Сорбент для связывания металлов и его получение - RU2676067C2
Код документа: RU2676067C2
Чертежи







Описание
Настоящее изобретение относится к сорбенту, который подходит для связывания металлов из растворов, к получению соответствующего сорбента, а также к применению сорбента для связывания металлов из растворов.
Удаление, или извлечение, или выделение металлов, в частности, тяжелых металлов, из промышленных сточных вод, например, в гальванических цехах, из каталитических остатков, образующихся на предприятиях нефтехимической или фармацевтической промышленности, из рудничной воды, например, из шахт, восстановление почв, загрязненных тяжелыми металлами, представляет собой все более важную проблему, поскольку тяжелые металлы, в частности, пагубно влияют на окружающую среду, а их выделение представляет собой экономический интерес. Другими словами, с одной стороны, приоритетными являются экологические аспекты, а с другой стороны - большой интерес представляет собой также получение ценных металлов, доступность которых становится все более сомнительной или цена которых растет. Дополнительной важной областью применения сорбентов для удаления, или извлечения, или выделения металлов или тяжелых металлов является их удаление при очистке питьевой воды, а также при обессоливании морской воды. Аналогично, удаление тяжелых металлов из концентрированных солевых растворов, таких как используют при хлорщелочном электролизе или в аналогичных процессах, также представляет собой большой интерес.
Для указанных областей применения известные ранее фазы/сорбенты зачастую не обладают необходимой связывающей способностью для связывания металлов в достаточной степени, например, из высококонцентрированных или низкоконцентрированных растворов или из сильнокислотных растворов, в частности, также в присутствии ионов щелочных или щелочноземельных металлов. Кроме того, известные ранее фазы зачастую не были стабильными во всем диапазоне от рН 0 до рН 14. Дополнительный недостаток многих известных ранее фаз заключается в том, что требуемый металл может быть в действительности связан, но не может быть выделен легко или не может быть выделен вовсе из использованного сорбента. Вследствие почти всегда неудовлетворительной связывающей способности известных сорбентов/фаз, зачастую необходим большой объем сорбента/фазы, в результате чего процессы связывания металла являются весьма трудоемкими и дорогостоящими. Кроме того, вследствие почти всегда низкой связывающей способности известных металлосвязывающих сорбентов, необходимо многократное повторение процесса, например, для обеспечения возможности получения воды, не содержащей тяжелых металлов, в качестве питьевой воды.
Таким образом, задача настоящего изобретения заключается в обеспечении нового сорбента, который частично или полностью не имеет вышеуказанных недостатков. В частности, задача настоящего изобретения заключается в обеспечении сорбента с высокой связывающей способностью в отношении металлов, в частности, тяжелых металлов и благородных металлов, на грамм или на миллилитр. Предпочтительно, сорбент, предложенный согласно настоящему изобретению, является, в частности, возобновляемым с помощью гидроксида натрия или обеспечивает возможность выделения металлов простым способом. Дополнительная задача настоящего изобретения заключается в обеспечении сорбента, который обладает относительно высокой связывающей способностью в отношении металлов даже в кислотных условиях.
Кроме того, по сравнению с сорбентами, связывающими металлы, известными из уровня техники, объем сорбента, используемого для связывания металла, должен быть снижен.
Задача настоящего изобретения решена с помощью сорбента, который содержит пористый материал подложки, покрытый полимером, содержащим аминогруппу, при этом концентрация аминогрупп сорбента, определенная посредством титрования, составляет по меньшей мере 600 мкмоль/мл относительно общего объема сорбента.
В дополнительном варианте реализации настоящего изобретения предпочтительно, что концентрация аминогрупп сорбента, определенная посредством титрования, составляет по меньшей мере 800 мкмоль/мл, более предпочтительно по меньшей мере 1000 мкмоль/мл, еще более предпочтительно по меньшей мере 1200 мкмоль/мл и наиболее предпочтительно по меньшей мере 1500 мкмоль/мл сорбента. Верхний предел концентрации аминогрупп сорбента согласно настоящему изобретению, определенной посредством титрования, ограничен пространственной возможностью или максимально возможной плотностью расположения аминогрупп в полимере, содержащем аминогруппы, и составляет не более примерно 4000 мкмоль/мл, более предпочтительно 3000 мкм/мл и наиболее предпочтительно примерно 2500 мкмоль/мл. Концентрация аминогрупп сорбента, определенная посредством титрования, означает концентрацию, которая получена в соответствии с аналитическим способом, указанным в разделе примеров настоящей заявки, посредством пробойного измерения с 4-толуолсульфоновой кислотой.
Кроме того, предпочтительно, что сорбент согласно настоящему изобретению имеет отношение массы полимера, содержащего аминогруппу, к общему объему пор пористого материала подложки более или равно 0,1 г/мл, более предпочтительно более или равно 0,125 г/мл, еще более предпочтительно более или равно 0,15 г/мл и наиболее предпочтительно более или равно 0,20 г/мл. И снова, для верхнего предела указанного отношения установлены физические ограничения, однако предпочтительно оно составляет не более примерно 0,5 г/мл, более предпочтительно не более 0,4 г/мл и наиболее предпочтительно не более 0,3 г/мл.
Масса полимера, содержащего аминогруппу, может быть определена по увеличению насыпной плотностью по сравнению с материалом подложки, в соответствии с DIN 53194. Общий объем пор [V] пористого материала подложки может быть определен по емкости абсорбции растворителя (WAC) пористого материала подложки. Объем пор [об. %] также может быть определен аналогичным образом. В данном контексте в каждом случае он представляет собой объем свободно доступных пор материала подложки, поскольку с помощью емкости абсорбции растворителя может быть определен только этот параметр. Емкость абсорбции растворителя определяет объем растворителя, необходимый для полного заполнения порового пространства одного грамма сухого сорбента (предпочтительно неподвижной фазы). При этом в качестве растворителя может быть использована чистая вода или водная среда, а также органические растворители, такие как диметилформамид. Если сорбент увеличивается в объеме (набухает) при увлажнении, то автоматически определяют количество растворителя, использованного для этого. Для измерения WAC точно взвешенное количество сухого сорбента тщательно смачивают избытком эффективно увлажняющего растворителя, а избыток растворителя удаляют из межчастичного объема посредством центрифугирования. При этом растворитель удерживается в порах сорбента. Массу удержанного растворителя определяют взвешиванием и пересчитывают в объем через плотность. WAC сорбента записывают как объем на грамм сухого сорбента (мл/г).
Покрытие из полимера, содержащего аминогруппу, на пористом материале подложке предпочтительно присутствует в форме гидрогеля. Это, в частности, обусловлено тем, что полимер, содержащий аминогруппу, имеет вышеуказанную высокую концентрацию аминогрупп. В данном контексте гидрогель означает содержащий растворитель (предпочтительно воду), но растворимый в растворителе полимер, молекулы которого химически, например, посредством ковалентных или ионных связей, или физически, например, посредством переплетения полимерных цепей, связаны в трехмерную сетчатую структуру. Вследствие встроенных полярных (предпочтительно гидрофильных) полимерных компонентов, они набухают в растворителе (предпочтительно воде) со значительным увеличением объема, однако не теряя когезию материала. Из уровня техники известно, что гидрогели необратимо до некоторой степени теряют свои свойства при высушивании. Однако в настоящей заявке гидрогели не теряют свои свойства, поскольку они химически и механически стабилизированы пористым материалом подложки. Затем покрытие, содержащее аминогруппу, присутствует, в частности, в виде гидрогеля в сорбенте согласно настоящему изобретению, если он набух в растворителе, т.е., в частности, при его использовании для связывания металлов из растворов, описанных ниже.
Пористый материал подложки предпочтительно является мезопористым или макропористым материалом подложки. Средний размер пор пористого материала подложки предпочтительно составляет от 6 нм до 400 нм, более предпочтительно от 10 до 300 нм и наиболее предпочтительно от 20 до 150 нм. Размер пор в указанном диапазоне является важным для обеспечения достаточно высокой связывающей способности. В случае слишком малого размера пор полимер, содержащий аминогруппу, на поверхности пористого материала подложки может блокировать поры, и внутренний объем пор не заполнен полимером, содержащим аминогруппу. Кроме того, предпочтительно, что пористый материал подложки имеет объем пор в диапазоне от 30 до 90 об. %, более предпочтительно от 40 до 80 об. % и наиболее предпочтительно от 60 до 70 об. %, в каждом случае относительно общего объема пористого материала подложки.
Средний размер пор пористого материала подложки может быть определен методом заполнения пор ртутью в соответствии с DIN 66133.
Пористый материал подложки может содержать органический полимер, неорганический материал или композиционный материал органических полимеров и неорганических материалов, или может состоять из них.
Для обеспечения возможности получения сорбента с высокой стабильностью сорбента в диапазоне рН от 0 до рН 14, предпочтительно, если пористый материал подложки представляет собой органический полимер.
Предпочтительно, органический полимер для пористого материала подложки выбран из группы, состоящей из полиалкила, предпочтительно с ароматическим звеном в боковой цепи (т.е. связанным с полиалкильной цепью), полиакрилата, полиметакрилата, полиакриламида, поливинилового спирта, полисахаридов (например, крахмала, целлюлозы, сложных эфиров целлюлозы, амилозы, агарозы, сефарозы, маннана, ксантана и декстрана), а также их смесей. Наиболее предпочтительно, органический полимер представляет собой полистирол или производное полистирола, которое предпочтительно представляет собой сополимер полистирола (или производного полистирола) и дивинилбензола. Если органический полимер содержит ароматическое звено, то оно предпочтительно является сульфонированным. В особенно предпочтительном варианте реализации настоящего изобретения органический полимер представляет собой сульфонированный сшитый поли(стирол-со-дивинилбензол) или его производное.
Если пористый материал подложки представляет собой неорганический материал или он содержит неорганический материал, то неорганический материал предпочтительно представляет собой неорганический минеральный оксид, выбранный из группы, состоящей из оксида кремния, оксида алюминия, оксида магния, оксида титана, оксида циркония, флоризила, магнетита, цеолитов, силикатов (например, кизельгура), слюды, гидроксиапатита, фторапатита, металлорганических структур, керамических материалов, стекла, пористого стекла (например, Trisoperl), металлов, например, алюминия, кремния, железа, титана, меди, серебра и золота, графита и аморфного углерода. Особенно предпочтительно, неорганический пористый материал подложки представляет собой диоксид кремния или оксид алюминия, в частности, диоксид кремния. Диоксид кремния предпочтительно представляет собой силикагель.
В частности, по соображениям применения в широком диапазоне рН, в частности, в щелочном диапазоне, пористый материал подложки предпочтительно представляет собой органический полимер.
Пористый материал подложки, используемый в соответствии с настоящим изобретением, может иметь гомогенный или гетерогенный состав и, следовательно, в частности, содержит материалы, которые состоят из одного или более вышеуказанных материалов, например, в многослойных композициях.
Пористый материал подложки предпочтительно представляет собой дисперсный материал со средним размером частиц в диапазоне от 5 до 2000 мкм, более предпочтительно в диапазоне от 10 до 1000 мкм. Пористый материал подложки также может представлять собой пластинчатый или волокнистый материал, такой как, например, мембрана или пена. Таким образом, внешняя поверхность пористого материала подложки может быть плоской (пластинки, пленки, диски, мембраны, волокнистый материал или неволокнистый материал) или искривленной (вогнутой или выпуклой: сферические частицы, гранулы, (полые) волокна, трубки или капилляры).
Как упомянуто выше, пористый материал подложки покрыт полимером, содержащим аминогруппу, который состоит из отдельных полимерных цепей или содержит их. Полимерные цепи предпочтительно ковалентно связаны друг с другом. Полимер, содержащий аминогруппу, предпочтительно ковалентно не связан с поверхностью пористого материала подложки.
Применение ковалентно не связанного с поверхностью сшитого полимера в качестве полимера, содержащего аминогруппу, на пористом материале подложки, также имеет три следующих преимущества: (1) гибкость полимера, поскольку он ковалентно не связан с поверхностью пористого материала подложки; (2) сшивание полимера, содержащего аминогруппу, обеспечивает сохранность пленки на поверхности пористого материала подложки и отсутствие ее потерь при использовании сорбента; (3) толщина полимера, содержащего аминогруппу, на материале подложки может быть выбрана в соответствии с подходящим размером, если полимер ковалентно не связан с материалом подложки.
Достаточная гибкость и проницаемость полимера, содержащего аминогруппу, важна, поскольку некоторые аминогруппы могут переходить в конформацию, которая обеспечивает возможность многократного координационного связывания металлов.
Высокая способность связывания металлов сорбентов согласно настоящему изобретению или сорбентов, полученных в соответствии со следующими способами согласно настоящему изобретению, была неожиданной для авторов изобретения последующим причинам:
- несмотря на почти полное заполнение пор материала подложки полимером, содержащим аминогруппу, поры доступны для металлов благодаря проницаемости полимера, в результате чего сорбент согласно настоящему изобретению обладает высокой способностью связывания металлов. Это было тем более удивительно, поскольку полимерные стандарты, полученные с помощью обращенной эксклюзионной хроматографии, не демонстрировали доступности или проницаемости. Такой эффект должен наблюдаться также для значений более 90%, даже в случае наименьших стандартов размером 450 Да.
- В отличие от нормальных хроматографических сорбентов и металлосвязывающих сорбентов, которые основаны на принципе функционализации поверхности, по высокой металлосвязывающей способности было неожиданно обнаружено, что в настоящем изобретении использован весь объем полимера, отвечающего за связывание, а не только его поверхность, т.е. полимер, содержащий аминогруппу, вместе с растворителем, содержащим металлы, образует так называемый гидрогель, в котором полимерная сетчатая структура демонстрирует нанопористость. В результате металлосвязывающая способность определена не только поверхностью материала подложки, а скорее всем объемом нанесенного полимера.
- Высокая металлосвязывающая способность сорбентов согласно настоящему изобретению или сорбентов, полученных в соответствии с настоящим изобретением, обусловлена образованием химических комплексов между группами полимера, содержащего аминогруппу, и металлами, подлежащими связыванию. Указанные группы могут представлять собой сами аминогруппы или могут представлять собой остатки, которые обладают свойствами основания Льюиса, которые связаны с полимером, содержащим аминогруппу (как описано ниже). Это, например, обеспечивает преимущество высокой переносимости солей и связывающей способности в кислотной среде по сравнению с классическими ионообменниками.
- Параллельно с образованием химических комплексов, приводящим к связыванию металлов, фаза также обладает очень высокой связывающей способностью в отношении анионов, например, сульфата, фосфата, нитрита, нитрата, хромата, арсената и т.д.
Полимер, содержащий аминогруппу, на сорбенте согласно настоящему изобретению предпочтительно представляет собой полимер, который содержит первичные и/или вторичные аминогруппы. Он может представлять собой полимер из одинаковых повторяющихся звеньев (полимеризованные мономеры), но он также может представлять собой сополимер, который предпочтительно содержит простые алкеновые мономеры или полярные, инертные мономеры, такие как винилпирролидон, в качестве сомономеров.
Примеры полимера, содержащего аминогруппу, представлены ниже: полиамины, такие как любые полиалкиламины, например, поливиниламин и полиаллиламин, полиэтиленимин, полилизин и т.д. Среди них предпочтительны полиалкиламины, а поливиниламин и полиаллиламин являются более предпочтительными, при этом особенно предпочтительным является поливиниламин.
В соответствии с предпочтительным вариантом реализации сорбента согласно настоящему изобретению, полимер, содержащий аминогруппу, имеет степень сшивания по меньшей мере 2% относительно общего количества сшиваемых групп в полимере, содержащем аминогруппу. Более предпочтительно, степень сшивания составляет от 2,5 до 60%, более предпочтительно от 5 до 50% и наиболее предпочтительно от 10 до 40%, в каждом случае относительно общего количества сшиваемых групп в полимере, содержащем аминогруппу. Степень сшивания может быть отрегулирована с помощью соответствующего необходимого количества сшивающего агента. В данном контексте принято, что 100 мол. % сшивающего агента вступает в реакцию и образует поперечные связи. Это можно проверить аналитическими методами, такими как спектроскопия ЯМР с вращением образца под магическим углом и количественное определение количества сшивающего агента относительно количества использованного полимера. Указанный способ является предпочтительным согласно настоящему изобретению. Однако степень сшивания может быть определена также ИК спектроскопией на основании, например, колебаний С-О-С или ОН с использованием калибровочной кривой. Оба способа представляют собой стандартные аналитические методы для специалистов в данной области техники. Если степень сшивания находится выше указанного верхнего предела, то полимерное покрытие из полимера, содержащего аминогруппу, не является достаточно гибким и обусловливает более низкую металлосвязывающую способность. Если степень сшивания ниже указанного нижнего предела, то полимерное покрытие не является достаточно стабильным на поверхности пористого материала подложки.
Сшивающий агент имеет две, три или более функциональных групп, посредством связывания которых с полимером происходит поперечное сшивание. Сшивающий агент, который используют для сшивания полимера, содержащего аминогруппу, предпочтительно выбран из группы, состоящей из дикарбоновых кислот, трикарбоновых кислот, мочевины, бис-эпоксидов или трис-эпоксидов, диизоцианатов или триизоцианатов, и дигалогеналкилена или тригалогеналкилена, при этом предпочтительны дикарбоновые кислоты из бис-эпоксиды, такие как, например, терефталевая кислота, бифенилдикарбоновая кислота, диглицидиловый эфир этиленгликоля и 1,12-бис(5-норборнен-2,3-дикарбоксимидо)декандикарбоновая кислота, при этом диглицидиловый эфир этиленгликоля и 1,12-бис(5-норборнен-2,3-дикарбоксимидо)декандикарбоновая кислота являются более предпочтительными. В одном из вариантов реализации настоящего изобретения сшивающий агент представляет собой предпочтительно линейную, конформационно гибкую молекулу с длиной от 4 до 20 атомов.
Предпочтительная молекулярная масса полимера, содержащего аминогруппу, в сорбенте согласно настоящему изобретению, предпочтительно составляет от 5000 до 50000 г/моль, что относится, в частности, к применению поливиниламина.
В дополнительном варианте реализации сорбент согласно настоящему изобретению может также иметь органические остатки, которые связаны на полимере, содержащем аминогруппу, и имеют природу основания Льюиса. В данном контексте особенно предпочтительно, что указанный органический остаток связан на аминогруппе полимера, содержащего аминогруппу. Также особенно предпочтительно, что аминогруппа, на которой связан органический остаток, после такого связывания представляет собой вторичную аминогруппу, так что она также все еще демонстрируют достаточную основность Льюиса, но без стерического затруднения.
В дополнительном варианте реализации настоящее изобретение относится также к способу получения сорбента, предпочтительно сорбента согласно настоящему изобретению, который включает следующие стадии (предпочтительно в указанном порядке):
(a) обеспечение пористого материала подложки;
(b) нанесение полимера, содержащего аминогруппу, на пористый материал подложки посредством способа заполнения пор;
(c) удаление растворителя, использованного в способе заполнения пор;
(d) повторение стадий (b) и (с); и
(e) сшивание полимера, содержащего аминогруппу.
В способе согласно настоящему изобретению для получения сорбента пористый материал подложки, обеспечиваемый на стадии (а), представляет собой материал, упомянутый выше в отношении сорбента согласно настоящему изобретению. Предпочтительные варианты реализации, упомянутые в указанном разделе, в равной степени применимы в данном контексте.
На стадии (b) способа согласно настоящему изобретения предпочтительно используют не сшитый полимер, содержащий аминогруппу, который является таким, как описано выше в отношении полимера, содержащего аминогруппу, сорбента согласно настоящему изобретению. Предпочтительные варианты реализации, упомянутые в указанном разделе, в равной степени применимы в данном контексте.
Способ заполнения пор на стадии (b) настоящей заявки полимера, содержащего аминогруппу, на пористом материале подложки в способе согласно настоящему изобретению имеет преимущество в сравнении с обычными способами пропитывания, заключающееся в том, что, в целом, может быть нанесено большее количество полимера, содержащего аминогруппу, на пористый материал подложки, в результате чего связывающая способность в отношении металлов увеличивается. Это обеспечивает вышеуказанные неожиданные преимущества.
Указанный способ заполнения пор, в целом, означает специальный способ нанесения покрытия, в котором раствор, который содержит полимер, содержащий аминогруппу, наносят на пористый материал подложки в количестве, которое соответствует общему объему пор пористого материала подложки. В данном контексте общий объем пор пористого материала подложки на стадии (b), т.е. при первом нанесении, определяют заранее, как описано выше.
На стадии (с) способа согласно настоящему изобретению растворитель, используемый для способа заполнения пор, предпочтительно удаляют высушиванием материала при температурах от 40°С до 90°С, более предпочтительно от 50°С до 70°С и наиболее предпочтительно от 50°С до 60°С. При этом на высушивание проводят, в частности, при давлении от 0,01 до 1 бар, более предпочтительно при давлении от 0,1 до 0,5 бар.
Принципиальная стадия способа согласно настоящему изобретению для получения сорбента заключается в том, что на стадии (d) после высушивания или удаления растворителя после первой стадии нанесения с помощью способа заполнения пор, повторяют стадии (b) и (с) нанесения полимера, содержащего аминогруппу, на пористый материал подложки с помощью способа заполнения пор. Для этого общий объем пор, доступный для повторного нанесения полимера, содержащего аминогруппу, на пористый материал подложки, определяют после стадии (b) с помощью дифференциального взвешивания влажного и сухого материала. В дополнительном варианте реализации способа согласно настоящему изобретению еще более предпочтительно, что стадии (b) и (с) повторяют по меньшей мере дважды. Общий объем пор, доступный для осуществления способа заполнения пор, также определяют дифференциальным взвешиванием влажного и сухого материала до второго повтора стадий (b) и (с). Повторение стадий (b) и (с) предпочтительно осуществляют в указанном порядке.
После стадий нанесения полимера, содержащего аминогруппу, на стадии (е) осуществляют сшивание полимера, содержащего аминогруппу, предпочтительно с помощью сшивающего агента, указанного в отношении сорбента согласно настоящему изобретению. Все признаки, указанные выше в связи с сорбентом согласно настоящему изобретению в отношении сшивания, применимы также к способу получения согласно настоящему изобретению.
Дополнительно предпочтительно, что между многочисленными стадиями нанесения полимера, содержащего аминогруппу, на пористый материал подложки посредством способа заполнения пор, не проводят сшивание полимера, содержащего аминогруппу.
Предпочтительно, удаление растворителя, используемого в способе заполнения пор, каждый раз осуществляют высушиванием в сошниковой сушилке, поскольку указанная стадия может быть заметно ускорена с помощью нее.
В дополнительном варианте реализации в способе согласно настоящему изобретению стадии (b) и (с) повторяют до стадии (е) достаточно часто, чтобы концентрация аминогрупп сорбента, определенная после стадии (е) посредством титрования, составляла по меньшей мере 600 мкмоль/мл, более предпочтительно по меньшей мере 800 мкмоль/мл, еще более предпочтительно по меньшей мере 1000 мкмоль/мл и наиболее предпочтительно по меньшей мере 1200 мкмоль/мл, в каждом случае относительно общего объема сорбента. Верхние пределы концентрации аминогрупп сорбента, указанные выше в отношении сорбента согласно настоящему изобретению, также представляют собой верхние пределы, предпочтительные в способе согласно настоящему изобретению.
В дополнительном варианте реализации способа согласно настоящему изобретению предпочтительно, что отношение массы полимера, содержащего аминогруппу, к общему объему пор пористого материала подложки после стадии (d) составляет более или равно 0,1 г/мл, более предпочтительно более или равно 0,125 г/мл и наиболее предпочтительно более или равно 0,15 г/мл. Верхний предел указанного отношения предпочтительно составляет не более 0,5 г/мл, более предпочтительно не более 0,4 г/мл и наиболее предпочтительно не более 0,3 г/мл.
В способе заполнения пор на стадии (b) способа согласно настоящему изобретению, в качестве растворителя для полимера, содержащего аминогруппу, предпочтительно используют растворитель, в котором растворим полимер, содержащий аминогруппу. Концентрация полимера, содержащего аминогруппу, в растворителе, используемая для способа заполнения пор на стадии (b) способа согласно настоящему изобретению, предпочтительно составляет от 5 г/л до 200 г/л, более предпочтительно от 10 г/л до 180 г/л, наиболее предпочтительно от 30 до 160 г/л. Концентрация ниже указанного нижнего предела имеет недостаток, заключающийся в том, что стадии (b) и (с) следует проводить слишком часто для достижения требуемой концентрации аминогрупп сорбента, определяемой титрованием, которая обеспечивает достаточную связывающую способность в отношении металлов. Концентрация выше указанного верхнего предела не обеспечивает возможность достаточной степени проникновения полимера в поры пористого материала подложки.
В дополнительном варианте реализации способа согласно настоящему изобретению предпочтительно, что на стадии (f), предпочтительно после стадии (е), органический остаток, который имеет природу основания Льюиса, связан на полимере, содержащем аминогруппу. В данном контексте особенно предпочтительно, что указанный органический остаток связан на аминогруппах полимера, содержащего аминогруппы. В данном контексте дополнительно предпочтительно, что после связывания органического остатка аминогруппы присутствуют в виде вторичных аминогрупп, поэтому их основность Льюиса не утрачена, и не возникает никакого стерического затруднения для связывания аминогрупп с металлами. Органический остаток, который имеет природу основания Льюиса, понимают, в частности, как обозначающий остатки, которые образуют комплексные связи с металлом, подлежащим связыванию.
Органические остатки, которые содержат основание Льюиса, представляют собой, например, остатки, которые содержат гетероатомы со свободными парами электронов, такие как N, О, Р, As или S.
Все предпочтительные варианты реализации, упомянутые выше в отношении сорбента согласно настоящему изобретению, в такой же степени относятся к сорбенту, полученному способом согласно настоящему изобретению, или к компонентам, используемым в способе согласно настоящему изобретению.
В дополнительном варианте реализации настоящее изобретение относится также к сорбенту, который может быть получен способом согласно настоящему изобретению, в частности, к сорбенту, который может быть получен способом согласно настоящему изобретению, в котором на стадии (f) органический остаток, который имеет природу основания Льюиса, связан на полимере, содержащем аминогруппу. Вследствие функционализации органическим остатком такой сорбент также может иметь концентрацию аминогрупп сорбента, определенную титрованием, менее указанного выше предела, но однако характеризуется, в частности, тем, что в результате однократного или многократного повторения стадий (b) и (с) в способе согласно настоящему изобретению, свободно доступные поры материала подложки по существу полностью заполнены полимером, содержащим аминогруппу (это достигается при WAC менее 0,5 мас. %), или отношение массы полимера, содержащего аминогруппу, к общему объему пористого материала подложки после стадии (d) находится в диапазоне, указанном выше. Такой сорбент, который содержит органический остаток, имеющий природу основания Льюиса, также включает сорбенты, которые после удаления органического остатка из полимера, содержащего аминогруппу, имеют концентрацию аминогрупп сорбента, определенную титрованием, по меньшей мере 600 мкмоль/мл относительно общего объема сорбента.
Дополнительный вариант реализации настоящего изобретения относится к применению сорбента для связывания металлов из растворов, при этом сорбент представляет собой либо пористый материал подложки, покрытый полимером, содержащим аминогруппу, причем концентрация аминогрупп сорбента, определенная посредством титрования, составляет по меньшей мере 300 мкмоль/мл, более предпочтительно по меньшей мере 400 мкмоль/мл и еще более предпочтительно 500 мкмоль/мл, либо сорбент представляет собой сорбент, который может быть получен способом согласно настоящему изобретению.
Другими словами, настоящее изобретение относится также к способу связывания металлов из растворов с применением сорбента, при этом сорбент представляет собой либо пористый материал подложки, покрытый полимером, содержащим аминогруппу, причем концентрация аминогрупп сорбента, определенная посредством титрования, составляет по меньшей мере 300 мкмоль/мл, более предпочтительно по меньшей мере 400 мкмоль/мл и еще более предпочтительно 500 мкмоль/мл, либо сорбент представляет собой сорбент, который может быть получен способом согласно настоящему изобретению.
Растворы, из которых могут быть связаны металлы, согласно настоящему изобретению могут быть концентрированными или разбавленными, водными или неводными, кислотными, основными или нейтральными растворами.
Металлы согласно настоящей заявке предпочтительно представляют собой металлы, которые присутствуют в указанных растворах в ионной форме или также в виде координационных соединений металла и лиганда в ионной форме. Металлы предпочтительно представляют собой комплексообразующие металлы, т.е. металлы, которые могут участвовать в координационной связи металла с лигандом. Более предпочтительно, металлы представляют собой переходные металлы или редкоземельные металлы, еще более предпочтительно благородные металлы или редкоземельные металлы. Особенно предпочтительно, металлы представляют собой медь, никель и хром.
В дополнительном варианте реализации применения согласно настоящему изобретению растворы, из которых связывают металлы, представляют собой растворы, имеющие содержание солей ионов щелочных металлов по меньшей мере 5 г/л.
Кроме того, растворы, из которых связывают металлы, предпочтительно представляют собой водные растворы, в частности также кислотные водные растворы с рН≤5, более предпочтительно ≤4 и еще более предпочтительно ≤3.
Для связывания металлов из растворов металлосодержащие растворы приводят в контакт с сорбентом согласно настоящему изобретению. Например, это может быть осуществлено в колонке с нормальными фазами. В то же время, сорбенты согласно настоящему изобретению, которые разработаны для связывания других металлов, также могут присутствовать в смеси друг с другом. Это, как правило, осуществляют посредством связывания различных органических остатков на полимере, содержащем аминогруппу.
Таким же образом, приведение в контакт сорбента согласно настоящему изобретению с раствором, содержащим металл, также может быть осуществлено периодическим способом, т.е. без пропускания раствора через емкость с сорбентом, а в форме суспензии сорбента в указанном растворе.
Далее настоящее изобретение иллюстрировано на основании следующих фигур и примеров, которые однако следует рассматривать лишь в качестве примера:
Описание графических материалов:
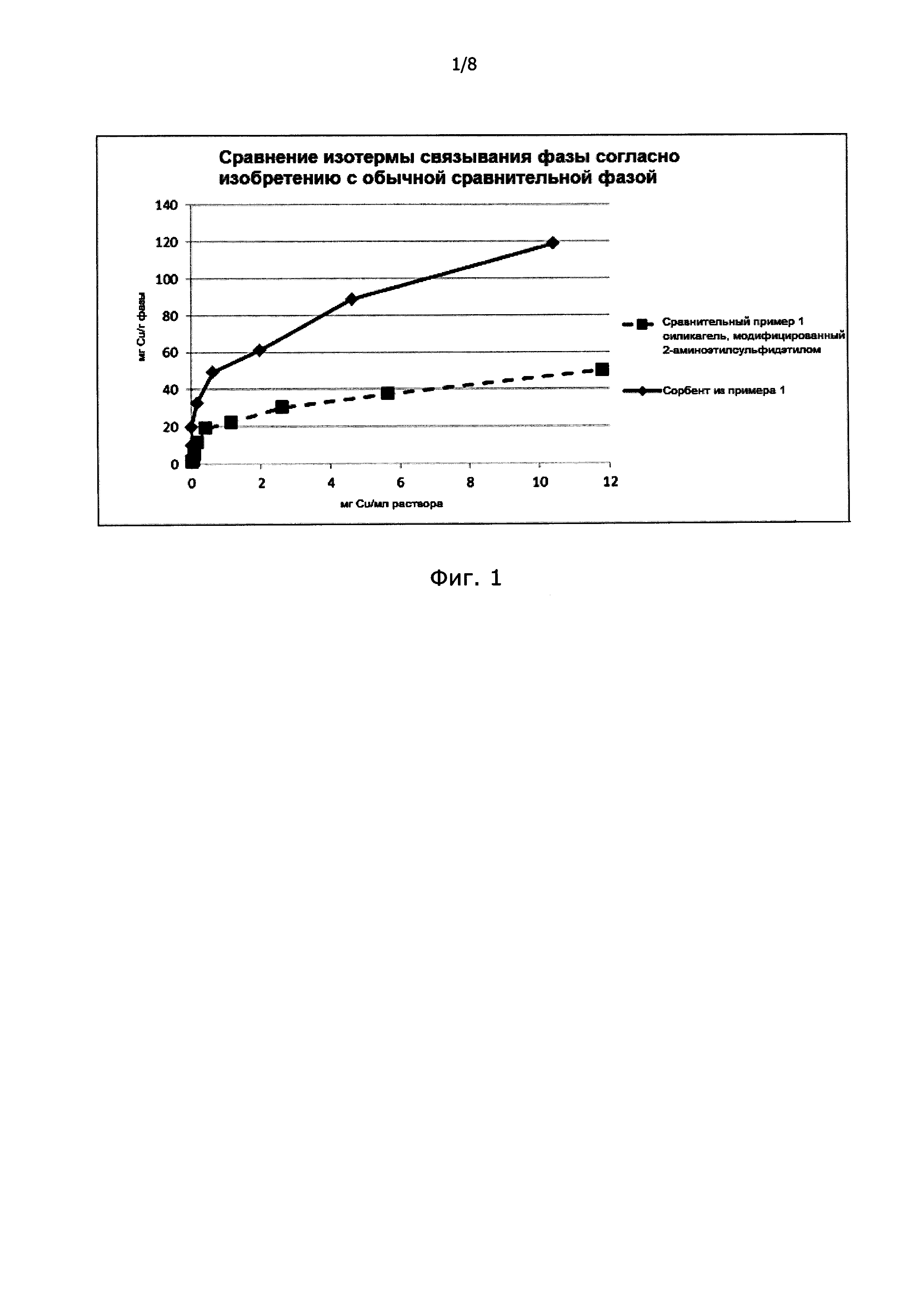
Фигуры 1 и 2: На фиг. 1 и 2 представлено сравнение изотерм сорбентов из примера 1 и сравнительного примера 1 для связывания меди из водных растворов в соответствии с примером 2.
Фигура 3: На фиг. 3 представлена емкость связывания металла сорбента из примера 1 для металлической меди, никеля и хрома в зависимости от концентрации металла в соответствии с примером 3.
Фигура 4: На фиг. 4 представлено количество абсорбированной меди [г] на количество сорбента [кг] в присутствии различных концентраций NaCl в соответствии с примером 4.
Фигуры 5 и 6: На фиг. 5 и 6 представлено изменение абсорбции меди сорбентом из примера 1 в зависимости от времени в соответствии с примером 5.
Фигура 7: На фиг. 7 представлена связывающая емкость в отношении меди после регенерации сорбента после различных циклов в соответствии с примером 6.
Фигура 8: На фиг. 8 представлено сравнение связывания меди многократно покрытыми сорбентами с однократно покрытым сорбентом в соответствии с примером 7.
Примеры:
Аналитические методы:
Определение концентрации аминогрупп сорбента посредством пробойного измерения с 4-толуолсульфоновой кислотой (титриметрический анализ):
Динамическую анионообменную емкость определяли с помощью колонки неподвижной фазы, подлежащей испытанию. Для этого сначала все обмениваемые анионы в колонке обменивали на трифторацетат. Затем колонку промывали водным раствором реагента толуол-4-сульфоновой кислоты до появления указанного раствора в той же концентрации в конце колонки (пробоя). По концентрации раствора толуол-4-сульфоновой кислоты, ее скорости потока и площади пробоя на хроматограмме рассчитывали количество толуол-4-сульфоновой кислоты, связанной материалом колонки. Количество толуол-4-сульфоновой кислоты, определенное таким образом, определяет концентрацию аминогрупп сорбента.
Динамическая анионообменная емкость в отношении толуол-4-сульфоновой кислоты в воде связана с объемом фазы, и ее записывают в ммоль на литр (мМ/л).
Пример 1: Получение сорбента согласно настоящему изобретению:
200 г материала подложки из сульфонированного полистирола/дивинилбензола (средний размер пор 30 нм) взвешивали в емкости. Указанный материал имел объем пор, определенны с помощью WAC, 1,48 мл/г. В первом покрытии объем пор должен быть заполнен на 95%. Получали раствор полимера для покрытия. 165,3 г раствора поливиниламина (содержание твердых веществ 12,1 мас. %) разбавляли в 108 г воды. рН раствора доводили до 9,5 с помощью 7 мл конц. хлористоводородной кислоты. Раствор полимера добавляли к подложке и перемешивали в течение 3 часов на подвесном шейкере. Затем подложку с нанесенным покрытие сушили в течение 48 часов при 50°С в вакуумном сушильном шкафу при 25 мбар. Потеря воды при высушивании составляла 197,7 г. На материал наносили покрытие второй раз. Для этого 165,0 г раствора поливиниламина (содержание твердых веществ 12,1 мас. %) доводили до рН 9,5 с помощью 6,8 мл конц. HCl и разбавляли в 20 г воды. Раствор полимера добавляли к подложке и перемешивали в течение 3 часов на подвесном шейкере. Затем подложку с нанесенным покрытие сушили в течение 48 часов при 50°С в вакуумном сушильном шкафу при 25 мбар. Потеря воды при высушивании составляла 181,2 г. На материал наносили покрытие третий раз. Для этого 165,2 г раствора поливиниламина (содержание твердых веществ 12,1%) доводили до рН 9,5 с помощью 7,1 мл конц. HCl и разбавляли в 5 г воды. Раствор полимера добавляли к подложке и перемешивали в течение 3 часов на подвесном шейкере. Затем фазу сушили до постоянной массы в вакуумном сушильном шкафу при 50°С и 25 мбар.
На материал наносили покрытие в 3 стадии с общим содержанием 0,20 г PVA на мл объема пор.
Высушенный материал суспендировали в 1,5 л изопропанола в реакторе с двойной рубашкой и подвергали сшиванию с 24,26 г диглицидилового эфира этилендигликоля при 55°С в течение 6 часов.
Материал с покрытием промывали следующими растворителями: 600 мл изопропанола, 3600 мл 0,1 М HCl, 1800 мл воды, 1800 мл 1 М NaOH, 1800 мл воды и 1800 мл метанола.
Затем материал сушили. Выход составил 275 г высушенного материала.
Анализ: концентрация аминогрупп, определенная титрованием, составила 963 мкмоль/мл.
Сравнительный пример 1: Получение обычного сорбента:
Обычный сорбент представляет собой силикагель, модифицированный 2-аминоэтилсульфидэтилом ((Si)-CH2-CH2-S-CH2-CH2-NH2) с размером частиц >45 мкм (производитель Phosphonics, поставщик Sigma-Aldrich, номер по каталогу: 743453-10G; содержание 0,8-1,3 ммоль/г).
Модификация может быть осуществлена посредством взаимодействия силикагеля с 3-меркапто-пропил-триметоксииланом с последующим взаимодействием с этилимином.
Пример 2: Применение сорбентов, полученных в соответствии с примером 1 и сравнительным примером 1, для связывания меди из водных растворов:
Для построения изотерм, представленных на фиг. 1 и 2, использовали следующую методику:
10 образцов сорбента, каждый массой примерно 100 мг, точно взвешивали и в каждом случае выдерживали с различными водными растворами Сu (II) (как CuSO4) разной концентрации в течение по меньшей мере 1,5 часов. Сорбент отфильтровывали и фотометрически определяли концентрацию Сu (II) в растворе. По оставшейся концентрации меди рассчитывали количество связанной меди и получали изотермы.
Сравнивая изотермы на фиг. 1 и 2, можно видеть, что сорбент согласно настоящему изобретению имеет существенно более высокую связывающую способность, чем обычный сорбент. По аппроксимированным прямоугольным изотермам для сорбента согласно настоящему изобретению можно видеть, что он демонстрирует очень прочное связывание без фактического установления равновесия. Это также обеспечивает возможности почти полного удаления тяжелых металлов из очень разбавленных растворов.
Пример 3: Применение сорбента, полученного в соответствии с примером 1, для связывания трех переходных металлов - никеля, меди и хрома - из раствора:
В каждом случае 10 образцов определенного количества сорбента точно взвешивали и в каждом случае выдерживали с различными водными растворами металлов разной концентрации в течение по меньшей мере 1,5 часов. Сорбент отфильтровывали и определяли концентрацию металла в растворе фотометрически или методом определения металла компании Hach-Lange (предпочтительно фотометрически). По оставшейся концентрации металла рассчитывали количество связанного металла и получали изотермы.
По данным, представленным на фиг. 3, можно видеть, что сорбент согласно настоящему изобретению в высокой степени связывает металлы никель (~70 мг/мг сорбента), медь (~120 мг/мг сорбента) и хром (~80 мг/мг сорбента). Такие же результаты можно наблюдать для растворов металлов палладия, свинца и иридия.
Пример 4: Применение сорбента, полученного в соответствии с примером 1, для связывания металлов из растворов с высоким содержанием соли:
5 образцов примерно по 100 мг сорбента точно взвешивали и каждый выдерживали в течение по меньшей мере 0,5 часа с раствором Сu (в виде CuSO4*5H2O=50 мг/мл воды), который содержал 0 М NaCl, 0,01 М NaCl, 0,1 М NaCl, 0,5 М NaCl и 1 М NaCl, соответственно. Затем сорбент отфильтровывали и фотометрически определяли концентрацию меди в фильтрате. По оставшейся концентрации металла рассчитывали количество связанной меди и получали изотермы.
Как можно видеть на фиг. 4, даже при общей концентрации солей до 1 М связывающая способность сорбента в отношении меди составляет >100 мг Сu/г сорбента. Это свидетельствует об участии неионного механизма связывания, который, следовательно, заметно отличается от ионообменников, «имеющихся в продаже».
Отсутствует конкуренция за связывающие сайты меди с натрием, который не участвует в комплексообразовании. Таким образом, сохраняется связывающая способность в отношении меди.
Такое свойство обеспечивает возможность применения указанной фазы для обработки питьевой, поверхностной, рудничной и сточной воды, для установок обессоливания морской воды, воды хлорщелочного электролиза и т.д., в которой повсеместно встречаются и присутствующие в большом избытке щелочные и щелочноземельные металлы не должны быть помехой.
Пример 5: Степень связывания сорбента, полученного в соответствии с примером V.
Образцы примерно по 100 мг фазы точно взвешивали и выдерживали с раствором Сu (в виде CuSO4*5H2O=50 мг/мл воды) в течение определенного периода. Затем сорбент отфильтровывали и фотометрически определяли концентрацию меди в фильтрате. По оставшейся концентрации металла рассчитывали количество связанной меди.
На фиг. 5 и 6 представлено связывание меди из растворов в зависимости от времени. Примерно через 90 минут все связывающие сайты сорбента заняты медью. Изменения концентрации не наблюдали даже через 48 часов.
Пример 6: Возможность повторного использования сорбента, полученного в соответствии с примером 1:
1 г сорбента из примера 1 взвешивали и обрабатывали следующим образом:
1. Промывали 1 М NaOH (3×5 мл)
2. Промывали водой (3×5 мл).
3. Добавляли 50 мл Сu (в виде CuSO4*5H2O, 50 мг/мл)
4. Выдерживали в течение 90 мин.
5. Фильтровали
6. Определяли концентрацию меди в фильтрате (фотометрически) и рассчитывали количество связанной меди
7. Повторяли процедуру
Как можно видеть на фиг. 7, связывающая емкость в отношении меди не ухудшается через 10 циклов регенерации и повторного использования сорбента через 10 циклов (за исключением циклов 5 и 6), даже при обработке 5 М HCl и 1 М NaOH.
Пример 7: Сравнение сорбентов с многократным покрытием с сорбентом с однократным покрытием:
Получение сорбента с однократным покрытием на подложке из диоксида кремния:
В емкости взвешивали 100 г силикагеля AGC D-50-120A (средний размер пор 12 нм). Этот материал имел объем пор, определенный с помощью WAC, 1,12 мл/г. Получали раствор полимера для покрытия. 79,6 г раствора поливиниламина (содержание твердых веществ 11,3 мас. %) разбавляли в 20 г воды. рН раствора доводили до 9,5 с помощью 3 мл конц. хлористоводородной кислоты. Раствор полимера добавляли к подложке и перемешивали в течение 6 часов на просеивающем устройстве. Затем подложку с нанесенным покрытие сушили в течение 48 часов при 50°С в вакуумном сушильном шкафу при 25 мбар.
Содержание покрытия в материале составляло 0,08 г PVA на мл объема пор.
Высушенный материал суспендировали в 0,5 л изопропанола в реакторе с двойной рубашкой и подвергали сшиванию с 3,64 г диглицидилового эфира этилендигликоля при 55°С в течение 6 часов.
Материал с покрытием промывали следующими растворителями: 400 мл изопропанола, 1200 мл 0,1 М HCl, 400 мл воды, 800 мл 0,5 М триэтиламина в воде, 600 мл воды и 600 мл метанола.
Затем материал сушили. Выход составил 108,0 г высушенного материала.
Анализ: концентрация аминогрупп, определенная титрованием, составила 593 мкмоль/мл.
Получение сорбента с двойным покрытием на подложке из диоксида кремния:
В емкости взвешивали 250 г силикагеля AGC D-50-120A (средний размер пор 12 нм). Этот материал имел объем пор, определенный с помощью WAC, 1,12 мл/г. Получали раствор полимера для покрытия. 200 г раствора поливиниламина (содержание твердых веществ 11,3 мас. %) разбавляли в 60 г воды. рН раствора доводили до 9,5 с помощью 7,5 мл конц. хлористоводородной кислоты. Раствор полимера добавляли к подложке и перемешивали с помощью вибрации в течение 6 часов на просеивающем устройстве. Затем подложку с нанесенным покрытие сушили в течение 48 часов при 50°С в вакуумном сушильном шкафу при 25 мбар. Потеря воды при высушивании составляла 230 г. На материал наносили покрытие второй раз. Для этого 200 г раствора поливиниламина (содержание твердых веществ 11,3 мас. %) доводили до рН 9,5 с помощью 6,8 мл конц. HCl и разбавляли в 23 г воды. Раствор полимера добавляли к подложке и снова перемешивали с помощью вибрации в течение 6 часов на просеивающем устройстве. Затем подложку с нанесенным покрытие сушили в течение 48 часов при 50°С в вакуумном сушильном шкафу при 25 мбар.
Содержание покрытия в материале составляло 0,16 г PVA на мл объема пор.
Высушенный материал суспендировали в 1,5 л изопропанола в реакторе с двойной рубашкой и подвергали сшиванию с 18,2 г диглицидилового эфира этилендигликоля при 55°С в течение 6 часов.
Материал с покрытием промывали следующими растворителями: 1000 мл изопропанола, 3000 мл 0,1 М HCl, 1000 мл воды, 2000 мл 0,5 М триэтиламина в воде, 1500 мл воды и 1500 мл метанола.
Затем материал сушили. Выход составил 308 г высушенного материала.
Анализ: концентрация аминогрупп, определенная титрованием, составила 1254 мкмоль/мл.
Получение сорбента стройным покрытием на подложке из диоксида кремния:
В емкости взвешивали 250 г силикагеля AGC D-50-120A (средний размер пор 12 нм). Этот материал имел объем пор, определенный с помощью WAC, 1,12 мл/г. Получали раствор полимера для покрытия. 199 г раствора поливиниламина (содержание твердых веществ 11,3 мас. %) разбавляли в 60 г воды. рН раствора доводили до 9,5 с помощью 7,6 мл конц. хлористоводородной кислоты. Раствор полимера добавляли к подложке и перемешивали с помощью вибрации в течение 6 часов на просеивающем устройстве. Затем подложку с нанесенным покрытие сушили в течение 48 часов при 50°С в вакуумном сушильном шкафу при 25 мбар. Потеря воды при высушивании составляла 231 г. На материал наносили покрытие второй раз. Для этого 200 г раствора поливиниламина (содержание твердых веществ 11,3 мас. %) доводили до рН 9,5 с помощью 7,0 мл конц. HCl и разбавляли в 24 г воды. Раствор полимера добавляли к подложке и снова перемешивали с помощью вибрации в течение 6 часов на просеивающем устройстве. Затем подложку с нанесенным покрытие сушили в течение 48 часов при 50°С в вакуумном сушильном шкафу при 25 мбар. Потеря воды при высушивании составляла 210 г. На материал наносили покрытие третий раз. Для этого 199 г раствора поливиниламина (содержание твердых веществ 11,3 мас. %) доводили до рН 9,5 с помощью 7,0 мл конц. HCl и разбавляли в 4 г воды. Раствор полимера добавляли к подложке и снова перемешивали с помощью вибрации в течение 6 часов на просеивающем устройстве. Затем подложку с нанесенным покрытие сушили в течение 48 часов при 50°С в вакуумном сушильном шкафу при 25 мбар.
Содержание покрытия в материале составляло 0,24 г PVA на мл объема пор.
Высушенный материал суспендировали в 1,5 л изопропанола в реакторе с двойной рубашкой и подвергали сшиванию с 27,3 г диглицидилового эфира этилендигликоля при 55°С в течение 6 часов.
Материал с покрытием промывали следующими растворителями: 1000 мл изопропанола, 3000 мл 0,1 М HCl, 1000 мл воды, 2000 мл 0,5 М триэтиламина в воде, 1500 мл воды и 1500 мл метанола.
Затем материал сушили. Выход составил 330 г высушенного материала.
Анализ: концентрация аминогрупп, определенная титрованием, составила 1818 мкмоль/мл.
На фиг. 8 однозначно показано, что связывающая емкость сорбента с двойным и тройным покрытием резко увеличивается по сравнению с однократно покрытым сорбентом.
Сравнительный пример 2: Получение сорбента в соответствии с примером 2 DE 102011107197 А1:
В качестве основы для сорбента использовали Amberchrom CG1000S компании Rohm & Haas. Его сульфонировали следующим образом: 165 мл конц. H2SO4 помещали в реактор объемом 250 мл с регулируемой температурой. 30,0 г материала подложки добавляли к серной кислоте, а затем стаканчик для взвешивания трижды промывали, используя в каждом случае 20 мл конц. серной кислоты. После добавления материала подложки суспензию перемешивали и поддерживали при 80°С. Через 3 часа времени реакции суспензию выгружали из реактора и распределяли на два шприца по 150 мл. Серную кислоту удаляли при пониженном давлении, а фазу последовательно промывали 200 мл разбавленной (62%) серной кислоты, 125 мл воды, 175 мл метанола, 125 мл воды и, наконец, 175 мл метанола. Фазу высушивали при пониженном давлении, а затем сушили при 50°С под вакуумом. Влагопоглощающую способность или объем пор полученного сульфонированного полистирола определяли взвешиванием высушенного сульфонированного полистирола, обработкой таким же объемом воды с последующим удалением избытка воды центрифугированием. При этом вода, содержащаяся в порах, оставалась на месте.
Для покрытия полученного полистирола получали водный раствор поливиниламина, который состоял из поливиниламина со средней молярной массой 35000 г/моль. рН доводили до 9,5. Количество поливиниламина в данном случае составляло 15% от полистирола, подлежащего нанесению покрытия, а объем раствора составлял 95% от измеренного объема пор полистирола. Раствор поливиниламина вместе с полистиролом помещали в плотно закрытую РЕ бутылку и встряхивали с высокой частотой в течение 6 часов на сортировочном шейкере. При этом следует уделять внимание достаточному перемешиванию. После этого раствор поливиниламина самопроизвольно занял поры в полистироле. Затем полистирол сушили до постоянной массы при 50°С вакуумном сушильном шкафу.
Для сшивания поливиниламина полистирол с нанесенным покрытием помещали в трехкратный объем изопропанола и обрабатывали 5% диглицидилового эфира диэтиленгликоля относительно содержания аминогрупп в поливиниламине. Реакционную смесь перемешивали в течение шести часов в реакторе при 55°С. Затем ее переносили на стеклянный всасывающий фильтр и промывали 2 объемами слоя изопропанола, 3 объемами слоя 0,5 М раствора ТФК, 2 объемами слоя воды, 4 объемами слоя 1 М раствора гидроксида натрия и, наконец, 8 объемами слоя воды.
Анализ: концентрация аминогрупп, определенная титрованием, составила 265 мкмоль/мл.
Реферат
Изобретение относится к сорбенту, который подходит для связывания металлов из растворов, к получению соответствующего сорбента, а также к применению сорбента для связывания металлов из растворов. Сорбент содержит пористый материал подложки, покрытый полимером, содержащим аминогруппу. Концентрация аминогрупп сорбента, определенная титрованием, составляет по меньшей мере 600 мкмоль/мл относительно общего объема сорбента. Полимер, содержащий аминогруппу, присутствует в нековалентно связанном виде с пористым материалом подложки. Объем пор пористого материала подложки составляет от 30 до 90 об. % относительно общего объема пористого материала подложки. Пористый материал подложки имеет средний размер пор от 6 нм до 400 нм. Изобретение обеспечивает получение сорбента с высокой связывающей способностью. 4 н. и 9 з.п. ф-лы, 8 ил.
Комментарии