Порошкообразный гидрофобный полимер и колонка длявысокоэффективной жидкостной хроматографии с обращенной фазой - RU2296137C2
Код документа: RU2296137C2
Описание
Перекрестная ссылка на родственные заявки
По настоящей заявке в соответствии с положениями статьи 111(а) 35-го раздела Кодекса законов США испрашивается приоритет по заявке на патент США №60/328794 от 15 октября 2001 г. согласно положениям статьи 111(b) 35-го раздела Кодекса законов США и согласно положениям статьи 119(е) (1) раздела 35 Кодекса законов США.
Область техники, к которой относится изобретение
Настоящее изобретение относится к порошкообразному гидрофобному полимеру, обладающему превосходной кислото/щелочестойкостью, который является оптимальным наполнительным материалом для жидкостной хроматографии с обращенной фазой; к способу его получения; к колонке для жидкостной хроматографии с обращенной фазой, заполненной этим порошкообразным полимером; и к способу анализа с использованием такой колонки.
Предпосылки создания изобретения
В качестве одной из разделительных систем в жидкостной хроматографии широко известна жидкостная хроматография с обращенной фазой, где разделение осуществляют, основываясь на разнице сил удерживания благодаря разнице в гидрофобности у наполнительного материала и вещества, которое необходимо разделить.
Обычно в качестве наполнительного материала для жидкостной хроматографии с обращенной фазой широко применяют наполнительный материал, получаемый введением октадецильной группы в силанольную группу силикагеля с использованием силанового связующего агента, обладающего октадецильной группой (этот наполнительный материал в дальнейшем в настоящем описании обозначен как "ОДС"). Это обусловлено тем, что относительно легко может быть получена тонкодисперсная частица, обладающая высокой механической прочностью.
Однако силикагель обладает низкой щелочестойкостью и, следовательно, круг пригодных для использования элюентов ограничен элюентом, значение рН которого составляет от 2 до 8. Более того, поскольку в действительности химически связать связующий агент со всеми силанольными группами невозможно, возникает серьезная проблема, состоящая в том, что непрореагировавшая силанольная группа легко адсорбирует основное соединение, такое как амин.
В последние годы с целью улучшить работоспособность в щелочных условиях созданы различные наполнительные материалы полимерного типа для жидкостной хроматографии с обращенной фазой, и некоторые из них технически доступны в виде колонки для жидкостной хроматографии с обращенной фазой. Их примеры включают следующие материалы:
(1) стирол-дивинилбензольные сополимерные частицы ((см., Analytical Chemistry, Vol.45, с.1383 (1973), технически доступный продукт Shodex (зарегистрированный товарный знак фирмы Showa Denko K.K.) RSpak RP18-413, выпускаемый фирмой Showa Denko K.K.);
(2) частицы из сетчатого полимера метакрилатного типа ((технически доступный продукт Shodex (зарегистрированный товарный знак фирмы Showa Denko K.K.) RSpak DE-413, выпускаемый фирмой Showa Denko K.K.);
(3) частицы из сетчатого полимера типа поливинилового спирта, обладающего химически связанной с ним длинноцепочечной ацильной группой ((технически доступный продукт Shodex (зарегистрированный товарный знак фирмы Showa Denko K.K.), Asahipak (зарегистрированный товарный знак фирмы Showa Denko K.K.) ODP-40 4D, выпускаемый фирмой Showa Denko K.K.);
(4) частицы из материала типа сополимера эфира метакриловой кислоты, обладающего длинноцепочечной алкильной группой (см., JP-A 2000-9707 (использованное в настоящем описании понятие "JP-A" означает "еще не прошедшая экспертизу опубликованная заявка на патент Японии"));
(5) частицы из сополимера глицидилметакрилата и эфира (мет)акриловой кислоты и многоатомного спирта, с которым химически связана длинноцепочечная алкильная группа (см., JP-A 61-272654); и
(6) частицы из сетчатого полимера метакрилатного типа, содержащего гидроксильную группу, обладающего химически связанной с ним длинноцепочечной ацильной группой (см., JP-A 4-58154).
Колонки для жидкостной хроматографии с обращенной фазой, заполненные этими частицами полимерного типа, создают некоторые проблемы, которыми нельзя пренебречь, несмотря на преимущество такой колонки, состоящее в том, что эффективный интервал значений рН более широк, чем эффективный интервал значений колонки с ОДС.
Однако колонки, заполненные порошкообразными полимерами с (1) по (5), неприемлемы для разделения и анализа природных продуктов или медицинских препаратов, молекулы которых обладают полициклическим ароматическим участком, поскольку хроматограмма полициклического ароматического соединения характеризуется широкими пиками.
В колонке, заполненной порошкообразным полимером (1), сам порошкообразный полимер характеризуется плохим набуханием или усадкой, зависящей от растворителя, и осуществление удовлетворительного разделения и анализа варьированием элюента в колонке сопряжено с затруднениями технологического порядка.
Колонки, заполненные порошкообразными полимерами со (2) по (6), создают проблему, состоящую в том, что когда колонку в течение длительного периода времени используют при рН 2 или ниже или при рН 11 или выше, то поскольку сложноэфирная связь, содержащаяся в их структуре, обладает низкой стойкостью к кислоте/щелочи, эффективность колонки серьезно понижается, форма пика основного вещества, такого как амин, меняется в худшую сторону, и осуществление непрерывного определения становится невозможным.
В качестве метода устранения такой проблемы было бы эффективным получение простой эфирной связи, которая обладает высокой стойкостью к кислоте/щелочи, реакцией эпоксисоединения с порошкообразным полимером, имеющим гидроксильную группу, при введении функциональной группы. В качестве примеров такого метода известны (А) метод проведения реакции в воде, содержащей основание, такое как гидроксид натрия, или в полярном органическом растворителе, таком как диметилформамид, и (Б) метод проведения реакции в растворителе типа простого эфира, такого как диоксан, в присутствии кислоты Льюиса, такой как комплекс трифторида бора и диэтилового эфира (см., например, заявку JP-A 61-272654, упомянутую выше в (5)). Однако осуществление этих методов сопряжено с проблемой, состоящей в том, что реакция занимает длительное время, а поскольку эпоксисоединение используют в избыточном количестве, последующая обработка, такая как фильтрование, оказывается очень обременительной. Более того, если субстратный гель представляет собой гель типа сложного полиэфира, как в случае, в котором используют вышеупомянутый материал (5), то когда проводят реакцию по методу (А) или (Б), гидролизом разрывают сложноэфирную связь самого порошкообразного полимера, и образуется карбоксильная группа, поскольку субстратный гель в течение длительного периода времени подвергается воздействию кислоты или щелочи. В случае наличия карбоксильной группы порошкообразный полимер адсорбирует основное вещество, такое как амин, и следовательно, для применения в качестве наполнительного материала для жидкостной хроматографии с обращенной фазой не подходит.
Наполнительный материал, применение которого позволяет устранить вышеописанные проблемы, и способ его получения неизвестны, поэтому их поиск является насущной потребностью.
Описание изобретения
Объектом настоящего изобретения является создание согласно новому способу получения наполнительного материала для жидкостной хроматографии с обращенной фазой, который обладает необычайно высокой кислото/щелочестойкостью, уменьшенной степенью набухания и усадки собственно порошкообразного полимера, способен сохранять эффективность колонки при применении различных растворителей, даже когда в колонке самым разнообразным путем меняют элюент, и обеспечивает получение хроматограммы с острыми пиками для полициклических ароматических соединений.
В результате широких исследований с целью разрешить вышеописанные проблемы на пути создания настоящего изобретения впервые было установлено, что (1) когда колонку, используемую для жидкостной хроматографии с обращенной фазой, заполняют порошкообразным полимером, полученным реакцией (А) порошкообразного сетчатого полимера, имеющего гидроксильную группу, с (Б) способным сшивать эпоксисоединением, гидролизом оксирановых колец после покрытия субстрата и затем его взаимодействия с (В) эпоксисоединением, содержащим в общей сложности от 6 до 40 углеродных атомов, могут быть достигнуты необычайно высокая устойчивость к воздействию кислоты и щелочи и к замене растворителей и может быть достигнута хроматограмма для полициклических ароматических соединений с острыми пиками, и что (2) когда реакцию способного сшивать эпоксисоединения (Б) и эпоксисоединения (В), содержащего в общей сложности от 6 до 40 углеродных атомов, проводят в присутствии кислоты Льюиса как катализатора в низкополярном растворителе, эта реакция протекает очень быстро. Эти открытия дают возможность добиться вышеописанной цели.
Более конкретно признаками настоящего изобретения являются следующие объекты.
(1) Порошкообразный гидрофобный полимер, полученный реакцией (А) порошкообразного сетчатого полимера, имеющего гидроксильную группу, с (Б) способным сшивать эпоксисоединением, гидролизом оксирановых колец, а затем реакцией полученного соединения, содержащего гидроксильную группу, с (В) эпоксисоединением, содержащим в общей сложности от 6 до 40 углеродных атомов.
(2) Порошкообразный гидрофобный полимер, как он представлен выше в разделе (1), где этот порошкообразный сетчатый полимер (А), содержащий гидроксильную группу, представляет собой сополимер двух или большего числа представителей, выбранных из группы, включающей (I) поли(мет)акрилат многоатомного спирта, в молекуле которого содержится по меньшей мере одна гидроксильная группа, (II) глицидил(мет)акрилат и (III) поли(мет)акрилат многоатомного спирта, в молекуле которого гидроксильная группа не содержится, или гомополимер поли(мет)акрилата многоатомного спирта (I).
(3) Порошкообразный гидрофобный полимер, как он представлен выше в разделе (2), где поли(мет)акрилат многоатомного спирта (I), в молекуле которого содержится по меньшей мере одна гидроксильная группа, представляет собой диметакрилат глицерина, глицидил(мет)акрилат (II) представляет собой глицидилметакрилат, а поли(мет)акрилат многоатомного спирта (III), в молекуле которого гидроксильная группа не содержится, представляет собой алкиленгликольдиметакрилат.
(4) Порошкообразный гидрофобный полимер, как он представлен выше в разделе (3), где алкиленгликольдиметакрилат представляет собой этиленгликольдиметакрилат.
(5) Порошкообразный гидрофобный полимер, как он представлен выше в разделе (1), где способное сшивать эпоксисоединение (Б) представляет собой соединение, выбранное из эпигалогидрина и эпоксисоединений, содержащих два или большее число оксирановых колец.
(6) Порошкообразный гидрофобный полимер, как он представлен выше в разделе (5), где эпоксисоединение, содержащее два или большее число оксирановых колец, представляет собой этиленгликольдиглицидиловый эфир, пропиленгликольдиглицидиловый эфир, бутиленгликольдиглицидиловый эфир, триметилолпропандиглицидиловый эфир, триметилолпропантриглицидиловый эфир, пентаэритриттриглицидиловый эфир или триглицидилизоцианурат.
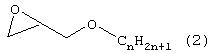
(7) Порошкообразный гидрофобный полимер, как он представлен выше в разделе (1), где эпоксисоединение (В), содержащее в общей сложности от 6 до 40 углеродных атомов, представляет собой соединение, выбранное из группы, включающей соединения, отвечающие следующим формулам с (1) по (4):

(в которой n обозначает целое число от 4 до 38);
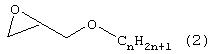
(в которой n обозначает целое число от 3 до 37);

(в которой n обозначает целое число от 0 до 32);

(в которой n обозначает целое число от 0 до 31).
(8) Порошкообразный гидрофобный полимер, как он представлен выше в разделе (7), где эпоксисоединение (В), содержащее в общей сложности от 6 до 40 углеродных атомов, представляет собой стеарилглицидиловый эфир.
(9) Порошкообразный гидрофобный полимер, как он представлен в любом одном из приведенных выше разделов с (1) по (8), средний размер частиц которого составляет от 1 до 2000 мкм.
(10) Способ получения порошкообразного гидрофобного полимера, включающий взаимодействие (А) порошкообразного сетчатого полимера, имеющего гидроксильную группу, с (Б) способным сшивать эпоксисоединением, гидролиз оксирановых колец и затем его взаимодействие с (В) эпоксисоединением, содержащим в общей сложности от 6 до 40 углеродных атомов.
(11) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (10), в котором этот порошкообразный сетчатый полимер (А), содержащий гидроксильную группу, представляет собой сополимер двух или большего числа представителей, выбранных из (I) поли(мет)акрилата многоатомного спирта, в молекуле которого содержится по меньшей мере одна гидроксильная группа, (II) глицидил(мет)акрилата и (III) поли(мет)акрилата многоатомного спирта, в молекуле которого гидроксильная группа не содержится, или гомополимер поли(мет)акрилата многоатомного спирта (I).
(12) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (10), в котором способное сшивать эпоксисоединение (Б) представляет собой соединение, выбранное из эпигалогидрина и эпоксисоединений, содержащих два или большее число оксирановых колец.
(13) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (11), в котором поли(мет)акрилат многоатомного спирта, в молекуле которого содержится по меньшей мере одна гидроксильная группа (I), представляет собой диметакрилат глицерина, глицидил(мет)акрилат (II) представляет собой глицидилметакрилат, а поли(мет)акрилат многоатомного спирта (III), в молекуле которого гидроксильная группа не содержится, представляет собой алкиленгликольдиметакрилат.
(14) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (13), в котором алкиленгликольдиметакрилат представляет собой этиленгликольдиметакрилат.
(15) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (12), в котором эпоксисоединение, содержащее два или большее число оксирановых колец, представляет собой этиленгликольдиглицидиловый эфир, пропиленгликольдиглицидиловый эфир, бутиленгликольдиглицидиловый эфир, триметилолпропандиглицидиловый эфир, триметилолпропантриглицидиловый эфир, пентаэритриттриглицидиловый эфир или триглицидилизоцианурат.
(16) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (10), в котором эпоксисоединение (В), содержащее в общей сложности от 6 до 40 углеродных атомов, представляет собой соединение, выбранное из соединений, отвечающих следующим формулам с (1) по (4), описанным выше в разделе (7).
(17) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (16), в котором эпоксисоединение (В), содержащее в общей сложности от 6 до 40 углеродных атомов, представляет собой стеарилглицидиловый эфир.
(18) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (10), в котором реакцию между порошкообразным сетчатым полимером (А), имеющим гидроксильную группу, и эпоксисоединением, содержащим два или большее число оксирановых колец, проводят в присутствии кислоты Льюиса в низкополярном растворителе.
(19) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (10), в котором реакцию с эпоксисоединением (В), содержащим в общей сложности от 6 до 40 углеродных атомов, проводят в присутствии кислоты Льюиса в низкополярном растворителе.
(20) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (18) или (19), в котором низкополярный растворитель представляет собой углеводород, содержащий в общей сложности от 5 до 10 углеродных атомов.
(21) Способ получения порошкообразного гидрофобного полимера, как он представлен выше в разделе (18) или (19), в котором концентрация кислоты Льюиса составляет от 1 до 70 мас.% в пересчете на порошкообразный сетчатый полимер.
(22) Способ получения порошкообразного гидрофобного полимера, как он представлен в любом одном из приведенных выше разделов с (10) по (21), в котором средний размер частиц порошкообразного гидрофобного полимера составляет от 1 до 2000 мкм.
(23) Колонка для жидкостной хроматографии с обращенной фазой, заполненная порошкообразным гидрофобным полимером, как он представлен в любом одном из приведенных выше разделов (1) по (9).
(24) Способ анализа образца, содержащего полициклическое ароматическое соединение, включающий применение колонки для жидкостной хроматографии с обращенной фазой так, как изложено в вышеприведенном разделе (23).
Подробное описание изобретения
Объектом настоящего изобретения являются порошкообразный гидрофобный полимер, обладающий превосходной кислото/щелочестойкостью, который является оптимальным в качестве наполнительного материала для жидкостной хроматографии с обращенной фазой; способ его получения; колонка для жидкостной хроматографии с обращенной фазой, заполненная этим порошкообразным полимером; и способ анализа с применением этой колонки.
Примеры поли(мет)акрилата (I) многоатомного спирта, в молекуле которого содержится по меньшей мере одна гидроксильная группа, пригодного для применения при выполнении настоящего изобретения, включают диакрилат глицерина, диметакрилат глицерина, пентаэритриттриакрилат, пентаэритритдиакрилат, пентаэритриттриметакрилат, пентаэритритдиметакрилат, триметилолпропандиакрилат и триметилолпропандиметакрилат. Среди них предпочтительны, учитывая легкодоступность и экономические выгоды, диакрилат глицерина и диметакрилат глицерина. Эти соединения можно использовать самостоятельно или в сочетании двух или большего их числа.
Предпочтительным глицидил(мет)акрилатом (II), учитывая химическую стойкость получаемого порошкообразного сетчатого полимера, является глицидилметакрилат.
Примеры поли(мет)акрилата (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, включают (мет)акрилат полиалкиленгликоля, такого как этиленгликольди(мет)акрилат, диэтиленгликольди(мет)акрилат, пропиленгликольди(мет)акрилат, полиэтиленгликольди(мет)акрилатиполипропиленгликольди(мет)акрилат; три(мет)акрилат глицерина и триметилолпропантри(мет)акрилат. Среди них предпочтителен, учитывая легкодоступность и экономические выгоды, этиленгликольдиметакрилат. Эти соединения можно использовать самостоятельно или в сочетании двух или большего их числа.
При выполнении настоящего изобретения для реакции порошкообразного сетчатого полимера, имеющего гидроксильную группу, с (Б) способным сшивать эпоксисоединением, порошкообразный гомополимер поли(мет)акрилата (I) многоатомного спирта, в молекуле которого содержится по меньшей мере одна гидроксильная группа, или порошкообразный сополимер поли(мет)акрилата (I) многоатомного спирта и поли(мет)акрилата (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, можно использовать, как это изложено в дальнейшем, для взаимодействия с эпоксисоединением. По другому варианту после сополимеризации поли(мет)акрилата (I) многоатомного спирта и глицидил(мет)акрилата (II), глицидил(мет)акрилата (II) и поли(мет)акрилата (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, или поли(мет)акрилата (I), глицидил(мет)акрилата (II) и поли(мет)акрилата (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, оксирановые кольца в сополимере можно гидролизовать, после чего, как это изложено в дальнейшем, можно осуществлять реакцию со способным сшивать эпоксисоединением.
При получении сополимера поли(мет)акрилата (I) многоатомного спирта и поли(мет)акрилата (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, реагент (I) в предпочтительном варианте используют в количестве 10 мас.% или больше, более предпочтительно 20 мас.% или больше. Если количество реагента (I) меньше 10 мас.%, порошкообразный полимер, получаемый в результате реакции с эпоксисоединением (В), содержащим в общей сложности от 6 до 40 углеродных атомов, обладает плохими разделительными эксплуатационными свойствами для применения в качестве наполнительного материала для жидкостной хроматографии с обращенной фазой, в частности пик полициклического ароматического соединения становится широким.
При получении сополимера поли(мет)акрилата (I) многоатомного спирта и глицидил(мет)акрилата (II) в предпочтительном варианте реагент (I) используют в количестве 30 мас.% или больше, более предпочтительно 50 мас.% или больше. Если количество реагента (I) меньше 30 мас.%, порошкообразный получаемый полимер обладает более низкой механической прочностью.
При получении сополимера глицидил(мет)акрилата (II) и поли(мет)акрилата (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, в предпочтительном варианте реагент (III) используют в интервале от 30 до 90 мас.%, более предпочтительно от 50 до 80 мас.%. Если количество реагента (III) меньше 30 мас.%, получаемый порошкообразный полимер обладает пониженной механической прочностью, тогда как если оно превышает 90 мас.%, порошкообразный полимер, получаемый в результате реакции с эпоксисоединением (В), содержащим в общей сложности от 6 до 40 углеродных атомов, обладает плохими разделительными эксплуатационными свойствами для применения в качестве наполнительного материала для жидкостной хроматографии с обращенной фазой, и пик полициклического ароматического соединения становится широким.
При получении сополимера поли(мет)акрилата (I) многоатомного спирта, глицидил(мет)акрилата (II) и поли(мет)акрилата (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, предпочтительное общее количество используемых реагентов (I) и (III) составляет 30 мас.% или больше, более предпочтительно 50 или больше. Если общее количество реагентов (I) и (III) составляет меньше 30 мас.%, получаемый порошкообразный полимер обладает пониженной механической прочностью. Одновременно с этим в предпочтительном варианте реагенты (I) и (II) используют в общем количестве 10 мас.% или больше, более предпочтительно 20 мас.% или больше. Если общее количество реагентов (I) и (II) составляет меньше 10 мас.%, порошкообразный полимер, получаемый в результате реакции с эпоксисоединением (В), содержащим в общей сложности от 6 до 40 углеродных атомов, обладает плохими разделительными эксплуатационными свойствами для применения в качестве наполнительного материала для жидкостной хроматографии с обращенной фазой, и пик полициклического ароматического соединения становится широким.
При выполнении настоящего изобретения поли(мет)акрилат (I) многоатомного спирта, в молекуле которого содержится по меньшей мере одна гидроксильная группа, глицидил(мет)акрилат (II) и поли(мет)акрилат (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, могут быть подвергнуты суспензионной полимеризации в воде в присутствии не смешивающегося с водой органического растворителя с получением порошкообразного сетчатого (со)полимера (в дальнейшем в настоящем описании каждый из (I), (II) и (III) упоминается как "мономер").
Список не смешивающихся с водой органических растворителей, пригодных для применения при выполнении настоящего изобретения, не особенно ограничен. Однако чем выше сродство к мономеру используемого органического растворителя, тем в большей степени уменьшен размер пор порошкообразного сетчатого (со)полимера и более высока физическая прочность. В связи с этим предпочтителен органический растворитель, главным образом включающий спирт, содержащий от 5 до 12 углеродных атомов, такой как изоамиловый спирт, 1-гексанол, 1-октанол, 2-этилгексанол, 1-деканол и 1-додеканол. Эти органические растворители можно использовать самостоятельно или в сочетании двух или большего их числа.
Органический растворитель можно использовать в интервале от 10 до 300 мас.% в пересчете на общее количество мономеров (I), (II) и (III), но в предпочтительном варианте используют в интервале от 25 до 100 мас.%, учитывая удельную площадь поверхности и физическую прочность порошкообразного сетчатого (со)полимера.
Примеры инициаторов полимеризации, приемлемых для применения при выполнении настоящего изобретения, включают органические пероксиды, такие как бензоилпероксид, дихлорбензоилпероксид, дикуменилпероксид и лауроилпероксид, и соединения типа азосоединений, такие как 2,2'-азобис(изобутиронитрил) и 2,2'-азобис(2,4-диметилвалеронитрил). Эти соединения можно использовать самостоятельно или в сочетании двух или большего их числа. Среди них предпочтительны, учитывая простоту в обращении, инициаторы полимеризации типа азосоединений.
Инициатор полимеризации используют в интервале от 0,1 до 4 мас.%, предпочтительно от 1 до 2 мас.%, в пересчете на общее количество мономеров. Если количество инициатора полимеризации меньше 0, 1 мас.%, для завершения полимеризации необходимо очень длительное время, тогда как если оно превышает 4 мас.%, полимеризация протекает с высокой скоростью, поэтому такой вариант с точки зрения безопасности не предпочтителен.
При выполнении настоящего изобретения в водную фазу можно добавлять диспергатор, такой как слаборастворимый фосфат или водорастворимое полимерное соединение. Примеры слаборастворимого фосфата включают фосфат кальция (трехосновный) и фосфат магния. Примеры водорастворимого полимерного соединения включают поливиниловый спирт, алкилцеллюлозу, гидроксиалкилцеллюлозу и карбоксиалкилцеллюлозу.
Диспергатором может служить слаборастворимый фосфат или водорастворимое полимерное соединение, однако предпочтительно водорастворимое полимерное соединение, поскольку это соединение может быть удалено промывкой в нейтральных условиях. В предпочтительном варианте водорастворимое полимерное соединение используют в интервале от 0,01 до 3 мас.% в пересчете на воду.
Кроме того, в водную фазу можно добавлять водорастворимую неорганическую соль с целью понизить растворимость в воде мономеров или не смешивающийся с водой органический растворитель. Примеры водорастворимой неорганической соли включают хлорид натрия, хлорид кальция и сульфат натрия. Эти водорастворимые неорганические соли можно использовать самостоятельно или в сочетании двух или большего их числа. Концентрация используемой соли не особенно ограниченна, но, например, хлорид натрия в предпочтительном варианте используют в интервале от 0,5 до 15 мас.% в пересчете на используемую воду.
Поли(мет)акрилат (I) многоатомного спирта, в молекуле которого содержится по меньшей мере одна гидроксильная группа, глицидил(мет)акрилат (II), поли(мет)акрилат (III) многоатомного спирта, в молекуле которого гидроксильная группа не содержится, и инициатор полимеризации предварительно смешивают и смесь добавляют и диспергируют в водном растворе, содержащем водорастворимую неорганическую соль. В этом случае в соответствии с целевым размером частиц можно применять диспергатор, такой как смеситель-гомогенизатор.
В предпочтительном варианте используемое количество воды превышает по массе от 1 до 50 раз общее количество мономеров и органического растворителя, однако, принимая во внимание стабильность дисперсии в растворе или время, необходимое для фильтрования на последней стадии, воду целесообразно использовать в количестве, которое превышает их по массе от 2 до 10 раз.
В случае, когда используют мономер (II), глицидильную группу получаемого порошкообразного сетчатого (со)полимера гидролизуют с использованием кислоты. Примеры кислотного катализатора включают серную кислоту, перхлорную кислоту, соляную кислоту, толуолсульфоновую кислоту и бензолсульфоновую кислоту. Среди них предпочтительна, учитывая простоту в обращении, соляная кислота.
Предпочтительная концентрация используемой кислоты составляет от 0,01 до 5 н., более предпочтительно от 0,05 до 2 н. Если концентрация ниже 0,01 н., реакция протекает с невысокой скоростью, тогда как если она превышает 5 н., вероятен процесс гидролиза сложноэфирной связи. Во время проведения этой реакции, если только в растворителе содержатся по меньшей мере 10 мас.% или больше воды, в сочетании можно использовать органический растворитель. Список используемых органических растворителей чем-либо конкретным не ограничен, лишь бы они смешивались как с кислотой, так и с водой и были неактивными в отношении кислоты и глицидильной группы. Их примеры включают ацетон, метилэтилкетон, 1,4-диоксан и тетрагидрофуран.
Количество используемого кислотного раствора может считаться достаточным, если его достаточно много для погружения в растворитель порошкообразного сетчатого полимера. Так, например, оно может быть равным или превышать массу геля. Реакционные условия не могут быть заданы неупорядоченно, однако в предпочтительном варианте эту реакцию осуществляют при температуре от 25 до 100°С в течение от 3 до 10 ч.
Получаемый таким образом порошкообразный сетчатый (со)полимер подвергают поверхностной сшивке с использованием способного сшивать эпоксисоединения (Б). Эта операция необходима с целью придать порошкообразному полимеру достаточно высокую механическую прочность и крайне высокую кислото/щелочестойкость.
Примеры способного сшивать эпоксисоединения (Б) охватывают соединения, выбранные из группы, включающей эпигалогидрин и эпоксисоединения, содержащие два или большее число оксирановых колец. Примеры эпигалогидринов включают эпихлоргидрин, эпибромгидрин и эпииодгидрин. Примеры эпоксисоединений, содержащих два или большее число оксирановых колец, включают этиленгликольдиглицидиловый эфир, пропиленгликольдиглицидиловый эфир, бутиленгликольдиглицидиловый эфир, триметилолпропандиглицидиловый эфир, триметилолпропантриглицидиловый эфир, пентаэритриттриглицидиловый эфир и триглицидилизоцианурат. Эти соединения можно использовать самостоятельно или в сочетании двух или большего их числа. Среди них предпочтительны, принимая во внимание баланс между достигаемой прочностью и экономическими выгодами, эпихлоргидрин или этиленгликольдиглицидиловый эфир.
В случае осуществления реакции поверхностной сшивки с использованием в качестве способного сшивать эпоксисоединения (Б) эпигалогидрина реакция протекает при одновременном высвобождении или удалении атома галогена и, следовательно, реакцию проводят в присутствии водного раствора щелочи. Примеры щелочи, которую обычно используют, включают гидроксид и карбонат щелочного металла, а также гидроксид и карбонат щелочноземельного металла. Эту реакцию можно также осуществлять в смешанном растворителе из воды и полярного органического растворителя, такого как диметилформамид, N-метилпирролидон и диметилсульфоксид.
В предпочтительном варианте количество используемого эпигалогидрина составляет от 5 до 200 мас.%, более предпочтительно от 50 до 150 мас.%, в пересчете на порошкообразный сетчатый полимер. Если используемое количество эпигалогидрина равно меньше 5 мас.%, реакция поверхностной сшивки протекает с низкой скоростью и занимает очень длительное время, тогда как если оно превышает 200 мас.%, очень обременительной оказывается постобработка после реакции.
Предпочтительная концентрация используемого водного раствора щелочи составляет от 5 до 40 мас.%, а с точки зрения простоты получения более предпочтительна от 10 до 30 мас.%. Количество водного раствора щелочи может считаться достаточным, если оно достаточно велико для погружения в растворитель порошкообразного полимера. Так, например, оно может превышать массу геля в 2 или большее число раз. Во время осуществления реакции добавлением полярного органического растворителя, такого как диметилформамид, N-метилпирролидон и диметилсульфоксид, в систему, в которую вводят водный раствор щелочи, в количестве, превышающем массу геля в два или большее число раз, добавляют полярный органический растворитель.
Количество добавляемого полярного органического растворителя чем-то конкретным не ограничено, но его можно добавлять в количестве, которое от 0,1 до 2 раз превышает количество водного раствора щелочи. В предпочтительном варианте эту реакцию осуществляют при температуре от 25 до 100°С в течение от 3 до 16 ч, более предпочтительно от 30 до 60°С в течение от 5 до 12 ч.
В случае использования в качестве способного сшивать эпоксисоединения (Б) такого эпоксисоединения, которое содержит два или большее число оксирановых колец, реакцию поверхностной сшивки можно осуществлять либо в щелочных условиях, либо в кислых условиях. Во время осуществления реакции поверхностной сшивки в щелочной среде реакционные условия могут быть совершенно такими же, как в случае использования эпигалогидрина в качестве способного сшиваться эпоксисоединения (Б).
С другой стороны, что касается метода осуществления реакции поверхностной сшивки в кислых условиях, то можно воспользоваться методом проведения реакции в присутствии в качестве катализатора кислоты Льюиса. Список кислот Льюиса чем-то конкретным не ограничен, но их примеры включают тетрахлорид олова, тетрахлорид титана и комплекс простого эфира и трифторида бора. Эти соединения можно использовать в сочетании двух или большего их числа. Среди них наиболее предпочтителен, учитывая простоту в обращении, комплекс простого эфира и трифторида бора.
Примеры растворителя, используемого во время осуществления поверхностной сшивки, включают растворители углеводородного типа, содержащие от 5 до 18 углеродных атомов, такие как бензол, толуол, о-ксилол, м-ксилол, п-ксилол, мезитилен, этилбензол, пропилбензол, кумол, бутилбензол, изобутилбензол, амилбензол, изоамилбензол, пентан, гексан, циклогексан, циклопентан, декалин, гептан, октан, изооктан, н-нонан, н-декан, н-ундекан, н-додекан, н-тридекан, н-тетрадекан, н-гексадекан и н-октадекан; растворители типа простого эфира, такие как анизол, этилизоамиловый эфир, этилтрет-бутиловый эфир, диизоамиловый эфир, диизопропиловый эфир, дифениловый эфир, дибутиловый эфир, дипропиловый эфир, дибензиловый эфир, тетрагидрофуран, метилтретбутиловый эфир, 1,4-диоксан, диэтиленгликольдиэтиловый эфир и триэтиленгликольдиметил; и галоидированные углеводороды, такие как хлороформ, тетрахлорид углерода, дихлорметан, 1,1-дихлорэтан, 1,2-дихлорэтан, хлорбензол, о-хлортолуол, п-хлортолуол, о-дихлорбензол, м-дихлорбензол, п-дихлорбензол, дибромэтан, дибромбутан, дибромпропан и дибромбензол.
Принимая во внимание экономические выгоды, простоту в обращении и т.п., предпочтительны растворители углеводородного типа или растворители типа простого эфира. Если учитывать скорость реакции, более предпочтительны растворители углеводородного типа, содержащие в общей сложности от 6 до 10 углеродных атомов, такие как бензол, толуол, о-ксилол, м-ксилол, п-ксилол, мезитилен, этилбензол, гексан, октан, изооктан, н-нонан и н-декан.
Используемое количество способного сшивать эпоксисоединения (Б) в предпочтительном варианте находится в интервале от 0,1 до 100 мас.%, более предпочтительно от 0,5 до 50 мас.%, в пересчете на порошкообразный сетчатый (со)полимер. Если используемое количество составляет меньше 0,1 мас.%, эффект поверхностной сшивки не проявляется и кислото/щелочестойкость понижается, тогда как если оно превышает 100 мас.%, постобработка после реакции оказывается очень обременительной.
Количество используемого растворителя может считаться достаточным, если оно достаточно велико для погружения в растворитель порошкообразного сетчатого полимера. Так, например, оно может в 2 или большее число раз превышать массу порошкообразного сетчатого полимера. В предпочтительном варианте, учитывая простоту в обращении, количество используемого растворителя от 3 до 10 раз, более предпочтительно от 4 до 7 раз, превышает массу порошкообразного сетчатого полимера.
Концентрация кислоты Льюиса, используемой в качестве катализатора, может составлять от 0,1 до 100 мас.% в пересчете на способное сшивать эпоксисоединение (Б), однако, принимая во внимание скорость реакции и экономические выгоды, предпочтительная концентрация составляет от 1 до 70 мас.%. В предпочтительном варианте эту реакцию осуществляют при температуре от 10 до 100°С в течение от 1 до 16 ч, более предпочтительно от 20 до 60°С в течение от 2 до 10 ч.
В дальнейшем эпоксигруппу получаемого порошкообразного поверхносшитого полимера гидролизуют с использованием кислоты. Примеры кислотного катализатора включают серную кислоту, перхлорную кислоту, соляную кислоту, толуолсульфоновую кислоту и бензолсульфоновую кислоту. Среди них предпочтительна, принимая во внимание простоту в обращении, соляная кислота.
В предпочтительном варианте концентрация используемой кислоты составляет от 0,01 до 5 н., более предпочтительно от 0,05 до 2 н. Если концентрация меньше 0,01 н., реакция протекает с невысокой скоростью, тогда как если она превышает 5 н., вероятен процесс гидролиза сложноэфирной связи. В это время, если только в виде растворителя содержатся по меньшей мере 10 мас.% или больше воды, в сочетании можно использовать органический растворитель.
Список используемых органических растворителей чем-либо конкретным не ограничен, лишь бы они смешивались как с кислотой, так и с водой и были неактивными в отношении кислоты и оксиранового кольца. Их примеры включают ацетон, метилэтилкетон, 1,4-диоксан и тетрагидрофуран.
Полученный таким образом порошкообразный поверхностносшитый (со)полимер с раскрытым эпоксидным циклом вводят в реакцию с эпоксисоединением (В), содержащим в общей сложности от 6 до 40 углеродных атомов, с получением порошкообразного гидрофобного полимера.
Примеры эпоксисоединения (В), содержащего в общей сложности от 6 до 40 углеродных атомов, включают соединения, отвечающие формулам с (1) по (4). Предпочтительны, учитывая легкодоступность, эпоксисоединения, отвечающие формуле (1) или (2) и содержащие в общей сложности от 6 до 22 углеродных атомов, а более предпочтительны соединения, содержащие в общей сложности от 8 до 18 углеродных атомов.
(в которой n обозначает целое число от 4 до 38);
(в которой n обозначает целое число от 3 до 37);
(в которой n обозначает целое число от 0 до 32);
(в которой n обозначает целое число от 0 до 31).
Реакцию эпоксисоединения и порошкообразного поверхностносшитого (со)полимера с раскрытым эпоксидным циклом можно осуществлять либо в щелочных условиях, либо в кислых условиях, однако, принимая во внимание скорость реакции и воспроизводимость реакции, в предпочтительном варианте эту реакцию осуществляют в кислых условиях. Типичным примером метода ее осуществления является метод проведения реакции в присутствии кислоты Льюиса в качестве катализатора. Примеры используемой при этом кислоты Льюиса включают тетрахлорид олова, тетрахлорид титана и комплекс простого эфира и трифторида бора. Список используемых кислот Льюиса чем-либо конкретным не ограничен, и можно использовать в сочетании два или большее число кислот Льюиса. Предпочтителен, учитывая легкодоступность, комплекс простого эфира и трифторида бора.
Примеры растворителей, используемых для реакции эпоксисоединения (В), содержащего в общей сложности от 6 до 40 углеродных атомов, и порошкообразного поверхностносшитого полимера с раскрытым эпоксидным циклом, включают растворители углеводородного типа, содержащие от 5 до 18 углеродных атомов, такие как бензол, толуол, о-ксилол, м-ксилол, п-ксилол, мезитилен, этилбензол, пропилбензол, кумол, бутилбензол, изобутилбензол, амилбензол, изоамилбензол, пентан, гексан, циклогексан, циклопентан, декалин, гептан, октан, изооктан, н-нонан, н-декан, н-ундекан, н-додекан, н-тридекан, н-тетрадекан, н-гексадекан и н-октадекан; растворители типа простого эфира, такие как анизол, этилизоамиловый эфир, этилтрет-бутиловый эфир, диизоамиловый эфир, диизопропиловый эфир, дифениловый эфир, дибутиловый эфир, дипропиловый эфир, дибензиловый эфир, тетрагидрофуран, метилтрет-бутиловый эфир, 1,4-диоксан, диэтиленгликольдиэтиловый эфир и триэтиленгликольдиметил; и галоидированные углеводороды, такие как хлороформ, тетрахлорид углерода, дихлорметан, 1,1-дихлорэтан, 1,2-дихлорэтан, хлорбензол, о-хлортолуол, п-хлортолуол, о-дихлорбензол, м-дихлорбензол, п-дихлорбензол, дибромэтан, дибромбутан, дибромпропан и дибромбензол.
Принимая во внимание экономические выгоды, простоту в обращении и т.п., предпочтительны растворители углеводородного типа или растворители типа простого эфира. Учитывая скорость реакции, более предпочтителен растворитель углеводородного типа, содержащий в общей сложности от 6 до 10 углеродных атомов, такой как бензол, толуол, о-ксилол, м-ксилол, п-ксилол, мезитилен, этилбензол, гексан, октан, изооктан, н-нонан и н-декан.
Эпоксисоединения (В) можно использовать самостоятельно или в сочетании двух или большего их числа.
Предпочтительное количество используемого эпоксисоединения (В) составляет от 10 до 2000 мас.%, более предпочтительно от 100 до 1000 мас.%, в пересчете на способное сшивать эпоксисоединение (В). Если используемое количество меньше 10 мас.%, воспроизводимость реакции становится плохой, тогда как если оно превышает 2000 мас.%, становится обременительной промывка на более поздней стадии.
Количество используемого растворителя может считаться достаточным, если оно достаточно велико для погружения в растворитель порошкообразных сетчатых полимеров. Так, например, оно может превышать в 2 или большее число раз массу порошкообразного сетчатого полимера. Предпочтительно, учитывая простоту в обращении, количество, которое от 3 до 10 раз, более предпочтительно от 4 до 7 раз, превышает его массу.
Концентрация кислоты Льюиса, используемой в качестве катализатора, может составлять от 0,1 до 100 мас.% в пересчете на способное сшивать эпоксисоединение (Б), однако, принимая во внимание скорость реакции и экономические выгоды, предпочтительная концентрация составляет от 1 до 70 мас.%. В предпочтительном варианте эту реакцию осуществляют при температуре от 10 до 100°С в течение от 1 до 16 ч, более предпочтительно от 20 до 60°С в течение от 2 до 10 ч.
Получаемые таким образом сферические частицы, размер которых составляет от 1 до 2000 мкм, предпочтительно от 3 до 25 мкм, при необходимости классифицируют, а затем их можно использовать в качестве наполнительного материала для жидкостной хроматографии с обращенной фазой. Примеры элюента включают воду/ацетонитрил, воду/метанол, ацетонитрил/(кислоту или водный раствор щелочи) и метанол/(кислоту или водный раствор щелочи).
Колонку, заполненную наполнительным материалом по настоящему изобретению, набухание/усадку которого в зависимости от растворителя удовлетворительным образом подавляют, можно использовать при рН от 1 до 13, и при этом она проявляет превосходную кислото/щелочестойкость. Более того, соотношение между числом теоретических тарелок для пирена и числом теоретических тарелок для нафталина составляет 0,7 или больше, и для полициклических ароматических соединений может быть получен острый пик.
Наилучший вариант выполнения изобретения
Настоящее изобретение более подробно изложено ниже со ссылками на примеры, однако этими примерами объем настоящего изобретения не ограничен.
Пример 1
Стадия 1: синтез субстратного порошкообразного сетчатого полимера
В смешанном растворе, содержавшем 2000 г диметакрилата глицерина и 1000 г 1-гексанола, растворяли 30 г 2,2'-азобис(изобутиронитрила) с получением масляной фазы. Отдельно в 3 л воды растворяли 120 г поливинилового спирта (Kuraray Poval PVA-224, выпускаемого фирмой Kuraray Co., Ltd.), в дальнейшем в раствор добавляли 7 л воды, а затем водный раствор (2 л), содержавший 240 г хлорида натрия, и перемешивали с получением водной фазы. Приготовленные таким образом масляную фазу и водную фазу смешивали в изготовленном из нержавеющей стали контейнере 20-литрового объема и смесь диспергировали в высокоскоростном диспергаторе (гомогенизаторе), одновременно регулируя число оборотов и время диспергирования, с получением масляных капелек, максимальный размер которых был равным 4 мкм.
После этого дисперсию перемешивали при 150 об/мин и давали возможность реакции протекать при 60°С в течение 7 ч. Полученный порошкообразный сетчатый полимер подвергали центрифугированию (при 2000 об/мин в течение 10 мин), верхний слой выбрасывали, осадок диспергировали в 12 л горячей воды при 70°С (с помощью ультразвукового промывного аппарата) и дисперсию перемешивали при 70°С в течение 3 ч, а затем фильтровали под вакуумом. Фильтровальный пирог на воронке промывали 60 л горячей воды при 70°С, после чего 18 л ацетона, сушили на воздухе распределением этого фильтровального пирога в изготовленной из нержавеющей стали ванне и дополнительно сушили под пониженным давлением при 60°С в течение 24 ч. Частицы классифицировали с помощью пневматического классификатора с получением 620 г порошкообразного сетчатого полимера, средний размер частиц которого составлял 4 мкм (в дальнейшем в настоящем описании упоминается под названием "субстратного геля").
Стадия 2: тщательная промывка
К 50 г субстратного геля, полученного на стадии 1, добавляли 500 мл чистой воды. Приготовленную смесь перемешивали с выдержкой при 60°С в течение 5 ч, а затем частицы собирали фильтрованием, последовательно промывали 2000 мл горячей воды при 70°С и 300 мл метанола, сушили на воздухе распределением частиц в изготовленной из нержавеющей стали ванне и дополнительно сушили под пониженным давлением при 70°С в течение 24 ч с получением 48 г тщательно промытого субстратного геля.
Стадия 3: поверхностная сшивка
В 100 мл толуола диспергировали 20 г тщательно промытого субстратного геля, полученного на стадии 2. После добавления в дисперсию при одновременном перемешивании 2 г этиленгликольдиглицидилового эфира смесь перемешивали при комнатной температуре в течение 30 мин и в нее по каплям добавляли раствор, приготовленный растворением 500 мг комплекса трифторида бора и диэтилового эфира в 5 мл толуола, и при 40°С проводили реакцию в течение 3 ч.
Нерастворимые вещества собирали фильтрованием и последовательно промывали 100 мл толуола и 100 мл тетрагидрофурана. Затем порошкообразный полимер собирали фильтрованием и переносили в химический стакан емкостью 300 мл. После добавления в него 100 мл тетрагидрофурана с помощью ультразвукового промывного аппарата смесь подвергали обработке ультразвуковыми волнами в течение 10 мин. Порошкообразный полимер вновь собирали фильтрованием и затем последовательно промывали 100 мл тетрагидрофурана и 100 мл ацетона, сушили на воздухе и дополнительно сушили под пониженным давлением при 60°С в течение 2 ч (выход: 21,2 г).
Стадия 4: реакция размыкания эпоксицикла
В 60 мл водного 0,05 н. раствора соляной кислоты диспергировали 15 г порошкообразного поверхностносшитого полимера, полученного на стадии 3, и перемешивали при 50°С в течение 1 ч. Полученный порошкообразный полимер с раскрытым эпоксидным циклом собирали фильтрованием, промывали 500 мл чистой воды, сушили на воздухе и дополнительно сушили под пониженным давлением при 60°С в течение 2 ч (выход: 15,5 г). В дальнейшем полученный таким образом порошкообразный полимер с раскрытым эпоксидным циклом в настоящем описании упоминается как "немодифицированный гель".
Стадия 5: реакция введения С18
В 100 мл толуола диспергировали 15 г немодифицированного геля, полученного на стадии 4. После добавления в него 105 г стеарилглицидилового эфира смесь перемешивали при 40°С в течение 0,5 ч, в нее добавляли раствор, приготовленный растворением 1 г комплекса трифторида бора и диэтилового эфира в 5 мл толуола, и при 40°С проводили реакцию в течение 5 ч. Далее в реактор добавляли 100 мл толуола, порошкообразный полимер собирали фильтрованием, промывали 200 мл тетрагидрофурана и затем переносили в химический стакан емкостью 300 мл. После добавления в него 100 мл тетрагидрофурана с помощью ультразвукового промывного аппарата смесь подвергали обработке ультразвуковыми волнами в течение 10 мин и порошкообразный полимер вновь собирали фильтрованием.
Этот порошкообразный полимер промывали 100 мл тетрагидрофурана, вновь переносили в химический стакан емкостью 300 мл и диспергировали в денатурированном спирте/водном 1%-ном растворе вторичного кислого фосфата калия (в объемном соотношении 50/50). Образовавшуюся дисперсию с помощью ультразвукового промывного аппарата подвергали обработке ультразвуковыми волнами в течение 10 мин и порошкообразный полимер вновь собирали фильтрованием. Собранный порошкообразный полимер промывали 500 мл чистой воды и 200 мл ацетона, сушили на воздухе и дополнительно сушили под пониженным давлением при 60°С в течение 2 ч (порошкообразный сшитый полимер с введенной С18 в дальнейшем в настоящем описании упоминается как "модифицированный гель"; выход: 17,5 г).
Степень введения группы С18
По соответствующим данным элементного анализа немодифицированного геля и модифицированного геля рассчитывали степень введения группы С18 и устанавливали, что она составляла 20 мас.%.
Заполнение модифицированным гелем
Модифицированным гелем, полученным на стадии 5, по суспензионному методу набивали изготовленную из нержавеющей стали колонку 4,6 мм (внутренний диаметр) × 150 мм (длина) для жидкостной хроматографии с обращенной фазой (в дальнейшем в настоящем описании упоминается как "колонка А").
Определение возможностей 1: соотношение чисел теоретических тарелок для нафталина и пирена
Колонку А испытывали для определения числа теоретических тарелок по пикам нафталина и пирена в следующих условиях определения посредством жидкостной хроматографии с обращенной фазой.
Условия определения посредством жидкостной хроматографии с обращенной фазой:
Элюент: СН3CN/вода в объемном соотношении 65/35
Скорость потока: 1,00 мл/мин
Температура в колонке: 40°С
Детектор: UV 254 нм
Образцы: нафталин (0,4 мг/мл)
пирен (0,3 мг/мл)
Инжектируемый объем: 5 мкл
В результате для каждого пика в виде числа теоретических тарелок получали следующие значения. В скобках приведено отношение к значению для нафталина, которое принимали за 1. Нафталин: 7300 (1,00), пирен: 6200 (0,85).
Определение возможностей 2: испытание на кислотостойкость
Колонку А испытывали на кислотостойкость в следующих условиях.
Элюент: МеОН/водный 1%-ный раствор трифторуксусной кислоты (с рН 1,2) в объемном соотношении 10/90
Скорость потока: 1,00 мл/мин
Температура в колонке: 60°С
Детектор: UV 254 нм
Образцы: п-гидроксибензойная кислота (0,5 мг/мл)
Инжектируемый объем: 10 мкл
Время испытания: 70 ч
В результате в виде времени удерживания п-гидроксибензойной кислоты получали следующие значения. В скобках приведено отношение ко времени удерживания п-гидроксибензойной кислоты сразу же после начала эксперимента, взятому за 1. Сразу же после начала испытания: 12,6 мин (1,00), после 70 ч от начала испытания: 12,3 мин (0,98).
Определение возможностей 3: испытание на щелочестойкость
Колонку А испытывали на щелочестойкость в приведенных ниже условиях и время удерживания пирена, определенное в таких же условиях, как при определении возможностей 1, сопоставляли до и после испытания.
Элюент: СН3CN/0,1 н. NaOH (рН: 13) в объемном соотношении 20/80
Скорость потока: 0,50 мл/мин
Температура в колонке: 25°С
Время испытания: 18 ч
В результате в виде времени удерживания пирена получали следующие значения. В скобках приведено отношение к значению до испытания, взятому за 1. Перед испытанием на щелочестойкость: 11,6 мин (1,00), после истечения 18 ч испытания на щелочестойкость: 11,6 мин (1,00).
Определение возможностей 4: испытание с заменой растворителя
Колонку А испытывали на смену растворителя в приведенных ниже условиях и каждое число теоретических тарелок для нафталина и пирена, которое определяли в таких же условиях, как при определении возможностей 1, сопоставляли до и после испытания. Каждые 60 мин заменяли друг другом чистую воду и метанол при скорости потока 0,5 мл/мин и для каждого образца повторяли 5 циклов.
В результате для каждого пика в виде числа теоретических тарелок получали следующее значение. В скобках приведены значение перед осуществлением испытания с заменой растворителя и соотношение между числом тарелок перед испытанием и числом тарелок после испытания. Нафталин: 7300 (7300, 1,00), пирен: 6200 (6200, 1,00).
Сравнительный пример 1
Стадия 1: синтез субстратного порошкообразного сшитого полимера
Субстратный порошкообразный сетчатый полимер (в дальнейшем в настоящем описании упоминается просто как "субстратный гель") получали проведением такого же процесса, как в примере 1.
Стадия 2: тщательная промывка
Тщательно промытый субстратный гель получали проведением такого же процесса, как в примере 1.
Стадия 3: поверхностная сшивка
20 г тщательно промытого субстратного геля, полученного на стадии 2, смешивали с 20 г этиленгликольдиглицидилового эфира и смесь перемешивали при комнатной температуре в течение 30 мин. Затем в нее добавляли 50 г 1 н. NaOH и при 30°С перемешивали в течение 3 ч.
После сбора фильтрованием нерастворимые вещества промывали 1000 мл чистой воды, а затем 100 мл ацетона и сушили под пониженным давлением при 60°С в течение 2 ч (выход: 20,7 г).
Стадия 4: реакция раскрытия эпоксицикла и
Стадия 5: реакция введения С18
Порошкообразный сшитый полимер с введенной С18 (в дальнейшем в настоящем описании упоминается как "модифицированный гель") получали проведением такого же процесса, как в примере 1 (выход: 18,1 г).
Степень введения группы С18
По соответствующим данным элементного анализа немодифицированного геля и модифицированного геля рассчитывали степень введения группы С18 и устанавливали, что она составляла 19 мас.%.
Заполнение модифицированным гелем
Заполнение осуществляли проведением такого же процесса, как в примере 1 (в дальнейшем в настоящем описании упоминается как "колонка Б").
Определение возможностей 1: соотношение чисел теоретических тарелок для нафталина и пирена
Колонку Б испытывали в таких же условиях, как в примере 1.
В результате для каждого пика в виде числа теоретических тарелок получали следующие значения. В скобках приведено отношение к значению для нафталина, взятому за 1. Нафталин: 6900 (1,00), пирен: 5800 (0,84).
Определение возможностей 2: испытание на кислотостойкость
Колонку Б испытывали в таких же условиях, как в примере 1.
В результате в виде времени удерживания п-гидроксибензойной кислоты получали следующие значения. В скобках приведено отношение ко времени удерживания п-гидроксибензойной кислоты сразу же после начала эксперимента, взятому за 1. Сразу же после начала испытания: 11,4 мин (1,00), после 70 ч от начала испытания: 10,8 мин (0,95).
Определение возможностей 3: испытание на щелочестойкость
Колонку Б испытывали в таких же условиях, как в примере 1.
В результате в виде времени удерживания пирена получали следующие значения. В скобках приведено отношение к значению перед испытанием, взятому за 1. Перед испытанием на щелочестойкость: 11,0 мин (1,00), после истечения 18 ч испытания на щелочестойкость: 9,9 мин (0,90).
Определение возможностей 4: испытание с заменой растворителя
Колонку Б испытывали в таких же условиях, как в примере 1.
В результате для каждого пика в виде числа теоретических тарелок получали следующие значения. В скобках приведены значение перед осуществлением испытания с заменой растворителя и соотношение между значениями до и после испытания. Нафталин: 6500 (6900, 0,94), пирен: 5000 (5800, 0,86).
Сравнительный пример 2
Стадия 1: синтез субстратного порошкообразного сшитого полимера
Субстратный порошкообразный сетчатый полимер (в дальнейшем в настоящем описании упоминается просто как "субстратный гель") получали проведением такого же процесса, как в примере 1.
Стадия 2: тщательная промывка
Тщательно промытый субстратный гель получали проведением такого же процесса, как в примере 1.
Стадия 3: реакция введения С18
В 110 мл толуола диспергировали 20 г тщательно промытого субстратного геля, полученного на стадии 2. После добавления к нему 3,9 г пиридина смесь подвергали обработке ультразвуковыми волнами в течение 3 мин. В нее по каплям в течение 15 мин при одновременном перемешивании добавляли 6,0 г стеароилхлорида и при 60°С проводили реакцию в течение 5 ч. Нерастворимые вещества собирали фильтрованием и последовательно промывали тетрагидрофураном (250 мл), денатурированным спиртом (250 мл), денатурированным спиртом/водой (в соотношении 1/1) (250 мл), тетрагидрофураном (250 мл) и метанолом (250 мл) с получением 35,57 г смоченного метанолом модифицированного геля.
Стадия 4: обработка блокированием
В 100 мл 2,2-диметоксипропана диспергировали 35,57 г смоченного метанолом модифицированного геля, полученного на стадии 3. После добавления в дисперсию 2,0 мл концентрированной соляной кислоты дисперсию подвергали обработке ультразвуковыми волнами в течение 3 мин, а затем перемешивали при 50°С в течение 2 ч. Нерастворимые вещества собирали фильтрованием и последовательно промывали метанолом (250 мл), метанолом/водой (в соотношении 1/1) (250 мл) и метанолом (250 мл). Затем гель сушили на воздухе и дополнительно сушили под пониженным давлением при 60°С в течение 24 ч с получением обработанного блокированием модифицированного геля (21,03 г).
Степень введения октадеканоильной группы
По соответствующим данным элементного анализа субстратного геля и модифицированного геля рассчитывали степень введения группы С18 в пересчете на все гидроксильные группы в субстратном геле и устанавливали, что она составляла 14 мас.%.
Заполнение модифицированным гелем
Заполнение осуществляли проведением такого же процесса, как в примере 1 (в дальнейшем в настоящем описании упоминается как "колонка В").
Определение возможностей 1: соотношение чисел теоретических тарелок для нафталина и пирена
Колонку В испытывали в таких же условиях, как в примере 1.
В результате в виде числа теоретических тарелок для каждого пика получали следующие значения. В скобках приведено отношение к значению для нафталина, взятому за 1. Нафталин: 15100 (1,00), пирен: 12100 (0,80).
Определение возможностей 2: испытание на кислотостойкость
Колонку В испытывали в таких же условиях, как в примере 1.
В результате в виде времени удерживания п-гидроксибензойной кислоты получали следующие значения. В скобках приведено отношение ко времени удерживания п-гидроксибензойной кислоты, взятому за 1 сразу же после начала эксперимента. Сразу же после начала испытания: 11,9 мин (1,00), после 70 ч от начала испытания: 7,1 мин (0,60).
Определение возможностей 3: испытание на щелочестойкость
Колонку В испытывали в таких же условиях, как в примере 1.
В результате в виде времени удерживания пирена получали следующие значения. В скобках приведено отношение к значению перед испытанием, взятому за 1. Перед испытанием на щелочестойкость: 11,0 мин (1,00), после истечения 18 ч испытания на щелочестойкость: 6,4 мин (0,58).
Определение возможностей 4: испытание с заменой растворителя
Колонку В испытывали в таких же условиях, как в примере 1.
В результате для каждого пика в виде числа теоретических тарелок получали следующие значения. В скобках приведены значение перед осуществлением испытания с заменой растворителя и соотношение между числом тарелок перед и после испытания. Нафталин: 11500 (15100, 0,76), пирен: 6200 (12100, 0,51).
Сравнительный пример 3
Синтез осуществляли аналогично примеру 1, кроме того, что исключали стадию 3. В результате получали 17,1 г модифицированного геля, который не подвергали обработке поверхностным сшиванием. На стадии 4 в качестве материала использовали 15 г тщательно промытого субстратного геля, полученного на стадии 2.
Степень введения группы С18
По соответствующим данным элементного анализа субстратного геля, который не подвергали обработке поверхностным сшиванием, и модифицированного геля рассчитывали степень введения группы С18 и устанавливали, что она составляла 17 мас.%.
Заполнение модифицированным гелем
Заполнение осуществляли проведением такого же процесса, как в примере 1 (в дальнейшем в настоящем описании упоминается как "колонка Г").
Определение возможностей 1: соотношение чисел теоретических тарелок для нафталина и пирена
Колонку Г испытывали в таких же условиях, как в примере 1.
В результате для каждого пика в виде числа теоретических тарелок получали следующие значения. В скобках приведено отношение к значению для нафталина, взятому за 1. Нафталин: 6900 (1,00), пирен: 5500 (0,80).
Определение возможностей 2: испытание на кислотостойкость
Колонку Г испытывали в таких же условиях, как в примере 1.
В результате в виде времени удерживания п-гидроксибензойной кислоты получали следующие значения. В скобках приведено отношение ко времени удерживания п-гидроксибензойной кислоты сразу же после начала эксперимента, взятому за 1. Сразу же после начала испытания: 10,6 мин (1,00), после 70 ч от начала испытания: 8,5 мин (0,80).
Определение возможностей 3: испытание на щелочестойкость
Колонку Г испытывали в таких же условиях, как в примере 1.
В результате в виде времени удерживания пирена получали следующие значения. В скобках приведено отношение к значению перед испытанием, взятому за 1. Перед испытанием на щелочестойкость: 9,5 мин (1,00), после истечения 18 ч испытания на щелочестойкость: 6,9 мин (0,69).
Определение возможностей 4: испытание с заменой растворителя
Колонку Г испытывали в таких же условиях, как в примере 1.
В результате для каждого пика в виде числа теоретических тарелок получали следующие значения. В скобках приведены значение перед осуществлением испытания с заменой растворителя и соотношение между числом тарелок перед и после испытания. Нафталин: 11500 (15100, 0,76), пирен: 5100 (6900, 0,74).
Результаты различных оценок, полученные в примерах 1 и 2 и сравнительных примерах с 1 по 3, совместно представлены в таблице 1.
В колонке А, подготовленной в примере 1, соотношение между числами тарелок для нафталина и пирена является высоким, а удерживание после испытания на кислото/щелочестойкость не уменьшается. Более того, даже если осуществляют смену растворителя с заменой друг другом чистой воды и метанола, то набухание и усадка, характерные для наполнительного материала полимерного типа, подавляются и число теоретических тарелок не уменьшается.
Видно, что кислото/щелочестойкость колонки В, заполненной модифицированным гелем, полученным осуществлением реакции поверхностной сшивки в щелочных условиях, в сравнении с колонкой А оказывается более низкой, а устойчивость против смены растворителя не является удовлетворительно высокой.
Возможность промышленного применения
В соответствии со способом получения наполнительного материала для жидкостной хроматографии с обращенной фазой по настоящему изобретению может быть получен высокоэффективный наполнительный материал для жидкостной хроматографии с обращенной фазой. Колонку для жидкостной хроматографии с обращенной фазой, заполненную наполнительным материалом для жидкостной хроматографии с обращенной фазой по настоящему изобретению, можно применять как в области очень низких значений рН, так и в области очень высоких значений рН.
Когда применяют способ анализа жидкостной хроматографией с обращенной фазой по настоящему изобретению, могут быть обеспечены высокоточные выделение/анализ, в особенности медицинских/сельскохозяйственных препаратов, пищевых добавок и промежуточных продуктов для них, природных или синтетических полимеров и добавок для них и загрязнителей окружающей среды. Таким образом, настоящее изобретение можно применять в данной области техники в широком диапазоне.
Реферат
Изобретение относится к наполнительным материалам для жидкостной хроматографии с обращенной фазой. Техническая задача - получение наполнительного материала с высокой кислото/щелочестойкостью, уменьшенной степенью набухания и усадки полимера и способностью сохранять эффективность колонки при применении различных растворителей. Предложены порошкообразный гидрофобный полимер, полученный реакцией (А) порошкообразного сшитого (мет)акрилатного полимера, имеющего гидроксильную группу, с (Б) способным сшивать эпоксисоединением, гидролизом оксирановых колец, а затем реакцией полученного соединения, содержащего гидроксильную группу, с эпоксисоединением (В), содержащим в общем от 6 до 40 углеродных атомов, и колонка для жидкостной хроматографии с обращенной фазой, заполненная заявленным полимером. Колонка по данному изобретению обеспечивает получение хроматограммы с острыми пиками для полициклических ароматических соединений. 2 н. и 8 з.п. ф-лы, 1 табл.
Формула
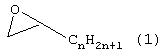
Комментарии