Способ получения акролеина, или акриловой кислоты, или их смеси из пропана - RU2391330C9
Код документа: RU2391330C9
Описание
Настоящее изобретение относится к способу получения акролеина или акриловой кислоты или их смеси из пропана, в соответствии с которым
A) в первую реакционную зону А вводят по меньшей мере два содержащих пропан газообразных питающих потока, по меньшей мере один из которых содержит свежий пропан, и введенный в эту реакционную зону пропан подвергают гетерогенно катализируемому дегидрированию, получая содержащую пропан и пропилен газообразную смесь продуктов А,
B) которую выводят из реакционной зоны А, в первой зоне разделения А выделяют из нее по меньшей мере часть содержащихся в ней компонентов, отличающихся от пропана и пропилена, а остающуюся при этом газообразную смесь продуктов А', содержащую пропан и пропилен,
C) используют во второй реакционной зоне В для питания по меньшей мере одного реактора окисления, и пропилен, содержащийся в газообразной смеси продуктов А′, по меньшей мере в одном реакторе окисления подвергают (селективному) гетерогенно катализируемому газофазному частичному окислению молекулярным кислородом в акролеин, акриловую кислоту или их смесь в качестве целевого продукта, а также в содержащую избыток молекулярного кислорода газообразную смесь продуктов В,
D) которую выводят из реакционной зоны В, во второй зоне разделения В выделяют содержащийся в ней целевой продукт, и по меньшей мере часть остающегося после этого остаточного газа, содержащего непревращенный пропан, молекулярный кислород, а также при необходимости непревращенный пропилен, рециркулируют в реакционную зону А в качестве по меньшей мере одного из двух содержащих пропан питающих потоков.
Акриловая кислота является важным основным химикатом, используемым, в частности, в качестве мономера для синтеза полимерных продуктов, которые, например, в виде дисперсий в водной среде применяют в качестве связующих. Акролеин является важнейшим промежуточным продуктом, используемым, например, для синтеза глутарового альдегида, метионина, фолиевой кислоты и акриловой кислоты.
Указанный в начале описания способ получения акролеина или акриловой кислоты или их смеси из пропана известен, например, из международной заявки WO 01/96270, патента США US 2003/0187299 А1, а также из немецких заявок на патент DE-A 10245585 и DE-A 10246119 и цитируемого в них уровня техники.
При этом методы выделения целевого продукта, содержащегося в газообразной смеси продуктов В, отличаются тем, что его переводят из газообразной фазы в конденсированную фазу, например, благодаря осуществлению абсорбции и/или конденсации. При этом в качестве абсорбентов пригодны, например, вода, водный раствор и/или органический растворитель. При подобной «конденсации» целевого продукта остаточный газ, содержащий сравнительно трудно конденсирующиеся компоненты газообразной смеси продуктов В, обычно не переходит в конденсированную фазу. Такими компонентами прежде всего обычно являются продукты с температурой кипения при нормальном давлении (1 бар), составляющей ≤-30°С (их общее содержание в остаточном газе, как правило, составляет ≥70% об., часто ≥80% об. и чаще всего ≥90% об.). К ним в первую очередь относятся непревращенный пропан, остающийся в реакционной зоне В избыточный молекулярный кислород, а также при необходимости непревращенный пропилен. Остаточный газ, как правило, дополнительно содержит инертные разбавляющие газы, например, такие как азот, диоксид углерода, благородные газы (гелий, неон, аргон и так далее), монооксид углерода, а также незначительные количества акриловой кислоты, акролеина и/или воды (содержание водяного пара в остаточном газе может составлять до 25% об., часто до 20% об. или до 10% об., а чаще всего также менее 10% об. или менее 5% об.). Подобный остаточный газ (в пересчете на содержащейся в нем пропан) образует основное количество (обычно по меньшей мере 80%, соответственно по меньшей мере 90% или по меньшей мере 95% или более) получаемого в разделительной зоне В остаточного газа, в связи с чем в настоящем описании его называют также, в частности, основным остаточным газом.
Прежде всего в том случае, если конденсацию целевого продукта осуществляют его абсорбцией органическим растворителем, в разделительной зоне В, как правило, образуется по меньшей мере один второй остаточный газ, содержащий непревращенный пропан, а также при необходимости непревращенный пропилен (количество второго остаточного газа в пересчете на содержащийся в нем пропан обычно значительно меньше количества основного остаточного газа). Образование второго остаточного газа обусловлено тем, что возникающая при абсорбции конденсированная фаза поглощает также определенное количество непревращенного пропана и при необходимости непревращенного пропилена. При последующем выделении целевого продукта из конденсированной фазы экстракцией, дистилляцией, кристаллизацией и/или десорбцией этот непревращенный пропан, а также при необходимости пропилен, обычно регенерируют в качестве компонента по меньшей мере другой газовой фазы и предпочтительно рециркулируют в реакционную зону А. Рециркуляцию можно осуществлять, например, в смеси с основным остаточным газом, которую в настоящем описании называют общим остаточным газом. Однако возможна также рециркуляция в реакционную зону А соответствующих индивидуальных газовых потоков. Подобные газовые потоки могут не содержать кислород или могут содержать его (могут быть побочным остаточным газом) (например, если кислород образуется вследствие отпаривания воздухом или продувки головной части ректификационной колонны воздухом, используемым в качестве ингибитора полимеризации).
Согласно настоящему изобретению основной остаточный газ, общий остаточный газ и побочный остаточный газ образуют рециркулируемый в реакционную зону А остаточный газ, содержащий непревращенный пропан, молекулярный кислород и при необходимости непревращенный пропилен. Образующийся в зоне разделения В остальной газ, содержащий непревращенный пропан, а также при необходимости непревращенный пропилен, но не содержащий молекулярного кислорода, согласно изобретению можно рециркулировать в реакционную зону А в смеси с основным остаточным газом и/или побочным остаточным газом (то есть, например, в качестве составной части общего остаточного газа) и/или также самостоятельно (в данном случае речь идет не о том остаточном газе, который рециркулируют в реакционную зону А в соответствии с изобретением). В последнем случае подобную рециркуляцию можно осуществлять без каких-либо ограничений, то есть, например, также в качестве составной части исходной реакционной газовой смеси реакционной зоны А. Согласно предлагаемому в изобретении способу в реакционную зону А предпочтительно рециркулируют все образующиеся в зоне разделения В газовые потоки, содержащие непревращенный пропан, а также при необходимости непревращенный пропилен. Однако, как более подробно поясняется ниже, часть этих газовых потоков при необходимости можно использовать также для других целей, например, для производства энергии, получения синтез-газа и/или в качестве разбавляющего газа в реакционной зоне В.
Согласно рассмотренному выше уровню техники рециркулируемый в реакционную зону А остаточный газ, содержащий непревращенный пропан, молекулярный кислород, а также при необходимости непревращенный пропилен, следует вводить в то же место реакционной зоны А, в которое в нее вводят другие, содержащие пропан питающие потоки (то есть в качестве составной части исходной реакционной газовой смеси; смотри, например, фиг.6 в заявке на патент США US-2003/0187299 А1). Недостаток подобной технологии состоит в том, что присутствие в остаточном газе молекулярного кислорода обусловливает снижение селективности образования пропилена в реакционной зоне А и повышение селективности образования побочных продуктов (монооксида и диоксида углерода, образующихся при сгорании части пропана и/или пропилена). Прежде всего это происходит в том случае, если подлежащим рециркуляции остаточным газом является основной остаточный газ.
В немецкой заявке на патент DE-A 10211275 указанный выше недостаток пытаются устранить, используя технологию, подобную описанной выше, но при этом разделяя газообразную смесь продуктов А, образующуюся в реакционной зоне А, на две части одинакового состава, одну из которых рециркулируют в реакционную зону А в качестве источника водорода. Однако недостаток предлагаемой в указанной публикации технологии состоит в том, что она не позволяет обеспечить достаточно низкий темп деактивирования катализаторов дегидрирования.
Учитывая вышеизложенное, в основу настоящего изобретения была положена задача предложить способ получения акролеина или акриловой кислоты или их смеси из пропана, улучшенный по сравнению с рассмотренным выше уровнем техники.
Указанная задача решается благодаря способу получения акролеина или акриловой кислоты или их смеси из пропана, в соответствии с которым
A) в первую реакционную зону А вводят по меньшей мере два содержащих пропан газообразных питающих потока, по меньшей мере один из которых содержит свежий пропан, и введенный в эту реакционную зону пропан подвергают гетерогенно катализируемому дегидрированию, получая содержащую пропан и пропилен газообразную смесь продуктов А (она, как правило, содержит также молекулярный водород),
B) которую выводят из реакционной зоны А, в первой зоне разделения А выделяют из нее по меньшей мере часть содержащихся в ней компонентов, отличающихся от пропана и пропилена, а остающуюся при этом газообразную смесь продуктов А′, содержащую пропан и пропилен,
C) используют во второй реакционной зоне В для питания по меньшей мере одного реактора окисления, и пропилен, содержащийся в газообразной смеси продуктов А′, по меньшей мере в одном реакторе окисления подвергают (селективному) гетерогенно катализируемому газофазному частичному окислению молекулярным кислородом в акролеин или акриловую кислоту или их смесь в качестве целевого продукта, а также в содержащую избыток молекулярного кислорода газообразную смесь продуктов В,
D) которую выводят из реакционной зоны В, во второй зоне разделения В выделяют содержащийся в ней целевой продукт, и по меньшей мере часть (основного и/или побочного, соответственно общего) остающегося после этого остаточного газа, содержащего непревращенный пропан, молекулярный кислород и при необходимости непревращенный пропилен (предпочтительно по меньшей мере 50% об. или по меньшей мере 75% об., еще более предпочтительно все количество в соответствующем индивидуальном пересчете на общий остаточный газ, основной остаточный газ и/или побочный остаточный газ) рециркулируют в реакционную зону А в качестве по меньшей мере одного из двух содержащих пропан питающих потоков,
отличающийся тем, что указанную рециркуляцию в реакционную зону А осуществляют вдоль пути гетерогенно катализируемого дегидрирования пропана в этой реакционной зоне таким образом, чтобы в месте ввода рециркулируемого газа в реакционной зоне А уже было подвергнуто дегидрированию по меньшей мере 5% мол. пропана (в пересчете на однократное пропускание через реакционную зону А), (суммарно) введенного (перед этим) в эту реакционную зону с другими питающими потоками.
В случае, если согласно предлагаемому в изобретении способу в реакционную зону А рециркулируют не весь образующийся в зоне разделения В (основной и/или побочный, соответственно общий) остаточный газ, остальной газ, как указано выше, можно использовать, например, для производства энергии, получения синтез-газа и/или в качестве разбавляющего газа в реакционной зоне В. Согласно изобретению в реакционную зону А, как правило, рециркулируют по меньшей мере половину или две трети (то есть 50 или 66,6% об.), предпочтительно по меньшей мере три четверти и еще более предпочтительно весь указанный выше образующийся в зоне разделения В остаточный газ (в соответствующем индивидуальном пересчете на основной и/или побочный, соответственно общий остаточный газ). В случае, если в зоне разделения В образуется только один поток остаточного газа, содержащий непревращенный пропан, молекулярный кислород и непревращенный пропилен (чаще всего происходит именно так), то согласно изобретению этот поток предпочтительно полностью рециркулируют в реакционную зону А (при необходимости за вычетом части газа идентичного состава, направляемого в качестве разбавляющего газа в реакционную зону В). Однако этот поток можно разделить также на две части одинакового состава и, как указано выше, рециркулировать в реакционную зону А только одну его часть, а другую часть использовать иным образом.
В случае, если в зоне разделения В образуется более одного подобного потока остаточного газа, то согласно изобретению эти потоки (как указано выше) можно рециркулировать в реакционную зону А совместно (например, предварительно объединив) или лишь по отдельности. Указанные в настоящем описании процентные данные, касающиеся рециркуляции остаточного газа, относятся прежде всего ко всему этому газу (то есть к сумме всех потоков остаточного газа). Как указано выше, (прежде всего основной) остаточный газ обычно содержит ≥70% об., часто ≥80% об. и чаще всего ≥90% мол., наиболее часто ≥95% мол. или ≥98% мол. компонентов, температура кипения которых при нормальном давлении (1 бар) составляет ≤-30°C. Согласно изобретению рециркуляцию образующегося в зоне разделения В содержащего молекулярный кислород (основного и/или побочного, соответственно общего) остаточного газа в реакционную зону А вдоль пути каталитического дегидрирования, очевидно, можно осуществлять, вводя его не только в одном единственном месте, но и в нескольких последовательно расположенных местах этой реакционной зоны.
Под свежим пропаном в настоящем изобретении подразумевают еще не пропущенный через реакционную зону А пропан. Речь при этом идет, как правило, о сыром пропане (предпочтительно удовлетворяющем требованиям спецификаций, приведенных в немецких заявках на патент DE-А 10246119 и DE-A 10245585), который содержит также незначительные количества компонентов, отличающихся от пропана.
Под путем реакции в реакционной зоне А в настоящем описании подразумевают путь течения пропана, вводимого в эту зону с потоками, отличающимися от потока остаточного газа из зоны разделения В, в зависимости от конверсии этого пропана при дегидрировании (степени его превращения при гетерогенно катализируемом дегидрировании).
Согласно настоящему изобретению вводимая в реакционную зону А исходная реакционная газовая смесь (питающая газовая смесь) должна являться суммой всех газов, вводимых в реакционную зону А в точке пути реакции, расположенной на одинаковом уровне с местом ввода свежего пропана.
Согласно настоящему изобретению рециркуляцию (основного и/или побочного, соответственно общего) остаточного газа в реакционную зону А вдоль пути реакции в этой реакционной зоне предпочтительно осуществляют таким образом, чтобы в месте ввода уже было подвергнуто дегидрированию по меньшей мере 10% мол. или по меньшей мере 15% мол., предпочтительно по меньшей мере 20% мол. или по меньшей мере 25% мол., еще более предпочтительно по меньшей мере 30% мол. или по меньшей мере 35% мол., лучше всего по меньшей мере 35% мол. или по меньшей мере 40% мол. пропана, (суммарно) введенного (перед этим) в реакционную зону А вместе с другими питающими потоками (конверсия ZU). Согласно предлагаемому в изобретении способу в месте ввода рециркулируемого (основного и/или побочного, соответственно общего) остаточного газа в реакционную зону А в ней, как правило, уже подвергнуто дегидрированию менее 70% мол., часто менее 60% мол. и чаще всего ≤50% мол. пропана, введенного в эту зону с другими питающими потоками (конверсия ZU).
При осуществлении предлагаемого в изобретении способа так, как рекомендовано в немецкой заявке на патент DE-A 10211275, то есть при разделении образующейся в реакционной зоне А газообразной смеси продуктов А на две части одинакового состава и рециркуляции одной из них в реакционную зону А (прежде всего в том случае, если эту часть рециркулируют в качестве составляющей исходной реакционной газовой смеси, вводимой в реакционную зону А), вместо всех указанных выше параметров конверсии ZU справедливыми предпочтительно будут параметры конверсии ZUKr, определяемые уравнением:

В этом уравнении KGV означает отношение рециркулируемой в реакционную зону А части газообразной смеси продуктов А ко всему количеству образующейся в реакционной зоне А газообразной смеси продуктов А.
Согласно предлагаемому в изобретении способу содержание молекулярного кислорода в образующемся в зоне разделения В, рециркулируемом в реакционную зону А (основном и/или побочном, соответственно общем) остаточном газе обычно составляет от 0,5 до 10% об., часто от 1 до 8% об. и чаще всего от 2 до 5% об. Присутствие кислорода в этом остаточном газе обычно обусловлено прежде всего тем, что избыток молекулярного кислорода в реакционной зоне В (по сравнению с необходимым для осуществления целевой реакции стехиометрическим количеством кислорода), как правило, благоприятно отражается на долговечности катализаторов окисления и кинетике протекающего на них селективного гетерогенно катализируемого газофазного частичного окисления пропилена в акролеин или акриловую кислоту или их смесь. В отличие от реакционных условий в предлагаемой в изобретении реакционной зоне А молярное отношение реагентов в реакционной зоне В преимущественно не оказывает влияния на соответствующую термодинамику, поскольку селективное гетерогенно катализируемое газофазное частичное окисление пропилена в акролеин или акриловую кислоту или их смесь является кинетически контролируемой реакцией.
Согласно предлагаемому в изобретении способу отношение количества пропана, вводимого в реакционную зону А с рециркулируемым из зоны разделения В (основным и/или побочным, соответственно общим) остаточным газом, к суммарному количеству пропана, вводимого в реакционную зону А с другими содержащими пропан потоками, как правило, составляет от 0,1 до 10 или от 0,5 до 5, предпочтительно от 0,5 до 1,5 или от 3 до 5.
В одном из вариантов осуществления предлагаемого в изобретении способа в реакционную зону А кроме рециркулируемого из зоны разделения В, содержащего пропан (основного и/или побочного, соответственно общего) остаточного газа дополнительно вводят лишь сырой пропан (содержащий кроме пропана незначительные количества примесей) в качестве второго питающего потока. Согласно изобретению подобный сырой пропан (пропан, используемый согласно настоящему изобретению в общем случае) предпочтительно соответствует спецификациям, рекомендуемым в описаниях немецких заявок на патент DE-А 10245585 и DE-A 10246119. То же касается и спецификации питающей газовой смеси, используемой согласно предлагаемому в изобретении способу для реакционной зоны В. Однако согласно предлагаемому в изобретении способу в качестве вводимых в реакционную зону А питающих потоков, очевидно, можно совместно использовать также содержащие пропан (а также при необходимости пропилен) потоки вторичного газа, отличающиеся от предлагаемого в настоящем изобретении способа (смотри международную заявку WO 02/00587; однако указанный в разделе с) этой заявки газовый поток можно вводить также только в реакционную зону В).
Для осуществления настоящего изобретения важно лишь, чтобы в месте реакционной зоны А (по высоте), в которой вводят (основной и/или побочный, соответственно общий) остаточный газ из зоны разделения В, уже было продегидрировано по меньшей мере 5% мол. введенного в реакционную зону А со всеми другими питающими потоками пропана, подлежащего каталитическому дегидрированию в этой реакционной зоне. Если указанное условие не выполняется, то в месте ввода остаточного газа уменьшается молярное отношение молекулярного водорода к молекулярному кислороду, что либо способствует полному сгоранию используемых углеводородов с тремя атомами углерода, либо обусловливает сокращение срока службы используемого для дегидрирования катализатора и/или вынуждает вводить в реакционную зону А молекулярный водород из внешнего источника. Подобные последствия, очевидно, обусловлены тем, что как используемые углеводороды с тремя атомами углерода, так и сам катализатор дегидрирования в атмосфере с повышенным молярным отношением молекулярного водорода к молекулярному кислороду лучше защищены от полного сгорания и термической (оказывающей деактивирующее действие) деструкции катализатора дегидрирования.
В отличие от протекающего благодаря присутствию кислорода экзотермического гетерогенно катализируемого окислительного дегидрирования, в процессе которого не происходит образования промежуточного свободного водорода (водород, отщепляемый от подлежащего дегидрированию углеводорода, непосредственно связывается, образуя воду), соответственно присутствия свободного водорода не обнаруживают, под предлагаемым в изобретении гетерогенно катализируемым дегидрированием следует подразумевать («обычное») дегидрирование, которое в отличие от окислительного дегидрирования протекает с эндотермическим тепловым эффектом (в качестве дополнительной стадии предлагаемого в изобретении способа можно предусмотреть экзотермическое сжигание водорода) и при котором образуется по меньшей мере промежуточный свободный молекулярный водород. Для осуществления подобного процесса, как правило, необходимы иные реакционные условия и катализаторы, чем для реализации процессов окислительного дегидрирования.
Таким образом, процесс, осуществляемый согласно предлагаемому в изобретении способу, обычно протекает с выделением водорода, происходящим прежде всего до рециркуляции остаточного газа из зоны разделения В в реакционную зону А, в связи с чем молярное содержание водорода в реакционной газовой смеси в месте ее ввода в реакционную зону А (в пересчете на содержащееся в этой смеси молярное количество пропана), обычно превышает молярное содержания водорода во вводимой в реакционную зону А исходной реакционной газовой смеси.
В соответствии с предлагаемым в изобретении способом последнее обстоятельство предпочтительно относится также к реакционной газовой смеси, которая образуется в месте ввода вследствие соединения рециркулируемого в это место остаточного газа с реакционной газовой смесью, имеющейся перед подобной рециркуляцией в месте ввода.
Кроме того, для предлагаемого в изобретении способа благоприятно, чтобы молярное отношение содержащегося в реакционной газовой смеси пропилена к содержащемуся в ней молекулярному водороду в пределах реакционной зоны А не превышало 10, предпочтительно 5, лучше 3 и еще лучше 2. Особенно предпочтительным является интервал указанных отношений от 0,5, соответственно 1 до 2.
Кроме того, принцип гетерогенно катализируемого дегидрирования в реакционной зоне А, осуществляемого предлагаемым в изобретении способом (а следовательно, и самой реакционной зоны А), можно реализовать аналогично международной заявке WO 01/96270, а также немецким заявкам на патент DE-A 3313573, DE-A 10131297, DE-A 10245585, DE-A 10246119 и DE-A 10211275. Таким образом, реакционную зону А (в пересчете на однократное пропускание вводимой в нее газовой смеси) можно реализовать в изотермическом режиме благодаря целенаправленному теплообмену с пропускаемыми снаружи этой зоны (текучими, то есть жидкими или газообразными) теплоносителями. Однако при одинаковой базе сравнения реакционную зону А можно реализовать также в адиабатическом режиме, то есть преимущественно без указанного выше целенаправленного теплообмена с пропускаемыми снаружи этой зоны теплоносителями. Благодаря рекомендованным в вышеизложенном описании и рассматриваемым ниже техническим мероприятиям адиабатический режим можно реализовать таким образом, чтобы тепловой брутто-эффект (в пересчете на однократное пропускание через реакционную зону А вводимой в нее исходной реакционной газовой смеси) был эндотермическим (отрицательным), автотермическим (преимущественно нулевым) или экзотермическим (положительным). Согласно изобретению для этого требуется лишь осуществление дополнительной рециркуляции остаточного газа из зоны разделения В в реакционную зону А, как предусматривается в изобретении. Катализаторы, рекомендуемые в указанных выше публикациях, можно использовать и для осуществления способа, предлагаемого в настоящем изобретении.
Для реализации типичного варианта гетерогенно катализируемого частичного дегидрирования пропана в пропилен требуются сравнительно высокие реакционные температуры. Достигаемая при этом конверсия пропана обычно бывает ограничена термодинамическим равновесием реакции дегидрирования. Типичные реакционные температуры составляют от 300 до 800°С, соответственно от 400 до 700°С. При этом на молекулу пропана, дегидрированного до пропилена, образуется одна молекула водорода.
Высокие температуры и удаление водорода как продукта реакции благоприятствуют смещению термодинамического равновесия реакции дегидрирования в сторону образования целевого продукта.
Поскольку гетерогенно катализируемая реакция дегидрирования протекает с увеличением объема, конверсию пропана можно повысить благодаря снижению парциального давления реакционных продуктов. Этого можно достичь простым методом, например, осуществляя дегидрирование при пониженном давлении и/или добавляя преимущественно инертные разбавляющие газы, которые в обычном случае инертны по отношению к реакции дегидрирования, например, такие как водяной пар. Разбавление водяным паром в качестве дополнительного преимущества, как правило, обусловливает уменьшение коксования используемого катализатора, поскольку водяной пар реагирует с образующимся коксом в соответствии с принципом газификации угля. Кроме того, водяной пар можно использовать в качестве разбавляющего газа в последующей реакционной зоне В. Вместе с тем водяной пар можно также простым методом (например, конденсацией) частично или полностью выделить из газообразной смеси продуктов дегидрирования (газообразной смеси продуктов А), что при дальнейшем использовании получаемой при этом модифицированной газообразной смеси продуктов (газообразной смеси продуктов А′) в реакционной зоне В позволяет повысить содержание азота, используемого в качестве разбавляющего газа. Кроме того, пригодными для гетерогенно катализируемого дегидрирования пропана разбавителями являются, например, монооксид углерода, метан, этан, диоксид углерода, азот и благородные газы, такие как гелий, неон и аргон. Любые из указанных разбавителей можно использовать как индивидуально, так и в виде смесей самого различного состава. Преимущество применения указанных разбавителей состоит в том, что они, как правило, являются также разбавителями, пригодными для использования в реакционной зоне В. Согласно предлагаемому в изобретении способу предпочтительными в общем случае являются разбавители, характеризующиеся инертным поведением в соответствующей реакционной зоне (то есть химическому изменению подвержено менее 5% мол., предпочтительно менее 3% мол. и еще лучше менее 1% мол. разбавителя).
Для гетерогенно катализируемого дегидрирования пропана в принципе пригодны любые известные из уровня техники катализаторы дегидрирования. Грубо их можно разделить на две группы. Одну из этих групп образуют катализаторы оксидной природы (например, оксид хрома и/или оксид алюминия), в то время как другую группу образуют катализаторы, состоящие по меньшей мере из одного, как правило, сравнительно благородного металла (например, платины), нанесенного, как правило, на оксидный носитель. В соответствии с этим можно использовать, в частности, любые катализаторы дегидрирования, рекомендуемые в международной заявке WO 01/96270, европейской заявке на патент ЕР-А 731077, немецких заявках на патент DE-A 10211275 и DE-A 10131297, международной заявке WO 99/46039, патенте США US-A 4788371, европейской заявке на патент ЕР-А-0705136, международной заявке WO 99/29420, патентах США US-A 4220091, US-A 5430220 и US-A 5877369, европейской заявке на патент ЕР-А-0117146 и немецких заявках на патент DE-A 19937196, DE-A 19937105 и DE-A 19937107. Особенно пригодным является катализатор из примера 1, примера 2, примера 3 и примера 4 согласно немецкой заявке на патент DE-A 19937107.
Речь при этом идет о катализаторах дегидрирования, которые содержат от 10 до 99,9% мас. оксида циркония, от 0 до 60% мас. оксида алюминия, диоксида кремния и/или диоксида титана и от 0,1 до 10% мас. по меньшей мере одного элемента первой или второй главной группы, одного элемента третьей побочной группы, одного элемента восьмой побочной группы периодической системы элементов, лантана и/или олова, при условии, что сумма указанных массовых процентов составляет 100% мас.
Особенно пригодным является также катализатор дегидрирования, используемый в примерах и сравнительных примерах, приведенных в настоящем описании.
Под катализаторами дегидрирования в общем случае подразумевают катализаторы в виде стерженьков (типичный диаметр от 1 до 10 мм, предпочтительно от 1,5 до 5 мм, типичная длина от 1 до 20 мм, предпочтительно от 3 до 10 мм), таблеток (их размеры предпочтительно аналогичны размерам стерженьков) и/или колец (типичные наружный диаметр и длина соответственно составляют от 2 мм до 30 или до 10 мм, целесообразная толщина стенок от 1 до 10 мм или до 5 мм или до 3 мм).
Катализаторы дегидрирования (прежде всего используемые в приведенных в настоящем описании примерах, а также рекомендуемые в немецкой заявке на патент DE-A 19937107, прежде всего в приведенных в ней примерах), как правило, создают таким образом, чтобы они были пригодны для катализирования как дегидрирования пропана, так и сгорания молекулярного водорода. При этом в случае конкуренции между сгоранием водорода и дегидрированием пропана на подобных катализаторах гораздо быстрее протекает сгорание водорода.
Для осуществления гетерогенно катализируемого дегидрирования пропана в принципе пригодны любые известные из уровня техники типы реакторов и варианты технологии. Описания соответствующих технологических вариантов содержатся, например, во всех цитируемых публикациях уровня техники, касающихся катализаторов дегидрирования, а также в цитируемом в начале настоящего описания уровне техники.
Пригодные согласно изобретению методы дегидрирования довольно подробно рассмотрены также в Catalytica® Studies Division, Oxidative Dehydrogenation and Alternative Dehydrogenation Processes, Study Number 4192 OD, 1993, 430 Ferguson Drive, Mountain View, Калифорния, 94043-5272, США.
Как указано выше, характерной особенностью частичного гетерогенно катализируемого дегидрирования пропана является эндотермичность этого процесса. Это означает, что теплота (энергия), необходимая для установления необходимой реакционной температуры, а также для протекания дегидрирования, должна поступать или вместе с исходной реакционной газовой смесью и/или в процессе гетерогенно катализируемого дегидрирования. Требуемая теплота реакции при необходимости должна высвобождаться самой реакционной газовой смесью.
Кроме того, особенностью гетерогенно катализируемого дегидрирования пропана, обусловленной необходимыми для его осуществления высокими реакционными температурами, является образование небольших количеств труднокипящих высокомолекулярных органических соединений, включая углерод, которые оседают на поверхности катализатора, вызывая его деактивирование. Для минимизации подобного отрицательного сопутствующего явления содержащую пропан реакционную газовую смесь, подлежащую пропусканию через поверхность катализатора с целью гетерогенно катализируемого при повышенной температуре дегидрирования, можно разбавлять водяным паром. В подобных условиях происходит частичное или полное удаление осаждающегося на поверхности катализатора углерода в соответствии с принципом газификации угля.
Другая возможность удаления образуемых соединениями углерода отложений состоит в осуществляемом время от времени при повышенной температуре пропускании через катализатор дегидрирования содержащего кислород газа (целесообразно в отсутствие углеводородов), что обеспечивает квазивыгорание отложений углерода. Существенное подавление образования отложений углерода возможно также благодаря тому, что к пропану, подлежащему гетерогенно катализируемому дегидрированию, перед его пропусканием при повышенной температуре через катализатор дегидрирования добавляют молекулярный водород.
К пропану, подлежащему гетерогенно катализируемому дегидрированию, очевидно, можно также добавлять смесь водяного пара с молекулярным водородом. Добавление молекулярного водорода к подлежащему гетерогенному катализу пропану уменьшает также нежелательное образование прежде всего (пропадиена), пропина и ацетилена в качестве побочных продуктов. Частичное окисление добавляемого подобным образом водорода способствует также выделению необходимой теплоты реакции.
Пригодной конструктивной формой реактора для гетерогенно катализируемого дегидрирования пропана в реакционной зоне А является трубчатый реактор со стационарным слоем катализатора, соответственно кожухотрубный реактор. Это означает, что катализатор дегидрирования находится в виде стационарного слоя в реакционной трубке или в пучке реакционных трубок. Согласно изобретению внутри контактной трубки может располагаться предпочтительно центрированная вторая трубка с находящимися на самых разных уровнях (или только на одном уровне) отверстиями (смотри международные заявки WO 01/85333 и WO 01/85330), через которые может поступать остаточный газ из зоны разделения В. В другом конструктивном варианте соединенные в донной части контактные трубки могут иметь прерванные участки (пространства), в которые можно вводить остаточный газ из зоны разделения В. Обогрев реакционных трубок осуществляют благодаря сжиганию в окружающем их пространстве газа, например углеводорода, такого как метан. Подобную форму прямого нагревания контактных трубок целесообразно использовать лишь для стационарной засыпки первого уровня (примерно от 20 до 30% по высоте), а остальную часть стационарной засыпки следует нагревать до требуемой реакционной температуры посредством выделяющейся при сгорании лучистой теплоты. Подобный метод нагревания позволяет обеспечивать примерно изотермический режим осуществления дегидрирования. Пригодный внутренний диаметр реакционной трубки составляет примерно от 10 до 15 см. Типичный кожухотрубный реактор дегидрирования содержит от 300 до 1000 реакционных трубок. Температура внутри реакционных трубок составляет от 300 до 700°С, предпочтительно от 400 до 700°С. Исходную реакционную газовую смесь предпочтительно вводят в трубчатый реактор предварительно нагретой до реакционной температуры. Газообразная смесь продуктов А на выходе из реакционной трубки может иметь температуру ниже указанной на величину, составляющую от 50 до 100°С. Однако температура газообразной смеси на выходе из трубок может быть также выше указанной или аналогичной ей. Для рассмотренного выше процесса дегидрирования целесообразно использовать оксидные катализаторы на основе оксида хрома и/или оксида алюминия. Какой-либо разбавляющий газ часто не используют, и исходным реакционным газом является в основном только сырой пропан. Катализатор дегидрирования в большинстве случаев также используют в неразбавленном состоянии.
При промышленном производстве в реакционной зоне А параллельно могут функционировать несколько (например, три) подобных кожухотрубных реактора. При этом согласно изобретению при необходимости один (или два) из них могут функционировать в режиме дегидрирования, в то время как во втором (третьем) реакторе выполняют не нарушающую функционирования реакционной зоны В регенерацию катализатора.
Подобную технологию целесообразно использовать, например, для осуществления известного из литературы способа дегидрирования пропана BASF-Linde. Однако для осуществления настоящего изобретения достаточно применения одного подобного кожухотрубного реактора.
Подобную технологию можно использовать также для осуществления разработанной фирмой Phillips Petroleum Co. конверсии с водяным паром (так называемого процесса STAR) (смотри, например, заявки на патент США US-A 4902849, US-A 4996387 и US-A 5389342). Катализатором дегидрирования для осуществления процесса STAR предпочтительно является содержащая промоторы платина на используемой в качестве носителя цинковой (магниевой) шпинели (смотри, например, заявку на патент США US-A 5073662). В отличие от способа дегидрирования пропана BASF-Linde пропан, подлежащий дегидрированию в соответствии с процессом STAR, разбавляют водяным паром. Типичное молярное отношение водяного пара к пропану находится в интервале от 4 до 6. Давление на входе в реактор чаще всего составляет от 3 до 8 бар, целесообразной реакционной температуре соответствует интервал от 480 до 620°С. Типичная среднечасовая скорость пропускания пропана через слой катализатора составляет от 200 до 4000 ч-1.
При этом под среднечасовой скоростью пропускания (исходной) смеси реакционных газов через слой катализатора подразумевают количество этой смеси в нормальных литрах (нл) (нл означает объем в литрах, который эта смесь занимала бы в нормальных условиях, то есть при температуре 0°С и давлении 1 атмосфера), которое протекает через литр слоя катализатора в течение часа. Среднечасовая скорость пропускания может относиться также только к одному компоненту (исходной) смеси реакционных газов. В подобном случае речь идет о количестве этого компонента в нормальных литрах, пропускаемом через литр слоя катализатора в течение часа (нл/л·ч). Вместо обозначения среднечасовой скорости в нормальных литрах в час (то есть вместо нл/л·ч) нередко используют соответствующее сокращенное обозначение «ч-1».
Гетерогенно катализируемое дегидрирование пропана предлагаемым в изобретении способом можно выполнять также в движущемся слое катализатора. Движущийся слой катализатора может быть помещен, например, в реактор с радиальным движением потока. Катализатор в подобном реакторе медленно опускается сверху вниз, в то время как реакционная газовая смесь перемещается в радиальном направлении. Подобную технологию используют, например, для осуществления так называемого способа дегидрирования UOP-Oleflex. Поскольку реакторы согласно этой технологии функционируют в квазиадиабатическом режиме, целесообразной является эксплуатация нескольких (в типичном случае до четырех) последовательно соединенных в каскад реакторов. В подобный каскад можно подавать остаточный газ из зоны разделения В. Благодаря этому можно избежать слишком большой разности температур реакционной газовой смеси на входе в реактор и выходе из него (в случае адиабатического режима исходная реакционная газовая смесь выполняет функцию теплоносителя, от теплосодержания которого зависит снижение реакционной температуры) и несмотря на это обеспечить приемлемую общую конверсию пропана.
Катализатор, выгруженный из реактора с движущимся слоем, направляют на регенерацию, а затем вновь используют. В качестве катализатора дегидрирования в данном случае можно использовать, например, катализатор сферической формы, состоящий преимущественно из платины на носителе в виде шариков оксида алюминия. При использовании технологии UOP во избежание преждевременного старения катализатора к подлежащему дегидрированию пропану добавляют водород. Рабочее давление обычно составляет от 2 до 5 атм. Целесообразное (молярное) отношение водорода к пропану находится в интервале от 0,1 до 1. Реакционная температура предпочтительно составляет от 550 до 650°С, время пребывания катализатора в реакторе находится в интервале примерно от 2 до 10 часов.
Для реализации рассмотренных выше процессов дегидрирования в стационарном слое можно использовать сферический катализатор, а также цилиндрический (полый или сплошной) катализатор или катализатор с частицами иной геометрической формы.
Другим возможным технологическим вариантом гетерогенно катализируемого дегидрирования пропана в соответствии с предлагаемым в изобретении способом является метод дегидрирования пропана в псевдоожиженном слое, описанный в Proceedings De Witt, Petrochem. Review, Houston, Texas, 1992 a, N1.
Согласно изобретению в этом случае могут функционировать, например, два последовательно соединенных псевдоожиженных слоя, один из которых периодически может находиться в состоянии регенерации, что не оказывает негативного влияния на протекание общего технологического процесса. При этом в качестве активной массы используют оксид хрома на оксиде алюминия. Рабочее давление обычно составляет от 1 до 2 бар, температура дегидрирования, как правило, находится в интервале от 550 до 600°С. Необходимое для дегидрирования тепло поступает в реакционную систему вместе с предварительно нагретым до реакционной температуры катализатором дегидрирования. Подобная технология известна из литературы также как способ Snamprogetti-Yarsintez.
Другой вариант рассмотренного выше гетерогенно катализируемого дегидрирования пропана в соответствии с предлагаемым в изобретении способом можно реализовать также, используя технологию, разработанную фирмой ABB Lummus Crest (смотри Proceedings De Witt, Petrochem. Review, Houston, Texas, 1992, P1).
Согласно изобретению остаточный газ при использовании обеих указанных выше технологий можно вводить в заявляемую точку пути реакции в виде одинарного питающего потока.
Общей особенностью рассмотренных выше методов гетерогенно катализируемого дегидрирования пропана является то, что конверсия пропана (в пересчете на однократное пропускание через реакционную зону и весь вводимый пропан) составляет ≥30% мол. (как правило, ≤60% мол.). Однако согласно изобретению предпочтительно ограничиться конверсией пропана, находящейся в интервале от ≥5% мол. до ≤30 или ≤25% мол. Это означает, что предлагаемое в изобретении гетерогенно катализируемое дегидрирование пропана в реакционной зоне А можно осуществлять также при его конверсиях от 10 до 20% мол. (указанные значения относятся к однократному пропусканию пропана через реакционную зону А). Подобное ограничение обусловлено, в частности, тем, что остающийся непревращенным пропан в последующей реакционной зоне В преимущественно выполняет функцию инертного разбавляющего газа и согласно изобретению в основном без потерь может быть рециркулирован в реакционную зону А в качестве компонента остаточного газа, образующегося в зоне разделения В.
Однако согласно изобретению предпочтительными являются повышенные конверсии пропана в реакционной зоне А, поскольку это обусловливает уменьшение количества пропионовой кислоты, образующейся в реакционной зоне В в качестве побочного продукта. Подобные предпочтительные конверсии пропана составляют, например, от 30 или 40% мол. до 50 или 60% мол. (в пересчете на однократное пропускание пропана через реакционную зону А и весь вводимый пропан).
Для реализации указанных выше конверсий гетерогенно катализируемое дегидрирование пропана целесообразно осуществлять при рабочем давлении от 0,3 до 3 бар. Кроме того, подлежащий гетерогенному каталитическому дегидрированию пропан целесообразно разбавлять водяным паром. Теплоемкость воды, с одной стороны, способствует частичной компенсации воздействия эндотермического эффекта дегидрирования, а с другой стороны, разбавление водяным паром снижает парциальное давление эдуктов и продуктов дегидрирования, что благоприятно отражается на положении равновесия этой реакции. Кроме того, совместное применение водяного пара, как указано выше, положительно влияет на долговечность содержащих благородный металл катализаторов дегидрирования. При необходимости в качестве дополнительного компонента можно добавлять также молекулярный водород. При этом молярное отношение молекулярного водорода к пропану, как правило, составляет ≤5. Молярное отношение водяного пара к пропану может составлять, например, от ≥0 до 30, в целесообразном варианте от 0,1 до 2, в оптимальном варианте от 0,5 до 1. Преимущество использования технологии с пониженной конверсией пропана состоит в том, что при однократном пропускании реакционного газа через реакционную зону А потребляется лишь сравнительно небольшое количество тепла, и для достижения превращения бывают достаточны сравнительно низкие реакционные температуры.
Дегидрирование пропана согласно изобретению, как указано выше, можно осуществлять также в (квази)адиабатическом режиме и при этом эндотермически. При этом исходную реакционную газовую смесь, как правило, сначала нагревают до температуры от 500 до 700°С (соответственно от 550 до 650°С) (например, благодаря прямому огневому обогреву окружающих ее стенок). В этом случае при адиабатическом пропускании по меньшей мере через один слой катализатора реакционная газовая смесь в зависимости от конверсии пропана и разбавления остывает до температуры, составляющей примерно от 30 до 200°С. После места реализуемой согласно изобретению подачи остаточного газа из зоны разделения В происходит определенное нагревание реакционной газовой смеси. Присутствие водяного пара в качестве теплоносителя благоприятно и при осуществлении дегидрирования в адиабатическом режиме. Пониженные реакционные температуры способствуют более длительному сроку службы слоя используемого катализатора. Повышенные реакционные температуры способствуют увеличению конверсии пропана.
Независимо от режима гетерогенно катализируемого дегидрирования пропана в реакционной зоне А (адиабатического или изотермического) этот процесс в принципе можно осуществлять как в реакторе со стационарным слоем катализатора, так и в реакторе с движущимся или псевдоожиженным слоем катализатора.
Следует подчеркнуть, что согласно предлагаемому в изобретении способу в качестве реакционной зоны А может быть достаточным использование единственного, функционирующего также в адиабатическом режиме шахтного реактора (реактора со стационарным слоем), реакционная газовая смесь через который проходит в осевом и/или радиальном направлениях.
В простейшем случае речь при этом идет о единственном замкнутом реакционном объеме, например, о резервуаре с внутренним диаметром от 0,1 до 10 м, возможно также от 0,5 до 5 м, стационарный слой катализатора в котором располагается на опорном приспособлении (например, колосниковой решетке). При этом через заполненный катализатором реакционный объем, который в случае адиабатического режима дегидрирования преимущественно снабжен теплоизоляцией, в осевом направлении пропускают содержащий пропан горячий реакционный газ. При этом катализатор может обладать как сферической, так и кольцеобразной или стержнеобразной формой. Остаточный газ из зоны разделения В может поступать через помещенные внутрь слоя катализатора питающие патрубки. Поскольку реакционный объем в рассматриваемом случае может быть выполнен в виде чрезвычайно экономичного аппарата, предпочтение можно отдать катализаторам любой геометрической формы, характеризующимся особенно низкими потерями напора. Прежде всего речь идет о катализаторах с геометрической формой, обусловливающей формирование крупных полостей или структур, например, таких как монолиты, соответственно сотовые тела. Для реализации радиального течения содержащего пропан реакционного газа реактор может состоять, например, из двух концентрически вставленных друг в друга цилиндрических колосниковых решеток, снабженных рубашкой в виде гильзы, в кольцевой зазор между которыми загружают катализатор. В случае адиабатического режима металлическая гильза при необходимости может быть снабжена теплоизоляцией.
Для предлагаемого в изобретении гетерогенно катализируемого дегидрирования пропана прежде всего пригодны также катализаторы, известные из немецкой заявки на патент DE-A 19937107, прежде всего приведенные в соответствующих примерах.
Указанные выше катализаторы после длительной эксплуатации можно подвергать регенерации, осуществляемой, например, простым методом, в соответствии с которым на первой ступени регенерации через слой катализатора пропускают (предпочтительно) разбавленный азотом и/или водяным паром воздух с температурой на входе от 300 до 600°С, часто от 400 до 550°С. При этом среднечасовая скорость подачи регенерирующего газа (например, воздуха) может составлять, например, от 50 до 10000 ч-1при содержании кислорода в регенерирующем газе от 0,1 или 0,5 до 20% об.
На следующей ступени регенерации в качестве регенерирующего газа можно использовать воздух, сохранив прочие условия регенерации неизменными. С производственно-технической точки зрения перед регенерацией катализатор целесообразно продуть инертным газом (например, азотом).
Затем, как правило, рекомендуется выполнить дополнительную регенерацию катализатора чистым молекулярным водородом или молекулярным водородом, разбавленным инертным газом (предпочтительно водяным паром) (содержание водорода в смеси с инертным газом должно составлять ≥1% об.), при сохранении остальных условий регенерации неизменными.
Гетерогенно катализируемое дегидрирование пропана при сравнительно незначительной конверсии (≤30% мол.) во всех случаях можно осуществлять с той же среднечасовой скоростью подачи (как всего количества реакционной газовой смеси, так и содержащегося в ней пропана), что и в вариантах дегидрирования, предусматривающих высокую конверсию пропана (>30% мол.). Соответствующая среднечасовая скорость подачи реакционного газа может составлять, например, от 100 ч-1 до 40000 или до 10000 ч-1, часто от 300 до 7000 ч-1, чаще всего примерно от 500 до 4000 ч-1.
С производственно-технической точки зрения реакционную зону А согласно предлагаемому в изобретении способу целесообразно выполнить в виде реактора с решетчатыми полками, что позволяет особенно простым образом вводить остаточный газ из зоны разделения В между двумя полками.
Подобный реактор содержит более одного слоя катализатора дегидрирования, которые пространственно расположены один над другим. Количество слоев катализатора может составлять от 1 до 20, целесообразно от 2 до 8, а также от 3 до 6. Увеличение количества решетчатых полок позволяет обеспечить определенное повышение конверсии пропана. Слои катализатора располагаются последовательно предпочтительно в радиальном или осевом направлении. С производственно-технической точки зрения в подобном полочном реакторе целесообразно использовать катализаторы, предназначенные для применения в стационарном слое.
В простейшем случае стационарные слои катализатора располагают аксиально в шахтном реакторе или в кольцевых зазорах между концентрически вставленными одна в другую цилиндрическими колосниковыми решетками. Однако кольцевые зазоры можно располагать также один над другим в виде сегментов, пропуская через них газ в радиальном направлении от одного сегмента к другому, находящемуся выше или ниже предыдущего.
В целесообразном варианте реакционную газовую смесь в реакторе с решетчатыми полками на пути от одного слоя катализатора к следующему подвергают промежуточному нагреванию, например, благодаря пропусканию над обогреваемыми горячими газами поверхностями (например, ребрами теплообменника) или пропусканию через трубы, обогреваемые горячими горючими газами.
При функционировании полочного реактора дегидрирования в адиабатическом режиме для обеспечения конверсии пропана ≤30% мол. прежде всего при использовании катализаторов, описанных в немецкой заявке на патент DE-A 19937107 (особенно в примерах осуществления этого изобретения), реакционную газовую смесь перед введением в реактор достаточно нагреть до температуры от 450 до 550°С и поддерживать эту температуру внутри реактора. Это означает, что общее дегидрирование пропана осуществляют при чрезвычайно низких температурах, что особенно благоприятно отражается на сроке службы стационарных слоев катализатора между двумя последовательно реализуемыми регенерациями. Для получения более высоких конверсии пропана реакционную газовую смесь целесообразно вводить в полочный реактор дегидрирования, предварительно нагрев ее до более высоких температур (которые могут достигать 700°С), и поддерживать эту повышенную температуру внутри реактора.
Еще более предпочтительным является выполнение рассмотренного выше промежуточного нагревания реакционной газовой смеси прямым методом (автотермический режим). Для этого в реакционную газовую смесь до ее пропускания через первый слой катализатора (в этом случае исходная реакционная газовая смесь должна содержать добавленный к ней молекулярный водород) и/или между последовательно расположенными слоями катализатора вводят ограниченное количество молекулярного кислорода. Подобным образом можно обеспечить (как правило, катализируемое самими катализаторами дегидрирования) ограниченное сгорание содержащегося в реакционной газовой смеси молекулярного водорода, образующегося в процессе гетерогенно катализируемого дегидрирования пропана и/или добавляемого к реакционной газовой смеси (с производственно-технической точки зрения может быть также целесообразной загрузка в полочный реактор слоев катализатора, особенно специфическим образом (селективно) катализирующего сгорание водорода (подобные катализаторы приведены, например, в описаниях патентов США US-A 4788371, US-A 4886928, US-A 5430209, US-A 5530171, US-A 5527979 и US-A 5563314; например, подобные слои катализаторов можно поместить в полочный реактор в чередующейся последовательности со слоями, содержащими катализатор дегидрирования). Выделяющееся вследствие сгорания водорода тепло оказывает квазиавтотермическое содействие реализации практически изотермического режима гетерогенно катализируемого дегидрирования пропана (тепловой брутто-эффект преимущественно равен нулю). Таким образом, с увеличением выбранного времени пребывания реакционного газа в слое катализатора дегидрирование пропана становится возможным при снижаемых или преимущественно постоянных температурах, что способствует особенно длительному периоду эксплуатации катализатора между двумя последовательно осуществляемыми процессами его регенерации.
Согласно изобретению описанную выше подачу кислорода в общем случае следует осуществлять таким образом, чтобы его содержание в реакционной газовой смеси в пересчете на содержащееся в ней количество молекулярного водорода составляло от 0,5% об. до 50, соответственно 30% об., предпочтительно от 10 до 25% об. При этом источником кислорода может быть как чистый молекулярный кислород, так и кислород, разбавленный инертным газом, например монооксидом углерода, диоксидом углерода, азотом и/или благородными газами, прежде всего также воздухом, а также оксид азота. Образующиеся в результате сгорания газы, как правило, оказывают дополнительный эффект разбавления, тем самым способствуя гетерогенно катализируемому дегидрированию пропана.
В соответствии с предлагаемым в изобретении способом рециркулируемый из зоны разделения В в реакционную зону А (основной и/или побочный, соответственно общий) остаточный газ является особенно богатым источником молекулярного кислорода, который согласно изобретению добавляют к реакционной газовой смеси самое раннее, как правило, после ее пропускания через первый слой находящегося в полочном реакторе катализатора.
Изотермический режим гетерогенно катализируемого дегидрирования пропана можно оптимизировать, поместив в пространства между слоями катализатора полочного реактора герметично закрытые встроенные элементы (например, в виде труб), которые перед их заполнением целесообразно (но необязательно) подвергнуть эвакуации. Подобные встроенные элементы можно монтировать также в соответствующем слое катализатора. В таких элементах находятся соответствующие твердые вещества или жидкости, которые испаряются или плавятся при превышающей определенное значение температуре, поглощая при этом тепло в тех зонах, в которых происходит это превышение температуры, и вновь конденсируются, высвобождая тепло.
Реакционную зону А для предлагаемого в изобретении способа, очевидно, можно выполнить также подобно немецкой заявке на патент DE-A 10211275, являющейся частью настоящей заявки (то есть в виде так называемого петлевого варианта).
Таким образом, один из вариантов предлагаемого в изобретении способа предусматривает введение в реакционную зону А по меньшей мере трех содержащих пропан газообразных питающих потоков, по меньшей мере один из которых содержит свежий пропан и по меньшей мере один из которых является рециркулируемым из зоны разделения В в реакционную зону А (основным и/или побочным, соответственно общим) остаточным газом, содержащим молекулярный кислород, непревращенный пропан и при необходимости непревращенный (в реакционной зоне В) пропилен, и рециркулируемый подобным образом в реакционную зону А пропан подвергают гетерогенно катализируемому (частичному) дегидрированию (в принципе для этой цели пригодны любые указанные в настоящем описании методы гетерогенно катализируемого дегидрирования), получая содержащую пропан и пропилен газообразную смесь продуктов А, при условии, что
a) газообразную смесь продуктов А разделяют на две части одинакового состава и одну из них возвращают (в виде циркуляционного газа дегидрирования) в реакционную зону А в качестве одного из по меньшей мере трех содержащих пропан питающих потоков (предпочтительно в качестве составной части вводимой в реакционную зону А газовой смеси (исходной реакционной газовой смеси), а другую часть газообразной смеси продуктов А (предпочтительно всю другую часть) подвергают осуществляемой согласно изобретению дальнейшей переработке в первой зоне разделения А, и
b) ввод (основного и/или побочного, соответственно общего) остаточного газа из зоны разделения В в реакционную зону А вдоль пути гетерогенно катализируемого дегидрирования пропана осуществляют таким образом, чтобы в месте ввода уже было подвергнуто дегидрированию по меньшей мере 5% мол. или по меньшей мере 10% мол., предпочтительно по меньшей мере 15% мол. или по меньшей мере 20% мол., особенно предпочтительно по меньшей мере 25% мол. или по меньшей мере 30% мол., еще более предпочтительно по меньшей мере 35% мол. или по меньшей мере 40% мол., лучше всего по меньшей мере 45% мол. или по меньшей мере 50% мол. (однако, как правило, менее 70% мол., часто менее 60% мол. и чаще всего ≤50% мол.) (суммарного) пропана, уже введенного в реакционную зону А вместе с другими питающими потоками (при этом особенно предпочтительными технологическими вариантами также являются такие, при которых указанные выше параметры конверсии ZU заменены на параметры, определяемые по уравнению ZUKr=(ZU/1+KGV)). При этом поступающий из зоны разделения В (основной и/или побочный, соответственно общий) остаточный газ вводят в одно или несколько последовательно расположенных мест реакционной зоны А.
Целесообразной является рециркуляция в реакционную зону А по меньшей мере 10% об., соответственно 20% об. газообразной смеси продуктов А. Однако количество газообразной смеси продуктов А, возвращаемой в реакционную зону А в качестве циркуляционного газа, предпочтительно составляет не более 90% об., соответственно не более 80% об. То есть часть газообразной смеси продуктов А, возвращаемая в реакционную зону А в качестве циркуляционного газа, может составлять, например, от 20 до 80% об., от 30 до 70% об., от 40% об. до 60 или 50% об. Особенно предпочтительно эта часть составляет от 50 до 70% об. газообразной смеси продуктов А.
Нагревание вводимой в реакционную зону А исходной реакционной газовой смеси до реакционной температуры, необходимой для осуществления гетерогенно катализируемого дегидрирования пропана в реакционной зоне А, возможно также благодаря добавлению молекулярного водорода, его сжиганию в присутствии молекулярного кислорода, например, на соответствующих (например, указанных в настоящем описании) специфических катализаторах сжигания (например, при простом пропускании над ними и/или через них) и выделяющемуся при этом теплу. Продукты сгорания, такие как диоксид углерода и вода, а также необходимый для сжигания водорода молекулярный кислород, при необходимости смешанный с азотом, образуют предпочтительно инертные разбавляющие газы.
В случае, если вдоль пути осуществляемого в реакционной зоне А гетерогенно катализируемого дегидрирования пропана уже рециркулируют содержащий другой молекулярный кислород остаточный газ из зоны разделения В, то независимо от конкретного конструктивного исполнения реакционной зоны А согласно изобретению целесообразно обращать внимание на то, чтобы вводимый подобным образом молекулярный кислород не вызывал слишком сильного снижения содержания водорода в реакционной газовой смеси в реакционной зоне А до места ввода рециркулируемого из зоны разделения В, содержащего молекулярный кислород остаточного газа.
Согласно изобретению остаточный газ, рециркулируемый из зоны разделения В в реакционную зону А, может быть также единственным вводимым в реакционную зону А газом, содержащим молекулярный кислород (независимо от конкретного конструктивного исполнения реакционной зоны А). Однако для осуществления предлагаемого в изобретении способа подобный вариант, как правило, не используют.
В предпочтительном варианте конструктивного исполнения и функционирования реакционная зона А состоит из первой и второй секций (ниже данный вариант называют двухсекционным), причем
I) в первую секцию реакционной зоны А вводят по меньшей мере один содержащий газообразный пропан питающий поток, который содержит свежий пропан, и введенный в первую секцию пропан подвергают в ней гетерогенно катализируемому дегидрированию, получая газообразную смесь продуктов А*, содержащую пропан, пропилен и молекулярный водород, причем дегидрированию подвергают по меньшей мере 5% мол. или по меньшей мере 10% мол., предпочтительно по меньшей мере 15% мол. или по меньшей мере 20% мол., особенно предпочтительно по меньшей мере 25% мол. или по меньшей мере 30% мол., еще более предпочтительно по меньшей мере 35% мол. или по меньшей мере 40% мол. и еще лучше по меньшей мере 45% мол. или по меньшей мере 50% мол. (однако, как правило, менее 70% мол., менее 60% мол. или ≤50% мол.) пропана, (суммарно) введенного в первую секцию, и образующаяся смесь продуктов А* содержит (в пересчете на содержащееся в ней молярное количество пропана) предпочтительно большее молярное количество молекулярного водорода, чем исходная реакционная газовая смесь, вводимая в первую секцию реакционной зоны А;
II) затем к газообразной смеси продуктов А* добавляют (основной и/или побочный, соответственно общий) остаточный газ из зоны разделения В, содержащий молекулярный кислород, непревращенный пропан и при необходимости непревращенный (в реакционной зоне В) пропилен (согласно изобретению предпочтительно по меньшей мере половину, предпочтительно по меньшей мере две трети или три четверти и еще более предпочтительно все количество (при необходимости за вычетом части газа идентичного состава, рециркулируемого в реакционную зону В в качестве разбавляющего газа) указанного остаточного газа, образующегося в зоне разделения В (в соответствующем индивидуальном пересчете на (основной и/или побочный, соответственно общий остаточный газ), и образующуюся при этом реакционную газовую смесь А* направляют во вторую секцию реакционной зоны А;
III) во второй секции реакционной зоны А при образовании содержащей пропан и пропилен газообразной смеси продуктов А содержащийся в реакционной газовой смеси А* молекулярный кислород (предпочтительно по меньшей мере 25% мол., предпочтительно по меньшей мере 50% мол., особенно предпочтительно по меньшей мере 75% мол., еще лучше по меньшей мере 90% мол. и лучше всего по меньшей мере 95% мол. содержащегося в реакционной газовой смеси А* молекулярного кислорода) участвует в сопровождаемом образованием воды гетерогенно катализируемом сжигании содержащегося в этой смеси молекулярного водорода, а содержащийся в реакционной газовой смеси А* пропан (как правило, менее 40% мол., соответственно менее 30% мол., часто менее 20% мол., нередко менее 10% мол. и чаще всего менее 5% мол. содержащегося в ней пропана) при необходимости подвергают гетерогенно катализируемому дегидрированию до пропилена. Указанные выше количественные характеристики можно регулировать, варьируя объем соответствующего слоя катализатора;
IV) затем газообразную смесь продуктов А выводят из реакционной зоны А и подвергают осуществляемой согласно изобретению, описанной выше переработке в первой зоне разделения А предлагаемого в изобретении способа (газообразную смесь продуктов А, не направляемую в первую зону разделения А, при необходимости можно подвергнуть, например, сжиганию с целью производства энергии и/или использовать для получения синтез-газа и тому подобное).
При этом в качестве катализаторов для второй секции реакционной зоны А пригодны любые катализаторы, которые рекомендованы в настоящем описании для гетерогенно катализируемого дегидрирования пропана и прежде всего пригодны также для применения в первой секции реакционной зоны А, поскольку они, как указано в начале настоящего описания, как правило, способны катализировать также сжигание молекулярного водорода (это относится прежде всего к катализаторам, приведенным в немецкой заявке на патент DE-A 19937107 (прежде всего в соответствующих примерах); в случае конкуренции между гетерогенно катализируемым дегидрированием пропана и гетерогенно катализируемым сжиганием водорода последняя реакция, как правило, протекает гораздо быстрее и доминирует над первой). Однако для применения во второй секции реакционной зоны А, очевидно, пригодны также специально разработанные катализаторы, специфичные для реакции селективного сжигания молекулярного водорода. Подобные катализаторы приведены, например, в описаниях заявок на патент США US-A 4788371, US-A 4886928, US-A 5430203, US-A 5530171, US-A 5527979 и US-A 5563314.
Первую секцию реакционной зоны А в принципе можно сконструировать пригодной для функционирования как в изотермическом, так и в адиабатическом режиме. В последнем случае в первой секции реакционной зоны А предпочтительным является тепловой брутто-эффект от эндотермического до автотермического (в пересчете на однократное пропускание через эту секцию добавляемой (вводимой) исходной реакционной газовой смеси).
Реакционные условия во второй секции реакционной зоны А (температура, например, от 400, соответственно 500°С, до 800, соответственно 700°С, давление, например, от 1 бар до 10 или до 5 или до 3 бар, и среднечасовая скорость подачи газовой смеси, например, от 500 (или менее) до 80000 (или более) нл/л·ч) в принципе могут быть аналогичны реакционным условиям в первой секции реакционной зоны А.
Вторую секцию реакционной зоны А в принципе также можно выполнить пригодной для функционирования как в изотермическом, так и в адиабатическом режиме. В последнем случае во второй секции реакционной зоны А предпочтительным является экзотермический тепловой брутто-эффект (в пересчете на однократное пропускание через эту секцию добавляемой (вводимой) исходной реакционной газовой смеси (реакционной газовой смеси А*).
Согласно изобретению как первую, так и вторую секции реакционной зоны А предпочтительно выполняют пригодными для функционирования в адиабатическом режиме, причем в первой реакционной секции особенно предпочтительным является тепловой брутто-эффект от эндотермического до автотермического, а во второй реакционной секции экзотермический тепловой брутто-эффект.
При этом особенно предпочтительным является конструктивное исполнение первой секции реакционной зоны А в виде полочной структуры, которая согласно изобретению функционирует предпочтительно в адиабатическом режиме от эндотермического до автотермического. Полочная структура включает один или несколько последовательных пространственно расположенных слоев катализатора дегидрирования. Количество слоев катализатора может составлять от 1 до 20, целесообразно от 2 до 8, прежде всего от 3 до 6 (для обеспечения высоких конверсий при дегидрировании пропана в первой секции реакционной зоны А предпочтительным является большое число слоев катализатора).
Реакционный газ пропускают через слои катализатора предпочтительно в радиальном или осевом направлении.
В простейшем случае стационарные слои катализатора последовательно располагают в реакторе аксиально или в кольцевых зазорах, образуемых концентрически вставленными друг в друга цилиндрическими колосниковыми решетками. При этом реактор может иметь конструкцию, например, шахтной печи. Возможна также реализация первой секции реакционной зоны А в отдельной шахтной печи, как и реализация всей реакционной зоны А в полочном реакторе.
Если к реакционному газу в каком-либо месте до и/или после его ввода в первую секцию реакционной зоны А не добавляют содержащий молекулярный кислород газ, то реакционный газ в структурированной полками первой секции реакционной зоны А на пути его перемещения от одного слоя катализатора к следующему целесообразно было бы подвергать промежуточному нагреванию, например благодаря пропусканию над обогреваемыми горячими газами поверхностями, например ребрами теплообменника, или через встроенные элементы, например обогреваемые горячим горючим газом трубы.
Однако в соответствии с предлагаемым в изобретении способом рассмотренное выше промежуточное нагревание по меньшей мере частично предпочтительно осуществляют прямым методом. Для этого к реакционному газу до его пропускания через первый слой катализатора и/или при его прохождении между последовательно расположенными слоями катализатора добавляют незначительное количество содержащего молекулярный кислород газа (предпочтительно воздуха или смеси кислорода с инертными газами, такими как диоксид углерода, азот и благородные газы) или чистого кислорода.
Таким образом, в одном, нескольких или при необходимости во всех слоях катализатора сжигают молекулярный водород, незначительное количество которого заранее добавляют к реакционному газу и/или он образуется в процессе дегидрирования. Выделяющееся при этом тепло допускает возможность адиабатической реализации преимущественно автотермического режима в первой секции реакционной зоны А. Количество добавляемого молекулярного кислорода в пересчете на содержащееся в реакционном газе количество молекулярного водорода должно составлять от 0,5 до 50% об., соответственно до 30% об. (предпочтительно от 10 до 25% об.). В связи со значительной величиной теплоты сгорания водорода для достижения указанной выше цели, как правило, оказывается достаточным частичное сжигание сравнительно небольшого количества этого газа.
В одном из вариантов осуществления изобретения содержащий кислород газ при необходимости вводят перед каждой полкой полочного реактора. В другом варианте содержащий кислород газ вводят перед каждой полкой, за исключением первой. Еще в одном варианте осуществления предлагаемого в изобретении способа после каждой точки ввода кислорода расположен слой специфического катализатора окисления, пригодного для окисления водорода, за которым следует слой катализатора дегидрирования. При необходимости перед каждой полкой из внешнего источника дополнительно вводят (чистый или разбавленный инертным газом) молекулярный водород. В менее предпочтительном варианте слои катализатора могут состоять также из смесей, образованных катализатором дегидрирования и катализатором окисления водорода.
Однако согласно изобретению осуществление предлагаемого в изобретении способа не требует обязательной подпитки внешним молекулярным водородом. Температура дегидрирования в полочной структуре (реакторе с решетчатыми полками), как правило, составляет от 400 до 800°С, давление в общем случае находится в интервале от 0,2 до 10 бар, предпочтительно от 0,5 до 4 бар и особенно предпочтительно от 1 до 3 бар. Среднечасовая скорость подачи газа, как правило, составляет от 500 до 10000 ч-1, а при высокой нагрузке также до 80000 ч-1 (как правило, от 30000 до 40000 ч-1).
Присутствие водяного пара позволяет увеличить срок службы катализатора.
Уменьшение парциального давления реагентов благодаря снижению давления, сжиганию образующегося при дегидрировании водорода и/или разбавлению инертными газами, а также повышение температуры и увеличение количества полок способствуют повышению степени превращения пропана при его дегидрировании в первой секции реакционной зоны А.
Для второй секции реакционной зоны А предпочтительно используют реактор аналогичного типа, как и для первой секции реакционной зоны А. Однако подобный реактор, как правило, содержит только один слой катализатора. В качестве реакторов обеих секций предпочтительно используют простые шахтные печи.
Молярное отношение молекулярного кислорода к молекулярному водороду (в реакционной газовой смеси А*) во второй секции реакционной зоны А может составлять от 1:2 до 1:10, предпочтительно от 1:2 до 1:4. Таким образом, во второй секции реакционной зоны А может происходить взаимодействие преимущественно всего содержащегося в реакционной смеси А* молекулярного кислорода с молекулярным водородом (горение водорода) с образованием воды, в связи с чем образующаяся газообразная смесь продуктов А характеризуется максимально полным отсутствием молекулярного кислорода. Так, например, она может содержать менее 5% об., менее 3% об. или менее 1% об., чаще всего даже менее 0,5 или менее 0,2% об. молекулярного кислорода. Предпочтительным является полное отсутствие молекулярного кислорода в газообразной смеси продуктов А.
Первая и вторая секции реакционной зоны А могут быть реализованы как в отделенных друг от друга реакторах, так и в одном отдельном реакторе (например, в полочном реакторе, например, в виде шахтной печи).
В более предпочтительном варианте осуществления изобретения выполнение и функционирование реакционной зоны А возможны в виде комбинации упомянутого выше «петлевого» и «двухсекционного» вариантов.
При этом газообразную смесь продуктов А, образующуюся во второй секции реакционной зоны А описанного выше «двухсекционного» варианта, дополнительно разделяют на две части идентичного состава, одну из которых (циркуляционный газ дегидрирования) возвращают в первую секцию реакционной зоны А в качестве другого содержащего пропан питающего потока (предпочтительно в качестве составной части исходной реакционной газовой смеси первой секции реакционной зоны А), а другую часть (предпочтительно всю другую часть) подвергают осуществляемой согласно изобретению дальнейшей переработке в первой зоне разделения А.
Согласно изобретению рециркуляция газообразной смеси продуктов А в первую секцию реакционной зоны А является предпочтительной по той причине, что образующаяся во второй секции реакционной зоны А газообразная смесь продуктов А вследствие взаимодействия молекулярного водорода с молекулярным кислородом (сжигания водорода), с одной стороны, обычно (по меньшей мере при адиабатическом экзотермическом функционировании второй секции реакционной зоны А) обладает повышенной температурой, а с другой стороны, вследствие расходования кислорода на сжигание водорода содержит незначительное количество кислорода и указанное выше количество водяного пара. Содержание водорода в этой смеси также, как правило, бывает незначительным. Для обеспечения во второй секции реакционной зоны А, например, необходимой температуры реакционной газовой смеси А (а также для исключения содержащегося в ней молекулярного кислорода) прежде всего при уменьшенных конверсиях пропана, дегидрируемого в первой секции реакционной зоны А, к реакционной газовой смеси А* перед подачей во вторую секцию реакционной зоны А можно дополнительно добавлять внешний молекулярный водород (речь идет о молекулярном водороде, который не является ни компонентом возвращаемого в реакционную зону А циркуляционного газа, ни водородом, образующимся в самой реакционной зоне А (или в одной из других реакционных зон/зон разделения предлагаемого в изобретении способа)).
Итак, рециркуляция газообразной смеси продуктов А в первую секцию реакционной зоны А в качестве составной части исходной реакционной газовой смеси, реализуемая в соответствии с рассмотренным выше особым вариантом, пригодна для нагревания исходной реакционной газовой смеси, вводимой в первую секцию реакционной зоны А, до реакционной температуры без одновременного негативного воздействия уже содержащегося в исходной реакционной газовой смеси молекулярного водорода на термодинамику дегидрирования, соответственно без негативного воздействия уже содержащегося молекулярного кислорода на кинетику дегидрирования. Водяной пар, содержащийся в рециркулируемой газообразной смеси продуктов А, дополнительно способствует гетерогенно катализируемому дегидрированию пропана в первой секции реакционной зоны А, и отдельная подача внешнего водяного пара, как правило, становится ненужной.
Таким образом, преимущество предлагаемой в изобретении технологии состоит, в частности, в том, что она позволяет реализовать предлагаемый в изобретении режим при удовлетворительном сроке службы катализатора без необходимости подачи внешнего водяного пара в ту или иную реакционную зону, соответственно зону разделения (речь идет о водяном паре, который не образуется ни в реакционной зоне, ни в зоне разделения предлагаемого в изобретении способа).
В связи с повышенной температурой направляемой в первую зону разделения А другой части образующейся во второй секции реакционной зоны А газообразной смеси продуктов А эта часть одновременно отлично пригодна для того, чтобы благодаря косвенному теплообмену в газовом холодильнике нагреть (преимущественно до необходимой реакционной температуры) как подаваемый в реакционную зону А свежий пропан, так и рециркулируемый в эту зону из зоны разделения В остаточный газ (или только один из двух этих газовых потоков), и при этом предпочтительно остыть самой до температуры, необходимой для первой зоны разделения А.
Согласно изобретению в первую секцию реакционной зоны А целесообразно рециркулировать по меньшей мере 10% об., соответственно 20% об. образующейся во второй секции газообразной смеси продуктов А. Однако часть газообразной смеси продуктов А, возвращаемая согласно изобретению в качестве циркуляционного газа в первую секцию реакционной зоны А, предпочтительно составляет не более 90% об., соответственно 80% об. Таким образом, в соответствии с рассмотренным выше технологическим вариантом часть газообразной смеси продуктов А, возвращаемая в качестве циркуляционного газа в первую секцию реакционной зоны А, может составлять, например, от 20 до 80% об., от 30 до 70% об., от 40 до 60% об. или также 50% об. образующейся газообразной смеси продуктов А. Особенно предпочтительное количество (возвращаемого на дегидрирование) циркуляционного газа составляет от 50 до 70% об.
В еще более предпочтительном варианте рециркуляцию указанной выше газообразной смеси продуктов А в первую секцию реакционной зоны А осуществляют следующим образом.
Сначала в газообразную смесь продуктов А* вводят (основной и/или побочный, соответственно общий) остаточный газ из зоны разделения В, содержащий молекулярный кислород, пропан и при необходимости непревращенный (в реакционной зоне В) пропилен, для чего используют принцип струйного насоса, функционирующего с помощью этого остаточного газа в качестве приводной струи (подобный принцип поясняется на фиг.1 немецкой заявки на патент DE-A 10211275; принцип эжектирующего сопла подлежит включению и в настоящее описание), причем перемещение дросселированной в рабочем сопле (1) приводной струи через зону смешения (2) и диффузор (3) во второй секции реакционной зоны А, а также обеспечиваемый всасывающим патрубком (4) эффект всасывания направлены в сторону первой секции реакционной зоны А, а сочленение «всасывающий патрубок - зона смешения - диффузор» образует единственное соединение между обеими секциями реакционной зоны А.
Таким образом, во всасывающем патрубке (4) возникает разрежение, благодаря которому газообразная смесь продуктов А* всасывается из первой секции реакционной зоны А и при одновременно происходящем в зоне смешения (2) (смесительной трубе) смешивании с приводной струей перемещается через диффузор (3) в качестве реакционной газовой смеси А* во вторую секцию реакционной зоны А (нумерация позиций соответствует нумерации, указанной на приведенной в настоящем описании фиг.1).
Напор приводной струи (обычно его регулируют с помощью радиального компрессора, соответственно (в более редких случаях) с помощью нагнетателя (обычно осевого компрессора с низким отношением конечного давления к начальному давлению)) необходимо подбирать таким образом, чтобы давление образующейся во второй секции реакционной зоны А газообразной смеси продуктов А превышало давление газообразной смеси продуктов А*, образующейся в первой секции реакционной зоны А. Использования простого трубного соединения между выходом из второй секции реакционной зоны А и входом в первую секцию реакционной зоны А, которое целесообразно выполнить в виде тройника с делителем расхода, предназначенного для подачи части газообразной смеси продуктов А в первую зону разделения А, обычно вполне достаточно для создания циркуляционного потока (естественной циркуляции) другой части смеси продуктов А, возвращаемой (в соответствии с перепадом давления) в первую секцию реакционной зоны А.
Учитывая вышеизложенное, в еще более предпочтительном варианте реакционную зону А (17) предлагаемого в изобретении способа выполняют описанным ниже образом в соответствии со схемой, приведенной на фиг.1 настоящего описания, и указанными на ней позициями.
При этом выполнение и функционирование реакционной зоны А (17) скомбинированы из рассмотренных выше «петлевого» и «двухсекционного» вариантов при дополнительной реализации следующих технических мероприятий.
I. В первую секцию (0) реакционной зоны А вводят по меньшей мере один содержащий свежий пропан (5) газообразный питающий поток (9), подвергая пропан гетерогенно катализируемому (частичному) дегидрированию с образованием газообразной смеси продуктов А*, содержащей пропан, пропилен и молекулярный водород, причем дегидрированию подлежит по меньшей мере 5% мол. или по меньшей мере 10% мол., предпочтительно по меньшей мере 15% мол. или по меньшей мере 20% мол., особенно предпочтительно по меньшей мере 25% мол. или по меньшей мере 30% мол., еще более предпочтительно по меньшей мере 35% мол. или по меньшей мере 40% мол. и еще лучше по меньшей мере 45% мол. или по меньшей мере 50% мол. (однако, как правило, менее 70% мол., менее 60% мол. или ≤50% мол.) (суммарно) подаваемого в эту первую секцию пропана (при «петлевом» варианте предпочтительными для реакционной зоны А являются такие технологические варианты, в соответствии с которыми указанные выше параметры конверсии ZU соответственно заменены на ZUKr=(ZU/1+KGV));
при этом первая секция реакционной зоны А функционирует в основном в адиабатическом режиме (то есть в этой секции преимущественно отсутствуют (целенаправленные) подача и отбор тепла посредством находящегося вне ее теплоносителя);
тепловой брутто-эффект в первой секции реакционной зоны А в пересчете на однократное пропускание вводимой в нее газовой смеси может изменяться от эндотермического (отрицательного) до автотермического (преимущественно нулевого);
первая секция реакционной зоны А предпочтительно обладает полочной структурой;
полочная структура (6) включает предпочтительно от 2 до 20, особенно предпочтительно от 2 до 10 и еще более предпочтительно от 2 до 6 слоев катализатора, расположенных предпочтительно аксиально (например, в шахтной печи) или радиально (например, в кольцевых зазорах, образуемых концентрически вставленными одна в другую цилиндрическими колосниковыми решетками), реакционный газ через которые пропускают соответственно в аксиальном или радиальном направлении;
полочную структуру можно реализовать, например, в одном отдельном реакторе или нескольких последовательно соединенных друг с другом реакторах;
слои катализаторов (например, приведенных в немецкой заявке на патент DE-A 19937107, прежде всего в соответствующих примерах) предпочтительно загружены таким образом, чтобы в начальном потоке реакционного газа, состав которого допускает возможность конкуренции между гетерогенно катализируемым сгоранием водорода и гетерогенно катализируемым дегидрированием пропана, быстрее происходило гетерогенно катализируемое сгорание водорода;
в направляемую в первую секцию реакционной зоны А исходную реакционную газовую смесь предпочтительно не добавляют ни внешний молекулярный кислород, ни внешний молекулярный водород, ни внешний молекулярный водяной пар (согласно изобретению под внешним газом, вводимым в какой-либо точке реакционной зоны А (или другой реакционной зоны/зоны разделения предлагаемого в изобретении способа) в общем случае подразумевают газ, который не образуется сам в реакционной зоне А (или в другой реакционной зоне/зоне разделения предлагаемого в изобретении способа) и который не возвращают непосредственно и/или опосредованно в реакционную зону А, если он образовался в этой зоне);
после прохождения (исходной) реакционной газовой смеси (предназначенной для питания первой секции реакционной зоны А) через первый слой катализатора до ее дальнейшего пропускания через каждый последующий в направлении течения слой катализатора к ней предпочтительно добавляют газ с небольшим содержанием молекулярного кислорода, при этом количество добавляемого кислорода ограничивают таким образом, чтобы результирующее содержание кислорода в реакционной газовой смеси (в пересчете на содержащийся в ней водород) составляло от 0,5 до 50% об., соответственно до 30% об., предпочтительно от 10 до 25% об.;
в качестве подобного содержащего кислород газа предпочтительно используют воздух (7), однако можно использовать также чистый кислород, смесь кислорода с инертными газами, такими как диоксид углерода, азот и/или благородные газы, или оксид азота;
после добавления содержащего кислород газа реакционный газ перед поступлением в следующий слой катализатора предпочтительно пропускают через статический смеситель (8);
температура в первой секции реакционной зоны А предпочтительно составляет от 400 до 700°С;
рабочее давление в первой секции реакционной зоны А предпочтительно составляет от 0,5 до 10 бар;
вводимая в первую секцию реакционной зоны А исходная реакционная газовая смесь (9) (ее температура предпочтительно составляет от 400 до 700°С, например, 560°С, давление предпочтительно находится в интервале от 0,5 до 10 бар) предпочтительно обладает следующим составом:
от 10 до 35% об. (например, 19 или 28% об.) пропана,
от 3 до 10% об. (например, 7,5 или 5,5% об.) пропена,
от 5 до 20% об. (например, 9 или 12% об.) водяного пара,
от 0,01 до 6% об. (например, 3,9 или 4,3% об.) молекулярного водорода,
от 0 до 0,02% об. молекулярного кислорода;
среднечасовая скорость пропускания через слои катализатора часто составляет от 250 до 5000 ч-1 (для процессов с высокой нагрузкой также до 40000 ч1), предпочтительно от 10000 до 25000 нл/л·ч, особенно предпочтительно от 15000 до 20000 нл/л·ч;
для нагревания вводимого в первую секцию реакционной зоны А свежего пропана до реакционной температуры его предпочтительно подвергают косвенному теплообмену (в газовом холодильнике обычной конструкции) с горячей газообразной смесью продуктов А, поступающей в зону разделения А из второй секции реакционной зоны А;
в первую секцию реакционной зоны А предпочтительно не вводят никаких внешних газов, кроме указанных в настоящем описании;
газообразная смесь продуктов А*, образующаяся в первой секции реакционной зоны А, предпочтительно обладает следующим составом:
от 8 до 32% об. (например, 16 или 24% об.) пропана,
от 3 до 12% об. (например, 9 или 6% об.) пропилена,
от 5 до 23% об. (например, 11 или 15% об.) водяного пара,
от 0,1 до 6% об. (например, 1 или 3% об.) молекулярного водорода,
от 0 до 0,05% об. молекулярного кислорода;
температура газообразной смеси продуктов А* предпочтительно составляет от 500 до 750°С (например, 580°С), рабочее давление предпочтительно находится в интервале от 0,05 до 10 бар.
II. Затем к газообразной смеси продуктов А* добавляют (основной и/или побочный, соответственно общий) остаточный газ из зоны разделения В, содержащий молекулярный кислород, пропан и при необходимости непревращенный (в реакционной зоне В) пропилен (согласно изобретению предпочтительно вводят по меньшей мере половину, предпочтительно по меньшей мере две трети или три четверти, еще более предпочтительно все количество образующегося в зоне разделения В остаточного газа), и образующуюся реакционную газовую смесь А* направляют во вторую секцию реакционной зоны А;
добавляемый к газообразной смеси продуктов А* остаточный газ, как правило, содержит от 0,5 до 10% об., часто от 1 до 8% об., предпочтительно от 2 до 5% об. молекулярного кислорода;
добавляемый к газообразной смеси продуктов А* (основной и/или побочный, соответственно общий) остаточный газ предпочтительно обладает следующим составом:
от 10 до 40% об. (например, 15 или 32% об.) пропана,
от 0 до 1% об. пропена,
от >0 до 5% об. (например, 3 или 4% об.) молекулярного кислорода,
от 1 до 10% об. (например, 2% об.) водяного пара,
от 0 до 0,5% об. (например, от 0 до 0,3% об. или от 0 до 0,1% об.) молекулярного водорода;
к газообразной смеси продуктов А* предпочтительно добавляют такое количество остаточного газа, чтобы содержание кислорода в образующейся после добавления реакционной газовой смеси А* в пересчете на содержащийся в ней водород составляло от 15 до 50% об., предпочтительно от 25 до 50% об., особенно предпочтительно от 30 или 40 до 50% об.;
реакционная газовая смесь А* (12) предпочтительно обладает следующим составом:
от 10 до 38% об. (например, 16 или 28% об.) пропана,
от 3 до 10% об. (например, 5% об.) пропилена,
от 0 до 0,05% об. молекулярного кислорода,
от 3 до 20% об. (например, 7 или 12% об.) водяного пара,
от 0,1 до 6% об. (например, 4% об.) молекулярного водорода;
температура реакционной газовой смеси А* (12) предпочтительно составляет от 300 до 600°С (например, от 400 до 500°С), давление предпочтительно находится в интервале от 0,5 бар, соответственно 0,6 бар, до 12 бар;
(основной и/или побочный, соответственно общий) остаточный газ из зоны разделения В добавляют к газообразной смеси продуктов А* (11), предпочтительно используя принцип струйного насоса, приводимого в действие посредством этого остаточного газа в качестве приводной струи, причем перемещение дросселированной в рабочем сопле (1) приводной струи через зону смешения (2) и диффузор (3) второй секции (13) реакционной зоны А, а также всасывание (всасывающее действие) всасывающего патрубка (4) направлены в сторону первой секции реакционной зоны А, и сочленение «всасывающий патрубок - зона смешения - диффузор» образует единственное соединение между двумя указанными секциями реакционной зоны А;
температура приводной струи предпочтительно составляет от 400°С, соответственно 500°С до 600°С, ее давление находится в интервале от 3 до 6 бар (предпочтительно от 4 до 5 бар);
остаточный газ (15), направляемый из зоны разделения В в реакционную зону А, предпочтительно нагревают до указанной температуры благодаря косвенному теплообмену (14) с направляемой в зону разделения А горячей газообразной смесью продуктов А (16) (предпочтительно после того, как эта газообразная смесь продуктов А нагреет свежий пропан);
остаточный газ, направляемый в реакционную зону А из зоны разделения В, сжимают до указанного выше давления предпочтительно посредством радиального, соответственно осевого компрессора;
кроме указанных в настоящем описании газовых потоков к реакционной газовой смеси А* предпочтительно не добавляют никаких других газовых потоков.
III. Во второй секции реакционной зоны А при формировании содержащей пропан и пропилен газообразной смеси продуктов А содержащийся в реакционной газовой смеси А* молекулярный кислород (предпочтительно по меньшей мере 25% мол., предпочтительно по меньшей мере 50% мол., особенно предпочтительно по меньшей мере 75% мол., еще лучше по меньшей мере 90% мол. и лучше всего по меньшей мере 95% мол. этого кислорода) расходуется на гетерогенно катализируемое сжигание содержащегося в этой смеси молекулярного водорода, сопровождаемое образованием воды, а содержащийся в реакционной газовой смеси А* пропан (как правило, менее 40% мол., соответственно менее 30% мол., наиболее часто менее 20% мол., чаще всего менее 10% мол. и часто менее 5% мол. содержащегося в ней пропана) при необходимости подвергают гетерогенно катализируемому дегидрированию до пропилена;
при этом вторая секция реакционной зоны А функционирует преимущественно в адиабатическом режиме (то есть преимущественно отсутствует как (целенаправленное) поступление тепла во вторую секцию реакционной зоны А от находящегося вне этого участка теплоносителя, так и соответствующий (целенаправленный) отбор тепла;
тепловой брутто-эффект во второй секции реакционной зоны А в пересчете на однократное пропускание через нее реакционной газовой смеси А* является экзотермическим (для обеспечения положительного теплового эффекта, прежде всего в случае сравнительно невысоких конверсий при дегидрировании пропана в первой секции реакционной зоны А, целесообразно повышать содержание водорода в реакционной газовой смеси А* благодаря введению внешнего водорода; с производственно-технической точки зрения молекулярный водород целесообразно вводить предпочтительно уже в газообразную смесь продуктов А*, хотя внешний молекулярный водород можно вводить также, например, в рециркулируемый из зоны разделения В остаточный газ (19) или в образующуюся при этом реакционную газовую смесь А*);
во вторую секцию реакционной зоны А предпочтительно не вводят никаких других внешних газов, кроме указанных в настоящем описании;
температуру во второй секции реакционной зоны А предпочтительно поддерживают в интервале от 400 до 750°С (соответственно от 500 до 700°С);
рабочее давление во второй секции реакционной зоны А предпочтительно составляет от 0,5 до 12 бар;
для использования во второй секции реакционной зоны А пригодны, например, катализаторы, которые в настоящем описании рекомендуются также для осуществления гетерогенно катализируемого дегидрирования пропана в первой секции реакционной зоны А, поскольку, как указано в начале настоящего описания, подобные катализаторы, как правило, способны катализировать также сжигание молекулярного водорода (прежде всего это относится к катализаторам, приведенным в немецкой заявке на патент DE-A 19937107 (прежде всего в соответствующих примерах); в связи с чем подобные катализаторы еще более предпочтительны для использования в обеих секциях реакционной зоны А; в случае конкуренции между гетерогенно катализируемым дегидрированием пропана и гетерогенно катализируемым сгоранием водорода последняя реакция, как правило, протекает гораздо быстрее и доминирует над первой); однако для использования во второй секции реакционной зоны А, очевидно, пригодны также специально разработанные катализаторы, специфичные для реакции селективного сжигания молекулярного водорода; подобные катализаторы приведены, например, в описаниях патентов США US-A 4788371, US-A 4886928, US-А 5430203, US-A 5530171, US-A 5527979 и US-A 5563314;
в качестве реакторов для второй секции реакционной зоны А в принципе пригодны реакторы, используемые в первой секции реакционной зоны А;
реактор, используемый во второй секции реакционной зоны А, предпочтительно содержит лишь один слой катализатора, а не множество подобных слоев;
с производственно-технической точки зрения подобный единственный слой катализатора целесообразно поместить в простую шахтную печь и пропускать через него реакционную газовую смесь в аксиальном или радиальном направлении;
среднечасовая скорость пропускания реакционной газовой смеси А* через этот слой катализатора, как правило, составляет от 500 до 80000 нл/л·ч, предпочтительно от 10000 до 50000 нл/л·ч, особенно предпочтительно от 20000 до 40000 нл/л·ч (при небольших степенях дегидрирования пропана во второй секции реакционной зоны А и преимущественно полном исчерпании молекулярного кислорода за счет сжигания молекулярного водорода указанные скорости могут также достигать 200000 нл/л·ч);
первую и вторую секции реакционной зоны А предпочтительно размещают в одном и том же реакторе;
с производственно-технической точки зрения предпочтительной является вторая секция реакционной зоны А в виде одной решетчатой полки с помещенным на нее катализатором, которая находится в полочном реакторе, остальные полки которого образуют первую секцию реакционной зоны А;
газообразная смесь продуктов А, образующаяся во второй секции реакционной зоны А, предпочтительно обладает следующим составом:
от 7 до 30% об. (например, 12 или 24% об.) пропана,
от 3 до 12% об. (например, 6 или 8% об.) пропена,
от 0 до 0,05% об. молекулярного кислорода,
от 5 до 22% об. (например, 10 или 13% об.) водяного пара,
от 0,1 до 6% об. (например, 4% об.) молекулярного водорода;
температура газообразной смеси продуктов А предпочтительно составляет от 400 до 700°С; давление реакционной газовой смеси предпочтительно находится в интервале от 0,4 до 10 бар и вместе с тем предпочтительно превышает давление газообразной смеси продуктов А*.
IV. Образующуюся во второй секции реакционной зоны А газообразную смесь продуктов А разделяют на две части одинакового состава, одну из которых (10) рециркулируют в первую секцию реакционной зоны А в качестве содержащего дополнительный пропан питающего потока (циркуляционного газа дегидрирования) (предпочтительно в качестве составляющей исходной реакционной газовой смеси для первой секции реакционной зоны А и предпочтительно благодаря перепаду давления в естественном контуре циркуляции, обеспечивающем напор на выходе из первой секции реакционной зоны А, смешивание со свежим пропаном предпочтительно осуществляют посредством статического смесителя перед первым в направлении потока слоем катализатора первой секции реакционной зоны А), а другую часть (18) газообразной смеси продуктов А (предпочтительно всю другую часть) подвергают осуществляемой согласно изобретению дальнейшей переработке в первой зоне разделения А;
другую часть (18) газообразной смеси продуктов А направляют в зону разделения А предпочтительно через косвенный теплообменник (14), обеспечивая нагревание (до необходимой температуры) свежего пропана и/или поступающего из зоны разделения В остаточного газа.
В соответствии с предлагаемым в изобретении гетерогенно катализируемым дегидрированием пропана газообразная смесь продуктов А, образующаяся в реакционной зоне А в результате осуществления предлагаемого в изобретении способа, как правило, содержит пропан, пропен, молекулярный водород, азот, воду, метан, этан, этилен, бутен-1, прочие бутены (например, изобутен) и другие углеводороды с четырьмя атомами углерода (н-бутан, изобутан, бутадиен и так далее), монооксид углерода, диоксид углерода, а также, как правило, кислородсодержащие соединения, такие как спирты, альдегиды и карбоновые кислоты (с числом атомов углерода обычно ≤9). Кроме того, газообразная смесь продуктов А может содержать незначительные количества компонентов, происходящих из остаточного газа, например, таких как образующиеся в реакционной зоне В побочные соединения, а также используемый в зонах разделения А и/или В абсорбент. Образующаяся газообразная смесь продуктов А, как правило, находится под давлением от 0,3 до 10 бар и зачастую имеет температуру от 400 до 650°С, соответственно до 500°С, в оптимальных случаях от 450 до 500°С.
Согласно европейской заявке на патент ЕР-А 117146, немецкой заявке на патент DE-A 3313573 и заявке на патент США US-A 3161670 газообразную смесь продуктов А, образующуюся в результате гетерогенно катализируемого дегидрирования пропана, рекомендуется использовать для подачи в реакционную зону В как таковую, в то время как согласно настоящему изобретению перед дальнейшим использованием газообразной смеси продуктов А для подачи в реакционную зону В необходимо отделить от содержащихся в ней пропана и пропилена по меньшей мере часть различных других компонентов, присутствующих в ней в качестве побочных продуктов. При этом следует учитывать требования немецкой заявки на патент DE-A 10211275. Если газообразная смесь продуктов А еще содержит существенные количества молекулярного водорода, то выделение части побочных продуктов осуществляют, как правило, при одновременном выделении по меньшей мере части водорода, или подобное выделение водорода предпочтительно выполняют предварительно.
Предварительно выделить водород можно, например, благодаря пропусканию газообразной смеси продуктов А через проницаемую только для молекулярного водорода мембрану, как правило, обладающую конструкцией трубы (пригоден также соответствующий пластинчатый или рулонный узел), которое при необходимости осуществляют после предшествующего охлаждения газообразной смеси продуктов А в косвенном теплообменнике (как указано выше, отводимое при этом тепло целесообразно использовать в реакционной зоне А для нагревания питающего газа, необходимого для осуществления предлагаемого в изобретении способа). Часть выделенного подобным образом молекулярного водорода при необходимости можно вернуть на стадию гетерогенно катализируемого дегидрирования пропана (то есть в реакционную зону А) и/или использовать иным образом. Так, например, возможно сжигание водорода в топливных элементах.
В другом варианте водород можно частично или полностью выделить также (осуществляемой предпочтительно под давлением) частичной конденсацией, адсорбцией и/или ректификацией. Частичное или полное выделение молекулярного водорода из газообразной смеси продуктов А согласно предлагаемому в изобретении способу можно выполнить также реализуемым вне реакционной зоны А селективным (например, гетерогенно катализируемым) сжиганием в присутствии молекулярного кислорода. Образующуюся при этом реакционную воду можно частично или полностью выделить или оставить в газовой смеси, поскольку она может выполнять функцию инертного разбавляющего газа в реакционной зоне В. Катализаторы, пригодные для селективного сжигания водорода, известны, например, из патентов США US-A 4788371, US-A 4886928, US-A 5430209, US-A 55530171, US-A 5527979 и US-A 5563314.
Согласно изобретению целесообразным является предварительное и/или одновременное выделение по меньшей мере 10% мол. или по меньшей мере 25% мол., часто по меньшей мере 35% мол. или по меньшей мере 50% мол., чаще всего по меньшей мере 75% мол. и часто всего молекулярного водорода, содержащегося в выводимой из реакционной зоны А газообразной смеси продуктов А, которое реализуют, прежде чем направить остающуюся газообразную смесь продуктов А' в реакционную зону В. Однако еще более предпочтительный отличительный признак рассмотренного выше еще более предпочтительного технологического варианта, используемого в реакционной зоне А, а именно комбинирования «петлевого» и «двухсекционного» вариантов с адиабатическим эндотермическим выполнением первой секции и адиабатическим экзотермическим выполнением второй секции (соответственно реакционной зоны А) состоит в том, что в этом случае образуется газообразная смесь продуктов А, уже содержащая минимальное количество молекулярного водорода, образующийся в реакционной зоне А молекулярный водород почти количественно предпочтительно используют уже в реакционной зоне А, и образующийся при этом водяной пар также предпочтительно используют в качестве составляющей той части газообразной смеси продуктов А, которую рециркулируют в первую секцию реакционной зоны А. Перед дальнейшей переработкой газообразной смеси продуктов А в реакционной зоне из нее при необходимости можно также частично или полностью выделить (например, конденсацией) возможно содержащуюся в ней воду. Очевидно, что в процессе выделения молекулярного водорода и/или водяного пара при необходимости можно также выделять другие, отличающиеся от пропана и пропилена, компоненты газообразной смеси продуктов А.
Для этой цели можно использовать простую технологию, которая, например, состоит в том, что реализуют контакт предпочтительно охлажденной (предпочтительно до температуры от 10 до 100°С, соответственно до 70°С), газообразной смеси продуктов А, например, при давлении от 0,1 до 50 бар, предпочтительно от 5 до 15 бар, и температуре, например, от 0 до 100°С, предпочтительно от 20 до 40°С, с (предпочтительно высококипящим и предпочтительно гидрофобным) органическим растворителем (например, реализуя простое пропускание газа через подобный растворитель), который абсорбирует пропан и пропилен (целесообразна преимущественная абсорбция пропана и пропилена по сравнению с другими компонентами газообразной смеси продуктов А). Благодаря последующей десорбции, ректификации и/или отпариванию газом с инертным поведением в реакционной зоне В и/или газом, присутствие которого как реагента необходимо в этой реакционной зоне (например, посредством воздуха или другой смеси, состоящей из молекулярного кислорода и инертного газа), осуществляют регенерацию смеси чистых пропана и пропилена, которую используют для подачи в реакционную зону В (предпочтительная последовательность технологических операций указана в сравнительном примере 1). Вторичный газ процесса абсорбции, при необходимости содержащий молекулярный водород, можно, например, вновь подвергнуть мембранному разделению, а выделенный при этом водород при необходимости можно использовать в реакционной зоне А для осуществления гетерогенно катализируемого дегидрирования пропана.
Впрочем, коэффициент разделения углеводородов с тремя атомами углерода (С3) и углеводородов с четырьмя атомами углерода (C4) в соответствии с рассмотренной выше технологией сравнительно мал и часто недостаточен для реализации поставленных в немецкой заявке на патент DE-А 10245585 задач.
Учитывая вышеизложенное, в качестве альтернативы рассмотренному выше разделению методом абсорбции предпочтительным методом достижения целей настоящего изобретения часто оказывается адсорбция при переменном давлении или ректификация под давлением.
В качестве абсорбентов для осуществления рассмотренного выше абсорбционного разделения в принципе пригодны любые абсорбенты, способные поглощать пропан и пропилен. Под абсорбентом предпочтительно подразумевают предпочтительно гидрофобный и/или высококипящий органический растворитель. Подобный растворитель предпочтительно обладает температурой кипения (при нормальном давлении 1 атм), составляющей по меньшей мере 120°С, предпочтительно по меньшей мере 180°С, предпочтительно от 200 до 350°С, прежде всего от 250 до 300°С, более предпочтительно от 260 до 290°С. Температура воспламенения абсорбента (при нормальном давлении 1 бар) в оптимальном случае превышает 110°С. Пригодными абсорбентами в общем случае являются неполярные органические растворители, например, такие как алифатические углеводороды, предпочтительно не содержащие полярных групп с внешним действием, а также ароматические углеводороды. В общем случае необходимо, чтобы абсорбент обладал возможно более высокой температурой кипения и одновременно возможно более высокой способностью растворять пропан и пропилен. Так, например, пригодными абсорбентами являются алифатические углеводороды, например алаканы или алкены с 8-20 атомами углерода, ароматические углеводороды, например средние нефтяные фракции перегонки парафинов, простые эфиры с блокированными (стерически затрудненными) группами, соединенными с атомом кислорода, или смеси указанных продуктов, причем к ним можно добавлять полярный растворитель, например, такой как известный из немецкой заявки на патент DE-A 4308087 1,2-диметилфталат. Кроме того, в качестве абсорбентов пригодны простые эфиры бензойной кислоты и фталевой кислоты и неразветвленных алканолов с 1-8 атомами углерода, такие как н-бутиловый эфир бензойной кислоты, метиловый эфир бензойной кислоты, этиловый эфир бензойной кислоты, диметиловый эфир фталевой кислоты, диэтиловый эфир фталевой кислоты, а также так называемые масляные теплоносители, такие как дифенил, дифениловый эфир и смеси дифенила с дифениловым эфиром или их хлорсодержащие производные и триарилалкены, например, 4-метил-4′-бензилдифенилметан и его изомеры: 2-метил-2′-бензилдифенилметан, 2-метил-4′-бензилдифенилметан и 4-метил-2′-бензилдифенилметан и смеси этих изомеров. Пригодным абсорбентом является смесь растворителей предпочтительно азеотропного состава, состоящая из дифенила и дифенилового эфира, которая содержит прежде всего около 25% мас. дифенила (бифенила) и около 75% мас. дифенилового эфира, например коммерчески доступный продукт Diphyl® (поставляемый, например, акционерным обществом Bayer). К подобной смеси растворителей часто добавляют от 0,1 до 25% мас. растворителя, такого как диметилфталат (в пересчете на общую смесь растворителей). Особенно пригодными абсорбентами являются также октаны, нонаны, деканы, ундеканы, додеканы, тридеканы, тетрадеканы, пентадеканы, гексадеканы, гептадеканы и октадеканы, причем особенно пригодными прежде всего являются тетрадеканы. Используемый абсорбент в оптимальном варианте, с одной стороны, обладает указанной выше температурой кипения, а с другой стороны, не слишком высокой молекулярной массой. Предпочтительная молекулярная масса абсорбента составляет ≤300 г/моль. Пригодными абсорбентами являются также описанные в немецкой заявке на патент DE-A 3313573 парафиновые масла с 8-16 атомами углерода. Примерами соответствующих пригодных торговых продуктов являются продукты, выпускаемые фирмой Haltermann, такие как Halpasole i, Halpasol 250/340 i и Halpasol 250/275 i, а также масла для печатных красок с торговым наименованием PKWF и Printosol. Предпочтительными являются торговые продукты, не содержащие ароматических соединений, например продукты типа PKWFaf.
Выполнение абсорбции не связано с какими-либо особыми ограничениями. Можно использовать любые известные специалистам методы и условия абсорбции. Контакт газовой смеси с абсорбентом предпочтительно реализуют при давлении от 1 до 50 бар, предпочтительно от 2 до 20 бар, более предпочтительно от 5 до 15 бар, и при температуре от 0 до 100°С, прежде всего от 20 до 50°С, соответственно до 40°С. Абсорбцию можно осуществлять как в колоннах, так и в аппаратах с резким охлаждением. При этом абсорбцию можно осуществлять как в режиме прямотока, так и (предпочтительно) в режиме противотока. Пригодными абсорбционными колоннами являются, например, тарельчатые колонны (с колпачковыми и/или сетчатыми тарелками), колонны со структурированными насадочными элементами (например, насадочными элементами из листового металла с удельной поверхностью от 100 до 1000 м2/м3 или до 750 м2/м3, например, Mellapak® 250 Y) и насадочные колонны (например, заполненные кольцами Рашига). Возможно также применение пленочных колонн с орошаемыми стенками, распылительных колонн, абсорберов с графитовыми блоками, поверхностных абсорберов, таких как толстопленочные и тонкопленочные абсорберы, а также тарельчатых скрубберов, горизонтальных скрубберов с механическим перемешиванием и ротационных скрубберов. Кроме того, оптимальным может быть осуществление абсорбции в барботажной колонне со встроенными элементами или без них.
Отделение пропана и пропилена от абсорбента можно осуществлять отпариванием, мгновенным испарением в выпарном аппарате и/или дистилляцией.
Отделение пропана и пропилена от абсорбента предпочтительно осуществляют отпариванием и/или десорбцией. Десорбцию можно выполнять обычным образом благодаря изменению давления и/или температуры, предпочтительно при давлении от 0,1 до 10 бар, прежде всего от 1 до 5 бар, более предпочтительно от 1 до 3 бар, и при температуре от 0 до 200°С, прежде всего от 20 до 100°С, более предпочтительно от 30 до 70°С, особенно предпочтительно от 30 до 50°С. Пригодным для отпаривания газом является, например, водяной пар, однако предпочтительными прежде всего являются смеси кислорода с азотом, например воздух. При использовании воздуха, соответственно смесей кислорода с азотом, содержащих более 10% об. кислорода, может оказаться целесообразным осуществляемое до и/или во время отпаривания добавление газа, обеспечивающее сужение области взрывного горения. Особенно пригодными для этой цели являются газы с удельной теплоемкостью при 20°С ≥ 29 Дж/моль·K, например, такие как метан, этан, пропан, пропилен, бензол, метанол, этанол, а также аммиак, диоксид углерода и вода. Однако согласно изобретению предпочтительно следует избегать использования в качестве подобных добавок углеводородов с четырьмя атомами углерода. Для отпаривания особенно пригодны также барботажные колонны со встроенными элементами или без них.
Отделение пропана и пропилена от абсорбента можно осуществлять также дистилляцией, соответственно ректификацией, причем можно использовать известные специалистам колонны с насадочными элементами, насадочными телами или соответствующими встроенными элементами. Предпочтительными условиями для осуществления дистилляции, соответственно ректификации, являются давление от 0,01 до 5 бар, прежде всего от 0,1 до 4 бар, более предпочтительно от 1 до 3 бар, и температура (в кубе) от 50 до 300°С, прежде всего от 150 до 250°С.
Полученную в результате отпаривания абсорбента газовую смесь перед подачей в реакционную зону В дополнительно направляют на другую технологическую операцию (например, сепарацию в каплеотбойниках и/или нутч-фильтрах), осуществляемую, например, для снижения потерь уносимого абсорбента и одновременной защиты реакционной зоны В от проникания в нее абсорбента или дополнительной оптимизации эффекта разделения углеводородов с тремя, соответственно четырьмя атомами углерода (С3/С4). Подобное отделение абсорбента можно осуществлять любыми известными специалистам методами. В соответствии с предлагаемым в изобретении способом предпочтительным методом отделения абсорбента является, например, быстрое охлаждение потока, выходящего из отпарного аппарата, водой. В этом случае происходит вымывание абсорбента из содержащего его выходящего потока водой и одновременное насыщение этого потока водой. Подобную промывку, соответственно быстрое охлаждение, можно осуществлять, например, реализуемым в головной части десорбционной колонны противоточным орошением водой с использованием тарелки для улавливания жидкостей или в индивидуальном аппарате.
Для оптимизации эффекта разделения в предназначенном для быстрого охлаждения пространстве монтируют известные специалистам в области ректификации, абсорбции и десорбции встроенные элементы, обеспечивающие увеличение площади контакта.
Вода является предпочтительным промывочным агентом, поскольку обычно она не создает помех по меньшей мере в одной последующей отдельной зоне. После выполненной водой отмывки абсорбента из содержащего пропан и пропилен выходящего потока смесь воды с абсорбентом направляют в узел разделение фаз, а подвергнутый подобной обработке выходящий поток непосредственно в реакционную зону В.
Отпаренный абсорбент, преимущественно не содержащий углеводородов с тремя атомами углерода, а также абсорбент, регенерированный в узле разделения фаз, можно вновь использовать для осуществления абсорбции.
Содержащую пропан и пропилен газовую смесь, полученную рассмотренными выше в качестве примера методами разделения, можно известным образом использовать для гетерогенно катализируемого газофазного частичного окисления пропилена в акролеин и/или акриловую кислоту, например, в соответствии с международной заявкой WO 01/96270 и описаниями немецких заявок на патент DE-A 10245585, DE-A 10246119, DE-A 10313210, DE-A 10313214, DE-A 10313213, DE-A 10313212, DE-A 10308824, DE-A 10313208 и DE-A 10211275. При этом в качестве окислительного агента можно использовать чистый молекулярный кислород, воздух, воздух, обогащенный кислородом, или любую иную смесь кислорода с инертным газом.
При этом согласно предлагаемому в изобретении способу состав направляемой в реакционную зону В газовой смеси (смотри немецкие заявки на патент DE-A 10245585 и DE-A 10246119) устанавливают таким образом, чтобы молярное отношение пропан:пропен:N2:O2:Н2O:прочее составляло (0,5-20):1:(0,1-40):(0,1-10):(0-20):(0-1).
Согласно изобретению молярное отношение указанных выше компонентов предпочтительно составляет (1 или 2-10):1:(0,5-20):(0,5-5):(0,01-10):(0-1).
Согласно изобретению благоприятным является также молярное отношение указанных выше компонентов, составляющее (3-6):1:(1-10):(1-3):(0,1-2):(0-0,5).
В известном варианте гетерогенно катализируемое частичное газофазное окисление пропилена в акриловую кислоту молекулярным кислородом осуществляют в две последовательно реализуемые вдоль реакционной координаты стадии, первая из которых обеспечивает образование акролеина, а вторая образование акриловой кислоты из акролеина.
Указанное протекание реакции в две последовательно осуществляемые во времени стадии открывает саму по себе известную возможность двухступенчатой реализации реакционной зоны В предлагаемого в изобретении способа, то есть в двух последовательно расположенных зонах окисления, причем для каждой из этих зон можно подобрать оптимальный оксидный катализатор (можно также прервать реакцию на стадии образования акролеина). Так, например, для первой зоны окисления (пропилен → акролеин), как правило, предпочтительно можно использовать мультиметаллоксидный катализатор, содержащий комбинацию молибден (Мо) - висмут (Bi) - железо (Fe), в то время как для второй зоны окисления (акролеин → акриловая кислота) предпочтительными обычно являются мультиметаллоксидные катализаторы, содержащие комбинацию молибден (Мо) - ванадий (V).
Соответствующие пригодные для обеих зон мультиметаллоксидные катализаторы окисления многократно описаны в литературе и известны специалистам. Ссылки на соответствующие патенты США приведены, например, на странице 5 европейской заявки на патент ЕР-А 253409.
Оптимальные катализаторы для обеих зон окисления известны также из немецких заявок на патент DE-A 4431957, DE-A 102004025445 и DE-А 4431949. Прежде всего это относится к приведенным в описаниях этих заявок катализаторам общей формулы I. Особенно предпочтительные катализаторы для обеих зон окисления приведены в описаниях немецких заявок на патент DE-A 10325488, DE-A 10325487, DE-A 10353954, DE-A 10344149, DE-A 10351269, DE-A 10350812 и DE-A 10350822.
В указанных выше описаниях приведены также пригодные согласно изобретению методы частичного газофазного окисления.
Как указано выше, для осуществления первой стадии частичного окисления (гетерогенно катализируемого газофазного частичного окисления пропилена в акролеин) в принципе пригодны любые активные мультиметаллоксидные массы, содержащие молибден (Мо), висмут (Bi) и железо (Fe).
Речь идет прежде всего об активных мультиметаллоксидных массах общей формулы I из немецкой заявки на патент DE-A 19955176, активных мультиметаллоксидных массах общей формулы I из немецкой заявки на патент DE-A 19948523, активных мультиметаллоксидных массах общих формул I, II и III из немецкой заявки на патент DE-A 10101695, активных мультиметаллоксидных массах общих формул I, II и III из немецкой заявки на патент DE-A 19948248 и активных мультиметаллоксидных массах общих формул I, II и III из немецкой заявки на патент DE-A 19955168, а также об активных мультиметаллоксидных массах, приведенных в европейской заявке на патент ЕР-А 700714.
Кроме того, для осуществления первой стадии окисления пригодны содержащие молибден (Мо), висмут (Bi) и железо (Fe) мультиметаллоксидные катализаторы, приведенные в описаниях немецких заявок на патент DE-A 10046957, DE-A 10063162, DE-C 3338380 и DE-А 19902562, европейской заявки на патент ЕР-А 15565, немецкой заявки на патент DE-C 2380765, европейских заявок на патент ЕР-А 807465 и ЕР-А 279374, немецкой заявки на патент DE-A 3300044, европейской заявки на патент ЕР-А 575897, патента США US-A 4438217, немецкой заявки на патент DE-A 19855913, международной заявки WO 98/24746, немецкой заявки на патент DE-A 19746210 (катализаторы общей формулы II), японской заявки на патент JP-A 91/294239 и европейских заявок на патент ЕР-А 293224 и ЕР-А 700714. При этом речь идет прежде всего о примерах осуществления изобретения, особенно предпочтительные из которых приведены в описаниях европейских заявок на патент ЕР-А 15565 и ЕР-А 575897 и немецких заявок на патент DE-A 19746210 и DE-A 19855913. В этой связи особо следует упомянуть катализатор согласно примеру 1с из европейской заявки на патент ЕР-А 15565, а также катализатор, получаемый соответствующим образом, но содержащий активную массу состава Mo12Ni6,5Zn2Fe2Bi1P0,0065K0,06Ox·10SiO2. Кроме того, особо следует выделить пример 3 из немецкой заявки на патент DE-A 19855913, в котором приведен сплошной катализатор стехиометрического состава Mo12Co7Fe3Bi0,6K0,08Si1,6Ox в виде полых цилиндров с размерами 5 мм × 3 мм × 2 мм (наружный диаметр × высота × внутренний диаметр), а также сплошной катализатор на основе мультиметаллоксида II согласно примеру 1 из немецкой заявки на патент DE-A 19746210. Кроме того, следует упомянуть мультиметаллоксидные катализаторы из патента США US-A 4438217. В последнем случае речь идет прежде всего о катализаторе в виде полых цилиндров с размерами 5,5 мм × 3 мм × 3,5 мм или 5 мм × 2 мм × 2 мм или 5 мм × 3 мм × 2 мм или 6 мм × 3 мм × 3 мм или 7 мм × 3 мм × 4 мм (соответственно наружный диаметр × высота × внутренний диаметр). Возможна другая геометрическая форма указанных катализаторов, а именно стерженьки, например, высотой 7,7 мм и диаметром 7 мм или высотой 6,4 мм и диаметром 5,7 мм.
Множество активных мультиметаллоксидных масс, пригодных для осуществления стадии превращения пропилена в акролеин, может быть описано общей формулой IV

переменные в которой означают следующее:
X1 означает никель и/или кобальт,
X2 означает таллий, щелочной металл и/или щелочноземельный металл,
X3 означает цинк, фосфор, мышьяк, бор, сурьму, олово, церий, свинец и/или вольфрам,
X4 означает кремний, алюминий, титан и/или цирконий,
а означает число от 0,5 до 5,
b означает число от 0,01 до 5, предпочтительно от 2 до 4,
с означает число от 0 до 10, предпочтительно от 3 до 10,
d означает число от 0 до 2, предпочтительно от 0,02 до 2,
е означает число от 0 до 8, предпочтительно от 0 до 5,
f означает число от 0 до 10 и
n означает число, которое определяется валентностью и повторяемостью отличающихся от кислорода элементов в общей формуле IV.
Указанные активные мультиметаллоксидные массы могут быть получены известными методами (смотри, например, немецкую заявку на патент DE-А 4023239), и их используют обычно в виде формованных в массе шариков, колец или цилиндров или в виде слоистых катализаторов, то есть покрытых активной массой инертных тел носителя. Активные массы в качестве катализаторов, очевидно, можно использовать также в порошкообразной форме.
В принципе активные массы общей формулы IV можно изготовить простым методом, состоящим в том, что, используя пригодные источники образующих их элементарных компонентов, получают как можно более тщательно перемешанную, предпочтительно мелкодисперсную сухую смесь соответствующего стехиометрического состава, которую подвергают кальцинированию при температуре от 350 до 650°С. Кальцинирование можно осуществлять как в атмосфере инертного газа, так и в окисляющей атмосфере, например, такой как воздух (смесь инертного газа и кислорода), а также в восстанавливающей атмосфере (например, в смеси инертного газа, аммиака, монооксида углерода и/или водорода). Длительность кальцинирования может составлять от нескольких минут до нескольких часов и может быть сокращена при повышении температуры этого процесса. Источниками элементарных компонентов активных мультиметаллоксидных масс общей формулы IV могут быть соединения, которые уже являются оксидами, и/или соединения, которые можно перевести в оксиды нагреванием, осуществляемым в присутствии по меньшей мере кислорода.
Наряду с оксидами подобными исходными соединениями могут быть прежде всего галогениды, нитраты, формиаты, оксалаты, цитраты, ацетаты, карбонаты, комплексы аминов, соли аммония и/или гидроксиды (в тщательно перемешанную сухую смесь можно дополнительно вводить такие соединения, как NH4OH, (NH4)2CO3, NН4NО3, NH4CHO2, СН3СООН, NН4СН3СO2 и/или оксалат аммония, которые способны диссоциировать и/или деструктировать с образованием газообразных улетучивающихся продуктов самое позднее при последующем кальцинировании).
Тщательному перемешиванию можно подвергать исходные соединения для получения активных мультиметаллоксидных масс общей формулы IV, находящиеся как в сухом, так и во влажном состоянии. При перемешивании в сухом состоянии исходные соединения целесообразно использовать в виде мелкодисперсных порошков, а после перемешивания и при необходимости осуществляемого уплотнения подвергать кальцинированию. Однако тщательное перемешивание исходных соединений предпочтительно осуществляют во влажном состоянии. При этом исходные соединения обычно смешивают друг с другом в виде водного раствора и/или суспензии. Особенно тщательно перемешанные указанным выше методом сухие смеси получают в том случае, если исходят из имеющихся источников элементарных компонентов, которые находятся исключительно в растворенном виде. В качестве растворителя предпочтительно используют воду. Полученную водную массу, в заключение, подвергают сушке, которую предпочтительно осуществляют методом распылительной сушки водной смеси при температуре на выходе, составляющей от 100 до 150°С.
Активные мультиметаллоксидные массы общей формулы IV, пригодные для реализации стадии «пропилен → акролеин», можно использовать как в порошкообразной форме, так и в виде получаемого формованием катализатора с определенными геометрическими параметрами, причем формование можно выполнять до или после заключительного кальцинирования. Так, например, уплотнением находящейся в порошкообразной форме активной массы или соответствующей не кальцинированной и/или частично кальцинированной исходной массы (например, таблетированием, экструдированием или штранг-прессованием) можно изготовить сплошные катализаторы требуемой геометрической формы, причем при необходимости могут быть добавлены вспомогательные средства, например, такие как графит или стеариновая кислота, в качестве улучшающих скольжение средств и/или средств облегчения формования, и армирующие средства, такие как микроволокна из стекла, асбеста, карбида кремния или титаната калия. Пригодной геометрической форме сплошных катализаторов соответствуют например, сплошные или полые цилиндры с наружным диаметром и высотой от 2 до 10 мм. Целесообразная толщина стенок полых цилиндров составляет от 1 до 3 мм. Сплошной катализатор, очевидно, может также обладать формой шариков, диаметр которых может составлять от 2 до 10 мм.
Особенно благоприятной геометрической форме полых цилиндров (прежде всего сплошных катализаторов) соответствуют размеры 5 мм × 3 мм × 2 мм (наружный диаметр × длина × внутренний диаметр).
Формование порошкообразной активной массы или соответствующей порошкообразной, еще не подвергнутой кальцинированию и/или частично кальцинированной исходной массы, очевидно, можно выполнить также ее нанесением на предварительно формованный инертный носитель для катализаторов. Нанесение активной массы на тела носителя с целью получения слоистых катализаторов, как правило, осуществляют в соответствующем вращающемся резервуаре, например, таком как описан в немецкой заявке на патент DE-A 2909671 или европейских заявках на патент ЕР-А 293859 и ЕР-А 714700. Наносимую на тела носителя порошкообразную массу целесообразно увлажнять, а после нанесения вновь подвергать сушке, например, горячим воздухом. Целесообразная толщина нанесенного на тела носителя слоя порошкообразной массы составляет от 10 до 1000 мкм, предпочтительно от 50 до 500 мкм и особенно предпочтительно от 150 до 250 мкм.
При этом материалами носителя могут быть обычные пористые или непористые оксиды алюминия, диоксид кремния, диоксид тория, диоксид циркония, карбид кремния или силикаты, такие как силикат магния или силикат алюминия. В условиях осуществления целевой реакции в соответствии с предлагаемым в изобретении способом указанные материалы ведут себя, как правило, преимущественно инертно. Телам носителя можно придать регулярную или нерегулярную форму, причем предпочтительными являются тела носителя регулярной формы, например шарики или полые цилиндры, с отчетливо выраженной шероховатостью поверхности. Пригодным является применение преимущественно непористых сферических тел носителя из стеатита, диаметр которых составляет от 1 до 10, соответственно 8 мм, предпочтительно от 4 до 5 мм, и которые обладают шероховатой поверхностью. Пригодным является также применение тел носителя в виде цилиндров высотой от 2 до 10 мм с наружным диаметром от 4 до 10 мм. В случае, если пригодными согласно изобретению телами носителя являются кольца, то, кроме того, они характеризуются толщиной стенок, обычно составляющей от 1 до 4 мм. Согласно изобретению подлежащие предпочтительному использованию кольцеобразные тела носителя обладают высотой от 2 до 6 мм, наружным диаметром от 4 до 8 мм и толщиной стенок от 1 до 2 мм. Согласно изобретению в качестве тел носителя прежде всего пригодны также кольца с размерами 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр). Толщину слоя каталитически активных оксидных масс, наносимого на поверхность тела носителя, очевидно, необходимо приводить в соответствие с требуемой толщиной оболочки (смотри европейскую заявку на патент ЕР-А 714700).
Кроме того, активными мультиметаллоксидными массами, подлежащими применению на стадии превращения пропилена в акролеин, являются массы общей формулы V

переменные в которой означают следующее:
Y1 означает только висмут или висмут и по меньшей мере один из следующих элементов: теллур, сурьма, олово или медь,
Y2 означает молибден или молибден и вольфрам,
Y3 означает щелочной металл, таллий и/или самарий,
Y4 означает щелочноземельный металл, никель, кобальт, медь, марганец, цинк, олово, кадмий и/или ртуть,
Y5 означает железо или железо и по меньшей мере один из следующих элементов: хром и церий,
Y6 означает фосфор, мышьяк, бор и/или сурьму,
Y7 означает редкоземельный металл, титан, цирконий, ниобий, тантал, рений, рутений, родий, серебро, золото, алюминий, галлий, индий, кремний, германий, свинец, торий и/или уран,
а′ означает число от 0,01 до 8,
b′ означает число от 0,1 до 30,
с′ означает число от 0 до 4,
d′ означает число от 0 до 20,
е′ означает число от >0 до 20,
f′ означает число от 0 до 6,
g′ означает число от 0 до 15,
h′ означает число от 8 до 16,
x′, y′ означают числа, которые определяются валентностью и повторяемостью отличающихся от кислорода элементов в общей формуле V, и
p, q означают числа, отношение которых (p/q) составляет от 0,1 до 10,
содержащие трехмерные области химического состава Y1a'Y2b'Ox', отграниченные от их локального окружения в связи с отличием их состава от состава локального окружения, причем максимальный диаметр подобных областей (наиболее длинный отрезок, проходящий через центр их тяжести и соединяющий две находящиеся на поверхности области (граничной поверхности) точки) составляет от 1 нм до 100 мкм, нередко от 10 до 500 нм или от 1 мкм до 50, соответственно 25 мкм.
Согласно изобретению особенно предпочтительными являются такие мультиметаллоксидные массы общей формулы V, в которых Y1 означает только висмут.
Из подобных масс предпочтительными, в свою очередь, являются мультиметаллоксидные массы общей формулы VI

переменные в которой означают следующее:
Z2 означает молибден или молибден и вольфрам,
Z3 означает никель и/или кобальт,
Z4 означает таллий, щелочной металл и/или щелочноземельный металл,
Z5 означает фосфор, мышьяк, бор, сурьму, олово, церий и/или свинец,
Z6 означает кремний, алюминий, титан и/или цирконий,
Z7 означает медь, серебро и/или золото,
а′′ означает число от 0,1 до 1,
b′′ означает число от 0,2 до 2,
с′′ означает число от 3 до 10,
d′′ означает число от 0,02 до 2,
е′′ означает число от 0,01 до 5, предпочтительно от 0,1 до 3,
f′′ означает число от 0 до 5,
g′′ означает число от 0 до 10,
h′′ означает число от 0 до 1,
x′′, y′′ означают числа, которые определяются валентностью и повторяемостью отличающихся от кислорода элементов в общей формуле VI,
p′′, q′′ означают числа, отношение которых (p′′/q′′) составляет от 0,1 до 5,
предпочтительно от 0,5 до 2,
причем еще более предпочтительными являются такие мультиметаллоксидные массы общей формулы VI, в которых Z2b′′ означает (вольфрам)b′′ и Z212 означает (молибден)12.
Кроме того, предпочтительно, если по меньшей мере 25% мол. (предпочтительно по меньшей мере 50% мол. и особенно предпочтительно 100% мол.) общего количества [Y1a′Y2b′Ox′]p (Biа′′Z2b′′Ох′′]p′′) в пригодных согласно изобретению мультиметаллоксидных массах общей формулы V (мультиметаллоксидных массах общей формулы VI) присутствует в виде трехмерных областей химического состава Y1a′Y2b′Ox′[Bia′′Z2b′′Ox′′], отграниченных от их локального окружения в связи с отличием их состава от состава локального окружения, причем максимальный диаметр подобных областей составляет от 1 нм до 100 мкм.
Формование катализаторов с мультиметаллоксидными массами общей формулы V осуществляют аналогично формованию катализаторов с мультиметаллоксидными массами общей формулы IV.
Приготовление активных мультиметаллоксидных масс общей формулы V описано, например, в европейской заявке на патент ЕР-А 575897, а также в немецкой заявке на патент DE-A 19855913.
Рекомендованные выше инертные материалы носителя, в частности, пригодны также в качестве инертных материалов для разбавления и/или отграничения соответствующих стационарных слоев катализатора, соответственно в качестве защиты этих слоев и/или предваряющей их засыпки, предназначенной для нагревания газовой смеси.
Следует подчеркнуть, что любые катализаторы и мультиметаллоксидные массы, рекомендованные выше в качестве пригодных для осуществления стадии превращения пропилена в акролеин, в принципе пригодны также для частичного окисления пропилена в акрилонитрил.
Как указано выше, в качестве активных масс для осуществления второй стадии (гетерогенно катализируемого газофазного частичного окисления акролеина в акриловую кислоту) в принципе пригодны любые содержащие молибден (Мо) и ванадий (V) мультиметаллоксидные массы, например, приведенные в немецкой заявке на патент DE-A 10046928.
Множество подобных мультиметаллоксидных масс, например, приведенных в немецкой заявке на патент DE-A 19815281, могут быть описаны общей формулой VII
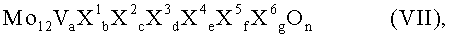
переменные в которой означают следующее:
X1 означает вольфрам (W), ниобий (Nb), тантал (Та), хром (Cr) и/или церий (Се),
X2 означает медь (Cu), никель (Ni), кобальт (Со), железо (Fe), марганец (Mn) и/или цинк (Zn),
X3 означает сурьму (Sb) и/или висмут (Bi),
X4 означает один или несколько щелочных металлов,
X5 означает один или несколько щелочноземельных металлов,
X6 означает кремний (Si), алюминий (Аl), титан (Ti) и/или цирконий (Zr),
а означает число от 1 до 6,
b означает число от 0,2 до 4,
с означает число от 0,5 до 18,
d означает число от 0 до 40,
е означает число от 0 до 2,
f означает число от 0 до 4,
g означает число от 0 до 40 и
n означает число, которое определяется валентностью и повторяемостью отличающихся от кислорода элементов в общей формуле VII.
Согласно изобретению предпочтительными являются активные мультиметаллоксидные массы общей формулы VII, переменные в которой означают следующее:
X1 означает вольфрам (W), ниобий (Nb) и/или хром (Cr),
X2 означает медь (Cu), никель (Ni), кобальт (Со) и/или железо (Fe),
X3 означает сурьму (Sb),
X4 означает натрий (Na) и/или калий (K),
X5 означает кальций (Са), стронций (Sr) и/или барий (Ва),
X6 означает кремний (Si), алюминий (Аl) и/или титан (Ti),
а означает число от 1,5 до 5,
b означает число от 0,5 до 2,
с означает число от 0,5 до 3,
d означает число от 0 до 2,
е означает число от 0 до 0,2,
f означает число от 0 до 1 и
n означает число, которое определяется валентностью и повторяемостью отличающихся от кислорода элементов в общей формуле VII.
Согласно изобретению еще более предпочтительными мультиметаллоксидными массами общей формулы VII являются такие, которые, в свою очередь, обладают общей формулой VIII

в которой Y1 означает вольфрам (W) и/или ниобий (Nb),
Y2 означает медь (Cu) и/или никель (Ni),
Y5 означает кальций (Са) и/или стронций (Sr),
Y6 означает кремний (Si) и/или алюминий (Al),
а′ означает число от 2 до 4,
b′ означает число от 1 до 1,5,
с′ означает число от 1 до 3,
f′ означает число от 0 до 0,5,
g′ означает число от 0 до 8 и
n′ означает число, которое определяется валентностью и повторяемостью отличающихся от кислорода элементов в общей формуле VIII.
Согласно изобретению пригодные активные мультиметаллоксидные массы общей формулы (VII) могут быть получены известными методами, например, приведенными в немецкой заявке на патент DE-A 4335973 или европейской заявке на патент ЕР-А 714700.
Активные мультиметаллоксидные массы, пригодные для реализации стадии «акролеин → акриловая кислота» и обладающие прежде всего общей формулой VII, в принципе можно получить известным методом, состоящим в том, что, используя пригодные источники образующих их элементарных компонентов, получают как можно более тщательно перемешанную, предпочтительно мелкодисперсную сухую смесь соответствующего стехиометрического состава, которую кальцинируют при температуре от 350 до 600°С. Кальцинирование можно осуществлять как в атмосфере инертного газа, так и в окисляющей атмосфере, например, такой как воздух (смесь инертного газа и кислорода), а также в восстанавливающей атмосфере (например, в смеси инертного газа с восстанавливающими газами, такими как водород, аммиак, монооксид углерода, метан и/или акролеин, или в самих газах с восстанавливающим действием). Длительность кальцинирования может составлять от нескольких минут до нескольких часов и может быть сокращена при повышении температуры этого процесса. Источниками элементарных компонентов активных мультиметаллоксидных масс общей формулы VII могут служить соединения, которые уже являются оксидами, и/или соединения, которые можно перевести в оксиды нагреванием, осуществляемым в присутствии по меньшей мере кислорода.
Тщательному перемешиванию можно подвергать исходные соединения для получения активных мультиметаллоксидных масс общей формулы VII, находящиеся как в сухом, так и во влажном состоянии. При перемешивании в сухом состоянии исходные соединения целесообразно использовать в виде мелкодисперсных порошков, а после перемешивания и при необходимости осуществляемого уплотнения подвергать кальцинированию. Однако тщательное перемешивание исходных соединений предпочтительно осуществляют во влажном состоянии.
При этом исходные соединения обычно смешивают друг с другом в виде водного раствора и/или суспензии. Особенно тщательно перемешанные указанным выше методом сухие смеси образуются в том случае, если исходят из имеющихся источников элементарных компонентов, которые находятся исключительно в растворенном состоянии. В качестве растворителя предпочтительно используют воду. Полученную водную массу, в заключение, подвергают сушке, которую предпочтительно осуществляют методом распылительной сушки водной смеси при температуре на выходе, составляющей от 100 до 150°С.
Получаемые в результате этого активные мультиметаллоксидные массы прежде всего общей формулы VII можно использовать для окисления акролеина как в порошкообразном виде, так и в виде получаемого формованием катализатора определенной геометрической формы, которое можно выполнять до или после заключительного кальцинирования. Так, например, уплотнением находящейся в порошкообразном виде активной массы или соответствующей не кальцинированной и/или частично кальцинированной предварительной массы (например, таблетированием, экструдированием или штранг-прессованием) можно изготовить сплошные катализаторы требуемой геометрической формы, причем при необходимости могут быть добавлены вспомогательные средства, например, такие как графит или стеариновая кислота, в качестве улучшающих скольжение средств и/или средств облегчения формования, и армирующие средства, такие как микроволокна из стекла, асбеста, карбида кремния или титаната калия. Пригодной геометрической форме сплошных катализаторов соответствуют например, сплошные или полые цилиндры с наружным диаметром и высотой от 2 до 10 мм. Целесообразная толщина стенок полых цилиндров составляет от 1 до 3 мм. Сплошной катализатор, очевидно, может также обладать формой шариков, диаметр которых может составлять от 2 до 10 мм (например, 8,2 мм или 5,1 мм).
Формование порошкообразной активной массы или соответствующей порошкообразной, не подвергнутой кальцинированию предварительной массы, очевидно, можно выполнять также ее нанесением на предварительно формованный инертный носитель для катализаторов. Нанесение активной массы на тела носителя с целью получения слоистых катализаторов, как правило, осуществляют в пригодном вращающемся резервуаре, например таком, как описан в немецкой заявке на патент DE-A 2909671 или европейских заявках на патент ЕР-А 293859 или ЕР-А 714700.
Порошкообразную массу, подлежащую нанесению на тела носителя, целесообразно увлажнить, а после нанесения вновь подвергнуть сушке, например, горячим воздухом. Целесообразная толщина нанесенного на тела носителя слоя порошкообразной массы составляет от 10 до 1000 мкм, предпочтительно от 50 до 500 мкм и особенно предпочтительно от 150 до 250 мкм.
При этом материалами носителя могут быть обычные пористые или непористые оксиды алюминия, диоксид кремния, диоксид тория, диоксид циркония, карбид кремния или силикаты, такие как силикат магния или силикат алюминия. Телам носителя можно придавать регулярную или нерегулярную форму, причем предпочтительными являются тела носителя регулярной формы с отчетливо выраженной шероховатостью поверхности, например шарики или полые цилиндры с расщепленной опорной поверхностью. Пригодным является применение преимущественно непористых сферических тел носителя из стеатита с шероховатой поверхностью, диаметр которых составляет от 1 до 10 мм, соответственно до 8 мм, предпочтительно от 4 до 5 мм. Таким образом, пригодные сферические тела носителя обладают диаметром 8,2 мм или 5,1 мм. Пригодным является также применение тел носителя в виде цилиндров высотой от 2 до 10 мм с наружным диаметром от 4 до 10 мм. При использовании в качестве тел носителя колец они, кроме того, характеризуются толщиной стенок, обычно составляющей от 1 до 4 мм. Подлежащие предпочтительному использованию кольцеобразные тела носителя обладают высотой от 2 до 6 мм, наружным диаметром от 4 до 8 мм и толщиной стенок от 1 до 2 мм. В качестве тел носителя прежде всего пригодны также кольца с размерами 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр). Толщину наносимого на поверхность тел носителя слоя каталитически активных оксидных масс, очевидно, следует приводить в соответствие с требуемой толщиной оболочки (смотри европейскую заявку на патент ЕР-А 714700).
Кроме того, подлежащие применению активные мультиметаллоксидные массы, оптимальные для реализации стадии «акролеин → акриловая кислота», обладают общей формулой IX

переменные в которой означают следующее:
D означает Мо12Va′′Z1b′′Z2c′′Z3d′′Z4e′′Z5f′′Z6g′′Oх′′,
Е означает Z712Cuh′′Hi′′Oy′′,
Z1 означает вольфрам (W), ниобий (Nb), тантал (Та), хром (Cr) и/или церий (Се),
Z2 означает медь (Cu), никель (Ni), кобальт (Со), железо (Fe), марганец (Mn) и/или цинк (Zn),
Z3 означает сурьму (Sb) и/или висмут (Bi),
Z4 означает литий (Li), натрий (Na), калий (K), рубидий (Rb), цезий (Cs) и/или водород (Н),
Z5 означает магний (Мg), кальций (Са), стронций (Sr) и/или барий (Ва),
Z6 означает кремний (Si), алюминий (Al), титан (Ti) и/или цирконий (Zr),
Z7 означает молибден (Мо), вольфрам (W), ванадий (V), ниобий (Nb) и/или тантал (Та), предпочтительно Мо и/или W,
а′′ означает число от 1 до 8,
b′′ означает число от 0,2 до 5,
с′′ означает число от 0 до 23,
d′′ означает число от 0 до 50,
е′′ означает число от 0 до 2,
f′′ означает число от 0 до 5,
g′′ означает число от 0 до 50,
h′′ означает число от 4 до 30,
i′′ означает число от 0 до 20,
x′′, y′′ означают числа, которые определяются валентностью и повторяемостью отличающихся от кислорода элементов в общей формуле IX, и
p, q означают отличающиеся от нуля числа, отношение которых (p/q) составляет от 160:1 до 1:1,
и могут быть получены отдельным предварительным формованием находящейся в порошкообразном состоянии твердой мультиметаллоксидной массы Е (исходной массы 1)

которую затем при необходимом количественном отношении р к q вводят в водный раствор, водную суспензию или мелкодисперсную сухую стехиометрическую смесь D источников элементов Мо, V, Z1, Z2, Z3, Z4, Z5, Z6 (исходную массу 2)

при необходимости получаемую при этом водную смесь сушат и сухую предварительную массу до или после сушки кальцинируют при температуре от 250 до 600°С, получая катализатор требуемой геометрической формы.
Предпочтительными являются мультиметаллоксидные массы общей формулы IX, которые получают, вводя предварительно сформованную твердую исходную массу 1 в водную исходную массу 2 при температуре ниже 70°С. Получение катализаторов с мультиметаллоксидными массами общей формулы VI подробно описано, например, в европейской заявке на патент ЕР-А 668104 и немецких заявках на патент DE-A 19736105, DE-A 10046928, DE-A 19740493 и DE-A 19528646.
Формование катализаторов с мультиметаллоксидными массами общей формулы IX осуществляют аналогично формованию катализаторов с мультиметаллоксидными массами общей формулы VII.
Отлично пригодные для реализации стадии «акролеин → акриловая кислота» мультиметаллоксидные катализаторы, прежде всего содержащие активные мультиметаллоксидные массы общей формулы I согласно настоящему описанию, приведены также в немецкой заявке на патент DE-A 19815281.
Для реализации стадии превращения пропилена в акролеин предпочтительно используют сплошные кольцеобразные катализаторы, в то время как для реализации стадии превращения акролеина в акриловую кислоту предпочтительно используют слоистые кольцеобразные катализаторы.
Первую стадию частичного окисления (превращения пропилена в акролеин) с помощью рассмотренных выше катализаторов можно осуществлять, например, в состоящем из множества контактных трубок (то есть кожухотрубном) однозонном реакторе со стационарным слоем катализатора, описанном в немецкой заявке на патент DE-A 4431957.
В качестве окислительного агента используют кислород. При выборе азота в качестве инертного разбавляющего газа особенно предпочтительным источником кислорода является воздух.
Объемное отношение (в нормальных литрах) пропилен:кислород:инертные газы (включая водяной пар) чаще всего составляет 1:(1,0-3,0):(5-25), предпочтительно 1:(1,7-2,3):(10-15). Реакционное давление обычно находится в интервале от 1 до 3 бар, среднечасовая скорость пропускания реакционного газа предпочтительно составляет от 1500 нл/л·ч до 4000, соответственно 6000 нл/л·ч или более. Среднечасовая скорость пропускания пропилена обычно составляет от 90 нл/л·ч до 200 или 300 нл/л·ч или более.
Исходная газовая смесь поступает в указанный выше однозонный кожухотрубный реактор со стационарным слоем катализатора предпочтительно сверху. В качестве теплоносителя целесообразно использовать солевой расплав, предпочтительно состоящий из 60% мас. нитрата калия (KNО3) и 40% мас. нитрита натрия (NaNO2) или из 53% мас. нитрата калия (KNО3), 40% мас. нитрита натрия (NaNO2) и 7% мас. нитрата натрия (NaNO3).
Возможно пропускание солевого расплава и реакционной газовой смеси через реактор как прямотоком, так и противотоком (если смотреть на реактор сверху). Солевой расплав омывает контактные трубки предпочтительно в виде извилистого потока.
При пропускании реакционной газовой смеси через контактные трубки в направлении сверху вниз их заполнение катализатором целесообразно осуществлять в следующей последовательности (в направлении снизу вверх) (в случае пропускания реакционной газовой смеси через контактные трубки в направлении снизу вверх целесообразной является обратная последовательность загрузки катализатора):
- нижние участки контактных трубок (от 40 до 80%, соответственно до 60% их общей высоты) заполняют либо только катализатором, либо смесью катализатора с инертным материалом, содержание которого в этой смеси составляет до 20% мас. (секция С реактора),
- следующие по уровню расположения участки контактных трубок (от 20 до 50%, соответственно до 40% их общей высоты) заполняют либо только катализатором, либо смесью катализатора с инертным материалом, содержание которого в этой смеси составляет до 40% мас. (секция В реактора), и
- самые верхние участки контактных трубок (от 10 до 20% их общей высоты) заполняют инертным материалом (секция А реактора) предпочтительно таким образом, чтобы в этой секции реактора была обеспечена максимально низкая потеря напора потока реакционной газовой смеси.
В секцию С предпочтительно загружают катализатор, не разбавленный инертным материалом.
Приведенный выше вариант прежде всего целесообразно использовать для заполнения реактора катализаторами, подобными указанным в примере 1 или 3 немецкой заявки на патент DE-A 10046957, и инертным материалом, представляющим собой кольца из стеатита с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × высота × внутренний диаметр). Температура солевой бани соответствует температуре, указанной в немецкой заявке на патент DE-A 4431957.
Первую стадию частичного окисления (превращения пропилена в акролеин) на рассмотренных выше катализаторах можно осуществлять также, например, в состоящем из множества контактных трубок (кожухотрубном) двухзонном реакторе со стационарным слоем, подобном описанному в немецкой заявке на патент DE-A 19910506. В обоих рассмотренных выше случаях конверсия пропилена при его однократном пропускании через реактор обычно составляет ≥90% мол., соответственно ≥95% мол. Вторую стадию частичного окисления (превращения акролеина в акриловую кислоту) на рассмотренных выше катализаторах можно осуществлять, например, в однозонном кожухотрубном реакторе со стационарным слоем катализатора, подобном описанному в немецкой заявке на патент DE-A 4431949.
Смесь продуктов частичного окисления пропилена в акролеин, как правило, направляют на стадию окисления акролеина в акриловую кислоту как таковую (при необходимости после промежуточного охлаждения), то есть без выделения побочных продуктов.
При этом кислород, необходимый для осуществления второй стадии частичного окисления, предпочтительно вводят в виде воздуха и, как правило, непосредственно добавляют к газообразной смеси продуктов частичного окисления пропилена (однако кислород уже может присутствовать в исходной реакционной газовой смеси, используемой для частичного окисления пропилена).
В этом случае составу газовой смеси, направляемой на частичное окисление акролеина, как правило, соответствует объемное отношение (в нормальных литрах) акролеин:кислород:водяной пар:инертный газ, составляющее 1:(1-3):(0-20):(3-30), предпочтительно 1:(1-3):(0,5-10):(7-18).
Реакционное давление в данном случае также, как правило, составляет от 1 до 3 бар, а среднечасовая скорость пропускания реакционного газа предпочтительно находится в интервале от 1000 нл/л·ч до 3800, соответственно 4800 нл/л·ч или более. Среднечасовая скорость пропускания акролеина обычно составляет от 80 нл/л·ч до 190 нл/л·ч, соответственно 290 нл/л·ч или более.
Исходную газовую смесь вводят в однозонный кожухотрубный реактор со стационарным слоем катализатора второй стадии синтеза также предпочтительно сверху. В качестве теплоносителя на второй стадии синтеза также целесообразно использовать солевой расплав, предпочтительно состоящий из 60% мас. нитрата калия (KNO3) и 40% мас. нитрита натрия (NaNO2) или из 53% мас. нитрата калия (KNO3), 40% мас. нитрита натрия (NaNO2) и 7% мас. нитрата натрия (NаNО3). Солевой расплав и реакционную газовую смесь можно пропускать через реактор как прямотоком, так и противотоком (если смотреть на реактор сверху). Солевой расплав омывает контактные трубки предпочтительно в виде извилистого потока.
В случае пропускания газовой смеси через контактные трубки в направлении сверху вниз их заполнение катализатором целесообразно осуществлять в следующей последовательности (в направлении снизу вверх):
- нижние участки контактных трубок (от 50 до 80%, соответственно до 70% их общей высоты) заполняют либо только катализатором, либо смесью катализатора с инертным материалом, содержание которого в этой смеси составляет до 20% мас. (секция С реактора),
- следующие по уровню расположения участки контактных трубок (от 20 до 40% их общей высоты) заполняют либо только катализатором, либо смесью катализатора с инертным материалом, содержание которого в этой смеси составляет до 50% мас., соответственно до 40% мас. (секция В реактора), и
- самые верхние участки контактных трубок (от 5 до 20% их общей высоты) заполняют инертным материалом (секция А реактора) предпочтительно таким образом, чтобы в этой секции реактора была обеспечена максимально низкая потеря напора потока газовой смеси.
В секцию С предпочтительно загружают катализатор, не разбавленный инертным материалом. Подобно тому, как принято в общем случае для гетерогенно катализируемого газофазного частичного окисления акролеина в акриловую кислоту (прежде всего при высокой среднечасовой скорости пропускания акролеина через слой катализатора и высоких содержаниях водяного пара в исходной газовой смеси), секция В может состоять также из двух последовательных слоев разбавленного катализатора (с целью минимализации температуры в горячих точках и чувствительности этой температуры). При этом в нижнюю часть секции В загружают катализатор, содержащий до 20% мас. инертного материала, а верхнюю часть этой секции заполняют катализатором, содержащим от >20 до 50% мас., соответственно до 40% мас. инертного материала. В этом случае загружаемый в секцию С катализатор предпочтительно не разбавляют инертным материалом.
В случае пропускания исходной газовой смеси через контактные трубки в направлении снизу вверх целесообразной является последовательность их заполнения катализатором, обратная вышеуказанной.
Приведенный выше вариант прежде всего целесообразно использовать для заполнения реактора катализаторами, подобными указанным в примере 5 немецкой заявки на патент DE-A 10046928 или в примерах из немецкой заявки на патент DE-A 19815281, и инертным материалом, представляющим собой кольца из стеатита с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × высота × внутренний диаметр). Температура солевого расплава соответствует температуре, указанной в немецкой заявке на патент DE-A 4431949. Ее, как правило, выбирают таким образом, чтобы при однократном пропускании акролеина через реактор его конверсия обычно составляла ≥90% мол., соответственно ≥95% мол. или ≥99% мол.
Вторую стадию частичного окисления (превращения акролеина в акриловую кислоту) на рассмотренных выше катализаторах можно осуществлять также, например, в состоящем из множества контактных трубок (кожухотрубном) двухзонном реакторе со стационарным слоем, подобным описанному в немецкой заявке на патент DE-A 19910506. Для достижения необходимой степени превращения акролеина справедливы указанные выше условия. В случае осуществления второй стадии в кожухотрубном двухзонном реакторе для приготовления подаваемой в него газовой смеси целесообразным является также непосредственное использование направляемой на первую стадию газообразной смеси продуктов частичного окисления (после при необходимости осуществляемого промежуточного охлаждения) (как это было описано выше). При этом кислород, необходимый для осуществления второй стадии частичного окисления, предпочтительно вводят в виде воздуха и во втором случае добавляют непосредственно к газообразной смеси продуктов первой стадии частичного окисления.
При двухступенчатой технологии с непосредственным использованием газообразной смеси продуктов первой стадии частичного окисления для питания второй стадии частичного окисления, как правило, два однозонных кожухотрубных реактора со стационарными слоями катализатора последовательно соединяют с двумя двухзонными кожухотрубными реакторами (при характерной в общем случае высокой скорости пропускания реагентов через катализатор предпочтительным считается прямоток реакционного газа и солевого расплава (теплоносителя) через кожухотрубный реактор). Возможным является также смешанное последовательное соединение реакторов (однозонный/двухзонный или наоборот).
Между реакторами может располагаться промежуточный охладитель, который при необходимости может содержать инертные сыпучие массы, способные выполнять функцию фильтрования. Температура солевого расплава в кожухотрубных реакторах первой стадии частичного окисления пропилена в акриловую кислоту, как правило, составляет от 300 до 400°С. Температура солевого расплава в кожухотрубных реакторах второй стадии частичного окисления пропилена в акриловую кислоту (частичного окисления акролеина в акриловую кислоту) чаще всего составляет от 200 до 350°С. Кроме того, теплоносители (предпочтительно солевые расплавы) обычно пропускают через соответствующие кожухотрубные реакторы со стационарным слоем катализатора с таким расходом, чтобы разница их температур на входе и выходе, как правило, составляла ≤5°С. Как сообщалось выше, обе стадии частичного окисления пропилена в акриловую кислоту можно осуществлять также в одном реакторе на одном загруженном в него катализаторе, как описано в немецкой заявке на патент DE-A 10121592.
Необходимо отметить также, что часть газовой смеси, подаваемой на первую стадию синтеза («пропилен → акролеин»), может являться циркуляционным (остаточным) газом частичного окисления.
Как указано выше, речь при этом идет о содержащем молекулярный кислород газе, который остается после выделения целевого продукта (акролеина и/или акриловой кислоты) из газообразной смеси продуктов частичного окисления и часть которого можно рециркулировать в качестве инертного разбавляющего газа для питания первой и/или при необходимости второй стадии частичного окисления пропилена в акролеин и/или акриловую кислоту.
Однако подобный циркуляционный (остаточный) газ, содержащий пропан, молекулярный кислород и при необходимости непревращенный пропилен, предпочтительно рециркулируют только в соответствии с предлагаемым в изобретении способом в реакционную зону А.
Необходимо отметить также, что предлагаемое в изобретении частичное окисление можно осуществлять, сначала пропуская через катализатор не содержащую кислород реакционную газовую смесь. В подобном случае необходимый для частичного окисления кислород предоставляет кристаллическая решетка. На последующей стадии регенерации слой катализатора подвергают регенерации содержащим кислород газом (например, воздухом, воздухом, обогащенным кислородом, или воздухом, обедненным кислородом), благодаря чему не содержащая кислород реакционная смесь вновь содержит его, и так далее.
Кожухотрубный реактор с загруженным в него катализатором, соответствующим образом изменяющимся вдоль отдельных контактных трубок с завершением первой реакционной стадии (подобные процессы частичного окисления пропилена, пригодные согласно изобретению в качестве реакционной зоны В, описаны, например, в европейских заявках на патент ЕР-А 911313, ЕР-А 979813 и ЕР-А 990636 и немецкой заявке на патент DE-A 2830765), представляет собой простейшую форму реализации двух зон обеих стадий частичного окисления пропилена в акриловую кислоту. При этом загруженный в контактные трубки катализатор при необходимости прерывается слоем инертного материала.
Однако обе зоны окисления предпочтительно реализуют в виде двух последовательно соединенных кожухотрубных систем. Подобные системы могут находиться в одном реакторе, в котором переход от одного пучка труб к другому образован загруженным не в контактные трубки (целесообразно доступным для прохода) инертным материалом. Теплоноситель, как правило, омывает контактные трубки, однако не контактирует с инертным материалом. В связи с этим оба пучка контактных трубок предпочтительно располагают пространственно изолированно друг от друга. При этом между обоими кожухотрубными реакторами, как правило, располагается промежуточный охладитель, предназначенный для минимизации в некоторых случаях происходящего вторичного окисления акролеина в газообразной смеси продуктов, выходящей из первой зоны окисления. Вместо кожухотрубных реакторов можно использовать также реакторы в виде пластинчатых теплообменников с солевым и/или испарительным охлаждением, например таких, как описаны в немецких заявках на патент DE-A 19929487 и DE-A 19952964.
Реакционная температура в первой зоне окисления, как правило, составляет от 300 до 450°С, предпочтительно от 320 до 390°С. Реакционная температура во второй зоне окисления, как правило, составляет от 200 до 370°С, часто от 220 до 330°С. Целесообразное давление в обеих зонах окисления находится в интервале от 0,5 до 5 бар, предпочтительно от 1 до 3 бар. Среднечасовая скорость пропускания реакционного газа (нл/л·ч) через катализаторы окисления обеих зон часто составляет от 1500 до 2500 ч-1, соответственно до 4000 ч-1. При этом среднечасовая скорость пропускания пропилена может составлять от 100 до 200 или 300 нл/л·ч и более.
Обе зоны окисления согласно предлагаемому в изобретении способу в принципе можно сконструировать, например, как описано, в немецких заявках на патент DE-A 19837517, DE-A 19910506, DE-A 19910508 и DE-A 19837519. Наружное термостатирование обеих зон окисления (при необходимости в системах многозонных реакторов) обычно известным образом согласуют со специфическим составом реакционной газовой смеси, а также загруженным катализатором.
В случае двухступенчатого частичного окисления пропилена в акриловую кислоту общий молекулярный кислород, необходимой в качестве окислительного агента для предлагаемой в изобретении реакционной зоны В, можно в полном объеме заранее добавлять к подаваемой на стадию окисления пропилена газовой смеси. Однако кислород, очевидно, можно добавлять и после стадии окисления пропилена. Предпочтительным является добавление кислорода на стадии синтеза акриловой кислоты.
При этом молярное отношение пропилена к молекулярному кислороду, предпочтительно устанавливаемое в первой зоне окисления (пропилен→акролеин), составляет 1:(1-3), часто 1:(1,5-2). Для второй зоны (частичного окисления акролеина в акриловую кислоту) пригодно аналогичное в численном выражении молярное отношение акролеина к молекулярному кислороду (предпочтительным было бы отношение 1:(0,5-1,5).
При этом избыток молекулярного кислорода, как правило, благоприятно отражается на кинетике реализуемого в обеих зонах газофазного окисления. В отличие от реакционной зоны А предлагаемого в изобретении способа молярное отношение реагентов преимущественно не оказывает влияния на термодинамические условия в реакционной зоне В, поскольку гетерогенно катализируемое газофазное частичное окисление пропилена в акриловую кислоту является кинетически контролируемым процессом. В связи с этим в первой зоне окисления в принципе можно использовать, например, также молярный избыток пропилена по отношению к молекулярному кислороду. В подобном случае избыточный пропилен фактически играет роль разбавляющего газа.
Гетерогенно катализируемое газофазное частичное окисление пропилена в акриловую кислоту в принципе можно реализовать также в единственной зоне окисления. В этом случае обе реакционные стадии осуществляют в одном реакторе окисления, заполненном катализатором, который способен катализировать обе реакции. Возможным, очевидно, является также непрерывное или резкое изменение загрузки катализатора вдоль реакционной координаты в пределах зоны окисления. В соответствии с одним из возможных вариантов осуществления изобретения при по меньшей мере одном частичном окислении, реализуемом в виде двух последовательно соединенных зон окисления, из газообразной смеси продуктов первой зоны окисления при необходимости, очевидно, можно частично или полностью выделять содержащийся в ней оксид углерода и водяной пар, которые образуются в этой зоне в качестве побочных продуктов, прежде чем направить полученную в первой зоне газообразную смесь во вторую зону окисления. Согласно изобретению предпочтительно выбирают технологию, которая не требует подобного выделения.
В качестве источника молекулярного кислорода, необходимого для реализации по меньшей мере одного частичного окисления и добавляемого, например, к газообразной смеси продуктов А′ перед ее использованием для подачи в реакционную зону В, пригоден как чистый молекулярный кислород, так и молекулярный кислород или оксид азота, разбавленный инертным газом, таким как диоксид углерода, монооксид углерода, благородные газы, азот и/или насыщенные углеводороды.
Для удовлетворения по меньшей мере частичной потребности в молекулярном кислороде в качестве его источника целесообразно использовать воздух.
Добавление холодного воздуха к горячей газообразной смеси продуктов А′ позволяет также обеспечивать ее непосредственное охлаждение при осуществлении предлагаемого в изобретении способа.
Длительная эксплуатация реакционных зон А и В обычно сопровождается ухудшением качества используемых в них катализаторов (прежде всего снижением активности и селективности). Этому можно противодействовать, во-первых, благодаря периодически осуществляемой регенерации соответствующего слоя катализатора (для этого в зонах окисления через соответствующий слой катализатора можно пропускать при повышенной температуре, например, также остаточный газ), например, как предложено в немецких заявках на патент DE-A 10351269, DE-A 10350812 и DE-A 10350822. Срок службы катализатора можно увеличить также благодаря повышению реакционной температуры, компенсирующему снижение активности катализатора. Однако неожиданно выяснилось, что дополнительное противодействие деактивированию катализаторов в реакционных зонах А, В в период их эксплуатации предпочтительно можно оказать благодаря повышению соответствующего рабочего давления в газовой фазе (в пересчете на одинаковую скорость пропускания реакционной газовой смеси через соответствующий слой катализатора в нл/л·ч), например, как это предложено в немецкой заявке на патент DE-A 102004025445. С этой целью после каждой реакционной зоны и зоны разделения, используемых согласно предлагаемому в изобретении способу, можно смонтировать, например, регулятор давления (в простейшем случае речь идет о дросселирующем устройстве, например дроссельном клапане или направляющем устройстве для закручивания потока) (например, также о снабженных отверстиями частично проницаемых диафрагмах с возможностью частичного или полного постепенного закрывания). Подобный регулятор давления, очевидно, можно смонтировать также только после одной из указанных выше зон. Таким образом, в принципе вполне достаточно смонтировать регулятор давления в каком-либо другом месте потока реакционного газа, откуда повышение давления благодаря обратному подпору может передаваться в реакционную зону. Рассмотренное выше повышение давления (которое можно реализовать непрерывно в соответствии с происходящим деактивированием катализатора, а также периодически) в течение эксплуатационного периода в типичном случае может составлять 3000 мбар и более.
Газообразная смесь продуктов В, выходящая из используемой согласно изобретению реакционной зоны В при синтезе акролеина и/или акриловой кислоты, как правило, преимущественно состоит из целевого продукта (акролеина, акриловой кислоты или смеси акролеина с акриловой кислотой), непревращенного молекулярного кислорода, пропана, непревращенного пропилена, молекулярного азота, водяного пара, образующегося в качестве побочного продукта и/или совместно используемого в качестве разбавляющего газа, оксидов углерода, образующихся в качестве побочных продуктов и/или совместно используемых в качестве разбавляющих газов, а также из незначительных количеств прочих низших альдегидов, низших алканкарбоновых кислот (например, уксусной кислоты, муравьиной кислоты и пропионовой кислоты), малеинового ангидрида, бензальдегида, ароматических карбоновых кислот и ангидридов ароматических карбоновых кислот (например, фталевого ангидрида и бензойной кислоты), в некоторых случаях других углеводородов, например, таких как углеводороды с четырьмя атомами углерода (например, бутен-1 и возможно другие бутены), а также других инертных разбавляющих газов.
Целевой продукт можно выделять из газообразной смеси продуктов В в зоне разделения В известными методами (например, частичной или полной, а также при необходимости фракционирующей конденсацией акриловой кислоты, абсорбцией акриловой кислоты водой или высококипящим гидрофобным органическим растворителем или абсорбцией акролеина водой или водными растворами низших карбоновых кислот, а также последующей переработкой конденсатов и/или абсорбатов; согласно изобретению предпочтительной является фракционирующая конденсация газообразной смеси продуктов В; смотри, например, европейские заявки на патент ЕР-А 1388533 и ЕР-А 1388532, немецкую заявку на патент DE-А 10235847, европейскую заявку на патент ЕР-А 792867, международную заявку WO 98/01415, европейские заявки на патент ЕР-А 1015411 и ЕР-А 1015410, международные заявки WO 99/50219, WO 00/53560 и WO 02/09839, немецкую заявку на патент DE-A 10235847, международную заявку WO 03/041833, немецкие заявки на патент DE-A 10223058, DE-А 10243625 и DE-A 10336386, европейскую заявку на патент ЕР-А 854129, патент США US-A 4317926, немецкие заявки на патент DE-A 19837520, DE-А 19606877, DE-A 190501325, DE-A 10247240 и DE-A 19740253, европейские заявки на патент ЕР-А 695736, ЕР-А 982287, ЕР-А 1041062 и ЕР-А 117146, немецкие заявки на патент DE-A 4308087, DE-A 4335172, DE-A 4436243, DE-A 19924532, DE-A 10332758 и DE-A 19924533). Акриловую кислоту можно выделять также, как предложено в европейских заявках на патент ЕР-А 982287 и ЕР-А 982289, немецких заявках на патент DE-А10336386, DE-A 10115277, DE-A 19606877, DE-A 19740252, DE-A 19627847 и DE-A 10053086 и европейской заявке на патент ЕР-А 982288. Предпочтительным является выделение, осуществляемое в соответствии с фиг.7 международной заявки WO/0196271 и приведенными в настоящем описании примерами.
Как сообщалось в начале настоящего описания, общая особенность указанных выше методов разделения состоит в том, что в головной части ректификационных колонн, которые содержат соответствующие встроенные элементы для повышения эффективности разделения и в нижнюю часть которых обычно после предварительного прямого и/или косвенного охлаждения вводят смесь продуктов В, обычно остается остаточный газовый поток (основной остаточный газ), который преимущественно содержит компоненты газообразной смеси продуктов В, температура кипения которых при нормальном давлении (1 бар) составляет ≤-30°С (то есть трудно конденсируемые, соответственно легколетучие компоненты).
В нижней части ректификационной колонны обычно накапливаются труднолетучие компоненты газообразной смеси продуктов В в виде конденсированной фазы, содержащей соответствующий целевой продукт.
Компонентами остаточной газовой смеси в первую очередь являются пропан, при необходимости непревращенный в реакционной зоне В пропилен, молекулярный кислород, а также часто совместно используемые в реакционных зонах А и В инертные разбавляющие газы, прежде всего предпочтительно азот и диоксид углерода. В зависимости от методов разделения остаточный газ может содержать лишь следы водяного пара или его содержание может составлять до 20% об. или более.
Согласно изобретению по меньшей мере часть этого (основного) остаточного газа (предпочтительно обладающая составом (основного) остаточного газа), в котором содержатся пропан, молекулярный кислород и при необходимости непревращенный (в реакционной зоне В) пропилен (предпочтительно все количество, но при необходимости также лишь половину, две трети или три четверти этого общего количества), рециркулируют в реакционную зону А в качестве содержащего пропан питающего потока. Однако, как указано выше, частичные количества (основного) остаточного газа можно также рециркулировать в реакционную зону В и/или подвергать сжиганию с целью производства энергии.
Как указано выше, согласно изобретению при переработке конденсированной фазы (для выделения целевого продукта) могут образоваться другие остаточные газы, поскольку обычно пытаются вернуть в реакционную зону А и регенерировать в рамках осуществления целевого разделения все количество непревращенного пропана, содержащегося в газообразной смеси продуктов А. Хотя подобные остаточные газы, как правило, и содержат пропан, а также при необходимости пропилен, однако в них зачастую отсутствует молекулярный кислород (если они содержат его, то в настоящем описании их называют побочными остаточными газами). В связи с этим после при необходимости выполненного объединения с основным остаточным газом, приводящего к образованию общего остаточного газа, их можно рециркулировать в предлагаемую в изобретении реакционную зону А. Однако возможно также независимое использование подобных других остаточных газов. Так, например, если они не содержат молекулярного кислорода, их можно также рециркулировать в реакционную зону А в качестве составной части исходной реакционной газовой смеси для этой зоны (как правило, после предварительного сжатия). В случае, если они в основном не содержат молекулярного кислорода, можно осуществить также их рециркуляцию в другие места реакционной зоны А.
Благодаря предпочтительно полной рециркуляции (основного и/или побочного, соответственно общего) остаточного газа из зоны разделения В, которая может включать несколько ректификационных колонн, а также при необходимости другое, отличающееся от них устройство для разделения, в реакционную зону А, при непрерывном осуществлении предлагаемого в изобретении способа происходит непрерывное превращение пропана в акриловую кислоту и/или акролеин.
При этом согласно изобретению важно, чтобы благодаря предлагаемой в изобретении рециркуляции (основного и/или побочного, соответственно общего) остаточного газа из зоны разделения В в реакционную зону А в последней можно было обеспечить превращение пропана в пропилен, протекающее с практически стопроцентной селективностью.
При этом преимущества предлагаемой в изобретении технологии проявляются как при низких (≤30% мол.), так и при высоких (≥30% мол.) степенях превращения пропана при его дегидрировании (в пересчете на однократное пропускание реакционной газовой смеси через реакционную зону А). Преимущества предлагаемой в изобретении технологии прежде всего обусловлены тем, что более высокие конверсии пропана при его дегидрировании в реакционной зоне А, как правило, сопровождаются повышенным содержанием пропилена в газообразной смеси продуктов А, а следовательно, также повышенным содержанием пропилена в газовой смеси, поступающей в реакционную зону В. Последнее обстоятельство обычно обусловливает повышенное содержание остаточного кислорода в (основном) остаточном газе, который, в свою очередь, становится пригоден для сжигания повышенных количеств водорода, образующихся вследствие повышения конверсии пропана в зоне его дегидрирования (реакционной зоне А). Таким образом, преимуществом предлагаемого в изобретении способа является присущий ему механизм оптимизирующего саморегулирования. Однако более высокие конверсии пропана могут обусловить более низкие среднечасовые скорости пропускания общего газа через загруженный в реакционной зоне В катализатор. Согласно изобретению в общем случае целесообразно, чтобы содержание водорода в реакционной газовой смеси реакционной зоны А в месте ввода рециркулируемого в эту зону остаточного газа находилось по меньшей мере в стехиометрическом соотношении с содержащимся в рециркулируемом остаточном газе кислородом.
Предлагаемый в изобретении способ пригоден для соответствующего применения, если в реакционной зоне В выполняют частичное аммоксидирование пропилена в акрилонитрил. Способ соответственно можно использовать также в том случае, если направляемый в реакционную зону А пропан заменяют изобутаном и образующийся при этом изобутилен подвергают в реакционной зоне В соответствующему частичному окислению в метакролеин и/или метакриловую кислоту.
Сравнительные примеры и примеры
Сравнительный пример 1
А) Конструкция реакционной зоны А (стадия дегидрирования)
Стадию дегидрирования осуществляли в трех одинаковых последовательно соединенных трубчатых реакторах, которые были одинаковым образом заполнены катализатором дегидрирования.
Каждый из трубчатых реакторов представлял собой стальную трубку из высококачественной стали (номер 1.4841 согласно стандарту DIN) длиной 1300 мм с толщиной стенок 3,6 мм и внутренним диаметром 53,1 мм. Реакционную газовую смесь пропускали через соответствующие трубчатые реакторы сверху вниз.
В нижнюю часть каждого трубчатого реактора помещена выполненная из той же высококачественной стали опорная решетка. На опорной решетке находился следующий загруженный в реактор материал (в направлении снизу вверх):
175 мм слой шариков диаметром 4-5 мм из стеатита С-220 фирмы CeramTec;
21 мм слой шариков диаметром 1,5-2,5 мм из стеатита С-220 фирмы CeramTec;
210 мм слой катализатора дегидрирования (сплав платины и олова, содержащий цезий (Cs), калий (K) и лантан (La) в виде оксидов, нанесенный на наружную и внутреннюю поверхности стерженьков носителя из смеси оксидов ZrO2·SiO2 (средняя высота 6 мм, гауссово распределение в интервале от 3 до 12 мм с максимумом около 6 мм; диаметр 2 мм) в количестве, обеспечивающем стехиометрическое массовое соотношение элементов (включая носитель) Рt0,3Sn0,6La3,0Сs0,5K0,2 (ZrO2)88,3(SiO2)7,1; приготовление предварительного катализатора и его активирование до образования активного катализатора выполняли аналогично примеру 4 из немецкой заявки на патент DE-A 10219879);
21 мм слой шариков диаметром 1,5-2,5 мм из стеатита С-220 фирмы CeramTec;
в верхнюю часть трубчатого реактора засыпаны шарики диаметром 4-5 мм из стеатита С-220 фирмы CeramTec.
Верхний участок каждого из реакторов (в направлении от входа к опорной решетке) длиной 500 мм представлял собой зону предварительного нагревания, окруженную двумя медными полуоболочками толщиной 200 мм, обеспечивающими равномерное распределение тепла, подводимого от наружной манжеты с электрическим нагревом (фирма Horst, Гейдельберг, Германия, длина 500 мм, внутренний диаметр 100 мм).
Для обеспечения адиабатического режима нижняя часть каждого из трубчатых реакторов длиной 600 мм помещена в две пары теплоизолирующих полуоболочек толщиной 25 мм (материал MPS-Super G фирмы Microtherm, Германия), расположенных одна над другой со смещением друг относительно друга на 90°. Теплоизолирующие оболочки, в свою очередь, находились внутри цилиндрической рубашки из высококачественной стали (наружный диаметр 173 мм, внутренний диаметр 167 мм), на которую с целью сопровождающего нагрева была надета обогревательная манжета фирмы Horst, Гейдельберг, Германия (длина 675 мм, внутренний диаметр 173 мм). На соответствующем адиабатическом участке удавалось минимизировать проникновение теплового потока из внешней среды в реакционную трубку и из реакционной трубки во внешнюю среду.
Кроме того, в центр каждой реакционной трубки помещена термогильза длиной 1370 мм (наружный диаметр 6 мм, внутренний диаметр 4 мм), в которую введен многоканальный термоэлемент толщиной 3,2 мм (всего десять точек измерения, расположенных через каждые 4 см в направлении от нижнего конца реактора вверх).
Перед каждым отдельным трубчатым реактором располагалась присоединенная к нему стальная трубка длиной 1300 мм, заполненная кольцами из стеатита С-220 фирмы CeramTec с размерами 7 мм × 3 мм × 3 мм (наружный диаметр × внутренний диаметр × высота), которую использовали в качестве нагревателя. В подобном нагревателе осуществляли предварительное нагревание реакционной газовой смеси до соответствующей температуры входа в последующий трубчатый реактор и одновременно обеспечивали идеальное перемешивание образующих эту смесь компонентов. Участок стальной трубки длиной 1200 мм (толщина стенок 3,6 мм, внутренний диаметр 53,1 мм, высококачественная сталь номер 1.4841 согласно стандарту DIN) нагревали надетой на него нагревательной манжетой фирмы Horst (Гейдельберг, Германия). Соединение между нагревательными трубками и трубчатыми реакторами осуществляли с помощью обычных теплоизоляционных материалов, термически изолированных трубками из специальной стали (высококачественная сталь номер 1.4841 согласно стандарту DIN, наружный диаметр 21,3 мм, внутренний диаметр 16,1 мм, длина около 700 мм).
Перед входом реакционной газовой смеси в соответствующий нагреватель смонтирован входной кран, с помощью которого в реактор можно было вводить реакционную газовую смесь, соответственно сжатый воздух. Ниже рассматривается стационарный режим функционирования трубчатых реакторов.
В первый реактор дегидрирования вводили исходную реакционную газовую смесь, состоящую из 395 г/ч сырого пропана (первый питающий поток, содержащий пропан), 500 г/ч воды и 3649 г/ч (общего) остаточного газа в качестве циркуляционного газа (второй, содержащий пропан питающий поток), с температурой 400°С и давлением 2,8 бар.
Состав сырого пропана:
Состав (общего) остаточного газа (циркуляционного газа):
Состав сырого пропана и всех других газов определяли методом газовой хроматографии [хроматограф HP 6890 с блоком химической подготовки; детекторы: пламенно-ионизационный (FID), WLD; колонки Al2O3/KCl (Chrompack), Carboxen 1010 (Supelco)]. Содержащийся в газовых смесях водяной пар перед хроматографическим анализом конденсировали в водоотделителе благодаря охлаждению и при необходимости осуществляемому снижению давления. Несконденсированный газ подвергали анализу и все данные указывали в пересчете на сухой газ (то есть не учитывали содержащееся в анализируемой газовой смеси количество водяного пара).
Исходную реакционную газовую смесь приготавливали в присоединенном к первому нагревателю испарителе. Испаритель обладал конструкцией, аналогичной конструкции нагревателя. В испаритель вводили 395 г/ч газообразного сырого пропана (65°С, 5 бар), 3649 г/ч (общего) остаточного газа (циркуляционного газа) (50°С, 2,8 бар) и 500 г/ч воды (20°С, 3 бар). Обогрев испарителя регулировали таким образом, чтобы температура выходящей из него газовой смеси составляла 200°С. Испаритель был соединен с первым нагревателем так же, как реакторы с нагревателями.
Обогрев первого нагревателя регулировали таким образом, чтобы температура вводимой в него из испарителя газовой смеси на выходе из первого нагревателя составляла 400°С (необходимая для этого температура стенок нагревателя составляла около 440°С). Затем исходная реакционная газовая смесь поступала в первый трубчатый реактор и дополнительно нагревалась на участке предварительного нагревания до температуры входа в реакционную зону (423,3°С).
Температура реакционной газовой смеси при пропускании через первый трубчатый реактор проходила через максимум при 549,1°С (так называемую горячую точку) (указанные в рассматриваемом случае количественные данные относятся к состоянию после 200 часов работы реактора; в процессе последующей эксплуатации реактора различные температуры выбирали таким образом, чтобы конверсия в пересчете на однократное пропускание реакционного газа и выход в пересчете на единицу объема/времени преимущественно оставались постоянными; аналогичным образом поступали уже в первые 200 часов работы реактора), который в процессе непрерывной эксплуатации опытной установки вследствие постепенного деактивирования катализатора смещался в направлении потока (скорость смещения составляла около 0,03 мм/ч).
Выходящая из первого реактора дегидрирования реакционная газовая смесь обладала следующим составом:
Температура реакционной газовой смеси составляла 509,1°С, давление 2,55 бар.
Перед входом в последующий нагреватель к реакционной газовой смеси добавляли 130 нл/ч сжатого воздуха (23°С, 4,2 бар).
Затем реакционную газовую смесь нагревали до 477°С (температуры входа во вторую реакционную зону) посредством электрически обогреваемого нагревателя (температура стенок около 540°С) и участка предварительного нагревания следующей (второй) реакционной трубки (температура стенок около 560°С). Давление реакционной газовой смеси при этом составляло 2,55 бар.
Температура реакционной газовой смеси при пропускании через второй трубчатый реактор проходила через максимум при 515,5°С, который в процессе непрерывной эксплуатации опытной установки вследствие постепенного деактивирования катализатора смещался в направлении потока (скорость смещения составляла около 0,1 мм/ч). Выходящая из второго реактора дегидрирования реакционная газовая смесь обладала следующим составом:
Температура реакционной газовой смеси составляла 493,8°С, давление 2,5 бар.
Перед входом в последующий нагреватель к реакционной газовой смеси добавляли 195 нл/ч сжатого воздуха (23°С, 4,2 бар).
Затем реакционную газовую смесь нагревали до 464°С (температуры входа в третью реакционную зону) посредством электрически обогреваемого нагревателя (температура стенок около 480°С) и участка предварительного нагревания следующей (третьей) реакционной трубки (температура стенок около 480°С). Давление реакционной газовой смеси при этом составляло 2,5 бар.
Температура реакционной газовой смеси при пропускании через третий трубчатый реактор проходила через максимум при 492,8°С, который в процессе непрерывной эксплуатации опытной установки вследствие постепенного деактивирования катализатора смещался в направлении потока (скорость смещения составляла около 0,1 мм/ч). Выходящая из третьего реактора дегидрирования реакционная газовая смесь обладала следующим составом:
Температура реакционной газовой смеси составляла 480,4°С, давление 2,45 бар.
Таким образом, общая конверсия пропана на стадии дегидрирования в пересчете на однократное пропускание исходной реакционной газовой смеси составляла 19,83% мол.
При прогрессирующем деактивировании катализатора дегидрирование прекращали и катализатор подвергали регенерации, как описано в немецкой заявке на патент DE-A 10028582. Этот момент преимущественно всегда наступал в том случае, когда температура горячей точки во всех трех трубчатых реакторах составляла около 580°С.
Неожиданным обстоятельством явилось то, что деактивирование катализатора дегидрирования при повышенном рабочем давлении происходило медленнее.
В) Конструкция зоны разделения А
Температуру выходящей из третьего реактора дегидрирования газообразной смеси продуктов (газообразной смеси продуктов А) снижали до 40°С в холодильнике прямого охлаждения за счет непосредственного охлаждения распыляемой охлаждающей водой с температурой 20°С (остающуюся при этом газообразную смесь выводили из холодильника в направлении, противоположном направлению вводимого в него потока газообразных продуктов). При прямом охлаждении газообразной смеси продуктов конденсировалось около 75% мас. содержащегося в ней водяного пара (водяной пар добавляли к исходной газовой смеси реакционной зоны А, и он образовывался на стадии дегидрирования вследствие сгорания водорода, а также возможных углеводородов в присутствии кислорода воздуха; теплота сгорания водорода обеспечивала поддержание постоянной температуры газовой смеси в реакционной зоне А). Образующийся при этом конденсат отбирали из холодильника с помощью системы управления уровнем и направляли на утилизацию. Использованную для прямого охлаждения воду подавали насосом в контур циркуляции, охлаждали за счет косвенного теплообмена и вновь распыляли в холодильнике прямого охлаждения.
Вместо описанного выше прямого охлаждения водой газообразную смесь продуктов стадии дегидрирования можно также сначала охладить, например, в косвенном прямоточном или противоточном кожухотрубном теплообменнике за счет теплообмена с подаваемой на стадию дегидрирования исходной реакционной газовой смесью, нагревающейся при этом до 400°С. Благодаря этому температуру газообразной смеси продуктов стадии дегидрирования (около 500°С) снижали до уровня, составляющего примерно от 200 до 300°С.
Дальнейшее охлаждение газообразной смеси продуктов стадии дегидрирования можно осуществлять в косвенном теплообменнике, используя эту смесь для нагревания исходной газовой смеси, подаваемой на рассматриваемую ниже стадию частичного окисления полученного при дегидрировании пропилена, и/или для уравновешивания рассматриваемого ниже охлаждения вторичного газа абсорбера при его предпочтительно двухступенчатом расширении в турбодетандерах, или для компенсирования тепла, необходимого для предварительного нагревания.
Благодаря этому температура газообразной смеси продуктов снижалась примерно до 180°С. Затем смесь можно было охладить до температуры от 30 до 60°С в воздушном и/или водяном холодильнике.
Образующийся при охлаждении водный конденсат накапливали во встроенных в холодильник или последовательно присоединенных к нему каплеотделителях и с помощью системы управления уровнем направляли на утилизацию.
Затем охлажденную и освобожденную от водяного пара газообразную смесь продуктов стадии дегидрирования, находящуюся под давлением около 2 бар, сжимали до давления, составляющего от 10 до 13 бар.
Во избежание чрезмерного повышения температуры при сжатии в целесообразном с производственно-технической точки зрения варианте сжатие осуществляли в две ступени (подобного повышения температуры избегали также благодаря предварительно выполненному охлаждению; предварительное выделение водяного пара позволяло дополнительно снизить потребляемую компрессором мощность). Давление на первой ступени сжатия повышали до величины, составляющей от 4 до 4,5 бар. Температура газовой смеси после первой ступени сжатия составляла около 115°С.
В последовательно присоединенном косвенном теплообменнике (воздушном или водяном холодильнике) газовую смесь вновь охлаждали до температуры от 40 до 60°С, при этом происходила дополнительная конденсация водяного пара. Конденсат накапливали в каплеотделителях, из которых его выводили с помощью системы управления уровнем.
На второй ступени сжатия давление поступающей в соответствующий компрессор газовой смеси (около 4 бар) повышали до конечной величины, составляющей от 10 до 13 бар. Температура газовой смеси на выходе из второго компрессора составляла около 126°С.
В двух последовательно соединенных косвенных теплообменниках (сначала в воздушном холодильнике (обычно использовали кожухотрубный теплообменник с целесообразным пропусканием подлежащего охлаждению газа через трубки), а затем в водяном холодильнике) сжатую газовую смесь охлаждали сначала до температуры от 40 до 60°С, а затем до 30°С. При этом сконденсированную воду вновь отделяли с помощью каплеотделителей и выводили из системы. Газовая смесь на выходе из второго компрессора содержала не более 0,2% мас. воды. Незначительное влагосодержание позволяло избежать образования двух фаз при последующей абсорбции, а повышенное давление позволяло сократить количество абсорбента, необходимого для осуществления абсорбции.
Для сжатия газовой смеси в производственных условиях используют промышленные турбокомпрессоры (бесконтактные компрессоры с сухим ходом без масляной смазки, например турбокомпрессоры типа 12 МН 4 В фирмы Mannesmann DEMAG, Германия), тогда как в рассматриваемом случае использовали мембранные компрессоры MV 3459 II фирмы Sera. В качестве привода подобных компрессоров в принципе можно использовать как электродвигатели, так и паровые или газовые турбины. Наиболее экономичными зачастую оказываются паровые турбины. Сжатую и охлажденную указанным образом газовую смесь (около 4220 г/ч) вводили в абсорбционную колонну непосредственно над ее кубом. Газовая смесь обладала следующим составом:
Абсорбционная колонна выполнена из высококачественной стали 1.4571. Диаметр колонны составлял 80 мм, толщина стенок 4 мм, высота 1,70 м.
Около 70% объема абсорбционной колонны заполнены насадочными элементами фирмы Montz (Montz BSH-750 с удельной поверхностью 750 м2/м3) в качестве повышающих эффективность разделения встроенных элементов. Расположенными непосредственно один над другим насадочными элементами была заполнена нижняя часть колонны (1/5 ее общей высоты). Наружный обогрев или охлаждение абсорбционной колонны отсутствовали. В качестве абсорбента в верхнюю часть колонны подавали технический тетрадекан типа PKWF 4/7 af (фирма Haltermann, Германия) с температурой 30°С; процентный состав исходного (свежего) тетрадекана, определенный методом газовой хроматографии (FID-детектор) по площадям соответствующих пиков:
Указанный состав в процессе непрерывной эксплуатации абсорбционной колонны (по истечении примерно 3000 часов) изменялся следующим образом:
Плотность орошения составляла 15 м3 абсорбента на 1 м2 площади свободного сечения в час (расход тетрадекана 25 кг/ч).
Направляемый на сжигание вторичный газ абсорбционной колонны содержал 1700 млн-1 (об.) пропана и 400 млн-1 (об.) пропилена. В условиях промышленного производства подобный поток вторичного газа направляют на сжигание через детандер (например, турбодетандер), чтобы рекуперировать большую часть затраченной на двухступенчатое сжатие газа энергии и вернуть ее на стадию двухступенчатой компрессии. В целесообразном варианте расширение вторичного газа также осуществляют в две ступени, что позволяет избежать нежелательной конденсации. Получаемую при расширении вторичного газа механическую энергию можно использовать непосредственно в качестве параллельного или главного привода одного из компрессоров и/или для выработки тока.
В промышленных условиях, прежде чем направить подвергнутый расширению вторичный газ абсорбера на сжигание, может оказаться целесообразным выделение содержащегося в нем водорода. Водород можно выделить, например, пропуская вторичный газ через выполненную, как правило, в виде трубы мембрану, проницаемую только для молекулярного водорода. Выделенный подобным образом молекулярный водород можно вернуть, например, на стадию гетерогенно катализируемого дегидрирования пропана или использовать иначе (например, в топливных элементах).
Выделение водорода из вторичного газа абсорбера в принципе можно осуществлять также частичной конденсаций, адсорбцией и/или ректификацией (предпочтительно под давлением, например в виде адсорбции с переменным давлением). В условиях промышленного производства с целью концентрирования вторичного газа абсорбера его, как правило, целесообразно также пропускать через рассмотренную ниже более подробно кислотную воду.
Абсорбат обладал следующим составом (в % мас. в пересчете на его массу):
Прежде чем направить абсорбат в верхнюю часть последующей десорбционной колонны, его нагревали в косвенном теплообменнике до температуры от 36 до 37°С. В качестве теплоносителя использовали жидкий слив из десорбционной колонны с температурой 37°С.
Затем давление абсорбата снижали до величины, составляющей от 2,5 до 3 бар (его можно было снизить, например, в инверсном насосе или посредством клапана, причем механическую энергию, высвобождающуюся в случае применения инверсного насоса, целесообразно использовать для декомпрессии абсорбента, освобожденного в десорбционной колонне от содержащихся в нем компонентов), и образующуюся при этом двухфазную смесь направляли в верхнюю часть десорбционной колонны.
В нижнюю часть десорбционной колонны (внутренний диаметр 80 мм, длина 1,70 м, толщина стенок 4 мм) противотоком по отношению к стекающему из верхней части абсорбату подавали воздух под давлением 3 бар (1340 нл/ч). Расход вводимого в верхнюю часть десорбционной колонны абсорбата составлял 35 л/ч. Выводимый из нижней части десорбционной колонны абсорбент, в основном не содержащий десорбированных компонентов, охлаждали за счет косвенного теплообмена с абсорбатом, сжимали до необходимого для подачи в абсорбционную колонну давления, охлаждали до 30°С в еще одном косвенном теплообменнике (в производственных условиях целесообразным является теплообмен с поверхностной водой) и возвращали в верхнюю часть абсорбционной колонны. Десорбционная колонна содержала в качестве повышающих эффективность разделения встроенных элементов структурированные насадочные элементы фирмы Montz из листового металла (Montz BSH-750 с удельной поверхностью 750 м2/м3). Выводимый из десорбционной колонны (при необходимости через механический каплеотделитель) газовый поток с целью улавливания абсорбента промывали водой. То есть этот газовый поток пропускали через насадочный элемент фирмы Montz (Montz BSH-750 с удельной поверхностью 750 м2/м3), подавая противотоком воду (70 л/ч) с температурой 18°С. Под насадочным элементом была помещена улавливающая тарелка, с которой можно было отбирать водную фазу. В разделителе фаз осуществляли разделение на водную и органическую фазы. Весьма незначительное количество органической фазы объединяли с потоком абсорбента, возвращаемого в верхнюю часть абсорбционной колонны. Водную фазу после повторного охлаждения и добавления свежей воды (для компенсации потерь при испарении) вновь направляли к насадочному элементу. Промывку осуществляли в верхней части десорбционной колонны.
Промытый газовый поток, выводимый из промывочной секции через механический каплеотделитель (отделенную в нем жидкую фазу возвращали на промывку), обладал следующим составом (если в реакционной зоне А при дегидрировании пропана достигают более высоких конверсий, указанное ниже содержание пропилена может составлять также от 8 до 12% об., например, промытый газовый поток может содержать также 15% об. пропана, 10% об. пропилена и 14,5% об. кислорода):
Температуру газовой смеси указанного выше состава за счет косвенного теплообмена повышали до 250°С и ее направляли в качестве новой исходной реакционной газовой смеси в последующее устройство для частичного окисления со среднечасовой скоростью пропускания 2260 нл/л·ч и начальным давлением 2,1 бар.
С) Конструкция реакционной зоны В (стадия частичного окисления)
1. Первый реактор со стационарным слоем для частичного окисления пропилена в акролеин
Реактор. Двустенный цилиндр из высококачественной стали (направляющая труба с толщиной стенок от 2 до 5 мм, окруженная наружным цилиндрическим резервуаром).
Внутренний диаметр наружного цилиндра 168 мм. Внутренний диаметр направляющей трубы около 60 мм.
Двухстенный цилиндр сверху закрывала крышка, снизу дно.
Контактная трубка помещена в окруженную цилиндрическим резервуаром направляющую трубу таким образом, чтобы она выступала с верхней и нижней сторон цилиндрического резервуара, уплотненных соответственно крышкой и дном, соответственно на 250 мм.
Теплоноситель находился в замкнутом объеме внутри цилиндрического резервуара. С целью создания как можно более однородных термических граничных условий на наружной поверхности участка контактной трубки, окруженного цилиндрическим резервуаром (длина этого участка составляла 3700 мм), через теплоноситель барботировал азот, обеспечивая его циркуляцию.
Барботирующий азот перемещал теплоноситель в направляющей трубе снизу вверх, после чего теплоноситель стекал через промежуток между направляющей трубой и цилиндрическим наружным резервуаром (аналогичную интенсивность циркуляции теплоносителя можно обеспечить также его перекачкой, например, посредством пропеллерного насоса). Необходимую температуру теплоносителя можно отрегулировать посредством надетого на наружный цилиндр электрического нагревателя. Для охлаждения теплоносителя использовали воздух.
Питание реактора. Солевой расплав и реакционная газовая смесь перемещались в реакторе противотоком. Реакционная газовая смесь поступала в реактор сверху. Ее температура на входе в реакционную трубку составляла 250°С.
Солевой расплав на входе в нижнюю часть направляющей трубы обладал температурой Твход 320°С, а на выходе из ее верхней части температурой Твыход. Разница между Твход и Твыход составляла около 2°С. Средняя температура расплава Тсред. составляла (Твход+Твыход)/2.
Заполнение контактной трубки (сверху вниз). Секция А высотой 50 см. Предварительный слой колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × длина × внутренний диаметр).
Секция В высотой 100 см.
Контактная трубка заполнена гомогенной смесью 30% мас. колец из стеатита С 220 фирмы CeramTec с размерами 5 мм × 3 мм × 2 мм (наружный диаметр × длина × внутренний диаметр) и 70% масс. сплошного катализатора, аналогичного используемому в секции С.
Секция С высотой 170 см.
Контактная трубка заполнена сплошным катализатором из примера 1 немецкой заявки на патент DE-A 10046957 в виде колец с размерами 5 мм × 3 мм × 2 мм (наружный диаметр × длина × внутренний диаметр).
Секция D высотой 50 см.
Завершающий слой колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × длина × внутренний диаметр).
2. Промежуточное охлаждение и промежуточная подача кислорода
Газообразную смесь реакционных продуктов из первого реактора со стационарным слоем катализатора с целью промежуточного охлаждения (косвенного охлаждения воздухом) пропускали через соединительную трубку из высококачественной стали (длина 400 мм, внутренний диаметр 26 мм, толщина стенок 2 мм) со средним участком длиной 200 мм, заполненным инертным слоем шариков из стеатита фирмы CeramTec диаметром от 5 до 6 мм, которая была прифланцована непосредственно к контактной трубке первого реактора со стационарным слоем.
Газовая смесь поступала в соединительную трубку с температурой более 310°С и выходила из нее с температурой около 140°С. Затем ее смешивали со сжатым воздухом (290 нл/ч), используемым в качестве источника кислорода.
После перемешивания в статическом смесителе газовую смесь с температурой 220°С вводили в реактор со стационарным слоем для реализации стадии частичного окисления акролеина в акриловую кислоту.
3. Второй реактор со стационарным слоем для частичного окисления акролеина в акриловую кислоту
Использовали реактор со стационарным слоем катализатора, конструкция которого аналогична конструкции реактора первой стадии. Солевой расплав и реакционная газовая смесь перемещались относительно друг друга в реакторе прямотоком. Солевой расплав и реакционная газовая смесь поступали в реактор снизу.
Заполнение контактной трубки (снизу вверх)
Секция А высотой 20 см.
Предварительный слой колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × длина × внутренний диаметр).
Секция В высотой 100 см.
Контактная трубка заполнена гомогенной смесью 30% мас. колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр) и 70% мас. слоистого катализатора, аналогичного используемому в секции С.
Секция С длиной 200 см.
Контактная трубка заполнена слоистым катализатором из примера получения 5 немецкой заявки на патент DE-А 10046928 в виде колец с размерами 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр) (секцию С можно также заполнить полученными соответствующим методом аналогичными слоистыми катализаторами, которые, однако, содержат активную массу со стехиометрическим составом Mo12V2,8W1,2Cu2,4Ox или Mo12V3,5W1,3Cu2,4Ox).
Секция D длиной 50 см.
Завершающий слой колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × длина × внутренний диаметр).
Среднечасовая скорость пропускания исходной газовой смеси через второй реактор составляла около 3850 г/ч. Средняя температура расплава (Tсред.), вычисляемая аналогично первому реактору, составляла 274°С.
Конверсия пропилена в первом реакторе составляла 97,7% мол., конверсия акролеина во втором реакторе 99,4% мол.
Газообразная смесь продуктов (газообразная смесь продуктов В), выходящая из второго реактора со стационарным слоем катализатора, обладала температурой 283°С, давлением 1,8 бар и следующим составом:
Используемые для осуществления обеих реакционных стадий катализаторы могут быть заменены также катализаторами из примеров, приведенных в немецкой заявке на патент DE-A 10351269. Катализатор, используемый на первой реакционной стадии, может быть заменен также катализаторами из примеров и сравнительных примеров, приведенных в немецкой заявке на патент DE-A 10344149. Катализатор, используемый на второй реакционной стадии, может быть заменен также катализаторами из примеров, приведенных в немецких заявках на патент DE-A 10360057 и DE-A 10360058.
D) Конструкция зоны разделения В (выделения целевой акриловой кислоты из газообразной смеси продуктов частичного окисления)
Горячую газообразную смесь продуктов (газообразную смесь продуктов В) охлаждали примерно до 180°С в холодильнике прямого охлаждения, орошаемом распыляемой через форсунки жидкостью с температурой от 140 до 150°С, состоящей из 57,4% мас. дифенилового эфира, 20,7% мас. дифенила и 20% мас. о-диметилфталата, которую использовали также в качестве абсорбента. Накапливаемую в нижней части холодильника охлаждающую жидкость отбирали и после охлаждения в теплообменнике повторно распыляли в верхней части холодильника. Газообразную смесь продуктов вводили непосредственно в накапливаемую в холодильнике охлаждающую жидкость. Охлажденная газообразная смесь продуктов поступала в последовательно присоединенную абсорбционную колонну. В циркуляционном контуре охлаждающей жидкости концентрировались подлежащие выведению высококипящие побочные продукты. В связи с этим периодически (раз в сутки) отбирали часть циркулирующей охлаждающей жидкости (1 кг) и время от времени подвергали ее переработке в ротационном испарителе. Очищенный абсорбент возвращали в контур циркуляции охлаждающей жидкости, потери которой восполняли, добавляя смесь дифенилового эфира, дифенила и о-диметилфталата.
Охлажденную газообразную смесь продуктов вводили в абсорбционную колонну в точке, находящейся выше ее куба. Длина абсорбционной колонны составляла 5200 мм, внутренний диаметр 50 мм, и она была снабжена насадочными элементами Kuehni (CH) Rombopak 9M. Абсорбционная колонна выполнена из стекла и снабжена предназначенной для термостатирования сегментированной стеклянной рубашкой. Над первым снизу насадочным элементом смонтирован редукционный клапан (регулируемый дроссель), позволяющий имитировать потерю напора в абсорбционной колонне, составляющую от 100 до 300 мбар (которая наблюдалась бы в промышленных условиях). Рабочее давление в верхней части абсорбционной колонны составляло 1,35 бар. Двойная стеклянная рубашка состояла из пяти функционирующих независимо друг от друга сегментов, посредством которых обеспечивали необходимый температурный профиль.
Сегменты имели следующую температуру (в направлении снизу вверх) (термостатирование подаваемой насосом водой):
Над самым верхним насадочным элементом располагалась улавливающая тарелка, с которой отбирали обогащенный водой конденсат (кислотную воду). Конденсат, охлажденный в теплообменнике до 20°С, возвращали в абсорбционную колонну выше самого верхнего насадочного элемента, а его избыточное количество (реакционную воду) выводили с помощью системы управления уровнем. Отбираемую кислотную воду направляли в разделитель фаз. Отделившуюся в нем органическую фазу возвращали в абсорбционную колонну выше второго насадочного элемента, водную фазу подвергали утилизации. Абсорбент с температурой 40°С 3,5 л/ч вводили в колонну непосредственно над улавливающей тарелкой.
Выводимый из верхней части абсорбционной колонны (основной) остаточный газ (температура 18°С, давление 1,35 бар) обладал следующим составом:
(Основной) остаточный газ за счет косвенного теплообмена нагревали до 40°С (для исключения нежелательной конденсации), сжимали до 2,75 бар компрессором KNF Neuberger типа РМ 17739-1200 (в производственных условиях турбокомпрессором с приводом от электродвигателя, например, типа 12МН4В фирмы Mannesmann DEMAG, Германия) и в качестве компонента циркуляционного газа (первого компонента (общего) остаточного газа) возвращали в реакционную зону А для подачи в первый реактор дегидрирования пропана.
Из куба абсорбционной колонны с помощью системы управления уровнем отбирали 3,9 л/ч кубовой жидкости. Второй менее интенсивный поток кубовой жидкости (около 0,5 л/ч) возвращали в холодильник прямого охлаждения для охлаждения горячей газообразной смеси продуктов.
Выгруженный из куба абсорбционной колонны продукт (сначала его можно было отпарить частичным количеством остаточного газа (предпочтительно предварительно отмыв в основном всю акриловую кислоту), как описано в немецкой заявке на патент DE-A 10336386, содержащий отпаренные компоненты газ затем можно было вернуть в абсорбционную колонну, как описано в немецкой заявке на патент DE-A 10336386) направляли в верхнюю часть перегонной колонны (внутренний диаметр 50 мм, длина 2000 мм, стекло, термостатирующая рубашка (150°С), 20 сетчатых тарелок с диаметром отверстий 6,5 мм и треугольным шагом). В перегонной колонне при давлении около 200 мбар и температуре 150°С испаряли все низкокипящие компоненты (прежде всего пропан и пропилен), основную часть акриловой кислоты, компоненты со средней температурой кипения (например, уксусную кислоту), а также часть абсорбента. Из куба перегонной колонны насосом (Hermetic, производительность 500 л/ч) отбирали циркуляционный поток (200 л/ч) с температурой 190°С, который возвращали в перегонную колонну, пропустив его через теплообменник (выпарной аппарат с принудительной циркуляцией). Выходящий из куба перегонной колонны поток растворителя с низким содержанием указанных выше компонентов возвращали в колонну для абсорбции акриловой кислоты, вводя его ниже улавливающей тарелки, с которой отбирали реакционную воду (кислотную воду).
Парогазовую смесь из перегонной колонны направляли в колонну без встроенных элементов для отделения конденсата. Из ее куба выгружали 100 л/ч конденсата, который для отбора теплоты конденсации пропускали через теплообменник и затем с температурой 20°С возвращали в верхнюю часть этой колонны.
Кроме того, из куба колонны без встроенных элементов выгружали 600 мл/ч продукта, содержащего целевую акриловую кислоту, воду и остаточное количество абсорбента, из которого при необходимости можно было известным методом выделить акриловую кислоту. В верхней части этой колонны вновь конденсировалась использованная для прямого охлаждения кислотная вода, которую выводили из колонны посредством приемного днища. Стекающий в приемное днище конденсат смешивали с отобранной из абсорбционной колонны (для газового потока продуктов частичного окисления) кислотной водой и после охлаждения до 18°С возвращали в используемую для прямого охлаждения кислотную воду. Отбирали избыток кислотной воды. Выделяющийся из кислотной воды газ в виде другого остаточного газа, содержащего пропан и непревращенный пропилен, после компрессии возвращали в реакционную зону А в качестве другой составной части циркуляционного газа (второго компонента (общего) остаточного газа) для подачи в первый реактор дегидрирования пропана.
Сравнительный пример 2
Пример выполняли аналогично сравнительному примеру 1. Однако газообразную смесь продуктов А из третьего реактора дегидрирования пропана посредством делителя потока разделяли на два потока идентичного состава, одну половину которого направляли в зону разделения А, а другую возвращали в реакционную зону А в качестве составной части исходной реакционной газовой смеси первого реактора дегидрирования.
Для этого давление (общего) остаточного газа повышали до 4 бар и сжатый остаточный газ использовали в качестве приводной струи струйного насоса, причем перемещение дросселированной в рабочем сопле до 3 бар приводной струи через зону смешения и диффузор в присоединенном к первому нагревателю испарителе, а также всасывающий патрубок были направлены в сторону возвращаемой половины потока смеси продуктов А, и сочленение «всасывающий патрубок - зона смешения - диффузор» являлось единственным соединением между подлежащей возвращению смесью продуктов А и присоединенным к первому нагревателю испарителем.
Катализаторы дегидрирования начинали терять активность уже после 10 часов непрерывной работы.
Сравнительный пример 3 (в отсутствие особых указаний все приведенные ниже значения давлений, как и в остальном описании, указаны в абсолютных единицах)
А) Конструкция реакционной зоны А (стадия дегидрирования)
Зона дегидрирования включала три одинаковых последовательно соединенных трубчатых реактора, которые были одинаковым образом заполнены катализатором дегидрирования.
Каждый из трубчатых реакторов представлял собой стальную трубку из высококачественной стали (номер 1.4841 согласно стандарту DIN) длиной 1300 мм с толщиной стенок 3,6 мм и внутренним диаметром 53,1 мм. Реакционную газовую смесь пропускали через соответствующие трубчатые реакторы сверху вниз.
В нижнюю часть каждого трубчатого реактора была помещена выполненная из той же высококачественной стали опорная решетка. На опорной решетке находился следующий загруженный материал (в направлении снизу вверх):
175 мм слой шариков диаметром 4-5 мм из стеатита С-220 фирмы CeramTec;
21 мм слой шариков диаметром 1,5-2,5 мм из стеатита С-220 фирмы CeramTec;
210 мм слой катализатора дегидрирования (сплав платины и олова, содержащий цезий (Cs), калий (K) и лантан (La) в виде оксидов, который нанесен на наружную и внутреннюю поверхности стерженьков носителя из смеси оксидов ZrO2·SiO2 со средней высотой 6 мм (гауссово распределение в интервале от 3 до 12 мм с максимумом около 6 мм) и диаметром 2 мм в количестве, обеспечивающем стехиометрическое массовое соотношение элементов (включая носитель) Рt0,3Sn0,6Lа3,0Сs0,5K0,2 (ZrO2)88,3(SiO2)7,1; приготовление предварительного катализатора и его активирование с образованием активного катализатора аналогично примеру 4 из немецкой заявки на патент DE-A 10219879);
21 мм слой шариков диаметром 1,5-2,5 мм из стеатита С-220 фирмы CeramTec;
остальная часть трубчатого реактора засыпана шариками диаметром 4-5 мм из стеатита С-220 фирмы CeramTec.
Верхний участок каждого из реакторов (в направлении от входа к опорной решетке) длиной 500 мм представлял собой зону предварительного нагревания, окруженную двумя медными полуоболочками толщиной 200 мм, обеспечивающими равномерное распределение тепла, подводимого от наружной манжеты с электрическим нагревом (фирма Horst, Гейдельберг, Германия, длина 500 мм, внутренний диаметр 100 мм).
Для обеспечения адиабатического режима нижняя часть каждого из трубчатых реакторов длиной 600 мм помещена в две пары теплоизолирующих полуоболочек толщиной 25 мм (материал MPS-Super G фирмы Microtherm, Германия), расположенных одна над другой со смещением друг относительно друга на 90°. Теплоизолирующие оболочки, в свою очередь, находились внутри цилиндрической рубашки из высококачественной стали (наружный диаметр 173 мм, внутренний диаметр 167 мм), на которую с целью сопровождающего нагрева была надета обогревательная манжета фирмы Horst, Гейдельберг, Германия (длина 675 мм, внутренний диаметр 173 мм). На соответствующем адиабатическом участке удавалось минимизировать проникновение теплового потока из внешней среды в реакционную трубку и из реакционной трубки во внешнюю среду.
Кроме того, в центр каждой реакционной трубки помещена термогильза длиной 1370 мм (наружный диаметр 6 мм, внутренний диаметр 4 мм), в которую был введен многоканальный термоэлемент толщиной 3,2 мм (всего десять точек измерения через каждые 4 см в направлении от нижнего конца реактора вверх).
Перед каждым отдельным трубчатым реактором располагалась присоединенная к нему стальная трубка длиной 1300 мм, заполненная кольцами из стеатита С-220 фирмы CeramTec с размерами 7 мм × 3 мм × 3 мм (наружный диаметр × внутренний диаметр × высота), которую использовали в качестве нагревателя. В подобном нагревателе осуществляли предварительное нагревание реакционной газовой смеси до соответствующей температуры входа в последующий трубчатый реактор и одновременно обеспечивали идеальное перемешивание образующих эту смесь компонентов. Участок стальной трубки длиной 1200 мм (толщина стенок 3,6 мм, внутренний диаметр 53,1 мм, высококачественная сталь номер 1.4841 согласно стандарту DIN) нагревали надетой на него нагревательной манжетой фирмы Horst (Гейдельберг, Германия). Соединение между нагревательными трубками и трубчатыми реакторами осуществляли с помощью обычных теплоизоляционных материалов, термически изолированных трубками из специальной стали (высококачественная сталь номер 1.4841 согласно стандарту DIN, наружный диаметр 21,3 мм, внутренний диаметр 16,1 мм, длина около 700 мм).
Перед входом реакционной газовой смеси в соответствующий нагреватель смонтирован входной кран, с помощью которого в реактор можно было вводить реакционную газовую смесь, соответственно сжатый воздух. Ниже рассматривается стационарный режим эксплуатации трубчатых реакторов.
В первый реактор дегидрирования подавали исходную реакционную газовую смесь, состоящую из 300 г/ч сырого пропана (первый питающий поток, содержащий пропан), 375 г/ч воды и 3768 г/ч (общего) остаточного газа в качестве циркуляционного газа (второй, содержащий пропан питающий поток) с температурой 400°С и давлением 2,6 бар (в производственных условиях для компенсации повышенной потери напора в реакционной зоне А, обусловленной более высокой скоростью течения, давление на входе целесообразно было бы увеличить примерно на 0,5 бар).
Состав сырого пропана:
Состав (общего) остаточного газа (циркуляционного газа):
Состав сырого пропана и всех других газов определяли методом газовой хроматографии [хроматограф HP 6890 с блоком химической подготовки; детекторы: пламенно-ионизационный (FID), WLD; колонки Al2O3/KCl (Chrompack), Carboxen 1010 (Supelco)]. Содержащийся в газовых смесях водяной пар перед хроматографическим анализом конденсировали в водоотделителе благодаря охлаждению и при необходимости осуществляемому снижению давления. Несконденсированный газ подвергали анализу и все данные указывали в пересчете на сухой газ (то есть не учитывали содержащееся в анализируемой газовой смеси количество водяного пара).
Приготовление исходной реакционной газовой смеси осуществляли в присоединенном к первому нагревателю испарителе. Испаритель обладал аналогичной нагревателю конструкцией. В испаритель вводили 300 г/ч газообразного сырого пропана (65°С, 5 бар), 3768 г/ч (общего) остаточного газа (циркуляционного газа) (50°С, 2,8 бар) и 375 г/ч воды (20°С, 3 бар). Обогрев испарителя регулировали таким образом, чтобы температура выходящей из него газовой смеси составляла 200°С. Испаритель соединен с первым нагревателем аналогично соединению реакторов с нагревателями.
Обогрев первого нагревателя регулировали таким образом, чтобы температура вводимой в него из испарителя газовой смеси на выходе из первого нагревателя составляла 400°С (необходимая для этого температура стенок нагревателя составляла около 440°С). Затем исходную реакционную газовую смесь направляли в первый трубчатый реактор и дополнительно нагревали на участке предварительного нагревания до температуры входа в реакционную зону (460°С).
Температура реакционной газовой смеси при пропускании через первый трубчатый реактор проходила через максимум при 549,1°С (так называемую горячую точку) (указанные в рассматриваемом случае количественные данные относятся к состоянию после 200 часов работы реактора; в процессе его последующей эксплуатации различные температуры выбирали таким образом, чтобы конверсия в пересчете на однократное пропускание реакционного газа и выход в пересчете на единицу объема/времени преимущественно оставались постоянными; аналогичным образом поступали уже в первые 200 часов работы реактора), который в процессе непрерывной эксплуатации опытной установки вследствие постепенного деактивирования катализатора смещался в направлении потока (скорость смещения составляла около 0,03 мм/ч).
Выходящая из первого реактора дегидрирования реакционная газовая смесь обладала следующим составом:
Температура реакционной газовой смеси составляла 509°С, давление около 2,56 бар.
Перед входом в последующий нагреватель к реакционной газовой смеси добавляли 80 нл/ч сжатого воздуха (23°С, 4,2 бар).
Затем реакционную газовую смесь нагревали до 465°С (температуры входа во вторую реакционную зону) посредством электрически обогреваемого нагревателя (температура стенок около 540°С) и участка предварительного нагревания следующей (второй) реакционной трубки (температура стенок около 560°С). Давление реакционной газовой смеси при этом составляло 2,56 бар.
Температура реакционной газовой смеси при ее пропускании через второй трубчатый реактор проходила через максимум при 560°С, который в процессе непрерывной эксплуатации опытной установки вследствие постепенного деактивирования катализатора смещался в направлении потока (скорость смещения составляла около 0,1 мм/ч). Выходящая из второго реактора дегидрирования реакционная газовая смесь обладала следующим составом:
Температура реакционной газовой смеси составляла около 493°С, давление около 2,52 бар.
Перед входом в последующий нагреватель к реакционной газовой смеси добавляли 98 нл/ч сжатого воздуха (23°С, 4,2 бар).
Затем реакционную газовую смесь посредством электрически обогреваемого нагревателя (температура стенок около 540°С) и участка предварительного нагревания следующей (третьей) реакционной трубки (температура стенок около 540°С) нагревали до 521°С (температуры входа в третью реакционную зону). Давление реакционной газовой смеси при этом составляло 2,52 бар.
Температура реакционной газовой смеси при пропускании через третий трубчатый реактор проходила через максимум при 570°С, который в процессе непрерывной эксплуатации опытной установки вследствие постепенного деактивирования катализатора смещался в направлении потока (скорость смещения составляла около 0,1 мм/ч). Выходящая из третьего реактора дегидрирования реакционная газовая смесь обладала следующим составом:
Температура реакционной газовой смеси составляла 480,4°С, давление 2,48 бар.
Таким образом, общая конверсия пропана на стадии дегидрирования в пересчете на однократное пропускание исходной реакционной газовой смеси составляла 19,91% мол.
При прогрессирующем деактивировании катализатора дегидрирование прекращали и катализатор подвергали регенерации, как описано в немецкой заявке на патент DE-A 10028582. Этот момент преимущественно всегда наступал в том случае, когда температура горячей точки во всех трех трубчатых реакторах составляла около 580°С.
Неожиданным обстоятельством явилось то, что деактивирование катализатора дегидрирования при повышенном рабочем давлении происходило медленнее.
В) Конструкция зоны разделения А
Температуру газообразной смеси продуктов (газообразной смеси продуктов А), выходящей из третьего реактора дегидрирования, снижали до 40°С в холодильнике прямого охлаждения за счет непосредственного охлаждения распыляемой форсунками охлаждающей воды с температурой 20°С (остающуюся при этом газообразную смесь выводили из холодильника в направлении, противоположном направлению вводимого в него потока газообразных продуктов). При прямом охлаждении газообразной смеси продуктов конденсировалось около 75% мас. содержащегося в ней водяного пара (водяной пар добавляли к исходной газовой смеси реакционной зоны А, и он образовывался на стадии дегидрирования вследствие сгорания водорода, а также возможных углеводородов в присутствии кислорода воздуха; теплота сгорания водорода обеспечивала поддержание постоянной реакционной температуры газовой смеси в реакционной зоне А). Образующийся при этом конденсат отбирали из холодильника с помощью системы управления уровнем и направляли на утилизацию. Использованную для прямого охлаждения воду подавали насосом в контур циркуляции, охлаждали за счет косвенного теплообмена и вновь распыляли в холодильнике прямого охлаждения.
Вместо описанного выше прямого охлаждения водой газообразную смесь реакционных продуктов стадии дегидрирования можно также сначала охладить, например, в косвенном прямоточном или противоточном кожухотрубном теплообменнике за счет теплообмена с подаваемой на стадию дегидрирования исходной реакционной газовой смесью, нагревающейся при этом до температуры, например, от 350 до 450°С. Благодаря этому температуру газообразной смеси реакционных продуктов стадии дегидрирования, составляющую, например, от 450 до 550°С, снижали до уровня, составляющего примерно от 200 до 300°С.
Дальнейшее охлаждение газообразной смеси реакционных продуктов стадии дегидрирования можно осуществлять в косвенном теплообменнике с использованием этой смеси для нагревания исходной газовой смеси, предназначенной для подачи на рассматриваемую ниже стадию частичного окисления полученного при дегидрировании пропилена, и/или для уравновешивания рассматриваемого ниже охлаждения вторичного газа абсорбера при его предпочтительно двухступенчатом расширении в турбодетандерах, или для компенсирования тепла, необходимого для предварительного нагревания.
Благодаря этому температура газообразной смеси продуктов снижалась примерно до 180°С. Затем смесь можно было охладить до температуры от 30 до 60°С в воздушном и/или водяном холодильнике.
Образующийся при охлаждении водный конденсат накапливали во встроенных в холодильник или последовательно присоединенных к нему каплеотделителях и с помощью системы управления уровнем направляли на утилизацию.
Затем охлажденную и освобожденную от водяного пара газообразную смесь продуктов стадии дегидрирования, находящуюся под давлением около 2 бар, сжимали до давления, составляющего от 10 до 13 бар.
Во избежание чрезмерного повышения температуры при сжатии в целесообразном с производственно-технической точки зрения варианте сжатие осуществляли в две ступени (подобного повышения температуры избегали также благодаря предварительно выполненному охлаждению; предварительное выделение водяного пара позволяло дополнительно снизить потребляемую компрессором мощность). Давление на первой ступени сжатия повышали до величины, составляющей от 4 до 4,5 бар. Температура газовой смеси после первой ступени сжатия составляла около 115°С.
В последовательно присоединенном косвенном теплообменнике (воздушном или водяном холодильнике) газовую смесь вновь охлаждали до температуры от 40 до 60°С, при этом происходила дополнительная конденсация водяного пара. Конденсат накапливали в каплеотделителях, из которых его выводили с помощью системы управления уровнем.
На второй ступени сжатия давление поступающей в соответствующий компрессор газовой смеси (около 4 бар) повышали до конечного давления, составляющего 10 бар (в данном случае сжатие можно осуществлять также до давления, составляющего 13 бар и более). Температура газовой смеси на выходе из второго компрессора составляла около 126°С.
В двух последовательно соединенных косвенных теплообменниках (сначала в воздушном холодильнике (обычно использовали кожухотрубный теплообменник с целесообразным пропусканием подлежащего охлаждению газа через трубки), а затем в водяном холодильнике) сжатую газовую смесь охлаждали сначала до температуры от 40 до 60°С, а затем до 30°С. При этом сконденсированную воду вновь отделяли с помощью каплеотделителей и выводили из системы. Газовая смесь на выходе из второго компрессора содержала не более 0,2% мас. воды. Незначительное влагосодержание обусловливало образование незначительного количества воды, позволяя при последующей абсорбции избежать производственных проблем, связанных с образованием двух жидких фаз, а повышенное давление обусловливало сокращение количество абсорбента, необходимого для осуществления абсорбции.
Для сжатия газовой смеси в производственных условиях используют промышленные турбокомпрессоры (бесконтактные компрессоры с сухим ходом, без масляной смазки, например турбокомпрессоры типа 12 МН 4В фирмы Mannesmann DEMAG, Германия), тогда как в рассматриваемом случае использовали мембранные компрессоры MV 3459 II фирмы Sera. В качестве привода подобных компрессоров в принципе можно использовать как электродвигатели, так и паровые или газовые турбины. Наиболее экономичными зачастую оказываются паровые турбины. Сжатую и охлажденную указанным образом газовую смесь (около 4650 г/ч) вводили в абсорбционную колонну непосредственно над ее кубом. Газовая смесь обладала следующим составом.
Абсорбционная колонна выполнена из высококачественной стали 1.4571. Диаметр колонны составлял 80 мм, толщина стенок 4 мм, длина 1,70 м.
Около 70% объема абсорбционной колонны заполнены насадочными элементами фирмы Montz (Montz BSH-750 с удельной поверхностью 750 м2/м3) в качестве встроенных элементов, повышающих эффективность разделения. Расположенными непосредственно один над другим насадочными элементами была заполнена нижняя часть колонны (1/5 ее общей высоты). Наружный обогрев или охлаждение абсорбционной колонны отсутствовали. В качестве абсорбента в верхнюю часть колонны подавали технический тетрадекан типа PKWF 4/7 af (фирмы Haltermann, Германия) с температурой 30°С; процентный состав исходного (свежего) тетрадекана, определенный методом газовой хроматографии (FID-детектор) по площадям соответствующих пиков:
Указанный состав в процессе непрерывной эксплуатации абсорбционной колонны (по истечении примерно 3000 часов) изменялся следующим образом:
Плотность орошения составляла 15 м3 абсорбента на 1 м2 площади свободного сечения в час (расход тетрадекана 28 кг/ч).
Направляемый на сжигание вторичный газ абсорбционной колонны содержал 950 млн-1 (об.) пропана и 250 млн-1 (об.) пропилена. В условиях промышленного производства подобный поток вторичного газа направляют на сжигание через детандер (например, турбодетандер), чтобы рекуперировать большую часть затраченной на двухступенчатое сжатие газа энергии и вернуть ее на стадию двухступенчатой компрессии. В целесообразном варианте расширение вторичного газа также осуществляют в две ступени, что позволяет избежать нежелательной конденсации. Получаемую при расширении вторичного газа механическую энергию можно использовать непосредственно в качестве параллельного или главного привода одного из компрессоров и/или для выработки тока.
В промышленных условиях, прежде чем направить подвергнутый расширению вторичный газ абсорбера на сжигание, может оказаться целесообразным выделение содержащегося в нем водорода. Водород можно выделить, например, пропуская вторичный газ через выполненную, как правило, в виде трубы мембрану, проницаемую только для молекулярного водорода. Выделенный подобным образом молекулярный водород можно вернуть, например, на стадию гетерогенно катализируемого дегидрирования пропана или использовать иначе (например, в топливных элементах).
Выделение водорода из вторичного газа абсорбера в принципе можно осуществлять также частичной конденсаций, адсорбцией и/или ректификацией (реализуемыми предпочтительно под давлением). В условиях промышленного производства с целью концентрирования вторичного газа абсорбера его, как правило, целесообразно также пропускать через рассмотренную ниже более подробно кислотную воду.
Абсорбат обладал следующим составом (в % мас. в пересчете на его массу):
Перед подачей абсорбата в верхнюю часть последующей десорбционной колонны его нагревали в косвенном теплообменнике до 60°С.
Затем давление абсорбата снижали до 2,7 бар (его можно было снизить, например, в инверсном насосе или посредством клапана, причем механическую энергию, высвобождающуюся в случае применения инверсного насоса, целесообразно использовать для повторного сжатия абсорбента, освобожденного в десорбционной колонне от содержащихся в нем компонентов), и образующуюся при этом двухфазную смесь направляли в верхнюю часть десорбционной колонны.
В нижнюю часть десорбционной колонны (внутренний диаметр 80 мм, длина 1,70 м, толщина стенок 4 мм) противотоком по отношению к стекающему из верхней части абсорбату подавали воздух под давлением 3 бар (1269 нл/ч). Расход абсорбата, подаваемого в верхнюю часть десорбционной колонны, составлял 33,7 л/ч. Выводимый из нижней части десорбционной колонны абсорбент, в основном не содержащий десорбированных компонентов, охлаждали за счет косвенного теплообмена с абсорбатом, сжимали до необходимого для подачи в адсорбционную колонну давления, охлаждали до 16°С в еще одном косвенном теплообменнике (в производственных условиях целесообразным является теплообмен с поверхностной водой) и рециркулировали в верхнюю часть абсорбционной колонны. Десорбционная колонна в качестве встроенных элементов для повышения эффективности разделения содержала структурированные насадочные элементы фирмы Montz из листового металла (Montz BSH-750 с удельной поверхностью 750 м2/м3). Выводимый из десорбционной колонны (при необходимости через механический каплеотделитель) газовый поток с целью улавливания абсорбента промывали водой. То есть этот газовый поток пропускали через насадочный элемент фирмы Montz (Montz BSH-750 с удельной поверхностью 750 м2/м3), подавая противотоком воду (70 л/ч) с температурой 18°С. Под насадочным элементом располагалась улавливающая тарелка, с которой можно было отбирать водную фазу. В разделителе фаз осуществляли разделение на водную и органическую фазы. Весьма незначительное количество органической фазы объединяли с потоком абсорбента, возвращаемого в верхнюю часть абсорбционной колонны. Водную фазу после повторного охлаждения и добавления свежей воды (для компенсации потерь при испарении) вновь направляли к насадочному элементу. Промывку осуществляли в верхней части десорбционной колонны.
Промытый газовый поток, выводимый из промывочной секции через механический каплеотделитель (отделенную в нем жидкую фазу возвращали на промывку), обладал следующим составом (если в реакционной зоне А при дегидрировании пропана достигают более высоких конверсий, указанное ниже содержание пропилена может составлять также от 8 до 12% об., например, промытый газовый поток может содержать также 15% об. пропана, 10% об. пропилена и 14,5% об. кислорода):
Температуру газовой смеси указанного выше состава за счет косвенного теплообмена повышали до 250°С, и смесь в качестве новой исходной реакционной газовой смеси направляли в последующее устройство для частичного окисления со среднечасовой скоростью пропускания 2128 нл/л·ч и начальным давлением 2,1 бар.
С) Конструкция реакционной зоны В (стадия частичного окисления)
1. Первый реактор со стационарным слоем для частичного окисления пропилена в акролеин
Используемый теплоноситель: солевой расплав, состоящий из 53% мас. нитрата калия, 40% мас. нитрита натрия и 7% мас. нитрата натрия.
Геометрические параметры контактной трубки: общая длина 4200 мм, внутренний диаметр 26 мм, наружный диаметр 30 мм, толщина стенок 2 мм.
Реактор: двустенный цилиндр из высококачественной стали (направляющая труба с толщиной стенок от 2 до 5 мм, окруженная наружным цилиндрическим резервуаром).
Внутренний диаметр наружного цилиндра 168 мм. Внутренний диаметр направляющей трубы около 60 мм.
Двухстенный цилиндр сверху закрывала крышка, снизу дно.
Контактная трубка помещена в окруженную цилиндрическим резервуаром направляющую трубу таким образом, чтобы она выступала с верхней и нижней сторон цилиндрического резервуара, уплотненных соответственно крышкой и дном, соответственно на 250 мм.
Теплоноситель находился в замкнутом объеме внутри цилиндрического резервуара. С целью создания как можно более однородных термических граничных условий на наружной поверхности участка контактной трубки, окруженного цилиндрическим резервуаром (длина этого участка составляла 3700 мм), через теплоноситель барботировали азот, обеспечивающий его циркуляцию.
Барботирующий азот перемещал теплоноситель в направляющей трубе снизу вверх, после чего теплоноситель стекал через промежуток между направляющей трубой и цилиндрическим наружным резервуаром (аналогичную интенсивность циркуляции теплоносителя можно обеспечить также его перекачкой, например, пропеллерным насосом). Необходимую температуру теплоносителя можно отрегулировать посредством надетого на наружный цилиндр электрического нагревателя. Для охлаждения теплоносителя использовали воздух.
Питание реактора. Солевой расплав и реакционная газовая смесь перемещались в реакторе относительно друг друга противотоком. Реакционная газовая смесь поступала в реактор сверху. Ее температура на входе в реакционную трубку составляла 250°С.
Солевой расплав на входе в нижнюю часть направляющей трубы обладал температурой Tвход 335°С, а на выходе из ее верхней части температурой Твыход. Разница между Твход и Твыход составляла около 2°С. Средняя температура расплава Тсред. составляла (Твход+Твыход)/2.
Заполнение контактной трубки (сверху вниз): секция А высотой 50 см.
Предварительный слой, образуемый кольцами из стеатита С 220 фирмы CeramTec с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × длина × внутренний диаметр).
Секция В высотой 100 см.
Контактная трубка заполнена гомогенной смесью 30% мас. колец из стеатита С 220 фирмы CeramTec с размерами 5 мм × 3 мм × 2 мм (наружный диаметр × длина × внутренний диаметр) и 70% мас. сплошного катализатора, аналогичного используемому в секции С.
Секция С высотой 170 см.
Контактная трубка заполнена сплошным катализатором из примера 1 немецкой заявки на патент DE-A 10046957 в виде колец с размерами 5 мм × 3 мм × 2 мм (наружный диаметр × длина × внутренний диаметр).
Секция D высотой 50 см.
Завершающий слой колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 7 мм × 4 мм (наружный диаметр х длина х внутренний диаметр).
2. Промежуточное охлаждение и промежуточная подача кислорода
Газообразную смесь реакционных продуктов из первого реактора со стационарным слоем катализатора с целью промежуточного охлаждения (косвенного охлаждения воздухом) пропускали через соединительную трубку из высококачественной стали (длина 400 мм, внутренний диаметр 26 мм, толщина стенок 2 мм) со средним участком длиной 200 мм, заполненным инертным слоем шариков из стеатита фирмы CeramTec диаметром от 5 до 6 мм, которая была прифланцована непосредственно к контактной трубке первого реактора со стационарным слоем.
Газовая смесь поступала в соединительную трубку с температурой более 310°С и выходила из нее с температурой около 140°С. Затем ее смешивали со сжатым воздухом (269 нл/ч), используемым в качестве источника кислорода.
Газовую смесь с температурой 220°С после перемешивания в статическом смесителе подавали в реактор со стационарным слоем для реализации стадии частичного окисления акролеина в акриловую кислоту.
3. Второй реактор со стационарным слоем для частичного окисления акролеина в акриловую кислоту
Использовали реактор со стационарным слоем катализатора, конструкция которого аналогична конструкции реактора первой стадии. Солевой расплав и реакционная газовая смесь перемещались в реакторе прямотоком по отношению друг к другу. Как солевой расплав, так и реакционная газовая смесь поступали в реактор снизу.
Заполнение контактной трубки (снизу вверх)
Секция А высотой 20 см.
Предварительный слой колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × длина × внутренний диаметр).
Секция В высотой 100 см.
Контактная трубка заполнена гомогенной смесью 30% мас. колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр) и 70% мас. слоистого катализатора, аналогичного используемому в секции С.
Секция С длиной 200 см.
Контактная трубка заполнена слоистым катализатором из примера получения 5 немецкой заявки на патент DE-А 10046928 в виде колец с размерами 7 мм × 3 мм × 4 мм (наружный диаметр × длина × внутренний диаметр) (секцию С можно также заполнять полученными соответствующим методом аналогичными слоистыми катализаторами, которые, однако, содержат активную массу со стехиометрическим составом Mo12V2,8W1,2Cu2,4Ox или Mo12V3,5W1,3Cu2,4Ox).
Секция D длиной 50 см.
Завершающий слой колец из стеатита С 220 фирмы CeramTec с размерами 7 мм × 7 мм × 4 мм (наружный диаметр × длина × внутренний диаметр).
Среднечасовая скорость пропускания питающей газовой смеси через второй реактор составляла около 3607 г/ч. Средняя температура расплава (Тсред.), вычисляемая аналогично первому реактору, составляла 284°С.
Конверсия пропилена в первом реакторе составляла 98,0% мол., конверсия акролеина во втором реакторе составляла 99,8% мол.
Газообразная смесь продуктов (газообразная смесь продуктов В), выходящая из второго реактора со стационарным слоем катализатора, обладала температурой 281°С, давлением 1,8 бар и следующим составом:
Используемые на обеих реакционных стадиях катализаторы могут быть заменены также катализаторами из примеров, приведенных в немецкой заявке на патент DE-A 10351269. Катализатор, используемый на первой реакционной стадии, может быть заменен также катализаторами из примеров и сравнительных примеров, приведенных в немецкой заявке на патент DE-A 10344149. Катализатор, используемый на второй реакционной стадии, может быть заменен также катализаторами из примеров, приведенных в немецких заявках на патент DE-A 10360057 и DE-A 10360058. Кроме того, частичное окисление можно реализовать в виде процесса с повышенной нагрузкой, предлагаемого в немецких заявках на патент DE-A 10313210, DE-A 10313213, DE-A 10313212, DE-A 10313211, DE-A 10313208, DE-A 10313209 и DE-A 10313214, а также указанного в настоящем описании при рассмотрении уровня техники.
D) Конструкция зоны разделения В (выделение целевой акриловой кислоты из газообразной смеси продуктов частичного окисления)
Поток горячей газообразной смеси продуктов (газообразной смеси продуктов В) (после соединения с описанным ниже насыщенным потоком отпаривающего газа) подвергали прямоточному охлаждению в холодильнике прямого охлаждения примерно до 180°С благодаря разбрызгиванию форсунками охлаждающей жидкости с температурой от 140 до 150°С, которую использовали также в качестве абсорбента (она содержала 57,4% мас. дифенилового эфира, 20,7% мас. дифенила, 20% мас. о-диметилфталата и до 1% мас. фенотиазина (дополняющего до 100% мас.), а также высококипящие компоненты (например, олигомерную акриловую кислоту)). В нижней части холодильника прямого охлаждения накапливалась неиспаренная охлаждающая жидкость, которую отбирали и после охлаждения в теплообменнике до исходной температуры вновь разбрызгивали в верхней части холодильника. Газообразную смесь продуктов В (соединенную с насыщенным отпаривающим газом) и разбрызгиваемую форсунками охлаждающую жидкость вводили в холодильник прямого охлаждения одновременно. Затем охлажденную газообразную смесь продуктов В направляли в последовательно присоединенную абсорбционную колонну. В циркуляционном контуре охлаждающей жидкости накапливались требующие выведения высококипящие побочные продукты. В связи с этим периодически (раз в сутки) отбирали часть циркулирующей охлаждающей жидкости (1 кг) и время от времени подвергали ее переработке в ротационном испарителе (8 мбар, 180°С). Полученный при этом очищенный конденсат абсорбента направляли в описанный ниже буферный резервуар для кубовой жидкости, выполненный из высококачественной стали колонны, предназначенной для отмывки остаточного газа под давлением, и при этом раз в неделю восполняли происходящую при переработке в ротационном испарителе потерю абсорбента (около 2,9 г/ч), добавляя к конденсату свежую смесь дифенилового эфира (21,2% мас.), дифенила (58,8% мас.) и о-диметилфталата (20% мас.) (смесь двух первых компонентов является продуктом Diphyl®).
Охлажденную газообразную смесь продуктов В вводили в абсорбционную колонну в точке выше ее куба и ниже первого насадочного элемента. Длина абсорбционной колонны (без куба) составляла 5420 мм, внутренний диаметр 50 мм, и колонна была снабжена насадочными элементами Kuehni (CH) Rombopak 9M с общей длиной 3,3 м. Абсорбционная колонна была выполнена из стекла и снабжена термостатирующей сегментированной стеклянной рубашкой. Непосредственно над вторым снизу насадочным элементом колонны смонтирован редукционный клапан (регулируемый дроссель), позволяющий имитировать потерю напора в абсорбционной колонне, составляющую от 100 до 300 мбар (которая наблюдалась бы в промышленных условиях). Рабочее давление в верхней части абсорбционной колонны составляло 1,30 бар, а в кубе 1,40 бар.
Температура в кубе составляла 113°С. Стеклянная рубашка состояла из пяти функционирующих независимо друг от друга сегментов, посредством которых обеспечивали необходимый температурный профиль абсорбционной колонны. Подобное сегментирование соответствовало также внутренней структуре абсорбционной колонны.
Ниже приведены температуры и конструктивное исполнение сегментов (снизу вверх), которых термостатировали подаваемой насосом водой (термостатирование первого сегмента осуществляли имеющим соответствующую температуру масляным теплоносителем):
1-й сегмент: длина 1020 мм (направлен вверх от точки ввода газообразной смеси продуктов В); 113°С; насадочный элемент Rhombopak (0,4 м) между местом ввода газообразной смеси продуктов В, находящейся непосредственно над кубом абсорбционной колонны, и точкой ввода непрерывно отбираемой из куба и возвращаемой в нее кубовой жидкости (с температурой около 113°С и расходом около 200 л/ч), и насадочный элемент Rhombopak (0,6 м) над местом ввода кубовой жидкости; расстояние между обоими насадочными элементами составляло около 2 см.
2-й сегмент: длина 1250 мм, 75°С; в нижней части сегмента расположен редукционный клапан, а над клапаном до места ввода потока конденсата низкокипящих продуктов из рассматриваемого ниже узла очистной дистилляции насадочный элемент Rhombopak (0,5 м).
3-й сегмент: длина 950 мм, 60°С; насадочный элемент Rhombopak (0,6 м) расположен непосредственно над местом ввода потока конденсата низкокипящих продуктов во 2-й сегмент.
4-й сегмент: длина 950 мм, 50-55°С, насадочный элемент Rhombopak (0,6 м), который заканчивается непосредственно ниже места ввода рассматриваемого ниже основного потока абсорбента.
5-й сегмент: длина 1250 мм, 20°С, насадочный элемент Rhombopak (0,6 м); расположен между рассматриваемой ниже улавливающей тарелкой для слива кислотной воды, смонтированной непосредственно над местом ввода абсорбента в 4-й сегмент, и местом ввода кислотной воды, рециркулируемой в верхнюю часть абсорбционной колонны.
С указанной выше улавливающей тарелки 5-го сегмента непрерывно отбирали около 70,2 л/ч обогащенного водой конденсата (кислотной воды). Конденсат подвергали в теплообменнике косвенному охлаждению до 20°С и рециркулировали в абсорбционную колонну, преимущественно вводя его выше самого верхнего насадочного элемента Rhombopak (70 л/ч). Нерециркулируемую часть отобранной кислотной воды (реакционную воду) выводили с помощью системы управления уровнем и направляли на стадию экстракции. На этой стадии удаленную кислотную воду соединяли и смешивали в снабженном мешалкой стеклянном сосуде (объемом 1 л, диаметром 80 мм), находящемся под атмосферным давлением и при температуре окружающей среды, с меньшим отдельным потоком (ниже рассмотренным более подробно) малонасыщенного (акриловой кислотой и побочными компонентами) абсорбента. Образующаяся смесь за счет свободного перелива непрерывно перетекала в предназначенный для разделения фаз стеклянный сосуд (объемом 7,5 л, диаметром 150 мм), также находящийся при температуре окружающей среды. В этом сосуде перетекающая жидкая смесь расслаивалась на две жидкие фазы, причем короткоцепные кислотные побочные компоненты (например, уксусная кислота) предпочтительно переходили в водную фазу с меньшим удельным весом, а акриловая кислота предпочтительно переходила в органическую фазу с большим удельным весом. Органическую фазу посредством мембранного дозирующего насоса с принудительной подачей соединяли с большим отдельным потоком малонасыщенного абсорбента, и общий поток (около 3,0 л/ч, 40°С), вводили, как указано выше, в качестве основного потока абсорбента в абсорбционную колонну непосредственно ниже улавливающей тарелки для кислотной воды.
Выходящий из верхней части абсорбционной колонны (основной) остаточный газ (температура 20°С, давление 1,30 бар) обладал следующим составом:
(Основной) остаточный газ (во избежание нежелательной конденсации) нагревали за счет косвенного теплообмена до 40°С, сжимали мембранным компрессором (в производственных условиях турбокомпрессором с приводом от электродвигателя, например турбокомпрессором типа 12МН4В фирмы Mannesmann DEMAG, Германия) до 2,70 бар и около 78% об. возвращали в качестве циркуляционного газа ((общего) остаточного газа) в первый реактор дегидрирования реакционной зоны А.
Около 22% об. сжатого (основного) остаточного газа после снижения давления до 1,6 бар отводили в колонну из высококачественной стали для промывки под давлением (сталь 1.4571, внутренний диаметр 30 мм, 2 м насадка Rhombopak 9M), в которой его подвергали противоточной промывке под давлением около 1,6 бар и при температуре около 51°С рассмотренным ниже общим потоком наименее насыщенного абсорбента (абсорбент подавали в верхнюю часть, а газ в нижнюю часть промывочной колонны).
Промытый газовый поток под давлением около 1,5 бар и при температуре от 119 до 125°С вводили в другую функционирующую под давлением колонну из высококачественной стали (1.4571, внутренний диаметр 30 мм, 3 м Rhombopak 9M, отпарная колонна с рубашкой для термостатирования масляным теплоносителем) для отпаривания легкокипящих компонентов (например, как описано в немецкой заявке на патент DE-A 10336386) из потока насыщенного абсорбента, непрерывно выводимого из куба абсорбционной колонны (около 3,5 л/ч, температура около 113°С). Образующийся при этом насыщенный низкокипящими компонентами (более летучими, чем акриловая кислота) газовый поток (насыщенный газ отпаривания) нагревали в термостатируемом при 170°С трубопроводе с рубашкой (диаметр 15 мм, наружная стенка рубашки также выполнена из высококачественной стали) и на входе в прямой холодильник соединяли с горячим потоком газообразной смеси продуктов В.
Поток малонасыщенного абсорбента с помощью системы управления уровнем из нижней части колонны для промывки под давлением из высококачественной стали сливали в указанный выше буферный стеклянный сосуд объемом 5 л (как указано выше, в этот сосуд поступал также образующийся в ротационном испарителе конденсат абсорбента).
Больший отдельный поток (2,5 л/ч) малонасыщенного абсорбента из буферного сосуда посредством мембранного насоса в качестве составной части основного потока абсорбента направляли, как описано выше, непосредственно под улавливающую тарелку 4-го сегмента абсорбционной колонны, предназначенную для кислотной воды. Меньший отдельный поток (460 мл/ч) малонасыщенного абсорбента посредством другого мембранного насоса направляли, как описано выше, в снабженный мешалкой стеклянный сосуд стадии экстракции кислотной воды.
Из кубовой жидкости отпарной колонны (около 100 л/ч), циркулирующей посредством приводимого в действие сжатым воздухом мембранного насоса, с помощью системы управления уровнем отбирали около 3,5 кг кубовой жидкости, пропускали через тканевый фильтр в корпусе из высококачественной стали и регулировочный клапан и для очистки направляли в узел вакуумной дистилляции.
Узел вакуумной дистилляции представлял собой стеклянную колонну с зеркальным покрытием и вакуумной изоляцией (очистную колонну) длиной 4000 мм с внутренним диаметром 50 мм. Температуру в кубе этой колонны благодаря циркуляции кубовой жидкости с принудительным расширением (около 250 л/ч, тангенциальный лопастный насос, давление 4 бар) поддерживали на уровне 191°С. Абсолютное давление в кубе составляло около 230 мбар, в верхней части колонны 100 мбар. Для ингибирования полимеризации выше уровня кубовой жидкости вводили воздух (52 нл/ч).
Между кубом колонны для вакуумной дистилляции и вводом в нее насыщенного потока из контура циркуляции куба отпарной колонны находились 6 колпачковых тарелок (с промежутком 5 см), а выше еще 15 колпачковых тарелок (с промежутком 5 см); над колпачковыми тарелками посредством дозирующего мини-насоса можно было осуществлять отбор проб.
Выше места отбора проб находились 10 сетчатых тарелок (каждая тарелка имела шесть равноудаленных друг от друга отверстий диаметром 6,5 мм; расстояние между тарелками составляло 5 см), над которыми располагалась улавливающая тарелка, с которой непрерывно отбирали около 364 г/ч очищенной акриловой кислоты (целевого продукта), которую после охлаждения направляли в соответствующий сборник.
Содержание побочных компонентов в целевом продукте составляло:
На самую верхнюю из сетчатых тарелок, расположенных ниже места отбора целевого продукта, рециркулировали другое количество (732 мл/ч) акриловой кислоты, отбираемой с улавливающей тарелки дистилляционной колонны при температуре около 75°С.
Выше места отбора целевого продукта находились еще 10 сетчатых тарелок, каждая из которых имела 6 отверстий диаметром 5,5 мм (расстояние между тарелками составляло 5 см), что позволяло концентрировать остающиеся низкокипящие компоненты в верхней части колонны. Над этими сетчатыми тарелками располагалась другая тарелка, предназначенная для улавливания конденсата низкокипящих компонентов, образующегося при косвенном охлаждении в верхней части колонны (26°С, абсолютное давление 100 мбар). Температура конденсата на этой улавливающей тарелке составляла 73°С. Конденсат содержал следующие низкокипящие компоненты, % мас.:
Основную часть (570 мл/ч) отбираемого с улавливающей тарелки общего потока конденсированных низкокипящих компонентов возвращали в дистилляционную колонну ниже предназначенной для улавливания этого конденсата тарелки. Флегму возвращали в дистилляционную колонну. Остальную часть (190 мл/ч) общего потока конденсированных низкокипящих компонентов охлаждали до 40°С и направляли в колонну для абсорбции акриловой кислоты (над 2-м сегментом). Для ингибирующего смачивания стенок в самую верхнюю область верхней части очистной колонны посредством полноструйной форсунки с четырьмя выходными отверстиями впрыскивали стабилизированный 0,5% мас. фенотиазина целевой продукт (51 мл/ч) с температурой 25°С.
Газовый поток, выводимый из верхней части очистной колонны посредством мембранного вакуумного насоса, в основном состоял из инертных газов и низкокипящих компонентов. В охлажденной до 8°С ловушке из этого газового потока удавалось выделить еще 4,6 г/ч конденсируемых низкокипящих остаточных компонентов. Образующийся при этом конденсат состоял из следующих остаточных компонентов, % мас.:
«Инертный газовый поток» после отбора образованного остаточными компонентами конденсата обладал следующим составом:
Этот поток рециркулировали в точку выше редукционного клапана и ниже находящегося над этим клапаном насадочного элемента Rhomopak 2-го сегмента абсорбционной колонны.
Выгружаемый из куба очистной колонны с помощью системы управления уровнем, в основном освобожденный от акриловой кислоты абсорбент направляли в виде общего потока малонасыщенного абсорбента в описанную выше колонну для промывки под давлением из высококачественной стали, причем для выгрузки абсорбента из вакуумированной колонны повышали давление нагнетания насоса, используемого для циркуляции кубовой жидкости очистной колонны.
Состав малонасыщенного абсорбента:
Для снижения потерь эдукта, а также для обеспечения повышенного выхода целевого продукта все насыщенные эдуктом или целевым продуктом проанализированные газовые потоки объединяли в стеклянном сосуде емкостью 0,5 л, посредством миниатюрного мембранного насоса подавали к компрессору первой ступени сжатия зоны разделения А, перед компрессором объединяли с охлажденной газообразной смесью продуктов стадии дегидрирования и затем сжимали. Выход акриловой кислоты в пересчете на использованный пропан составлял 78,9% мол.
Полученную в качестве целевого продукта сырую акриловую кислоту можно дополнительно очистить перекристаллизацией или ректификацией, как описано в европейской заявке на патент ЕР-А 616998 (при соблюдении условий, приведенных в европейской заявке на патент ЕР-А 912486), немецкой заявке на патент DE-A 19606877 (маточный щелок можно рециркулировать в абсорбционную и/или очистную колонну) или европейской заявке на патент ЕР-А 648732, получая чистую акриловую кислоту, пригодную для осуществляемого известными методами синтеза суперабсорбирующих воду полимеров. Как сырая, так и чистая акриловая кислота отлично пригодны для синтеза сложных эфиров акриловой кислоты, например алкилакрилатов.
Сравнительный пример 4
Подобно сравнительным примерам 1 и 3 в реакционной зоне А с последовательно присоединенной к ней зоне разделения А получают исходную реакционную газовую смесь для реакционной зоны В, которая обладает следующим составом:
Реакционная зона В состоит из двух параллельно функционирующих систем сдвоенных реакторов (групп реакторов), каждая из которых выполнена подобно системе сдвоенных реакторов из примера, приведенного в немецкой заявке на патент DE-A 10351269, и заполнена катализатором. В каждую из обеих групп реакторов подают 61022 нм3/ч указанной выше исходной реакционной газовой смеси. Расход воздуха, подаваемого между обеими стадиями частичного окисления, составляет 11200 нм3/ч (для каждой из групп реакторов). Реакционный газ и солевой расплав на обеих стадиях частичного окисления пропускают противотоком относительно друг друга.
Расход подаваемого на первую реакционную стадию пропилена составляет около 110 нл/л·ч. Температура входа солевого расплава на первую реакционную стадию составляет 346°С, температура его выхода 349°С. Температура входа солевого расплава на вторую реакционную стадию составляет 272°С. Температура выхода солевого расплава со второй реакционной стадии составляет 275°С. Исходная реакционная газовая смесь поступает в реактор первой ступени с температурой 300°С. Температура газа, поступающего на вторую реакционную стадию, составляет 230°С.
Производительность насосов для циркуляции солевого расплава аналогична указанной в немецкой заявке на патент DE-A 10351269. Давление входа на первую реакционную стадию составляет 1,9 бар. Давление входа на вторую реакционную стадию составляет 1,4 бар. Конверсия пропилена на первой реакционной стадии составляет 97% мол. (в пересчете на однократное пропускание). Степень превращения акролеина на второй реакционной стадии составляет 99,3% мол. (в пересчете на однократное пропускание).
Температура газообразных смесей продуктов на выходе с обеих вторых стадий частичного окисления составляет 275°С. Газообразные смеси продуктов объединяют в общий газовый поток (145000 нм3/ч), который обладает следующим составом (в пересчете на общее количество этого газового потока), % мас.:
Охлаждение газообразной смеси продуктов В
Газообразную смесь продуктов В направляют в выполненный из аустенитной стали (1.4571) прямой холодильник обычной конструкции диаметром 2,6 м и высотой 14,5 м. Газообразную смесь продуктов охлаждают до температуры 165,5°С за счет частичного испарения отбираемого из куба абсорбционной колонны абсорбента. При этом охлаждающая среда перемещается относительно подвергаемой охлаждению газообразной смеси продуктов В прямотоком и поступает в точку прямого конденсатора, расположенную выше форсунки с отражательным щитком.
Охлажденная газообразная смесь продуктов В вместе с неиспаренным абсорбентом поступает в куб последующего узла абсорбции.
Узел абсорбции
Узел абсорбции представляет собой тарельчатую колонну с 45 клапанными тарелками (без монтажа крышек отдельных клапанов) и тремя улавливающими тарелками, давление в верхней части которой составляет 1,2 бар. Тарельчатая колонна может быть снабжена сетчатыми тарелками или провальными сетчатыми тарелками, используемыми в качестве эффективно разделяющих встроенных элементов.
Самая нижняя тарелка абсорбционной колонны выполнена в виде двухстенной улавливающей тарелки. Под этой тарелкой происходит отделение выходящей из прямого холодильника газовой смеси от неиспаренного в холодильном устройстве абсорбента.
Выходящая из системы прямого охлаждения смесь неиспаренного абсорбента с газообразной смесью продуктов поступает в абсорбционную колонну с тангенциальным импульсом. Пропускание смеси через циклон препятствует непосредственному уносу капель в самую нижнюю улавливающую тарелку.
Температура в кубе абсорбционной колонны составляет 162,7°С. Из куба выгружают 1067 м3/ч кубовой жидкости следующего состава, % мас.:
Кубовую жидкость (жидкий слив из абсорбционной колонны) возвращают с помощью центробежного насоса преимущественно в качестве охлаждающей среды в систему прямого охлаждения газообразной смеси продуктов В. Часть кубовой жидкости (0,3% от общего количества) отбирают и направляют в узел дистилляции.
Сырой, соответственно малонасыщенный абсорбент подают в абсорбционную колонну на 34-ю тарелку снизу, находящуюся непосредственно под 3-ей улавливающей тарелкой. Вводимый в абсорбционную колонну малонасыщенный абсорбент состоит из содержащего ингибитор полимеризации отдельного потока (86,6% мас. от общего вводимого количества), который поступает из рассматриваемого ниже узла промывки циркуляционного газа, и отдельного потока (13,4% мас. от общего вводимого количества), поступающего из узла экстракции кислотной воды. Температура вводимых в абсорбционную колонну потоков составляет 51,9°С.
С самой нижней тарелки абсорбционной колонны, выполненной в виде улавливающей тарелки, отбирают (в виде другого жидкого слива абсорбционной колонны) 1728 м3/ч насыщенного акриловой кислотой абсорбента (абсорбата). Его температура составляет 119,6°С. Отбираемый центробежным насосом абсорбат содержит следующие компоненты (в пересчете на общее количество), % мас.:
11,4% мас. от общего количества абсорбата направляют в узел десорбции, 3,1% мас. от общего количества абсорбата направляют непосредственно в куб абсорбционной колонны и 85,5% мас. возвращают на шестую тарелку абсорбционной колонны снизу, предварительно охладив в серии теплообменников на 15 К.
Отводимое в первом (кожухотрубном) теплообменнике тепло используют для частичного испарения образующейся в описанном выше узле конденсации кислотной воды. Температура абсорбата на выходе из теплообменника составляет 113,3°С. Дальнейшее охлаждение абсорбата до необходимой для рециркуляции температуры осуществляют в воздушных холодильниках.
Тарелки, находящиеся между первой улавливающей тарелкой и тарелкой, на которую поступает абсорбат, выполнены в виде четырехканальных клапанных тарелок (сталь VV12, кроме крышек). Расстояние между двумя тарелками составляет 0,7 м. Тарелки, от той, на которую поступает абсорбат, до 13-й снизу, выполнены в виде двухканальных клапанных тарелок (сталь VV12, кроме крышек) и смонтированы на расстоянии 0,5 м друг от друга.
Над 13-й тарелкой снизу располагается другая двухстенная улавливающая тарелка, с которой центробежным насосом отбирают жидкий поток с температурой 71,8°С. 9% мас. от общего количества отбираемой с этой тарелки жидкости рециркулируют непосредственно ниже этой второй улавливающей тарелки, причем от 1 до 10% мас. этого рециркулируемого количества используют для орошения форсунками второй улавливающей тарелки. Остальное отбираемое количество жидкости охлаждают с помощью воздушных холодильников до 59,4°С и направляют на 19-ю тарелку снизу. Тарелки, находящиеся между второй улавливающей тарелкой и 19-й тарелкой, выполнены в виде четырехканальных клапанных тарелок (сталь VV12, кроме крышек), причем расстояние между тарелками составляет 0,6 м.
В отбираемый со второй улавливающей тарелки поток вводят также поток содержащих акриловую кислоту низкокипящих компонентов, отбираемый из верхней части рассматриваемой ниже ректификационной колонны. Кроме того, при осуществлении процесса в промышленном масштабе вторая улавливающая тарелка пригодна также для подачи содержащих акриловую кислоту материальных потоков, например, сырой акриловой кислоты, не отвечающей требованиям спецификации, или содержащих акриловую кислоту материальных потоков других технологических ступеней, например, из узла очистки акриловой кислоты дистилляцией или кристаллизацией.
Тарелки, расположенные между 19-й тарелкой снизу и местом подачи в абсорбционную колонну сырого, соответственно малонасыщенного абсорбента, являются двухканальными клапанными тарелками (сталь VV12, кроме крышек), расстояние между которыми составляет соответственно 0,5 м. Над 34-й тарелкой снизу расположена третья двустенная улавливающая тарелка.
Остающуюся непоглощенной абсорбентом смесь остаточных газообразных продуктов, прежде всего содержащую низкокипящие и неконденсируемые компоненты, охлаждают в узле конденсации, расположенном непосредственно над третьей улавливающей тарелкой. Узел конденсации выполнен в виде системы прямого охлаждения и включает десять двухканальных клапанных тарелок (сталь VV12, кроме крышек), расстояние между которыми составляет 0,5 м.
Водную фракцию конденсата низкокипящих компонентов, отобранную с третьей улавливающей тарелки, используют для непосредственного охлаждения. 2% мас. (от общего количества конденсата кислотной воды, отбираемого с третьей улавливающей тарелки), выгружают с температурой 45°С и направляют на экстракцию кислотной воды. Выгруженная кислотная вода в основном обладает следующим составом, % мас.:
Невыведенный конденсат кислотной воды охлаждают в кожухотрубном теплообменнике до 33°С. 79% мас. (от количества охлажденного до 33°С конденсата кислотной воды) подают на 40-ю тарелку снизу. Остальное количество конденсата кислотной воды охлаждают в другом теплообменнике до 16°С. Этот теплообменник энергетически связан с испарителем, необходимым для испарения жидкого пропана.
Остающаяся после конденсации кислотной воды газовая смесь, выходящая из узла абсорбции с температурой 29°С, преимущественно содержит следующие компоненты, % мас.:
Остаточный газовый поток (циркуляционный газ) после конденсации кислотной воды выводят из колонны через каплеотбойник и нагревают в кожухотрубном теплообменнике на 6K. Благодаря этому избегают возможной конденсации в трубопроводах вторичного газа. Остаточный газовый поток посредством турбокомпрессора с электроприводом сжимают до 3,3 бар.
20% об. этого остаточного газового потока (от его общего количества) используют в качестве отпаривающего газа в рассматриваемой ниже промывочной колонне. Остальной остаточный газовый поток возвращают в реакционную зону А для гетерогенно катализируемого дегидрирования пропана.
Узел десорбции
Абсорбат, эффективно выводимый с самой нижней улавливающей тарелки абсорбционной колонны, направляют в узел десорбции для выделения еще содержащихся в нем низкокипящих компонентов. Сначала его нагревают до 130°С в обогреваемом водяным паром кожухотрубном теплообменнике, после чего подают в верхнюю часть десорбционной колонны. При этом 5% мас. впрыскиваемого через форсунки абсорбата (от общего количества вводимого в десорбционную колонну абсорбата) используют для увлажнения стенок, а также люка и крышки колонны.
В качестве встроенных элементов в десорбционной колонне используют 38 провальных сетчатых тарелок, смонтированных на соответствующих волноломах. При этом диаметр отверстий тарелок от 1-ой до 9-ой (снизу) составляет 30 мм, диаметр отверстий 10-й тарелки составляет 25 мм, а от 11-й тарелки и выше 20 мм. Расстояние между отдельными тарелками составляет 0,5 м. Давление в верхней части десорбционной колонны составляет 1,83 бар.
Подачу тепла в куб десорбционной колонны осуществляют посредством двух параллельно функционирующих испарителей с принудительной циркуляцией.
В качестве отпаривающего газа непосредственно в куб десорбционной колонны вводят часть остаточного газового потока из верхней части абсорбционной колонны, предварительно подвергнутого рассматриваемой ниже промывке в промывочной колонне. При этом отпаривающий газ перед подачей в десорбционную колонну смешивают с частью отбираемой из куба десорбционной колонны, нагретой в испарителе кубовой жидкости, и смесь нагревают в кожухотрубном теплообменнике до 120°С. Смешиваемый с отпаривающим газом отдельный поток кубовой жидкости составляет 9% мас. от общего количества отбираемой кубовой жидкости. При необходимости можно также отказаться от указанного смешивания с нагретой кубовой жидкостью.
Содержащий акриловую кислоту абсорбент, образующийся в кубе десорбционной колонны, включает следующие компоненты, % мас.:
Кубовую жидкость с температурой 131,5°С, отбираемую из десорбционной колонны для подачи в испарители с принудительной циркуляцией, нагревают до 150°С.
Часть общего количества отбираемой кубовой жидкости, составляющую 63% мас., возвращают на 8-ю тарелку десорбционной колонны снизу.
Образующийся в верхней части десорбционной колонны отпаривающий газ, насыщенный низкокипящими компонентами, рециркулируют в холодильник прямого охлаждения для охлаждения газообразной смеси продуктов В. Рециркулируемый насыщенный отпаривающий газ включает также жидкость, уносимую с самой верхней тарелки десорбционной колонны, и преимущественно обладает следующим составом, % мас.:
Узел ректификации
Отдельный поток, отбираемый из узла десорбции после испарителей с принудительной циркуляцией, который состоит главным образом из акриловой кислоты, дифила и диметилфталата, направляют в узел ректификации, включающий укрепляющую часть и исчерпывающую часть.
Узел ректификации содержит 43 смонтированные на соответствующих волноломах провальные сетчатые тарелки с различным диаметром отверстий, составляющим (снизу вверх) для тарелок от 1-ой до 10-й 50 мм, от 11-й до 13-й 25 мм и от 14-й до самой верхней тарелки 14 мм. Расстояние между отдельными тарелками составляет 0,4 м. Расстояние между 8-й и 9-й тарелками составляет 1 м.
Давление в верхней части ректификационной колонны составляет 106 мбар. Насыщенный абсорбент с частично выделенными из него в узле десорбции низкокипящими компонентами подают на 8-ю тарелку (снизу) через снабженный несколькими патрубками кольцевой трубопровод.
В несколько точек ректификационной колонны, находящихся ниже 1-й тарелки, через кольцевой трубопровод подают 430 нм3/ч воздуха.
Подачу тепла в куб ректификационной колонны осуществляют посредством двух параллельно функционирующих внешних испарителей с принудительной циркуляцией. Температура в кубе ректификационной колонны обычно составляет 188°С. Конденсирующаяся в кубе фракция тяжелокипящих продуктов преимущественно содержит следующие компоненты, % мас.:
Выводят до 83% мас. отбираемой из узла ректификации кубовой жидкости (в пересчете на поступающий в узел ректификации поток), содержащей конденсат труднокипящего абсорбента, и частично рециркулируют его в кубовую часть ректификационной колонны через теплообменник. Выведенную фракцию труднокипящих компонентов направляют в узел промывки, пропустив ее через сепаратор для отделения твердых веществ (циклон) и при необходимости добавив сырой абсорбент (дифил и диметил-фталат). Меньший отдельный поток (1,2% мас. от общего количества выведенной кубовой жидкости) направляют в область куба узла абсорбции.
С 27-й тарелки выше ввода в ректификационную колонну через боковой штуцер отбирают сырую акриловую кислоту. Отбор сырой акриловой кислоты осуществляют посредством провальной сетчатой тарелки с встроенной средней вытяжной чашкой. Отбираемая сырая акриловая кислота преимущественно содержит следующие компоненты, % мас.:
Отбираемую сырую акриловую кислоту охлаждают до 25°С в двух последовательно соединенных теплообменниках. 15,5% мас. отбираемой сырой акриловой кислоты (в пересчете на вводимый в ректификационную колонну продукт) выводят и меньшую часть используют для растворения ингибитора полимеризации.
Выделенные в верхней части ректификационной колонны низкокипящие компоненты подвергают двухступенчатому охлаждению в холодильнике прямого охлаждения. Охлаждающую жидкость пропускают через обе ступени охлаждения противотоком, причем температура конденсата первой ступени составляет 52°С, а второй ступени 24,5°С. Паровую фазу второй ступени конденсации направляют в вакуумный узел. При этом параллельно функционируют два жидкостных секционных насоса, и согласно немецкой заявке на патент DE-A-10143565 в качестве уплотняющей жидкости используют конденсат второй ступени конденсации. В последующем резервуаре жидкую фазу (в первую очередь циркулирующую жидкость) отделяют от неконденсируемых компонентов, которые образуют вторичный газ узла ректификации. Этот газ преимущественно обладает следующим составом, % мас.:
Вторичный газ узла ректификации подвергают сжиганию вместе с другими производственными остатками. Однако при высоком содержании пропана этот вторичный газ можно также рециркулировать на стадию абсорбции.
Выделенная в верхней части ректификационной колонны фракция низкокипящих компонентов в основном содержит, % мас.:
Часть отобранной в качестве низкокипящей фракции жидкости (47,6% мас. в пересчете на вводимый в ректификационную колонну продукт) используют в качестве флегмы, подаваемой в колонну посредством нескольких форсунок. При этом небольшую часть жидкости используют также для осуществляемого с помощью форсунок обрызгивания крышки колонны, а также выпарного трубопровода содержащим ингибитор конденсатом. 1,3% мас. фракции низкокипящих компонентов (в пересчете на вводимый в ректификационную колонну продукт) выводят и, как описано выше, возвращают в узел абсорбции.
В качестве ингибитора полимеризации используют фенотиазин. Ингибитор готовят в виде раствора концентрацией 1,1% мас. в выводимой через боковой штуцер, как описано выше, сырой акриловой кислоте и непрерывно добавляют к флегме, состоящей из конденсированных низкокипящих компонентов, а также к конденсатам обеих ступеней прямого охлаждения узла ректификации. Твердый стабилизатор в виде чешуек или крошки дозируют в резервуар для приготовления его раствора посредством шнеков.
Узел промывки
Давление в верхней части промывочной колонны составляет 3 бар. В качестве повышающих эффективность разделения встроенных элементов используют провальные сетчатые тарелки с диаметром отверстий 30 мм. 30 сетчатых тарелок монтируют на расстоянии друг от друга 0,4 м.
В качестве промывочной жидкости используют кубовую жидкость узла ректификации.
Промывочную жидкость подают в верхнюю часть промывочной колонны при температуре 50°С. В точку, расположенную под самой нижней тарелкой, вводят циркуляционный газ узла абсорбции, предварительно охлажденный до температуры входа (50°С). Расход циркуляционного газа составляет 37% мас. от расхода подаваемой промывочной жидкости.
Образующаяся в кубе промывочной колонны жидкость преимущественно содержит следующие компоненты, % мас.:
Образующуюся в узле промывки кубовую жидкость выводят и разделяют на две части, находящиеся в массовом отношении 7,2:1. Большую часть вводят, как описано выше, в абсорбционную колонну, а меньшую часть направляют на экстракцию кислотной воды.
Промытый циркуляционный газ преимущественно содержит следующие компоненты, % мас.:
Промытый циркуляционный газ направляют в рассмотренный выше узел десорбции в качестве отпаривающего газа.
Экстракция кислотной воды
Конденсат кислотной воды из узла абсорбции экстрагируют в узле экстракции частью поступающего из промывочного узла жидкого слива.
Узел экстракции состоит из емкости с двухступенчатой турбинной мешалкой, а также горизонтального отстойного резервуара. Вводимый в отстойный резервуар и выводимый из него потоки отделяют друг от друга благодаря встроенным элементам, расположенным в направлении, поперечном течению. Температура на входе конденсата кислотной воды в узел экстракции составляет 45°С. Массовое отношение выводимого из куба узла промывки потока к кислотной воде из узла ее конденсации составляет 0,8:1.
Образующийся в узле экстракции водный экстракт поглощает выводимые из его куба полярные компоненты, такие как диакриловая кислота (аддукт Майкла) и малеиновый ангидрид, при этом последний гидролизуется. Экстракт преимущественно содержит следующие компоненты, % мас.:
Выведенную водную фазу сжигают вместе с другими остаточными продуктами. Перед их сжиганием часть кислотной воды подвергают упариванию. Необходимую для упаривания теплоту получают в кожухотрубном теплообменнике узла абсорбции благодаря теплообмену с охлаждаемым в нем, как описано выше, образующимся на первой улавливающей тарелке конденсатом.
Образующаяся в узле экстракции органическая фаза (очищенный продукт) преимущественно содержит следующие компоненты, % мас.:
Выводимую органическую фазу, как описано выше, направляют в узел абсорбции.
Дистилляция труднокипящих компонентов
Указанное выше частичное количество кубовой жидкости из узла абсорбции направляют в узел дистилляции, в котором благодаря нагреванию осуществляют выделение содержащейся в кубовой жидкости фракции труднолетучих и фракции легколетучих компонентов. Узел дистилляции состоит из одной ступени и функционирует под давлением 90 мбар. Возможно также техническое исполнение дистилляционной колонны, предусматривающее использование известных специалистам встроенных элементов, повышающих эффективность разделения.
Подвод тепла осуществляют посредством наружного испарителя с принудительной циркуляцией. Перегрев циркулирующей жидкости в испарителе составляет 5 K. Устанавливаемая в узле дистилляции температура составляет 188°С.
Часть жидкости, циркулирующей в испарителе с принудительной циркуляцией, отбирают и после разбавления пригодным разбавителем (например, диметилфорамидом или метанолом) вместе с другими производственными остатками подвергают сжиганию.
Фракция труднокипящих компонентов преимущественно содержит следующие продукты,% мас.:
1,7% мас. фракции труднокипящих компонентов (в пересчете на вводимый в узел дистилляции продукт) выводят и подвергают сжиганию.
Фракцию легкокипящих компонентов, переводимую в узле дистилляции в паровую фазу, охлаждают и конденсируют благодаря комбинированию прямого и косвенного охлаждения. Для этого уже подвергнутую конденсации фракцию низкокипящих компонентов используют для орошения паровой фазы узла дистилляции, охлаждают в кожухотрубном теплообменнике совместно с неподвергнутой конденсации паровой фазой, и охлажденную жидкость используют для орошения паровой фазы выше теплообменника.
Выходящий из холодильника материальный поток с температурой 52°С разделяют в резервуаре на газообразную и жидкую фазы.
Образующаяся в узле дистилляции жидкая фракция низкокипящих компонентов преимущественно содержит следующие продукты, % мас.:
Жидкую фракцию низкокипящих компонентов разбрызгивают центробежным насосом, как описано выше, над кожухотрубным теплообменником. Выводят 98,2% мас. жидкости, отбираемой в качестве фракции низкокипящих компонентов (в пересчете на подаваемое в узел дистилляции количество продукта), и рециркулируют ее в узел абсорбции ниже первой улавливающей тарелки.
Выводимый из узла дистилляции газообразный вторичный газ преимущественно содержит следующие компоненты, % мас.:
Указанный вторичный газ подвергают сжиганию вместе с другими производственными остатками.
Полученную сырую акриловую кислоту можно подвергать дальнейшей переработке на других технологических стадиях в чистую акриловую кислоту известными методами. Для этой цели пригоден одноступенчатый или, в случае сырой акриловой кислоты со значительным содержанием легкокипящих побочных компонентов, двухступенчатый дистилляционный метод, предусматривающий предварительную обработку альдегида содержащим по меньшей мере одну первичную аминогруппу соединением (например, гидразином или аминогуанидингидрокарбонатом) согласно европейским заявкам на патент ЕР-А 270999 и ЕР-А 648732, а также метод кристаллизации согласно европейским заявкам на патент ЕР-А 616998, ЕР-А 792867 и ЕР-А 1189861 и международной заявке WO 98/01404.
При получении чистой акриловой кислоты методом кристаллизации, например, слоевой кристаллизации, наряду с чистой акриловой кислотой образуется также содержащий ее поток маточного щелока/кислоты, который, как указано выше, можно рециркулировать в узел абсорбции для получения сырой акриловой кислоты.
Пример 1
Данный пример выполняли аналогично сравнительному примеру 1. Однако (общий) остаточный газ подавали не в присоединенный к первому нагревателю испаритель, а совместно со сжатым воздухом (195 нл/ч) направляли его под соответствующим давлением в нагреватель, присоединенный к третьему реактору дегидрирования.
Вследствие этого содержание диоксида углерода в выходящей из третьего реактора реакционной газовой смеси (3,25% об.) снижалось до 1,6% об.
Это означает, что предлагаемый в изобретении способ позволяет менее чем наполовину сократить выгорание углеводородов с тремя атомами углерода.
Кроме того, предлагаемый в изобретении способ позволяет осуществлять технологический процесс в течение 7500 часов в основном без заметного ухудшения его показателей.
Пример 2
Ниже рассматривается осуществление технологического процесса также в стационарном режиме.
В шахтную печь в виде полочного адиабатического реактора загружены два последовательно расположенных в направлении реакционного потока слоя катализатора (стационарные слои катализатора дегидрирования из сравнительного примера 1), образующие первую секцию реакционной зоны А. Перед каждым стационарным слоем находится статический газовый смеситель.
В первый в направлении потока слой катализатора подают следующую исходную реакционную газовую смесь:
а) рециркулируемая газообразная смесь продуктов А (132872 нм3/ч) с температурой 617°С и давлением 2,75 бар, содержащая
26,4% об. пропана,
6,6% об. пропилена,
0,1% об. Н2,
8,8% об. Н2O и
0% об. O2,
и
b) сырой пропан из сравнительного примера (8758 нм3/ч).
Температуру сырого пропана регулируют таким образом, чтобы исходная реакционная газовая смесь имела температуру 560°С.
Исходная реакционная газовая смесь (141630 нм3/ч) состоит из
31,0% об. пропана,
6,2% об. пропилена,
0,1% об. Н2,
8,2% об. Н2O и
0% об. O2.
Давление исходной реакционной газовой смеси на входе в первый слой катализатора составляет 2,75 бар.
Высоту первого стационарного слоя катализатора, предназначенного для пропускания исходной реакционной газовой смеси, устанавливают таким образом, чтобы на выходе из него реакционная газовая смесь обладала следующим составом:
26,4% об. пропана,
9,5% об. пропилена,
3,6% об. Н2,
7,9% об. Н2O и
0% об. O2.
Расход реакционной газовой смеси на выходе из первого слоя катализатора составляет 146533 нм3/ч. Ее температура составляет 500°С, давление 2,65 бар. Конверсия пропана, пропускаемого через первый слой катализатора, составляет 11,8% мол.
К выходящей из первого стационарного слоя катализатора реакционной газовой смеси указанного выше состава добавляют 6250 нм3/ч воздуха. Воздух предварительно нагревают до 500°С, а его давление повышают таким образом, чтобы реакционная газовая смесь после смешивания с воздухом обладала давлением 2,65 бар.
При пропускании реакционной газовой смеси через второй стационарный слой катализатора сначала происходит сгорание половины содержащегося в ней молекулярного водорода в присутствии добавляемого в виде воздуха молекулярного кислорода, которое сопровождается образованием воды. При этом реакционная газовая смесь нагревается до 550°С. При дальнейшем пропускании реакционной газовой смеси через второй стационарный слой катализатора происходит гетерогенно катализируемое дегидрирование пропана. При этом высоту второго стационарного слоя устанавливают таким образом, чтобы выходящая из него реакционная газовая смесь (154746 нм3/ч) имела температуру 513°С, давление 2,55 бар и следующий состав:
22,9% об. пропана,
11,9% об. пропилена,
3,9% об. Н2,
9,2% об. Н2O и
0% об. O2.
К этой газообразной смеси продуктов А* сначала добавляют 761,6 нм3/ч молекулярного водорода. Затем добавляют 112792 нм3/ч общего остаточного газа с температурой 567°С из зоны разделения В. Общий остаточный газ обладает следующим составом:
31,1% об. пропана,
0% об. пропилена,
0% об. Н2,
1,9% об. Н2O и
3,0% об. O2.
Подачу общего остаточного газа осуществляют в соответствии с принципом струйного насоса, общий остаточный газ в котором выполняет функцию приводной струи, причем перемещение дросселируемой через рабочее сопло приводной струи через зону смешивания и диффузор во второй секции реакционной зоны А, а также эффект всасывания всасывающего патрубка направлены в сторону смеси молекулярного водорода с газообразной смесью продуктов А*.
Образующаяся при этом реакционная газовая смесь А* (267538 нм3/ч) с температурой 536°С и давлением 2,85 бар поступает во второй реактор в виде шахтной печи, образующий вторую секцию реакционной зоны А, которая функционирует также в адиабатическом режиме и содержит стационарный слой катализатора, также являющийся катализатором дегидрирования из сравнительного примера 1. Выходящая из него реакционная газовая смесь А* обладает следующим составом:
26,3% об. пропана,
6,4% об. пропилена,
2,6% об. Н2,
6,1% об. Н2O и
1,3% об. O2.
При пропускании реакционной газовой смеси А* через третий стационарный слой катализатора сначала происходит в основном полное превращение содержащегося в ней молекулярного кислорода вследствие сгорания содержащегося в ней молекулярного водорода, которое сопровождается образованием воды, причем реакционная газовая смесь нагревается до 620°С.
Высоту третьего стационарного слоя катализатора устанавливают таким образом, чтобы до выхода реакционной газовой смеси из этого слоя происходило гетерогенно катализируемое дегидрирование еще присутствующего в смеси незначительного количества пропана.
В итоге из третьего стационарного слоя катализатора выходит 265745 нм3/ч газообразной смеси продуктов А с температурой 617°С и давлением 2,75 бар. Реакционная смесь обладает следующим составом:
26,4% об. пропана,
6,6% об. пропилена,
0,1% об. Н2,
8,8% об. Н2O и
0% об. O2.
Газообразную смесь продуктов А разделяют на две половины одинакового состава.
Одну из половин рециркулируют в первую секцию реакционной зоны А в качестве компонента исходной реакционной газовой смеси.
Другую половину направляют в косвенный теплообменник W для газов зоны разделения А. В этом теплообменнике остаточный газ, рециркулируемый в реакционную зону А из зоны разделения В, нагревается до 567°С.
В зоне разделения В аналогично сравнительному примеру 1 осуществляют абсорбционное выделение пропана и пропилена, содержащегося во второй половине газообразной смеси продуктов А, а в реакционной зоне В пропилен аналогично сравнительному примеру 1 подвергают двухступенчатому гетерогенно катализируемому частичному окислению в акриловую кислоту.
Из газообразной смеси продуктов В в зоне разделения В выделяют акриловую кислоту аналогично сравнительному примеру 1, остающийся общий остаточный газ сжимают (примерно до 4 бар), направляют, как указано выше, через теплообменник W в струйный насос, в котором общий остаточный газ используют в качестве приводной струи, и соединяют его с газообразной смесью продуктов А*, содержащей добавленный к ней молекулярный водород. В зоне разделения В выделяют 332 кмоль/ч акриловой кислоты. Остаточный газ, содержащийся в рассмотренных выше газовых смесях, состоит преимущественно из молекулярного азота и незначительных количеств оксидов углерода. Рассмотренный выше способ пригоден для применения в основном в течение длительного эксплуатационного периода.
Пример 3
Ниже рассматривается осуществление технологического процесса также в стационарном режиме.
В шахтную печь в виде полочного адиабатического реактора загружены два последовательно расположенных в направлении реакционного потока слоя катализатора (стационарные слои катализатора дегидрирования из сравнительного примера 1), образующие первую секцию реакционной зоны А. Перед каждым стационарным слоем находится статический газовый смеситель.
В первый в направлении потока слой катализатора вводят исходную реакционную газовую смесь следующего состава:
а) рециркулируемая газообразная смесь продуктов А с температурой 676°С и давлением 1,75 бар (106510 нм3/ч), содержащая
12,3% об. пропана,
8,2% об. пропилена,
2,2% об. Н2,
7,7% об. Н2O и
0% об. O2,
и
b) сырой пропан из сравнительного примера 1 (8758 нм3/ч).
Температуру сырого пропана регулируют таким образом, чтобы температура исходной реакционной газовой смеси составляла 652°С.
Исходная реакционная газовая смесь (115268 нм3/ч) обладает следующим составом
19,0% об. пропана,
7,6% об. пропилена,
2,0% об. H2,
7,1% об. Н2О и
0% об. O2.
Давление исходной реакционной газовой смеси на входе в первый слой катализатора составляет 1,75 бар.
Высоту первого стационарного слоя катализатора, через который пропускают исходную реакционную газовую смесь, устанавливают таким образом, чтобы реакционная газовая смесь на выходе из этого слоя обладала следующим составом:
15,1% об. пропана,
10,6% об. пропилена,
5,1% об. H2,
6,9% об. H2O и
0% об. O2.
Из первого слоя катализатора выходит 119308 нм3/ч реакционной газовой смеси. Ее температура составляет 590°С, давление 1,65 бар. Конверсия пропана, пропущенного через первый слой катализатора, составляет 17,5% мол.
К реакционной газовой смеси указанного выше состава, выходящей из первого стационарного слоя катализатора, добавляют 2598 нм3/ч воздуха. Воздух предварительно нагревают до 590°С, а его давление повышают таким образом, чтобы давление реакционной газовой смеси после смешивания с воздухом составляло 1,65 бар.
При пропускании реакционной газовой смеси через второй стационарный слой катализатора сначала происходит сгорание 18% содержащегося в ней молекулярного водорода в присутствии добавляемого в виде воздуха молекулярного кислорода, которое сопровождается образованием воды. При этом реакционная газовая смесь нагревается до 618°С. При дальнейшем пропускании реакционной газовой смеси через второй стационарный слой катализатора происходит гетерогенно катализируемое дегидрирование пропана. При этом высоту второго стационарного слоя катализатора устанавливают таким образом, чтобы выходящая из второго стационарного слоя катализатора реакционная газовая смесь (124387 нм3/ч) обладала температурой 546°С, давлением 1,55 бар и следующим составом:
12,0% об. пропана,
12,6% об. пропилена,
6,6% об. H2,
7,5% об. H2O и
0% об. O2.
К этой газообразной смеси продуктов А* добавляют 89715 нм3/ч общего остаточного газа с температурой 627°С из зоны разделения В. Общий остаточный газ обладает следующим составом:
14,6% об. пропана,
0 % об. пропилена,
0 % об. Н2,
1,9% об. Н2O и
3,0% об. O2.
Подачу общего остаточного газа осуществляют в соответствии с принципом струйного насоса, общий остаточный газ в котором выполняет функцию приводной струи, причем перемещение дросселируемой через рабочее сопло приводной струи через зону смешивания и диффузор во второй секции реакционной зоны А, а также эффект всасывания всасывающего патрубка направлены в сторону смеси молекулярного водорода с газообразной смесью продуктов А*.
Образующаяся при этом реакционная газовая смесь А* (214102 нм3/ч) с температурой 580°С и давлением 1,85 бар поступает во второй реактор в виде шахтной печи, образующий вторую секцию реакционной зоны А, которая функционирует также в адиабатическом режиме и содержит стационарный слой катализатора, также являющийся катализатором дегидрирования из сравнительного примера 1. Выходящая из этой зоны реакционная газовая смесь А* обладает следующим составом:
13,1% об. пропана,
7,3% об. пропилена,
3,8% об. H2,
5,2% об. H2O и
1,3% об. O2.
При пропускании реакционной газовой смеси А* через третий стационарный слой катализатора сначала происходит в основном полное превращение содержащегося в этой смеси молекулярного кислорода при сгорании также содержащегося в ней молекулярного водорода, сопровождаемое образованием воды, причем реакционная газовая смесь нагревается до 692°С.
Высоту третьего стационарного слоя катализатора устанавливают таким образом, чтобы до выхода реакционной газовой смеси из этого слоя происходило гетерогенно катализируемое дегидрирование еще присутствующего незначительного количества пропана.
В итоге из третьего стационарного слоя катализатора выходит 213021 нм3/ч газообразной смеси продуктов А с температурой 677°С и давлением 1,75 бар. Эта реакционная смесь обладает следующим составом:
12,3% об. пропана,
8,2% об. пропилена,
2,2% об. Н2,
7,7% об. Н2O и
0% об. O2.
Газообразную смесь продуктов А разделяют на две половины идентичного состава.
Одну половину рециркулируют в первую секцию реакционной зоны А в качестве составной части исходной реакционной газовой смеси.
Другую половину направляют в косвенный теплообменник W для газов зоны разделения А. В этом теплообменнике остаточный газ, рециркулируемый в реакционную зону А из зоны разделения В, нагревается до 627°С.
В зоне разделения В аналогично сравнительному примеру 1 осуществляют абсорбционное выделение пропана и пропилена, содержащегося во второй половине газообразной смеси продуктов А, а в реакционной зоне В пропилен аналогично сравнительному примеру 1 подвергают двухступенчатому гетерогенно катализируемому частичному окислению в акриловую кислоту.
Из полученной газообразной смеси продуктов В в зоне разделения В аналогично сравнительному примеру 1 выделяют акриловую кислоту, остающийся общий остаточный газ сжимают (примерно до 4 бар), направляют, как указано выше, через теплообменник W в струйный насос, в котором общий остаточный газ используют в качестве приводной струи, и соединяют его с газообразной смесью продуктов А*, в которую добавлен молекулярный водород. В зоне разделения В выделяют 332 кмоль/ч акриловой кислоты. Остаточный газ, содержащийся в рассмотренных выше газовых смесях, состоит преимущественно из молекулярного азота и незначительных количеств оксидов углерода. Рассмотренный выше способ пригоден для применения в основном в течение длительного эксплуатационного периода.
Предварительные описания патентов США No. 60/584,469 (дата подачи 01.07.2004) и 60/662,804 (дата подачи 18.03.2005) включены в настоящую заявку в качестве ссылок. С учетом указанных документов возможны многочисленные изменения и отклонения от настоящего изобретения. В связи с этим возможно выполнение изобретения согласно прилагаемой формуле, отличающееся от представленного выше специфического описания.
Реферат
Изобретение относится к усовершенствованному способу получения акролеина, акриловой кислоты или их смеси из пропана. В соответствии со способом А) в первую реакционную зону А вводят по меньшей мере два содержащих пропан газообразных питающих потока, по меньшей мере один из которых содержит свежий пропан, и введенный в эту реакционную зону пропан подвергают гетерогенно катализируемому дегидрированию со стационарным слоем катализатора, получая содержащую пропан и пропилен газообразную смесь продуктов А, В, которую выводят из реакционной зоны А, в первой зоне разделения А выделяют из нее по меньшей мере часть содержащихся в ней компонентов, отличающихся от пропана и пропилена, а остающуюся при этом газообразную смесь продуктов А′, содержащую пропан и пропилен, С) используют во второй реакционной зоне В для питания по меньшей мере одного реактора окисления, и пропилен, содержащийся в газообразной смеси продуктов А′, по меньшей мере в одном реакторе окисления подвергают гетерогенно катализируемому двухступенчатому газофазному частичному окислению молекулярным кислородом в акриловую кислоту или смесь акролеина и акриловой кислоты, в качестве целевого продукта, а также в содержащую избыток молекулярного кислорода газообразную смесь продуктов В, D), которую выводят из реакционной зоны В, во второй зоне разделения В выделяют содержащийся в ней целевой продукт методом абсорбции или фракционирующей конденсации, и по меньшей мере часть остающегося после этого остаточного газа, содержащего непревращенный пропан, молекулярный кислород, а также при необходимости непревращенный пропилен, рециркулируют в реакци
Формула
A) в первую реакционную зону А вводят по меньшей мере два содержащих пропан газообразных питающих потока, по меньшей мере один из которых содержит свежий пропан, и введенный в эту реакционную зону пропан подвергают гетерогенно катализируемому дегидрированию со стационарным слоем катализатора, получая содержащую пропан и пропилен газообразную смесь продуктов А,
B) которую выводят из реакционной зоны А, в первой зоне разделения А выделяют из нее по меньшей мере часть содержащихся в ней компонентов, отличающихся от пропана и пропилена, а остающуюся при этом газообразную смесь продуктов А′, содержащую пропан и пропилен,
C) используют во второй реакционной зоне В для питания по меньшей мере одного реактора окисления, и пропилен, содержащийся в газообразной смеси продуктов А′, по меньшей мере в одном реакторе окисления подвергают гетерогенно катализируемому двухступенчатому газофазному частичному окислению молекулярным кислородом в акриловую кислоту или смесь акролеина и акриловой кислоты, в качестве целевого продукта, а также в содержащую избыток молекулярного кислорода газообразную смесь продуктов В,
D) которую выводят из реакционной зоны В, во второй зоне разделения В выделяют содержащийся в ней целевой продукт методом абсорбции или фракционирующей конденсации, и по меньшей мере часть остающегося после этого остаточного газа, содержащего непревращенный пропан, молекулярный кислород, а также, при необходимости, непревращенный пропилен, рециркулируют в реакционную зону А в качестве по меньшей мере одного из двух содержащих пропан питающих потоков, отличающийся тем, что указанную рециркуляцию в реакционную зону А осуществляют вдоль пути гетерогенно катализируемого дегидрирования пропана в этой реакционной зоне таким образом, чтобы в месте ввода рециркулируемого газа в реакционной зоне А уже было подвергнуто дегидрированию по меньшей мере 5 мол.% пропана, введенного в эту реакционную зону с другими питающими потоками, причем молярное отношение содержащегося в реакционной газовой смеси пропилена к содержащемуся в ней молекулярному водороду в пределах реакционной зоны А не превышает 10.
i) в первую секцию реакционной зоны А вводят по меньшей мере один содержащий газообразный пропан питающий поток, который содержит свежий пропан, и введенный в эту первую секцию пропан подвергают в ней гетерогенно катализируемому дегидрированию, получая газообразную смесь продуктов А*, содержащую пропан, пропилен и молекулярный водород, причем дегидрированию подвергают по меньшей мере 5 мол.% введенного в первую секцию пропана, и получаемая смесь продуктов А* в пересчете на содержащееся в ней молярное количество пропана содержит большее молярное количество молекулярного водорода, чем исходная реакционная газовая смесь, вводимая в первую секцию реакционной зоны А;
ii) затем к газообразной смеси продуктов А* добавляют остаточный газ из зоны разделения В, содержащий молекулярный кислород, непревращенный пропан и при необходимости непревращенный пропилен, и образующуюся при этом реакционную газовую смесь А* направляют во вторую секцию реакционной зоны А, и
iii) во второй секции реакционной зоны А при образовании газообразной смеси продуктов А, содержащей пропан и пропилен, содержащийся в реакционной газовой смеси А* молекулярный кислород участвует в сопровождаемом образованием воды гетерогенно катализируемом сгорании содержащегося в этой смеси молекулярного водорода, а содержащийся в реакционной газовой смеси А* пропан, при необходимости, гетерогенно каталитически дегидрируют до пропилена.
тепловой брутто-эффект, отнесенный к однократному пропусканию исходной реакционной газовой смеси через первую секцию реакционной зоны А, может изменяться от эндотермического до автотермического, и тепловой брутто-эффект, отнесенный к однократному пропусканию реакционной газовой смеси А* через вторую секцию реакционной зоны А является экзотермическим.
от 10 до 40 об.% пропана,
от 0 до 1 об.% пропилена,
от >0 до 5 об.% молекулярного кислорода,
от 1 до 10 об.% водяного пара и
от 0 до 0,5 об.% молекулярного водорода.
Комментарии