Согласно одному варианту носитель добавляется к суспензии осадка (то есть после получения суспензии осадка). Согласно другому варианту носитель соединяется с исходным соединением для молибдена до объединения исходного соединения для молибдена с исходными соединениями для введения остальных элементов в катализатор с образованием "второй смеси", описанной выше.
Суспензию предшественника катализатора высушивают и подвергают денитрификации (то есть удаляют нитраты) для получения предшественника катализатора. Предпочтительно высушивать суспензию предшественника катализатора при распылении при температуре между 110°С и 180°С. Температура денитрификации может находиться в пределах от 100°С до 500°С, предпочтительно от 250°С до 450°С.
В конце процесса высушенный предшественник катализатора подвергают прокаливанию. Согласно одному из вариантов прокаливание проводится на воздухе. Согласно другому варианту прокаливание проводится в инертной атмосфере, такой как азот. Предпочтительные условия прокаливания включают температуры в пределах от примерно 300°С до примерно 700°С, более предпочтительно от примерно 350°С до примерно 650°С, и согласно некоторым вариантам прокаливание может проводиться при температуре около 600°С.
Катализаторы согласно данному изобретению могут быть получены любым из многочисленных способов получения катализаторов, которые известны специалистам в данной области. Согласно одному из вариантов компоненты катализатора можно смешивать с носителем в виде суспензии с последующей сушкой или компоненты катализатора могут быть введены на диоксиде кремния или на других носителях.
Процесс аммоксидирования
Способ и катализатор по настоящему изобретению могут применяться для конверсии пропилена в акрилонитрил, цианистый водород и ацетонитрил путем взаимодействия в паровой фазе пропилена с газом, содержащим молекулярный кислород, и аммиаком в присутствии катализатора при повышенной температуре и давлении.
Предпочтительно, если реакцию аммоксидирования осуществляют в реакторе с псевдоожиженным слоем, хотя предусмотрены и другие типы реакторов, такие как реакторы с транспортными линиями. Реакторы с псевдоожиженным слоем для получения акрилонитрила хорошо известны из уровня техники. Например, пригоден реактор, описанный в патенте США №3230246, который включен в данную заявку посредством отсылки.
Условия для осуществления реакции аммоксидирования также хорошо известны в уровне техники, например, они описаны в патентах США №№5093299, 4863891, 4767878 и 4503001, включенных в данную заявку посредством отсылок. Обычно процесс аммоксидирования осуществляют путем контактирования пропилена в присутствии аммиака и кислорода в псевдоожиженном слое катализатора при повышенной температуре с получением акрилонитрила, цианистого водорода и ацетонитрила. Может быть использован любой источник кислорода. Однако по экономическим причинам предпочтительно применять воздух. Типичное мольное отношение кислорода к олефину в сырье должно быть в пределах от 0,5:1 до 4:1, предпочтительно от 1:1 до 3:1.
Мольное отношение аммиака к пропилену в сырье для проведения реакции может меняться и находится между 0,5:1 и 2:1. В действительности не существует верхнего предела отношения аммиака к пропилену, но обычно нет причины превышать отношение 2:1 по экономическим соображениям. Подходящие отношения исходных соединений, применяемые в способе и катализаторе по изобретению для получения акрилонитрила, цианистого водорода и ацетонитрила из пропилена, включают мольное отношение аммиака к пропилену в пределах от 0,9:1 до 1,3:1 и мольное отношение воздуха к пропилену в пределах от 8,0:1 до 12,0:1. Способ и катализатор согласно данному изобретению способны обеспечивать высокий выход акрилонитрила, цианистого водорода и ацетонитрила при сравнительно низких исходных отношениях аммиака к пропилену от примерно 1:1 до примерно 1,05:1. Эти "условия с небольшим количеством аммиака" помогают снизить количество непрореагировавшего аммиака в потоке, выходящем из реактора, это условие, известное как "шаг вперед" в применении аммиака, затем способствует снижению количества отходов. Непрореагировавший аммиак должен быть удален из потока, выходящего из реактора, до выделения акрилонитрила, цианистого водорода и ацетонитрила. Непрореагировавший аммиак обычно удаляется путем контактирования потока, выходящего из реактора, с серной кислотой с получением сульфата аммония или путем контактирования потока, выходящего из реактора, с акриловой кислотой с получением акрилата аммония, что в обоих случаях позволяет обработать поток отходов и/или ликвидировать его.
Реакцию проводят при температуре в пределах от примерно 260°С до примерно 600°С, предпочтительно от 310°С до 500°С, особенно предпочтительно от 350°С до 480°С. Время контактирования, хотя и не является критическим, обычно находится в пределах от 0,1 до 50 с, предпочтительно от 1 до 15 с.
Полученные продукты реакции могут быть выделены и очищены любым из методов, известных специалисту в данной области. Один такой метод включает промывку выходящих из реактора газов холодной водой или подходящим растворителем для удаления продуктов реакции и затем очистки продукта реакции путем перегонки. Катализаторы, полученные способом согласно данному изобретению, в основном применяются для аммоксидирования пропилена с получением акрилонитрила. Другое применение включает аммоксидирование пропана с получением акрилонитрила и аммоксидирование этанола с получением ацетонитрила. Катализатор, полученный способом согласно данному изобретению, может быть также использован для окисления пропилена до акриловой кислоты. Такие способы являются обычно двухстадийными процессами, где пропилен превращается в присутствии катализатора сначала в акролеин и акролеин превращается в присутствии катализатора в акриловую кислоту на второй стадии. Катализатор, описанный в данной заявке, пригоден для применения на первой стадии окисления пропилена в акролеин.
КОНКРЕТНЫЕ ВАРИАНТЫ
Для иллюстрации настоящего изобретения проводили оценку катализаторов, полученных в соответствии с настоящим изобретением, и сравнивали их с подобными катализаторами, полученными известными способами при похожих условиях реакции и не входящими в объем данного изобретения. Эти примеры приведены только с целью иллюстрации.
Катализаторы, имеющие состав Cs0,1K0,1Ni5Mg2Na0,05Fe1,8Bi0,45Ce1,1Mo12,55O50,35+45 вес.% Na SiO2, были получены различными препаративными способами, которые описаны ниже, и испытаны при аммоксидировании пропилена в лабораторном реакторе с получением акрилонитрила. Испытание проводили в реакторе с псевдоожиженным слоем объемом 40 куб. см. Пропилен подавали в реактор со скоростью 0,06 ВВЧ (то есть вес пропилена/вес катализатора/ч). Давление в реакторе поддерживалось равным 10 ф/дюйм2. Мольное отношение пропилен/аммиак/воздух составляло примерно 1/1,2/9,5. Температура реакции была равна 430°С. После периода стабилизации, равного примерно 20 ч, отбирали образцы продуктов реакции. Поток, выходящий из реактора, собирали в скрубберах барботажного типа, содержащих холодный раствор НСl. Скорость отходящего газа измеряли при помощи расходомера с мыльной пленкой, и состав отходящего газа определяли в конце опыта с помощью газового хроматографа (ГХ), снабженного анализатором для разделенного на выходе из колонки газа. В конце выделения всю жидкость в скруббере разбавляли до примерно 200 г дистиллированной водой. Взвешенное количество 2-бутанона использовали в качестве внутреннего стандарта в виде аликвоты разбавленного раствора, равной примерно 50 г. Образец объемом 2 мкл анализировали в ГХ, снабженном плазменным детектором и колонкой Carbowax. Количество аммиака определяли путем титрования избытка свободной НС1 с помощью раствора NaOH.
Сравнительный пример С1: общепринятый способ
Реакционную смесь А получали путем нагревания 224 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (203,75 г) в течение 30 мин при перемешивании с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (625 г, 32,5 вес.% диоксида кремния). Реакционную смесь В готовили путем нагревания 30 мл деионизированной воды до 55°С и последующего добавления при перемешивании Fе(NО3)3×9Н2O (66,9 г), Ni(NO3)2×6Н2O (133,7 г), Mg(NO3)2×6H2O (47,2 г), Вi(NО3)2×5H2O (20,1 г), CsNO3 (1,79 г), KNO3 (0,93 г) и NaNO3 (0,39 г). Затем при перемешивании добавляли 110,0 г 50 вес.% водного раствора (NН4)2Се(NО3)6.
Реакционную смесь В затем добавляли к реакционной смеси А при перемешивании для получения суспензии предшественника катализатора. Суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Затем ее гомогенизировали в смесителе в течение 3 мин с окружной скоростью 5000 об/мин. Суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации путем тепловой обработки в течение 3 ч на воздухе при температуре 290°С и затем при температуре 425°С на воздухе еще в течение 3 ч. Затем полученный порошок подвергали прокаливанию на воздухе при 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора аммоксидирования пропилена в микрореакторе объемом 40 куб. см. Результаты испытаний приведены в Таблице 1.
Сравнительный пример С2: проводили, как описано в патенте США №4212766 (то есть не осуществляли получение суспензии осадка Bi-Ce-Mo на отдельной стадии).
Реакционную смесь А получали путем нагревания 233 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (212,1 г) в течение 30 мин при перемешивании с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (692 г, 32,5 вес.% диоксида кремния).
Реакционную смесь В готовили путем нагревания 33 мл деионизированной воды до 55°С и последующего добавления при перемешивании Fе(NО3)3×9Н2О (73,6 г), Ni(NO3)2×6Н2О (147,1 г), Mg(NO3)2×6H2O (51,9 г), CsNO3 (1,97 г), KNO3 (1,02 г) и NaNO3 (0,43 г) и 122,0 г 50 вес.% водного раствора (NH4)2Ce(NO3)6.
Реакционную смесь С получали путем нагревания 152 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (12,1 г)при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D получали путем растворения Вi(NО3)3×5Н2O (22,1 г) в 160 г 10 вес.% раствора НNО3.
Реакционную смесь Е получали путем добавления реакционной смеси В к реакционной смеси А при перемешивании.
Реакционную смесь F получали путем добавления реакционной смеси D к реакционной смеси С при перемешивании. Это приводило к осаждению бесцветного твердого продукта. Перемешивание продолжали в течение 15 мин при поддержании температуры, равной 50-55°С.
Затем добавляли реакционную смесь F к реакционной смеси Е с получением суспензии предшественника катализатора.
Суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Затем ее гомогенизировали в смесителе в течение 3 мин при окружной скорости 5000 об/мин. Суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации путем тепловой обработки в течение 3 ч при температуре 290°С на воздухе и затем еще в течение 3 ч при температуре, равной 425°С. Затем полученный порошок подвергали прокаливанию на воздухе при температуре 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора аммоксидирования пропилена в микрореакторе объемом 40 куб. см. Результаты испытаний приведены в Таблице 1.
Пример 1: осуществляли процесс согласно данному изобретению.
Реакционную смесь А получали путем нагревания 198 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (180,4 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (692 г, 32,5 вес.% диоксида кремния). Реакционную смесь В готовили путем нагревания 33 мл деионизированной воды до 55°С с последующим добавлением при перемешивании Fе(NО3)3×9H2O (73,6 г), Ni(NO3)2×6H2O (147,1 г) и Mg(NO3)2×6H2O (51,9 г).
Реакционную смесь С получали путем нагревания 48 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (43,75 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания 122,0 г 50 вес.% водного раствора (NН4)2Се(NO3)6 до 55°С и (ii) и последовательного добавления Bi(NO3)3×5H2O (22,1 г), CsNO3 (1,97 г), KNO3 (1,02 г) и NaNO3 (0,43 г) при нагревании и перемешивании полученного раствора, что приводило к образованию прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления при перемешивании реакционной смеси В к реакционной смеси А.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D. Это приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Затем ее гомогенизировали в смесителе в течение 3 мин при окружной скорости 5000 об/мин. Суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке в течение 3 ч на воздухе при температуре 290°С и затем при температуре 425°С еще в течение 3 ч на воздухе. Затем полученный порошок подвергали прокаливанию на воздухе при температуре 560°С в течение 3 ч. Полученный прокаленный порошок впоследствии испытывали в качестве катализатора в процессе аммоксидирования пропилена в микрореакторе объемом 40 куб. см. Результаты испытаний приведены в Таблице 1.
Пример 2: получение согласно данному изобретению.
Реакционную смесь А получали путем нагревания 198 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (180,4 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь В готовили путем нагревания 33 мл деионизированной воды до 55°С с последующим добавлением при перемешивании Fе(NО3)3×9Н2О (73,6 г), Ni(NО3)2×6Н2O (147,1 г) и Mg(NO3)2×6Н2O (51,9 г).
Реакционную смесь С получали путем нагревания 48 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (43,75 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания 122,0 г 50 вес.% водного раствора (NH4)2Се(NO3)6 до 55°С и (ii) и последовательного добавления Bi(NO3)3×5Н2O (22,1 г), CsNO3 (1,97 г), КНО3 (1,02 г) и NaNO3 (0,43 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления при перемешивании реакционной смеси В к реакционной смеси А.
Реакционную смесь F получали путем (i) добавления реакционной смеси С к реакционной смеси D, что приводило к осаждению твердого продукта оранжевого цвета (полученная смесь представляла собой суспензию осадка), (ii) перемешивания суспензии осадка в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С и (III) добавления при перемешивании золя диоксида кремния (692 г, 32,5 вес.% диоксида кремния).
Затем реакционную смесь Е добавляли к реакционной смеси F с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Затем ее гомогенизировали в смесителе в течение 3 мин при окружной скорости 5000 об/мин. Суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке в течение 3 ч на воздухе при температуре 290°С и затем при температуре 425°С на воздухе еще в течение 3 ч. Затем полученный порошок подвергали прокаливании на воздухе при температуре 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора процесса аммоксидирования пропилена в микрореакторе объемом 40 куб. см. Результаты испытаний приведены в Таблице 1.
Таблица 1
Сравнение свойств катализатора состава Cs0,1K0,1Ni5Mg2Na0,05Fe1,8Bi0,45Ce1,1Mo12,55O50,35+45 вес.% SiO2, полученного разными способами
Примечания:
1. Все испытанные составы катализаторов содержали 55 вес.% активной фазы и 45 вес.% SiO2 с частицами размером 22 нм и с низким содержанием Na.
2. "HOS" обозначает "время работы реактора в ч”, то есть количество времени, в течение которого проводили испытание катализатора.
3. "Величина общей конверсии С3-“ означает конверсию пропилена (в мольных процентах) во все продукты за один проход в расчете на пропущенный пропилен.
4. "Степень конверсии в AN” означает конверсию пропилена (в мольных процентах) в акрилонитрил за один проход.
5. "Степень конверсии в HCN” означает конверсию пропилена (в мольных процентах) в цианистый водород за один проход.
6. "Степень конверсии в AN и HCN” означает общую конверсию пропилена (в мольных процентах) в акрилонитрил и цианистый водород за один проход.
Как видно из Таблицы 1, композиции катализатора, полученные согласно данному изобретению, обеспечивают более высокие величины степени конверсии в акрилонитрил и HCN, когда осуществляли аммоксидирование пропилена над таким катализатором при повышенных температурах в присутствии аммиака и воздуха, по сравнению с идентичными катализаторами, полученными способами, не входящими в объем данного изобретения.
Для дальнейшей иллюстрации настоящего изобретения получали катализаторы с составами по изобретению (Примеры 3-7), испытывали их и сравнивали с подобными катализаторами, имеющими состав не по изобретению и полученными при похожих условиях реакции (Сравнительные примеры С3-С7). Эти примеры приводятся только с целью иллюстрации.
Сравнительный пример 3 - катализатор С-49МС для получения акрилонитрила Результаты испытаний и другие данные для катализатора С-49МС для получения акрилонитрила приведены в Таблице 2. Катализатор "С-49МС" представляет собой промышленный катализатор, который производится и продается компанией INEOS USA LLC. Состав катализатора С-49МС является коммерческим секретом компании INEOS USA LLC. Обозначение "С-49МС" является товарным знаком INEOS USA LLC. Сравнительный пример С4 - катализатор Ni7,5Mg3Fe1,8Rb0,l2Cr0,1Bi0,45Ce1,1Mo15,85O63,835+50 вес.% SiO2 с размером частиц 39 нм, содержащей 27 м. д. Na,
Реакционную смесь А получали путем нагревания 225,1 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (204,6 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (595,2 г, 42 вес.% диоксида кремния, 27 м. д. Na, средний размер частиц 39 нм).
Реакционную смесь В готовили путем нагревания 32,2 мл деионизированной воды до 55°С и последующего добавления Fe(NO3)3×9H2O (53,2 г), Ni(NO3)2×6H2O (159,5 г), Mg(NO3)2×6H2O (56,2 г), Cr(NO3)2×9H2O (2,9 г) и RbNО3 (1,3 г) при перемешивании.
К раствору В добавляли 88,2 г 50 вес.% водного раствора (NH4)2Ce(NO3)6 с последующими добавлением этой полученной смеси к раствору А при перемешивании в течение 1 ч при температуре около 55°С и охлаждением до 40°С. Полученную суспензию катализатора затем подвергали гомогенизации в смесителе в течение 3 мин при окружной скорости 5000 об/мин. Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке в течение 3 ч на воздухе при температуре 290°С и затем при температуре 425°С на воздухе в течение 3 ч. Затем полученный порошок подвергали прокаливанию на воздухе при 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора аммоксидирования пропилена в микрореакторе объемом 40 куб. см. Результаты испытаний приведены в Таблице 2.
Сравнительный пример С5: катализатор Ni7,5Mg3Fe1,8 Rb0,12Cr0,1Bi0,45Ce1,1Mo15,85O63,835+50 вес.% SiO2.
Реакционную смесь А получали путем нагревания 191,1 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (173,8 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (27 м. д. Na, 595,2 г, 42 вес.% диоксида кремния, средний размер частиц 39 нм).
Реакционную смесь В готовили путем нагревания 32,1 мл деионизированной воды до 55°С и последующего добавления при перемешивании Fе(NО3)3×9Н2О (53,2 г), Ni(NO3)2×6Н2O (159,5 г), Mg(NO3)2×6H2O (56,2 г), Сr(NО3)2×9H2O (2,9 г).
Реакционную смесь С получали путем нагревания 33,9 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (30,8 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания 88,2 г 50 вес.% водного раствора (НN4)2Се(NО3)6 до 55 С и (ii) последовательного добавления Вi((NО3)3×5Н2O (16,0 г) и RbNO3 (1,3 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления при перемешивании реакционной смеси В к реакционной смеси А.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D, что приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Эту суспензию затем подвергали гомогенизации в смесителе в течение 3 мин при окружной скорости 5000 об/мин. Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке в течение 3 ч на воздухе при температуре 290°С и затем в течение 3 ч на воздухе при температуре 425°С. Затем полученный порошок подвергали прокаливанию на воздухе при 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора в процессе аммоксидирования пропилена в микрореакторе объемом 40 куб. см. Результаты испытаний приведены в Таблице 2.
Пример 6А: катализатор Ni5Mg2Fe1,8 Rb0,12Cr0,1Bi0,45Ce1,1Mo12,35O49,835+50 вес.% SiO2.
Реакционную смесь А получали путем нагревания 1619,7 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (1472,5 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (5357,1 г, 42 вес.% диоксида кремния, 27 м. д. Na, средний размер частиц 39 нм).
Реакционную смесь В готовили путем нагревания 276,2 мл деионизированной воды до 55°С и последующего добавления Fе(NО3)3×9H2O (608,6 г), Ni(NO3)2×6H2O (1216,8 г), Mg(NO3)2×6H2O (429,2 г) и Сr(NO3)2×9Н2О (33,5 г) при перемешивании.
Реакционную смесь С получали путем нагревания 387,7 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (352,4 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания до 55°С 1009,4 г 50 вес.% водного раствора (НN4)2Се(NО3)6 и (in) последовательного добавления Вi((NО3)3×5Н2О (182,7 г) и RbNO3 (14,8 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления реакционной смеси В к реакционной смеси А при перемешивании.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D. Это приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Затем эту суспензию подвергали гомогенизации в смесителе в течение 3 мин при окружной скорости 5000 об/мин. Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке на воздухе в течение 3 ч при температуре 290°С и затем на воздухе при температуре 425°С в течение 3 ч. Затем полученный порошок подвергали прокаливанию на воздухе при температуре 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора в процессе аммоксидирования пропилена. Результаты испытаний приведены в Таблице 2.
Пример 7А: катализатор Ni4Mg3Fe1,8Rb0,192Ci0,1Bi0,45Ce1,1Mo12,386O49,979+50 вес.% SiO2.
Реакционную смесь А получали путем нагревания 181,5 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (165 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (606,8 г, 41,2 вес.% диоксида кремния, 90 м. д. Na, средний размер частиц 39,2 нм).
Реакционную смесь В готовили путем нагревания 30,7 мл деионизированной воды до 55°С и последующего добавления Fе(NO3)3×9H2O (68,2 г), Ni(NO3)2×6Н2О (109,1 г), Mg(NO3)2×6H2O (72,1 г), Сr(NО3)2×9H2O (3,8 г) при перемешивании.
Реакционную смесь С получали путем нагревания 44,1 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (40,1 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания 113,1 г 50 вес.% водного раствора (NН4)2Се(NО3)6 до температуры 55°С и (ii) последовательного добавления Вi(NО3)3×5Н2O (20,5 г) и RbNО3 (2,7 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления реакционной смеси В к реакционной смеси А при перемешивании.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D, что приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Затем эту суспензию подвергали гомогенизации в смесителе в течение 3 мин при окружной скорости, равной 5000 об/мин. Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке при температуре 290°С в течение 3 ч на воздухе и затем на воздухе при температуре 425°С в течение 3 ч. Затем полученный порошок подвергали прокаливанию на воздухе при температуре 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора в процессе аммоксидирования пропилена. Результаты испытаний приведены в Таблице 2.
Пример 3: катализатор Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,72Ce1,76Mo12,806O51,806+50 вес.% SiO2.
Реакционную смесь А получали путем нагревания 154,5 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (140,4 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (595,2 г, 42 вес.% диоксида кремния, 27 м. д. Na, средний размер частиц 39,0 нм).
Реакционную смесь В готовили путем нагревания 26,5 мл деионизированной воды до 55°С и последующего добавления при перемешивании Fе(NO3)3×9H2O (32,2 г), Ni(NO3)2×6Н2О (102,9 г), Mg(NO3)2×6H2O (68 г), Сr(NО3)2×9H2O (1,8 г).
Реакционную смесь С получали путем нагревания 65,5 мл деионизированной воды до температуры 65°С и последующего добавления гептамолибдата аммония (59,6 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания до 55°С 170,6 г 50 вес.% водного раствора (NH4)2Ce(NO3)6 и (ii) последовательного добавления Вi(NО3)3×5Н2О (30,9 г) и RbNO3 (2,5 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления реакционной смеси В к реакционной смеси А при перемешивании.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D. Это приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Эту суспензию затем подвергали гомогенизации в смесителе в течение 3 мин при скорости 5000 об/мин. Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке при температуре 290°С в течение 3 ч на воздухе и затем при температуре 425°С еще в течение 3 ч также на воздухе. Затем полученный порошок подвергали прокаливанию на воздухе при 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора в процессе аммоксидирования пропилена. Результаты испытаний приведены в Таблице 2.
Пример 4: катализатор Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,72Ce1,76Mo12,335O50,126+50 вес.% SiO2.
Реакционную смесь А получали путем нагревания 149,9 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (136,3 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (595,2 г, 42 вес.% диоксида кремния, 27 м. д. Na, средний размер частиц 39,0 нм).
Реакционную смесь В готовили путем нагревания 27,1 мл деионизированной воды до 55°С и последующего добавления Fе(NО3)3×9Н2O (32,9 г), Ni(NO3)2×6H2O (105,4 г), Mg(NO3)2×6Н2О (69,7 г), Сr(NО3)2×9Н2O (1,8 г) при перемешивании.
Реакционную смесь С получали путем нагревания 67,1 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (61 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания 174,8 г 50 вес.% водного раствора (NН4)2Се(NО3)6 до 55°С и (ii) последовательного добавления Вi(NО3)3×5Н2О (31,6 г) и RbNO3 (2,6 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления реакционной смеси В к реакционной смеси А при перемешивании.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D. Это приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Эту суспензию затем подвергали гомогенизации в смесителе в течение 3 мин при окружной скорости 5000 об/мин. Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке при температуре 290°С в течение 3 ч на воздухе С и затем при температуре 425°С еще в течение 3 ч также на воздухе. Затем полученный порошок подвергали прокаливанию на воздухе при 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора в процессе аммоксидирования пропилена. Результаты испытаний приведены в Таблице 2.
Пример 5: катализатор Ni4,26Mg3,195Fe0,9Rb0,192Cr0,05Bi0,72Ce1,76Mo12,806O51,994+50 вес.% SiO2.
Реакционную смесь А получали путем нагревания 152,9 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (139,0 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (595,2 г, 42 вес.% диоксида кремния, 27 м. д. Na, средний размер частиц 39,0 нм).
Реакционную смесь В готовили путем нагревания 27,4 мл деионизированной воды до 55°С и последующего добавления Fе(NO3)3×9Н2O (31,8 г), Ni(NO3)2×6H2O (108,5 г), Mg(NO3)2×6H2O (71,7 г), Сr(NO3)2×9Н2O (1,8 г) при перемешивании.
Реакционную смесь С получали путем нагревания 64,9 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (59 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания 169 г 50 вес.% водного раствора (NН4)2Се(NО3)6 до температуры 55°С и (ii) последовательного добавления Вi(NО3)3×5Н2O (30,6 г) и RbNO3 (2,5 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления реакционной смеси В к реакционной смеси А при перемешивании.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D, что приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой конечную суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Эту суспензию затем подвергали гомогенизации в смесителе в течение 3 мин при окружной скорости, равной 5000 об/мин. Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке при температуре 290°С в течение 3 ч на воздухе С и затем при температуре 425°С в течение 3 ч также на воздухе. Затем полученный порошок подвергали прокаливанию на воздухе при 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора в процессе аммоксидирования пропилена. Результаты испытаний приведены в Таблице 2.
Пример 6 В: катализатор Ni4Mg3Fe0,9 Rb0,192Cr0,05Bi0,72Ce1,76Mo12,502O50,627+50 вес.% SiO2.
Реакционную смесь А получали путем нагревания 1363,6 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (1239,6 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (5461,2 г, 41,2 вес.% диоксида кремния, 90 м. д. Na, средний размер частиц 39,0 нм).
Реакционную смесь В готовили путем нагревания 241,9 мл деионизированной воды до температуры 55°С с последующим добавлением Fе(NО3)3×9Н2О (293,9 г), Ni(NO3)2×6Н2O (940,2 г), Mg(NO3)2×6Н2O (621,8 г) и Сr(NО3)2×9Н2O (16,2 г) при перемешивании.
Реакционную смесь С получали путем нагревания 599,1 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (544,6 г) при перемешивании в течение 30 мин, что приводило к образованию прозрачного бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания 1559,9 г 50 вес.% водного раствора (NН4)2Се(NО3)6 до 55°С и (и) последовательного добавления Вi(NО3)3×5Н2O (282,3 г) и RbNO3 (22,9 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления реакционной смеси В к реакционной смеси А при перемешивании.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D, что приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Эту суспензию затем подвергали гомогенизации в смесителе в течение 3 мин при скорости 5000 об/мин. Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке при температуре 290°С в течение 3 ч на воздухе и затем при температуре 425°С в течение 3 ч также на воздухе. Затем полученный порошок подвергали прокаливанию на воздухе при 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора в процессе аммоксидирования пропилена. Результаты испытаний приведены в Таблице 2.
Пример 7 В: катализатор Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,72Ce1,76Mo12,806O51,539+50 вес.% SiO2 с размером частиц 22 нм.
Реакционную смесь А получали путем нагревания 154,4 мл деионизированной воды до 65°С и последующего добавления гептамолибдата аммония (140,4 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора. Затем при перемешивании добавляли золь диоксида кремния (568 м. д. Na, 625,0 г, 40,0 вес.% диоксида кремния, средний размер частиц 22,0 нм).
Реакционную смесь В готовили путем нагревания 26,5 мл деионизированной воды до 55°С и последующего добавления Fe(NO3)3×9H2O (32,2 г), Ni(NO3)2×6Н2О (102,9 г), Mg(NO3)2×6Н2О (68 г) и Сr(NO3)2×9H2O (1,8 г) при перемешивании.
Реакционную смесь С получали путем нагревания 65,5 мл деионизированной воды до 65°С с последующим добавлением гептамолибдата аммония (59,6 г) при перемешивании в течение 30 мин с образованием чистого бесцветного раствора.
Реакционную смесь D готовили путем (i) нагревания до 55°С 170,6 г 50 вес.% водного раствора (NН4)2Се(NО3)6 и (ii) последовательного добавления Вi((NO3)3×5H2O (30,9 г) и RbNO3 (2,5 г) при нагревании и перемешивании полученного раствора, что приводило к получению прозрачного раствора оранжевого цвета.
Реакционную смесь Е получали путем добавления реакционной смеси В к реакционной смеси А при перемешивании.
Реакционную смесь F получали путем добавления реакционной смеси С к реакционной смеси D, что приводило к осаждению твердого продукта оранжевого цвета. Полученная смесь представляла собой суспензию осадка. Перемешивание реакционной смеси F продолжали в течение 15 мин, в то время как температуру поддерживали в пределах 50-55°С.
Затем реакционную смесь F добавляли к реакционной смеси Е с получением конечной суспензии предшественника катализатора.
Полученную суспензию предшественника катализатора перемешивали в течение 1 ч, в то время как она охлаждалась до примерно 40°С. Эту суспензию затем подвергали гомогенизации в смесителе в течение 3 мин при окружной скорости, равной 5000 об/мин.
Затем суспензию сушили при распылении в сушилке с распылением при температурах на входе/выходе, составляющих 325/140°С. Полученный порошок подвергали денитрификации при тепловой обработке при температуре 290°С в течение 3 ч на воздухе и затем при температуре 425°С в течение 3 ч также на воздухе. Затем полученный порошок подвергали прокаливанию на воздухе при 560°С в течение 3 ч. Полученный прокаленный порошок затем испытывали в качестве катализатора в процессе аммоксидирования пропилена. Результаты испытаний приведены в Таблице 2 и в Таблице 3.
Примечания к Таблице 2 и к Таблице 3:
1. "Отношение X/Y" означает отношение интенсивностей на XRD, описанной выше.
2. "HOS" означает "время работы реактора в ч".
3. "Конв.С3-, %" или "% PC" означают степень конверсии пропилена.
4. "Выход AN, %" означает выход акрилонитрила.
5. "Выход HCN, %" означает выход цианистого водорода.
6. "Выход ACN, %" означает выход ацетонитрила.
7. α вычисляют следующим образом: [(% AN+(3 х % HCN)+(1,5 х % ACN))÷% PC] x 100.
8. "Селект.по AN, %" означает селективность по акрилонитрилу в % .
9. "Выход -CN, %" означает общий выход акрилонитрила, цианистого водорода и ацетонитрила в %%.
10. Указанные катализаторы описаны как составы на основе Мо12 (то есть нижний индекс у молибдена равен 12), для превращения любой из указанной композиций в состав на основе Мо12 нужно просто разделить каждый нижний индекс в композиции на указанный у молибдена нижний индекс и затем умножить на 12. Например, в Примере 3 композиция Ni4Mg3Fe0,9Rb0,192Cr0,05Bi0,72Ce1,1Mo12,806O51,539 эквивалентна композиции Ni3,748Mg2,811Fe0,843Rb0,180Cr0,047Bi0,675Ce1,65Mo12O48,295 на основе Mo12.
Примечания:
1. "Потеря массы" определяли при проведении теста на истирание при действии затопленной струи, указанные числа означают потерю массы катализатора (износ) в % в течение промежутка времени от 0 до 20 ч. Это мера прочности всех частиц катализатора. Желательными являются меньшие величины потерь. Величины более примерно 8,0 являются нежелательными для промышленных катализаторов из-за высоких скоростей потерь массы катализатора. Устройство для проведения этого испытания описано в ASTM D5757-00 Standard Test Method for Determination of Attrition and Abrasion of Powdered Catalysts by Air Jets.
Как можно видеть из Таблиц 2, 3 и 4, каталитическая композиция согласно данному изобретению (следует иметь в виду отношение X/Y и величину α) приводит (i) к высоким выходам акрилонитрила, цианистого водорода и ацетонитрила в процессе аммоксидирования пропилена при повышенных температурах в присутствии этой каталитической композиции, аммиака и воздуха и (ii) к меньшим потерям массы при истирании (большей прочности частиц) по сравнению с похожими катализаторами, которые не входят в объем данного изобретения.
Сравнительные пример С8 и примеры 8-11
С применением способов, описанных в данной заявке, были получены различные составы катализаторов. В Таблице 5 показано, что эти катализаторы с отношениями Ce/Fe менее 0,7 имеют большие потери массы при истирании (большую степень износа), чем катализаторы с большими отношениями Ce/Fe.
В то время как приведенные выше описание и варианты данного изобретения являются типичными для осуществлении данного изобретения, в свете данной заявки, для специалиста в данной области очевидны его многие альтернативы, модификации и изменения. Соответственно подразумевается, что такие альтернативы, модификации и изменения охвачены данным изобретением и входят в его объем, определяемый прилагаемой формулой изобретения.
Реферат
Изобретение относится к способу получения акрилонитрила, ацетонитрила и цианистого водорода. Способ включает приведение в контакт при повышенной температуре пропилена, аммиака и кислорода в паровой фазе в присутствии катализатора. Указанный катализатор представляет собой комплекс оксидов металлов, в котором отношение элементов выражено формулой MoBiFeADEFGCeO. В формуле A обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия; D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария; E обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура; F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца; G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; а имеет значение от 0,05 до 7, b - от 0,1 до 7, с - от 0,01 до 5, d - от 0,1 до 12, е - от 0 до 5, f - от 0 до 5, g - от 0 до 0,2, h - от 0,01 до 5 и х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов. При этом 0.15≤(a+h)/d≤1 и 0.8≤h/b≤5, а относительные выходы акрилонитрила, ацетонитрила и цианистого водорода, полученных этим способом, определяются равенством α=[(%AN+(3×%HCN)+(1.5×%ACN))÷%PC]×100, где %AN обозначает выход акрилонитрила и %AN≥82, %HCN означает выход цианистого водорода, %ACN означает выход ацетонитрила, %PC означает конверсию пропилена и где α больше 100. Предлагаемый способ позволяет повысить выхо
Формула
Mo12BiaFebAcDdEeFfGgCehOx,
где А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и
а имеет значение от 0,05 до 7,
b имеет значение от 0,1 до 7,
с имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов;
где 0.15≤(a+h)/d≤1 и 0.8≤h/b≤5; и
при этом относительные выходы акрилонитрила, ацетонитрила и цианистого водорода, полученных этим способом, определяются следующим равенством:
α=[(%AN+(3×%HCN)+(1.5×%ACN))÷%PC]×100,
где %AN обозначает выход акрилонитрила и %AN≥82,
%HCN означает выход цианистого водорода,
%ACN означает выход ацетонитрила и %PC означает конверсию пропилена, и
где α больше 100.
Mo12BiaFebAcDdEeFfGgCehOx,
где А обозначает по меньшей мере один элемент, выбранный из группы, состоящей из натрия, калия, рубидия и цезия;
D обозначает по меньшей мере один элемент, выбранный из группы, состоящей из никеля, кобальта, марганца, цинка, магния, кальция, стронция, кадмия и бария;
Е обозначает по меньшей мере один элемент, выбранный из группы, состоящей из хрома, вольфрама, бора, алюминия, галлия, индия, фосфора, мышьяка, сурьмы, ванадия и теллура;
F обозначает по меньшей мере один элемент, выбранный из группы, состоящей из редкоземельного элемента, титана, циркония, гафния, ниобия, тантала, алюминия, галлия, индия, таллия, кремния, германия и свинца;
G обозначает по меньшей мере один элемент, выбранный из группы, состоящей из серебра, золота, рутения, родия, палладия, осмия, иридия, платины и ртути; и где
а имеет значение от 0,05 до 7,
b имеет значение от 0,1 до 7,
c имеет значение от 0,01 до 5,
d имеет значение от 0,1 до 12,
е имеет значение от 0 до 5,
f имеет значение от 0 до 5,
g имеет значение от 0 до 0,2,
h имеет значение от 0,01 до 5 и
х обозначает число атомов кислорода, необходимое для насыщения валентности других имеющихся элементов;
где 0.15≤(a+h)/d≤1 и 0.8≤h/b≤5; и
при этом при применении этой каталитической композиции для получения акрилонитрила, ацетонитрила и цианистого водорода в способе, включающем приведение в контакт пропилена, аммиака и кислорода в паровой фазе при повышенной температуре в присутствии катализатора, относительные величины выходов акрилонитрила, ацетонитрила и цианистого водорода определяются следующим равенством:
α=[(%AN+(3×%HCN)+(1.5×%ACN))÷%РС]×100,
где %AN обозначает выход акрилонитрила и %AN≥82,
%HCN означает выход цианистого водорода,
%ACN означает выход ацетонитрила и
%PC означает конверсию пропилена, и
где α больше 100.

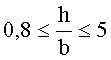
Документы, цитированные в отчёте о поиске
Катализатор производства акрилонитрила
Катализатор для производства акрилонитрила
Комментарии