Способ получения производных пирипиропена - RU2578605C2
Код документа: RU2578605C2
Чертежи

Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННУЮ ЗАЯВКУ
Данная заявка на патент является заявкой, по которой запрашивается приоритет по предшествующей патентной заявке Японии, заявке на патент Японии № 44416/2010 (дата подачи: 1 марта 2010 г.). Полное раскрытие предшествующей заявки включено в данный документ посредством ссылки.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения производных пирипиропена, применимых в качестве средств борьбы с сельскохозяйственными вредителями, и, более конкретно, к получению производных пирипиропена, которые замещены ацилоксигруппой в его 1-положении и 11-положении и гидроксильной группой в 7-положении.
Предшествующий уровень техники
Производные пирипиропена, имеющие ацилоксигруппу в его 1-положении и 11-положении и гидроксильную группу в 7-положении, представляют собой соединения, которые эффективны в борьбе против сельскохозяйственных вредителей, как описано в WO 2006/129714.
WO 2006/129714 и выложенная заявка на патент Японии № 259569/1996 раскрывают способ получения производных пирипиропена, замещенных ацилоксигруппой в его 1-положении и 11-положении и гидроксильной группой в 7-положении. В соответствии со способом получения производные пирипиропена очищают или выделяют из множества продуктов, полученных посредством неселективного гидролиза ацилоксигруппы с использованием 1,7,11-триацилоксисоединения в качестве исходного соединения.
Дополнительно, выложенная заявка на патент Японии № 259569/1996 описывает применение комбинации защитных групп для синтеза производных пирипиропена. Journal of Antibiotics Vol. 49, No. 11, p. 1149 (1996), Bioorganic Medicinal Chemistry Letter Vol. 5, No. 22, p. 2683 (1995), и выложенная заявка на патент Японии № 269065/1996 раскрывают пример синтеза, в котором вводят ацил в 7-положение посредством использования защитной группы.
WO 2009/022702 раскрывает способ получения 1,11-диацил-7-деацетилпирипиропена из 1,7,11-тридеацетилпирипиропена с использованием защитной группы.
Производные пирипиропена, имеющие ацилоксигруппу в 1-положении и 11-положении и гидроксильную группу в 7-положении до этого времени получали через множество стадий с использованием неселективного гидролиза 1,7,11-триацилоксисоединения и с использованием защитной группы. Соответственно, при получении производных пирипиропена в промышленном масштабе желательными являются дополнительное улучшение эффективности при получении, например через снижение производственных затрат, улучшение выхода, упрощение очистки и выделения или снижение числа стадий.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения добились успеха при получении рассматриваемого применимого 1,11-диацилоксисоединения через короткий способ посредством селективного ацилирования, либо непосредственного, либо постадийного, гидроксильной группы в 1-положении и 11-положении тридеацилсоединения (выложенная заявка на патент Японии № 259569/1996 и Journal of Technical Disclosure No. 500997/2008), легко получаемого из пирипиропена А (природного вещества) и его аналога (Pure Appl. Chem., vol. 71, No. 6, pp.1059-1064, 1999; WO 94/09147; выложенная заявка на патент Японии № 239385/1996, выложенная заявка на патент Японии № 259569/1996, Bioorganic Medicinal Chemistry Letter Vol. 5, No. 22, p. 2683 (1995); и WO 2004/060065), что привело к завершению настоящего изобретения.
1. В соответствии с настоящим изобретением предоставлен способ получения соединения C, представленного формулой C:
[Химическая формула 1]

где R представляет собой прямую цепь, разветвленную цепь или циклический C2-6 алкилкарбонил, при условии, что когда алкильный фрагмент в алкилкарбонильной группе является разветвленной цепью или циклическим радикалом, R представляет собой С3-6алкилкарбонил, включающий
одно-трехстадийное селективное ацилирование гидроксильных групп в 1-положении и 11-положении соединения B1, представленного формулой B1:
[Химическая формула 2]

ацилирующим агентом в присутствии или в отсутствие основания.
2. В соответствии с настоящим изобретением предоставлен способ, в соответствии с указанным выше параграфом 1, отличающийся тем, что соединение C одностадийно ацилируют из соединения B1. То есть, в соответствии с этим вариантом осуществления в способе, в соответствии с указанным выше параграфом 1, соединение C получают посредством одностадийного ацилирования гидроксильных групп в 1-положении и 11-положении соединения B1.
3. В соответствии с настоящим изобретением предоставлен способ в соответствии с указанным выше параграфом 1, отличающийся тем, что осуществляют двустадийное ацилирование, состоящее из стадий:
ацилирования гидроксильной группы в 11-положении соединения B1 ацилирующим агентом с получением соединения B2, представленного формулой B2:
[Химическая формула 3]

где R такой, как определено в формуле C в указанном выше параграфе 1;
и
дополнительного ацилирования гидроксильной группы в 1-положении соединения B2. То есть, в соответствии с этим вариантом осуществления, в способе в соответствии с указанным выше параграфом 1, соединение C получают посредством двустадийного ацилирования, состоящего из стадий ацилирования гидроксильной группы в 11-положении соединения B1 ацилирующим агентом с получением соединения B2; и дополнительного ацилирования гидроксильной группы в 1-положении соединения B2.
4. В соответствии с другим аспектом настоящего изобретения предоставлен способ в соответствии с указанным выше параграфом 1, отличающийся тем, что осуществляют трехстадийное ацилирование, состоящее из стадий ацилирования гидроксильной группы в 11-положении соединения B1 с получением соединения B2; переноса ацильной группы в 11-положении соединения B2 к гидроксильной группе в 1-положении с получением соединения B3, представленного формулой B3:
[Химическая формула 4]
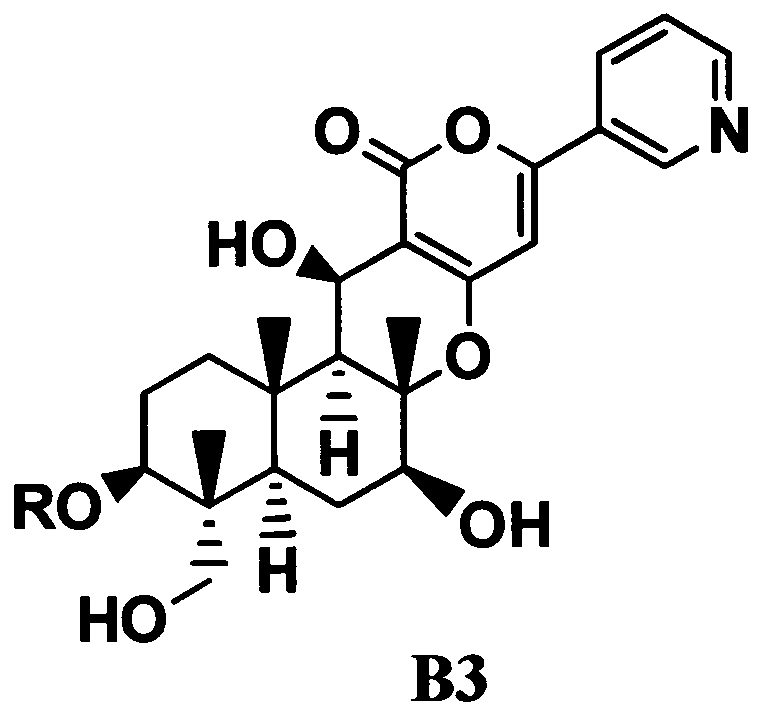
где R такой, как определено в формуле C, в указанном выше параграфе 1; и
ацилирования гидроксильной группы в 11-положении соединения B3. То есть, в соответствии с этим вариантом осуществления, в способе в соответствии с указанным выше параграфом 1, соединение C получают посредством трехстадийного ацилирования, состоящего из стадий
ацилирования гидроксильной группы в 11-положении соединения B1 с получением соединения B2; переноса ацильной группы в 11-положении соединения B2 к гидроксильной группе в 1-положении с получением соединения B3; и ацилирования гидроксильной группы в 11-положении соединения B3.
5. Дополнительно, в соответствии с настоящим изобретением предоставлен способ в соответствии с любым из указанных выше параграфов от 1 до 4, включающий в качестве стадии получения соединения B1 гидролиз ацильных групп в 1-положении, 7-положении и 11-положении соединения A1, представленного формулой A1 в присутствии основания:
[Химическая формула 5]
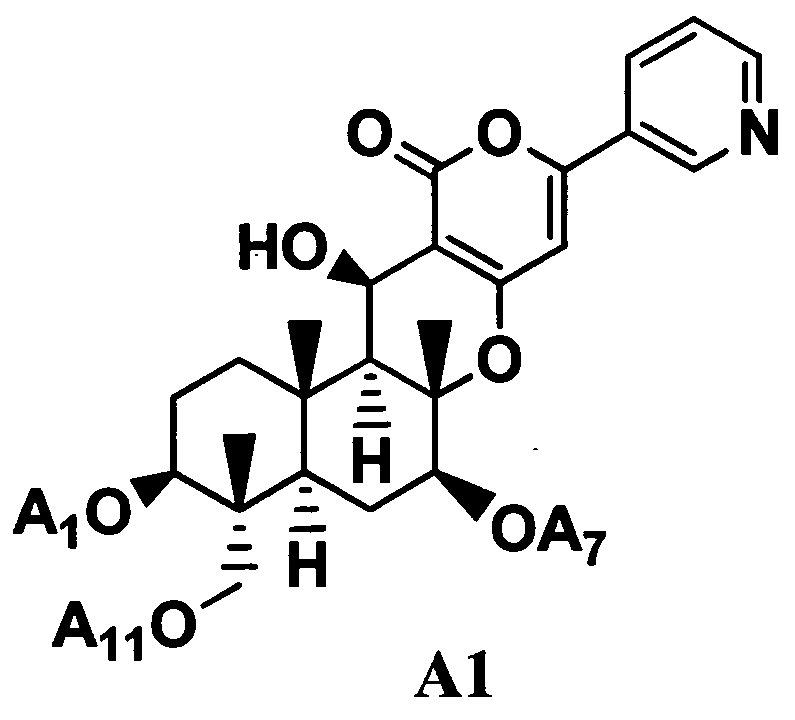
где A1, A7 и A11, которые могут быть одинаковыми или различными, представляют ацетил или пропионил. То есть, в соответствии с этим вариантом осуществления, способ в соответствии с указанными выше параграфами от 1 до 4, дополнительно включает, в качестве стадии получения соединения B1 гидролиз ацильных групп в 1-положении, 7-положении и 11-положении соединения A1 в присутствии основания.
6. В соответствии с еще одним другим аспектом настоящего изобретения предоставлен способ получения соединения C, который включает стадии ацилирования гидроксильных групп в 1-положении, 11-положении и 7-положении соединения B1 с получением соединения B4, представленного формулой B4:
[Химическая формула 6]

где R такой, как определено выше; и затем селективного деацилирования гидроксильной группы в 7-положении.
В соответствии с дополнительным аспектом настоящего изобретения предоставлен способ выделения и очистки кристаллов сольвата соединения C, полученных в соответствии с любым из указанных выше параграфов от 1 до 5, способ, включающий добавление подходящего растворителя к неочищенному продукту соединения C, полученному посредством концентрирования реакционного раствора, содержащего соединение C, полученного посредством любого из указанных выше параграфов от 1 до 5, при пониженном давлении; концентрирование этилацетатного экстракта реакционного раствора, содержащего соединение C, полученное посредством способа в соответствии с любым из указанных выше параграфов от 1 до 5; или дополнительное добавление выбранного растворителя к концентрату для осаждения кристаллов сольвата соединения C.
В соответствии с еще одним аспектом настоящего изобретения предоставлен способ выделения и очистки кристаллов сольвата соединения C,
способ, включающий стадии
(a) экстракции реакционного раствора, содержащего соединение C, органическим растворителем, выбранным из группы, состоящей из метилацетата, этилацетата, бутилацетата, толуола, этилбензола, хлорбензола, хлороформа, дихлорметана, диэтилового эфира, диизопропилового эфира, тетрагидрофурана и диоксана, и концентрирования экстракта после или без сушки;
(b) упаривания реакционного раствора, содержащего соединение C, досуха с получением неочищенного продукта и затем растворения неочищенного продукта в органическом растворителе, выбранном из группы, состоящей из метилацетата, этилацетата, бутилацетата, толуола, этилбензола, хлорбензола, хлороформа, дихлорметана, диэтилового эфира, диизопропилового эфира, тетрагидрофурана, диоксана, метанола и этанола при комнатной температуре или при нагреве; или
(c) упаривания реакционного раствора, содержащего соединение C, досуха с получением неочищенного продукта, растворения неочищенного продукта в органическом растворителе, выбранном из группы, состоящей из метилацетата, этилацетата, бутилацетата, толуола, этилбензола, хлорбензола, хлороформа, дихлорметана, диэтилового эфира, диизопропилового эфира, тетрагидрофурана, диоксана, метанола и этанола при комнатной температуре или при нагреве, и добавления слабого растворителя, выбранного из группы, состоящей из гептана, гексана и циклогексана, к раствору. В предпочтительном варианте осуществления настоящего изобретения указанная стадия (a) должна быть стадией (a') экстракции реакционного раствора, содержащего соединение C, этилацетатом, и концентрирования экстракта после или без осушки. В еще одном предпочтительном варианте осуществления настоящего изобретения указанная стадия (b) должна быть стадией (b') упаривания реакционного раствора, содержащего соединение C, досуха с получением неочищенного продукта и затем растворения неочищенного продукта в этилацетате при комнатной температуре или при нагреве. В другом предпочтительном варианте осуществления настоящего изобретения указанная стадия (c) должна быть стадией (c') упаривания реакционного раствора, содержащего соединение C, досуха с получением неочищенного продукта, растворения неочищенного продукта в этилацетате при комнатной температуре или при нагреве и добавления гексана к раствору.
В соответствии с еще одним другим аспектом настоящего изобретения предоставлен способ получения соединения C из соединения B1 в соответствии с любым из указанных выше параграфов от 1 до 5, который включает стадию выделения и очистки соединения C посредством кристаллизации из реакционного раствора, содержащего соединение C. То есть, в соответствии с этим вариантом осуществления способ в соответствии с любым из указанных выше параграфов от 1 до 5 дополнительно включает стадию выделения и очистки соединения C посредством кристаллизации из реакционного раствора, содержащего соединение C.
В соответствии с настоящим изобретением производные пирипиропена, которые замещены ацилоксигруппой в 1-положении и 11-положении и гидроксильной группой в 7-положении и являются применимыми в качестве средств уничтожения сельскохозяйственных насекомых-вредителей, могут быть эффективно получены через короткий способ.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 представляет собой порошковую рентгенограмму, измеренную для кристаллов этилацетатного сольвата 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Способ получения
Термин "алкил", как используется в настоящем документе, в качестве заместителя или части заместителя означает алкил, то есть, с прямой цепью, разветвленной цепью или циклического типа или типа их комбинации, если не указано иначе.
Символ "Ca-b", присоединенный к заместителю, как используется в настоящем документе, означает, что число атомов углерода, содержащихся в заместителе, как используется в настоящем документе, равно от a до b. Дополнительно, "Ca-b" в "Ca-b алкилкарбонил" означает, что число атомов углерода в алкильном фрагменте, исключая атом углерода в карбонильном фрагменте равно от a до b.
Конкретные примеры C2-6 алкилкарбонила с прямой, разветвленной цепью или циклического, представленного R, при условии, что когда алкильный фрагмент в алкилкарбонильной группе представляет собой разветвленную цепь или циклическую группу, R представляет собой C3-6 алкилкарбонил, включают циклопропанкарбонил и пропионил.
В соответствии с еще одним предпочтительным вариантом осуществления настоящего изобретения в способе в соответствии с любым из указанных выше параграфов от 1 до 5 ацилирование осуществляют в отсутствие основания.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения в способе в соответствии с любым из указанных выше параграфов от 1 до 5 основание, используемое при ацилировании гидроксильной группы в 1-положении и 11-положении соединения B1, представляет собой 2,4,6-коллидин или 2,6-лутидин.
В соответствии с еще одним предпочтительным вариантом осуществления настоящего изобретения в способе в соответствии с указанным выше параграфом 2 ацилирующий агент используют в количестве от 2,0 до 5,0 эквивалентов в расчете на соединение B1.
В соответствии с дополнительным предпочтительным вариантом осуществления настоящего изобретения способ в соответствии с указанным выше параграфом 3 отличается тем, что растворитель, применяемый на стадии получения соединения B2, отличается от растворителя, применяемого на стадии дополнительного ацилирования гидроксильной группы в 1-положении соединения B2.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения способ в соответствии с указанным выше параграфом 4 отличается тем, что стадию получения соединения B3 из соединения B2 осуществляют в присутствии основания.
В соответствии с еще одним предпочтительным вариантом осуществления настоящего изобретения способ в соответствии с указанным выше параграфом 4 отличается тем, что стадию получения соединения B3 из соединения B2 осуществляют в присутствии 1,8-диазабицикло[5.4.0]ундека-7-ена (DBU) в качестве основания.
В соответствии с дополнительным предпочтительным вариантом осуществления настоящего изобретения C2-6 алкилкарбонил, представленный R, является циклическим C3-6 алкилкарбонилом, более предпочтительно циклопропанкарбонилом.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения в способе в соответствии с указанным выше параграфом 3 основание применяют на стадии получения соединения B2 и на стадии дополнительного ацилирования гидроксильной группы в 1-положении соединения B2, количество основания, применяемого на стадии получения соединения B2, составляет от 1,0 до 3,0 эквивалентов, в расчете на соединение B1, общее количество основания, применяемого на стадии получения соединения B2, и основания, применяемого на стадии дополнительного ацилирования гидроксила в 1-положении соединения B2, составляет от 2,0 до 4,5 эквивалентов, более предпочтительно от 2,0 до 3,0 эквивалентов.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения в способе в соответствии с любым из указанных выше параграфов от 1 до 4 ацилирующий агент используют в количестве от 2,0 до 5,0 эквивалентов в расчете на соединение B1.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения в способе в соответствии с указанным выше параграфом 3. ацилирующий агент используют на стадии получения соединения B2 и на стадии дополнительного ацилирования гидроксильной группы в 1-положении соединения B2, количество ацилирующего агента, используемое на стадии получения соединения B2, составляет от 1,0 до 3,5 эквивалентов в расчете на соединение B1, общее количество ацилирующего агента, применяемого на стадии получения соединения B2 и ацилирующего агента, применяемого на стадии дополнительного ацилирования гидроксильной группы в 1-положении соединения B2, составляет от 2,0 до 4,5 эквивалентов.
В соответствии с еще одним предпочтительным вариантом осуществления настоящего изобретения предоставлено применение соединения B2 в качестве промежуточного соединения при получении соединения C из соединения B1. То есть, в варианте осуществления предоставлено применение соединения B2 при получении соединения C.
В соответствии с еще одним предпочтительным вариантом осуществления настоящего изобретения предоставлено применение соединения B2 и соединения B3 в качестве промежуточного соединения при получении соединения C из соединения B1. То есть, в этом варианте осуществления предоставлено применение соединения B3 при получении соединения C.
Настоящее изобретение будет описано более подробно в соответствии со следующей схемой.
[Химическая формула 7]

где А1, A7, A11 и R являются такими, как определено выше.
Продукт, получаемый на каждой стадии схемы, может также применяться без последующей обработки или выделения на следующей стадии.
1-1: Получение соединения B1 из соединения A1
Соединение A1 может быть получено посредством способа, описанного, например, в Pure Appl. Chem., vol. 71, No. 6, pp. 1059-1064, 1999.; выложенной заявке на патент Японии № 239385/1996, выложенной заявке на патент Японии № 184158/1994, WO 2004/060065, выложенной заявке на патент Японии № 259569/1996 или Bioorganic Medicinal Chemistry Letter vol. 5, No. 22, p. 2683.
Когда соединение A1 в качестве исходного соединения представляет пирипиропен A, пирипиропен А может быть один получен посредством способа, описанного в Journal Synthetic Organic Chemistry (1998), Vol. 56, No. 6, p. 478-488 или WO 94/09147.
Соединение B1 может также являться производным, полученным посредством способа, описанного, например, в выложенной заявке на патент Японии № 259569/1996 или Journal of Technical Disclosure No. 50997/2008.
Способ, описанный в WO 2009/022702, может быть упомянут как способ получения соединения B1 из соединения A1, и соединение B1 может быть получено посредством гидролиза ацильной группы в 1-положении, 7-положении и 11-положении соединения A1 в присутствии основания.
Более конкретно, растворители, применяемые в данном изобретении, включают спиртовые растворители, имеющие от 1 до 4 атомов углерода, такие как метанол; растворители из простых эфиров, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан; апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, и ацетонитрил; галогенированные растворители, такие как дихлорметан и хлороформ; или вода; и смешанные растворители, составленные из двух или более из этих растворителей.
Основания, применяемые в данном изобретении, включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития и гидроксид бария; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия; алкоксиды щелочноземельных металлов; и органические основания, такие как 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гидразин и гуанидин. Предпочтительными являются 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия и гидроксид калия.
Количество применяемого основания составляет предпочтительно от 0,01 до 4,5 эквивалентов в расчете на количество соединения A1. Температура реакции составляет предпочтительно от -20°C до 50°C. Время реакции составляет предпочтительно от 0,5 час до 72 час.
2-1: Получение соединения C из соединения B1
(1) Стадия получения соединения C непосредственно из соединения B1
Растворители, применяемые в способе получения соединения C из соединения B1 в указанном выше параграфе 2., включают растворители из простых эфиров, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан; апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, ацетонитрил, N-метил-2-пирролидинон, N-метил-2-пиперазинон и N,N-диметил-2-имидазолидинон; галогенированные растворители, такие как дихлорметан и хлороформ; или ароматические углеводородные растворители, такие как толуол; и смешанные растворители, составленные из двух или более из этих растворителей. Предпочтительными являются апротонные полярные органические растворители. Более предпочтительными являются N-метил-2-пирролидинон, N,N-диметил-2-имидазолидинон и N,N-диметилацетамид. Особенно предпочтительным является N-метил-2-пирролидинон и N,N-диметилацетамид.
Способ в соответствии с указанным выше параграфом 2 предпочтительно осуществляют в отсутствие основания. Однако, когда способ осуществляют в присутствии основания, примеры применимых оснований включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития, и гидроксид бария; и органические основания, такие как 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гуанидин, лутидин, коллидин, 2,2'-бипиридил, трифениламин, хинолин, N,N-диметиланилин и N,N-диэтиланилин. Предпочтительными являются пиридин, 2,6-лутидин, 2,4,6-коллидин, 2,2'-бипиридил, трифениламин, N,N-диметиланилин, N,N-диэтиланилин и т.п. Более предпочтительными являются 2,6-лутидин, 2,4,6-коллидин, трифениламин, N,N-диметиланилин и N,N-диэтиланилин. Особенно предпочтительными являются 2,6-лутидин и 2,4,6-коллидин.
Когда применяют основание, количество основания составляет предпочтительно от 2,0 до 4,5 эквивалентов, более предпочтительно от 2,0 до 3,0 эквивалентов, в расчете на количество соединения B1.
Группа R может быть введена в 1-положение и 11-положение с использованием ROH, RCl, (R)2O или смешанного кислотного ангидрида, предпочтительно RCl или (R)2О, в качестве ацилирующего агента, соответствующего рассматриваемому R. Реакция может осуществляться в присутствии или отсутствии основания или с использованием конденсирующего агента, такого как дициклогексилкарбодиимид, гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида, карбонилдиимидазол, дипиридилдисульфид, диимидазолилдисульфид, 1,3,5-трихлорбензоилхлорид, 1,3,5-трихлорбензоил ангидрид, PyBop или PyBrop. Предпочтительно, реакцию осуществляют с использованием RCl или (R)2О в присутствии или отсутствии основания.
Более предпочтительные ацилирующие агенты включают циклопропанкарбонилхлорид, бутирилхлорид, и ангидрид циклопропанкарбоновой кислоты.
Количество применяемого ацилирующего агента составляет предпочтительно от 2,0 до 5,0 эквивалентов, более предпочтительно от 2,2 до 4,5 эквивалентов, в расчете на количество соединения B1. Это количество применяют сразу или двумя-пятью разделенными частями.
Температура реакции составляет предпочтительно от -20°C до 50°C, более предпочтительно от -10°C до 50°C, еще более предпочтительно от -10°C до комнатной температуры. Время реакции составляет предпочтительно от 0,1 час до 7 дней, более предпочтительно от 3 час до 4 дней.
В соответствии с этим способом соединение C может быть получено из соединения B1 через единственную стадию при выходе не меньше, чем 40%.
(2) Стадия получения соединения B2 из соединения B1
Растворители, применяемые в способе получения соединения B2 из соединения B1 в указанном выше параграфе 3 или 4, включают растворители из простых эфиров, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан; апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, ацетонитрил, N-метил-2-пирролидинон, N-метил-2-пиперазинон и N,N-диметил-2-имидазолидинон; галогенированные растворители, такие как дихлорметан и хлороформ; или ароматические углеводородные растворители, такие как толуол; и смешанные растворители, составленные из двух или более из этих растворителей. Предпочтительными являются апротонные полярные органические растворители. Особенно предпочтительными является N-метил-2-пирролидинон и N,N-диметилацетамид.
Реакция может осуществляться без применения основания. Однако когда применяют основание, примеры применимых оснований включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития, гидроксид бария, трет-бутилат натрия (NaOt-Bu), метилат калия (KOMe), ацетат калия (KOAc), метилат натрия (NaOMe), моногидрат гидроксида цезия (CsOH.H2O), метилат лития (LiOMe) и трет-бутилат лития (LiOt-Bu); и органические основания, такие как 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гуанидин, лутидин, коллидин, 2,2'-бипиридил, трифениламин, хинолин, N,N-диметиланилин и N,N-диэтиланилин. Предпочтительными являются пиридин, 2,6-лутидин, 2,4,6-коллидин, 2,2'-бипиридил, трифениламин, N,N-диметиланилин, N,N-диэтиланилин и т.п. Более предпочтительными являются триэтиламин, 2,6-лутидин, 2,4,6-коллидин, трифениламин, N,N-диметиланилин и N,N-диэтиланилин. Особенно предпочтительными являются триэтиламин и 2,6-лутидин.
ROH, RCl, (R)2О или смешанный кислотный ангидрид, предпочтительно RCl, (R)2O или смешанный кислотный ангидрид, применяют в качестве ацилирующего агента для введения в качестве группы R. Реакция может осуществляться в присутствии или отсутствии основания или с использованием конденсирующего агента, такого как дициклогексилкарбодиимид, гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида, карбонилдиимидазол, дипиридилдисульфид, диимидазолилдисульфид, 1,3,5-трихлорбензоилхлорид, 1,3,5-трихлорбензоилангидрид, PyBop или PyBrop. Предпочтительно, реакцию осуществляют с использованием RCl или (R)2O в присутствии или в отсутствие основания.
Более предпочтительные ацилирующие агенты включают циклопропанкарбонилхлорид, ангидрид циклопропанкарбоновой кислоты и пивалевый ангидрид циклопропанкарбоновой кислоты.
Количество применяемого ацилирующего агента составляет предпочтительно от 1,0 до 3,5 эквивалентов, более предпочтительно от 1,1 до 3,0 эквивалентов, в расчете на количество соединения B1.
Когда применяют основание, количество основания составляет предпочтительно от 1,0 до 3,0 эквивалентов, более предпочтительно от 1,1 до 2,5 эквивалентов, в расчете на количество соединения B1.
Температура реакции составляет предпочтительно от -20°C до 60°C, более предпочтительно от -10°C до 60°C. Время реакции составляет предпочтительно от 0,1 час до 7 дней, более предпочтительно от 45 мин до 48 час.
(3) Стадия получения соединения C из соединения B2
Растворители, применяемые в способе получения соединения C из соединения B2 в указанном выше параграфе 3, включают растворители из простых эфиров, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан; апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, ацетонитрил, N-метил-2-пирролидинон, N-метил-2-пиперазинон и N,N-диметил-2-имидазолидинон; галогенированные растворители, такие как дихлорметан и хлороформ; или ароматические углеводородные растворители, такие как толуол; и смешанные растворители, составленные из двух или более из этих растворителей. Предпочтительными являются апротонные полярные органические растворители. Особенно предпочтительным является N-метил-2-пирролидинон.
Реакция может осуществляться без применения основания. Однако, когда применяют основание, примеры применимых оснований включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития и гидроксид бария; и органические основания, такие как 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гуанидин, лутидин, коллидин, 2,2'-бипиридил, трифениламин, хинолин, N,N-диметиланилин и N,N-диэтиланилин. Предпочтительными являются пиридин, 2,6-лутидин, 2,4,6-коллидин, 2,2'-бипиридил, трифениламин, N,N-диметиланилин, N,N-диэтиланилин и т.п. Более предпочтительными являются триэтиламин, 2,6-лутидин, 2,4,6-коллидин, трифениламин, N,N-диметиланилин и N,N-диэтиланилин. Особенно предпочтительными являются триэтиламин и 2,6-лутидин.
ROH, RCl, (R)2O или смешанный кислотный ангидрид, предпочтительно RCl или (R)2O, применяют в качестве ацилирующего агента для введения в качестве группы R. Реакция может осуществляться в присутствии или отсутствии основания или с использованием конденсирующего агента, такого как дициклогексилкарбодиимид, гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида, карбонилдиимидазол, дипиридилдисульфид, диимидазолилдисульфид, 1,3,5-трихлорбензоилхлорид, 1,3,5-трихлорбензоил ангидрид, PyBop или PyBrop. Предпочтительно, реакцию осуществляют с использованием RCl или (R)2O в присутствии или в отсутствие основания.
Более предпочтительные ацилирующие агенты включают циклопропанкарбонилхлорид и ангидрид циклопропанкарбоновой кислоты.
Когда применяют основание, количество основания составляет предпочтительно от 0,1 до 5,0 эквивалентов, более предпочтительно от 0,1 до 3,0 эквивалентов в расчете на количество соединения B2. В более предпочтительном варианте осуществления общее количество основания, используемого на этой стадии и на стадии, описанной в указанном выше параграфе (2), составляет от 2,0 до 4,5 эквивалентов, более предпочтительно от 2,0 до 3,0 эквивалентов.
Количество используемого ацилирующего агента составляет предпочтительно от 1,0 до 3,0 эквивалентов в расчете на количество соединения B1, более предпочтительно от 2,0 до 4,5 эквивалентов с учетом общего количества ацилирующего агента, используемого на этой стадии и на стадии, описанной в указанном выше параграфе (2).
Температура реакции составляет предпочтительно от -20°C до 60°C. Время реакции составляет предпочтительно от 0,1 час до 7 дней.
Эта стадия может также осуществляться непрерывно без отбора продукта, полученного на стадии, описанной в указанном выше параграфе (2).
(4) Стадия получения соединения B3 из соединения B2
Растворители, применяемые в способе получения соединения B3 из соединения B2 в указанном выше параграфе 4, включают растворители из простых эфиров, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан; апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, ацетонитрил, N-метил-2-пирролидинон, N-метил-2-пиперазинон и N,N-диметил-2-имидазолидинон; галогенированные растворители, такие, как дихлорметан и хлороформ; или ароматические углеводородные растворители, такие как толуол, хлорбензол и дихлорбензол; и смешанные растворители, составленные из двух или более из этих растворителей. Предпочтительными являются апротонные полярные органические растворители.
Основания, применяемые в данном изобретении, включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития, гидроксид бария, и трет-бутоксид калия; и органические основания, такие как 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гуанидин, лутидин, коллидин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, фосфазен. Предпочтительными являются карбонат калия, карбонат цезия, трет-бутоксид калия, 1,8-диазабицикло[5.4.0]ундека-7-ен и 1,5-диазабицикло[4.3.0]нона-5-ен и т.п. Более предпочтительным являются 1,8-диазабицикло[5.4.0]ундека-7-ен и 1,5-диазабицикло[4.3.0]нона-5-ен.
Количество применяемого основания составляет предпочтительно от 0,1 до 3,0 эквивалентов, более предпочтительно от 0,1 до 2,0 эквивалентов, в расчете на количество соединения B2.
Температура реакции составляет предпочтительно от 0°C до 150°C. Время реакции составляет предпочтительно от 0,1 час до 7 дней.
(5) Стадия получения соединения C из соединения B3
Растворители, применяемые в способе получения соединения C из соединения B3 в указанном выше параграфе 4, включают растворители из простых эфиров, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан; апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, ацетонитрил, N-метил-2-пирролидинон, N-метил-2-пиперазинон и N,N-диметил-2-имидазолидинон; галогенированные растворители, такие как дихлорметан и хлороформ; или ароматические углеводородные растворители, такие как толуол; и смешанные растворители, составленные из двух или более из этих растворителей. Предпочтительными являются апротонные полярные органические растворители. Особенно предпочтительным является N-метил-2-пирролидинон.
Реакция может осуществляться без применения основания. Однако когда применяют основание, примеры применимых оснований включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития, и гидроксид бария; и органические основания, такие как 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гуанидин, лутидин, коллидин, 2,2'-бипиридил, трифениламин, хинолин, N,N-диметиланилин и N,N-диэтиланилин. Предпочтительными являются пиридин, 2,6-лутидин, 2,4,6-коллидин, 2,2'-бипиридил, трифениламин, N,N-диметиланилин, N,N-диэтиланилин и т.п. Более предпочтительными являются 2,6-лутидин, 2,4,6-коллидин, трифениламин, N,N-диметиланилин и N,N-диэтиланилин.
ROH, RCl, (R)2O или смешанный кислотный ангидрид, предпочтительно RCl или (R)2O, применяют в качестве ацилирующего агента для введения R в качестве группы. Реакция может осуществляться в присутствии или отсутствии основания или с использованием конденсирующего агента, такого как дициклогексилкарбодиимид, гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида, карбонилдиимидазол, дипиридилдисульфид, диимидазолилдисульфид, 1,3,5-трихлорбензоилхлорид, 1,3,5-трихлорбензоилангидрид, PyBop или PyBrop. Предпочтительно, реакцию осуществляют с использованием RCl или (R)2O в присутствии или в отсутствие основания.
Более предпочтительные ацилирующие агенты включают циклопропанкарбонилхлорид и ангидрид циклопропанкарбоновой кислоты.
Когда применяют основание, количество основания составляет предпочтительно от 1,0 до 3,0 эквивалентов в расчете на количество соединения B2.
Количество применяемого ацилирующего агента составляет предпочтительно от 1,0 до 2,5 эквивалентов в расчете на количество соединения B1.
Температура реакции составляет предпочтительно от -20°C до 60°C. Время реакции составляет предпочтительно от 0,1 час до 7 дней.
(6) Способ очистки и выделения соединения C из неочищенного продукта
Способ получения соединения C посредством кристаллизации предпочтительно упомянут как способ очистки и выделения соединения C из реакционного раствора или неочищенного продукта соединения C, полученного в способе, описанном в указанном выше параграфе (1), (3) или (5). Кристаллы могут быть получены в виде кристаллов сольвата, содержащих растворитель, включенный в кристаллическую решетку. Альтернативно, соединение C, не содержащее какого-либо растворителя или воды, может быть получено посредством сушки кристаллов сольвата или посредством получения осадков, например, посредством растворения кристаллов сольвата в метаноле и добавления воды к раствору, собирая осадки фильтрацией, и сушки собранных осадков посредством нагрева при пониженном давлении.
В соответствии с предпочтительным вариантом осуществления для получения кристаллов соединения C предоставлен способ, который включает экстракцию реакционного раствора, содержащего соединение C, полученное посредством способа в соответствии с любым из указанных выше параграфов от 1 до 5, органическим растворителем, выбранным из группы, состоящей из метилацетата, этилацетата, бутилацетата, толуола, этилбензола, хлорбензола, хлороформа, дихлорметана и простого эфира, концентрирование экстракта после или без сушки и, в этом состоянии, обеспечение протекания кристаллизации, или способ, который включает упаривание реакционного раствора, содержащего соединение C, досуха с получением неочищенного продукта, растворение неочищенного продукта в органическом растворителе, выбранном из группы, состоящей из метилацетата, этилацетата, бутилацетата, толуола, этилбензола, хлорбензола, хлороформа, дихлорметана, эфира, метанола, и этанола, при комнатной температуре или при нагреве, и добавление слабого растворителя, выбранного из группы, состоящей из гептана, гексана и циклогексана, к раствору, чтобы вызвать кристаллизацию. Простой эфир, применяемый в способе, предпочтительно выбирают из диэтилового эфира, диизопропилового эфира, тетрагидрофурана и диоксана.
Более конкретный пример способа получения кристаллов соединения C включает либо стадию добавления растворителя к реакционному раствору, удаления растворителя посредством перегонки с получением неочищенного продукта и добавления этилацетата к неочищенному продукту, или стадию концентрирования этилацетатного экстракта реакционного раствора; и выделение этилацетатных сольватов кристаллов после отстаивания при комнатной температуре или, необязательно, после нагрева. Если необходимо, пентан, гексан или циклогексан, предпочтительно гексан, добавляют к этилацетатному экстракту или концентрату этилацетатного экстракта для получения этилацетатных сольватов кристаллов. Соединение C может быть получено посредством растворения этилацетатных сольватов кристаллов в метаноле, добавления воды к раствору, сбора полученных в результате осадков посредством фильтрации, и сушки собранных осадков посредством нагрева при пониженном давлении.
2-2: Получение соединения C из соединения B1 через соединение B4
Стадия получения соединения B4 из соединения B1 в способе согласно вышеуказанному параграфу 6. может также осуществляться в отсутствие растворителя. Однако когда стадию осуществляют в присутствии растворителя, примеры применимых растворителей включают кетоновые растворители, такие как ацетон и диэтилкетон; растворители из простых эфиров, такие как диэтиловый эфир, диизопропиловыйэфир и тетрагидрофуран; сложноэфирные растворители, такие как этилацетат и бутилацетат; апротонные полярные органические растворители, такие как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, ацетонитрил, N-метил-2-пирролидинон и N-метил-2-пиперазинон; галогенированные углеводородные растворители, такие как дихлорметан и хлороформ; или ароматические углеводородные растворители, такие как толуол; и смешанные растворители, составленные из двух или более таких растворителей.
ROH, RCl, (R)2O или смешанный кислотный ангидрид можно упомянуть в качестве ацилирующего агента, для введения группы R. Ацилирующий агент представляет собой предпочтительно RCl или (R)2O. Реакция может осуществляться в присутствии или отсутствии основания или с использованием конденсирующего агента, такого как дициклогексилкарбодиимид, 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид, карбонилдиимидазол, дипиридилдисульфид, диимидазолилдисульфид, 1,3,5-трихлорбензоилхлорид, 1,3,5-трихлорбензоил ангидрид, PyBop или PyBrop.
Основания, применяемые в данном изобретении, включают карбонат натрия, карбонат калия, гидрид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, пиридин, лутидин, 4-диметиламинопиридин, имидазол, 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, триэтиламин и диизопропилэтиламин.
Температура реакции составляет предпочтительно от -20°C до 50°C. Время реакции составляет предпочтительно от 0,5 час до 48 час.
Растворители, применяемые на стадии получения соединения C из соединения B4, в способе согласно вышеуказанному параграфу 6 включают спиртовые растворители, имеющие от 1 до 4 атомов углерода, такие как метанол; растворители из простых эфиров, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран и диоксан; апротонные полярные органические растворители, такие как N,N-диметилформамид, диметилсульфоксид, N,N-диметилацетамид, ацетонитрил, N-метил-2-пирролидинон и N-метил-2-пиперазинон; галогенированные растворители, такие как дихлорметан и хлороформ; или воду; и смешанные растворители, составленные из двух или более из этих растворителей.
Основания, применяемые в данном изобретении, включают неорганические основания, такие как карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия, гидроксид калия, гидрид натрия, гидрид калия, цианид натрия, цианид калия, гидроксид магния, гидроксид кальция, гидроксид лития и гидроксид бария; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия; алкоксиды щелочноземельных металлов; и органические основания, такие как 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, триэтиламин, диизопропилэтиламин, пиридин, гидразин и гуанидин. Предпочтительными являются 1,8-диазабицикло[5.4.0]ундека-7-ен, 1,5-диазабицикло[4.3.0]нона-5-ен, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, гидроксид натрия и гидроксид калия.
Количество, используемого основания составляет предпочтительно от 0,01 до 24 эквивалентов в расчете от количества соединение B4. Температура реакции составляет предпочтительно от -20°C до 50°C. Время реакции составляет предпочтительно от 0,5 час до 14 дней.
ПРИМЕРЫ
Настоящее изобретение дополнительно иллюстрируется посредством следующих Примеров, которые не подразумевают ограничение изобретения.
Чистота, описанная в Экспериментальных Примерах, означает процентную долю площади рассматриваемого вещества, измеренную при следующих условиях ВЭЖХ, если не обозначено иначе.
Условия измерения для ВЭЖХ
Колонка: Inertsil ODS-2 или ODS-4 (5 мкм); 4,6 мм диаметр × 150 мм.
(ODS-2 применяли в Примерах 1-13, а ODS-4 применяли в Примерах 14-20)
Темп. колонки: 30°C
Подвижная фаза: Вода-ацетонитрил
Условия для подвижной фазы: Как показано в Таблице 1 ниже
[Таблица 1]
Объемная скорость потока: 1,0 мл/мин
Длина волны детектирования: УФ 320 нм
Пример 1
Синтез 11-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (1,00 г), синтезированный в соответствии со способом, описанным в WO2006/129714, суспендировали в 5 мл N-метил-2-пирролидинона, к суспензии добавляли 0,55 мл (2,2 эквивалента) 2,6-лутидина, и 0,44 мл (2,2 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии при комнатной температуре. После одного часа добавления по каплям реакционный раствор добавляли по каплям к 200 мл воды. Смесь перемешивали в течение 5 час и полученный в результате осадок затем собирали посредством фильтрации, промывали водой и сушили с получением 0,816 г порошка, состоящего, в основном, из 11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A. Отдельно, к фильтрату добавляли 25 г хлорида натрия, и смесь экстрагировали 20 мл этилацетата. Этилацетатный слой промывали водой, этилацетат удаляли посредством перегонки, и остаток сушили с получением 0,27 г пенистого вещества состоящего, в основном, из 11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена А. Порошок и пенистое вещество объединяли вместе, с последующей хроматографией на силикагеле (100 мл силикагеля C-60 производство Merck Ltd.; этилацетат-метанол (50:1 (об./об.); объемная скорость потока 10 мл/мин) с получением 532 мг 11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (выход: 46,3%) (чистота: 95,6%).
FAB-МС; m/z 526 (M+H)+;1H-ЯМР (CDCl3) δ 2,15 (1H, дт, J=3,4, 9,5 Гц), 2,42 (1H, шир.с), 2,96 (1H, с), 3,41 (1H, дд, J=5,1, 11,0 Гц), 3,75 (1H, д, J=11,9 Гц), 3,83 (1H, дд, J=4,9, 11,9 Гц), 4,29 (1H, д, J=11,7 Гц), 5,00 (1H, д, J=3,2 Гц), 6,52 (1H, с), 7,42 (1H, дд, J=4,9, 8,1 Гц), 8,11 (1H, дт, J=2,0, 8,3 Гц), 8,69 (1H, дд, J=1,3, 4,8 Гц), 9,00 (1H, д, J=1,7 Гц).
Пример 2
Синтез 11-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (1,00 г) суспендировали в 5 мл N-метил-2-пирролидинона, 0,50 мл (2,0 эквивалента) 2,6-лутидина добавляли к суспензии, и 0,33 мл (1,7 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии при комнатной температуре. После 45 мин покапельного добавления реакционный раствор добавляли по каплям к 100 мл воды. К ним добавляли хлорид натрия (5 г), и смесь перемешивали в течение ночи. Полученный в результате осадок затем собирали посредством фильтрации, промывали водой и сушили с получением 1,053 г порошка, состоящего, в основном, из 11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A. Порошок (526 мг; половинное количество) очищали посредством хроматографии на силикагеле (100 мл силикагеля C-60N (40-50 мкм), производство KANTO CHEMICAL CO., INC.; этилацетат-метанол (50:1 (об./об.); объемная скорость потока 5 мл/мин) с получением 366 мг 11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (выход: 63,7%) (чистота: 95,1%).
Пример 3
Синтез 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (1,00 г) суспендировали в 5 мл N-метил-2-пирролидинона, 0,76 мл (2,6 эквивалента) 2,4,6-коллидина добавляли к суспензии и смесь добавляли по каплям к 0,50 мл (2,5 эквивалента) циклопропанкарбонилхлорида при комнатной температуре. Обеспечивали протекание реакции в течение 8,5 час. Реакционный раствор затем добавляли по каплям к 200 мл воды. Смесь перемешивали в течение ночи и полученный в результате осадок затем собирали посредством фильтрации и сушили с получением 1,135 г порошка, состоящего, в основном, из 1,11-ди-О-циклопропанкарбонил-l,7,11-тридеацетилпирипиропена A. Отдельно, 25 г хлорида натрия добавляли к фильтрату, и смесь экстрагировали 20 мл этилацетата. Этилацетатный слой промывали водой, этилацетат удаляли посредством перегонки и остаток сушили с получением 0,12 г пенистого вещества, состоящего в основном из 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A. Порошок и пенистое вещество объединяли вместе с последующей хроматографией на силикагеле (150 мл силикагеля C-60, производство Merck Ltd.; только этилацетат; объемная скорость потока 10 мл/мин) с получением 743 мг 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (выход: 57,2%) (чистота: 80,8%). Для соединения, полученного таким образом, измеряют FAB-МС и1Н-ЯМР, и в результате было обнаружено, что данные соответствовали данным для соединения 261, описанного в WO2006/129714.
FAB-MC; m/z 594 (M+H)+;1H-ЯМР (CDCl3) δ 3,75 (1H, д, J=12,0 Гц), 3,79 (1H, дд, J=4,6, 11,7 Гц), 3,87 (1H, д, J=11,7 Гц), 4,82 (1H, дд, J=4,9, 11,2 Гц), 4,99 (1H, с), 6,52 (1H, с), 7,42 (1H, дд, J=4,8, 7,9 Гц), 8,10 (1H, д, J=7,8 Гц), 8,69 (1H, д, J=3,9 Гц), 9,00 (1H, с).
Пример 4
Синтез 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (1,00 г) суспендировали в 4 мл N-метил-2-пирролидинона, 0,75 мл (3,0 эквивалента) 2,6-лутидина добавляли к суспензии и 0,54 мл (2,7 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии при комнатной температуре. Обеспечивали протекание реакции в течение трех часов. Реакционный раствор добавляли по каплям к 100 мл воды. Смесь перемешивали в течение двух часов и затем к ней добавляли 10 г хлорида натрия. Смесь затем перемешивали в течение ночи, и полученный в результате осадок собирали посредством фильтрации, промывали водой и сушили с получением 1,276 г порошка, состоящего в основном из 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A. 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропен A, полученный таким образом, очищали посредством хроматографии на силикагеле (силикагель C-60 производство Merck Ltd.; 50 мл для первого раза, 150 мл в собранных основных фракциях для второго раза и только этилацетат; объемная скорость потока 5 мл/мин) с получением 576 мг 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (выход: 44,4%) (чистота: 88,6%) и 115 мг (выход: 8,8%) (чистота: 74,9%).
Пример 5
Синтез 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (500 мг) суспендировали в 2,5 мл N-метил-2-пирролидинона и 0,25 мл (2,5 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии при комнатной температуре. Обеспечивали протекание реакции в течение 24 час. Реакционный раствор добавляли по каплям к 50 мл воды. Смесь доводили до pH 7,5 посредством добавления 8% водного бикарбоната натрия. К ней затем добавляли хлорид натрия (5 г) и смесь перемешивали в течение ночи. Полученный в результате осадок затем собирали посредством фильтрации и промывали водой с получением порошка. Порошок сушили с получением 604 мг порошка,состоящего, в основном, из 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A. 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропен A, полученный таким образом, очищали посредством хроматографии на силикагеле (100 мл силикагеля C-60N производство KANTO CHEMICAL CO., INC.; только этилацетат; объемная скорость потока 5 мл/мин) с получением 338 мг 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (выход: 52,0%) (чистота: 93,2%).
Пример 6
Синтез 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (500 мг) суспендировали в 2,5 мл N-метил-2-пирролидинона, суспензию охлаждали до 0°C и 0,15 мл (1,5 эквивалента) циклопропанкарбонилхлорида добавляли к ней по каплям. Смесь перемешивали при 0°C в течение 20 час и 0,1 мл (1,0 эквивалент) циклопропанкарбонилхлорида затем добавляли дополнительно. Смесь перемешивали в течение 66 час и 0,1 мл (1,0 эквивалент) циклопропанкарбонилхлорида далее добавляли дополнительно. Смесь перемешивали в течение 95 час и добавляли по каплям к 50 мл воды со льдом. Смесь доводили до pH 7,5 посредством добавления 8% водного бикарбоната натрия. Затем к ней добавляли хлорид натрия (5 г) и смесь перемешивали. Полученный в результате осадок затем собирали посредством фильтрации и промывали водой. Фильтрат экстрагировали этилацетатом, и этилацетатный слой затем промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель затем удаляли посредством перегонки при пониженном давлении. Остаток и осадок объединяли вместе с последующей очисткой посредством хроматографии на силикагеле (150 мл силикагеля C-60N, производство KANTO CHEMICAL CO., INC.; только этилацетат; объемная скорость потока 5 мл/мин) с получением 396 мг 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (выход: 60,9%) (чистота: 95,3%).
Пример 7
Синтез 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропен A (200 мг, чистота: 95,6%), полученный в Примере 1, суспендировали в 1,0 мл N-метил-2-пирролидинона и 0,06 мл (1,5 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии при комнатной температуре. Обеспечивали протекание реакции в течение 21,5 час и 20 мл воды добавляли к реакционному раствору. Смесь доводили до pH 7,5 посредством добавления 8% водного бикарбоната натрия и 10 мл этилацетата и к ней добавляли 3 г хлорида натрия. Смесь экстрагировали и затем промывали водой. Этилацетат (10 мл) далее добавляли к водному слою и смесь экстрагировали. Экстракт затем промывали водой и объединяли с этилацетатным слоем, полученным выше. Этилацетат удаляли посредством перегонки при пониженном давлении с получением порошка (295 мг) состоящего, в основном, из 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A. Порошок очищали посредством хроматографии на силикагеле (100 мл силикагеля C-60N, производство KANTO CHEMICAL CO., INC.; только этилацетат; объемная скорость потока 5 мл/мин) с получением 119 мг 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (выход: 55,0%) (чистота: 96,5%).
Пример 8
Синтез 1,7,11-три-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (500 мг) суспендировали в 2,5 мл N-метил-2-пирролидинона, 0,44 мл (5 экв.) пиридина добавляли к суспензии и 0,45 мл (4,5 экв) циклопропанкарбонилхлорида добавляли по каплям к суспензии при комнатной температуре. Обеспечивали протекание реакции в течение 1,5 час. Реакционный раствор добавляли по каплямк 50 мл воды. Смесь перемешивали в течение трех часов и затем к ней добавляли 5 г хлорида натрия. Затем реакционный раствор перемешивали в течение 1,5 час, и полученный в результате осадок затем собирали посредством фильтрации и промывали водой. Порошок, полученный таким образом, сушили с получением 721 мг 1,7,11-три-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A в виде порошка (выход: 99,4%) (чистота: 89,6%). Для соединения, полученного таким образом, измеряли FAB-МС и1Н-ЯМР, и в результате было обнаружено, что данные соответствовали соединению 218, описанному в WO 2006/129714.
FAB-MC; m/z 662 (M+H)+;1H-ЯМР (CDCl3) δ 2,89 (1H, с), 3,72 (1H, д, J=11,7 Гц), 3,82 (1H, д, J=11,7 Гц), 4,79 (1H, дд, J=4,9, 11,5 Гц), 5,01 (1H, шир.с), 5,02 (1H, дд, J=4,9, 11,2 Гц), 6,46 (1H, с), 7,41 (1H, дд, J=4,8, 7,9 Гц), 8,10 (1H, дт, J=1,7, 6,4 Гц), 8,69 (1H, шир.с), 9,02 (1H, с).
Пример 9
Синтез 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-O-трициклопропанкарбонил-1,7,11-тридеацетилпирипиропен A (1,0 г), синтезированный в Примере 8, растворяли в 95% растворе водного метанола (30 мл) и к нему добавляли трет-бутоксид калия (85 мг) при комнатной температуре. Смесь перемешивали при этой температуре в течение 16 час и к ней затем добавляли уксусную кислоту. Метанол удаляли посредством перегонки при пониженном давлении и остаток экстрагировали хлороформом. Хлороформный слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель затем удаляли посредством перегонки при пониженном давлении с получением неочищенного продукта 1,11-O-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (724 мг, чистота: 50%). Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (Merck силикагель 60F254 0,5 мм; гексан:ацетон=10:5,5)с получением 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (370 мг, выход: 41%).
Пример 10
Синтез 1,11-O-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (способ с использованием кристаллизации)
1,7,11-О-трициклопропанкарбонил-1,7,11-тридеацетилпирипиропен A (4 г), синтезированный в Примере 8, растворяли посредством нагрева в метаноле (100 мл) и к нему добавляли карбонат калия (420 мг) при комнатной температуре. Смесь перемешивали при этой температуре в течение 6 час, к ней добавляли уксусную кислоту (370 мг) и воду (100 мл) и смеси давали отстояться в течение 23 час. Осажденное исходное вещество удаляли посредством фильтрации, затем добавляли воду (50 мл) и смеси давали отстояться в течение 20 час. Метанол удаляли посредством перегонки при пониженном давлении и остатку давали отстояться в течение 7 час. В результате 1,11-O-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропен A был осажден и осажденный 1,11-O-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропен A собирали посредством фильтрации (900 мг, выход: 25,1%, чистота: 81%).
Пример 11
Синтез 11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропен A
1,7,11-Тридеацетилпирипиропен A (4,53 г) суспендировали в 22,5 г N-метил-2-пирролидинона, и к суспензии добавляли 1,51 г (1,51 эквивалента) триэтиламина и 2,25 г ангидрида (1,47 эквивалента) циклопропанкарбоновой кислоты, и смесь нагревали при перемешивании при 60°C в течение 23 час. Затем нагретую смесь концентрировали при пониженном давлении при температуре бани, равной 70°C. Воду (10 мл) добавляли к маслу, полученному таким образом, для отверждения. Твердое вещество промывали трижды 10 мл воды и собирали посредством фильтрации. Порошок, полученный таким образом, промывали 5 млводы и сушили при пониженном давлении при 40°C в течение одного дня с получением 11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (4,73 г, выход: 91,4%, чистота: 76,2%).
Пример 12
Синтез 1-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
11-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропен A (199,7 мг, чистота: 95,6%), полученный тем же образом, что в Примере 1, суспендировали в 2,0 мл хлорбензола. DBU (0,02 мл, приблизительно 0,4 эквивалента) добавляли к суспензии и смесь нагревали при перемешивании при 80°C в течение 9 час. Затем реакционный раствор постепенно охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение двух дней. К нему добавляли этилацетат (20 мл) и 5 мл воды, и органический слой отделяли и концентрировали при пониженном давлении. Кристаллы осаждали в таком состоянии, что хлорбензол оставался в системе. Соответственно, кристаллы собирали посредством фильтрации и промывали толуолом. Кристаллы сушили при пониженном давлении при 60°C в течение ночи с получением 1-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (153,4 мг, выход: 76,8%, чистота: 94,5%).
Пример 13
Синтез 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропен A (500 мг) суспендировали в 3,0 мл N-метил-2-пирролидинона и суспензию добавляли по каплям к 0,10 мл (1,0 эквивалент) циклопропанкарбонилхлорида при 0°C. Обеспечивали протекание реакции в течение одного дня и к ней добавляли 0,025 мл (0,25 эквивалента) циклопропанкарбонилхлорида. Далее, через 41 час после добавления циклопропанкарбонилхлорида, 1,0 мл N-метил-2-пирролидинона и 0,025 мл (0,25 эквивалента) циклопропанкарбонилхлорида добавляли к реакционному раствору и обеспечивали протекание реакции в течение 65 час. Реакционный раствор затем выливали в 30 мл воды со льдом и 50 мл этилацетата. Смесь нейтрализовали 8% водного бикарбоната натрия, к ней добавляли 3 г хлорида натрия, и смесь перемешивали, с последующим разделением. Органический слой промывали дважды 10 мл воды и растворитель удаляли посредством перегонки при пониженном давлении. Порошок (678 мг), полученный таким образом, подвергали хроматографии на силикагеле (силикагель C-60 (80 мл) производство Merck Ltd.; этилацетат-метанол (50:1 (об./об.)) для извлечения 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (479 мг, выход: 83,3%, чистота: 95,2%) и 51 мг (10,2%) 1-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A.
Пример 14
Синтез 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (1,00 г) суспендировали в 7,0 мл N-метил-2-пирролидинона, суспензию охлаждали до 0°C и 0,4 мл (2,0 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии.
Затем 0,1 мл (0,5 эквивалента) циклопропанкарбонилхлорида дополнительно добавляли к ней по каплям при 0°C, после прохождения каждого из периодов, равных 7 часам, 23 часам и 26 часам, после завершения покапельного добавления. Через 4 дня после добавления по каплям реакционный раствор выливали в 50 мл этилацетата и 50 мл воды со льдом. Далее, смесь нейтрализовали 0,7 г бикарбоната натрия и к ней добавляли 8% водного бикарбоната натрия и 5,0 г хлорида натрия. Смесь перемешивали и ей давали возможность отстояться с последующим разделением. Органический слой промывали дважды 20 мл воды и концентрировали при пониженном давлении. Этилацетат (8,0 мл) добавляли к пенистому порошку, полученному таким образом, смесь нагревали до 60°C и к ней добавляли 8,0 мл н-гексана. Смесь охлаждали до 50°C и добавляли очень малое количество затравочных кристаллов. После осаждения кристаллов добавляли 2,0 мл н-гексана и смесь перемешивали в течение ночи. Кристаллы собирали посредством фильтрации и собранные кристаллы промывали 10 мл н-гексан-этилацетат (1:1 (об./об.)). Кристаллы, полученные таким образом, сушили в течение ночи при 60°C с получением 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (787 мг, выход: 60,5%, чистота: 87,5%).
Пример 15
Синтез 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (10,0 г) суспендировали в 40,0 мл N-метил-2-пирролидинона, суспензию охлаждали до 0°C, и 3,0 мл (1,5 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии.
Затем к ней дополнительно добавляли циклопропанкарбонилхлорид по каплям при 0°C после прохождения каждого из периодов, равного 6 часам (2,0 мл, 1,0 эквивалент), 24 часам (1,0 мл, 0,5 эквивалента), 32 часам (0,50 мл, 0,25 эквивалента) и 48 часам (1,0 мл, 0,5 эквивалента) после завершения покапельного добавления. Дополнительно, добавляли N-метил-2-пирролидинон через 6 часов (20,0 мл) и 48 часов (10,0 мл) после завершения покапельного добавления. Через 96 часов после покапельного добавления реакционный раствор выливали в 100 мл этилацетата и 200 мл воды со льдом и перемешивали с последующим разделением.
Этилацетат (170 мл) добавляли к водному слою, смесь далее нейтрализовали 10,1 г бикарбоната натрия и к ней добавляли 20,0 г хлорида натрия. Смесь перемешивали и ей давали возможность отстояться с последующим разделением. Органический слой промывали однократно 50 мл 5% солевым раствором и дважды 30 мл воды и концентрировали при пониженном давлении. Этилацетат добавляли к остатку до полного объема, равного 110 мл. Смесь далее нагревали до 60°C и к ней добавляли 100,0 мл н-гексана. Смесь охлаждали до 50°C и добавляли очень малое количество затравочных кристаллов. После трехчасового осаждения кристаллов добавляли 20 мл н-гексана, и смесь перемешивали в течение двух дней. Кристаллы собирали посредством фильтрации и собранные кристаллы промывали 50 мл смеси н-гексан-этилацетат (1:1 (об./об.)). Кристаллы, полученные таким образом, сушили при 60°C в течение одного дня с получением 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (8,83 г, массовый выход: 67,9%, чистота: 86,4%).
Затем часть массой 8,70 г от 8,83 г 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A растворяли в 43,5 мл метанола, и 29,0 мл воды добавляли к ней при комнатной температуре. В результате раствор становился млечным и, следовательно, его нагревали до 30°C. К нему добавляли метанол (1,0 мл) и очень малое количество затравочных кристаллов. После осаждения кристаллов смешанный раствор, состоящий из 15,0 мл воды и 3,0 мл метанола, добавляли двумя разделенными порциями. Смесь перемешивали при комнатной температуре в течение ночи и отфильтровывали. Кристаллы промывали смешанным раствором, состоящим из 16,0 мл воды и 4,0 мл метанола. Кристаллы сушили при 80°C при пониженном давлении с получением 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (6,22 г, массовый выход всего: 48,5%, чистота: 94,5%).
Пример 16
Синтез 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (10,0 г) суспендировали в 40,0 мл N-метил-2-пирролидинона, суспензию охлаждали до 0°C и к суспензии добавляли по каплям 7,0 мл (3,5 эквивалента) циклопропанкарбонилхлорида.
Затем обеспечили протекание реакции при 0°C в течение 53 часов и реакционный раствор выливали в 50 мл этилацетата и 80 мл воды со льдом. Смесь перемешивали при 7°C или ниже с последующим разделением. Этилацетат (30 мл) добавляли к водному слою и смесь перемешивали с последующим разделением. Этилацетат (100 мл) добавляли к водному слою, полученному таким образом, и смесь нейтрализовали 72 мл 1N гидроксида натрия и небольшим количеством 8% водного бикарбоната натрия. Хлорид натрия (15,0 г) добавляли к смеси при 10-15°C, и смесь перемешивали, и ей давали возможность отстояться с последующим разделением. Органический слой промывали однократно 30 мл 5% солевым раствором и дважды 30 мл воды, и смесь концентрировали при пониженном давлении. Этилацетат (20 мл) добавляли к остатку, смесь нагревали до 60°C и к ней добавляли 14 мл н-гексана. В результате раствор становился млечным и поэтому 4,0 мл этилацетата добавляли для растворения. Раствор затем охлаждали до 50°C и добавляли очень малое количество затравочных кристаллов. Через 1,5 часа после осаждения кристаллов добавляли 10 мл н-гексана и смесь перемешивали в течение ночи. Кристаллы собирали посредством фильтрации и промывали 30 мл смеси н-гексан-этилацетат (1:1 (об./об.)). Кристаллы сушили при 80°C в течение двух дней с получением 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (8,48 г, массовый выход: 65,2%, чистота: 83,4%).
Этилацетатный раствор, полученный при последующей обработке, нейтрализовали, промывали насыщенным солевым раствором и водой и сушили при пониженном давлении. Отдельно, фильтрат, полученный при сборе кристаллов, и промывки концентрировали и сушили. Эти два вещества, полученные таким образом, объединяли вместе (5,71 г) и смесь растворяли в метаноле (30,0 мл). Затем 5,16 мл 5N раствора гидроксида натрия добавляли по каплям при комнатной температуре. 5N раствор гидроксида натрия (2,0 мл) далее добавляли по каплям через 1,5 часа после добавления по каплям 5N раствора гидроксида натрия. Смесь перемешивали при комнатной температуре в течение 18 часов, отфильтровывали и промывали 22 мл смеси метанол-вода (1:1 (об./об.)).
Кристаллы, полученные таким образом, сушили при 80°C в течение одного дня с получением исходного вещества, т.е., 1,7,11-тридеацетилпирипиропена A (1,96 г, извлечение: 19,6%, чистота: 94,5%).
Когда извлечение было учтено, выход 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A составил 81,0%.
Пример 17
Синтез 1,1l-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (10,0 г) суспендировали в 40,0 мл N-метил-2-пирролидинона, суспензию охлаждали до 3°C и 7,0 мл (3,5 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии. Затем обеспечивали протекание реакции при 0°C в течение 48 час и реакционный раствор выливали в 50 мл этилацетата и 80 мл воды со льдом. Смесь перемешивали при 10°C или ниже с последующим разделением. Этилацетат (100 мл) добавляли к водному слою, смесь нейтрализовали 25 мл 5N гидроксида натрия и небольшим количеством 8% водного бикарбоната натрия и к ней добавляли 8 г хлорида натрия при от 10 до 15°C. Смесь перемешивали для растворения и ей давали возможность отстояться с последующим разделением. Органический слой промывали однократно 30 мл 5% солевого раствора и дважды 30 мл воды, концентрировали до 40 мл при пониженном давлении и перемешивали при комнатной температуре в течение 5 часовдля осаждения кристаллов. Затем добавляли 20 мл н-гексана в течение двух часов и смесь перемешивали в течение ночи. Кристаллы отфильтровывали и промывали 30 мл смеси н-гексан-этилацетат (1:1 (об./об.)). Кристаллы сушили при комнатной температуре при пониженном давлении в течение 30 мин с получением 7,31 г кристаллов 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A. ЯМР-спектр (прибор: Lambda-400, растворитель: CDCl3, отношение между интегральным значением для двух протонов CH3COOCH2CH3 при δ 4,12 и интегральное значение для одного протона 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A) кристаллов, полученных таким образом, показал, что содержание этилацетата составило 0,96 мол. в расчете от 1,0 мол. 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена А (массовый выход (в виде этилацетатного сольвата): 49,1%, чистота: 88,9%).
Дифракционная рентгенограмма порошка кристаллов имела следующие значения.
Дифракционная рентгенограмма порошка
Прибор: RINT 2200 (производство Rigaku Denki Co., Ltd.)
Условия измерения: рентгеновский источник: CuKα/40 кВ/20 мА, зазор для образца: 0,020°, скорость сканирования: 0,500°/мин, зазор сканирования: 2θ/θ и диапазон сканирования: от 3,0 до 40,0°.
Характеристические пики проявлялись при следующих дифракционных углах [2θ (°)].
Дифракционные углы (2θ): 7,4±0,1°, 12,0±0,1°, 17,0±0,1°, 18,3±0,1° и 19,1±0,1°.
Дифракционная рентгенограмма порошка показана на Фиг.1.
Пример 18
Синтез 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (10,0 г) суспендировали в 40,0 мл N-метил-2-пирролидинона, суспензию охлаждали до 0°C и 3,0 мл (1,5 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии. Через 4 часа после добавления по каплям к ней по каплям добавляли циклопропанкарбонилхлорид (2,0 мл (1,0 эквивалент)) при 0°C.
Обеспечивали протекание реакции при 0°C в течение 69 часов, и реакционный раствор затем выливали в 100 мл этилацетата и 120 мл воды со льдом и перемешивали с последующим разделением. Этилацетат (100 мл) добавляли к водному слою, смесь далее нейтрализовали 9,5 г бикарбоната натрия и к ней добавляли 8,0 г хлорида натрия. Смесь перемешивали и ей давали возможность отстояться с последующим разделением. Органический слой промывали однократно 30 мл 5% солевого раствора и дважды 30 мл воды и смесь концентрировали при пониженном давлении. Этилацетат (35,0 мл) добавляли к остатку и смесь затем перемешивали при комнатной температуре в течение 1,5 час. Затем к ней по каплям добавляли 35,0 мл н-гексана в течение двух часов. Смесь перемешивали при комнатной температуре в течение ночи. Осажденные кристаллы затем собирали посредством фильтрации, промывали 30 мл смеси н-гексан-этилацетат (1:1 (об./об.)) и сушили при пониженном давлении в течение четырех часов с получением 9,39 г кристаллов, содержащих 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропен A. Кристаллы, полученные таким образом, анализировали посредством метода, описанного в Примере 17, и было обнаружено, что они содержат 1 моль этилацетата в расчете на 1,0 мол. 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (массовый выход (в виде этилацетатного сольвата): 63,0%) (чистота: 85,5%).
Дифракционная рентгенограмма порошка кристаллов соответствовала приведенной в Примере 17.
Этилацетатный сольват (порция в 8,00 г в 9,39 г) 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A, полученный таким образом, растворяли в 16,0 мл метанола. Раствор нагревали до 35°C, и добавляли 10,0 мл воды. В результате раствор становился млечным и поэтому к нему добавляли 1,0 мл метанола. Через один час после добавления метанола смесь охлаждали до 25°C, и смешанный раствор, состоящий из 16,8 мл воды и 7,2 мл метанола добавляли к ней по каплям при от 20 до 25°C в течение двух часов. Смесь перемешивали при комнатной температуре в течение ночи. Полученный в результате осадок собирали посредством фильтрации и промывали смешанным раствором, состоящим из 7,0 мл воды и 3,0 мл метанола. Образец (500 мг) экстрагировали из твердого вещества, полученного таким образом, и оставшуюся часть сушили при 80°C при пониженном давлении с получением 5,68 г 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (6,01 г, с учетом количества экстрагированного образца) (общий массовый выход от 1,7,11-тридеацетилпирипиропена A: 54,2%) (чистота: 92,3%).
Пример 19
Синтез 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (20,0 г) суспендировали в 80,0 мл N-метил-2-пирролидинона, суспензию охлаждали до -10°C и 12,0 мл (3,0 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии. Обеспечивали протекание реакции при -10°C в течение 4 часов и 4,0 мл (1,0 эквивалент) циклопропанкарбонилхлорида добавляли к ней по каплям дополнительно. Затем обеспечивали протекание реакции при -10°C в течение 72 час и реакционный раствор выливали в 200 мл этилацетата и 180 мл 8% водного бикарбоната натрия при 5°C или ниже. Смесь нейтрализовали 20 мл 8% водного бикарбоната натрия, затем добавляли 20 мл 15% насыщенного солевого раствора и смесь перемешивали при 10°C с последующим разделением. Органический слой промывали трижды 60 мл воды и концентрировали до 60 мл при пониженном давлении. Затем к ней добавляли 100 мл этилацетата и смесь концентрировали до 80 млпри пониженном давлении. Смесь перемешивали при комнатной температуре в течение ночи, и осажденные кристаллы затем собирали посредством фильтрации и промывали смешанным раствором, состоящим из 10 мл н-гексана и 20 мл этилацетата. Кристаллы, полученные таким образом, сушили при пониженном давлении при 80°C в течение ночи с получением 17,80 г 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A. Кристаллы анализировали посредством метода, описанного в Примере 17, и было обнаружено, что они содержат 0,75 мол. этилацетата в расчете на 1,0 мол. 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (массовый выход: 61,8% (в виде этилацетатного сольвата)) (чистота: 87,5%).
Пример 20
Синтез 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (50,0 г) суспендировали в 200 мл N-метил-2-пирролидинона, суспензию охлаждали до -10°C и 15,0 мл (1,5 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии. Затем циклопропанкарбонилхлорид добавляли по каплям к смеси в количестве, равном 15,0 мл (1,5 эквивалента), 3 часа после добавления по каплям и в количестве, равном 10,0 мл (1,0 эквивалент), 5 часов после добавления по каплям при -10°C. Обеспечивали протекание реакции при -10°C в течение 72 час. Реакционный раствор затем выливали в 500 мл этилацетата и 500 мл 8% водного бикарбоната натрия при 5°C или ниже. Смесь нейтрализовали небольшим количеством 8% водного бикарбоната натрия и к ней добавляли 300 мл 15% насыщенного солевого раствора при 10°C или выше с последующим разделением. Органический слой промывали трижды 100 мл воды и концентрировали до 150 мл при пониженном давлении. Затем к ней добавляли 250 мл этилацетата и смесь снова концентрировали до 200 мл при пониженном давлении. К ней добавляли этилацетат (50 мл) и смесь перемешивали при комнатной температуре в течение ночи. Осажденные кристаллысобирали посредством фильтрации и промывали 80 мл этилацетата. Кристаллы, полученные таким образом, сушили при пониженном давлении при 50°C в течение двух часов с получением 44,90 г кристаллов, содержащих 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропен A. Кристаллы анализировали посредством метода, описанного в Примере 17, и было обнаружено, что они содержат 0,99 моль этилацетата в расчете на 1,0 мол. 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (массовый выход: 60,2% (в виде этилацетатного сольвата)) (чистота: 87,5%).
Пример 21
Синтез 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A
1,7,11-Тридеацетилпирипиропен A (50,0 г) суспендировали в 200 мл N-метил-2-пирролидинона, суспензию охлаждали до -10°C, и 15,0 мл (1,5 эквивалента) циклопропанкарбонилхлорида добавляли по каплям к суспензии. Затем циклопропанкарбонилхлорид добавляли по каплям к смеси в количестве, равном 15,0 мл (1,5 эквивалента), 3 часа после добавления по каплям и в количестве, равном 10,0 мл (1,0 эквивалент), через 5 час после добавления по каплям при -10°C. Обеспечивали протекание реакции при -10°C в течение 75 час. Реакционный раствор затем выливали в смешанный раствор, состоящий из 500 мл этилацетата, 500 мл воды со льдом и 40,0 г бикарбоната натрия при 5°C или ниже. Смесь нейтрализовали небольшим количеством 8% водного бикарбоната натрия и 300 мл 15% солевого раствора добавляли при 10°C или выше с последующим разделением. Органический слой промывали трижды 150 мл воды и концентрировали до 100 мл при пониженном давлении. Затем к ней добавляли 200 мл этилацетата и смесь снова концентрировали до 150 мл при пониженном давлении. Дополнительно, затем к ней добавляли 50 мл этилацетата, и смесь перемешивали при комнатной температуре в течение ночи. Осажденные кристаллы собирали посредством фильтрации и промывали 60 мл этилацетата. Кристаллы, полученные таким образом, сушили при пониженном давлении при 40°C в течение одного часа и сушили при комнатной температуре в течение двух часов с получением 49,10 г кристаллов, содержащих 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A (массовый выход: 65,8% (в виде этилацетатного сольвата)) (чистота: 84,7%).
Кристаллы анализировали таким же образом, как в Примере 17, и было обнаружено, что они содержат 0,98 мол. этилацетата в расчете на 1,0 мол. 1,11-ди-О-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A.
Порцию массой 24,0 г кристаллов, полученных таким образом, суспендировали в 48,0 мл этилацетата, и суспензию перемешивали при 70°C в течение одного часа и перемешивали при комнатной температуре в течение ночи. Затем реакционный раствор отфильтровывали с последующей промывкой 30 мл этилацетата. Промытый продукт сушили при комнатной температуре в течение 5 часов с получением 20,54 г рассматриваемого продукта (массовый выход: 56,4% (в виде этилацетатного сольвата; общий выход от 1,7,11-тридеацетилпирипиропена A) (чистота: 93,2%).
Кристаллы, полученные таким образом, содержали 1,00 моль этилацетата в расчете на 1,0 мол. 1,11-ди-O-циклопропанкарбонил-1,7,11-тридеацетилпирипиропена A.
Пример 22
1,11-O-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропен A синтезировали из 1,7,11-О-трициклопропанкарбонил-1,7,11-тридеацетилпирипиропена A, синтезированного в Примере 8, при реагентах, растворителях, времени и температурных условиях, описанных в Таблице 3 ниже. После завершения реакции, реакционный раствор анализировали посредством высокоэффективной жидкостной хроматографии при следующих аналитических условиях для определения количества 1,11-O-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена A, полученного в реакционном растворе. Результаты показаны в Таблице 3.
Аналитические условия
Детектор: детектор поглощения в ультрафиолетовом диапазоне или детектор с фотодиодной матрицей (длина волны измерения: 254 нм)
Колонка: CAPCELL PAK C18; 2,0 мм вн. диам. × 150 мм внутренний диаметр; 5 мкм
Темп колонки.: 40°C
Подвижная фаза A: Вода
Подвижная фаза B: Ацетонитрил для жидкостной хроматографии
Подача подвижной фазы: Градиент концентрации регулируется посредством изменения отношения между подвижной фазой A и подвижной фазой B следующим образом.
Объемная скорость потока: 0,2 мл/мин
Условия для подвижной фазы: как показано в Таблице 2 ниже
[Таблица 2]
[Таблица 3]
Пример 23
Синтез 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетил-пирипиропена А
1,7,11-тридеацетилпирипиропен А (45,0 г) суспендировали в N,N-диметилацетамиде (185,0 г), суспензию охлаждали до -10°С и 34,4 г (3,5 эквивалентов) циклопропанкарбонилхлорида добавляли к суспензии в течение 15 мин. В реакционной смеси обеспечивали протекание реакции при -10°С в течение 72 ч. Реакционный раствор затем вводили по частям в течение 15 мин в смешанный раствор, составленный из толуола (640 г) и 8% бикарбоната натрия (500 г), при 20°С. После прекращения выделения СО2 смесь нагревали до 60°С и фазы разделяли. Органическую фазу промывали три раза водой (135 г) и затем толуол (595 г) отгоняли. После добавления толуола (160 г) раствор затравливали кристаллами 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена А (0,06 г), медленно охлаждали до комнатной температуры и перемешивали в течение ночи. Осажденные кристаллы собирали посредством фильтрации и промывали толуолом (25 г). Кристаллы, полученные таким образом, сушили при пониженном давлении при 100°С в течение 3 дней с получением 29,3 г 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена А (выход: 46,9%, чистота: 87,8%).
Пример 24
Синтез 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена А
1,7,11-тридеацетилпирипиропен А (45,0 г) суспендировали в N,N-диметилацетамиде (185,0 г), суспензию охлаждали до -10°С и 34,4 г (3,5 эквивалентов) циклопропанкарбонилхлорида добавляли к суспензии в течение 15 мин. В реакционной смеси обеспечивали протекание реакции при -10°С в течение 88 ч. Реакционный раствор затем вводили по частям в течение 15 мин в смешанный раствор, составленный из толуола (640 г) и 8% бикарбоната натрия (500 г), при 20°С. После прекращения выделения СО2 смесь нагревали до 40°С и фазы разделяли. Органическую фазу промывали три раза водой (135 г), и затем толуол (630 г) отгоняли. После добавления толуола (100 г) раствор затравливали кристаллами 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена А (0,05 г), медленно охлаждали до комнатной температуры и перемешивали в течение ночи. Осажденные кристаллы собирали посредством фильтрации и промывали толуолом (25 г). Кристаллы, полученные таким образом, сушили при пониженном давлении при 120°С в течение 1 дня с получением 32,7 г 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена А (выход: 50,9%, чистота: 85,2%).
Пример 25
Синтез 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена А
1,7,11-тридеацетилпирипиропен А (45,0 г) суспендировали в N,N-диметилацетамиде (185,0 г), суспензию охлаждали до 0°С и 34,4 г (3,5 эквивалентов) циклопропанкарбонилхлорида добавляли к суспензии в течение 15 мин. В реакционной смеси обеспечивали протекание реакции при -10°С в течение 50 ч. Реакционный раствор затем вводили по частям в течение 15 мин в смешанный раствор, составленный из толуола (640 г) и 8% бикарбоната натрия (500 г), при 20°С. После прекращения выделения СО2 смесь нагревали до 40°С и фазы разделяли. Органическую фазу промывали три раза водой (135 г), и затем толуол добавляли по частям (всего 350 г) и отгоняли (всего 912 г). Раствор затравливали кристаллами 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена А (0,05 г), медленно охлаждали до комнатной температуры и перемешивали в течение ночи. Осажденные кристаллы собирали посредством фильтрации и промывали толуолом (33 г). Кристаллы, полученные таким образом, сушили при пониженном давлении при 120°С в течение 1 дня с получением 27,6 г 1,11-О-дициклопропанкарбонил-1,7,11-тридеацетилпирипиропена А (выход: 47,4%, чистота: 93,9%).
Пример 26
Прямое ацилирование кислотным ангидридом
[Химическая формула 8]

1,7,11-тридеацетилпирипиропен А (1,0 г) и основание (~20% мол.) суспендировали в N,N-диметилацетамиде (4,0 г) и к суспензии добавляли 0,81 г (2,1 эквивалента) пивалевого ангидрида циклопропанкарбоновой кислоты. Реакционную смесь перемешивали в течение приблизительно 5 ч, затем нагревали до 60°С и перемешивали в течение 18 ч.
Конверсия реакции сопровождалась качественным ВЭЖХ-анализом. Условия, используемые для ВЭЖХ, представлены в обобщенном виде в Таблице 4. Данные обобщены в Таблице 5.
[Таблица 4]
[Таблица 5]
Реферат
Изобретение относится к способу получения соединения С, который включает одно-трехстадийное селективное ацилирование гидроксильной группы в 1-положении и 11-положении соединения, представленного формулой B1, ацилирующим агентом в растворителе, выбранном из N-метил-2-пирролидинона и N,N-диметилацетамида, в присутствии основания, выбранного из 2,4,6-коллидина и 2,6-лутидина, или в отсутствие основания. Способ позволяет селективно получать соединение С с высоким выходом. 3 н. и 10 з.п. ф-лы, 1 ил., 5 табл., 25 пр.
Формула

где R представляет собой С3-6 алкилкарбонил,
включающий одно-трехстадийное селективное ацилирование гидроксильных групп в 1-положении и 11-положении соединения В1, представленного формулой В1

ацилирующим агентом в растворителе, выбранном из N-метил-2-пирролидинона и N,N-диметилацетамида, в присутствии основания, выбранного из 2,4,6-коллидина и 2,6-лутидина, или в отсутствие основания и
(i) где соединение С получают посредством одностадийного ацилирования гидроксильных групп в 1-положении и 11-положении соединения B1;
(ii) способ включает получение соединения С посредством двустадийного ацилирования, состоящего из стадий:
ацилирования гидроксильной группы в 11-положении соединения В1 ацилирующим агентом с получением соединения В2, представленного формулой В2

где R такой, как определено в формуле С; и
дополнительного ацилирования гидроксильной группы в 1-положении соединения В2; или
(iii) способ включает получение соединения С посредством трехстадийного ацилирования, состоящего из стадий:
ацилирования гидроксильной группы в 11-положении соединения В1 с получением соединения В2, представленного формулой В2

где R такой, как определено в формуле С;
переноса ацильной группы в 11-положении соединения В2 к гидроксильной группе в 1-положении с получением соединения В3, представленного формулой В3

где R такой, как определено в формуле С; и
ацилирования гидроксильной группы в 11-положении соединения В3.
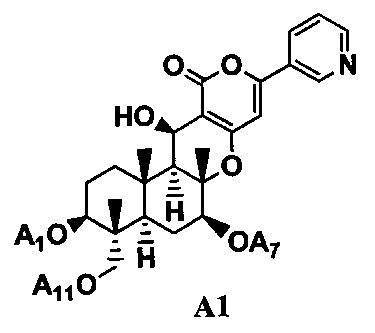
где А1, А7 и А11, которые могут быть одинаковыми или различными, представляют собой ацетил или пропионил в присутствии основания.
(a) экстракции реакционного раствора, содержащего соединение С, органическим растворителем, выбранным из группы, состоящей из метилацетата, этилацетата, бутилацетата, толуола, этилбензола и хлорбензола, и концентрирования экстракта после или без осушки;
(b) упаривания реакционного раствора, содержащего соединение С, досуха с получением неочищенного продукта и затем растворения неочищенного продукта в органическом растворителе, выбранном из группы, состоящей из метилацетата, этилацетата, бутилацетата, толуола, этилбензола и хлорбензола, при комнатной температуре или при нагревании, или
(c) упаривания реакционного раствора, содержащего соединение С, досуха с получением неочищенного продукта, растворения неочищенного продукта в органическом растворителе, выбранном из группы, состоящей из метилацетата, этилацетата, бутилацетата, толуола, этилбензола и хлорбензола, при комнатной температуре или при нагреве и добавления к раствору слабого растворителя, выбранного из группы, состоящей из гептана, гексана и циклогексана.

где R такой, как определено в формуле С,
в получении соединения С, представленного формулой С

где R представляет собой С3-6 алкилкарбонил.

где R такой, как определено в формуле С по п. 1,
в получении соединения С, представленного формулой С

где R представляет собой С3-6 алкилкарбонил.
Комментарии