Способ получения акролеина и/или акриловой кислоты - RU2285690C2
Код документа: RU2285690C2
Чертежи
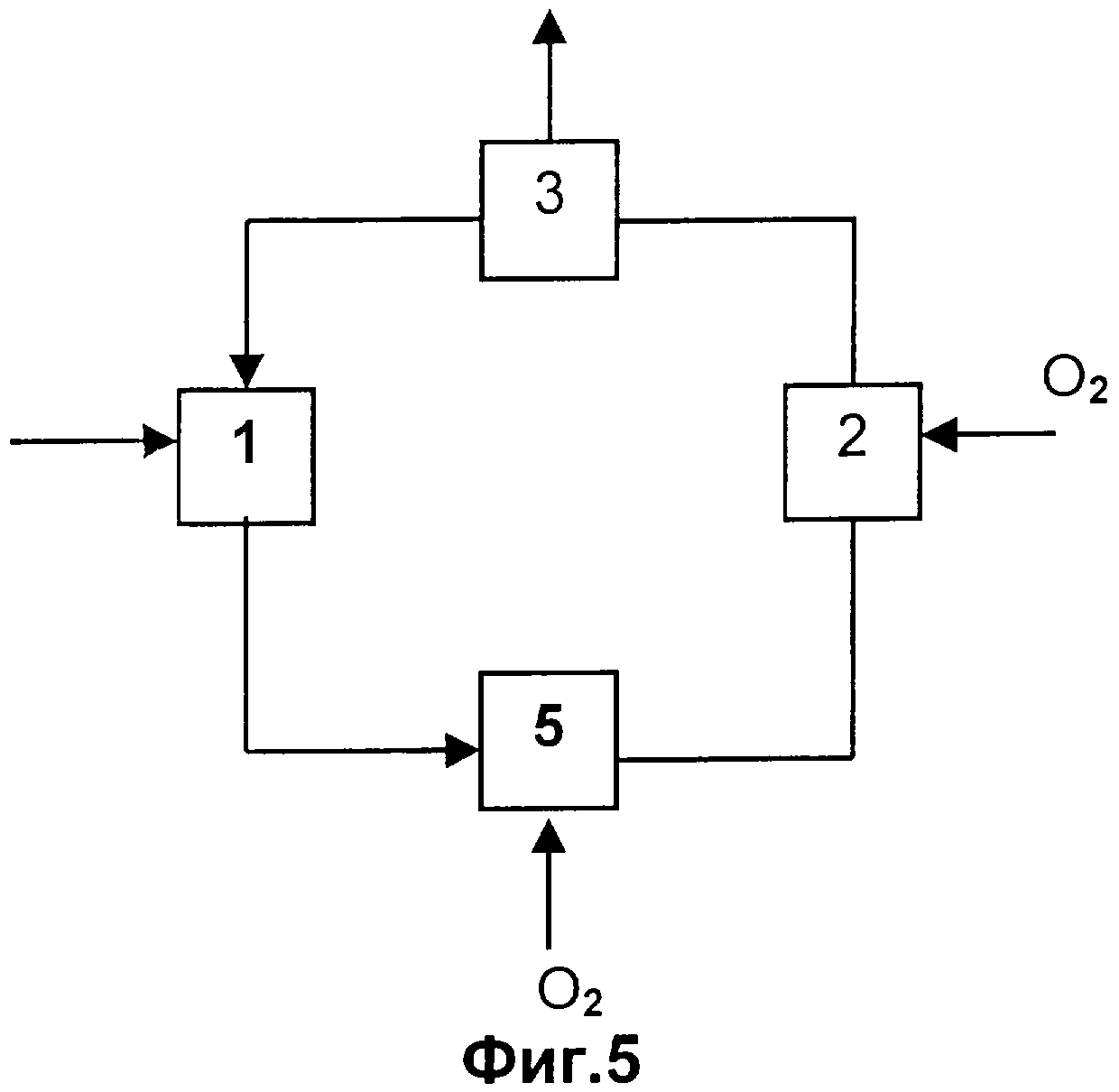
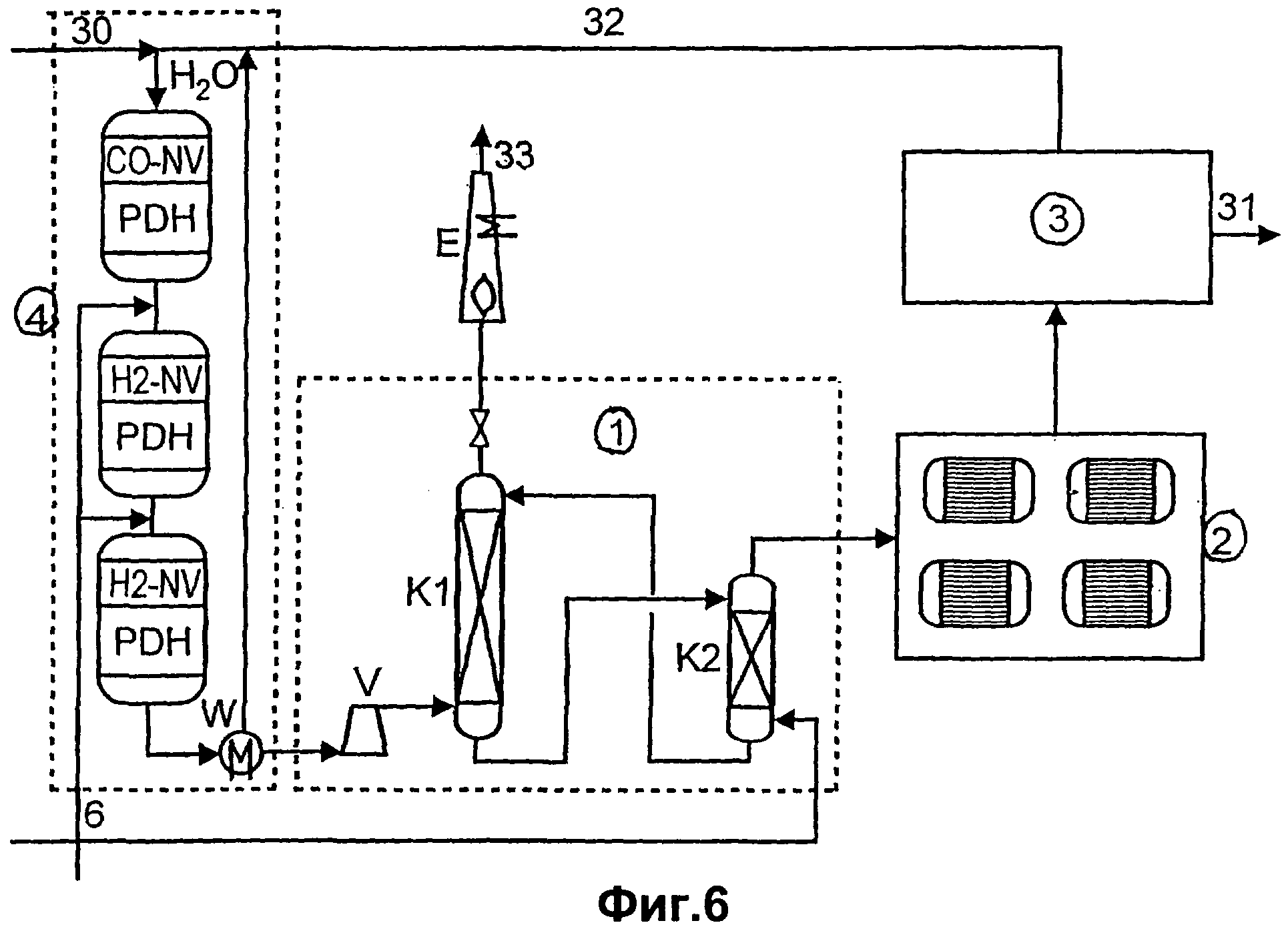
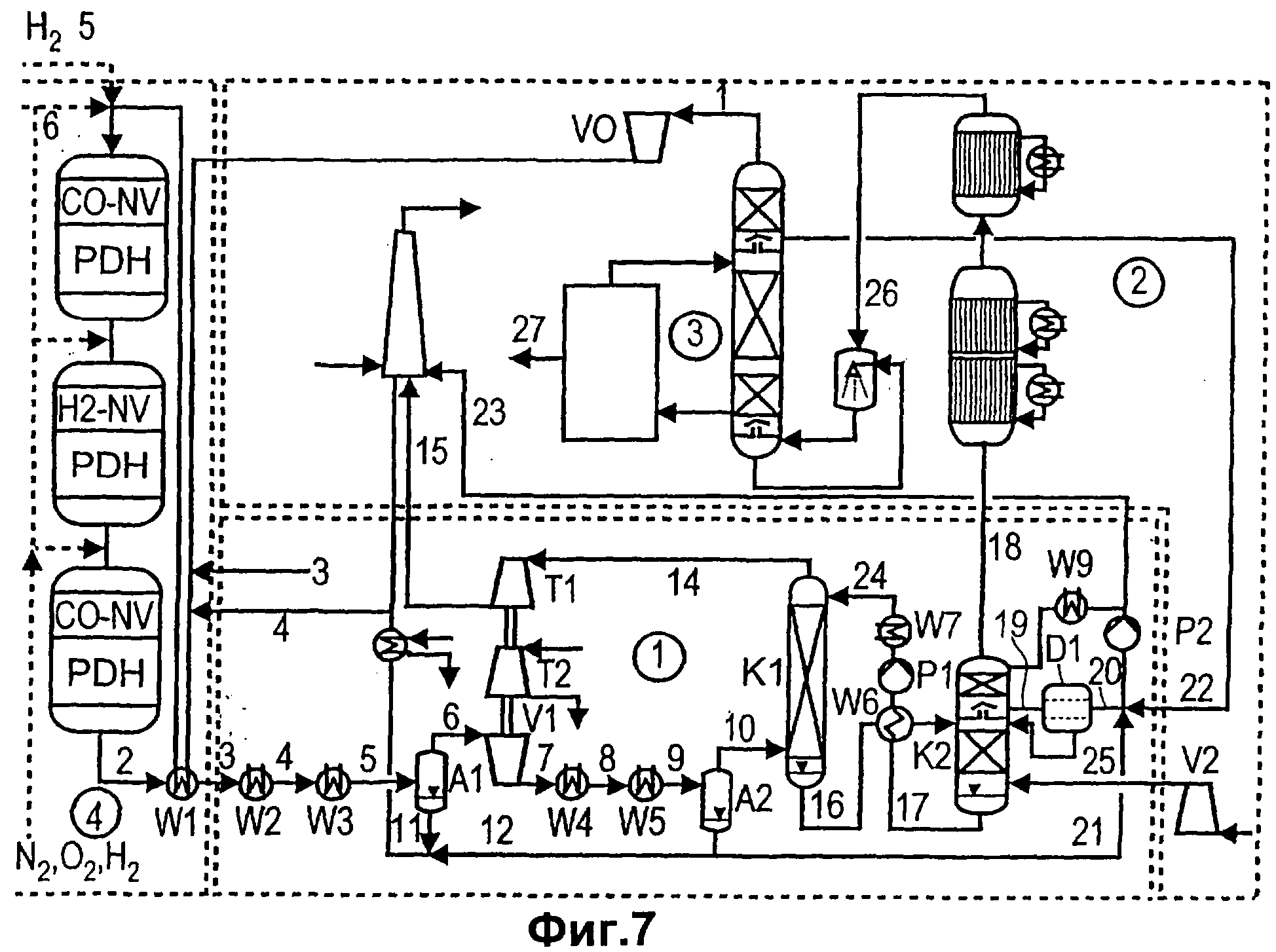
Описание
Настоящее изобретение касается способа получения акролеина и/или акриловой кислоты из пропана и/или пропена.
Акролеин и акриловая кислота являются важными химическими продуктами. Так, акриловая кислота находит применение, в частности, в качестве исходного мономера для получения полимеров, водные дисперсии которых используют, например, в качестве связующих. В зависимости от сферы применения этих полимеров акриловая кислота перед полимеризацией может быть подвергнута этерификации. Акролеин является важным полупродуктом, используемым, например, для получения глутарового альдегида, метионина, фолевой и акриловой кислот.
Согласно известным способам исходными продуктами для получения акролеина и/или акриловой кислоты являются пропан и/или пропен. Из немецкой заявки на патент DE-A 3313573 и европейской заявки на патент ЕР-А-0117146 известен двух- или трехстадийный способ превращения пропана в акролеин и/или акриловую кислоту, на первой стадии которого осуществляют дегидрирование пропана до пропена, а на второй стадии окисление пропена до акролеина. Важная особенность данного способа заключается в том, что пропан не отделяют между первой и второй стадиями от образующихся при его дегидрировании побочных компонентов, например от молекулярного водорода. Окисление пропена осуществляют в условиях, исключающих заметное окисление водорода. На третьей стадии акролеин может быть подвергнут окислению до акриловой кислоты. Кроме того, предусматривается возможность выделения непревращенных на второй или третьей стадии пропана и пропена путем их абсорбции и возвращения на первую стадию (стадию дегидрирования) после отделения от абсорбента.
В японской заявке на патент JP-A-10-36311 описан способ получения α,β-ненасыщенных карбоновых кислот, в частности акриловой кислоты, путем газофазного окисления пропана в присутствии композиционного катализатора на основе оксида металла, причем для достижения высокого выхода целевого продукта отношение пропана к кислороду и, при необходимости, газообразному разбавителю в составе исходной смеси поддерживают в определенном диапазоне, одновременно обеспечивая определенную степень превращения пропана. Непревращенный пропан может быть выделен из продуктов реакции с помощью селективного сепаратора, включающего устройство для адсорбции при переменном давлении (Pressure-Swing-Adsorption), и вновь подвергнут газофазному окислению.
В заявке на патент Великобритании GB 1378178 описан способ, согласно которому непревращенный в процессе окисления углеводород поглощают абсорбентом, подвергаемым последующему отпариванию с использованием соответствующего отпаривающего средства, которое добавляют к подлежащему выделению углеводороду в таком количестве, чтобы состав смеси находился вне пределов воспламенения.
Задача настоящего изобретения состояла в создании способа газофазного каталитического получения акролеина и/или акриловой кислоты из пропана и/или пропена, отличающегося экономичностью и максимально возможной продолжительностью эксплуатации используемого катализатора без регенерации.
Согласно изобретению эту задачу решают путем поглощения абсорбентом пропана и/или пропена из содержащей эти углеводороды смеси, выделения пропана и/или пропена из абсорбента и использования пропана и/или пропена для последующего окисления до акролеина и/или акриловой кислоты.
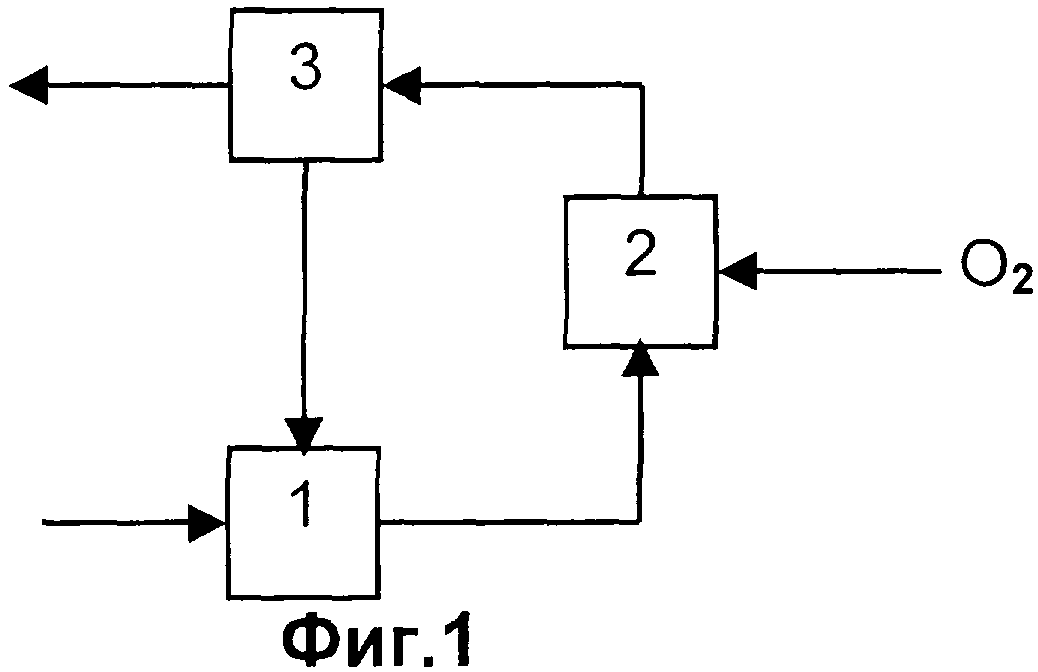
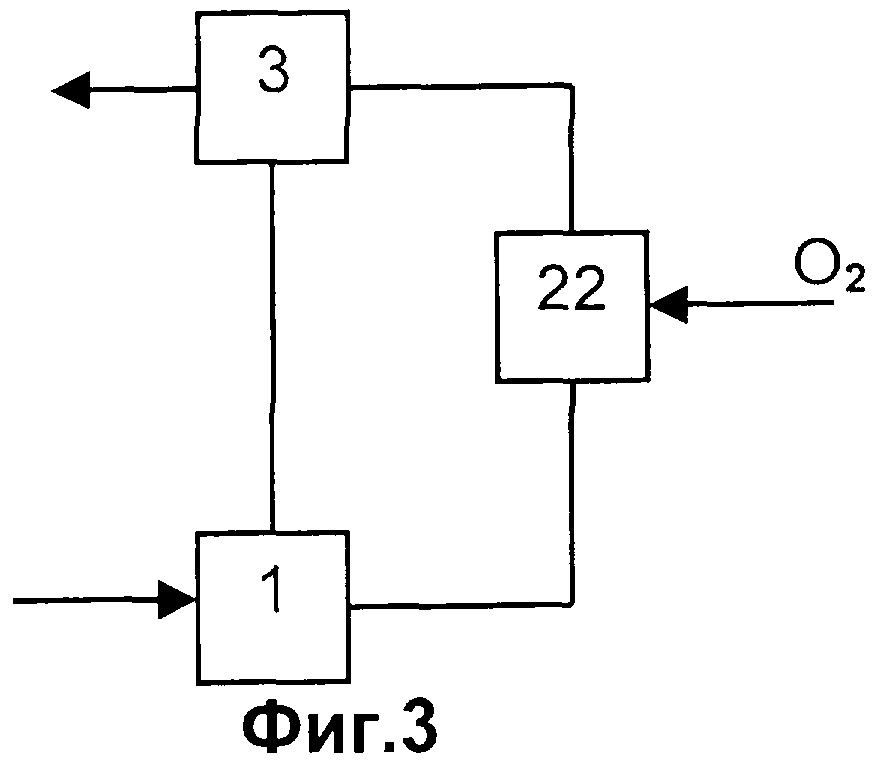
Таким образом, изобретение касается способа получения акролеина и/или акриловой кислоты из пропана и/или пропена, включающего следующие стадии:
a) выделение пропана и/или пропена из содержащей пропан и/или пропен газовой смеси А путем их поглощения абсорбентом,
b) выделение пропана и/или пропена из абсорбента с получением содержащего пропан и/или пропен газа В и
c) использование полученного на стадии (b) газа В для окисления пропана и/или пропена до акролеина и/или акриловой кислоты,
причем между стадиями (b) и (с) не производят гетерогенное каталитическое дегидрирование пропана без подачи кислорода. Предпочтительные варианты осуществления изобретения приведены в представленном ниже описании и на соответствующих чертежах.
Поскольку пропан и/или пропен перед стадией их окисления выделяют абсорбцией, газ В, как правило, содержит остаточные количества абсорбента. Неожиданно оказалось, что несмотря на это окисление протекает без каких-либо затруднений. В частности, не наблюдалось сколько-нибудь существенное снижение активности катализатора окисления, который можно было эксплуатировать в течение длительного периода без регенерации. Кроме того, не наблюдалось никаких проблем, обусловленных, при необходимости, происходящим на стадии окисления пропана и/или пропена образованием каких-либо продуктов окисления абсорбента. Если наличие остатков абсорбента и создает определенные трудности, которые, как правило, отсутствуют, если в качестве абсорбентов используют обладающие высокой температурой кипения углеводороды, абсорбент может быть удален, например, путем водной закалки или адсорбции.
Согласно немецкой заявке на патент DE-A 3313573 рекуперированные (выделенные) путем абсорбции пропан и пропен возвращают на стадию гетерогенного каталитического дегидрирования пропана, при осуществлении которого может произойти дезактивация соответствующего катализатора, например, в результате коксования, в связи с чем катализаторы дегидрирования нуждаются в частой регенерации. Наличие абсорбента в направляемом на дегидрирование газовом потоке не создает никаких проблем, поскольку абсорбент способен сгорать вместе с продуктами коксования. Катализаторы, используемые для окисления пропена до акролеина и/или акриловой кислоты, обычно не регенерируют так часто, поэтому дополнительные расходы на регенерацию, обусловленные присутствием в используемом газе абсорбента, бывают более существенны, чем при дегидрировании. Преимущество способа согласно изобретению состоит в том, что катализатор окисления в течение длительного времени может эксплуатироваться без регенерации.
Способ согласно изобретению отличается от способа согласно немецкой заявке на патент DE-A 3313573 тем, что выделенный путем абсорбции пропан и/или пропен направляют на стадию окисления. Другое отличие способа согласно изобретению состоит в том, что между стадиями выделения пропана и/или пропена из абсорбента и их окисления до акролеина и/или акриловой кислоты не производят гетерогенное каталитическое дегидрирование пропана без подачи кислорода.
В рамках настоящего изобретения под газом В может также подразумеваться газовая смесь.
На стадии (а) может использоваться газовая смесь А с любым содержанием пропана и/или пропена. Предпочтительное молярное соотношение пропана и пропена в газовой смеси А составляет от 0:100 до 100:0, в частности от 10:90 до 90:10, часто от 80:20 до 40:60.
Газовая смесь А предпочтительно содержит, по меньшей мере, один дополнительный, отличающийся от пропана и/или пропена компонент, тип которого не подлежит никаким специальным ограничениям и, как правило, определяется происхождением газовой смеси. В частности, речь идет, по меньшей мере, об одном компоненте, выбранном из группы, включающей кислород, водород, оксиды углерода, в частности монооксид или диоксид углерода, побочные компоненты, образующиеся при дегидрировании пропана, газофазном окислении пропена до акролеина и/или акриловой кислоты или окислении пропана до акролеина и/или акриловой кислоты. Дополнительным компонентом часто является, по меньшей мере, водород, кислород, оксид углерода или смесь этих газов.
В качестве используемых на стадии (а) абсорбентов в принципе пригодны любые, способные поглощать пропан и/или пропен абсорбенты. Под абсорбентом подразумевают предпочтительно органический растворитель, который предпочтительно является гидрофобным и/или высококипящим соединением. Его точка кипения (при нормальном давлении, составляющем 1 атм), предпочтительно составляет, по меньшей мере, 120°С, предпочтительно, по меньшей мере, 180°С, предпочтительно находится в интервале от 200 до 350°С, в частности от 250 до 300°С, более предпочтительно от 260 до 290°С. Целесообразным является использование растворителей, температура вспышки которых при нормальном давлении, составляющем 1 атм, превышает 110°С. В общем случае в качестве абсорбентов пригодны слабополярные органические растворители, например алифатические углеводороды, которые предпочтительно не содержат оказывающих внешнее воздействие полярных групп, а также ароматических углеводородов. В общем случае желательно, чтобы абсорбент обладал возможно более высокой точкой кипения и вместе с тем, возможно, более полно растворял пропан и/или пропен. Пригодными абсорбентами являются, в частности, алифатические углеводороды, например алканы или алкены с 8-20 атомами углерода, ароматические углеводороды, например, образующиеся при перегонке парафинов средние фракции, простые эфиры с объемистыми, присоединенными к атому кислорода группам или смеси указанных соединений, причем в их состав может быть дополнительно введен 1,2-диметилфталат, как, например, описано в немецкой заявке на патент DE-A 4308087. Кроме того, пригодны сложные эфиры, образованные бензойной и фталевой кислотами и неразветвленными алканолами с 1-8 атомами углерода, в частности н-бутиловый, метиловый или этиловый эфир бензойной кислоты, диметиловый или диэтиловый эфир фталевой кислоты, а также так называемые масляные теплоносители, в частности дифенил, дифениловый эфир, смеси дифенила с дифениловым эфиром или соответствующие хлорсодержащие производные и триарилалкены, например 4-метил-4'-бензилдифенилметан и его изомеры: 2-метил-2'-бензилдифенилметан, 2-метил-4'-бензилдифенилметан, 4-метил-2'-бензилдифенилметан, а также их смеси. Пригодным абсорбентом является смесь дифенила и дифенилового эфира предпочтительно азеотропного состава, в частности, состоящая из 25 мас.% дифенила (бифенила) и 75 мас.% дифенилового эфира, выпускаемая под торговым наименованием дифил. Иногда к ней добавляют дополнительный растворитель, например диметилфталат, в количестве от 0,1 до 25 мас.% (в расчете на общую смесь растворителей). Особенно пригодными абсорбентами являются также октаны, нонаны, деканы, ундеканы, додеканы, тридеканы, тетрадеканы, пентадеканы, гексадеканы, гептадеканы и октадеканы, причем особенно пригодными являются, в частности, тетрадеканы. Предпочтительно, чтобы используемый абсорбент, с одной стороны, обладал указанной выше температурой кипения, а, с другой стороны, имел не слишком высокую молекулярную массу. Предпочтительными являются абсорбенты, молекулярная масса которых меньше или равна 300 г/моль. Кроме того, пригодны парафиновые масла с 8-10 атомами углерода, описанные в немецкой заявке на патент DE-A 3313573. Например, пригодны продукты серии Haplasol i, поставляемые на рынок фирмой Haltermann, в частности Haplasol 250/340 i и Haplasol 250/275 i, а также масла PKWF и Printosol, используемые в составе печатных красок.
Абсорбцию можно осуществлять, без каких-либо ограничений, используя любые, известные специалистам способы и условия. Предпочтительно контактирование газовой смеси с абсорбентом осуществляют под давлением от 1 до 50 бар, предпочтительно от 2 до 20 бар, более предпочтительно от 5 до 10 бар, при температуре от 0 до 100°С, в частности от 30 до 50°С. Абсорбцию можно осуществлять как в соответствующих колоннах, так и в аппаратах для водной закалки при одностороннем перемещении потоков или их противотоке. Пригодными абсорбционными колоннами являются, например, тарельчатые колонны с колпачковыми и/или сетчатыми тарелками, колонны со структурированными насадками (например, с листовой насадкой, обладающей удельной поверхностью от 100 до 500 м2/м3, в частности насадкой Mellapak® 250 Y) и колонны, заполненные насадочными телами, например кольцами Рашига. Кроме того, могут использоваться пленочные и распылительные колонны, абсорберы с графитовыми блоками, поверхностные, в частности, тонко- и толстослойные абсорберы, а также тарельчатые скрубберы, горизонтальные скрубберы с механическим перемешиванием и ротационные скрубберы. Кроме того, предпочтительной может оказаться абсорбция в барботажной колонне со встроенными элементами или без них.
Выделение пропана и/или пропена из абсорбента можно осуществлять путем отпаривания, мгновенного испарения (Flashen) и/или дистилляции.
Пропан и/или пропен выделяют из абсорбента предпочтительно путем осуществляемого на стадии (b) отпаривания или десорбции посредством газа, обладающего инертным поведением на стадии (с) согласно изобретению, и/или посредством молекулярного кислорода (например, воздуха). Отпаривание можно осуществлять обычным способом, изменяя давление и/или температуру, предпочтительно при давлении от 0,1 до 10 бар, в частности от 1 до 5 бар, более предпочтительно от 1 до 2 бар, и температуре от 0 до 200°С, в частности от 20 до 200°С, более предпочтительно от 30 до 50°С. Другим газом, пригодным для осуществления отпаривания, является, например, водяной пар, однако предпочтительно использование, в частности, смесей кислорода с азотом, например воздуха. Если для отпаривания используют воздух или смеси кислорода и азота, содержащие свыше 10 об.% кислорода, то до отпаривания или в процессе его осуществления может оказаться целесообразным добавление газа, ограничивающего область взрыва. При этом особенно пригодными являются газы, теплоемкость которых при 20°С превышает 29 Дж/моль·К, в частности метан, этан, пропан, бутан, пентан, гексан, бензол, метанол, этанол, а также аммиак, диоксид углерода и вода. Для осуществления отпаривания особенно пригодны барботажные колонны со встроенными элементами или без них.
Пропан и/или пропен можно также выделять из абсорбента путем дистилляции, для чего могут использоваться хорошо известные специалистам колонны с насадками, насадочными телами или соответствующими встроенными элементами. Предпочтительными являются следующие условия дистилляции: давление от 0,01 до 5 бар, в частности от 0,1 до 3 бар, более предпочтительно от 1 до 2 бар, температура в кубе колонны от 50 до 300°С, в частности от 150 до 250°С.
Если в состав газовой смеси А входит вода, предпочтительным является комбинирование абсорбции с конденсацией влаги (так называемая водная закалка, Wasserquench). Предпочтительным является также осуществление водной закалки после стадии десорбции, что позволяет свести к минимуму потери абсорбента.
Стадию (с) нередко реализуют непосредственно после стадии (b), то есть без осуществления промежуточных технологических операций. Однако не исключается возможность дополнительного выделения абсорбента между стадиями (b) и (с), осуществляемого, например, путем водной закалки.
Окисление пропана и/или пропена до акролеина и/или акриловой кислоты может быть реализовано на стадии (с) любым известным специалистам способом без каких-либо ограничений, причем возможно одно- или двухступенчатое окисление пропена до акролеина и/или акриловой кислоты, окисление пропана до акролеина и/или акриловой кислоты или одновременное окисление пропана и пропена до акролеина и/или акриловой кислоты. Целесообразным является гетерогенное каталитическое газофазное окисление молекулярным кислородом, приводящее к получению смеси газообразных продуктов реакции, содержащих акролеин и/или акриловую кислоту. При необходимости подаваемый на стадию окисления пропан и/или пропен предварительно нагревают до необходимой для осуществления этой реакции температуры путем косвенного теплообмена.
Предпочтительный вариант осуществления стадии (с) способа согласно изобретению предусматривает окисление пропена до акролеина и/или акриловой кислоты.
Гетерогенное каталитическое газофазное окисление пропена до акролеина и/или акриловой кислоты молекулярным кислородом протекает в две следующие друг за другом реакционные ступени, первая из которых приводит к образованию акролеина, а вторая к образованию акриловой кислоты. Протекание реакции в две последовательно реализуемые ступени предоставляет возможность, используя, в сущности, известный способ, осуществлять стадию (с) способа согласно изобретению в двух последовательно расположенных зонах окисления, причем используемый в каждой из них оксидный катализатор может быть оптимизирован соответствующим образом. Так, в первой зоне окисления (пропен → акролеин) предпочтительным, как правило, является использование катализатора на основе оксидов металлов, содержащего комбинацию молибдена, висмута и железа (Mo-Bi-Fe), тогда как для второй зоны окисления (акролеин → акриловая кислота) предпочтительными являются обычные катализаторы на основе оксидов металлов, содержащие комбинацию молибдена и ванадия (Mo-V). Используемые в обеих реакционных зонах катализаторы на основе оксидов металлов неоднократно описаны и хорошо известны специалистам. Например, ссылки на соответствующие патенты США приведены на странице 5 европейской заявки на патент ЕР-А-0253409. Катализаторы, пригодные для использования в обеих зонах окисления, описаны в немецких заявках на патент DE-A 4431957 и DE-A 4431949. Речь в этих документах идет, в частности, о соединениях общей формулы I. Смесь продуктов, образующихся в первой зоне окисления, направляют во вторую зону окисления, как правило, без промежуточной обработки.
Наиболее простая форма реализации обеих зон окисления предусматривает использование кожухотрубного реактора, причем загрузку катализатора в отдельные контактные трубки в месте завершения первой ступени реакции соответствующим образом изменяют (смотри, например, европейские заявки на патент ЕР-А-0911313, ЕР-А-0979813, ЕР-А-0990636 и немецкую заявку на патент DE-A 2830765). Например, в соответствующую часть трубок вместо катализатора, при необходимости, загружают инертный материал.
Предпочтительным, однако, является конструктивное исполнение обеих зон окисления в виде двух последовательно соединенных систем, образованных пучками трубок, которые могут находиться в общем реакторе, причем зону перехода от одного пучка контактных трубок к другому образует инертный, находящийся вне трубок, доступный для прохода реакционных компонентов насыпной материал. Контактные трубки, как правило, омываются теплоносителем, в то время как смывание им находящегося вне контактных трубок инертного материала отсутствует. Поэтому оба пучка контактных трубок предпочтительно помещают в пространственно отделенные друг от друга реакторы. Между этими реакторами, как правило, помещают промежуточный холодильник, чтобы свести к минимуму, при необходимости, происходящее вторичное окисление акролеина, содержащегося в смеси выходящих из первой зоны окисления газообразных продуктов. Вместо трубчатых реакторов могут использоваться также пластинчатые реакторы-теплообменники с солевым и/или испарительным охлаждением, например, описанные в немецких заявках на патент DE-A 19929487 и DE-A 19952964.
Реакционная температура в первой зоне окисления, как правило, составляет от 300 до 450°С, предпочтительно от 320 до 390°С. Реакционная температура во второй зоне окисления, как правило, составляет от 200 до 300°С, часто от 220 до 290°С. Давление в обеих зонах окисления целесообразно поддерживать в интервале от 0,5 до 5 атм, предпочтительно от 1 до 3 атм. Скорость пропускания реакционного газа через катализаторы окисления в обеих зонах реактора, измеряемая в нл/л·час, часто составляет от 1500 до 2500 час-1 или до 4000 час-1. Скорость пропускания пропена в нл/л·час часто составляет от 50 до 300 час-1, в частности от 100 до 200 час-1.
В принципе обе зоны окисления могут иметь конструктивное исполнение, описанное, например, в немецких заявках на патент DE-A 19837517, DE-A 19910506, DE-A 19910508 и DE-A 19837519. Обычно внешнее термостатирование обеих зон окисления, при необходимости, входящих в состав многозонных систем реакторов, известными способами приводят в соответствие с особым составом смеси газообразных реакционных продуктов и загружаемым катализатором.
Согласно изобретению предпочтительно, чтобы при осуществлении способа согласно изобретению пропан, смешанный с пропеном, при гетерогенном каталитическом окислении вел себя подобно газообразному, предпочтительно инертному разбавителю.
Общее количество молекулярного кислорода, необходимого для окисления, может быть сразу добавлено к газу В, однако не исключается возможность дополнительной подачи кислорода в конце первой зоны окисления.
Молярное отношение пропена к молекулярному кислороду в первой зоне окисления предпочтительно устанавливают в интервале от 1:1 до 1:3, часто от 1:1,5 до 1:2. Предпочтительное молярное отношение акролеина к молекулярному кислороду во второй зоне окисления составляет от 1:0,5 до 1:2.
Избыток молекулярного кислорода, как правило, оказывает предпочтительное влияние на кинетику газофазного окисления в той и другой реакционной зоне. Поскольку гетерогенное каталитическое газофазное окисление пропена до акриловой кислоты подлежит кинетическому контролю, пропен в принципе можно использовать в молярном избытке по отношению к молекулярному кислороду, что, например, касается и первой зоны окисления, причем избыточное количество пропена фактически играет роль газообразного разбавителя.
В принципе гетерогенное каталитическое газофазное окисление пропена до акриловой кислоты можно осуществить и в одной единственной зоне окисления. В этом случае обе ступени окисления реализуют в одном реакторе, заполненном катализатором, способным катализировать ту и другую ступени окисления, причем загрузка катализатора вдоль реакционной координаты может изменяться плавно или резко. При варианте осуществления стадии (с) в виде двух последовательно соединенных зон окисления, при необходимости, можно частично или полностью выделять оксид углерода и водяной пар, образующиеся в качестве побочных продуктов в первой зоне окисления, из смеси покидающих эту зону газообразных продуктов, прежде чем подать их во вторую зону окисления. Предпочтительно выбирают способ окисления, не требующий такого выделения.
Для осуществления реакционной стадии (с) наряду с чистым молекулярным кислородом может использоваться молекулярный кислород, разбавленный инертным газом, в частности диоксидом углерода, монооксидом углерода, благородными газами, азотом и/или насыщенными углеводородами.
Целесообразно, по меньшей мере, часть потребности в молекулярном кислороде компенсировать за счет использования воздуха в качестве его источника. В предпочтительном варианте газ В, подаваемый на стадию (с) способа согласно изобретению, по существу состоит только из пропана и пропена, а в качестве источника молекулярного кислорода, необходимого для окисления, используют исключительно воздух. При необходимости путем добавления к подаваемому на стадию (с) газу В холодного воздуха можно способствовать непосредственному охлаждению этого газа.
Если целевым продуктом является акролеин, использовать вторую зону окисления при осуществлении стадии (с) представляется нецелесообразным.
Окисление пропена до акролеина и/или акриловой кислоты на стадии (с) может быть осуществлено так же, как описано в европейской заявке на патент ЕР-А-0117146, патентах США US-A-5198578 и US-A-5183936, или аналогично немецкой заявке на патент DE-A 3313573, патенту Канады СА-А-1217502, патентам США US-A-3161670, US-A-4532365 и международной заявке WO 97/36849. Пригодные способы окисления пропена описаны также в европейских заявках на патент ЕР-А-0293224, ЕР-А-0253409, немецких заявках на патент DE-A 4431957, DE-A 4132263 или DE 19508532, причем предпочтительными являются, в частности, способы окисления, предусматривающие использование газообразных разбавителей.
Окисление акролеина до акриловой кислоты может быть осуществлено в реакторе с псевдоожиженным слоем, как описано в международной заявке WO 00/39065.
Окисление пропена до акролеина и/или акриловой кислоты может быть осуществлено также в пластинчатых реакторах-теплообменниках, описанных в немецкой заявке на патент DE-A 19952964.
Согласно другому предпочтительному варианту стадию (с) способа согласно изобретению осуществляют путем окисления пропана до акролеина и/или акриловой кислоты в одну или несколько ступеней в присутствии соответствующего катализатора, причем пригодными являются любые, известные специалистам способы, один из которых описан, например, в японской заявке на патент JP-A-1036311.
Катализаторами, пригодными для гетерогенного каталитического газофазного окисления пропана до акролеина и/или акриловой кислоты, являются смеси оксидов металлов, общей формулы (I)
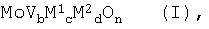
в которой М1 является теллуром (Те) и/или сурьмой (Sb),
М2 является, по меньшей мере, одним элементом из группы, включающей ниобий (Nb), тантал (Та), вольфрам (W), титан (Ti), алюминий (AI), цирконий (Zr), хром (Cr), марганец (Mn), галлий (Ga), железо (Fe), рутений (Ru), кобальт (Со), рений (Rh), никель (Ni), палладий (Pd), платину (Pt), лантан (La), висмут (Bi), бор (В), цезий (Cs), олово (Sn), цинк (Zn), кремний (Si) и индий (In),
b равно от 0,01 до 1,
с равно от больше 0 до 1, предпочтительно от 0,01 до 1,
d равно от больше 0 до 1, предпочтительно от 0,01 до 1,
n является числом, определяемым валентностью и повторяемостью отличающихся от кислорода элементов в формуле (I).
Смеси оксидов металлов, стехиометрический состав которых соответствует формуле (I), известны (смотри, например, европейские заявки на патент ЕР-А-0608838, ЕР-А-0529853, японские заявки на патент JP-A 7-232071, JP-A 10-57813, JP-A 2000-37632, JP-A 10-36311, международную заявку WO 00/29105, Proceedings ISO'99, Sept. 10-11, 1999, G.Centi and S.Perathoner Ed., SCI Pub. 1999, европейскую заявку на патент ЕР-А-0767164, Catalysis Today 49 (1999), S.141-153, европейскую заявку на патент ЕР-А-0962253, Applied Catalysis A: General 194-195 (2000), S.479-485, японскую заявку на патент JP-A 11/169716, европейскую заявку на патент ЕР-А-0895809, немецкую заявку на патент DE-A 19835257, японские заявки на патент JP-A 8-57319, JP-A 10-28862, JP-A-11-43314, JP-A 11-57479, международную заявку на патент WO 00/29106, японские заявки на патент JP-A 10-330343, JP-A 11-285637, JP-A 10-310539, JP-A 11-42434, JP-A 11-343261, JP-A 11-343262, международную заявку на патент WO 99/03825, японские заявки на патент JP-A 7-53448, JP-A 2000-51693 и JP-A 11-263745).
Особенно пригодными являются описанные ниже смеси оксидов металлов (I), (II) и (III).
В смесях оксидов металлов (I) формулы (I) М1 является теллуром (Те) и/или сурьмой (Sb); M2 является, по меньшей мере, одним элементом из группы, включающей ниобий (Nb), тантал (Та), вольфрам (W), титан (Ti), алюминий (AI), цирконий (Zr), хром (Cr), марганец (Mn), железо (Fe), рутений (Ru), кобальт (Со), рений (Rh), никель (Ni), палладий (Pd), платину (Pt), висмут (Bi), бор (В) и цезий (Cs); b равно от 0,01 до 1; с равно от 0,01 до 1; d равно от 0,01 до 1; n является числом, определяемым валентностью и повторяемостью отличающихся от кислорода элементов в составе смеси (I).
Смесь оксидов металлов (I) предпочтительно получают следующим образом. Смесь источников элементарных составляющих смеси оксидов металлов (I) подвергают гидротермальной обработке, выделяют вновь образующийся твердый продукт и путем тепловой обработки переводят его в активный оксид. Предпочтительной является смесь оксидов металлов (I) следующего состава: М1 является теллуром (Те), М2 является ниобием (Nb), b равно 0,1-0,6, с равно 0,05-0,4 и d равно 0,01-0,6. Температура тепловой обработки предпочтительно составляет от 350 до 700°С, причем начальную стадию тепловой обработки осуществляют, в частности, при температуре от 150 до 400°С в кислородсодержащей атмосфере, а заключительную стадию при температуре от 350 до 700°С в атмосфере инертного газа. Пригодные стехиометрические составы смеси оксидов металлов (I) аналогичны приведенным в европейской заявке на патент ЕР-А-0608838, международной заявке на патент WO 00/29106, японской заявке на патент JP-A 11/169716 и европейской заявке на патент ЕР-А-0962253 составам.
О гидротермальном получении предварительных (неактивироавнных) смесей оксидов металлов хорошо известно специалистам (смотри, например, Applied Catalysis A: 194-195 (2000) 479-485, Kinetics and Catalysis, Vol.40, No.3, 1999, pp.401-404, Chem. Commun., 1999, 517-518, японские заявки на патент JP-A 6/227819 и JP-A 2000/26123).
Под гидротермальной обработкой подразумевают, в частности, тепловую обработку предпочтительно тщательно смешанных источников элементарных составляющих желаемой смеси оксидов металлов (I), которую производят в эксплуатируемой под избыточным давлением емкости (автоклаве) в присутствии находящегося под избыточным давлением водяного пара при температуре, обычно составляющей от больше 100 до 600°С. Давление обычно составляет до 500 атм, предпочтительно до 250 атм. Температура может превышать 600°С, а давление может быть выше 500 атм, однако, с технологической точки зрения использование такого режима малоцелесообразно. Особенно предпочтительной является гидротермальная обработка, осуществляемая при одновременном присутствии водяного пара и конденсированной воды. Такая обработка может быть произведена при температуре от больше 100 до 374,15°С (критическая температура воды) и соответствующем давлении. При этом целесообразно использовать такое количество воды, чтобы происходило полное поглощение исходных соединений жидкой фазой с образованием суспензии и/или раствора.
Возможен также такой способ осуществления гидротермальной обработки, согласно которому тщательно перемешанная смесь исходных соединений полностью абсорбирует водный конденсат, находящийся в равновесии с водяным паром.
Гидротермальную обработку предпочтительно осуществляют при температурах больше 100 - 300°С, предпочтительно 150 - 250°С (например, 160 - 180°С). Содержание источников элементарных составляющих желаемой смеси оксидов металлов (I) в смеси воды с этими источниками, находящейся в автоклаве, составляет, как правило, не менее 1 мас.% и обычно не превышает 90 мас.%. Типичное содержание источников элементарных составляющих составляет от 3 до 60 мас.% или от 5 до 30 мас.%, часто от 5 до 15 мас.%.
Гидротермальную обработку можно осуществлять как при перемешивании, так и без перемешивания. В качестве исходных соединений (источников элементарных составляющих смеси оксидов металлов) для гидротермальной обработки пригодны, в частности, любые соединения, которые при нагревании под избыточным давлении в присутствии воды способны образовывать соответствующие оксиды и/или гидроксиды. Для гидротермальной обработки могут совместно использоваться уже готовые оксиды и/или гидроксиды элементарных составляющих или только одни такие оксиды и/или гидроксиды. Источники элементарных составляющих, как правило, используют в тонкодисперсном состоянии.
В качестве источников элементарных составляющих пригодны любые соединения, которые при нагревании, при необходимости, осуществляемом в присутствии воздуха, способны образовывать оксиды и/или гидроксиды. В качестве таких исходных соединений могут совместно использоваться уже готовые оксиды и/или гидроксиды элементарных составляющих или только одни такие оксиды и/или гидроксиды.
Пригодными источниками элементарной составляющей, содержащей молибден (Мо), являются, например, оксиды молибдена, в частности триоксид молибдена, соли молибденовой кислоты, в частности гептамолибдотетрагидрат аммония, и галогениды молибдена, в частности хлорид молибдена.
Пригодными источниками элементарной составляющей, содержащей ванадий (V), являются, например, ванадилацетилацетонат, соли ванадиевой кислоты, в частности метаванадат аммония, оксиды ванадия, в частности пентоксид ванадия (V2O5), галогениды ванадия, в частности тетрахлорид ванадия (VCl4), и оксигалогениды ванадия, в частности оксихлорид ванадия (VOCl3). В качестве исходных соединений целесообразно совместно использовать ванадийсодержащие соединения, в которых ванадий обладает степенью окисления +4.
Пригодными источниками элементарной составляющей, содержащей теллур, являются оксиды теллура, в частности диоксид теллура, металлический теллур, галогениды теллура, в частности дихлорид теллура (TeCl2), а также теллуровые кислоты, в частности ортотеллуровая кислота (H6TeO6).
Предпочтительными исходными соединениями, содержащими сурьму, являются галогениды сурьмы, в частности трихлорид сурьмы (SbCl3), оксиды сурьмы, в частности триоксид сурьмы (Sb2О3), сурьмяные кислоты, в частности HSb(ОН)6, а также сульфат оксида сурьмы ((SbO2)SO4).
Пригодными источниками элементарной составляющей, содержащей ниобий, являются, например, оксиды ниобия, в частности пентоксид ниобия (Nb2O5), оксигалогениды ниобия, в частности оксихлорид ниобия (NbOCl3), галогениды ниобия, в частности хлорид ниобия (NbCl5), а также комплексные соединения ниобия и органических карбоновых и/или дикарбоновых кислот, например оксалаты и алкоголяты ниобия. В качестве источника ниобия пригодны также используемые согласно европейской заявке на патент ЕР-А-0895809 растворы, содержащие ниобий.
Что касается возможных исходных соединений, содержащих другие элементы М2, речь, прежде всего, идет о соответствующих галогенидах, нитратах, формиатах, оксалатах, ацетатах, карбонатах и/или гидроксидах. Пригодными исходными соединениями нередко являются также оксопроизводные, например вольфраматы или соответствующие им кислоты, а также соли аммония.
Пригодными исходными соединениями, кроме того, являются полианионы типа Андерсона, описанные, например, в Polyhedron. Vol.6, No.2, pp.213-218, 1987, которые были использованы для получения соответствующих оксидов металлов (I), например, в Applied Catalysis A: General 194-195 (2000) 479-485, или полианионы, приведенные в цитируемых в этих публикациях литературных источниках. Полианионы типа Андерсона описаны также в Kinetics and Catalysis. Vol.40, No.1999, pp.401-404.
Другими пригодными исходными соединениями являются, например, полианионы типа Давсона или Кеггина. Предпочтительными являются такие исходные соединения, которые при повышенных температурах в присутствии или в отсутствие кислорода превращаются в оксиды, при необходимости, с выделением газообразных продуктов.
Для осуществления гидротермальной обработки, как правило, необходимо длительное время, составляющее от нескольких часов до нескольких дней. Типичная продолжительность гидротермальной обработки составляет 48 часов. С технологической точки зрения такую обработку целесообразно производить в автоклаве с внутренней фторопластовой облицовкой. Автоклав перед гидротермальной обработкой может быть эвакуирован, при необходимости, с одновременным удалением находящейся в нем водной смеси. Перед повышением температуры автоклав может быть заполнен инертным газом (азотом, благородным газом). Можно также отказаться от осуществления обоих указанных мероприятий. Водная смесь перед гидротермальной обработкой может быть инертизирована путем дополнительной продувки инертного газа. Использование указанных инертных газов может оказаться целесообразным и с технологической точки зрения, создавая в автоклаве избыточное давление перед гидротермальной обработкой.
Тепловую обработку твердого продукта, выделенного по завершении гидротермальной обработки, целесообразно производить при температуре от 350 до 700°С, часто при температуре от 400 до 650°С или от 400 до 600°С, причем автоклав по завершении гидротермальной обработки может быть охлажден до комнатной температуры быстро или медленно, то есть в течение длительного промежутка времени (например, без использования принудительного охлаждения). Тепловую обработку можно осуществлять в оксидирующей, восстанавливающей или инертной атмосфере. Для создания оксидирующей атмосферы можно, например, использовать обычный воздух, а также воздух, обогащенный или обедненный молекулярным кислородом. Тепловую обработку предпочтительно осуществляют в инертной атмосфере, то есть, например, в атмосфере молекулярного азота и/или благородного газа. Разумеется, ее можно проводить и под вакуумом.
Если тепловую обработку осуществляют в газообразной атмосфере, обрабатываемый продукт может находиться в виде стационарного или псевдоожиженного слоя.
Общая продолжительность тепловой обработки может составлять 24 часа или более длительное время.
Тепловую обработку предпочтительно начинают в оксидирующей кислородсодержащей атмосфере (например, атмосфере воздуха) при температуре от 150 до 400°С или от 250 до 350°С. По завершении этой начальной стадии целесообразно продолжить тепловую обработку, осуществляя ее в атмосфере инертного газа при температуре от 350 до 700°С, от 400 до 650°С или от 400 до 600°С. Тепловая обработка предварительных катализаторов, полученных в результате гидротермальной обработки, может быть также осуществлена путем таблетирования с последующей тепловой обработкой и измельчением.
С технологической точки зрения полученный в результате гидротермальной обработки твердый продукт перед последующей тепловой обработкой целесообразно подвергнуть измельчению.
Если смеси оксидов металлов (I) получают из исходных соединений, аналогичных используемым в обычных способах получения таких смесей, и тепловую обработку полученной обычным способом, тщательно перемешанной сухой смеси производят аналогично тепловой обработке твердого продукта, полученного путем предварительной гидротермальной обработки, то при соблюдении прочих равных условиях смеси оксидов металлов (I), подвергнутые гидротермальной обработке, при гетерогенном каталитическом газофазном окислении пропана, как правило, обеспечивают более селективный выход акриловой кислоты и обладают более высокой каталитической активностью.
Смеси оксидов металлов (I) могут использоваться в качестве катализаторов как таковые (например, измельченные в порошок или размолотые) или в виде формованных изделий. Катализ можно осуществлять в стационарном, движущемся или псведоожиженном слое.
Рентгеновская дифрактограмма смеси оксидов металлов (I), как правило, в основном аналогична соответствующим дифрактограммам, представленным в европейских заявках на патент ЕР-А-0529853, ЕР-А-0608838 и немецкой заявке на патент DE-A 19835247.
Активные смеси оксидов металлов (I) можно также использовать в сочетании с тонкодисперсными, например, коллоидными материалами, в частности диоксидом кремния, диоксидом титана, оксидом алюминия, оксидом цинка, оксидом ниобия, играющими роль разбавителей.
Массовое отношение разбавителя к активной смеси может достигать 9:1, то есть может составлять также 6:1 и 3:1. Активную смесь можно совмещать с разбавителем перед тепловой обработкой (прокаливанием) и/или после нее. Разбавитель, как правило, вводят перед гидротермальной обработкой. Если его вводят перед тепловой обработкой, он должен оставаться в составе прокаленной смеси, что справедливо и при введении разбавителя перед гидротермальной обработкой. Этому условию, например, как правило, удовлетворяют прокаленные при высоких температурах оксиды.
Другими пригодными для окисления пропана катализаторами являются смеси оксидов металлов (II), обладающие приведенной выше формулой (I), с дифракционными рефлексами h, i и k на соответствующих рентгеновских дифрактограммах, пики которых лежат при углах дифракции (2θ) 2,22±0,4° (h), 27,3±0,4° (i) и 28, 2±0,4° (k), причем
- дифракционный рефлекс h является наиболее сильным на дифрактограмме и обладает максимальной полушириной 0,5°,
- интенсивность Pi дифракционного рефлекса i и интенсивность Pk дифракционного рефлекса k удовлетворяют соотношению 0,65≤R≤0,85, в котором R представляет собой отношение интенсивностей, определенное по формуле R=Рi/(Рi+Pk), и
- полуширина дифракционных рефлексов i и k составляет ≤1.
Предпочтительным является значение R, удовлетворяющее соотношению 0,67≤R≤0,75, еще более предпочтительно значение R, равное 0,70-0,75, или R, равное 0,72.
Предпочтительным является использование смеси оксидов металлов (II), где М1 является теллуром (Те). Кроме того, благоприятно использование смесей оксидов (II), где М2 является ниобием (Nb), танталом (Та), вольфрамом (W) и/или титаном (Ti). Предпочтительными являются смеси, где М2является ниобием (Nb).
Стехиометрический коэффициент «b» в смесях оксидов металлов (II) предпочтительно составляет от 0,1 до 0,6. Предпочтительный диапазон значений стехиометрического коэффициента «с» составляет соответственно от 0,01 до 1 или от 0,05 до 0,4, а предпочтительное значение коэффициента «d» находится в интервале от 0,01 до 1 или от 0,1 до 0,6. Особенно предпочтительным является использование таких смесей оксидов металлов (II), значения стехиометрических коэффициентов «b», «с» и «d» которых находятся в пределах указанных выше предпочтительных интервалов. Другие пригодные стехиометрические составы смесей оксидов металлов (II) приводятся в цитируемых выше документах, соответствующих уровню техники, в частности в японской заявке на патент JP-A 7-53448.
Целенаправленный способ получения смесей оксидов металлов (II) описан, например, в японской заявке на патент JP-A 11-43314, согласно которой такие смеси рекомендуется использовать в качестве катализаторов гетерогенного окислительного дегидрирования этана до этена.
Согласно известному способу, описанному в цитируемых выше документах, соответствующих уровню техники, сначала получают смесь диоксидов металлов формулы (I), состоящую из i-фазы и других фаз (например, k-фазы). Содержание i-фазы в полученной смеси может быть повышено, например, в результате отбора других фаз, например, k-фазы, осуществляемого под микроскопом, или ее промывки соответствующими жидкостями, которыми могут быть, например, водные растворы органических кислот, в частности щавелевой, уксусной, лимонной или винной, неорганических кислот, в частности азотной кислоты, спирты и водные растворы пероксида водорода. Способ получения смесей диоксидов металлов (II) описан также в японской заявке на патент JP-A 7-232071.
Смеси оксидов металлов (II) могут быть получены способом, описанным в немецкой заявке на патент DE-A 198 35247. Согласно этому способу из соответствующих источников элементарных составляющих получают возможно более тщательно перемешанную, предпочтительно тонкодисперсную сухую смесь, которую подвергают тепловой обработке при температуре от 350 до 700°С, от 400 до 650°С или от 400 до 600°С.Тепловую обработку можно осуществлять в оксидирующей, восстанавливающей или инертной атмосфере. Для создания оксидирующей атмосферы используют, например, обычный воздух, а также воздух, обогащенный или обедненный молекулярным кислородом. Тепловую обработку предпочтительно осуществляют в инертной атмосфере, например в атмосфере молекулярного азота и/или благородного газа. Обычно ее осуществляют при нормальном давлении (1 атм). Разумеется, тепловая обработка может быть осуществлена под вакуумом или при пониженном давлении.
Если для тепловой обработки используют газообразную атмосферу, ее можно осуществлять в стационарном или псевдоожиженном слое. Тепловая обработка в общей сложности может занимать 24 часа или более продолжительное время.
Тепловую обработку предпочтительно начинают в оксидирующей кислородсодержащей атмосфере (например, в атмосфере воздуха) при температуре от 150 до 400°С или от 250 до 350°С. По завершении начальной стадии целесообразно продолжить тепловую обработку в атмосфере инертного газа при температуре от 350 до 700°С, от 400 до 650°С или от 400 до 600°С.Тепловая обработка предварительных катализаторов, полученных в результате гидротермальной обработки, может быть также осуществлена следующим образом. Сначала их таблетируют, при необходимости, предварительно измельчив до порошкообразного состояния (таблетирование производят, при необходимости, с добавлением от 0,5 до 2 мас.% тонкодисперсного графита), а затем подвергают тепловой обработке и повторному измельчению.
Исходные соединения для получения смесей оксидов металлов (II) могут быть перемешаны в сухом или мокром состоянии. Если осуществляют сухое перемешивание, исходные соединения целесообразно использовать в виде тонкодисперсного порошка и после перемешивания и, при необходимости, уплотнения подвергнуть прокаливанию (тепловой обработке). Предпочтительным, однако, является перемешивание в мокром состоянии, причем исходные соединения обычно находятся в виде водного раствора и/или суспензии. Полученную в результате перемешивания водную смесь сушат и прокаливают. Под водной смесью предпочтительно подразумевают водный раствор или водную суспензию. Сушку предпочтительно осуществляют непосредственно после получения водной смеси в прямоточной или противоточной распылительной сушилке (температура на выходе из сушилки, как правило, составляет от 100 до 150°С), причем особенно тщательно перемешанную сухую смесь получают, прежде всего, в том случае, если поступающая в сушилку водная смесь представляет собой водный раствор или водную суспензию.
В качестве исходных соединений для получения смеси оксидов металлов (II) (источников элементарных составляющих) пригодны описанные выше соединения, предназначенные для получения смесей оксидов (I).
Использовать смеси оксидов металлов (II) и получать соответствующие формованные изделия можно аналогично описанным выше смесям оксидов металлов (I). Смеси оксидов металлов (II) могут быть подвергнуты формованию, например, путем нанесения на подложку, как описано ниже для смесей оксидов металлов (III), экструзии и/или таблетирования в тонкодисперсном состоянии, которому могут быть подвергнуты также и тонкодисперсные предварительные смеси оксидов (II).
Смеси оксидов металлов (II) можно использовать также подобно смесям оксидов (I) в разбавленном тонкодисперсными материалами состоянии.
Катализаторам на основе смесей оксидов металлов (II) может быть придана форма сферических частиц, полнотелых цилиндров или колец (полых цилиндров), продольный размер которых, как правило, составляет от 1 до 10 мм. Длина цилиндров предпочтительно составляет от 2 до 10 мм, наружный диаметр от 4 до 10 мм. Толщина стенок колец обычно составляет от 1 до 4 мм. Пригодные кольцеообразные частицы катализатора могут обладать также следующими геометрическими параметрами: длиной от 3 до 6 мм, наружным диаметром от 4 до 8 мм и толщиной стенок от 1 до 2 мм. Наряду с этим не исключается возможность использования кольцеобразных частиц, обладающих параметрами 7 мм × 3 мм × 4 мм или 5 мм × 3 мм × 2 мм (наружный диаметр × длина × внутренний диаметр).
Интенсивности дифракционных рефлексов на соответствующей рентгеновской дифрактограмме описаны в немецких заявках на патент DE-A 19835247, DE-A 10051419 и DE-A 10046672.
То есть, если А1 означает пик дифракционного рефлекса 1, В1 означает ближайший минимум, проявляющийся слева от пика А1 на линии рентгеновской дифрактограммы, если смотреть вдоль оси интенсивности, перпендикулярной оси 2θ (плечи дифракционных рефлексов не учитываются), В2 означает ближайший минимум, проявляющийся справа от пика А1, а С1означает точку, в которой прямая, проведенная из пика А1 перпендикулярно оси 2θ, пересекает прямую, соединяющую точки В1 и В2, то интенсивность дифракционного рефлекса 1 определяется длиной отрезка прямой А1, соединяющего пик А1 с точкой С1. Минимум в данном случае означает точку, в которой градиент наклона касательной к кривой в области основания дифракционного рефлекса 1 изменяет значение от отрицательного до положительного, или точку, в которой градиент наклона проходит через ноль, причем для определения градиентов наклона используют координаты оси 2θ и оси интенсивности.
Полушириной в данном случае является длина отрезка прямой, образуемого между точками пересечения Н1 и Н2, если в середине отрезка А1C1провести прямую, параллельную оси 2θ, причем Н1 и Н2 соответственно считаются первыми точками пересечения этой параллельной прямой с указанной выше линией рентгеновской дифрактограммы слева и справа от пика А1.
Пример определения полуширины и интенсивности представлен на фиг.6 немецкой заявки на патент DE-A 10046672.
Наряду с дифракционными рефлексами h, i и k на рентгеновской дифрактограмме предпочтительных каталитически активных смесей оксидов металлов (II), как правило, можно обнаружить дополнительные рефлексы, пикам которых соответствуют следующие углы дифракции (2θ):
9,0±0,4° (l),
6,7±0,4° (о) и
7,9±0,4° (р).
Предпочтительно, если на рентгеновской дифрактограмме каталитически активных смесей оксидов общей формулы (I) присутствует дополнительный дифракционный рефлекс, вершине которого соответствует угол дифракции (2θ):45,2±0,4° (q).
На рентгеновской дифрактограмме смесей оксидов металлов (II) часто можно обнаружить также рефлексы 29,2±0, 4° (m) и 35,4±0,4° (n).
На рентгеновской дифрактограмме некоторых смесей оксидов металлов (II) может отсутствовать дифракционный рефлекс, пику которого соответствует угол 2θ=50,0±0,3°, что свидетельствует об отсутствии в таких смесях k-фазы.
Тем не менее смеси оксидов металлов (II) могут содержать и k-фазу, причем на рентгеновской дифрактограмме, как правило, проявляются и другие рефлексы, пикам которых соответствуют углы дифракции (2θ):
36,2±0,4° и
50, 0±0,4°.
Если принять интенсивность дифракционного рефлекса h за 100, предпочтительно, чтобы указанные ниже дифракционные рефлексы i, I, m, n, о, р, q согласно соответствующей шкале обладали следующими значениями интенсивности:
i: от 5 до 95, часто от 5 до 80, в частности от 10 до 60;
l: от 1 до 30;
m: от 1 до 40;
n: от 1 до 40;
о: от 1 до 30;
р: от 1 до 30 и
q: от 5 до 60.
Если на рентгеновской дифрактограмме присутствуют указанные выше дополнительные рефлексы, их полуширина, как правило, составляет меньше или равна 1°.
Все приведенные выше данные относятся к рентгеновским дифрактограммам, полученным с использованием рентгеновского излучения Cu-Ka (дифрактометр Siemens Theta-Theta D-2000, напряжение на трубке 40 кВ, ток трубки 40 мА, апертурная диафрагма V20 (сменная), диафрагма рассеяния излучения V20 (сменная), диафрагма вторичного монохроматора (0,1 мм), диафрагма детектора (0,6 мм), интервал измерения (2θ) 0,02°, время измерения (в расчете на шаг) 2,4 сек, сцинцилляционный счетчик в качестве детектора).
Удельная поверхность смеси оксидов металлов (II) часто составляет от 1 до 30 м2/г (метод БЭТ, атмосфера азота).
Для окисления пропана пригоден также катализатор (III), представляющий собой каталитически активную смесь оксидов металлов приведенной выше формулы (I), нанесенную на поверхность подложки.
Предпочтительным является использование смесей оксидов общей формулы (I), где М1 является теллуром (Те). В качестве М2 предпочтительны ниобий (Nb), тантал (Та), вольфрам (W) и/или титан (Ti). Предпочтительным является использование в качестве М2 ниобия (Nb).
Предпочтительны следующие интервалы значений стехиометрических коэффициентов для смеси оксидов общей формулы (I) в составе катализатора (III): «b» от 0,1 до 0,6, «с» от 0,01 до 1 или от 0,05 до 0,4 и «d» от 0,01 до 1 или от 0,1 до 0,6. Особенно предпочтительными являются смеси оксидов, стехиометрические коэффициенты «b», «с» и «d» которых одновременно находятся в пределах указанных выше предпочтительных интервалов.
Другие пригодные стехиометрические составы смесей оксидов общей формулы (I) приведены в указанных выше документах, в частности в европейских заявках на патент ЕР-А-0608838, ЕР-А-0962253, международной заявке на патент WO 00/29106 и японской заявке на патент JP-A 11/169716.
Особенно предпочтительным является также получение катализатора (III) путем нанесения описанных выше смесей оксидов металлов (II) в качестве смесей оксидов, обладающих формулой (I), на соответствующий носитель.
Носители являются предпочтительно химически инертными материалами, то есть они преимущественно не участвуют в протекании каталитического газофазного окисления пропана до акриловой кислоты. В качестве материала носителя могут использоваться, например, оксид алюминия, диоксид кремния, силикаты, в частности глина, каолин, стеатит, пемза, алюмосиликат и силикат магния, карбид кремния, диоксид цинка, диоксид тория.
Поверхность частиц носителя может быть гладкой или шероховатой. Предпочтительными являются носители, обладающие шероховатой поверхностью, поскольку шероховатость обеспечивает повышение адгезии нанесенной на нее активной смеси оксидов.
Шероховатость поверхности Rz носителя часто находится в интервале от 2 до 200 мк, в частности в интервале от 20 до 100 мкм (определение шероховатости по DIN 4768, лист 1, с использованием тестера Хоммеля для измерения параметров поверхности по DIN-ISO, производство фирмы Hommelwerke, Германия).
Наряду с этим носитель может обладать пористой или монолитной структурой. Целесообразным является использование монолитного носителя с отношением общего объема пор к объему носителя, меньшим или равным 1 об.%.
Толщина оболочки, образованной на поверхности носителя активной смесью оксидов металла, предпочтительно составляет от 10 до 1000 мкм. Она может составлять также от 50 до 700, 100 до 600, 300 до 500 или 150 до 400 мкм. Возможной является также толщина оболочки от 10 до 500, 100 до 500 или 150 до 300 мкм.
В принципе частицы носителя могут обладать любыми геометрическими параметрами, причем продольный размер, как правило, составляет от 1 до 10 мм. Предпочтительными являются носители в виде сферических или цилиндрических частиц, в частности в виде полых цилиндров (колец). Предпочтительный диаметр сферических частиц носителя составляет от 1,5 до 4 мм. Если частицы носителя обладают цилиндрической формой, их длина предпочтительно составляет от 2 до 10 мм, наружный диаметр предпочтительно от 4 до 10 мм. Толщина стенок кольцеобразных частиц носителя обычно составляет от 1 до 4 мм. Пригодные кольцеобразные частицы могут обладать длиной от 3 до 6 мм, наружным диаметром от 4 до 8 мм и толщиной стенок от 1 до 2 мм. Может использоваться также носитель в виде частиц с геометрическими параметрами 7 мм × 3 мм × 4 мм или 5 мм × 3 мм × 2 мм (наружный диаметр × длина × внутренний диаметр).
Простейший способ получения катализаторов (III) заключается в приготовлении активных смесей оксидов общей формулы (I), их переводе в тонкодисперсное состояние и нанесении на поверхность частиц носителя посредством жидкого связующего. Для этого поверхность частиц увлажняют жидким связующим, на увлажненной поверхности бесконтактным способом фиксируют слой тонкодисперсной активной смеси оксидов общей формулы (I), после чего осуществляют сушку частиц носителя с покрытием. Неоднократно повторяя описанный процесс, можно наносить все новые и новые слои, получая носитель с повышенной толщиной покрытия.
Дисперсность наносимой на поверхность частиц носителя каталитически активной смеси оксидов общей формулы (I) подбирают в зависимости от желаемой толщины покрытия. Для оболочек толщиной от 100 до 500 мкм пригодны, например, порошкообразные активные смеси, по меньшей мере, 50% частиц от общего количества которых проходят через сито с размером отверстий от 1 до 20 мкм, а содержание частиц с продольным размером, превышающим 50 мкм, составляет менее 10%. Дисперсность частиц порошкообразных смесей оксидов, определяемая величиной продольного размера, как правило, описывается распределением Гаусса, что обусловлено способом их получения.
Описанный выше способ нанесения покрытия на носитель в техническом масштабе рекомендуется осуществлять, используя, например, изложенный в немецкой заявке на патент DE-A 2909671 технологический принцип. Это означает, что носители, на которые должно быть нанесено покрытие, загружают во вращающуюся предпочтительно в наклонном положении емкость, например в ротационный дисковый резервуар или дражировочный барабан, причем угол наклона (угол между центральной осью вращающейся емкости и горизонтальной плоскостью), как правило, составляет от 0 до 90°, чаще всего от 30 до 90°. Находящийся во вращающемся резервуаре носитель в виде частиц, обладающих, например, сферической или цилиндрической формой, проходит под двумя расположенными на определенном расстоянии друг от друга дозирующими устройствами. Целесообразно, чтобы посредством первого из дозаторов, представляющего собой форсунку (например, приводимое в действие сжатым воздухом распылительное сопло), осуществлялось напыление жидкого связующего на поверхность частиц носителя, перекатывающихся на вращающихся тарелках, с одновременным контролем степени увлажнения.
Второе дозирующее устройство расположено вне конуса распыления жидкого связующего и служит для подачи тонкодисперсной активной смеси оксидов, например, посредством качающегося желоба или шнека. Частицы носителя, например, сферической или цилиндрической формы, увлажненные контролируемым количеством жидкого связующего, поглощают дозируемую порошкообразную активную смесь, которая уплотняется на наружной поверхности частиц, образуя прочно присоединенную к ней оболочку.
При необходимости частицы носителя с созданным описанным выше способом слоем покрытия при продолжающемся вращении резервуара вновь проходят под распылительной форсункой, где их подвергают контролируемому увлажнению, чтобы в процессе дальнейшего перемещения можно было нанести дополнительный слой тонкодисперсной активной смеси оксидов, причем этот процесс может быть повторен неоднократно (промежуточная сушка носителя, как правило, не требуется). Тонкодисперсную смесь оксидов и жидкое связующее при этом, как правило, подают непрерывно и синхронно.
Избыток жидкого связующего можно удалить по завершении нанесения покрытия, например, путем продувки носителя горячими газами, в частности горячим азотом или воздухом. Следует отметить, что описанный способ нанесения покрытий обеспечивает отличную взаимную адгезию последовательно наносимых слоев и отличное сцепление основного слоя с поверхностью носителя.
Важная особенность описанного выше способа нанесения покрытия состоит в том, что увлажнение поверхности частиц носителя осуществляют контролируемым образом. То есть поверхность частиц носителя рекомендуется увлажнять таким образом, чтобы жидкое связующее адсорбировалось на ней без образования жидкой фазы, которая была бы заметна при визуальном наблюдении. Слишком сильное увлажнение поверхности носителя приводит к объединению частиц тонкодисперсной каталитически активной смеси оксидов в агломераты вместо их равномерного распределения по поверхности частиц. Более подробная информация по этому поводу приводится в немецкой заявке на патент DE-A 2909671.
Упомянутое выше удаление жидкого связующего может быть осуществлено контролируемым образом, например, путем испарения и/или сублимирования. В наиболее простом случае связующее может быть удалено путем продувки носителя газом, подогретым до соответствующей температуры, часто составляющей от 50 до 300°С, в частности 150°С. Благодаря воздействию горячих газов может быть осуществлена и предварительная сушка носителя. Окончательная же сушка носителя может быть произведена в сушильных печах любого типа, например в ленточных сушилках, при этом воздействующая на носитель температура не должна превышать температуру, при которой осуществляют прокаливание (тепловую обработку) при получении активной смеси оксидов. Сушку также можно произвести с использованием только сушильной печи.
Тип связующего, используемого для нанесения покрытия, не зависит от типа и геометрических параметров частиц носителя. В качестве связующего можно использовать воду, одновалентные спирты, в частности этанол, метанол, пропанол и бутанол, поливалентные спирты, в частности этиленгликоль, 1,4-бутандиол, 1,6-гександиол или глицерин, одно- или поливалентные карбоновые кислоты, в частности пропионовую, щавелевую, малоновую, глутаровую или малеиновую кислоту, аимноспирты, в частности этаноламин или диэтаноламин, а также одно- или поливалентные органические амиды, в частности формамид. Предпочтительные связующие представляют собой также растворы, состоящие из воды (от 20 до 90 мас.%) и растворенного в ней органического соединения (от 10 до 80 мас.%), температура кипения или сублимации которого при нормальном давлении (1 атм) превышает 100°С, предпочтительно 150°С. Органическое соединение предпочтительно выбирают из приведенного выше перечня возможных органических связующих. Предпочтительное содержание органического соединения в указанных водных растворах связующего составляет от 10 до 50 мас.%, особенно предпочтительно от 20 до 30 мас.%. В качестве органических компонентов пригодны также моносахариды и олигосахариды, в частности глюкозы, фруктозы, сахарозы или лактозы, а также полиэтиленоксиды и полиакрилаты.
Каталитически активные смеси оксидов общей формулы (I) могут быть получены известными способами, описанными в цитированных выше документах согласно уровню техники. Это означает, что они могут быть получены, например, как гидротермальным, так и обычным способами.
В последнем случае каталитически активные смеси оксидов общей формулы (I) получают, готовя из соответствующих источников элементарных составляющих возможно более тщательно перемешанную, предпочтительно тонкодисперсную сухую смесь и подвергая ее тепловой обработке при температуре от 350 до 700°С, от 400 до 650°С или от 400 до 600°С. Тепловую обработку и тщательное перемешивание исходных соединений можно производить аналогично описанному выше способу получения смеси оксидов металлов (II).
В рамках описанного выше способа получения каталитически активных смесей оксидов металлов общей формулы (I) в качестве источников элементарных составляющих пригодны исходные соединения, которые используют для получения описанной выше смеси оксидов металлов (I).
Особенно предпочтительными катализаторами являются носители с покрытием, состоящим из смеси оксидов металлов (II) в качестве каталитически активной смеси оксидов общей формулы (I).
Для получения катализаторов с покрытием пригодны также активные смеси оксидов общей формулы (I), описанные в международной заявке на патент WO 00/29106 и обладающие преимущественно аморфной структурой, которая обнаруживается на рентгеновской дифрактограмме в виде очень широких дифракционных рефлексов с пиками в области углов дифракции (2θ) 22° и 27°.
Пригодными являются также активные смеси оксидов общей формулы (I), описанные в европейских заявках на патент ЕР-А-0529853 и ЕР-А-0608838, на рентгеновских дифрактограммах которых обнаруживаются очень узкие дифракционные рефлексы с пиками в области углов дифракции (2θ) 22,1±0,3°, 28,2±0,3°, 36,2±0,3°, 45,2±0,3° и 50,0±0,3°.
Катализаторы в виде носителей с покрытием могут быть получены не только путем нанесения на увлажненную поверхность частиц носителя готовых тонкодисперсных активных смесей оксидов общей формулы (I), но и путем нанесения на подготовленную аналогичным образом поверхность тонкодисперсных предварительных смесей оксидов, осуществляемого аналогичными способами с использованием аналогичных связующих, и прокаливания носителя с нанесенным на него покрытием после предварительной сушки. Тонкодисперсная предварительная смесь оксидов может быть получена, например, следующим образом. Сначала из источников элементарных составляющих активной смеси оксидов общей формулы (I) желаемого состава получают возможно более тщательно перемешанную тонкодисперсную сухую смесь (например, путем распылительной сушки водной суспензии или раствора источников элементарных составляющих), а затем эту смесь в течение нескольких часов подвергают тепловой обработке (при необходимости, после таблетирования, осуществляемого с добавлением тонкодисперсного графита в количестве от 0,5 до 2 мас.%) при температуре от 150 до 350°С, предпочтительно от 250 до 350°С, в оксидирующей кислородсодержащей атмосфере (например, атмосфере воздуха) и, при необходимости, размалыванию. После нанесения на частицы носителя предварительной смеси их прокаливают предпочтительно в атмосфере инертного газа (можно использовать любую другую атмосферу) при температуре от 360 до 700°С, от 400 до 650°С или от 400 до 600°С.
Описанные выше смеси оксидов металлов (II) или катализаторы (III) в сочетании со смесью оксидов металлов (II) в качестве каталитически активной смеси также можно использовать для окисления пропена, которое можно осуществлять в присутствии пропана, играющего роль газообразного разбавителя, хотя часть его и может окисляться до акриловой кислоты.
Окисление пропана может быть осуществлено любыми, известными специалистам способами без каких-либо ограничений, например способом, описанным в европейской заявке на патент ЕР-А-0608838 или международной заявке на патент WO 00/29106. Это означает, что газ В, подаваемый на стадию (с) для каталитического окисления, осуществляемого при температуре, например, от 200 до 550°С, от 230 до 480°С или от 300 до 440°С, может обладать, например, следующим составом:
Другими возможными составами газовой смеси В, подаваемой на стадию (с) являются, в частности, следующие:
Для окисления пропана пригодны также описанные в немецкой заявке на патент DE-A 199 52964 пластинчатые реакторы-теплообменники. Согласно другому варианту выполнения изобретения окисление пропана осуществляют способами, описанными в немецких заявках на патент DE-A 19837517, DE-A 19837518, DE-A 19837519 и DE-A 19837520.
В состав реакционной смеси, образующейся в результате окисления пропена и/или пропана на стадии (с) по способу согласно изобретению, наряду с преимущественно содержащимися в ней целевыми продуктами, то есть акролеином и/или акриловой кислотой, как правило, входят непревращенные молекулярный кислород, пропан и пропен, молекулярный азот, водяной пар и диоксид углерода, образующиеся в качестве побочных продуктов и/или используемые в качестве газообразных разбавителей, а также небольшие количества низкомолекулярных альдегидов, углеводородов и других инертных газоообразных разбавителей.
Целевые продукты (акролеин и/или акриловая кислота) могут быть выделены из смеси продуктов окисления известными способами, например путем азеотропной отгонки, фракционированной дистилляции, осуществляемой в присутствии растворителя или без него, или кристаллизации.
Например, пригодными способами выделения являются частичная конденсация акриловой кислоты, ее абсорбция водой или высококипящим гидрофобным органическим растворителем или абсорбция акролеина водой или водными растворами низкомолекулярных карбоновых кислот с последующей обработкой абсорбата. Альтернативным способом выделения целевых продуктов из реакционной смеси является фракционированная конденсация (смотри, например, европейскую заявку на патент ЕР-А-0117146, немецкие заявки на патент DE-A 4308087, DE-A 4335172, DE-А 4436243, DE-A 19924532 и DE-A 19924533.
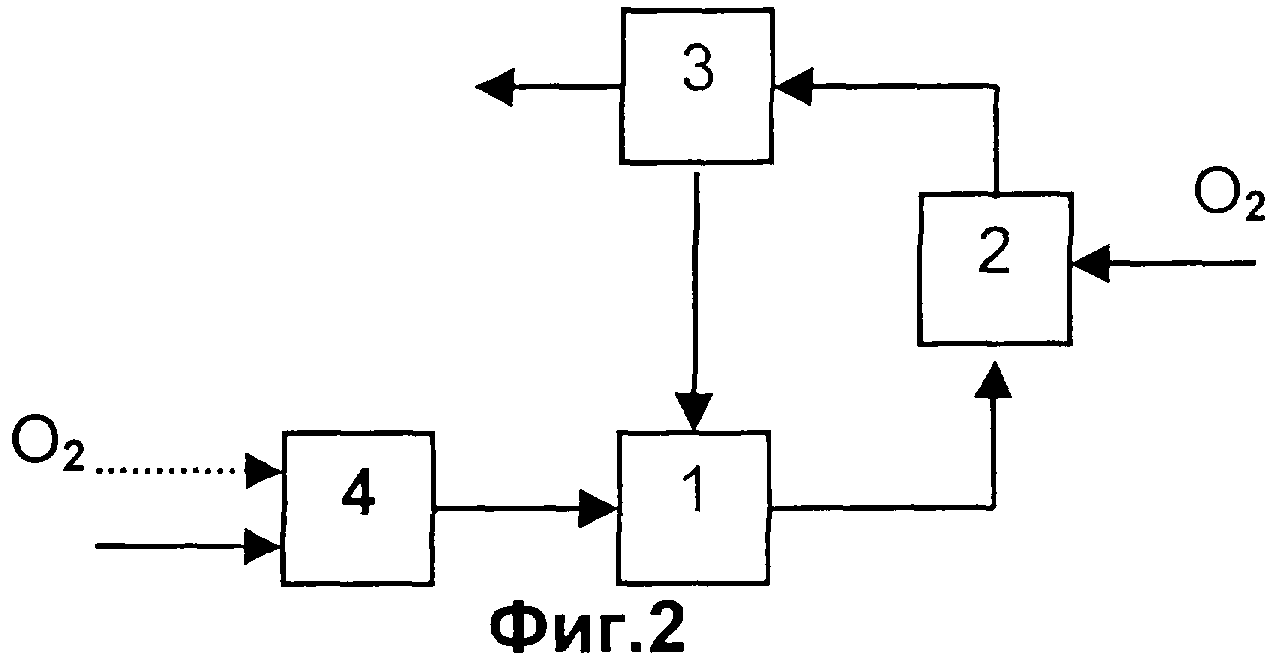
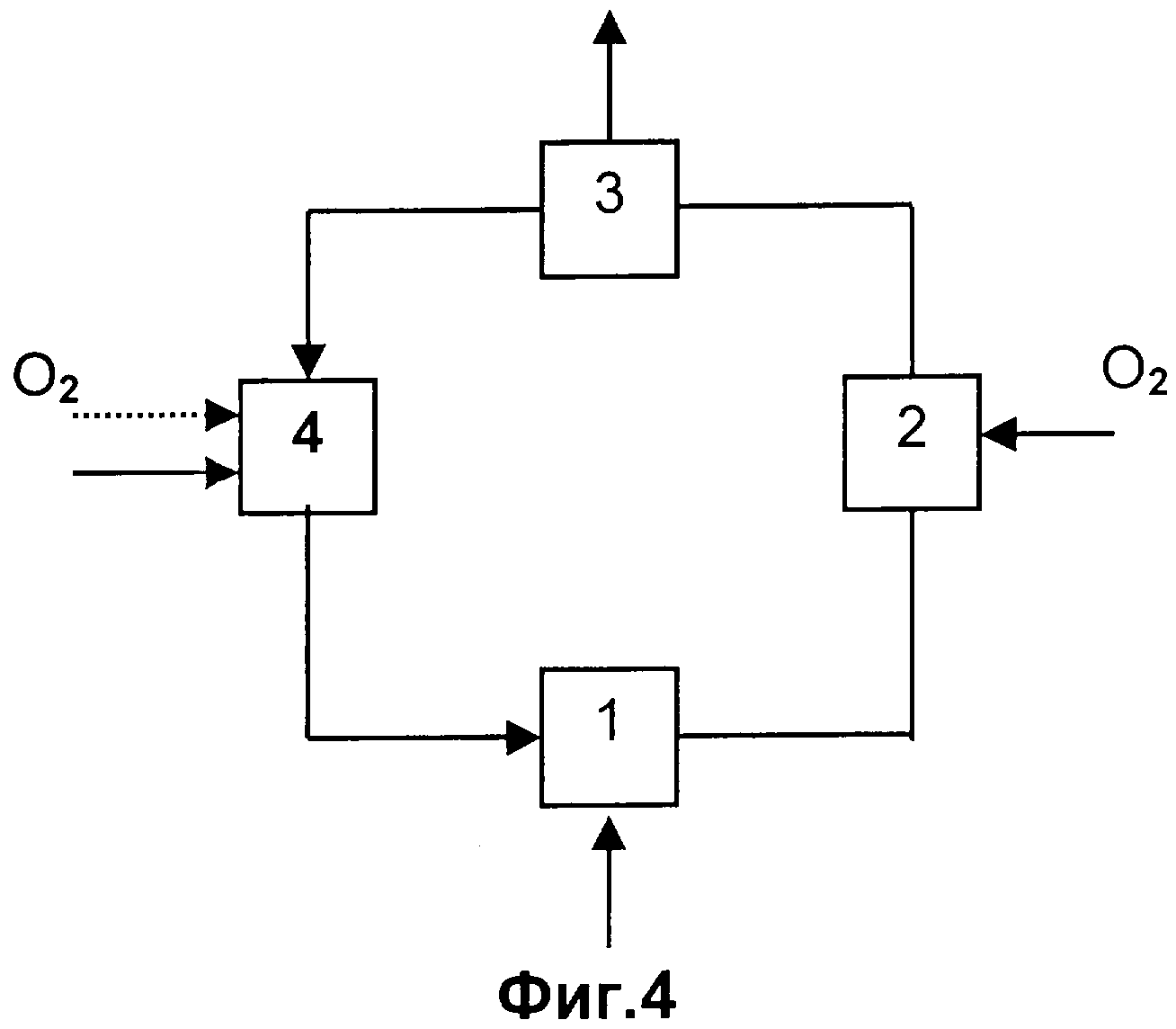
Согласно особенно предпочтительному варианту выполнения способа согласно изобретению непревращенные на стадии (с) пропан и/или пропен выделяют из остающейся после выделения целевого продукта газовой смеси, реализуя стадии (а) и (b), и вновь возвращают их на стадию (с).
Газовая смесь А, используемая на стадии (а) способа согласно изобретению, может иметь состав, аналогичный составу газовой смеси, образующейся в результате каталитического дегидрирования пропана до пропена. Причем дегидрирование пропана может быть окислительным, то есть осуществляемым с добавлением кислорода, или без добавления кислорода, в частности, преимущественно без добавления кислорода. Если речь идет о дегидрировании с добавлением кислорода, возможны два следующих варианта. В первом варианте весь образующийся водород окисляется за счет использования избыточного количества кислорода, в результате чего водород полностью отсутствует в газообразных продуктах дегидрирования, но они содержат избыточный кислород (окислительное дегидрирование). Во втором варианте кислород подают лишь в том количестве, которое необходимо, чтобы компенсировать тепловой эффект реакции, поэтому кислород в газообразных продуктах реакции полностью отсутствует, однако они содержат водород (автотермический процесс). Дегидрирование пропана может осуществляться каталитическим или некаталитическим (гомогенным) способом.
Дегидрирование пропана может быть осуществлено, например, так, как описано в немецкой заявке на патент DE-A 3313573 и европейской заявке на патент ЕР-А-0117146.
В принципе окислительное дегидрирование может быть осуществлено в виде гомогенного и/или гетерогенного оксидегидрирования пропана до пропена с использованием молекулярного кислорода.
Если эта первая ступень реакции состоит в гомогенном окислительном дегидрировании, в принципе оно может быть реализовано, как описано в патенте США US-A-3798 283, китайской заявке на патент CN-A-1105352, Applied Catalysis, 70(2), 1991, S.175-187, Catalysis Today 13, 1992, S.673-678, и немецкой заявке на патент DE-A-19622331, причем в качестве источника кислорода можно использовать также воздух.
Целесообразный температурный интервал гомогенного окислительного дегидрирования составляет от 300 до 700°С, предпочтительно от 400 до 600°С, особенно предпочтительно от 400 до 500°С. Рабочее давление может составлять от 0,5 до 100 бар, в частности от 1 до 10 бар. Время пребывания обычно составляет от 0,1 или 0,5 до 20 сек, предпочтительно от 0,1 или 0,5 до 5 сек. В качестве реактора можно использовать, например, трубчатую печь или кожухотрубный реактор, например противоточную трубчатую печь с дымовым газом в качестве теплоносителя или кожухотрубный реактор с солевым расплавом в качестве теплоносителя. Соотношение пропан : кислород в газовой смеси, выходящей из реактора, предпочтительно составляет от 0,5:1 до 40:1, в частности от 1:1 до 6:1, более предпочтительно от 2:1 до 5:1. Газовая смесь может содержать и другие, преимущественно инертные компоненты, в частности воду, диоксид углерода, монооксид углерода, азот, благородные газы и/или пропен, при этом речь может идти и о возвращаемых на стадию (а) компонентах, которые в общем случае обозначают как рециркулирующий газ.
Если дегидрирование пропана осуществляют в виде гетерогенного каталитического окислительного дегидрирования, то в принципе оно может быть реализовано, как описано, например, в патенте США US-A 4788371, китайской заявке на патент CN-A 1073893, Catalysis Letters 23 (1994), 103-106, W.Zhang, Gaodeng Xuexiao Huaxue Xuebao, 14 (1993) 566, Z.Huang, Shiyou Huagong, 21 (1992), 592, международной заявке на патент WO 97/36849, немецкой заявке на патент DE-A 19753817, патентах США US-A 3862256 и US-A 3887631, немецкой заявке на патент DE-A 19530454, патенте США US-A 4341664, J. of Catalysis 167, 560-569 (1997), J. of Catalysis 167, 550-559 (1997), Topics in Catalysis 3 (1996), 265-275, патенте США 5086032, Catalysis Letters 10 (1991), 181-192, Ind. Eng. Chem. Res. 1996, 35, 14-18, патенте США US-А 4255284, Applied Catalysis A.: General, 100 (1993), 111-130, J. of Catalysis 148, 56-67 (1994), V.Cortes Corberan und S.Vic.Bellon (Ed.), New Developments in Selective Oxidation II, 1994, Elsevier Science B.V., S.305-313, 3rd World Congress on Oxidation Catalysis, R.K. Grasselli, S.T. Oyama, A.M.Gaffney and J.E.Lyons (Ed.), 1997, Elsevier Science B.V., S.375 ff. В качестве источника кислорода можно использовать воздух, однако предпочтительным является источник, содержащий, по меньшей мере, 90 мол.% кислорода, более предпочтительно 95 мол.% кислорода (относительно источника, содержащего 100 мол.% кислорода).
Для гетерогенного каталитического окислительного дегидрирования без каких-либо особых ограничений пригодны любые, известные специалистам в этой области химической технологии катализаторы, способные катализировать окисление пропана до пропена. В частности, могут быть использованы любые, приведенные в перечисленных выше документах катализаторы окислительного дегидрирования. Предпочтительными являются, например, смеси оксидов молибдена, ванадия и ниобия или ванадилпирофосфат, используемые совместно с промотором. Особенно пригодным катализатором является, например, смесь оксидов металлов, содержащая в качестве основных компонентов молибден (Мо), ванадий (V), теллур (Те), кислород (О) и X, причем Х представляет собой, по меньшей мере, один элемент, выбранный из группы, включающей ниобий, тантал, вольфрам, титан, алюминий, цирконий, хром, марганец, железо, рутений, кобальт, родий, никель, палладий, платину, сурьму, висмут, бор, индий и церий. Кроме того, особенно пригодными катализаторами окислительного дегидрирования являются смеси нескольких оксидов металлов или катализаторы А, описанные в немецкой заявке на патент DE-A-19753817, причем смеси оксидов металлов или катализаторы А, указанные в данной заявке в качестве предпочтительных, обладают особенно благоприятным каталитическим действием. Это означает, что в качестве активных смесей используют, в частности, смеси нескольких оксидов металлов (IV) общей формулы IV
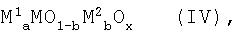
в которой М1 является кобальтом (Со), никелем (Ni), магнием (Mg), цинком (Zn), марганцем (Mn) и/или медью (Cu),
М2 является вольфрамом (W), ванадием (V), теллуром (Те), ниобием (Nb), фосфором (Р), хромом (Cr), железом (Fe), сурьмой (Sb), цезием (Cs), оловом (Sn) и/или лантаном (La),
а равно 0,5-1,5,
b равно 0-0,5,
x является числом, определяемым валентностью и повторяемостью отличающихся от кислорода элементов в формуле (IV).
Пригодные активные смеси (IV) в принципе могут быть получены следующим простым способом. Из подходящих источников элементарных составляющих получают возможно более тщательно перемешанную, предпочтительно тонкодисперсную сухую смесь необходимого стехиометрического состава и прокаливают ее при температуре от 450 до 1000°С. В качестве источников элементарных составляющих активных смесей оксидов металлов (IV) используют оксиды и/или соединения, которые путем нагревания, осуществляемого, по меньшей мере, в присутствии кислорода, способны превращаться в оксиды. Речь при этом, прежде всего, идет о галогенидах, нитратах, формиатах, оксалатах, цитратах, карбонатах, комплексных аминосолях, солях аммония и/или гидроксидах. Тщательное перемешивание исходных соединений для получения смесей оксидов металлов (IV) можно осуществлять сухим способом, например, используя соответствующие тонкодисперсные порошки, или мокрым способом, например, в присутствии воды в качестве растворителя. Смеси оксидов металлов (IV) могут использоваться в порошкообразном состоянии или в виде формованных частиц, обладающих определенными геометрическими параметрами, причем формование может быть произведено до или после прокаливания. Могут использоваться полные катализаторы или порошкообразной активной или предварительной смеси оксидов может быть придана определенная форма путем их нанесения на предварительно сформованный инертный носитель. Материалами носителя могут быть обычные пористые или монолитные оксиды алюминия, диоксид кремния, диоксид тория, диоксид цинка, карбид кремния или силикаты, причем частицы носителя могут обладать правильной или неправильной формой.
Температура гетерогенного каталитического окислительного дегидрирования пропана предпочтительно лежит в интервале от 200 до 600°С, в частности в интервале от 250 до 500°С, более предпочтительно в интервале от 350 до 440° С. Рабочее давление предпочтительно составляет от 0,5 до 10 бар, в частности от 1 до 10 бар, более предпочтительно от 1 до 5 бар. Особенно предпочтительным является рабочее давление более 1 бар, например от 1,5 до 10 бар. Гетерогенное окислительное дегидрирование пропана производят, используя, как правило, стационарный слой катализатора, который согласно целесообразному варианту осуществления способа загружают в трубки кожухотрубного реактора, описанного, например, в европейских заявках на патент ЕР-А-0700893 и ЕР-А-0700714, а также в цитируемой в этих заявках литературе. Целесообразное среднее время пребывания реакционной смеси в слое катализатора составляет от 0,5 до 20 сек. Отношение пропана к кислороду варьируют в зависимости от желаемой степени превращения пропана и селективности катализатора, причем целесообразное отношение составляет от 0,5:1 до 40:1, в частности от 1:1 до 6:1, более предпочтительно от 2:1 до 5:1. Селективность выхода пропена с увеличением степени превращения пропана, как правило, уменьшается, в связи с чем дегидрирование пропана предпочтительно осуществляют таким образом, чтобы обеспечить высокую селективность выхода пропена при относительно невысоких степенях превращения пропана. Особенно предпочтительной степени превращения пропана соответствует интервал от 5 до 40%, более предпочтительно от 10 до 30%, при этом под степенью превращения подразумевают ту часть пропана от его общего вводимого количества, которая претерпевает превращение. Особенно предпочтительной селективности соответствует интервал от 50 до 98%, более предпочтительно от 80 до 98%, причем термин «селективность» означает процентное отношение числа молей полученного пропена к числу молей превращенного пропана.
В 100 мас.% исходной смеси, используемой для окислительного дегидрирования пропана, предпочтительно содержится от 5 до 95 мас.% пропана. Исходная смесь наряду с пропаном и кислородом может содержать и другие, в частности, инертные составляющие, например воду, диоксид углерода, монооксид углерода, азот, инертные газы и/или пропен. Гетерогенное окислительное дегидрирование можно осуществлять также в присутствии разбавителей, например водяного пара.
Как гомогенное, так и гетерогенное каталитическое окислительное дегидрирование может быть осуществлено в любой, известной специалистам последовательности. Например, дегидрирование может происходить в одну, две или несколько стадий, в промежутки между которыми осуществляют подачу кислорода. Кроме того, предоставляется возможность комбинирования гомогенного и гетерогенного каталитического окислительного дегидрирования.
Возможными составляющими реакционной смеси, образующейся в результате окислительного дегидрирования пропана, являются, например, следующие соединения: пропен, пропан, диоксид углерода, монооксид углерода, вода, азот, кислород, этан, этен, метан, акролеин, акриловая кислота, этиленоксид, бутан, уксусная кислота, формальдегид, муравьиная кислота, пропиленоксид и бутен. В 100 мас.% смеси продуктов, получаемых в результате окислительного дегидрирования пропана, предпочтительно содержится от 5 до 10 мас.% пропена, от 1 до 2 мас.% монооксида углерода, от 1 до 3% мас, диоксида углерода, от 4 до 10 мас.% воды, от 0 до 1 мас.% азота, от 0,1 до 0,5 мас.% акролеина, от 0 до 1 мас.% акриловой кислоты, от 0,05 до 0,2 мас.% уксусной кислоты, от 0,01 до 0,05 мас.% формальдегида, от 1 до 5 мас.% кислорода, от 0,1 до 1,0 мас.% других, приведенных выше компонентов, в частности остаточного пропана.
В общем случае газовая смесь А может быть получена также путем гетерогенного каталитического дегидрирования пропана, преимущественно осуществляемого в присутствии избыточного количества кислорода, как описано в немецкой заявке на патент DE-A 3313573 или изложено ниже.
Поскольку дегидрирование сопровождается увеличением объема, степень превращения пропана можно повысить путем снижения его парциального давления. Простейшим способом достижения этой цели может быть, например, осуществление дегидрирования пропана при пониженном давлении и/или введение в состав исходного газа преимущественно инертных газообразных разбавителей, например водяного пара, который при обычном дегидрировании ведет себя подобно инертному газу. Дополнительным преимуществом разбавления пропана водяным паром является, как правило, уменьшение коксования катализатора дегидрирования, поскольку водяной пар реагирует с образующимся коксом по принципу газификации угля. Кроме того, водяной пар может служить газообразным разбавителем смеси, подаваемой на последующее окисление (стадию (с)). Наряду с этим водяной пар можно частично или полностью выделять из газовой смеси, подаваемой на стадию (с), например, путем конденсации, что предоставляет возможность увеличения содержания в этой смеси азота, используемого в качестве газообразного разбавителя. Другими пригодными для дегидрирования пропана газообразными разбавителями являются, например, монооксид углерода, диоксид углерода, азот и благородные газы, в частности ксенон, неон и аргон. Указанные газообразные разбавители можно использовать как по отдельности, так и в смеси друг с другом. В предпочтительном варианте они, как правило, пригодны и для разбавления подаваемой на стадию (с) газовой смеси. В общем случае предпочтительным является использование для осуществления соответствующих реакций инертного газообразного разбавителя (допустимое химическое изменение разбавителя не превышает 5 мол.%, предпочтительно составляет не более 3 мол.% и более предпочтительно не более 1 мол.%). Для дегидрирования пропана в принципе пригодны любые катализаторы дегидрирования согласно уровню техники, которые грубо можно разделить на две следующие группы. Первую группу составляют катализаторы, обладающие оксидной природой, например оксид хрома и/или оксид алюминия. Вторую группу образуют катализаторы, представляющие собой, по меньшей мере, один, как правило, благородный металл (например, платину), нанесенный на носитель, как правило, представляющий собой оксид.
Наряду с прочими могут использоваться катализаторы дегидрирования, рекомендованные в международной заявке на патент WO 99/46039, патенте США US-A 4788371, европейской заявке на патент ЕР-А-0705136, международном патенте WO 99/29420, патентах США US-A 4220091, US-A 5430220 и US-A 5877369, европейской заявке на патент ЕР-А-0 117146, немецких заявках на патент DE-A 19937196, DE-A 19937105 и DE-A 19937107. В частности, могут использоваться катализаторы, полученные согласно примерам 1, 2, 3 и 4, представленным в немецкой заявке на патент DE-A 19937107.
Речь при этом идет о катализаторах дегидрирования, содержащих от 10 до 99,9 мас.% диоксида цинка, от 0 до 60 мас.% оксида алюминия, диоксида кремния и/или диоксида титана и от 0,1 до 10 мас.%, по меньшей мере, одного элемента из первой и второй главных групп, одного элемента из третьей подгруппы, одного элемента из восьмой подгруппы периодической системы элементов, лантана и/или олова, при условии, что соответствующая сумма составляет 100 мас.%.
Для дегидрирования пропана в принципе пригодны любые, соответствующие уровню техники типы реакторов и варианты осуществления способа. Описания таких вариантов приводятся, например, во всех документах, касающихся катализаторов дегидрирования согласно уровню техники.
Достаточно подробное описание пригодного способа дегидрирования согласно изобретению приводится также в «Catalytical Studies Division, Oxidative Dehydrogenation and Alternative Dehydrogenation Processes, Study Number 4192 OD, 1993, 430 Ferguson Drive, Mountain View, California, 94043-5272, USA».
Характерной особенностью частичного гетерогенного каталитического дегидрирования пропана является эндотермичность. Это означает, что тепло (тепловая энергия), необходимое для установления требуемой температуры реакции, должно подводиться к реакционному газу до осуществления каталитического дегидрирования и/или в его ходе.
Наряду с этим характерной особенностью гетерогенного каталитического дегидрирования пропана, требующего использования высоких температур, является образование небольших количеств тяжелокипящих высокомолекулярных органических соединений, вплоть до углерода, которые образуют отложения на поверхности катализатора, что приводит к его дезактивации. Чтобы свести к минимуму этот нежелательный побочный эффект, пропан, подаваемый к поверхности катализатора дегидрирования, осуществляемого при повышенной температуре, может быть разбавлен водяным паром. В этих условиях происходит частичное или полное элиминирование отложений углерода согласно принципу, лежащему в основе газификации угля.
Другая возможность удаления отложений, образуемых углеводородными соединениями, состоит в том, что через катализатор дегидрирования при повышенной температуре периодически пропускают кислородсодержащий газ, благодаря чему определенная часть отложений углерода выгорает.
Уменьшить образование отложений углерода можно также, вводя в состав подлежащего каталитическому дегидрированию пропана молекулярный водород, прежде чем пропустить пропан через нагретый до высокой температуры катализатор дегидрирования.
Кроме того, существует возможность добавления к подлежащему каталитическому дегидрированию пропану смеси водяного пара и молекулярного водорода. Добавление молекулярного водорода способствует также подавлению нежелательного образования таких побочных продуктов каталитического дегидрирования пропана, как аллен и ацетилен.
Пригодным реактором для осуществления дегидрирования пропана является кожухотрубный реактор с неподвижным слоем катализатора, загружаемого в одну трубку или пучок трубок. Обогрев реакционных трубок осуществляют, сжигая в окружающем их объеме горючий газ, например углеводород, в частности метан. Этим прямым способом предпочтительно следует обогревать лишь первые 20-30% неподвижного слоя катализатора, помещенного в контактные трубки. Необходимую реакционную температуру в остальной части трубок следует поддерживать за счет использования выделяемой при сжигании горючего газа лучистой теплоты. Благодаря этому способу обогрева могут быть обеспечены практически изотермические условия дегидрирования. Пригодный диаметр реакционных трубок составляет от 10 до 15 см. Типичный кожухотрубный реактор дегидрирования содержит от 300 до 1000 реакционных трубок. Температура внутри трубок составляет от 300 до 700°С, предпочтительно от 400 до 700°С.
Предпочтительным является подогрев подаваемого в трубчатый реактор реакционного газа до температуры реакции. Температура смеси выходящих из реактора продуктов часто бывает на 50-100°С ниже температуры реакции. Для осуществления указанного выше способа окислительного дегидрирования целесообразно использование катализаторов на основе оксида хрома и/или оксида алюминия. Нередко в качестве исходного реакционного газа используют преимущественно чистый, не содержащий газообразных разбавителей пропан. Катализатор дегидрирования в большинстве случаев также не содержит разбавителей.
Для крупномасштабного производства оказалось бы целесообразным использование трех кожухотрубных реакторов, два из которых эксплуатировались бы в режиме дегидрирования, а третий реактор в режиме регенерации катализатора.
Для осуществления описанного выше дегидрирования пропана используют, например, известный из литературы способ BASF-Linde.
Наряду с ним используют и так называемый процесс активной конверсии с водяным паром («steam active reforming (STAR) process»), разработанный фирмой Phillips Petroleum Co. (смотри, например, патенты США US-А 4902849, US-A 4996387 и US-A 5389342). В качестве катализатора дегидрирования в этом случае предпочтительно используют содержащую промоторы платину на цинковой (магниевой) шпинели в качестве носителя (смотри, например, патент США US-A 5073662). В отличие от способа BASF-Linde пропан, дегидрируемый согласно процессу STAR, разбавляют водяным паром. Типичное мольное отношение водяной пар : пропан составляет от 4:1 до 6:1. Рабочее давление чаще всего находится в интервале от 3 до 8 атм, температуру реакции целесообразно поддерживать в интервале от 480 до 620°С. Типичная нагрузка общей реакционной смеси на катализатор составляет от 0,5 до 10 час.-1.
Дегидрирование пропана можно осуществлять также в реакторе с движущимся слоем катализатора. Катализатор может быть помещен, например, в реактор с радиальным течением газового потока, в котором катализатор медленно опускается сверху вниз, в то время как смесь реакционных газов перемещается в радиальном направлении. Подобный принцип используют, например, для дегидрирования так называемым способом UOP-Oleflex. Поскольку реакторы, используемые для осуществления этого способа, работают в квазиадиабатическом режиме, целесообразна эксплуатация нескольких последовательно соединенных реакторов (в типичном случае их количество достигает четырех). Благодаря этому можно избежать слишком большой температурной разницы между смесью реакционных газов на входе в реактор и выходе из него (при адиабатическом режиме дегидрирования исходная смесь реакционных газов выполняет функцию теплоносителя, и от ее способности удерживать тепло зависит температура реакции) и достичь необходимых степеней превращения.
Слой катализатора, покидающего реактор, направляют на регенерацию и последующее повторное использование. Для осуществления данного способа можно использовать, например, катализатор дегидрирования со сферической формой частиц, преимущественно состоящий из платины, нанесенной на поверхность круглых частиц оксида алюминия, используемых в качестве носителя. В соответствии со способом UOP к подаваемому на дегидрирование пропану добавляют водород, чтобы предотвратить преждевременное старение катализатора. В типичном случае рабочее давление составляет от 2 до 5 бар. Отношение водород : пропан целесообразно поддерживать в интервале от 0,1:1 до 1:1. Температура реакции предпочтительно составляет от 550 до 650°С, время контакта катализатора с реакционной газовой смесью составляет от 2 до 6 час-1.
В способах дегидрирования, предусматривающих использование стационарного слоя катализатора, частицы катализатора могут обладать как сферической, так и цилиндрической формой (полые или полнотелые цилиндры).
Согласно другому варианту, описанному в Proceedings De Witt, Petrochem. Review, Houston, Texas, 1992 a, N1, гетерогенное каталитическое дегидрирование пропана может быть осуществлено в псевдоожиженном слое без разбавления пропана.
При этом целесообразно использовать два псевдоожиженные слоя, в одном из которых катализатор, как правило, подвергают регенерации. В качестве активной смеси используют оксид хрома на носителе, представляющем собой оксид алюминия. В типичном случае рабочее давление составляет от 1 до 1,5 атм, а температура дегидрирования, как правило, находится в интервале от 550 до 600°С. Необходимое для осуществления дегидрирования тепло поступает в реакционную систему благодаря предварительному разогреву катализатора до реакционной температуры. Рабочее давление составляет от 1 до 2 атм, типичная температура реакции от 550 до 600°С. Рассмотренный способ дегидрирования известен из литературы как способ Snamprogetti-Yarsintez.
Альтернативой описанным выше может служить способ гетерогенного каталитического дегидрирования пропана, осуществляемого при практически полном исключении кислорода, разработанный ABB Lummus Crest (смотри Proceedings De Witt, Petrochem. Review, Houston, Texas, 1992, P1).
Общей особенностью до сих пор известных способов гетерогенного каталитического дегидрирования пропана, осуществляемых при практически полном исключении кислорода, является то, что степень превращения пропана за однократный проход через реактор превышает 30 мол.%, как правило, меньше или равно 60 мол.%. Можно достичь преимуществ, ограничив степень превращения интервалом больше или равно 5 мол.%, но меньше или равно 30 мол.% или меньше или равно 25 мол.%. Это означает, что дегидрирование пропана может быть реализовано до степени его превращения за однократный проход, составляющей от 10 до 20 мол.%. Это приводит, в частности, к тому, что непревращенный пропан на последующей стадии (с) разбавляется молекулярным кислородом, благодаря чему уменьшается выход таких побочных продуктов, как пропионовый альдегид и/или пропионовая кислота.
Для реализации указанных выше степеней превращения пропана предпочтительным является дегидрирование при рабочем давлении от 0,3 до 3 атм. Наряду с этим благоприятную роль играет разбавление подвергаемого дегидрированию пропана водяным паром. Благодаря высокой теплоемкости воды, с одной стороны, удается частично компенсировать эндотермический эффект дегидрирования, а, с другой стороны, разбавление водяным паром обеспечивает снижение парциального давления продукта дегидрирования, что благоприятно отражается на положении равновесия. Кроме того, как сообщалось выше, использование водяного пара оказывает благоприятное воздействие на долговечность катализатора. При необходимости в качестве дополнительного компонента в исходную смесь можно добавлять молекулярный водород, причем его мольное отношение к пропану, как правило, меньше или равно 5:1. Учитывая сравнительно невысокую степень превращения пропана, мольное отношение водяной пар : пропан может составлять от больше или равно 0:1 до 30:1, целесообразно от 0,1:1 до 2:1, предпочтительно от 0,5:1 до 1:1. Благоприятной особенностью способа дегидрирования пропана с низкой степенью его превращения является также то, что при однократном пропускании реакционного газа через реактор расходуется сравнительно небольшое количество тепла и для достижения превращения достаточна сравнительно низкая температура реакции.
Может оказаться целесообразным осуществление дегидрирования пропана до сравнительно низкой степени его превращения в (квази)адиабатических условиях. Это значит, что исходную газообразную реакционную смесь нагревают, как правило, до температуры от 500 до 700°С (например, путем прямого огневого отопления стенок реактора) или от 550 до 650°С. В обычной ситуации хватает единственного адиабатического пропускания исходного газа через слой катализатора, чтобы достичь желаемой степени превращения, при этом температура реакционной смеси снижается на 30-200°С (в зависимости от степени превращения). Осуществлению дегидрирования в адиабатическом режиме способствует также использование в качестве теплоносителя водяного пара. Пониженная температура реакции позволяет продлить срок службы используемого для дегидрирования слоя катализатора.
Дегидрирование пропана при сравнительно низкой степени его превращения независимо от режима (адиабатического или изотермического) в принципе может быть реализовано в реакторе как с неподвижным, так и с движущимся или псевдоожиженным слоем катализатора.
Примечательно, что для осуществления дегидрирования, в частности, в адиабатическом режиме достаточно использовать в качестве реактора с неподвижным слоем катализатора шахтную печь, причем реакционную смесь пропускают в аксиальном и/или радиальном направлениях.
В наиболее простом случае речь идет об отдельном, замкнутом реакционном объеме, например о резервуаре, внутренний диаметр которого составляет от 0,1 до 10 м, возможно также от 0,5 до 5 м, причем неподвижный слой катализатора помещают на несущее приспособление, например колосниковую решетку, и горячий, содержащий пропан реакционный газ пропускают в аксиальном направлении через заполненный катализатором, снабженный теплоизоляцией реакционный объем, чтобы оказалась возможной эксплуатация реактора в адиабатическом режиме. Катализатор может иметь форму сфер, колец или стержней. Поскольку речь в данном случае идет об использовании чрезвычайно экономичного реакционного устройства, частицы катализатора могут обладать любыми геометрическими параметрами, способными обеспечить низкую потерю напора реакционного газа. В первую очередь используют катализаторы с геометрическими параметрами, позволяющими создать большое полое пространство или структуру, например, подобную сотовой. Для реализации радиального потока содержащего пропан реакционного газа реактор может, например, состоять из двух цилиндрических, коаксиально расположенных друг относительно друга, помещенных в обечайку колосниковых решеток, причем катализатор загружают в кольцевой зазор между ними. Для осуществления дегидрирования в адиабатическом режиме металлическую обечайку следует снабдить теплоизоляцией.
В качестве катализаторов дегидрирования пропана до сравнительно ограниченной глубины его превращения, достигаемой при однократном проходе через реактор, пригодны, в частности, катализаторы, описанные в немецкой заявке на патент DE-A 19937107, прежде всего, приведенные в немецкой заявке на патент DE-A 19937107, прежде всего, приведенные в соответствующих примерах.
После продолжительной эксплуатации данные катализаторы могут быть регенерированы простым способом, согласно которому на первой стадии, осуществляемой при температуре от 300 до 600°С, в частности от 400 до 500°С, через слой катализатора пропускают разбавленный азотом воздух. Нагрузка регенерирующей газовой смеси на катализатор может составлять, например, от 50 до 10000 час-1, а содержание кислорода от 0,5 до 20 об.%.
Следующую стадию регенерации осуществляют в аналогичных условиях, но в качестве регенерирующего газа используют воздух. С технологической точки зрения целесообразно перед регенерацией продуть катализатор инертным газом, например азотом.
В заключение, как правило, рекомендуется произвести окончательную регенерацию катализатора, пропуская через него чистый молекулярный водород или разбавленный инертным газом молекулярный водород (содержание водорода должно быть больше или равно 1 об.%), при этом остальные условия регенерации сохраняются неизменными.
Дегидрирование пропана до сравнительно небольшой глубины превращения (меньше или равно 30 мол.%) во всех случаях можно осуществлять при соблюдении тех же нагрузок на катализатор (касающихся как общего реакционного газа, так и содержащегося в нем пропана), как и в вариантах с высокой степенью превращения (более 30 мол.%). Нагрузка реакционного газа на катализатор может составлять, например, от 100 до 10000 час-1, в частности от 100 до 3000 час-1, то есть часто составляет от 100 до 2000 час-1.
Особенно привлекательным является осуществление способа дегидрирования пропана до сравнительно небольшой глубины превращения в хордовом реакторе.
В хордовый реактор помещают несколько последовательно располагаемых предпочтительно в радиальном или аксиальном направлении слоев катализатора дегидрирования, количество которых может составлять от 1 до 20, целесообразно от 2 до 8, а также от 3 до 6. С технологической точки зрения в хордовых реакторах целесообразно использовать неподвижные слои катализатора.
В наиболее простом случае слои неподвижного катализатора расположены в шахтовой печи аксиально или в кольцевых зазорах между коаксиально вставленными друг в друга цилиндрическими колосниковыми решетками. Возможен также вариант, согласно которому катализатор помещают в сегменты кольцевых зазоров, расположенные на разных уровнях, и подаваемый в радиальном направлении газ поступает из одного сегмента в другой, расположенный ниже или выше первого.
Целесообразным является промежуточный подогрев смеси реакционных газов в хордовом реакторе, для чего на пути от одного слоя катализатора к следующему эту смесь пропускают, например, через обогреваемый горячими газами ребристый теплообменник или через трубу, обогреваемую горячими горючими газами.
Если хордовый реактор эксплуатируют в адиабатическом режиме, для желаемой степени превращения пропана (меньшей или равной 30 об.%), осуществляемого, в частности, с использованием катализаторов, описанных в немецкой заявке на патент DE-A 19937107, в частности, в вариантах исполнения, представленных в соответствующих примерах, достаточно ввести в хордовый реактор предварительно нагретую до 450-550°С смесь реакционных газов и поддерживать внутри него эту температуру. Таким образом, дегидрирование пропана может быть осуществлено при чрезвычайно низких температурах, что обеспечивает особенно продолжительный срок службы неподвижных слоев катализатора между двумя последовательно осуществляемыми циклами регенерации.
Более эффективным является непосредственное промежуточное нагревание реакционных газов (автотермический способ нагревания). Согласно данному способу к смеси реакционных газов, выходящих из первого слоя катализатора и/или проходящих между последующими слоями, добавляют небольшое количество молекулярного кислорода. В зависимости от особенностей используемого для дегидрирования катализатора это приводит к ограниченному сгоранию содержащихся в смеси реакционных газов, а также, при необходимости, уже образовавшихся на поверхности катализатора отложений угля или углеподобных соединений и/или водорода, образующегося в процессе дегидрирования пропана и/или вводимого в состав смеси реакционных газов. С технологической точки зрения может оказаться целесообразным введение в хордовый реактор слоев дополнительного катализатора, способствующего специфическому (селективному) горению водорода (и/или углеводородов). О таких дополнительных катализаторах сообщается, например, в патентах США US-A 4788371, US-A 4886928, US-A 5430209, US-A 5530171, US-A 5527979 и US-A 5563314. Например, образованные ими слои могут чередоваться в хордовом реакторе со слоями катализатора дегидрирования. Тепло, выделяемое благодаря использованию квазиавтотермического способа, позволяет осуществлять гетерогенное каталитическое дегидрирование пропана практически в изотермическом режиме. При этом с увеличением времени пребывания реакционного газа в слое катализатора становится возможным дегидрирование пропана при снижающейся или преимущественно постоянной температуре, что позволяет обеспечить особенно продолжительный период эксплуатации катализатора между двумя последовательно осуществляемыми циклами регенерации.
Указанную выше подпитку кислородом следует осуществлять, как правило, таким образом, чтобы содержание кислорода в смеси реакционных газов составляло от 0,5 до 10 об.% по отношению к входящим в состав этой смеси пропану и пропену. Источником кислорода может быть как чистый молекулярный кислород, так и кислород, разбавленный инертными газами, например монооксидом углерода, диоксидом углерода, азотом, благородными газами, и, в частности, воздух. Образующиеся при горении газы, как правило, играют роль дополнительных разбавителей, благодаря чему они способствуют протеканию гетерогенного каталитического дегидрирования пропана.
Более полное приближение к изотермическому режиму при гетерогенном каталитическом дегидрировании пропана можно обеспечить, помещая в хордовый реактор, а именно в свободное пространство между слоями катализатора, замкнутые, при необходимости, предварительно эвакуированные, а, возможно, и неэвакуированные внутренние элементы, например, в виде труб. Такие элементы могут быть помещены также непосредственно в слой катализатора. Они содержат соответствующим образом подобранные твердые или жидкие вещества, которые при определенной температуре испаряются или плавятся, на что расходуется соответствующая тепловая энергия, а там, где температура ниже точки испарения или плавления, происходит сопровождаемая выделением тепла конденсация этих веществ.
Другая возможность нагревания смеси реакционных газов состоит в сжигании определенной части содержащегося в ней пропана и/или водорода благодаря добавлению необходимого для этого количества молекулярного кислорода (например, путем простого перепуска и/или пропускания реакционных газов через обладающие специфическим действием катализаторы горения) и использовании выделяющейся в результате сжигании тепловой энергии для нагревания газа до желаемой температуры. Возникающие при горении продукты, в частности диоксид углерода, вода, а также азот, при необходимости, подаваемый вместе с используемым для сжигания молекулярным кислородом, играют роль предпочтительно инертных газообразных разбавителей.
Согласно изобретению непревращенный на стадии (с) пропан (при необходимости, вместе с пропеном) после выделения целевых продуктов (акролеина и/или акриловой кислоты) может быть подвергнут дегидрированию, осуществляемому, как описано выше, и получаемую в результате этого смесь газообразных продуктов подвергают переработке на стадии (а).
В общем случае непревращенный при дегидрировании пропан играет на стадии (с) роль газообразного разбавителя.
Кроме того, если, в частности, производят дегидрирование пропана, существует возможность добавления к подаваемому на стадию (с) газу В чистого пропана и/или пропена.
Если стадия (с) заключается в превращении пропена в акриловую кислоту, то соответствующий абгаз наряду с непревращенными в процессе окисления компонентами, а именно пропаном, азотом и остаточным кислородом, содержит также способные окисляться побочные компоненты, например монооксид углерода, муравьиную кислоту, формальдегид, уксусную кислоту, а также небольшие количества акриловой кислоты. В соответствии с особенно предпочтительным вариантом осуществления изобретения эти побочные продукты перед дегидрированием пропана могут быть подвергнуты каталитическому окислению остаточным кислородом и, при необходимости, дополнительным количеством молекулярного кислорода, чтобы нагреть газ, подаваемый на дегидрирование. Окисление побочных продуктов может быть осуществлено в присутствии катализатора дожигания, в частности палладия, нанесенного на подложку из оксида алюминия, например катализатора RO-20/13 или RO-20/25 (BASF).
Предпочтительные варианты осуществления изобретения представлены на фиг.1-7, которые более подробно поясняют изобретение, не ограничивая его объем.
На фиг.1-5 схематически изображены предпочтительные способы, причем для простоты не показаны все подводимые и отводимые потоки. На фиг.1 приведены следующие стадии технологического процесса: 1 - абсорбция и десорбция, 2 - окисление пропена до акролеина и/или акриловой кислоты и 3 - выделение. На стадии 1 из смеси, содержащей пропан, пропен, водород, монооксид и диоксид углерода, а также, возможно, азот и другие углеводороды, путем абсорбции соответствующим абсорбентом выделяют пропан и пропен, которые затем десорбируют путем отпаривания воздухом. Благодаря осуществлению этой стадии удаляют водород, оксиды углерода, другие углеводороды и азот. Затем газовый поток, содержащий пропен и, при необходимости, пропан, направляют на стадию окисления 2, где пропен окисляют до акролеина и/или акриловой кислоты. Полученный на стадии 2 продукт направляют для переработки на стадию 3, где из него выделяют акролеин или акриловую кислоту в качестве целевых продуктов. Непревращенный пропен, а также пропан, оксиды углерода и, при необходимости, остаточный азот и кислород направляют на стадию абсорбции и десорбции 1.
Цифры, приведенные на остальных чертежах, означают то же, что и на фиг.1.
В отличие от фиг.1 на фиг.2 приведена стадия дегидрирования пропана 4, которое может быть осуществлено с подачей кислорода или без нее. Газовую смесь, полученную при дегидрировании пропана и содержащую наряду с пропаном и пропеном водород, оксиды углерода и возможные остаточные количества азота и углеводородов, направляют на стадию абсорбции/десорбции 1.
В отличие от фиг.1 на фиг.3 вместо стадии окисления пропена 2 представлена стадия 22, на которой осуществляют окисление пропана до акролеина и/или акриловой кислоты.
Согласно фиг.4 продукты, выделенные на стадии 3, направляют на стадию дегидрирования пропана 4, которое осуществляют с подачей кислорода или без нее, и образующуюся на этой стадии газовую смесь подают на стадию абсорбции/десорбции 1.
На фиг.5 показан другой предпочтительный вариант осуществления изобретения, согласно которому после стадии абсорбции и десорбции 1 осуществляют дегидрирование пропана 5 с подачей кислорода.
На фиг.6 и 7 показаны другие предпочтительные способы. Способу, изображенному на фиг.6, соответствует технологическая схема, представленная на фиг.4. Как видно из приведенной на фиг.6 схемы, для осуществления дегидрирования пропана (стадии 4) используют три реактора, в первом из которых перед дегидрированием пропана (PDH) осуществляют дожигание монооксида углерода (CO-NV), а в двух следующих реакторах перед дегидрированием пропана (PDH) дожигают водород (Н2-NV). Благодаря дожиганию указанных продуктов получают необходимую для дегидрирования пропана тепловую энергию. Количество реакторов, предназначенных для дегидрирования пропана, не ограничивают тремя. Пропан подают на стадию дегидрирования 4 по трубопроводу (30), а по трубопроводу (6) может поступать воздух. Полученную в результате дегидрирования пропана газовую смесь подают через теплообменник W и компрессор V на абсорбционную колонну К1 и десорбционную колонну К2. Абсорбент после десорбции в колонне К2 возвращают на абсорбционную колонну К1. Не поглощенный абсорбентом газ удаляют из процесса в виде абгаза (33), при необходимости, предварительно пропускаемого через устройство для дожигания Е. Газовый поток, содержащий выделенный пропан и/или пропен, поступает на стадию окисления 2, в состав которой входят четыре реактора. Количество предназначенных для окисления реакторов, однако, не ограничивается четырьмя. После этого на стадии 3 выделяют акролеин и/или акриловую кислоту в качестве целевых продуктов и отводят их через трубопровод (31). Непревращенные пропан и/или пропен в виде рециркуляционного газа возвращают по трубопроводу (32) на стадию дегидрирования 4 вместе с другими, не выделенными абсорбцией газообразными составными частями.
Как показано на фиг.7, рециркуляционный газ (1), выходящий со стадии выделения 3 при температуре от 10 до 90°С и давлении от 0,8 до 5 бар, который может быть подвергнут дополнительному сжатию, например, посредством компрессора VO до давления от 2 до 10 бар, нагревают до температуры от 100 до 650°С, пропуская через теплообменник W1, в который противотоком подают реакционный газ (2), полученный в результате дегидрирования пропана (PDH) на стадии 4. Здесь и в дальнейшем описании приведенной на фиг.7 схемы значения давления указываются в абсолютных единицах.
Сжатие газовых смесей может осуществляться посредством любых пригодных для этой цели, известных специалистам компрессоров, конструктивное исполнение которых будет описано ниже более подробно.
Рециркуляционный газовый поток (1) содержит от 40 до 80 об.% азота, от 1 до 5 об.% диоксида углерода, от 0,1 до 2 об.% монооксида углерода, от 2 до 5 об.% кислорода, от 0,5 до 25 об.% влаги, другие побочные продукты окисления, от 5 до 40 об.% непревращенного пропана и от 0,1 до 3 об.% непревращенного пропена. До или после нагревания этот газовый поток смешивают со свежим пропаном (3) и предпочтительно водой и водяным паром (4), прежде чем направить его на стадию дегидрирования пропана 4. В качестве источника свежего пропана пригодны любые доступные газы или жидкости, содержащие пропан. Предпочтительным источником свежего пропана является, однако, технический пропан (содержание основного продукта более 90%, в частности более 95%, особенно предпочтительно более 98% при ограниченном содержании C4+) или чистый пропан (содержание основного продукта более 99%). Мольное отношение воды или водяного пара к пропану в газовом потоке составляет от 0,05:1 до 5:1, предпочтительно от 0,1:1 до 2:1. Благоприятную роль может сыграть дополнительное введение в состав этого газового потока водорода (5), воздуха (6) или кислородсодержащего газа, а также других компонентов, превращающихся в процессе дегидрирования пропана с экзотермическим эффектом, например, монооксида углерода или его смесей с водородом, в частности синтез-газа. К степени чистоты этих газов не предъявляется никаких претензий. Экзотермичность окисления горючих компонентов в процессе дегидрирования пропана позволяет скомпенсировать эндотермический эффект, сопровождающий дегидрирование, поэтому может оказаться необходимым подведение извне лишь ограниченного количества тепловой энергии, а в наиболее оптимальной ситуации оно может не потребоваться вовсе.
Давление при дегидрировании пропана составляет от 0,3 до 10 бар, предпочтительно от 1 до 5 бар, температура от 350 до 700°С, предпочтительно от 400 до 600°С. Для дегидрирования пропана могут использоваться реакторы любого, известного специалистам конструктивного типа, например аппараты с аксиальным направлением газового потока, в частности хордовые реакторы, а также аппараты с несколькими, упорядоченными в виде полых цилиндров слоями катализатора и радиальным направлением газового потока или несколько отдельных аппаратов, например, имеющих форму колонн, тортов (Tortenform) или сфер. Количество используемых для дегидрирования пропана реакторов не ограничивается тремя. Несколько отдельных аппаратов используют потому, что это обеспечивает простоту промежуточной подачи дополнительных газов. Кроме того, это позволяет в ходе технологического процесса осуществлять обработку находящегося в том или ином реакторе катализатора, например его регенерацию, независимо от того, что происходит в других реакторах. Для этого, например, реактор с подлежащим регенерации катализатором изолируют от основного газового потока посредством соответствующих запорных органов, например заслонок, вентилей или клапанов, расположенных на соединяющих отдельные реакторы трубопроводах, пропускают через слой катализатора необходимые для его регенерации газы, например азот, водород, обедненный кислородом воздух или обогащенные воздухом или кислородом газы и удаляют образовавшиеся на поверхности катализатора отложения. В остальные реакторы, общее количество которых может составлять от 1 до 20, предпочтительно от 2 до 5, продолжают подавать основной газовый поток, получая в качестве целевого продукта пропен.
В предназначенных для дегидрирования пропана реакторах слои катализатора могут быть уложены на решетки, сыпучие массы из инертного материала или иные, известные специалистам опорные устройства. Частицы катализатора могут обладать любой формой без каких-либо ограничений, включая, например, раздробленные частицы, сферы, короткие стержни, кольца, цилиндры, а также структурированные насадки или монолиты. Предпочтительными являются геометрические параметры, которым соответствует минимальная потеря напора.
Для равномерного распределения подводимого к катализатору газа могут использоваться известные специалистам устройства, например сетчатые тарелки, кольцевые или трубчатые распределители газа, а также беспорядочные засыпки или структурированные насадки, например статические смесители.
В реакторы, предназначенные для дегидрирования пропана, может быть помещено несколько слоев катализатора, каждый из которых выполняет особую функцию. Если используют несколько таких слоев, то перед слоем катализатора дегидрирования предпочтительно помещают один или несколько слоев, состоящих из катализаторов, под влиянием которых может происходить окисление веществ, не являющихся пропаном или пропеном, например водорода (катализатор Н2-NV), монооксида углерода (катализатор CO-NV) и/или другого, способного окисляться компонента. Однако если эти функции выполняет сам катализатор дегидрирования пропана или потери пропана в результате его окисления являются экономически оправданными, следует, по возможности, отказаться от использования дополнительных катализаторов.
Удельная производительность катализаторов дегидрирования пропана может составлять от 100 до 20000 нормальных литров (нл) пропана на литр катализатора в час. Предпочтительно этот показатель находится в интервале от 500 до 10000, более предпочтительно от 1000 до 10000. Удельная производительность катализаторов, преимущественно окисляющих, например монооксид углерода или водород при одновременном незначительном окислении пропана или пропена, обычно находится в интервале от 5000 до 30000 нл на литр катализатора в час.
Степень превращения пропана при дегидрировании составляет от 10 до 60%, предпочтительно от 20 до 50%, в частности от 88 до 96%. Предпочтительным является полное превращение вводимых в реактор газообразных монооксида углерода, водорода или других горючих газов. Степень превращения водорода, образующегося в процессе дегидрирования пропана, в зависимости от степени превращения самого пропана составляет от 1 до 99%, чаще всего от 10 до 80%, в частности от 30 до 70%.
Реакционный газ (2), образующийся в результате дегидрирования пропана, содержит от 20 до 60 об.% азота, от 1 до 5 об.% диоксида углерода, от 0,5 до 45 об.% воды, от 5 до 45 об.% пропана, от 1 до 20 об.% пропена от 1 до 20 об.% водорода, а также другие побочные продукты, например этан, этен, метан и С4+.
Образующийся при дегидрировании пропана реакционный газ (2) обладает температурой от 400 до 650°С, более предпочтительно от 450 до 600°С, и давлением от 0,3 до 10 бар, более предпочтительно от 1 до 5 бар. Газ (2) благодаря теплообмену в противоточном теплообменнике W1 с рециркуляционным газом (1) охлаждается до температуры, по меньшей мере, на 5°С, лучше, по меньшей мере, на 50°С и предпочтительно, по меньшей мере, на 100°С более высокой, чем температура рециркуляционного газа (1) на входе в теплообменник. Затем газовый поток (3) в зависимости от температуры на выходе из противоточного теплообменника W1 подвергается дополнительному охлаждению на 10-60°С, осуществляемому в одну или несколько ступеней. При использовании нескольких ступеней в зависимости от уровня температуры охлаждение в теплообменнике W2 может происходить за счет генерирования пара или подачи охлаждающего воздуха, а охлаждение в теплообменнике W3 за счет подачи охлаждающего воздуха, воды или рассола. При этом в зависимости от давления, температуры и влагосодержания газовых потоков (3)-(5) происходит конденсация влаги, которую отделяют от газового потока (5) в сепараторе А1. Сепаратор может иметь любое, известное специалистам и пригодное для отделения газа конструктивное исполнение.
Затем охлажденный и, при необходимости, частично обезвоженный газовый поток (6) подвергают сжатию, причем степень сжатия находится в интервале, ограниченном величиной давления газа на выходе из сепаратора А1, с одной стороны, и 50 барами, с другой. Сжатие может осуществляться посредством одноступенчатого или многоступенчатого компрессора с промежуточным охлаждением или без него. Компрессор V1 может иметь любое, известное специалистам конструктивное исполнение. Например, можно использовать поршневые, ротационные, винтовые, мембранные, пластинчатые, турбинные и центробежные компрессоры, а также ротационные и центробежные воздуходувки, причем предпочтительными являются турбинные и центробежные компрессоры. Критериями при выборе типа компрессора служат степень сжатия и количество подвергаемого сжатию газа. При многоступенчатом сжатии с промежуточным охлаждением происходит конденсация влаги и, при необходимости, других, способных конденсироваться компонентов, которые могут быть выделены из газового потока, как описано выше, после промежуточного охлаждения или во время его осуществления, прежде чем направить газовый поток на следующую ступень сжатия. Сжатый до необходимого конечного давления газовый поток (7) может быть подвернут одно- или многоступенчатому охлаждению, как описано выше, причем влага и, при необходимости, другие, способные конденсироваться компоненты могут быть отделены от газовых потоков (7)-(9).
В качестве привода компрессора V1 могут использоваться электродвигатели, а также паро- или газотурбинные установки. Выбор типа привода зависит от производственной инфраструктуры, однако чаще всего в качестве привода используют паровые турбины, отличающиеся наиболее высокой экономичностью.
Если необходимо повысить отношение воды к подаваемому на дегидрирование пропану, совокупность конденсированных потоков, например потока (11) (после повышения давления) и потока (12) возвращают на стадию дегидрирования пропана, а остаток шлюзуют и, при необходимости, сжигают. Поток конденсата (13) перед рециркуляцией или шлюзованием испаряют или подвергают какой-либо другой обработке, например очистке.
Газовый поток (10) направляют в абсорбционную колонну К1, где контактируемый с ним абсорбент поглощает С3-углеводороды (пропан и/или пропен) и, при необходимости, другие компоненты. В качестве абсорбента используют любые, известные специалистам вещества, причем предпочтительными являются указанные выше абсорбенты. Предпочтительным является многоступенчатое противоточное контактирование газового потока (10) с абсорбентом. Температура абсорбции может составлять от 10 до 150°С, лучше от 20 до 80°С, предпочтительно от 30 до 60°С, а давление от 1 до 50 бар, лучше от 3 до 30 бар, предпочтительно от 5 до 20 бар.
Пригодными являются известные специалистам конструктивные варианты исполнения абсорбера К1, например, описанные в «Thermische Trennver-fahren», Klaus Sattel, VCH, 1988, (ISBN 3-527-28636-5). Предпочтительными являются колонны с внутренними элементами также известного специалистам конструктивного исполнения, например, абсорберы с сетчатыми тарелками, тарелками с двойным потоком, колпачковыми, туннельными, решетчатыми или клапанными тарелками, абсорберы с беспорядочной засыпкой, например, колец Рашига, колец Паля, седел типа Intalox, седел Берли, седел типа Super или Torus, внутренних насадочных тел или колец из металлической ткани, а также абсорберы со структурированной насадкой (например, типа Sulzer-Kerapak, Sulzer-Packung BX или CY, или, например, насадками фирмы Montz и других изготовителей). Особенно пригодной является насадка Ralu-Pak 250. YC фирмы Raschig. При этом к предпочтительным относятся встроенные элементы, которые позволяют эксплуатировать абсорбер с повышенным расходом жидкости (высокой плотностью орошения), например неструктурированные засыпки или структурированные насадки. В предпочтительном варианте плотность орошения должна составлять более 50 м3, еще лучше более 80 м3 жидкости в расчете на квадратный метр свободной поверхности в час. Встроенные элементы могут быть выполнены из металла и керамики, а также из полимеров или сочетаний нескольких материалов. Основным принципом при выборе материала, пригодного для использования в качестве засыпки или насадки, является хорошее смачивание абсорбентом.
Соотношение между потоками подаваемого в абсорбер абсорбента (24) и газа (10) устанавливают на основании соответствующих требований термодинамики. Оно зависит от количества ступеней выделения, температуры, давления, поглощающей способности абсорбента и требуемой степени выделения абсорбируемого продукта. Указанное соотношение обычно составляет от 1:1 до 50:1, в частности от 1:1 до 30:1, предпочтительно от 1:1 до 20:1 (кг/кг) при числе теоретических ступеней от 1 до 40, в частности от 2 до 30, предпочтительно от 5 до 20. Теоретическое число ступеней может быть рассчитано, используя специальную литературу, например «Thermische Trennverfahren», Klaus Sattel, VCH, 1988 (ISBN 3-527-28636-5).
Чтобы, при необходимости, сократить потери абсорбента, освобожденный от пропана и/или пропена газовый поток (14) может быть направлен на стадию закалки. Соответствующий принцип более подробно объясняется при нижеследующем описании стадии десорбции.
После возможной закалки газовый поток (14) может быть подвергнут декомпрессии, которая может быть осуществлена путем одно- или многоступенчатого дросселирования без регенерации энергии или путем одно- или многоступенчатой регенерации механической энергии в газовой турбине Т1. В последнем случае газовый поток (14) перед подачей в турбину иногда бывает необходимо нагреть. Нагревание может быть осуществлено непосредственно путем катализируемого или некатализируемого окисления содержащихся в составе этого газового потока или вводимых извне горючих и окисляющихся компонентов или путем косвенного подвода тепла посредством пара или наружного огневого отопления. Получаемая в результате декомпрессии механическая энергия может быть использована для приведения в действие привода одного из компрессоров, предпочтительно компрессора V1 в качестве дополнительного или основного источника энергии и/или для выработки электрического тока.
В зависимости от степени чистоты получаемый в результате декомпрессии поток абгаза (15) может быть направлен на катализируемое или некатализируемое сжигание или сброшен в атмосферу.
Поток абсорбента (16), преимущественно состоящий из пропана (от 2 до 30 об.%) и/или пропена (от 2 до 30 об.%) и, при необходимости, содержащий другие компоненты (например, диоксид углерода, С2-, С4+, воду), при необходимости, подвергают декомпрессии, после чего направляют в десорбционную колонну К2. Декомпрессия абсорбента может быть осуществлена без регенерации механической энергии путем одно- или многоступенчатого дросселирования, а также с регенерацией механической энергии, например, в турбине или во вращающемся в обратном направлении центробежном насосе. Может возникнуть необходимость нагревания абсорбента (16) перед десорбцией, которое предпочтительно осуществляют благодаря теплообмену в противоточном теплообменнике W6 с потоком (17). Не исключается необходимость дополнительного нагревания потока (16), выходящего из этого теплообменника.
Десорбция пропана и/или пропена в десорбере К2 может быть реализована путем дистилляции (однократного простого испарения) или отпаривания, причем десорбции способствует снижение давления до 0,1-10 бар, в частности до 1-5 бар, предпочтительно до 1,5-3 бар.
Если речь идет о десорбции путем дистилляции, она может быть осуществлена любым, известным специалистам способом. Особенно простой формой осуществления десорбции является одноступенчатое простое (мгновенное) испарение абсорбента в предназначенном для этого аппарате. Перед данной операцией целесообразно нагреть поток (16) до температуры от 20 до 300°С, в частности от 40 до 200°С, предпочтительно от 50 до 150°С. Соответствующий аппарат должен иметь конструкцию, позволяющую обеспечить надлежащие термодинамические условия выделения пропана или пропена из абсорбента и надлежащие гидродинамические условия отделения газа от жидкости. В частности, аппарат может иметь форму цилиндра или сферы, а также иную, известную специалистам конструктивную форму. Если он обладает цилиндрической формой, его можно расположить как в вертикальной, так и в горизонтальной плоскости. Подводящая линия, как правило, находится между штуцерами, предназначенными для отвода, соответственно, газа и жидкости. В простейшем случае в аппарате отсутствуют какие-либо дополнительные встроенные элементы. Для создания оптимальных термодинамических условий десорбции внутри аппарата могут быть смонтированы встроенные элементы, обычно используемые в технике дистилляции, абсорбции и отпаривания, в частности элементы, приведенные выше при описании абсорбции. Для обеспечения оптимальных гидродинамических условий отделения газа от жидкости внутри аппарата могут быть интегрированы соответствующие, известные специалистам дополнительные встроенные элементы, например проволочная ткань, перегородки, изменяющие направление движения, или аналогичные элементы. Кроме того, в состав аппарата могут быть включены устройства, обеспечивающие подведение тепла, например нагреваемые змеевики или экраны.
Если в рассматриваемом случае в распоряжении имеется воздух или аналогичная отпаривающая среда, например водяной пар, азот, свежий пропан или другой, необходимый для использования в технологическом процессе газ, целесообразно воспользоваться ими для поддержания процесса мгновенного испарения абсорбента.
При этом особой формой осуществления процесса является многоступенчатое отпаривание летучих компонентов (пропана и/или пропена) посредством исходного газового потока (25), предназначенного для окисления (в этом случае, как и при одноступенчатом отпаривании, происходит одновременное отпаривание из потока (10) всех дополнительно абсорбированных компонентов, а также других веществ, которые могли образоваться благодаря их летучести). В простейшем случае исходным газовым потоком (25) является воздух, необходимый для окисления пропана до акролеина или акриловой кислоты. При этом сжатие воздуха или исходного газового потока может осуществляться как перед десорбцией, так и после нее. Исходный газовый поток (25) наряду с воздухом может содержать также рециркуляционный газ со стадии получения акриловой кислоты, а также водяной пар, свежий пропан или другие инертизированные газообразные компоненты. Особенно благоприятным для процесса десорбции является противоток или перекрестный ток исходного газового потока (25) по отношению к жидкому абсорбенту. В случае противотока десорбер может быть выполнен аналогично описанной выше абсорбционной колонне.
Перекрестный ток целесообразно использовать, если в процессе десорбции переходят границу области взрыва. Такая ситуация возникает в том случае, если с точки зрения склонности к воспламенению исходный газовый поток (25) представляет собой «тощую» газовую смесь, а выходящий из десорбера, содержащий пропан и/или пропен газовый поток (18) является «жирной» газовой смесью. Газовую смесь называют «тощей», если она содержит слишком мало горючих веществ, чтобы обладать способностью к воспламенению, а «жирной», если она содержит слишком много горючих веществ, чтобы обладать такой способностью.
При использовании перекрестного тока общий газовый поток подают не в куб десорбционной колонны, а в виде отдельных потоков вводят в несколько расположенных вдоль нее точек таким образом, чтобы ни в одной части десорбера не присутствовала способная к воспламенению газовая смесь. При этом десорбер может быть расположен как в вертикальной, так и в горизонтальной плоскости.
Другая возможность решения проблемы взрывоопасности при десорбции способных к воспламенению компонентов, входящих в состав кислородсодержащих газовых потоков, заключается в смешивании исходного газового потока перед входом в десорбционную колонну с веществом (например, пропаном, пропеном, метаном, этаном, бутаном, водой и так далее), благодаря присутствию которого подаваемая в десорбер газовая смесь становится «жирной». Существует также возможность разделить исходный газовый поток и вводить в куб десорбционной колонны исходный газ, не содержащий пропана или пропена, чтобы обеспечить максимально низкое содержание пропана и/или пропена в потоке абсорбента (17), а исходный газ, «жирность» которого повышена, например, благодаря добавлению пропана и/или пропена, подавать в ту часть десорбционной колонны, где может образоваться способная к воспламенению газовая смесь.
Поток абсорбента (17) с низким содержанием пропана и/или пропена после возможного пропускания через противоточный теплообменник W6 и повышения давления, осуществляемого посредством насоса Р1, может быть подвергнут дополнительному одно- или многоступенчатому охлаждению (например, в теплообменнике W7) и через трубопровод (24) вновь направлен в абсорбер К1.
В общем случае многоступенчатую десорбцию можно осуществлять при любых давлениях и температурах. Предпочтительными, однако, являются более низкие давления и более высокие температуры по сравнению с абсорбцией. В рассматриваемом случае давление составляет от 1 до 5 бар, в частности от 2 до 3 бар, температура от 20 до 200°С, в частности от 30 до 100°С, особенно предпочтительно от 35 до 70°С.
Отношение потока абсорбента (17) к потоку исходного газа (25) должно соответствовать требованиям термодинамики, и оно зависит от количества ступеней разделения, температуры, давления, способности абсорбента десорбировать поглощенные вещества и требуемой степени разделения. Обычно оно составляет от 1:1 до 50:1, в частности от 5:1 до 40:1, предпочтительно от 10:1 до 30:1 (кг/кг) при теоретическом количестве ступеней разделения от 1 до 20, в частности от 2 до 15, предпочтительно от 3 до 10.
В общем случае исходный, содержащий пропан и/или пропен газовый поток без дополнительной обработки может быть направлен на стадию окисления 2. Однако может оказаться целесообразным направить его перед окислением на дополнительную технологическую стадию, например, с целью сокращения потерь отпаренного абсорбента. Абсорбент может быть отделен от подаваемого на окисление газового потока любыми, известными специалистам способами, одним из которых является резкое охлаждение (закалка) газового потока водой. Водную промывку (закалку) можно производить в верхней части десорбционной колонны над улавливающей жидкость тарелкой или в отдельном аппарате. Для усиления эффекта разделения внутри устройства для закалки могут быть смонтированы встроенные элементы, аналогичные обычно используемым при дистилляции, абсорбции и десорбции и указанные выше при описании абсорбции. То же относится и к отдельному аппарату для закалки.
Смесь воды и абсорбента, образующаяся в результате выделения абсорбента из содержащего пропан и/или пропен газового потока путем промывки водой (19), может быть подвергнута фазовому разделению в устройстве D1, и освобожденный от воды газовый поток (18) после возможного подогрева направляют на стадию окисления пропана (2).
Разделение фаз может быть реализовано в любой, известной специалистам форме, например, путем жидкостной экстракции. В наиболее простом случае органический абсорбент отделяют от использованной для закалки воды в горизонтально или вертикально установленных аппаратах продолговатой формы, снабженных встроенными элементами или без них. Отношение диаметра таких аппаратов к длине может составлять от 1:1 до 1:100, в частности от 1:1 до 1:10, предпочтительно от 1:1 до 1:3. Аппарат может быть полностью заполнен жидкостью или в нем может находиться газовая подушка. Для лучшего отделения органической фазы его можно оснастить куполом, из которого может производиться слив абсорбента. Более качественному разделению фаз может способствовать использование известных специалистам встроенных элементов, например проволочных тканей, свечей с обмоткой (Wickelkerzen) или перегородок, изменяющих направление движения. Для разделения фаз могут использоваться также вращающиеся аппараты, например центрифуги.
Полученный в результате разделения фаз абсорбент (20) может быть возвращен в десорбер. Закалочная вода перед возвращением в закалочный аппарат, при необходимости, может быть охлаждена или подогрета в теплообменнике W9. Большие количества закалочной воды более предпочтительно перекачивать соответствующим насосом Р2. Плотность орошения в закалочном аппарате превышает 30 м3, в частности 50 м3, предпочтительно 80 м3, однако составляет менее 1000 м3, в частности менее 500 м3, предпочтительно менее 300 м3 воды на квадратный метр площади свободного поперечного сечения закалочного аппарата в час.
Потери воды при закалке могут быть компенсированы подачей водного конденсата (21) или содержащей кислоту воды (22), образующейся на стадии получения акриловой кислоты. Чтобы избежать переполнения, часть оборотной закалочной воды шлюзуют в виде очищающего потока (23) и направляют на установку для сжигания или устраняют иным способом (например, направляют на установку для очистки сточных вод).
Если подаваемый на стадию окисления 2 газовый поток (18) обладает температурой ниже 90°С, в частности ниже 70°С, и давлением от 1 до 3 бар, то к нему целесообразно добавить воду, что осуществляют путем его смешивания с водяным паром или насыщения водой в сатураторе известного специалистам конструктивного типа.
Газовый поток (18) содержит от 30 до 70 об.% азота, от 5 до 20 об.% кислорода, от 2 до 15 об.% пропена, от 2 до 40 об.% пропана, от 0,5 до 25 об.% воды и другие компоненты, в частности диоксид углерода, метан, этан, этен, C4+. Он может быть направлен на стадию окисления 2, осуществляемую, как описано выше, или известными из патентной литературы способами. Окисление пропена или акролеина может быть осуществлено в реакторах согласно уровню техники с солевой баней, например, производства судостроительного завода Деггендорфера или в реакторах другого типа. Между первой (получение акролеина) и второй (получение акриловой кислоты) ступенями окисления может быть осуществлена дополнительная подпитка реакционного газа воздухом или водяным паром. Повышенное содержание С3-углеводородов в подаваемом на стадию окисления 2 газовом потоке (18) в любом случае способствует отводу тепла реакции из реакционного объема. Пригодной является подача пропена в количестве от 50 до 350, в частности от 90 до 300, предпочтительно от 100 до 250, нл на литр катализатора в час.
Выделение на стадии 3 акриловой кислоты из реакционного газа (26), поступающего со стадии окисления 2, может быть осуществлено, как описано выше, например, используя высококипящие растворители, в частности смесь дифила с диметилфталатом, а также путем абсорбции водой или фракционированной конденсации.
Акриловая кислота может быть очищена от примесей отпариванием и дистилляцией, азеотропной дистилляцией или кристаллизацией.
Способ, осуществляемый в соответствии с изображенной на фиг.7 технологической схемой, пригоден как для переоснащения любых существующих установок, предназначенных для получения акролеина и/или акриловой кислоты, так и для комбинирования с новыми установками синтеза акриловой кислоты.
Неожиданно было обнаружено, что, как правило, ожидаемое присутствие остатков абсорбента в составе газа В не приводит к какому-либо отрицательному воздействию на процесс окисления и свойства используемого для его осуществления катализатора. Не наблюдалось также никаких проблем, обусловленных, при необходимости, происходящим на стадии окисления пропана и/или пропена образованием каких-либо продуктов окисления абсорбента. Если все же и возникали какие-либо проблемы, связанные с наличием остатков абсорбента, которых, как правило, удается избежать при использовании в качестве абсорбентов высококипящих углеводородов, в частности парафинов, то такие проблемы могут быть решены благодаря использованию водной закалки или адсорбции.
Таким образом, неожиданно оказалось, что в способе согласно изобретению может быть использована абсорбция. В отличие от адсорбции, используемой в соответствии с японской заявкой на патент JP-A-1036311, абсорбция пропана и/или пропена согласно настоящему изобретению предоставляет возможность гораздо более легкого и экономичного управления.
Кроме того, преимущество настоящего изобретения состоит в том, что установки, на которых в качестве исходного продукта для получения акролеина и/или акриловой кислоты используют пропен, могут быть удобным способом переоборудованы с целью использования в качестве исходного сырья более дешевого пропана.
Изобретение поясняет приведенный ниже пример, касающийся предпочтительного варианта осуществления способа согласно изобретению.
Пример
Акриловую кислоту получают способом, схематически изображенным на фиг.7, поэтому ниже используются те же цифровые обозначения, что и на фиг.7.
2090 нл/час рециркуляционного газа (1) после технологической стадии 3, обладающего температурой 30°С и давлением 1,2 бар, подают на компрессор VO, где его подвергают сжатию до 2,0 бар, и далее в противоточный теплообменник W1, в котором его нагревают до температуры 450°С за счет теплообмена с реакционным газом (2), поступающим со стадии дегидрирования пропана. В данном случае и при нижеследующем описании примера указывается абсолютная величина давления в барах.
Рециркуляционный газ (1) содержит 60,3 об.% азота, 1,2 об.% диоксида углерода, 0,5 об.% монооксида углерода, 3,4 об.% кислорода, 1,9 об.% воды, 32,2 об.% пропана и 0,4 об.% пропена, а также побочные продукты окисления. Перед поступлением в теплообменник рециркуляционный газ смешивают со свежим пропаном (3), подаваемым в количестве 170 нл/час, и водяным паром (4). В качестве свежего пропана используют технический пропан с содержанием основного продукта более 98% и C4+, равным 100 млн.-1 Мольное отношение водяного пара к пропану в подаваемом на дегидрирование газовом потоке составляет 0,5:1.
Газовую смесь вводят в первый из четырех реакторов, внутренний диаметр которых составляет 50 мм. Конструкция реакторов допускает возможность их эксплуатации в автотермическом режиме. В каждом реакторе содержится слой катализатора высотой 110 мм. Катализатор представляет собой короткие стержни диаметром 3 мм и высотой 5 мм.
Степень превращения пропана в четырех реакторах составляет 20% при селективности образования пропена 92%.
Выходящий из реакторов дегидрирования газ (2) содержит 44,9 об.% азота, 2,7 об.% диоксида углерода, 16,9 об.% воды, 24,0 об.% пропана, 5,8 об.% пропена, 5,5 об.% водорода, а также небольшое количество побочных продуктов, в частности этана, этена, метана и С4+.
Реакционный газ (2) после дегидрирования с температурой 520°С и давлением 1,5 бар охлаждают до 30°С. Охлаждение газа сопровождается конденсацией около 350 г/час воды (11).
Охлажденный газовый поток (6) подвергают одноступенчатому сжатию поршневым компрессором до 7,5 бар, после чего вновь охлаждают до 30°С.Образующийся при охлаждении водный конденсат (12) объединяют с конденсатом (11), частично испаряют и смешивают с рециркуляционным потоком (1). Другую часть конденсата (21) направляют в устройство для закалки, а остаток подвергают шлюзованию.
2340 нл/час газового потока (10) подают в куб абсорбционной колонны К1 (металлический корпус, внутренний диаметр 80 мм, длина 3 м). Внутренний объем абсорбера на 60% заполнен насадочными элементами фирмы Montz (Montz-Pak, тип В1).
Газовый поток (10) содержит 53,9 об.% азота, 3,3 об.% диоксида углерода, 0,4 об.% воды, 28,8 об.% пропана, 7,0 об.% пропена, 6, 6 об.% водорода и небольшие количества других побочных продуктов, в частности этана, этена, метана и C4+.
В верхнюю часть абсорбционной колонны К1 из десорбера К2 подают 35 кг/час тетрадекана (24), обладающего температурой 30°С и содержащего небольшое количество С3-углеводородов.
Абгаз (14), содержащий 1150 млн.-1 об. пропана и 750 млн.-1 об. пропена, пропускают через поддерживающий напор клапан для снижения температуры и давления до соответствующих атмосферных значений, после чего сжигают.
Поток абсорбента (16) из куба колонны К1 пропускают через поддерживающий напор клапан, снижая давление до 2,4 бар, и подают в верхнюю часть десорбционной колонный К2.
Размеры десорбционной колонны К2 и тип заполняющей ее насадки аналогичны абсорбционной колонне К1.
В куб десорбционной колонны подают 1310 нл/час сжатого до 2,45 бар воздуха с температурой 30°С. Колонну термостатируют при температуре 40°С.
Абгаз, выходящий из десорбционной колонны (2190 нл/час) и содержащий 30,7 об.% пропана, 7,4 об.% пропена, 12,3 об.% кислорода, 46,4 об.% азота, 1,5 об.% воды, 1,6 об.% диоксида углерода и незначительное количество тетрадекана, направляют в закалочный аппарат, находящийся в верхней части десорбционной колонны К2.
Абсорбент из куба десорбционной колонны К2 насосом Р1 подают в теплообменник W7 и далее в верхнюю часть абсорбционной колонны К1.
Закалочный аппарат представляет собой металлическую колонну с внутренним диаметром 80 мм, подобно абсорбционной колонне К1 содержащую встроенные элементы. Закалку водой осуществляют при температуре 30°С, расход орошающей воды составляет 120 л/час. Отбираемую из куба закалочного аппарата двухфазную жидкую смесь направляют в устройство для разделения фаз D1, которое представляет собой находящийся в горизонтальном положении резервуар диаметром 200 мм и длиной 500 мм, первая треть внутреннего объема которого (в направлении перемещения потока) заполнена особым образом расположенной тонкой проволочной тканью. Водная фаза, выделенная в устройстве D1, подается насосом в верхнюю часть закалочного аппарата, а выделенный тетрадекан (его среднее количество составляет около 1 г/час) направляют в соответствующий сборник. Потеря воды при закалке восполняется за счет добавления водного конденсата (21).
Абгаз из аппарата для закалки подогревают до температуры 200°С и направляют на двухступенчатое окисление.
Окисление осуществляют в моделирующих трубках длиной 4 м с внутренним диаметром 26 мм. 2,7 м первой моделирующей трубки заполнены катализатором, описанным в европейской заявке на патент ЕР-А-0575879, а 3,0 м второй моделирующей трубки катализатором, описанным в европейской заявке на патент ЕР-А-0017000. Между первой и второй ступенями окисления подают дополнительный воздух в количестве 315 нл/час.
Выделение акриловой кислоты из реакционного газа (26), получаемого в результате окисления, и ее очистку от примесей осуществляют в соответствии с европейской заявкой на патент ЕР-А-0982289.
Согласно данному способу в среднем получают 440 г/час сырой акриловой кислоты (27) с содержанием основного продукта более 99,5%.
Реферат
Настоящее изобретение относится к способу получения акролеина и/или акриловой кислоты из пропана и/или пропена, включающему следующие стадии: а) выделение пропана и/или пропена из содержащей пропан и/или пропен газовой смеси А путем их поглощения абсорбентом, b) выделение пропана и/или пропена из абсорбента с получением содержащего пропан и/или пропен газа В и с) использование полученного на стадии (b) газа В для окисления пропана и/или пропена до акролеина и/или акриловой кислоты, причем между стадиями (b) и (с) не осуществляют гетерогенное каталитическое дегидрирование пропана без подачи кислорода. Способ отличается экономичностью и максимально возможной продолжительностью эксплуатации используемого катализатора без регенерации. 12 з.п. ф-лы, 7 ил.
Формула
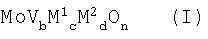
Комментарии