Способ получения фторидных пирохлоров индия cscuinf6 или галлия cscugaf6 - RU2781423C1
Код документа: RU2781423C1
Чертежи
Описание
Изобретение относится к получению неорганических соединений фторидных пирохлоров индия CsCuInF6 и галлия CsCuGaF6 и может найти применение при изготовлении материалов для электронной промышленности, в качестве матриц для фиксации радионуклидов, индикаторов геохимических процессов.
Фторидные пирохлоры, представляющие собой обширный класс соединений общей формулы AIBIIMIIIF6(AI = K, Rb, Cs; ВII = Cu, Zn, Ni, Со, Mn, Pd и МIII = Al, Ga, In, Tl, Sc, Fe, Cr, Ti, V, Mn, Rh), демонстрируют широкий спектр физических свойств, зависящий от присутствия в них различных элементов и степеней их окисления. Наличие обширного ряда доступных электронных состояний пирохлоров обеспечивает набор присущих им различных свойств: изолирующих (Gd2Ti2О7), полупроводниковых свойств керамики с металлическим характером (Sm2Мо2О7). В некоторых фазах пирохлоров наблюдаются сверхпроводниковые свойства, впервые обнаруженные у фазы a-Cd2Re2O7, а затем у AOs2O6 (А = К, Rb, Cs). Современные исследования в химии пирохлоров в основном сосредоточены на сегнето- и пироэлектрических, магнитных и электронных свойствах членов этого семейства, в первую очередь, из-за их возможного применения в современной технике.
Сведения о фторидных пирохлорах индия CsCuInF6 и галлия CsCuGaF6, в том числе о способах их получения, ограничены, известно получение крайне немногочисленных представителей этого класса соединений.
Известен способ получения пирохлоров различного химического состава, описанный в работе Редькина А.Ф., Бородулина Г.П. «Вестник ОНЗ РАН», т. 3, NZ6082, DOI. 10.2205/2011NZ000212, 2011, осуществляемый путем гидротермального синтеза в присутствии кислородных буферов в течение 7 суток в платиновых ампулах при температуре 800°С и давлении 2000 бар в среде насыщенного при 22°С раствора фторида натрия NaF с использованием в качестве исходных материалов для синтеза хорошо перетертых смесей химических реактивов NaF (или Na2CO3), СаСО3, Nb2O5, Ta2O5, TiO2, ZrO2, V2O5, а также U3O8и UO4⋅2H2O. Однако при этом качественный и количественный состав получаемой с его помощью целевой продукции сильно варьирует в зависимости от числа участвующих в синтезе известного способа исходных компонентов и соотношений между их содержанием, из-за чего непосредственно с помощью известного способа трудно однозначно добиться ожидаемого результата, который был запланирован. Проведенные в известной работе эксперименты, главным образом, подтверждают принципиальную возможность получения соединений со структурой пирохлора. Для успешного практического применения известного способа необходима его самостоятельная доработка.
Наиболее близким к предлагаемому является описанный в работе R. Норре, R. Jesse «Quaternare Fluoridemitzweiwertigem Kupfer: MICuIIMIIIF6 (MI: Cs, Rb, Al, Ga, In, Tl, Sc, Fe, Cr, Ti, V, Mn, Rh) // Z. Anorg. Allg. Chem. 1973. V. 402, N 1, pp.29-38» способ получения фторидных пирохлоров, в том числе фторидных пирохлоров индия MCuInF6 и галлия MCuGaF6 (М=K, Rb, Cs), путем высокотемпературного синтеза и фторирования синтезированного продуктагазообразным фтором, в котором в качестве исходных компонентов используют предварительно синтезированные соединения MCuCl3 (М=K, Rb, Cs), полученные взаимодействием ацетата щелочного метала и CuCl2 в водном растворе, и фториды индия InF3⋅3H2O или галлия GaF3⋅3H2O, полученные многократной обработкой соответствующих оксидов 40% раствором фтористоводородной кислоты с последующим фторированием образцов при температуре 450°С в токе газообразного фтора F2.
Подготовленную реакционную смесь подвергают термообработке в запаянной золотой ампуле при температуре 450-530°С в атмосфере газообразного фтора в течение примерно 50 часов с получением целевого продукта.
Известный способ является энергоемким, продолжительным по времени, требует специального оборудования и особых мер безопасности для защиты персонала и окружающей среды от высокотоксичного и легко распространяющегося газообразного фтора. Кроме того, полученная с его помощью целевая продукция представляет собой соединения, неустойчивые к длительному воздействию влаги, в том числе, атмосферной, и разбавленных кислот.
Задачей изобретения является разработка способа получения фторидных пирохлоров индия и галлия, не требующего чрезвычайно высоких энергетических затрат, непродолжительного по времени, при этом не создающего повышенной опасности для организма человека и окружающей среды, не требующего соблюдения особых мер безопасности и использования специального дорогостоящего оборудования.
Технический результат предлагаемого способа заключается в снижении его энергоемкости за счет проведения синтеза при более низкой температуре и сокращения его продолжительности при одновременном уменьшении опасного воздействия на здоровье человека и окружающую среду, упрощении удешевлении аппаратурного оформления, а также в повышении устойчивости получаемой продукции к воздействию влаги, в том числе, атмосферной, и разбавленных кислот.
Указанный технический результат достигают способом получения фторидных пирохлоров индия CsCuInF6 или галлия CsCuGaF6, путем синтеза исходной реакционной из соединений, содержащих одновалентный катион щелочного металла, двухвалентный катион меди, анионы фтора, а также трехвалентные катионы индия или галлия, с одновременным фторированием, в котором, в отличие от известного, в качестве соединений, содержащих указанные катионы и анионы используют фторид цезия CsF, фторид двухвалентной меди CuF2⋅2H2O, фторид индия InF3⋅3H2O или галлия GaF3⋅3H2O в мольном соотношении 1-2:2:1, при этом для приготовления исходной реакционной смеси к раствору расчетного количества фторида индия или фторида галлия в 40% фтористоводородной кислоте, разбавленной в объемном отношении 1:1, добавляют водный раствор, содержащий фторид цезия и фторид двухвалентной меди, в количествах, обеспечивающих упомянутое мольное соотношение, упаривают полученный смешанный раствор для уменьшения объема на 1/3 и оставляют при комнатной температуре на 1-2 часа до выпадения осадка, который отфильтровывают под вакуумом, промывают охлажденной до 5-10°С дистиллированной водой и высушивают на воздухе, а затем подвергают дегидратации путем нагревания от 20 до 220°С в течение 40-90 мин.
Способ осуществляют следующим образом.
Синтез проводят путем приведения во взаимодействие исходных компонентов: фторида цезия CsF, фторида двухвалентной меди CuF2⋅2H2O и фторида индия InF3⋅3H2O или галлия GaF3⋅3H2O, взятых в количествах, рассчитанных в соответствии с мольными соотношениями, экспериментально установленными между этими компонентами, а именно 1-2:2:1.
Расчетное количество фторида индия InF3⋅3H2O или фторида галлия GaF3⋅3H2O растворяют в емкости из фторопласта-4 в небольшом количестве разбавленной 1:1 40%-фтористоводородной кислоты при нагревании на водяной бане и перемешивании. К полученному прозрачному раствору добавляют водный раствор, содержащий расчетные количества фторида цезия CsF и фторида меди CuF2⋅2H2O при мольном соотношении 1-2:2.
Полученный многокомпонентный раствор упаривают на 1/3 и оставляют на 1-2 часа при комнатной температуре для кристаллизации. Образовавшийся мелкокристаллический осадок отделяют от маточного раствора фильтрованием под вакуумом, промывают небольшим количеством охлажденной до 5-10°С дистиллированной воды и высушивают на воздухе в течение нескольких часов до постоянного веса.
Затем выполняют дегидратацию синтезированных соединений Cs[Cu(H2O)4]InF6 (I) и Cs[Cu(H2O)4]GaF6 (II) путем нагревания одним из известных приемов: на дериватографе в статичной атмосфере воздуха либо в токе осушенного газообразного азота или гелия, либо в сушильном шкафу, осуществляя нагрев от 20 до 220°С в течение 40-90 мин.
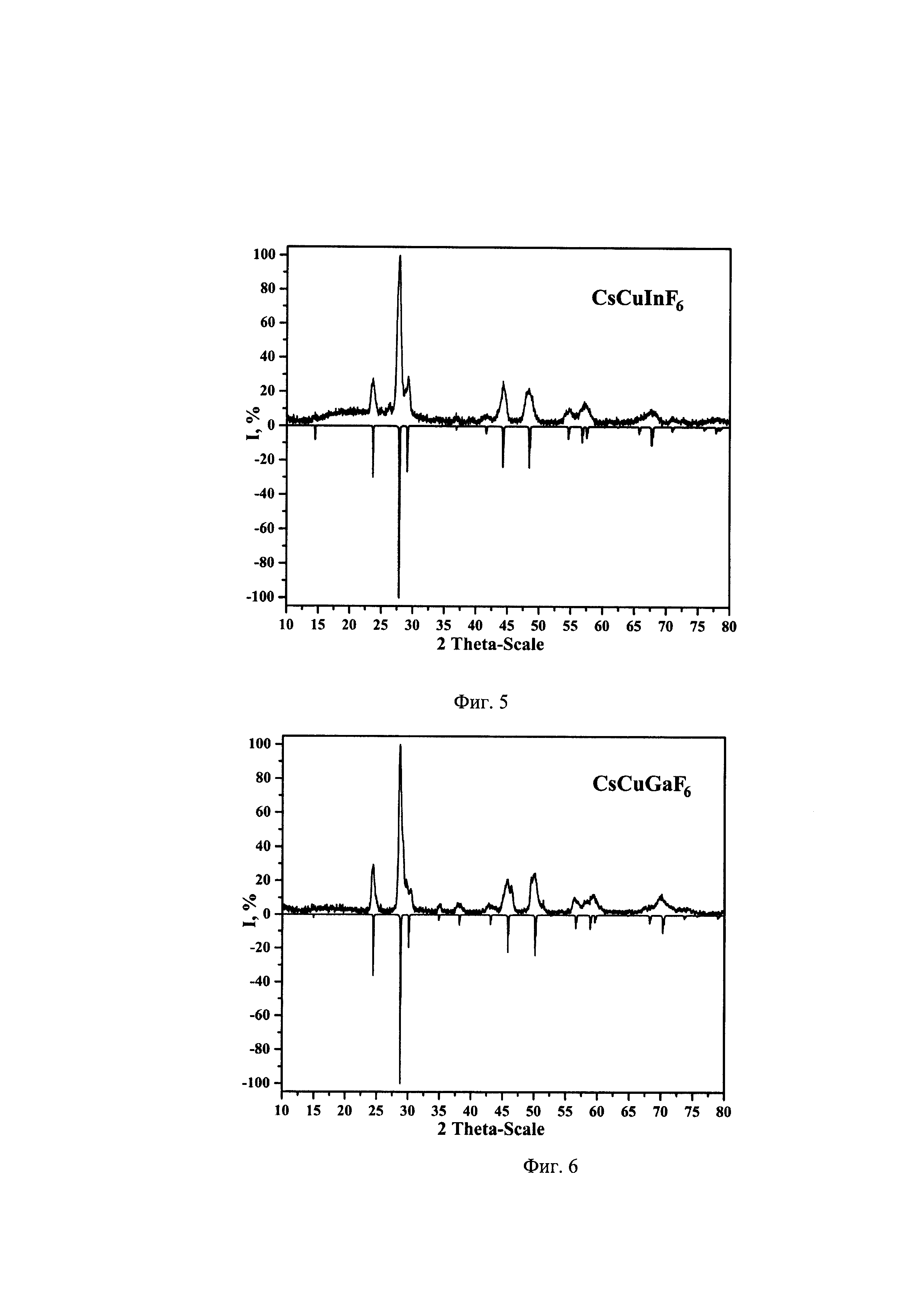
Характер дегидратации соединений Cs[Cu(H2O)4]InF6 (I) и Cs[Cu(H2O)4]GaF6(II) наглядно представлен дериватограммами соединений, синтезированных по примерам 1-4 конкретного осуществления предлагаемого способа (на фиг. 3 представлена дериватограмма соединения (I), на фиг. 4 - дериватограмма соединения (II).
Примеры конкретного осуществления способа.
Синтез фторидных соединений (I) и (II) проводили в вытяжном шкафу. Для их синтеза были использованы стандартные химические реактивы, в частности, фторид цезия CsF, фторид двухвалентной меди CuF2⋅2H2O, 40% фтористоводородная кислота, а также фторид индия InF3⋅3H2O и фторид галлия GaF3⋅3H2O. В случае необходимости фторид индия и фторид галлия получали по известной реакции между соответствующим оксидом In2O3 или Ga2O3 и 40% раствором фтористоводородной кислоты с последующей перекристаллизацией полученного продукта из воды.
Индивидуальность полученных соединений Cs[Cu(H2O)4]InF6 (соединение I) и Cs[Cu(H2O)4]GaF6 (соединение II) устанавливали методами рентгенофазового анализа и ИК спектроскопии.
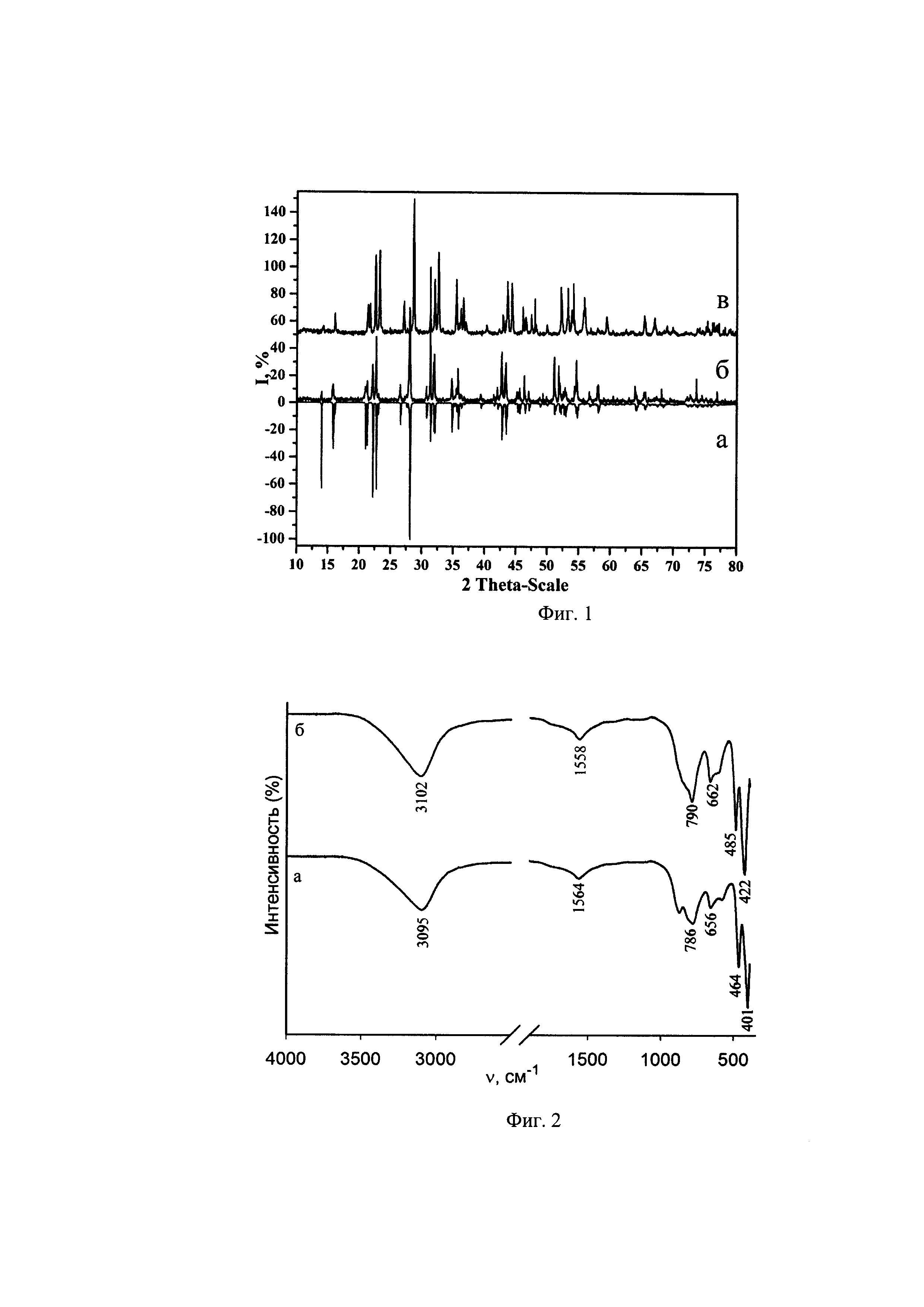
На фиг. 1 приведены: (а) теоретическая рентгенограмма, полученная расчетным путем; и две рентгенограммы синтезированных соединений (I) и (II): (б) Cs[Cu(H2O)4]InF6 (I); (в) Cs[Cu(H2O)4]GaF6 (II).
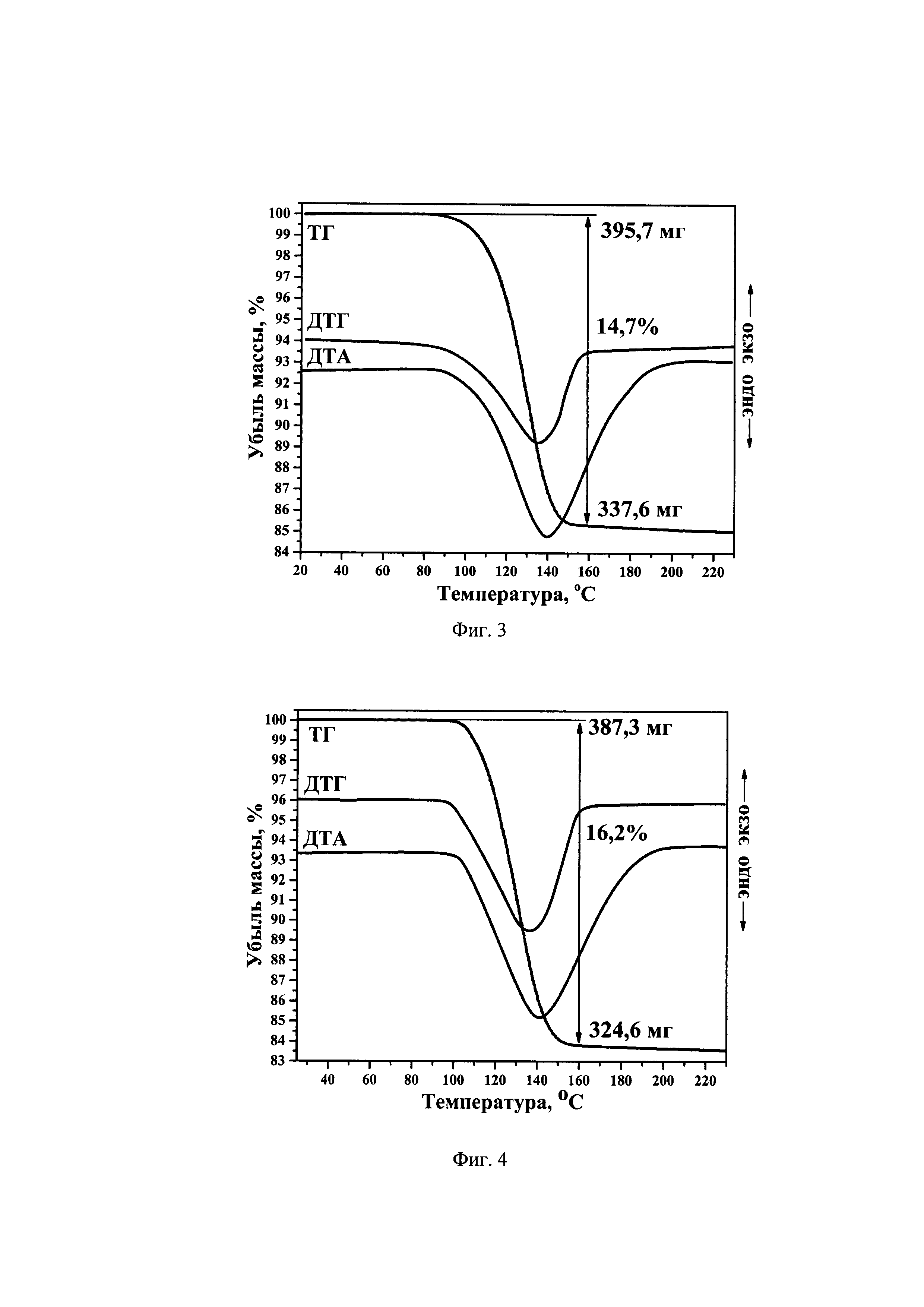
На фиг. 2 представлены ИК спектры: (a) Cs[Cu(H2O)4]InF6; (б) Cs[Cu(H2O)4]GaF6.
Фазовый состав полученных соединений определяли методом рентгенофазового анализа образцов на рентгеновском дифрактометре Bracer D8 ADVANCE в CuKα-излучении; идентификацию полученных рентгенограмм выполняли по программе EVA с банком порошковых данных PDF-2.
ИК спектры снимали с помощью ИК-спектрометра Shimadzu IRTracer-100 с НПВО-приставкой Quest. Разрешение 2 см-1, диапазон 390-4000 см-1.
Характер термического разложения полученных соединений и ход процесса дегидратации изучали по дериватограммам, которые снимали на дериватографе Q1500 в открытом платиновом тигле.
Пример 1
Соединение (I) Cs[Cu(H2O)4]InF6) получали следующим образом.
Пять граммов фторида индия InF3⋅3Н2О растворяли в чашке из фторопласта-4 объемом 100 мл в 30 мл разбавленной 1:1 фтористоводородной кислоты (15 мл Н2О: 15 мл 40% HF) при нагревании на водяной бане и перемешивании. К полученному прозрачному раствору добавляли совместно растворенные в воде фторид цезия CsF (3,63 г) и фторид меди CuF2⋅2H2O (6,09 г), обеспечивая мольное соотношение исходных компонентов CsF:CuF2⋅2H2O:InF3⋅3H2O=1:2:1.
Полученный раствор упаривали до небольшого объема (примерно на 1/3) и оставляли для кристаллизации при комнатной температуре. Образовавшиеся через 1 ч мелкокристаллический осадок отделяли от маточного раствора фильтрованием под вакуумом, промывали небольшим количеством охлажденной до 5°С дистиллированной воды и сушили на воздухе часов до постоянной массы в течение 6 часов до постоянной массы.
Пример 2
Пять граммов фторида индия InF3⋅3H2O растворяли в чашке из фторопласта-4 в условиях примера 1. К полученному прозрачному раствору добавляли совместно растворенные в воде фторид цезия CsF (7,26 г) и фторид меди CuF2⋅2H2O (6,09 г), обеспечивая мольное соотношение исходных компонентов CsF:CuF2⋅2H2O:InF3⋅3H2O=2:2:1.
Получено соединение (I) Cs[Cu(H2O)4]InF6), дальнейшую обработку которого проводили аналогично примеру 1. Осадок отстаивали 2 часа, дистиллят охлаждали до 10°С.
Пример 3
Соединение (И) Cs[Cu(H2O)4]GaF6 получали аналогичным образом. Пять граммов фторида галлия GaF3⋅3H2O растворяли в чашке из фторопласта-4 в 30 мл разбавленной 1:1 фтористоводородной кислоты (15 мл Н2О : 15 мл 40% HF) при нагревании на водяной бане и перемешивании. К полученному прозрачному раствору добавляли совместно растворенные в воде фторид цезия F (4,20 г) и фторид меди CuF2⋅2H2O (7,61 г), обеспечивая мольное соотношение исходных компонентов CsF:CuF2⋅2H2O:GaF3⋅3H2O=1:2:1.
Полученный раствор упарили примерно на 1/3 и оставили для кристаллизации при комнатной температуре. Образовавшиеся через неполные 2 часа кристаллические осадки отделяли фильтрованием под вакуумом, промывали небольшим количеством охлажденной до 5°С дистиллированной воды и сушили на воздухе часов до постоянной массы в течение 6 часов до постоянной массы.
Пример 4
Пять граммов фторида индия GaF3⋅3H2O растворяли в чашке из фторопласта-4 в 30 мл разбавленной 1:1 фтористоводородной кислоты (15 мл Н2О: 15 мл 40% HF) при нагревании на водяной бане и перемешивании. К полученному прозрачному раствору добавляли совместно растворенные в воде фторид цезия CsF (8,40 г) и фторид меди CuF2⋅2H2O (7,61 г), обеспечивая мольное соотношение исходных компонентов CsF:CuF2⋅2H2O:GaF3⋅3H2O=2:2:1.
Дальнейшую обработку полученного материала проводили как в предшествующих примерах.
Характер термического разложения синтезированных соединений (I) и (II) представлен на дериватограммах (фиг.3 и 4). Дегидратация соединений происходит в одну стадию, начало разложения Cs[Cu(H2O)4]InF6 - 80°С, Cs[Cu(H2O)4]GaF6 - 100°С. Эндотермические эффекты, наблюдаемые На дериватограммах соединений (I) и (II), связаны с удалением четырех молекул воды, из координационной сферы атома углерода С, как представлено уравнениями (1) и (2):
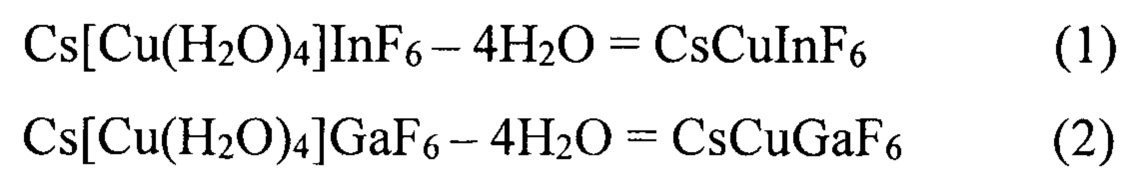
Термогравиметрическое исследование соединений Cs[Cu(H2O)4]InF6 и Cs[Cu(H2O)4]GaF6 проводили на дериватографе Q1500 в открытом платиновом тигле. Масса навески соединений составляла 300-400 мг, скорость нагревания 5°С/мин.
Пример 5
Навеску Cs[Cu(H2O)4]InF6, полученного по примерам 1-2 в количестве 395,7 мг, нагревали на дериватографе со скоростью 5°С/мин. Экспериментально найденная в температурном интервале 80-220°С убыль массы составила 14,7%, что согласуется с расчетными значениями убыли массы для четырех молекул Н2О из соединения (I) - 14,48% (фиг. 3 Дериватограмма Cs[Cu(H2O)4]InF6).
Пример 6
Навеску Cs[Cu(H2O)4]GaF6, полученного по примерам 3-4 в количестве 387,3 мг нагревали на дериватографе со скоростью 5°С/мин. Экспериментально найденная в температурном интервале 100-220°С убыль массы составила 16,2%, что согласуется с расчетными значениями убыли массы для четырех молекул Н2О из соединения (II) - 15,92% (фиг. 4 Дериватограмма Cs[Cu(H2O)4]GaF6).
На кривых убыли массы дериватограмм исследованных соединений (I) и (II) (фиг. 3, 4) в области 160-230°С (после удаления молекул воды) присутствуют площадки, отвечающие образованию соединений CsCuInF6 и CsCuGaF6.
Для рентгенофазового анализа и ИК- спектроскопии соединения состава CsCuInF6 и CsCuGaF6 получали путем дегидратации исходных Cs[Cu(H2O)4]InF6и Cs[Cu(H2O)4]GaF6 при 230°С до прекращения изменения массы (40 мин). Убыль массы совпадает с данными термогравиметрического исследования.
Пример 7
Навеску 250 мг Cs[Cu(H2O)4]InF6, полученного по примерам 1-2, нагревали в сушильном шкафу при 110°С 90 мин, вес остатка составил 214,25 мг. Убыль массы - 35,75 мг, что составляет 14,3%.
Пример 8
Навеску 280 мг Cs[Cu(H2O)4]GaF6, полученного по примерам 3-4, нагревали в сушильном шкафу при 110°С 90 мин, вес остатка составил 234,92 мг. Убыль массы - 45,08 мг, что составляет 16,1%.
Полученные соединения CsCuInF6 и CsCuGaF6 идентифицировали методами рентгенофазового анализа и ИК - спектроскопии.
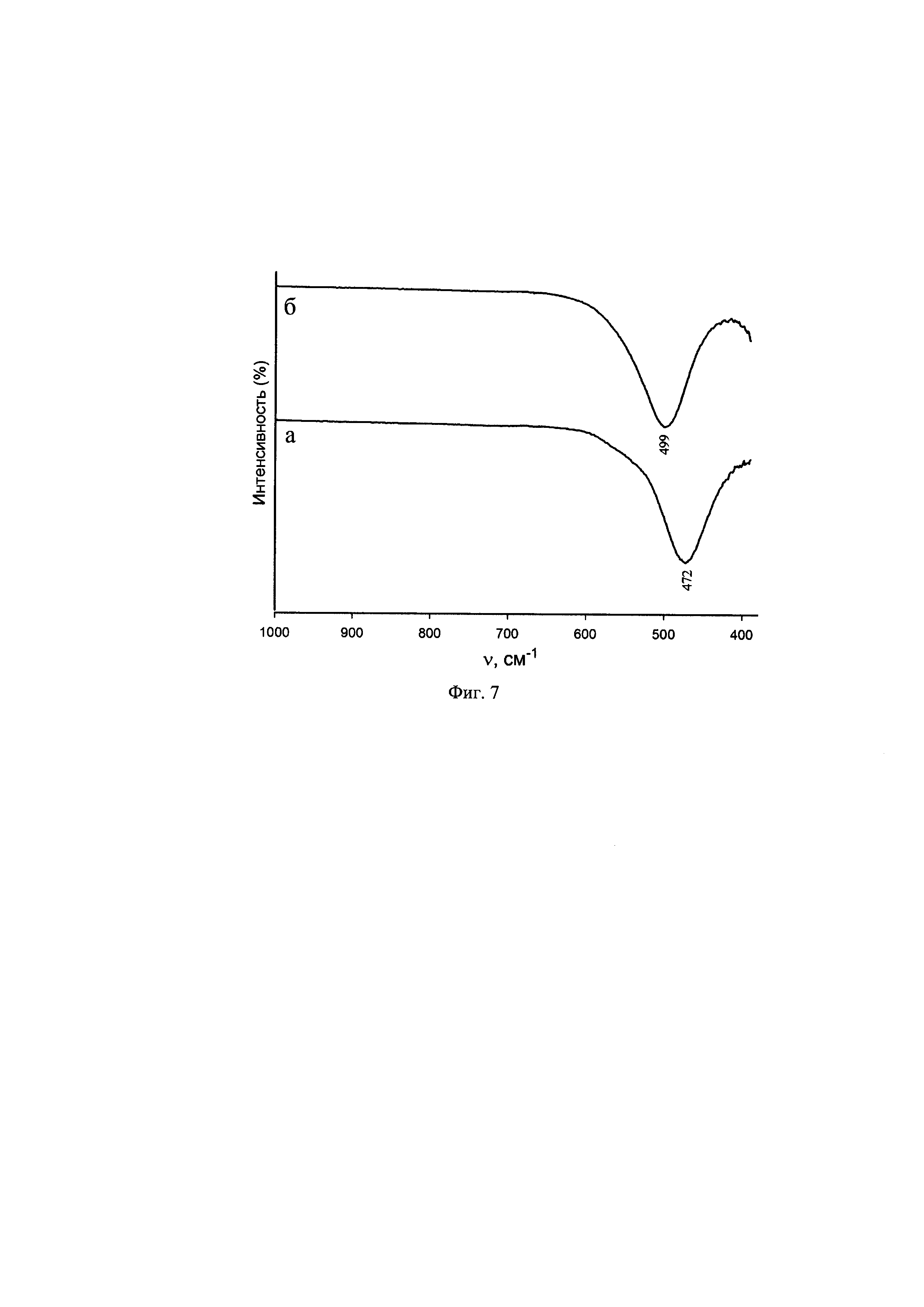
На фиг. 5 и фиг. 6 приведены экспериментальные и теоретические рентгенограммы продуктов дегидратации исходных соединений Cs[Cu(H2O)4]InF6 и Cs[Cu(H2O)4]GaF6 соответственно.
На основании результатов сравнения экспериментальных дифрактограмм образцов, полученных дегидратацией соединений Cs[Cu(H2O)4]InF6 и Cs[Cu(H2O)4]GaF6 (I и II) в интервале температур 20-220°С в течение 40-90 мин (фиг. 5, 6) с теоретическими рентгенограммам и кубических пирохлоров CsCuInF6(CIF файл CCDC1526354, а=10,62 Å) и CsCuGaF6 (CIF файл CCDC 7221421 а=10,28 Å), исходя из их сходства и полученных данных дегидратации соединений (I) и (II) (убыль массы см. выше) сделано заключение, что образующиеся в результате дегидратации соединений (I) и (II) образцы являются кубическими фторидными пирохлорами CsCuInF6 и CsCuGaF6.
Образование безводных соединений CsCuInF6 и CsCuGaF6 согласно приведенным выше уравнениям 1 и 2 подтверждено также и ИК спектрами продуктов дегидратации соединений (I) и (II) (фиг. 7 ИК спектры соединений: CsCuInF6 (а) и CsCuGaF6 (б)), на которых отсутствуют характеристические полосы поглощения молекул Н2О (сравнение с фиг. 2).
Реферат
Изобретение относится к получению неорганических соединений фторидных пирохлоров индия(III) CsCuInF6 или галлия(III) CsCuGaF6 и может найти применение при изготовлении материалов для электронной промышленности, в качестве матриц для фиксации радионуклидов, индикаторов геохимических процессов. Фторидные пирохлоры CsCuInF6 или CsCuGaF6 получают путем синтеза исходной реакционной смеси из соединений, содержащих фторид цезия CsF, фторид двухвалентной меди CuF2⋅2H2O, фторид индия InF3⋅3Н2О или галлия GaF3⋅3H2O в мольном соотношении 1-2:2:1 с одновременным фторированием, при этом для приготовления исходной реакционной смеси к раствору расчетного количества фторида индия или фторида галлия в 40% фтористоводородной кислоте, разбавленной в объемном отношении 1:1, добавляют водный раствор, содержащий фторид цезия и фторид двухвалентной меди, в количествах, обеспечивающих упомянутое мольное соотношение, упаривают полученный смешанный раствор для уменьшения объема на 1/3 и оставляют при комнатной температуре на 1-2 ч до выпадения осадка, который отфильтровывают под вакуумом, промывают охлажденной до 5-10°С дистиллированной водой и высушивают на воздухе, а затем подвергают дегидратации путем нагревания от 20 до 220°С в течение 40-90 мин. Технический результат - снижение энергоемкости способа за счет проведения синтеза при более низкой температуре и сокращения его продолжительности при одновременном уменьшении опасного воздействия на организм человека и окружающую среду, упрощении и удешевлении аппаратурного оформления способа, а также повышение устойчивости получаемой продукции к воздействию влаги и разбавленных кислот. 8 пр., 7 ил.
Комментарии