Фермент с активностью эндо-1,3(4)-β-глюканазы, кодирующая его днк и способ его получения - RU2215034C2
Код документа: RU2215034C2
Чертежи









Описание
Область техники, к которой относится изобретение
Предметом настоящего изобретения является фермент с активностью эндо-1,3(4)-β-глюканазы,
конструкция ДНК, кодирующая этот фермент, способ получения данного фермента и ферментный препарат на его основе, использование этого фермента или ферментного препарата для разрушения или модификации
материалов, содержащих β-глюкан.
В объем настоящего изобретения также входит выделенная чистая культура Saccharomyces cerevisiae NN049006, трансформированная последовательностью ДНК, полученной из Acremonium sp. (CBS 265.95).
Известный уровень техники
Эндо-1,3(4)-β-глюканазы (No. 3.2.1.6 классификации ферментов) образуют группу гидролаз,
которые
служат катализаторами для эндогидролиза 1,3-β-D-гликозидных связей в таких β-1,3-глюканах, как курдлан, лихенан и ламинарин, которые являются основным компонентом клеточных
стенок грибов
(включая дрожжи) и β-1,4-связей в смешанных β-1,3-1,4-глюканах, таких как β-D-глюканы зерновых культур. Рассматриваемое соединение имеет название 1,3 (1,3:1,
4)-β
-D-глюкан-3(4)-глюкангидролаза, общепринятым названием является ламинариназа, но в настоящем описании изобретения используется сокращенный термин эндо-1,3(4)-β-глюканаза.
Клеточные стенки микроорганизмов, таких как дрожжи и грибы, представляют собой комплексные структуры, которые помимо β-глюкана содержат ряд других компонентов. Например, клеточные стенки дрожжей помимо слоя глюкана (Andrews and Asenjo, 1987) включает слой маннано-белкового комплекса, а клеточные стенки нитевидных грибов дополнительно включают разные количества хитина и хитозана (Hudson H.J, 1986).
Хорошо известно, что микроорганизмы образуют ряд ценных продуктов, таких как красители, ароматизаторы, витамины, которые необходимо выделить из клеток. Чтобы выделить продукты, образуемые внутри клеток, нужно разрушить или лизировать клеточные стенки микробных продуцентов.
Из-за сложного состава клеточных стенок микробов их разрушение или лизис обычно осуществляется с помощью сильных химических веществ и/или механических средств.
В качестве альтернативы химическому или механическому разрушению в процессе получения дрожжевых экстрактов или других продуктов, образуемых внутриклеточно (Andrew and Asenjo, 1987; Phaff, 1977), предлагается ферментативный лизис и разрушение микробных клеток. Кроме того, ферментативный лизис предлагается использовать для получения протопластов из грибов или дрожжей (Hamlyn и др. , 1981). Промышленность выпускает ряд ферментных препаратов, которые можно использовать для ферментативного лизиса клеток дрожжей и грибов. Такие продукты, которые включают β-1,3- и/или β-1,6-глюканазу, протеазу, хитиназу, манназу и другие ферменты, обычно обладают различной ферментативной активностью и способны расщеплять компоненты клеточных стенок.
Питсон и др. (1993) считают, что такие нитевидные грибы, как Rhizopus arrhizus, Trichoderma longibranchiatum и Penicillum funiculosium, образуют ферменты с активностью эндо-1,3(4)-β-глюконазы.
Целью настоящего изобретения является новая эндо-β-глюконаза и способ получения эндо-β-глюконазы с более высоким выходом и степенью чистоты по сравнению с достигаемыми в настоящее время, а также использование эндо-1,3(4)-β-глюконазы в отдельности или в сочетании с другими ферментами для разрушения клеточных стенок растений или микробов. Кроме того, целью настоящего изобретения является создание новых продуктов с большим содержанием эндо-1, 3(4)-β-глюконазы по сравнению с исходным веществом.
Существует необходимость в увеличении способности таких ферментных препаратов разрушать или модифицировать клеточные стенки и с большей степенью точности расщеплять или модифицировать определенные компоненты клеточных стенок растений или микробов.
Краткое изложение сущности изобретения
Авторы
настоящего изобретения успешно выделили и охарактеризовали последовательность ДНК, кодирующую фермент с активностью эндо-1,3(4)-β-глюконазы, что сделало возможным получение однокомпонентных
эндо-1,3(4)-β-глюконаз.
Поэтому первым аспектом настоящего изобретения является конструкция ДНК, представляющая собой последовательность ДНК, кодирующую фермент с активностью
эндо-β-глюконазы, которая является:
а) последовательностью ДНК с идентификационным No.3 или
б) аналогом последовательности ДНК с идентификационным No.3, который
i)
гомологичен последовательностям ДНК с идентификационным No.3 и/или
ii) образует гибрид с аналогичным олигонуклеотидным зондом в виде последовательности ДНК с идентификационным No.3, и/или
iii) кодирует полипептид, который гомологичен полипептиду, закодированному последовательностью ДНК с идентификационным No.3, и/или
iv) кодирует полипептид, который обладает иммунной
реактивностью в отношении антитела, индуцированного против очищенной эндо-1,3(4)-β-глюконазы, закодированной последовательностью No.4, которую выделяют из Trichoderma harzianum, CBS 243.71.
Одним из вариантов осуществления настоящего изобретения является указанная выше последовательность ДНК, кодирующая фермент с активностью эндо-1,3(4)-β-глюконазы.
В
контексте настоящего изобретения "аналог" последовательности ДНК с идентификационным No. 3 означает любую последовательность ДНК, кодирующую фермент с активностью эндо-β-глюконазы (такой как
эндо-1,3-β-глюканаза и эндо-1,3(4)-β-глюканаза), которая обладает свойствами, указанными выше в пунктах i) - iv). Аналогичную последовательность ДНК обычно
- выделяют из
другого
или родственного (в частности, такого же) микроорганизма, который, как известно, способен образовывать фермент с активностью эндо-β-глюканазы на основе последовательности ДНК No.3 или
любой
другой последовательности ДНК с идентификационным No.1 или 2, в частности, посредством описанных здесь процедур;
- конструируют на основе последовательности ДНК No.3 или любой другой
последовательности ДНК с идентификационным No.1 или 2, например, путем введения нуклеотидных заместителей, которые не способны образовывать другую амнокислотную последовательность эндо-β
-глюканазы, закодированной данной последовательностью ДНК, но соответствуют кодону организма-хозяина, служащему для образования фермента, или путем введения нуклеотидных заместителей, которые
способны
образовывать другую аминокислотную последовательность, а следовательно, и другую белковую структуру, формирующую мутант эндо-β-глюкозы, свойства которого отличаются от природного
фермента.
Другие примеры возможных модификаций включают вставку одного или нескольких нуклеотидов в последовательность, добавление одного или нескольких нуклеотидов к концу последовательности, такой
как область
связывания целлюлозы, или удаление одного или нескольких нуклеотидов в конце или внутри последовательности. Например, аналогичная последовательность ДНК может представлять собой
последовательность ДНК
с идентификационным No.3 или с номерами 1 или 2.
Вполне понятно, что последовательности ДНК или их части с идентификационными номерами 1 и 2 представляют собой последовательности, которые можно использовать для выделения всей последовательности ДНК, кодирующей фермент с активностью эндоглюканазы. Термин "аналог" означает всю последовательность ДНК с идентификационным No.3, которая включает по крайней мере часть последовательностей, обозначаемых идентификационными номерами 1 или 2, либо их части.
Гибридизация, рассматриваемая выше в пункте i), означает, что аналогичная последовательность ДНК служит для образования гибрида с таким же зондом, что и последовательность ДНК, кодирующая фермент эндо-1,3(4)-β-глюканазы в особых условиях, которые подробно описываются в приводимом ниже разделе "Материалы и методы". Аналогичная последовательность ДНК, как правило, гомологична последовательности ДНК на 40-50%, гораздо предпочтительнее, если она гомологична на 60-70% любой из приведенных выше последовательностей, кодирующих эндо-β-глюканазу по настоящему изобретению, в частности на 75, 80, 85, 90% или даже на 95%.
Степень гомологии, рассматриваемая выше в пункте i), определяется как степень идентичности двух последовательностей, свидетельствующая о выведении первой последовательности из второй. Гомологию можно определить с помощью существующих вычислительных программ. Последовательность ДНК, как правило, характеризуется степенью идентичности, равной по крайней мере 40-50%, предпочтительнее 60-70%, еще лучше 80 или 90%, в отношении конструкции ДНК, представляющей собой последовательность ДНК с идентификационным No.3 или по крайней мере части последовательности ДНК с идентификационным No.l и/или 2.
Степень гомологии, рассматриваемая выше в пункте iii), определяется как степень идентичности двух последовательностей, свидетельствующая о выведении первой последовательности из второй. Гомологию можно определить с помощью существующих вычислительных программ. Полипептид, закодированный аналогичной последовательностью ДНК, обладает степенью гомологии, равной по крайней мере 40-50%, предпочтительнее 60-70%, еще лучше 80 или 90%, в отношении фермента, закодированного конструкцией ДНК, представляющей собой последовательность ДНК с идентификационным No. 3 или любую часть последовательности ДНК с идентификационным No.1 и/или 2.
Термин "выделенный из" относительно свойства, рассматриваемого выше в пункте iii), означает не только эндо-1,3(4)-β -глюканазу, полученную с помощью штамма CBS 243.71, но и эндо-1,3(4)-β-глюканазу, закодированную последовательностью ДНК, выделенной из штамма CBS 243.71, и образованную в организме-хозяине, трансформированном с помощью указанной последовательности ДНК. Иммунную реактивность можно определить по методу, описанному в приводимом ниже разделе "Материалы и методы".
Другие аспекты настоящего изобретения включают экспрессирующий вектор, определяющий конструкцию ДНК по этому изобретению, клетку, имеющую такую конструкцию ДНК или экспрессирующий вектор, и способ получения фермента с активностью эндо-1,3(4)-β-глюканазы, который включает культивирование указанной клетки в условиях, делающих возможным получение такого фермента, и выделение фермента из этой культуры.
Еще одним аспектом настоящего изобретения является фермент с активностью эндо-β-глюканазы, который
а) закодирован
конструкцией ДНК по настоящему изобретению;
b) получен по способу, предлагаемому настоящим изобретением;
с) обладает иммунной реактивностью в отношении антитела, индуцированного
против очищенной эндо-1,3(4)-β-глюканазы
с идентификационным No.4, выделенной из Trichoderma harzianum, штамм CBS 243.71.
Выделенный фермент по настоящему изобретению имеет характеристики, описанные в приведенных ниже примерах. Например, с идентификационным No. 3 или любую часть последовательности ДНК с идентификационным No. 1 и/или 2.
Термин "выделенный из" относительно свойства, рассматриваемого выше в пункте iii), означает не только эндо-1,3(4)-β-глюканазу, полученную с помощью штамма CBS 243.71, но и эндо-1,3(4)-β-глюканазу, закодированную последовательностью ДНК, выделенной из штамма CBS 243.71, и образованную в организме-хозяине, трансформированном с помощью указанной последовательности ДНК. Иммунную реактивность можно определить по методу, описанному в приводимом ниже разделе "Материалы и методы".
Другие аспекты настоящего изобретения включают экспрессирующий вектор, определяющий конструкцию ДНК по этому изобретению, клетку, имеющую такую конструкцию ДНК или экспрессирующий вектор, и способ получения фермента с активностью эндо-1,3(4)-β-глюканазы, который включает культивирование указанной клетки в условиях, делающих возможным получение такого фермента, и выделение фермента из этой культуры.
Еще одним аспектом настоящего изобретения является фермент с
активностью эндо-β-глюканазы, который
a)
закодирован конструкцией ДНК по настоящему изобретению;
b) получен по способу, предлагаемому настоящим изобретением;
c)
обладает иммунной реактивностью в отношении антитела,
индуцированного против очищенной эндо-1,3(4)-β-глюканазы с идентификационным No.4, выделенной из Trichoderma harzianum, штамм CBS
243.71.
Выделенный фермент по настоящему изобретению имеет характеристики, описанные в приведенных ниже примерах. Например, было установлено, что средняя молекулярная масса (Mw), определяемая посредством электрофореза в полиакриламидном геле с додецилсульфатом натрия, составляет 31 кДа. Средняя изоэлектрическая точка (рI) равнялась 5,1.
Краткое
описание чертежей
Фиг. 1 - активность
эндо-β-1,3(4)-глюканазы по настоящему изобретению в зависимости от показателя рН при температуре 30oС.
Фиг. 2 - активность эндо-β-1,3(4)-глюканазы по настоящему изобретению в зависимости от температуры при рН 5,0.
Фиг. 3 - деполимеризация ламинарина посредством эндо-β-1, 3(4)-глюканазы по настоящему изобретению.
Фиг. 4 - деполимеризация β-1,3-1,4-глюкана посредством эндо-1,3(4)-β-глюканазы по настоящему изобретению.
Фиг. 5 - деполимеризация курдлана посредством эндо-1, 3(4)-β-глюканазы по настоящему изобретению.
Фиг. 6 - спектры13С ЯМР-глюкана, полученные во время и после обработки эндо-1,3(4)-β-глюканазой по настоящему изобретению.
Подробное описание изобретения
Последовательность ДНК по настоящему изобретению, кодирующая фермент с активностью
эндо-β-глюканазы, можно выделить с
помощью обычного метода, включающего
клонирование в приемлемых векторах библиотеки ДНК из Trichodernaa harzianum;
трансформацию приемлемых
дрожжевых клеток-хозяев с помощью указанных
векторов;
культивирование клеток-хозяев в соответствующих условиях с целью экспрессии любого представляющего интерес фермента, закодированного
клоном в библиотеке ДНК;
скрининг
положительных клонов путем определения активности эндо-β-глюканазы фермента, образуемого такими клонами;
выделение из таких клонов
кодирующей фермент ДНК.
Этот метод рассматривается в заявке на патент WO 93/11249, включенной в настоящее описание изобретения в качестве противопоставленного материала. Более подробное описание скрининг-метода дано в приведенном ниже примере 1.
Последовательность ДНК, кодирующую фермент, можно выделить путем скрининга библиотеки кДНК Trichoderma harzianum, в частности штамма CBS 243.71, который может быть предоставлен центром Centraalbureau voor Schimmelcultures, г. Делфт, Нидерланды, и выбора клонов, проявляющих соответствующую ферментативную активность (то есть активность эндо-1,3(4)-β -глюканазы, определяемую по способности фермента гидролизовать связи β-1,3(4)-глюкана приемлемого субстрата, такого как AZCL-курдлан и AZCL-глюкан, как это описывается в приводимом ниже разделе "Материалы и методы"). Необходимую последовательность ДНК можно выделить из клона с помощью стандартных процедур, например, так, как это описано в примере 1.
Предполагается, что последовательность ДНК, кодирующая гомологичный фермент, то есть аналогичную последовательность ДНК, можно получить из других микроорганизмов. Последовательность ДНК можно выделить путем аналогичного скрининга библиотеки кДНК другого микроорганизма, например такого гриба, как штамм Aspergillus sp., в частности A. aculeatus или A. niger, штамм другого вида Trichoderma sp., в частности T. reesie, Т. viride, Т. longibrachiatum или Т. koningii, или штамм Fusarium sp., в частности F. oxysporum, или штамм Humicola sp. , штамм Rhizopus sp., в частности Rhizopus arrhizus, штамм Acremonium sp. , штамм Botrytis sp., штамм Penicillium sp. и штамм Scleotium sp.
Типичным примером такой последовательности ДНК, кодирующей гомологиченый фермент, может служить последовательность, выделенная из Acremonium sp. (CBS 265.95). Две частичные последовательности ДНК, кодирующие такой фермент, известны под идентификационными номерами 5 и 6.
Изолят Saccharomyces cerevisiae, трансформированный с помощью экспрессирующей плазмиды pYES 2.0 (инвитроген), представляющий собой последовательности кДНК с идентификационными номерами 5 и 6, был зарегистрирован в Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Mascheroder Weg 1b, D-3300 Брауншвейг, ФРГ, (DSM), в соответствии с процедурой выдачи патента, дата которой указана ниже. Поскольку DSM является международным банком данных в соответствии с Будапештским договором, то здесь возможно хранение данных на основании положения 9 данного договора.
Дата регистрации - 11.05.95
Регистрационный номер депозитора - NN049006
Обозначение DSM - DSM 9970
ДНК, кодирующую эндо-1,3(4)-β
-глюканазу по настоящему изобретению можно альтернативно выделить из ДНК
любых вышеуказанных организмов посредством известных процедур с использованием синтетических олигонуклеотидных зондов,
полученных на основе описываемой здесь последовательности ДНК. Например,
приемлемый олигонуклеотидный зонд можно получить на основе нуклеотидной последовательности с идентификационным номером 3 или
по крайней мере части нуклеотидных последовательностей с идентификационными
номерами 1 и 2.
Последовательность ДНК можно затем вставить в рекомбинантный экспрессирующий вектор. Им может быть любой вектор, пригодный для выполнения процедур с использованием рекомбинантной ДНК, причем выбор вектора часто зависит от клетки-хозяина, в которую он должен вводиться. Так, вектором может быть автономно реплицирующий вектор, то есть вектор, который существует в виде внехромосомной части, репликация которого не зависит от хромосомной репликации, в частности плазмиды. Альтернативно таким вектором может быть вектор, который при вводе в клетку-хозяина внедряется в геном клетки-хозяина и реплицируется вместе с хромосомой (хромосомами), в которую он был внедрен.
В векторе последовательность ДНК, кодирующая эндо-β-глюканазу, должна быть присоединена к приемлемой последовательности промотора и терминатора. Промотором может быть любая последовательность ДНК, которая проявляет транскрипционную активность в выбранной клетке-хозяине и может быть выделена из генов, кодирующих гомологичные или гетерологичные белки в клетке-хозяине. Процедуры, используемые для лигирования последовательностей ДНК, кодирующих эндо-β/-глюконазу, соответственно промотора и терминатора, и вставки их в соответствующие векторы, хорошо известны специалистам в этой области (см., например, Sambrook и др. , Molecular Cloning. A Laboratory Manual, Cold Spring Harbor, Нью-Йорк, 1989).
Клетка-хозяин, которая трансформируется с помощью последовательности ДНК, кодирующей фермент по настоящему изобретению, предпочтительно является эукариотической клеткой, в частности клеткой такого гриба, как дрожжи, или клеткой нитевидного гриба. В частности, эта клетка может относиться к виду Aspergillus, предпочтительнее к виду Aspergillus oryzae или Aspergillus niger. Клетки грибов можно трансформировать с помощью процесса, включающего образование протопластов и трансформацию таких протопластов с последующей регенерацией клеточной стенки в соответствии с известными способами. Использование вида Aspergillus в качестве микроорганизма-хозяина описывается в европейском патенте No 238023 (Novo Nordisk A/S), который включен в это описание изобретения в виде противопоставленного материала. Клеткой-хозяином может быть также клетка дрожжей, например, штамма Saccharomyces sp., в частности Saccharomyces cerevisiae, Saccharomyces kluyveri или Saccharomyces uvarum, штамма Schizosaccharomyces sp. , такого как Schizosaccharomyces pombe, штамма Hansenula sp., штамма Pichia sp., штамма Yarrowia sp., такого как Yarrowia lipolytica, или штамм kluyveromyces sp., такого как Kluyveromyces lactis.
Еще одним аспектом настоящего изобретения предусматривается способ получения фермента, в соответствии с которым клетка-хозяин, трансформированная с помощью последовательности ДНК, кодирующей фермент, культивируется в условиях, отвечающих требованиям получения фермента, после чего полученный фермент выделяют из этой культуры.
В качестве среды для культивирования трансформированных клеток-хозяев можно использовать любую среду, пригодную для выращивания таких клеток-хозяев. Выраженную эндо-β-глюканазу можно секретировать в культурную среду и выделять из нее с помощью хорошо известных процедур, которые включают отделение клеток от среды центрифугированием или фильтрованием, осаждение белковых компонентов среды под действием такой соли, как сульфат аммония, с последующим хроматографированием посредством ионообменной хроматографии, аффинной хроматографии или подобной процедуры.
Еще одним аспектом настоящего изобретения является ферментный препарат, пригодный для модификации или разрушения содержащих β-глюкан материалов, причем указанный препарат обогащают ферментом с активностью эндо-1,3(4)-β-глюканазы, описываемым выше.
Ферментный препарат, обогащенный ферментом по настоящему изобретению, может обладать различной ферментативной активностью, в частности такой, которая необходима для модификации или разрушения клеточных стенок микробов. Примеры такого ферментного препарата включают литические ферментные системы, в частности микробного (грибного или бактериального) происхождения, например полученные из штамма Trichoderma, такого как Trichoderma harzianum, Trichoderma viride или Trichoderma reesie, штамма Oerskovia sp., такого как Oerskovia xanthineolytica, штамма Arthrobacter sp., такого как Arthrobacter luteus, штамма Rhizoctonia sp. или Cytophaga sp., штамма Staphylococcus sp. или штамма Streptomyces sp. Выпускаемыми промышленностью ферментными препаратами, которые можно подвергнуть повторной иммунизации ферментом по настоящему изобретению, являются Novozyme 234, Cereflo 200L и Glucanex фирмы Novo A/S, Дания, Cellulase (фирмы Merck), Cellulase CP и Cellulfse CT (фирмы Sturge) и/или Chitinase (фирмы Sigma).
Термин "обогащенный", используемый в настоящем описании изобретения, означает усиление активности эндо-1,3(4)-β-глюканазы данного ферментного препарата, например, на коэффициент обогащения, равный по крайней мере 1,1, что обычно достигается путем добавления фермента по настоящему изобретению, полученного в соответствии с описанным выше методом.
Альтернативно ферментным препаратом, обогащенным ферментом с активностью эндо-1,3(4)-β-глюканазы, может быть препарат, который содержит фермент по настоящему изобретению в качестве основного компонента, например, однокомпонентный ферментный препарат.
Ферментный препарат можно получить в соответствии с известными методами в жидком или сухом виде. Например, ферментный препарат может быть в форме гранул или микрогранул. Фермент, включаемый в препарат, может быть стабилизирован известными методами.
Ферментный препарат по настоящему изобретению помимо эндо-1,3(4)-β-глюканазы может содержать еще один или несколько других ферментов, разрушающих клеточные стенки, например, таких, которые способны разлагать целлюлозу, маннан, хитин или белок; ими, в частности, являются целлюлоза, эндоглюканаза, β-глюкозидаза, α-1,6-глюканаза, манназа, эндо- или экзохитиназа, протеаза, β- или β-маннозидаза или мутаназа. Дополнительный фермент можно получить с помощью микроорганизмов, относящихся к виду Aspergillus, предпочтительно Aspergillus niger, Aspergillus aculeatus, Aspergillus awamori или Aspergillus oryzae, или к виду Trichoderma, либо с помощью микроорганизмов, рассмотренных выше в связи с выпускаемым промышленностью ферментными препаратами.
Ниже приводятся примеры предпочтительного использования ферментных препаратов по настоящему изобретению. Дозирование ферментного препарата и другие условия его применения можно определить с помощью известных методов.
Ферментный препарат по настоящему изобретению можно предпочтительно использовать в качестве средства для разрушения или модификации содержащего β -глюкан материала, такого как клеточные стенки микробов. Ферментный препарат по настоящему изобретению можно использовать для разрушения или лизиса клеточных стенок микроорганизмов, благодаря чему высвобождаются продукты, вырабатываемые микроорганизмами.
Вполне понятно, что состав ферментного препарата должен соответствовать составу клеточных стенок, подлежащих разрушению или лизису. Например, было установлено, что клеточные стенки дрожжей состоят из двух основных слоев: наружного слоя, представляющего собой комплекс белка и манна, и внутреннего слоя, содержащего глюкан. Для эффективного разрушения клеточных стенок дрожжей ферментный препарат должен включать протеазу, манназу и β -глюканазу.
Экстракт, выделяемый в результате разрушения клеточных стенок микробов, обычно включает несколько разных компонентов, таких как витамины, красители, ароматизаторы, эмульгаторы и стабилизаторы. Экстракты, получаемые при разрушении дрожжей, то есть дрожжевые экстракты, используют в том виде, как есть, например, в пищевой или кормовой отраслях промышленности. Кроме того, можно выделить их компоненты, которые при необходимости подвергают дальнейшей обработке.
Примерами таких продуктов являются эмульгаторы, стабилизаторы, витамины, красители (например, каратиноиды, Q-10 и астаксантин), ферменты, белки и ароматизаторы или усилители аромата (например, MSG, 5-GMP и IMP). Получаемыми продуктами являются свойственные данным микроорганизмам продукты либо это могут быть продукты, для выработки которых микроорганизмы были специально созданы, в частности рекомбинантные продукты.
Кроме того, эндо-1,3(4)-β-глюканазу по настоящему изобретению можно использовать для экстракции маннано-белкового комплекса из наружного слоя клеточных стенок дрожжей, таких как Saccharomyces cerevisiae. Маннано-белковый комплекс можно использовать в качестве эффективного биоэмульгатора.
Ферментный препарат по настоящему изобретению можно также использовать для получения протопласта из дрожжей (например, Saccharomyces sp. или Schizosaccharomyces sp.) или из грибов (например, Aspergillus sp. или Penicillium sp.). Получение и регенерация протопласта из таких микроорганизмов имеет важное значение в исследованиях по слиянию, трансформации и клонированию. Протопласты могут быть получены в соответствии с известными методами.
Ферментный препарат по настоящему изобретению можно использовать для модификации β-глюканов, таких как курдлан, ламинарин и лихенан.
Еще одним аспектом настоящего изобретения является ферментный препарат, предназначенный для разрушения или модификации клеточных стенок растений, причем указанный препарат обогащают ферментом с эндо-1,3(4)-β-глюканазой по настоящему изобретению.
Ферментным препаратом, обогащенным ферментом по настоящему изобретению и предназначенным для разрушения или модификации клеточных стенок растений, может быть. например, ферментный препарат, обладающий различной ферментативной активностью, в частности, содержащий несколько ферментов, разрушающих клеточные стенки растений, таких как Pectinex, Pectinex Ultra SP, Celluclast или Celluzyme (фирмы Novo Nordisk A/S).
Ферментный препарат по настоящему изобретению может помимо эндо-1,3(4)-β-глюканазы содержать еще один или несколько других ферментов, разрушающих клеточные стенки растений, например, таких, которые способны разлагать целлюлозу, ксилан или пектин; ими, в частности, являются ксиланаза, арабиназа, галактаназа, рамногалактуроназа, ацетилэстераза, галактаназа, полигалактуроназа, пектинлиаза, пектатлиаза, эндоглюканаза (например, обладающая другой специфичностью по сравнению с описываемой здесь эндо-1,3(4)-β-глюканазой) или пектинмети-лэстераза. Эти дополнительные ферменты можно получить с помощью микроорганизмов, относящихся к виду Aspergillus, предпочтительно Aspergillus niger, Aspergillus aculeatus, Aspergillus awamori или Aspergillus oryzae.
Эндо-1,3(4)-β-глюканазу по настоящему изобретению можно
получить без каких-либо других ферментов, разрушающих клеточные стенки. Это
позволяет использовать данный
фермент отдельно или вместе с другими однокомпонентными ферментами с целью достижения
оптимального сочетания ферментов для определенного применения. Таким образом можно создавать комбинации ферментов,
которые способны разрушать лишь определенные части клетки. Такое специфическое
разрушение ранее было не возможно при использовании выпускаемых промышленностью препаратов на основе целлюлозы, глюкана,
хитиназы, мутаназы,
гемицеллюлазы и/или пектиназы.
Было установлено, что эндо-1,3(4)-β-глюканаза по настоящему изобретению обладает высокой специфичностью в отношении β-1,3-глюканов, а также в отношении β-1,3-1,4-глюканов со смешанными связями.
Активность, проявляемая в отношении смешанных β-1,3-1,4-глюканов, делает эндо-1, 3(4)-β-глюканазу и ее гомологи полезными в пивоварении, виноделии и при изготовлении соков.
В пивоварении ферменты разрушают β-глюкан ячменя, благодаря чему уменьшается вязкость и улучшается фильтруемость сусла. В пивоварении преимуществом этого фермента является более высокая специфичность по сравнению с другими эндоглюканазами, так как вязкость, обуславливаемую β-глюканом, можно уменьшить без одновременного разрушения целлюлозных структур, которые имеют важное значение для фильтруемости сусла, когда использованное зерно служит в качестве дополнительного фильтра.
В виноделии и изготовлении соков эндо-1,3(4)-β-глюканаза помогает улучшить фильтруемость, предотвращая размножение микроорганизмов, таких как Botrytis cinerea, которые инфицируют виноград.
Кроме того, активность, проявляемая в отношении смешанных β-1,3-1,4-глюканов делают этот фермент полезным в пищевой и кормовой отраслях промышленности, позволяя улучшить усвоение и/или переваривание пищи. Помимо эндо-1,3(4)-β-глюканазу можно использовать для улучшения качества печеных изделий и других хлебных продуктов.
Эндо-1,3(4)-β-глюканаза по настоящему изобретению оказывается весьма полезной для получения олигосахаридов из растительных материалов со смесью β-1,3-1,4-глюканов. Полученные олигосахариды можно использовать в качестве наполнителей при изготовлении пищевых продуктов.
Эндо-1,3(4)-β-глюканазу можно также использовать для экстракции ароматических соединений из растительных материалов.
Дозирование ферментного препарата по настоящему изобретению в вышеуказанных областях применения и создание других условий его получения следует определять на основании известных методов.
В объем настоящего изобретения входит также использование эндо-1,3(4)-β-глюканазы или ферментного препарата на ее основе в качестве активного ингредиента, входящего в состав средств для очистки зубных протезов. Такой состав способен эффективно удалять микроорганизмы с поверхности таких протезов.
Этот фермент можно также использовать в составах для удаления зубного налета и в зубных пастах. Налет, образующийся на поверхности зубов, состоит в основном из полисахаридов. Они скапливаются на поверхности зубов и в местах микроорганизмов во рту. Эндо-1,3(4)-β-глюканазу по настоящему изобретению можно использовать вместе с другими глюканазами, такими как мутаназа и декстраназа.
С помощью разных глюканаз, в том числе эндо-1,3(4)-β-глюканазы по настоящему изобретению, можно удалять биопленки, образующиеся на поверхности контактных линз.
С помощью составов, содержащих эндо-1,3(4)-β-глюканазу по настоящему изобретению, можно удалять плесень на покрытиях.
В объем настоящего изобретения также входит использование эндо-1,3(4)-β -глюканазы в качестве противогрибкового средства.
Эндо-1,3(4)-β-глюканазу по настоящему изобретению можно также использовать для удаления избытка красителя с тканей.
Это изобретение более подробно описывается в следующих примерах, которые ни в коем случае не ограничивают его объем.
Материалы и методы
Донорский микроорганизм: мРНК
выделяли из Trichoderma harzianum, CBS 243.71, культивируемого
при перемешивании с целью улучшения аэрации в сбраживаемой среде с кукурузной крупой. Мицелий собирали после культивирования в течение
3-5 дней, сразу же замораживали в жидком азоте и хранили при
температуре -80oС.
Штаммы дрожжей: использовали штамм Saccharomyces cerevisiae yNG231 (MAT alpha, leu 2, ura 3-52, his4-539, pep 4-delta 1, cir+) или JG169 (MAT; ura 3-52; leu 2-3, 112; his 3-D200; pep 4-1137; prcl::HIS3; prb 1:: LEU2; cir+).
Зарегистрированный микроорганизм: Saccharomyces cerevisiae NN049006 трансформировали с помощью последовательности ДНК, включающей две последовательности ДНК с идентифицированными номерами 5 и 6, входящие в состав экспрессирующей плазмиды pYES 2.0. Трансформированная последовательность ДНК гомологична последовательности с идентификационным номером 3.
Выделение последовательности ДНК с идентификационными номерами 5 и 6: экспрессирующий вектор дрожжей pYES 2.0, содержащий последовательность кДНК из Acremonium sp. с идентификационными номерами 5 и 6, можно выделить из зарегистрированного микроорганизма Saccharomyces cerevisiae NN049006 путем экстракции кДНК плазмиды с помощью известных методов.
Зарегистрированный микроорганизм можно культивировать на планшетах с агаровой средой, содержащей хлорид натрия (SC)+2% галактозы, и инкубировать при температуре 30oС в течение 3-5 дней так, как описывается ниже.
Плазмиды: экспрессирующую плазмиду pYHD17, содержащую промотор дрожжей TPI, получали из выпускаемой промышленностью плазмиды pYES 2,0 (инвитроген). Эта плазмида и ее структура далее описывается в заявке на патент WO 93/11249, которая включена в это описание изобретения в качестве противопоставленного материала.
Экспрессирующий вектор pHD414 вида Aspergillus является производным плазмиды р755 (описываемой в европейском патенте No. 238023). Структура вектора pHD414 далее описывается в заявке на патент WO 94/14952.
Плазмида pYES 2.0 (инвитроген).
Экстракция полной РНК: полную РНК получали путем экстракции с помощью гуанидинтиоцианата с последующим ультрацентрифугированием через слой 5,7 моля CsCl, как описывается Чиргвином и др., 1979 г., а также в заявке на патент WO 94/14952.
Выделение поли(А) РНК: поли(А) РНК выделяли с помощью аффинной хроматографии на основе олиго(тимидин)целлюлозы в соответствии с процедурой, описанной в заявке на патент WO 93/11249.
Синтез и модификация кДНК: двунитевую кДНК синтезировали из 5 мкг поли(А) РНК с помощью метода на основе РНКазы Н (Gubler & Hoffman, 1983, Sambrook и др. , 1989) с использованием модификации шпильки. Синтез двунитевой кДНК выполняли в соответствии с описанием, приведенным в заявке на патент WO 95/02044, за исключением того, что к реакционной смеси для синтеза первой нити добавляли 25 нг произвольной гексануклеотидной затравки (Gibco BRL, США). После обработки нуклеазой золотистой фасоли (Bethesda Reseach Laboratories) двунитевую кДНК дефосфолировали полимераой ДНК Т4 (инвитроген), после чего кДНК лигировали до образования непалиндромной последовательности BstX I (интервал) в соответствии с инструкциями изготовителей.
Создание библиотек кДНК: адаптированную двухнитевую кДНК выделяли центрифугированием, промывали 70% этиловым спиртом и вновь суспендировали в 25 мл дистиллированной воды. До массового лигирования библиотеки выполняли четыре тестовых лигирования в 10 мкл буфера для лигирования (представленного выше), каждый из которых содержал 1 мкл двунитевой кДНК (реакционные пробирки # 1-# 3), 2 единицы лигазы Т4 (инвитроген) и 50 нг (пробирка #1), 100 нг (пробирка #2) и 200 нг (пробирки #3 и #4) экспрессирующего вектора гидролизованных дрожжей Bst XI (вектор pYES 2.0 инвитроген или уНD13). Лигирование выполняли путем инкубирования при температуре 16oС в течение 12 часов с последующим нагревом реакционной смеси при температуре 70oС в течение 5 минут, после чего 1 мкл продукта каждого лигирования подвергали электропорации (200 Ом, 2,5 кВ, 25 мкФ) до образования 40 мкл компетентных клеток МС1061 E.coli (оптическая плотность OD600=0,9 в 1 литре питательного бульона, дважды промывали в холодной дистиллированной воде, один раз в 20 мл 10% глицерина, вновь суспендировали в 2 мл 10% глицерина). После добавления 1 мл SOC в каждую трансформационную смесь клетки культивировали при температуре 37oС в течение 1 часа, 50 мкл пассировали на планшетах, содержащих буфер для лигирования и ампициллин (100 мкг/мл), и культивировали при температуре 37oС в течение 12 часов.
Массовое лигирование выполняли в оптимальных условиях в 40 мкл буфера для лигирования, содержащего 9 единиц лигазы Т4, и инкубировали реакционную смесь при температуре 16oС в течение 12 часов. Лигирование прекращали путем нагрева при температуре 70oС в течение 5 минут, этанол осаждали при температуре -20oС в течение 12 часов, удаляли центрифугированием и вновь суспендировали в 10 мкл дистиллированной воды. Аликвоты объемом 1 мкл трансформировали в электрокомпетентные клетки E. coli 1061 в представленных выше условиях электропорации, трансформированные клетки титровали, а библиотеку пассировали на планшетах, содержащих буфер для лигирования + ампициллин с 5000 - 7000 КОЕ/планшет. На каждый планшет дополнительно наносили 3 мл питательной среды. Бактерии соскабливали, добавляли 1 мл глицерина и хранили в виде пулов при температуре -80oС. Остальные 2 мл использовали для выделения ДНК. Если выделенное количество ДНК оказывалось недостаточным для получения необходимого числа дрожжевых трансформантов массовую ДНК получали из 500 мл питательной среды (ТВ), инкубируемой с 50 мкл бактериальной культуры, культивированной в течение ночи при температуре -80oС.
Создание библиотек дрожжей: для гарантии тестирования в дрожжах всех бактериальных клонов, дрожжевые трансформанты использовали в количестве, которое в 5 раз превышало число бактериальных клонов в исходных пулах.
Аликвоты очищенной плазмидной ДНК (100 нг/мкл) обьемом 1 мкл, взятые из отдельных пулов, подвергали электропорации (200 Ом, 1,5 кВ, 25 мкФ) в 40 мкл компетентных клеток JG 169 вида S. cerevisiae (оптическая плотность OD600= 1,5 в 500 мл в среде YPD, дважды промывали в холодной дистиллированной воде, один раз в холодном 1 М растворе сорбита, вновь суспендировали в 0,5 мл 1 М раствора сорбита, Becker & Guarante, 1991). После добавления 1 мл 1 М холодного раствора сорбита 80 мкл аликвоты пассировали на планшетах с урациловой агаровой средой, содержащей хлорид натрия (SC) + глюкозу, с образованием 250-500 КОЕ/планшет и инкубировали при температуре 30o С в течение 3-5 дней.
Идентификация положительных колоний: после культивирования в течение 3-5 дней планшеты с агаровой средой повторно пассировали на нескольких чашках с урациловой агаровой средой, содержащих хлорид натрия (SC) + галактозу. Один комплект планшетов, содержащих 0,1% AZCL-курдлана или AZCL-β-глюкана, инкубировали в течение 3-5 дней при температуре 30oС с целью выявления активности эндо-1,3(4)-β-глюканазы. Положительные колонии идентифицировали в виде колоний, окруженных зоной синего цвета.
Клетки из ферментоположительных колоний помещали на агар для выделения единичных колоний, после чего изо всех идентифицированных колоний, образующих эндо-1,3(4)-β-глюканазу, выбирали единичную ферментообразующую колонию.
Характеристика положительных клонов: положительные клоны получали в виде единичных колоний, вставки кДНК амплифицировали непосредственно из колонии дрожжей с использованием затравки в виде биотинилированного полилинкера, очищенного системой магнитных шариков (Dynabead M-280, Dynal), и характеризовали по отдельности путем секвенирования 5'-конца каждого клона кДНК по методу терминации цепи (Sanger и др., 1977) и с помощью системы на основе секвеназы (United States Biochemical).
Выделение гена кДНК для экспрессии в виде Aspergillus: одну или несколько колоний, образующих эндо-1,3(4)-β-глюканазу, инкубировали в 20 мл пептон-дрожжевого бульона с глюкозой (YPD), помещенного в 50 мл стеклянную пробирку. Эту пробирку в течение 2 дней встряхивали при температуре 30oС. Клетки собирали путем центрифугирования в течение 10 минут со скоростью 3000 оборотов в минуту.
Клетки вновь суспендировали в 1 мл раствора 0,9 моля сорбита, 0,1 моля этилендиаминтетрауксусной кислоты с рН 7,5. Осадок переносили в пробирку Эппендорфа и центрифугировали с максимальной скоростью. Клетки вновь суспендировали в 0,4 мл раствора 0,9 моля сорбита, 0,1 моля этилендиаминтетрауксусной кислоты, 14 ммолях β-меркатоэтанола. Добавляли 100 мкл раствора 2 мг/мл зимолазы, 30 минут инкубировали эту суспензию при температуре 37oС и центрифугировали в течение 30 секунд. Осадок (сферопласты) вновь суспендировали в 0,4 мл триэтилена. Добавляли 90 мкл (1,5 мл 0,5 М раствора этилендиаминтетрауксусной кислоты с рН 8,0, 0,6 мл 2 М раствора трис-Cl буфера с рН 8,0, 0,6 мл 10% додецилсульфата натрия) и 30 минут инкубировали суспензию при температуре 65oС. Добавляли 80 мкл 5 М раствора КОАс, не менее 60 минут инкубирвали суспензию в ледяной бане и центрифугировали с максимальной скоростью в течение 15 минут. Надосадочную жидкость переносили в чистую пробирку, заполненную этиловым спиртом (при комнатной температуре), после чего эту смесь тщательно, но осторожно перемешивали и центрифугировали в течение 30 секунд. Осадок 30 секунд промывали холодным 70% этиловым спиртом и сушили при комнатной температуре. Осадок вновь суспендировали в 50 мкл триэтилена и центрифугировали в течение 15 минут. Надосадочную жидкость переносили в чистую пробирку. Добавляли 2,5 мкл раствора 10 мг/мл РНКазы, инкубировали при температуре 37oС в течение 30 минут и при осторожном перемешивании добавляли 500 мкл изопропанола. Эту смесь центрифугировали в течение 30 cекунд, после чего удаляли надосадочную жидкость. Осадок промывали хододным 96% этиловым спиртом и сушили при комнатной температуре. ДНК растворяли в 50 мкл воды до конечной концентрации, равной 100 мкг/мл.
ДНК трансформировали в E.coli с помощью стандартных процедур. Плазмидную ДНК выделяли из E.coli обычными методами и анализировали посредством рестрикционного аналиа. кДНК-вставку выщепляли с помощью соответствующих рестрикционных ферментов и лигировали в экспрессирующий вектор Aspergillus.
Трансформация Aspergillus oryzae или Aspergillus niger (общая процедура): 10 мл пептон-дрожжевого бульона с глюкозой (YPD) (Sherman и др.. Methods in Yeast Genetics, Cold Spring Harbor Laboratory, 1981) инокулировали спорами A.oryzae или A.niger и инкубировали, встряхивая при температуре 37oС в течение 2 дней. Мицелий собирали фильтрованием через фильтровальную ткань, промывали 200 мл 0,6 М раствора MgSO4 и суспендировали в 15 мл 1,2 М раствора MgSO4 с 10 ммолями NaH2PO4, рН=5,8. Суспензию охлаждали в ледяной бане и добавляли 1 мл буфера, содержащего 120 мг Novozym® 234, партия 1687. Через 5 минут добавляли 1 мл раствора 12 мг/мл бычьего сывороточного альбумина (Sigma type H25) и инкубировали при слабом перемешивании в течение 1,5-2,5 часов при температуре 37oС до тех пор, пока в пробе, исследуемой под микроскопом, не появлялось большое количество протопластов.
Эту суспензию фильтровали через фильтровальную ткань, фильтрат переносили в стерильную пробирку и доливали в нее 5 мл раствора 0,6 моля сорбита, 100 ммолей трис-НСl буфера с рН 7,0. Центрифугировали в течение 15 минут при градиенте скорости, равном 100, после чего с верхней части слоя MgSO4 собирали протопласты. К суспензии протопластов добавляли 2 объема STC (1,2 моля сорбита, 10 ммолей трис-НСl буфера с рН 7,5, 10 ммолей СаСl2) и центрифугировали эту смесь в течение 5 минут при градиенте скорости, равнoм 1000. Осадок протопластов вновь суспендировали в 3 мл раствора STC и еще раз осаждали. Эту процедуру повторяли. И, наконец, протопласты еще раз суспендировали в 0,2-1 мл раствора STC.
100 мкл суспензии протоплаcтов смешивали с 5-25 мкг соответствующей ДНК в 10 мкл раствора STC. Протопласты смешивали с вектором p3SR2 (плазмида с геном amdS вида A. nidulans). Эту смесь оставляли при комнатной температуре на 25 минут. Добавляли 0,2 мл раствора 60% полиэтиленгликоля 4000 (BDH 29576), 10 ммолей СаСl2 и 10 ммолей трис-HCl буфера с рН 7,5, тщательно перемешивали (дважды), после чего добавляли 0,85 мл аналогичного раствора и тщательно перемешивали. Эту смесь оставляли при комнатной температуре на 25 минут, центрифугировали при градиенте скорости, равном 2500, в течение 15 минут и вновь суспендировали полученный осадок в 2 мл 1,2 М раствора сорбита. После вторичного осаждения протопласты переносили на соответствующие планшеты. Протопласты наносили на планшеты минимального размера (Cove Biochem. Biophys. Acta, 113 (1966), 51-56) с раствором 1,0 моля сахарозы, рН 7,0, 10 ммолей ацетамида в качестве источника азота и 20 ммолей CsCl для ингибирования роста фоновой среды. После инкубации в течение 4-7 дней при температуре 37oС споры собирали и распределяли на единичные колонии. Эту процедуру повторяли и после повторного выделения споры единичной колонии хранили в виде идентифицированного трансформанта.
Тестирование трансформантов A. oryzae: все трансформанты инокулировали в 10 мл пептон-дрожжевого бульона с мальтодекстрином (YPM) и культивировали в этой среде. После инкубации в течение 2-5 дней при температуре 37oС удаляли 10 мл надосадочной жидкости. Активность эндо-1,3(4)-β-глюканазы идентифицированы с помощью AZCL-курдлана или AZCL-β-глюкана.
Условия гибридизации (создаваемые для оценки свойства конструкции ДНК по настоящему изобретению, описанного в пункте 1)): приемлемые условия для определения гибридизации между олигонуклеотидным зондом и "аналогичной" последовательностью ДНК включают предварительное вымачивание фильтра, содержащего последовательности ДНК, предназначенные для гибридизации, в 5-кратном обьеме раствора хлорида и цитрата натрия; предварительную гибридизацию в течение 1 часа при температуре ~50oС в 5-кратном обьеме раствора хлорида и цитрата натрия, 5-кратном объеме раствора Денхардта, 50 ммолях фосфата натрия, рН 6,8, и 50 мкг денатурированной и обработанной ультразвуком ДНК тимуса теленка; последующую гибридизацию в аналогичном растворе, дополненном меченым зондом 50 мкСi 32-P-dCTP, в течение 18 часов при температуре ~50oС и трехкратную промывку в 2 объемах раствора хлорида и цитрата натрия, 0,2% додецилсульфата натрия, выполняемую в течение 30 минут при температуре 50oС.
Приемлемый олигонуклеотидный зонд, предназначенный для использования в процессе гибридизации, можно получить на основе любой последовательности ДНК с идентификационным номером 3 или по крайней мере части последовательности ДНК с идентификационными номерами 1 или 2 либо с идентификационными номерами 5 или 6.
Иммунная перекрестная реактивность: антитела, предназначенные для использования при определении иммунной перекрестной реактивности, можно получить с помощью очищенной эндо-1,3(4)-β-глюканазы. В частности, антисыворотку против эндо-1, 3(4)-β-глюканазы по настоящему изобретению можно получить путем иммунизации кроликов (или других грызунов) в соответствии с процедурой, описанной в работе Н.Аксельсена и др. "A Manual of Quantitative Immunoelectrophoresis", Blackwell Scientific Publications, 1973, глава 23, или в работе А. Джонстоуна и Р.Торпе "Immunochemistry in Practice", Blackwell Scientific Publications, 1982 (стр.27-31). Очищенные иммуноглобулины можно получить из антисывороток, например, путем осаждения соли (NH4)2SO4 с последующим выполнением диализа и ионообменной хроматографии, например, на диэтиламиноэтилен-сефадексе. Иммунохимическую характеристику белков можно произвести с помощью анализа по методу двойной диффузии Оучерлони (O.Oucherlony, Handbook of Experimental Immunology (D.M.Weir, ed.), Blackwell Scientific Publications, 1967, стр.655-706), выполняемого посредством перекрестного иммуноэлектрофореза (N.Axelsen и др., см. выше, главы 3 и 4) или ракетного иммуноэлектрофореза (N.Axelsen и др., глава 2).
Окрашивание красителем кумасси: гель осторожно удаляли со стеклянных пластин и инкубировали на медленно вращающемся и
качающемся столе в 100 мл следующих растворов:
1) 30 минут в растворе 40%
(в объемном отношении) этанола, 5% (в объемном отношении) уксусной кислоты;
2) 30 минут в растворе 40% (в
объемном отношении) этанола, 5% (в объемном отношении) уксусной кислоты + 0,1%
кумасси бриллиантового голубого R250;
3) обесцвечивание производили в течение 30 минут в растворе 40% (в
объемном отношении) этанола, 5% (в объемном отношении) уксусной кислоты до достаточно
полного устранения фона;
4) наконец, гель инкубировали в закрепляющем растворе, содержащем 5% (в
объемном отношении) уксусной кислоты, 10% (в объемном отношении) этанола, 5% (в объемном
отношении) глицерина, и подвергали воздушной сушке между двумя листами целлофановой мембраны.
Окрашивание серебром: гель осторожно удаляли со стеклянных пластин и инкубировали на
медленно вращающемся и качающемся столе в 100 мл следующих растворов:
1) 30 минут в растворе 40% (в
объемном отношении) этанола, 5% (в объемном отношении) уксусной кислоты;
2) 20
минут в растворе 10% (в объемном отношении) этанола, 5% (в объемном отношении) уксусной кислоты;
3) 20
минут в растворе 0,0057% (в отношении веса к объему) персульфата аммония (0,25 ммоля);
4) 60 минут в растворе 0,1% (в отношении веса к объему) AgNO3.
5) Для проявления гель на 30-60 секунд погружали в проявитель, содержащий 0,015% формальдегида, 2% (в отношении веса к объему) Na2CO3. Затем гель инкубировали во второй порции проявителя до достижения удовлетворительного окрашивания белков (5-15 минут). И, наконец, гель инкубировали в закрепляющем растворе, содержащем 5% (в объемном отношении) уксусной кислоты, 10% (в объемном отношении) этанола, 5% (в объемном отношении) глицерина, и подвергали воздушной сушке между листами целлофановой мембраны.
Среды
YPD: 10 г дрожжевого экстракта, 20
г пептона, Н2О до 900 мл. Эту смесь автоклавировали с добавлением 100 мл 20% раствора
глюкозы (стерильно профильтрованной).
YPM: 10 г дрожжевого экстракта, 20 г пептона, Н2О до 900 мл. Эту смесь автоклавировали с добавлением 100 мл 20% раствора мальтодекстрина (стерильно профильтрованного).
10-кратный объем основной соли: 75 г азотно-дрожжевой основы, 113 г янтарной кислоты, 68 г NaOH, H2O до 1000 мл. Эту смесь стерильно фильтровали.
SC-URA: 100 мл 10-кратного объема основной соли, 28 мл 20% раствора казаминокислот без витаминов, 10 мл 1% раствора триптофана, Н2О до 900 мл. Эту смесь автоклавировали с добавлением 3,6 мл 5% раствора треонина и 100 мл 20% раствора глюкозы или 20% раствора галактозы.
Агар на основе SC-URA: cреда SC-URA с добавлением 20 г/л агара.
AZCL-курдлан (Megazyme, Австралия).
AZCL-β-глюкан (Megazyme, Австралия).
Очищенная плазмидная ДНК фирмы Qiagen (Qiagen, США).
Комплект β (U.S.Biochemical corp., США).
Произвольные гексануклеотидные затравки (Gibco BRL, США).
Пример 1.
Была создана библиотека E.coli из вида Т.harzianum, включающая примерно 106 отдельных клонов в 150 пулах.
ДНК выделяли из 20 отдельных клонов библиотеки и анализировали в отношении наличия кДНК-вставок. Частота вставок составила >90%. ДНК из некоторых пулов трансформировали в дрожжи, причем из каждого получали 50-100 планшетов, содержащих 200-500 колоний дрожжей.
Идентифицировали положительные колонии и помещали их на агар. кДНК-вставки амплифицировали прямо из колонии дрожжей и характеризовали так, как описывалось выше в разделе "Материалы и методы". Последовательности кДНК, кодирующей эндо-1,3(4)-Sequenase®-глюканазу, был присвоен идентификационный No.3.
Пример 2.
Из колонии дрожжей выделяли полную ДНК и посредством описанной выше трансформации E. coli определяли плазмидную ДНК. Для экспрессии эндо-1,3(4)-β-глюканазы в Aspergillus ДНК гидролизовали с помощью HindIII/XbaI, фракционировали по размеру на геле и очищали фрагмент, соответствующий гену эндо-1,3(4)-β -глюканазы. Этот ген затем сшивали с вектором pHD414, гидролизованным с помощью HindIII/XbaI.
После амплификации ДНК в E.coli плазмидный вектор pA2CU3 трансформировали в Aspergillus oryzae, как это описывалось выше.
Тестирование трансформантов A.oryzae
Все
трансформанты тестировали в отношении активности эндо-1,3(4)-β-глюканазы в соответствии с
приведенным выше описанием. Некоторые трансформанты обладали более высокой активностью эндо-1,
3(4)-β-глюканазы, чем фон Aspergillus oryzae. Это свидетельствует о значительной экспрессии эндо-1,
3(4)-β-глюканазы в Aspergillus oryzae. Выбирали трансформант с наибольшей активностью
эндо-1,3(4)-β-глюканазы и инкубировали его в 500 мл качающейся колбе со средой YPM. После
ферментации в течение 3-5 дней при достаточно сильном перемешивании для обеспечения хорошей аэрации
культуральный бульон центрифугировали в течение 10 минут с градиентом скорости, равным 2000, и
удаляли надосадочную жидкость.
Пример 3.
Очистка эндо-1,3(4)-β
-глюканазы
Надосадочную жидкость, полученную в результате ферментации Aspergillus
oryzae или A. niger и экспрессирующую эндо-1,3(4)-β-глюканазу, центрифугировали и фильтровали через 0,
2 мкм фильтр с целью удаления мицелия.
35-50 мл фильтровальной надосадочной жидкости (5-50 мг рекомбинантного фермента) ультрафильтровали в ультрафильтрующем аппарате "Амикон" с мембраной 10 кДа до достижения 10-кратной концентрации.
Этот концентрат 100-кратно разбавляли в 25 мM триc-буфера с рН 8,0 и выполняли два последовательных цикла фильтрации в том же аппарате. Ультрофильтрованную пробу загружали со скоростью 1,5 мл/мин в анионообменник "Pharmacia XK16/10 Fast Flow Q Sepharose", уравновешенный 25 мМ трис-буфера с рН 8,0. После введения пробы колонку промывали двумя объемами 25 мМ трис-буфера с рН 8,0 и связанные белки элюировали возрастающим линейным градиентом раствора NaCl, содержащим от 0 до 0,5 мМ NaCl в 25 мМ трис-буфера с рН 8,0.
Фракции объемом 5 мл собирали и анализировали в отношении активности эндо-1,3(4)-β-глюкaнaзы в соответствии с приводимым описанием. Эндо-1,3(4)-β-глюканаза, элюированная из колонки с использованием 0,1 М раствора NaCl, не была полностью очищена.
Таким образом, фракции, содержащие эндо-1,3(4)-β-глюканазу, группировали и концентрировали посредством ультрафильтрации в ультрафильтрирующем аппарате "Амикон" с мембраной 10 кДа до достижения 10-кратной концентрации. Этот концентрат 100-кратно разбавляли в 10 мМ фосфата натрия с рН 6,8 и выполняли два последовательных цикла ультрафильтрации в том же аппарате. Ультрафильтрованную пробу загружали со скоростью 1 мл/мин в колонку Pharmacia XK10/20, заполненную BioGel (BioBad, США) и уравновешенную 10 мМ фосфата натрия с рН 6,8. После введения пробы колонку промывали двумя объемами 10 мМ фосфата натрия с рН 6,8 и элюировали связанные белки фосфатом натрия в количестве от 0,01 до 0,5 М с линейно увеличивающейся концентрацией. Фракции объемом 5 мл собирали и анализировали в отношении активности эндо-1,3(4)-β-глюканазы. Эндо-1,3(4)-β-глюканаза не сохранялась в колонке при этих условиях, поэтому фракции, содержащие эндо-1, 3(4)-β-глюканазу, концентрировали в устройстве "Filtron Macrosep", рассчитанном на 10 кДа, до конечного объема, равного 3 мл. Эту пробу загружали в колонку Pharmacia Superdex 075, уравновешенную 0,25 М раствором ацетата аммония с рН 5,5 со скоростью, равной 1 мл/мин. 5 м фракции собирали и анализировали в отношении активности эндо-1,3(4)-β -глюканазы. Эндо-1,3(4)-β -глюканаза, элюированная из этой колонки, характеризовалась 50% чистотой, определяемой посредством электрофореза в полиакриламидном геле с додецилсульфатом натрия, после чего ее использовали для дальнейшего определения характеристик.
Пример 4.
Определение молекулярной массы посредством электрофореза в полиакриламидном геле с
додецилсульфатом натрия
Электрофорез в полиакриламидном геле с додецилсульфатом натрия выполняли в устройстве для электрофореза Mini-Leak 4 (Kem-En-Tec, Копенгаген) в виде модифицированной
процедуры Лаемли (Laemli 1970;
Christgau, Schierbeck и др., 1991). Эту процедуру можно кратко описать следующим образом: разделяющий гель формовали из 12% акриламида, 0,2% BIS-акриламида, 0,1%
додецилсульфата натрия, 0,375 М
трис-буфера с рН 8,8, 0,04% персульфата аммония и 0,04% триэтилэтилендиамина (TEMED). После полимеризации в течение 6-15 часов формовали концентрирующий гель с
использованием 4,5% (в весовом
отношении) акриламида, 0,075% BIS-акриламида, 0,1% додецилсульфата натрия, 66,5 мМ трис-буфера с рН 6,8, 0,4% (в весовом отношении) персульфата аммония и 0,4%
триэтилметилэтилендиамина (TEMED).
Электродные камеры заполняли буфером, содержащим 25 мМ трис-основания, 0,192 М глицина и 0,05% додецилсульфата натрия, после чего загружали пробы и пропускали через
гель ток величиной 2-4 мА/гель для
обработки в течение ночи и 10-30 мА/гель для быстрой обработки, после чего гель удаляли и окрашивали кумасси голубым или серебром, как описывалось выше в разделе
"Материалы и методы".
Было установлено, что средняя молекулярная масса равна 31 кДа.
Пример 5.
Определение изоэлектрической точки посредством
изоэлектрического фокусирования
Изоэлектрическое фокусирование выполняли на пластинах, покрытых полиакриламидным гелем с амфолином, имеющим показатель рН 3,5-9,5 (Pharmacia, Upsala), в
аппарате для электрофореза Multophor в
соответствии с инструкциями изготовителей. После электрофореза гель окрашивали красителем кумасси или серебром, как это описывалось выше, за исключением того,
что перед окрашиванием гель инкубировали
в течение 20 минут в 20% трихлоруксусной кислоте.
Среднее значение изоэлектрической точки (рI) равнялось 5,1.
Пример 6.
Оптимальное значение рН
Буферы со значениями рН от 2,5 до 8,0 готовили путем смешивания 0,1 М тринатрийфосфата с 0,1 М лимонной кислотой. Эндо-1,3(4)-β-глюканазу разбавляли
с тем, чтобы результат анализа
находился в пределах линейного диапазона. Субстрат представлял собой 0,4% суспензию AZCL-β-глюкана (MegaZyme), смешанного в отношении 1:1 с цитрат-фосфатным
буфером до конечной концентрации
субстрата, равной 0,2% AZCL-глюкана. К 1 мл субстрата в 1,5 мл полипропиленовых пробирках Eppendorf добавляли 10 мкл раствора фермента и инкубировали в течение 15
минут в термосмесителях Eppendorf с
регулируемой температурой, прежде чем в течение 20 минут произвести термоинактивацию ферментов в отдельном термосмесителе при температуре 95oС. Пробирки
центрифугировали, надосадочную
жидкость объемом 200 мкл из каждой пробирки переносили в лунки на 96-луночном планшете для микротитрования и измеряли оптическую плотность при длине волны 620 нм в
аппарате для прочтения планшетов
ELISA (Labsystems Multiskan MCC/340).
Для определения оптимального значения рН инкубацию производили при температуре 30oС. Для каждого значения рН в три пробирки добавляли фермент и инкубировали до выполнения термоинактивации, при этом в одну пробирку (контрольный анализ) добавляли фермент и сразу же производили термоинактивацию. Высчитывали среднее значение для трех инкубированных проб и вычитали из него значение контрольного анализа. Результат при оптимальном показателе рН составил 100%.
Активность эндо-β-1,3(4)-глюканазы в зависимости от показателя рН при температуре 30oС изображена на фиг.1, из которой видно, что активность эндо-β-1,3(4)-глюканазы составляет более 60% при рН 4,0-6,5, причем оптимальная активность достигается при рН 4,5-5,5.
Пример 7.
Оптимальная температура
Для определения оптимальной температуры
инкубацию производили, как описывалось
выше, в цитрат-фосфатном буфере с показателем рН 5,0. Температуру измеряли в интервале от 30 до 80oС. Для каждой температуры выполняли три инкубации и
высчитывали среднее значение. Было
произведено три контрольных анализа, в соответствии с которыми сразу же производили терминактивацию ферментов, при этом среднее значение вычитали из величин,
полученных для инкубированных проб.
Активность при оптимальной температуре равнялась 100%.
Активность эндо-

Пример 8.
Кинетические параметры
Константу Михаэлиса (Km) и удельную активность определяли, производя инкубацию при температуре 30oС в течение 15 минут при концентрациях
(S) β-1,3-1,4-глюкана (MegaZyme) в диапазоне от 0,05 до 1,5% в цитрат-фосфатном буфере с рН 5,0. Скорость реакции (v) измеряли в виде функции
образования восстанавливающего сахара/мин.
Концентрацию восстанавливающихся сахаров после инактивации фермента определяли по взаимодействию на планшетах для микротитрования с реагентом, содержащим 0,
15 г гидразида пара-гидроксибензойной
кислоты (Sigma Н-9882), 0,50 г тартрата калия и натрия (Merck 8087) и 2% раствор NaOH, добавляемый до 10,0 мл. В качестве эталона использовали глюкозу, при этом
результаты контрольных анализов
вычитали. Отношение S/v выражали в виде функции S и выполняли линейный регрессионный анализ. Угол наклона кривой (=l/Vmax) и отрезок (Km/Vmax) использовали для
вычисления константы Михаэлиса(Km) и
удельной активности (=Vmax/E), где величина Е соответствовала количеству добавленного фермента. Удельную активность измеряли в виде ед/мг фермента, где 1 единица =
1 мкмолю восстанавливающего
сахара/мин.
Удельная активность эндо-β-1,3(4)-глюканазы при температуре 30oС и pH 5,0 составляла 100-200 ед/мг фермента, а константа Михаэлиса (Km) соответствовала 0,02-0,06% β-1,3-1,4-глюкана.
Пример 9.
Деполимеризация β-глюканов
Для выполнения гель-фильтрационной
хроматографии 1 мл аликвоты 1% растворов
постулана (Roth), ламинарина (Sigma), курдлана (MegaZyme) или β-1,3-1,4-глюкана (MegaZyme) инкубировали до термоинактивации с 10 мкл 0,2 мг/мл раствора
фермента при температуре 30oС
в течение 0, 1, 2, 4 и 24 часов. 25 мкл пробы последовательно вводили в три TSK-колонки (TosoHaas) (PW G4000, PW G3000, PW G2500) и элюировали сахариды 0,4 М
раствором ацетатного буфера с рН 3,0 со
скоростью 0,8 мл/мин. Элюируемые сахариды определяли с помощью детектора преломления Шимадзу, причем данные начинали получать через пятнадцать минут после ввода
пробы. Данные обрабатывали с помощью
программного обеспечения Dionex. В качестве эталонов молекулярной массы использовали декстраны (предоставленные фирмой Serva).
В соответствии с определением активности эндо-β -1,3(4)-глюканазы этот фермент легко разрушал ламинарин (см.фиг.3), β-1,3-глюкан (курдлан) (фиг.4) и β-глюкан, полученный из овса, (β-1,3-1, 4-глюкан) (фиг.5), в то время как разрушения пустулана не происходило.
Пример 10.
Гидролитическое разрушение β-глюкана эндо-1,3(4)-β-глюканазой
Спектры13С ЯМР
регистрировали спектрометром Brucker AC300 с частотой 75,47 МГц. Спектры регистрировали при 330 кДа, собирая данные в течение ночи.
β-1,3-1,4-Глюкан (10 мг) растворяли в D2О (1000 мкл, 99,95% D (Merck)) и регистрировали спектры13С ЯМР (фиг.6, нижний спектр). Добавляли раствор эндо-β-l,3(4)-глюканазы (10 мкл 0,070 мг белка/мл) и после выдерживания этой смеси при комнатной температуре в течение 5 часов снова регистрировали спектр13С ЯМР (фиг.6, средний спектр). Добавляли вторую часть ферментного раствора (10 мк) и выдерживали реакционную смесь 3 дня при комнатной температуре, после этого регистрировали ее спектр13С ЯМР (фиг.6, верхний спектр).
Спектр, полученный для β -глюкана, был идентичен спектру, описанному в литературе (Bock и др.,1991, Carbohydrate Research, 211, 219-233). Спектр, полученный после пятичасовой инкубации с эндо-β-1, 3(4)-глюканазой показал, что произошло лишь незначительное гидролитическое разрушение субстрата. Однако через три дня в спектре присутствовали сигналы, соответствующие аномерным атомам углерода, резонирующим при 96, 7 (C-1β) и 93,0 частях на миллион (С-1α). Кроме того, сигнал, относящийся к С-3 в 3-глюкозилированных единицах оставался без изменения, свидетельствуя о том, что 1, 3-связи не были гидролизованы этим ферментом. Полученные результаты подтверждают что ферментом является эндо-1,3(4)-β-глюканаза (Е. С.3.2.1.6), которая помимо β-1,3-связи в β-1, 3-глюкане гидролизует также β-1,4-связь в β-1,3-1,4-глюканах со смешанными связями.
Пример 11.
Создание и скрининг библиотеки кДНК Acremonium sp.
Полную РНК получали из замороженного порошкообразного мицелия Acremonium sp. , штамм CDS 265.95, путем экстракции гуанидинтиоцианатом с последующим ультрацентрифугированием через слой 5,7 М CsCl (Chirgwin и др., 1979, Biochemistry, 18, 5294-5299). Поли(А)+РНК выделяли посредством аффинной хроматографии на олиго(тимидин)целлюлозе (Aviv и Leder, 1972, Proc. Natl. Acad. Sci. , США, 69, 1408-1412). Двунитевую кДНК синтезировали из 5 мкг поли(А)+РНК в соответствии с описанным методом (Gubler и Hoffman, 1983, Gene, 25, 263-269; Sambrook и др., 1989, Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, Нью-Йорк. Cold Spring Harbor Laboratory) за исключением того, что во время синтеза первой нити использовали 25 нг произвольных гексануклеотидных затравок (Gibco BRL, США). Библиотека кДНК включающая 1,5 х 106 клонов, была создана в экспрессирующем векторе дрожжей pYES 2,0 в соответствии с описанием, известным из литературы (Kofod и др., 1994, G.Biol. Chem. , 261, 8407-8413). Плазмидную ДНК из пула библиотеки кДНК трансформировали в S. cerevisiae W3124 (van den Hazel и др., 1992, Eur. J. Biochem., 207, 277-283) посредством электропорации (Becker и Guarente, 1991, Methods Enzymol. , 194, 182-187) и культивировали трансформанты на агаре с хлоридом натрия (SC) (Sherman, 1991). Methods Enzymol., 194, 3-21), содержащем 2% глюкозы после инкубации при температуре 30oС в течение 3-4 дней колонии вновь помещали на планшеты с агаровой средой с хлоридом натрия, содержащей 2% галактозы, и инкубировали в течение 3 дней при температуре 30oС. Ферментную активность трансформированного штамма S. cerevisiae проверяли на AZCL-β-глюкане и AZCL-курдлане. Были идентифицированы положительные клоны эндо-1,3(4)-глюканазы.
Выделяли полную ДНК из положительных колоний дрожжей и сохраняли вставку, содержащую клоны pYES 2,0 путем трансформации E.coli МС 1061 (Meissner и др., (1987), Proc. Natl. Acad. Sci., США, 84, 4171-4176) до резистентности ампициллина.
Анализ нуклеотидной последовательности
Нуклеотидную последовательность кДНК-вставки определяли в двух нитях по методу
терминации
дидезокси-цепи (Sanger и др., 1977, Proc. Natl. Sci., США, 74, 5463-5467) с использованием очищенной плазмидной ДНК фирмы Qiagen, комплекта Sequenase® или синтетических
олигонуклеотидных затравок. Анализ последовательности выполняли в соответствии с методом Деверекса и др., 1984, Nucleic Acids Res. , 12, 387-395). Двум частичным последовательностям кДНК были
присвоены идентификационные номера 5 и 6.
Пример 12.
Гидролизные исследования материала выделенных клеточных стенок дрожжей
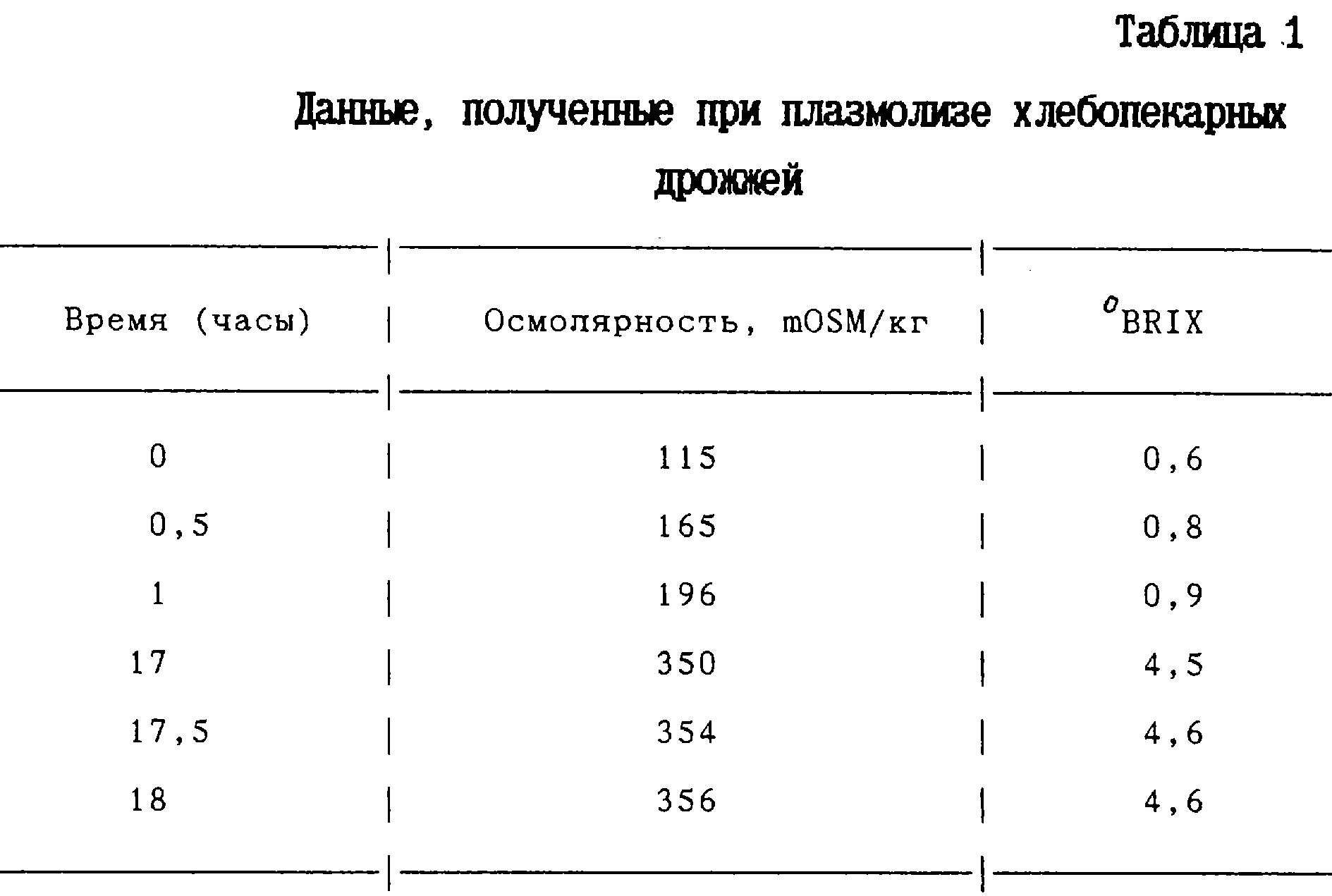
450 г хлебопекарных дрожжей,
Saccharomyces cerevisiae, суспендировали в 1350 г воды. Плазмолиз осуществляли в условиях перемешивания при температуре 50oС в течение 18 часов. Пробы объемом 10 мл отбирали через 0,5, 1,
17, 17,5 и 18 часов. Эти пробы центрифугироваля в центрифуге "Labofuge" фирмы Heraus со скоростью 3400 х градиент (что эквивалентно 4500 оборотам в минуту применяемой центрифуги) в течение 5 минут.
После завершения реакции с помощью усовершенствованного широкодиапазонного осмометра 3W2 (Advanced Instruments) измеряли осмолярность надосадочной жидкости. Содержание сухих веществ в продуктах
центрифугирования определяли электронным рефректометром DUR (Schmidt Haensch). Полученные данные приведены в таблице 1.
Через 18 часов все содержимое центрифугировали 30 минут со скоростью 4500 оборотов в минуту в центрифуге с охлаждением RC-3G Sorvall. Было получено 1475,8 г надосадочной жидкости и 227,3 г твердого компонента. Твердое вещество суспендировали в 1475,8 г воды и еще раз центрифугировали 30 минут со скоростью 4500 оборотов в минуту в центрифуге с охлаждением RC-3G Sorvall. После этого было получено 266,1 г промытого материала клеточных стенок.
Определение свойств сухого вещества производили при температуре 105oС в течение ночи. Содержание сухого вещества составило 22,8% в весовом отношении. Это вещество определяли как клеточные стенки, выделенные из дрожжей.
Контрольный гидролиз выполняли с использованием 21,9 г смоченного материала выделенных клеточных стенок, которые суспендировали в 70 мл воды в 250 мл колбе Эрленмейера. Показатель рН доводили до 5,0 с использованием 6 н. раствора НСl. Общее содержимое колбы Эрленмейера доводили до 100 мл. Пробы отбирали через t = 1, 5, 10, 20, 30, 60, 90, 120, 180, 240 минут. Эти пробы сразу же 5 минут центрифугировали в центрифуге "Labofuge" фирмы Heraeus со скоростью 3400 х градиент (что эквивалентно 4500 оборотам в минуту на применяемой центрифуге). После завершения реакции с помощью усовершенствованного широкодиапазонного осмометра 3W2 (Advanced Instruments) измеряли осмолярность надосадочной жидкости. Содержание сухих веществ в продуктах центрифугирования определяли электронным рефрактометром DUR.
Гидролиз осуществляли так же, как описывалось выше, но после регулирования показателя рН содержимое колбы Эрленмейера доводили до 95 мл. После обработки в термостате добавляли 2,5 мг эндо-1,3(4)-β-глюканазы. Гидролиз осуществляли в условиях перемешивания магнитной мешалкой при температуре 50oС, при этом колбу Эрленмейера помещали в водяную баню. Пробы отбирали и измеряли, так, как описывалось выше.
В таблице 2 приведены данные, полученные для контрольного гидролиза и материала клеточных стенок дрожжей, обработанного эндо-1,3(4)-β-глюканазой.
Противопоставленные материалы
Aviv H. & Leder, P. 1972. Proc.Natl.
Acad. Sci., USA, 69:
1408-1412.
Becker D.M. & Guarante L. 1991. Methods Enzymol., 194: 181-187.
Chirgwin J.M., Przybya A.E., MacDonald, R.J. & Putter, W.J. 1979. Biochemistry 18: 5294-5299.
Gubler U. & Hoffman B.J. 1983. Gene 25: 263-269.
Sambrook J., Frisch E.F. & Maniatis T., 1989. Molecular Cloning: F Laboratory Manual. Cold Spring Harbor, NY.
Sanger F., Nicklen S. & Coulson A.R. 1977. Proc. Natl. Acad. Sci., USA, 74: 5463-5467.
Pitson et al., Noncellolytic fungal β-glucanases: Their physiology and regulation. In Enzyme Microb. Technol., 1993, vol.15, March.
Reed and Nagodawithana, in Yeast Technology, Second Edition, pp.372-380.
Phaff H.J., Enzymatic Yeast Cell Wall Degradation. In Food Proteins, Improvement through Chemical and Enzymatic Modification, edited by Feeney and Whitaker, Waschington, 1977.
Hamlyn et al., Efficient protoplast isolation from fungi using commtrcial enzymes. In Enzyme Microb. Technol., 1981, vol.3, October.
Andrews and Asenjo, Enzymatic lysis and disruption of microbial cells. In Tibtech, October 1987, vol.5.
Hudson H.J. in "Fungal Biology", eds. Willis A.J. and Sleigh M.A., 1986, Edward Arnold (Publishers), Ltd.
Kofod et al., 1994, J. Biol. Chem., 261, 8407-8413 van den Hazel et al., 1992, Eur. J.Biochem., 207. 277-283.
Becker and Guarente, 1991, Methods Enzymol., 194, 182-187.
Sherman, 1991, Methods Enzymol. 194, 3-21.
Meissner et al., 1987, Proc. Matl. Acad. Sci. USA, 84, 4171-4176.
Devereux et al., 1984, Nucleic Acids Res., 12, 387-395.
Laemmli et al., 1970. Nature, vol.227, p.680-685.
Christgau, Schierbeck et al., 1991, J.Biol. Chem., vol.266, p.21157-21164.
Реферат
Изобретение относится к области получения ферментных препаратов методами генной инженерии и может быть использовано в биотехнологических процессах и микробиологической промышленности. В результате скрининга кДНК-библиотеки Trichoderma harzianum получена и секвенирована последовательность ДНК, кодирующая фермент эндо-1,3(4)-β-глюканазу, обладающий способностью расщеплять 1,3-β -D-гликозидные связи в структуре β-глюканов. Предложены рекомбинантные векторы, содержащие полученную последовательность ДНК и обеспечивающие ее экспрессию в дрожжевых клетках. При культивировании штаммов Aspergillus sp., трансформированных сконструированным вектором экспрессии, получена рекомбинантная эндо-1,3(4)-β-глюканаза. Определены физико-химические и каталитические характеристики фермента, в соответствии с которыми он может применяться в процессах, предусматривающих разрушение клеточных стенок многих микробных и растительных продуцентов. 3 с. и 12 з.п. ф-лы, 6 ил., 2 табл.


Комментарии