Замещенные полициклические пиразольные ингибиторы киназной активности и их применение - RU2655921C2
Код документа: RU2655921C2
Чертежи

Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к области медицинской химии и относится к производным 4-(замещенный пятичленный гетероциклический пиримидин/пиридин)амино-1H-3-пиразолкарбоксамида, способам их получения и фармацевтической композиции, содержащей указанные соединения, а также к их применению в медицине, особенно при противоопухолевой терапии в качестве ингибитора протеинкиназы.
Предпосылки создания изобретения
В нормальных условиях клеточный цикл регулируется группой связанных протеаз, имеющих различные биологические функции, включая ингибирование или активирование клеточного цикла, где большинство белков, которые способствуют клеточному циклу, принадлежат к киназам. Киназы играют важную роль в регуляции белка для активирования важных физиологических функций. Их основная функция in vivo заключается в передаче фосфата из высокоэнергетических молекул аденозинтрифосфата (ATP) рецепторам, с тем, чтобы регулировать активацию или деактивацию рецепторного белка и, наконец, регулировать клеточный цикл. Однако во многих раковых клетках было обнаружено, что эти регулирующие нормальный клеточный цикл киназы внезапно исчезают из-под контроля. Таким образом, считается, что если эти нерегулируемые киназы могут быть подавлены, пролиферация раковых клеток будет под контролем. В последние годы, циклинзависимые киназы (CDK), киназы Aurora, поло-подобные киназы (PLK), кинезин (кинезины веретенообразного белка, KSP) и киназа контрольной точки клеточного цикла (CHK) и некоторые другие новые мишени находятся в тесной связи с клеточным циклом.
Среди них, соактивация киназы Aurora в центросоме и CDK имеет важное значение для инициации митоза. Они связаны друг с другом и взаимно способствуют регуляции клеточного цикла и процесса митоза. Соответствующие ингибиторы этих двух ингибиторов были исследованы, и различные соединения подвергались клиническим исследованиям, демонстрируя хорошие перспективы для разработки противораковых лекарственных средств.
Было обнаружено, что почти все опухоли связаны с расстройством регуляции клеточного цикла, которое может вызвать бесконтрольный рост клеток, нарушение дифференцировки клеток и аномальный апоптоз, и чрезмерная активация CDK (циклинзависимых киназ, CDK) является одной из важных причин для этих условий. CDK являются важными серин/треонин протеинкиназами, которые не проявляют биологической активности как таковой, до тех пор, пока они комбинированы с циклинами. После активации, CDK могут катализировать фосфорилирование субстрата, стимулируя каждую фазу клеточного цикла, выполнять последовательно синтез ДНК и митоз, и, наконец, индуцировать рост клеток и клеточную пролиферацию. Тем временем, CDK можно также связать с CDK ингибиторами (CDI) для оказания негативной роли на прогрессию регуляции, с тем, чтобы ингибировать клеточный цикл и предотвратить деление клеток. Так как CDK имеют решающее значение в регуляции пролиферации клеток опухоли и апоптоза, избирательное ингибирование активности CDK в тканях опухоли может играть позитивную роль в лечении опухолей и злокачественных заболеваний. Таким образом, исследование и скрининг низкомолекулярных ингибиторов для CDK является одной из главных областей для лечения рака и разработки новых химиотерапевтических лекарственных средств.
CDK1, CDK2, CDK4 и CDK6 являются более важными подтипами CDK в регуляции прогрессирования клеточного цикла. Благодаря тому, что дисрегуляция клеточного цикла является одной из основных причин рака, если клеточный цикл можно предотвратить от вступления в S-фазу, аберрантной репликации ДНК не произойдет. Процесс от G1 к S фазе регулируется главным образом CDK2/циклин E, поэтому CDK2 ингибиторы клеточного цикла могут предотвратить вступление в S-фазу для дальнейшей репликации ДНК. Кроме того, в дополнение к контролю от G1 в S-фазу, CDK2/циклин A также может контролировать прогрессирование S и G2 фазы через клеточный цикл. Можно увидеть, что CDK2 играет весьма значительную роль в клеточном цикле, и поэтому, если можно эффективно ингибировать активность CDK2, клеточный цикл будет контролируемым и неконтролируемая пролиферация клеток опухоли будет подавлена.
В последние годы ряд низкомолекулярных ингибиторов CDK были раскрыты и большинство из них показывают хорошую ингибирующую активность против CDK2. Они оказывают ингибирующее действие главным образом конкурентным связыванием с ATP активным сайтом CDK.
Семейство Aurora представляет собой серин/треонин-киназу. Существует три вида подтипов киназы Aurora, которые весьма существены в структуре и функциях в клетках человека: Aurora A, B и C. Она вовлечена в регуляцию клеточного митоза, включая дублирование центросома, формирование биполярного веретена и перегруппировку хромосомы в веретене, и т.д., и можно точно мониторить контрольную точку веретена, прерывать неправильную прогрессию клеточного цикла и полный процесс восстановления. Во время прогрессии клеточного цикла, киназы Aurora главным образом действуют на фазе М, и начинает серию биохимических событий митоза в сочетании с CDK.
Aurora А и Aurora В тесно связаны с опухолями. Во-первых, Aurora A расположена на 20q13.2, в то время как Aurora B расположена на 17p13. Обе из них расположены в хромосомных сегментах активных транслокаций, делеций или амплификаций, означая, что они обладают природной нестабильностью. Эти исследования показывают, что, когда Aurora A сверхэкспрессирована, она является потенциальным онкогеном. Амплификация этих двух хромосомных областей преобладает в тканях опухоли рака молочной железы и колоректального рака, и клеточной линии рака молочной железы, рака яичников, рака толстой кишки, рака простаты, рака шейки матки и нейробластомы. В настоящее время существует несколько исследований на канцерогенное действие Aurora C.
Aurora A, B и C являются высоко гомологичными в каталитической области только с различными короткими аминокислотными последовательностями в конечной области регуляции и каталитического домена. Активные сайты, где ингибиторы связываются, расположены в области петли. Пуриновое кольцо ATP может быть размещено в гидрофобном кармане киназы Aurora и образует водородную связь с аминокислотными остатками в области петли. Ингибитор киназы Aurora может конкурентно связываться с сайтом связывания ATP киназы Aurora, а также принадлежит к ATP конкурентным ингибиторам.
Сообщается, что на конечной стадии G2 микроинъекция антитела киназы Aurora может значительно задержать митотическую инициацию. В настоящее время полагают, что существует механизм участия киназы Aurora A в качестве последующего эффектора для активированных комплексов CDK/циклин в серии биохимических событий для инициации митоза. Он формирует положительную обратную связь интерактивной активации с комплексами CDK/циклин, таким образом, что комплексы CDK/циклин сначала активируют киназы Aurora, и киназы Aurora, в свою очередь, способствуют полной активации CDK и облегчению позиционирования в ядре комплекса. Эти два события являются важными для инициации митоза. Короче говоря, в ходе клеточного цикла соактивация киназы Aurora и CDK на центросоме является одним из необходимых условий для начала митоза клеток, при котором они взаимно связаны друг с другом в регуляции процесса клеточного цикла и митоза. Таким образом, если активности киназы Aurora и CDK могут ингибироваться одновременно, сдерживание разрастания опухолевых клеток может двойственно тормозится. Поэтому это имеет большое значение при разработке новых мульти-нацеленных ингибиторов CDK/Aurora.
На протяжении клеточного цикла, в добавление к контролю G1 в S-фазе, CDK2 также контролирует клеточные процессы в S и G2 фазах. Таким образом, путем ингибирования CDK2 можно препятствовать нормальной репликации ДНК в клеточном цикле. Вместе с тем, в фазе М регулирование митоза клеток главным образом зависит от Aurora A, которая играет незаменимую роль в дублировании центросомы, формировании биполярного веретена, хромосомных перестройках и тому подобное. Таким образом, полагают, что ингибирование Aurora A может предотвращать клеточный митоз. Поэтому, поиск низкомолекулярных мульти-мишеней, одновременно нацеливающих на CDK и киназы Aurora и затрагивающих клеточный цикл раковых клеток во множественных направлениях, будет наилучшим путем достижения цели лечения рака.
До сих пор многое, касающееся кристаллических структур CDK2 и Aurora A, было решено, и ингибирующее действие ингибиторов и мишеней стало ясным, что служит основой для создания на основе структуры лекарственных средств мульти-нацеленных ингибиторов. Для сравнения, ингибиторы CDK2 и киназы Aurora A конкурентно связываются с ATP-связывающим карманом, главным образом связываются с ферментом посредством водородных связей и гидрофобных взаимодействий. Существуют некоторые общие признаки для режимов действий низкомолекулярных ингибиторов с этими двумя киназами, и область их совместной эффективности заключаются в следующем: трехмерной структуре и физико-химических свойствах, таких как водородные связи, аналогичности гидрофобного и гидрофильного пространственного распределения с CDK2 и Aurora A: 1) шарнирное (шарнир): шарнирная область является самой важной областью для всех ATP-конкурентных ингибиторов, и в этой области зачастую имеются две или три критические водородные связи; и остатками в этих двух киназах, вовлеченными в формирование водородных связей, являются Glu81 и Leu83 для CDK2, и Glu211 и Ala213 для Aurora A. Более того, ряд гидрофобных плоских сегментов часто располагаются в шарнирной области, с тем чтобы обеспечить некоторые гидрофобные эффекты; 2) гидрофобная область A: эта область относится к гидрофобной полости, образованной между шарнирной областью и аспарагиновой кислотой (Asp145 для CDK2 и Asp274 для Aurora), которая расположена недалеко от области мотива киназы DFG. Поскольку область петли имеет определенную гибкость, выбор гидрофобных структурных фрагментов показывает некоторое разнообразие; 3) гидрофильная область: оба активных сайта двух киназ содержат гидрофильные области, где введение гидрофильных групп в эту область будет иметь большое значение для модуляции физико-химических свойств соединений. Гидрофильные области CDK2 расположены вблизи Gln85, в то время как Aurora A расположена около Leu215. Эти области высоко перекрывающихся фармакодинамических эффектов дают возможность конструирования мульти-нацеленных ингибиторов CDK/Aurora. Мульти-нацеленные лекарственные средства образованные перекрывающимися молекулами лигандов, как правило, имеют небольшую молекулярную массу, хорошие физические и химические свойства и значительно улучшенную лекарственную устойчивость.
В последние годы с развитием исследования в отношении протеинкиназ на основе структуры и последовательности гена, концепция семейства белка постоянно представляется как семейство ферментов с аналогичной структурностью и функциональностью и вовлеченное в различную передачу сигнала и клеточную регуляцию. Протеинкиназы могут быть классифицированы на множество субсемейств, основанных на различных фосфорилированных субстратах, таких как протеин-тирозин, протеин-серин/треонин, липиды и др. Протеинкиназы характеризуются их механизмами регуляции, включая аутофосфорилирование, трансфосфорилирование с другими киназами, белок-белковые взаимодействия, белок-липидные взаимодействия и взаимодействия белок-полинуклеотид. Единственная протеинкиназа может быть вовлечена в различные механизмы регуляции. Протеинкиназы катализируют δ-фосфат на конце ATP и фосфорилируют боковую цепь гидроксила остатков серина, треонина или тирозина, которые регулируют их субстратную активность, опосредуя большинство путей передачи клеточного сигнала, и регулируют множество различных клеточных процессов. Клеточные процессы включают, но, не ограничиваясь ими, пролиферацию, дифференцировку, апоптоз, моторику, транскрипцию, передачу и другие процессы передачи сигнала. Фосфорилирование действует в качестве молекулярного переключателя, регулирующего биологические функции белка-мишени. Фосфорилирование белков-мишеней происходит в ответ на разнообразные внеклеточные сигналы, события клеточного цикла, экологические или пищевые стрессы и т.д. Внеклеточные сигналы включают гормоны, нейромедиаторы, факторы роста и дифференцировки, и т.д. Специфическая протеинкиназа играет определенную роль в путях передачи сигналов, которые прямо или косвенно активируют или деактивируют метаболические ферменты, регуляторные белки, рецепторы, цитоскелетные белки, ионные каналы или насосы, или факторы транскрипции. Неконтролируемая передача сигналов, вызванная недостатком контролируемого фосфорилирования белков, имеет отношение ко многим заболеваниям, включая воспаление, рак, диабет, астмы/аллергии, заболевания и расстройства иммунной системы, расстройства и заболевания центральной нервной системы, а также ангиогенез при заболеваниях и расстройствах.
Кином человеческого белка содержит 518 членов протеинкиназ, включая 90 тирозинкиназ, 388 серин/треонин-киназ и 40 неклассических киназ. На основании филогенетического анализа, Хэнкс и Хантер разделили протеинкиназы человека на несколько категорий. С увеличением числа клонированных членов протеинкиназ, их классификация является более и более систематизированной и подробной. На основании филогенетического древа, они предложили классификацию на каталитический домен всех членов протеинкиназы, как опубликовано до июня 1993 года. Филогенетическое древо содержит четыре крупных семейства киназ: (a) семейство AGC, включающее семейство cAMP-зависимой протеинкиназы (PKA и PKG семейство), семейство протеинкиназы C, семейство B-адренергических рецепторов киназы (BARK), семейство рибосомной S6 киназы и других связанных киназ; (b) семейство CaMK, включающее семейство Ca2+/- кальмодулин регулируемой протеинкиназы, семейство Snfl/AMPK и других связанных протеинкиназ; (c) семейство CMGC, включая семейство CDK, семейство Erk (MAP) киназы, семейство гликоген-синтаза-киназы 3 (GSK3), семейство казеин-киназы II, семейство Clk и других связанных киназ; и (d) семейство протеин-тирозин-киназы (PTK). Филогенетическое древо также включает количество протеинкиназ, которые не принадлежат ни к одному из четырех семейств. Каждое большое семейство может далее подразделяться на подсемейства, и иметь по меньшей мере один из примеров, подобно Абельсон-киназе (ABL), Akt/протеинкиназе B (Akt/PKB), рецептору эпидермального фактора роста (EGFR), рецептору фактора роста фибробластов (FGFR), смешанной линии киназ (MLK), рецептору тромбоцитарного фактора роста (PDGFR), тирозин-киназе с иммуноглобулин-подобным и EGF-подобным доменами (TIE), рецептору васкулярно-эндотелиального фактора роста (VEGFR). В дополнение к основной структуре члены одного и того же семейства высоко согласуются в топологии, в виде регуляции и субстратной специфичности. Эволюционно подобные члены обладают схожей функциональностью.
CMGC представляет собой серин/треонин-киназу, и сайты фосфорилирования в основном расположены на серине или треонине в пролин-обогащенной среде. Члены этого семейства имеют большой нитрон в функциональностях X и XI субдоменов. Поскольку Dyrk (MNB), Dyrk2, Dyrk3 имеют высокую гомологию с Yakl, они организованы в семейство. В качестве члена семейства CMGC, CDK была приведена ранее. CDK1, CDK2, CDK4 и CDK6 главным образом вовлечены в регуляцию всего клеточного цикла, тогда как другие CDK связаны с другими биохимическими процессами. Для примера, для надлежащего развития нервной системы, требуется CDK5 и причастные к фосфорилированию различные нейрональные белки, такие как Tau, NUDE-1, синапсин 1, DARPP32 и комплекс Munc18/синтаксин 1A. Как правило, нейрональная CDK5 активируется путем связывания с p35/p39 белком. Однако активность может быть разупорядочена путем связывания с p25 (укороченная форма p35). Преобразование p35 в p25 и разупорядочение активности CDK5 может быть вызвана ишемией, эксцитотоксичностью и β-амилоидным пептидом. Таким образом, p25 относится к патогенезу при нейродегенеративных заболеваниях, таких как болезнь Альцгеймера, и имеет отношение к лечению таких заболеваний в качестве прямой мишени.
CDK7 представляет собой нуклеопротеин, который обладает cdc2CAK активностью и связан с циклином H. CDK7 является компонентом TFIIH транскрипции комплекса с РНК полимеразной II активностью C-концевого домена (CTD). Ее биохимические пути, при опосредовании Tat, связаны с регуляцией ВИЧ-1 транскрипции. CDK8 связывается с циклином C и имеет отношение к фосфорилированию РНК-полимеразы II CTD. Аналогично, комплекс CDK9/циклин-T1 (P-TEFb комплекс) имеет отношение к длительному контролю РНК-полимеразы II. PTEF-b также требует взаимодействия генома ВИЧ-1 с циклином T1, через которое он транскрибируется и активируется вирусным трансактивированным белком Tat. Таким образом, CDK7, CDK8, CDK9 и комплекс P-TEFb являются потенциальными мишенями для антивирусной обработки.
Регулирование активности комплекса CDK/циклин на молекулярном уровне требует серии стимуляции и ингибирования фосфорилирования и дефосфорилирования. Фосфорилирование CDK осуществляется группой активирующих киназ CDK (CAK) и/или некоторых киназ, таких как wee1, Myt1 и Mik1. Дефосфорилирование проводится фосфатазами, такими как cdc25 (a и c), pp2a, или KAP.
В то же время, было обнаружено, что гиперэкспрессия циклин D1 связана с раком пищевода, раком молочной железы, плоскоклеточным раком и немелкоклеточным раком легких. В патогенезе рака легких инактивация генов-супрессоров опухолей в настоящее время находится в "горячей точке". Ген-супрессор опухолей p15/MTS2, который обозначен как p15 ген и кодирует белок p15, принадлежит к семейству белков INK4. Он действует на CDK и циклиновый комплекс, специально ингибирующих активность CDK4 и CDK16 киназы, предотвращая фосфорилирование белков R6, ограничивая клеточный процесс от G1 к S-фазе и, таким образом, уменьшая пролиферацию клеток.
Митоген-активированная протеинкиназа (MAPK) является еще одним членом семейства CMGG киназы и рода серин/треонин-киназы. Она может передавать внеклеточные сигналы в клетки и ядро и регулировать экспрессию генов активацией факторов транскрипции через консервативные трехуровневые каскады (MAPKKK-MAPKK-MAPK). Этот путь существует у большинства клеток и принимает участие в различных клеточных функциях, таких как клеточное движение, апоптоз, дифференцировка и пролиферация клеток, и многих других физиологических процессах. Были идентифицированы четыре пути передачи сигнала MAPK, и каждый из них является весьма специфичным с индивидуальными функциями. В некоторой степени эти пути сигнала имеют некоторые перекрестные помехи. Исследования с ингибиторами и активаторами на различных путях сигнала могут не только содействовать пониманию механизма пути сигнала, но также создают новые возможности для диагностики и лечения заболеваний. ERK путь сигнала является одним из наиболее тщательно изученных путей, где Мек является ключевым ферментом в Ras-Raf-МЕК-ERK пути передачи сигнала, регулирующим клеточные ответы на различные сигналы роста. МЕК имеет семь подтипов, которые фосфорилируют и активируют последовательно MAPK, соответственно. MEK1 и MEK2 активируют "ERK"; MEK3 и MEK4 активируют p38; и MEK5 и MEK6 активируют JNK. Таким образом, MEK1/MEK2 обычно используется в качестве мишени при лечении рака для разработки перспективных противораковых лекарственных средств при исследовании пути сигнала ERK. Путь сигнала P38/MAPK представляет собой важную ветвь пути MAPK, который может быть активирован стрессорами (например, осмотическим шоком, УФ, гипоксией), цитокинами, факторами роста и инсулином, и даже может быть активирован в нормальной иммунной и воспалительной реакциях. Между тем, изучение другого пути сигнала p38, главной мишенью для лечения ревматоидного артрита в клинических исследованиях, также становится горячей точкой в последние годы. c-Jun N-концевой киназы (JNK)/путь сигнала стресс-активированного белка (SAPK) является важным членом семейства MAPK, в котором c-Jun является членом AP-1 фактора транскрипции комплекса, вовлеченного в контроле пролиферации, трансформации, выживания и апоптоза клеток. JNK также фосфорилирует p53 и некоторые ненуклеопротеинов. Фосфорилирование белка-мишени, опосредованное JNK, является очень важным, что может индуцировать экспрессию генов IL, VEGF, COX-2, MMP-9, гемциклооксигеназы-1 ICAM-1, NCX1, GnRHR и других цитокинов. Путь сигнала JNK вовлечен в воспалительные и аутоиммунные заболевания, такие как ревматоидный артрит, синдром раздраженного кишечника и атеросклероз. ERK5/BMK1, путь сигнала K5/большая митоген-активированная протеинкиназа (BMK1), является последним обнаруженным путем в семействе MAPK. Внеклеточные стрессоры включают высокий уровень сахара, низкий кислород, касательное напряжение кровяного потока, реактивнооксигенные виды (ROS), осмотическое давление и разнообразные митогены, подобно EGF, NGF и т.д. ERK5/BMK1 также следует за MAPK каскадом, MEKK 2/3 (MAPK-KK)-MEK5 (MAPKK)-BMK1/ERK5 (MAPK). После активации, ERK5 перемещается из цитоплазмы в ядро и фосфорилирует большое количество последовательных мишеней, которые включают MEF2C, c-Myc, Bim, AP-1 и т.д. ERK5 играет важную роль в выживании, пролиферации и дифференцировке клеток. Текущее исследование показало, что она тесно связана с патологическими процессами, диабетом, заболеваниями почек, фиброзом печени и опухолями.
Гликоген-синтаза-киназа-3 (GSK-3) представляет собой серин/треонин-киназу, существующую в виде двух повсеместно экспрессированных изоформах в организме человека (GSK-3α и GSK-3β). GSK-3 вовлечена в эмбриональное развитие, синтез белков, пролиферацию клеток, дифференцировку клеток, динамику микротрубочек, подвижность клеток и апоптоз клеток. Аналогичным образом, GSK-3 вовлечена в прогрессирование болезненных состояний, таких как диабет, рак, болезнь Альцгеймера, инсульт, эпилепсия, заболевание двигательных нейронов и/или травма головы. Филогенетически GSK-3 наиболее тесно связана с CDK.
При формировании части пути ответа на инсулин у млекопитающих, GSK-3 способна фосфорилировать, и тем самым инактивировать гликогенсинтазу. Разрегулирование активности гликогенсинтазы и, таким образом, синтеза гликогена через ингибирование GSK-3 рассматривается в качестве потенциального средства для борьбы с сахарным диабетом типа II или инсулин-независимым сахарным диабетом (NIDDM): состоянием, в котором тело ткани становится устойчивым к стимуляции инсулином. Ингибирование GSK-3, например, путем инактивации белка «мишени рапамицина в клетках млекопитающих» (mTOR), можно разрегулировать биосинтез белка. Наконец, имеются некоторые свидетельства для регуляции активности GSK-3 посредством MAPK пути через фосфорилирование GSK-3 киназами, такими как митоген-активированная протеинкиназа, активированная протеинкиназа 1 (MAPKAP-K1 или RSK). Эти данные позволяют предположить, что активность GSK-3 может быть модулирована митогенной, инсулиновой и/или аминокислотной стимуляцией.
Кроме того, GSK-3β является ключевым компонентом в сигнальном пути у позвоночных Wnt. Этот биохимический путь был показан, как имеющий критически важное значение для нормального эмбрионального развития и регуляции пролиферации клеток в нормальных тканях. В ответ на стимуляцию Wnt, GSK-3 становится ингибированной, что может вызвать дефосфорилирование субстрата GSK-3 (например, Axin, генный продукт аденоматозного полипоза (APC) и β-катенина). Аберрантный Wnt путь регуляции связан со многими типами рака. Мутации APC и/или β-катенина очень распространены при колоректальном раке и других опухолях, которые показывают, что β-катенин играет очень важную роль в клеточной адгезии. Таким образом, GSK-3 может также в некоторой степени модулировать процессы клеточной адгезии. Отдельно от уже описанных биохимических путей, существуют также данные, показывающие, что GSK-3 регулирует деление клеток посредством фосфорилирования циклина-D1, и фосфорилирует факторы транскрипции, такие как c-Jun, CCAAT/усилитель связывания белка α (C/EBPα), c-Myc и/или другие субстраты, такие как ядерный фактор активированных Т-клеток (NFATc), фактор теплового шока-1 (HSF-1) и c-AMP-ответный элемент активирующего белка (CREB). Независимо от специфики ткани, GSK-3 также играет роль в регуляции клеточного апоптоза. Роль GSK-3 в модуляции клеточного апоптоза через проапоптический механизм может иметь особое значение при медицинских состояниях, при которых может произойти апоптоз нейронов. Примерами являются травма головы, инсульт, эпилепсия, болезнь Альцгеймера и заболевание двигательных нейронов, прогрессивный надъядерный паралич, кортико-базальная дегенерация и болезнь Пика. Как было показано in vitro, GSK-3 способна гиперфосфорилировать микротрубочки ассоциированного белка Tau. Гиперфосфорилирование Tau разрушает его нормальное связывание с микротрубочками, а также может привести к образованию внутриклеточных нитей Tau. Считается, что постепенное накопление этих нитей приводит к возможной нейрональной дисфункции и дегенерации. Ингибирование Tau фосфорилирования путем ингибирования GSK-3 может, таким образом, обеспечить средства ограничения или предотвращения нейродегенеративных эффектов.
Протеин-тирозин-киназа (PTK), другое важное семейство протеинкиназы, катализированная γ-фосфатной группой ATP тирозиновых остатков многих важных белков, и поэтому фосфорилирует гидроксильные группы фенола. В нормальных клетках (за исключением нервных клеток) редко происходит фосфорилирование тирозина. Хотя фосфорилированный тирозин составляет только 0,5% фосфорилированных аминокислот в организме, он продемонстрировал, что фосфорилирование тирозина играет важную роль в регуляции многих клеточных процессов. Это является важным фактором в передаче сигнала путем передачи сигналов клетками. Кроме того, PTK вовлечены в серию клеточных функций и тесно связаны с ростом клеток, дифференцировкой и пролиферацией. PTK также играют весьма важную роль в росте и пролиферации злокачественных клеток. Расстройства функций тирозинкиназы могут привести к активации его последующих путей передачи сигнала и спровоцировать нарушения пролиферации клеток, в конечном итоге, приводя к образованию опухоли. Таким образом, ингибиторы тирозинкиназы полезны в лечении и профилактике рака.
PTK могут быть классифицированы на нерецепторные тирозинкиназы (NRTK) и рецепторные тирозинкиназы (RTK) в соответствии с тем, могут ли они существовать в рецепторах клеточной мембраны. До настоящего времени было обнаружено 58 видов RTK. Эти протеинкиназы структурно очень похожи на каталитическую область, которая состоит из около 270 аминокислотных остатков. RTK являются трансмембранными белками, и, как правило, состоящих из внеклеточного домена, трансмембранного домена и внутриклеточного киназного домена. Клинические исследования предполагают, что эти рецепторы и их лиганды имеют весьма прямое отношение ко многим видам рака. Гиперэкспрессия соответствующих факторов роста, которые вовлечены в рак, может привести к избыточным сигналам фосфорилирования тирозина, передаваемым в клетки. Такие факторы роста, подобно PDGF рецепторам (PDGF рецептор α и β), рецептору колониестимулирующего фактора (CSF-I) (CSF-1R, c-Fms), FLT-3 и c-kit и др., имеют отношение ко многим заболеваниям, таким как воспаление, и пролиферации клеток. Среди них ген FLT3, расположенный на хромосоме 13q12, является ранним гемопоэтическим фактором роста, обнаруженным в 1991 году, и закодированный рецептор FLT3 принадлежит к третьему типу семейства рецепторов RTK. Когда внеклеточный домен рецептора FLT3 связывается с его эндогенным лигандом, гомо- или гетеродимерный комплекс будет сформирован, что приведет к активации тирозинкиназы, открытию активной петли и подготовке белкового субстрата к связыванию с сайтом связывания ATP. Следовательно, белковый субстрат фосфорилируется, что приводит к передаче серии последовательных сигналов, вызывая пролиферацию и дифференцировку клеток. FLT3 рецепторы широко распространены в гемопоэтических стволовых/прогениторных клетках, тимусе, лимфатических, плацентных, мозговых, гонадных и многих других тканях. Однако мутация гена FLT3 (главным образом, включая внутренние тандемные дублированные мутации околомембранного домена и точечные мутации домена тирозинкиназы) и гиперэкспрессия могут привести к различным гематологическим злокачественным заболеваниям, таким как острый миелобластный лейкоз. В результате, FLT-3 становится "горячей точкой" в лечении рака, в частности в лечении гемобластозом. Гиперэкспрессия или мутация FLT-3 приводит к индукции неконтролируемых рецепторов FLT3 и впоследствии каналов, которые могут привести к активации Ras. Гемобластозомы включают лейкемию, лимфому (NHL), болезнь Ходжкина (также известный как лимфома Ходжкина) и миелому, подобно острому лимфобластному лейкозу (ALL), острому миелобластному лейкозу (AML), острому промиелоцитарному лейкозу (APL), хроническому лимфолейкозу (CLL), хроническому миелолейкозу (CML), хроническому нейтрофильному лейкозу (CNL), острому недифференцированному лейкозу (AUL), анапластической крупноклеточной лимфоме (ALCL), незрелому лимфолейкозу (PML), ювенильному миеломоноцитарному лейкозу (JMML), возрастному Т-клеточному острому лимфобластному лейкозу, трехлинейной миелодисплазии (AML/TMDS), лейкемии смешанного происхождения (MLL), миелодиспластическим синдромам (MDS), миелодисплазии (MPD), множественной миеломе (MM) и саркоме спинного мозга.
Тем временем, внеклеточный домен RTK можно связать с конкретными лигандами, такими как факторы роста, в то время как их внутриклеточные домены фосфорилированы. Сигнальные пути и биологические процессы, опосредованные RTK, расположены в ангиогенезе. Показано, что путь, активированный RTK, выбран в ангиогенезе. Активация путей, таких как через VEGFR или PDGFR, может привести к различным процедурам по ангиогенезу, подобно пролиферации клеток, миграции, выживанию и сосудистой проницаемости, которые тесно связаны с сериями сосудистых заболеваний.
В настоящее время существует 32 нерецепторные тирозинкиназы (nRTK), которые постоянно или временно существуют в цитоплазме, или связаны с трансмембранным белком в клетке. Таким образом, они также известны как цитоплазматические тирозинкиназы. В тканях опухоли nPTK часто являются активированными, способствующими пролиферации клеток, сопротивляющимися апоптозу и способствующими развитию и прогрессии опухоли. nRTK главным образом содержат 10 семейств: SRC, ABL, JAK, ACK, CSK, FAK, FES, FRK, TEC, SYK и др. Цитокины могут передавать сигналы через различные каналы для участия в регуляции роста клеток, дифференцировке и апоптозе. Как правило, рецепторы цитокина не содержат доменов RTK в цитоплазме, но сигнал опосредованно передается цитокином, когда связывание с его рецептором существуют в цитокин-нацеленной клетке. Среди них, Янус-киназа (JAK) и ее последующий STAT составляют важный путь передачи сигнала, и многие цитокины могут активировать JAK/STAT путь передачи сигнала. Когда цитокины связаны с их рецепторами, конформационные изменения будут происходить в цитоплазматических рецепторах, тем самым активизируя ассоциированные с семейством рецепторов JAK киназы. JAK киназы вызывает активацию STAT путем содействия ее соответствующему фосфорилированию. Активированный STAT затем диссоциирует от его рецептора, формирует димер, вступает в ядро и связывается с расширенным семейством GAS, таким образом, активируя транскрипцию, вызывая клеточную трансформацию и регуляцию некоторых генов экспрессии, связанных с пролиферацией клеток и их выживанием, что играет важную роль в возникновении опухолей.
В настоящее время рецепторы тирозинкиназ, такие как VEGFR и EGFR, главным образом исследуются, и ингибиторы ангиогенеза были разработаны для создания стратегия лечения системных типов рака. Ранее продаваемые ингибиторы протеинкиназы являются главным образом однонацеленными ингибиторами против одной мишени. Хотя они добились замечательных предварительных успехов в терапии рака, по мере увеличения времени их использования и случаев, все чаще встречаются недостатки. В противоположность этому, мульти-нацеленные ингибиторы протеинкиназы показали некоторые преимущества. Одновременное нацеливание на множественные мишени и множественные киназные пути передачи сигналов, ингибиторы киназ, мульти-нацеленные ингибиторы киназы могут не только предотвратить лекарственную устойчивость, вызванную мутацией одной мишени, но также значительно расширить их противоопухолевый спектр. Недостаток одно-нацеленных ингибиторов SU5416 и SU6668 указывает, что мульти-нацеленные ингибиторы киназ станут преобладающими в развитии ингибиторов киназ в будущем. SU5416 и SU6668 мишени KDR и PDGFR-β, соответственно, были приостановлены из-за плохой эффективности на клинической стадии III и II. Однако мульти-нацеленный ингибитор сунитиниб, который нацелен на множественные киназы, подобно KDR и т.д., в конечном итоге, остается успешным на рынке. Большинство изучаемых в настоящее время соединений являются мульти-нацеленными ингибиторами, так как они показывают лучшую ингибирующую активность и переносимость пациентами по сравнению с одно-нацеленными ингибиторами. Низкомолекулярные ингибиторы тирозина, которые в настоящее время присутствуют на рынке или в клинических испытаниях, в основном делятся на следующие категории в зависимости от химической структуры: хиназолины, индолиноны, пиридазины, цианохинолины, пирролопиримидины и др. Три антиангиогенных TKI, включая сунитиниб, сорафениб и пазопаниб с различной способностью к связыванию с ангиогенными киназами, недавно одобрены для успешного лечения рака у пациентов (с клеточной карциномой почки, стромальной опухолью желудочно-кишечного тракта и гепатоцеллюлярной карциномой). Многие другие антиангиогенные TKI в настоящее время находятся на клинических испытаниях от фазы I до фазы III. В дополнение к выгодной противоопухолевой активности эти лекарственные средства также были показаны, как обладающие клинической переносимостью и токсичностью.
Длительное и высокодозное применение инъекций таксанов привело к лекарственной устойчивости у пациентов и к снижению эффективности. Все свидетельства показывают, что резистентность может ограничить эффективность таргетирующего рецептора TKI. Таким образом, она имеет важное значение для разработки нового поколения противораковых лекарственных средств. В то же время, исследования показывают, что связанные с киназой заболевания являются эндогенно-родственными, делая однонацеленные ингибиторы затруднительными в приложении их ингибирующей активности.
Из-за существенно важной роли CDK и Auraro A и их родственных белков в пролиферирующих клетках для координации и промотирования клеточного цикла, их соответствующие ингибиторы могут быть использованы для лечения пролиферативных расстройств, таких как рак (применения, как правило, нацеленные на CDK или CDK специфическую терапию), а также для лечения других заболеваний, таких как вирусные инфекции, аутоиммунные заболевания, нейродегенеративные заболевания. При использовании в сочетании с существующими или новыми терапевтическими средствами, CDK-нацеленная терапия может также предоставлять клинические преимущества в лечении заболеваний, описанных выше. По сравнению со многими существующими противоопухолевыми средствами и в отношении приведенных выше тирозинкиназ, мутации CDK и лекарственная устойчивость ее ингибиторов происходят относительно меньше. Таким образом, CDK нацеленная противоопухолевая терапия может потенциально иметь преимущества перед многими существующими в настоящее время противоопухолевыми средствами, так как они не будут непосредственно взаимодействовать с ДНК и поэтому должны будут уменьшать риск развития вторичных опухолей.
Низкомолекулярные мульти-нацеленные ингибиторы CDK, такие как флавопиридол и UCN201, уже показали хорошую противоопухолевую активность в клинических испытаниях I и II. Тем не менее, большинство ингибиторов являются одно-нацеленными и многие компании провели исследования в этом аспекте. Для примера, новый низкомолекулярный ингибитор CDK AT7519, который в настоящее время на клинических испытаниях I/II действует на множественные мишени, такие как CDK1/циклин B, CDK2/циклин А, CDK3/циклин E и др. В то же время, AT7519 может вызвать активацию белка, члена его семейства GSK-3β ослабленной регуляцией уровня его фосфорилирования, ведущей к апоптозу клеток. Относительно типа структуры семейства ингибиторов кросс-протеинкиназ, в настоящее время редко сообщалось, и таким образом разработка ингибиторов киназы, которые могут избирательно действовать на множественные мишени конкретных заболеваний, будет иметь большое значение.
Сущность изобретения
На основе изучения низкомолекулярных ингибиторов CDK2 и Aurora A, согласно кристаллическим структурам CDK2 и Aurora A, авторы сконструировали модели взаимозависимости структура-активность (SAR) и модели виртуального скрининга средствами компьютерной разработки лекарственных средств. Кроме того, авторы создали библиотеку целенаправленных соединений методами фрагмент-выращивания. Через виртуальный скрининг, авторы идентифицировали и синтезировали ряд новых соединений с родительской структурой 4-(замещенных пятичленных гетероциклических пиримидин/пиридин)амино-1H-3-пиразолкарбоксамидов. Фармакологические испытания показали, что соединения настоящего изобретения являются не только хорошими двойными ингибиторами CDK2 и Aurora A, но также проявляют ингибирующую активность против различных киназ семейства CMGC и семейства TK. Они также показали мощную ингибирующую активность против множества опухолевых клеточных линий, и некоторые из них обладают преимуществом над известным ингибитором CDK2 AT7519, ингибитором Aurora A AT-9283 и мульти-нацеленным ингибитором стауроспорином.
Настоящее изобретение относится к соединениям, определенным формулой (I):

или их фармацевтически приемлемым солям или таутомерам,
где:
R1, R2 и R3, каждый независимо, представляет собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил, диарилалкил, арил или Het;
X и Y, каждый независимо, представляет собой атом N или группу CH, где группа CH может быть необязательно замещена R4, и R4 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил, диарилалкил, арил или Het;
Z и M, каждый независимо, представляет собой NH, O, S или группу CH, при условии, что один из Z и M представляет собой NH, O или S, где группа CH или NH, каждая необязательно и независимо, может быть замещена R5, и R5 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил, диарилалкил, арил или Het;
A1 независимо представляет собой NH, O, S или алкиленовую группу, где NH или алкиленовая группа, каждая необязательно и независимо, может быть замещена R6, и R6 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил, диарилалкил, арил или Het;
A2 независимо представляет собой алкилен, C(O)NH, C(O), NHC(O), алкилен-C(O), C(O)-алкилен, алкилен-C(O)-алкилен или NHC(O)NH, где указанные выше группы, каждая необязательно и независимо, может быть замещена R7, и R7 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил, диарилалкил, арил или Het;
Q1 выбран из арила или Het, где арил или Het, каждый необязательно и независимо, может быть замещен одним или несколькими R8, и R8 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил, диарилалкил, арил или Het;
Q2 выбран из арила или Het, где арил или Het, каждый необязательно и независимо, может быть замещен одним или несколькими R9, и R9 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил, диарилалкил, арил или Het;
термин "алкил" относится к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, которая присоединена к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода;
термин "алкилен" относится к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, которая присоединена к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода; где один атом водорода отсутствует;
термин "алкоксил" относится к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, которая присоединена к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода; где каждый атом углерода необязательно замещен кислородом;
термин "алкилтиол" относится к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, которая присоединена к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода; где каждый атом углерода необязательно замещен серой;
термин "алкоксилалкил" относится к алкильной группе, как определено выше, которая присоединена к алкоксильной группе, как определено выше;
термин "арил" относится к углеродному кольцу, выбранному из фенила, нафтила, аценафтенила или тетралила, каждый из которых может быть необязательно замещен 1, 2 или 3 заместителями, каждый независимо, выбранный из H, алкила, циано, галогена, галогеналкила, гидроксила, тиола, алкоксила, алкилтиола, алкоксилалкила, аралкила, диарилалкила, арила или Het;
термин "аралкил" или "диарилалкил" относится к алкильной группе, как определено выше, которая присоединена к арильной группе, как определено выше;
термин "Het" относится к моноциклической гетероциклильной группе, выбранной из пиперидила, пирролила, пиразолила, имидазолила, фурила, тиенила, оксазолила, изоксазолила, тиазолила, изотиазолила, пиридила, пиримидинила, пиразинила или пиридазинила, или бициклической гетероциклильной группе, выбранной из хинолила, хиноксалинила, индолила, бензимидазолила, бензоксазолила, бензизоксазолила, бензотиазолила, бензизотиазолила, бензофурила, бензотиенила, 2,3-дигидро-1,4-бензoдиоксинила или 1,3-бензодиоксолила; где моноциклическая или бициклическая группа, каждая необязательно замещена 1, 2 или 3 заместителями, каждый независимо выбранный из галогена, галогеналкила, гидроксила, алкила или алкоксила;
термин "галоген" относится к заместителю, выбранному из фтора (F), хлора (Cl), брома (Br), или иода (I);
термин "галогеналкил" относится к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, или к насыщенной циклической углеводородной группе, имеющей 3-6 атомов углерода, которая присоединена к насыщенной углеводородной группе с прямой или разветвленной цепью, имеющей 1-6 атомов углерода; где один или несколько атомов углерода замещены одним или несколькими галогенами.
В предпочтительном варианте осуществления
R1, R2 и R3, каждый независимо, представляет собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил или арил;
X и Y, каждый независимо, представляет собой атом N или группу CH, где группа CH может быть необязательно замещена R4, и R4 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил или арил;
Z и M, каждый независимо, представляет собой NH, O, S или группу CH, при условии, что один из Z и M представляет собой NH, O или S, где группа CH или NH, каждая необязательно и независимо, может быть замещена R5, и R5 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил или арил;
A1 независимо представляет собой NH, O, S или алкиленовую группу, где NH или алкиленовая группа, каждая необязательно и независимо, может быть замещена R6, и R6 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил или арил;
A2 независимо представляет собой алкилен, C(O)NH, C(O), NHC(O), алкилен-C(O), C(O)-алкилен, алкилен-C(O)-алкилен или NHC(O)NH, где указанные выше группы, каждая необязательно и независимо, может быть замещена R7, и R7 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил или арил;
Q1 выбран из арила или Het, где арил или Het, каждый необязательно и независимо, может быть замещен одним или несколькими R8, и R8 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил или арил;
Q2 выбран из арила или Het, где арил или Het, каждый необязательно и независимо, может быть замещен одним или несколькими R9, и R9 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол, алкоксилалкил, аралкил или арил.
В другом предпочтительном варианте осуществления
R1, R2 и R3, каждый независимо, представляет собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
X и Y, каждый независимо, представляет собой атом N или группу CH, где группа CH может быть необязательно замещена R4, и R4 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
Z и M, каждый независимо, представляет собой NH, O, S или группу CH, при условии, что один из Z и M представляет собой NH, O или S, где группа CH или NH, каждая необязательно и независимо, может быть замещена R5, и R5 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
A1 независимо представляет собой NH, O, S или алкиленовую группу, где NH или алкиленовая группа, каждая необязательно и независимо, может быть замещена R6, и R6 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
A2 независимо представляет собой алкилен, C(O)NH, C(O), NHC(O), алкилен-C(O), C(O)-алкилен, алкилен-C(O)-алкилен или NHC(O)NH, где указанные выше группы, каждая необязательно и независимо, может быть замещена R7, и R7 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
Q1 выбран из арила или Het, где арил или Het, каждый необязательно и независимо, может быть замещен одним или несколькими R8, и R8 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
Q2 выбран из арила или Het, где арил или Het, каждый необязательно и независимо, может быть замещен одним или несколькими R9, и R9 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил.
В остальных предпочтительных вариантах осуществления
R1, R2 и R3, каждый независимо, представляет собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
X и Y, каждый независимо, представляет собой атом N или группу CH, где группа CH может быть необязательно замещена R4, и R4 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
Z и M, каждый независимо, представляет собой NH, O, S или группу CH, при условии, что один из Z и M представляет собой NH, O или S, где группа CH или NH, каждая необязательно и независимо, может быть замещена R5, и R5 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
A1 независимо представляет собой NH, O, S или алкиленовую группу, где NH или алкиленовая группа, каждая необязательно и независимо, может быть замещена R6, и R6 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
A2 независимо представляет собой алкилен, C(O)NH, C(O), NHC(O), алкилен-C(O), C(O)-алкилен, алкилен-C(O)-алкилен или NHC(O)NH, где указанные выше группы, каждая необязательно и независимо, может быть замещена R7, и R7 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
Q1 представляет собой ненасыщенное или насыщенное ароматическое кольцо, выбранное из фенила, нафтила, пирролила, фурила, тиенила, пиридила, пиразинила или пиримидинила, и указанные выше группы, каждая необязательно и независимо, может быть замещена одним или несколькими R8, и R8 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
Q2 представляет собой ароматическое кольцо, выбранное из фенила, нафтила, пиразолила, фурила, тиенила, пиридила, пиразинила, пиримидинила; или C3-C8 алифатическое углеродное кольцо или алифатическое гетероциклическое кольцо, выбранное из тетрагидропирролила, пиперидила, морфолинила, метилпиперазинила; и указанные выше группы, каждая необязательно и независимо, может быть замещена одним или несколькими R8, и R8 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил.
В дополнительном предпочтительном варианте осуществления
R1, R2 и R3, каждый независимо, представляет собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
X и Y, каждый независимо, представляет собой атом N или группу CH, где группа CH может быть необязательно замещена R4, и R4 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
Z и M, каждый независимо, представляет собой NH, O, S или группу CH, при условии, что один из Z и M представляет собой NH, O или S, где группа CH или NH, каждая необязательно и независимо, может быть замещена R5, и R5 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
A1 независимо представляет собой NH, O, S или алкиленовую группу, где NH или алкиленовая группа, каждая необязательно и независимо, может быть замещена R6, и R6 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
A2 независимо представляет собой алкилен, C(O)NH, C(O), NHC(O), алкилен-C(O), C(O)-алкилен, алкилен-C(O)-алкилен или NHC(O)NH, где указанные выше группы, каждая необязательно и независимо, может быть замещена R7, и R7 может представлять собой H, алкил, циано, галоген, галогеналкил, гидроксил, тиол, алкоксил, алкилтиол или алкоксилалкил;
Q1 представляет собой ненасыщенное или насыщенное ароматическое кольцо, выбранное из фенила, нафтила, пирролила, фурила, тиенила, пиридила, пиразинила или пиримидинила, и указанными заместителями могут быть 1-2 галогена или трифторметил;
Q2 представляет собой ароматическое кольцо, выбранное из фенила, нафтила, пиразолила, фурила, тиенила, пиридила, пиразинила, пиримидинила; или C3-C8 алифатическое углеродное кольцо или алифатическое гетероциклическое кольцо, выбранное из тетрагидропирролила, пиперидила, морфолинила, метилпиперазинила.
В остальных предпочтительных вариантах осуществления
R1, R2 и R3, каждый независимо, представляет собой H или C1-4алкил;
X и Y, каждый независимо, представляет собой атом N или группу CH;
Z и M, каждый независимо, представляет собой NH, O, S или группу CH, при условии, что один из Z и M представляет собой NH, O или S;
A1 независимо представляет собой NH, O, S или группу CH2;
A2 независимо представляет собой цепь, подобную C1-4алкилену, C(O)NH, C(O) или NHC(O);
Q1 представляет собой ненасыщенное или насыщенное ароматическое кольцо, выбранное из фенила, нафтила, пирролила, фурила, тиенила, пиридила, пиразинила или пиримидинила, и указанными заместителями могут быть 1-2 галогена или трифторметил;
Q2 представляет собой ароматическое кольцо, выбранное из фенила, нафтила, пиразолила, фурила, тиенила, пиридила, пиразинила, пиримидинила; или C3-C8 алифатическое углеродное кольцо или алифатическое гетероциклическое кольцо, выбранное из тетрагидропирролила, пиперидила, морфолинила, метилпиперазинила.
В дополнительном предпочтительном варианте осуществления
R1, R2 и R3, каждый независимо, представляет собой H или метил;
A1 представляет собой NH;
A2 представляет собой CH2;
Q1 представляет собой фенил;
Q2 представляет собой морфолинил или метилпиперазинил.
Согласно настоящему изобретению, фармацевтически приемлемые соли соединений настоящего изобретения включают кислотно-аддитивные соли, образованные соединением формулы I со следующими кислотами: хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, метансульфоновой кислотой, бензолсульфоновой кислотой, п-толуолсульфоновой кислотой, нафталинсульфоновой кислотой, лимонной кислотой, винной кислотой, молочной кислотой, пировиноградной кислотой, уксусной кислотой, малеиновой кислотой или янтарной кислотой, фумаровой кислотой, салициловой кислотой, фенилуксусной кислотой, миндальной кислотой. Кроме того, кислотные соли неорганических оснований включают, например, соли, содержащие катион щелочного металла, катион щелочноземельного металла или катион аммония. Когда присутствует сера, и когда природа соседних атомов и групп является приемлемой, она может существовать в форме -S-, -S(O)- или -S(O)2-.
Среди соединений формулы I, следующие соединения являются предпочтительными:
4-(4-тиено[2,3-d]пиримидиниламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-1);
4-(4-тиено[2,3-d]пиримидиниламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-2);
4-(4-(6-метилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-3);
4-(4-(6-метилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-4);
4-(4-(5-метилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-5);
4-(4-(5-метилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-6);
4-(4-(5,6-диметилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-7);
4-(4-(5,6-диметилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-8);
4-(4-тиено[3,2-d]пиримидиниламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-9);
4-(4-тиено[3,2-d]пиримидиниламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-10);
4-(4-(7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-11);
4-(4-(7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-12);
4-(4-(6-метил-7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-13);
4-(4-(6-метил-7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-14);
4-(4-(5-метил-7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-15);
4-(4-(5-метил-7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-16);
4-(4-(5H-пирроло[3,2-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-17);
4-(4-(5H-пирроло[3,2-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-18);
4-(4-(6-метил-5H-пирроло[3,2-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-19);
4-(4-(6-метил-5H-пирроло[3,2-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-20);
4-(4-фуро[2,3-d]пиримидиниламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-21);
4-(4-фуро[2,3-d]пиримидиниламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-22);
4-(4-фуро[3,2-d]пиримидиниламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-23);
4-(4-фуро[3,2-d]пиримидиниламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-24);
4-(4-тиено[3,2-c]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-25);
4-(4-тиено[3,2-c]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-26);
4-(4-(2-метилтиено[3,2-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-27);
4-(4-(2-метилтиено[3,2-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-28);
4-(7-тиено[2,3-c]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-29);
4-(7-тиено[2,3-c]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-30);
4-(7-(3-метилтиено[2,3-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-31);
4-(7-(3-метилтиено[2,3-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-32);
4-(4-фуро[3,2-c]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-33);
4-(4-фуро[3,2-c]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-34);
4-(4-(2-метилфуро[3,2-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-35);
4-(4-(2-метилфуро[3,2-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-36);
4-(7-фуро[2,3-c]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-37);
4-(7-фуро[2,3-c]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-38);
4-(7-фуро[3,2-b]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-39);
4-(7-фуро[3,2-b]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-40);
4-(4-фуро[2,3-b]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-41);
4-(4-фуро[2,3-b]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-42);
4-(7-(1H-пирроло[2,3-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-43);
4-(7-(1H-пирроло[2,3-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-44);
4-(7-(2-метил-1H-пирроло[2,3-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-45);
4-(7-(2-метил-1H-пирроло[2,3-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-46);
4-(4-(2-метилтиено[3,2-d]пиримидин)иламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-47);
4-(4-(2-метилтиено[3,2-d]пиримидин)иламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-48);
Соединения настоящего изобретения могут быть получены согласно следующим методикам:
Схема 1

Схема 2

Схема 3

Схема 4

Соединения настоящего изобретения могут быть получены приведенными выше способами или аналогичными способами с использованием соответствующих исходных веществ согласно выбранным заместителям и их положению.
В соединениях формулы (I) пиразольное кольцо может существовать в двух таутомерных формах A и B, ниже:

Другие примеры таутомерных форм включают, например, кето-, енол- и енолятные формы, например, как следующие таутомерные пары: кето/енол (как проиллюстрировано ниже), имин/енамин, амид/иминоспирт, амидин/амидин, нитрозо/оксим, тиокетон/енетиол и нитро/асинитро.
Соединения формулы (I) и их субгруппы являются ингибиторами семейства CMGC киназ, в частности, они выбраны из циклинзависимых киназ, гликогенсинтаз-киназ (GSK), митоген активированной протеинкиназы (MAPK) и CDK-подобной киназы (CLK). Предпочтительные соединения могут ингибировать одну или более циклинзависимых киназ, гликогенсинтаз-киназ (GSK) и митоген активированных протеинкиназ (MAPK), и киназа выбрана из CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK9, GSK3, CHK2, ERK7, FGFR, VEGFR, JAK, JNK, KDR, PDGFR, C-SCR, Aurora и FLT3.
Соединения настоящего изобретения могут быть использованы в качестве ингибиторов семейства TK киназ, в частности они выбраны из ингибиторов семейства рецепторных тирозинкиназ (в частности, семейства рецептора эпидермального фактора роста (EGFR), рецептора фактора роста тромбоцитов (PDGFR), рецептора фактора роста нервной ткани (NGFR), рецепторов фактора роста фибробластов (FGFR), рецептора фактора роста гепатоцитов (HGFR), рецептора сосудистого эндотелиального фактора роста (VGFR)) и ингибиторов семейства нерецепторных тирозинкиназ (включающего 10 семейств, таких как SRC, ABL, JAK, ACK, CSK, FAK, FES, FRK, TEC, SYK и т.д.). Соединения могут модулировать или ингибировать активности семейства CMGC и TK киназ, и, таким образом, ожидается, что будут полезны при обеспечении средств для задерживания или восстановления пролиферации клеток, дифференциации и связанной с аномалией передачи сигнала. Поэтому предполагается, что соединения будут полезны при лечении или предотвращении пролиферативных расстройств, таких как рак. Также предполагается, что соединения настоящего изобретения будут полезны при лечении состояний, таких как воспаление, вирусная инфекция, сахарный диабет типа II или инсулин-независимый сахарный диабет, аутоиммунные заболевания, травма головы, инсульт, эпилепсия, неврологические заболевания (такие как болезнь Альцгеймера), заболевание двигательных нейронов.
Соединения настоящего изобретения также могут быть использованы в качестве ингибиторов GSK3. Вследствие их активности в модулировании или ингибировании CDK киназ и гликогенсинтаза-киназ, они, как ожидается, будут полезны в обеспечении средств для задерживания или восстановления аномальной дифференциации клеток. Поэтому предполагается, что соединения будут полезны при лечении или предотвращении пролиферативных расстройств, таких как разновидности рака. Предполагается также, что соединения настоящего изобретения будут полезны при лечении состояний, таких как вирусные инфекции, сахарный диабет типа II или инсулин-независимый сахарный диабет, аутоиммунные заболевания, травма головы, инсульт, эпилепсия, нейродегенеративные заболевания (такие как болезнь Альцгеймера), заболевание двигательных нейронов, прогрессирующий надъядерный паралич, кортико-базальная дегенерация и болезнь Пика. Одна подгруппа болезненных состояний и расстройств, где предполагается, что соединения настоящего изобретения будут полезны, включает вирусные инфекции, аутоиммунные заболевания и нейродегенеративные заболевания. Примеры разновидностей рака, которые могут быть ингибированы, включают, но, не ограничиваясь ими, карциному, например рак мочевого пузыря, рак молочной железы, рак толстой кишки (например, колоректальный рак), рак легких. GSK-3b могут модулировать пролиферацию и апоптоз раковых клеток регуляцией белковых факторов, подобных гликогенсинтазе, p27 или подобных, и участвовать в классическом внутриклеточном пути передачи сигнала, играть важную роль в патогенезе нервно-психических заболеваний, принимая участие в модуляции нейроцептора моноаминооксидазы, и опосредовать другие факторы и пути возникновения нейродегенеративных заболеваний. Таким образом, она становится горячей ингибиторной мишенью в лечении различных заболеваний.
Изобретение включает применение соединения настоящего изобретения для ингибирования активности FLT3 киназ в клетках или у субъекта, или при лечении состояний, связанных с активностью или экспрессией FLT3 киназы.
Примеры разновидностей рака, которые могут быть ингибированы, включают, но, не ограничиваясь ими, карциному, например, рак мочевого пузыря, рак молочной железы, рак толстой кишки (например, колоректальный рак, такой как аденокарцинома толстой кишки и аденома толстой кишки), рак почки, эпидермальный рак, рак печени, рак легких (например, аденокарциному, мелкоклеточный рак легких и немелкоклеточный рак легких), рак пищевода, рак желчного пузыря, рак яичников, рак поджелудочной железы (например, экзокринный рак поджелудочной железы), рак желудка, рак шейки матки, рак щитовидной железы, рак простаты или рак кожи (например, плоскоклеточный рак); гемопоэтическую опухоль лимфоидного ростка (например, лейкоз, острый лимфобластный лейкоз, B-клеточную лимфому, T-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, лимфому волосатых клеток или лимфому Беркетта); гемопоэтическую опухоль миелоидного ростка (например, острый или хронический миелобластный лейкоз, миелодиспластичекий синдром или промиелоцитарную лейкемию); фолликулярный рак щитовидной железы; опухоль мезенхимального происхождения (например, фибросаркому или рабдомиосаркому); опухоль центрально или периферической нервной системы (например, астроцитому, нейробластому, глиому или шванному); меланому; семиному; тератокарциному; остеосаркому; пигментную ксеродерму; кератоктантому; фолликулярный рак щитовидной железы; саркому Капоши, B-клеточную лимфому и хронический лимфоцитарный лейкоз.
Применение соединений настоящего изобретения в качестве ингибитора для обоих семейств CMGC и TK киназ может быть определено по методикам следующих примеров, и уровни активности соединений могут быть определены значениями IC50.
Фармакологические тесты и результаты кратко изложены следующим образом.
(1) Анализ на ингибирующую активность таргетирующего соединения в отношении CDK2
Ингибирующую активность синтезированных соединений в отношении CDK2/A определяли методом резонансного переноса энергии флюоресценции (FRET) и сравнивали с положительным контролем, лекарственным средством, таким как скринированное соединение с лучшей активностью. CDK2/A приобретали в виде набора или получали посредством очистки.
Методики: CDK2/A разводили до подходящей концентрации киназным разбавителем. Киназная реакционная смесь содержала CDK2/A, пептидный субстрат, HEPES (pH 7,5), BRIJ-35, MgCl2 и EDTA. Фосфопептидный субстрат CDK2 использовали в качестве контроля 100% фосфорилирования и ATP-свободный в качестве контроля 0% фосфорилирования. Спустя 1 час, разведенный разрабатываемый реагент A добавляли в реакционную систему. Затем реакции давали возможность протекать в течение 1 часа и затем гасили стоп-реагентом. Длина волны возбуждения составляла 400 нм, и интенсивность флюоресценции детектировали при 445 нм (кумарин) и 520 нм (флуоресцеин). Ингибирующее действие тестируемых соединений вычисляли в соответствии с формулой.
(2) Анализ на ингибирующую активность таргетирующего соединения в отношении Aurora A
Ингибирующую активность синтезированных соединений в отношении Aurora A определяли методом резонансного переноса энергии флюоресценции (FRET) и сравнивали с положительным контролем, лекарственным средством, таким как скринированное соединение с лучшей активностью. Aurora A приобретали в виде набора или получали посредством очистки.
Методики: Aurora A разводили до подходящей концентрации киназным разбавителем. Киназная реакционная смесь содержала Aurora A, пептидный субстрат, HEPES (pH 7,5), BRIJ-35, MgCl2 и EDTA. Aurora A фосфопептидный субстрат использовали в качестве контроля 100% фосфорилирования и ATP-свободный в качестве контроля 0% фосфорилирования. Спустя 1 час, разведенный разрабатываемый реагент A добавляли в реакционную систему. Затем реакции давали возможность протекать в течение 1 часа и затем гасили стоп-реагентом. Длина волны возбуждения составляла 400 нм, и интенсивность флюоресценции детектировали при 445 нм (кумарин) и 520 нм (флуоресцеин). Ингибирующее действие тестируемых соединений вычисляли в соответствии с формулой.
(3) Результаты ингибирования CDK2, Aurora A киназ
(4) Мультинаправленный скрининг тестированных соединений
Эксперимент основан на базе платформы хот-спот для скрин киназы представлена RCB с помощью метода стандартно изотопно-меченных киназ. Киназу (которая клонирована в домен бацилловидного вируса выраженной киназы и у которой определено значение IC50, и система FastBac бацилловидного вируса использована в качестве бацилловидного вируса), субстраты и процессы (субстрат+[33P]-ATP 33P-субстрат+ADP) использовали для детекции взаимодействия тестируемого соединения и заболевания, связанного с 342 киназами или родственными мутантами. Данная система является наиболее всестороннеразвитой системой для высокопроизводительного скрининга соединений, воздействующих на киназы человека. В данном эксперименте использовали 10 мкМ ATP, [33P]ATP и биотинилированные пептиды, и SA-флэш панель использовали для измерения скорости включения 33P. Использовали различные концентрации при серийном разведении маточного раствора в ДМСО. Значения IC50 для соединений определяли регрессионным анализом с использованием данных, соответствующих различным концентрациям, или одиночные концентрации использовали для определения коэффициента ингибирования. Синтезированные соединения скринировали против 342 киназ в единичной дозе 10 мкМ в два прохода в соответствии со стандартным процессом скрининга. Стауроспорин использовали в начальной концентрации 10 мкМ, в 4-кратном разведении для 10 доз в качестве положительного контроля. Другие положительные контроли использовали в начальной концентрации 20 мкМ, в 3-кратном разведении для 10 доз. Протестированные 342 киназы обеспечивались биологическими реакциями в Пенсильвании. ДМСО получали от Sigma, USA.
Тестированные киназы: ABL1(1), ABL2/ARG(2), ACK1(3), AKT1(4), AKT2(5), AKT3(6), ALK(7), ALK1/ACVRL1(8), ALK2/ACVR1(9), ALK3/BMPR1A(10), ALK4/ACVR1B(11), ALK5/TGFBR1(12), ALK6/BMPR1B(13), ARAF(14), ARK5/NUAK1(15), ASK1/MAP3K5(16), Aurora A(17), Aurora B(18), Aurora C(19), AXL(20), BLK(21), BMPR2(22), BMX/ETK(23), BRAF(24), BRK(25), BRSK1(26), BRSK2(27), BTK(28), c-Kit(29), c-MER(30), c-MET(31), c-Src(32), CAMK1a(33), CAMK1b(34), CAMK1d(35), CAMK1g(36), CAMK2a(37), CAMK2b(38), CAMK2d(39), CAMK2g(40), CAMK4(41), CAMKK1(42), CAMKK2(43), CDC7/DBF4(44), CDK1/циклин A(45), CDK1/циклин B(46), CDK1/циклин E(47), CDK16/циклин Y (PCTAIRE)(48), CDK2/циклин A(49), CDK2/циклин A1(50), CDK2/циклин E(51), CDK3/циклин E(52), CDK4/циклин D1(53), CDK4/циклин D3(54), CDK5/p25(55), CDK5/p35(56), CDK6/циклин D1(57), CDK6/циклин D3(58), CDK7/циклин H(59), CDK9/циклин K(60), CDK9/циклин T1(61), CHK1(62), CHK2(63), CK1a1(64), CK1d(65), CK1эпсилон(66), CK1g1(67), CK1g2(68), CK1g3(69), CK2a(70), CK2a2(71), CLK1(72), CLK2(73), CLK3(74), CLK4(75), COT1/MAP3K8(76), CSK(77), CTK/MATK(78), DAPK1(79), DAPK2(80), DCAMKL1(81), DCAMKL2(82), DDR1(83), DDR2(84), DLK/MAP3K12(85), DMPK(86), DMPK2(87), DRAK1/STK17A(88), DYRK1/DYRK1A(89), DYRK1B(90), DYRK2(91), DYRK3(92), DYRK4(93), EGFR(94), EPHA1(95), EPHA2(96), EPHA3(97), EPHA4(98), EPHA5(99), EPHA6(100), EPHA7(101), EPHA8(102), EPHB1(103), EPHB2(104), EPHB3(105), EPHB4(106), ERBB2/HER2(107), ERBB4/HER4(108), ERK1(109), ERK2/MAPK1(110), ERK5/MAPK7(111), ERK7/MAPK15(112), FAK/PTK2(113), FER(114), FES/FPS(115), FGFR1(116), FGFR2(117), FGFR3(118), FGFR4(119), FGR(120), FLT1/VEGFR1(121), FLT3(122), FLT4/VEGFR3(123), FMS(124), FRK/PTK5(125), FYN(126), GCK/MAP4K2(127), GLK/MAP4K3(128), GRK1(129), GRK2(130), GRK3(131), GRK4(132), GRK5(133), GRK6(134), GRK7(135), GSK3a(136), GSK3b(137), Haspin(138), HCK(139), HGK/MAP4K4(140), HIPK1(141), HIPK2(142), HIPK3(143), HIPK4(144), HPK1/MAP4K1(145), IGF1R(146), IKKa/CHUK(147), IKKb/IKBKB(148), IKKe/IKBKE(149), IR(150), IRAK1(151), IRAK4(152), IRR/INSRR(153), ITK(154), JAK1(155), JAK2(156), JAK3(157), JNK1(158), JNK2(159), JNK3(160), KDR/VEGFR2(161), KHS/MAP4K5(162), LATS1(163), LATS2(164), LCK(165), LCK2/ICK(166), LIMK1(167), LIMK2(168), LKB1(169), LOK/STK10(170), LRRK2(171), LYN(172), LYN B(173), MAPKAPK2(174), MAPKAPK3(175), MAPKAPK5/PRAK(176), MARK1(177), MARK2/PAR-1Ba(178), MARK3(179), MARK4(180), MEK1(181), MEK2(182), MEK3(183), MEKK1(184), MEKK2(185), MEKK3(186), MELK(187), MINK/MINK1(188), MKK4(189), MKK6(190), MLCK/MYLK(191), MLCK2/MYLK2(192), MLK1/MAP3K9(193), MLK2/MAP3K10(194), MLK3/MAP3K11(195), MNK1(196), MNK2(197), MRCKa/CDC42BPA(198), MRCKb/CDC42BPB(199), MSK1/RPS6KA5(200), MSK2/RPS6KA4(201), MSSK1/STK23(202), MST1/STK4(203), MST2/STK3(204), MST3/STK24(205), MST4(206), MUSK(207), MYLK3(208), MYO3b(209), NEK1(210), NEK11(211), NEK2(212), NEK3(213), NEK4(214), NEK5(215), NEK6(216), NEK7(217), NEK9(218), NLK(219), OSR1/OXSR1(220), P38a/MAPK14(221), P38b/MAPK11(222), P38d/MAPK13(223), P38g(224), p70S6K/RPS6KB1(225), p70S6Kb/RPS6KB2(226), PAK1(227), PAK2(228), PAK3(229), PAK4(230), PAK5(231), PAK6(232), PASK(233), PBK/TOPK(234), PDGFRa(235), PDGFRb(236), PDK1/PDPK1(237), PHKg1(238), PHKg2(239), PIM1(240), PIM2(241), PIM3(242), PKA(243), PKAcb(244), PKAcg(245), PKCa(246), PKCb1(247), PKCb2(248), PKCd(249), PKCэпсилон(250), PKCeta(251), PKCg(252), PKCiota(253), PKCmu/PRKD1(254), PKCnu/PRKD3(255), PKCтэта(256), PKCзэта(257), PKD2/PRKD2(258), PKG1a(259), PKG1b(260), PKG2/PRKG2(261), PKN1/PRK1(262), PKN2/PRK2(263), PKN3/PRK3(264), PLK1(265), PLK2(266), PLK3(267), PLK4/SAK(268), PRKX(269), PYK2(270), RAF1(271), RET(272), RIPK2(273), RIPK3(274), RIPK5(275), ROCK1(276), ROCK2(277), RON/MST1R(278), ROS/ROS1(279), RSK1(280), RSK2(281), RSK3(282), RSK4(283), SGK1(284), SGK2(285), SGK3/SGKL(286), SIK1(287), SIK2(288), SIK3(289), SLK/STK2(290), SNARK/NUAK2(291), SRMS(292), SRPK1(293), SRPK2(294), SSTK/TSSK6(295), STK16(296), STK22D/TSSK1(297), STK25/YSK1(298), STK32B/YANK2(299), STK32C/YANK3(300), STK33(301), STK38/NDR1(302), STK38L/NDR2(303), STK39/STLK3(304), SYK(305), TAK1(306), TAOK1(307), TAOK2/TAO1(308), TAOK3/JIK(309), TBK1(310), TEC(311), TESK1(312), TGFBR2(313), TIE2/TEK(314), TLK1(315), TLK2(316), TNIK(317), TNK1(318), TRKA(319), TRKB(320), TRKC(321), TSSK2(322), TSSK3/STK22C(323), TTBK1(324), TTBK2(325), TXK(326), TYK1/LTK(327), TYK2(328), TYRO3/SKY(329), ULK1(330), ULK2(331), ULK3(332), VRK1(333), VRK2(334), WEE1(335), WNK1(336), WNK2(337), WNK3(338), YES/YES1(339), ZAK/mLTK(340), ZAP70(341), ZIPK/DAPK3(342).
Результаты для некоторых соединений представлены на фиг.1: соединения показывают 90% ингибирование активностей почти для 200 киназ (число киназ отмечено снаружи) и показана селективность в отношении семейства GMGC: семейства CDK киназ CDK6/циклин D1(57), CDK6/циклин D3(58), CDK4/циклин D1(53), CDK4/циклин D3(54), CDK5/p35(56), GSK3b киназа(137), CDK5/p25(55), CDK16/циклин Y PIM1(48), DAPK2(98), ERK7/MAPK15(112); и семейства TK: KDR/VEGFR2(161), FLT1/VEGFR1(121), FLT4/VEGR3(123), FLT3(122). Ингибирующая активность составляла более чем 99%.
Соединения изобретения тестировали на IC50 против семейства GMGC и семейства ТЗ киназ. Начальная концентрация составляла 1 мкм. Серийные разведения готовили в растворе 100% ДМСО, в 10 точках для каждого соединения 3-кратно. Реакционная смесь содержала 20 мкМ ATP, киназу и субстрат биотинилированного пептида. Попадания скринировали количественным, точечным, чувствительным к 33P, меченным изотопной меткой, методом детектирования. % trl=[(сигнал тестируемого соединения-сигнал положительного контроля)/(сигнал отрицательного контроля-сигнал положительного контроля)]%. Отрицательный контроль=ДМСО (100% контроль); положительный контроль=контрольное соединение (0% контроль). Значения IC50 рассчитываются с использованием уравнения Хилла и стандартной кривой доза-ответ. Ниже перечислены некоторые результаты IC50, вычисленные на Prism Graphpad 5.
Результаты указывают на то, что синтезированные соединения показывают активность и селективность в отношении рецепторов семейства тирозинкиназ (RTK), таких как FGFR1, FGFR2, KDR/VEGFR2, FLT1/VEGFR1, FLT3, FLT4/VEGFR3; и семейства киназ CGCM, таких как CDK, GSK3b, JAK, ERK7/MAPK15 и подобных, в частности, высокую активность и селективность в отношении киназ CDK, GSK3β и FLT3. Соединения показывают ингибирующую активность против VEGFR и CDK, одновременно с высокой достоверностью.
(4) Анализ на противоопухолевую активность таргетирующего соединения in vitro
Ингибирующую активность против клеточных линий различных типов рака, таких как клеточная линия рака молочной железы MDA231, клеточная линия рака желудка MGC803, клеточная линия рака желудка BSG823, клеточная линия лейкоза K562, клеточная линия рака молочной железы MCF-7, клеточная линия резистентного рака молочной железы MCF-7, клеточная линия лейкоза NB4, клеточная линия рака печени HEPG2, линия эндотелиальных клеток пуповинной вены HUVEC, клеточная линия рака легких A549, клеточная линия рака толстой кишки HCT116, клеточная линия крупноклеточного рака легких H460, клеточная линия рака печени 7721, клеточная линия рака легких H1229 и подобных, определяли методом MTT.
Метод MTT: Дегидрогеназа, связанная с NADP в митохондриях живых клеток, способна к снижению внешнего влияния MTT на нерастворимые голубовато-фиолетовые кристаллы (формазаны), которые осаждаются в клетке. Мертвые клетки не обладают такой функцией. ДМСО или тройную жидкость (10% SDS-5% изобутанол-0,01 моль/л HCl) использовали для растворения кристаллов в клетке. Оптическая плотность (OD), определенная при 570 нм на микропланшет-ридере, может косвенно отражать количество живых клеток.
Методики: опухолевые клетки в фазе логарифмического роста размещали в 96-луночный планшет и инкубировали в течение 24 часов, куда добавляли образец для скрининга (для суспендированных клеток образцы могут быть добавлены непосредственно). Клетки дополнительно инкубировали в 5% CO2 при 37°C в течение 48 часов и затем добавляли MTT, и клетки инкубировали в течение дополнительных 4 часов. Кристаллы растворяли в ДМСО, и детектирование осуществляли на микропланшет-ридере.
Противоопухолевая активность некоторых таргетирующих соединений in vitro против линии клеток рака толстой кишки HCT116, линии клеток рака печени 7721 и линии клеток рака легких H1229 приведена ниже:
Противоопухолевая активность таргетирующего соединения (I-1) против клеточной линии рака молочной железы MDA231, клеточной линии рака желудка MGC803, клеточной линии рака желудка BSG823, клеточной линии лейкоза K562, клеточной линии рака молочной железы MCF-7, клеточной линии резистентного рака молочной железы MCF-7, клеточной линии лейкоза NB4, клеточной линии рака печени HEPG2, линии эндотелиальных клеток пуповинной вены HUVEC, клеточной линии рака легких A549, клеточной линии рака толстой кишки HCT116, клеточной линии крупноклеточного рака легких H460 приведены ниже.
Результаты фармакологического теста демонстрируют, что соединения настоящего изобретения, обладающие мульти-киназной ингибирующей активностью, могут быть использованы для лечения или предотвращения клинических заболеваний, связанных с ингибированием киназы, таких как меланома, рак печени, рак почки, острый лейкоз, немелкоклеточный рак легких, рак простаты, рак щитовидной железы, рак кожи, колоректальный рак, рак поджелудочной железы, рак яичников, рак молочной железы, миелодиспластичекий синдром, рак пищевода, рак желудочно-кишечного тракта или мезотелиома.
Фармацевтические составы
Активное соединение может быть введено как таковое или в форме фармацевтической композиции (например, состава). Композиция содержит по меньшей мере одно активное соединение настоящего изобретения и один или более фармацевтически приемлемых носителей, адъювантов, эксципиентов, разбавителей, наполнителей, стабилизаторов, консервантов. Соответственно, в дополнительном аспекте настоящее изобретение обеспечивает синтезированное соединение и его подгруппу в форме фармацевтической композиции, например, соединение формулы (I), как определено выше, и его подгруппу. Фармацевтические композиции могут быть в любой форме, подходящей для перорального, парентерального, местного, интраназального, офтальмологического, ушного, ректального, интравагинального или трансдермального введения. Когда композиция предназначена для парентерального введения, она может быть сформулирована для внутривенного, внутримышечного, внутрибрюшинного, подкожного введения или для прямой доставки в орган-мишень или ткань инъекцией, инфузией или другими средствами доставки.
Некоторые фармацевтические составы получают следующим образом.
1) Лиофилизированные составы: Аликвоты формулируемого соединения формулы (I) и его подгрупп, как определено выше, помещают в 50 мл флаконы и подвергают лиофилизации. В ходе лиофилизации композиции замораживают, с использованием одноступенчатого протокола замораживания при -45°C. Температуру повышают до -10°C для нормализации, затем снижают до -45°C для замораживания, с последующей первой сушкой при 25°C в течение приблизительно 3400 минут, и затем второй сушкой при 50°C. Давление в ходе первой и второй сушки составляет 80 миллиторр. 2) Составы в форме таблетки: композицию в форме таблетки, содержащую (252 мг) синтезированного соединения, получают смешиванием 50 мг данного соединения с 197 мг лактозы (BP) в качестве разбавителя и 3 мг стеарата магния в качестве лубриканта, и прессуют в форму таблетки известными способами. 3) Состав в форме капсул: композицию в форме капсул получают смешиванием 100 мг синтезированного соединения с 100 мг лактозы и заполняют полученной смесью стандартные непрозрачные твердые желатиновые капсулы. 4) Инъецируемые составы I: композиция для парентерального введения инъекцией может быть получена разведением синтезированного соединения (например, в форме соли) в воде, содержащей 10% пропиленгликоля, с получением концентрации активного соединения 1,5% по массе. Полученный раствор стерилизуют фильтрованием, и флакон затем герметизируют. 5) Инъецируемые составы II: композиция для парентерального введения может быть получена разведением синтезированного соединения (например, в форме соли) (2 мг/мл) и маннита (50 мг/мл) в воде. Полученный раствор стерилизуют фильтрованием и заполняют им герметичные 1 мл флаконы или ампулы. 6) Составы для подкожной инъекции: композицию для подкожного введения получают смешиванием синтезированного соединения с фармацевтической степенью очистки кукурузным маслом, с получением концентрации 5 мг/мл. Полученную композицию стерилизуют и помещают в подходящий контейнер.
В дополнительном аспекте настоящее изобретение обеспечивает применение соединения формулы (I) и его подгрупп, как определено выше, в качестве противогрибкового средства. Данное соединение может быть использовано в терапии животных (например, при лечении млекопитающих, таких как человек), или при обработке растений (например, в сельском хозяйстве и садоводстве), или в качестве общего противогрибкового средства, например в качестве консерванта и дезинфицирующего средства. В другом аспекте настоящее изобретение предоставляет противогрибковую композицию для использования в сельском хозяйстве (включая садоводство), содержащую соединение формулы (I) или его подгруппу, как определено выше, и аграрно приемлемый разбавитель или носитель.
Например, противогрибковую активность синтезированного соединения определяли с применением следующего протокола, включающего Candida parpsilosis, Candida tropicalis, Candida albicans-ATCC 36082 и Cryptococcus neoformans. Тестируемые микроорганизмы выдерживали на сабуро-дикстрозном скошенном агаре при 4°C. Синглетную суспензию каждого микроорганизма получали путем выращивания дрожжей в течение ночи при 27°C во вращающемся барабане в основе азотного агара для дрожжей (YNB) с аминокислотами (Difco, Detroit, Mich.), (pH 7,0) и 0,05 морфолинпропансульфоновой кислотой (MOPS). Суспензию затем центрифугировали и дважды промывали 0,85% NaCl перед ультразвуковым диспергированием суспензии промытых клеток в течение 4 секунд (Branson Sonifier, model 350, Danbury, Conn.). Синглетные бластоспоры подсчитывали в счетной камере и доводили до требуемой концентрации в 0,85% NaCl. Активность тестируемых соединений определяли с использованием модифицированной технологии микроразведения бульона. Тестируемое соединение разбавляли в ДМСО до соотношения 1,0 мг/мл, затем разбавляли до 64 мкм/мл в бульоне YNB (pH 7,0) MOPS (флуконазол использовали в качестве контроля) до получения рабочего раствора каждого из соединений. При использовании 96-луночного планшета, в лунках 1 и 3-12 получали бульон YNB, 10-кратно разбавленный раствор соединения получали в лунках 2-11 (интервал концентраций составлял 64-0,125 мкм/мл). Лунка 1431 служила в качестве стерильного контроля и холостой пробы для спектрофотометрического анализа. Лунка 12 служила в качестве контроля роста. Микротитровальный планшет инокулировали 10 мкл в каждую лунку 2-11 (конечная инокуляция составляла 104 организмы/мл). Инокулированный планшет инкубировали в течение 48 часов при 35°C. Значения MIC определяли спектрофотометрически при измерении поглощения при 420 нм (автоматический микропланшет-ридер, DuPont Instruments, Wilmington, Del.) после возбуждения планшетов в течение 2 минут на вихревом смесителе (Vorte-Genie 2 Mixer, Scientific Industries, Inc., Bolemia, N.Y.). Конечную точку MIC определяли как низшую концентрацию лекарственного средства, составляющую приблизительно 50% (или более) снижение роста по сравнению с контрольной лункой. Что касается анализа на мутность, ее определяли как низшую концентрацию лекарственного средства, при которой мутность в лунке составляла <50% от контроля (IC50). Минимальную цитолитическую концентрацию (MCC) определяли субкультивированием во всех лунках 96-луночного планшета на сабуро-дикстрозном агаре (SDA), инкубированием в течение 1-2 дней при 35°C, и затем проверкой на жизнеспособность.
Некоторые результаты представлены ниже.
Изобретение также предоставляет способ лечения грибковой инфекции растений или семян, включающий обработку растений или семян эффективным противогрибковым количеством фунгицидной композиции, как определено выше.
Соединение растворяли в ацетоне, с последующим серийным разведением в ацетоне с получением ряда желаемых концентраций. Окончательную обработку проводили путем добавления 9 объемов 0,05% водного раствора Tween-20™ или 0,01% TritonX-100™, в зависимости от патогена. Композицию затем использовали для тестирования активности соединения изобретения против ожога листьев томата (возбудителем фитофтороза), применяя следующий протокол. Семенам томата (сорта Rutgers) давали расти в горшочной смеси гидропоники на основе торфа до тех пор, пока всходы не достигнут высоты 10-20 см. Растения затем опрыскивали со стоком тестируемым соединением при норме 100 ч./млн. Спустя 24 часа, тестовые растения инокулировали распылением из спорового мешочка водной суспензии возбудителя фитофтороза и выдерживали в орошаемой камере в течение ночи. Затем растения переносили в теплицу до тех пор, пока болезнь не разовьется на необработанных контролем растениях.
Некоторые результаты тестирования соединений представлены ниже.
Краткое описание фигур
На фиг.1 показан уровень ингибирования некоторыми соединениями 342 киназ, где киназы показаны под различными номерами (см. выше).
Подробное описание изобретения
Температуры плавления определяли в трубках типа b для точек плавления, где средой являлось метилсиликоновое масло и температура не корректировалась. ИК спектры снимали на инфракрасном спектрометре Nicolet Impact 410, где компрессию проводили с KBr.1H ЯМР проводили на ЯМР спектрометре JEOL FX90Q Fourier-Transform, ЯМР спектрометре BRUKER ACF-300 и ЯМР спектрометре BRUKER AM-500 (внутренний стандарт TMS). МС проводили на масс-спектрометре Nicolet 2000 Fourier-Transform и масс-спектрометре MAT-212. Реакцию при микроволновом облучении осуществляли в одномодовой микроволновой печи CEM Discover.
ПРИМЕР 1
4-метил-1-(4-нитробензил)пиперазин (I-a)
п-Нитробензилбромид (10 г, 46,3 ммоль) и дихлорметан (100 мл) добавляли в 500 мл одногорлую колбу, в которую медленно и по каплям добавляли смесь N-метилпиперазина (4,7 г, 47,0 ммоль) и триэтиламина (7,1 г, 70,3 ммоль) в дихлорметане (20 мл) на ледяной бане. Реакционную смесь кипятили с обратным холодильником в течение 1 часа. Расходование исходных веществ подтверждали ТСХ (этилацетат:петролейный эфир = 1:2). 150 мл хлороформа и 100 мл насыщенного раствора бикарбоната натрия добавляли в реакционную смесь, которую энергично перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь экстрагировали хлороформом (100 мл × 3). Органические слои объединяли и промывали водой и насыщенным хлоридом натрия по одному разу, соответственно (100 мл × 1). Сушку осуществляли сухим сульфатом магния с последующим фильтрованием. После удаления растворителя при пониженном давлении, получали 8,5 г желтоватого твердого вещества; выход: 78,1%. Продукт использовали для следующей реакции без дополнительной очистки.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,15 (3H, с, -CH3), 2,3-2,5 (8H, м, -CH2-×4), 3,5 (2H, с, -CH2-), 7,5 (2H, д, J=8,7 Гц, ArH), 8,1 (2H, д, J=8,7 Гц, ArH).
ПРИМЕР 2
4-(4-нитробензил)морфолин (I-b)
п-Нитробензилбромид (10 г, 46,5 ммоль) и дихлорметан (100 мл) добавляли в 500 мл одногорлую колбу, в которую медленно и по каплям добавляли смесь морфолина 4,1 г (47,1 ммоль) и триэтиламина (7,1 г, 70,3 ммоль) в дихлорметане (20 мл) на ледяной бане. Реакционную смесь кипятили с обратным холодильником в течение 1 часа. Расходование исходных веществ подтверждали ТСХ (этилацетат:петролейный эфир = 1:2). 150 мл хлороформа и 100 мл насыщенного раствора бикарбоната натрия добавляли в реакционную смесь, которую энергично перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь экстрагировали хлороформом (100 мл × 3). Органические слои объединяли и промывали водой и насыщенным хлоридом натрия по одному разу, соответственно (100 мл × 1). Сушку осуществляли сухим сульфатом магния с последующим фильтрованием. После удаления растворителя при пониженном давлении, получали 8,7 г желтоватого твердого вещества (I-b); выход: 84,5%%. Продукт использовали для следующей реакции без дополнительной очистки.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, м, -NCH2-×2), 3,3-3,5 (6H, м, -OCH2-×2, -CH2-), 6,9 (2H, д, J=8,7 Гц, ArH), 7,6 (2H, д, J=8,7 Гц, ArH).
ПРИМЕР 3
4-((4-метил-1-пиперазинил)метил)анилин (I-c)
Неочищенное соединение I-a (8,5 г, 36,2 ммоль), FeO(OH)/C, 2,0 г в качестве катализатора и 95% этанол (100 мл) добавляли в 500 мл одногорлую колбу и кипятили с обратным холодильником. В реакционную систему медленно и по каплям добавляли смесь 25 мл гидразингидрата и 20 мл 95% этанола. Расходование исходных веществ подтверждали ТСХ (метанол:хлороформ = 1:15). Фильтрование с отсасыванием осуществляли, пока реакционная смесь была горячей. Осадок на фильтре дважды промывали теплым этанолом (30 мл × 2). После удаления растворителя при пониженном давлении, получали белое твердое вещество, которое сушили в вакууме, с получением 6,7 г соединения (I-c); выход: 90,3%. Продукт использовали для следующей реакции без дополнительной очистки.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,3-2,5 (8H, м, -CH2-×4), 3,5 (2H, с, -CH2-), 4,0 (2H, с, -NH2), 7,5 (2H, д, J=8,7 Гц, ArH), 8,1 (2H, д, J=8,7 Гц, ArH).
ПРИМЕР 4
4-((4-морфолино)метил)анилин (I-d)
Неочищенное соединение I-b (8,5 г, 38,3 ммоль), FeO(OH)/C, 2,0 г в качестве катализатора и 95% этанол (100 мл) добавляли в 500 мл одногорлую колбу и кипятили с обратным холодильником. В реакционную систему медленно и по каплям добавляли смесь 25 мл гидразингидрата и 20 мл 95% этанола. Расходование исходных веществ подтверждали ТСХ (метанол:хлороформ = 1:20). Фильтрование с отсасыванием осуществляли, пока реакционная смесь была горячей. Осадок на фильтре дважды промывали теплым этанолом (30 мл × 2). После удаления растворителя при пониженном давлении, получали белое твердое вещество, которое сушили в вакууме, с получением 6,6 г соединения (I-d); выход: 89,7%. Продукт использовали для следующей реакции без дополнительной очистки
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, м, -NCH2-×2), 3,2 (4H, м, -OCH2-×2), 3,5 (2H, с, -CH2-), 4,9 (2H, с, -NH2), 6,5 (2H, д, J=8,4 Гц, ArH), 6,9 (2H, д, J=8,4 Гц, ArH).
ПРИМЕР 5
N-(4-((4-метил-1-пиперазинил)метил)фенил)-4-нитро-1H-3-пиразолкарбоксамид (I-e)
Неочищенное соединение I-c (7,5 г 36,6 ммоль), 4-нитро-1H-пиразол-3-карбоновую кислоту (6,3 г, 40,1 ммоль), EDC·HCl (8,4 г, 44,0 ммоль), HOBT (6,0 г, 44,4 ммоль) и безводный ДМФА (100 мл) добавляли в 250 мл круглодонную колбу и перемешивали в течение 24 часов при комнатной температуре. Расходование исходных веществ подтверждали ТСХ (метанол:хлороформ = 1:10). Реакционную смесь выливали 200 мл ледяной воды, и образовывалось большое количество желтого осадка твердого вещества, которому давали отстояться и фильтровали с отсасыванием, с получением желтого твердого вещества. Неочищенное вещество перекристаллизовывали из смешанных растворителей из этилацетата и метанола, с получением 11,1 г соединения (I-e); выход: 88,2%; т.пл.: 194-196°C; МС [M+H]+ 345,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,2 (3H, с, -CH3), 2,3-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (2H, д, J=8,4 Гц, ArH), 8,8 (1H, с, ArH), 10,6 (1H, с, -NHCO-), 14,2 (1H, с, -NH-, пиразол).
ПРИМЕР 6
N-(4-((4-морфолинил)метил)фенил)-4-нитро-1H-3-пиразолкарбоксамид (I-f)
Неочищенное соединение I-d (7,5 г, 39,0 ммоль), 4-нитро-1H-пиразол-3-карбоновую кислоту (6,3 г, 40,1 ммоль), EDC·HCl (8,4 г, 44,0 ммоль), HOBT (6,0 г, 44,4 ммоль) и безводный ДМФА (100 мл) добавляли в 250 мл круглодонную колбу и перемешивали в течение 24 часов при комнатной температуре. Расходование исходных веществ подтверждали ТСХ (метанол:хлороформ = 1:20). Реакционную смесь выливали 200 мл ледяной воды, и образовывалось большое количество желтого осадка твердого вещества, которому давали отстояться и фильтровали с отсасыванием, с получением желтого твердого вещества. Неочищенное вещество перекристаллизовывали из смешанных растворителей из этилацетата и метанола, с получением 11,6 г соединения (I-f); выход: 89,7%; т.пл.: 208-210°C; МС [M+H]+ 332,4.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,4 (4H, т, J=4,1 Гц, -NCH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,1 Гц, -OCH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (2H, д, J=8,4 Гц, ArH), 8,9 (1H, с, ArH), 10,7 (1H, с, -NHCO-), 14,2 (1H, с, пиразол).
ПРИМЕР 7
N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамид (I-g)
Неочищенное соединение I-e (6,0 г, 17,4 ммоль), FeO(OH)/C, 2 г в качестве катализатора и 95% этанол (100 мл) добавляли в 250 мл одногорлую колбу и кипятили с обратным холодильником. В реакционную систему медленно и по каплям добавляли смесь 25 мл гидразингидрата и 20 мл 95% этанола. Расходование исходных веществ подтверждали ТСХ (метанол:хлороформ = 1:10). Фильтрование с отсасыванием осуществляли, пока реакционная смесь была горячей. Осадок на фильтре дважды промывали теплым этанолом (30 мл × 2). После удаления растворителя при пониженном давлении, получали не совсем белое твердое вещество. Неочищенное вещество перекристаллизовывали из смешанных растворителей из этилацетата и метанола, с получением 3,5 г соединения (I-g); выход: 63,9%. т.пл.: 199-201°C, МС [M+H]+ 315,8.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,3-2,5 (8H, м, -CH2-×4), 3,3 (2H, с, -CH2-), 4,7 (2H, с, -NH2), 7,1-7,2 (3H, м, ArH), 7,7 (2H, д, ArH), 9,7 (1H, с, -NHCO-), 12,7 (1H, с, пиразол).
ПРИМЕР 8
N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамид (I-h)
Неочищенное соединение I-f (6,0 г, 18,1 ммоль), FeO(OH)/C, 2 г в качестве катализатора и 95% этанол (100 мл) добавляли в 250 мл одногорлую колбу и кипятили с обратным холодильником. В реакционную систему медленно и по каплям добавляли смесь 25 мл гидразингидрата и 20 мл 95% этанола. Расходование исходных веществ подтверждали ТСХ (метанол:хлороформ = 1:10). Фильтрование с отсасыванием осуществляли, пока реакционная смесь была горячей. Осадок на фильтре дважды промывали теплым этанолом (30 мл × 2). После удаления растворителя при пониженном давлении, получали не совсем белое твердое вещество. Неочищенное вещество перекристаллизовывали из смешанных растворителей из этилацетата и метанола, с получением 3,2 г соединения (I-h); выход: 58,6%. Т.пл.: 216-218°C, МС [M+H]+ 302,0.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,5 (4H, м, -NCH2-×2), 3,3 (2H, с, -CH2-), 3,6 (4H, м, -OCH2-×2), 4,7 (2H, с, -NH2), 7,2 (3H, м, ArH), 7,7 (2H, д, J=8,4 Гц, ArH), 9,7 (1H, с, -NHCO-), 12,7 (1H, с, пиразол).
ПРИМЕР 9
4-(4-тиено[2,3-d]пиримидиниламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-1)
129 мг (0,41 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида, 70 мг (0,41 ммоль) 4-хлортиено[2,3-d]пиримидина и 25 мл 50% водной уксусной кислоты добавляли в 50 мл одногорлую колбу и кипятили с обратным холодильником. Расходование исходных веществ подтверждали ТСХ (метанол:хлороформ = 1:10). Реакционную смесь охлаждали до комнатной температуры и гасили насыщенным водным раствором NaOH до pH 8-9, и экстрагировали этилацетатом три раза (50 мл × 3). Экстракты объединяли, сушили над сухим сульфатом магния. После фильтрования с отсасыванием и удаления растворителя при пониженном давлении, получали желтоватое твердое вещество. Неочищенное вещество подвергали колоночной хроматографии (подвижная фаза: метанол:хлороформ = 1:15), с получением 70 мг соединения (I-1). Выход: 37,8%; т.пл.: 285-287°C; [M++H]+ 449,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,5 (1H, д, J=5,4 Гц, ArH), 7,7-7,8 (3H, м, ArH), 8,5 (1H, с, ArH), 8,6 (1H, с, ArH), 10,0 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 10
4-(4-тиено[2,3-d]пиримидиниламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-2)
Соединение I-2 (78 мг) получали аналогично соединению I-1 с использованием 124 мг (0,41 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,41 ммоль) 4-хлортиено[2,3-d]пиримидина в качестве исходных веществ. Выход: 43,6%; т.пл.: 262-265°C; МС [M+H]+ 436,2.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,5 (4H, т, J=4,2 Гц, -CH2-×2), 7,3 (2H, д, J=8,4, ArH), 7,5 (1H, д,J=6,0 Гц, ArH), 7,7-7,8 (3H, м, ArH), 8,5 (1H, с, ArH), 8,6 (1H, с, ArH), 9,9 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 11
4-(4-(6-метилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-3)
Соединение I-3 (75 мг) получали аналогично соединению I-1 с использованием 120 мг (0,38 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 120 мг (0,38 ммоль) 4-хлор-6-метилтиено[2,3-d]пиримидина в качестве исходных веществ. Выход: 42,9%; т.пл.: 235-238°C, МС [M+H]+ 463,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1(3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 7,2 (1H, с, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,5 (2H, с, ArH), 9,8 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 12
4-(4-(6-метилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-4)
Соединение I-4 (80 мг) получали аналогично соединению I-1 с использованием 118 мг (0,38 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 4-хлор-6-метилтиено[2,3-d]пиримидина в качестве исходных веществ. Выход: 47,0%; т.пл.: >280°C, МС [M+H]+ 450,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,4 (4H, т, J=4,2 Гц, -CH2-×2), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 7,2 (1H, с, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,5 (2H, с, ArH), 9,8 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 13
4-(4-(5-метилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-5)
Соединение I-5 (72 мг) получали аналогично соединению I-1 с использованием 120 мг (0,38 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 4-хлор-5-метилтиено[2,3-d]пиримидина в качестве исходных веществ. Выход: 41,1%; т.пл.: 245-247°C, МС [M+H]+ 463,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 2,8 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,6 (1H, с, ArH), 8,7 (1H, с, ArH), 10,2 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 14
4-(4-(5-метилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-6)
Соединение I-6 (64 мг) получали аналогично соединению I-1 с использованием 118 мг (0,38 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 4-хлор-5-метилтиено[2,3-d]пиримидина в качестве исходных веществ. Выход: 37,6%; т.пл.: >280°C, МС [M+H]+ 450,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,4 (4H, т, J=4,2 Гц, -CH2-×2), 2,8 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,6 (1H, с, ArH), 8,7 (1H, с, ArH), 10,2 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 15
4-(4-(5,6-диметилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-7)
Соединение I-7 (66 мг) получали аналогично соединению I-1 с использованием 111 мг (0,35 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,35 ммоль) 4-хлор-5,6-диметилтиено[2,3-d]пиримидина в качестве исходных веществ. Выход: 39,3%; т.пл.: 264-267°C; МС [M+H]+ 477,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 2,6 (3H, с, -CH3), 2,8 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,6 (1H, с, ArH), 8,7 (1H, с, ArH), 10,2 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 16
4-(4-(5,6-диметилтиено[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-8)
Соединение I-8 (73 мг) получали аналогично соединению I-1 с использованием 110 мг (0,35 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,35 ммоль) 4-хлор-5,6-диметилтиено[2,3-d]пиримидина в качестве исходных веществ. Выход: 44,5%; т.пл.: 254-256°C, МС [M+H]+ 464,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,4 (4H, т, J=4,2 Гц, -CH2-×2), 2,6 (3H, с, -CH3), 2,8 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,6 (1H, с, ArH), 8,7 (1H, с, ArH), 10,2 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 17
4-(4-тиено[3,2-d]пиримидиниламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-9)
Соединение I-9 (88 мг) получали аналогично соединению I-1 с использованием 129 мг (0,41 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,41 ммоль) 4-хлортиено[3,2-d]пиримидина в качестве исходных веществ. Выход: 47,8%; mp:>280°C, МС [M+H]+ 449,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,2 (3H, с, -CH3), 2,3-2,5 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,5 (1H, д, J=5,3 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,2 (1H, д, J=5,3 Гц, ArH), 8,5 (1H, с, ArH), 8,7 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 18
4-(4-тиено[3,2-d]пиримидиниламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-10)
Соединение I-10 (93 мг) получали аналогично соединению I-1 с использованием 128 мг (0,41 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,41 ммоль) 4-хлортиено[3,2-d]пиримидина в качестве исходных веществ. Выход: 52,0%; т.пл.: 275-277°C; МС [M+H]+ 436,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,4 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,5 (1H, д, J=5,2 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,3 (1H, д, J=5,2 Гц, ArH), 8,5 (1H, с, ArH), 8,7 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 19
4-(4-(7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-11)
Соединение I-11 (63 мг) получали аналогично соединению I-1 с использованием 144 мг (0,46 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 4-хлор-7H-пирроло[2,3-d]пиримидина в качестве исходных веществ. Выход: 32,0%; т.пл.: 229-230°C, МС [M+H]+ 432,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 6,5 (1H, с, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 20
4-(4-(7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-12)
Соединение I-12 (70 мг) получали аналогично соединению I-1 с использованием 142 мг (0,46 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 4-хлор-7H-пирроло[2,3-d]пиримидина в качестве исходных веществ. Выход: 36,6%; т.пл.: 213-214°C, МС [M+H]+ 419,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,4 Гц, -CH2-×2), 2,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,4 Гц, -CH2-×2), 6,5 (1H, с, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 21
4-(4-(6-метил-7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-13)
Соединение I-13 (56 мг) получали аналогично соединению I-1 с использованием 132 мг (0,42 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,42 ммоль) 4-хлор-6-метил-7H-пирроло[2,3-d]пиримидина в качестве исходных веществ. Выход: 23,0%; т.пл.: 268-270°C, МС [M+H]+ 446,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (11H, м, -CH2-×4, -CH3), 3,4 (2H, с, -CH2-), 6,5 (1H, с, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 22
4-(4-(6-метил-7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-14)
Соединение I-14 (61 мг) получали аналогично соединению I-1 с использованием 130 мг (0,42 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,42 ммоль) 4-хлор-6-метил-7H-пирроло[2,3-d]пиримидина в качестве исходных веществ. Выход: 33,7%; т.пл.: 271-273°C, МС [M+H]+ 433,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (7H, м, -CH2-×2, -CH3), 2,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,4 Гц, -CH2-×2), 6,5 (1H, с, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 23
4-(4-(5-метил-7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-15)
Соединение I-15 (53 мг) получали аналогично соединению I-1 с использованием 132 мг (0,45 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,45 ммоль) 4-хлор-5-метил-7H-пирроло[2,3-d]пиримидина в качестве исходных веществ. Выход: 28,3%; т.пл.: 258-261°C, МС [M+H]+ 446,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 24
4-(4-(5-метил-7H-пирроло[2,3-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-16)
Соединение I-16 (62 мг) получали аналогично соединению I-1 с использованием 130 мг (0,45 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,45 ммоль) 4-хлор-5-метил-7H-пирроло[2,3-d]пиримидина в качестве исходных веществ. Выход: 34,3%; т.пл.: 267-269°C, МС [M+H]+ 433,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,4 Гц, -CH2-×2), 2,4 (2H, с, -CH2-), 2,6 (3H, с, -CH3), 3,6 (4H, т, J=4,4 Гц, -CH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 25
4-(4-(5H-пирроло[3,2-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-17)
Соединение I-17 (50 мг) получали аналогично соединению I-1 с использованием 144 мг (0,46 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 4-хлор-5H-пирроло[3,2-d]пиримидина в качестве исходных веществ. Выход: 25,4%; т.пл.: 261-263°C, МС [M+H]+ 432,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,2 (1H, с, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 26
4-(4-(5H-пирроло[3,2-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-18)
Соединение I-18 (67 мг) получали аналогично соединению I-1 с использованием 142 мг (0,46 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 4-хлор-5H-пирроло[3,2-d]пиримидина в качестве исходных веществ. Выход: 35,1%; т.пл.: 258-260°C, МС [M+H]+ 419,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,4 Гц, -CH2-×2), 2,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,4 Гц, -CH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,2 (1H, с, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 27
4-(4-(6-метил-5H-пирроло[3,2-d]пиримидинил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-19)
Соединение I-19 (55 мг) получали аналогично соединению I-1 с использованием 132 мг (0,42 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,42 ммоль) 4-хлор-6-метил-5H-пирроло[3,2-d]пиримидина в качестве исходных веществ. Выход: 29,4%; т.пл.: 265-267°C, МС [M+H]+ 446,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 28
4-(4-(6-метил-5H-пирроло[3,2-d]пиримидинил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-20)
Соединение I-20 (69 мг) получали аналогично соединению I-1 с использованием 130 мг (0,42 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,42 ммоль) 4-хлор-6-метил-5H-пирроло[3,2-d]пиримидина в качестве исходных веществ. Выход: 38,1%; т.пл.: 268-270°C, МС [M+H]+ 433,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,4 Гц, -CH2-×2), 2,4 (2H, с, -CH2-), 2,6 (3H, с, -CH3), 3,6 (4H, т, J=4,4 Гц, -CH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,4 (1H, с, ArH), 8,6 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 29
4-(4-фуро[2,3-d]пиримидиниламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-21)
Соединение I-21 (45 мг) получали аналогично соединению I-1 с использованием 143 мг (0,45 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,45 ммоль) 4-хлорфуро[2,3-d]пиримидина в качестве исходных веществ. Выход: 23,0%; т.пл.: 255-257°C, МС [M+H]+ 433,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 7,1 (1H, д, J=2,5 Гц, ArH), 7,2 (2H, д, J=8,4 Гц, ArH), 7,7 (2H, д, J=8,4 Гц, ArH), 8,0 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 30
4-(4-фуро[2,3-d]пиримидиниламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-22)
Соединение I-22 (53 мг) получали аналогично соединению I-1 с использованием 141 мг (0,45 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,45 ммоль) 4-хлорфуро[2,3-d]пиримидина в качестве исходных веществ. Выход: 27,9%; т.пл.: >280°C, МС [M+H]+ 420,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 7,1 (1H, д, J=2,5 Гц, ArH), 7,3 (2H, д, J=8,2 Гц, ArH), 7,8 (2H, д, J=8,2 Гц, ArH), 8,0 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 31
4-(4-фуро[3,2-d]пиримидиниламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-23)
Соединение I-23 (71 мг) получали аналогично соединению I-1 с использованием 143 мг (0,45 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,45 ммоль) 4-хлорфуро[3,2-d]пиримидина в качестве исходных веществ. Выход: 36,2%; т.пл.: 277-279°C, МС [M+H]+ 433,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 7,2 (2H, д, J=8,4 Гц, ArH), 7,7 (2H, д, J=8,4 Гц, ArH), 7,8 (1H, д, J=2,5 Гц, ArH), 8,2 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 32
4-(4-фуро[3,2-d]пиримидиниламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-24)
Соединение I-24 (80 мг) получали аналогично соединению I-1 с использованием 141 мг (0,45 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,45 ммоль) 4-хлорфуро[3,2-d]пиримидина в качестве исходных веществ. Выход: 42,1%; т.пл.: 271-273°C, МС [M+H]+ 420,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 7,3 (2H, д, J=8,2 Гц, ArH), 7,7 (2H, д, J=8,2 Гц, ArH), 7,8 (1H, д, J=2,5 Гц, ArH), 8,2 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 33
4-(4-тиено[3,2-c]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-25)
129 мг (0,41 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида, 70 мг (0,41 ммоль) 4-хлортиено[3,2-c]пиридина и 1 мл ледяной уксусной кислоты растворяли в изопропаноле (8 мл). Реакционную смесь нагревали в микроволновой печи (300 W) при 190°C в течение 30 мин. Изопропанол отгоняли при пониженном давлении и полученное твердое вещество растворяли в дистиллированной воде. Использовали насыщенный водный раствор гидроксида натрия для доведения pH до 8~9 и полученную смесь экстрагировали этилацетатом три раза (50 мл × 3). Экстракты объединяли и сушили над сухим сульфатом магния. После фильтрования с отсасыванием, растворитель отгоняли при пониженном давлении, с получением желтоватого твердого вещества. Неочищенное вещество подвергали колоночной хроматографии (подвижная фаза: метанол:хлороформ = 1:15), с получением соединения I-25 (67 мг). Выход: 36,4%; т.пл.: 268-270°C, МС [M+H]+ 448,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,5 (1H, д, J=5,4 Гц, ArH), 7,7-7,8 (3H, м, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,5 (1H, с, ArH), 10,0 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 34
4-(4-тиено[3,2-c]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-26)
Соединение I-26 (71 мг) получали аналогично соединению I-25 с использованием 128 мг (0,41 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,41 ммоль) 4-хлортиено[3,2-c]пиридина в качестве исходных веществ. Выход: 39,7%; т.пл.: 269-271°C, МС [M+H]+ 435,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,5 (4H, т, J=4,2 Гц, -CH2-×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,5 (1H, д, J=5,4 Гц, ArH), 7,7-7,8 (3H, м, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,5 (1H, с, ArH), 9,9 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 35
4-(4-(2-метилтиено[3,2-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-27)
Соединение I-27 (68 мг) получали аналогично соединению I-25 с использованием 121 мг (0,38 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 4-хлор-2-метилтиено[3,2-c]пиридина в качестве исходных веществ. Выход: 38,6%; т.пл.: 267-269°C, МС [M+H]+ 462,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,6 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,2 (1H, с, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,5 (1H, с, ArH), 10,0 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 36
4-(4-(2-метилтиено[3,2-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-28)
Соединение I-28 (59 мг) получали аналогично соединению I-25 с использованием 119 мг (0,38 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 4-хлор-2-метилтиено[3,2-c]пиридина в качестве исходных веществ. Выход: 34,5%; т.пл.: 265-267°C, МС [M+H]+ 449,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 3,5 (4H, т, J=4,2 Гц, -CH2-×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,2 (1H, с, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,5 (1H, с, ArH), 9,9 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 37
4-(7-тиено[2,3-c]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-29)
Соединение I-29 (59 мг) получали аналогично соединению I-25 с использованием 129 мг (0,41 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,41 ммоль) 7-хлортиено[2,3-c]пиридина в качестве исходных веществ. Выход: 30,4%; т.пл.: 274-276°C, МС [M+H]+ 448,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,2 (3H, с, -CH3), 2,3-2,5 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,5 (1H, д, J=5,3 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,2 (1H, д, J=5,3 Гц, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 38
4-(7-тиено[2,3-c]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-30)
Соединение I-30 (81 мг) получали аналогично соединению I-25 с использованием 128 мг (0,41 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,41 ммоль) 7-хлортиено[2,3-c]пиридина в качестве исходных веществ. Выход: 45,3%; т.пл.: 271-273°C, МС [M+H]+ 435,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,4 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,5 (1H, д, J=5,2 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,3 (1H, д, J=5,2 Гц, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 39
4-(7-(3-метилтиено[2,3-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-31)
Соединение I-31 (71 мг) получали аналогично соединению I-25 с использованием 121 мг (0,38 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 7-хлор-3-метилтиено[2,3-c]пиридина в качестве исходных веществ. Выход: 40,3%; т.пл.: 258-260°C, МС [M+H]+ 462,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,2 (3H, с, -CH3), 82,3-2,5 (8H, м, -CH2-×4), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,2 (1H, с, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 40
4-(7-(3-метилтиено[2,3-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-32)
Соединение I-32 (71 мг) получали аналогично соединению I-25 с использованием 119 мг (0,38 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 7-хлор-3-метилтиено[2,3-c]пиридина в качестве исходных веществ. Выход: 42,7%; т.пл.: 275-277°C, МС [M+H]+ 449,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,4 (4H, т, J=4,2 Гц, -CH2-×2), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,3 (2H, д, J=8,4 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,3 (1H, с, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 41
4-(4-фуро[3,2-c]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-33)
Соединение I-33 (75 мг) получали аналогично соединению I-25 с использованием 120 мг (0,46 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 4-хлорфуро[3,2-c]пиридина в качестве исходных веществ. Выход: 38,1%; т.пл.: 268-270°C, МС [M+H]+ 432,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,1(1H, д, J=2,5 Гц, ArH), 7,2 (2H, д, J=8,4 Гц, ArH), 7,7 (2H, д, J=8,4 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,0 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 42
4-(4-фуро[3,2-c]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-34)
Соединение I-34 (68 мг) получали аналогично соединению I-25 с использованием 119 мг (0,46 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 4-хлорфуро[3,2-c]пиридина в качестве исходных веществ. Выход: 35,6%; т.пл.: 268-271°C, МС [M+H]+ 419,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,1 (1H, д, J=2,5 Гц, ArH), 7,3 (2H, д, J=8,2 Гц, ArH), 7,8 (2H, д, J=8,2 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,0 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 43
4-(4-(2-метилфуро[3,2-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-35)
Соединение I-35 (47 мг) получали аналогично соединению I-25 с использованием 144 мг (0,42 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,42 ммоль) 4-хлор-2-метилфуро[3,2-c]пиридина в качестве исходных веществ. Выход: 25,1%; т.пл.: 274-276°C, МС [M+H]+ 446,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,1(1H, с, ArH), 7,2 (2H, д, J=8,4 Гц, ArH), 7,7 (2H, д, J=8,4 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 44
4-(4-(2-метилфуро[3,2-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-36)
Соединение I-36 (63 мг) получали аналогично соединению I-25 с использованием 142 мг (0,42 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,42 ммоль) 4-хлор-2-метилфуро[3,2-c]пиридина в качестве исходных веществ. Выход: 34,8%; т.пл.: 275-277°C, МС [M+H]+ 433,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,1 (1H, с, ArH), 7,3 (2H, д, J=8,2 Гц, ArH), 7,8 (2H, д, J=8,2 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 45
4-(7-фуро[2,3-c]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-37)
Соединение I-37 (45 мг) получали аналогично соединению I-25 с использованием 132 мг (0,46 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 7-хлорфуро[2,3-c]пиридина в качестве исходных веществ. Выход: 22,8%; т.пл.: 258-261°C, МС [M+H]+ 432,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,2 (2H, д, J=8,4 Гц, ArH), 7,7 (2H, д, J=8,4 Гц, ArH), 7,8 (1H, д, J=2,5 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,3 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 46
4-(7-фуро[2,3-c]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-38)
Соединение I-38 (47 мг) получали аналогично соединению I-25 с использованием 130 мг (0,46 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 7-хлорфуро[2,3-c]пиридина в качестве исходных веществ. Выход: 24,6%; т.пл.: 268-272°C, МС [M+H]+ 419,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,3 (2H, д, J=8,2 Гц, ArH), 7,7 (2H, д, J=8,2 Гц, ArH), 7,8 (1H, д, J=2,5 Гц, ArH), 7,9 (1H, д, J=8,0 Гц, ArH), 8,3 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 47
4-(7-фуро[3,2-b]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-39)
Соединение I-39 (48 мг) получали аналогично соединению I-25 с использованием 144 мг (0,46 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 7-хлорфуро[3,2-b]пиридина в качестве исходных веществ. Выход: 24,4%; т.пл.: 268-270°C, МС [M+H]+ 432,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,2 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, с, ArH), 7,7 (2H, д, J=8,4 Гц, ArH), 7,8 (1H, д, J=2,5 Гц, ArH), 8,2 (1H, д, J=8,0 Гц, ArH), 8,3 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 48
4-(7-фуро[3,2-b]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-40)
Соединение I-40 (53 мг) получали аналогично соединению I-25 с использованием 142 мг (0,46 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 7-хлорфуро[3,2-b]пиридина в качестве исходных веществ. Выход: 27,7%; т.пл.: 275-278°C, МС [M+H]+ 419,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH2-×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,3 (2H, д, J=8,2 Гц, ArH), 7,7 (2H, д, J=8,2 Гц, ArH), 7,8 (1H, д, J=2,5 Гц, ArH), 8,2 (1H, д, J=8,0 Гц, ArH), 8,3 (1H, д, J=2,5 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 49
4-(4-фуро[2,3-b]пиридиламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-41)
Соединение I-41 (64 мг) получали аналогично соединению I-25 с использованием 144 мг (0,46 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 4-хлорфуро[2,3-b]пиридина в качестве исходных веществ. Выход: 32,5%; т.пл.: 273-276°C, МС [M+H]+ 432,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 6,7 (1H, д, J=8,0 Гц, ArH), 7,1 (1H, д, J=2,5 Гц, ArH), 7,2 (2H, д, J=8,4 Гц, ArH), 7,7 (2H, д, J=8,4 Гц, ArH), 8,0 (1H, д, J=2,5 Гц, ArH), 8,2 (1H, д, J=8,0 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 50
4-(4-фуро[2,3-b]пиридиламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-42)
Соединение I-42 (56 мг) получали аналогично соединению I-25 с использованием 142 мг (0,46 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 4-хлорфуро[2,3-b]пиридина в качестве исходных веществ. Выход: 29,3%; т.пл.: 269-271°C, МС [M+H]+ 419,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц,-CH3×2), 6,7 (1H, д, J=8,0 Гц, ArH), 7,1 (1H, д, J=2,5 Гц, ArH), 7,3 (2H, д, J=8,2 Гц, ArH), 7,8 (2H, д, J=8,2 Гц, ArH), 8,0 (1H, д, J=2,5 Гц, ArH), 8,2 (1H, д, J=8,0 Гц, ArH), 8,4 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 13,4 (1H, с, пиразол).
ПРИМЕР 51
4-(7-(1H-пирроло[2,3-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-43)
Соединение I-43 (64 мг) получали аналогично соединению I-25 с использованием 145 мг (0,46 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 7-хлор-1H-пирроло[2,3-c]пиридина в качестве исходных веществ. Выход: 32,3%; т.пл.: 279-282°C, МС [M+H]+ 431,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, д, J=8,0 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,0 (1H, д, J=8,0 Гц, ArH), 8,2 (1H, с, ArH), 8,4 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 52
4-(7-(1H-пирроло[2,3-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-44)
Соединение I-44 (52 мг) получали аналогично соединению I-25 с использованием 143 мг (0,46 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,46 ммоль) 7-хлор-1H-пирроло[2,3-c]пиридина в качестве исходных веществ. Выход: 27,1%; т.пл.: 265-267°C, МС [M+H]+ 420,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,4 Гц, -CH2-×2), 2,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,4 Гц, -CH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, д, J=8,0 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,0 (1H, д, J=8,0 Гц, ArH), 8,2 (1H, с, ArH), 8,4 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 53
4-(7-(2-метил-1H-пирроло[2,3-c]пиридил)амино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-45)
Соединение I-45 (49 мг) получали аналогично соединению I-25 с использованием 132 мг (0,42 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,42 ммоль) 7-хлор-2-метил-1H-пирроло[2,3-c]пиридина в качестве исходных веществ. Выход: 26,2%; т.пл.: 276-278°C, МС [M+H]+ 445,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,4 (8H, м, -CH2-×4), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, д, J=8,0 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,0 (1H, д, J=8,0 Гц, ArH), 8,4 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 54
4-(7-(2-метил-1H-пирроло[2,3-c]пиридил)амино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-46)
Соединение I-46 (73 мг) получали аналогично соединению I-25 с использованием 131 мг (0,42 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,42 ммоль) 7-хлор-2-метил-1H-пирроло[2,3-c]пиридина в качестве исходных веществ. Выход: 40,1%; т.пл.: 254-258°C, МС [M+H]+ 432,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,4 Гц, -CH2-×2), 2,4 (2H, с, -CH2-), 2,6 (3H, с, -CH3), 3,6 (4H, т, J=4,4 Гц, -CH2-×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, д, J=8,0 Гц, ArH), 7,6 (1H, с, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,0 (1H, д, J=8,0 Гц, ArH), 8,4 (1H, с, ArH), 9,2 (1H, с, -NHCO-), 10,2 (1H, с, -NH-), 12,0 (1H, с, пиррол), 13,4 (1H, с, пиразол).
ПРИМЕР 55
4-(4-(2-метилтиено[3,2-d]пиримидин)иламино)-N-(4-((4-метил-1-пиперазинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-47)
Соединение I-47 (85 мг) получали аналогично соединению I-1 с использованием 120 мг (0,38 ммоль) N-(4-((4-метил-1-пиперазинил)метил)фенил-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 4-хлор-2-метилтиено[3,2-d]пиримидина в качестве исходных веществ. Выход: 49,7%; т.пл.: >280°C, МС [M+H]+ 463,3.
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,1 (3H, с, -CH3), 2,2-2,5 (8H, м, -CH2-×4), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 7,3 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, д, J=4,1 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,2 (1H, д, J=4,1 Гц, ArH), 8,6 (1H, с, ArH), 9,6 (1H, с, -NHCO-), 10,3 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 56
4-(4-(2-метилтиено[3,2-d]пиримидин)иламино)-N-(4-((4-морфолинил)метил)фенил)-1H-3-пиразолкарбоксамид (I-48)
Соединение I-48 (93 мг) получали аналогично соединению I-1 с использованием 119 мг (0,38 ммоль) N-(4-((4-морфолинил)метил)фенил)-4-амино-1H-3-пиразолкарбоксамида и 70 мг (0,38 ммоль) 4-хлор-2-метилтиено[3,2-d]пиримидина в качестве исходных веществ. Выход: 54,4%; т.пл.: 259-262°C, МС [M+H]+ 450,2,
1H-ЯМР [300 МГц, ДМСО-d6]: δ 2,3 (4H, т, J=4,2 Гц, -CH2-×2), 2,6 (3H, с, -CH3), 3,4 (2H, с, -CH2-), 3,6 (4H, т, J=4,2 Гц, -CH3×2), 7,3 (2H, д, J=8,4 Гц, ArH), 7,4 (1H, д, J=4,1 Гц, ArH), 7,8 (2H, д, J=8,4 Гц, ArH), 8,2 (1H, д, J=4,1 Гц, ArH), 8,5 (1H, с, ArH), 9,7 (1H, с, -NHCO-), 10,4 (1H, с, -NH-), 13,5 (1H, с, пиразол).
ПРИМЕР 57
1. Экспериментальные материалы
Реагенты и материалы: соединение 1, мезилат соединения 1 (IS), соединение 2, мезилат соединения 2 (2S), ацетонитрил, этилацетат, метанол, хроматографическая колонка Hypersil ODS (4,6 мм × 200 мм, 5 мм), центрифужная пробирка, пробирка EP, наконечник пипетки, резиновые перчатки, шприц (1 мл) и т.д.
2. Инструменты
Agilent 1200 HPLC (Agilent Technologies Co., ltd., USA), вибраторная водяная баня с постоянной температурой Model SHZ-88 (Jintan Instrument Co., ltd., China), ультразвуковой очиститель Model KQ3200DB (Kunshan Ultrasonic Instrument Co., ltd., China), фотометр ультрафиолетовой спектрометрии Model UV-2102PCS (Shanghai Longnike Instrument Co., ltd., China), настольная центрифуга Model TGL-16 (Shanghai Anting Science Instrument Co., ltd., China), вихревой миксер Model XW-80A (Shanghai Jinke, China), электронные весы Model PL203 Mettler Toledo (Switzerland).
Животные: крысы Male Wistar (массой 200±20 г) (China Pharmaceutical University, China).
3. Измерение растворимости в воде
Избыточное количество образца добавляли в 50 мл трехгорлую колбу, в которую добавляли 10 мл дистиллированной воды, и затем колбу подвергали вибрации на вибраторе с постоянной температурой при 25°C в течение 72 часов. Полученный раствор центрифугировали при 10000 об./мин. в течение 15 мин, и затем супернатант фильтровали через 0,22 мкМ микрофильтровальную мембрану для удаления нерастворившегося лекарственного средства. 2 мл фильтрата отмеряли в 10 мл метанола. Содержание лекарственного средства определяли инъекцией 20 мкл образца. Результаты показаны в таблице 3.
Результаты в таблице 3 показывают, что растворимость в воде соединений I-1 и I-2 увеличивается, когда они находятся в форме мезилатов (I-1-S) и (I-2-S).
4. Измерение коэффициента разделения липид-вода (LogP)
н-Октанолу и дистиллированной воде давали возможность взаимного насыщения в течение 24 ч. Количество образца точно отвешивали в 50 мл объемную колбу и заполняли н-октанолом, который был насыщен водой (образец растворялся полностью). 10 мл раствора образца в н-октаноле помещали в 50 мл трехгорлую колбу, в которую добавляли 10 мл дистиллированной воды, которая была насыщена н-октанолом. Смесь уравновешивали (125 об./мин, 72 ч) при 25°C при встряхивании на вибраторе с постоянной температурой. Коэффициент разделения липид-вода рассчитывали по концентрациям лекарственного средства в н-октанольном исходном растворе перед экспериментом и в н-октанольном слое после эксперимента. Результаты показаны в таблице 4.
Результаты в таблице 4 показывают, что коэффициент разделения липид-вода (LogP) соединений I-1 и I-2 не имеют существенных отличий в сравнении с мезилатами (I-1-S) и (I-2-S).
3. Анализы на стабильность
3.1 Обработка образца плазмы
300 мкл крови, отобранной у крыс, смешивали 30 мкл метанола и 2 мл этилацетата. Образцы перемешивали на вихревой мешалке в течение 3 мин и центрифугировали при 4000 об./мин. в течение 15 мин. Супернатант переносили в другую центрифужную пробирку. Нижний слой повторно экстрагировали и затем супернатанты объединяли и сушили в атмосфере азота. После повторного растворения в 150 мкл метанола, их фильтровали через 0,22 мкМ мембрану и затем подвергали анализам в инъецируемом объеме 20 мкл.
3.2 Стабильность в плазме
Стандартный раствор соединения (I-1 и I-2) разводили плазмой и отбирали через 0, 1, 2, 4, 6, 8, 12 и 24 ч. Образцы получали в соответствии с методикой, описанной в 3.1. 20 мкл вводили в систему ВЭЖХ и область пика записывали для анализов для определения стабильности соединений 1 и 2 в плазме. Результаты показаны в таблице 5.
Можно видеть, что соединения (I-1 и I-2) показывают хорошую плазменную стабильность в течение 24 часов.
4. Эксперимент на животных
4.1 Постановка эксперимента и данные по концентрации в плазме в зависимости от времени
Количество соединения точно отбирали для получения CMC-Na водного раствора, содержащего 3 мг/мл лекарственного средства. 12 здоровых самцов крыс Wister произвольно распределяли на две группы, каждая из которых включала по 6 крыс. Первой группе перорально вводили соединение 1. Второй группе перорально вводили соединение 2. Дозирование в обеих группах составляло 30 мг/кг (соответственно, по 2 мл для каждой крысы). Крыс не кормили в течение 12 часов перед введением, и был свободный доступ к воде. 0,6 мл образцы крови отбирали из венозных синусов каждые 0,5, 1, 1,5, 2, 2,5, 3, 5, 8, 12 и 24 часа после введения лекарственного средства и добавляли в EP пробирки, которые заранее были промыты гепарином натрия. Образцы центрифугировали при 4000 об./мин. в течение 15 мин и получали верхнюю плазму. 300 мкл плазмы подвергали анализам, и записывали хроматограммы и области пиков. Вычисляли концентрацию соединений 1 и 2 в плазме с получением кривой зависимости концентрации от времени. В таблицах 6 и 7 суммированы все результаты.
4.2 Данные анализов
Использовали программу DAS 2,0 для анализа зависимости концентрации в плазме от времени после перорального введения доз (нг*мл/л) (таблицы 6, 7). Фитинг и методы AIC использовали для определения модели. Анализы основывались на принципах, чем больше фитинг и меньше AIC, лучше модель. Пероральное введение согласуется с двухкомпонентной моделью. Статистические расхождения параметров использовали для сравнения фармакокинетических параметров 2 соединений. Результаты представлены в таблице 8.
Как можно видеть из таблицы 8, фармакокинетические параметры соединений 1 и 2 являются приемлемыми.
ПРИМЕР 58
Ингибирование пересаженной животному опухоли S180 лекарственным средством
1. Экспериментальные материалы и животные
Тестируемое лекарственное средство: соединение I-1; положительное лекарственное средство: AT7519, получено от Jinan Great Chemical Co., Ltd.
Тестовые животные: мыши ICR, со степенью чистоты, полученные из центра Yangzhou University Medical, лицензия No: SCXK(Su)2007-0001; 18-22 г, самки; грануляторы с плоской матрицей, полученные от Jiangsu Xietong Organism Co., ltd.; условия питания: комната с воздушным кондиционированием, 18-24°C, относительная влажность 70%.
Опухоль: S180, полученная от исследовательского Института Опухолевых Лекарственных средств Цзянсу.
Инструмент: медицинский микростол YJ-875 (Suzhou Medical Instrument).
2. Методики
ICR мышам инокулировали солидную опухоль в соответствии процессом пересадки опухолей (образец опухоли взвешивали в стерильных условиях, гомогенизировали в стеклянном гомогенизаторе для тканей, помещали в стерильный контейнер, в который добавляли физиологический раствор, с получением клеточной суспензии 1:3. Контейнер помещали на лед и аспирировали, и клетки гомогенно смешивали перед каждым аспирированием. Каждую мышь инокулировали подкожно 0,2 мл в правую переднюю подмышечную впадину). 24 часа после инокуляции, мышей взвешивали и произвольно распределяли на 5 групп, каждая группа включала 10 мышей. Каждую группу лекарственных средств вводили первый раз через 24 часа после инокуляции (d1). Мышам вводили внутривенно каждый день, в общем 7 введений. Объем введения составил 0,4 мл/20 г. Мышей, имеющих опухоль, приносили в жертву через 8 дней после инокуляции (d8). Опухоль ткани отделяли и взвешивали. Данные анализировали статистическим методом (t-тест).
Распределение доз: в общей сложности на 5 групп.
Группа модельного контроля; группа положительного контроля: AT7519 15 мг/кг; тестируемое лекарственное средство: 30 мг/кг; тестируемое лекарственное средство: 15 мг/кг; тестируемое лекарственное средство: 7,5 мг/кг
4. Результаты

5. Заключение
Результаты указывают на то, что в сравнении с группой модельного контроля, тестируемое лекарственное средство (30 мг/кг и 15 мг/кг) обладает весьма существенным ингибирующим эффектом на рост опухоли S180 (P<0,01), и тестируемое лекарственное средство (7,5 мг/кг) обладает существенным ингибирующим эффектом на рост опухоли S180 (P<0,05). Тестируемое лекарственное средство (30 мг/кг и 15 мг/кг) обладает существенным ингибирующим эффектом на массу тела экспериментальных животных (P<0,05).
Реферат
Настоящее изобретение относится к области медицинской химии и, в частности, относится к производным 4-(замещенного пятичленного гетероциклического пиримидин/пиридин)амино-1H-3-пиразолкарбоксамида формулы (I), в которой радикалы и символы определены в формуле изобретения. Соединения формулы (I) являются ингибиторами протеинкиназ. Изобретение также относится к фармацевтической композиции, содержащей эти соединения, и к их применению в медицине в качестве противоопухолевого средства. 6 н. и 6 з.п. ф-лы, 1 ил., 7 табл., 58 пр.
Формула
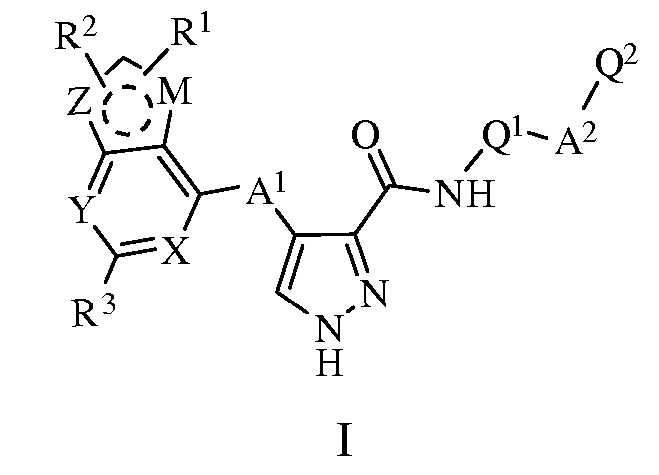
Комментарии