Способ получения нанодисперсных оксидов металлов - RU2633582C1
Код документа: RU2633582C1
Описание
Изобретение относится к области неорганической химии, а именно к способам получения модифицированных нанодисперсных порошкообразных оксидов металлов, в частности оксидов 3d-металлов (скандий, титан, ванадий, хром, марганец, железо, кобальт, никель, медь, цинк), 4d-металлов (иттрий, цирконий), металлов второй и третьей групп главных подгрупп (магний, алюминий, галлий, индий). Модифицированные нанодисперсные оксиды металлов могут быть использованы в качестве исходных веществ для получения материалов конструкционных слоев и покрытий, необходимым условием эффективного функционирования которых является отсутствие агломератов, способных значительно исказить заданные характеристики изделия или параметры рабочего режима системы, в частности для получения покрытий со специальными свойствами, применяемых в таких отраслях, как радиоэлектроника, техника средств связи, специальное приборостроение, медицинское приборостроение, техника средств сигнализации и контроля.
Известны способы получения микрочастиц соединений иттрия и европия путем микроволнового нагревания микроэмульсий [1]. Способы являются специфическими и технически трудновыполнимыми вариантами решения задач получения соединений наноструктуры узкого и специального применения с привлечением труднодоступных технических средств, а получаемые конечные продукты не обладают антиагломеризационной способностью.
Известно применение различных вариантов гидротермального синтеза для получения кристаллов силикатов, боратов, ванадатов и фосфатов различных элементов, в том числе при получении новых структурных типов [2]. Однако из указанного источника информации следует, что простое сочетание обычных гидротермальных условий, высоких температур и давлений не обеспечивает возможность получения продуктов нанодисперсного уровня.
Известен способ получения в условиях гидротермального синтеза силикатов калия и ниобия, которые применяют в качестве затравок при дальнейшем выращивании крупных монокристаллических образцов для нелинейной оптики [3]. Гидротермальные процессы ведут с применением высоких температур и давлений, они отличаются длительностью и трудоемкостью. При этом получаемые монокристаллы имеют размер не менее 1 мм, то есть целевые продукты не относятся к наночастицам.
Известен способ получения оксидных катализаторов [4], согласно которому нерастворимые в воде гидроксиды металлов получают из нитратов и бихроматов в водных растворах при соотношениях компонентов соль : карбамид как 1 моль : (5-14) моль. Указанный способ представляет собой многоэтапный и технически сложный процесс, включающий высокотемпературную обработку гидроксидов металлов на поверхности носителей, который достаточно эффективен при получении каталитических систем, но мало пригоден для получения чистых нанодисперсных оксидов таких металлов, как никель, кобальт, железо и некоторых других. Целевые продукты представляют собой осажденные на поверхностях носителей агломераты, пригодные только для использования в специальных каталитических системах.
Известны способы, основанные на применении проточных реакторов с противотоком различных по концентрациям и исходным температурам растворов исходных компонентов [5]. Таким путем удается обеспечить, в частности, получение оксидов металлов из растворов их нитратов. Однако целевые продукты не обладают антиагломеризационной способностью. Кроме того, получение наноразмерных оксидов связано с применением сложных технических устройств и необходимостью регулирования процессов реакционного противотока нескольких исходных растворов.
Известен способ получения фотокатализатора на основе диоксида титана [6], заключающийся в приготовлении водного раствора сульфата титанила и его гидротермальной обработке, отличающийся тем, что исходный продукт подвергают гидролизу в течение 0,5-24 ч при температурах 100-250°С. Способ включает центрифугирование продукта, очистку его от маточного раствора дистиллированной водой и высушивание на воздухе при 60-70°С. При этом, согласно описанию способа, получают нанодисперсный продукт с частицами размером 11-25 нм. Однако полученный целевой продукт не обладает антиагломеризационной активностью. Как свидетельствуют результаты экспериментальных исследований авторов заявленного способа, при таких мягких условиях проведения процесса и последующем низкотемпературном высушивании (60-70°С) невозможно обеспечить полную конверсию исходного продукта, полное удаление влаги и деструктуризацию агломератов, что обусловливает невозможность проведения гидрофобизационной обработки. Фактически, продукт, состоящий из отдельных наночастиц, в условиях рассматриваемого способа получить невозможно. Кроме того, известный способ характеризуется длительностью и трудоемкостью выполнения.
Известен способ получения фотокатализатора на основе нанокристаллического диоксида титана [7]. Способ отличается от предыдущего тем, что обработка раствора, проводимая при тех же параметрах, дополнительно включает микроволновое излучение мощностью от 500 до 2000 Вт. Условия дальнейшей обработки полученного оксида титана аналогичны описанным в источнике [6], а дисперсность полученных по обоим способам наночастиц диоксида титана и их свойства близки. Таким образом, рассматриваемый способ обладает всеми недостатками известного способа [6].
Известен способ получения порошка диоксида циркония для изготовления керамики [8]. Осаждение гидроксида циркония, стабилизированного иттрием, проводят в непрерывном режиме (каскад реакторов) при ультраозвучивании. После сушки и прокаливания продукт измельчают воздействием ультразвукового поля с диапазоном частоты 20-50 кГц. Однако наложение ультразвукового поля вызывает обратный процесс, а именно укрупнение оксидного порошка, размер частиц которого лежит в диапазоне от 100-4000 нм, то есть в субмикронном диапазоне. Кроме того, полученный целевой продукт не обладает антиагломеризационными свойствами.
Наиболее близким по совокупности существенных признаков к заявленному решению является способ получения нанодисперсных оксидов металлов из водных растворов нитратов металлов [9]. Указанный способ, принятый за прототип, может быть использован для получения оксидов 3d-металлов (скандий, титан, ванадий, хром, марганец, железо, кобальт, никель, медь, цинк), 4d-металлов (иттрий, цирконий), а также металлов главной подгруппы третьей группы (алюминий, галлий, индий). Согласно способу-прототипу проводят непрерывное взаимодействие нитратов металлов с карбамидом в молярном соотношении 2 моль нитратных групп соли металла к 1 моль карбамида. При этом реакционную среду подвергают воздействию микроволнового излучения. Карбамид вводят в исходный водный раствор нитратов металлов при концентрации до 70 г/л. Процесс ведут в устройстве, включающем смеситель, камеру электромагнитного излучения, камеру приема продуктов. Реакционная смесь под действием электромагнитного излучения превращается в нанодисперсные оксиды металлов с образованием паров воды и газообразных продуктов, которые необходимо вывести из камеры электромагнитного излучения. Таким образом, использование электромагнитного излучения позволяет проводить термическую обработку в одну стадию. При этом в описании известного решения не приведены сведения о дальнейших условиях обработки полученных оксидов после выделения агломератов частиц целевого продукта в реакционном пространстве.
Однако способ-прототип не обеспечивает возможность получения технического результата, достигаемого при реализации заявленного способа. Это обусловлено следующим. В способе-прототипе применен реактор гидротермального синтеза, состоящий из цилиндрической емкости, помещенной в микроволновый излучатель, причем цилиндрическая емкость целиком располагалась в объеме микроволнового излучателя таким образом, что ее торцы были выведены за пределы объема излучателя. Один из торцов снабжен устройством для ввода компонентов, другой устройством для выгрузки полученных нанопродуктов. Однако такое конструкционное оформление способа прототипа, когда скорость процесса практически определяется заданной скоростью перемещения шнека, не предохраняет от смешения между растворами компонентов, в разное время попавших в общий реакционный объем. Это обусловливает неравномерность протекания процессов в общем неразделенном объеме реакции. Поэтому длительность микроволнового воздействия на раствор в различных зонах реактора гидротермального синтеза неодинакова. При описываемом процессе имеет место только механическое перемешивание компонентов раствора, находящегося в различных локальных зонах реакционного пространства. При этом указанное перемешивание может осуществляться только в узком зазоре между внешней поверхностью шнека и внутренней поверхностью стенок реактора. Конвекционное перемешивание в системе практически отсутствует. Следует заметить, что именно вследствие непрерывности процесса образование водяных паров, а также газообразных продуктов окислительно-восстановительных реакций происходит непрерывно. В этих условиях постоянно создается переменное давление в различных зонах реакционного пространства, препятствующее равномерному прохождению раствора под воздействием шнека. При этом могут возникать противоток, нерегламентированное перемещение раствора и газов в объеме, задержка ритмичного поступления исходных продуктов в реактор и вывода конечных продуктов из него. В целом процесс протекает спонтанно в различных зонах реактора, а ограниченность реакционного объема и размеров входных и выходных отверстий не может не создавать технических и технологических трудностей при выделении азотсодержащих газов и испаряемой воды. Таким образом, при осуществлении процесса согласно способу-прототипу исключается возможность достижения полной конверсии нитратов металлов в их оксиды, что, с одной стороны, с неизбежностью приводит к наличию в целевых продуктах следов не вступивших в реакции исходных компонентов (нитратов металлов и карбамида), а с другой стороны, значимо снижает выход целевых продуктов. Кроме того, как следует из описания способа-прототипа, окислительно-восстановительные взаимодействия проводятся в растворах при низкой концентрации и с небольшими количествами компонентов, что также значительно снижает выход целевых продуктов.
Отсутствие в способе-прототипе гидрофобизационной обработки исключает возможность получения целевых продуктов с антиагломеризационными свойствами.
Задачей изобретения является создание способа, обеспечивающего получение нанодисперсных оксидов металлов, обладающих пролонгированной устойчивостью против агломеризации при отсутствии в целевых продуктах следов не вступивших в реакции исходных компонентов и при повышении выхода целевых продуктов.
Технический результат заключается в обеспечении полной конверсии нитратов металлов в оксиды в условиях конвекционного перемешивания реакционной смеси, а также в создании оптимальных условий для контакта поверхности наночастиц с абсорбционными агентами и обволакивания поверхности указанных частиц гидрофобной пленкой.
Технический результат достигается тем, что в способе получения нанодисперсных оксидов металлов из водных растворов нитратов металлов, включающем формирование реакционной смеси путем внесения нитратов металлов и карбамида в водную среду в стехиометрическом соотношении и воздействия микроволнового излучения на реакционную среду в реакционном объеме, согласно изобретению реакционную смесь формируют непосредственно в реакционном объеме при следующем соотношении компонентов в реакционной среде, мас. %:
а воздействие микроволнового излучения осуществляют при открытом доступе к реакционной среде в реакционном объеме. После воздействия микроволнового излучения промежуточный продукт реакций подвергают сушке при температуре не менее 200°С, измельчают высушенный продукт до размеров частиц не более 20 нм, при этом в ходе измельчения высушенного продукта параллельно осуществляют его гидрофобизационную обработку гидрофобизирующей смесью силанов и силиконовых олигомеров, взятых в соотношении, мас. %: силан - 17-33, силиконовый олигомер - 67-83. Эффективным является использование в качестве реакционного объема термостойкого реактора гидротермального синтеза открытого типа из проницаемого для микроволнового излучения материала. Предпочтительным является воздействие микроволнового излучения мощностью не менее 1400 Вт. Эффективным является использование гидрофобизирующей смеси, состоящей из силанов с молекулярной массой не более 200 у.е. (углеродных единиц) и силиконовых олигомеров с молекулярной массой не более 1000 у.е. и кинематической вязкостью при температуре 20°С не более 500 мм2/с, имеющих значения рН не более 7,0 и обладающих разной абсорбционной способностью к нанодисперсным частицам, причем абсорбционная способность силанов выше, чем абсорбционная способность силиконовых олигомеров.
Обоснование оптимальных режимов и условий реализации способа проводили следующим образом.
Для определения оптимальной мощности микроволнового излучения исследовали условия протекания окислительно-восстановительного взаимодействия нитратов трех различных металлов: нитрата цинка Zn(NO3)2, нитрата магния Mg(NO3)2 и нитрата алюминия Al(NO3)3 с карбамидом CO(NH2)2 в водной среде. В качестве реакционных объемов использовали 24 термостойких емкости объемом 1 л каждая, в которые вносили:
в емкости №№1-8 - 37,2 г нитрата цинка и 12,0 г карбамида;
в емкости №№9-16 - 29,6 г нитрата магния и 12,0 г карбамида;
в емкости №№17-24 - 21,3 г нитрата алюминия и 9,0 г карбамида.
Затем в емкости №№1-16 добавляли по 200 мл дистиллированной воды, а в емкости №№17-24 - 400 мл дистиллированной воды. Перемешивали содержимое во всех емкостях до полного растворения. После этого емкости помещали последовательно в установку микроволнового излучения МХР5203 (АСР Inc., США) и воздействовали на емкости микроволновым излучением с мощностью, варьировавшей в диапазоне 600-2000 Вт. Устанавливали путем визуального наблюдения момент начала интенсивного парообразования в реакционном объеме в каждой из емкостей, что соотносили с началом активного конвекционного перемешивания, и фиксировали с помощью секундомера время от начала воздействия микроволнового излучения до наступления указанного момента.
Результаты измерений представлены в табл. 1.

Из данных табл. 1 видно, что начиная со значения мощности микроволнового излучения 1400 Вт время от начала микроволнового воздействия до начала интенсивного парообразования в реакционном объеме (начала активного конвекционного перемешивания) становится минимальным (2 мин) и не изменяется при дальнейшем увеличении мощности.
Для определения времени воздействия микроволнового излучения, обеспечивающего максимальную степень конверсии нитратов металлов в соответствующие оксиды определяли степень конверсии в оксидах металлов с валентностью II (оксид цинка ZnO, оксид магния MgO), III (оксид алюминия Al2O3, оксид железа Fe2O3), IV (оксид титана TiO2, оксид циркония ZrO2), полученных в результате окислительно-восстановительного взаимодействия из нитратов, соответственно нитрата цинка Zn(NO3)2 и нитрата магния Mg(NO3)2, нитрата алюминия Al(NO3)3 и нитрата железа Fe(NO3)3, нитрата титана Ti(NO3)4 и нитрата циркония Zr(NO3)4 при воздействии микроволнового излучения фиксированной мощности и варьировании времени указанного воздействия. В качестве реакционных объемов использовали 36 термостойких емкостей объемом 1 л каждая, в которые вносили:
в емкости №№1-6 - 37,2 г нитрата цинка и 12,0 г карбамида;
в емкости №№7-12 - 29,6 г нитрата магния и 12,0 г карбамида;
в емкости №№13-18 - 21,3 г нитрата алюминия и 9,0 г карбамида;
в емкости №№18-24 - 22,4 г нитрата железа и 9,0 г карбамида;
в емкости №№25-30 - 29,4 г нитрата титана и 12,0 г карбамида;
в емкости №№31-36 - 33,9 г нитрата циркония и 12,0 г карбамида.
Затем в емкости №№1-12 добавляли по 200 мл дистиллированной воды, а в емкости №№13-36 - 400 мл дистиллированной воды. Перемешивали содержимое во всех емкостях до полного растворения. После этого емкости помещали последовательно в установку микроволнового излучения МХР5203 (АСР Inc., США) и воздействовали на емкости микроволновым излучением мощностью 1400 Вт при времени воздействия указанного излучения, варьировавшем в диапазоне 3-13 мин. Степень конверсии определяли путем взвешивания полученного промежуточного продукта реакций на аналитических весах, вычисления, исходя из результатов взвешивания, количества прореагировавшего нитрата, с последующим количественным сопоставлением прореагировавшего нитрата металла с его исходным количеством в реакционной смеси.
Результаты определений представлены в табл. 2.

Из данных табл. 2 следует, что полная конверсия для нитратов металлов с валентностью II достигалась через 7 мин от начала воздействия микроволнового излучения мощностью 1400 Вт, а для нитратов металлов с валентностью III и IV - через 9 мин от начала указанного воздействия.
Для установления оптимального соотношения компонентов в реакционной смеси определяли средние размеры частиц оксидов металлов с валентностью II (оксид цинка ZnO, оксид магния MgO), III (оксид алюминия Al2O3, оксид железа Fe2O3), IV (оксид титана TiO2, оксид циркония ZrO2), как промежуточных продуктов окислительно-восстановительных превращений, при воздействии микроволнового излучения мощностью 1400 Вт при времени воздействия, обеспечивающем полную конверсию исходных нитратов металлов (7 мин - для нитратов металлов с валентностью II и 9 мин для нитратов металлов с валентностью III и IV), а также выход указанных оксидов при различных количественных соотношениях компонентов в реакционной смеси. Размеры частиц определяли на основе данных просвечивающей электронной микроскопии с использованием электронного микроскопа JEOL JEM-1400 (JEOL Ltd., Япония). Выход промежуточных продуктов реакций определяли по результатам взвешивания на аналитических весах продукта, подвергнутого сушке при температуре 250°С в течение 1,5 ч (контрольная сушка).
Результаты определений выборочно (для оксида цинка, оксида алюминия и оксида титана) представлены в табл. 3.

Данные табл. 3 показывают, что соотношение суммарного количества нитрата металла с карбамидом и воды, варьирующих в диапазонах соответственно (10-20):(80-90), обеспечивало получение частиц оксидов металлов с размерами 10-16 нм, которые являются оптимальными для проведения дальнейших стадий заявленного способа (в частности, для стадии гидрофобизационной обработки) при максимально возможном (или приближающемся к нему) выходе указанных промежуточных продуктов реакций (57-74 г/л), что позволило принять этот диапазон в качестве диапазона оптимальных значений. Увеличение доли суммарного количества нитрата металла с карбамидом в рассматриваемом соотношении свыше верхней границы диапазона оптимальных значений не приводило к дальнейшему увеличению выхода промежуточного продукта, но способствовало значимому укрупнению частиц. Снижение доли суммарного количества нитрата металла с карбамидом в рассматриваемом соотношении за пределы нижней границы диапазона оптимальных значений приводило к неоправданно низкому выходу промежуточного продукта (4-12 г/ч).
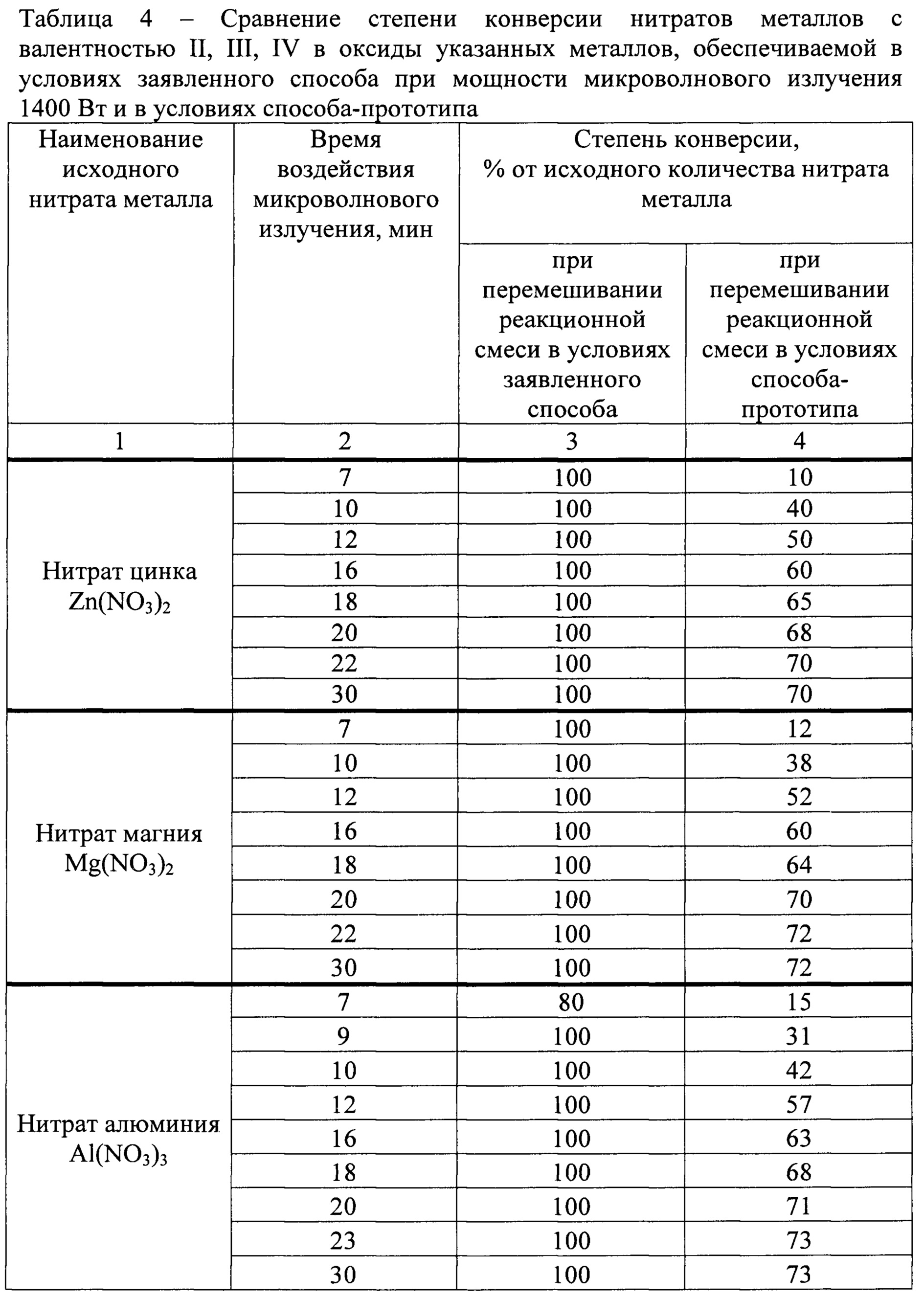
Для сравнения эффективности условий осуществления заявленного способа на стадии окислительно-восстановительных превращений и условий, воспроизводящих условия способа прототипа, определяли степень конверсии в оксидах металлов с валентностью II (оксид цинка ZnO, оксид магния MgO), III (оксид алюминия Al2O3, оксид железа Fe2O3), IV (оксид титана TiO2, оксид циркония ZrO2), полученных в результате окислительно-восстановительного взаимодействия из исходных нитратов, соответственно нитрата цинка Zn(NO3)2 и нитрата магния Mg(NO3)2, нитрата алюминия Al(NO3)3 и нитрата железа Fe(NO3)3, нитрата титана Ti(NO3)4 и нитрата циркония Zr(NO3)4. Заявленный способ на стадии окислительно-восстановительных превращений осуществляли при следующих условиях: в качестве реакционных объемов использовали 48 термостойких емкостей объемом 1 л каждая; мощность микроволнового излучения 1400 Вт; время воздействия микроволнового излучения варьировало в диапазоне 7-30 мин; соотношение анализируемых нитратов металлов и карбамида: на один моль каждой из нитратных групп соли металла 0,5 моля карбамида; соотношение суммарного количества нитрата металла с карбамидом и воды 15:75. Способ-прототип осуществляли в условиях, описанных в патенте РФ №2538585 [9], с использованием реактора гидротермального синтеза, конструкция которого описана в патенте РФ №124590 [10]. При этом соотношения анализируемых нитратов металлов и карбамида, а также суммарного количества нитратов металлов с карбамидом и воды, мощность микроволнового излучения и время его воздействия соответствовали описанным выше для условий заявленного способа. Степень конверсии в условиях заявленного способа и способа-прототипа определяли путем взвешивания полученного промежуточного продукта реакций на аналитических весах, вычисления, исходя из результатов взвешивания, количества прореагировавшего нитрата, с последующим количественным сопоставлением прореагировавшего нитрата металла с его исходным количеством в реакционной смеси.
Результаты определений приведены в табл. 4.
Результаты, представленные в табл. 4, свидетельствуют о том, что условия способа-прототипа не позволяли добиться полной конверсии нитратов металлов даже в случаях, когда время воздействия микроволновым излучением на реакционную смесь значимо превышало время, указанное в примерах реализации способа-прототипа [9]. В условиях заявленного способа полная конверсия достигалась при времени воздействия микроволнового излучения 7-9 мин.

Для определения оптимальной температуры сушки определяли размеры частиц промежуточного продукта, подвергнутого сушке, и содержание воды в указанном продукте при варьировании температуры сушки в диапазоне 18-220°С. Размеры частиц определяли на основе данных просвечивающей электронной микроскопии с использованием электронного микроскопа JEOL JEM-1400 (JEOL Ltd., Япония). Содержание воды в 100 г промежуточного продукта определяли по результатам взвешивания на аналитических весах продукта, подвергнутого сушке при исследуемой температуре, и того же продукта после повторной (контрольной) сушки при температуре 250°С в течение 1,5 ч с последующим вычислением разницы результатов указанных определений..
Результаты определений приведены в табл. 5.
Из данных табл. 5, видно, что температура сушки, равная 200°С и выше, обеспечивала получение частиц высушенных промежуточных продуктов с размерами 17-28 нм, которые находились в пределах экспериментально установленного авторами изобретения диапазона значений указанного параметра, являющихся оптимальными для стадии гидрофобизационной обработки. Указанная температура позволяла также минимизировать содержание воды в промежуточных высушенных продуктах (до значений (0,07-0,15) г/100 г промежуточного продукта, снизив этот показатель за пределы пороговых значений, превышение которых оказывало (по результатам экспериментов авторов изобретения) негативное влияние на эффективность гидрофобизационной обработки.

Достижение технического результата, обеспечиваемого при реализации заявленного способа, обусловлено следующим.
Осуществление воздействия микроволнового излучения при открытом доступе к находящейся в реакционном объеме смеси исходных компонентов, приготовленной в этом же объеме, обеспечивает равномерное распределение всех реагентов в реакционном пространстве. При этом происходит равномерное и беспрепятственное поступление энергии микроволнового излучения во все локальные зоны реакционного пространства, что способствует, с одной стороны, ускоренному аккумулированию лучевой энергии реагентами, непосредственно участвующими в окислительно-восстановительных взаимодействиях, а, с другой стороны, активации энергоаккумулирующих и энергопередающих функций воды. Энергия, воспринятая водой, трансформируется в энергию конвекционного перемешивания реагентов, которое играет ключевую роль в формировании в реакционном пространстве энергетического баланса, обусловливающего быстрый переход процесса конверсии в автокаталитическую фазу. Таким образом, возникновение конвекционного перемешивания является своеобразным индикатором вступления процесса конверсии в автокаталитическую фазу, а функционирование конвекционного перемешивания, как такового, служит поддерживающим фактором автокаталитического режима окислительно-восстановительных взаимодействий. В этих условиях все реакционно-способные компоненты обеспечиваются количеством энергии, необходимым и достаточным не только для преодоления энергетического порога причинно-значимых окислительно-восстановительных реакций, но и для поддержания энергетического уровня, превышающего пороговый, на протяжении всего периода воздействия микроволновым излучением. Это обеспечивает, по результатам экспериментальных исследований авторов изобретения, возможность полной конверсии нитратов металлов в их оксиды в рамках стехиометрических соотношений при любых значениях мощности микроволнового излучения в пределах рабочих диапазонов, используемых в процессах гидротермального синтеза солей и оксидов металлов. При этом воздействие микроволнового излучения мощностью 1400 Вт и более позволяет минимизировать время достижения полной конверсии, что дополнительно способствует повышению эффективности окислительно-восстановительных взаимодействий. Достижение полной конверсии нитратов металлов в их оксиды обусловливает, с одной стороны, отсутствие в целевых продуктах следов не вступивших в реакции исходных компонентов (нитратов металлов и карбамида), а с другой стороны, - повышение выхода целевых продуктов.
Проведение окислительно-восстановительных реакций при заявленном соотношении компонентов реакционной смеси (смесь нитрата и карбамида и вода) обеспечивает возможность протекания указанных реакций в реальном соответствии с существующими стехиометрическими соотношениями, что при энергетической насыщенности реакционно-способных компонентов в условиях автокаталитической фазы процесса конверсии минимизирует возможность агломеризации как промежуточных продуктов окислительно-восстановительных реакций, так и непрореагировавших компонентов, что в дальнейшем вносит свой вклад в формирование наночастиц, способных к гидрофобизации.
Осуществление процесса сушки при заявленной температуре позволяет гарантированно избавиться от следов воды, молекулы которой обладают способностью абсорбироваться на поверхности наночастиц оксидов металлов, препятствуя тем самым образованию абсорбционных контактов между наночастицами и абсорбционными агентами в составе гидрофобизирующей смеси.
Гидрофобизационная обработка высушенного продукта смесью силанов и сииконовых олигомеров, проводимая одновременно с измельчением указанного продукта, позволяет сформировать динамическую трехкомпонентную абсорбционную систему, обеспечивающую двухуровневое абсорбционное взаимодействие свободных индивидуальных наночастиц оксидов металлов с абсорбционно активными гидрофобизирующими агентами (силанами), способными к реализации первичного контакта и удержания наночастиц, и с абсорбционно индифферентными гидрофобизирующими агентами (силиконовыми олигомерами), обладающими сродством к молекулам абсорбированных силанов, способных к формированию обволакивающего контура, охватывающего наночастицы с абсорбированными силанами, а также поддерживать в указанной системе состояние устойчивого динамического равновесия причинно-значимых агентов как в процессе гидрофобизационной обработки, так и после завершения процесса с получением целевых продуктов. Результатом динамических взаимодействий в указанной системе является получение покрытых защитной пленкой наночастиц, обладающих пролонгированной антиагломеризационной способностью. Использование в составе гидрофобизационной смеси компонентов, обладающих заявленными стерическими и кинематическими свойствами, химическим сродством, физической совместимостью и химической инертностью к химическому составу наночастиц, обеспечивает дополнительную оптимизацию динамических взаимодействий в абсорбционной системе.
Способ осуществляют следующим образом.
Вносят в реакционный объем термостойкого реактора гидротермального синтеза открытого типа из проницаемого для микроволнового излучения материала, в качестве которого может быть использован, например термостойкий стакан В-2-150 или В-1-50 объемом 1000, 2000 или 3000 л (ПАО «Химлаборприбор», Россия), нитрат металла и карбамид в соотношении: на один моль каждой из нитратных групп соли металла 0,5 моля карбамида, добавляют в реакционный объем реактора дистиллированную воду при соотношении компонентов в реакционной среде, мас. %: смесь нитрата металла и карбамида - 10-20, вода - 80-90. Перемешивают реакционную смесь до полного растворения нитрата и карбамида в воде. Помещают реактор гидротермального синтеза в установку микроволнового излучения закрытого типа, например марки МХР5203 или МХР5201 (АСР Inc., США) или в микроволновую печь Kuppersberg RMW 969 ANT (Португалия) и воздействуют микроволновым излучением мощностью 1400 Вт в течение 7-9 мин. После завершения воздействия микроволнового излучения промежуточный продукт реакций двукратно промывают дистиллированной водой, затем подвергают сушке при температуре 200°С, например в сушильном шкафу ШС-80-01-СПУ (ОАО «Смоленское СКТБ СПУ», Россия) в течение 1-2 ч. Высушенный промежуточный продукт переносят в устройство для измельчения, например в бисерную мельницу из лабораторной диспергирующей установки ЛДУ-3 МПР (Лаботекс, Россия) или в высокоскоростную шаровую мельницу Emax (RETSCH, Германия). Параллельно готовят гидрофобизирующую смесь на основе силанов и силиконовых олигомеров в соотношении, мас. %: силан 17-33, силиконовый олигомер 67-83, путем перемешивания указанных компонентов при температуре (16±2)°С. В качестве силанов используют, например триметилвинилсилан, триметилдиэтиламиносилан, тетраэтоксисилан, триэтоксисилан, а в качестве силиконовых олигомеров, например полиметилсилоксан-5 (ПМС-5), полиметилсилоксан-10 (ПМС-10), полиэтилсилоксан-5 (ПЭС-5), этилгидридсилоксан (ГКЖ-94). Вносят полученную гидрофобизирующую смесь в устройство для измельчения и в ходе измельчения высушенного продукта параллельно осуществляют его гидрофобизационную обработку гидрофобизирующей смесью. Выход целевого продукта - модифицированного нанодисперсного оксида металла - 60-110 г/ч.
Изобретение иллюстрируется следующими примерами.
Пример 1. Получение оксида цинка
Процесс описывается уравнением:
Zn(NO3)2+(NH2)2CO=ZnO+CO2+2H2O+2N2O.
Вносили в реакционный объем реактора гидротермального синтеза открытого типа, в качестве которого использовали термостойкий стакан В-1-50 объемом 1000 мл (ПАО «Химлаборприбор», Россия), 18,9 г твердого нитрата цинка и 6,0 г карбамида (в соотношении 0,2 моль нитратных групп:0,1 моль карбамида). Добавляли в реакционный объем реактора 223 мл дистиллированной воды (в соотношении смесь нитрата металла и карбамида, мас. %:вода - 10:90). Перемешивали реакционную смесь до полного растворения нитрата и карбамида в воде. Помещали реактор в установку микроволнового излучения закрытого типа марки МХР5203 (АСР Inc., США), включали установку и осуществляли воздействие микроволновым излучением мощностью 1400 Вт в течение 7 мин. Извлекали реактор с полученным агломератом оксида цинка (промежуточным продуктом реакций) из установки. Переносили промежуточный продукт в термостойкую емкость и дважды промывали дистиллированной водой. Затем помещали емкость в сушильный шкаф ШС-80-01-СПУ (ОАО «Смоленское СКТБ СПУ», Россия) и подвергали сушке при температуре 200°С в течение 1,5 ч. Высушенный продукт переносили в бисерную мельницу из лабораторной диспергирующей установки ЛДУ-3 МПР (Лаботекс, Россия) и размалывали. В процессе размола высушенный продукт подвергали гидрофобизации смесью триметилвинилсилана в ПМС-5 (массовое соотношение 1:3), которую вносили на начальном этапе размола. Выгружали полученный нанодисперсный оксид цинка (целевой продукт) из бисерной мельницы и взвешивали на аналитических весах. Выход целевого продукта составил 67 г. Содержание в целевом продукте примесей нитратов и карбамида, определенное с помощью инструментальных методов, составило (0,1±0,01) % от массы целевого продукта (по каждой из примесей). Данные просвечивающей электронной микроскопии с использованием электронного микроскопа JEOL JEM-1400 (JEOL Ltd., Япония) свидетельствовали о том, что размеры наночастиц оксида цинка находились в пределах 7-15 нм. По результатам просвечивающей электронной микроскопии с помощью указанного выше электронного микроскопа и рентгеновской дифрактометрии с использованием автоматического рентгеновского дифрактометра ДРОН-3М (Буревестник, Россия) было установлено, что содержание агломератов составляло (0,05±0,01) % от массы целевого продукта в течение срока наблюдения продолжительностью 30 сут.
Пример 2. Получение оксида алюминия
Процесс описывается уравнением:
2Al(NO3)3+3(NH2)2CO=Al2O3+3CO2+6H2O+6N2O.
Вносили в реакционный объем реактора гидротермального синтеза открытого типа, в качестве которого использовали термостойкий стакан В-2-150 объемом 1000 мл (ПАО «Химлаборприбор», Россия), 21,3 г твердого нитрата алюминия и 9,0 г карбамида (в соотношении 0,3 моль нитратных групп:0,15 моль карбамида). Добавляли в реакционный объем реактора 169,7 мл дистиллированной воды (в соотношении смесь нитрата металла и карбамида:вода, мас. % - 15:85). Далее процесс осуществляли аналогично примеру 1. Воздействие микроволновым излучением мощностью 1400 Вт в течение 9 мин. Сушка при температуре 210°С в течение 2 ч. Гидрофобизирующая смесь состояла из триметилдиэтиламиносилана в ПЭС-5 (массовое соотношение 1:4). Выход целевого продукта составил 61 г/ч. Содержание в целевом продукте примесей нитратов и карбамида, составило (0,1±0,01) % от массы целевого продукта (по каждой из примесей). По результатам просвечивающей электронной микроскопии и рентгеновской дифрактометрии, осуществляемых на тех же приборах, что и в примере 1, размеры наночастиц оксида магния находились в пределах 11-13 нм, содержание агломератов составляло (0,05±0,01) % от массы целевого продукта в течение срока наблюдения продолжительностью 30 сут.
Пример 3. Получение оксида магния
Процесс описывается уравнением:
Mg(NO3)2+(NH2)2CO=MgO+CO2+2H2O+2N2O.
Вносили в реакционный объем реактора гидротермального синтеза открытого типа, в качестве которого использовали термостойкий стакан В-2-150 объемом 1000 мл (ПАО «Химлаборприбор», Россия), 14,8 г твердого нитрата магния и 6,0 г карбамида (в соотношении 0,2 моль нитратных групп:0,1 моль карбамида). Добавляли в реакционный объем реактора 150,0 мл дистиллированной воды (в соотношении смесь нитрата металла и карбамида:вода, мас. % - 14:86). Далее процесс осуществляли аналогично примеру 1. Воздействие микроволновым излучением мощностью 1400 Вт в течение 7 мин. Сушка при температуре 200°С в течение 1,5 ч. Гидрофобизирующая смесь состояла из тетраэтоксисилана в ПМС-10 (массовое соотношение 1:3). Выход целевого продукта составил 63 г/ч. Содержание в целевом продукте примесей нитратов и карбамида (0,1±0,01) % от массы целевого продукта (по каждой из примесей). По результатам просвечивающей электронной микроскопии и рентгеновской дифрактометрии: размеры наночастиц оксида алюминия находились в пределах 11-13 нм, содержание агломератов составляло (0,05±0,01) % от массы целевого продукта в течение срока наблюдения продолжительностью 30 сут.
Пример 4. Получение оксида меди
Процесс описывается уравнением:
Cu(NO3)2+(NH2)2CO=CuO+CO2+2H2O+2N2O.
Вносили в реакционный объем реактора гидротермального синтеза открытого типа, в качестве которого использовали термостойкий стакан В-1-50 объемом 1000 мл (ПАО «Химлаборприбор», Россия), 18,8 г твердого нитрата меди и 6,0 г карбамида (в соотношении 0,2 моль нитратных групп:0,1 моль карбамида). Добавляли в реакционный объем реактора 190,0 мл дистиллированной воды (в соотношении смесь нитрата металла и карбамида:вода, мас. % - 12:88). Далее процесс осуществляли аналогично примеру 1. Воздействие микроволновым излучением мощностью 1400 Вт в течение 7 мин. Сушка при температуре 200°С в течение 1 ч. Гидрофобизирующая смесь состояла из триэтоксисилана в ГКЖ-94 (массовое соотношение 1:5).Выход целевого продукта составил 60 г/ч. Содержание в целевом продукте примесей нитратов и карбамида (0,1±0,01) % от массы целевого продукта (по каждой из примесей). По результатам просвечивающей электронной микроскопии и рентгеновской дифрактометрии: размеры наночастиц оксида меди в пределах 10-12 нм, содержание агломератов составляло (0,04±0,01) % от массы целевого продукта в течение срока наблюдения продолжительностью 30 сут.
Пример 5. Получение диоксида титана
Процесс описывается уравнением:
Ti(NO3)4+2(NH2)2CO=TiO2+2CO2+4H2O+4N2O.
Вносили в реакционный объем реактора гидротермального синтеза открытого типа, в качестве которого использовали термостойкий стакан В-1-50 объемом 2000 мл (ПАО «Химлаборприбор», Россия), 59,2 г твердого нитрата титана и 24,0 г карбамида (в соотношении 0,4 моль нитратных групп:0,2 моль карбамида). Добавляли в реакционный объем реактора 333,0 мл дистиллированной воды (в соотношении смесь нитрата металла и карбамида:вода, мас. % - 20:80). Далее процесс осуществляли аналогично примеру 1. Воздействие микроволновым излучением мощностью 1500 Вт в течение 8 мин. Сушка при температуре 210°С в течение 1,5 ч. Гидрофобизирующая смесь состояла из тетраэтоксисилан в ПМС-10 (массовое соотношение 1:2). Выход целевого продукта составил 75 г/ч. Содержание в целевом продукте примесей нитратов и карбамида (0,1±0,01) % от массы целевого продукта (по каждой из примесей). По результатам просвечивающей электронной микроскопии и рентгеновской дифрактометрии: размеры наночастиц оксида титана в пределах 13-17 нм, содержание агломератов составляло (0,05±0,01) % от массы целевого продукта в течение срока наблюдения продолжительностью 30 сут.
Пример 6. Получение оксида никеля
Процесс описывается уравнением:
Ni(NO3)2+(NH2)2CO=NiO+CO2+2H2O+2N2O.
Вносили в реакционный объем реактора гидротермального синтеза открытого типа, в качестве которого использовали термостойкий стакан В-1-50 объемом 1000 мл (ПАО «Химлаборприбор», Россия), 18,4 г твердого нитрата никеля и 6,0 г карбамида (в соотношении 0,2 моль нитратных групп:0,1 моль карбамида). Добавляли в реакционный объем реактора 160,0 мл дистиллированной воды (в соотношении смесь нитрата металла и карбамида:вода, мас. % - 13:87). Далее процесс осуществляли аналогично примеру 1. Воздействие микроволновым излучением мощностью 1400 Вт в течение 7 мин. Сушка при температуре 200°С в течение 1,0 ч. Гидрофобизирующая смесь состояла из триметилвинилсилана в ПМС-5 (массовое соотношение 1:4). Выход целевого продукта составил 88 г/ч. Содержание в целевом продукте примесей нитратов и карбамида (0,1±0,01) % от массы целевого продукта (по каждой из примесей). По результатам просвечивающей электронной микроскопии и рентгеновской дифрактометрии: размеры наночастиц оксида титана в пределах 11-14 нм, содержание агломератов составляло (0,04±0,01) % от массы целевого продукта в течение срока наблюдения продолжительностью 30 сут.
Пример 7. Получение оксида циркония
Процесс описывается уравнением:
Zr(NO3)4+2(NH2)2CO=ZrO2+2CO2+4H2O+4N2O.
Вносили в реакционный объем реактора гидротермального синтеза открытого типа, в качестве которого использовали термостойкий стакан В-1-50 объемом 3000 мл (ПАО «Химлаборприбор», Россия), 67,8 г твердого нитрата циркония и 24,0 г карбамида (в соотношении 0,4 моль нитратных групп:0,2 моль карбамида). Добавляли в реакционный объем реактора 500,0 мл дистиллированной воды (в соотношении смесь нитрата металла и карбамида:вода, мас. % - 16:84). Далее процесс осуществляли аналогично примеру 1. Воздействие микроволновым излучением мощностью 1500 Вт в течение 9 мин. Сушка при температуре 210°С в течение 1,0 ч. Гидрофобизирующая смесь состояла из триэтоксисилана в ГКЖ-94 (массовое соотношение 1:4). Выход целевого продукта составил 114 г/ч. Содержание в целевом продукте примесей нитратов и карбамида (0,1±0,01) % от массы целевого продукта (по каждой из примесей). По результатам просвечивающей электронной микроскопии и рентгеновской дифрактометрии: размеры наночастиц оксида циркония в пределах 14-16 нм, содержание агломератов составляло (0,06±0,01) % от массы целевого продукта в течение срока наблюдения продолжительностью 30 сут.
Пример 8. Получение оксида железа
Процесс описывается уравнением:
2Fe(NO3)3+3(NH2)2CO=Fe2O3+3CO2+6H2O+6NO2.
Вносили в реакционный объем реактора гидротермального синтеза открытого типа, в качестве которого использовали термостойкий стакан В-1-50 объемом 1000 мл (ПАО «Химлаборприбор», Россия), 24,2 г твердого нитрата железа и 9,0 г карбамида (в соотношении 0,3 моль нитратных групп:0,15 моль карбамида). Добавляли в реакционный объем реактора 200,0 мл дистиллированной воды (в соотношении смесь нитрата металла и карбамида:вода, мас. % - 14:86). Далее процесс осуществляли аналогично примеру 1. Воздействие микроволновым излучением мощностью 1400 Вт в течение 9 мин. Сушка при температуре 220°С в течение 1,5 ч. Гидрофобизирующая смесь состояла из тетраэтоксисилана в ПМС-10 (массовое соотношение 1:4). Выход целевого продукта составил 71 г/ч. Содержание в целевом продукте примесей нитратов и карбамида (0,1±0,01) % от массы целевого продукта (по каждой из примесей). По результатам просвечивающей электронной микроскопии и рентгеновской дифрактометрии: размеры наночастиц оксида железа в пределах 13-15 нм, содержание агломератов составляло (0,06±0,01) % от массы целевого продукта в течение срока наблюдения продолжительностью 30 сут.
Сравнение заявленного способа и способа-прототипа производили путем сопоставительной оценки времени агломеризации, целевых характеристик (относительное содержание в целевых продуктах агломератов и примесей - нитратов металлов и карбамида) и выхода целевых продуктов - нанодисперсных оксидов металлов, полученных с использованием указанных способов. В качестве целевых продуктов анализировали оксид цинка ZnO, оксид магния MgO, оксид циркония ZrO2, оксид меди CuO, оксид никеля NiO, оксид алюминия Al2O3, оксид железа Fe2O3 и оксид титана TiO2. Относительное содержание агломератов в целевых продуктах и время агломеризации определяли по результатам просвечивающей электронной микроскопии с помощью электронного микроскопа JEOL JEM-1400 (JEOL Ltd., Япония) и рентгеновской дифрактометрии с использованием автоматического рентгеновского дифрактометра ДРОН-3М (Буревестник, Россия).
Результаты определения представлены в табл. 6.


Источники информации
1. Qi Pang et all, A novel approach for preparation of Y2O3:Eu nanoparticles by microemulsion-microwawe heating, Materials Science and Engineering: В, V 103, 2003, P. 57-61.
2. Димитрова О.В. Рост и морфология кристаллов. Гидротермальный синтез кристаллов силикатов, боратов, ванадатов и фосфатов. РУДН М, 2015. - 107 с.
3. Способ получения монокристаллов K2(NBO)22SI4O12: заявка RU на изобретение №94021160, C01G 15/00, заявл. 07.06.1994, опубл. 27.04.1996.
4. Способ приготовления оксидных катализаторов: патент RU 2055638 на изобретение, B01J 23/74, B01J 23/34, B01J 37/02, B01J 23/74, B01J 101:36, B01J 101:40, заявл. 29.11.1993, опубл. 10.03.1996.
5. Противоточный реактор смешения: патент US 7566436 на изобретение, C01F 17/00, C01G 3/02, C01G 5/00, C01G 11/00, C01G 23/047, C01G 25/02, C01G 49/02, C01G 55/00, C01G 57/00, В82В 1/00, В82В 3/00, заявл. 11.02.2005, опубл. 25.08.2007.
6. Способ получения фотокатализатора на основе диоксида титана: патент RU 2408427 на изобретение, B01J 37/08, C01G 23/053, B01D 53/86, B01J 21/06, C02F 1/30, В82В 3/00, заявл. 20.07.2009, опубл. 10.01.2011.
7. Способ получения фотокатализатора на основе нанокристаллического диоксида титана: патент RU 2408428 на изобретение, B01J 37/34, B01J 21/06, C02F 1/30, В82В 3/00, B01J 37/08, C01G 23/053, B01D 53/86, заявл. 20.07.2009, опубл. 10.01.2011.
8. Способ получения порошка диоксида циркония для изготовления керамики: патент RU 2058939 на изобретение, C01G 25/02, заявл. 26.06.1991, опубл. 27.04.1996.
9. Способ получения нанодисперсных оксидов металлов: патент RU 2538585 на изобретение, C01G 25/02, B01J 19/12, заявл. 31.08.2012, опубл. 10.01.2015.
10. Реактор гидротермального синтеза: патент RU 124590 на полезную модель, B01J 19/08, заявл. 31.08.2012, опубл. 10.02.2013.
Реферат
Изобретение может быть использовано в неорганической химии. Способ получения нанодисперсных оксидов металлов включает формирование реакционной смеси путем внесения нитратов металлов и карбамида в водную среду в стехиометрическом соотношении. На реакционную среду воздействуют микроволновым излучением. Реакционную смесь формируют непосредственно в реакционном объеме при следующем соотношении компонентов, мас. %: смесь нитрата и карбамида 10-20, вода - остальное. Воздействие микроволновым излучением осуществляют при открытом доступе к реакционной среде в реакционном объеме. Промежуточный продукт реакций подвергают сушке при температуре не менее 200°С. Высушенный продукт измельчают до размеров частиц не более 20 нм. В ходе измельчения высушенного продукта параллельно осуществляют гидрофобизационную обработку гидрофобизирующей смесью, состоящей из силанов и силиконовых олигомеров, взятых в соотношении, мас. %: силан 17-33, силиконовый олигомер 67-83. Изобретение позволяет обеспечить полную конверсию нитратов металлов в оксиды, обладающие пролонгированной устойчивостью к агломерации, повысить выход продуктов, исключить наличие следов исходных компонентов в продуктах. 3 з.п. ф-лы, 6 табл., 8 пр.
Формула
Документы, цитированные в отчёте о поиске
Способ получения нанодисперсных оксидов металлов
Комментарии