Хиральные оптические активные адсорбенты, способы их получения, сетчатый полимер, производные винной кислоты и способы их получения - RU2121395C1
Код документа: RU2121395C1
Чертежи
Описание
Изобретение относится к новым хиральным адсорбентам и способам их получения. Изобретение относится также к некоторым новым соединениям, на которых основаны хиральные адсорбенты, и получению этих новых соединений.
Оптические изомеры можно разделить путем образования диастереомеров с применением хиральных реагентов и последующего разделения их жидкостной или газовой хроматографией или перекристаллизацией или путем прямого хроматографического разделения с применением систем с хиральной фазой. Растущий интерес к разделению фармацевтических веществ и определению их энантиомерной чистоты вызывает повышенную потребность в способе прямого хроматографического разделения энантиомеров. В этом способе разделения применяют хиральное селективное вещество в подвижной фазе или хиральную стационарную фазу. В последние годы большое внимание уделялось прямому хроматографическому разделению энантиомеров с применением хиральных стационарных фаз. Предложен ряд различных хиральных адсорбентов, но только несколько из них, например те, которые основаны на производных целлюлозы и производных аминокислот, оказались заметно коммерчески удачными в препаративной хроматографии. Это в значительной степени зависит от строгих требований, которые предъявляются к хиральным стационарным фазам, чтобы они были пригодны для препаративного разделения, т. е. разделения в больших количествах, главным образом (ЖХВР) жидкостной хроматографией высокого разрешения. Для такого разделения колонки должны обладать высокой степенью энантиоселективности, высокой адсорбционной емкостью, т.е. допускают присоединение относительно большого количества рацемата, высокой эффективностью, т.е. дают небольшое расширение пиков на хроматограмме, а также высокой универсальностью, т.е. позволяют проводить разделение как можно больше структурно различных типов химических соединений.
В соответствии с настоящим изобретением найдено, что хиральные стационарные фазы, которые основаны на сетчатых полимеризованных производных дикарбоновых кислот, диаминов, диолов или гидроксикислот, которые химически связаны с твердым носителем, полностью удовлетворяют требованиям, предъявляемым таким фазам при использовании их как для аналитического, так и препаративного разделения. Одним примером такого производного является винная кислота как таковая, которая является одним из менее дорогих оптически активных органических исходных материалов, доступных сегодня на рынке, что делает настоящее изобретение в его различных аспектах экономически привлекательным.
Оптически активный адсорбент в соответствии с настоящим изобретением, характеризуется оптически активным сетчатым полимером, ковалентно связанным с носителем.
Оптически активный сетчатый полимер представляет собой оптически активные производные дикарбоновых кислот, диаминов, диолов или гидроксикислот.
Каждая функциональная группа оптически активных производных дикарбоновых кислот, диаминов или диолов содержит по меньшей мере один алифатический углеродный остаток, имеющий вплоть до 15 атомов углерода и содержащий по меньшей мере одну концевую ненасыщенную связь.
Производные диолов являются алифатическими сложными эфирами, карбонатами или карбаматами, имеющими вплоть до 15 атомов углерода в углеродной цепи и концевую ненасыщенную связь.
Производные диаминов являются амидами, карбаматами и карбамидами, имеющими вплоть до 15 атомов углерода в углеродной цепи и концевую ненасыщенную связь.
Производные дикарбоновых кислот являются эфирами и амидами, имеющими вплоть до 15 атомов углерода в углеродной цепи и концевую ненасыщенную связь.
Наиболее предпочтительным производным гидроксикислот является винная кислота.
Примерами соединений, представляющих интерес, являются:
D - или L-винная кислота,
(1R,2R)-(-)-1,2-диаминоциклогексан,
(+)-2,2'-диаминодинафтил-(1,1'),
(1R,2R)-(-)-1,2-циклогександиол,
(+)-(2R,3R)-1,4-диметокси-2,3-бутандиол,
D-(-)-цитраяблочная кислота,
D-(+)-яблочная кислота.
Изобретение определяется более подробно в прилагаемой форме изобретения.
Адсорбенты в соответствии с одним предпочтительным вариантом настоящего изобретения основаны на сетчатых полимеризованных производных винной кислоты, которые связаны с носителем, например силикагелем (гелем SiO2). Как известно в настоящей области знаний, некоторые производные винной кислоты, связанные с силикагелем, можно применять в качестве хиральных стационарных фаз. Такие фазы с неполимерными производными, связанными с силикагелем (так называемые типа щетки), а также ряд хиральных применений таких производных винной кислоты описаны W. Lindner и I.Hirschbock в J.Pharm. Biomed. Anal., 1984, 2,2, 183-189. Хиральные стационарные фазы, основанные на простом неполимеризованном производном винной кислоты, описаны также Y.Dobashi и S.Hara в J.Org. Chem., 1987, 52, 2490-2496. Преимущества производного винной кислоты, являющегося частью сетчатой полимерной фазы, как в настоящем изобретении, заключаются в том, что на носителе получают несколько хиральных центров, а это приводит к повышенной хроматографической емкости, и что получают носитель с более защищенной поверхностью. Для случаев, когда носителем является диоксид кремния, это приводит к уменьшению числа доступных свободных силанольных групп, что означает снижение ахиральных полярных взаимодействий, которые ослабляют энантиоселективность. Более высокую энантиоселективность по сравнению с мономерной фазой получают также при применении полимерной фазы, вероятно из-за того, что полимер может образовать трехмерную структуру, которая может иметь хиральные полости.
Производные винной кислоты, которые
полимеризуют, сами являются оптически гомохиральными производными и содержат по меньшей мере два стереогенных центра. Эти производные можно характеризовать общей формулой:
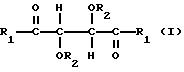
в которой
R1 представляет собой группу RNH-, RO-, RR'N- или HO- и R2 представляет собой группу RNHCO-, RCO-, ROCO-, H- или R-, где R является алифатическим углеводородным остатком, имеющим вплоть до 15 атомов углерода, арильной группой, аралкильной группой, нафтильной группой или антрильной группой и R' является водородом или алкильной группой, имеющей вплоть до 7 атомов углерода, причем эти производные содержат по меньшей мере две группы R1 и R2, имеющие алифатическую ненасыщенную связь. Группы R1 и R2 могут содержать один или несколько хиральных центров. Когда R представляет собой алифатический углеводородный остаток, он может быть алкилом, циклоалкилом, алкенилом или алкинилом. В таком случае пригоден R, содержащий вплоть до 10 атомов углерода и являющийся алкилом или алкенилом, предпочтительно алкенилом. R может быть арильной группой или аралкильной группой. Эти группы могут содержать 1, 2 или 3 ядра и незамещены или замещены одним или несколькими заместителями у ядра или ядер. Примерами таких заместителей являются алкильные группы, гидроксигруппы, атомы галогена, нитрогруппы и алкенильные группы. Пригодны R', являющиеся водородом или алкильной группой, имеющей 1 или 2 атома углерода. Пригодны R1, являющиеся группой RNH-, RO-, или RR'N-, предпочтительно группой RNH-. В таком случае пригодны R, являющиеся аллильной группой, α-фенилэтильной группой или нафтильной группой, наиболее предпочтительно любыми из первых двух указанных групп. Пригодны R2, являющиеся группой RNHCO-, RCO- или H-, предпочтительно группой RNHCO- или RCO-. В таком случае пригодны R, являющиеся фенилом, аллилом, 3,5-динитрофенилом, нафтилом, метакрилом, α-фенилэтилом, 3,5-диметилфенилом, трет-бутилом или изопропилом.
Предпочтительно R является фенилом, аллилом, 3,5-динитрофенилом, нафтилом, метакрилом или α-фенилэтилом. Две группы R1 в производных должны быть одинаковыми и две группы R2 должны быть также одинаковыми.
Особенно пригодны производные винной кислоты формулы I, которые можно характеризовать формулами:
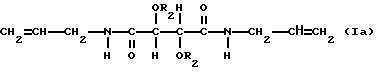
и
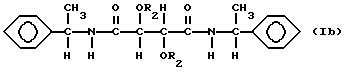
В соединениях формулы Ia R1 таким образом является аллиламиногруппой и в соединениях формулы Ib R1 является фенилэтиламиногруппой и R2 имеет значения, указанные выше.
Соединения формулы Ia включают диаллилдиамид винной кислоты (R, R или S, S), который коммерчески доступен, и его производные. В соединениях формулы Ia пригодны R2, которые являются группой RNHCO-, RCO- или H-, где R имеет значения, указанные выше. R может быть, например объемной алкильной группой, например изопропилом или третичным бутилом, бензильной группой, фенильной группой, нафтильной группой или антрильной группой. Любые заместители у ароматического ядра могут быть заместителями, указанными выше. Наиболее предпочтительно R2 является группой RNHCO или RCO-, где R содержит арильную группу, которая возможно замещена. Благоприятны соединения, содержащие ароматическое ядро, поскольку в таком случае возникает π,π - взаимодействие с ароматическими рацематами, которое способствует разделению. Примерами некоторых конкретных подходящих групп R2 для соединений формулы Ia являются: фенилкарбамоил, α- фенилэтилакарбамоил, 3,5-диметилфенилкарбамоил, нафтилкарбамоил, α- нафтилэтилкарбамоил, бензоил и 3,5-динитробензоил и 3,5-диметилбензоил.
Соединения формулы Ia можно получить обычными реакциями ацилирования и карбамоилирования. Сложные эфиры диаллилдиамида винной кислоты можно таким образом получить реакцией диамида с соответствующим хлорангидридом или ангидридом. В соответствии с подходящим способом диамид растворяют в растворителе, который действует так же, как основание, например пиридине, после чего добавляют соответствующий хлорангидрид, который можно применять по меньшей мере в эквимолярном количестве. После окончания реакции, которую можно проводить при комнатной температуре, полученные продукты обрабатывают обычным образом, например экстракцией, выпариванием и кристаллизацией. Карбаматы диаллилдиамида винной кислоты можно получить реакцией амида с соответствующим изоцианатом. Амид можно растворить в походящем растворителе, например тетрагидрофуране, и обработать изоцианатом в присутствии каталитического количества основания, например 4-диметиламинопиридина, или катализатора, например соли олова. Реакцию можно проводить при кипячении с обратным холодильником и после окончания реакции продукт выделяют обычным способом.
Соединения формулы Ib можно получить из продукта реакции эфира, R,R- или S, S-винной кислоты, такого как алкилтартрат, например
диметилтартрата, и оптически активного α-фенилэтиламина. В соединениях формулы Ib подходящий R2 является группой RNHCO- или RCO- и тогда R таким образом должен содержать
алифатическую двойную связь, предпочтительно концевую двойную связь. Особенно подходящими группами R2 являются
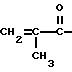
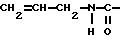
Соединения, имеющие такие группы R2, можно получить известными реакциями ацилирования из ангидрида и известными реакциями карбамоилирования соответственно. Для получения соединений, соответствующих формуле Ib, где R2 представляет собой остаток метакриловой кислоты, диамид обрабатывают ангидридом метакриловой кислоты. Диамид можно растворить в подходящем растворителе, например тетрагидрофуране или хлорированном углеводороде, и обработать диамидом в присутствии основания, например 4-диметиламинопиридина, при комнатной температуре. Для получения соединений формулы Ib, которые являются карбаматами, можно применять ту же методику, которую применяют для получения карбаматов формулы Ia.
Полимеризованные производные, ковалентно связанные с материалом-носителем, и сам сетчатый полимеризат могут быть гомо- или сополимерами указанных производных винной кислоты или такими полимерами, которые были получены реакциями гидросилилирования.
В качестве носителя применяют органический или
неорганический материал. Примерами органических носителей являются полимер стирол-дивинилбензола. Примерами неорганических носителей являются диоксид кремния, оксид алюминия и оксид циркония, которые
модифицируют силанами. Полимеризованные производные связывают с органическими носителями C-C-связью и с неорганическими носителями Si-C или Si-O-Si-связью. Материалы-носители должны иметь высокую
удельную поверхность и удовлетворительную механическую устойчивость. Поверхность материала-носителя должна иметь реакционноспособную функциональную группу, которая содержит концевую двойную связь,
гидроксилильную группу или силанольную группу, чтобы производные винной кислоты могли связываться с носителем. Примерами подходящих групп, содержащих двойную связь, являются винил, гексенил, октенил,
акриловая и метакриловая группы. Такие группы, а также гидросилильные группы, могут соединяться с поверхностью материала-носителя, например диоксида кремния, известными реакциями модифицирования
поверхности. Структурно несколько различных подходящих гидросилилмодифицированных поверхностей диоксида кремния можно схематически представить следующим образом:
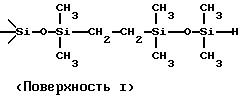
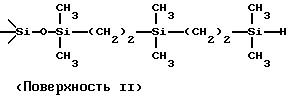

Поверхности I и II получали модификацией виниловой поверхности 1,1,3,3-тетраметилдисилоксаном и 1,1,4, 4-тетраметилдисилилэтиленом соответственно. Поверхность III получали модификацией исходного (недериватизированного) диоксида кремния 1,3,5,7-тетраметилциклотетрасилоксаном. Вариант поверхности III можно получить с помощью 1,3,5,7-тетравинилтетраметилциклотетрасилоксана и полимеризацией его, модифицирующей поверхность диоксида кремния. Этот вариант предпочтителен, поскольку в этом случае обеспечивается оптимальное покрытие поверхности.
Оптически активные адсорбенты соответственно настоящему изобретению можно получить сетчатой полимеризацией производных винной кислоты в присутствии материала-носителя или сначала полимеризацией этих производных и затем закреплением сетчатого полимера на материале-носителе ковалентным связыванием.
Для некоторых целей может быть пригодно также применение производных винных кислот формулы 1 в качестве мономеров для получения линейных полимеров винной кислоты. В таких случаях полимеризацию производного винной кислоты, содержащего две концевые ненасыщенные группы, проводят путем радикальной полимеризации или путем полимеризации с применением дифункционального гидросилана или гидросилоксана.
Сетчатую полимеризацию производных винной кислоты, которые могут существовать в R,R- или S,S-форме, можно проводить реакцией радикальной полимеризации или реакцией полимеризации через гидросилирование. Первоначальная хиральность производных поддерживается при полимеризации. Радикальную полимеризацию можно проводить обычным способом. Применяют инициаторы, образующие свободные радикалы, например азосоединения и пероксиды. Реакцию проводят при повышенных температурах от 50 до 150oC. Время реакции от 1 до 24 час. Полимеризацию проводят в органическом растворителе, например толуоле, хлороформе или диоксане.
Полимеризацию через гидросилирование проводят с применением гидросиланов или гидросилоксанов. Пригодные гидросиланы и гидросилоксаны можно представить общей
формулой:
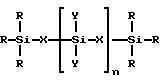
в которой
R представляет собой алкильную группу, имеющую 1-4 атома углерода, или H или их смесь, X представляет собой группу (CH2)m или O и Y представляет собой R или группу
-O-Si(R)3
и
n является целым числом от 0 до 3000, M является целым числом от 0 до 10. Полимеризация через гидросилирование известна, она описана например в J. Chromatogr. , 1992, 594, 283-290. Основную методику, указанную в этом описании, можно применять для получения хиральных адсорбентов настоящего изобретения. Реакцию можно проводить с применением в качестве катализатора комплекса металла, например комплекса платины или родия, при температуре от около 50 до 180oC, наиболее предпочтительно при температуре выше 100oC. Для полимеризации применяют растворители, которые инертны в условиях гидросилирования. Примерами таких растворителей являются толуол, диоксан, смеси толуола и диоксана, хлороформ, тетрагидрофуран и ксилол. Поскольку полимеризация через гидросилирование является относительно медленной реакцией, для ее проведения может требоваться период времени от 1 до 48 час.
Радикальную полимеризацию проводят в присутствии материала-носителя, наиболее эффективно она проходит, когда материалы-носители имеют поверхность указанного выше типа стирила, метакрилоила, метакриламида или акриламида и производные винной кислоты также содержат эти группы. Сетчатая полимеризация через гидросилирование, однако, предпочтительна. Такая полимеризация проходит очень эффективно со всеми указанными выше типами поверхностей. Гидросиланы можно включать не только для изменения содержания сомономеров в полимеризатах производных винной кислоты, но также для осуществления связывания с материалом-носителем. Сетчатую полимеризацию через гидросилирование можно проводить в присутствии материала-носителя или в его отсутствие. В последнем случае прикрепление к поверхности носителя проводят контактированием носителя и полимера друг с другом. Это можно осуществить добавлением материала-носителя непосредственно в раствор полимера. Свободные гидросилильные группы на сетчатом полимере затем связываются с модифицированной поверхностью носителя в присутствии катализатора при повышенных температурах, применяемых при полимеризации.
Можно применять от 1 до 30 мкмоль мономерного производного винной кислоты на 1 м2 поверхности носителя и от 1 до 30 мкмоль гидросилана на 1 м2 поверхности носителя. Такая высокая степень покрытия в мкмолях на м2 поверхности диоксида кремния, конечно, желательна. Настоящий способ позволяет достичь удовлетворительную степень покрытия по меньшей мере около 0,70 мкмоль/м2.
Настоящее изобретение относится также к оптически активному адсорбенту, который получают сетчатой полимеризацией через гидросилирование производных винной кислоты формулы 1 в присутствии гидросилана или гидросилоксана и материала-носителя, поверхность которого модифицирована таким образом, чтобы она имела одну концевую двойную связь или являлась гидросилильной группой, а также к адсорбенту, полученному сетчатой полимеризацией через гидросилирование производных винной кислоты формулы (1) в присутствии гидросилана или гидросилоксана с последующим добавлением в раствор полученного полимера материала-носителя, поверхность которого модифицирована таким образом, чтобы она содержала одну реакционноспособную функциональную группу, которая имеет концевую двойную связь или является гидросилильной группой.
Продукты, полученные как указано выше, т.е. материалы-носители, покрытые полимеризатом, фильтруют и промывают растворителем и сушат. Сушку можно проводить при 80-90oC, пригодно проводить ее в вакууме. Полученными таким образом хиральными адсорбентами наполняют хроматографические колонки под давлением по известной методике.
Хиральные адсорбенты настоящего изобретения при применении в хроматографии проявляют прекрасные свойства в отношении универсальности, энантиоселективности и емкости. Их можно применять для непосредственного энантиомерного разделения, они хорошо пригодны для применения в ЖХВР. Хиральные адсорбенты можно применять как для аналитических, так и для препаративных целей, они пригодны для разделения очень большого числа рацематов различного химического строения при очень хорошей селективности. Примерами различных типов рацемических фармацевтических веществ, которые можно разделить с применением настоящих хиральных адсорбентов, могут служить бензодиазепиноны, бензотиадиазины, дигидропиридины и лактамы.
Некоторые из производных винной кислоты, применяемых для получения хиральных адсорбентов, являются новыми
соединениями и изобретение предлагает также новые соединения, которые можно характеризовать формулой:
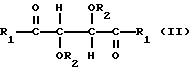
в которой
R1 представляет собой группу RNH-, RO-, RR'N- или HO- и R2 представляет собой группу RNHCO-, RCO-, ROCO-, H- или R-, где R является алифатическим углеводородным остатком, имеющим вплоть до 15 атомов углерода, арильной группой, аралкильной группой или полиароматической группой и R' является водородом или алкильной группой, имеющей вплоть до 7 атомов углерода, причем соединения содержат по меньшей мере две группы R1 или R2, имеющие алифатическую ненасыщенную связь, R1 не может быть фенилэтиламиногруппой, когда R2 является водородом. Для R1 и R2 R, R', пригодные и предпочтительные группы, соответствуют группам, которые были указаны ранее для соединений формулы I.
Особенно предпочтительны соединения формул
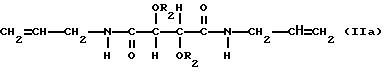
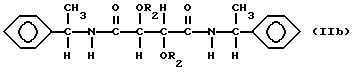
в которых
R2 представляет собой группу RNHCO-, RCO- или R-, где R имеет указанные выше значения. Для соединений формулы IIb R является, однако, алифатическим углеводородным остатком, имеющим вплоть до 15 атомов углерода и содержащим двойную связь. В остальном пригодные и предпочтительные соединения формул IIa и IIb соответствуют указанным выше соединениям формул Ia и Ib.
Новые соединения можно получить общими способами, которые описаны выше и будут описаны ниже более подробно.
Изобретение будет описано более полно в следующих ниже, не ограничивающих изобретение примерах. В примерах части и проценты обозначают массовые части и массовые проценты соответственно, если не оговорено особо.
Пример 1.
В этом примере описано получение хиральных производных винной кислоты.
1a) получение (+)-N,N'-бис-(α --фенилэтил)диамина-L-винной кислоты.
(+)-Диметил-L-тартрат (20,0 г, 0,112 моль) растворяли в метаноле (200 мл), после чего добавляли D-(+)-альфа-фенилэтиламин (135 мл, 1,058 моль). Раствор кипятили с обратным холодильников в течение 3 дней. Раствор в метаноле выпаривали досуха в вакууме. Остаток растворяли в хлористом метилене (2 л). Фазу хлористого метилена экстрагировали HCl (10%, 3 x 400 мл), раствором NaHCO3 (5%, 2 x 200 мл) и водой (1 x 200 мл). Фазу хлористого метилена сушили над Na2SO4 (безводным), после чего раствор выпаривали досуха в вакууме. Остаток перекристаллизовывали два раза в ацетонитриле (2 x 200 мл), после чего получали белые кристаллы (20,9 г, выход 52%).
Продукт анализировали и получали следующие результаты: чистоту по ЖХВР (220 нм) выше 99%, температуру плавления 131 - 132oC, [α] + 16,0o (MeOH, c = 1,05).
1H ЯМР-спектр (60 МГц, DMSO-D6):δ 1,40 (д, 6H), 4,27 (д, 2H), 4,99 (м, 2H), 5,64 (д, 2H), 7,31 (м, 10H), 7,92 (д, 2H).
1b) Получение (+(-N, N'-бис(α-фенилэтил)диамида O,O'-диметакрилоил-L-винной кислоты.
(+)-N, N'-Бис(α-фенилэтил)диамид-L-винной кислоты (14,0 г, 39,3 ммоль) растворяли в диоксане (280 мл) при комнатной температуре. Затем добавляли метакриловый ангидрид (12,9 мл, 86,5 ммоль) и 4-диметиламинопиридин (10,6 г, 86,5 ммоль). Раствор перемешивали при комнатной температуре 4 часа. Раствор диоксана выпаривали досуха при 30oC в вакууме. Остаток растворяли в хлористом метилене (350 мл). Фазу хлористого метилена экстрагировали HCl (10%, 3 x 200 мл), раствором NaHCO3 (1 x 200 мл, 5%) и водой (1 x 200 мл). Фазу хлористого метилена сушили Na2SO4 (безводным) и затем выпаривали досуха при 30oC в вакууме. Получали 20,9 г продукта в виде масла. Это масло очищали препаративной жидкостной хроматографией: колонка 5 x 25 см с кромасилом ® -CI8, 16 мкм. После такой очистки получали белый кристаллический продукт (11,5 г, выход 60%).
Продукт анализировали и получили следующие результаты: чистоту по ЖХВР (220 нм) выше 99%, температуру плавления 129 - 130oC, [α] + 60,4o (MeOH, c = 1,0).
1H ЯМР-спектр (60 МГц, CDCl3) :δ 1,43 (д, 6H), 1,92 (с, 6H), 5,06 (м, 2H), 5,70 (м, 4H), 6,16 (с, 2H), 6,51 (6м, 2H), 7,24 (м, 10H).
1c) Получение (+)-N,N'-бис-(α-фенилэтил)диамида(O,
O'-ди-(аллилкарбамоил)-L-винной кислоты
(+)-N, N'-Бис-(α-фенилэтил)диамид-L-винной кислоты (10,0 г, 28,0 ммоль) растворяли в тетрагидрофуране (300 мл). Затем добавляли
4-диметиламинопиридин (7,9 г, 64,6 ммоль) и аллилизоцианат (11,4 мл, 129 ммоль). Раствор кипятили с обратным холодильником при перемешивании в течение 24 час. Продукт, который выпал в осадок в
тетрагидрофуране через 24 час. Продукт, который выпал в осадок в тетрагидрофуране через 24 час, отделяли фильтрованием и промывали тетрагидрофураном и петролейным эфиром (температура кипения 30
- 40oC). Получали 12,9 г белого кристаллического продукта. Продукт перекристаллизовывали из диметилформамида (30 мл), фильтровали и промывали тетрагидрофураном. После перекристаллизации
получали 11,0 г продукта (выход 75%).
Продукт анализировали и получили следующие результаты: чистоту по ЖХВР (220 нм) выше 99%, температуру плавления 225oC, [α] + 7,6o (DMSO, c = 1,02).
1H ЯМР-спектр (400 МГц, DMSO-D6 ):δ 1,38 (д, 6H), 3,61 (д, 4H), 4,93 (м, 2H), 5,04 (д, 2H), 5,13 (д, 2H), 5,51 (с, 2H), 5,75 (м, 2H), 7,23 (м, 10H), 7,37 (т, 2H), 8,03 (д, 2H).
1d) Получение N, N'-Диаллилдиамида-О,О'-ди-(3,5-динитробензоил)-L-винной кислоты.
N, N'-Диаллилдиамид-L-винной кислоты (14,6 г, 63,95 ммоль) растворяли в пиридине (50 мл). Затем при охлаждении льдом добавляли хлористый 3,5-динитробензоил (30,18 г, 130,9 ммоль). Раствор перемешивали 3 часа при комнатной температуре. В раствор в пиридине добавляли хлористый метилен (1,0 л) и затем фазу хлористого метилена экстрагировали HCl (10%, 3 x 300 мл), NaHCO3 (5%, 2 x 200 мл) и водой (1 x 200 мл). Фазу хлористого метилена сушили Na2SO4 (безводным) и выпаривали досуха. Получали желто-белый кристаллический остаток. Остаток перекристаллизовывали в диметилформамиде (70 мл) и получали белый кристаллический продукт (32,0 г, выход 81%).
Продукт анализировали и получили следующие результаты: чистоту по ЖХВР (220 нм) выше 99%, температуру плавления 232 - 233oC, [α]- 75o (DMSO-D6), c = 1,02).
1H ЯМР-спектр (60 МГц, DMSO-D6):δ 3,71 (м, 4H), 4,94 (м, 4H), 5,65 (м, 2H), 5,99 (с, 2H), 8,85 (д, 2H), 9,0 (м, 6H).
1e) Получение N, N'-диаллилдиамида O,O'-ди(R)-α--фенилэтил)карбамоил-L-винной кислоты.
N, N'-Диаллилдиамид-L-винной кислоты (4,6 г, 20 ммоль) растворяли в сухом тетрагидрофуране (100 мл) при перемешивании. Затем добавляли 4 капли триэтиламина и по каплям (+)-фенилэтилизоцианат (6,8 мл, 48 ммоль). После добавления всего количества изоцианата реакционную смесь кипятили с обратным холодильником в течение 36 час. Реакционный раствор выпаривали и остаток растворяли в хлористом метилене и экстрагировали разбавленной H2SO4, раствором NaHCO3 и H2O. Органическую фазу сушили MgSO4, выпаривали и остаток перекристаллизовывали из смеси диметилформамид/метанол. Получили продукт в виде белых игл с выходом 54%.
Продукт анализировали и получили следующие результаты: температуру плавления 268,6 - 269,7oC, [α] + 20o (DMSO, c = 1).
1H ЯМР-спектр (400 МГц, DMSO-D6) :δ 1,36 (д, 6H), 3,64 (м, 4H), 4,62 (м, 2H), 4,92 (д, 2H), 5,05 (д, 2H), 5,34 (с, 2H), 5,68 (м, 2H), 7,29 (м, 10H), 7,69 (д, 2H), 7,94 (м, 2H).
1f) Получение N,N'-диаллилдиамида O,O'-дибензоил-L-винной кислоты.
N,N'-Диаллилдиамид-L-винной кислоты (1 г) растворяли в пиридине (4 мл) и раствор перемешивали при около 5oC. Затем по каплям добавляли хлористый бензоил (1,26 г). Реакционную смесь после этого перемешивали около 1 часа при комнатной температуре, затем добавляли хлористый метилен (50 мл). Органическую фазу экстрагировали 1M H2SO4, водой, насыщенным раствором NaHCO3 и водой. Органическую фазу сушили над Na2SO4. Хлористый метилен выпаривали и остаток перекристаллизовывали из смеси ацетона и гексана.
Продукт анализировали и получили следующие результаты: температуру плавления 200 - 201oC, [α]- 120o ± 2o (ацетон, c = 0,5).
1H ЯМР-спектр (60 МГц, DMSO-D6) :δ 3,68 (4H, м), 4,92 (4H, м), 5,58 (2H, м), 5,84 (2H, с), 7,64 (6H, м), 8,08 (4H, м), 8,64 (2H, т).
1g) Получение N,N'-диаллилдиамида O, O'-дифенилкарбамоил-L-винной кислоты.
N, N'-Диаллилдиамид-L-винной кислоты (4,6 г, 20 ммоль) суспендировали в 150 мл сухого CHCl3. При перемешивании добавляли 4 капли триэтиламина. Смесь кипятили с обратным холодильником до полного растворения диамида. Затем в смесь добавляли по каплям фенилизоцианат (5,2 мл, 48 ммоль). Реакционную смесь кипятили с обратным холодильником при перемешивании в течение 12 час. Охлажденный раствор экстрагировали 50 мл 1M H2SO4, 50 мл насыщенного раствора NaHCO3 и 2 x 50 мл H2O. Органическую фазу сушили MgSO4, выпаривали и остаток перекристаллизовали из смеси тетрагидрофурана и метанола. Продукт получали в виде белых игл с выходом 82%.
Продукт
анализировали и получили следующие результаты:
температуру плавления 252,2-255oC, [α]- 83,4o (DMSO, c = 0,5), [α]- 60,8o (c = 1,0 в
ТГФ).
1H ЯМР-спектр (60 МГц, DMSO-D6):δ 3,72 (4H, м), 5,04 (4H, м), 5,62 (2H, с), 5,76 (м, 2H), 6,92 (2H, м), 7,00 (2H, м), 7,28 (4H, м), 7,46 (4H, м), 8,30 (2H, т).
1h) Получение N,N'-диаллилдиамина O,O'-динафтилкарбамоил-L-винной кислоты.
N, N'-Диаллиилдиамид-L-винной кислоты (0,46 г, 2 ммоль) растворяли в 200 мл сухого тетрагидрофурана. Добавляли 1 каплю триэтиламина, затем добавляли по каплям 1-нафтилизоцианат (0,69 мл, 4,8 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 36 час. Получали плотный красно-белый осадок, который отделяли фильтрованием, промывали 50 мл метанола и перекристаллизовали из смеси диметилформамида и метанола. Получали продукт в виде белых игл с выходом 33%.
Продукт анализировали и получали следующие результаты:
[α]- 24o (DMSO, c = 1).
1H ЯМР-спектр (400 МГц, DMSO-D6):δ 3,82 (м, 4H), 5,03 (д, 2H), 5,21 (д, 2H), 5,65 (с, 2H), 5,82 (м, 2H), 7,54 (м, 8H), 7,77 (м, 2H), 7,92 (м, 2H), 8,07 (м, 2H), 8,36 (т, 2H), 8,63 (м, 2H).
Пример 2.
Этот пример иллюстрирует модификацию поверхности исходного материала-носителя для введения функциональных групп.
1. Модификация поверхности для введения функциональной группы, содержащей концевую двойную связь.
10 г КромасилаR, представляющего собой диоксид кремния и полученного Eka Nobel AB, Швеция, и имеющего средний размер частиц 5 мкм, удельную площадь поверхности 256 м2/г и средний диаметр пор 150
Диметилвинилхлорсилан,
Тривинилхлорсилан,
М,п-стирилэтилдиметилхлорсилан,
6-Гекс-1-енилдиметилхлорсилан,
7-Окт-1-енилдиметилхлорсилан,
3-Метакрилоксипропилдиметилхлорсилан.
Применяли также другой способ введения на поверхность виниловых групп. Для модификации такого же, как описанный выше диоксид кремния, применяли винилсодержащий циклический тетрасилоксан. Диоксид кремния (10 г) перемешивали в 5 мг толуола. Затем добавляли тетравинилтетраметилциклотетрасилоксан (8,0 мкмоль/м2 SiO2) и трифторметансульфокислоту (10 мг, каталитическое количество). Раствор кипятили с обратным холодильником в атмосфере азота при перемешивании в течение 18 час. Раствор затем фильтровали и модифицированный диоксид кремния промывали хлористым метиленом, тетрагидрофураном и метанолом. Диоксид кремния с модифицированной полимерной виниловой поверхностью затем сушили при 80-90oC в течение 24 час.
II. Модификация поверхности для введения гидросилильной группы.
IIa) 5 г диоксида кремния (кромасил ® ), поверхность которого была модифицирована винилдиметилхлорсиланом, суспендировали в 25 мл хлороформа, после чего добавляли раствор H2PtCl6 (0,15 мл, концентрация 55 мг/мл изопропанола) и затем 1,1,3,3-тетраметилдисилоксан (8,0 мкмоль/м2 SiO2). Раствор кипятили с обратным холодильником в атмосфере азота в течение 18 час. Дериватизированный диоксид кремния промывали и затем сушили, как указано выше. Этим способом достигали степень покрытия по гидросилану 1,72 мкмоль/м2 SiO2. δC: 2,0%.
IIb) Модификацию поверхности проводили таким же образом, как в IIa, но отличие был в том, что вместо хлороформа применяли толуол и силановым реагентом был 1,1,4,4-тетраметилдисилилэтилен. Степень покрытия по гидросилану была 1,64 мкмоль/м2 и δC: 2,35%.
IIc) В этой методике проведения модификации в качестве основного материала применяли немодифицированный кромасил ® . 5,0 г диоксида кремния диспергировали в 25 мл толуола. Затем добавляли 1,3,5,7-тетраметилциклотетрасилоксан (8,0 мкмоль/м2 SiO2, 2,50 мл) и трифторметансульфокислоту (10 мг). Раствор кипятили с обратным холодильником в атмосфере азота в течение 18 час. Степень покрытия была 8,80 мкмоль/м2 SiO2, δC: 2,35%.
Пример .3
Следующий пример иллюстрирует полимеризацию через гидросилилирование производных винной кислоты на носителях, в качестве которых применяют диоксид кремния. Во всех случаях применяемым диоксидом
кремния был кромасил ®.
a) 5,0 г диоксида кремния, модифицированного винилом, суспендировали в 30 мл смеси (1:1) толуола и диоксана, после чего добавляли раствор H2PtCl6 (0,10 мл, концентрация 60 мг/мл изопропанола), затем полиметилгидросилоксан (Mw 360-420, 2,8 мл). Раствор кипятили с обратным холодильником в атмосфере азота в течение 2 час. Затем добавляли N,
N'-диаллилдиамид-O-O'-дибензоил-L-винной кислоты (10 ммоля). Раствор опять кипятили с обратным холодильником в атмосфере азота еще 18 час. Обработанный таким образом диоксид кремния фильтровали и
промывали диоксаном, ацетонитрилом и тетрагидрофураном. Диоксид кремния затем сушили при 90oC в вакууме 24 час.
Элементный анализ дал следующие результаты (в мас.%): C 16, 15%, (δC: 11,5%), N 0,38% )0,56 мкмоль/м2 по отношению к диаллилдиамиду дибензоилвинной кислоты).
b) N, N'-Диаллилдиамид-O-O'-ди-(1-нафтоил)-L-винной кислоты (8,9 ммоля, 4,79 г) растворяли в смеси толуол/диоксан (1:1, 45 мл), после чего добавляли раствор H2PtCl6 (0,15 мл, концентрация 55 мг/мл изопропанола), а также тетра(диметилсилокси)силан (6,7 ммоля, 2,50 мл). Раствор кипятили с обратным холодильником в атмосфере азота в течение 24 час. Затем в раствор добавляли 5,0 г материала-носителя (кромасил ®, модифицированный винилом). Реакционную смесь далее кипятили с обратным холодильником в атмосфере азота еще 24 час. Продукт фильтровали и промывали тетрагидрофураном, толуолом и дихлорметаном и сушили при 90oC в вакууме 24 часа. Анализ на углерод и азот дал их содержание 9,1 и 0,30 соответственно в мас.%, что соответствует 0,44 мкмоль/м2 SiO2.
c) 5,0 г Материала-носителя (кромасил ®, модифицированный винилом) суспендировали в 45 мл тетрагидрофурана, затем добавляли раствор H2PtCl6 (0,15 мл, концентрация 55 мг/мл изопропанола), тетра(диметилсилокси)силан (7,5 ммоля, 2,8 мл) и N,N'-диаллилдиамид-O,O'-дифенилкарбамоил-L-винной кислоты (10,25 мкмоль, 4,8 г). Раствор помещали в автоклав. Реакционную смесь выдерживали при 125oC в течение 18 час в атмосфере азота. Продукт фильтровали и промывали диметилформамидом и тетрагидрофураном. Анализ на углерод и азот дал 12,1 и 0,95 мас.% соответственно, что соответствует 0,72 мкмоль/м2 SiO2.
d) N,N'-Диаллилдиамид-O,O'-дибензоил-L-винной кислоты (10,0 ммоль, 4,36 г) растворяли в смеси толуол/диоксан (1:1, 30 мл), после чего добавляли раствор H2PtCl6 (0,15 мл, концентрация 55 мг/мл изопропанола), затем тетра(диметилсилокси)силан (7,5 ммоля, 2,8 мл). Раствор кипятили с обратным холодильником в атмосфере азота в течение 24 час. Затем в раствор добавляли 5,0 г материала-носителя (кромасил ®, модифицированный винилом) и реакционную смесь кипятили с обратным холодильником в атмосфере азота еще 24 час. Продукт фильтровали и промывали тетрагидрофураном, толуолом и дихлорметаном и сушили 24 часа при 90oC в вакууме. Анализ на углерод и азот дал содержание их 11,85 мас.% и 0,50 мас.% соответственно, что соответствует 0,76 мкмоль/м2 SiO2.
Пример 4.
Этот пример иллюстрирует хроматографию с применением хиральной стационарной фазы в соответствии с изобретением.
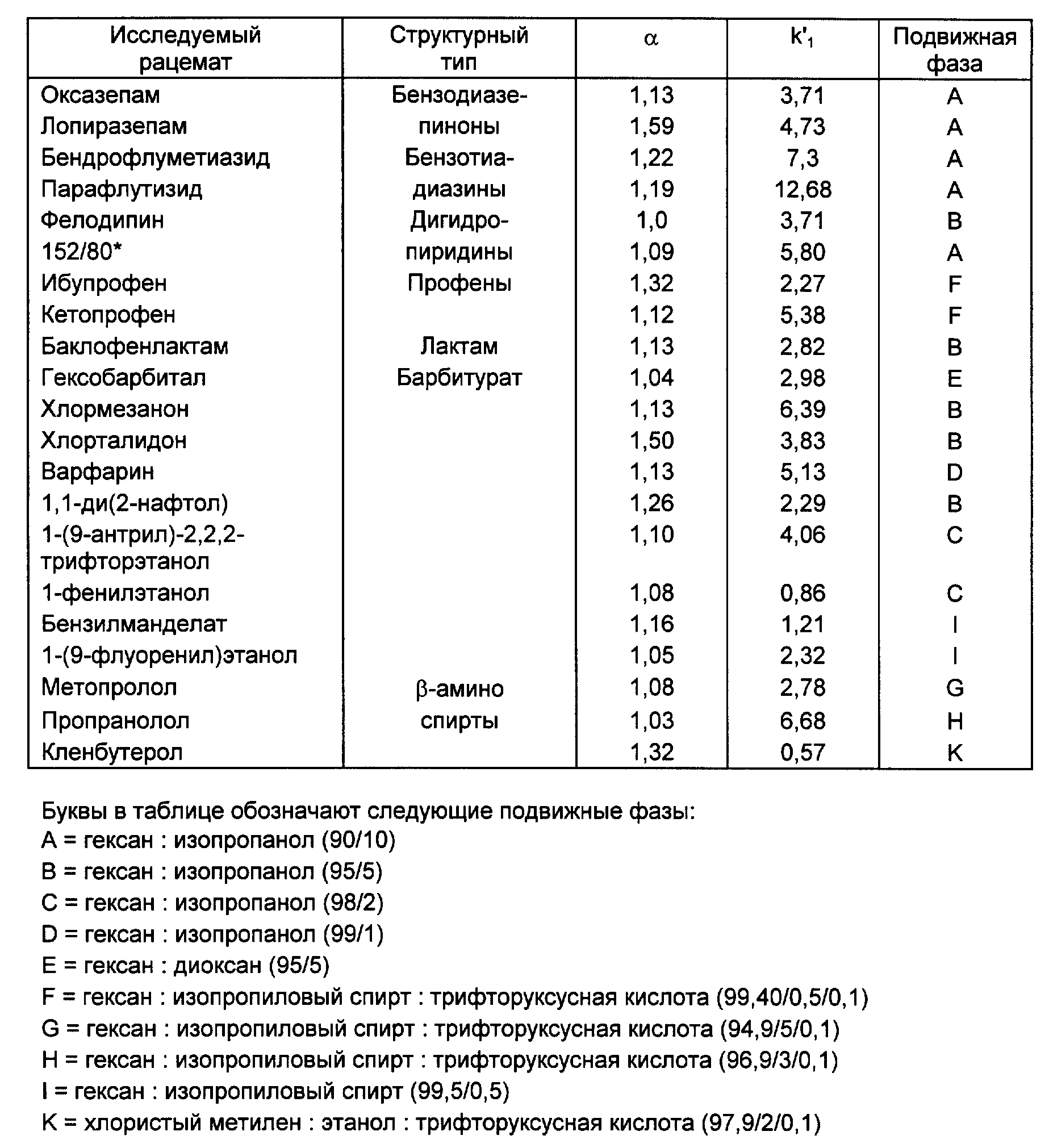
Диоксидом кремния с сетчатым полимеризованным производным винной кислоты (соответственно примеру 3d) наполняли (обычным способом наполнения взвесью) стальную (нержавеющую) колонку для ЖХВР (4,6х250 мм). Исследовали энантиоселективность ряда рацематов. Обследуемые рацематы были фармацевтическими веществами, которые приводятся в следующей таблице под их зарегистрированными товарными знаками с указанием структурного типа или химического или родового названия. Энантиоселективность (в таблице она представлена буквой α) обозначает соотношение между коэффициентом емкости энантиомеров.
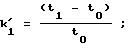
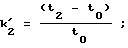
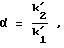
где
t1 и t2 = время удерживания энантиомеров, элюированных первым и последним соответственно, t0 = время удерживания незадержанного соединения,
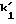
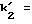
Указанные соотношения смесей приведены в объемных процентах.
Из приведенных в таблице результатов видно, что хиральные стационарные фазы, которые основаны на сетчатых полимерах производных винной кислоты, обладают общей энантиоселективностью для большинства типов фармацевтических веществ.
Реферат
Предложены оптически активные адсорбенты на основе сетчатых полимеризованных производных дикарбоновых кислот, диаминов или диолов, которые химически связаны с носителем. Производные этих соединений можно полимеризовать радикальной полимеризацией или через гидросилирование в присутствии твердого носителя. Оптически активные адсорбенты эффективны для хроматографического разделения рацемических смесей энантиомеров. 9 с. и 13 з.п. ф-лы.
Формула
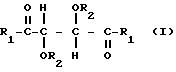
где R1 представляют собой группу RNH-, RO-, RR'N- или HO-,
R2 представляют собой группу RNHCO-, RCO-, ROCO, R- или H-, где R является алифатическим углеводородным остатком, имеющим вплоть до 15 атомов углерода, арильной группой, аралкильной группой, нафтильной группой или антрильной группы и R' является водородом или алкильной группой, имеющей вплоть до 7 атомов углерода, причем эти производные содержит по меньшей мере две группы R1 или R2, содержащие алифатическую ненасыщенную связь,
и сетчатые полимеризованные производные винной кислоты ковалентно соединены с поверхностью твердого материала-носителя.
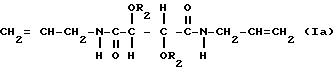
где R2 представляет собой группу RNHCO-, RCO- или H и R является алифатическим углеводородным остатком, содержащим вплоть до 15 атомов углерода, арильной группой, аралкильной группой, нафтильной группой или антрильной группой.
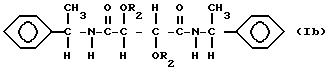
где R2 представляет собой группу RNHCO- или RCO-, где R является алифатическим углеводородным остатком, имеющим вплоть до 15 атомов углерода и содержащим алифатическую двойную связь.
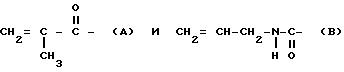
8. Адсорбент по любому из пп.1 - 7, отличающийся тем, что твердым материалом-носителем является диоксид кремния.
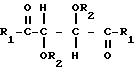
где R1 представляет собой группу RNH-, RO-, RR'N- или HO-;
R2 представляет собой группу RNHCO-, RCO-, ROCO-, R- или H-, причем R представляет собой алифатический углеводородный остаток, содержащий до 15 атомов углерода, арильную группу, аралкильную группу, нафтильную группу или антрильную группу, а R' представляет собой водород или алкильную группу, содержащую до 7 атомов углерода, причем указанные производные содержат по меньшей мере две группы R1 или R2, содержащие алифатическую ненасыщенную связь,
подвергают сетевой полимеризации в присутствии твердого материала носителя.
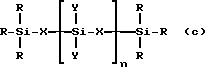
где R представляет собой алкильную группу, имеющую от 1 до 4 атомов углерода, или H, или их смесь;
X представляет собой группу (CH2)m или O;
Y представляет собой R или группу -O-Si(R)3;
n является целым числом от 0 до 3000;
m является целым числом от 1 до 10.
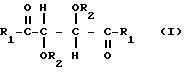
где R1 представляет собой группу RNH-, RO-, или RR'N или HO-;
R2 представляет собой группу RNHCO-, RCO-, ROCO-, R- или H-, причем R представляет собой алифатический углеводородный остаток, содержащий до 15 атомов углерода, арильную группу, аралкильную группу, нафтильную группу или антрильную группу, а R' представляет собой водород или алкильную группу, содержащую до 7 атомов углерода, причем указанные производные содержат по меньшей мере две группы R1 или R2, содержащие алифатическую ненасыщенную связь,
подвергают сетевой полимеризации через гидроксилирование в присутствии гидросилана и гидросилоксана общей формулы
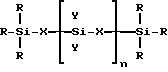
где R представляет собой алкильную группу, содержащую от 1 до 4 атомов углерода или H, или их смесь;
X представляет собой (CH2)m или o;
Y представляет собой R или группу -O-Si(R)3;
n представляет собой целое число от 0 до 3000;
m представляет собой целое число от 1 до 10,
и полученный сетевой полимер после этого закрепляют на поверхности твердого материала носителя в присутствии катализатора и при температуре полимеризации.
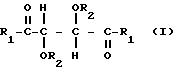
где R1 представляет собой группу RNH-, RO-, RR'N или HO-;
R2 представляет собой группу RNHCO-, RCO-, ROCO-, R- или H-, причем R представляет собой алифатический углеводородный остаток, содержащий до 15 атомов углерода, арильную группу, аралкильную группу, нафтильную группу или антрильную группу, а R' представляет собой водород или алкильную группу, содержащую от 7 атомов углерода, причем указанные производные содержат по меньшей мере две группы R1 или R2, содержащие алифатическую ненасыщенную связь,
в присутствии твердого материала носителя.
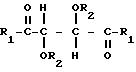
где R1 представляет собой группу RNH-, RO-, RR'N- или HO-;
R2 представляет собой группу RNHCO-, RCO-, ROCO-, R- или H-, причем R представляет собой алифатический углеводородный остаток, содержащий до 15 атомов углерода, арильную группу, аралкильную группу, нафтильную группу или антрильную группу, а R' представляет собой водород или алкильную группу, содержащую до 7 атомов углерода, причем указанные производные содержат по меньшей мере две группы R1 или R2, содержащие алифатическую ненасыщенную связь,
в присутствии гидросилана или гидросилоксана общей формулы
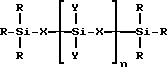
где R представляет собой алкильную группу, содержащую от 1 до 4 атомов углерода, или H, или их смесь;
X представляет собой (CH2)m или O;
Y представляет собой R или группу -O-Si-(R)3;
n представляет собой целое число от 0 до 3000;
m представляет собой целое число от 1 до 10,
с последующим закреплением полученного сетевого полимера на поверхности твердого материала носителя в присутствии катализатора и при температуре полимеризации.
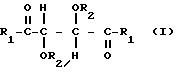
где R1 представляет собой группу RNH-, RO-, RR'N- или HO-;
R2 представляет собой группу RNHCO-, RCO-, ROCO-, R- или H-, причем R представляет собой алифатический углеводородный остаток, содержащий до 15 атомов углерода, арильную группу, аралкильную группу, нафтильную группу или антрильную группу, а R' представляет собой водород или алкильную группу, содержащую до 7 атомов углерода, причем указанные производные содержат по меньшей мере две группы R1 или R2, содержащие алифатическую ненасыщенную связь,
в присутствии гидросилана или гидросилоксана общей формулы:
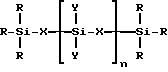
где R представляет собой алкильную группу, содержащую от 1 до 4 атомов углерода, или H, или их смесь;
X представляет собой (CH2)m или O;
Y представляет собой R или группу -O-Si-(R)3;
n представляет собой целое число от 0 до 3000;
n представляет собой целое число от 1 до 10.
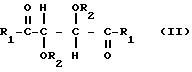
где R1 представляет собой группу RNH-, RO-, RR'N или HO-;
R2 представляет собой группу RNHCO-, RCO-, ROCO-, R- или H-, где R является алифатическим углеводородным остатком, имеющим вплоть до 15 атомов углерода, арильной группой, аралкильной группой, нафтильной группой или антрильной группой и R' является водородом или алкильной группой, имеющей вплоть до 7 атомов углерода, причем эти производные содержат по меньшей мере две группы R1 или R2, имеющие алифатическую ненасыщенную связь, однако R1 не может быть фенилэтиламиногруппой, если R2 является водородом.
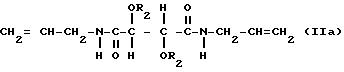
где R2 представляет собой группу RNHCO-, или RCO-, где R является алифатическим углеводородным остатком, имеющим вплоть до 15 атомов углерода, арильной группой, аралкильной группой, нафтильной группой или антрильной группой.
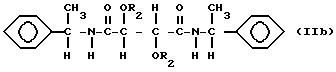
где R2 представляет собой группу RNHCO-, или RCO-, где R является алифатическим углеводородным остатком, имеющим вплоть до 15 атомов углерода и содержащим алифатическую двойную связь.
Комментарии