Способ получения гексафторизопропанола и фторметилгексафторизопропилового эфира (севофлурана) - RU2629366C2
Код документа: RU2629366C2
Описание
Область техники
Настоящее изобретение относится к способу получения гексафторизопропанола и фторметилгексафторизопропилового эфира, известного как ингаляционный анестетик «севофлуран».
Уровень техники
Гексафторизопропанол (далее в данном описании гексафторизопропанол может быть обозначен «ГФИП») получают в больших количествах в качестве растворителя, демонстрирующего особую способность к растворению полимеров, и в качестве промежуточного соединения для получения ингаляционного анестетика «севофлурана». ГФИП обычно получают посредством гидрирования гексафторацетона (далее в данном описании он может быть обозначен «ГФА»), и были предложены различные способы, в зависимости от сочетания формы исходного материала ГФА, режима проведения реакции, типов восстанавливающего агента и катализатора и т.д.
В качестве газофазного способа известен способ гидрирования ГФА водородом (H2) в присутствии нанесенного на оксид алюминия палладиевого катализатора (Pd/Al2O3) (US 3468964) или в присутствии нанесенного на активированный уголь палладиевого катализатора (Pd/C) (US 3702872) и способ гидрирования гидрата ГФА в присутствии никелевого катализатора или нанесенного на оксид алюминия палладиевого катализатора (JP S57-81424).
Что касается способа получения ГФИП с помощью газообразного водорода в жидкой фазе, известен способ с использованием гидрата ГФА и способ с использованием ангидрида ГФА. В качестве способа с использованием ангидрида, в US 3418337 описан способ, включающий проведение его реакции в течение 6 ч при температуре от 110 до 150°C при давлении от 20,0 до 90,0 МПа (от 200 до 900 атмосфер) с использованием оксида платины в качестве катализатора.
С другой стороны, способ гидрирования гидрата ГФА в жидкой фазе включает проведение его реакции в течение 3,5 ч при температуре от 70 до 75°C при давлении от 0,35 до 0,7 МПа (от 3,5 до 7 кг/см2) с использованием палладия/углерода в качестве катализатора (JP S59-204142) и способ проведения его реакции в течение 6 ч при температуре 100°C при давлении 0,5 МПа (5 кг/см2) с использованием палладия/Al2O3 в качестве катализатора (JP Н1-301631).
В качестве способа с использованием восстанавливающего агента, отличного от водорода (H2), в US 3418337 описан способ восстановления ангидрида ГФА с использованием боргидрида натрия в качестве восстанавливающего агента в растворителе метаноле, а также способ с использованием алюмогидрида лития, гидрида кальция, гидрида натрия или т.п. в качестве восстанавливающего агента в кислородсодержащем растворителе, таком как диэтиловый эфир, метанол, изопропанол, тетрагидрофуран или т.п.
В соответствии с JP Н6-184025, гидрирование гидрата ГФА посредством контакта с водородом в присутствии палладиевого катализатора дает чрезмерно гидрированный продукт, 1,1,1-трифторацетон (ТФА), помимо целевого продукта ГФИП. Указано, что ТФА трудно отделить путем перегонки, поскольку температура его кипения близка к температуре кипения целевого продукта ГФИП. Однако в JP Н6-184025 отмечено, что при использовании «комбинированного катализатора, состоящего из палладия и рутения» в качестве катализатора гидрирования, целевой продукт ГФИП может быть получен достаточно избирательно, что даже когда получают ТФА в небольшом количестве, данное соединение можно легко превратить в 1,1,1-трифторизопропанол (легко отделяемое соединение, далее может быть обозначено ТФИП) и что, соответственно, после реакции становится значительно легче получить гексафторизопропанол высокой чистоты (см. далее).

С другой стороны, в JP Н6-184026 указано, что когда неочищенный ГФИП, полученный посредством гидрирования ГФА в присутствии катализатора, обрабатывают органическим аминовым соединением, трудно отделяемый ТФА может быть удален из системы в форме «ассоциата с аминовым соединением» и посредством последующей перегонки может быть получен ГФИП, по существу не содержащий ТФА.
Кроме того, в JP 2009-051798 указано, что когда ГФА гидрируют посредством контакта с газообразным водородом при температуре от -20 до 60°C в присутствии металлического катализатора, такого как палладий, рутений или т.п., или несущего металл катализатора в растворителе фториде водорода, может быть получен ГФИП, по существу не содержащий избыточно гидрированного 1,1,1-трифторацетона.
Что касается получения гексафторацетона (ГФА), т.е. исходного материала для получения ГФИП, известен способ эпоксидирования гексафторпропена (US 3321515) с последующей изомеризацией полученного эпоксидного соединения в присутствии катализатора с получением ГФА (US 3213134). Также известен способ хлорирования ацетона с получением гексахлорацетона (JP S56-139436), который впоследствии подвергают заместительному фторированию фторидом водорода в присутствии хромового катализатора, нанесенного на активированный уголь, или т.п. (JP S39-13060).
Как описано выше, ГФИП является чрезвычайно важным в качестве исходного материала для получения ингаляционного анестетика севофлурана (химическое название фторметил-1,1,1,3,3,3-гексафторизопропиловый эфир). Более конкретно, как представлено в US 4250334, ингаляционный анестетик севофлуран может быть получен посредством добавления концентрированной серной кислоты и фторида водорода к параформальдегиду, последующего нагревания полученной смеси при определенной температуре и добавления в нее по капле ГФИП.
Краткое описание изобретения
В соответствии со способами, раскрытыми в US 3468964, US 3702872, JP S57-81424, JP S59-204142, JP H1-301631, JP H6-184025, JP H6-184026 и JP 2009-051798, ГФИП, который является важным органическим промежуточным соединением, можно производить в массовом масштабе посредством каталитической реакции с использованием ГФА в качестве исходного материала. Металлический катализатор, необходимый для реакции гидрирования, содержит благородный металл, такой как палладий, платина или т.п., в качестве активного компонента. Его важной характеристикой является то, что он имеет высокую каталитическую активность, и даже при его использовании в очень небольшом количестве по сравнению с исходным материалом ГФА, ГФА можно преобразовать ГФИП с высокой степенью превращения (другими словами, катализатор имеет высокий оборот).
Однако способ получения ГФИП посредством каталитического гидрирования имеет проблему, которой нельзя пренебречь и которая состоит в том, что «скорость реакции уменьшается по мере развития реакции». Более конкретно, в течение почти 3 часов после начала гидрирования гексафторацетона скорость реакции высока, но затем она постепенно снижается. Реакция не останавливается, и никакого воздействия на качество продукта не происходит, но скорость реакции несоразмерно снижается в середине реакции и далее, по сравнению со скоростью в начале реакции. В данном случае, гексафторацетон (ГФА) является дорогостоящим реагентом. Соответственно, желательно, чтобы степень превращения гексафторацетона посредством реакции гидрирования достигала от 80 до 100% (предпочтительно от 90 до 100%, особенно предпочтительно от 98 до 100%). Как описано выше, скорость реакции ближе к концу реакции является чрезвычайно низкой, и поэтому для достижения такой высокой степени превращения реакции требуется относительно большое время реакции (см. представленные далее сравнительные примеры) и существует потребность в усовершенствовании (в данном описании фраза «начиная со середины до конца реакции гидрирования» предназначена, хотя и не всегда этим ограничена, для указания стадии, на которой степень превращения ГФА достигает приблизительно 70% или более от начала реакции.)
Как описано ниже в разделе «стадия 4», при «гидрировании ГФА» кислотные компоненты, такие как хлорид водорода, фторид водорода и т.п., образуются по мере развития реакции, и pH реакционной системы постепенно снижается. В данном случае понятно, что когда в реакционную систему предварительно добавлено небольшое количество «акцептора кислоты (основного вещества)», кислотные вещества, образующиеся в качестве побочных продуктов в ходе реакции гидрирования, можно сразу же нейтрализовать. Известно, что в результате этого скорость реакции значительно увеличивается (например, см. JP H6-184025. Фактически, как показано в приведенных ниже примерах, когда реакцию гидрирования осуществляют с добавлением акцептора кислоты, реакция заканчивается за значительно более короткий период времени по сравнению со случаем без добавления акцептора кислоты.) Однако даже «в случае добавления акцептора кислоты» нельзя избежать того, что «скорость реакции уменьшается по мере развития реакции». Даже когда количество акцептора кислоты увеличивают, это явление нельзя предотвратить в достаточной степени (см. представленные ниже примеры и сравнительные примеры).
Целью настоящего изобретения является обеспечение нового способа получения гексафторизопропанола из гексафторацетона в качестве исходного материала, в котором предотвращают снижение скорости реакции от середины до конца реакции гидрирования.
В свете вышеуказанных проблем, были проведены тщательные исследования. В результате было обнаружено, что содержание 1,1,1-трифтор-2,2-дихлорэтана (CF3CHCl2; далее в данном описании может быть обозначен «HCFC-123») среди примесей, содержащихся в гексафторацетоне в момент начала гидрирования, имеет тесную взаимосвязь со «скоростью реакции в середине и после середины реакции» гидрирования.
Более конкретно, неожиданно было обнаружено, что когда реакцию гидрирования начинают с использованием гексафторацетона, в котором концентрация HCFC-123 снижена до 120 ppm (частей на млн) или менее, вышеуказанное «явление снижения скорости реакции с развитием реакции» может быть существенно подавлено. В результате, даже если количество катализатора гидрирования и количество акцептора кислоты являются такими же, реакция гидрирования достигает уровня требуемой степени превращения реакции за более короткий период времени.
Данный HCFC-123 представляет собой соединение, которое обнаруживают в количестве более 120 ppm в качестве побочного продукта при получении ГФА посредством взаимодействия гексафторацетона и фторида водорода (сразу после синтеза ГФА посредством фторирования гексахлорацетона соединение содержится в реакционной смеси в количестве нескольких тысяч ppm, и как показано далее в данном документе, соединение все еще содержится в количестве от 1000 до 2000 ppm или т.п. даже после его простой перегонки.) В общем, исходный материал ацетон не содержит такое соединение, содержащее «два атома углерода». Поэтому полагают, что в жестких условиях хлорирования или фторирования углерод-углеродная связь разрывается, отчасти с образованием «соединения с двумя атомами углерода» (даже когда ГФА синтезируют другим способом, этот способ обязательно должен включать стадию хлорирования и стадию фторирования, и поэтому можно предполагать образование побочного продукта HCFC-123.)
HCFC-123 имеет температуру кипения 28°C, которая отличается от температур кипения ГФА и тригидрата ГФА. Однако при простой перегонке тригидрата ГФА HCFC-123 не может быть полностью удален из тригидрата ГФА (см. сравнительные примеры, представленные далее в данном документе). Кроме того, HCFC-123 само по себе не содержит активной функциональной группы, способной реагировать с ГФА и ГФИП. После получения ГФИП в результате гидрирования соединение может быть успешно отделено и удалено из ГФИП посредством перегонки. Следовательно, до настоящего времени не делали попыток преднамеренного отделения и удаления HCFC-123 в высокой степени из ГФА или тригидрата ГФА.
Причина того, почему присутствие HCFC-123 в количестве более 120 ppm увеличивает время реакции неясна. Как очевидно из примера 1, представленного далее в данном документе, пик «HCFC-123» исчезает на относительно ранней стадии после начала гидрирования. С другой стороны, установлено, что «явление снижения скорости реакции» возникает значительно позднее. Исходя из этого, можно с хорошей вероятностью полагать, что сам по себе «HCFC-123» напрямую не препятствует гидрированию.
В связи с этим, в случае, когда HCFC-123 содержится в ГФА и когда HCFC-123 подвергается гидрированию, можно полагать, что образуется 1,1,1-трифтор-2-хлорэтан (CF3CH2Cl; далее в данном документе это соединение может быть обозначено «HCFC-133a»), и когда это соединение в свою очередь подвергается гидрированию, образуется 1,1,1-трифторэтан (CF3H3). Обе реакции сопровождаются образованием хлорида водорода (HCl) в качестве побочного продукта.
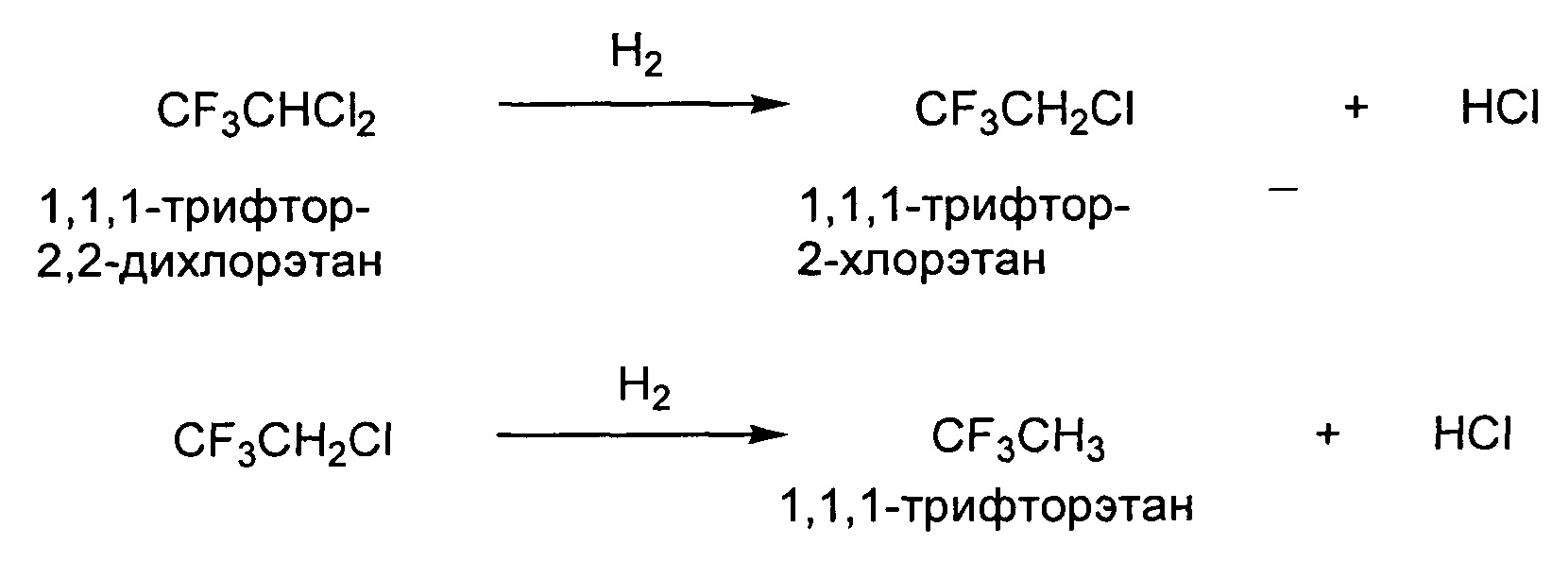
Может существовать возможность, что хлорид водорода, полученный в результате данной реакции, действует как каталитический яд, снижая активность металлического катализатора, что вызывает «увеличение времени реакции». Однако, как показано в примерах и сравнительных примерах, представленных далее в данном документе, в случае, когда HCFC-123 присутствует в количестве более 120 ppm, и когда реакцию начинают при увеличении количества акцептора кислоты, скорость реакции обычно возрастает, но «явление снижения скорости реакции в середине и после середины реакции» не исчезает. Исходя из этого предполагают, что существуют какие-то другие факторы, которые нельзя выявить на основании вышеизложенной гипотезы.
Было обнаружено, что явление, состоящее в том, что «снижение скорости реакции в середине и после середины реакции» может быть предотвращено посредством «регулирования содержания HCFC-123», не зависит от наличия или отсутствия акцептора кислоты (см. примеры и сравнительные примеры, представленные далее в данном документе).
Исходя из этого полагают, что эффект «снижения количества HCFC-123» в настоящем изобретении (предотвращение снижения скорости реакции в середине и после середины реакции) является отличным и не зависит от эффекта «добавления акцептора кислоты в ходе реакции гидрирования» (общее увеличение скорости реакции).
Вместе с тем было обнаружено, что гидрирование в настоящем изобретении можно осуществлять особенно благоприятным образом только в случае, когда соблюдены два требования, а именно «добавление акцептора кислоты» и «снижение содержания HCFC-123 до определенного количества или менее».
Так или иначе, «количество HCFC-123 в момент начала гидрирования» является важным показателем. Когда его доводят до величины, составляющей 120 ppm или менее, можно предотвратить снижение скорости реакции в середине и после середины реакции гидрирования, и это является неожиданным и полезным открытием. На основании этого открытия удалось получить ГФИП более экономично, чем когда-либо прежде.
В соответствии с настоящим изобретением, стало возможно синтезировать гексафторизопропанол за более короткий период времени. Соответственно, при синтезе севофлурана с применением гексафторизопропанола, полученного в настоящем изобретении, ингаляционный анестетик «севофлуран» получают с гораздо большими преимуществами, чем когда-либо ранее, с точки зрения широкой перспективы. Здесь можно сказать, что настоящее изобретения имеет большие преимущества.
Более конкретно, в настоящем изобретении предложен способ получения гексафторизопропанола и севофлурана, признаки которого изложены в пп. 1-12 формулы изобретения.
Стадии (a), (b) и (c), указанные в п. 1 формулы изобретения, соответствуют упоминаемым впоследствии стадиям 3, 4 и 5, соответственно. Стадии (m), (n), (o)-(q), (r) и (s), указанные в п. 12 формулы изобретения, соответствуют упоминаемым впоследствии стадиям 1, 2, 3a, 4a и 5, соответственно.
Согласно изобретению по п. 1, предложен способ получения фторметилгексафторизопропилового эфира (севофлурана), включающий следующие стадии:
(a) очистка смеси, содержащей гексафторацетон и более 120 ppm 1,1,1-трифтор-2,2-дихлорэтана в качестве примеси, с получением очищенного гексафторацетона, содержащего 120 ppm или менее 1,1,1-трифтор-2,2-дихлорэтана;
(b) приведение водорода (H2) в контакт с очищенным гексафторацетоном в присутствии катализатора, посредством чего осуществляют гидрирование гексафторацетона с получением гексафторизопропанола, и
(c) проведение реакции гексафторизопропанола, формальдегида и фторида водорода в присутствии кислоты Льюиса или кислоты Бренстеда.
Согласно изобретению по п. 2, предложен способ по п. 1, в котором стадию (а) осуществляют посредством перегонки.
Согласно изобретению по п. 3, предложен способ по п. 1 или 2, в котором стадия (а) включает следующие стадии:
(d) приведение воды в контакт со смесью, содержащей гексафторацетон и более 120 ppm 1,1,1-трифтор-2,2-дихлорэтана в качестве примеси, посредством чего гексафторацетон, содержащийся в смеси, превращают в тригидрат гексафторацетона, и
(e) перегонка смеси, полученной на стадии (d).
Согласно изобретению по п. 4, предложен способ по п. 3, в котором стадия (e) включает следующие стадии:
(f) подача смеси, полученной на стадии (d), в ректификационную колонну и
(g) проведение перегонки при числе теоретических тарелок от 2 до 50 и флегмовом числе 0,5-8,0.
Согласно изобретению по п. 5, предложен способ по любому из пп. 1-4, в котором стадия (a) включает стадию
(h) проведения количественного анализа гексафторацетона с использованием газовой хроматографии в ходе очистки, чтобы определить, составляет ли содержание 1,1,1-трифтор-2,2-дихлорэтана в смеси 120 ppm или менее.
Согласно изобретению по п. 6, предложен способ по любому из пп. 1-5, в котором катализатор, используемый на стадии (b), представляет собой первый катализатор, включающий по меньшей мере один металл, выбранный из группы, состоящей из палладия, платины, рутения, родия и никеля, или второй катализатор, включающий по меньшей мере один из указанных металлов, нанесенный на носитель.
Согласно изобретению по п. 7, предложен способ по любому из предшествующих пп. 1-5, в котором катализатор, используемый на стадии (b), представляет собой по меньшей мере один катализатор, выбранный из группы, состоящей из третьего катализатора, включающего палладий и рутений, которые нанесены на один и тот же носитель, и четвертого катализатора, включающего смесь катализатора, содержащего палладий, нанесенный на носитель, и катализатора, содержащего рутений, нанесенный на носитель.
Согласно изобретению по п. 8, предложен способ по любому из пп. 1-7, в котором гидрирование на стадии (b) осуществляют в присутствии акцептора кислоты в реакционной системе.
Согласно изобретению по п. 9, предложен способ по п. 8, в котором карбонат или гидрокарбонат щелочного металла и гидроксид металла 13 группы Периодической таблицы элементов используют совместно в качестве акцептора кислоты на стадии (b).
Согласно изобретению по п. 10, предложен способ по любому из пп. 1-9, в котором смесь, содержащую гексафторацетон и более 120 ppm 1,1,1-трифтор-2,2-дихлорэтана в качестве примеси, предназначенную для очистки на стадии (a), получают способом, включающим следующие стадии:
(i) хлорирование ацетона хлором (Cl2) с получением смеси, содержащей гексахлорацетон, и
(j) фторирование гексахлорацетона посредством приведения фторида водорода в контакт со смесью, полученной на стадии (i), с получением смеси, содержащей гексафторацетон и более 120 ppm 1,1,1-трифтор-2,2-дихлорэтана в качестве примеси.
Согласно изобретению по п. 11, предложен способ по любому из пп. 1-10, в котором гексафторизопропанол, полученный на стадии (b), отделяют способом, включающим следующие стадии:
(k) отделение катализатора стадии (b) от реакционной смеси, полученной на стадии (b), с получением жидкого компонента и
(l) перегонка жидкого компонента, в результате которой отделяют гексафторизопропанол.
Согласно изобретению по п. 12, предложен способ получения фторметилгексафторизопропилового эфира (севофлурана), включающий следующие стадии:
(m) хлорирование ацетона хлором (Cl2) с получением смеси, содержащей гексахлорацетон;
(n) фторирование гексахлорацетона посредством приведения фторида водорода в контакт со смесью, полученной на стадии (m), с получением смеси, содержащей гексафторацетон и более 120 ppm 1,1,1-трифтор-2,2-дихлорэтана в качестве примеси;
(o) приведение воды в контакт со смесью, полученной на стадии (n), посредством чего гексафторацетон, содержащийся в смеси, превращают в тригидрат гексафторацетона;
(p) подача смеси, полученной на стадии (o), в ректификационную колонну;
(q) проведение перегонки смеси, полученной на стадии (o), в ректификационной колонне при числе теоретических тарелок от 2 до 50 и флегмовом числе 0,5-8,0 до достижения содержания 1,1,1-трифтор-2,2-дихлорэтана в смеси 120 ppm или менее, посредством чего получают очищенный тригидрат гексафторацетона, содержащий 120 ppm или менее 1,1,1-трифтор-2,2-дихлорэтана;
(r) приведение водорода (H2) в контакт с очищенным тригидратом гексафторацетона в присутствии акцептора кислоты и в присутствии по меньшей мере одного катализатора, выбранного из группы, состоящей из первого катализатора, включающего палладий и рутений, которые нанесены на один и тот же носитель, и второго катализатора, включающего смесь катализатора, содержащего палладий, нанесенный на носитель, и катализатора, содержащего рутений, нанесенный на носитель, с получением гексафторизопропанола, и
(s) осуществление реакции гексафторизопропанола, полученного на стадии (r), с формальдегидом и фторидом водородом в присутствии кислоты Льюиса или кислоты Брэнстеда, с получением фторметилгексафторизопропилового эфира (севофлурана).
Способ получения гексафторизопропанола с использованием гексафторацетона в качестве исходного материала согласно настоящему изобретению позволяет предотвратить снижение скорости реакции от середины до конца реакции гидрирования. Соответственно, гексафторизопропанол можно синтезировать за более короткий период времени. Таким образом, другой эффект настоящего изобретения заключается в том, что когда севофлуран синтезируют с использованием этого гексафторизопропанола, севофлуран можно получить с большими преимуществами.
Подробное описание изобретения
Далее настоящее изобретение описано подробно. Объем защиты настоящего изобретения не ограничен этим описанием. Помимо примеров, представленных далее в данном документе, изобретение может быть подходящим образом изменено и модифицировано в пределах объема защиты без отступления от сущности изобретения с достижением его технического результата. Все публикации, перечисленные в данном описании, например, документы известного уровня техники и публикации патентных заявок, публикации патентов и другие патентные документы, включены в данное описание посредством ссылки.
В данном описании гидрат гексафторацетона относится к гидрату с неограниченным количеством молекул воды или его водному раствору и включает тригидрат гексафторацетона. В данном описании гексафторацетон тригидрат может быть обозначен «ГФА-3В».
Как раскрыто в публикациях известного уровня техники, гексафторацетон может быть представлен рядом химических веществ, как таковой. Например, в водном растворе соединение существует в форме «тригидрата гексафторацетона».
Гексафторацетон имеет температуру кипения -28°C (при атмосферном давлении) и находится в состоянии пара (газа) при комнатной температуре и нормальном давлении. Для удобства в обращении, тригидрат гексафторацетона, полученный в виде композиции с постоянной температурой кипения от 106 до 108°C, используют в качестве исходного материала во многих реакциях или для хранения. В настоящем изобретении, термин «гексафторацетон» включает тригидрат гексафторацетона.
Для более наглядного описания настоящего изобретения, далее приведены стадии, относящиеся к настоящему изобретению.
[Стадия 1]
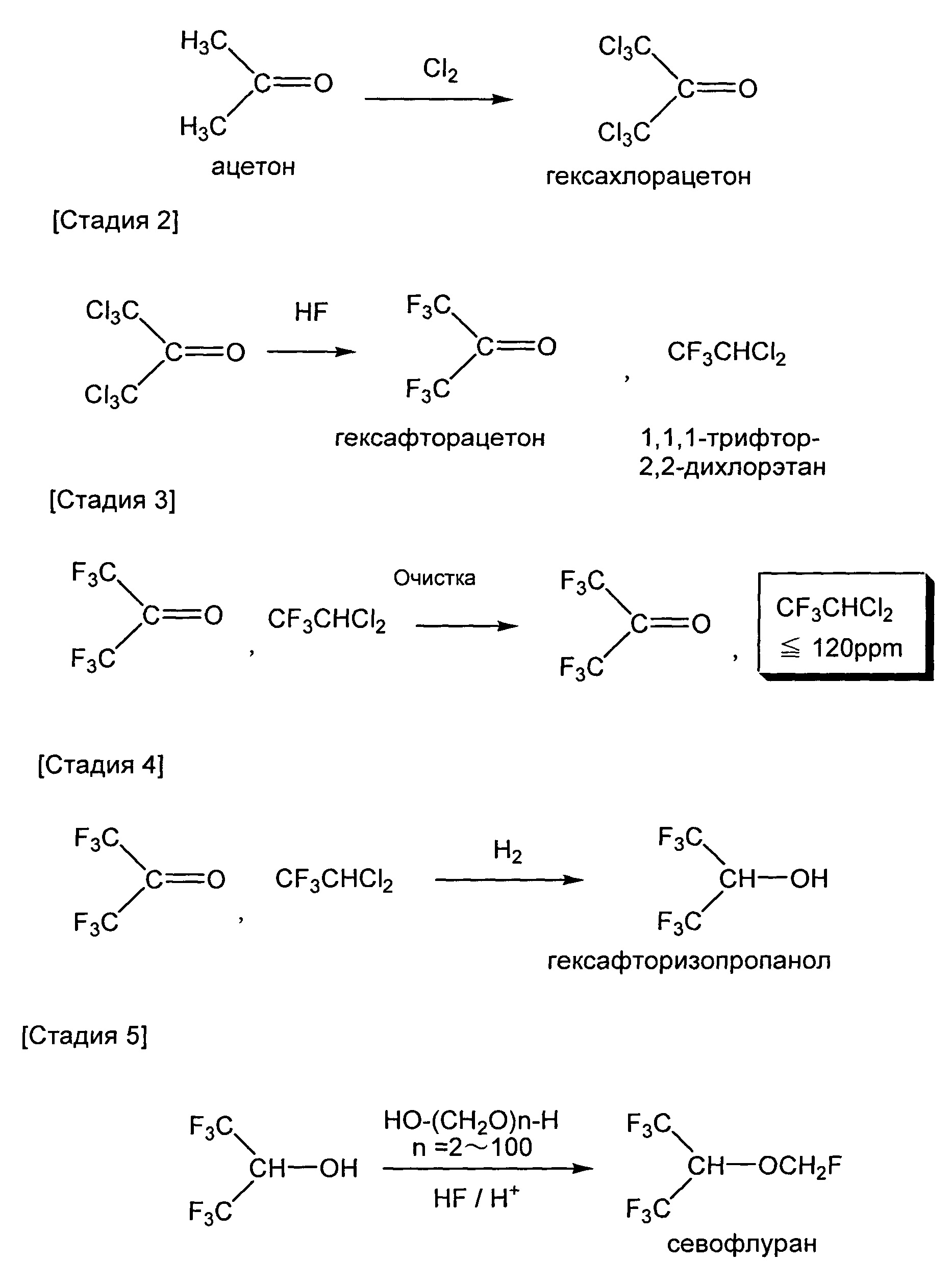
Среди вышеуказанных, стадия 3 и стадия 4 являются обязательными стадиями настоящего изобретения. В соответствии с настоящим изобретением, к ним могут быть добавлены стадия 5 или стадии 1 и 2. Стадия 3 в одном особенно предпочтительном воплощении может быть обозначена как «стадия 3a». Стадия 4 в одном особенно предпочтительном воплощении может быть обозначена как «стадия 4а». Далее приведено описание стадий.
Стадия 1
Стадия 1 представляет собой стадию хлорирования ацетона хлором (Cl2) с получением смеси, содержащей гексахлорацетон.
Данная стадия и последующая стадия 2 являются традиционными стадиями, но очень важны для понимания настоящего изобретения и описаны далее.
Данную стадию осуществляют посредством приведения хлора (газообразного хлора) в контакт с ацетоном. Хлорирование на данной стадии предпочтительно осуществляют в присутствии катализатора. В качестве катализатора используют катализатор, известный как так называемый катализатор хлорирования. Более конкретно, возможно использовать радикальный инициатор, включая азосоединения, такие как азобисизобутиронитрил, азобисвалеронитрил и т.д., и пероксиды, такие как бензоилпероксид, додеканоилпероксид, дилаурилпероксид, трет-бутилпероксипивалат и т.д., и соединения фосфора, такие как красный фосфор, пентахлорид фосфора, трихлорид фосфора, трифенилфосфин, трифенилфосфит и т.д., гетероциклические ароматические соединения, такие как пиридин, хинолин и т.д., и триэтиламин и т.д. Кроме того, хлорирование протекает с помощью фотооблучения. Среди указанных катализаторов хинолин является особенно предпочтительным.
Количество катализатора может составлять приблизительно от 0,001 до 0,5 эквивалентов относительно исходного материала ацетона, но специалист в данной области техники может подходящим образом регулировать это количество.
По мере протекания хлорирования, постепенно образуются хлорацетоны, содержащие от одного до пяти атомов хлора, которые являются частично хлорированными ацетонами (монохлорацетон, дихлорацетон и т.д.; в данном описании они могут быть упомянуты как «хлорированные соединения низкого порядка»). Предпочтительно реакцию осуществляют при использовании нижеуказанных давления реакции, температуры реакции и других условий, до тех пор, пока все углеродные атомы ацетона не будут замещены атомами хлора, и с использованием аналитических средств газовой хроматографии или т.п. для проверки степени превращения ацетона и степени хлорирования в течение реакции. «Степень хлорирования» в данном описании показывает среднее количество атомов хлора, введенных на одну молекулу ацетона, рассчитанное исходя из состава реакционной смеси в определенный момент времени.
Более конкретно, на одну молекулу ацетона, теоретическое количество молей хлора (Cl2), необходимое для замены всех атомов водорода в ацетоне атомами хлора (то есть для превращения ацетона в гексахлорацетон) составляет 6. Соответственно, при продолжении хлорирования с поддержанием температуры в интервале приблизительно от 20 до 260°C, при контроле степени хлорирования методом газовой хроматографии, хлорсодержащие соединения низкого порядка постепенно хлорируются с получением гексахлорацетона с высокой селективностью.
На данной стадии избыточный хлор выводят из реакционной системы, тогда как он остался непрореагировавшим и, следовательно, может быть извлечен для повторного использования.
В общем, давление реакции предпочтительно составляет от 0,05 до 5,0 МПа (абсолютное давление, то же самое относится к другим величинам давления, указанным далее в данном описании). Диапазон от нормального давления (0,1 МПа) до слегка повышенного давления 0,3 МПа или т.п. является простым в эксплуатации и предпочтительным. Настоящее изобретение не исключает реакции под давлением свыше 5,0 МПа, но слишком избыточное давление будет оказывать нагрузку на оборудование. Следовательно, вышеуказанный диапазон является предпочтительным.
Температура реакции обычно составляет от 20 до 260°C. Температуру предпочтительно контролируют при отслеживании степени хлорирования с помощью газовой хроматографии. Когда температура реакции составляет менее 20°C, это в некоторой степени влияет на скорость реакции с увеличением доли хлорированных соединений низкого порядка, и время превращения в гексахлорацетон увеличивается. В результате, производительность может снижаться. С другой стороны, при температуре более 260°C гексахлорацетон испаряется, поскольку его температура кипения при комнатной температуре/нормальном давлении составляет 204°C. Таким образом, увеличатся расходы, связанные с его выведением из системы. В этом случае необходимо оборудование для предотвращения испарения, что промышленно и экономически невыгодно, и использование такой высокой температуры может не давать никаких преимуществ.
Что касается режима подачи для хлорирования, может быть использован любой способ в непрерывном режиме или в периодическом режиме, без ограничений.
Необходимо только, чтобы реактор был изготовлен из материала, обладающего термостойкостью и коррозионной стойкостью к воздействию хлора, хлорида водорода и т.д. Возможно использовать реакторы, достаточно стойкие для проведения реакции при нормальном давлении или повышенном давлении, такие как металлические контейнеры, изготовленные из нержавеющей стали, Hastelloy™, Monel™, никеля, платины или т.п., и реакторы, облицованные тетрафторэтиленовой смолой, хлортрифторэтиленовой смолой, винилиденфторидной смолой, ПФА смолой, полипропиленовой смолой, полиэтиленовой смолой, стеклом и т.п.
Реакционная смесь, содержащая гексахлорацетон, который получают на данной стадии, может содержать хлорацетоны, содержащие от одного до пяти атомов хлора, хлорид водорода, хлор и др., помимо гексахлорацетона. Таким образом, для получения гексахлорацетона высокой чистоты предпочтительно используют средства очистки перегонкой или т.п. Перед началом перегонки предпочтительно заранее отделяют хлор и хлорид водорода для снижения нагрузки, такой как коррозия или т.п., на устройство в целом.
В соответствии с данным способом, можно получить гексахлорацетон высокой чистоты и его можно использовать в качестве исходного материала на последующей стадии фторирования. Хлорацетоны, содержащие от одного до пяти атомов хлора, которые отделяют и извлекают в качестве начальной фракции при перегонке, можно снова использовать в качестве исходных материалов для хлорирования.
Устройство для перегонки не ограничено особым образом, при условии, что оно изготовлено из материала, стойкого к хлору и хлориду водорода.
Стадия 2
Далее описана стадия 2. Стадия 2 представляет собой стадию фторирования смеси, содержащей гексахлорацетон, полученной на стадии 1, фтористым водородом с получением смеси, содержащей гексафторацетон и 1,1,1-трифтор-2,2-дихлорэтан (HCFC-123), количество которого превосходит 120 ppm.
Фторирование включает два режима, парофазный способ и жидкофазный способ. Как описано далее, данная реакция благоприятно протекает при относительно высокой температуре. Таким образом, предпочтительно использовать парофазный способ, который легко осуществлять при высокой температуре.
Предпочтительно стадию 2 осуществляют в присутствии так называемого «катализатора фторирования». Катализатором фторирования, используемым на данной стадии, является оксид, фторид, хлорид, фторхлорид, оксифторид, оксихлорид или оксифторхлорид металла. Более конкретно, металл представляет собой по меньшей мере один металл, выбранный из группы, состоящей из хрома, титана, алюминия, марганца, никеля, кобальта и циркония. Фторид алюминия, оксид алюминия, вторичный хлорид хрома и т.п. являются особенно предпочтительными катализаторами фторирования. Также возможно использование их сочетаний.
Количество катализатора составляет приблизительно от 0,001 до 0,5 молей на 1 моль гексахлорацетона, и специалист в данной области техники может подходящим образом его регулировать.
Температура реакции при фторировании составляет от 250 до 450°C, предпочтительно от 260 до 400°C, более предпочтительно от 260 до 350°C. Когда температура составляет менее 250°C, скорость реакции гексахлорацетона может быть низкой. Когда температура составляет более 450°C, может увеличиваться количество избыточно фторированных продуктов, и возрастает нагрузка на оборудование Таким образом, эти два условия не являются предпочтительными.
В качестве реактора, используемого на данной стадии, предпочтительно используют реактор, достаточно стойкий для проведения реакции при нормальном давлении или повышенном давлении, например, металлический контейнер, изготовленный из нержавеющей стали, Hastelloy™, Monel™, никеля или т.п., и такой реактор, который облицован тетрафторэтиленовой смолой, хлортрифторэтиленовой смолой, винилиденфторидной смолой, ПФА смолой, полипропиленовой смолой, полиэтиленовой смолой.
На данной стадии стехиометрическое молярное отношение фторида водорода к гексахлорацетону составляет 6, но для эффективного получения гексафторацетона при высокой степени превращения и с высоким выходом предпочтительно использовать фторид водорода в количестве, превышающем стехиометрическое количество. Соответственно, обычно реакцию осуществляют с использованием 8 молей или более, предпочтительно 10 молей или более, более предпочтительно 12 молей или более фторида водорода на 1 моль гексахлорацетона. Не существует верхнего предела количества фторида водорода. Хотя, если его используют в количестве 50 молей или более, скорость фторирования в общем не может быть увеличена еще больше. Кроме того, возрастают трудозатраты для извлечения непрореагировавшего фторида водорода. Соответственно, использование слишком большого количества фторида водорода является неблагоприятным. После реакции непрореагировавший фторид водорода отделяют от органического слоя, и его можно подавать рециклом в реакционную систему.
Давление реакции на данной стадии обычно составляет от 0,05 до 10 МПа и более предпочтительно от 0,08 до 1 МПа, что является близким к нормальному давлению.
В случае, когда стадию 2 осуществляют посредством парофазной реакции, время контакта реакции обычно составляет от 1 до 300 с, предпочтительно от 10 до 60 с. Однако оптимальное время контакта изменяют в зависимости от количества фторида водорода и условий реакции, таких как температура реакции, количество катализатора, давление реакции и т.д. Таким образом, специалист в данной области техники может регулировать его подходящим образом.
Также на стадии фторирования, аналогично вышеуказанной стадии хлорирования, гексахлорацетон постепенно превращается в гексафторацетон. Поэтому предпочтительно осуществлять реакцию при отслеживании степени превращения гексахлорацетона и степени протекания фторирования с использованием аналитических средств газовой хроматографии и т.д.
Как описано выше, реакционная смесь, полученная на стадии 2, содержит побочный продукт HCFC-123 в количестве более 120 ppm (обычно несколько тысяч ppm), помимо целевого продукта ГФА. Даже после простой перегонки смеси соединение все еще может оставаться в ГФА в количестве от 1000 до 2000 ppm.
Стадия 3
Далее описана стадия 3. Стадия 3 представляет собой стадию получения «очищенного гексафторацетона» посредством очистки «смеси, содержащей гексафторацетон и 1,1,1-трифтор-2,2-дихлорэтан в количестве более 120 ppm», полученной на стадии 2, в результате которой снижают содержание 1,1,1-трифтор-2,2-дихлорэтана, содержащегося в смеси, до не более 120 ppm. Средство очистки не ограничено особым образом, но перегонка (прецизионная перегонка) является особенно предпочтительной.
Гексафторацетон, полученный на стадии 2, обычно содержит остаточный фторид водорода и хлорид водорода. ГФА даже после простой перегонки все еще может содержать HCFC-123, который является проблемой в настоящем изобретении, обычно в количестве от 1000 до 2000 ppm, и образуется смесь из этих соединений. Стадия 3 представляет собой стадию снижения количества HCFC-123 до 120 ppm или менее. После данной стадии 3 можно существенно предотвратить явление «снижения скорости реакции в середине и после середины реакции» на стадии 4 (стадия гидрирования).
В конкретном воплощении стадии 3 операцию перегонки (прецизионной перегонки) предпочтительно осуществляют после того, как гексафторацетон поглощен водой с превращением его в гидрат (тригидрат ГФА с температурой кипения 106°C), так как с этим соединением легко обращаться.
При получении гидрата гексафторацетона реакционные условия не ограничены особым образом. Гексафторацетон в форме газа можно смешивать с водой (водой со льдом), содержащейся в реакторе, так что он поглощается водой. Предпочтительно превращать гексафторацетон в «тригидрат гексафторацетона», с которым легко обращаться. Для этого превращения предпочтительно ГФА и воду смешивают так, что количество воды может составлять 3 моля или более на 1 моль ГФА (молярное отношение воды к ГФА может составлять, например, от 3,0:1 до 20:1).
Когда смешивают более 3 молей воды с 1 молем гексафторацетона, получают смесь «тригидрата гексафторацетона (температура кипения 106°C)» и «воды (температура кипения 100°C)». Даже когда добавляют избыточную воду в данной операции, ее удаляют на последующей «операции перегонки». Даже когда остается избыточная вода, она не оказывает никакого влияния на «реакцию гидрирования» на стадии 4. С другой стороны, когда количество воды составляет менее 3 молей на 1 моль ГФА, получают смесь чистых химических веществ «ангидрида ГФА», «моногидрата ГФА» и «тригидрата ГФА», которые отличаются по температуре кипения. Такой случай является неблагоприятным, поскольку на последующей стадии могут возникнуть трудности в обращении.
Далее описана «перегонка (прецизионная перегонка)», которая особенно подходит в качестве «средства очистки для снижения количества HCFC-123 до 120 ppm или менее» на стадии 3, со ссылками на пример перегонки вышеуказанного «тригидрата ГФА».
Данная «перегонка» представляет собой операцию подачи тригидрата гексафторацетона в ректификационную колонну и осуществления фракционной перегонки предпочтительно при числе теоретических тарелок и флегмовом числе, указанных ниже (при описании «фракционной перегонки» в данном документе ее могут для удобства называть «перегонка» или «прецизионная перегонка»). С помощью такой перегонки количество HCFC-123 можно эффективно снижать с получением «тригидрата ГФА, в котором HCFC-123 составляет не более 120 ppm (очищенный тригидрат ГФА)».
В соответствии с данной стадией перегонки, HCFC-123, который представляет собой проблему в настоящем изобретении, может быть удален в виде фракции из тригидрата ГФА, наряду с «присутствующей в избытке водой», температура кипения которой меньше, чем температура кипения «тригидрата ГФА». Однако, как описано выше, трудно полностью отделить и удалить HCFC-123 из тригидрата ГФА. Когда стараются осуществить слишком полное удаление, на систему накладывают избыточную нагрузку при перегонке. Как также описано далее, поскольку количество HCFC-123 можно снизить до 120 ppm или менее, может быть в достаточной степени достигнут технический результат настоящего изобретения (то есть «предотвращение снижения скорости реакции в середине и после середины реакции» на стадии 4 гидрирования). Когда количество HCFC-123 составляет 110 ppm или менее, эффект может быть больше. Более предпочтительно, когда количество составляет 60 ppm или менее. Когда количество составляет 40 ppm или менее, может быть достигнут еще более благоприятный эффект. Напротив, хотя и возможно слишком намного снизить количество HCFC-123, нельзя достичь никакого дополнительного эффекта. Соответственно, обычно необязательно снижать количество «HCFC-123», например, до менее 3 ppm, особенно, менее 1 ppm.
В соответствии с вышеизложенным, в качестве «очистки» на стадии 3, режим с завершением операции очистки в момент, когда достигают целевого уровня содержания HCFC-123, который заранее определяют в интервале, например, от 5 до 110 ppm, может быть одним из приемлемых и предпочтительных воплощений. Целевой уровень может составлять от 10 до 60 ppm или от 5 до 40 ppm. При учете рабочих характеристик при операции очистки (особенно перегонки) специалист в данной области техники может подходящим образом установить целевой уровень.
Число тарелок ректификационной колонны можно изменять в зависимости от целевого количества HCFC-123. Оно может составлять, например, 2 или более и 50 или менее. Кроме того, число тарелок предпочтительно составляет 3 или более и 30 или менее, более предпочтительно 5 или более и 20 или менее.
Наполнитель, которым заполняют в ректификационную колонну, может представлять собой любой наполнитель из регулярной насадки и нерегулярной насадки. Регулярная насадка может представлять собой любую обычную насадку, включая, например, насадку Зульцера, Меллапак, Технопак, Флексипак и т.д. Нерегулярная насадка также может представлять собой любую обычную насадку, включая, например, насадку Хелипак, кольца Рашига, насадку Диксона и т.д.
Флегмовое число составляет от 0,5 до 8,0, предпочтительно от 0,5 до 7,0, более предпочтительно от 0,5 до 6,0.
В случае, когда тригидрат ГФА используют в качестве гексафторацетона, данный компонент становится нижним компонентом. Таким образом, предпочтительно очистку осуществляют посредством перегонки или т.п. при отслеживании количества HCFC-123, содержащегося в нижнем компоненте, с помощью количественного анализа посредством газовой хроматографии, и операцию очистки осуществляют до снижения содержания HCFC-123 до заданного уровня 120 ppm или менее.
Что касается типа колонки при осуществлении количественного анализа посредством газовой хроматографии, рекомендуется использовать «набивную колонку», подготовленную посредством помещения твердого адсорбента, такого как силикагель, активированный уголь, цеолит или т.п., в колонку в качестве наполнителя колонки, или посредством помещения твердой фазы (синтетический диоксид кремния или т.п., пропитанный нелетучей жидкостью) в колонку. Альтернативно, рекомендуется использование «капиллярной колонки», подготовленной нанесением твердой фазы адсорбента или т.п. на внутреннюю окружную поверхность полого капилляра из плавленого диоксида кремния или т.п. путем нанесения покрытия или образования химической связи.
В случае, когда на данной стадии используют набивную колонку, предпочтительно используют силикагель, активированный уголь или цеолит в качестве наполнителя колонки. В случае, когда используют капиллярную колонку, предпочтительно используют неполярную твердую фазу полидиметилсилоксана или т.п. или сильно полярную твердую фазу полиэтиленгликоля или т.п.
Содержание HCFC-123 рассчитывают исходя из «величины площади пика», определяемой посредством количественного анализа с помощью колоночной хроматографии. Например, компонент исследуют с использованием газового хроматографа, оборудованного пламенно-ионизационным детектором (ПИД), и отношение (%) площади пика HCFC-123 к общей (100%) площади всех пиков представляет собой содержание этого соединения.
При количественном анализе посредством газовой хроматографии используют обычный газ (азот, аргон, водород, гелий и т.д.) в качестве подвижной фазы. Температура колонки, давление подвижной газовой фазы, скорость потока подвижной фазы, длина колонки и другие факторы не ограничены особым образом, и специалист в данной области техники может регулировать их подходящим образом.
Приведенное выше в данном документе описание, относящееся к стадии 3, представляет способ прецизионной перегонки после превращения (обработки гидратацией) ГФА в «тригидрат ГФА», с которым проще обращаться. Однако не следует исключать из объема защиты настоящего изобретения воплощение, в котором удаление HCFC-123 проводят из безводного ГФА как такового, без обработки гидратацией. В этом случае, однако, следует обратить особое внимание на то, что безводный ГФА имеет температуру кипения (-28°C), которая ниже, чем температура кипения (28°C) HCFC-123. Другими словами, в случае осуществления разделения безводного ГФА и HCFC-123 посредством перегонки, отгоняемая фракция является целевым продуктом (безводный ГФА), а HCFC-123 концентрируется в нижней части. Если такие соединения с низкой температурой кипения необходимо разделить посредством перегонки, требуется выполнять особую операцию, такую как перегонка в условиях повышенного давления или перегонка в условиях регулируемой низкой температуры. Другими словами, нельзя сказать, что воплощение, осуществляемое на стадии 3 с использованием безводного ГФА, является наиболее подходящим воплощением настоящего изобретения, поскольку оно требует больших затрат на эксплуатацию и оборудование.
В настоящем изобретении наиболее предпочтительное воплощение стадии 3 обозначено как стадия 3a. Стадия 3a описана ниже.
Стадия 3a
Стадия 3a представляет собой стадию, на которой смесь, полученную на стадии 2, приводят в контакт с водой, чтобы тем самым превратить гексафторацетон, содержащийся в смеси, в тригидрат гексафторацетона, и затем смесь вводят в ректификационную колонку и там перегоняют при числе теоретических тарелок 2 или более и 50 или менее и флегмовом числе от 0,5 до 8,0, и перегонку осуществляют до достижения содержания 1,1,1-трифтор-2,2-дихлорэтана в смеси не более 120 ppm, которое определяют посредством газовой хроматографии, в результате чего получают очищенный тригидрат гексафторацетона, в котором содержание 1,1,1-трифтор-2,2-дихлорэтана составляет 120 ppm или менее.
Стадия 4
Далее описана стадия 4. Стадия 4 представляет собой стадию синтеза гексафторизопропанола посредством гидрирования очищенного гексафторацетона, полученного на стадии 3, посредством приведения его в контакт с водородом (H2) в присутствии катализатора.
На стадии 4 гидрирования можно непосредственно использовать традиционно известные операции и условия как таковые для проведения реакции. Отличительным признаком на данной стадии является то, что HCFC-123 содержится в исходном материале ГФА только в количестве 120 ppm или менее. Однако при этом снижение скорости реакции, начиная со середины и до конца реакции, можно в значительной степени предотвратить, хотя другие условия являются полностью такими же. Следовательно, в частности, когда реакцию продолжают до достижения высокой степени превращения ГФА, в настоящем изобретении обеспечивают преимущественный эффект, состоящий в том, что значительно сокращают время, необходимое для реакции.
На данной стадии предпочтительно реакцию осуществляют в жидкой фазе с использованием тригидрата гексафторацетона в качестве исходного материала, поскольку реакцию можно проводить плавно и активность катализатора можно сохранить на высоком уровне. Вышеуказанная стадия 3 описана при допущении, что ГФА также может быть «безводным ГФА». Однако на стадии 4, если «очищенный ГФА» является безводным ГФА, необходимо ангидрид преобразовать в тригидрат посредством способа, описанного на стадии 3, и затем полученный гидрат гидрируют на стадии 4.
В последующем описании «очищенный гексафторацетон», используемый в качестве исходного материала, представляет собой «тригидрат ГФА», и он вступает в реакцию в режиме жидкофазной реакции.
На данной стадии в качестве катализатора можно использовать различные катализаторы, известные как так называемые «катализаторы гидрирования». Более конкретно, особенно предпочтительным является «нанесенный катализатор (также называют гетерогенным катализатором или твердофазным катализатором»), в котором по меньшей мере один металл, выбранный из группы, состоящей из палладия (Pd), платины (Pt), рутения (Ru), родия (Rh) и никеля, нанесен на носитель, поскольку он обладает высокой каталитической активностью. Также можно использовать так называемый «гомогенный катализатор», в котором такой металлический компонент растворен или диспергирован во взвешенном состоянии в жидкой фазе, но использование «нанесенного катализатора» является более предпочтительным. «Нанесенный катализатор», когда его используют, позволяет обеспечить другое преимущество, состоящее в том, что катализатор (твердое вещество) можно легко отделить для повторного использования от реакционной смеси (жидкость) после реакции.
В качестве металла особенно предпочтительными являются палладий и рутений, поскольку они обладают высокой активностью. Химические формы металла включают металлы с нулевой валентностью, или оксиды, гидроксиды, хлориды и другие соединения, и также можно использовать комплексы, содержащие эти металлы. Степень окисления металла не ограничена.
В качестве носителя предпочтительными являются активированный уголь и оксид алюминия, поскольку с ними легко обращаться, и они являются легко доступными.
Нанесенное количество составляет от 0,0001 до 30 масс. %, предпочтительно от 0,01 до 20 масс. %, более предпочтительно от 0,1 до 10 масс. %, в показателях количества нанесенного металла относительно массы катализатора. Более конкретно, в качестве особенно предпочтительного катализатора можно отметить промышленно выпускаемый палладиевый катализатор, нанесенный на активированный уголь, в котором нанесенное количество составляет примерно от 0,1 до 5 масс. %, или катализатор, включающий от 0,1 до 5 масс. % рутения, нанесенного на активированный уголь.
Размер частиц катализатора не ограничен особым образом. Форма, подходящая для суспендирования катализатора, является предпочтительной. Применение мелкоизмельченного катализатора является предпочтительным, поскольку облегчается гомогенизация распределения катализатора в системе и улучшается контакт между реагентами и катализатором. Мелкие частицы также являются предпочтительными для извлечения и повторного использования катализатора после реакции и для отделения катализатора от продукта. Для получения катализатора можно использовать известные способы. Промышленно выпускаемый катализатор можно использовать непосредственно в состоянии поставки или после сушки или активирующей обработки (например, обработка, включающая приведение газообразного H2 в контакт с полученным катализатором при температуре от 25 до 200°C).
Количество катализатора не ограничено особым образом. В общем, количество катализатора (общее количество металлического компонента и носителя, или когда используют множество катализаторов, общее их количество) составляет от 0,00001 до 0,1 масс. ч., предпочтительно от 0,0005 до 0,03 масс. ч., более предпочтительно от 0,001 до 0,01 масс. ч. на 1 масс. ч. ангидрида ГФА.
На данной стадии количество (мольн. % относительно гексафторацетона) активного металлического компонента, такого как Pd, Ru или т.п., является более важным фактором. Однако в данной реакции гидрирования полной дезактивации катализатора практически не происходит. Таким образом, количество активного металлического компонента не ограничено особым образом. Количество может быть определено специалистом в данной области техники с учетом требуемого времени реакции. Например, количество металла относительно гексафторацетона (когда используют множество различных металлов, их общее количество) составляет от 0,0001 до 50 мольн. %, более предпочтительно от 0,001 до 1 мольн. %, еще более предпочтительно от 0,002 до 0,5 мольн. %.
Как описано выше, чтобы осуществлять реакцию гидрирования без образования пергидрированного побочного продукта ТФА, желательно использовать катализатор, содержащий как палладий, так и рутений в качестве активных компонентов. Более конкретно, желательно осуществлять гидрирование на стадии 4 в присутствии по меньшей мере одного типа катализатора, выбранного из группы, состоящей из «катализатора, в котором как палладий, так и рутений нанесены на один и тот же носитель» и «катализатора, полученного смешиванием катализатора, включающего палладий, нанесенный на носитель, и катализатора, включающего рутений, нанесенный на носитель». Например, одним особенно предпочтительным воплощением является применение смеси Pd/оксид алюминия нанесенного катализатора (или, возможно, Pd/активированный уголь нанесенного катализатора) и Ru/оксид алюминия нанесенного катализатора (или, возможно, Ru/активированный уголь нанесенного катализатора), при этом количество каждого из Pd и Ru доводят до количества от 0,001 до 0,25 мольн. % относительно ГФА (см. приведенные ниже примеры).
Однако «оптимизация типа катализатора» и «оптимизация количества катализатора» и «количество HCFC-123» являются совершенно различными факторами, и они независимы друг от друга. Даже когда тип и количество катализатора оптимизированы, может существовать значительная разница в «скорости реакции от середины до конца реакции» между случаем, когда количество HCFC-123 в начале гидрирования составляет более 120 ppm, и случаем, когда это количество составляет не более 120 ppm (это проиллюстрировано приведенными ниже примерами и сравнительными примерами).
На данной стадии предварительное добавление небольшого количества «акцептора кислоты (основного вещества)» в реакционную систему и запуск реакции после этого является предпочтительным, поскольку скорость реакции можно значительно увеличить. Полагают, что по мере протекания гидрирования ГФА, количество кислотных компонентов, таких как хлорид водорода, фторид водорода и т.п. (они могут быть каталитическими ядами), увеличивается в реакционной жидкости, но акцептор кислоты может быстро их нейтрализовать, и поэтому вряд ли будет происходить дезактивация катализатора, и тем самым скорость реакции может быть увеличена.
Акцептор кислоты, способный эффективно восстанавливать хлорид-ионы, включает карбонаты или гидрокарбонаты щелочных металлов, гидроксиды щелочных металлов или щелочноземельных металлов и т.д. Среди них карбонаты или гидрокарбонаты щелочных металлов являются предпочтительными.
Для восстановления фторид-ионов можно использовать гидроксиды, карбонаты, гидрокарбонаты и т.п. металлов 13 группы Периодической таблицы элементов.
Конкретные примеры карбонатов и гидрокарбонатов щелочных металлов включают карбонат натрия, карбонат калия, карбонат лития, гидрокарбонат натрия, гидрокарбонат калия, гидрокарбонат лития и т.п. Конкретные примеры гидроксидов щелочных металлов или щелочноземельных металлов, включают гидроксид натрия, гидроксид калия, гидроксид лития, гидроксид кальция, гидроксид магния и т.д. Среди них карбонат натрия, карбонат калия, гидрокарбонат натрия и гидрокарбонат калия являются предпочтительными. Гидрокарбонат натрия является особенно предпочтительным.
С другой стороны, конкретные примеры гидроксидов, карбонатов и гидрокарбонатов металлов 13 группы Периодической таблицы элементов включают гидроксид алюминия, гидроксид галлия, гидроксид индия, карбонат алюминия, гидрокарбонат алюминия, карбонат галлия, гидрокарбонат галлия и т.д.
В качестве способа эффективного восстановления фторид-ионов, также эффективным является применение комплексного гидроксида металлов, такого как алюминат магния или т.п. (например, соединение, представленное химической формулой «MgxAly(OH)2⋅X⋅nH2O» (где X является источником анионов, n является положительным целым числом, x, y и z представляют собой количество ионов)). Например, Halogen Killer (зарегистрированная торговая марка; производитель Horyu Co., Ltd.) доступен на рынке и прост в использовании.
На данной стадии одним предпочтительным воплощением является использование «карбоната или гидрокарбоната щелочного металла» и «гидроксида металла 13 группы Периодической таблицы элементов» в сочетании в качестве акцептора кислоты.
Добавляемое количество этих акцепторов кислоты не ограничено особым образом. При отслеживании развития реакции или в соответствии с количеством HCFC-123, содержащимся в исходном материале гексафторацетоне, специалист в данной области техники может регулировать количество акцептора кислоты подходящим образом. Предпочтительно это количество составляет от 0,005 до 0,1 моль на 1 моль гексафторацетона. Их можно использовать отдельно или в сочетании. (В случае, когда используют несколько типов акцепторов кислоты, вышеуказанное «добавляемое количество» означает общее количество всех акцепторов кислоты).
Предпочтительно акцептор кислоты добавляют постепенно при перемешивании в жидкий тригидрат гексафторацетона для однородного проведения реакции и для предотвращения повышения температуры вследствие выделения теплоты.
Как указано выше, эффект увеличения скорости реакции благодаря «наличию акцептора кислоты» и «эффект предотвращения снижения скорости реакции от середины до конца реакции» благодаря «снижению количества HCFC-123», который обнаружен настоящими авторами изобретения, являются совершенно различными и не зависят друг от друга. Только один из них может быть эффективен для увеличения скорости реакции. Однако сочетание обоих эффектов является более предпочтительным, поскольку позволяет дополнительно увеличить скорость реакции (см. примеры и сравнительные примеры, представленные далее в данном документе).
Температура реакции на данной стадии обычно составляет от 80 до 110°C и особенно предпочтительно составляет от 85 до 105°C.
Когда температура составляет менее 80°C, скорость реакции является низкой. Когда она составляет более 110°C, могут протекать побочные реакции и срок службы катализатора может сокращаться.
Давление реакции на данной стадии составляет от 0,05 до 5 МПа, предпочтительно от 0,1 до 1 МПа, более предпочтительно от 0,1 до 0,5 МПа. Когда давление составляет менее 0,05 МПа, скорость реакции может быть низкой. С другой стороны, когда реакцию осуществляют при давлении более 5 МПа, это накладывает ограничения на конструкцию реактора.
На данной стадии реакцию можно осуществлять с использованием растворителя. Растворитель не ограничен особым образом, при условии, что он не вступает в реакцию с исходным материалом и продуктом на стадии. Примеры растворителя включают гексафторизопропанол, который является продуктом в способе по настоящему изобретению, а также воду, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и т.д.; спирты, такие как метанол, этанол, 2-пропанол и т.д. Можно использовать один растворитель или два или более из этих растворителей можно использовать в сочетании.
Однако, как показано в примерах, представленных далее в данном документе, когда используют тригидрат гексафторацетона в качестве исходного материала на стадии 4, сам по себе тригидрат гексафторацетона является стабильной жидкостью, обладающей высокой текучестью. Таким образом, гидрирование может протекать в достаточной степени, даже когда растворитель не добавляют отдельно. Поэтому, как правило, стадию 4 осуществляют без добавления растворителя. Даже если не добавляют растворитель, может протекать гидрирование. В то время, как тригидрат гексафторацетона превращается в гексафторизопропанол, молекулы воды тригидрата гексафторацетона высвобождаются в систему. В результате, в системе появляется вода.
Синтез гексафторизопропанола на данной стадии можно осуществлять в любом режиме из периодической, полупериодической, непрерывной работы или в проточной системе. Что касается материала устройства, возможно, использовать металлический материал, такой как нержавеющая сталь, никелевая сталь, серебро; фтор-каучук, углерод или полиуретан, или используют устройства, футерованные или облицованные любым из этих материалов.
Предпочтительно, но не обязательно, реактор снабжен мешалкой. Обычно желательно снабжать реактор нагревательным устройством и/или охлаждающим устройством для регулирования температуры. Особенно предпочтительно снабжать реактор охлаждающим устройством.
Порядок размещения исходных материалов в реакторе не ограничен особым образом. Достаточно ввести вышеуказанные гексафторацетон и катализатор, затем добавить карбонат или гидрокарбонат щелочного металла в реакционную систему, и затем ввести газообразный водород при перемешивании для поддержания давления на заданном уровне, и продолжать введение газообразного водорода при поддержании температуры в пределах вышеуказанного диапазона.
Окончание реакции может быть подтверждено потреблением заданного количества водорода или прекращением поглощения водорода. Однако, как описано ранее, исходный материал ГФА в данной реакции является чрезвычайно дорогостоящим реагентом. Следовательно, особенно желательно, чтобы реакция продолжалась до достижения степени превращения ГФА от 98 до 100%. На такой «конечной стадии» значительное снижение скорости реакции неизбежно, даже при наилучших условиях. Другими словами, только очевидное «значительно более медленное потребление водорода» не может служить абсолютным подтверждением «завершения реакции». Следовательно, при проведении стадии 4 в особо крупном масштабе более предпочтительно периодически отбирать пробу из реакционной смеси по мере необходимости и отслеживать степень превращения ГФА с помощью газовой хроматографии или т.п., и когда степень превращения достигает заданного уровня, останавливать реакцию.
После проведения реакции содержимое в реакторе представляет собой реакционную смесь, содержащую целевой продукт гексафторизопропанол и другие органические вещества, а также катализатор. Жидкий компонент, не содержащий катализатор, можно извлечь из смеси, а гексафторизопропанол можно отделить и извлечь из жидкого компонента посредством перегонки.
Отделение катализатора от реакционной смеси предпочтительно осуществляют посредством фильтрации, когда катализатор является «нанесенным катализатором», который описан более подробно ранее. Обычно катализатор используют повторно. Поэтому является эффективным и предпочтительным оставлять катализатор в реакторе, когда содержащуюся в нем жидкость перемещают из реактора.
На стадии 4 одно особенно предпочтительное воплощение названо «стадия 4a». Она подробно описана ниже.
Стадия 4a
Это стадия, на которой «очищенный тригидрат гексафторацетона», полученный на стадии 3a, гидрируют посредством контакта с водородом (H2) в присутствии по меньшей одного катализатора, выбранного из группы, состоящей из «катализатора, содержащего палладий и рутений, нанесенные на один и тот же носитель», и «катализатора, полученного смешиванием катализатора, содержащего палладий, нанесенный на носитель, и катализатора, содержащего рутений, нанесенный на носитель», и в присутствии карбоната или гидрокарбоната щелочного металла в количестве от 0,005 до 0,1 моль на 1 моль тригидрата гексафторацетона, в результате чего получают гексафторизопропанол.
Стадия 5
Далее описана стадия 5. Стадия 5 представляет собой проведение реакции гексафторизопропанола, полученного на стадии 4, с формальдегидом и фторидом водорода в присутствии кислоты Бренстеда или кислоты Льюиса с получением фторметилгексафторизопропилового эфира (севофлурана).
Формальдегид, используемый на данной стадии, относится к понятию, включающему такие равноценные понятия, как параформальдегид (полиформальдегид, HO-(СH2O)n-Н, где n = от 2 до 100) и триоксан (полученный полимеризацией трех молекул формальдегида, 1,3,5-триоксан) и т.д.
Формальдегид как таковой находится в состоянии пара (газа) при комнатной температуре и при нормальном давлении, и при обращении с ним существуют значительные ограничения, например, его мгновенная полимеризация в присутствии небольшого количества примесей. Соответственно, предпочтительно используют параформальдегид или триоксан, с которыми легче обращаться. Параформальдегид является особенно предпочтительным.
Кислота Бренстеда, используемая в настоящем изобретении, представляет собой донор протонов (H+) и имеет константу кислотной диссоциации (pКa) приблизительно 3 или менее, предпочтительно 2 или менее.
Более конкретно, кислота включает серную кислоту, олеум, азотную кислоту, безводный сульфат, бромид водорода, йодид водорода, пропионовую кислоту, п-толуолсульфоновую кислоту, трихлоруксусную кислоту, трибромуксусную кислоту, метансульфоновую кислоту, трифторуксусную кислоту, трифторметансульфоновю кислоту и т.д. Среди них серная кислота, олеум, безводный сульфат, бромид водород, йодид водорода, трифторуксусная кислота и трифторметансульфоновая кислота являются предпочтительными. Серная кислота, олеум и трифторметансульфоновая кислота являются более предпочтительными.
Кислота Льюиса, используемая в настоящем изобретении, представляет собой акцептор электронной пары, включающий тетрахлорид титана, пентафторид фосфора, трифторид бора, трибромид бора, пентафторид сурьмы, хлорид алюминия, комплекс трифторида бора с диэтиловым эфиром и т.д.
Что касается количества молей формальдегида, фторида водорода и кислоты Бренстеда или кислоты Льюиса на 1 моль гексафторизопропанола, количество формальдегида составляет от 0,5 до 2,0 молей, количество фторида водорода составляет от 3,0 до 12,0 молей и количество кислоты Бренстеда или кислоты Льюиса составляет от 0,7 до 3,0 молей. Для увеличения выхода целевого продукта и предотвращения побочных реакций предпочтительно осуществлять реакцию с использованием от 0,78 до 1,65 молей формальдегида, от 6,05 до 9,50 молей фторида водорода и от 0,90 до 1,74 молей кислоты Бренстеда или кислоты Льюиса.
Среди этих кислот Бренстеда и кислот Льюиса, множество типов кислот можно использовать в сочетании.
Процесс получения фторметилгексафторизопропилового эфира осуществляют в любом из периодического, полунепрерывного, непрерывного или проточного режимов.
В качестве реактора, используемого на данной стадии, возможно использовать реакторы, подходящие для проведения реакции при нормальном давлении или повышенном давлении, такие как металлические контейнеры, изготовленные из нержавеющей стали, Monel™, Hastelloy™, никеля или т.п., и такие реакторы, которые облицованы тетрафторэтиленовой смолой, хлортрифторэтиленовой смолой, винилиденфторидной смолой, смолой ПФА, полипропиленовой смолой, полиэтиленовой смолой.
На данной стадии можно использовать реактор, оборудованный мешалкой, и его использование является предпочтительным.
Давление реактора на данной стадии составляет приблизительно от 0,05 до 5,0 МПа. Предпочтительно реакцию осуществляют при приблизительно нормальном давлении, которое не накладывает нагрузку на оборудование. Более конкретно, в вышеуказанном диапазоне давления, предпочтительным является давление от 0,05 до 2,0 МПа и более предпочтительным является давление от 0,08 до 0,5 МПа. Реакция в открытой системе (то есть при нормальном давлении) является предпочтительным воплощением данной стадии, поскольку это особенно просто реализовать.
Температура реакции на данной стадии составляет приблизительно от 40 до 100°C, предпочтительно от 50 до 80°C, более предпочтительно от 50 до 70°C.
В случае, когда данную стадию осуществляют в открытой системе при нормальном давлении, трудно значительно увеличить температуру реакции до температуры, которая намного больше температуры кипения (от 57 до 58°C) ГФИП и севофлурана, за исключением периода, очень близкого к концу реакции, поскольку температуры кипения ГФИП и севофлурана при нормальном давлении составляют от 57 до 58°C. Как представлено в примерах, из ГФИП и севофлурана в первую очередь севофлуран переходит в продукт перегонки. Соответственно, когда стадию 5 осуществляют в условиях открытой системы при нормальном давлении, предпочтительным является способ, в котором реакцию начинают от приблизительно комнатной температуры и затем систему постепенно нагревают до поддерживаемой «температуры (в пределах диапазона приблизительно от 50 до 60°C, хотя ее изменяют в зависимости от развития реакции), при которой севофлуран отгоняется, а ГФИП нет», и севофлуран как продукт перегонки, образующийся в течение реакции, собирают в ловушке. Приблизительно в конце реакции температуру постепенно повышают до значения выше этого и вплоть до температуры от 70 до 75°C. При этом степень превращения реакции может быть дополнительно увеличена. Естественно, заданную температуру реакции специалист в данной области техники может регулировать подходящим образом.
При подаче реагентов реакции или сразу после реакции может выделяться большое количество теплоты. Поэтому предпочтительно иметь нагревательное устройство и/или охлаждающее устройство для регулирования температуры. В частности, предпочтительно иметь охлаждающее устройство.
Время реакции на данной стадии обычно составляет от 2 до 24 часов. Поскольку оно зависит от температуры реакции и эквивалентного количества реагентов реакции, время не всегда ограничено этим диапазоном. Как описано выше, когда реакцию осуществляют в условиях открытой системы при нормальном давлении, реакцию можно прерывать в тот момент времени, когда прекращается испарение севофлурана. С другой стороны, при использовании аналитического прибора, такого как аппарат для ядерного магнитного резонанса (ЯМР), газовый хроматограф (ГХ) или т.п., момент времени, когда степень превращения реакции достигает заданного уровня, считают конечной точкой реакции.
На данной стадии полученный фторметилгексафторизопропиловый эфир предпочтительно очищают с применением операции перегонки. Более конкретно, полученную реакционную смесь промывают водой с получением двухслойной смеси жидкостей, которая содержит водный слой, содержащий 1,1,1,3,3,3-гексафторизопропанол, и органический слой, содержащий фторметилгексафторизопропиловый эфир. Затем водный слой, содержащий 1,1,1,3,3,3-гексафторизопропанол, отделяют от смеси жидкостей, а органический слой, содержащий фторметилгексафторизопропиловый эфир, подвергают перегонке с выделением фторметилгексафторизопропилового эфира высокой чистоты.
ПРИМЕРЫ
Далее настоящее изобретение описано более подробно со ссылками на примеры, представленные ниже. Однако настоящее изобретение не ограничено этими примерами. Здесь «%», указывающие значения, полученные при анализе состава, означают «% площади» композиции, полученные при анализе реакционной смеси посредством прямой газовой хроматографии (если конкретно не указано иное, детектором является плазменно-ионизационный детектор (ПИД)). В примерах и таблицах, представленных ниже, «HCFC-123» представляет собой «1,1,1-трифтор-2,2-дихлорэтан (CF3CHCl2)», «ТФИП» представляет собой «1,1,1-трифторизопропанол», «ГФА» представляет собой «гексафторацетон», «ГФИП» представляет собой «гексафторизопропанол», «ТеФИП» представляет собой «тетрафторизопропанол» и «ПФИП» представляет собой «пентафторизопропанол».
Пример получения - получение тригидрата гексафторацетона
В реактор объемом 1 дм3, облицованный стеклом (OC) и снабженный рубашкой, помещали 300 г (5,17 моль) ацетона и 1,85 г (0,01431 моль) хинолина. В то время как в реактор постепенно нагнетали 2262 г (31,90 моль) газообразного хлора, его постепенно нагревали до температуры 185°C и затем содержимое перемешивали в течение нескольких часов. Полученную реакционную жидкость перегоняли с получением 1268 г (4,79 моль) гексахлорацетона (чистота 99,5%; выход реакции хлорирования 92,6%; помимо ацетона, было обнаружено небольшое количество пентахлорацетона, но реакционную жидкость непосредственно использовали на следующей стадии фторирования).
Затем катализатор оксид хрома (III) (Cr2O3) помещали в реактор из нержавеющей стали, оборудованный рубашкой. В него вводили 1268 г (4,79 моль) гексахлорацетона, полученного как описано выше, и 1013 г (50,65 моль) безводной фтористоводородной кислоты и проводили фторирование в течение 40 ч при температуре 360°C. Полученный исходный гексафторацетон поглощали водой с получением 959 г (4,36 моль) исходного тригидрата гексафторацетона (выход реакции фторирования: 91,0%). В этот момент содержание HCFC-123 в исходном тригидрате гексафторацетона составляло 3550 ppm.
Неочищенный тригидрат гексафторацетона, полученный в данном примере получения, использовали в качестве исходного материла в примерах и сравнительных примерах, описанных ниже. Поэтому данный синтез осуществляли несколько раз.
Затем полученный как указано выше исходный тригидрат гексафторацетона подвергали простой перегонке и прецизионной перегонке. Что касается условий прецизионной перегонки, число теоретических тарелок составляло 10, а флегмовое число составляло от 0,2 до 5,0, чтобы оценить влияние HCFC-123, содержащегося в исходном тригидрате гексафторацетона на скорость реакции в течение реакции гидрирования. Как показано в примерах и сравнительных примерах, операцию перегонки проводили совместно с количественным анализом посредством газовой хроматографии до тех пор, пока не было подтверждено, что количество HCFC-123 снижено до заданного целевого уровня, в результате чего получали «тригидрат гексафторацетона, содержащий заданное количество (от 7 ppm до 198 ppm) HCFC-123».
Флегмовое число при прецизионной перегонке и содержание HCFC-123 в примерах и сравнительных примерах представлены в приведенной ниже таблице 1.

Все вещества, полученные в примерах и сравнительных примерах, использовали в качестве исходных материалов при синтезе гексафторизопропанола.
Пример 1
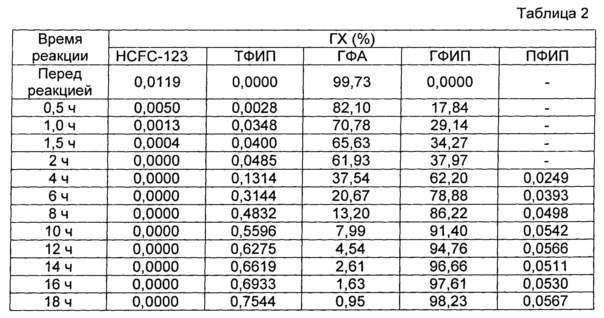
4,1 кг (18,63 моль) гидрата (тригидрата) гексафторацетона (состав гидрата и другие примеси представлены в таблице 2 ниже, и содержание HCFC-123 перед началом реакции составляло 119 ppm) помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,195 масс. % (масс. % относительно гидрата ГФА, то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (8 г), 0,122 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (5 г), 0,122 масс. % гидроксида алюминия (5 г) и 0,0146 масс. % гидрокарбоната натрия (0,6 г). Реактор продували водородом. Перемешивание начинали при нагревании вплоть до температуры 95°C горячей водой, давление водорода поддерживали равным 0,7 МПа. При этом начиналось поглощение водорода.
По прошествии 12 ч исследовали реакционную жидкость. При этом степень превращения ГФА гидрата составляла 95,46%. Что касается чистоты ГФИП, определенной посредством ГХ, она составляла 94,76%, количество ТФИП составляло 0,6275% и количество ГФА составляло 4,54%.
Затем реакцию продолжали, при этом исследовали реакционную жидкость. По прошествии 18 ч от начала реакции чистота ГФА, определенная посредством ГХ, достигала 0,95% (степень превращения гидрата ГФА составляла 99,05%). Поэтому нагрев и перемешивание останавливали для прекращения реакции. В этот момент времени чистота ГФИП, определенная посредством ГХ, составляла 98,23%, количество ТФИП составляло 0,7544% и количество ГФА составляло 0,95%. В примерах 1 и 2 и сравнительном примере 1 реакцию осуществляли при тех же условиях, за исключением количества HCFC-123 и того, что в тот момент времени, когда подтверждалась «степень превращения реакции ГФА 99%», реакцию прекращали и сравнивали время, требующееся для реакции.
Результаты представлены в нижеследующей таблице 2. Поскольку в данном примере количество HCFC-123 было немного меньше, чем критическая величина 120 ppm, время, необходимое для достижения конечной точки реакции, составляло 18 ч. По сравнению с временем примера 2, указанным ниже, время реакции в данном случае было больше. Однако при сравнении со временем сравнительного примера 1 (26 ч), также указанным ниже, понятно, что время реакции в данном случае было значительно сокращено.
Неочищенный ГФИП, полученный в этом примере, перегоняли при нормальном давлении, при этом собирали 2974 г ГФИП чистотой 99,99% или более. При этом общий выход составлял 95,0%.
Пример 2
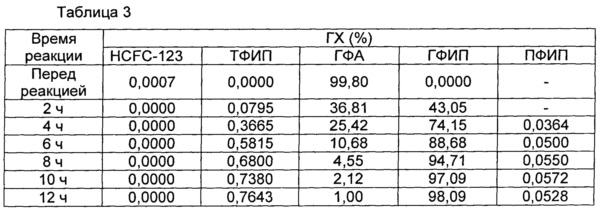
4,1 кг (18,63) гидрата (тригидрата) гексафторацетона (состав гидрата и другие примеси (включая HCFC-123) представлены в таблице 3 ниже, содержание HCFC-123 перед началом реакции составляло 7 ppm) помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Затем туда же добавляли 0,195 масс. % (масс. % относительно гидрата ГФА, тоже самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (8 г), 0,122 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (5 г), 0,122 масс. % гидроксида алюминия (5 г) и 0,0488 масс. % гидрокарбоната натрия (0,2 г). Реактор продували водородом и перемешивание начинали при нагревании вплоть до температуры 95°C горячей водой, давление водорода поддерживали равным 0,7 МПа. При этом начиналось поглощение водорода. По прошествии 8 ч исследовали реакционную жидкость. При этом степень превращения гидрата ГФА составляла 95,45%. Что касается чистоты ГФИП, определенной посредством ГХ, она составляла 94,71%, количество ТФА составляло 0,0039%, количество ТФИП составляло 0,6800% и количество ГФА составляло 4,55%.
Затем реакцию продолжали, при этом исследовали реакционную жидкость. В тот момент времени, когда чистота ГФА, определенная посредством ГХ, достигала 1,00%, нагрев и перемешивание останавливали для прекращения реакции. Время реакции до этого момента (от начала до конца реакции) составляло 12 ч, и степень превращения гидрата ГФА составляла 99,00%. Чистота ГФИП, определенная посредством ГХ, составляла 98,09%, количество ТФИП составляло 0,7643% и количество ГФА составляло 1,00%.
Результаты представлены в нижеследующей таблице 3. Когда количество HCFC-123, содержащегося в гидрате ГФА, составляло 7 ppm, «снижение скорости реакции на конечной стадии реакции» было более заметно предотвращено благодаря снижению содержания HCFC-123, по сравнению с приведенным выше примером 1. В результате, реакцию завершали за короткий период времени 12 ч. Результаты показывают, что в случае удовлетворения обоих требований, состоящих в том, что «гидрирование начинают в условиях содержания HCFC-123, составляющего более 120 ppm» и «акцептор кислоты должен присутствовать в системе» реализуют особо заметное увеличение скорости реакции.
Неочищенный ГФИП, полученный в этом примере, подвергали перегонке при нормальном давлении, при этом извлекали 2985 г ГФИП чистотой 99,99%. В этот момент общий выход составлял 95,35%.
Далее в последующих примерах 3-8 реакцию заканчивали через 8 ч. Эффект в данных примерах сравнивали с эффектом в сравнительных примерах 2-3, представленных далее, исходя из чистоты (%), определенной посредством ГХ, после реакции.
Пример 3
200 г ((0,91 моль), содержащий 12 ppm HCFC-123) гидрата (тригидрата) гексафторацетона помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,2 масс. % (масс. % относительно гидрата ГФА, и то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (0,4 г) и 0,12 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (0,24 г). Реактор продували водородом. При нагревании до температуры от 95 до 99°C на масляной бане начинали перемешивание, при этом поддерживали давление водорода от 0,70 до 0,71 МПа. При этом начиналось поглощение водорода.
По прошествии 8 ч от начала реакции нагрев и перемешивание останавливали и реакцию прекращали путем охлаждения. Степень превращения гидрата ГФА составляла 95,5%. Что касается чистоты реакционной жидкости, определенной посредством ГХ, количество ТФИП составляло 0,366%, количество ГФИП составляло 92,584%, количество ПФИП составляло 0,108%, количество ТеФИП составляло 0,040% и количество ГФА составляло 4,49%. Селективность по ГФИП в данной реакции составляла 99,4%.
Пример 3 представляет собой пример гидрирования в отсутствие акцептора кислоты. При сравнении со сравнительными примерами 2 и 3, приведенными ниже, понятно, что в случае, когда содержание HCFC-123 в гидрате гексафторацетона составляло 12 ppm, была получена повышенная степень превращения гидрата ГФА. Другими словами, подтверждено, что даже без добавления акцептора кислоты «снижение скорости реакции на конечной стадии реакции» может быть предотвращено посредством снижения содержания HCFC-123.
Пример 4
200 г ((0,91 моль), содержащий 12 ppm HCFC-123) гидрата (тригидрата) гексафторацетона помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,2 масс. % (масс. % относительно ГФА гидрата, и то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (0,4 г), 0,12 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (0,24 г), 0,12 масс. % гидроксида алюминия (0,24 г) и 0,002 масс. % гидрокарбоната натрия (0,004 г). Реактор продували водородом. При нагревании до температуры от 94 до 97°C на масляной бане начинали перемешивание, при этом поддерживали давление водорода от 0,70 до 0,71 МПа. При этом начиналось поглощение водорода.
По прошествии 8 ч от начала реакции нагрев и перемешивание останавливали и реакцию прекращали путем охлаждения. Степень превращения гидрата ГФА составляла 96,4%. Что касается чистоты реакционной жидкости, определенной посредством ГХ, количество ТФИП составляло 0,573%, количество ГФИП составляло 95,662%, количество ПФИП составляло 0,140%, количество ТеФИП составляло 0,066% и количество ГФА составляло 3,559%. Селективность по ГФИП в данной реакции составляла 99,2%.
Как отмечено выше, подтверждено, что степень превращения гидрата ГФА является чрезвычайно высокой по сравнению с этой величиной в сравнительных примерах 3 и 2, приведенных ниже.
Данный пример 4 отличается от примера 3 тем, что в нем используют акцептор кислоты. Обнаружено, что благодаря присутствию акцептора кислоты скорость реакции возрастает в большей степени, чем в примере 3 (не говоря уже о том, что по сравнению с примерами 5-8 степень превращения гидрата ГФА в данном случае чрезвычайно высока). Это дает веские основания полагать, что «увеличенная скорость реакции» при добавлении акцептора кислоты и «предотвращение снижение скорости реакции на конечной стадии реакции» посредством снижения содержания HCFC-123 до 12 ppm являются различными и независимым эффектами.
Пример 5
200 г ((0,91 моль), содержащий 52 ppm HCFC-123) гидрата (тригидрата) гексафторацетона помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,2 масс. % (масс. % относительно гидрата ГФА, и то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (0,4 г) и 0,12 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (0,24 г). Реактор продували водородом. При нагревании до температуры от 95 до 97°C на масляной бане, начинали перемешивание, при этом поддерживали давление водорода от 0,70 до 0,71 МПа. При этом начиналось поглощение водорода.
По прошествии 8 ч от начала реакции нагрев и перемешивание останавливали и реакцию прекращали путем охлаждения. Степень превращения гидрата ГФА составляла 93,6%. Что касается чистоты реакционной жидкости, определенной посредством ГХ, количество ТФИП составляло 0,331%, количество ГФИП составляло 93,061%, количество ПФИП составляло 0,112%, количество ТеФИП составляло 0,042% и количество ГФА составляло 6,449%. Селективность по ГФИП в данной реакции составляла 99,5%.
Пример 6
197,52 г ((0,90 моль), содержащий 46 ppm HCFC-123) гидрата (тригидрата) гексафторацетона помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,2 масс. % (масс. % относительно ГФА гидрата, и то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (0,4 г), 0,12 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (0,24 г), 0,12 масс. % гидроксида алюминия (0,24 г) и 0,002 масс. % гидрокарбоната натрия (0,004 г). Реактор продували водородом. При нагревании до температуры от 94 до 98°C на масляной бане начинали перемешивание, при этом поддерживали давление водорода от 0,71 до 0,72 МПа. При этом начиналось поглощение водорода.
По прошествии 8 ч от начала реакции нагрев и перемешивание останавливали и реакцию прекращали путем охлаждения. Степень превращения гидрата ГФА составляла 94,6%. Что касается чистоты реакционной жидкости, определенной посредством ГХ, количество ТФИП составляло 0,655%, количество ГФИП составляло 93,775%, количество ПФИП составляло 0,129%, количество ТеФИП составляло 0,063% и количество ГФА составляло 5,378%. Селективность по ГФИП в данной реакции составляла 99,1%.
Пример 7
200,02 г ((0,91 моль), содержащий 106 ppm HCFC-123) гидрата (тригидрата) гексафторацетона помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,2 масс. % (масс. % относительно гидрата ГФА, и то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (0,4 г), 0,12 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (0,24 г). Реактор продували водородом. При нагревании до температуры от 95 до 96°C на масляной бане начинали перемешивание, при этом поддерживали давление водорода от 0,70 до 0,71 МПа. При этом начиналось поглощение водорода.
По прошествии 8 ч от начала реакции нагрев и перемешивание останавливали и реакцию прекращали путем охлаждения. Степень превращения гидрата ГФА составляла 91,9%. Что касается чистоты реакционной жидкости, определенной посредством ГХ, количество ТФИП составляло 0,332%, количество ГФИП составляло 91,420%, количество ПФИП составляло 0,109%, количество ТеФИП составляло 0,041% и количество ГФА составляло 8,097%. Селективность по ГФИП в данной реакции составляла 99,5%.
Пример 8
200,02 г ((0,91 моль), содержащий 106 ppm HCFC-123) гидрата (тригидрата) гексафторацетона помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,2 масс. % (масс. % относительно ГФА гидрата, и то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (0,4 г), 0,12 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (0,24 г), 0,12 масс. % гидроксида алюминия (0,24 г) и 0,002 масс. % гидрокарбоната натрия (0,004 г). Реактор продували водородом. При нагревании до температуры от 95 до 97°C на масляной бане начинали перемешивание, при этом поддерживали давление водорода от 0,70 до 0,71 МПа. При этом начиналось поглощение водорода.
По прошествии 8 ч от начала реакции нагрев и перемешивание останавливали и реакцию прекращали путем охлаждения. Степень превращения гидрата ГФА составляла 94,9%. Что касается чистоты реакционной жидкости, определенной посредством ГХ, количество ТФИП составляло 0,515%, количество ГФИП составляло 94,196%, количество ПФИП составляло 0,126%, количество ТеФИП составляло 0,057% и количество ГФА составляло 5,108%. Селективность по ГФИП в данной реакции составляла 99,3%.
Как отмечено выше, подтверждено, что степень превращения гидрата ГФА является высокой в примерах 3-8 по сравнению со сравнительными примерами 2 и 3, приведенными ниже. При сравнении примеров 3 и 4 с примерами 5-8 видно, что степень превращения гидрата ГФА в примерах 3 и 4 выше, чем в примерах 5-8. Полагают, что путем соблюдение условий «добавления акцептора кислоты» и «снижения содержания HCFC-123 до величины, не превышающей заданное количество» и путем снижения количества HCFC-123 до величины, попадающей в интервал более предпочтительного воплощения (от 5 ppm до 40 ppm), можно эффективно предотвратить снижение скорости реакции на конечной стадии реакции.
Сравнительный пример 1
Осуществляли ту же операцию, как и в примере 1, при тех же условиях, за исключением того, что качестве исходного материала использовали гидрат (тригидрат) ГФА с содержанием HCFC-123, составляющим 144 ppm, и в реакционную систему добавляли 0,2 г гидрокарбоната натрия.
Реакцию осуществляли при анализе реакционной жидкости. В момент времени, когда чистота ГФА, определенная посредством ГХ, достигала 1,00% (степень превращения реакции=99%), нагрев и перемешивание прекращали и реакцию прекращали путем охлаждения. До этого момента время реакции (от начала до конца реакции) составило 26 часов. Хотя другие условия, кроме количества HCFC-123, были такими же, что и в примерах 1 и 2, в сравнительном примере 1 время реакции было значительно больше. Что касается чистоты, определенной посредством ГХ, количество ГФИП составляло 98,38%, количество ТФИП составляло 0,5473%, и количество ГФА составляло 1,00%.
Результаты представлены в таблице 4 ниже. Как отмечено выше, понятно, что даже в случае добавления акцептора кислоты, когда количество HCFC-123 составляет более 120 ppm, время реакции больше, чем в примере 1 и примере 2. Это означает, что «снижение скорости реакции на конечной стадии реакции» не коррелирует с «добавлением акцептора кислоты при гидрировании», а зависит от содержания HCFC-123.

Далее, в приведенных ниже сравнительном примере 2 и сравнительном примере 3 реакцию прекращали через 8 ч, так же как в приведенных выше примерах 3-8, и сравнивали эффекты исходя из определенной посредством ГХ чистоты (%) после реакции.
Сравнительный пример 2
200 г ((0,91 моль), содержащий 130 ppm HCFC-123) гидрата (тригидрата) гексафторацетона помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,2 масс. % (масс. % относительно гидрата ГФА, и то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (0,4 г) и 0,12 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (0,24 г). Реактор продували водородом. При нагреве до температуры 96°C на масляной бане начинали перемешивание, при этом давление водорода поддерживали равным 0,7 МПа. При этом начиналось поглощение водорода.
По прошествии 8 ч от начала реакции нагрев и перемешивание останавливали и реакцию прекращали путем охлаждения. Степень превращения гидрата ГФА составляла 86,78%. Что касается чистоты реакционной жидкости, определенной посредством ГХ, количество ТФИП составляло 0,285%, количество ГФИП составляло 86,30%, и количество ГФА составляло 13,22%.
Как отмечено выше, понятно, что когда содержание HCFC-123 составляет 130 ppm, степень превращения гидрата ГФА, исходя из определенной посредством ГХ чистоты реакционной жидкости, ниже через 8 ч реакции, чем степень превращения в примерах 3-8. Это позволяет предположить, что независимо от «добавления акцептора кислоты при гидрировании», скорость реакции снижается на конечной стадии реакции, когда содержание HCFC-123 составляет более 120 ppm.
Сравнительный пример 3
196,55 г ((0,91 моль), содержащий 198 ppm HCFC-123) гидрата (тригидрата) гексафторацетона помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. Туда же добавляли 0,2 масс. % (масс. % относительно ГФА гидрата, и то же самое относится к дальнейшему описанию) 5%-Pd/оксид алюминия нанесенного катализатора (0,4 г), 0,12 масс. % 5%-Ru/оксид алюминия нанесенного катализатора (0,24 г), 0,12 масс. % гидроксида алюминия (0,24 г) и 0,002 масс. % гидрокарбоната натрия (0,004 г). Реактор продували водородом. При нагреве до температуры от 93 до 96°C на масляной бане начинали перемешивание, при этом давление водорода поддерживали равным 0,7 МПа. При этом начиналось поглощение водорода.
По прошествии 8 ч от начала реакции нагрев и перемешивание останавливали и реакцию прекращали путем охлаждения. Степень превращения гидрата ГФА составляла 84,50%. Что касается чистоты реакционной жидкости, определенной посредством ГХ, количество ТФИП составляло 0,505%, количество ГФИП составляло 83,85%, и количество ГФА составляло 15,50%.
Как отмечено выше, понятно, что когда содержание HCFC-123 составляет 198 ppm, степень превращения гидрата ГФА, исходя из определенной посредством ГХ чистоты реакционной жидкости, ниже через 8 ч реакции, чем степень превращения в примерах 3-8, даже несмотря на то, что добавляют акцептор кислоты. Это позволяет предположить, что когда содержание HCFC-123 сильно превышает 120 ppm, скорость реакции снижается в большей степени на конечной стадии реакции.
Сравнительный пример. Получение тригидрата гексафторацетона (пример получения посредством простой перегонки неочищенного тригидрата гексафторацетона)
200 г (0,91 моль) неочищенного гидрата (тригидрата) гексафторацетона, полученного в приведенном выше примере получения, помещали в автоклав из нержавеющей стали (SUS-316) объемом 5 дм3, оборудованный мешалкой. В устройстве для простой перегонки, присоединенном к автоклаву, осуществляли простую перегонку при давлении 0,05 МПа и при внутренней температуре от 70 до 75°C.
В результате получали 170 г тригидрата гексафторацетона в качестве продукта перегонки. В полученном продукте перегонки определяли количество фторид-ионов посредством газовой хроматографии. При этом количество HCFC-123, содержащегося в тригидрате гексафторацетона, составляло 1504 ppm.
Исходя из вышеизложенного понятно, что даже если после синтеза тригидрата гексафторацетона проводят простую перегонку, содержание HCFC-123 не может быть менее 120 ppm.
Пример 9
14,13 г (0,42 моль) 95% параформальдегида, 132,4 г (1,35 моль) 97% серной кислоты, 53,02 г (2,65 моль) фторида водорода и 43,69 г (0,26 моль) гексафторизопропанола, полученного в примере 1, помещали в реактор, к которому присоединена ловушка, охлаждаемая до температуры примерно -15°C. Перемешивание осуществляли при температуре 20°C в течение от 2 до 3 ч. Затем реактор нагревали в течение от 5 до 6 ч, чтобы в конце температура составляла от 65 до 75°C, при этом осуществляли перемешивание. В ходе этого, при перемешивании, продукт в реакторе постепенно отгонялся. Поэтому продукт собирали в ловушку снаружи системы.
В тот момент времени, когда продукт перегонки больше не образовывался, реакцию останавливали. Ловушку, в которой был собран продукт (органическая фаза, содержащая фторметилгексафторизопропиловый эфир), промывали водой, затем кислым водным раствором и затем основным водным раствором с получением двухслойной смеси жидкостей, состоящей из водного слоя, содержащего непрореагировавший гексафторизопропанол и фторид водорода, и органического слоя, содержащего фторметилгексафторизопропиловый эфир. Затем водный слой, содержащий 1,1,1,3,3,3-гексафторизопропанол, отделяли и удаляли из смеси жидкостей. Органический слой, содержащий фторметилгексафторизопропиловый эфир, подвергали перегонке с получением 34 г (0,17 моль) фторметилгексафторизопропилового эфира, имеющего чистоту более 99%, при выходе 65,06%. Степень превращения ГФИП составляла 72,51%,
Промышленное применение
Гексафторизопропанол, который является целевым соединением настоящего изобретения, можно использовать в качестве промежуточного соединения для получения лекарственных средств и сельскохозяйственных химикатов и в качестве растворителя для анализа и растворителя для промывки материалов для электронных приборов и т.д. Севофлуран, получаемый из гексафторизопропанола, можно использовать в качестве лекарственного препарата, такого как ингаляционный анестетик и т.д.
Реферат
Настоящее изобретение относится к способу получения фторметилгексафторизопропилового эфира (севофлурана), применяемого в качестве ингаляционного анастетика, а также к способу получения гексафторизопропанола. Один из вариантов способа получения севофлурана включает следующие стадии: (a) очистка смеси, содержащей гексафторацетон и более 120 ppm 1,1,1-трифтор-2,2-дихлорэтана в качестве примеси, с получением очищенного гексафторацетона, содержащего 120 ppm или менее 1,1,1-трифтор-2,2-дихлорэтана; (b) приведение водорода (Н) в контакт с очищенным гексафторацетоном в присутствии катализатора, посредством чего осуществляют гидрирование гексафторацетона с получением гексафторизопропанола, и (c) проведение реакции гексафторизопропанола, формальдегида и фторида водорода в присутствии кислоты Льюиса или кислоты Бренстеда. Настоящее изобретение позволяет получить целевые продукты за более короткий период времени. 3 н. и 20 з.п. ф-лы, 4 табл., 25 пр.
Комментарии