Способы получения и очистки фторгидроолефинов - RU2446140C2
Код документа: RU2446140C2
Чертежи
Описание
Уровень техники
1. Область техники, к которой относится изобретение
Настоящее изобретение относится к области получения и очистки фторгидроолефиновых соединений. Настоящее изобретение относится также к способам использования азеотропных композиций для отделения фторгидроолефинов от фторсодержащих углеводородов и фторида водорода.
2. Описание предшествующего уровня техники
Хлорсодержащие соединения, такие как хлорфторуглеводороды (CFC), считаются вредными для озонового слоя Земли. Было установлено, что многие из фторсодержащих углеводородов, которые используют для замены CFC, вносят вклад в глобальное потепление. Поэтому необходимы новые соединения, которые не наносят вред окружающей среде, но обладают свойствами, необходимыми для того, чтобы их можно было использовать в качестве хладагентов, растворителей, очищающих средств, вспенивающих средств, газов-вытеснителей для аэрозолей, теплоносителей, диэлектриков, средств тушения огня, стерилизаторов и рабочих сред энергетического цикла. Фторсодержащие олефины, в частности фторсодержащие олефины, которые содержат в молекуле один или несколько атомов водорода (в настоящем описании их называют фторгидроолефинами), рассматривают на предмет их использования в указанных применениях, таких как охлаждение, а также в способах получения фторсодержащих полимеров.
1,1,3,3,3-Пентафторпропен представляет собой мономер, включающий способный к термоотверждению участок, который используют для полимеризации при получении фторсодержащих эластомеров. В патентах США № 6703533, 6548720, 6476281, 6369284, 6093859 и 6031141, а также в опубликованных патентных заявках Японии JP 09095459 и JP 09067281 и WIPO публикации WO 2004/018093 раскрываются способы, в которых 1,1,1,3,3,3-гексафторпропан нагревают до температур ниже 500°С в присутствии катализатора с образованием 1,1,3,3,3-пентафторпропена. Указанные низкотемпературные каталитические способы выбраны постольку, поскольку хорошо известно, что фторсодержащие углеводороды проявляют тенденцию к фрагментации при более высоких температурах, в частности, при температурах выше 500°С. Сказанное поясняется в монографии Chemistry of Organic Fluorine Compounds, Milos Hudlicky, 2nd Revised Edition, Ellis Horwood PTR Prentice Hall [1992] p. 515: “Полифторсодержащие парафины и, в частности, фторзамещенные углеводороды и другие перфторзамещенные производные, обладают значительной термостабильностью. Они обычно не разлагаются при температурах ниже 300°С. Однако их преднамеренное разложение, осуществляемое при температурах 500-800°С, вызывает разнообразные расщепления молекул фторзамещенных углеводородов и приводит к образованию комплексных смесей, которые трудно разделить”.
В патентной заявке США 2002/0032356 раскрывается способ получения перфторзамещенных мономеров тетрафторэтилена и гексафторпропилена в реакторе пиролиза, покрытом золотом.
Каталитический способ имеет ряд недостатков, включая получение катализатора, ввод в действие свежего катализатора, дезактивацию катализатора, потенциальное закупоривание заполненных катализатором реакторов побочными полимерными продуктами, утилизацию или реактивацию катализатора и длительные времена проведения реакции, которые приводят к снижению выхода на единицу объема катализатора в реакторе. Необходим высокопроизводительный некаталитический способ получения 1,1,3,3,3-пентафторпропена из 1,1,1,3,3,3-гексафторпропана.
Сущность изобретения
Один аспект настоящего изобретения касается способа получения фторгидроолефинов, включающего дегидрофторирование фторсодержащего углеводорода, который имеет у соседних атомов углерода, по крайней мере, один атом водорода и, по крайней мере, один атом фтора, с образованием смеси продуктов, содержащей указанный фторгидроолефин, не прореагировавший фторсодержащий углеводород и фторид водорода, где, по крайней мере, один из указанного фторгидроолефина и указанного фторсодержащего углеводорода присутствует в указанной смеси продуктов в виде азеотропной композиции с фторидом водорода.
Другой аспект настоящего изобретения касается способа отделения фторгидроолефина от фторсодержащего углеводорода, где указанный фторгидроолефин содержит на один атом водорода меньше и на один атом фтора меньше, чем указанный фторсодержащий углеводород, при этом указанный способ включает: а) образование смеси, включающей фторгидроолефин, фторсодержащий углеводород и фторид водорода; и b) стадию дистилляции, которой подвергают указанную смесь, с образованием в колонне композиции дистиллята, представляющей собой азеотропную или близкую к азеотропной композицию фторида водорода и фторгидроолефина, в которой практически отсутствует указанный фторсодержащий углеводород.
Еще один аспект настоящего изобретения касается способа выделения фторгидроолефина из смеси, которая представляет собой азеотропную или близкую к азеотропной композицию указанного фторгидроолефина и фторида водорода, при этом указанный способ включает: а) стадию первой дистилляции, которой подвергают указанную смесь, где композиция, обогащенная либо (i) фторидом водорода, либо (ii) фторгидроолефином, выделяется в виде композиции первого дистиллята, а композиция первых кубовых остатков обогащается другим из указанных компонентов (i) или (ii); и b) вторую стадию дистилляции, которой подвергают указанную композицию первого дистиллята, осуществляемую при другом давлении, во время которой компонент, обогащенный в виде композиции первых кубовых остатков на стадии (а), выделяется в виде композиции второго дистиллята, а композиция вторых кубовых остатков на второй стадии дистилляции обогащается тем же самым компонентом, которым была обогащена указанная композиция первого дистиллята.
Еще один аспект настоящего изобретения касается способа очистки фторгидроолефина из смеси фторгидроолефина, фторсодержащего углеводорода и фторида водорода, при этом указанный способ включает: а) стадию первой дистилляции, которой подвергают указанную смесь, с образованием композиции первого дистиллята, представляющей собой азеотропную или близкую к азеотропной композицию, содержащую фторгидроолефин и фторид водорода, и композиции первых кубовых остатков, представляющей собой фторсодержащий углеводород; b) вторую стадию дистилляции, которой подвергают указанный первый дистиллят, после которой композиция, обогащенная либо (i) фторидом водорода, либо (ii) фторгидроолефином, выделяется в виде композиции второго дистиллята, а композиция вторых кубовых остатков обогащается другим из указанных компонентов (i) или (ii); и с) третью стадию дистилляции, которой подвергают указанную композицию второго дистиллята, осуществляемую под давлением, отличным от давления на второй стадии дистилляции, где компонент, которым обогащена композиция вторых кубовых остатков на стадии (b), выделяется в виде композиции третьего дистиллята, а композиция третьих кубовых остатков на третьей стадии дистилляции обогащается тем же самым компонентом, которым была обогащена композиция второго дистиллята.
Еще один аспект настоящего изобретения касается способа получения фторгидроолефина, который включает: а) подачу фторсодержащего углеводорода, который содержит у соседних атомов углеводорода, по крайней мере, один атом водорода и, по крайней мере, один атом фтора, в зону реакции дегидрофторирования с образованием композиции продуктов реакции, содержащей фторгидроолефин, не прореагировавший фторсодержащий углеводород и фторид водорода; b) стадию первой дистилляции, которой подвергают указанную композицию продуктов реакции, с образованием композиции первого дистиллята, представляющей собой азеотропную или близкую к азеотропной композицию, содержащую фторгидроолефин и фторид водорода, и композиции первых кубовых остатков, которая представляет собой фторсодержащий углеводород; с) вторую стадию дистилляции, которой подвергают указанную композицию первого дистиллята, после которой композиция, обогащенная либо (i) фторидом водорода, либо (ii) фторгидроолефином, удаляется в виде композиции второго дистиллята, а композиция вторых кубовых остатков обогащается другим из указанных компонентов (i) или (ii); и d) третью стадию дистилляции, которой подвергают указанную композицию второго дистиллята, осуществляемую под давлением, отличным от давления на второй стадии дистилляции, где компонент, которым обогащена композиция вторых кубовых остатков на стадии (с), удаляется в виде композиции третьего дистиллята, а композиция третьих кубовых остатков на третьей стадии дистилляции обогащается тем же самым компонентом, которым была обогащена композиция второго дистиллята.
Еще один аспект настоящего изобретения касается фторгидроолефина, выбранного из группы, которая включает CF2=C(CHF2)2, CHF=C(CHF2)2, CH2=C(CH2F)CF3, CH2=CFCF2CF2CF3, CHF2CF2CF=CFCH3, CF3CF2CF=CHCH3, (CF3)2C=CFCHF2, (CF3)2CFCF=CHCH3, (CF3)2C=C(CH3)2, (CН3)2C=CFCF2CF3, C2F5CH=CHCF2C2F5, CF3CH=CHCF2CF2C2F5, CF3CF2CF2CF2CF=CHCH3, CF3CF2CF2CF=CHCH2CH3, (CF3)2C=CFCF2CF2CF3 и CF3CH=CFCH2CF3.
Еще один аспект настоящего изобретения касается способа получения CF3CH=CF2 в отсутствие катализатора дегидрофторирования. В частности, данный аспект включает пиролиз CF3CH2CF3 с образованием CF3CH=CF2. Пиролиз сопровождается термическим разложением CF3CH2CF3 при температуре больше чем 700°С.
Указанное селективное получение CF3CH=CF2 включает несколько неожиданных (неочевидных) результатов. Во-первых, неочевидно, что подводимое тепло процесса пиролиза не вызывает фрагментацию реагента CF3CH2CF3 с образованием соединения С-1, т.е. метана и соединений С-2, т.е. этана и этилена. Во-вторых, неочевидно, что продукт CF3CH=CF2 устойчив в условиях пиролиза и не претерпевает дальнейшее превращение в продукты перегруппировки или продукты, содержащие меньше атомов водорода и/или меньше атомов фтора. В-третьих, неочевидно, что пиролиз с образованием CF3CH=CF2 протекает с высокой селективностью.
Краткое описание чертежей
На фиг.1 приведена блок-схема, поясняющая один из способов осуществления процесса дистилляции азеотропной смеси в двух колоннах.
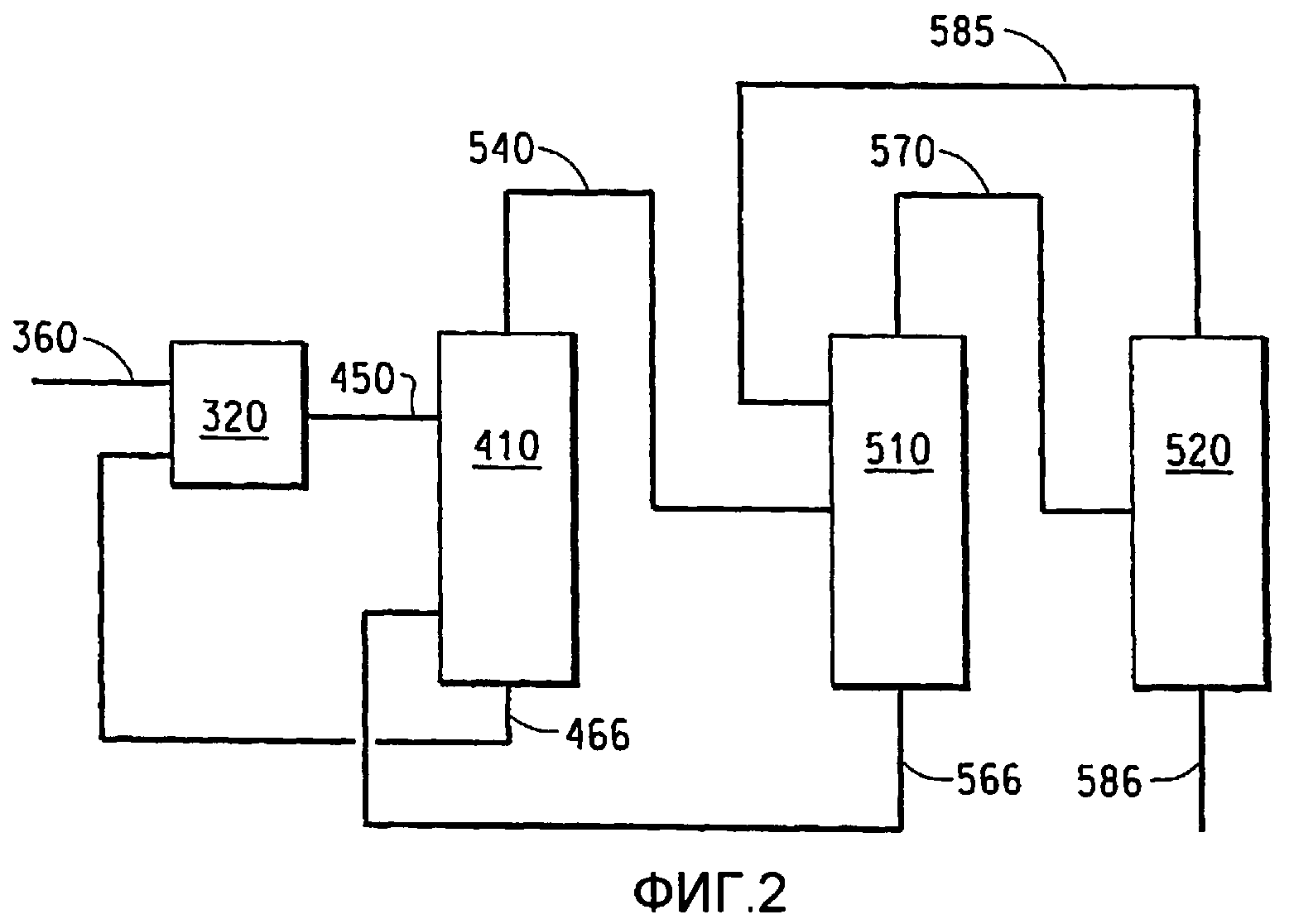
На фиг.2 приведена блок-схема, поясняющая один из способов осуществления способа получения фторгидроолефина.
Подробное описание изобретения
Один аспект настоящего изобретения касается способов получения и способов очистки фторгидроолефинов. Способы очистки включают способы отделения фторгидроолефинов от фторсодержащих углеводородов и фторида водорода, при этом в указанных способах применяют азеотропные или близкие к азеотропным композиции.
Другой аспект настоящего изобретения касается способа получения фторгидроолефина, включающего дегидрофторирование фторсодержащего углеводорода, который имеет у соседних атомов углерода, по крайней мере, один атом водорода и, по крайней мере, один атом фтора, при этом образуется смесь продуктов, содержащая указанный фторгидроолефин, не прореагировавший фторсодержащий углеводород и фторид водорода, где, по крайней мере, один из указанного фторгидроолефина и указанного фторсодержащего углеводорода присутствует в указанной смеси продуктов в виде азеотропной композиции с фторидом водорода.
Фторгидроолефины представляют собой ациклические соединения, содержащие от 3 до 8 атомов углерода, атомы фтора и водорода. Фторгидроолефины должны также содержать, по крайней мере, одну двойную связь, и они могут быть линейными или разветвленными. Ациклические фторгидроолефины могут быть представлены формулой, CxHa-1Fb-1, где х обозначает целое число от 3 до 8, а обозначает целое число от 2 до (х+1), b обозначает целое число от (х+1) до (2х), и где a+b=2x+2. Отдельные фторгидроолефины включают, однако этим не ограничиваются, трифторпропены, тетрафторпропены, пентафторпропены, тетрафторбутены, пентафторбутены, гексафторбутены, гептафторбутены, пентафторпентены, гексафторпентены, гептафторпентены, октафторпентены, нонафторпентены, гексафторгексены, гептафторгексены, октафторгексены, нонафторгексены, декафторгексены, ундекафторгексены, гептафторгептены, октафторгептены, нонафторгептены, декафторгептены, ундекафторгептены, додекафторгептены, тридекафторгептены, октафтороктены, нонафтороктены, декафтороктены, ундекафтороктены, додекафтороктены, тридекафтороктены, тетрадекафтороктены и пентадекафтороктены.
Включены также циклические фторгидроолефины, содержащие в общей сложности от 4 до 8 атомов углерода, в том числе от 4 до 6 атомов углерода в кольце. Циклические фторгидроолефины должны также содержать, по крайней мере, одну двойную связь и могут иметь разветвления в цикле. Циклические олефины могут быть представлены формулой CyHc-1Fd-1, где y обозначает целое число от 4 до 8, c обозначает целое число от 2 до y, d обозначает целое число от y до (2y-2), и где c+d=2y. Отдельные циклические фторгидроолефины включают, однако этим не ограничиваются, дифторциклопропены, трифторциклопропены, трифторциклобутены, тетрафторциклобутены, пентафторциклобутены, трифторметилциклопропены, тетрафторметилциклопропены, пентафторметилциклопропены, тетрафторциклопентены, пентафторциклопентены, гексафторциклопентены, гептафторциклопентены, тетрафтордиметилциклопропены, пентафтордиметилциклопропены, гексафтордиметилциклопропены, гептафтордиметилциклопропены, тетрафторэтилциклопропены, пентафторэтилциклопропены, гексафторэтилциклопропены, гептафторэтилциклопропены, тетрафторметилциклобутены, пентафторметилциклобутены, гексафторметилциклобутены, гептафторметилциклобутены, пентафторциклогексены, гексафторциклогексены, гептафторциклогексены, октафторциклогексены, нонафторциклогексены, пентафторметилциклопентены, гексафторметилциклопентены, гептафторметилциклопентены, октафторметилциклопентены, нонафторметилциклопентены, пентафтордиметилциклобутены, гексафтордиметилциклобутены, гептафтордиметилциклобутены, октафтордиметилциклобутены, нонафтордиметилциклобутены, пентафторэтилциклобутены, гексафторэтилциклобутены, гептафторэтилциклобутены, октафторэтилциклобутены, нонафторэтилциклобутены, пентафтортриметилциклопропены, гексафтортриметилциклопропены, гептафтортриметилциклопропены, октафтортриметилциклопропены и нонафтортриметилциклопропены.
Фторгидроолефины могут существовать в виде различных конфигурационных изомеров или стереоизомеров. Включены все индивидуальные изомеры, индивидуальные стереоизомеры или любые их комбинации. Например, HFC-1234ze (CF3CH=CHF) обозначает Е-изомер, Z-изомер или любую комбинацию или смесь обоих изомеров, взятых в любом соотношении. Другим примером является HFC-1336mzz (CF3CH=CHF3), которые обозначает Е-изомер, Z-изомер или любую комбинацию или смесь обоих изомеров, взятых в любом соотношении.
Фторсодержащие углеводороды являются ациклическими, линейными или разветвленными соединениями, содержащими от 3 до 8 атомов углерода, атомы водорода и фтора. По крайней мере, один атом водорода и, по крайней мере, один атом фтора должны располагаться у соседних атомов углерода. Фторсодержащие углеводороды могут быть представлены формулой CxHaFb, где х обозначает целое число от 3 до 8, а обозначает целое число от 2 до (х+1), b обозначает целое число от (х+1) до (2х), и где a+b=2x+2. Фторсодержащие углеводороды являются соединениями-предшественниками фторгидроолефинов. Фторсодержащие углеводороды могут подвергаться дегидрофторированию в паровой фазе с образованием фторгидроолефинов по настоящему изобретению вместе с фторидом водорода в качестве побочного продукта.
Включены также циклические фторсодержащие углеводороды, содержащие в общей сложности от 4 до 8 атомов углерода, при этом в цикле содержится от 4 до 6 атомов углерода. По крайней мере, один атом водорода и, по крайней мере, один атом фтора должны располагаться у соседних атомов углерода. Циклические фторсодержащие углеводороды могут быть представлены формулой CyHc1Fd1, где y обозначает целое число от 4 до 8, c обозначает целое число от 2 до y, d обозначает целое число от y до (2y-2) и где c+d=2y. Циклические фторсодержащие углеводороды являются соединениями-предшественниками циклических фторгидроолефинов. Циклические фторсодержащие углеводороды могут подвергаться дегидрофторированию в паровой фазе с образованием циклических фторгидроолефинов по настоящему изобретению вместе с фторидом водорода в качестве побочного продукта.
В таблице 1 в виде перечня приведены отдельные фторгидроолефины и соответствующие фторсодержащие углеводороды, которые являются их предшественниками. В таблице 2 в виде перечня приведены отдельные циклические фторгидроолефины и соответствующие циклические фторсодержащие углеводороды, которые являются их предшественниками. В зависимости от количества и расположения атомов водорода и фтора в соответствующем фторсодержащем углеводороде-предшественнике и при условии, что, по крайней мере, один атом водорода и, по крайней мере, один атом фтора должны находиться у соседних атомов углерода, может быть получено более одного фторгидроолефина. В таблице 1 и 2 указан один отдельный фторгидроолефин, получаемый из фторсодержащего углеводорода-предшественника, а в некоторых случаях - несколько фторгидроолефинов, получаемых из фторсодержащего углеводорода-предшественника.
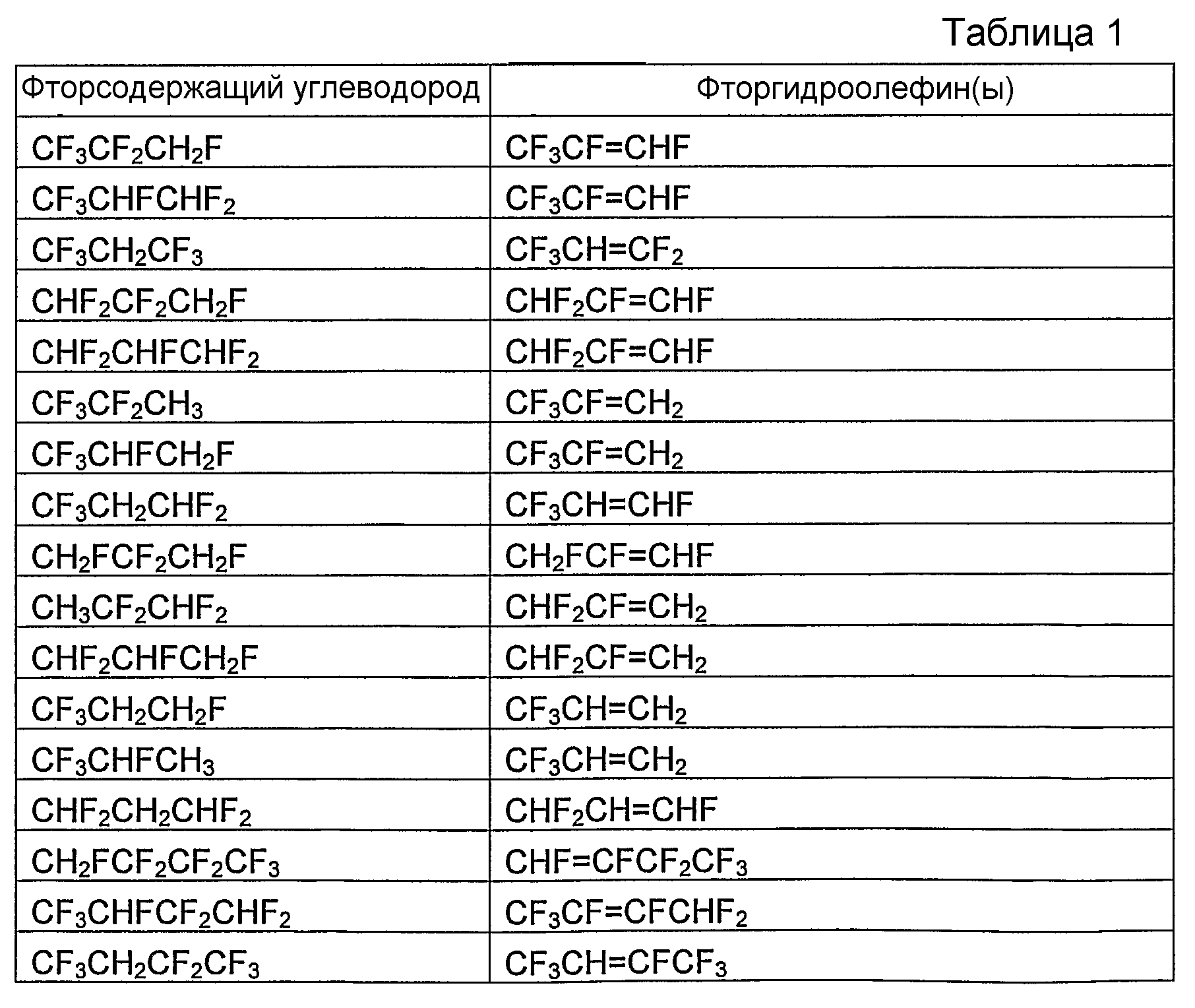
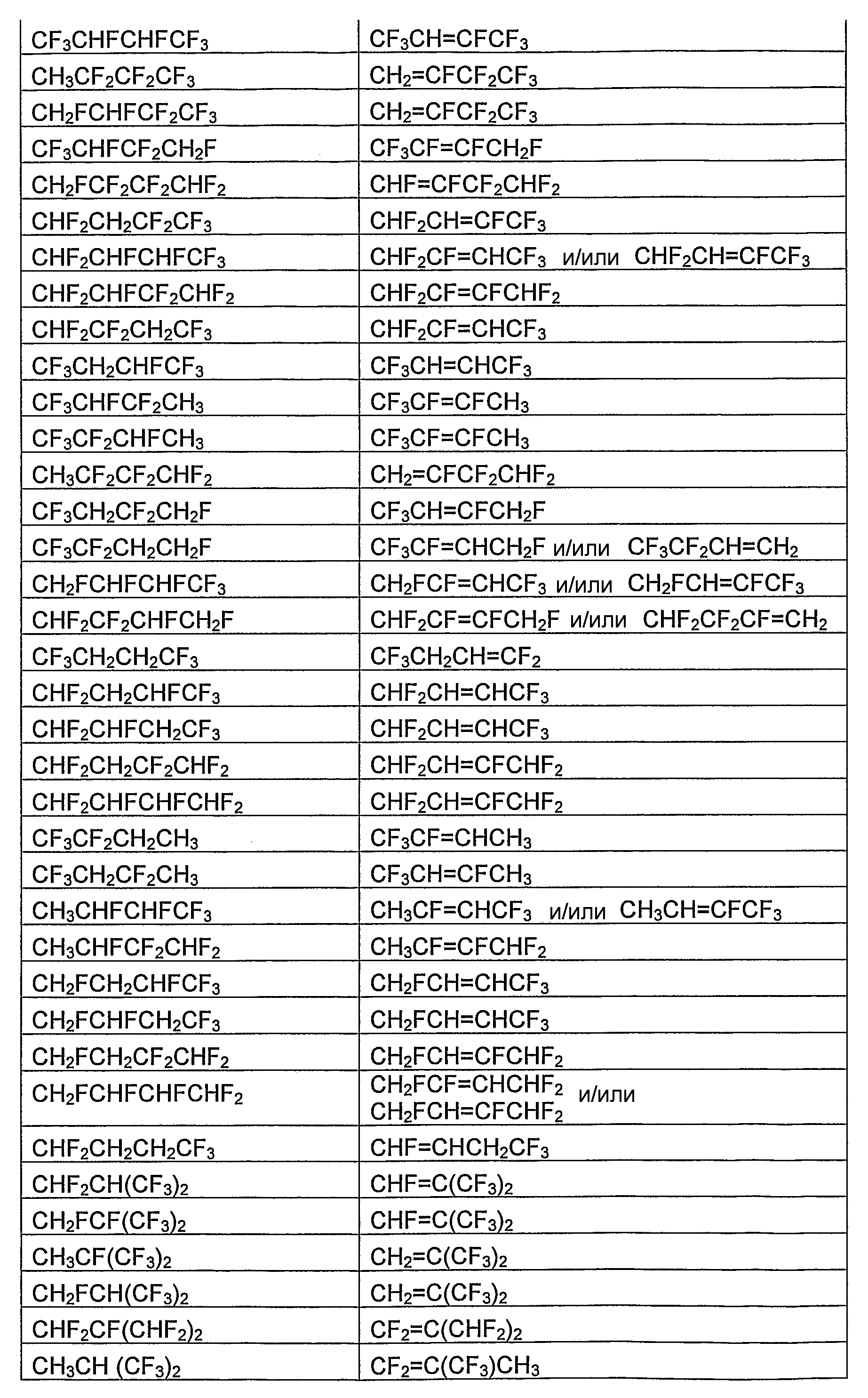

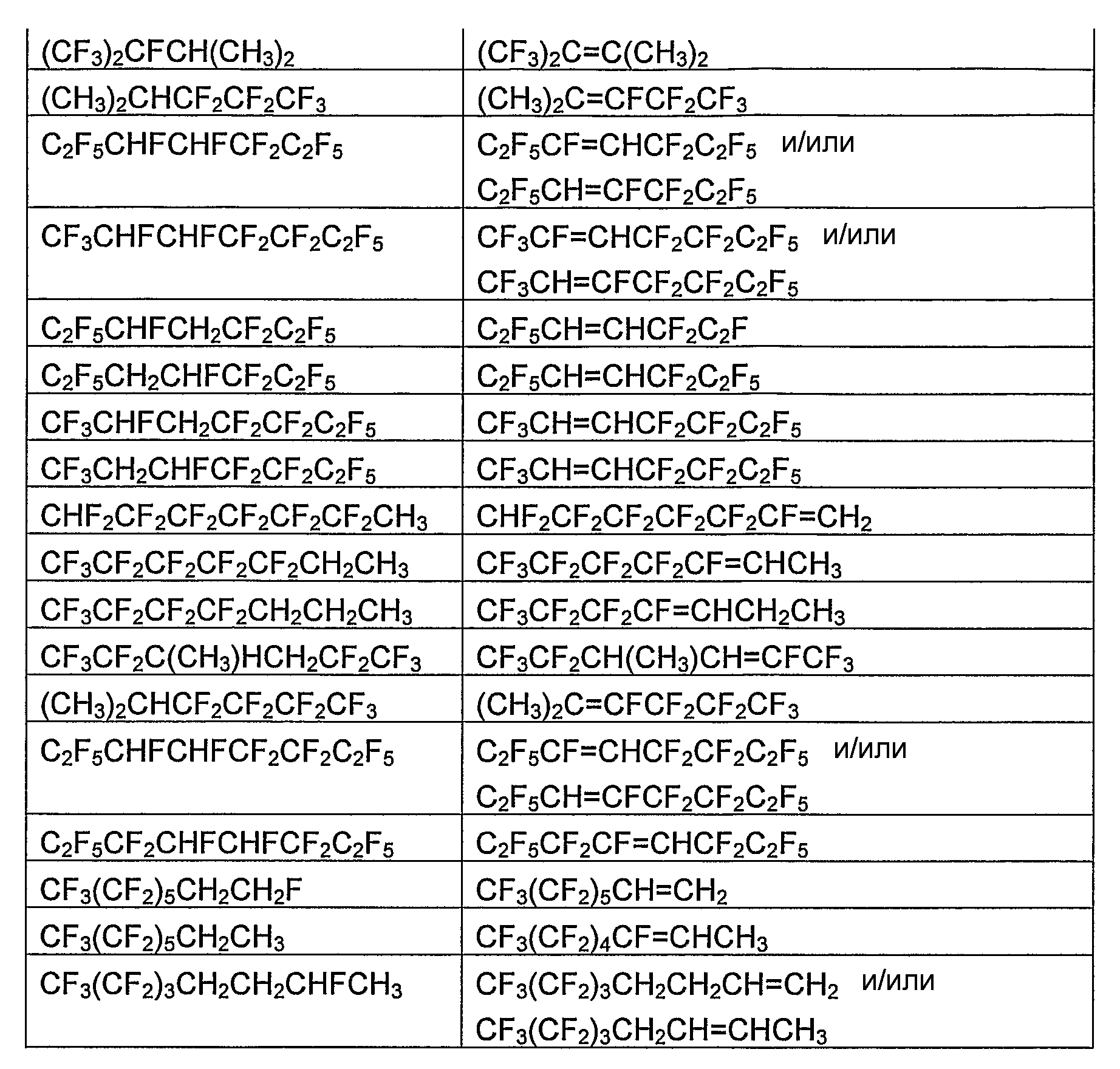
Фторсодержащие углеводороды коммерчески доступны, могут быть получены известными из области техники способами или могут быть получены, как указано в данном описании.
1,1,1,2,3,3-Гексафтор-2-(трифторметил)пентан ((CF3)2CFCF2CH2CH3) может быть получен взаимодействием 1,1,1,2,3,3-гексафтор-2-(трифторметил)-3-йодпропана с этиленом с образованием 1,1,1,2,3,3-гексафтор-2-(трифторметил)-5-йодпентана и последующим восстановлением цинком в таких кислотах, как HCl или уксусная кислота.
1,1,1,2-Тетрафтор-2-(трифторметил)пентан ((CF3)2CFCH2CH2CH3) может быть получен взаимодействием метил перфторизобутирата с этилмагнийбромидом с последующим гидролизом и образованием 1,1,1,2-тетрафтор-2-(трифторметил)-3-пентанола. Указанный пентанол превращают в 4,5,5,5-тетрафтор-4-(трифторметил)-2-пентен дегидратацией с помощью пентоксида фосфора, и двойную связь насыщают гидрированием с использованием палладия на угле в качестве катализатора.
1,1,1,2,2,3,3,4,4,5,5-Ундекафторгептан (CF3CF2CF2CF2CF2CH2CH3) может быть получен взаимодействием 1,1,1,2,2,3,3,4,4,5,5-ундекафтор-5-йодпентана с этиленом с образованием 1,1,1,2,2,3,3,4,4,5,5-ундекафтор-7-йодгептана и последующим восстановлением цинком в кислоте, такой как HCl или уксусная кислота.
Как указано ранее, некоторые фторсодержащие углеводороды-предшественники, которые приведены в таблице 1, а также в таблице 2, в результате реакции дегидрофторирования могут образовать единственный фторгидроолефин или же смесь двух или большего количества фторгидроолефинов. Приведены пары, составленные фторсодержащим углеводородом и каждым индивидуальным фторгидроолефином или же фторсодержащим углеводородом и смесью двух или большего количества фторгидроолефиновых соединений, которые могут быть получены в результате любой конкретной реакции.
Безводный фторид водорода (HF) также используется в способах по настоящему изобретению и является коммерчески доступным.
Один аспект настоящего изобретения касается способа получения фторгидроолефина, включающего дегидрофторирование фторсодержащего углеводорода, который имеет у соседних атомов углерода, по крайней мере, один атом водорода и, по крайней мере, один атом фтора, при этом образуется смесь продуктов, содержащая указанный фторгидроолефин, не прореагировавший фторсодержащий углеводород и фторид водорода, где, по крайней мере, один из указанного фторгидроолефина и указанного фторсодержащего углеводорода присутствует в указанной смеси продуктов в виде азеотропной композиции с фторидом водорода.
Дегидрофторирование фторсодержащего углеводорода можно осуществить в паровой фазе. Дегидрофторирование фторсодержащего углеводорода в паровой фазе можно осуществить с помощью типичных катализаторов дегидрофторирования. В общем случае дегидрофторирование по настоящему изобретению можно проводить с помощью любого катализатора дегидрофторирования, известного из области техники. Указанные катализаторы включают, однако этим не ограничиваются, фторид алюминия; фторированный оксид алюминия; металлы на фториде алюминия; металлы на фторированном оксиде алюминия; оксиды, фториды и оксифториды магния, цинка и смеси магния и цинка и/или алюминия; оксид лантана и фторированный оксид лантана; оксиды хрома, фторированные оксиды хрома и кубический трифторид хрома; углерод, промытый кислотой углерод, активированный уголь, углеродистые вещества с трехмерной матрицей; и соединения металлов, нанесенные на уголь. Соединениями металлов являются оксиды, фториды и оксифториды, по крайней мере, одного металла, выбранного из группы, которая включает натрий, калий, рубидий, цезий, иттрий, лантан, церий, празеодим, неодим, самарий, хром, железо, кобальт, родий, никель, медь, цинк и их смеси.
Катализаторы дегидрофторирования включают фторид алюминия, фторированный оксид алюминия, металлы на фториде алюминия и металлы на фторированном оксиде алюминия, как раскрыто в патенте США № 5396000, который включен в настоящее описание посредством ссылки. Фторированный оксид алюминия и фторид алюминия может быть получен, как описано в патенте США № 4902838, который включен в настоящее описание посредством ссылки. Подходящие металлы включают хром, магний (например, фторид магния), металлы группы VIIB (в частности, марганец), металлы группы IIIB (в частности, лантан) и цинк. На практике подобные металлы обычно присутствуют в виде галогенидов (в частности, фторидов), в виде оксидов и/или оксигалогенидов. Металлы на фториде алюминия и металлы на фторированном оксиде алюминия могут быть получены в соответствии с методиками, как описано в патенте США № 4766260, который включен в настоящее описание посредством ссылки. В одном из вариантов осуществления настоящего изобретения, когда используют металлы на носителе, общее содержание металла в катализаторе составляет от приблизительно 0,1 до 20 мас.%, обычно приблизительно 0,1 до 10 мас.%. Предпочтительные катализаторы включают катализаторы, которые в основном состоят из фторида алюминия и/или фторированного оксида алюминия.
Кроме того, катализаторы дегидрофторирования включают оксиды, фториды и оксифториды магния, цинка и смеси магния и цинка и/или алюминия. Подходящий катализатор может быть получен, например, путем сушки оксида магния до тех пор, пока вода практически полностью не удалится, в частности, сушки приблизительно 18 час при температуре приблизительно 100°С. Высушенное вещество затем помещают в реактор, в котором оно будет использоваться. Далее температуру постепенно повышают приблизительно до 400°С, при этом в реакторе поддерживают ток азота, с целью удаления из оксида магния и реактора любых оставшихся следов воды. Затем температуру понижают до приблизительно 200°С и через реактор пропускают фторирующий агент, такой как HF или же способные переходить в парообразное состояние фторсодержащие соединения, такие как HF, SF4, CCl3F, CCl2F3, CHF3, CHClF2 или CCl2FCClF2, которые необязательно разбавляют инертным газом, таким как азот. Количество инертного газа или азота можно постепенно снизить, пока через реактор не будет подаваться лишь HF или другие способные переходить в парообразное состояние фторсодержащие соединения. В этот момент температуру можно повысить приблизительно до 450°С и выдержать оксид магния при этой температуре, так чтобы превратить его в вещество, содержание фторида в котором составляет, по крайней мере, 40 мас.%, например, выдержать в течение от 15 до 300 мин в зависимости от расхода фторирующего агента и объема катализатора. Фториды представляют собой фторид магния или оксифторид магния; оставшуюся часть катализатора составляет оксид магния. Из области техники известно, что условия фторирования, такие как время и температура, можно подобрать таким образом, чтобы содержание фторсодержащего вещества составило более чем 40 мас.%.
Другая подходящая методика получения катализатора заключается в добавлении гидроксида аммония к раствору нитрата магния и, если они присутствуют, нитрата цинка и/или нитрата алюминия. Гидроксид аммония добавляют к раствору нитратов при рН в диапазоне от 9,0 до 9,5. По окончании добавления раствор отфильтровывают, полученное твердое вещество промывают водой, сушат и медленно нагревают до температуры 500°С, при которой проводят кальцинирование. Отожженный продукт затем обрабатывают подходящим фторсодержащим соединением, как описано выше.
Наконец, еще одна методика получения катализатора на основе фторида металла (в частности, магния, необязательно содержащего также цинк и/или алюминий), который содержит один или несколько фторидов металлов, заключается в обработке водного раствора галогенида(ов) металла(ов) в деионизованной воде при перемешивании 48%-ным водным раствором HF. Оставляют перемешиваться на ночь и суспензию упаривают досуха на водяной бане. Высушенное твердое вещество затем кальцинируют на воздухе при 400°С в течение приблизительно четырех часов, охлаждают до комнатной температуры, размалывают и просеивают, получая вещество, которое используют при проведении испытаний катализаторов.
Кроме того, катализаторы дегидрофторирования включают оксид лантана и фторированный оксид лантана.
Подходящие композиции фторированного оксида лантана могут быть получены любым способом, аналогичным известным из области техники способам получения фторированного оксида алюминия. Например, композицию катализатора можно получить фторированием оксида лантана.
Подходящие композиции катализатора можно также приготовить осаждением лантана в виде гидроксида, который затем сушат и кальцинируют, получая оксид; эта методика также хорошо известна из области техники. Полученный оксид затем может быть подвергнут обработке, которая приводится в настоящем описании.
Композицию катализатора можно фторировать до получения требуемого содержания фторида предварительной обработкой фторсодержащим соединением при повышенных температурах, в частности, в диапазоне от приблизительно 200°С до приблизительно 450°С. Предварительную обработку способным переходить в парообразное состояние фторсодержащим соединением, таким как HF, SF4, CCl3F, CCl2F3, CHF3, CHClF2 или CCl2FCClF2, можно осуществить любым удобным способом, в том числе в реакторе, который предполагается использовать для проведения реакции дегидрофторирования. Под способным переходить в парообразное состояние фторсодержащим соединением понимают фторсодержащее соединение, которое при его пропускании над катализатором в указанных условиях будет фторировать катализатор до требуемого уровня.
Подходящий катализатор может быть получен, например, сушкой La2O3 до тех пор, пока практически вся влага не будет удалена, например, в течение приблизительно 18 час при температуре приблизительно 400°С. Высушенный катализатор затем помещают в реактор, который предполагается использовать. Затем температуру постепенно повышают до приблизительно 400°С, поддерживая в реакторе ток N2, с целью удаления из катализатора и реактора любых оставшихся следов влаги. После этого температуру понижают до 200°С и пропускают через реактор способное переходить в парообразное состояние фторсодержащее соединение. В случае необходимости в качестве разбавителей используют азот или другие инертные газы. Количество N2 или других инертных разбавителей можно постепенно снизить, пока через реактор не будет проходить лишь способное переходить в парообразное состояние фторсодержащее соединение. В этот момент температуру можно повысить приблизительно до 450°С и выдержать La2O3 при этой температуре таким образом, чтобы превратить его в вещество, содержание фтора в котором соответствует, по крайней мере, 80 мас.% LaF3, например, в течение от 15 до 300 мин в зависимости от расхода фторсодержащего соединения и объема катализатора.
Другая подходящая методика получения катализатора заключается в добавлении гидроксида аммония к раствору La(NO3)3·6Н2О. Гидроксид аммония добавляют к раствору нитрата при величине рН, равной приблизительно от 9,0 до 9,5. По окончании добавления раствор отфильтровывают, полученное твердое вещество промывают водой, сушат и медленно нагревают приблизительно до температуры 500°С, при которой проводят кальцинирование. Отожженный продукт затем обрабатывают подходящим, способным переходить в парообразное состояние фторсодержащим соединением, как описано выше.
Кроме того, катализатор дегидрофторирования включает оксиды хрома, фторированные оксиды хрома и кубический трифторид хрома. Кубический трифторид хрома можно получить из CrF3ХН2О, где Х равно от 3 до 9, преимущественно равно 4, путем нагревания на воздухе или в инертной атмосфере (в частности, в азоте или аргоне) при температуре от приблизительно 350°С до приблизительно 400°С в течение от 3 до 12 час, преимущественно от 3 до 6 час.
Кубический трифторид хрома может применяться самостоятельно или же вместе с другими соединениями хрома в качестве катализатора дегидрофторирования. Получение кубического трифторид хрома описано в патенте США № 6031141, который включен в настоящее описание посредством ссылки. Следует отметить композиции катализатора, включающие хром, в которых, по крайней мере, 10 мас.% хрома находится в форме кубического трифторида хрома, предпочтительно композиции катализатора, в которых, по крайней мере, 25 мас.% хрома находится в форме кубического трифторида хрома и наиболее предпочтительно композиции катализатора, в которых, по крайней мере, 60 мас.% хрома находится в форме кубического трифторида хрома. Хром, в том числе кубический трифторид хрома, может быть нанесен на носитель и/или физически смешан с такими веществами, как уголь, фторид алюминия, фторированный оксид алюминия, фторид лантана, фторид магния, фторид кальция, фторид цинка и т.п. Предпочтительными являются комбинации, включающие кубический трифторид хрома в сочетании с фторидом магния и/или фторидом цинка.
Кроме того, катализаторы дегидрофторирования включают активированный уголь или углеродистые вещества с трехмерной матрицей, как раскрывается в патенте США № 6369284, который полностью включен в настоящее описание в качестве ссылки; или включают уголь или металлы, такие как натрий, калий, рубидий, цезий, иттрий, лантан, церий, празеодим, неодим, самарий, хром, железо, кобальт, родий, никель, медь, цинк и их смеси, нанесенные на уголь, как раскрывается в патенте США № 5268122, который включен в настоящее описание посредством ссылки. В способе по настоящему изобретению пригоден углерод, полученный из любого из следующих источников: дерева, торфа, угля, оболочки кокосовых орехов, глинистого угля, бурого угля, остатков нефтепереработки и сахара. Коммерчески доступными типами углерода, которые могут использоваться по настоящему изобретению, включают типы углерода, выпускаемые под следующими торговыми знаками: Barneby & Sutcliffe™, Darco™, Nucharm, Columbia JXN™, Columbia LCK™, Calgon PCB, Calgon BPL™, Westvaco™, Norit™ и Barnaby Cheny NB™.
Углерод включает промытый кислотой углерод (в частности, углерод, который обрабатывают соляной кислотой или хлористоводородной кислотой с последующей обработкой фтористоводородной кислотой). Обработка кислотой, как правило, достаточна для получения углерода, который содержит меньше чем 1000 м.д. зольного остатка. Пригодная к применению обработка углерода описывается в патенте США № 5136113, который включен в настоящее описание посредством ссылки. Углерод включает также углеродистые вещества с трехмерной матрицей. Соответствующие примеры приведены в патенте США № 4978649, который включен в настоящее описание посредством ссылки. Следует отметить углеродистые вещества с трехмерной матрицей, которые получают путем пропускания газообразных или парообразных углеродсодержащих веществ (в частности, углеводородов) через массу гранул углеродистого вещества (например, сажи); разложение углеродсодержащих соединений с целью осаждения углерода на поверхности гранул; и обработки полученного вещества активирующим газом, представляющим собой водяной пар, с целью получения пористого углеродистого вещества. Указанным способом получают композитное вещество углерод-углерод.
Физическая форма катализатора не является критической и может, например, включать шарики, порошки или гранулы. Кроме того, для катализаторов, нанесенных на уголь, уголь может быть в форме порошка, гранул, шариков и т.п. Хотя это и не существенно, катализаторы, которые не были фторированы, могут быть обработаны перед использованием фторидом водорода. Как полагают, при этом часть оксидов на поверхности превращается в оксифториды. Указанную предварительную обработку можно осуществить, поместив катализатор в подходящий контейнер (он может быть реактором, который предполагают использовать для проведения реакции по настоящему изобретению) и пропуская HF над высушенным катализатором таким образом, чтобы частично насытить катализатор фторидом водорода. Указанный процесс обычно осуществляют, пропуская HF над катализатором в течение определенного времени (в частности, от приблизительно 15 до 300 мин) при температуре, например, от приблизительно 200°С до приблизительно 450°С.
Каталитическое дегидрофторирование удобно проводить при температуре в диапазоне от приблизительно 200°С до приблизительно 500°С, в другом варианте осуществления настоящего изобретения - в диапазоне от приблизительно 300°С до приблизительно 450°С. Время контактирования, как правило, составляет от приблизительно 1 до приблизительно 450 сек, а в другом варианте осуществления настоящего изобретения составляет от приблизительно 10 до приблизительно 120 сек.
Давление реакции может быть меньше атмосферного, атмосферным или больше атмосферного. В общем случае предпочтительным является давление, близкое к атмосферному. Тем не менее, дегидрофторирование с успехом можно осуществлять при пониженном давлении (т.е. при давлении меньше одной атмосферы).
Каталитическое дегидрофторирование необязательно можно проводить в присутствии инертного газа, такого как азот, гелий или аргон. Введение инертного газа можно использовать для увеличения степени дегидрофторирования. Следует отметить процессы, в которых мольное отношение инертного газа к фторсодержащему углеводороду, который подвергают дегидрофторированию, составляет от приблизительно 5:1 до приблизительно 1:1. Предпочтительным инертным газом является азот.
Другой аспект настоящего изобретения касается способа получения фторгидроолефина дегидрофторированием фторсодержащего углеводорода в зоне реакции при повышенной температуре в отсутствие катализатора.
В варианте осуществления настоящего изобретения дегидрофторирование фторсодержащего углеводорода может быть осуществлено в зоне реакции при повышенной температуре в отсутствие катализатора, как указано ниже при описании пиролиза CF3CH2CF3 с образованием CF2=CHCF3 и HF. Подходящая температура может составлять в диапазоне от приблизительно 350°С до приблизительно 900°С, а в другом варианте осуществления настоящего изобретения - в диапазоне от приблизительно 450°С до приблизительно 900°С. Время контактирования газов в зоне реакции, как правило, составляет от приблизительно 0,5 до приблизительно 60 сек, а в другом варианте осуществления настоящего изобретения - от приблизительно 2 сек до приблизительно 20 сек.
Давление реакции при проведении реакции дегидрофторирования при повышенной температуре в отсутствие катализатора может быть меньше атмосферного, атмосферным или больше атмосферного. В общем случае предпочтительным является давление, близкое к атмосферному. Тем не менее, дегидрофторирование с успехом можно осуществлять при пониженном давлении (т.е. при давлении меньше одной атмосферы).
Дегидрофторирование при повышенной температуре в отсутствие катализатора необязательно можно проводить в присутствии таких инертных газов, как азот, гелий или аргон. Введение инертного газа можно использовать для увеличения степени дегидрофторирования. Следует отметить процессы, в которых мольное отношение инертного газа к фторсодержащему углеводороду, который подвергают дегидрофторированию, составляет от приблизительно 5:1 до приблизительно 1:1. Предпочтительным инертным газом является азот.
Зона проведения реакции как катализируемого, так и не катализируемого дегидрофторирования может представлять собой реактор, изготовленный из никеля, железа, титана или их сплавов, как описано в патенте США № 6540933, который полностью включен в настоящее описание в качестве ссылки. Может также использоваться реактор, изготовленный из указанных материалов (в частности, металлическая труба), необязательно содержащий металлическую насадку подходящей формы. Что касается сплавов, то под ними подразумевают сплав никеля, содержащий от приблизительно 1 до приблизительно 99,9 мас.% никеля, сплав железа, содержащий от приблизительно 0,2 до приблизительно 99,8 мас.% железа, и сплав титана, содержащий от приблизительно 72 до приблизительно 99,8 мас.% титана. Следует отметить использование незаполненного (без насадки) реактора, изготовленного из никеля или сплавов никеля, например, содержащих от приблизительно 40 мас.% до приблизительно 80 мас.% никеля, в частности, никелевого сплава Inconel™ 600, никелевого сплава Hastelloy™ C617 или никелевого сплава Hastelloy™ C276.
Если их используют в качестве насадки, то металл или металлические сплавы могут быть в виде частиц или сформованы, например, в виде перфорированных пластинок, колец, проволоки, сетки, стружки, трубки, дроби, проволочной ткани или ваты.
Смесь продуктов, получаемая в результате осуществления дегидрофторирования фторсодержащего углеводорода, содержит фторгидроолефин, не прореагировавший фторсодержащий углеводород и фторид водорода. Количество не прореагировавшего фторсодержащего углеводорода зависит от степени превращения, которая может быть достигнута при проведении реакции.
Было обнаружено и в совместно поданных и ожидающих решение экспертизы заявках на патент США, имеющих в книге записей поверенного номера FL-1158, FL-1160, FL-1161, FL-1169, FL-1188 и FL-1195, указано, что фторгидроолефины могут образовывать азеотропные или близкие к азеотропным композиции с фторидом водорода. Указанные азеотропные или близкие к азеотропным композиции пригодны для использования в способах получения фторгидроолефинов и в способах отделения фторгидроолефинов от фторида водорода и фторсодержащих углеводородов.
Как известно из области техники, азеотропная или близкая к азеотропной композиция представляет собой смесь двух или большего количества различных компонентов, которая, если композиция является жидкостью при данном давлении, кипит при практически постоянной температуре, при этом температура может быть выше или ниже чем температура кипения индивидуальных компонентов, и образует пар, состав которого практически идентичен составу жидкости, которую кипятят.
В данном описании близкая к азеотропной композиция (ее обычно также называют “композицией, подобной азеотропной”) означает композицию, которая ведет себя как азеотроп (т.е. имеет постоянные характеристики кипения или проявляет тенденцию не разделяться на фракции при кипячении или при испарении). Так, состав пара, образовавшегося при кипячении или при испарении, такой же или практически такой же, как и состав исходной жидкости. Таким образом, при кипячении или при испарении состав жидкости, если он вообще изменяется, изменяется лишь в минимальной степени или изменяется незначительно. Такое поведение в корне отличается от композиций, не являющихся близкими к азеотропным композициям, в которых при кипячении или при испарении состав жидкости в значительной степени меняется.
Кроме того, для композиций, близких к азеотропным, давление точки росы и давление температуры начала кипения практически не отличаются. Другими словами, разница между давлением точки росы и давлением температуры начала кипения при данной температуре представляет собой малую величину. Можно утверждать, что композиция с разницей в давлении точки росы и давлении температуры начала кипения, меньшей или равной 3% (по сравнению с давлением температуры начала кипения), может рассматриваться как композиция, близкая к азеотропной.
Таким образом, существенными особенностями азеотропной композиции или близкой к азеотропной композиции являются следующие: при данной величине давления температура кипения жидкой композиции фиксирована и состав пара над кипящей композицией практически такой же, что и состав кипящей композиции (т.е. не происходит фракционирования компонентов жидкой композиции). Из области техники известно также, что как температура кипения, так и массовые проценты каждого компонента азеотропной композиции могут изменяться, если азеотропную или близкую к азеотропной композицию кипятят при различных давлениях. Таким образом, азеотропную или близкую к азеотропной композицию можно определить либо в терминах уникального взаимоотношения, которое существует между компонентами, либо в терминах композиционных диапазонов компонентов, либо в терминах точных массовых процентов каждого компонента в композиции, которая характеризуется фиксированной температурой кипения при указанной величине давления.
Следует понимать, что хотя азеотропная или близкая к азеотропной композиция может существовать с конкретным соотношением компонентов при заданных температурах и давлениях, азеотропная композиция может также существовать в составах, которые содержат другие компоненты. Указанные дополнительные компоненты включают индивидуальные компоненты азеотропной композиции, при этом указанные компоненты присутствуют в виде избытка сверх количества, которое присутствует в азеотропной композиции. Например, азеотроп фторгидроолефина и фторида водорода может присутствовать в композиции, которая содержит избыток указанного фторгидроолефина, и это означает, что присутствует азеотропная композиция и также присутствует дополнительный фторгидроолефин. Кроме того, например, азеотроп фторгидроолефина и фторида водорода, а также азеотроп исходного фторсодержащего углеводорода и фторида водорода может присутствовать в композиции, которая имеет избыток исходного фторсодержащего углеводорода, используемого для проведения реакции дегидрофторирования при получении указанного фторгидроолефина, и это означает, что присутствует азеотропная композиция и также присутствует дополнительный исходный фторсодержащий углеводород. Например, азеотроп HFC-1225ye (CF3CF=CHF) и фторида водорода может присутствовать в композиции, которая имеет избыток HFC-1225ye, и это означает, что присутствует азеотропная композиции, но также присутствует и HFC-1225ye. Кроме того, например, азеотроп HFC-1225ye (CF3CF=CHF) и фторида водорода может присутствовать в композиции, которая содержит избыток соответствующего исходного фторсодержащего углеводорода, который используют при проведении реакции дегидрофторирования, такого как HFC-236ea, и это означает, что присутствует азеотропная композиции, но также присутствует и HFC-236ea.
Было обнаружено, что как фторгидроолефины, так и фторсодержащие углеводороды могут образовывать азеотропную композицию с фторидом водорода. В общем случае азеотропная композиция фторгидроолефин/фторид водорода кипит при более низкой температуре, чем соответствующий предшествующий фторсодержащий углеводород/фторид водорода. Таким образом, есть возможность отделить азеотроп фторгидроолефин/фторид водорода от азеотропа фторсодержащий углеводород/фторид водорода или отделить азеотроп фторгидроолефин/фторид водорода от фторсодержащего углеводорода азеотропной перегонкой.
Способ получения фторгидроолефина по настоящему изобретению может также включать стадию дистилляции смеси продуктов, с целью получения продукции композиции дистиллята, которая представляет собой азеотропную композицию, содержащую фторгидроолефин и фторид водорода.
На стадии дистилляции может быть дополнительно получена композиция кубовых остатков, представляющих собой не подвергнутый превращению фторсодержащий углеводород, который используют в реакции дегидрофторирования. В композиции кубовых остатков дополнительно может содержаться некоторое количество фторида водорода. Количество фторида водорода во фторсодержащем углеводороде из куба дистилляционной колонны может варьировать от приблизительно 50 мол.% до меньше чем 1 миллионной доли (м.д. в мольном отношении) в зависимости от способа проведения реакции дегидрофторирования.
Один вариант осуществления дистилляции по настоящему изобретению включает добавление избытка фторгидроолефина в дистилляционную колонну. Если в колонну загружают нужное количество фторгидроолефина, то весь фторид водорода может быть отобран сверху в виде азеотропной композиции, содержащей фторгидроолефин и фторид водорода. Таким образом, фторсодержащий углеводород, который извлекают из кубовых остатков, практически не содержит фторид водорода.
В настоящем описании выражение “практически не содержит фторид водорода” означает, что композиция содержит меньше чем приблизительно 100 м.д. (в мольном отношении), предпочтительно меньше чем приблизительно 10 м.д. и наиболее предпочтительно меньше чем приблизительно 1 м.д. фторида водорода.
Другой аспект настоящего изобретения касается способа отделения фторгидроолефина от фторсодержащего углеводорода, где указанный фторгидроолефин содержит на один атом фтора меньше и на один атом водорода меньше, чем указанный фторсодержащий углеводород, при этом указанный способ включает образование смеси, которая содержит фторгидроолефин, фторсодержащий углеводород и фторид водорода; и стадию дистилляции указанной смеси с образованием в колонне композиции дистиллята, представляющей собой азеотропную или близкую к азеотропной композицию фторида водорода и фторгидроолефина, которая практически не содержит указанный фторсодержащий углеводород.
В настоящем описании выражение “практически не содержит указанный фторсодержащий углеводород” означает, что композиция содержит меньше чем приблизительно 100 м.д. (в мольном отношении), предпочтительно меньше чем приблизительно 10 м.д. и наиболее предпочтительно меньше чем приблизительно 1 м.д. фторсодержащего углеводорода.
В способе отделения фторгидроолефина от фторсодержащего углеводорода может быть дополнительно получена композиция кубовых остатков, представляющих собой фторсодержащий углеводород, который используют в реакции дегидрофторирования. Композиция кубовых остатков, которая включает фторсодержащий углеводород, может содержать некоторое количество фторида водорода. Количество фторида водорода во фторсодержащем углеводороде из куба дистилляционной колонны может варьировать от приблизительно 50 мол.% до меньше чем 1 миллионной доли (м.д. в мольном отношении) в зависимости от способа проведения реакции дегидрофторирования.
Один вариант осуществления дистилляции по настоящему изобретению включает добавление избытка фторгидроолефина в дистилляционную колонну. Если в колонну загружают нужное количество фторгидроолефина, то весь фторид водорода может быть отобран сверху в виде азеотропной композиции, содержащей фторгидроолефин и фторид водорода. Таким образом, фторсодержащий углеводород, который извлекают из кубовых остатков, практически не содержит фторид водорода.
На стадии дистилляции композицию дистиллята, которая выводится через верхнюю часть дистилляционной колонны и содержит фторид водорода и фторгидроолефин, можно конденсировать с помощью, например, стандартных дефлегматоров. По крайней мере, часть указанного конденсированного потока может быть возвращена в верхнюю часть колонны в виде флегмы. Отношение конденсированного вещества, которое возвращают в верхнюю часть дистилляционной колонны в виде флегмы, к веществу, выделяемому в виде дистиллята, обычно называют флегмовым числом. Конкретные условия, которые используют для осуществления стадии дистилляции, зависят от ряда параметров, таких, среди прочих, как диаметр дистилляционной колонны, точки ввода исходных веществ и количество стадий разделения в колонне. Рабочее давление в колонне может составлять в диапазоне от приблизительно 10 фунтов на квадратный дюйм до приблизительно 300 фунтов на квадратный дюйм (1380 кПа), обычно от приблизительно 20 фунтов на квадратный дюйм до приблизительно 75 фунтов на квадратный дюйм. Дистилляционная колонна, как правило, работает под давлением приблизительно 25 фунтов на квадратный дюйм (172 кПа). Обычно увеличение флегмового числа приводит к повышению чистоты потока дистиллята, однако в общем случае флегмовое число составляет диапазоне от 0,2/1 до 200/1. Температура холодильника, который расположен вблизи верха колонны, обычно достаточна для того, чтобы практически полностью конденсировать дистиллят, который выводится из верхней части колонны, или же она соответствует температуре, необходимой для получения требуемого флегмового числа за счет частичной конденсации.
Композицию дистиллята в колонне, представляющую собой азеотропную или близкую к азеотропной композицию фторида водорода и фторгидроолефина, которая практически не содержит свободный фторсодержащий углеводород, можно подвергнуть обработке, с целью удаления фторида водорода и получения продукта в виде чистого фторгидроолефина. Указанную обработку можно осуществить, например, путем нейтрализации. Однако образование значительных количеств сливов после мокрой очистки газа может вызвать проблемы, связанные с утилизацией отходов. Таким образом, необходимы более эффективные, экономичные и безвредные для окружающей среды способы удаления фторида водорода из указанной смеси.
Было обнаружено, что фторгидроолефины можно выделить из смесей фторгидроолефинов и фторида водорода способом дистилляции, в котором используют преимущество изменения концентраций азеотропа при различных температурах и давлениях.
Другой аспект настоящего изобретения касается способа выделения фторгидроолефина из смеси, которая представляет собой азеотропную или близкую к азеотропной композицию указанного фторгидроолефина и фторида водорода, при этом указанный способ включает: а) стадию первой дистилляции, которой подвергают указанную смесь, где композиция, обогащенная либо (i) фторидом водорода, либо (ii) фторгидроолефином, удаляется в виде композиции первого дистиллята, а композиция первых кубовых остатков обогащается другим из указанных компонентов (i) или (ii); и b) вторую стадию дистилляции, которой подвергают указанную композицию первого дистиллята, осуществляемую при другом давлении, во время которой компонент, обогащенный в виде композиции первых кубовых остатков на стадии (а), удаляется в виде композиции второго дистиллята, а композиция вторых кубовых остатков на второй стадии дистилляции обогащается тем же самым компонентом, которым была обогащена указанная композиция первого дистиллята.
Может быть получена композиция первых кубовых остатков, представляющая собой фторгидроолефин, которая практически не содержит фторид водорода. Кроме того, может быть получена композиция вторых кубовых остатков, представляющая собой фторид водорода, которая практически не содержит фторгидроолефин.
Как указано в настоящем описании, выражение “практически не содержит фторгидроолефин” означает, что композиция содержит меньше чем приблизительно 100 м.д. (в мольном отношении), предпочтительно меньше чем приблизительно 10 м.д. и наиболее предпочтительно меньше чем приблизительно 1 м.д. фторгидроолефина.
В вышеуказанном способе используют преимущество изменения азеотропной композиции при различных давлениях с тем, чтобы осуществить разделение фторгидроолефина и фторида водорода. Первую стадию дистилляции можно проводить при более высоком давлении, по сравнению со второй стадией дистилляции. При более высоких давлениях азеотроп фторид водорода/фторгидроолефин содержит меньше фторгидроолефина. Таким образом, в результате указанной стадии дистилляции, которую проводят при высоком давлении, образуется избыток фторгидроолефина, который, поскольку он кипит выше, чем азеотроп, выходит из колонны в виде композиции кубовых остатков, представляющих собой фторгидроолефин. Композицию образующегося в колонне первого дистиллята затем направляют на вторую стадию дистилляции, которую проводят при меньшем давлении. При меньшем давлении состав азеотропа фторид водорода/фторгидроолефин смещается в сторону более низких концентраций фторида водорода. Таким образом, на указанной второй стадии дистилляции образуется избыток фторида водорода. Избыток фторида водорода, температура кипения которого выше, чем у азеотропа, выходит из второй дистилляционной колонны в виде композиции кубовых остатков, представляющих собой фторид водорода.
Возможен также обратный порядок осуществления процесса, который описан в предыдущем параграфе. Первую стадию дистилляции можно проводить при более низком давлении, по сравнению со второй стадией дистилляции. При более низком давлении азеотроп фторид водорода/фторгидроолефин содержит меньше фторида водорода. Таким образом, на указанной стадии дистилляции, которую проводят при низком давлении, образуется избыток фторида водорода, который, поскольку он кипит выше, чем азеотроп, выходит из колонны в виде композиции кубовых остатков, представляющих собой фторид водорода. Композицию образующегося в колонне первого дистиллята затем подают на вторую стадию дистилляции, которую проводят при более высоком давлении. При более высоком давлении состав азеотропа фторид водорода/фторгидроолефин смещается в сторону более высоких концентраций фторида водорода. Таким образом, на указанной второй стадии дистилляции образуется избыток фторгидроолефина. Избыток фторгидроолефина, температура кипения которого выше, чем у азеотропа, выходит из второй дистилляционной колонны в виде композиции кубовых остатков.
В общем случае дегидрофторирование фторсодержащих углеводородов представляет собой эндотермическую реакцию и, таким образом, может проводиться в трубчатом реакторе, при этом катализатор размещается в трубках, а теплоноситель находится в межтрубной зоне реактора. В качестве альтернативы теплоноситель может применяться с целью осуществления адиабатического процесса. Как практически чистый фторсодержащий углеводород, так и практически чистый фторгидроолефин, которые образуются в приведенном в настоящем описании процессе дистилляции, могут быть рециклированы обратно в реактор и служить в качестве теплоносителя. Предпочтительным теплоносителем является фторсодержащий углеводород, поскольку введение фторгидроолефина в реактор дегидрофторирования приведет к снижению степени прямоточного превращения фторсодержащего углеводорода.
Как на первой, так и на второй стадиях дистилляции можно конденсировать дистиллят, который выводится через верхнюю часть дистилляционной колонны и содержит фторид водорода и фторгидроолефин, с помощью, например, стандартных дефлегматоров. По крайней мере, часть указанного конденсированного потока может быть возвращена в верхнюю часть колонны в виде флегмы. Конкретные условия, которые используют для осуществления стадий дистилляции, зависят от ряда параметров, таких, среди прочих, как диаметр дистилляционной колонны, точки ввода исходных веществ и количество стадий разделения в колонне. Рабочее давление в первой колонне может составлять в диапазоне от приблизительно 50 фунтов на квадратный дюйм (345 кПа) до приблизительно 225 фунтов на квадратный дюйм (1550 кПа), обычно от приблизительно 50 фунтов на квадратный дюйм (345 кПа) до приблизительно 100 фунтов на квадратный дюйм (690 кПа). Первая дистилляционная колонна, как правило, работает под давлением приблизительно 75 фунтов на квадратный дюйм (520 кПа). Обычно увеличение флегмового числа приводит к повышению чистоты потока дистиллята, однако в общем случае флегмовое число составляет диапазоне от 0,1/1 до 100/1. Температура холодильника, который расположен вблизи верха колонны, обычно достаточна для того, чтобы практически полностью конденсировать дистиллят, который выводится из верхней части колонны, или же она соответствует температуре, необходимой для получения требуемого флегмового числа за счет частичной конденсации.
Рабочее давление во второй колонне может составлять в диапазоне от приблизительно 5 фунтов на квадратный дюйм (34 кПа) до приблизительно 50 фунтов на квадратный дюйм (345 кПа), обычно от приблизительно 5 фунтов на квадратный дюйм (34 кПа) до приблизительно 20 фунтов на квадратный дюйм (138 кПа). Вторая дистилляционная колонна, как правило, работает под давлением приблизительно 17 фунтов на квадратный дюйм (117 кПа). Обычно увеличение флегмового числа приводит к повышению чистоты потока дистиллята, однако в общем случае флегмовое число составляет в диапазоне от 0,1/1 до 50/1. Температура холодильника, который расположен вблизи верха колонны, обычно достаточна для того, чтобы практически полностью конденсировать дистиллят, который выводится из верхней части колонны, или же она соответствует температуре, необходимой для получения требуемого флегмового числа за счет частичной конденсации.
Фиг.1 иллюстрирует один из вариантов осуществления способа двухстадийной дистилляции по настоящему изобретению для разделения фторгидроолефина и фторида водорода. Если обратиться к фиг.1, то исходную смесь, полученную в результате предазеотропной дистилляции и содержащую фторид водорода и фторгидроолефин, подают по трубопроводу (540) в многотарельчатую дистилляционную колонну (510), рабочая температура в которой составляет приблизительно 77°С, а давление составляет приблизительно 335 фунтов на квадратный дюйм (2310 кПа). Кубовые остатки в дистилляционной колонне (510), представляющие собой практически чистый фторгидроолефин с температурой приблизительно 86°С и давлением приблизительно 337 фунтов на квадратный дюйм (2320 кПа), удаляются из нижней части колонны (510) по трубопроводу (566). Дистиллят из колонны (510), содержащий азеотроп фторид водорода/фторгидроолефин с температурой приблизительно 77°С и давлением приблизительно 335 фунтов на квадратный дюйм (2310 кПа), выводится через верх колонны (510) и направляется по трубопроводу (570) в многотарельчатую дистилляционную колонну (520). Дистиллят из колонны (520), содержащий азеотроп фторид водорода/фторгидроолефин с температурой приблизительно -19°С и давлением приблизительно 17 фунтов на квадратный дюйм (117 кПа), выводится из колонны (520) по трубопроводу (585) и рециклируется обратно в колонну (510). Кубовые остатки из колонны (520), содержащие практически чистый фторид водорода с температурой приблизительно 26°С и давлением приблизительно 19 фунтов на квадратный дюйм (131 кПа), извлекаются через трубопровод (586).
Еще один аспект настоящего изобретения касается способа очистки фторгидроолефина из смеси фторгидроолефина, фторсодержащего углеводорода и фторида водорода, при этом указанный способ включает: а) стадию первой дистилляции, которой подвергают указанную смесь, с образованием композиции первого дистиллята, которая представляет собой азеотропную или близкую к азеотропной композицию, содержащую фторгидроолефин и фторид водорода, и композиции первых кубовых остатков, представляющей собой фторсодержащий углеводород; b) вторую стадию дистилляции, которой подвергают указанный первый дистиллят, после которой композиция, обогащенная либо (i) фторидом водорода, либо (ii) фторгидроолефином, удаляется в виде композиции второго дистиллята, а композиция вторых кубовых остатков обогащается другим из указанных компонентов (i) или (ii); и с) третью стадию дистилляции, которой подвергают указанную композицию второго дистиллята, осуществляемую с давлением, отличным от давления на второй стадии дистилляции, где компонент, которым обогащена композиция вторых кубовых остатков на стадии (b), удаляется в виде композиции третьего дистиллята, а композиция третьих кубовых остатков на третьей стадии дистилляции обогащается тем же самым компонентом, которым была обогащена композиция второго дистиллята.
Способ по настоящему изобретению необязательно может включать также рециклирование, по крайней мере, части указанной композиции вторых кубовых остатков (фторгидроолефина) на указанную первую стадию дистилляции. Рециклирование фторгидроолефина способствует тому, что весь фторид водорода выводится через верх в виде азеотропной композиции с фторгидроолефином. Таким образом, может быть получен фторсодержащий углеводород, который выводится из процесса в виде композиции первых кубовых остатков, практически не содержащей фторид водорода и фторгидроолефин. Может быть получен фторгидроолефин, который выводится из процесса в виде композиции вторых кубовых остатков, практически не содержащей фторид водорода. Может быть получен фторид водорода, который выводится из процесса в виде композиции третьих кубовых остатков, практически не содержащей фторгидроолефин.
В настоящем описании выражение “практически не содержащий фторид водорода и фторгидроолефин” означает, что композиция содержит меньше чем приблизительно 100 м.д. (в мольном отношении), предпочтительно меньше чем приблизительно 10 м.д. и наиболее предпочтительно меньше чем приблизительно 1 м.д. каждого из фторида водорода и фторгидроолефина.
Условия проведения первой стадии дистилляции такие же, что и для процесса азеотропной дистилляции при отделении фторгидроолефина от фторсодержащего углеводорода, который приведен ранее в настоящем описании. Условия проведения второй и третьей стадий дистилляции такие же, что и условия при проведении процесса в двух колоннах при отделении фторгидроолефина от фторида водорода, которые также приведены ранее в настоящем описании.
Еще один аспект настоящего изобретения касается способа получения фторгидроолефина, который включает: а) подачу фторсодержащего углеводорода, который содержит у соседних атомов углеводорода, по крайней мере, один атом водорода и, по крайней мере, один атом фтора, в зону проведения реакции дегидрофторирования с образованием композиции продуктов реакции, содержащей фторгидроолефин, не прореагировавший фторсодержащий углеводород и фторид водорода; b) первую стадию дистилляции, которой подвергают указанную композицию продуктов реакции, с образованием композиции первого дистиллята, представляющей собой азеотропную или близкую к азеотропной композицию, содержащую фторгидроолефин и фторид водорода, и композиции первых кубовых остатков, которая представляет собой фторсодержащий углеводород; с) вторую стадию дистилляции, которой подвергают указанную композицию первого дистиллята, после которой композиция, обогащенная либо (i) фторидом водорода, либо (ii) фторгидроолефином, удаляется в виде композиции второго дистиллята, а композиция вторых кубовых остатков обогащается другим из указанных компонентов (i) или (ii); и d) третью стадию дистилляции, которой подвергают указанную композицию второго дистиллята, осуществляемую под давлением, отличным от давления на второй стадии дистилляции, где компонент, которым обогащена композиция вторых кубовых остатков на стадии (с), удаляется в виде композиции третьего дистиллята, а композиция третьих кубовых остатков на третьей стадии дистилляции обогащается тем же самым компонентом, которым была обогащена композиция второго дистиллята.
Способ необязательно может включать также рециклирование, по крайней мере, некоторой части указанной композиции первых кубовых остатков (фторсодержащего углеводорода) в указанную зону реакции. Кроме того, способ необязательно может включать рециклирование, по крайней мере, некоторой части указанной композиции вторых кубовых остатков или указанной композиции третьих кубовых остатков (т.е. композиций, представляющих собой фторгидроолефин) в указанную зону реакции. Кроме того, способ необязательно может включать рециклирование, по крайней мере, некоторой части указанной композиции вторых кубовых остатков или указанной композиции третьих кубовых остатков (т.е. композиций, представляющих собой фторгидроолефин) на указанную стадию первой дистилляции. Кроме того, способ необязательно может включать извлечение, по крайней мере, некоторой части указанной композиции вторых кубовых остатков или указанной композиции третьих кубовых остатков в виде фторгидроолефина, который практически свободен от фторсодержащего углеводорода и фторида водорода.
В настоящем описании выражение “практически свободен от фторсодержащего углеводорода и фторида водорода” означает, что композиция содержит меньше чем приблизительно 100 м.д. (в мольном отношении), предпочтительно меньше чем приблизительно 10 м.д. и наиболее предпочтительно меньше чем приблизительно 1 м.д. каждого из фторсодержащего углеводорода и фторида водорода.
Фиг.2 иллюстрирует один из вариантов осуществления способа получения фторгидроолефина по настоящему изобретению. Фторсодержащий углеводород загружают через трубопровод (360) в реактор (320). Покидающая реактор смесь, содержащая фторид водорода, фторсодержащий углеводород и фторгидроолефин, выводится из реактора по трубопроводу (450) и подается в многотарельчатую дистилляционную колонну (410). Кубовые остатки из дистилляционной колонны (410), состоящие практически из чистого фторсодержащего углеводорода, выводятся из нижней части колонны (410) по трубопроводу (466) и могут рециклироваться обратно в реактор. Дистиллят из колонны (410), содержащий азеотроп фторид водорода/фторгидроолефин, выводится через верхнюю часть колонны (410) и направляется по трубопроводу (540) во вторую многотарельчатую дистилляционную колонну (510). Кубовые остатки из дистилляционной колонны (510), которые представляют собой практически чистый фторгидроолефин, выводятся из колонны (510) по трубопроводу (566) и могут быть рециклированы обратно в реактор (320) в качестве теплоносителя. Дистиллят из колонны (510), содержащий азеотроп фторид водорода/фторгидроолефин, по трубопроводу (570) подают в третью многотарельчатую дистилляционную колонну (520). Дистиллят из колонны (520), содержащий фторид водорода/фторгидроолефин, выводится через трубопровод (585) и может быть рециклирован во вторую дистилляционную колонну (510). Композиция кубовых остатков из колонны (520) является практически чистым фторидом водорода, и ее удаляют из колонны (520) по трубопроводу (586). Получаемый в указанном способе практически чистый фторид водорода может быть использован соответствующим способом, таким как подача его в реактор фторирования для получения фторсодержащих соединений, или же он может быть нейтрализован с целью утилизации.
Хотя это и не показано на фигурах, должно быть понятно, что некоторые единицы технологического оборудования могут, с целью оптимизации процесса, использоваться в приведенных в настоящем описании способах. Например, там, где необходимо, могут применяться насосы, нагреватели или холодильники. Например, желательно, чтобы поступающее в дистилляционную колонну исходное сырье имело бы такую же температуру, что и температура в той точке колонне, через которую осуществляется загрузка исходного сырья. Таким образом, для выравнивания температуры может потребоваться нагревание или охлаждение потока реагентов.
Еще один аспект настоящего изобретения касается способа получения CF3CH=CF2 пиролизом CF3CH2CF3. Процесс может быть описан следующим образом:
CF3CH2CF3+Δ→CF3CH=CF2+HF,
где Δ обозначает тепло, а HF обозначает фторид водорода.
Термин пиролиз, который используют в настоящем описании, означает химическое изменение, которое осуществляют нагреванием в отсутствие катализатора. Реакторы пиролиза в общем случае имеют три зоны: а) зону предварительного нагрева, в которой реагенты доводят до температуры, близкой к температуре реакции; b) зону реакции, в которой реагенты достигают температуры реакции и, по крайней мере, частично подвергаются пиролизу с образованием продуктов и побочных продуктов; с) зону охлаждения, в которой поток, покидающий зону реакции, охлаждается, с целью прекращения реакции пиролиза. Лабораторные реакторы включают зону реакции, а зона предварительного нагрева и зона охлаждения могут отсутствовать.
В этом варианте осуществления настоящего изобретения реактор может иметь любую форму, которая совместима с процессом, однако, преимущественно, реактор имеет форму цилиндрической трубы как прямой, так и спиралеобразной. Хотя это и не критично, подобные реакторы, как правило, имеют внутренний диаметр от приблизительно 1,3 до приблизительно 5,1 см (от приблизительно 0,5 до приблизительно 2 дюйма). Тепло подводят с внешней стороны трубы, а химическая реакция протекает внутри трубы. Реактор и подсоединенные к нему подводящие трубопроводы, отводящие трубопроводы и необходимые узлы должны быть изготовлены, по крайней мере, их поверхности, которые подвергаются воздействию реагентов и продуктов реакции, должны быть изготовлены из материалов, которые устойчивы к действию фторида водорода. Типичные конструкционные материалы, хорошо известные из области фторирования, включают нержавеющую сталь, в частности, аустенитную нержавеющую сталь, хорошо известные сплавы с высоким содержанием никеля, такие как никель-медные сплавы Monel®, сплавы на основе хастеллоя и никель-хромовые сплавы Inconel®, а также сталь с медным покрытием. В тех случаях, когда реактор подвергается воздействию высокой температуры, реактор может быть сконструирован из нескольких материалов. Например, слой на внешней поверхности реактора должен быть выбран по принципу его способности сохранять структурную целостность и сопротивляться коррозии при температуре пиролиза, а слой на внутренней поверхности должен быть выбран из веществ, стойких к воздействию, т.е. инертных к действию реагентов и продуктов. В способе по настоящему изобретению образующийся в процессе фторид водорода обладает коррозионным действием на определенные вещества. Другими словами реактор может быть сконструирован таким образом, что материал внешнего слоя выбирают по его физической прочности при высокой температуре, а материал внутреннего слоя выбирают по стойкости к коррозии под действием реагентов и продуктов при температуре пиролиза.
В способе по настоящему изобретению слой на внутренней поверхности преимущественно изготавливают из сплава с большим содержанием никеля, т.е. сплава, который содержит, по крайней мере, приблизительно 50 мас.% никеля, предпочтительно сплава, который содержит, по крайней мере, приблизительно 75 мас.% никеля, более предпочтительно сплава, который содержит меньше чем приблизительно 8 мас.% хрома, еще более предпочтительно сплава, который содержит, по крайней мере, приблизительно 98 мас.% никеля и наиболее предпочтительно содержит практически чистый никель, такого как коммерческий сплав, известный как Nickel 200. Более предпочтительным веществом, по сравнению с никелем или его сплавами, для слоя на внутренней поверхности реактора является золото. Толщина слоя на внутренней поверхности не оказывает значительного влияния на пиролиз и не является критическим, при условии, что сохраняется целостность слоя на внутренней поверхности. Толщина слоя на внутренней поверхности, как правило, составляет от приблизительно 10 до приблизительно 100 мил (от 0,25 до 2,5 мм). Толщина слоя на внутренней поверхности может определяться способом изготовления, стоимостью материала и требуемым временем службы реактора.
Слой на внешней поверхности реактора устойчив к окислению или другому коррозионному воздействию и сохраняет значительную прочность при температуре проведения реакции, так что охраняет корпус реактора от разрушения или деформации. Этот слой предпочтительно представляет собой сплав Inconel® и более предпочтительно Inconel® 600.
Рассматриваемый пиролиз CF3CH2CF3 с образованием CF3CH=CF2 и HF проводят в отсутствие катализатора в практически пустом реакторе. Под отсутствием катализатора понимают, что в дополнение к пиролизу не вводится какое-либо вещество и не проводится какая-либо обработка, которая повышает скорость реакции за счет снижения энергии активации процесса пиролиза. Следует понимать, что, несмотря на то, что поверхности, которые неизбежно имеются у сосуда с защитной оболочкой, такого как реактор пиролиза, случайно могут оказывать каталитическое или антикаталитическое воздействие на процесс пиролиза, это воздействие вносит незначительный вклад, если вообще вносит какой-либо вклад, в скорость пиролиза. Более конкретно, отсутствие катализатора означает отсутствие обычных катализаторов, обладающих большой площадью поверхности в форме частиц, гранул, волокон или в виде материала на носителе, которые пригодны для ускорения элиминирования фторида водорода из фторсодержащего углеводорода (т.е. для дегидрофторирования). Примеры катализаторов дегидрофторирования включают: оксид хрома, необязательно содержащий другие металлы, оксиды металлов или галогениды металлов; фторид хрома в свободной форме и на носителе; и активированный уголь, необязательно содержащий другие металлы, оксиды металлов или галогениды металлов.
Практически незаполненные реакторы, пригодные для осуществления способа по настоящему изобретению, представляют собой трубки, включающие вышеуказанные конструктивные материалы. Практически пустые реакторы включают такие реакторы, в которых поток газов через реактор частичное затруднен, что вызывает противоточное смешение, т.е. турбулентность, и, таким образом, способствует смешиванию газов и хорошей теплопередаче. Указанное частичное препятствие можно создать специально путем размещения насадки внутри реактора, заполнения его сечения или используя перфорированные перегородки. Насадка реактора может быть в виде частиц или волокна, преимущественно в виде патрона, с целью облегчения ее размещения и удаления, может иметь открытую структуру наподобие колец Рашига или другой насадки с большим свободным объемом, с целью избежать накопления углеродистых отложений и минимизировать падение давления, и позволяет свободно протекать газу. Внешняя поверхность подобной насадки внутри реактора предпочтительно включает вещества, идентичные тем, из которых изготовлен слой на внутренней поверхности реактора; вещества, которые не катализируют дегидрофторирование фторсодержащих углеводородов и устойчивы к действию фторида водорода. Свободный объем представляет собой объем реакционной зоны за исключением объема вещества, которое составляет насадку реактора. Свободный объем составляет, по крайней мере, приблизительно 80%, предпочтительно приблизительно 90% и более предпочтительно приблизительно 95%.
Пиролиз, в результате которого CF3CH2CF3 превращается в CF3CH=CF2, обычно проводят при температуре, равной, по крайней мере, приблизительно 700°С, предпочтительно при температуре, по крайней мере, приблизительно 750°С и наиболее предпочтительно при температуре, по крайней мере, приблизительно 800°С. Максимальная температура составляет не более чем приблизительно 1000°С, предпочтительно не более чем приблизительно 950°С и наиболее предпочтительно не более чем приблизительно 900°С. Температура пиролиза представляет собой температуру газов приблизительно в средней части зоны реакции.
Продолжительность нахождения газов в зоне реакции, как правило, составляет от приблизительно 0,5 до приблизительно 60 сек, более предпочтительно от приблизительно 2 сек до приблизительно 20 сек при температуре от приблизительно 700 до 900°С и атмосферном давлении. Продолжительность нахождения в зоне реакции определяется общим объемом зоны реакции и объемной скоростью подачи газообразных исходных веществ в зону реакции при заданной температуре и давлении реакции и представляет собой среднее количество времени, в течение которого какой-либо объем газа остается внутри зоны реакции.
Пиролиз предпочтительно проводят до степени превращения CF3CH2CF3, равной, по крайней мере, приблизительно 25%, более предпочтительно до степени превращения, по крайней мере, приблизительно 35% и наиболее предпочтительно до степени превращения, по крайней мере, приблизительно 45%. Под степенью превращения понимают часть реагентов, которая потребляется при однократном проходе через реактор. Пиролиз предпочтительно осуществляют до тех пор, пока выход CF3CH=CF2 не составит, по крайней мере, приблизительно 50%, более предпочтительно, по крайней мере, приблизительно 60% и наиболее предпочтительно, по крайней мере, приблизительно 75%. Под выходом понимают количество моль CF3CH=CF2, которые получают на один моль потребленного CF3CH2CF3.
Реакцию преимущественно проводят при давлении меньше атмосферного или при общем давлении, равном атмосферному. Это означает, что реагенты плюс другие ингредиенты находятся при давлении меньше атмосферного или при атмосферном давлении. (Если в качестве других ингредиентов присутствуют инертные газы, как указано ниже, то сумма парциальных давлений реагентов плюс другие ингредиенты составляет давление меньше атмосферного или атмосферное давление.) Наиболее предпочтительным является общее давление, близкое к атмосферному. Реакцию удобно проводить при уменьшенном общем давлении (т.е. общее давление меньше одной атмосферы).
Реакцию в соответствии с вариантом осуществления настоящего изобретения можно проводить в присутствии одного или нескольких нереакционноспособных газов-разбавителей, т.е. газов-разбавителей, которые не принимают участие в реакции в условиях пиролиза. Подобные нереакционноспособные газы-разбавители включают инертные газы азот, аргон и гелий. Фторсодержащие углеводороды, которые стабильны в условиях пиролиза, например, трифторметан и перфторированные углеводороды, также могут использоваться в качестве нереакционноспособных газов-разбавителей. Было показано, что инертные газы могут использоваться для повышения степени превращения CF3CH2CF3 в CF3CH=CF2. Следует отметить процессы, где мольное отношение инертного газа к CF3CH2CF3, подаваемому в реактор пиролиза, составляет от приблизительно 5:1 до 1:1. Азот является предпочтительным инертным газом благодаря его относительно низкой стоимости.
В способе по настоящему изобретению в покидающем реактор потоке получают отношение HF и CF3CH=CF2, которое составляет 1:1. Покидающий реактор поток может также содержать не вступивший в реакцию реагент, CF3CH2CF3. Компоненты, которые составляют покидающий реактор поток, могут быть разделены обычными способами, такими как дистилляция. Фторид водорода и CF3CH=CF2 образуют гомогенный низкокипящий азеотроп, содержащий приблизительно 60 мол.% CF3CH=CF2. Покидающий реактор поток по настоящему изобретению можно подвергнуть дистилляции, и низкокипящий азеотроп HF и CF3CH=CF2 может быть отобран в виде потока из верхней части дистилляционной колонны, а качестве потока из нижней части колонны отбирают практически чистый CF3CH2CF3. Выделенный реагент CF3CH2CF3 можно рециклировать обратно в реактор. CF3CH=CF2 можно выделить из его азеотропа с НF обычными способами, такими как дистилляция под переменным давлением или нейтрализация HF с помощью щелочи.
Авторы настоящего изобретения полагают, что без дальнейших уточнений опытный специалист, используя приведенное описание, сможет использовать раскрытые композиции и в полной мере осуществить настоящее изобретение. Приведенные ниже примерные варианты осуществления настоящего изобретения следует рассматривать как иллюстративные, которые никак не ограничивают приведенное описание изобретения.
ПРИМЕРЫ
Пример 1
Дегидрофторирование CF3CH2CHF2 с образованием CF3CH=CHF (E и Z изомеров) над углеродистым катализатором
В реактор из никелевого сплава Hastelloy™ (внешний диаметр 2,54 см, внутренний диаметр 2,17 см, длина 24,1 см) помещают 14,32 г (25 мл) сферических частиц (8 меш) углеродистого вещества с трехмерной матрицей, получаемого практически в соответствии с методикой, приведенной в патенте США № 4978649, который полностью включен в настоящее описание в качестве ссылки. Заполненную насадкой часть реактора нагревают с помощью керамического ленточного нагревателя 5”x1”, который с помощью клемм закреплен снаружи реактора. Температуру в реакторе измеряют с помощью термопары, которая размещена между стенкой реактора и нагревателем. После загрузки в реактор углеродистого вещества через реактор пропускают азот (10 мл/мин, 1,7×10-7 м3/сек), температуру в течение одного часа повышают до 200°С и поддерживают указанную температуру еще в течение 4 час. Затем температуру в реакторе повышают до требуемой рабочей температуры и через реактор пускают поток CF3CH2CHF2 и азота.
Непрерывно отбирают пробу из общего исходящего из реактора газового потока для анализа органических продуктов с помощью газового хроматографа, оснащенного масс-селективным детектором (GC-MS); полученные результаты собраны в таблице 3. Весь исходящий из реактора газовый поток, содержащий органические продукты, а также неорганическую кислоту, такую как HF, обрабатывают водным раствором щелочи с целью нейтрализации.
Пример 2
Дегидрофторирование CF3CH2CHF2 с образованием CF3CH=CHF (E и Z изомеров) над катализатором из фторированного оксида алюминия.
В трубку из хастеллоя с габаритами 15 дюймов (38,1 см)×3/8 дюйма (0,95 см) помещают 7,96 г (13 куб.см) гамма-оксида алюминия, измельченного до размера зерен 12-20 меш (от 0,84 до 1,68 мм). Катализатор активируют нагреванием до 200°С в течение 15 мин в токе азота (50 станд. куб. см/мин, 8,3×10-7 м3/сек). Температуру повышают до 325°С в течение 10 мин, до 400°С в течение 20 мин, а затем снижают до 300°С в течение 60 мин. Расход азота снижают до 35 станд. куб. см/мин (5,8×10-7 м3/сек) и в течение 35 мин пропускают ток паров HF с расходом 12 станд. куб. см/мин (2,0×10-7 м3/сек). Затем температуру повышают до 325°С в течение 60 мин, до 350°С в течение 60 мин, до 375°С в течение 90 мин, до 400°С в течение 30 мин и до 425°С в течение 40 мин. После этого в течение 20 мин расход азота снижают до 25 станд. куб. см/мин (4,2×10-7 м3/сек), а расход HF повышают до 20 станд. куб. см/мин (3,3×10-7 м3/сек). Затем в течение 20 мин расход азота снижают до 15 станд. куб. см/мин (2,5×10-7 м3/сек), а расход HF повышают до 28 станд. куб. см/мин (4,7×10-7 м3/сек). Затем в течение 20 мин расход азота снижают до 5 станд. куб. см/мин (8,3×10-8 м3/сек), а расход HF повышают до 36 станд. куб. см/мин (6,0×10-7 м3/сек). После этого подачу азота прекращают, а расход HF в течение 121 мин повышают до 40 станд. куб. см/мин (6,7×10-7 м3/сек).
Устанавливают температуру реактора 375°С и подают CF3CH2CHF2 с расходом 5,46 мл/час (20,80 станд. куб. см/мин, 3,5×10-7 м3/сек) вместе а азотом, расход которого составляет 5,2 станд. куб. см/мин (8,7×10-8 м3/сек). Отходящие газы анализируют методом газовой хроматографии (ГХ); полученные результаты собраны в таблице 4.
Пример 3
Дегидрофторирование CF3CHFCHF2 с образованием CF3CF=CHF (E и Z изомеров) над углеродистым катализатором
Смесь CF3CHFCHF2 с азотом пропускают через реактор в соответствии с методикой, приведенной в примере 1. Результаты ГХ анализа исходящего из реактора потока собраны в таблице 5.
Пример 4
Синтез CF3CF=CH2 дегидрофторированием с использованием катализатора из фторированного оксида алюминия
Трубчатый реактор из сплава Hastelloy™ (внешний диаметр 2,54 см, внутренний диаметр 2,17 см, длина 24,1 см) помещают 25 куб. см. гамма-оксида алюминия, измельченного до размера зерен 12-20 меш (от 0,84 до 1,68 мм). Катализатор активируют нагреванием до 200°С в течение 15 мин в токе азота, а затем вводят во взаимодействие со смесью HF/N2, нагретой до 425°С, и получают 16,7 г активированного фторированного оксида алюминия.
При температуре 350°С смешивают 10 станд. куб. см/мин азота (1,7×10-7 м3/сек) и 15 станд. куб. см/мин (2,5×10-7 м3/сек) CF3CF2CH3 и пропускают через реактор. Затем температуру повышают до 400°С, при этом расход реагентов остается постоянным. Отходящие газы, образующиеся при обеих температурах, анализируют методом ГХ, чтобы определить концентрации, как указано в таблице 6.
Пример 5
Синтез CF3CF=CH2 с использованием угольного катализатора
В соответствии с методикой, приведенной в примере 3, смесь 10 станд. куб. см/мин (1,7×10-7 м3/сек) азота и 15 станд. куб. см/мин (2,5×10-7 м3/сек) CF3CF2CH3 пропускают через реактор таким образом, чтобы время контактирования было постоянным и составляло 60 сек. Скорости потоков снижают до 5 станд. куб. см/мин (8,3×10-8 м3/сек) азота и 7,5 станд. куб. см/мин (1,3×10-7 м3/сек) CF3CF2CH3 таким образом, чтобы время контактирования составило 120 сек. Берут образцы отходящих газов, образующихся при обоих наборах условий, и анализируют методом19F ЯМР спектроскопии. Состав отходящих газов, который определяют методом ГХ, приведен в таблице 7.
Пример 6
Синтез CHF2CF=CHF из CHF2CF2CH2F
В трубку из никелевого сплава Hastelloy™ с внутренним диаметром 0,375 дюймов (0,95 см) помещают 7,0 г (10 куб. см) гамма-оксида алюминия, измельченного до размера зерен 12/20 меш (от 0,84 до 1,68 мм). Трубу продувают азотом (50 станд. куб. см/мин, 8,3×10-7 м3/сек) в течение двадцати минут и одновременно повышают температуру от 40°С до 175°С. Продувку азотом продолжают и в реактор вводят фторид водорода (50 станд. куб. см/мин, 8,3×10-7 м3/сек) в течение 1,5 час. Расход азота снижают до 20 станд. куб. см/мин (3,3×10-7 м3/сек), а расход фторида водорода повышают до 80 станд. куб. см/мин (1,3×10-6 м3/сек) по мере того, как температуру в трубе в течение 3,7 час повышают со 174°С до 373°С. Затем расход азота снижают до 10 станд. куб. см/мин (1,7×10-7 м3/сек), а расход фторида водорода поддерживают на уровне 80 станд. куб. см/мин (1,3×10-6 м3/сек) в течение одного часа при температуре 400°С. Затем устанавливают температуру в реакторе 290°С и продувают реактор азотом.
CHF2CF2CH2F испаряют и подают в реактор с такой скоростью, чтобы время контактирования с катализатором составило 120 сек. Азот в процессе не используют. Данные ГХ анализа отходящих из реактора газов при трех температурах приведены в таблице 8.
Пример 7
Синтез CF3CН=CFCF3
В реактор из никелевого сплава Hastelloy™ (внешний диаметр 2,54 см, внутренний диаметр 2,17 см, длина 24,1 см) помещают 13,5 г (25 мл) сферических частиц (8 меш) углеродистого вещества с трехмерной матрицей, как описано в примере 1.
При температуре 300°С смешивают поток азота с расходом 12,5 станд. куб. см/мин (2,1×10-7 м3/сек) и поток 12,5 станд. куб. см/мин (2,1×10-7 м3/сек) CF3CНFCHFCH3 и пропускают через реактор. Температуру в реакторе поднимают до 350°С и, наконец, до 400°С, и отходящие из реактора газы при каждой температуре анализируют методом GC/MS. Состав отходящих реактора газов приведен в таблице 9.
Пример 8
Синтез CН2=CFCH2CF3 и E/Z-CF3CH=CFCH3
В реактор из никелевого сплава Hastelloy™ (внешний диаметр 2,54 см, внутренний диаметр 2,17 см, длина 24,1 см) помещают 13,5 г (25 мл) сферических частиц (8 меш) углеродистого вещества с трехмерной матрицей, как описано в примере 1.
При температуре 350°С смешивают 25 станд. куб. см/мин N2 (4,2×10-7 м3/сек) и 25 станд. куб. см/мин (4,2×10-7 м3/сек) CF3CН2CF2CH3 и пропускают через реактор. Отходящие из реактора газы анализируют методом GC/MS, и результаты приведены в таблице 10.
Пример 9
Дегидрофторирование CF3CHFCHFCF2CF3 с образованием CF3CH=CFCF2CF3 и CF3CF=CHCF2CF3 над углеродистым катализатором
В соответствии с методикой, приведенной в примере 3, смесь азота (10 мл/мин, 1,7×10-7 м3/сек) и CF3CHFCHFCF2CF3 (5 мл жидкости в час) пропускают через реактор. Результаты ГХ анализа отходящих из реактора газов в различных условиях собраны в таблице 11. В таблице 11 другие HFC включают CHF3, CF3CHF2 и CF3CH2F.
Пример 10
Исследование фазового состава смесей HF и E-CF3CH=CHF
Исследование фазового состава проводят для композиции, которая состоит практически из E-CF3CH=CHF и HF, при этом состав композиции варьируют и измеряют давление паров как при температуре 20°С, так и при температуре 70°С. На основании результатов, полученных при проведении исследования фазового состава, рассчитывают составы азеотропных композиций при других температурах и давлениях.
В таблице 12 собраны экспериментальные и расчетные азеотропные композиции для HF и E-CF3CH=CHF при указанных температурах и давлениях.
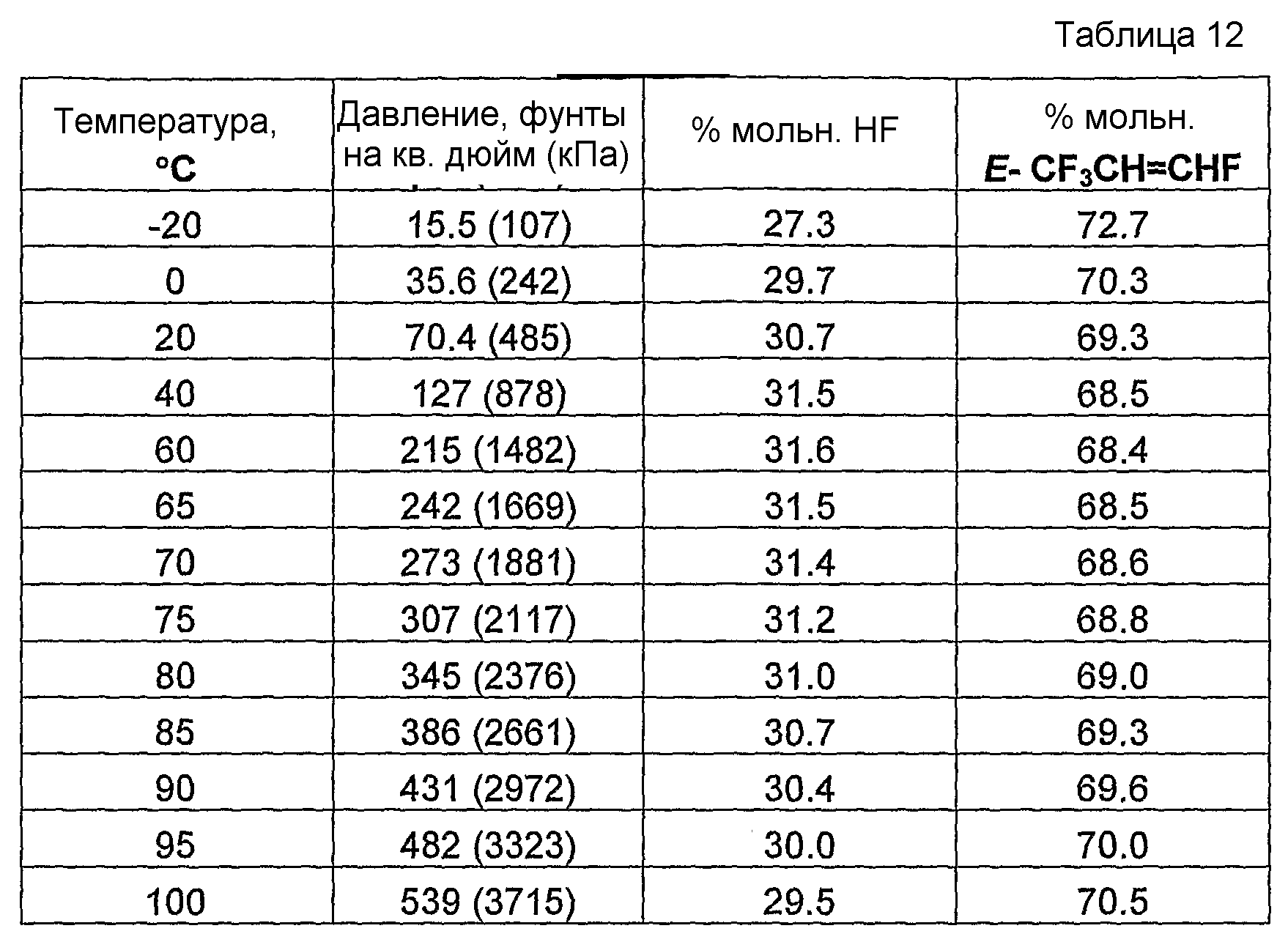
Пример 11
Исследование фазового состава смесей HF и Z-CF3CF=CHF
Исследование фазового состава проводят для композиции, которая состоит практически из Z-CF3CF=CHF и HF, при этом состав композиции варьируют и измеряют давление паров как при температуре 19,5°С, так и при температуре 70°С. На основании результатов, полученных при проведении исследования фазового состава, рассчитывают составы азеотропных композиций при других температурах и давлениях.
В таблице 13 собраны экспериментальные и расчетные азеотропные композиции для HF и Z-CF3CF=CHF при указанных температурах и давлениях.
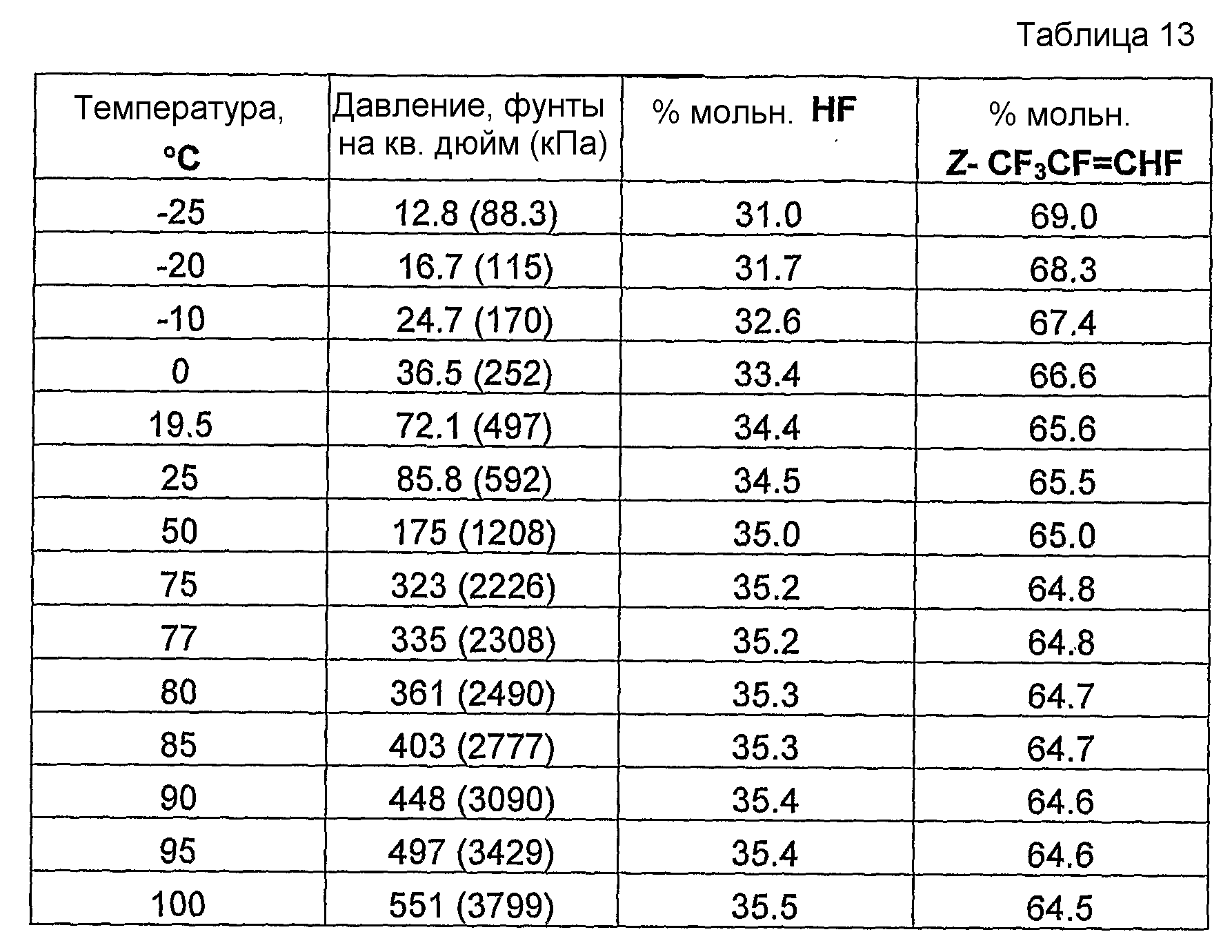
Пример 12
Исследование фазового состава смесей HF и смеси Z-CF3CH=CFCF2CF3 и Z-CF3CF=CHCF2CF3
Исследование фазового состава проводят для композиции, которая состоит практически из смеси Z-CF3CH=CFCF2CF3 и Z-CF3CF=CHCF2CF3 и HF, при этом состав композиции варьируют и измеряют давление паров как при температуре 20°С, так и при температуре 70°С. На основании результатов, полученных при проведении исследования фазового состава, рассчитывают составы азеотропных композиций при других температурах и давлениях.
В таблице 14 собраны экспериментальные и расчетные азеотропные композиции для HF и смеси Z-CF3CH=CFCF2CF3 и Z-CF3CF=CHCF2CF3 при указанных температурах и давлениях.
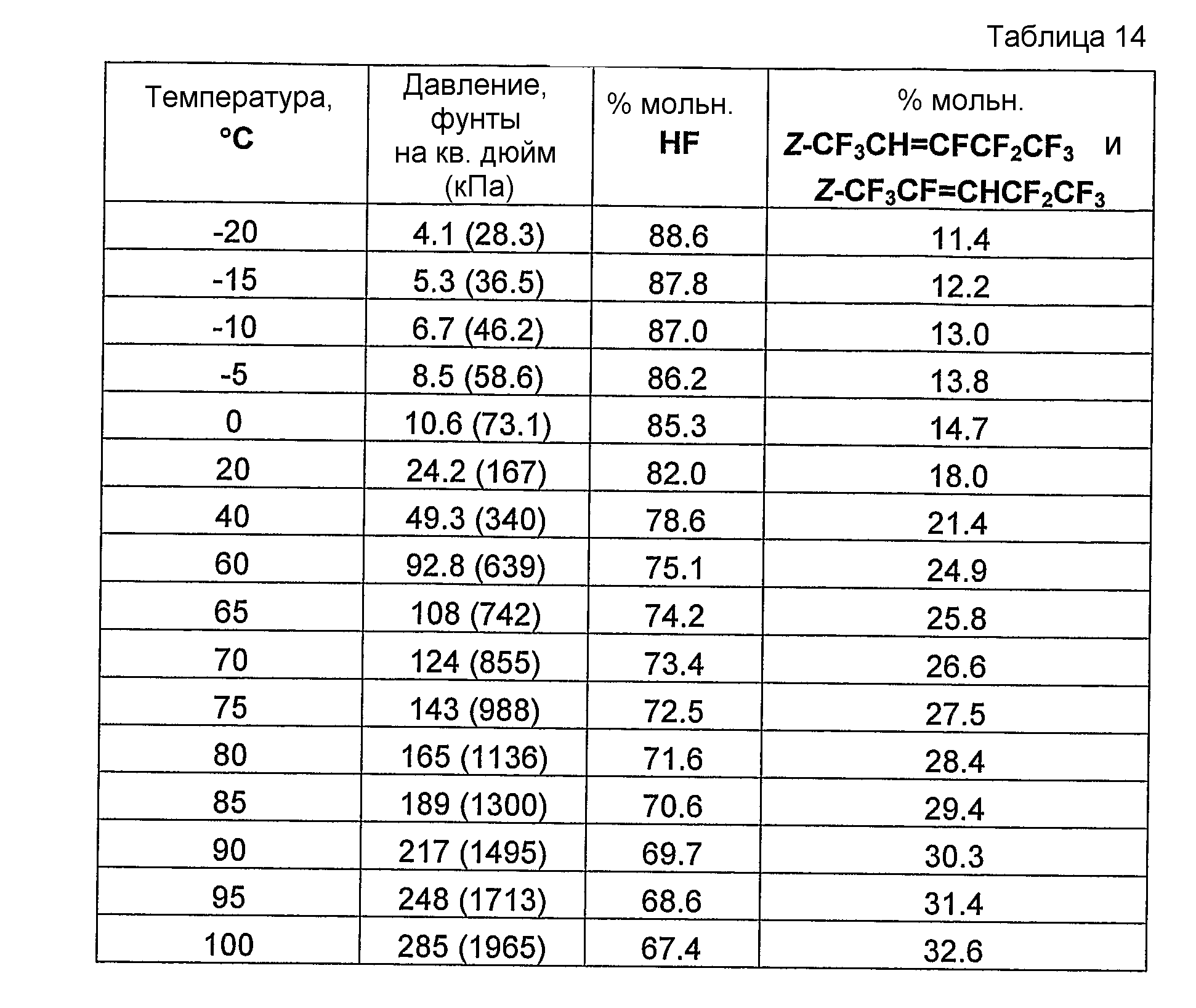
Пример 13
Исследование фазового состава смесей HF и CF3CHFCHFCF2CF3
Исследование фазового состава проводят для композиции, которая состоит практически из CF3CHFCHFCF2CF3 и HF, при этом состав композиции варьируют и измеряют давление паров как при температуре 30°С, так и при температуре 80°С. На основании результатов, полученных при проведении исследования фазового состава, рассчитывают составы азеотропных композиций при других температурах и давлениях.
В таблице 15 собраны экспериментальные и расчетные азеотропные композиции для HF и CF3CHFCHFCF2CF3 при указанных температурах и давлениях.
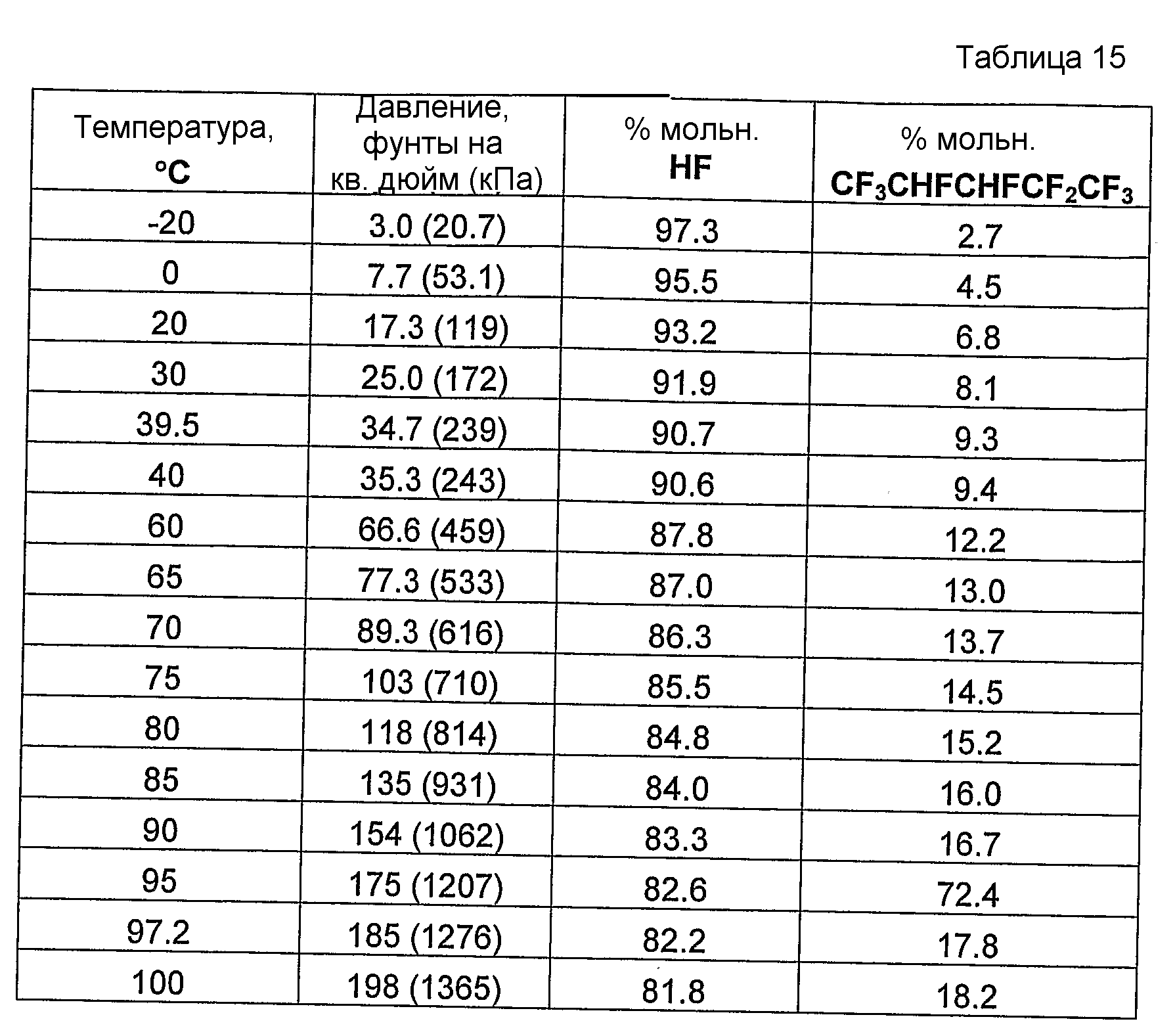
Пример 14
Исследование фазового состава смесей HF и CF3CF=CH2
Исследование фазового состава проводят для композиции, которая состоит практически из CF3CF=CH2 и HF, при этом состав композиции варьируют и измеряют давление паров как при температуре 9,3°С, так и при температуре 44,4°С. На основании результатов, полученных при проведении исследования фазового состава, рассчитывают составы азеотропных композиций при других температурах и давлениях.
В таблице 16 собраны экспериментальные и расчетные азеотропные композиции для HF и CF3CF=CH2 при указанных температурах и давлениях.
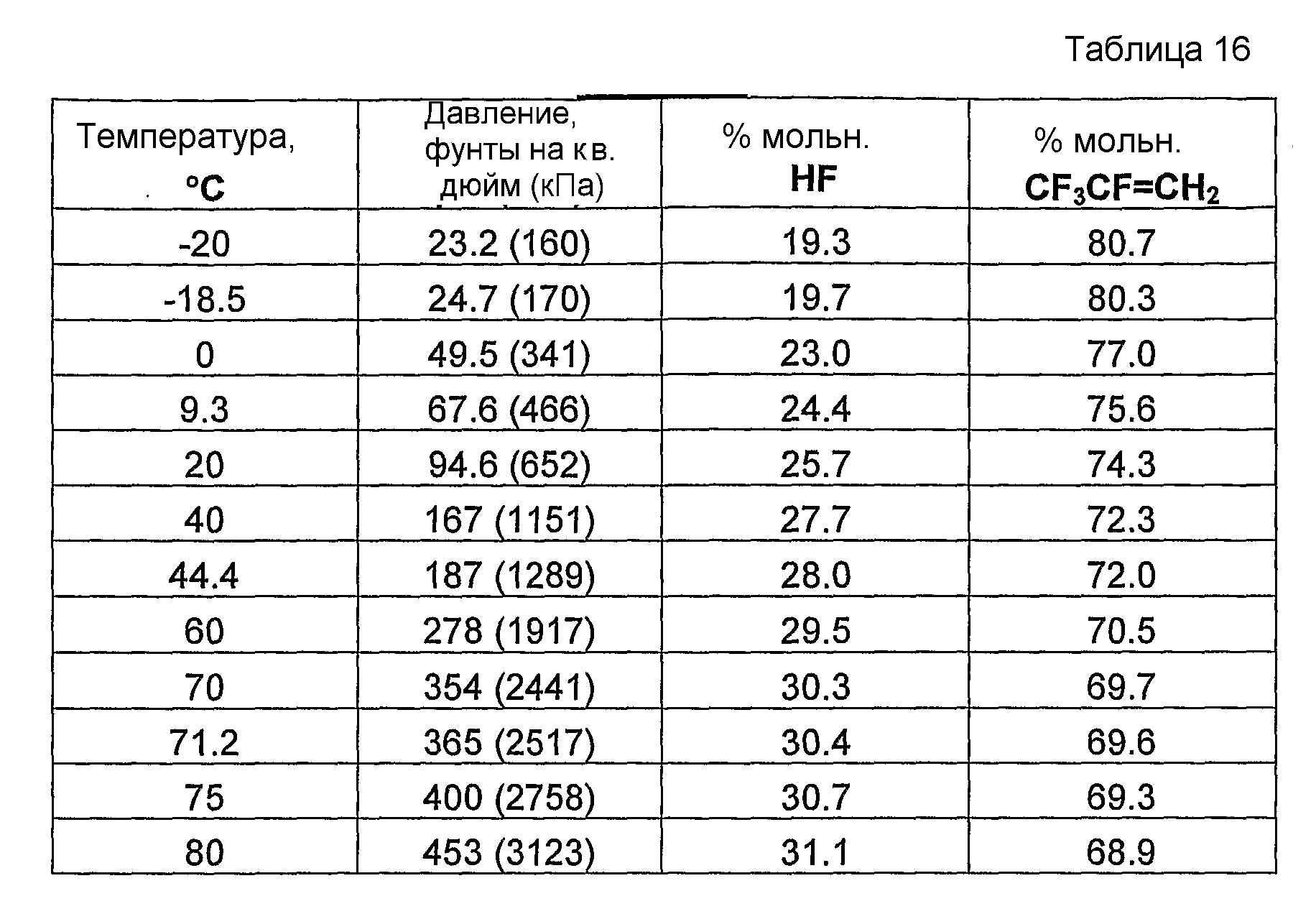
Пример 15
Исследование фазового состава смесей HF и CF3CH=CF2
Исследование фазового состава проводят для композиции, которая состоит практически из CF3CH=CF2 и HF, при этом состав композиции варьируют и измеряют давление паров как при температуре 0,3°С, так и при температуре 50,1°С. На основании результатов, полученных при проведении исследования фазового состава, рассчитывают составы азеотропных композиций при других температурах и давлениях.
В таблице 17 собраны экспериментальные и расчетные азеотропные композиции для HF и CF3CH=CF2 при указанных температурах и давлениях.
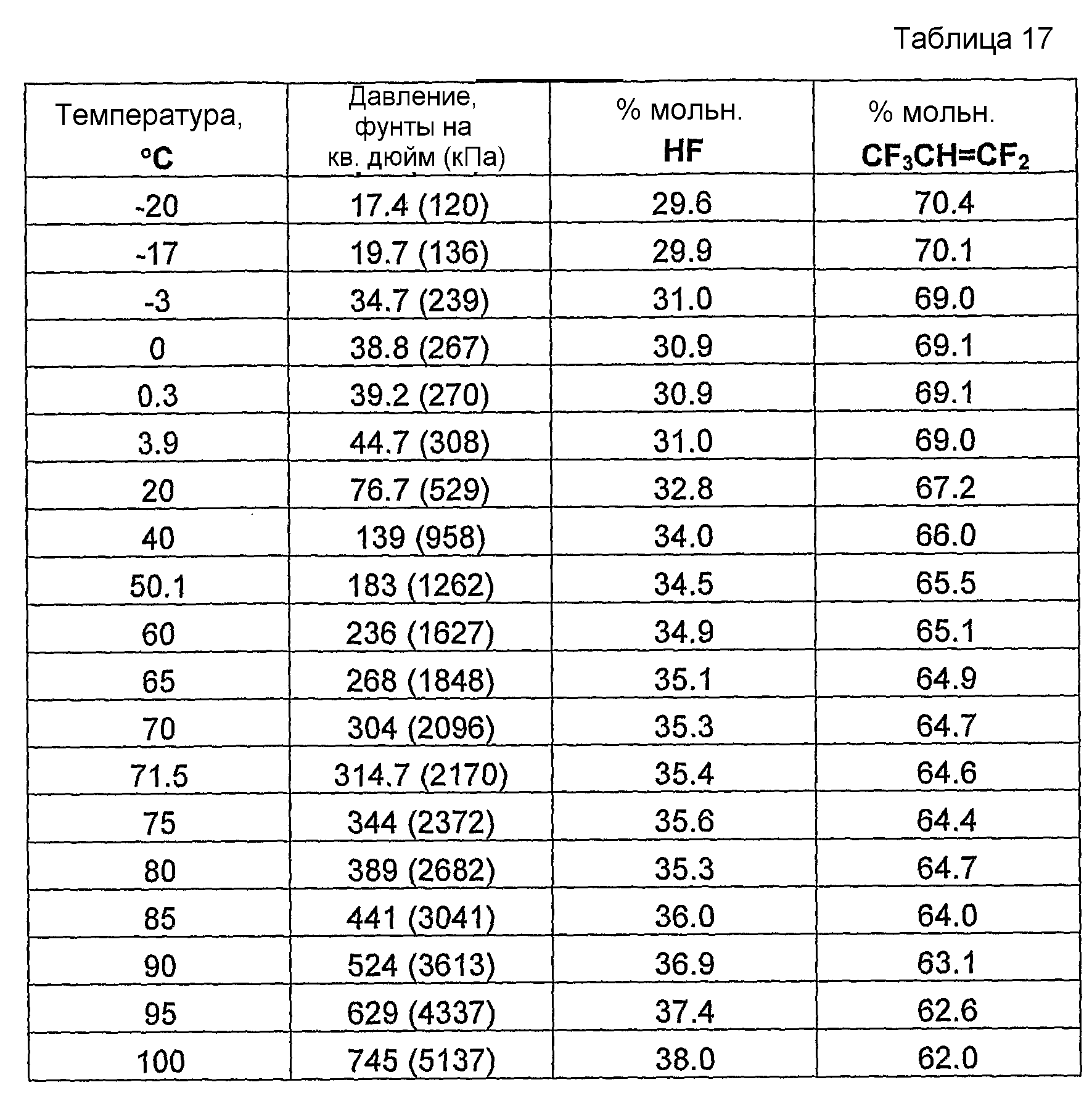
Пример 16
Исследование фазового состава смесей HF и смеси Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5
Исследование фазового состава проводят для композиции, которая состоит практически из HF и смеси Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5, при этом состав композиции варьируют и измеряют давление паров как при температуре 19,9°С, так и при температуре 69,6°С. На основании результатов, полученных при проведении исследования фазового состава, рассчитывают составы азеотропных композиций при других температурах и давлениях.
В таблице 18 собраны экспериментальные и расчетные азеотропные композиции для HF и смеси Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5 при указанных температурах и давлениях.
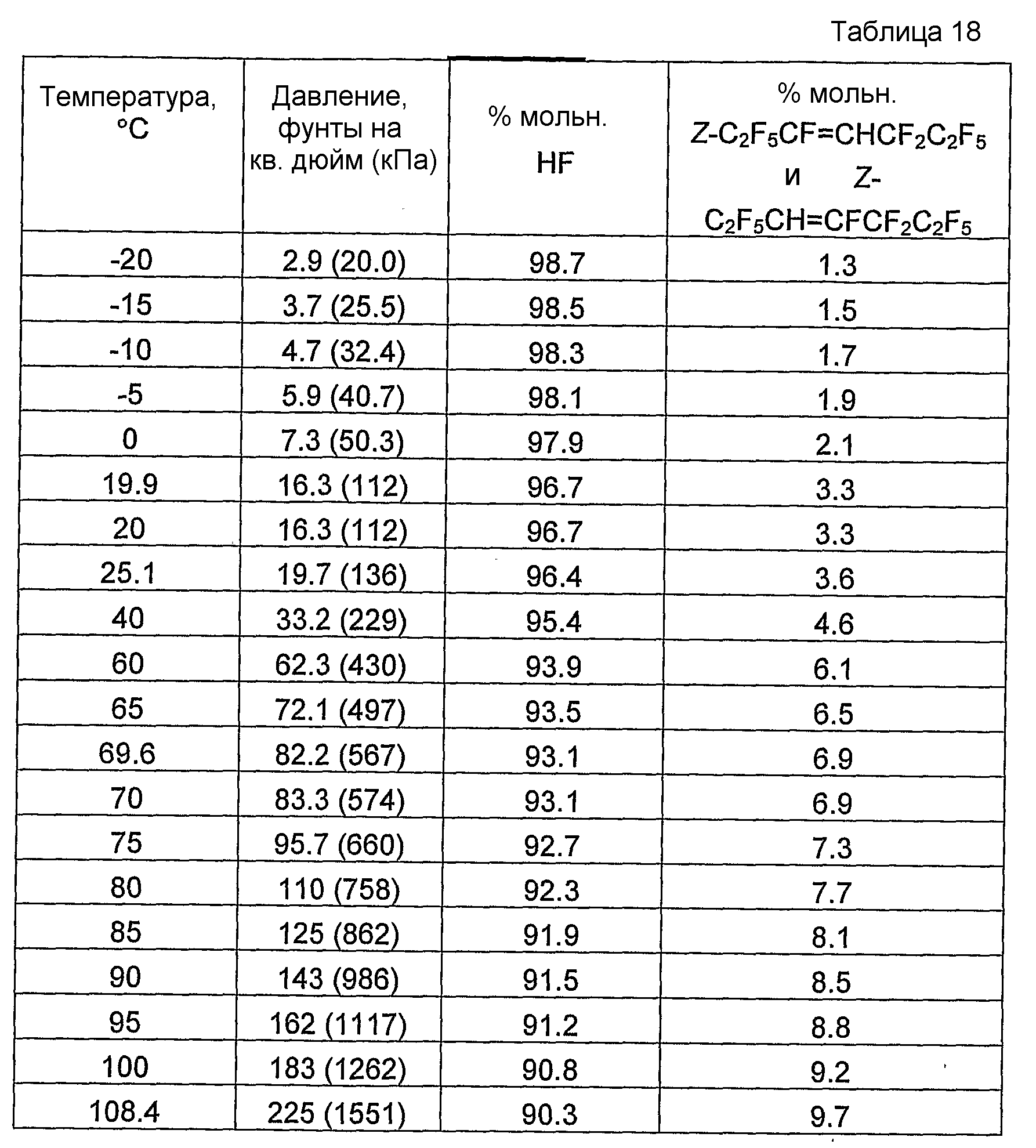
Пример 17
Исследование фазового состава смесей HF и C2F5CHFCHFCF2C2F5
Исследование фазового состава проводят для композиции, которая состоит практически из C2F5CHFCHFCF2C2F5 и HF, при этом состав композиции варьируют и измеряют давление паров как при температуре 30,8°С, так и при температуре 80,2°С. На основании результатов, полученных при проведении исследования фазового состава, рассчитывают составы азеотропных композиций при других температурах и давлениях.
В таблице 19 собраны экспериментальные и расчетные азеотропные композиции для HF и C2F5CHFCHFCF2C2F5 при указанных температурах и давлениях.
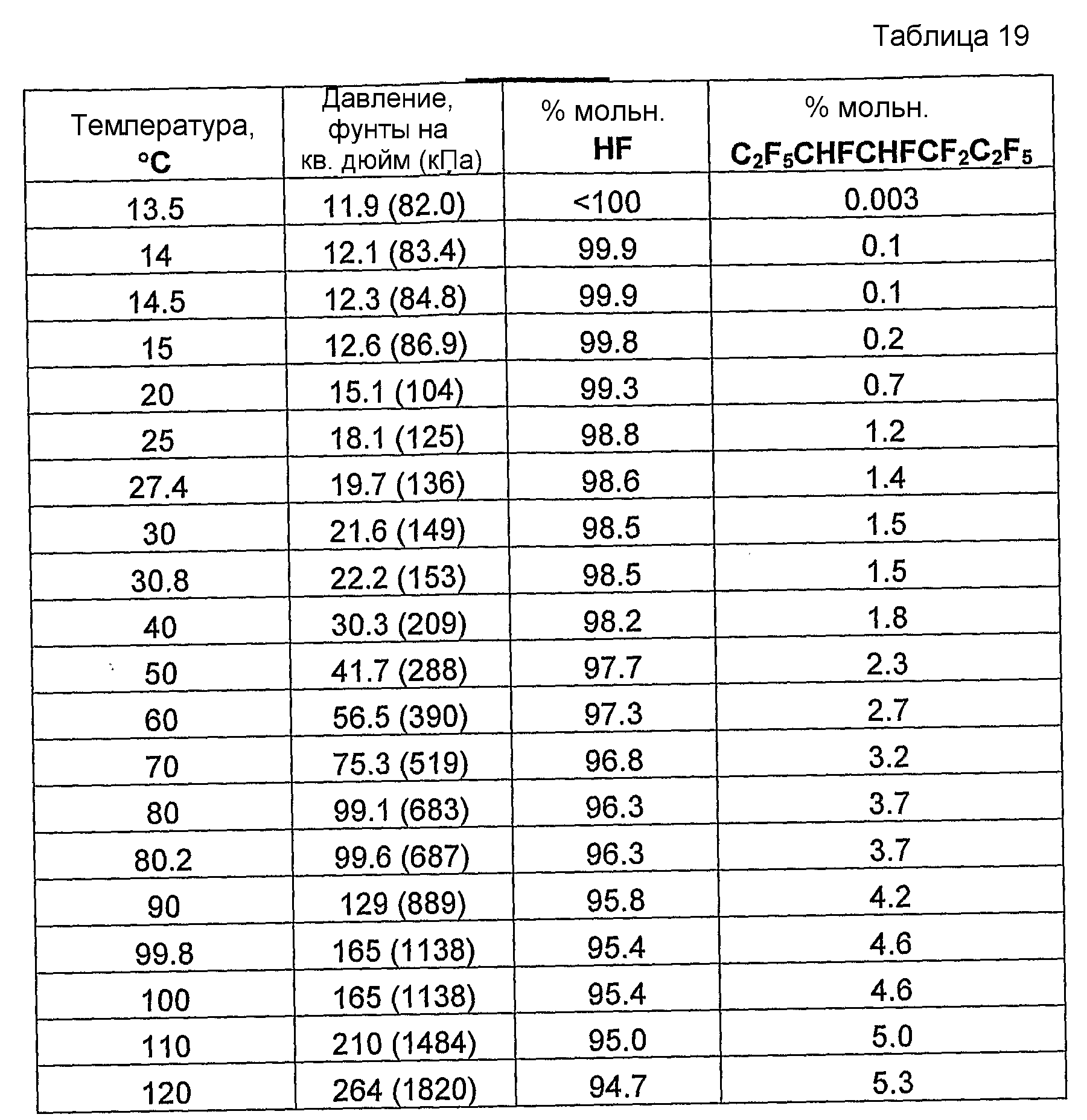
Пример 18
Азеотропная перегонка с целью отделения E-CF3CH=CHF от CF3CH2CHF2
Смесь HF, E-CF3CH=CHF и CF3CH2CHF2 загружают в дистилляционную колонну для очистки E-CF3CH=CHF. Данные в таблице 20 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров.
Пример 19
Азеотропная перегонка с целью отделения Z-CF3CF=CHF от CF3CHFCHF2
Смесь HF, Z-CF3CF=CHF и CF3CHFCHF2 загружают в дистилляционную колонну для очистки Z-CF3CF=CHF. Данные в таблице 21 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров.
Пример 20
Азеотропная перегонка с целью отделения Z-CF3CH=CFCF2CF3 и Z- CF3CF=CHCF2CF3 от CF3CHFCHFCF2CF3
Смесь HF, Z-CF3CH=CFCF2CF3 и Z-CF3CF=CHCF2CF3 и CF3CHFCHFCF2CF3 загружают в дистилляционную колонну для очистки Z-CF3CH=CFCF2CF3 и Z-CF3CF=CHCF2CF3. Данные в таблице 22 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров.
Пример 21
Азеотропная перегонка с целью отделения CF3CF=CH2 от CF3CF2CH3
Смесь HF, CF3CF=CH2 и CF3CF2CH3 загружают в дистилляционную колонну для очистки CF3CF=CH2. Данные в таблице 23 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров.
Пример 22
Азеотропная перегонка с целью отделения CF3CF=CF2 от CF3CH2CF3
Смесь HF, CF3CH=CF2 и CF3CH2CF3 загружают в дистилляционную колонну для очистки CF3CH=CF2. Данные в таблице 24 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров.
Пример 23
Азеотропная перегонка с целью отделения Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5 от C2F5CHFCHFCF2C2F5
Смесь HF, Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5 и C2F5CHFCHFCF2C2F5 загружают в дистилляционную колонну для очистки Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5. Данные в таблице 25 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров.
Пример 24
Азеотропная перегонка в двух колоннах с целью отделения E-CF3CH=CHF от HF
Смесь HF и E-CF3CH=CHF загружают в дистилляционную установку, состоящую из 2 колонн, размещенных последовательно, из которых первая работает под высоким давлением, а вторая работает под низким давлением. Данные в таблице 26 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров. Цифры, приведенные вверху столбцов, относятся к фиг.1.
Пример 25
Азеотропная перегонка в двух колоннах с целью отделения Z-CF3CF=CHF от HF
Смесь HF и Z-HFC-1225ye загружают в процесс дистилляции, с целью очистки Z-HFC-1225ye. Данные в таблице 27 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров. Цифры, приведенные вверху столбцов, относятся к фиг.1.
Пример 26
Азеотропная перегонка в двух колоннах с целью отделения Z-CF3CH=CFCF2CF3 и Z-CF3CF=CHCF2CF3 от HF
Смесь HF и Z-CF3CH=CFCF2CF3 и Z-CF3CF=CHCF2CF3 загружают в процесс дистилляции, с целью очистки Z-CF3CH=CFCF2CF3 и Z-CF3CF=CHCF2CF3. Данные в таблице 28 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров. Цифры, приведенные вверху столбцов, относятся к фиг.1.
Пример 27
Азеотропная перегонка в двух колоннах с целью отделения CF3CF=CH2 от HF
Смесь HF и CF3CF=CH2 загружают в процесс дистилляции с целью очистки CF3CF=CH2. Данные в таблице 29 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров. Цифры, приведенные вверху столбцов, относятся к фиг.1.
Пример 28
Азеотропная перегонка в двух колоннах с целью отделения CF3CH=CF2 от HF
Смесь HF и CF3CH=CF2 загружают в процесс дистилляции с целью очистки CF3CH=CF2. Данные в таблице 30 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров. Цифры, приведенные вверху столбцов, относятся к фиг.1.
Пример 29
Азеотропная перегонка в двух колоннах с целью отделения Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5 от HF
Смесь HF и Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5 загружают в процесс дистилляции с целью очистки Z-C2F5CF=CHCF2C2F5 и Z-C2F5CH=CFCF2C2F5. Данные в таблице 31 получены расчетным путем с использованием замеренных и рассчитанных термодинамических параметров. Цифры, приведенные вверху столбцов, относятся к фигуре 1.
Примеры 30-33
В примерах 30-33 используют один из трех реакторов:
Реактор А: Труба из Inconel® 600 (указанный сплав содержит приблизительно 76 мас.% никеля) с длиной 18 дюймов (45,7 см), внешним диаметром 1,0 дюйм (2,5 см) и внутренним диаметром 0,84 дюйма (2,1 см). Толщина стенки трубы 0,16 дюймов (0,41 см). Длина зоны предварительного нагрева 7 дюймов (17,8 см). Длина зоны реакции 2 дюйма (5,1 см). Длина зоны охлаждения 7 дюймов (17,8 см). Трубу нагревают с помощью ленточных керамических нагревателей диаметром 1 дюйм (2,5 см). Выводы 7-точечной термопары распределены по всей длине трубы, при этом некоторые из них находятся в середине зоны реакции (для измерения температуры газа).
Реактор В: Труба из Nickel 200 типоразмера Schedule 80 с покрытием Inconel® 617; длина реактора 18 дюймов (45,7 см), внешний диаметр 1,5 дюйма (3,8 см) и внутренний диаметр 0,84 дюйма (2,1 см). Длина зоны реакции составляет 2 дюйма (5,1 см). Зона реакции нагревается трубчатым нагревателем длиной 8,5 дюймов (21,6 см) с разрезами 2,5 дюймов (6,35 см). Выводы 7-точечной термопары распределены по всей длине трубы, при этом некоторые из них находятся в середине зоны реакции (для измерения температуры газа).
Реактор С: Hastelloy® С276 с золотым покрытием. Длина составляет 5 дюймов (12,7 см), внешний диаметр 0,50 дюйма (1,3 см), внутренний диаметр 0,35 дюйма (0,89 см). Толщина стенки трубы составляет 0,15 дюймов (3,8 мм). Толщина золотого покрытия составляет 0,03 дюйма (0,08 см). Длина зоны реакции составляет 2 дюйма (5,1 см), и она нагревается керамическим ленточным нагревателем.
Пример 30
Используют реактор А (поверхность в зоне реакции из Inconel® 600). Температура газа на входе в реактор (“Т газа на входе в реактор” в таблице 1) обозначает температуру реакции. Проводят два процесса при температуре реакции 724°С и 725°С, соответственно. В процессе А реагенты вводят, не разбавляя инертным газом. В процессе В гелий и реагенты вводят в соотношении 1,4:1. Польза от использования разбавления инертным газом заметна по повышению выхода в процессе В (80%) по сравнению с процессом А (71%). В процессе В меньше концентрация побочных фторсодержащих углеводородных продуктов. Результаты приведены в таблице 32.
Пример 31
В данном примере при изучении влияния температуры на степень превращения и выход используют реактор А (поверхность в зоне реакции из Inconel® 600). Процесс А проводят при температуре в реакторе 600°С. Процессы В и С проводят при температуре 699°С и 692°С, соответственно. Процессы А и В разбавляют гелием в соотношении 4:1. Процесс С проводят без разбавления. В процессе А (600°С) степень превращения низкая и составляет 0,3%. В процессах В и С (690-700°С) степень превращения выше, однако все еще остается низкой, по сравнению со степенью превращения в процессе по примеру 30, который проводят при 725°С и для значительно большей продолжительности нахождения реагентов в зоне реакции. Приведенные выходы не являются достоверными при столь низких степенях превращения. Из указанных экспериментов становится очевидна зависимость степени превращения от температуры и продолжительности нахождения реагентов в зоне реакции. Результаты собраны в таблице 33.
Пример 32
Используют реактор В (поверхность в зоне реакции из Nickel 200). В указанном реакторе температура реакции представляет собой температуру газа в средней части реактора (“Т газа в средней части реактора” в таблице 3). Процессы А, В и С проводят при температуре 800°С с постоянными отношениями гелий: реагент, равными 0:1, 1:1 и 2:1, соответственно. При указанной температуре, которая больше, чем температура в примере 30, и при сопоставимой продолжительности нахождения реагентов в зоне реакции на никелевой поверхности степень превращения высока и выходы выше. При проведении пиролиза более высокая температура обычно приводит к более низким выходам, поскольку возрастают скорости протекания нежелательных побочных реакций, дающих ненужные продукты. Их отсутствие в примере 32 свидетельствует о превосходстве никелевой поверхности в зоне реакции над поверхностью в зоне реакции из сплава никеля по примеру 30. Дальнейшим подтверждением этого вывода является процесс D, который проводят при температуре 850°С и при разбавлении гелием 4:1. Степень превращения высока и составляет 76,9%, а выход составляет 90,5%, что является лучшим из всех процессов по примеру 32. Результаты собраны в таблице 34.
Пример 33
Используют реактор C (поверхность в зоне реакции из золота). Подобно никелю поверхность из золота дает высокие выходы и, таким образом, уменьшает побочные реакции, дающие ненужные продукты. Влияние газа-разбавителя (уменьшение концентрации) на степень превращения не столь заметно, как на поверхностях из никеля или сплава никеля. При температуре 800°С (процессы А и В) степень превращения ниже, чем в процессах В и С по примеру 32, однако средний выход выше. Результаты представлены в таблице 35.
В примерах 30-33 приведены конкретные условия проведения пиролиза по настоящему изобретению, дающие с высоким выходом продукт CF3CH=CF2 с хорошей степенью превращения и лишь небольшим количеством ненужных побочных продуктов. Никель обладает преимуществом над сплавом никеля, поскольку позволяет получить продукт с бóльшим выходом. Золото обладает преимуществом над никелем.
Степень превращения низка при температуре приблизительно 700°С, становится хорошей при температуре 725°С и выше, не ухудшаясь вплоть до температуры 850°С.
Реферат
В изобретении раскрывается способ получения азеотропной композиции, содержащей фторгидроолефин, имеющий от 3 до 8 атомов углерода, дегидрофторированием фторсодержащего углеводорода. Раскрываются также способы отделения фторгидроолефинов от фторсодержащих углеводородов и от фторида водорода. Настоящее изобретение позволяет получать фторгидроолефины без использования катализатора. 3 н. и 6 з.п. ф-лы, 33 пр., 35 табл., 2 ил.
Формула
CF3CF2CH2F и CF3CF=CHF;
CHF2CF2CH2F и CHF2CF=CHF;
CHF2CHFCHF2 и CHF2CF=CHF;
CH2FCF2CH2F и CH2FCF=CHF;
CH3CF2CHF2 и CHF2CF=CH2;
CHF2CHFCH2F и CHF2CF=CH2;
CF3CHFCH3 и CF3CH=СН2;
CF3CH2CH2F и CF3CH=СН2;
CHF2CH2CHF2 и CHF2CH=CHF;
CH2FCF2CF2CF3 и CHF=CFCF2CF3;
CF3CHFCF2CHF2 и CF3CF=CFCHF2;
CF3CH2CF2CF3 и CF3CH=CFCF3;
CF3CHFCHFCF3 и CF3CH=CFCF3;
CH3CF2CF2CF3 и CH2=CFCF2CF3;
CF3CH2CH2CF3 и CF2=CHCH2CF3;
CF3CHFCF2CH2F и CF3CF=CFCH2F;
CH2FCF2CF2CHF2 и CHF=CFCF2CHF2;
CHF2CH2CF2CF3 и CHF2CH=CFCF3;
CHF2CF2CH2CF3 и CHF2CF=CHCF3;
CHF2CHFCHFCF3 и CHF2CF=CHCF3, CHF2CH=CFCF3 или их смеси;
CHF2CHFCF2CHF2 и CHF2CF=CFCHF2;
CF3CH2CHFCF3 и CF3CH=CHCF3;
CF3CHFCF2CH3 и CF3CF=CFCH3;
CF3CF2CHFCH3 и CF3CF=CFCH3;
CH3CF2CF2CHF2 и CH2=CFCF2CHF2;
CF3CF2CH2CH2F и CF3CF=CHCH2F, CF3CF2CH=CH2 или их смеси;
CH2FCHFCHFCF3 и CH2FCF=CHCF3, CH2FCH=CFCF3 или их смеси;
CHF2CF2CHFCH2F и CHF2CF=CFCH2F, CHF2CF2CF=CH2 или их смеси;
CF3CH2CF2CH2F и CF3CH=CFCH2F;
CHF2CH2CHFCF3 и CHF2CH=CHCF3;
CHF2CHFCH2CF3 и CHF2CH=CHCF3;
CHF2CH2CF2CHF2 и CHF2CH=CFCHF2;
CHF2CHFCHFCHF2 и CHF2CH=CFCHF2;
CHF2CH(CF3)2 и CHF=C(CF3)2;
CH2FCF(CF3)2 и CHF=С(CF3)2;
CF3CF2CH2CF2CF3 и CF3CF2CH=CFCF3;
CF3CH2CF2CF2CF3 и CF3CH=CFCF2CF3;
CF3CH2CHFCF2CF3 и CF3CH=CHCF2CF3;
CF3CHFCH2CF2CF3 и CF3CH=CHCF2CF3;
CH3CF2CF2CF2CHF2 и CH2=CFCF2CF2CHF2;
CF3CH2CF2CH2CF3;
CF3CF2CHFCHFCF2CF3 и CF3CF2CH=CFCF2CF3;
CH2FCHFCF2CF2CF2CF3 и CH2=CFCF2CF2CF2CF3;
CF3CH2CHFCF2CF2CF3 и CF3CH=CHCF2CF2CF3;
CF3CHFCH2CF2CF2CF3 и CF3CH=CHCF2CF2CF3;
CF3CH2CF2CF2CH2CF3 и CF3CH=CFCF2CH2CF3;
CF3CF2CH2CH2CF2CF3 и CF3CF=CHCH2CF2CF3;
CF3CHFCHFCF2CF2C2F5 и CF3CF=CHCF2CF2C2F5,
или их смеси;
C2F5CHFCHFCF2CF2C2F5 и C2F5CF=CHCF2CF2C2F5,
C2F5CH=CFCF2CF2C2F5 или их смеси;
C2F5CF2CHFCHFCF2C2F5 и C2F5CF2CF=CHCF2C2F5;
цикло-CF2CF2CF2CH2- и цикло-CF2CF2CF=CH-;
цикло-CF2CF2CHFCHF- и цикло-CF2CF2CF=CH-;
цикло-CF2CF2CH2CH2- и цикло-CF2CH2CH=CF-;
цикло-CF2CF2CF2CHFCHF- и цикло-CF2CF2CF2CF=СН-;
цикло-CF2CF2CF2CF2CHFCHF- и цикло-CF2CF2CF2CF2CF=СН-;
CF3CF2CH2CH3 и CF3CF=СНСН3;
CF3CH2CF2CH3 и CF3CH=CFCH3;
CH2FCH2CHFCF3 и CH2FCH=HCF3;
CH2FCHFCH2CF3 и CH2FCH=CHCF3;
CH3CF(CF3)2 и СН2=С(CF3)2;
CH2FCH(CF3)2 и СН2=С(CF3)2;
CHF2CF(CHF2)2;
CHF2CH(CHF2)2;
CH3CF(CHF2)2 и CH2=C(CHF2)2;
CH2FCH(CH2F)CF3;
CH2FCH(CHF2)2 и CH2=C(CHF2)2;
CH3CF2CF2CF2CF3;
CHF2CF2CF2CHFCH3;
CF3CF2CF2CH2CH3;
(CF3)2CFCH2CF3 и (CF3)2С=CHCF3;
(CF3)2CHCHFCF3 и (CF3)2С=CHCF3;
(CF3)2CFCHFCHF2;
CF3CF2CH2CHFCF2CF3 и CF3CF2CH=CHCF2CF3;
CH3CH2CF2CF2CF2CF3 и CH3CH=CFCF2CF2CF3;
CF3CF2CF2CH2CHFCH3 и CF3CF2CF2CH=СНСН3;
(CF3)2CFCF2CH2CH3;
(CF3)2CFCH2CH2CH3 и (CF3)2С=СНСН2СН3;
(CF3)2CHCHFCF2CF3 и (CF3)2C=CHCF2CF3;
(CF3)2CFCHFCHFCF3 и (CF3)2С=CFCHFCF3, (CF3)2CFCF=CHCF3,
(CF3)2CFCH=CFCF3 или их смеси;
(CF3)2CHCH2CF2CF3 и (CF3)2CHCH=CFCF3;
(CF3)2CFCH(СН3)2;
(СН3)2CHCF2CF2CF3;
C2F5CHFCH2CF2C2F5;
C2F5CH2CHFCF2C2F5;
CF3CHFCH2CF2CF2C2F5;
CF3CH2CHFCF2CF2C2F5;
CF3CF2CF2CF2CF2CH2CH3;
CF3CF2CF2CF2CH2CH2CH3 и
(CH3)2CHCF2CF2CF2CF3.
a) образование смеси, включающей фторгидроолефин, фторсодержащий углеводород и фторид водорода;
b) стадию дистилляции, которой подвергают указанную смесь, с образованием в колонне композиции дистиллята, представляющей собой азеотропную или близкую к азеотропной композицию фторида водорода и фторгидроолефина, в которой практически отсутствует указанный фторсодержащий углеводород.
a. CHF2CF=CHCF3, CHF2CH=CFCF3 и их смеси;
b. CF3CF=CHCH2F, CF3CF2CH=CH2 и их смеси;
c. CH2FCF=CHCF3, CH2FCH=CFCF3 и их смеси;
d. CHF2CF=CFCH2F, CHF2CF2CF=CH2 и их смеси;
е. C2F5CF=CHCF2CF2C2F5, C2F5CH=CFCF2CF2C2F5 и их смеси.
a) стадию первой дистилляции, на которой выделяют либо (i) фторид водорода/фторгидроолефин в качестве дистиллята, а из нижней части колонны выделяют чистый фторгидроолефин, либо (ii) фторид водорода/фторгидроолефин в качестве дистиллята, а из нижней части колонны выделяют чистый фторид водорода; и
b) вторую стадию дистилляции, на которой осуществляют воздействие на первую композицию дистиллята со стадии (а) при другом давлении, при которой для (i) в качестве дистиллята выделяют фторгидроолефин, по существу, не содержащий фторид водорода, а из нижней части колонны выделяют фторид водорода, по существу, не содержащий фторгидроолефин, либо для (ii) в качестве дистиллята выделяют фторид водорода, по существу, не содержащий фторгидроолефин, а из нижней части колонны выделяют фторгидроолефин, по существу, не содержащий фторид водорода.
Документы, цитированные в отчёте о поиске
Способ получения фторолефинов
Комментарии