Способ эпоксидирования - RU2742302C2
Код документа: RU2742302C2
Чертежи

Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА СВЯЗАННЫЕ ЗАЯВКИ
[0001] Настоящее изобретение испрашивает приоритет предварительной американской патентной заявки № 62/372653, поданной 9 августа 2016 г., которая включена в настоящий документ посредством ссылки во всей ее полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0002] Настоящее изобретение относится к способу для окисления этилена в оксид этилена.
УРОВЕНЬ ТЕХНИКИ
[0003] Хотя оксид этилена присутствует в естественных условиях в незначительных количествах, он был впервые синтезирован в лабораторных условиях в 1859 году французским химиком Шарлем-Адольфом Вюртцем с использованием так называемого «хлоргидринового» процесса. Однако полезность оксида этилена в качестве промышленного химиката не была полностью понята во время Вюртца; и таким образом промышленное производство оксида этилена с использованием хлоргидринового процесса не начиналось вплоть до кануна первой мировой войны, когда в нем появилась необходимость, по меньшей мере частично, из-за быстрого роста спроса на этиленгликоль (для которого оксид этилена является промежуточным продуктом) в качестве антифриза для использования на быстрорастущем автомобильном рынке. Но даже тогда хлоргидриновый процесс производил оксид этилена в относительно небольших количествах и был весьма неэкономичным.
[0004] Хлоргидриновый процесс был в конечном итоге вытеснен другим процессом, прямым каталитическим окислением этилена кислородом, который стал результатом второго прорыва в синтезе оксида этилена, открытого в 1931 г. другим французским химиком Теодором Лефортом. Лефорт использовал твердый серебряный катализатор с газофазным питанием, которое включало в себя этилен, и использовал воздух в качестве источника кислорода.
[0005] За восемьдесят лет, прошедших с момента разработки метода прямого окисления, производство оксида этилена возросло настолько значительно, что сегодня он является одним из крупнейших по объему продуктов химической промышленности, составляя, по некоторым оценкам, до половины общей стоимости органических химических веществ, получаемых в результате гетерогенного окисления. Мировое производство в 2000 году составляло приблизительно 15 миллиардов тонн. (Приблизительно две трети производимого оксида этилена перерабатываются в этиленгликоль, в то время как приблизительно десять процентов производимого оксида этилена используется напрямую в таких приложениях, как стерилизация паром).
[0006] Рост производства оксида этилена сопровождался продолжающимися интенсивными исследованиями катализа и переработки оксида этилена, которые остаются предметом внимания для исследователей, как в промышленности, так и в научных кругах. В последние годы особый интерес представляют правильные рабочие и технологические параметры для производства оксида этилена с использованием так называемых «высокоселективных катализаторов», то есть катализаторов эпоксидирования на основе Ag, которые особенно эффективны при катализе желаемой продуктовой реакции этилена и кислорода с образованием оксида этилена, а не побочной реакции этилена и кислорода, которая производит побочный продукт в виде диоксида углерода (а также воду).
[0007] Однако, хотя высокоселективные катализаторы уменьшают образование побочного продукта диоксида углерода, они также могут увеличивать производство других нежелательных побочных продуктов, в частности альдегидных примесей, таких как ацетальдегиды и формальдегиды и связанные с ними кислоты, а также диссоциированных ионов. Ацетальдегид и формальдегид издавна известны как побочные продукты, образующиеся в процессе эксплуатации установок по производству оксида этилена. Уксусный альдегид образуется в результате изомеризации оксида этилена, в то время как формальдегид образуется реакцией оксида этилена с кислородом. Соответствующие кислоты, уксусная кислота и муравьиная кислота, производятся путем окисления соответственно уксусного альдегида и формальдегида. Присутствие альдегидов и связанных с ними кислот может отрицательно влиять на стойкость раствора этиленгликоля к ультрафиолету, и тем самым вызывать разложение этиленгликоля, предназначенного для производства волокна. Дополнительно к этому, образование связанных с ними кислот (а также их альдегидных реагентов) может снизить рН до уровней, достаточно низких для того, чтобы вызвать коррозию в установке. Эти соображения являются еще более серьезными для установок, которые производят этиленгликоль, предназначенный для производства волокна. Кроме того, важно отметить, что хотя примеси, такие как диоксид углерода, образуются почти исключительно на слое катализатора в реакторе получения оксида этилена, ацетальдегиды, формальдегиды и связанные с ними кислоты образуются как на катализаторе, так и после слоя катализатора.
[0008] Один возможный способ предотвращения или уменьшения коррозии, вызываемой кислотными уровнями pH, заключается в замене компонентов из углеродистой стали компонентами из нержавеющей стали. В то время как это является очень эффективным по меньшей мере для снижения скорости коррозии, если не ее полного предотвращения, использование компонентов из нержавеющей стали увеличивает затраты и, как правило, не может быть применено к уже существующей установке. Кроме того, это, конечно же, не решает проблему низкого качества продукта этиленгликоля в результате загрязнения примесями.
[0009] Другое возможное решение раскрыто в патенте US № 4822926, в котором реакторный поток продукта подается в секцию охлаждения (расположенную в абсорбере оксида этилена), и в этой секции охлаждения реакторный поток продукта контактирует с содержащим основание рециркулирующим водным раствором для того, чтобы нейтрализовать pH и устранить некоторые органические вещества. Добавление основания, такого как, например, гидроксид натрия, действительно увеличивает значение pH (и как следствие уменьшает коррозию в установке), а также предотвращает формирование некоторых из органических веществ и альдегидных примесей. Однако добавление каустической соды также часто вызывает разложение продукта этиленгликоля, особенно для более тяжелых этиленгликолей, таких как триэтиленгликоль, который часто не может производиться с соответствием минимальным стандартам качества в процессе, использующем каустическую соду. Таким образом, в конечном итоге добавление каустической соды просто заменяет одну проблему (коррозии и образования примесей) на другую (плохое качество продукта).
[0010] В попытках уменьшить образование уксусного альдегида и формальдегида и связанных примесей были также разработаны другие технологии. Например, давно было известно, то изомеризация оксида этилена в различные альдегидные соединения происходит легче при более высоких температурах. Эта проблема может быть особенно явной, когда продукт выходит из реактора при высоких температурах и в основном поддерживается при таких температурах до его входа в теплообменник для охлаждения газа перед его подачей в абсорбционную секцию.
[0011] Таким образом, были разработаны технологии и конструкции оборудования для максимально быстрого снижения температуры этиленсодержащего газа. Патент US № 4376209 раскрывает использование инертных газов в зоне охлаждения реактора для охлаждения газа, однако, как ясно из патента, эта методика дает смешанные результаты, и возможно фактически увеличивает содержание ацетальдегида настолько же, насколько подавляет его образование.
[0012] Другим подходом является интегрированный узел реактора-охладителя, раскрытый в патенте US № 7294317, который предназначен для того, чтобы вызвать резкое падение температуры этиленсодержащего газа. Однако в то время как интегрированный узел реактора-охладителя показал себя чрезвычайно успешным в плане уменьшения образования побочных продуктов, он не в состоянии устранить те примеси, которые образуются на более поздних стадиях обработки. Однако для размещения узла реактора-охладителя, описанного в вышеупомянутом патенте, необходима обширная модернизация и реконструкция.
[0013] Использование ионообменных смол требует меньших переделок для уже существующих установок, чем узла реактора-охладителя, и является очень эффективным для удаления альдегидных и других примесей из рециркулирующей воды. Подходящие ионообменные смолы раскрыты в патенте US № 6187973. Эти ионообменные смолы являются чрезвычайно эффективными при удалении примесей из рециркулирующей воды, не вызывая упомянутых выше негативных последствий от щелочной обработки. Тем не менее, в то время как ионообменные смолы обеспечивают превосходную эффективность удаления примесей, остаются трудности с их использованием. Например, ионообменные смолы требуют частой регенерации для восстановления способности смолы захватывать и адсорбировать ионы и примеси. Эти циклы регенерации должны повторяться более часто при увеличении количества побочных продуктов. Эти циклы регенерации значительно усложняют процесс получения оксида этилена и уменьшают его эффективность, потому что они требуют, чтобы отработанный или израсходованный ионообменный слой был удален из эксплуатации, а вместо него использовался резервный слой. Эта увеличивающаяся частота в цикле регенерации является нежелательной, потому что каждый такой цикл требует значительного количества деминерализованной воды и химикатов для регенерации, и одновременно производит сточные воды, которые требуют очистки и утилизации. Учитывая затраты на деминерализованную воду и химикаты, а также затраты на очистку и утилизацию сточных вод, есть таким образом императив, существует настоятельная потребность в уменьшении частоты циклов регенерации.
[0014] Учитывая вышесказанное, в данной области техники существует постоянная потребность в способах снижения частоты регенерации ионообменных смол.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0015] Настоящее изобретение относится к способу окисления этилена с образованием оксида этилена, содержащему: обеспечение водного потока, содержащего этиленгликоль и примеси; введение этого водного потока в первый ионообменный слой для снижения содержания этих примесей; определение, имеет ли выход первого ионообменного слоя удельную электропроводность, превышающую примерно 5 мкСм/см; при определении того, что выход первого ионообменного слоя имеет удельную электропроводность, больше чем приблизительно 5 мкСм/см, введение выхода первого ионообменного слоя во второй ионообменный слой; и при определении того, что выход первого ионообменного слоя имеет удельную электропроводность, больше чем приблизительно 20 мкСм/см, предпочтительно больше чем приблизительно 100 мкСм/см, перенаправление подачи водного потока во второй ионообменный слой и регенерацию первого ионообменного слоя.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0016] Предыдущий раздел, а также последующее подробное описание предпочтительных вариантов осуществления настоящего изобретения будут лучше поняты при прочтении вместе с приложенными чертежами. С целью иллюстрирования настоящего изобретения данные чертежи показывают те варианты осуществления, которые являются предпочтительными в настоящее время. Следует понимать, однако, что настоящее изобретение не ограничивается показанными точными компоновками и инструментальными средствами. В прилагаемых чертежах:
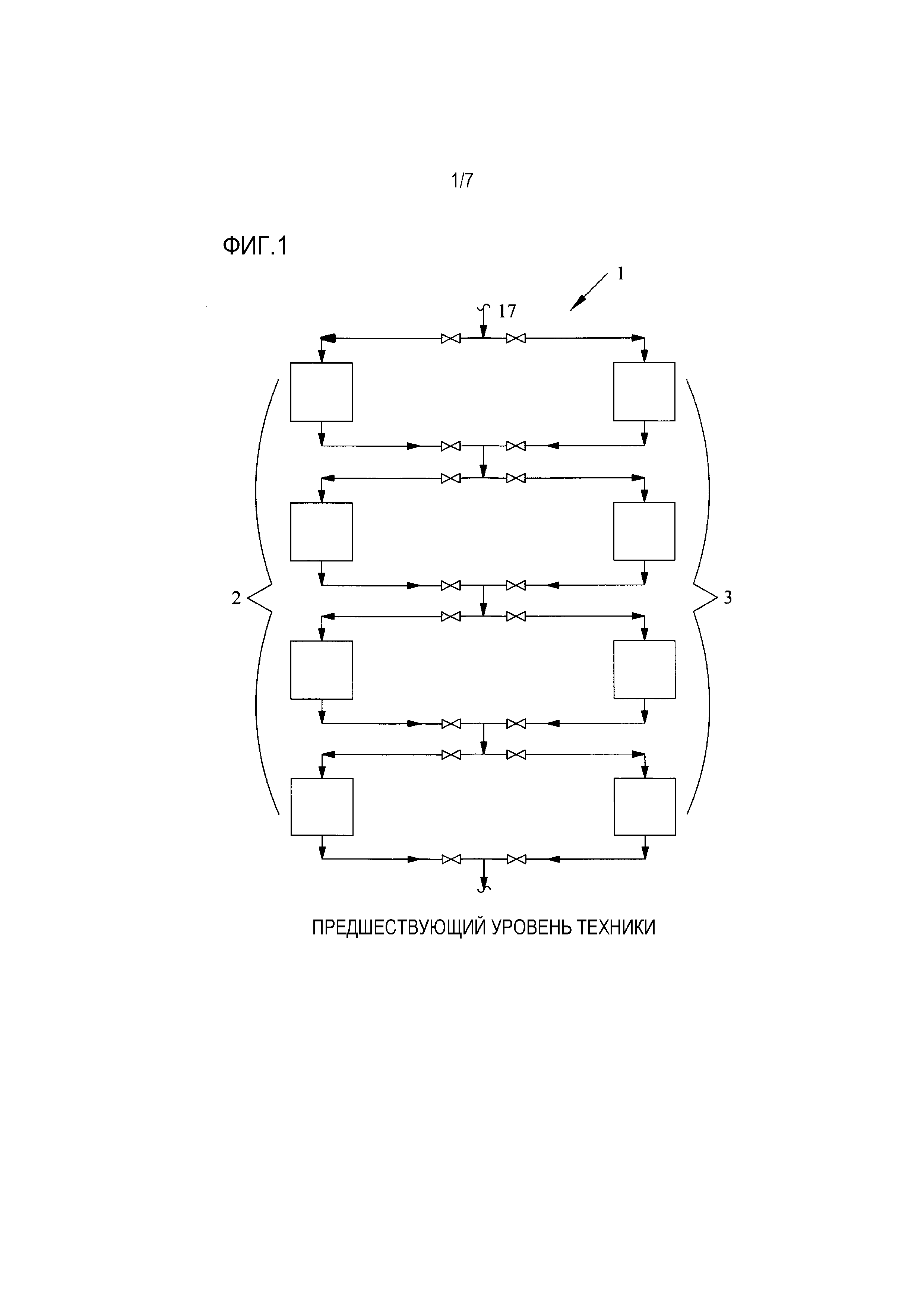
[0017] Фиг. 1 представляет собой упрощенную технологическую схему, показывающую два ряда слоев ионообменной обработки, выполненных в соответствии с предшествующим уровнем техники;
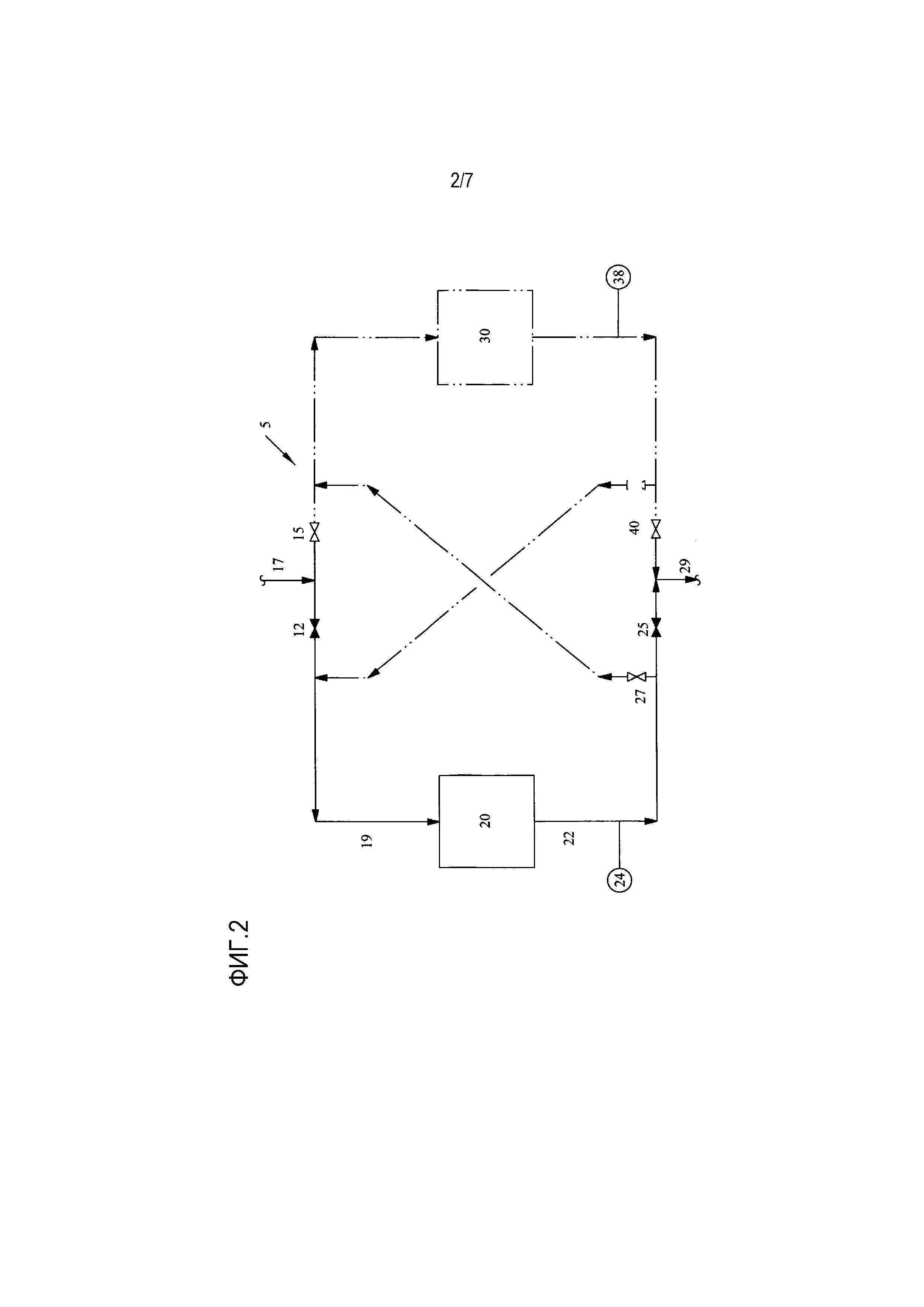
[0018] Фиг. 2 представляет собой упрощенную технологическую схему одного варианта осуществления способа для обработки водного потока с помощью слоя ионообменной обработки;
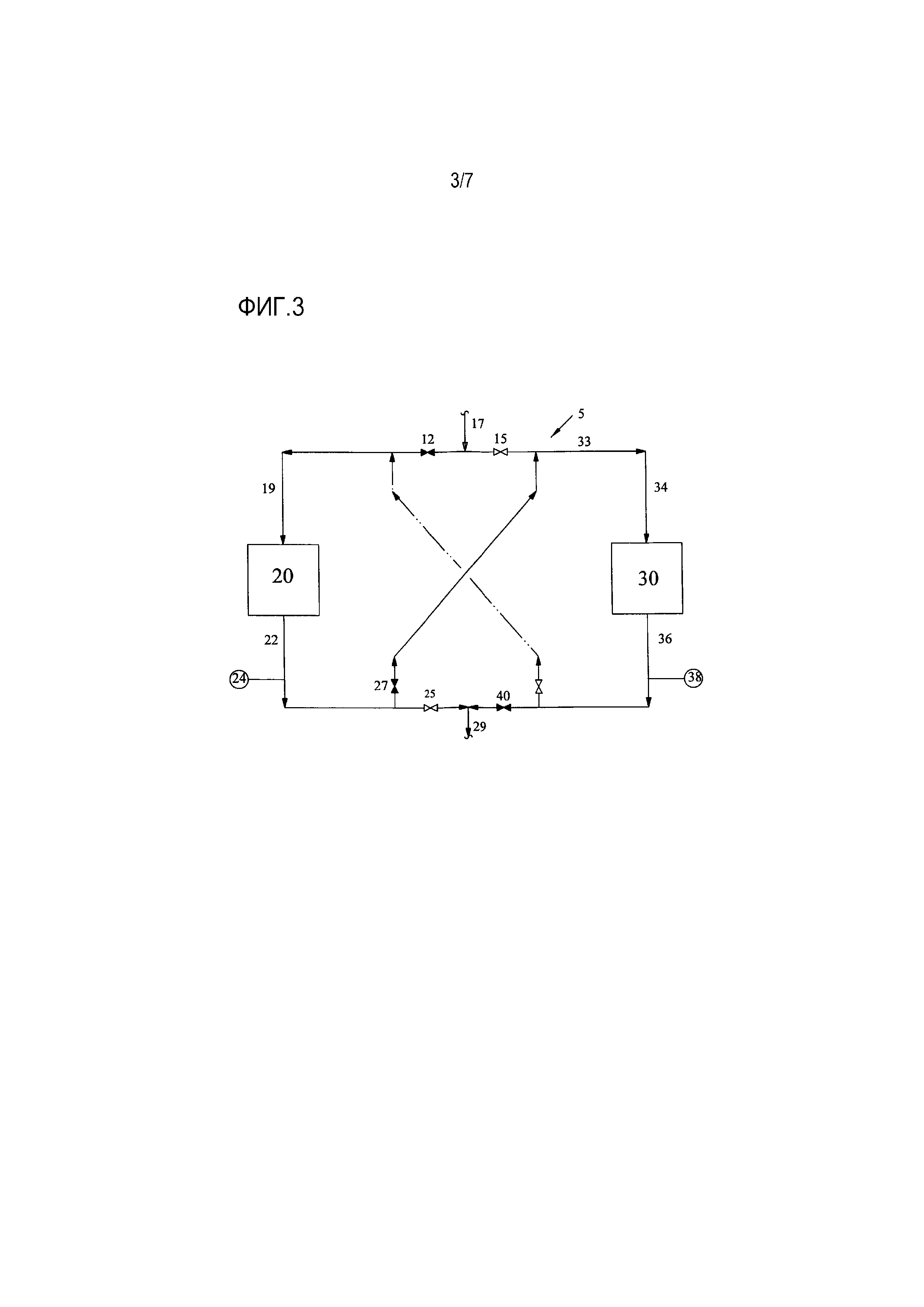
[0019] Фиг. 3 представляет собой упрощенную технологическую схему еще одного варианта осуществления способа для обработки водного потока с помощью двух слоев ионообменной обработки;
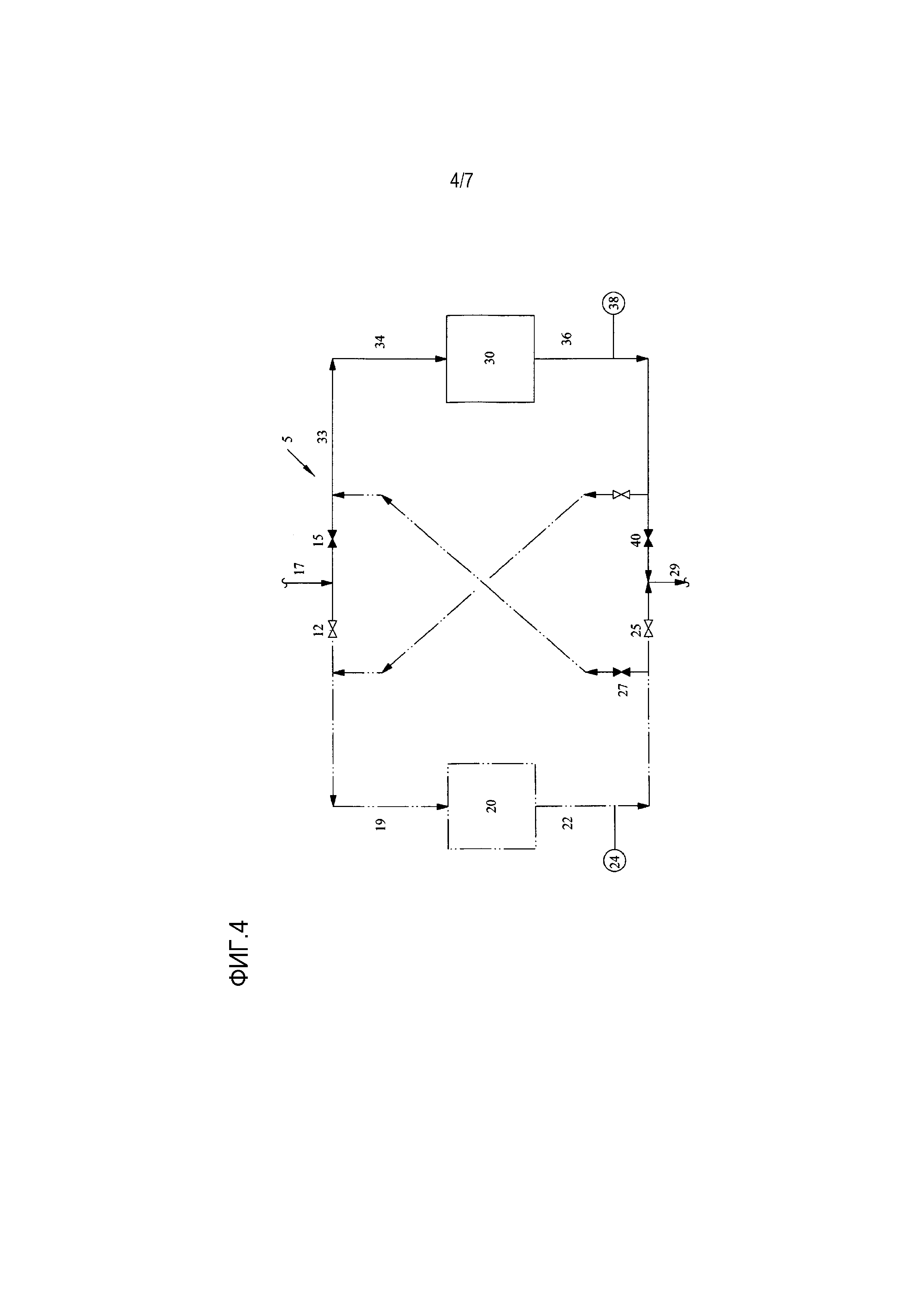
[0020] Фиг. 4 представляет собой упрощенную технологическую схему одного варианта осуществления способа для обработки водного потока с помощью одного слоя ионообменной обработки;
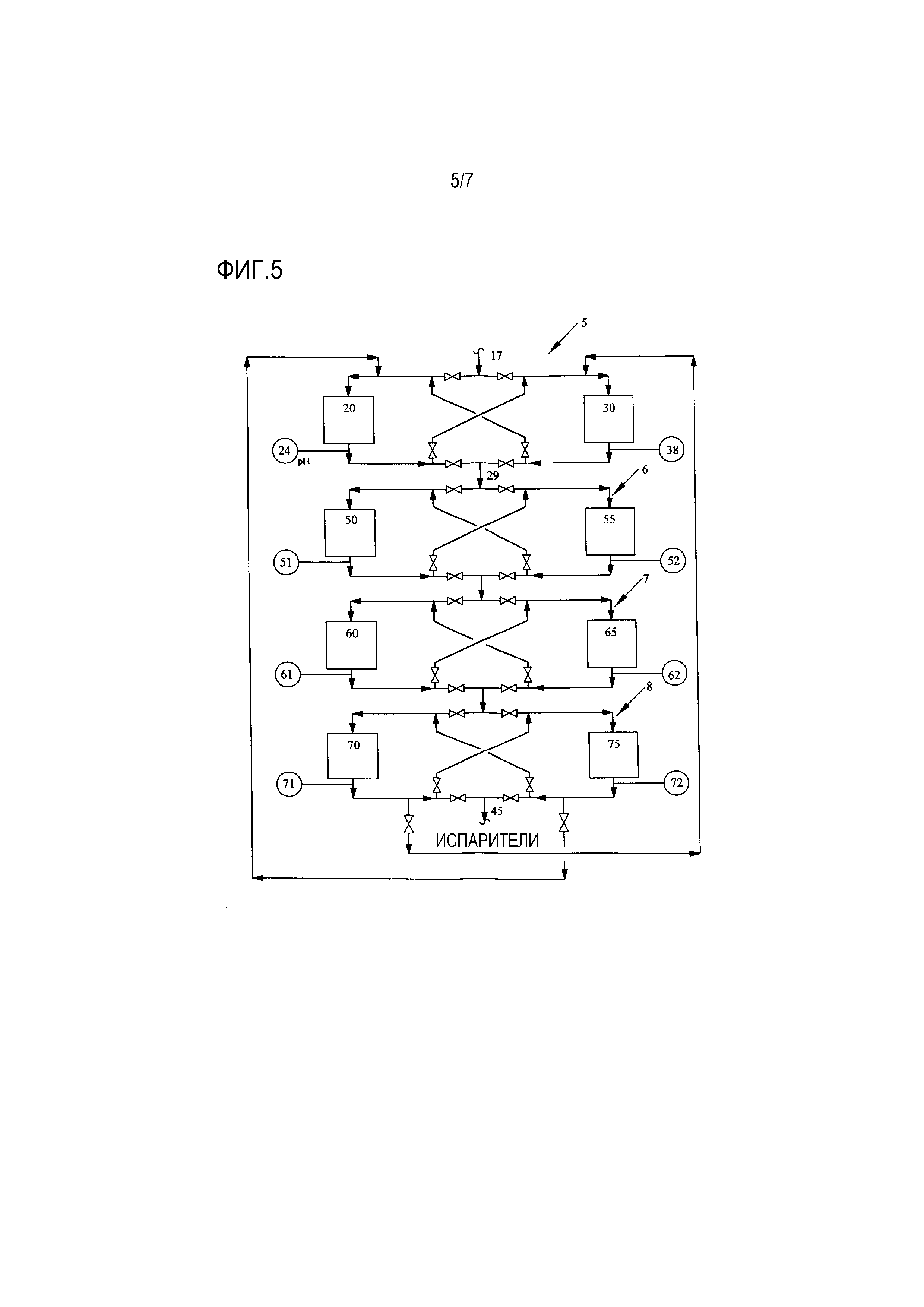
[0021] Фиг. 5 представляет собой упрощенную технологическую схему, показывающую два ряда слоев ионообменной обработки, выполненных в соответствии с настоящим изобретением, обеспечивающую связь по текучей среде между параллельными слоями ионообменной обработки;
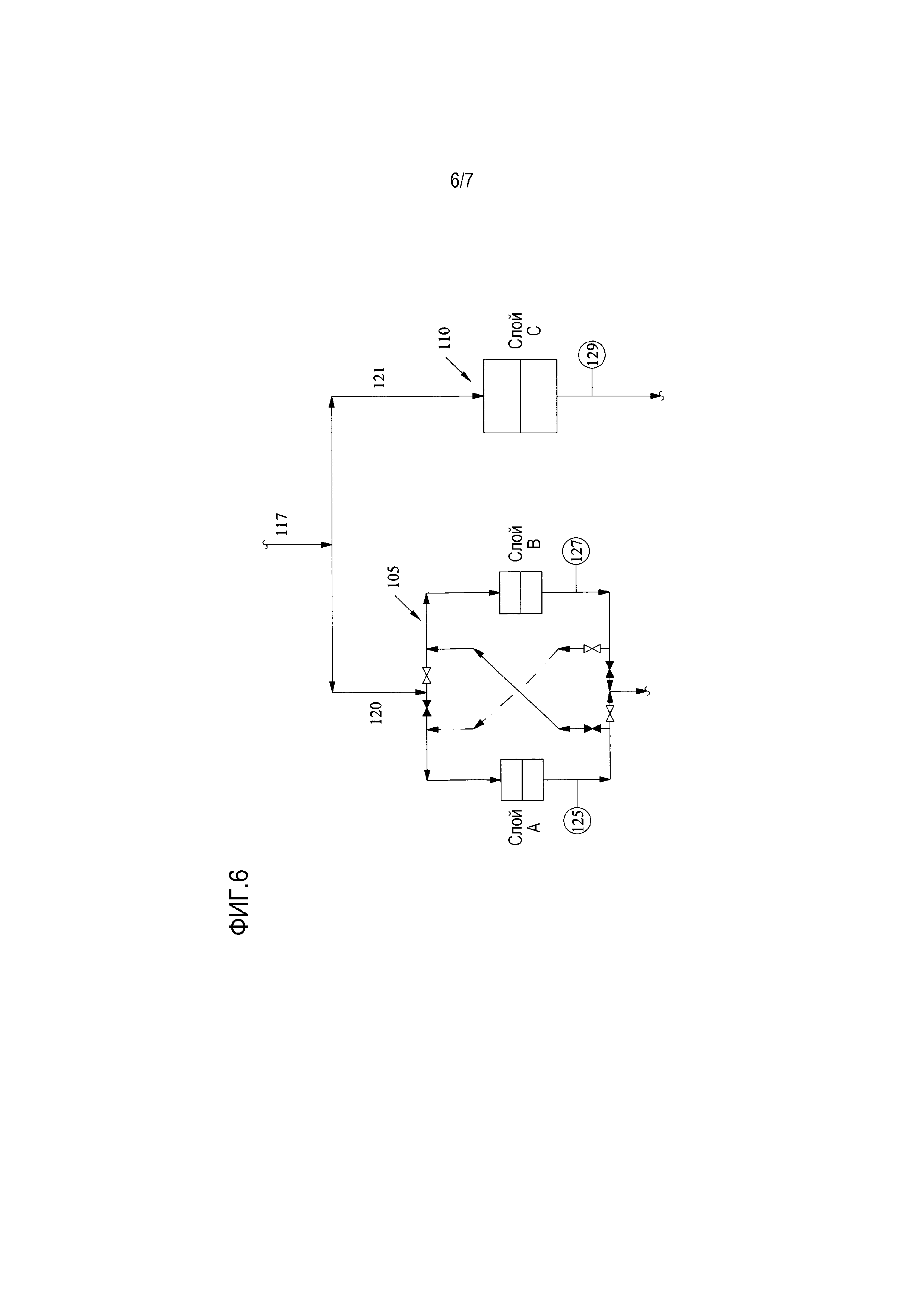
[0022] Фиг. 6 представляет собой упрощенную технологическую схему, показывающую примерные способы в соответствии с предшествующим уровнем техники и в соответствии с настоящим изобретением для обработки водного потока с помощью слоев ионообменной обработки; и
[0023] Фиг. 7 демонстрирует улучшение срока службы блока ионообменной обработки в соответствии с настоящим изобретением с графиками водного раствора и обработанного водного раствора в зависимости от времени и объема слоя для водного раствора, который был обработан каждым блоком обработки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0024] Все части, проценты и доли, используемые в настоящем документе, являются объемными, если явно не указано иное. Все документы, процитированные в настоящем документе, включены в него посредством ссылки.
[0025] Под «ионообменной смолой» подразумевается любая обычная ионообменная смола, известная специалисту в данной области техники и являющаяся легко доступным товаром.
[0026] В настоящем изобретении было обнаружено, что частота циклов регенерации может быть уменьшена, а качество продукта улучшено за счет одновременной работы первого и второго слоев ионообменной обработки. При обычной работе в соответствии с предшествующим уровнем техники слой ионообменной обработки эксплуатировался до «истощения» (как описано ниже), после чего он отключался и регенерировался, в то время как второй слой ионообменной обработки помещался в реальном масштабе времени на его место. Регенерированный слой оставался отключенным вплоть до истощения второго слоя.
[0027] В отличие от этого, в настоящем изобретении первый и второй слои ионообменной обработки эксплуатируются одновременно. Один вариант осуществления этого включает в себя первый и второй слои ионообменной обработки в конфигурации «опережения/запаздывания». Как только «проскок» (как описано ниже) наблюдается на выходе первого слоя ионообменной обработки («опережающего» слоя), подключается второй слой ионообменной обработки («запаздывающий» слой) (см. Фиг. 3), причем содержащий примеси раствор проходит через оба слоя ионообменной обработки. Это значительно снижает частоту регенерации, и таким образом уменьшает расход деминерализованной воды и регенерационных химикатов, производя при этом меньше сточных вод.
[0028] Альтернативно первый слой ионообменной обработки и второй слой ионообменной обработки могут работать одновременно до проскока на выходе первого слоя ионообменной обработки. В этом случае небольшие утечки из первого слоя ионообменной обработки, которые могут произойти даже перед проскоком, наблюдаются, будут улавливаться вторым слоем ионообменной обработки, в то время как емкость второго слоя ионообменной обработки будет в значительной степени сохранена для более позднего использования в цикле обслуживания после того, как первый слой ионообменной обработки приблизится к истощению.
[0029] В дополнение к описанной выше эффективности процесса одновременное использование первого и второго слоев ионообменной обработки приводит к важному эффекту. В настоящем изобретении было определено, что поддержание уровней примесей в водном потоке (предпочтительно этот водный поток представляет собой отводной поток из бедной рециркуляционной воды) ниже концентрации примесей, измеряемой по удельной электропроводности менее 5 мкСм/см, приводит к превосходному качеству продукта и уменьшает порчу оборудования, например, за счет коррозии, вызываемой высоким содержанием примесей в водном потоке.
[0030] Использование настоящего изобретения будет теперь описано более подробно в качестве компонента процесса производства оксида этилена.
[0031] Оксид этилена производится путем непрерывного контакта кислородсодержащего газа с олефином, предпочтительно этиленом, в присутствии катализатора оксида этилена («эпоксидирования») (описываемого более подробно ниже). Кислород может подаваться в реакцию по существу в чистой молекулярной форме или в смеси, такой как воздух. В качестве примера, типичные смеси исходных реагентов в рабочих условиях могут содержать от приблизительно 0,5% до приблизительно 45%, предпочтительно от приблизительно 5% до приблизительно 30% этилена и от приблизительно 3% до приблизительно 15% кислорода, с остатком, содержащим сравнительно инертные материалы, включая такие вещества, как диоксид углерода, вода, инертные газы, другие углеводороды и описанные в настоящем документе замедлители реакции. Неограничивающие примеры инертных газов включают в себя азот, аргон, гелий, а также их смеси. Неограничивающие примеры других углеводородов включают в себя метан, этан, пропан, а также их смеси. Диоксид углерода и вода являются побочными продуктами процесса эпоксидирования, а также обычными загрязняющими примесями в исходных газах. Оба они оказывают неблагоприятное воздействие на катализатор, поэтому концентрации этих компонентов обычно поддерживаются на минимальном уровне.
[0032] Как упоминалось ранее, в реакции также присутствуют один или несколько замедлителей реакции, неограничивающие примеры которых включают в себя органические галогенсодержащие соединения, такие как галогензамещенные углеводороды C1 - C8; причем особенно предпочтительными являются хлорсодержащие замедлители, такие как метилхлорид, этилхлорид, этилендихлорид, винилхлорид или их смеси. Контроль уровня концентрации хлорида особенно важен для ренийсодержащих катализаторов.
[0033] Как было упомянуто выше, обычный способ для процесса эпоксидирования этилена содержит парофазное окисление этилена молекулярным кислородом в присутствии катализатора эпоксидирования в трубчатом реакторе неподвижного слоя. Коммерческие этиленоксидные реакторы с неподвижным слоем обычно имеют форму множества параллельных удлиненных трубок (в подходящей оболочке) с наружным диаметром приблизительно от 0,7 до 2,7 дюймов, внутренним диаметром от 0,5 до 2,5 дюймов и длиной 15-53 фута, каждая из которых заполнена катализатором. Исходная реакционная смесь (описанная выше) вводится в эти трубы, и получаемый вытекающий реакторный газ содержит оксид этилена, неиспользованные реагенты и побочные продукты.
[0034] Только в целях иллюстрации ниже приведены условия, которые часто используются в современных коммерческих реакторных установках для получения оксида этилена: часовая объемная скорость газа (GHSV) 1500-10000 час-1, давление на входе в реактор 150-400 фунт/кв.дюйм изб., температура охладителя 180-315°C, степень конверсии кислорода 10-60%, и производительность по оксиду этилена (рабочая скорость) 7-20 фунтов/куб.фут катализатора/час. Состав питания на входе в реактор после завершения запуска и во время нормального функционирования обычно содержит (в об.%) 1-40% этилена, 3-12% O2; 0,3-20%, предпочтительно 0,3-5%, более предпочтительно 0,3-1% CO2; 0-3% этана, некоторое количество одного или более хлоридных замедлителей, которые описаны в настоящем документе; и остаток, состоящий из аргона, метана, азота или их смесей.
[0035] Типичные эксплуатационные условия для процесса эпоксидирования этилена включают в себя температуры в диапазоне от приблизительно 180°C до приблизительно 330°C, предпочтительно от приблизительно 200°C до приблизительно 325°C и более предпочтительно от приблизительно 225°C до приблизительно 280°C. Рабочее давление может варьироваться от приблизительно атмосферного давления до приблизительно 30 атм в зависимости от желаемой массовой скорости и производительности. В рамках настоящего изобретения могут использоваться более высокие давления. Время пребывания в реакторах промышленного масштаба обычно составляет от приблизительно 2 до приблизительно 20 с.
[0036] Прореагировавшая смесь вытекает из выходного отверстия реактора, охлаждается и течет к скрубберной колонне оксида этилена, где она контактирует с бедной рециркуляционной водой для абсорбции оксида этилена. Выходящая из скрубберной колонны жидкость (богатая рециркуляционная вода) подается затем в отпарную колонну для извлечения оксида этилена. В этой отпарной колонне оксид этилена отгоняется, и пары оксида этилена из верха колонны направляются во второй абсорбер оксида этилена. Жидкая вода вытекает из куба отпарной колонны («бедная рециркуляционная вода»), и отводимый поток отбирается из бедной рециркуляционной воды до того, как бедная рециркуляционная вода будет возвращена в скруббер оксида этилена, где она используется для абсорбирования оксида этилена. Этот отводимый поток содержит приблизительно 0,2-20 мас.% этиленгликоля, приблизительно 80-99,7 мас.% воды и приблизительно от 100 частей на миллион до 1,0 мас.% примесей. Примеси могут включать в себя, среди прочего, альдегидные примеси, такие как формальдегид, уксусный альдегид, гликолевый альдегид, их связанные кислоты и ионы, а также продукты их реакции, такие как сложные эфиры с длинными цепочками, которые получаются в результате реакции альдегидных примесей с оксидом этилена и/или этиленгликолем. Этот водный поток отправляется затем в блок очистки бедной рециркуляционной воды. Фиг. 1 показывает блок 1 очистки бедной рециркуляционной воды предшествующего уровня техники, содержащий два ряда 2, 3, каждый из которых содержит множество слоев ионообменной обработки (показанных как прямоугольники на Фиг. 1), расположенных последовательно для обработки водного потока 17, который представляет собой отводимый поток, описанный выше. Каждый ионообменный слой выбирается из анионного или катионного ионообменного слоя, или возможно вместо этого замещается другим слоем адсорбента из другого материала, как описано в настоящем документе. В процессе предшествующего уровня техники, показанном на Фиг. 1, ионообменные слои эксплуатируются строго как ряд, а не индивидуально. Таким образом, в то время как все ионообменные слои одного ряда находятся в активной работе, другие слои отключены, и либо регенерируются, либо находятся в резерве.
[0037] Фиг. 2 показывает блок 5 обработки в соответствии с настоящим изобретением с двумя слоями 20, 30 ионообменной обработки. На Фиг. 2 клапан 12 открыт, в то время как клапан 15 закрыт, так что водный поток 17, содержащий этиленгликоль и вышеописанные примеси, проходит через трубопровод 19 и входит в первый слой 20 ионообменной обработки, где он контактирует с ионообменной смолой для того, чтобы уменьшить концентрацию примесей. Водный поток контактирует с этой ионообменной смолой при температурах от приблизительно 30°C до приблизительно 50°C, хотя могут использоваться более высокие или более низкие температуры. Атмосферное давление является предпочтительным, но более высокие давления также могут использоваться в зависимости от того, является ли желательным перепад давления в следующем обрабатывающем блоке. Иллюстративные объемные скорости потока составляют приблизительно 1-10 объемов раствора на объем смолы в час, хотя они могут значительно различаться. Анализатор 24, расположенный на выходе 22 первого слоя 20 ионообменной обработки, измеряет удельную электропроводность, значение pH или коэффициент пропускания УФ-излучения водного потока 22 из первого слоя ионообменной обработки, чтобы определить приблизительную концентрацию ионов и примесей на выходе 22. Эти методики, которые более подробно обсуждаются ниже, используются для определения того, произошел ли «проскок» примесей. Проскок происходит, когда многие или большинство функциональных групп на активных центрах ионообменной смолы заменяются целевыми ионами или примесями, и таким образом слой ионообменной обработки больше не обладает достаточной способностью поглощать все примеси из водного потока, оставляя некоторую измеримую концентрацию «незахваченных» примесей для прохождения через ионообменную смолу в выходной поток. Поскольку некоторая небольшая концентрация примесей всегда может проходить через слой ионообменной обработки, в настоящем изобретении проскок определяется не при нулевой концентрации примесей или ионов или нулевой измеренной удельной электропроводности, а тогда, когда через слой ионообменной обработки проходят достаточные концентрации примесей, приводящие к удельной электропроводности в водном потоке более чем 5 мкСм/см.
[0038] Фиг. 2 показывает случай, когда такой проскок не произошел; в этом случае после определения того, что выход первого слоя ионообменной обработки имеет удельную электропроводность меньше чем приблизительно 5 мкСм/см (альтернативно, для определения того, произошел ли проскок, можно использовать измерение пропускания УФ-излучения или рН); клапан 25 остается открытым, а клапан 27 закрытым, так что водный поток проходит через выпускное отверстие 22 из первого слоя 20 ионообменной обработки и вытекает как выходной поток 29 из блока 5 обработки (пунктирные линии на прилагаемых чертежах показывают трубопроводы и блоки ионообменной обработки, которые отключаются, когда один или более клапанов являются закрытыми для того, чтобы предотвратить связь по текучей среде). После выхода из блока 5 обработки водный поток может быть подвергнут дальнейшей обработке или предпочтительно направлен в секцию очистки этиленгликоля установки для получения оксида этилена/этиленгликоля. Например, если слой 20 ионообменной обработки блока 5 обработки содержит катионную смолу, водный поток может затем течь к блоку обработки, содержащему анионные смолы. Если, с другой стороны, содержание примесей было в достаточной степени уменьшено, так что водный поток затем может быть дополнительно обработан без ущерба для установки или качества продукта, то этот водный поток может быть отправлен обратно к установке, например, к секции очистки этиленгликоля. Как показано на Фиг. 2, второй слой ионообменной обработки обходится и отсоединен от любого циркулирующего сырья или потоков, и в этом состоянии второй слой ионообменной обработки может просто находиться в режиме ожидания, будучи готовым для возможного использования, или, альтернативно, может находиться в процессе регенерации после истощения предыдущим использованием.
[0039] В отличие от Фиг. 2, Фиг. 3 показывает конфигурацию блока 5 обработки после того, как измеренная удельная электропроводность на выходе 22 превысила приблизительно 5 мкСм/см, указывая на то, что произошел проскок. Поскольку проскок указывает на то, что одного только первого слоя ионообменной обработки больше недостаточно для того, чтобы уменьшить концентрацию ионов и примесей до желаемых уровней, клапан 25 закрывается, и клапан 27 открывается, так что водный поток проходит через выход 22 первого слоя ионообменной обработки, течет через трубопроводы 32-33, а затем поступает через вход 34 во второй слой 30 ионообменной обработки, где он контактирует с ионообменной смолой для того, чтобы уменьшить концентрацию примесей во входном потоке и получить обработанный поток 36. Давление, температура и объемная скорость потока, с которыми водный поток контактирует с ионообменной смолой во втором слое 20 ионообменной обработки, находятся в тех же самых диапазонах, которые были указаны выше для первого слоя 30 ионообменной обработки.
[0040] Обработанный поток измеряется вторым анализатором 38, который так же как и анализатор 24 может выполнять измерения пропускания ультрафиолетового излучения, значения pH, удельной электропроводности, или любое другое подходящее измерение для определения концентрации обработанного потока 36. Затем поток течет через открытый клапан 40 на выход 29. После прохождения через выход 29, как было описано выше, в зависимости от измеренного содержания примесей и предпочтительного порядка ионообменной обработки, поток может течь к другому блоку обработки или может быть возвращен в установку.
[0041] Как было упомянуто выше, Фиг. 3 показывает ситуацию, в которой произошел проскок в первом слое 20 ионообменной обработки, но этот слой еще не истощен и не нуждается в регенерации благодаря доступности второго слоя для захвата примесей. Когда измеренная удельная электропроводность (или эквивалентное значение pH или пропускания ультрафиолетового излучения) в любом анализаторе 24, 38 составляет больше чем приблизительно 20 мкСм/см, предпочтительно больше чем приблизительно 100 мкСм/см (или, альтернативно, когда уменьшение измеренной удельной электропроводности составляет 30-70% относительно начальной удельной электропроводности), тогда первый ионообменный слой 20 достиг истощения и должен быть регенерирован. Это показано на Фиг. 4.
[0042] Фиг. 4 представляет по существу зеркальное отражение Фиг. 2. В блоке 5 обработки, изображенном на Фиг. 4, клапан 12 закрыт, а клапан 15 открыт, так что первый слой 20 ионообменной обработки обходится, и поток воды течет через 17, через входное отверстие 32, а затем входит во второй ионообменный слой 30, где контактирует с ионообменной смолой для уменьшения концентрации примесей. Удельная электропроводность (или значение pH, или пропускание ультрафиолетового излучения) водного потока на выходе 36 второго слоя ионообменной обработки измеряется вторым анализатором 38, и при условии, что это измерение не показывает проскока на выходе, поток направляется через открытый клапан 40 и через выход 29 для дальнейшей обработки или дальнейшего использования в процессе производства оксида этилена/этиленгликоля, как было описано выше.
[0043] В то время как первый слой 20 ионообменной обработки обходится в блоке 5 обработки, как показано на Фиг. 4, он может одновременно регенерироваться или просто находиться в резерве.
[0044] Фиг. 3 может также представлять альтернативный вариант осуществления. В этом варианте осуществления блок обработки конфигурируется так, чтобы водный поток проходил как через первый, так и через второй слои ионообменной обработки 20, 30. (В дополнение к этим явно показанным двум слоям ионообменной обработки, также могут использоваться один или более дополнительных слоев ионообменной обработки). В этом случае слой 30 ионообменной обработки эффективно действует как блок доочистки для улавливания небольших утечек примесей из первого слоя ионообменной обработки даже до проскока первого слоя ионообменной обработки. В этой конфигурации клапан 25 закрывается, а клапан 27 открывается, так что водный поток проходит через выход 22 первого слоя ионообменной обработки, течет через трубопроводы 32-33, а затем поступает через вход 34 во второй слой 30 ионообменной обработки, где он контактирует с ионообменной смолой для того, чтобы уменьшить концентрацию примесей во входном потоке и получить обработанный поток 36. Давление, температура и объемная скорость потока, с которыми водный поток контактирует с ионообменной смолой во втором слое 30 ионообменной обработки, находятся в тех же самых диапазонах, которые были указаны выше для первого слоя 30 ионообменной обработки. Использование первого и второго слоев ионообменной обработки вместе увеличивает время между регенерациями, потому что когда емкость первого слоя ионообменной обработки достигнута и он приближается к исчерпанию, второй слой ионообменной обработки все еще является относительно «свежим» и имеет достаточную емкость для того, чтобы абсорбировать дополнительные ионы и примеси. Когда измеренная удельная электропроводность превышает приблизительно 5 мкСм/см, блок 5 может быть отключен и регенерирован.
[0045] Как было упомянуто выше, после того, как обработанный поток выйдет из блока обработки через выход 29, будучи обработанным в первом ионообменном слое 20, втором ионообменном слое 30, или в обоих, поток может быть дополнительно обработан. В одном предпочтительном варианте осуществления настоящего изобретения первый ионообменный слой 20 и второй ионообменный слой 30 являются частями отдельных рядов слоев ионообменной обработки, расположенных последовательно, как показано на Фиг. 5, которая соответствует настоящему изобретению. Как видно на Фиг. 5, имеется множество блоков обработки, 5, 6, 7, 8 для обработки отводимого водного потока 17, как описано выше, состоящих из дополнительных слоев 50, 55, 60, 65, 70 и 75 ионообменной обработки и их соответствующих анализаторов 51, 52, 61, 62, 71 и 72. После прохождения через блоки обработки, показанные на Фиг. 5, поток наконец выходит через трубу 45 для дальнейшего использования в процессе производства оксида этилена/этиленгликоля, как описано выше.
[0046] На Фиг. 5 можно заметить, что конфигурация трубопровода, который обеспечивает связь по текучей среде между первым и вторым слоями ионообменной обработки, была расширена для обеспечения возможности связи по текучей среде между параллельными слоями ионообменной обработки. Фиг. 5 показывает, как конфигурация и взаимосвязь двух параллельных ионообменных слоев может быть расширена для того, чтобы связать два полностью отдельных ряда. Следует отметить, что Фиг. 5 не показывает блок в состоянии фактической работы, поскольку клапаны не открываются и не закрываются, чтобы направлять водные потоки через выбранные слои обработки.
[0047] Ионообменные смолы
[0048] Как было обсуждено выше, настоящее изобретение может включать в себя одну или более смол для ионообменной обработки.Ионообменные смолы имеют полимерную матрицу, которая содержит на своей поверхности ионообменные площадки, заполненные ионными функциональными группами. Ионообменные смолы обычно делятся на катионообменные или анионообменные, хотя также доступны другие типы ионообменных смол.
[0049] Подходящие полимерные матрицы для ионообменного слоя включают в себя матрицу из полистирола, полиакриловую матрицу, полиалкиламиновую смолу, а также другие полимерные материалы. Предпочтительно, чтобы полимерная матрица была сшита с дивинилбензолом в достаточной степени для того, чтобы увеличить ее рабочую емкость, при этом также не увеличивая плотность ионообменного материала до такой степени, что он становится слишком физически твердым и слишком химически стойким к химической обработке. Предпочтительно матрица представляет собой сополимер стирола и дивинилбензола. В дополнение к вышеупомянутым структурам, также могут использоваться альтернативные материалы и структуры, такие как макропористые смолы и естественные ионообменные материалы, такие как глинистые и цеолитные минералы.
[0050] На активных центрах ионообменной смолы располагаются ионные функциональные группы, которые определяют, будет ли функционировать смола как катионообменный или анионообменный слой. Сильно кислотные катионообменные смолы обычно включают в себя сульфоновые группы. Примеры сильно кислотных сульфоновых катионообменных смол включают в себя среди прочих Amberlite IR 120, Dowex HCR, Lewatit S 100 и Amberlyst 15. Слабо кислотные катионообменные смолы обычно включают в себя карбоксильные группы. Примеры слабо кислотных катионообменных смол включают в себя среди прочих Amberlite IRC 86 и Lewatit CNP. Дополнительные примеры подходящих катионообменных смол включают в себя Tulsion T56MP и TG 057 производства компании Thermax LTD, Пуна, Индия.
[0051] Подходящие анионообменные смолы включают в себя хлорметилированный полистирол, который может иметь различные степени основности и включить в себя различные марки Amberlite, такие как IRA 402, IRA 410 и IRA 96. Слабые анионообменные смолы могут также включать в себя полиакриловые смолы, снабженные функциональными группами путем реакции с полифункциональным амином с образованием анионообменных смол, таких как третичные слабоосновные IRA Amberlite 67 и Amberlyst A21, или затем дополнительно обработанные хлорметаном или диметилсульфатом, чтобы получить четвертичноаминовую сильноосновную смолу IRA 458. Дополнительные примеры подходящих анионообменных смол включают в себя Tulsion A8X MP и A9X MP производства компании Thermax LTD, а также анионообменные смолы, раскрытые в патенте US № 6187973, включенный в настоящий документ посредством ссылки.
[0052] В дополнение к указанным выше ионообменным материалам может быть использована любая другая подходящая катионообменная или анионообменная смола, такая как катионо- и анионообменные смолы, описанные в публикации de Dardel, F. and Arden, T. V. 2008, Ion Exchanger, in Ullman's Encyclopedia of Industrial Chemistry.
[0053] Регенерация ионообменного слоя
[0054] Как было упомянуто выше, когда они не используются, слои ионообменной обработки могут либо находиться в режиме резервирования, либо подвергаться регенерации в случае истощения. Регенерация осуществляется путем промывки твердого адсорбента и ионообменной смолы регенератором для удаления и вытеснения примесей и ионов, адсорбированных смолой, и пополнения активных центров на смоле соответствующими противоионами, выделенными смолой в раствор, восстанавливая таким образом способность смолы улавливать и адсорбировать ионы и примеси. Во время регенерации эти примеси, ионы и другие органические вещества элюируют и высвобождаются в жидкий регенератор. Регенератор должен быть специально выбран для пополнения ионов на поверхности ионообменной смолы - например, регенератор из гидроксида натрия является предпочтительным для регенерации анионного слоя с гидроксидными функциональными группами, в частности для пополнения гидроксида, который высвобождается смолой во время работы. Точно так же для катионной смолы с функциональными группами иона водорода регенерация обычно достигается путем использования HCl или серной кислоты в качестве регенератора. Другие типичные регенераторы включают в себя сульфат натрия.
[0055] Обычно регенерация сопровождается циклом промывки деминерализованной водой для вытеснения регенератора из ионообменного слоя. Эта промывка может выполняться в одном или более циклах, причем каждый цикл может выполняться при различных параметрах, таких как объемная скорость потока. По завершении стадии промывки ионообменный слой не должен содержать регенератора.
[0056] В дополнение к вышеупомянутым слоям ионообменной обработки, вакуумный дегазатор (не показан) может использоваться для уменьшения содержания свободного диоксида углерода в потоке до уровней в несколько частей на миллион.
[0057] Дегазатор
[0058] Система может быть спроектирована с вакуумным дегазатором или без него в зависимости от предпочтений пользователя. Дегазатор удаляет диоксид углерода из водного потока. Поскольку диоксид углерода является относительно слабой кислотой, при его удалении из водного потока можно использовать слабое основание для удаления остальной части примесей вместо сильного основания, поскольку не нужно удалять диоксид углерода из водного потока. Использование смолы слабого основания, а не смолы сильного основания уменьшает количество химикатов, необходимых для регенерации. Таким образом, дегазатор увеличивает капитальные затраты (за счет стоимости дегазатора), но уменьшает эксплуатационные расходы (за счет меньшего количества химикатов для регенерации). Таким образом, выбор одного из этих вариантов остается за индивидуальным конечным пользователем.
[0059] Блоки анализатора
[0060] Анализаторы размещаются в различных выходных потоках, чтобы обеспечить непрерывное измерение в реальном масштабе времени эффективности удаления примесей слоями ионообменной обработки. Как было упомянуто выше, любая подходящая аналитическая методика может использоваться в настоящей заявке, включая измерение удельной электропроводности, значения pH или УФ-пропускания выходного потока.
[0061] Удельная электропроводность может использоваться для измерения концентрации примесей в потоке, потому что вода, как неэлектролит, обычно является плохим проводником. Однако в результате электролитической диссоциации кислоты, основания и другие примеси диссоциируют в воде на ионы, что позволяет определять содержание электролита в воде. Таким образом, сильные и слабые электролиты можно дифференцировать по степени их диссоциации. В общих чертах, удельная электропроводность раствора электролита зависит от: (1) концентрации ионов в растворе; (2) валентности иона; и (3) скорости миграции или мобильности ионов. Для отдельных видов электролитов при постоянной температуре удельная электропроводность является линейной функцией концентрации электролита, поскольку валентность и мобильность ионов являются постоянными. Таким образом, использование этих известных принципов для измерения концентрации примесей в водном потоке находится в пределах компетенции специалиста в данной области техники.
[0062] Пропускание ультрафиолетового излучения также может использоваться для измерения концентрации примесей в водном растворе этиленгликоля. Если водный раствор этиленгликоля не соответствует определенному минимальному проценту пропускания УФ-излучения, то его качество будет недостаточным для производства волокна, и, следовательно, его ценность будет значительно снижена. Значение пропускания УФ-излучения в водном растворе гликоля при проскоке является следующим:
Таблица I
[0063] Когда коэффициент пропускания УФ-излучения обработанного водного раствора этиленгликоля начинает приближаться к этим минимальным значениям, это указывает на то, что способность ионообменных смол установки очистки бедной рециркуляционной воды адсорбировать примеси из водного раствора этиленгликоля исчерпана, и они должны быть регенерированы. Как и измерение значения pH, измерение пропускания УФ-излучения является полезным для измерения концентрации неионных примесей, таких как альдегидные примеси, которые обязательно должны удаляться. Измерение пропускания УФ-излучения также является полезным измерением, поскольку оно является типичной спецификацией продукта для этиленгликоля - выходящий из блока обработки поток, содержащий этиленгликоль, который соответствует требуемому уровню пропускания УФ-излучения, будет положительно влиять на конечный очищенный продукт этиленгликоля, соответствующий этой спецификации.
[0064] После обработки в установке для очистки бедной рециркуляционной воды обработанный водный поток предпочтительно направляется в секцию очистки гликоля, где этиленгликоль отделяется от очищенной воды (и очищенного гликоля), и не содержащая гликоля вода направляется в качестве рециркуляционной воды в гликолевые реакторы.
[0065] Катализатор эпоксидирования на основе серебра
[0066] Катализатор эпоксидирования на основе серебра включает в себя носитель и по меньшей мере каталитически эффективное количество серебра или содержащего серебро соединения; также опционально присутствует каталитическое количество рения или содержащего рений соединения; также опционально присутствует каталитическое количество одного или более щелочных металлов или содержащих щелочной металл соединений. Носитель, используемый в настоящем изобретении, может быть выбран из большого количества твердых огнеупорных носителей, которые могут быть пористыми и могут обеспечить предпочтительную пористую структуру. Хорошо известно, что глинозем полезен в качестве носителя катализатора для эпоксидирования олефина и является предпочтительным носителем.
[0067] Независимо от характера используемого носителя он обычно имеет форму частиц, кусков, кусочков, гранул, колец, сфер, колес, полых цилиндров с поперечными перегородками и т.п., с размерами, подходящими для его использования в реакторе эпоксидирования неподвижного слоя. Частицы носителя предпочтительно будут иметь эквивалентные диаметры в диапазоне от приблизительно 3 мм до приблизительно 12 мм, и более предпочтительно в диапазоне от приблизительно 5 мм до приблизительно 10 мм (эквивалентный диаметр - это диаметр сферы, имеющей такое же отношение внешней поверхности (без учета поверхности в порах частицы) к объему, что и у используемых частиц носителя). Подходящие носители доступны от компаний Saint-Gobain Norpro Co., Sud Chemie AG, Noritake Co., CeramTec AG и Industrie Bitossi S.p. A. Не ограничиваясь приведенными конкретными композициями и составами, дополнительную информацию о композициях носителей и способах их изготовления можно найти в патентной публикации US №2007/0037991.
[0068] Для того, чтобы произвести катализатор для окисления олефина до оксида олефина, носитель, имеющий вышеупомянутые характеристики, снабжается затем каталитически эффективным количеством серебра на его поверхности. В одном варианте осуществления каталитическое эффективное количество серебра составляет от 10 мас.% до 45 мас.%. Катализатор готовится путем пропитки носителя соединением серебра, комплексным соединением или солью серебра, растворенными в подходящем растворителе в количестве, достаточном для того, чтобы вызвать осаждение соединения прекурсора серебра на носителе. Предпочтительно используется водный раствор серебра.
[0069] Каталитическое количество рениевого компонента, который может быть содержащим рений соединением или содержащим рений комплексным соединением, также может быть осаждено на носитель до, одновременно или после осаждения серебра. Рениевый катализатор может присутствовать в количестве от приблизительно 0,001 мас.% до приблизительно 1 мас.%, предпочтительно от приблизительно 0,005 мас.% до приблизительно 0,5 мас.%, и более предпочтительно от приблизительно 0,01 мас.% до приблизительно 0,1 мас.% по полной массе катализатора, включая носитель, в пересчете на металлический рений.
[0070] Другие компоненты, которые также могут быть осаждены на носитель до, одновременно или после осаждения серебра и рения, представляют собой каталитические количества щелочного металла или смеси двух или более щелочных металлов, а также необязательное каталитическое количество компонента щелочноземельного металла Группы IIA или смеси двух или более компонентов щелочноземельного металла Группы IIA, и/или компонента переходного металла или смеси двух или более компонентов переходного металла, все из которых могут иметь форму ионов металлов, соединений металлов, комплексных соединений металлов и/или солей металлов, растворенных в подходящем растворителе. Носитель может пропитываться различными катализаторами одновременно или на отдельных стадиях. Конкретная комбинация носителя, серебра, катализатора (катализаторов) из щелочного металла, рениевого компонента и опционального дополнительного катализатора (катализаторов) настоящего изобретения будет обеспечивать улучшение одного или более каталитических свойств по сравнению с той же самой комбинацией серебра и основания, не содержащей ни одного, или содержащей только один из этих катализаторов.
[0071] Используемый в настоящем документе термин «каталитическое количество» некоторого компонента катализатора относится к такому количеству этого компонента, которое улучшает эффективность катализатора по сравнению с отсутствием этого компонента. Точные используемые концентрации, конечно же, будут зависеть, среди других факторов, от желаемого содержания серебра, природы носителя, вязкости жидкости и растворимости конкретного соединения, используемого для доставки этого катализатора в пропиточный раствор. Примеры каталитических свойств включают в себя, среди прочего, работоспособность (устойчивость к уносу), селективность, активность, конверсию, стабильность и выход. Специалисту в данной области техники должно быть понятно, что одно или более индивидуальных каталитических свойств могут быть улучшены «каталитическим количеством», в то время как другие каталитические свойства могут или не могут быть улучшены или даже могут ухудшиться.
[0072] Подходящие катализаторы из щелочного металла могут быть выбраны из лития, натрия, калия, рубидия, цезия или их комбинаций, причем цезий является предпочтительным, и комбинации цезия с другими щелочными металлами являются особенно предпочтительными. Количество щелочного металла, осажденного или присутствующего на носителе, должно быть каталитическим количеством. Предпочтительно это количество варьируется от приблизительно 10 частей на миллион до приблизительно 3000 частей на миллион, более предпочтительно от приблизительно 15 частей на миллион до приблизительно 2000 частей на миллион, еще более предпочтительно от приблизительно 20 частей на миллион до приблизительно 1500 частей на миллион, и особенно предпочтительно от приблизительно 50 частей на миллион до приблизительно 1000 частей на миллион по полной массе катализатора в пересчете на металл.
[0073] Подходящие катализаторы из щелочноземельного металла содержат элементы Группы IIA Периодической таблицы элементов, которые могут быть бериллием, магнием, кальцием, стронцием и барием или их комбинациями. Подходящие катализаторы из переходного металла могут содержать элементы Групп IVA, VA, VIA, VIIA и VIIIA Периодической таблицы элементов, а также их комбинации.
[0074] Количество катализатора (катализаторов) из щелочноземельного металла и/или переходного металла, осажденного на носителе, является каталитическим количеством. Катализатор из переходного металла обычно может присутствовать в количестве от приблизительно 0,1 мкмоль/г до приблизительно 10 мкмоль/г, предпочтительно от приблизительно 0,2 мкмоль/г до приблизительно 5 мкмоль/г.
[0075] Раствор серебра, используемый для пропитки носителя, может также содержать необязательный растворитель или комплексообразующий/солюбилизирующий агент, известный в данной области техники. Большое разнообразие растворителей или комплексообразующих/солюбилизирующих агентов может быть использовано для растворения серебра до желаемой концентрации в пропитывающей среде. Полезные комплексообразующие/солюбилизирующие агенты включают в себя амины, аммиак, щавелевую кислоту, молочную кислоту, а также их комбинации. Амины включают в себя алкилендиамин, имеющий от 1 до 5 атомов углерода. В одном предпочтительном варианте осуществления раствор представляет собой водный раствор оксалата серебра и этилендиамина. Комплексообразующий/солюбилизирующий агент может присутствовать в пропиточном растворе в количестве от приблизительно 0,1 до приблизительно 5,0 моль на моль серебра, предпочтительно от приблизительно 0,2 до приблизительно 4,0 моль, и более предпочтительно от приблизительно 0,3 до приблизительно 3,0 моль на каждый моль серебра.
[0076] Когда используется растворитель, он может быть органическим растворителем или водой, и может быть полярным или по существу или полностью неполярным. Как правило, растворитель должен обладать достаточной сольватирующей способностью для растворения компонентов раствора. В то же время предпочтительно, чтобы растворитель был выбран таким образом, чтобы избежать чрезмерного влияния или взаимодействия с сольватированными катализаторами. Предпочтительными являются органические растворители, которые имеют от 1 до приблизительно 8 атомов углерода в молекуле. Смеси из нескольких органических растворителей или смеси органического растворителя (растворителей) с водой могут использоваться при условии, что такие смешанные растворители функционируют в соответствии с требованиями настоящего изобретения.
[0077] Концентрация серебра в пропиточном растворе обычно находится в диапазоне от приблизительно 0,1 мас.% до максимальной растворимости, которую обеспечивает конкретная используемая комбинация растворитель/солюбилизирующий агент. Как правило, очень удобно использовать растворы, содержащие от 0,5% до приблизительно 45 мас.% серебра, причем предпочтительными являются концентрации серебра от 5 до 35 мас.%.
[0078] Пропитка выбранного носителя достигается с помощью любого из общепринятых способов; например, с помощью избыточной пропитки раствором, начальной пропитки влагой, нанесения покрытия распылением и т.д. Как правило, материал носителя контактирует с содержащим серебро раствором до тех пор, пока не поглотит достаточное количество раствора. Предпочтительно количество содержащего серебро раствора, используемого для пропитки пористого носителя, составляет не больше необходимого для того, чтобы заполнить поры носителя. Можно использовать одну пропитку или серию пропиток с промежуточной сушкой или без нее, в зависимости, в частности, от концентрации компонента серебра в растворе. Процедуры пропитки описаны, например, в патентах US №№ 4761394, 4766105, 4908343, 5057481, 5187140, 5102848, 5011807, 5099041 и 5407888. Можно использовать известные из предшествующего уровня техники процедуры предварительного осаждения, совместного осаждения и последующего осаждения различных катализаторов.
[0079] После пропитки носителя содержащим серебро соединением, то есть прекурсором серебра, рениевым компонентом, компонентом щелочного металла и необязательными другими промоторами, пропитанный носитель прокаливают в течение времени, достаточного для превращения содержащего серебро соединения в активные частицы серебра и удаления летучих компонентов из пропитанного носителя, что приводит к получению прекурсора катализатора. Кальцинирование может быть выполнено путем нагрева пропитанного носителя, предпочтительно постепенно, до температуры в диапазоне от приблизительно 200°C до приблизительно 600°C при давлении в диапазоне от приблизительно 0,5 до приблизительно 35 бар. Как правило, чем выше температура, тем короче требуемая продолжительность нагрева. Широкий диапазон продолжительностей нагрева был предложен в данной области техники; например, патент US № 3563914 раскрывает нагревание в течение менее 300 с, а патент US № 3702259 раскрывает нагревание в течение от 2 до 8 час при температуре от 100°C до 375°C, обычно в течение от приблизительно 0,5 до приблизительно 8 час. Однако важно только, чтобы время нагревания коррелировало с температурой, чтобы по существу все содержащееся серебро преобразовывалось в активные частицы серебра. С этой целью может использоваться непрерывное или ступенчатое нагревание.
[0080] Во время кальцинирования пропитанный носитель может подвергаться воздействию газовой атмосферы, содержащей инертный газ или смесь инертного газа с кислородсодержащим окисляющим компонентом в количестве от приблизительно 10 частей на миллион до 21% по объему. Для целей настоящего изобретения инертный газ определяется как газ, который по существу не реагирует с прекурсором катализатора или катализатором при условиях, выбранных для кальцинирования. Дополнительная информация о производстве катализатора может быть найдена в вышеупомянутой патентной публикации US № 2007/0037991.
[0081] ПРИМЕРЫ
[0082] Настоящее изобретение будет теперь описано более подробно с помощью следующих неограничивающих примеров.
[0083] Блок 105 обработки в соответствии с настоящим изобретением и сравнительный блок 110 обработки в соответствии с предшествующим уровнем техники показаны на Фиг. 6. Блок 105 обработки в соответствии с настоящим изобретением содержит слой A и слой B, соединенные последовательно, в то время как сравнительный блок 110 обработки предшествующего уровня техники имеет только один слой C. Все три слоя A, B, C имеют одну и ту же конструкцию с первой и второй камерами. Первая камера содержит катионообменную смолу Tulsion T56 MP, а вторая камера содержит анионообменную смолу Tuslion A8X MP.
[0084] Для того, чтобы проверить относительную эффективность этих блоков обработки для удаления примесей, водный поток 117, взятый в качестве отводного потока (как описано выше), получается с установки по производству оксида этилена. Этот водный поток содержит различные органические примеси, найденные в рециркуляционной воде процесса производства оксида этилена, такие как муравьиная кислота, уксусная кислота, формальдегид и т.п. Концентрация примесей в этом водном потоке, измеренные по удельной электропроводности, показаны крестиками на Фиг. 7. Как видно на Фиг. 7, средняя концентрация примесей, измеренная по удельной электропроводности в водном потоке перед обработкой, составляет приблизительно 102 мкСм/см (показано горизонтальной линией), с максимальным значением 144 мкСм/см и минимальным 60 мкСм/см. Водный поток 117 затем делится на идентичные первый и второй расщепленные потоки 120, 121, как показано на Фиг. 6, и первый расщепленный поток 120 направляется в блок обработки в соответствии с настоящим изобретением, в то время как второй расщепленный поток 122 направляется в сравнительный блок обработки в соответствии с предшествующим уровнем техники.
[0085] Блоки 105, 110 обработки удаляют примеси из первого и второго расщепленных потоков 120, 121. Концентрация примесей (измеренная по удельной электропроводности потоков) после абсорбции примесей на выходе из слоев измеряется анализаторами 125, 127 и 129, как показано на Фиг. 6. Измеренная концентрация (измеренная по удельной электропроводности) показана на Фиг. 7 для слоя А квадратиками (данные анализатора 125 на выходе из слоя A), для слоя B кружками (данные анализатора 127 на выходе из слоя B), и для слоя C треугольничками (данные анализатора 129 на выходе из слоя C). Так, например, по прошествии 76 час работы (в этот момент в процессе было пропущено приблизительно 205 объемов слоя водного отбираемого потока через каждый из слоев A, B и C) водный отбираемый поток, подаваемый из установки, имел удельную электропроводность 104 мкСм/см после разделения на расщепленные потоки 120, 121 и ввода в блоки обработки. Примеси удаляются из потоков в каждом блоке 105, 110 обработки. Так, на выходе из слоя А удельная электропроводность падала от 104 мкСм/см на входе до менее чем 1 мкСм/см. Поток, подаваемый к слою B, имеет такое низкое содержание примесей после обработки в слое А, что слой B должен выполнить лишь небольшую абсорбцию примесей или ионов (либо вообще нулевую), сохраняя таким образом свою абсорбционную емкость для более позднего использования в процессе. Соответственно, удельная электропроводность слабо изменяется после обработки в слое B, и расщепленный поток, выходящий из слоя B, покидает блок 105 обработки с окончательной измеренной удельной электропроводностью меньше чем 1 мкСм/см.
[0086] В то же самое время поток выходит из слоя C и покидает блок 110 обработки, имея удельную электропроводность, уменьшившуюся со 104 мкСм/см до 1,4 мкСм/см.
[0087] После приблизительно 106 час (когда количество обработанных объемов слоя составило приблизительно 285-300), ситуация значительно изменилась. Поток выходит из слоя А с удельной электропроводностью приблизительно 5 мкСм/см, что указывает на то, что слой A является теперь лишь частично эффективным для удаления примесей из потока, потому что его емкость уменьшилась и приближается к исчерпанию. Однако слой B продолжает уменьшать удельную электропроводность потока, выходящего из слоя B и блока 105 обработки до значения меньше чем 1, что означает, что блок обработки по настоящему изобретению не потерял эффективности удаления примесей из первого расщепленного потока 120.
[0088] Слой C и блок обработки предшествующего уровня техники работают аналогичным образом. Удельная электропроводность также составляет приблизительно 5 мкСм/см в этот момент времени. Но, как можно также заметить на Фиг. 7, слой C явно достиг исчерпания после 120 час работы (после обработки 320 объемов слоя в слое C), и удельная электропроводность обработанных потоков, проходящих через слой C, начинает резко расти - удельная электропроводность увеличивается и пересекает значение удельной электропроводности 20. В этой точке слой C больше не выполняет эффективного удаления примесей, и блок обработки предшествующего уровня техники теперь должен быть отключен и регенерирован.
[0089] Блок обработки в соответствии с настоящим изобретением, однако, продолжает эффективно удалять примеси. При более длинных периодах работы активные центры слоя А значительно насыщаются, и слой A становится неэффективным для удаления примесей, как показано высокой электропроводностью водных потоков, покидающих слой А (см. Фиг. 7). Например, после приблизительно 148 час работы (после обработки 400 объемов слоя в слое A), поток, входящий в слой A, имеет относительно высокое содержание примесей, на что указывает измеренная удельная электропроводность, равная почти 133. Слой A значительно уменьшает это содержание, но тем не менее измеренная удельная электропроводность потока, выходящего из слоя A, все еще остается неприемлемо высокой, будучи равной 75. Значения удельной электропроводности потока, выходящего из слоя A, увеличиваются примерно линейно на протяжении следующих нескольких дней, как показано аппроксимированной (штрихпунктирной) линией тренда. Однако слой B имеет достаточную емкость для абсорбирования примесей, которые не смог удержать слой A, и таким образом последовательно уменьшает значение удельной электропроводности получаемого потока до уровней ниже 5 мкСм/см, который, как было описано выше, был определен в настоящем изобретении как критический параметр для обеспечения эффективной работы установки и качества продукта. Слой A в состоянии уменьшить примеси так, чтобы измеренная удельная электропроводность потока, покидающего слой A, оставалась ниже 5 мкСм/см в течение всего экспериментального прогона, т.е. когда пример был завершен после обработки 580 объемов слоя.
[0090] В результате нет никакой необходимости отключать блок обработки в соответствии с настоящим изобретением для регенерации еще долгое время после того, как блок предшествующего уровня техники уже был отключен. Блок обработки предшествующего уровня техники должен быть отключен после приблизительно 120 час работы (после обработки 320 объемов слоя). В отличие от этого, блок обработки в соответствии с настоящим изобретением, все еще уменьшал содержание примесей в потоке в соответствии с конструктивным решением даже после обработки 580 объемов слоя. Таким образом, блок обработки в соответствии с настоящим изобретением может работать дольше между регенерациями ионообменного слоя. Как было упомянуто выше, увеличение времени между регенерациями значительно увеличивает эффективность процесса, одновременно снижая стоимость его эксплуатации.
[0091] Для специалиста в данной области техники будет очевидно, что различные модификации и вариации могут быть сделаны к вариантам осуществления, описанным в настоящем документе, без отступлений от духа или области охвата настоящего изобретения. Поэтому подразумевается, что настоящее изобретение не ограничивается конкретными раскрытыми вариантами осуществления, но покрывает все модификации в пределах духа и области охвата настоящего изобретения, определяемой приложенной формулой изобретения.
Реферат
Настоящее изобретение относится к вариантам способа окисления этилена для получения оксида этилена. Один из вариантов способа включает следующие стадии: обеспечение водного потока, содержащего этиленгликоль и примеси; введение этого водного потока в первый слой ионообменной обработки для уменьшения содержания примесей; определение того, имеет ли выход первого слоя ионообменной обработки удельную электропроводность больше чем 5 мкСм/см; при определении того, что выход первого слоя ионообменной обработки имеет удельную электропроводность больше чем 5 мкСм/см, подачу выхода первого слоя ионообменной обработки во второй слой ионообменной обработки; и при определении того, что выход первого слоя ионообменной обработки имеет удельную электропроводность от более чем 20 до 100 мкСм/см, перенаправление водного потока ко второму слою ионообменной обработки и регенерацию первого ионообменного слоя. Предлагаемое изобретение позволяет увеличить время между регенерациями ионообменного слоя и тем самым значительно увеличить эффективность процесса. 2 н. и 11 з.п. ф-лы, 7 ил.
Комментарии