Способ получения моногидроперфторалканов, бис(перфторалкил)фосфинатов и перфторалкилфосфонатов - RU2319705C2
Код документа: RU2319705C2
Описание
Представленное изобретение касается способа получения моногидроперфторалканов, бис(перфторалкил)фосфинатов и перфторалкилфосфонатов, который включает, по крайней мере, обработку, по крайней мере, одного перфторалкилфосфорана, по крайней мере, одним основанием в пригодной реакционной среде.
Моногидроперфторалканы известны некоторое время и нашли широкое применение в различных областях, inter alia, как озонбезопасные хладагенты (WO 01/40400, WO 01/23494, WO 01/23491, WO 99/36485, WO 98/08913), как моющие средства (WO 01/32323), как протравители в микроэлектронике (US 2001/0005637, US 6228775), в огнетушителях (WO 01/05468, Combust. Flame, 121, №3 (2000) с.с.471-487, CN 1218702), как вспенивающие агенты для пен (US 6225365, WO 01/18098) и для получения полимерных материалов и как сильные обезболивающие средства (Anesth. Analg (N.Y.), 79, №2 (1994), с.245-251, Т.Hudlicky et al., J. of Fluorine Chem., 59, №1 (1992), с.9-14).
Некоторые из этих моногидроперфторалканов, такие как, например, пентафторэтан, легко получить в промышленных масштабах с тонными выходами, получение обычно проводят путем каталитического гидрофторирования хлорированных углеводородов (WO 01/77048, ЕР 1052235).
Недостатком упомянутых способов является, во-первых, риск связанный с применением фтороводорода при относительно высоких температурах. Кроме того, способы требуют особенных катализаторов, которые получают перед сравнительно сложными процессами. Следующим недостатком этих способов является то, что при получении хлорированных углеводородов используется хлор, что является экологически опасным, и дополнительно увеличивается стоимость производства. В конце концов, известные способы получения пентафторэтана не пригодны для получения длинноцепочечных моногидроперфторалканов, таких как, например, 1-гидрононафторбутаны.
Кроме того, известны некоторые другие способы, в которых получают пентафторэтан, используя специальные фторирующие агенты, такие как, например, BrF3 (R.A.Devis, J. Org. Chem. 32 (1967), page 3478), XeF2 (JP 2000/119201), SF4 (G.Siegemund, Liebigs Ann. Chem., 1979, page 1280, E.R.Bissell, J. of Organic Chem., 29, (1964), page 1591), SbF5 (Г.Г.Беленький и др., Изв. Акад. Наук СССР, Сер. Хим., 1972, страницы 983, Chem. Abstr. 77 (1972) 75296, А.Ф.Ермолов и др., Ж. Орг. Хим., 17 (1981), страница 2239, J. Org. Chem. USSR (Engl. Translation), 17 (1981), page 1999, US 2426172), MoF6 (Л.Д.Шустов и др., Ж. Общей Хим., 53 (1983), страница 103, J. Gen. Chem. USSR (Engl. translation), 53 (1983), page 85) и CoF3 (US 6162955).
Однако упомянутые выше способы не имеют промышленной значимости, поскольку и соответствующие исходные соединения, и фторирующие агенты является сами по себе очень дорогими.
В противоположность, известно только несколько способов получения длинноцепочечных моногидроперфторалканов.
В соответствии с первым способом моногидроперфторалканы получают путем декарбоксилирования солей перфторированных карбоновых кислот (J.D.LaZerte et al., J. Am. Chem. Soc, 75 (1953), page 4525; R.N.Haszeldine, J. Chem. Soc. 1953, page 1548) или соответствующих сложных эфиров (Е.Bergman, J.Org. Chem., 23, (1958) page 476) путем обработки сильным основанием, таким как, например, этоксид натрия.
В соответствии с другим способом моногидроперфторалканы получают путем обработки перфорированных кетонов, имеющих трифторметильную группу на карбонильном атоме углерода, водной щелочью (Л.В.Салоутина и др., Изв. Акад. Наук СССР, Сер. Хим., 1984, №5, страницы 1114-1116, Chem. Abstr. 101 (1984) 210504х). Эти способы также имеют недостатки, состоящие в необходимости использования дорогих исходных материалов и высоких температур.
Кроме того, 1-гидро-н-нонафторбутан получают путем восстановления перфторбутилйодида, используя разные восстанавливающие агенты, такие как, например, цинковая пыль в метаноле (Т.Hudlicky et al., J. of Fluorine Chem., 59, №1 (1992), с.с.9-14), метоксид натрия (J.L.Howell et al., J. of Fluorine Chem., 72, №1 (1995), с.с.61-68), с помощью водорода в газовой фазе при высоких температурах (ЕР 632001) и с помощью комплекса таллия [ТаСр2(С2 Н4)Н] (Р.Н.Russel et al., Polyhedron 17, №7 (1998), с.с.1037-1043).
Однако эти способы так же имеют недостаток, который состоит в использовании как исходного соединения перфторбутилйодида, который можно получить только с использованием сравнительно дорогих способов.
Целью представленного изобретения было обеспечение способа, пригодного для простого и недорогого получения моногидроперфторалканов с хорошими выходами. Моногидроперфторалканы предпочтительно должны получаться с высокой чистотой. Следующей целью являлось получение бис(перфторалкил)фосфинатов и перфторалкилфосфонатов.
Эта цель достигается с помощью способа в соответствии с изобретением получения моногидроперфторалканов общей формулы Cn HF2n+1, в которой 1≤n≤8, предпочтительно 1≤n≤4, бис(перфторалкил)фосфинатов и перфторалкилфосфонатов, который включает, по крайней мере, обработку, по крайней мере, одного перфторалкилфосфорана, по крайней мере, одним основанием в пригодной реакционной среде.
В соответствии с изобретением получение моногидроперфторалканов с помощью способа в соответствии с изобретением может в каждом случае проводится с применением перфторалкилфосфорана или смеси двух или более перфторалкилфосфоранов. Предпочтительно только один перфторалкилфосфоран в каждом случае взаимодействует в способе в соответствии с изобретением.
Перфторалкилфосфораны, используемые в способе в соответствии с изобретением, можно получить с помощью обычных методов, известных среднему специалисту в данной области техники.
Перфторалкилфосфораны предпочтительно получают путем электрохимического фторирования пригодных исходных соединений, как описано В.Я.Семений и др., Жур. Об. Хим., 55, №12 (1985), страницы 2716-2720; N.Ignatiev, J. of Fluorine Chem., 103 (2000), c.c.57-61 и WO 00/21969. Соответствующие описания, таким образом, включены сюда как ссылки и рассматриваются как часть описания.
В предпочтительном воплощении способа в соответствии с изобретением применяется, по крайней мере, один перфторалкилфосфоран общей формулы I
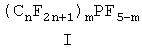
в которой 1≤n≤8, предпочтительно 1≤n≤4, и m в каждом случае означает 1, 2 или 3.
Особенно предпочтительно перфторалкилфосфорановые соединения выбирают из группы, которая включает дифтортрис(пентафторэтил)фосфоран, дифтортрис(н-нонафторбутил)фосфоран, дифтортрис(н-гептафторпропил)фосфоран и трифторбис(н-нонафторбутил)фосфоран.
Обработку перфторалкилфосфоранового соединения(й) с помощью способа в соответствии с изобретением предпочтительно в каждом случае, проводят, используя только одно основание. Однако, несомненно, также можно использовать смесь двух или большего количества оснований в способе в соответствии с изобретением. Соответствующие основания также можно использовать в форме соответствующих сольватов, предпочтительно в форме соответствующих гидратов, или в форме обычных аддуктов, известных среднему специалисту в данной области техники.
В следующем предпочтительном воплощении способа в соответствии с изобретением при получении моногидроперфторалканов обычно используется основание (а), предпочтительно неорганическое основание (б) или органическое основание (в). Неорганическое основание (б) предпочтительно выбирают из группы, включающей гидроксиды щелочных металлов и гидроксиды щелочноземельных металлов.
Если как основание (б) в способе в соответствии с изобретением используется гидроксид щелочного металла, его можно предпочтительно выбрать из группы, включающей гидроксид лития, моногидрат гидроксида лития, гидроксид натрия и гидроксид калия.
Если как основание (б) в способе в соответствии с изобретением используется гидроксид щелочноземельного металла, его можно предпочтительно выбрать из группы, включающей гидроксид бария, октагидрат гидроксида бария и гидроксид кальция.
Способ получения моногидроперфторалканов в соответствии с изобретением также предпочтительно можно провести, используя органическое основание (в) или органо-металлические соединения. Основания (в) предпочтительно можно выбрать из группы, включающей гидроксиды алкиламмония, гидроксиды ариламмония, гидроксиды алкилариламмония, гидроксиды алкилфосфония, гидроксиды арилфосфония, гидроксиды алкиларилфосфония, алкиламины, ариламины, алкилариламины, алкилфосфины, арилфосфины и алкиларилфосфины.
Предпочтительные органо-металлические соединения можно выбрать из группы, включающей алкоксиды металлов, предпочтительно алкоксиды щелочных металлов, арилоксиды металлов, алкилтиооксиды металлов, арилтиооксиды металлов, алкилметаллические соединения, арилметаллические соединения и реагенты Гриньяра.
Если один из упомянутых выше классов оснований содержит алкильный радикал, он может предпочтительно содержать от 1 до 4 атомов углерода. Если соответствующее основание содержит два или более алкильных радикала, они могут в каждом случае быть одинаковыми или разными, одинаковые алкильные радикалы являются предпочтительными.
Если один из упомянутых выше классов оснований содержит арильный радикал, он может предпочтительно быть незамещенным или, по крайней мере, монозамещенным фенильным радикалом.
Если алкоксид щелочного металла используется в способе в соответствии с изобретением, он может предпочтительно быть производным натрия и может предпочтительно иметь от 1 до 3 атомов углерода.
Пригодной реакционной средой для использования в способе в соответствии с изобретением является обычная реакционная среда, которая известна среднему специалисту в данной области техники, при условии, что она не подвергается необратимому химическому взаимодействию с соответствующим основанием или с соответствующим получаемым моногидроперфторалканом.
В следующем предпочтительном воплощении способа в соответствии с изобретением реакционной средой является вода, при желании смешанная с одним или большим количеством органических растворителей, где также в соответствии с изобретением включены двухфазные системы, такие как, например, смеси воды и углеводородов.
Способ получения моногидроперфторалканов в соответствии с изобретением также предпочтительно можно провести, используя один или большее количество органических растворителей, где, в случае, когда используются, по крайней мере, два растворителя, они при желании могут быть в форме двухфазной системы.
Пригодные органические растворители, которые используются в способе в соответствии с изобретением, в каждом случае индивидуально или в какой-либо желаемой комбинации друг с другом при желании также смешивают с водой, можно предпочтительно выбрать из группы, включающей спирты, простые эфиры, ациламиды, сульфоксиды, сульфоны, нитрилы и углеводороды.
Предпочтительными спиртами являются соединения, которые имеют от 1 до 4 атомов углерода в алкильной части. Соответствующие спирты можно предпочтительно выбрать из группы, включающей метанол, этанол, изопропанол и смеси, по крайней мере, двух этих упомянутых выше спиртов.
Количество моногидроперфторалкана, образующегося из соответствующего используемого перфторалкилфосфорана(ов) и тип дополнительных продуктов реакции можно контролировать в целевом методе в соответствии с способом в соответствии с изобретением, например, через температуру и/или давление во время реакции или через молярное соотношение перфторалкилфосфорана и алкана.
Например, можно путем выбора параметров специфично отщепить одну, две или три перфторалкильные группы в соответствующем используемом дифтортрисперфторалкилфосфоране.
При удалении одной перфторалкильной группы из соответствующего дифтортрисперфторалкилфосфорана, также inter alia, образуется соответствующий бис(перфторалкил)фосфинат, в дополнение к желаемому моногидроперфторалкану.
При удалении двух перфторалкильных групп из соответствующего дифтортрисперфторалкилфосфорана, также inter alia, образуется соответствующий перфторалкилфосфонат, в дополнение к желаемому моногидроперфторалкану.
Если удаляются три перфторалкильные группы в соответствующем дифтортрисперфторалкилфосфоране, также inter alia, образуется соответствующий фосфат, в дополнение к желаемому моногидроперфторалкану.
Соответствующий выбор оптимальных параметров для желаемой комбинации соответствующего моногидроперфторалкана, его количество и соответствующие дополнительные продукты реакции может быть определено средним специалистом в данной области техники с помощью простых предварительных экспериментов.
Если, например, планируется удаление одной перфторалкильной группы в соответствующем используемом дифтортрисперфторалкилфосфоране, способ в соответствии с изобретением можно предпочтительно проводить при температурах от -10 до 100°С и моль-эквивалентном соотношении дифтортрисперфторалкилфосфорана к основанию 1:3.
Если, например, планируется удаление двух перфторалкильных групп в соответствующем используемом дифтортрисперфторалкилфосфоране, способ в соответствии с изобретением можно предпочтительно проводить при температурах от 50 до 150°С и моль-эквивалентном соотношении дифтортрисперфторалкилфосфорана и основания 1:4.
Если, например, планируется удаление трех перфторалкильных групп в соответствующем используемом дифтортрисперфторалкилфосфоране, способ в соответствии с изобретением можно предпочтительно проводить при температурах от 100 до 250°С и моль-эквивалентном соотношении дифтортрисперфторалкилфосфорана и основания 1:5.
Моногидроперфторалканы, полученные с помощью способа в соответствии с изобретением, можно, если необходимо, выделить и, если необходимо, очистить с помощью обычных методов, известных среднему специалисту в данной области техники.
Если они являются легколетучими соединениями, они могут быть выделены из реакционной смеси путем, например, конденсирования в одной или большем количестве холодных ловушек, которые предпочтительно охлаждаются жидким азотом или сухим льдом.
Любое выделение и очистку дополнительных продуктов реакции также проводят с помощью обычных методов, известных среднему специалисту в данной области техники, таких как, например, с помощью фракционной кристаллизации или экстракции пригодными растворителями.
Если перфторалкилфосфоран реагирует с неорганическим основанием (б), образованные таким образом бис(перфторалкил)фосфинаты и перфторалкилфосфонаты можно превратить, непосредственно или после выделения, используя кислоту, предпочтительно используя серную кислоту, в соответствующие бис(перфторалкил)фосфиновые кислоты и перфторалкилфосфоновые кислоты.
Бис(перфторалкил)фосфиновые кислоты и перфторалкилфосфоновые кислоты, полученные этим путем, можно превратить в соли путем нейтрализации, предпочтительно используя органические основания (в).
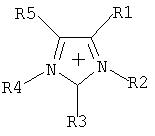
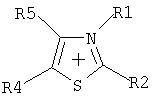
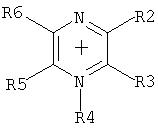
Путем выбора пригодных оснований предпочтительно получают частично алкилированные и пералкилированные аммониевые, фосфониевые, сульфониевые, пиридиниевые, пиридазиниевые, пиримидиниевые, пиразиниевые, имидазолиевые, пирразолиевые, тиазолиевые, оксазолиевые и триазолиевые соли.
Особое предпочтение отдается получению солей, имеющих катионы, которые выбирают из группы, включающей
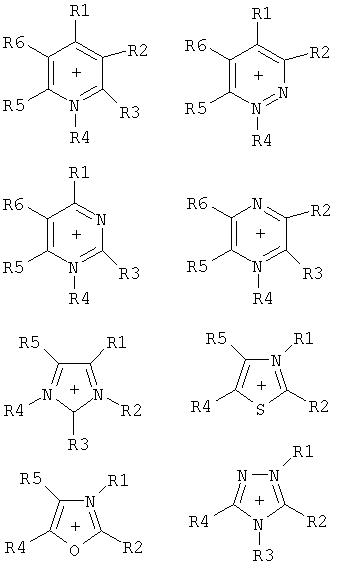
где R1-R5 являются одинаковыми или разными, необязательно связанными друг с другом с помощью простой или двойной связи, и каждый, индивидуально или вместе, имеет значение, приведенное далее:
-Н,
- галоген, где галогены не присоединены непосредственно к атому N,
- алкильный радикал
(C1-C8), который может быть частично или полностью замещен предпочтительно следующими группами: F, Cl, N(CnF(2n+1-x)Нx)2, O(CnF(2n+1-x)Hx), SO2(CnF(2n+1-x)Hx), CnF(2n+1-x)Hx, где 1 Эти соли также можно получить, если соль, полученную после реакции перфторалкилфосфорана с неорганическим основанием (б), подвергнуть обмену соли, непосредственно или после
выделения. Обмен солей можно провести с арил-, алкил- или алкиларил-аммониевыми или -фосфониевыми солями. Предпочтение отдается использованию гексафторфосфатов, тетрафторборатов,
гексафторарсенатов, сульфатов, фторидов, хлоридов или бромидов. Соли, полученные таким образом, можно обработать с помощью обычной методики известной среднему специалисту в данной
области техники. Способ получения моногидроперфторалканов в соответствии с изобретением дает возможность просто, недорого и надежно получить эти соединения с очень хорошими выходами. В
частности, перфторалкилфосфораны, используемые как исходные соединения, можно получить в неожиданно больших количествах. Кроме того, преимуществом является то, что побочные продукты,
полученные в способе в соответствии с изобретением, такие как, например, бис(перфторалкил)фосфинаты и перфторалкилфосфонаты, сам по себе являются сырьевыми материалами, которые пригодны, inter alia,
для получения соответствующих бис(перфторалкил)фосфиновых кислот и перфторалкилфосфоновых кислот и, таким образом, могут быть экономически полезны. Нейтрализация с использованием пригодных оснований
дает возможность получить из них, например, бис(перфторалкил)фосфинаты и перфторалкилфосфонаты, которые пригодны для использования как ионных жидкостей, поверхностно-активных веществ или катализаторов
фазового переноса. Также преимуществом является то, что влияние на окружающую среду в реакции способа в соответствии с изобретением является минимальным, что также имеет позитивное
влияние на стоимость производства получаемых моногидроперфторалканов с помощью способа в соответствии с изобретением. Кроме того, соответствующие моногидроперфторалканы получают с
высокой чистотой немедленно после проведения реакции по их получению, то есть без сложных стадий очистки. Изобретение объясняется ниже со ссылками на примеры. Эти примеры служат только
для объяснения изобретения и не ограничивают общий изобретательский замысел. ПРИМЕРЫ Пример 1 10,40 г (185,4 ммоль) гидроксида калия растворяют в 330
см3 воды в колбе и полученный раствор охлаждают при температуре бани -5°С. Впоследствии через капельную воронку на протяжении 15 минут при перемешивании прибавляют 25,53 г (59,9
ммоль) дифтортрис(пентафторэтил)фосфорана. Реакционную смесь впоследствии доводят до комнатной температуры. Газообразный пентафторэтан, образующийся вследствие щелочного гидролиза
дифтортрис(пентафторэтил)фосфорана, собирают в две последующие ловушки, каждую из которых охлаждают жидким азотом. В охлажденных ловушках получают 6,67 г твердого пентафторэтана, имеющего температуру
кипения -48°С. Это значение соответствует значению, указанному в литературе (L.Conte et al. in J. Fluor. Chem., 38 (1988), c.319-326. Выход пентафторэтана составляет 92,8%,
исходя из пентафторэтильной группы, удаленной в дифтортрис(пентафторэтил)фосфоране при этих условиях. Реакционная смесь в колбе также содержит раствор бис(пентафторэтил)фосфината калия
((С2F5)2Р(O)ОК) и фторида калия. Для того чтобы выделить бис(пентафторэтил)фосфинат калия, сначала нейтрализуют избыток гидроксида калия, используя несколько капель
водного раствора фтороводорода, и воду удаляют при пониженном давлении. Полученный твердый остаток сушат при пониженном давлении 120 Па и температуре бани 100°С на протяжении двух часов. Бис(пентафторэтил)фосфинат калия экстрагируют из высушенного остатка, используя 150 см3 метанола. Метанол впоследствии отгоняют при пониженном давлении 120 Па и твердый остаток
бис(пентафторэтил)фосфината калия сушат. Выход составляет 19,0 г, что соответствует 93,2%, исходя из использованного дифтортрис(пентафторэтил)фосфорана. Пентафторэтан характеризуют с
помощью1Н- и19F-ЯМР спектроскопии и бис(пентафторэтил)фосфинат калия с помощью19F- и31Р-ЯМР спектроскопии. Пентафторэтан 1Н- и19F-ЯМР спектры снимают на спектрометре Bruker WP 80 SY при частотах 80,1 МГц для1Н и 75,4 МГц для19F и температуре -70°С. С этой
целью используют ФЕП (фторэтиленовый полимер) пробирку, внутри которой находится тонкостенная 5 мм ЯМР пробирка с ацетон-D6 пленкой как внешний затвор и ТМС или CCl3F,
растворенный в ацетон-D6 пленке, как внешний стандарт. 1H-ЯМР спектр (ацетон-D6 пленка, относительно ТМС в пленке, δ, м.ч.): 5,80 тк;2
JH, F=52,3 Гц;3JH, F=2,1 Гц. 19F-ЯМР спектр (ацетон-D6 пленка, относительно CCl3F в пленке, δ, м.ч.): -86,54 с (CF3); -138,55 д (CHF2);2JH, F=52,5 Гц. Полученные данные соответствуют значениям, описанным в литературе. M.D.Bartberger
et al. in Tetrahedron, 53, №29 (1997), с.9857-9880, и N.Ignatiev et al. in Acta Chem. Scand. 53, №12 (1999), с.1110-1116. Бис(пентафторэтил)фосфинат калия ((C2F5
)2P(O)OK) 19F- и31Р-ЯМР спектры снимают на спектрометре Broker Avance 300 при частотах 282,4 МГц для19F и 121,5 МГц для31Р. 19F-ЯМР спектр (растворитель ацетон-D6, внутренний стандарт CCl3F, δ, м.ч.): -80,38 м (CF3); -125,12 дм (CF2);2
JH,F=67,3 Гц. 31P-ЯМР спектр (растворитель ацетон-D6, относительно 85 вес.% Н3PO4 в D2O, δ, м.ч.): 0,72 квин;2JP, F=67,2 Гц. Пример 2 5,99 г (142,8 ммоль) моногидрата гидроксида лития растворяют в 150 см3 воды в колбе и полученный раствор
охлаждают при температуре бани -10°С. Впоследствии через капельную воронку на протяжении 15 минут при перемешивании прибавляют 19,30 г (45,3 ммоль) дифтортрис(пентафторэтил)фосфорана.
Реакционную смесь впоследствии доводят до комнатной температуры. Газообразный пентафторэтан, образующийся вследствие гидролиза дифтортрис(пентафторэтил)фосфорана, собирают в две последующие ловушки,
каждую из которых охлаждают жидким азотом. В охлажденных ловушках получают 4,95 г пентафторэтана как твердое вещество. Выход пентафторэтана составляет 91,2%, исходя из пентафторэтильной группы,
удаленной в дифтортрис(пентафторэтил)фосфоране при этих условиях. Кроме того, реакционная смесь в колбе содержит раствор бис(пентафторэтил)фосфината лития ((C2F5
)2P(O)OLi) и фторид лития. Для того чтобы выделить бис(пентафторэтил)фосфинат лития, сначала нейтрализуют избыток гидроксида лития, используя несколько капель водного раствора фтороводорода,
осадок фторида лития отфильтровывают и воду удаляют при пониженном давлении. Полученное белое твердое вещество, бис(пентафторэтил)фосфинат лития, сушат при пониженном давлении 120 Па и температуре
бани 100°С на протяжении двух часов. Получают 13,1 г бис(пентафторэтил)фосфината лития, содержащего приблизительно 2 вес.% фторида лития, что соответствует выходу 93,7%, исходя из
использованного дифтортрис(пентафторэтил)фосфорана. Пентафторэтан характеризуют с помощью1Н- и19F-ЯМР спектроскопии и бис(пентафторэтил)фосфинат лития с помощью19F- и31Р-ЯМР спектроскопии. Химические сдвиги, определенные для пентафторэтана, соответствуют значениям указанным в Примере 1.
Бис(пентафторэтил)фосфинат лития 19F- и31Р-ЯМР спектры снимают на спектрометре Bruker Avance 300 при частотах 282,4 МГц для19F и 121,5 МГц для31Р. 19F-ЯМР спектр (растворитель ацетон-D6, внутренний стандарт CCl3F, δ, м.ч.): -80,32 м (CF3); -125,08 дм
(CF2);2JP, F=72,6 Гц. 31Р-ЯМР спектр (растворитель ацетон-D6, относительно 85 вес.% Н3PO4 - 15 вес.% D2O в ацетоне-D6, δ, м.ч.): 0,27 квин;2JP, F=72,7 Гц. Пример 3 4,1 г (73,1 ммоль) гидроксида калия растворяют в 150 см3 воды в колбе и полученный раствор охлаждают при температуре бани 0°С. Впоследствии через капельную воронку на протяжении 3 минут при перемешивании прибавляют 16,87 г (23,2 ммоль)
дифтортрис(н-нонафторбутил)фосфорана. Реакционную смесь впоследствии доводят до комнатной температуры, перемешивают при этой температуре на протяжении восьми часов и впоследствии нагревают с обратным
холодильником на протяжении еще восьми часов. Газообразный 1H-нонафтор-н-бутан, образующийся вследствие гидролиза дифтортрис(н-нонафторбутил)фосфорана, собирают в последующую ловушку, охлаждаемую
сухим льдом. В охлажденной ловушке получают 3,63 г жидкого 1H-нонафтор-н-бутана, имеющего температуру кипения 14°С. Выход 1H-н-нонафторбутана составляет 71,2%, исходя из
н-нонафторбутильной группы, удаленной в дифтортрис(н-нонафторбутил)фосфоране при этих условиях. Раствор, оставшийся в колбе, отделяют от вязкого остатка, оставшегося в колбе, и
нейтрализуют, используя хлородородную кислоту. Для того чтобы выделить бис(н-нонафторбутил)фосфинат калия, воду удаляют при пониженном давлении. Полученный твердый остаток сушат при пониженном
давлении 120 Па и температуре бани 100°С на протяжении двух часов. Высушенный остаток впоследствии экстрагируют тремя порциями метанола по 50 см3 каждая, фракции объединяют,
впоследствии перегоняют при пониженном давлении 125 Па и твердый остаток сушат. Выход бис(н-нонафторбутил)фосфината калия составляет 7,88 г, что соответствует 62,9%, исходя из использованного
дифтортрис(н-нонафторбутил)фосфорана. 1H-н-Нонафторбутан характеризуют с помощью1Н- и19F-ЯМР спектроскопии и бис(н-нонафторбутил)фосфинат калия с помощью19F- и31Р-ЯМР спектроскопии. 1Н-Нонафторбутан 1Н- и19F-ЯМР спектры снимают на спектрометре Bruker WP 80 SY при частотах 80,1 МГц
для1Н и 75,4 МГц для19F и температуре -60°С. С этой целью используют ФЕП (фторэтиленовый полимер) пробирку, внутри которой находится тонкостенная 5 мм ЯМР пробирка с
ацетон-D6 пленкой как внешний затвор и ТМС или CCl3F, растворенный в ацетон-D6 пленке как внешний стандарт. 1H-ЯМР спектр (ацетон-D6 пленка, относительно ТМС в пленке, δ, м.ч.): 6,14 тт;2JH, F=52,0 Гц;3JH, F=5,0 Гц. 19Р-ЯМР спектр (ацетон-D6 пленка, CCl3F в пленке, δ, м.ч.): -81,31 т (CF3); -127,93 м (CF2); -131,06 м (CF2); -137,92 дм (CF2);2JH, F
=52,0 Гц. Полученные данные соответствуют значениям, описанным в литературе (Т.Hudlicky et al. in J. of Fluorine Chem., 59, №1 (1992), c.9-14.
Бис(н-нонафторбутил)фосфинат калия 19F- и31Р-ЯМР спектры снимают на спектрометре Bruker Avance 300 при частотах 282,4 МГц для19F и 121,5 МГц для31Р. 19F-ЯМР спектр (растворитель D2O, относительно CF3СООН в D2O=76,53 м.ч., δ, м.ч.): -82,69 тт (CF3); -122,33 м (CF2); -123,31 дм (CF2); -127,46 тм (CF2);2JH,F=79,5 Гц;4JF,F=9,6 Гц;4JF,F=12,
0 Гц; JF,F=1,5 Гц. 31Р-ЯМР спектр (растворитель D2O, внутренний стандарт 85 вес.% Н3PO4, м.ч.): 4,81 квин;2JP,
F=78,9 Гц. Пример 4 7,0 г (124,8 ммоль) гидроксида калия растворяют в 10 см3 воды в колбе и полученный раствор нагревают при температуре бани
70-80°С. Впоследствии через капельную воронку на протяжении 20 минут при перемешивании прибавляют 12,18 г (16,8 ммоль) дифтортрис(н-нонафторбутил)фосфорана. Реакционную смесь впоследствии
нагревают при температуре бани 150°С и перемешивают при этой температуре на протяжении еще двух часов. Газообразный 1H-н-нонафторбутан, образующийся вследствие гидролиза
дифтортрис(н-нонафторбутил)фосфорана, собирают в последующую ловушку, охлаждаемую сухим льдом. В охлажденной ловушке получают 6,12 г жидкого 1H-н-нонафторбутана. Выход 1H-н-нонафторбутана составляет
82,9%, исходя из н-нонафторбутильной группы, удаленной в дифтортрис(н-нонафторбутил)фосфоране при этих условиях. Остаток, оставшийся в колбе, растворяют в 50 см воды и избыток
гидроксида калия нейтрализуют, используя водный раствор фтороводорода. Для того чтобы выделить дикалий(н-нонафторбутил)фосфонат, воду удаляют при пониженном давлении. Полученный твердый остаток сушат
при пониженном давлении 120 Па и температуре бани 100°С на протяжении двух часов. Дикалий(н-нонафторбутил)фосфонат, C4F9P(O)(OK)2, впоследствии экстрагируют из
высушенного остатка, используя две порции метанола по 50 см3 каждая, фракции объединяют и метанол отгоняют. Твердый остаток впоследствии сушат при пониженном давлении 125 Па. Выход
дикалий(н-нонафторбутил)фосфоната составляет 5,0 г, что соответствует выходу 79,2%, исходя из использованного дифтортрис(н-нонафторбутил)фосфорана. 1H-н-Нонафторбутан характеризуют с
помощью1Н- и19F-ЯМР спектроскопии и дикалий (н-нонафторбутил)фосфонат с помощью19F- и31Р-ЯМР спектроскопии. Химические сдвиги,
определенные для 1H-н-нонафторбутана, соответствуют значениям приведенным в Примере 3. Дикалий(н-нонафторбутил)фосфонат, С4F9Р(O)(ОК)2 19F- и31Р-ЯМР спектры снимают на спектрометре Bruker Avance 300 при частотах 282,4 МГц для19F и 121,5 МГц для31Р. 19Р-ЯМР
спектр (растворитель D2O, относительно CF3СООН в D2O=76,53 м.ч., δ, м.ч.): -81,64 тт (CF3); -121,94 м (CF2); -122,86 дм (CF2);
-126,66 тм (CF2);2JP, F=68,9 Гц;4JF, F=9,6 Гц;4JF, F=13,4 Гц; JF, F=3,9 Гц. 31
Р-ЯМР спектр (растворитель D2O, относительно 85 вес.% Н3PO4 в D2O, δ, м.ч.): 4,00 тт;2JP, F=68,8 Гц;3JP,
F=3,4 Гц. Пример 5 8,0 г (190,5 ммоль) моногидрата гидроксида лития суспендируют в 15 см3 воды в колбе и полученную суспензию нагревают при температуре
бани 70-80 °C. Впоследствии через капельную воронку на протяжении 30 минут при перемешивании прибавляют 21,21 г (29,2 ммоль) дифтортрис(н-нонафторбутил)фосфорана. Реакционную смесь впоследствии
нагревают до температуры бани 150°С и перемешивают при этой температуре на протяжении еще двух часов. Газообразный 1H-н-нонафторбутан, образующийся вследствие гидролиза
дифтортрис(н-нонафторбутил)фосфорана, собирают в последующую ловушку, охлаждаемую сухим льдом. В охлажденной ловушке собирают 7,24 г жидкого 1H-н-нонафторбутана. Выход 1H-н-нонафторбутана составляет
56,3%, исходя из н-нонафторбутильной группы, удаленной в дифтортрис(н-нонафторбутил)фосфоране при этих условиях. Остаток, оставшийся в колбе, растворяют в 50 см3 воды и
избыток гидроксида калия нейтрализуют, используя водный раствор фтороводорода, и образовавшийся осадок фторида лития отфильтровывают. Для того чтобы выделить дилитий(н-нонафторбутил)фосфонат, C4F9P(O)(OLi)2, воду удаляют при пониженном давлении. Полученное белое твердое вещество сушат при пониженном давлении (120 Па) и температуре бани 100°С на протяжении
двух часов. Получают 8,0 г дилитий-нонафторбутилфосфоната, что соответствует выходу 87,8%, исходя из использованного дифтортрис(н-нонафторбутил)фосфорана. 1H-н-Нонафторбутан
характеризуют с помощью1Н- и19F-ЯМР спектроскопии и дилитий(н-нонафторбутил)фосфонат с помощью19F- и31Р-ЯМР спектроскопии. Химические
сдвиги, определенные для 1H-н-нонафторбутана, соответствуют значениям, указанным в Примере 3. Дилитий-нонафторбутилфосфонат 19F- и31Р-ЯМР спектры
снимают на спектрометре Bruker Avance 300 при частотах 282,4 МГц для19F и 121,5 МГц для31Р. 19F-ЯМР спектр (растворитель D2O, относительно
CF3СООН в D2O=76,53 м.ч., δ, м.ч.): -81,85 тт (CF3); -122,03 м (CF2); -123,06 дм (CF2); -126,79 тм (CF2);2JP,
F=70,1 Гц;4JF, F=9,5 Гц;4JF, F=14,2 Гц; JF, F=3,9 Гц; (растворитель ацетон-D6, внутренний стандарт CCl3F, δ, м.ч.): -80,92 м (CF3); -120,66 м (CF2); -122,70 дм (CF2); -125,62 тм (CF2);2JP, F=78,6 Гц;4JF,
F=9,9 Гц;4JF, F=14,5 Гц; JF, F=3,2 Гц. 31Р-ЯМР спектр (растворитель D2O, относительно 85 вес.% Н3PO4
в D2O, δ, м.ч.): 3,81 тт;2JP, F=70,1 Гц;3JP, F=3,3 Гц; (растворитель ацетон-D6, относительно 85 вес.% Н3PO4 -15% D2O в ацетоне-D6, δ, м.ч.): -0,28 т;2JP, F=78,1 Гц. Пример 6 10,24 г (182,5 ммоль)
гидроксида калия растворяют в 10 см3 воды в колбе и полученный раствор нагревают при температуре бани 65-70°С. Впоследствии через капельную воронку на протяжении 60 минут при
перемешивании прибавляют 18,70 г (43,9 ммоль) дифтортрис(пентафторэтил)фосфорана. Реакционную смесь впоследствии нагревают при температуре бани 120°С и перемешивают при этой температуре на
протяжении еще одного часа. Газообразный пентафторэтан, образующийся вследствие гидролиза дифтортрис(пентафторэтил)фосфорана, собирают в последующую ловушку, охлаждаемую жидким
азотом. Получают 9,99 г твердого пентафторэтана в охлаждаемой ловушке. Выход пентафторэтана составляет 94,8%, исходя из двух пентафторэтильных групп, удаленных в
дифтортрис(пентафторэтил)фосфоране при этих условиях. Остаток, оставшийся в колбе, растворяют в 40 см3 воды и избыток гидроксида калия нейтрализуют, используя несколько
капель водного раствора фтороводорода. Для того чтобы выделить дикалийпентафторэтилфосфонат, воду удаляют при пониженном давлении. Полученное твердое вещество сушат при пониженном
давлении (120 Па) и температуре бани 100°С на протяжении одного часа. Дикалийпентафторэтилфосфонат впоследствии экстрагируют из твердого остатка, используя две порции метанола по 50 см3 каждая, фракции объединяют, метанол отгоняют и полученный остаток сушат при пониженном давлении (120 Па). Получают 16,54 г ди(фторида калия) дикалийпентафторэтилфосфоната,
(C2F5P(O)(OK)2)·2KF, что соответствует выходу 96,1%, исходя из использованного дифтортрис(пентафторэтил)фосфорана. Пентафторэтан характеризуют с
помощью1Н- и19F-ЯМР спектроскопии и ди(фторид калия) дикалийпентафторэтилфосфонат с помощью19F- и31Р-ЯМР спектроскопии. Химические
сдвиги, определенные для пентафторэтана, соответствуют значениям указанным в Примере 1. Ди(фторид калия) дикалийпентафторэтилфосфонат 19F-ЯМР спектр (растворитель D2O, относительно CF3СООН в D2O=76,53 м.ч., δ, м.ч.): -81,86 т (CF3); -125,91 к (CF2); -122,70 с
(2KF);2JP, F=68,4 Гц;3JF, F=1,6 Гц. 31Р-ЯМР спектр (растворитель D2O, относительно 85 вес.% Н3PO4 в D2O, δ, м.ч.): 3,17 т;2JP, F=68,4 Гц. Пример 7 8,50 г (151,5 ммоль) гидроксида калия растворяют в 8,8 см3
воды в колбе и полученный раствор нагревают при температуре бани 70-80°С. Впоследствии через капельную воронку на протяжении 90 минут при перемешивании прибавляют 15,77 г (37,0 ммоль)
дифтортрис(пентафторэтил)фосфорана. Газообразный пентафторэтан, образующийся вследствие гидролиза дифтортрис(пентафторэтил)фосфорана, собирают в последующую ловушку, охлаждаемую жидким
азотом. Получают 8,30 г твердого пентафторэтана в охлаждаемой ловушке. Выход пентафторэтана составляет 93,4%, исходя из двух пентафторэтильных групп, удаленных в
дифтортрис(пентафторэтил)фосфоране при этих условиях. Химические сдвиги, определенные для пентафторэтана, соответствуют значениям, указанным в Примере 1. Пример 8 6,23 г (111,0 ммоль) гидроксида калия растворяют в 12,18 г смеси этанол/вода (1:1 весовых частей) в колбе и полученный раствор нагревают при температуре бани 55-60°С. Впоследствии
через капельную воронку на протяжении 45 минут при перемешивании прибавляют 11,43 г (26,8 ммоль) дифтортрис(пентафторэтил)фосфорана и реакционную смесь нагревают при 80°С на протяжении 10
минут. Газообразный пентафторэтан, образующийся вследствие гидролиза дифтортрис(пентафторэтил)фосфорана, собирают в последующую ловушку, охлаждаемую жидким азотом.
Получают 5,23 г твердого пентафторэтана в охлаждаемой ловушке. Выход пентафторэтана составляет 81,3%, исходя из двух пентафторэтильных групп, удаленных в дифтортрис(пентафторэтил)фосфоране при этих
условиях. Химические сдвиги, определенные для пентафторэтана, соответствуют значениям указанным в Примере 1. Пример 9 13,46 г (31,6 ммоль)
дифтортрис(пентафторэтил)фосфорана прибавляют через капельную воронку при перемешивании на протяжении одного часа при комнатной температуре к 96,5 г (131,1 ммоль) 20 вес.% водного раствора гидроксида
тетраэтиламмония. Во время этой операции наблюдается нагревание реакционной смеси. Реакционную смесь впоследствии нагревают при 80°С на протяжении 30 минут. Газообразный пентафторэтан,
образующийся вследствие гидролиза дифтортрис(пентафторэтил)фосфорана, собирают в последующую ловушку, охлаждаемую жидким азотом. Получают 7,49 г твердого пентафторэтана в охлаждаемой ловушке. Выход
пентафторэтана составляет 98,8%, исходя из двух удаленных пентафторэтильных групп. Химические сдвиги, определенные для пентафторэтана, соответствуют значениям, указанным в Примере
1. Раствор, оставшийся в колбе, упаривают на роторном испарителе и полученное твердое вещество сушат при пониженном давлении (120 Па) и температуре 100°С, 24,67 г белого
кристаллического [(C2H5)4N]2[C2F5PO3]•2[(C2H5)4N]F•8H2O. [(C2H5)4N]2[C2F5PO3]•2[(C2H5)4N]F•8H2O характеризуют с
помощью1H-,19F- и19F31Р-ЯМР спектроскопии и элементного анализа. 19F-,1Н- и31Р-ЯМР спектры снимают на
спектрометре Bruker Avance 300 при частотах 282,4 МГц для19F и 121,5 МГц для31Р. 19Р-ЯМР спектр (растворитель ацетонитрил-D3, относительно
CCl3F, δ, м.ч.): -79,41 дт (CF3); -126,74 дк (CF2); -111,74 (2F-);2JP, F=54,0 Гц;3JP,F=1,1 Гц;3JF, F=1,0 Гц. 1Н-ЯМР спектр (растворитель ацетонитрил-D3, относительно ТМС, δ, м.ч.): 1,21 тм (СН3); 3,28 к (СН2);3JH, H=7,3 Гц. Имеет место обмен протона в молекуле H2O на дейтерий растворителя. 31Р-ЯМР спектр
(растворитель ацетонитрил-D3, относительно 85 вес.% Н3PO4 - 15% D2O в ацетонитриле-D3, δ, м.ч.): -1,77 т;2JP, F=54,
2 Гц. Элементный анализ: рассчитано для С34Н96F5N4O11Р: С 47,31%; Н 11,21%; N 6,49%; найдено: С
47,37%; Н 10,80%; N 6,40%. Пример 10 50,38 г (159,7 ммоль) октагидрата гидроксида бария суспендируют в 100 см3 воды в колбе и полученную суспензию нагревают
при температуре бани 65-70°С. Впоследствии через капельную воронку на протяжении 30 минут при перемешивании прибавляют 22,68 г (53,2 ммоль) дифтортрис(пентафторэтил)фосфорана. Реакционную смесь
впоследствии нагревают до температуры 150°С и перемешивают при этой температуре на протяжении двух часов. Газообразный пентафторэтан, образующийся вследствие гидролиза
дифтортрис(пентафторэтил)фосфорана, собирают в последующую ловушку, охлаждаемую сухим льдом. Получают 10,00 г жидкого пентафторэтана в охлаждаемой ловушке. Выход пентафторэтана
составляет 78,3%, исходя из двух пентафторэтильных групп, удаленных в дифтортрис(пентафторэтил)фосфоране при этих условиях. Остаток оставшийся в колбе переносят в 50 см3
воды и нейтрализуют, используя водный раствор фтороводорода. Отфильтровывают образовавшийся осадок фторида бария. Для того чтобы выделить пентафторэтилфосфонат бария, воду удаляют при
пониженном давлении. Полученное белое вещество сушат при пониженном давлении (120 Па) и температуре бани 100°С на протяжении одного часа. Получают 10,6 г пентафторфосфоната бария ([C2
F5P(O)O2]Ba), который содержит приблизително 2 вес.% фторида бария, что соответствует выходу 59,2%, исходя из использованного дифтортрис(пентафторэтил)фосфорана.
Пентафторэтан характеризуют с помощью1Н- и19F-ЯМР спектроскопии и пентафторфосфонат бария с помощью19F- и31Р-ЯМР спектроскопии.
Химические сдвиги, определенные для пентафторэтана, соответствуют значениям, указанным в Примере 1. Пентафторэтилфосфонат бария 19F-,1Н- и31Р-ЯМР спектры снимают на спектрометре Bruker Avance 300 при частотах 282,4 МГц для19F и 121,5 МГц для31Р. 19F-ЯМР спектр (растворитель D2O, относительно CF3СООН в D2О=76,53 м.ч., δ, м.ч.): -81,99 тд (CF3); -126,25 дк (CF2);2JP, F=70,5 Гц;3JF, F=1,8 Гц;3JP, F=0,5 Гц. 31Р-ЯМР спектр (растворитель D2O, относительно 85 вес.% Н3PO4 в D2O,
δ, м.ч.): 2,88 т;2JP, F=70,3 Гц. Пример 11 16,70 г (52,9 ммоль) октагидрата гидроксида бария суспендируют в 20 см3 воды в колбе и полученную суспензию нагревают при температуре бани 70-80°С. Впоследствии через капельную воронку на протяжении 30 минут при перемешивании прибавляют 17,79 г (24,5 ммоль)
дифтортрис(н-нонафторбутил)фосфорана. Реакционную смесь впоследствии нагревают при температуре бани 120°С и перемешивают при этой температуре на протяжении одного часа.
Газообразный 1H-н-нонафторбутан, образующийся вследствие гидролиза дифтортрис(н-нонафторбутил)фосфорана, собирают в последующей ловушке, охлаждаемой жидким азотом. Получают 7,72 г
твердого 1H-н-нонафторбутана в охлаждаемой ловушке. Выход 1H-н-нонафторбутана составляет 71,6%, исходя из двух н-нонафторбутильных групп, удаляемых в дифтортрис(н-нонафторбутил)фосфоране при этих
условиях. Остаток, оставшийся в колбе, переносят в 50 см3 воды и нейтрализуют, используя водный раствор фтороводорода. Отфильтровывают образовавшийся осадок фторида
бария. Для того чтобы выделить н-нонафторбутилфосфонат бария, воду удаляют при пониженном давлении. Полученное белое вещество сушат при пониженном давлении (120 Па) и температуре бани
100°С на протяжении одного часа. Получают 7,0 г н-нонафторбутилфосфоната бария ([н-С4F9Р(O)O2]Ва), который содержит приблизительно 2 вес.% фторида бария, что
соответствует выходу 64,87%, исходя из используемого дифтортрис(пентафторэтил)фосфорана. 1H-н-Нонафторбутан характеризуют с помощью1Н- и19F-ЯМР спектроскопии и
н-нонафторбутилфосфонат бария с помощью19F- и31Р-ЯМР спектроскопии. Химические сдвиги, определенные для 1H-нонафторбутана, соответствуют значениям указанным в
Примере 3. н-Нонафторбутилфосфонат бария 19F-ЯМР спектр (растворитель D2O, относительно CF3СООН в D2O=76,53 м.ч., δ,
м.ч.): -81,77 тт (CF3); -122,29 м (CF2); -123,66 дтм (CF2); -126,76 тм (CF2);2JP, F=75,8 Гц;4JF, F=9,7 Гц;4JF, F=13,8 Гц; JF, F=3,6 Гц. 31Р-ЯМР спектр (растворитель D2O, относительно 85 вес.% Н3PO4 в D2
O, δ, м.ч.): 2,22 т;2JP, F=76,1 Гц. Пример 12 10,32 г (183,9 ммоль) гидроксида калия и 20 см3 воды вносят в автоклав, имеющий
объем 100 см3. Автоклав охлаждают до -30°С и прибавляют 9,70 г (22,8 ммоль) дифтортрис(пентафторэтил)фосфорана. Автоклав впоследствии закрывают и нагревают при 200-210°С на
протяжении восьми часов с помощью масляной бани. Автоклав потом доводят до комнатной температуры и выход автоклава присоединяют к холодной ловушке, охлаждаемой жидким азотом. Получают
7,57 г чистого пентафторэтана, что соответствует выходу 92,2%, исходя из трех пентафторэтильных групп, удаляемых в используемом дифтортрис(пентафторэтил)фосфоране при этих условиях.
Химические сдвиги, определенные для пентафторэтана, соответствуют значениям, указанным в Примере 1. Пример 13 51,0 г гидроксида калия и 50 см3 воды вносят в
автоклав, имеющий объем 350 см3. Автоклав охлаждают до -30°С и прибавляют 95,9 г смеси трифторбис(н-нонафторбутил)фосфорана (60 мол.%) и дифтортрис(н-нонафторбутил)фосфорана (40
мол.%). Автоклав впоследствии закрывают и нагревают при 200-210°С на протяжении 18 часов с помощью масляной бани. Автоклав потом доводят до комнатной температуры и выход автоклава присоединяют
к холодной ловушке, охлаждаемой сухим льдом. Получают 68,0 г чистого 1H-нонафтор-н-бутана, что соответствует выходу 95,2%, исходя из двух н-нонафторбутильных групп, удаляемых в
используемых трифторбис(н-нонафторбутил)фосфоране и дифтортрис(н-нонафторбутил)фосфоране при этих условиях. 1H-Нонафтор-н-бутан характеризуют с помощью1Н- и19
F-ЯМР спектроскопии. Химические сдвиги, определенные для 1H-нонафтор-н-бутана, соответствуют значениям, указанным в Примере 3. Пример 14
Бис(пентафторэтил)фосфиновая кислота 4,09 г (12,0 ммоль) бис(пентафторэтил)фосфината калия вводят в дистиляционную колбу с 8,71 г (88,9 ммоль) 100% серной кислоты, H2SO4, и полученную бис(пентафторэтил)фосфиновую кислоту перегоняют при пониженном давлении (400 Па) и температуре масляной бани 90-120°С. Получают 3,25 г прозрачной и бесцветной жидкой
бис(пентафторэтил)фосфиновой кислоты, (C2F5)2P(O)OH, что соответствует выходу 89,5%. Найденные значения химических сдвигов соответствуют значениям,
описанным в публикации Т.Mahmood, Inorganic Chemistry, 25 (1986), с.3128-3131. Пример 15 1,0 г (10,2 ммоль) 100% серной кислоты, H2SO4, прибавляют
к перемешиваемому раствору 3,42 г (10,2 ммоль) пентафторэтилфосфоната бария в 50 см3 воды. Образуется осадок сульфата бария, который отделяют фильтрованием. Полученный фильтрат полностью
упаривают при пониженном давлении и сушат при 125 Па и температуре масляной бани 100°С на протяжении последующих 6 часов. Получают 1,75 г высоковязкой жидкой пентафторэтилфосфоновой кислоты
C2F5P(O)(OH)2, что соответствует выходу 83,8%. 19F-ЯМР спектр (растворитель: ацетонитрил-D3, относительно CCl3F,
δ, м.ч.): -81,03 т (CF3); -126,74 дк (CF2); J2P, F=89,4 Гц, J3F, F=1,6 Гц. 1Н-ЯМР
спектр (растворитель: ацетонитрил-D3, относительно ТМС, δ, м.ч.): 11,26 ш. c (ОН). 31Р-ЯМР спектр (растворитель: ацетонитрил-D3; относительно 85
вес.% Н3PO4 -15 вес.% D2O в ацетонитриле-D3): -3,40 т, J2P, F=89,6 Гц. Эти данные отвечают значениям,
описанным в литературе (Т.Mahmood and J.M.Shreeve, in Inorg. Chem., 25 (1986), c.3128-3131). Пример 16 Раствор 0,492 г (2,46 ммоль) пентафторэтилфосфоновой кислоты
получали, как описано в Примере 15, в 10 см3 воды нейтрализуют путем медленного прибавления при комнатной температуре при перемешивании 3,015 г 20 вес.% водного гидроксида тетраэтиламмония.
Воду упаривают при пониженном давлении и полученный остаток сушат при пониженном давлении (120 Па) и температуре бани 50°С на протяжении 2 часов. Получают 1,115 г белого
твердого бис(тетраэтиламмоний)пентафторэтилфосфоната. Выход составляет 99,0%, исходя из использованной пентафторэтилфосфоновой кислоты. Бис(тетраэтиламмоний)пентафторэтилфосфонат
характеризуют с помощью19F,31P и1H-ЯМР спектроскопии. 19F ЯМР спектр, м.ч. (растворитель: ацетонитрил-D3; относительно CCl3F): -79,49 с (CF3); -122,10 д (CF2); J2P, F=54,6 Гц. 1Н ЯМР спектр, м.ч. (растворитель: ацетонитрил-D3;
относительно ТМС): 1,20 тм (12Н, 4СН3); 3,29 к (8Н, 4СН2); J3H, H=7,3 Гц. 31Р ЯМР спектр, м.ч. (растворитель:
ацетонитрил-D3; относительно 85% Н3PO4): -2,28 т; J2P, F=54,9 Гц. Пример 17 Раствор
нонафтор-н-бутилфосфоновой кислоты, получали, как описано в Примере 15, из 3,73 г (8,57 ммоль) нонафтор-н-бутилфосфоната бария и 0,839 г 15 100 вес.% серной кислоты в 20 см3 воды,
нейтрализуют (рН=7) путем медленного прибавления при комнатной температуре при перемешивании 20 вес.% водного гидроксида тетраэтиламмония. Воду упаривают при пониженном давлении и полученный остаток
сушат при пониженном давлении (120 Па) и температуре бани 60°С на протяжении 2 часов. Получают 4,59 г твердого нонафтор-н-бутилфосфоната бис(тетраэтиламмония). Выход составляет
96,0%, исходя из используемого нонафтор-н-бутилфосфоната бария. Нонафтор-н-бутилфосфонат бис(тетраэтиламмония) характеризуют с помощью19F,31Р и1
Н-ЯМР спектроскопии. 19F ЯМР спектр, м.ч. (растворитель: ацетонитрил-D3; относительно CCl3F): -80,37 тт (CF3); -119,57 м (CF2);
-119,72 дм (CF2); -124,80 м (CF2); J2P, F=55,6 Гц; J3F, F=4,3 Гц; J4F, F=9,5 Гц. 1Н ЯМР спектр, м.ч. (растворитель: ацетонитрил-D3; относительно ТМС): 1,23 тм (12Н, 4СН3); 3,27 к (8Н, 4СН2); J3H,H
=7,4 Гц. 31Р ЯМР спектр, м.ч. (растворитель: ацетонитрил-D3; относительно 85% Н3PO4): -2,06 т; J2P, F=56,5
Гц. Пример 18 1,43 г пентафторэтилфосфоновой кислоты, получали, как описано в Примере 15, растворяют в 15 см3 воды и нейтрализуют (рН=7) путем медленного
прибавления при комнатной температуре при перемешивании 10 вес.% водного гидроксида калия. К полученному водному раствору дикалий пентафторэтилфосфоната при постоянном перемешивании при комнатной
температуре прибавляют раствор 2,09 г (11,9 ммоль) хлорида 1-этил-З-метилимидазолия в 3 см3 воды. Воду упаривают при пониженном давлении и полученный остаток сушат при пониженном давлении
(120 Па) и температуре бани 60°С на протяжении 1 часа. Впоследствии к остатку прибавляют 10 см3 изопропилового спирта, отфильтровывают белый осадок и дважды промывают 5 см3
изопропилового спирта. Изопропиловый спирт упаривают при пониженном давлении и полученный остаток сушат при пониженном давлении (1,4 Па) и температуре бани 80°С на протяжении 1,5 часа. Получают 2,56 г маслянистой жидкости пентафторэтилфосфоната ди(1-этил-3-метилимидазолия). Выход составляет 85,0%, исходя из использованной пентафторэтилфосфоновой кислоты.
Пентафторэтилфосфонат ди(1-этил-3-метилимидазолия) характеризуют с помощью19F,31Р и1Н-ЯМР спектроскопии. 19F ЯМР спектр, м.ч.
(растворитель: ацетонитрил-D3; относительно CCl3F): -79,68 с (CF3); -123,22 д (CF2); J2P, F=57,9 Гц. 1Н ЯМР спектр, м.ч. (растворитель: ацетонитрил-D3; относительно ТМС): 1,38 т (3Н, СН3); 3,94 с (3Н, СН3); 4,29 к (2Н, CH2); 7,70 с (1Н); 7,75 с (1Н);
10,82 с (1H); J3H, H=7,2 Гц. 31P ЯМР спектр, м.ч. (растворитель: ацетонитрил-D3; относительно 85% Н3PO4): -1,
28 т; J3P, F=57,4 Гц. Пример 19 Раствор 2,4 г (12,0 ммоль) пентафторэтилфосфоновой кислоты, получали, как описано в Примере 15, в 13
см3 воды нейтрализуют (рН=7) путем медленного прибавления при комнатной температуре при перемешивании 14,86 г приблизительно 40 вес.% водного гидроксида тетрабутилфосфония. Воду упаривают
при пониженном давлении и полученный остаток сушат при пониженном давлении (1,4 Па) и температуре бани 70°С на протяжении 2 часов. Получают 7,95 г высоковязкой жидкости, которая
медленно кристаллизуется в белый твердый пентафторэтилфосфонат бис(тетрабутилфосфония). Выход составляет 92,4%, исходя из использованной пентафторэтилфосфоновой кислоты. Температура
плавления 76-79°С. Пентафторэтилфосфонат бис(тетрабутилфосфония), [(C4H9)4P+]2C2F5
P(O)O22-, характеризуют с помощью19F,31P и1H-ЯМР спектроскопии. 19F ЯМР спектр, м.ч. (растворитель:
ацетонитрил-D3; относительно CCl3F): -79,39 с (CF3); -121,98 д (CF2); J2P, F=54,2 Гц. 1H ЯМР спектр,
м.ч. (растворитель: ацетонитрил-D3; относительно ТМС): 0,93 т (12Н, 4СН3); 1,45 м (16Н, 8CH2); 2,37 м (8Н, 4СН2); J3H, H=7,1
Гц. 31Р ЯМР спектр, м.ч. (растворитель: ацетонитрил-D3; относительно 85% Н3PO4): -1,84 т (1P); 32,73 м (2Р); J2P, F=54,6 Гц.
Реферат
Изобретение описывает способ получения моногидроперфторалканов, бис(перфторалкил)фосфинатов и перфторалкилфосфонатов, включающий, по крайней мере, обработку, по крайней мере, одного перфторалкилфосфорана, по крайней мере, одним основанием, где основание(я) выбирают из группы, состоящей из гидроксидов щелочноземельных металлов, органо-металлического соединения в пригодном растворителе или, по крайней мере, одного органического основания и при желании кислотой в пригодной реакционной среде. Также описываются новые перфторалкилфосфонаты и бис(перфторалкил)фосфинаты, применение новых перфторалкилфосфонатов и бис(перфторалкил)фосфинатов как ионных жидкостей, как катализаторов фазового переноса или поверхностно-активных веществ. 4 н. и 14 з.п. ф-лы.
Формула



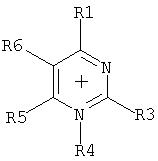
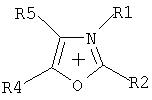

Комментарии