Нитроимидазооксазины и их применения при противотуберкулезной терапии - RU2542988C2
Код документа: RU2542988C2
Чертежи






















Описание
Предшествующий уровень техники
По данной заявке испрашивается приоритет на основании предварительной заявки на патент США с регистрационным № 61/230396, озаглавленной “Нитроимидазооксазины и их применения при противотуберкулезной терапии”, поданной 31 июля 2009 г., полное содержание которой таким образом включено посредством ссылки.
Изобретение относится к новым нитроимидазооксазинам, их получению и их применению в качестве лекарственных средств для лечения заболеваний, вызванных Mycobacterium tuberculosis и другими микробными инфекциями, либо отдельно, либо в комбинации с другими противоинфекционными лечениями.
Туберкулез остается основной инфекционной причиной смерти во всем мире (подсчитано, что смертность составляла 1,3 миллиона в 2008 г.) с недавним возрождением интереса к этому заболеванию в связи с повышенной восприимчивостью к нему у ВИЧ-пациентов, возрастающим распространением штаммов с множественной лекарственной резистентностью и появлением экстенсивно резистентных к лекарственным средствам штаммов. Современная лекарственная терапия туберкулеза является длительной и сложной, включающей комбинации многих лекарственных средств (обычно изониазида, рифампина, пиразинамида и этамбутола), даваемых пациенту ежедневно в течение свыше 6 месяцев. Кроме того, эти лекарственные средства являются относительно неэффективными против устойчивой формы данного заболевания, которая, как предполагается, имеет место в значительной части случаев (Ferrara et al., 2006). Лекарственные средства второго ряда, применяемые в продолжительных комбинированных терапиях для заболевания с множественной лекарственной резистентностью (обычно на протяжении 2 лет), большей частью имеют пониженную эффективность или более высокую токсичность, чем существующие средства первого ряда. Часто проводят незавершенное лечение, что приводит к высоким показателям рецидива и повышенной резистентности к лекарственному средству, поэтому существует настоятельная потребность в новых, более эффективных лекарственных средствах.
Задачей настоящего изобретения является обеспечение новых нитроимидазооксазинов с неожиданно высокой эффективностью против как аэробных (реплицирующих), так и гипоксических (латентных или персистентных) культур Mycobacterium tuberculosis и неожиданно высокой эффективностью на мышиных моделях инфицирования Mycobacterium tuberculosis для применения в качестве противотуберкулезных лекарственных средств и для лечения других болезней, вызванных микробными инфекциями.
Сущность изобретения
Данное изобретение относится к соединениям нитроимидазооксазина, способам их получения и применениям соединений для лечения туберкулеза и других болезней, вызванных микробными инфекциями.
Недавнее введение нитроимидазооксазина РА-824 в клиническое испытание является значимым, поскольку данное соединение проявляет хорошую активность in vitro и in vivo против Mycobacterium tuberculosis как в его активной, так и устойчивой формах (Tyagi et al., 2005). Родственный 2-нитроимидазо[2,1-b]оксазол, ОРС-67683, также находится в процессе клинического испытания (Sasaki et al., 2006). Структуры этих соединений показаны на фигуре 1. Без желания быть связанным с теорией, предполагают, что механизм действия РА-824 включает высвобождение оксида азота (Singh et al., 2008), последующую восстановительную стадию в процессе, зависящем от бактериальной глюкоза-6-фосфатдегидрогеназы (FGD1) и ее кофактора F420 (Stover et al., 2000). Исследования с микроанализами на мутантных штаммах дикого типа как FGD1, так и F420 показывают, что, как оказывается, белок с 151 аминокислотами (17,37 кДа) неизвестной функции, Rv3547, является критическим для этой активации (Manjunatha et al., 2006). Недавние механистические исследования восстановительной химии РА-824 поддерживают это утверждение (Anderson et al., 2008). Аналоги нитроимидазооксазина и их применение при туберкулезе были ранее описаны (патенты США №№ 5668127 (1997) и 6087358 (2000); Jiricek et al., WO 2007075872A2 (2007); Li et al., 2008; Kim et al., 2009).
В первом аспекте настоящее изобретение относится к соединению, имеющему общую структуру формулы I

где Х представляет собой O, OCH2, OCH2CH=CH или OCH2C≡C;
Y представляет собой группу любой из формул IIa-IId, показанных ниже, где


Z в формуле IIa представляет собой CH2, CH=CH, С≡С или прямую связь; и
каждый из R1 и R2 в формулах I и IIa представляет собой один, два или три заместителя из H, F, Cl, CF3, OCF2H, OCF3, аза (-CH= заменен на -N=), или диаза (-CH=CH- заменен на -N=N-, -CH=CH-CH= заменен на -N=CH-N= или -CH=CH-CH=CH- заменен на -N=CH-CH=N-) в любом из доступных положений кольца.
Предпочтительный подкласс соединений имеет общую структуру указанной выше формулы I, где
Х представляет собой O, OCH2, OCH2CH=CH или OCH2C≡C;
Y представляет собой группу любой из формул IIa-IId, показанных ниже, где

Z в формуле IIa представляет собой CH2, CH=CH, С≡С или прямую связь;
R1 в формуле I представляет собой 4-F или 4-OCF3 или 2-Cl, 4-OCF3 или 3-Cl, 4-OCF3 или 3-F, 4-OCF3 или 2-аза, 4-CF3 или 3-аза, 4-CF3 или 2-аза, 4-F;
R2 в формуле IIa представляет собой любую одну или две группы из Н, F или аза (-CH= заменен на -N=) в любом из доступных положений кольца.
Указанные соединения, а также их смеси, их изомеры, физиологические функциональные производные в форме солей и пролекарства являются применимыми при предотвращении или терапии для лечения заболеваний, вызванных Mycobacterium tuberculosis и другими микробными инфекциями.
Краткое описание чертежей
На фигуре 1 показаны структуры соединений РА-824 и ОРС-67683.
На фигуре 2 показаны общие структуры репрезентативных соединений, относящихся к таблице 1.
На фигуре 3 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 4 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 5 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 6 показана общая синтетическая схема получения репрезентативных соединений.
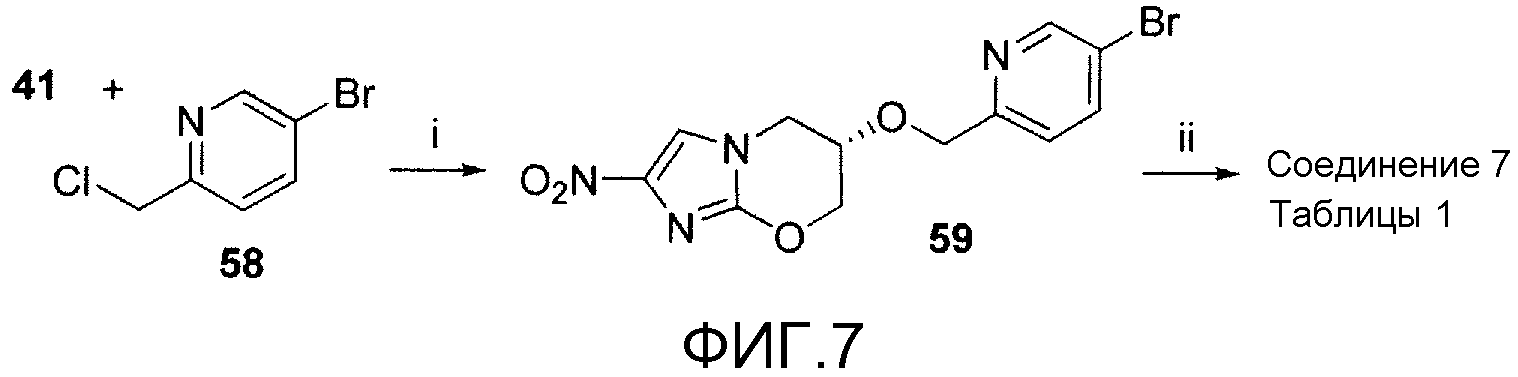
На фигуре 7 показана общая синтетическая схема получения репрезентативных соединений.
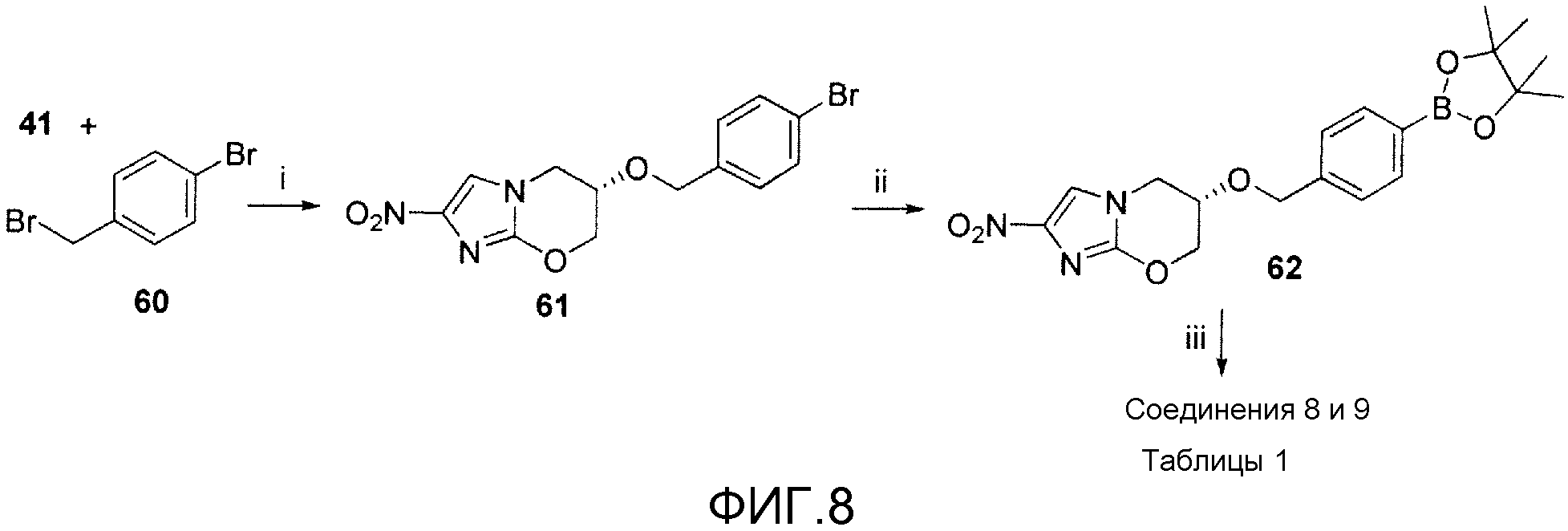
На фигуре 8 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 9 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 10 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 11 показана общая синтетическая схема получения репрезентативных соединений.
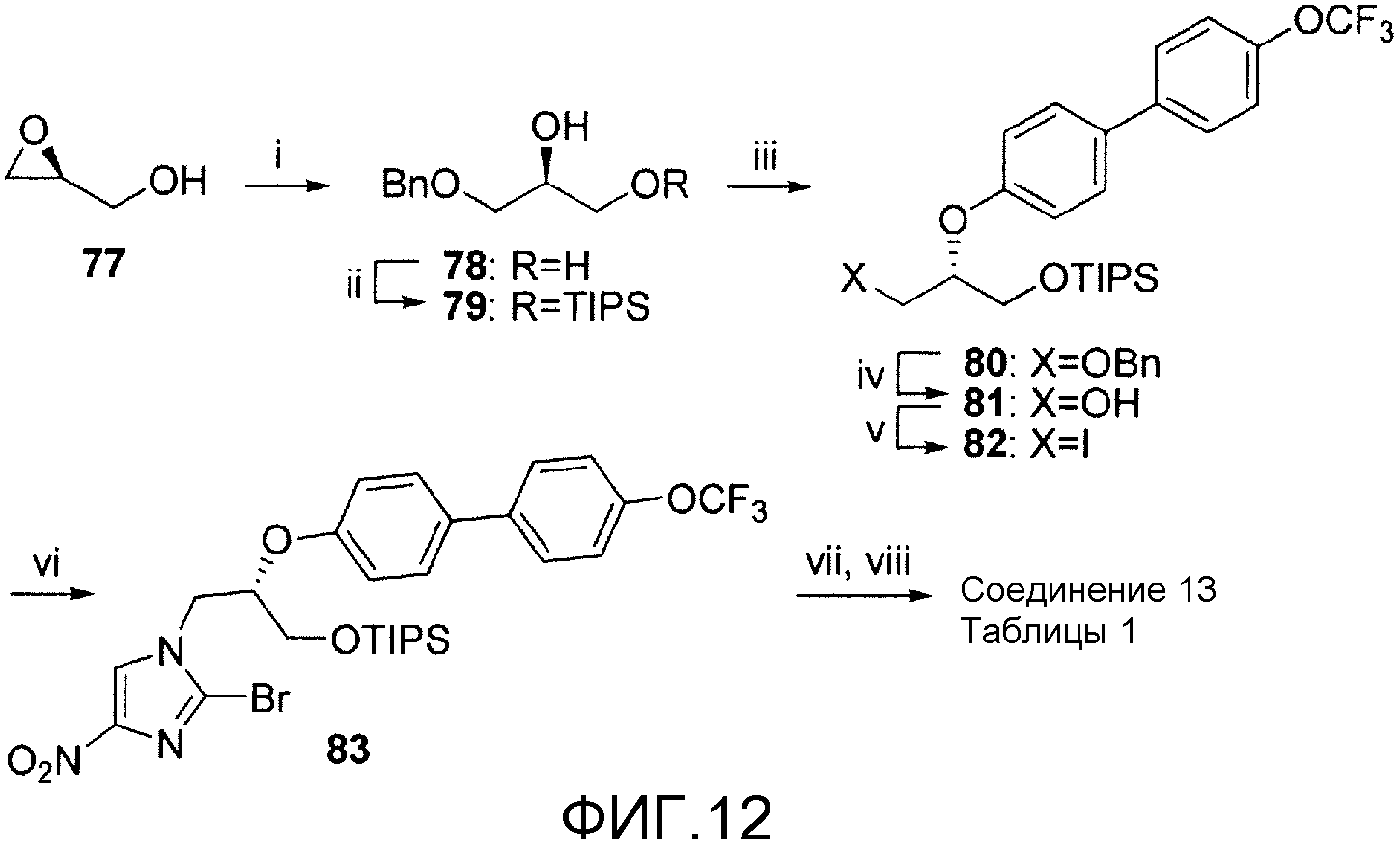
На фигуре 12 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 13 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 14 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 15 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 16 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 17 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 18 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 19 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 20 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 21 показана общая синтетическая схема получения репрезентативных соединений.
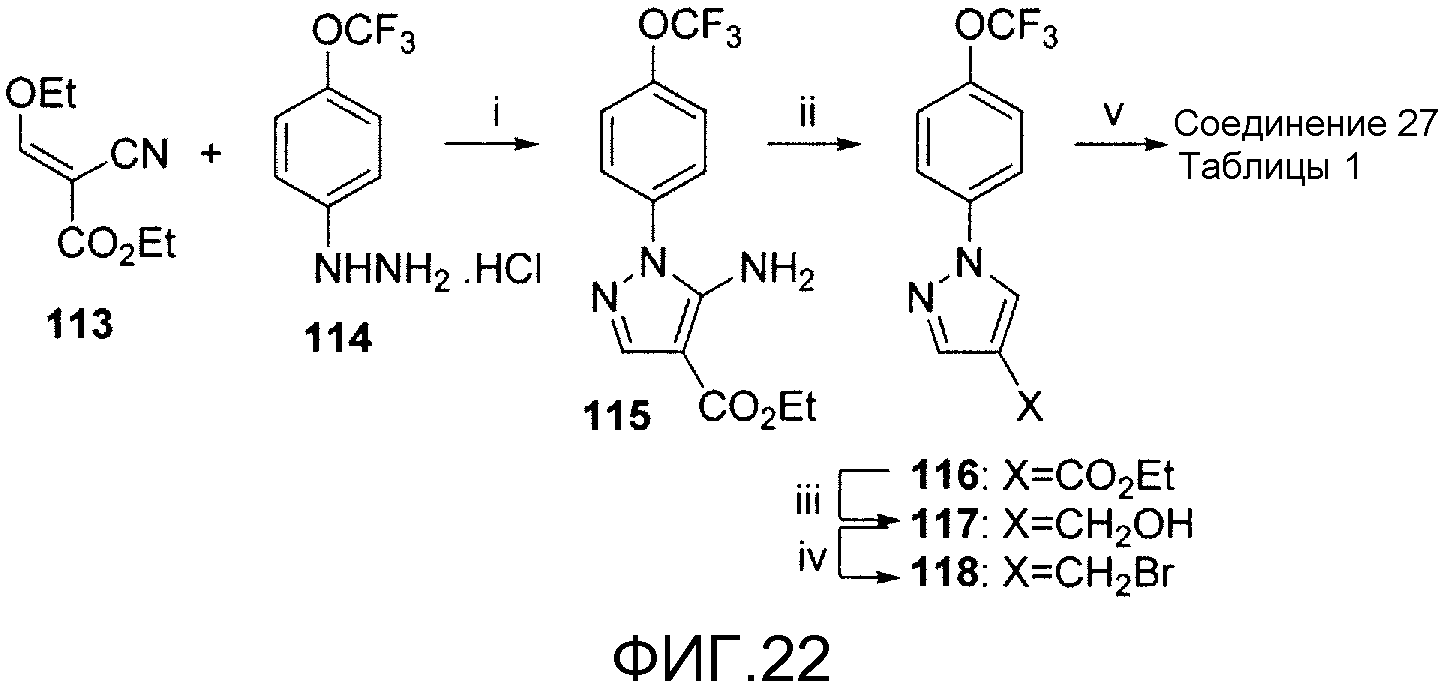
На фигуре 22 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 23 показана общая синтетическая схема получения репрезентативных соединений.
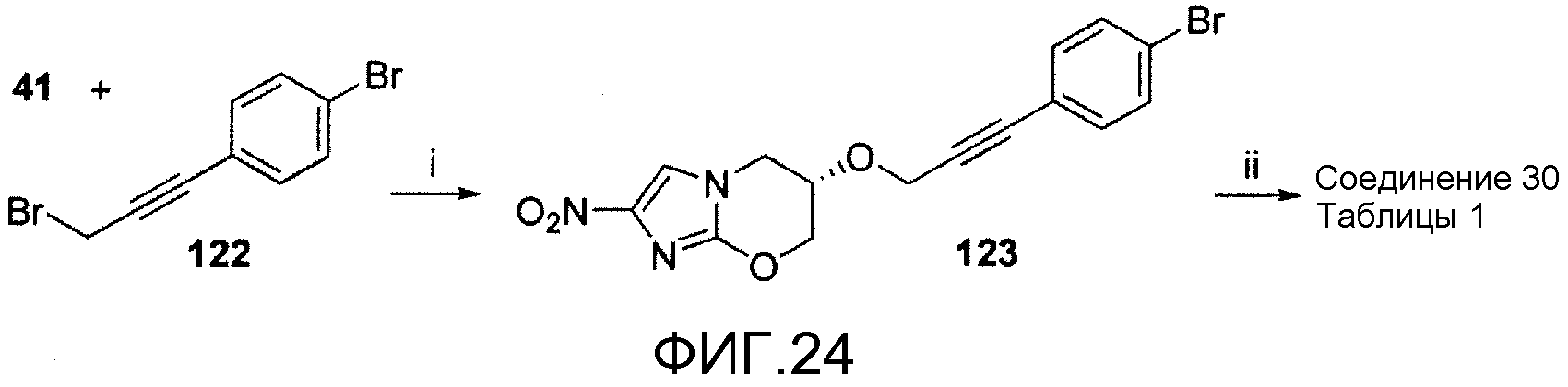
На фигуре 24 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 25 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 26 показана общая синтетическая схема получения репрезентативных соединений.
На фигуре 27 показаны структуры репрезентативных соединений 1-18, относящихся к таблице 1 и примерам 1-3.
На фигуре 28 показаны структуры репрезентативных соединений 19-33, относящихся к таблице 1 и примерам 1-3.
Подробное описание
Настоящее изобретение относится к соединениям нитроимидазооксазола, способам их получения и применению соединений для лечения туберкулеза и других микробных инфекций.
В первом аспекте настоящее изобретение относится к соединению, имеющему общую структуру формулы I

где Х представляет собой O, OCH2, OCH2CH=CH или OCH2C≡C;
Y представляет собой группу любой из формул IIa-IId, показанных ниже, где

Z в формуле IIa представляет собой CH2, CH=CH, С≡С или прямую связь; и
каждый из R1 и R2 в формулах I и IIa представляет собой любой один, два или три заместителя из H, F, Cl, CF3, OCF2H, OCF3, аза (-CH= заменен на -N=), или диаза (-CH=CH- заменен на -N=N-, -CH=CH-CH= заменен на -N=CH-N= или -CH=CH-CH=CH- заменен на -N=CH-CH=N-) в любом из доступных положений кольца.
Предпочтительный подкласс соединений имеет общую структуру указанной выше формулы I, где
Х представляет собой O, OCH2, OCH2CH=CH или OCH2C≡C;
Y представляет собой группу любой из формул IIa-IId, показанных ниже, где

Z в формуле IIa представляет собой CH2, CH=CH, С≡С или прямую связь;
R1 в формуле I представляет собой 4-F или 4-OCF3 или 2-Cl, 4-OCF3 или 3-Cl, 4-OCF3 или 3-F, 4-OCF3 или 2-аза, 4-CF3 или 3-аза, 4-CF3 или 2-аза, 4-F;
R2 в формуле IIa представляет собой любую одну или две группы из Н, F или аза (-CH= заменен на -N=) в любом из доступных положений кольца.
Наиболее предпочтительными соединениями, описываемыми формулой I, являются
А. (6S)-6-{[2'-хлор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 1 таблицы 1 и фигуры 27);
В. (6S)-6-{[3'-фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 2 таблицы 1 и фигуры 27);
С. (6S)-2-нитро-6-{[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 3 таблицы 1 и фигуры 27);
D. (6S)-2-нитро-6-({5-[4-(трифторметокси)фенил]-2-пиразинил}метокси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 4 таблицы 1 и фигуры 27);
E. (6S)-6-{[6-(4-фторфенил)-3-пиридинил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 5 таблицы 1 и фигуры 27);
F. (6S)-2-нитро-6-({6-[4-(трифторметокси)фенил]-3-пиридинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 6 таблицы 1 и фигуры 27);
G. (6S)-2-нитро-6({5-[4-(трифторметокси)фенил]-2-пиридинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 7 таблицы 1 и фигуры 27);
H. (6S)-2-нитро-6-({4-[5-(трифторметил)-2-пиридинил]бензил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 8 таблицы 1 и фигуры 27);
I. (6S)-2-нитро-6-({4-[6-(трифторметил)-3-пиридинил]бензил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 9 таблицы 1 и фигуры 27);
J. (6S)-2-нитро-6-({1-[4-(трифторметокси)фенил]-1H-пиразол-3-ил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 10 таблицы 1 и фигуры 27);
K. (6S)-6-({1-метил-3-[4-(трифторметокси)фенил]-1H-пиразол-5-ил}метокси)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 11 таблицы 1 и фигуры 27);
L. (6S)-6-{[3-Фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 12 таблицы 1 и фигуры 27);
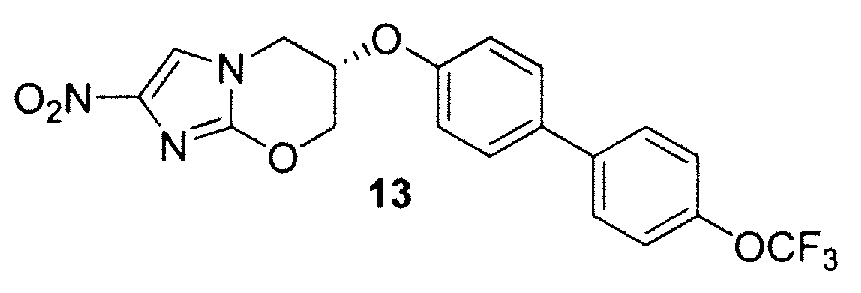
M. (6S)-2-нитро-6-{[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 13 таблицы 1 и фигуры 27);
N. (6S)-6-({2-фтор-4-[5-(трифторметил)-2-пиридинил]бензил}окси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 14 таблицы 1 и фигуры 27);
O. (6S)-6-{[2-фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 15 таблицы 1 и фигуры 27);
P. (6S)-2-нитро-6-({2-[4-(трифторметокси)фенил]-5-пиримидинил}метокси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 16 таблицы 1 и фигуры 27);
Q. (6S)-2-нитро-6-({4-[4-(трифторметокси)бензил]бензил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 17 таблицы 1 и фигуры 27);
R. (6S)-2-нитро-6-[(5-{[4-(трифторметокси)фенил]этинил}-2-пиридинил)метокси]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 18 таблицы 1 и фигуры 27);
S. (6S)-2-нитро-6-({(2E)-3-[4'-(трифторметокси)[1,1'-бифенил]-4-ил]-2-пропенил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 19 таблицы 1 и фигуры 28);
T. (6S)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин-6-ил-4-[4-(трифторметокси)фенил]-1-пиперазинкарбоксилат (соединение 20 таблицы 1 и фигуры 28);
U. (6S)-6-({6-[3-фтор-4-(трифторметокси)фенил]-3-пиридинил}метокси)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 21 таблицы 1 и фигуры 28);
V. (6S)-6-({5-[3-фтор-4-(трифторметокси)фенил]-2-пиридинил}метокси)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 22 таблицы 1 и фигуры 28);
W. (6S)-6-({2-фтор-4-[6-(трифторметил)-3-пиридинил]бензил}окси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 23 таблицы 1 и фигуры 28);
X. (6S)-2-нитро-6-({6-[4-(трифторметокси)фенил]-3-пиридазинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 24 таблицы 1 и фигуры 28);
Y. (6S)-2-нитро-6-[(5-{[6-(трифторметил)-3-пиридинил]этинил}-2-пиридинил)метокси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 25 таблицы 1 и фигуры 28);
Z. (6S)-6-{[4-(5-фтор-2-пиридинил)бензил]окси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 26 таблицы 1 и фигуры 28);
AA. (6S)-2-нитро-6-({1-[4-(трифторметокси)фенил]-1H-пиразол-4-ил}метокси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 27 таблицы 1 и фигуры 28);
BB. (6S)-6-({6-[3-хлор-4-(трифторметокси)фенил]-3-пиридинил}метокси)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 28 таблицы 1 и фигуры 28);
CC. (6S)-2-нитро-6-({5-[4-(трифторметокси)фенил]-2-пиримидинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 29 таблицы 1 и фигуры 28);
DD. (6S)-2-нитро-6-({3-[4'-(трифторметокси)[1,1'-бифенил]-4-ил]-2-пропинил}окси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 30 таблицы 1 и фигуры 28);
EE. (6S)-2-нитро-6-[(4-{(E)-2-[4-(трифторметокси)фенил]этенил}бензил)окси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 31 таблицы 1 и фигуры 28);
FF. (6S)-2-нитро-6-[(4-{[4-(трифторметокси)фенил]этинил}бензил)окси]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (соединение 32 таблицы 1 и фигуры 28) и
GG. (6S)-2-нитро-6-[(6-{[4-(трифторметокси)фенил]этинил}-3-пиридинил)метокси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (соединение 33 таблицы 1 и фигуры 28).
Соединения формулы I могут существовать в различных геометрических и энантиомерных формах, и как чистые формы, так и смеси этих отдельных изомеров включены в объем данного изобретения, а также включены их любые физиологически функциональные или фармацевтически приемлемые производные в виде солей или пролекарства. Получение этих альтернативных форм хорошо известно специалистам в данной области.
Настоящее изобретение относится также к способам предотвращения или терапии заболеваний, вызванных микробными инфекциями, такими как Mycobacterium tuberculosis, включающим стадию введения соединения формулы I.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I), определенной выше, и фармацевтически приемлемый эксципиент, адъювант, носитель, буфер или стабилизатор. “Терапевтически эффективное количество” следует понимать как количество соединения формулы I, которое достаточно для проявления противобактериальных или противомикробных эффектов. Действительное количество, частота и курс по времени введения будет зависеть от природы и тяжести подвергаемого лечению заболевания. Указание по лечению находится в пределах обязанности обычных практикующих врачей и других врачей в области медицины. Фармацевтически приемлемый эксципиент, адъювант, носитель, буфер и стабилизатор должны быть нетоксичными и не должны препятствовать эффективности активного ингредиента. Точная природа носителя или другого вещества будет зависеть от пути введения, который может быть пероральным, или пути введения посредством инъекции, таким как кожная, подкожная или внутривенная инъекция, или пути введения посредством ингалятора с сухим порошком.
Фармацевтические композиции для перорального введения могут быть в форме таблетки, капсулы, порошка или жидкости. Таблетка может содержать твердый носитель или адъювант. Жидкие фармацевтические композиции обычно содержат жидкий носитель, такой как вода, нефтепродукт, животные или растительные масла, минеральное масло или синтетическое масло. Может быть включен физиологический солевой раствор, раствор декстрозы или другого сахарида или гликоли, такие как этиленгликоль, пропиленгликоль или полиэтиленгликоль. Капсула может содержать твердый носитель, такой как желатин. Для внутривенной, кожной или подкожной инъекции активный ингредиент может быть в форме парентерально приемлемого водного раствора, который свободен от пирогенов и имеет подходящий рН, изотоничность и стабильность. Соответствующие специалисты в данной области вполне способны приготовить подходящие растворы с применением, например, изотонических носителей, таких как раствор для инъекции хлорида натрия, раствор для инъекции Рингера, содержащий лактат раствор для инъекции Рингера. При необходимости можно включать консерванты, стабилизаторы, буферы, антиоксиданты и/или другие добавки.
Фармацевтическая композиция может дополнительно содержать одно или несколько дополнительных противоинфекционных лекарственных средств. Этими противоинфекционными лекарственными средствами могут быть любые подходящие лекарственные средства, доступные из коммерческих источников или из других источников, которые, как известно, эффективно предотвращают или лечат заболевания, вызванные микробными инфекциями, такими как заболевания, вызванные Mycobacterium tuberculosis.
В другом аспекте при изготовлении лекарственного средства предложено применение терапевтически эффективного количества соединения формулы I, определенной выше, для введения субъекту. Предложен также способ получения соединения формулы I.
Термин “фармакологически приемлемая соль”, применяемый на всем протяжении описания, следует принимать как значение любой являющейся производным кислоты или основания соли, образованной из хлористоводородной, серной, фосфорной, уксусной, лимонной, щавелевой, малоновой, салициловой, молочной, фумаровой, янтарной, аскорбиновой, малеиновой, метансульфоновой, изоэтоновой кислот и тому подобных и карбоната калия, гидроксида натрия или калия, аммиака, триэтиламина, триэтаноламина и тому подобных.
Термин “пролекарство” означает фармакологическое вещество, которое вводят в неактивной или значительно менее активной форме. После введения пролекарство в результате метаболизма in vivo преобразуется в активный метаболит.
Термин “терапевтически эффективное количество” означает нетоксичное, но достаточное количество лекарственного средства для обеспечения требуемого терапевтического действия. Количество, которое является “эффективным”, изменяется от субъекта к субъекту в зависимости от возраста и общего состояния индивидуума, конкретной концентрации, вводимой композиции и тому подобного. Поэтому не всегда можно указать точное эффективное количество. Однако подходящее эффективное количество в любом отдельном случае может определить средний специалист в данной области с применением обычного экспериментирования. Кроме того, эффективным количеством является концентрация, которая находится в диапазоне, достаточном для того, чтобы позволить легко применять препарат, так чтобы доставить количество лекарственного средства, которое находится в пределах терапевтически эффективного диапазона.
Термин “аза” означает группу -СН=, замененную в соединении группой -N=. Термин “диаза” означает в соединении группу -СН=СН, замененную на -N=N-, группу -СН=СН-СН=, замененную на -N=CH-N=, или -СН=СН-СН=СН-, замененную на -N=CH-CH=N-.
Дополнительные аспекты настоящего изобретения станут очевидными из нижеследующего описания, представленного посредством только примера и со ссылкой на сопутствующие синтетические схемы.
Пример 1. Общие синтетические схемы
Соединения можно получить общими способами, указанными на схемах 1-24, показанных на фигурах 3-26, или любым другим подходящим способом. В описании схем 1-24 ниже дана ссылка на репрезентативные соединения, показанные в таблице 1 ниже и на фигурах 2 и 27-28.

На схеме 1, показанной на фигуре 3, реагенты и условия были следующими: (i) 2 М Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 88°С, 1-2,5 час; (ii) 30% HBr/AcOH, 20°С, 6-11 час; (iii) NaH, ДМФА, 0-20°С, 3 час. Сочетания Сузуки 4-(гидроксиметил)фенилбороновой кислоты (34) с галогенидами 35 и 36 в присутствии Pd(dppf)Cl2 давали бифениловые спирты 37 и 38, которые превращали в соответствующие бромметилсоединения 39 и 40. Сочетание их с известным спиртом 41 (описано получение его в патенте США № 5668127 посредством 4 стадий, исходя из 2,4-динитроимидазола и трет-бутилдиметилсилил-(S)-глицидилового простого эфира) давало требуемые соединения 1 и 2 таблицы 1.
На схеме 2, показанной на фигуре 4, реагенты и условия были следующими: (i) NaH, ДМФА, 5-20°С, 2 час, (ii) 2 М K2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, кипячение с обратным холодильником, 30 мин. Аналогичное сочетание с помощью NaH спирта 41 с 4-иодбензилбромидом (42) давало известный 4-иодбензиловый простой эфир 43 (описано получение в патенте США № 6087358 посредством такой же процедуры), который подвергали сочетанию Сузуки, как на схеме 1, с 4-(трифторметокси)фенилбороновой кислотой (44) с образованием соединения 3 таблицы 1.
На схеме 3, показанной на фигуре 5, реагенты и условия были следующими: (i) MsCl, Et3N, ТГФ, 0°С, 30 мин, затем NaI, ацетон, кипячение с обратным холодильником, 1 час; (ii) NaH, ДМФА, от -78 до 0°С, 1 час; (iii) 2 M K2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, кипячение с обратным холодильником, 30 мин. Сочетание с помощью NaH спирта 41 с 2-хлор-5-(иодметил)пиразином (46) (получен из известного (5-хлор-2-пиразинил)метанола (45) (получен хлорированием и восстановлением 5-гидроксипиразин-2-карбоновой кислоты, как описано Kiener et al., 1994) реакцией с MsCl с последующей реакцией с NaI) давало хлорид 47. Это соединение подвергали сочетанию Сузуки с 4-(трифторметокси)фенилбороновой кислотой (44), получая при этом соединение 4 таблицы 1.
На схеме 4, показанной на фигуре 6, реагенты и условия были следующими: (i) NaH, ДМФА, 5-20°С, 16 час; (ii) 44, 2 М K2CO3, ДМФА, Pd(dppf)Cl2 в атмосфере N2, 90°С, 2 дня; (iii) NBS, PPh3, CH2Cl2, 20°С, 3,5 час; (iv) 41, NaH, ДМФА, 0-20°С, 2,5 час; (v) 55-57, 2 М Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 90°С, 20-120 мин; (vi) водн. NaNO2, 25% H2SO4, 0°С, 12 мин, затем водн. KI, 20°С, 10 мин, затем 52°С, 2 час; (vii) n-BuLi, B(OiPr)3, толуол, ТГФ, от -78 до -20°С, 5 час, затем 2 н. HCl. Сочетание с помощью NaH 2-хлор-5-(хлорметил)пиридина 48 со спиртом 41 давало хлорид 49, который подвергали сочетанию Сузуки с 4-(трифторметокси)фенилбороновой кислотой (44), получая при этом соединение 6 таблицы 1. Бромирование коммерчески доступного (6-бром-3-пиридинил)метанола (50) с NBS/PPh3 давало бромметилпиридин 51, который аналогично сочетали с применением NaH со спиртом 41, получая при этом бромид 52. Это соединение подвергали сочетанию Сузуки с бороновыми кислотами 55 (полученными из анилина 53 посредством нового иодида 54), 56 или 57, получая при этом соответствующие соединения 28, 21 и 5 таблицы 1.
На схеме 5, показанной на фигуре 7, реагенты и условия были следующими: (i) NaH, ДМФА, 5-20°С, 2 час; (ii) 44, 2 М K2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 90°С, 30 мин. Сочетание с помощью NaH 5-бром-2-(хлорметил)пиридина (58) (получен хлорированием (5-бром-2-пиридинил)метанола, как описано van den Heuvel et al., 2004) со спиртом 41 давало бромид 59, который подвергали сочетанию Сузуки с 4-(трифторметокси)фенилбороновой кислотой (44), получая при этом соединение 7 таблицы 1.
На схеме 6, показанной на фигуре 8, реагенты и условия были следующими: (i) NaH, ДМФА, 20°С, 1 час; (ii) бис(пинаколято)диборон, Pd(dppf)Cl2 в атмосфере N2, KOAc, ДМСО, 90°С, 1 час; (iii) 2-хлор-5-(трифторметил)пиридин или 5-бром-2-(трифторметил)пиридин, 2 М К2СО3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, кипячение с обратным холодильником, 30 мин. Бромид 61 получали сочетанием с помощью NaH спирта 41 с 4-бромбензилбромидом (60). Реакция 61 с бис(пинаколято)дибороном давала 4-боронатный эфир 62, который подвергали сочетанию Сузуки с 2-хлор-5-(трифторметил)пиридином или 5-бром-2-(трифторметил)пиридином, получая при этом соответственно соединения 8 и 9 таблицы 1.
На схеме 7, показанной на фигуре 9, реагенты и условия были следующими: (i) водный пиридин, -5°С, 30 мин; (ii) бицикло[2.2.1]гепта-2,5-диен, Et3N, толуол, 70°С, 1 час, затем ксилол, кипячение с обратным холодильником, 2 час; (iii) LiAlH4, Et2O, 0-20°С, 1 час, (iv) PBr3, Et2O, 20°С, 17 час; (v) 41, NaH, ДМФА, 0°С, 2 час. Этил-(2Z)-хлор-{[4-(трифторметокси)фенил]гидразоно}этаноат (65) [получен из тетрафторбората 4-(трифторметокси)бензолдиазония (63) и этил-2-хлорацетоацетата (64)] подвергали реакции с бицикло[2.2.1]гепта-2,5-диеном, получая при этом карбоксилат 66. Его восстанавливали (LiAlH4) в спирт 67, который затем бромировали PBr3, получая при этом бромид 68. Сочетание с помощью NaH со спиртом 41 затем давало соединение 10 таблицы 1.
На схеме 8, показанной на фигуре 10, реагенты и условия были следующими: (i) CuI, PdCl2(PPh3), сульфат метилгидразина, водный NaHCO3, ТГФ, 20°С, 2 дня в атмосфере СО; (ii) 4 н. HCl, ТГФ, 80°С, 16 час; (iii) PBr3, Et2O, 0-20°С, 16 час; (iv) 41, NaH, ДМФА, 0°С, 2 час. Пиразол 71 получали реакцией 2-(2-пропинилокси)тетрагидро-2H-пирана (69), 1-иод-4-(трифторметокси)бензола (70) и метилгидразина в присутствии CuI и PdCl2(PPh3)2 и в атмосфере СО. Гидролизом простого эфира ТНР 71 в спирт 72 с последующим бромированием при помощи PBr3 получали бромид 73, который подвергали сочетанию при помощи NaH со спиртом 41, получая при этом соединение 11 таблицы 1.
На схеме 9, показанной на фигуре 11, реагенты и условия были следующими: (i) NaH, ДМФА, 0-20°С, 3 час; (ii) ArB(OH)2, 2 М Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 85-90°С, 1-3 час; (iii) бис(пинаколято)диборон, Pd(dppf)Cl2 в атмосфере N2; KOAc, ДМСО, 89°С, 5 час; (iv) 2-хлор-5-(трифторметил)пиридин, 2 М Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 90°С, 120 мин. Сочетание при помощи NaH 4-бром-2-фторбензилбромида (74) со спиртом 41 давало бромид 75, который подвергали сочетанию Сузуки с подходящими арилбороновыми кислотами, получая при этом соединения 12 и 23 таблицы 1. Реакция бромида 75 с бис(пинаколято)дибороном давала боронатный эфир 76, сочетанием Сузуки которого с 2-хлор-5-(трифторметил)пиридином получали соединение 14 таблицы 1.
На схеме 10, показанной на фигуре 12, реагенты и условия были следующими: (i) кат. CsF, PhCH2OH, 120°С, 16 час; (ii) TIPSCl, имидазол, ДМФА, 20°С, 16 час; (iii) 4'-(трифторметокси)[1,1'-бифенил]-4-ол, DIAD, PPh3, бензол, 5-20°С, 18 час; (iv) Н2, 5% Pd-C, EtOAc, EtOH, 413685,6 Па (60 фунт/кв. дюйм), 4 час; (v) I2, PPh3, имидазол, бензол, 20°С, 1 час; (vi) 2-бром-4(5)-нитроимидазол, К2СО3, ДМФА, 87°С, 20 час; (vii) TBAF, ТГФ, 20°С, 1 час; (viii) NaH, ДМФА, 5-20°С, 30 мин. Реакция (S)-глицидного спирта (77) и бензилового спирта в присутствии CsF давала диол 78, который монозащищали TIPS-хлоридом, и образовавшийся спирт 79 подвергали сочетанию Мицунобу с 4'-(трифторметокси)[1,1'-бифенил]-4-олом (Edsall et al., 2003, описано получение его посредством сочетания Сузуки 4-бромфенола и бороновой кислоты 44), получая при этом простой эфир 80. Его дебензилировали гидрогенолизом и образовавшийся спирт 81 иодировали I2/PPh3, получая при этом 82. Это соединение сочетали с 2-бром-4(5)-нитроимидазолом и образовавшееся соединение 83 дебензилировали TBAF и кольцо замыкали при помощи NaH, получая при этом соединение 13 таблицы 1.
На схеме 11, показанной на фигуре 13, реагенты и условия были следующими: (i) NaBH4, I2, ТГФ, 0-20°С, 14 час; (ii) 30% HBr/AcOH, 20°С, 20 час; (iii) NaH, ДМФА, 0-20°С, 3,5 час; (iv) 2 M Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 90°С, 6 час. Сочетание при помощи NaH 4-бром-3-фторбензилбромида (86) (получен из кислоты 84 с помощью известного спирта 85 (описан deSolms et al., 2003, получен посредством восстановления бораном 84)) со спиртовым производным оксазина 41 давало бромид 87, который подвергали сочетанию Сузуки с 4-(трифторметокси)фенилбороновой кислотой (44), получая при этом соединение 15 таблицы 1.
На схеме 12, показанной на фигуре 14, реагенты и условия были следующими: (i) 44, водный Na2CO3, толуол, EtOH, Pd(PPh3)4 в атмосфере N2, кипячение с обратным холодильником, 18 час; (ii) n-BuLi, ТГФ, -95°С, 0,5 мин, затем ДМФА, -90°С, 20 мин; (iii) NaBH4, MeOH, 0°С, 1 час; (iv) MsCl, Et3N, ТГФ, 0°С, 1 час, затем LiBr, Me2CO, кипячение с обратным холодильником, 1 час; (v) 41, NaH, ДМФА, от -78 до 0°С, 1 час. Сочетание Сузуки бороновой кислоты 44 и 2-иод-5-бромпиримидина (88) давало бромид 89, который обрабатывали n-BuLi и ДМФА, получая при этом альдегид 90. Его восстанавливали NaBH4 в спирт 91, который подвергали реакции с MsCl с последующей реакцией с LiBr, получая при этом бромид 92. Сочетание 92 со спиртом 41 давало соединение 16 таблицы 1.
На схеме 13, показанной на фигуре 15, реагенты и условия были следующими: (i) 44, 2 М K2CO3, DME, Pd(PPh3)4 в атмосфере N2, 105°С, 24 час; (ii) LiAlH4, Et2O, 20°С, 3 час; (iii) PBr3, CH2Cl2, 20°С, 2 час; (iv) 41, NaH, ДМФА, 20°С, 3 час. Сочетание Сузуки метил-4-(бромметил)бензоата (93) и 4-(трифторметокси)фенилбороновой кислоты (44) давало метилбензоат 94. Его восстанавливали LiAlH4 в спирт 95, который давал бромид 96 при обработке PBr3. Сочетание данного бромида со спиртом 41 затем давало соединение 17 таблицы 1.
На схеме 14, показанной на фигуре 16, реагенты и условия были следующими: (i) НС≡CTMS, Et3N, ДМФА, CuI, PdCl2(PPh3)2 в атмосфере N2, 50°С, 18 час, затем TBAF, ТГФ, 0-20°С, 2 час; (ii) 70 или 98, Et3N, ДМФА, CuI, PdCl2(PPh3)2 в атмосфере N2, от 20 до 50°С, 0,5 час. Сочетание Соногашира бромида 59 (см. схему 5) с этинил-TMS в присутствии Et3N, CuI и PdCl2(PPh3)2 с последующим десилилированием при помощи TBAF давало ацетилен 97, который аналогично сочетали с 1-иод-4-(трифторметокси)бензолом (70) или 5-бромо-2-(трифторметил)пиридином (98), получая при этом соответственно соединения 18 и 25 таблицы 1.
На схеме 15, показанной на фигуре 17, реагенты и условия были следующими: (i) 44, диоксан, 2 М К2СО3, Pd(dppf)Cl2 в атмосфере N2, кипячение с обратным холодильником, 1 час; (ii) DIBAL-H, толуол, от -78 до 20°С, 1 час; (iii) PBr3, Et2O, 0-20°С, 1 час; (iv) NaH, ДМФА, от -78 до 0°С, 1 час. Бромид 99 подвергали сочетанию Сузуки с бороновой кислотой 44 (см. схему 3), получая при этом сложный эфир 100, который восстанавливали с помощью DIBAL-H в толуоле с получением спирта 101. Бромирование 101 с помощью PBr3 давало соединение 102, которое подвергали с помощью NaH сочетанию со спиртом 41, получая при этом соединение 19 таблицы 1.
На схеме 16, показанной на фигуре 18, реагенты и условия были следующими: (i) трифосген, Et3N, 0-20°С, 105 мин; (ii) ТГФ, 20°С, 2 час. Спирт 41 обрабатывали трифосгеном и сырой карбонилхлорид 103 подвергали непосредственно реакции с 1-[4-(трифторметокси)фенил]пиперазином (104), получая при этом соединение 20 таблицы 1.
На схеме 17, показанной на фигуре 19, реагенты и условия были следующими: (i) 56, 2 М Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 89°С, 2 час; (ii) NBS, PPh3, CH2Cl2, 20°С, 3 час; (iii) NaH, ДМФА, 0-20°С, 2,5 час. Сочетание Сузуки бромида 105 с бороновой кислотой 56 (см. схему 4) давало спирт 106, который бромировали NBS/PPh3, получая при этом соединение 107. Его сочетали с помощью NaH со спиртом 41 с получением соединения 22 таблицы 1.
На схеме 18, показанной на фигуре 20, реагенты и условия были следующими: (i) NaH, ДМФА, -42°С, 1 час; (ii) 44, 2 М К2СО3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, кипячение с обратным холодильником, 0,5 час. Реакция при низкой температуре спиртового оксазина 41 и 3-(бромметил)-6-хлорпиридазина (108) (получен посредством радикальной реакции бромирования 3-хлор-6-метилпиридазина, как описано Ohshita J., EP 1555259, 2005) давала хлорид 109, сочетанием Сузуки которого с бороновой кислотой 44 (см. схему 3) получали соединение 24 таблицы 1.
На схеме 19, показанной на фигуре 21, реагенты и условия были следующими: (i) 2 М Na2CO3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, 89°С, 200 мин; (ii) NBS, PPh3, CH2Cl2, 20°С, 3 час; (iii) 41, NaH, ДМФА, 0-20°С, 135 мин. Сочетание Сузуки бромида 110 с бороновой кислотой 34 давало спирт 111, который бромировали с получением соединения 112, которое затем сочетали со спиртом 41, получая при этом соединение 26 таблицы 1.
На схеме 20, показанной на фигуре 22, реагенты и условия были следующими: (i) водный NaOAc, AcOH, 100°С, 15 час; (ii) (СН3)2СН(СН2)2ONO, ТГФ, кипячение с обратным холодильником, 20 час; (iii) LiAlH4, Et2O, кипячение с обратным холодильником, 2 час; (iv) PBr3, Et2O, 0°С, 2 час; (v) 41, NaH, ДМФА, 0°С, 2 час. Реакция этил-(2Е)-2-циано-3-этокси-2-пропеноата 113 и гидразина 114 давала пиразолкарбоксилат 115, который деаминировали изоамилнитритом. Образовавшийся карбоксилат 116 восстанавливали в спирт 117, который затем бромировали PBr3, получая при этом соединение 118. Это соединение с помощью NaH подвергали сочетанию со спиртом 41, получая при этом соединение 27 таблицы 1.
На схеме 21, показанной на фигуре 23, реагенты и условия были следующими: (i) NBS, AIBN, CCl4, 60°С, 3 час; (ii) NaH, ДМФА, от -78 до 0°С, 0,5 час; (iii) 44, 2 М К2СО3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, кипячение с обратным холодильником, 0,5 час. Бромирование 5-бром-2-метилпиримидина (119) давало соединение 120, которое с помощью NaH сочетали со спиртом 41, получая при этом бромид 121. Сочетание Сузуки 121 с бороновой кислотой 44 (см. схему 3) давало соединение 29 таблицы 1.
На схеме 22, показанной на фигуре 24, реагенты и условия были следующими: (i) NaH, ДМФА, 0°С, 1 час; (ii) 44, 2 М К2СО3, толуол, EtOH, Pd(dppf)Cl2 в атмосфере N2, кипячение с обратным холодильником, 0,5 час. Сочетание с помощью NaH спирта 41 и 1-бром-4-(3-бром-1-пропинил)бензола (122) (получен в две стадии из 1-бром-4-иодбензола и пропаргилового спирта, как описано в WO 9524400) давало бромид 123, который сочетали реакцией Сузуки с бороновой кислотой 44, получая при этом соединение 30 таблицы 1.
На схеме 23, показанной на фигуре 25, реагенты и условия были следующими: (i) К2СО3, 18-краун-6, ТГФ, CH2Cl2, кипячение с обратным холодильником, 18 час; (ii) LiAlH4, Et2O, 0-20°С, 0,5 час; (iii) PBr3, CH2Cl2, 0-20°С, 1 час; (iv) 41, NaH, ДМФА, от -78 до 0°С, 1 час. Реакция Виттига альдегида 125 и фосфониевой соли 124 давала сложный эфир 126, который восстанавливали LiAlH4, получая при этом спирт 127. Бромирование 127 PBr3 давало соединение 128, сочетание которого при помощи NaH со спиртом 41 давало соединение 31 таблицы 1.
На схеме 24, показанной на фигуре 26, реагенты и условия были следующими: (i) НС≡CTMS, Et3N, ДМФА, CuI, PdCl2(PPh3)2 в атмосфере N2, 20°С, 0,5-18 час, затем TBAF, ТГФ, 0-20°С, 2 час; (ii) 70, Et3N, ДМФА, CuI, PdCl2(PPh3)2 в атмосфере N2, 20°С, 0,5 час. Сочетания Соногашира иодида 43 (см. схему 2) или бромида 52 (см. схему 4) с этинил-TMS в присутствии Et3N, CuI и PdCl2(PPh3)2 с последующим дебензилированием при помощи TBAF давали ацетилены 129 или 130, соответственно, которые аналогично сочетали с 1-иод-4-(трифторметокси)бензолом (70), получая при этом соединения 32 и 33 таблицы 1.
Пример 2. Способы получения
А. Синтез (6S)-6-{[2'-хлор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (1) способом схемы 1

Перемешиваемую смесь 4-(гидроксиметил)фенилбороновой кислоты (34) (308 мг, 2,03 ммоль) и Pd(dppf)Cl2 (191 мг, 0,261 ммоль) в толуоле (22 мл) и EtOH (11 мл) дегазировали в течение 8 мин (вакуумный насос) и затем вводили N2. Добавляли водный раствор 2 M Na2CO3 (4,4 мл, 8,8 ммоль) посредством шприца и перемешиваемую смесь снова дегазировали в течение 8 мин и затем вводили N2. При помощи шприца добавляли 2-хлор-1-иод-4-(трифторметокси)бензол (35) (585 мг, 1,81 ммоль) и образовавшуюся смесь перемешивали при 88°С в течение 60 мин. Охлажденную смесь затем разбавляли водным NaHCO3 (100 мл) и экстрагировали CH2Cl2 (5×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-50% CH2Cl2/петролейный эфир сначала давало предварительные (не содержащие требуемое соединение) фракции, и затем дополнительное элюирование смесью 50% CH2Cl2/петролейный эфир давало 2'-хлор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метанол (37) (537 мг, 98%) в виде белого твердого вещества: т. пл. (пентан) 38-39°С;1H ЯМР (CDCl3) δ 7,46 (ушир. д, J=8,2 Гц, 2 H), 7,42 (дт, J=8,3, 2,0 Гц, 2 H), 7,37 (ушир. с, 1 H), 7,36 (д, J=8,5 Гц, 1 H), 7,19 (м, 1 H), 4,77 (д, J=5,9 Гц, 2 H), 1,70 (т, J=5,9 Гц, 1 H);HREIMS (МС высокого разрешения с ионизацией электрораспылением), вычислено для C14H10ClF3O2 m/z (М+) 304,0292, 302,0321, найдено 304,0294, 302,0317.
HBr в AcOH (5 мл 33% масс./масс. раствора) добавляли к раствору спирта 37 (618 мг, 2,04 ммоль) в ледяной AcOH (2,5 мл) и смесь перемешивали при комнатной температуре в течение 11 час. Образовавшийся оранжевый раствор медленно добавляли к смеси лед/вода (50 мл) при перемешивании и затем смесь экстрагировали пентаном (6×50 мл). Экстракты промывали смесью лед-вода (50 мл) и затем упаривали, получая при этом 4-(бромметил)-2'-хлор-4'-(трифторметокси)-1,1'-бифенил (39) (743 мг, 100%) в виде масла;1H ЯМР (CDCl3) δ 7,47 (дт, J=8,3, 1,9 Гц, 2 H), 7,39 (дт, J=8,3, 1,9 Гц, 2 H), 7,37 (м, 1 H), 7,35 (д, J=8,5 Гц, 1 H), 7,19 (м, 1 H), 4,55 (с, 2 H); HREIMS, вычислено для C14H9BrClF3O m/z (М+) 367,9427, 365,9457, 363,9477, найдено 367,9428, 365,9453, 363,9485.
Перемешиваемый раствор (6S)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-6-ола (41) (описан в патенте США No. 5668127, получен посредством 4 стадий, исходя из 2,4-динитроимидазола и трет-бутилдиметилсилил-(S)-глицидилового эфира) (342 мг, 1,85 ммоль) и бромида 39 (741 мг, 2,03 ммоль) в безводном ДМФА (7 мл) в атмосфере N2 при 0°С обрабатывали 60% NaH (111 мг, 2,78 ммоль), затем быстро дегазировали и герметизировали в атмосфере N2. После перемешивания при комнатной температуре в течение 3 час реакционную смесь охлаждали (СО2/ацетон), гасили смесью лед/водный NaHCO3 (20 мл), добавляли к насыщенному раствору соли (80 мл) и экстрагировали CH2Cl2 (6×80 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле с элюированием CH2Cl2, получая при этом 1 (694 мг, 80%) в виде бледно-желтого твердого вещества: т. пл. (CH2Cl2/пентан) 80-82°С;1H ЯМР (CDCl3) δ 7,45-7,35 (м, 6 H), 7,34 (д, J=8,5 Гц, 1 H), 7,20 (м, 1 H), 4,79 (д, J=12,0 Гц, 1 H), 4,68 (д, J=12,1 Гц, 1 H), 4,65 (ддд, J=12,1, 2,9, 2,5 Гц, 1 H), 4,37 (ушир. д, J=11,8 Гц, 1 H), 4,24-4,12 (м, 3 H).Анализ (C20H15ClF3N3O5) C, H, N.
B. Синтез (6S)-6-{[3'-фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (2) способом схемы 1
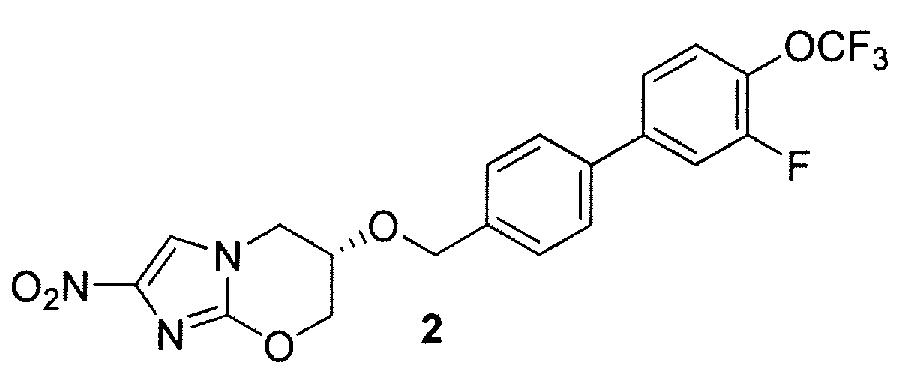
Сочетание Сузуки 4-(гидроксиметил)фенилбороновой кислоты (34) и 4-бром-2-фтор-1-(трифторметокси)бензола (36), как в примере 2A, в течение 2,5 час с последующей хроматографией продукта на силикагеле с элюированием смесью 0-40% CH2Cl2/петролейный эфир (предварительные фракции) и затем смесью 40% CH2Cl2/петролейный эфир давало 3'-фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метанол (38) (73%) в виде кремового твердого вещества: т. пл. (CH2Cl2/пентан) 70-71°С;1H ЯМР (CDCl3) δ 7,54 (дт, J=8,3, 1,8 Гц, 2 H), 7,46 (ушир. д, J=8,2 Гц, 2 H), 7,41 (ушир. д, J=1 1,2 Гц, 1 H), 7,40-7,32 (м, 2 H), 4,76 (д, J=5,9 Гц, 2 H), 1,69 (т, J=5,9 Гц, 1 H); HREIMS, вычислено для C14H10F4O2 m/z (М+) 286,0617, найдено 286,0616.
Бромирование спирта 38, как в примере 2А, в течение 6 час давало 4-(бромметил)-3'-фтор-4'-(трифторметокси)-1,1'-бифенил (40) (100%) в виде кремового твердого вещества, которое применяли непосредственно в следующей стадии;1H ЯМР (CDCl3) δ 7,52 (дт, J=8,5, 2,2 Гц, 2 H), 7,48 (дт, J=8,5, 2,2 Гц, 2 H), 7,43-7,32 (м, 3 H), 4,54 (с, 2 H);HRAPCIMS (масс-спектр высокого разрешения с химической ионизацией при атмосферном давлении), вычислено для C14H9F4O m/z [M-Br]+ 269,0584, найдено 269,0572.
Реакция бромида 40 (0,99 эквив.) со спиртом 41, как в примере 2А, с последующей хроматографией продукта на силикагеле с элюированием смесью 0-2% EtOAc/CH2Cl2 (предварительные фракции) и затем 2% EtOAc/CH2Cl2 давала 2 (76%) в виде кремового твердого вещества: т. пл. (CH2Cl2/пентан) 169-171°С;1H ЯМР (CDCl3) δ 7,54 (дт, J=8,3, 1,8 Гц, 2 H), 7,43-7,32 (м, 6 H), 4,78 (д, J=12,0 Гц, 1 H), 4,67 (д, J=11,9 Гц, 1 H), 4,64 (ддд, J=12,1, 3,7, 2,1 Гц, 1 H), 4,37 (дд, J=12,1, 1,3 Гц, 1 H), 4,23-4,12 (м, 3 H). Анализ (C20H15F4N3O5) C, H, N.
C. Синтез (6S)-2-нитро-6-{[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (3) способом схемы 2

Реакция спирта 41 с 4-иодбензилбромидом (42) и NaH в ДМФА при комнатной температуре в течение 2 час давала (6S)-6-[(4-иодбензил)окси]-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (43) (описано получение в патенте США No. 6087358 посредством такой же процедуры) (97%) в виде бледно-желтого твердого вещества: т. пл. (EtOAc/петролейный эфир) 210-212°С;1H ЯМР [(CD3)2SO] δ 8,01 (с, 1 H), 7,71 (дт, J=8,3, 2,0 Гц, 2 H), 7,13 (ушир. д, J=8,3 Гц, 2 H), 4,67-4,60 (м, 2 H), 4,59 (д, J=2,2 Гц, 1 H), 4,46 (д, J=12,0 Гц, 1 H), 4,27-4,19 (м, 3 H). Анализ (C13H12IN3O4) C, H, N.
Сочетание Сузуки иодида 43 и 4-(трифторметокси)фенилбороновой кислоты (44), как ниже в примере 2D, давало 3 (86%) в виде кремового твердого вещества: т. пл. (CH2Cl2/гексан) 199-201°С;1H ЯМР [(CD3)2SO] δ 8,03 (с, 1 H), 7,78 (дт, J=8,8, 2,6 Гц, 2 H), 7,66 (ушир. д, J=8,3 Гц, 2 H), 7,43 (ушир. т, J=8,5 Гц, 4 H), 4,72 (д, J=12,2 Гц, 1 H), 4,70-4,66 (м, 2 H), 4,49 (д, J=1 1,9 Гц, 1 H), 4,31-4,21 (м, 3 H). Анализ (C20H16F3N3O5) C, H, N.
D. Синтез (6S)-2-нитро-6-({5-[4-(трифторметокси)фенил]-2-пиразинил}метокси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (4) способом схемы 3

Et3N (4,17 мл, 29,9 ммоль) и мезилхлорид (1,57 мл, 20,3 ммоль) добавляли к раствору (5-хлор-2-пиразинил)метанола (45) (получен хлорированием и восстановлением 5-гидрокспиразин-2-карбоновой кислоты, как описано Kiener et al., 1994) (1,443 г, 9,98 ммоль) в безводном ТГФ (20 мл) при 0°С. Смесь перемешивали при 0°С в течение 0,5 час, затем распределяли между EtOAc и водой. Органическую фракцию сушили (MgSO4) и растворитель удаляли при пониженном давлении, получая при этом сырой мезилат. Мезилат растворяли в ацетоне (40 мл), добавляли иодид натрия (7,5 г, 50 ммоль) и смесь кипятили с обратным холодильником в течение 1 час. Растворитель удаляли при пониженном давлении и остаток распределяли между EtOAc и водой. Органическую фракцию концентрировали при пониженном давлении и остаток хроматографировали на силикагеле (элюирование CH2Cl2), получая при этом 2-хлор-5-(иодметил)пиразин (46) (1,54 г, 61%), который применяли сразу вследствие его нестабильности.
NaH (60% масс./масс., 0,36 г, 9,0 ммоль) добавляли к спиртовому производному оксазина 41 (0,93 г, 5,02 ммоль) и иоду 46 (1,54 г, 6,05 ммоль) в ДМФА (10 мл) при -78°С. Смесь перемешивали при 0°С в течение 1 час и затем гасили льдом. Добавляли EtOAc (200 мл), органический слой сушили (MgSO4) и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле, элюируя с градиентом смеси 0-5% MeOH/EtOAc, получая при этом (6S)-6-[(5-хлор-2-пиразинил)метокси]-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (47) (1,015 г, 65%) в виде белого твердого вещества: т. пл. 181-183°С;1H ЯМР [(CD3)2SO] δ 8,76 (д, J=1,4 Гц, 1 H), 8,50 (д, J=1,4 Гц, 1 H), 8,02 (с, 1 H), 4,85 (д, J=13,7 Гц, I H), 4,81 (д, J=13,7 Гц, 1 H), 4,70 (дт, J=12,1, 2,6 Гц, 1 H), 4,49 (ушир. д, J=12,0 Гц, 1 H), 4,29-4,38 (м, 2 H), 4,25 (дд, J=13,5, 3,3 Гц, 1 H).Анализ (C11H10ClN5O4) C, H, N.
Перемешиваемую смесь хлорида 47 (0,100 г, 0,32 ммоль) и 4-(трифторметокси)фенилбороновой кислоты (44) (0,080 г, 0,39 ммоль) в водном K2CO3 (1 мл, 2 M), EtOH (3 мл) и толуоле (5 мл) продували N2 в течение 5 мин. Добавляли Pd(dppf)Cl2 (5 мг, 6,25 мкмоль) и смесь кипятили с обратным холодильником в атмосфере N2 в течение 0,5 час. Раствор распределяли между EtOAc и водой и органический слой сушили (MgSO4) и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле, сначала элюируя EtOAc для удаления предварительных фракций, и затем элюировали смесью EtOAc:MeOH (95:5), получая при этом 4 (0,115 г, 82%) в виде белого твердого вещества: т. пл. 182-184°С;1H ЯМР [(CD3)2SO] δ 9,23 (д, J=1,4 Гц, 1 H), 8,72 (д, J=1,4 Гц, 1 H), 8,25 (д, J=8,9 Гц, 2 H), 8,03 (с, 1 H), 7,53 (д, J=8,9 Гц, 2 H), 4,89 (д, J=13,3 Гц, 1 H), 4,85 (д, J=13,3 Гц, 1 H), 4,74 (дт, J=12,0, 2,6 Гц, 1 H), 4,52 (ушир. д, J=11,9 Гц, 1 H), 4,33-4,43 (м, 2 H), 4,27 (дд, J=13,5, 3,2 Гц, 1 H).Анализ (C18H14F3N5O5) C, H, N.
E. Синтез (6S)-2-нитро-6-({6-[4-(трифторметокси)фенил]-3-пиридинил}метокси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (6) способом схемы 4

NaH (60% масс./масс., 0,584 г, 14,6 ммоль) добавляли к раствору спиртового оксазина 41 (2,073 г, 11,2 ммоль) и 2-хлор-5-(хлорметил)пиридина (48) (2,0 г, 12,3 ммоль) в безводном ДМФА (40 мл) при 5°С. Образовавшуюся смесь перемешивали при комнатной температуре в течение 16 час и затем гасили водой (150 мл). Осадок отделяли фильтрованием, промывали водой и сушили, получая при этом (6S)-6-[(6-хлор-3-пиридинил)метокси]-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (49) (3,39 г, 97%) в виде бледно-желтого твердого вещества: т. пл. 191-193°С;1H ЯМР [(CD3)2SO] δ 8,37 (д, J=2,3 Гц, 1 H), 8,02 (с, 1 H), 7,79 (дд, J=8,3, 2,4 Гц, 1 H), 7,51 (ушир. д, J=8,2 Гц, 1 H), 4,74 (д, J=12,4 Гц, 1 H), 4,69-4,64 (м, 2 H), 4,47 (д, J=11,8 Гц, 1 H), 4,29-4,21 (м, 3 H). HRESIMS, вычислено для C12H12ClN4O4 m/z [М+H]+ 313,0513, 311,0542, найдено 313,0518, 311,0545.
Хлорид 49 (1,0 г, 3,22 ммоль) и 4-(трифторметокси)фенилбороновую кислоту (44) (0,788 г, 3,82 ммоль) суспендировали в DME (50 мл) и добавляли водный раствор K2CO3 (2 M, 10 мл). Смесь продували N2 и затем обрабатывали Pd(dppf)Cl2 (50 мг, 0,068 ммоль) и перемешивали при 85°С в атмосфере N2 в течение 1 дня, проводя мониторинг посредством МС. Добавляли дополнительную кислоту 44 (0,150 г, 0,728 ммоль) и смесь перемешивали при 85°С в атмосфере N2 в течение 1 дня. Образовавшуюся смесь разбавляли водой (50 мл) и экстрагировали EtOAc (3×100 мл). Высушенные (MgSO4) органические слои адсорбировали на силикагеле и хроматографировали на силикагеле, элюируя EtOAc. Растирание продукта в Et2O давало 6 (0,942 г, 67%) в виде белого порошка: т. пл. 217-219°С;1H ЯМР [(CD3)2SO] δ 8,63 (д, J=1,7 Гц, 1 H), 8,20 (дт, J=8,9, 2,1 Гц, 2 H), 8,03 (с, 1 H), 7,99 (дд, J=8,2, 0,5 Гц, 1 H), 7,84 (дд, J=8,2, 2,2 Гц, 1 H), 7,47 (дд, J=8,8, 0,8 Гц, 2 H), 4,77 (д, J=12,3 Гц, 1 H), 4,71-4,68 (м, 2 H), 4,49 (д, J=11,7 Гц, 1 H), 4,31-4,26 (м, 3 H).Анализ (C19H15F3N4O5) C, H, N. Чистота по ВЭЖХ: 98,9%.
F. Синтез (6S)-6-{[6-(4-фторфенил)-3-пиридинил]метокси}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (5) способом схемы 4

Раствор (6-бром-3-пиридинил)метанола (50) (2,503 г, 13,3 ммоль) и трифенилфосфина (4,026 г, 15,4 ммоль) в безводном CH2Cl2 (100 мл) осторожно обрабатывали перекристаллизованным N-бромсукцинимидом (2,732 г, 15,4 ммоль) (охлаждение водяной баней) и смесь перемешивали при комнатной температуре в течение 3,5 час. Образовавшийся раствор концентрировали и затем добавляли к избыточному петролейному эфиру с верха колонки силикагеля (100 г в петролейном эфире), споласкивая минимальным дополнительным количеством CH2Cl2. Элюирование петролейным эфиром сначала давало предварительные фракции, и затем дальнейшее элюирование смесью 15-25% Et2O/пентан давало чистый 2-бром-5-(бромметил)пиридин (51) (Schubert et al., 1999) (3,045 г, 91%) в виде слезоточивого белого твердого вещества, которое применяли непосредственно в следующей стадии;1H ЯМР (CDCl3) δ 8,38 (д, J=2,5 Гц, 1 H), 7,59 (дд, J=8,2, 2,6 Гц, 1 H), 7,48 (д, J=8,2 Гц, 1 H), 4,42 (с, 2 H).
Раствор спиртового оксазина 41 (2,224 г, 12,0 ммоль) и бромида 51 (3,045 г, 12,1 ммоль) в безводном ДМФА (46 мл) в атмосфере N2 при 0°С обрабатывали 60% NaH (639 мг, 16,0 ммоль), затем быстро дегазировали и снова вводили N2. После перемешивания при комнатной температуре в течение 2,5 час, реакционную смесь охлаждали (CO2/ацетон), гасили смесью лед/водный NaHCO3(50 мл), добавляли к насыщенному раствору соли (250 мл) и экстрагировали CH2Cl2 (12×200 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% MeOH/CH2Cl2 сначала давало предварительные фракции, и затем дальнейшее элюирование смесью 1-1,5% MeOH/CH2Cl2 давало (6S)-6-[(6-бром-3-пиридинил)метокси]-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (52) (3,739 г, 88%) в виде кремового твердого вещества: т. пл. (MeOH/CH2Cl2/гексан) 200-203°С;1H ЯМР [(CD3)2SO] δ 8,35 (дд, J=2,3, 0,4 Гц, 1 H), 8,02 (с, 1 H), 7,69 (дд, J=8,2, 2,5 Гц, 1 H), 7,63 (дд, J=8,1, 0,5 Гц, 1 H), 4,72-4,62 (м, 3 H), 4,47 (ушир. д, J=11,8 Гц, 1 H), 4,31-4,19 (м, 3 H).Анализ (C12H11BrN4O4) C, H, N. Чистота по ВЭЖХ: 100%.
Бромид 52 (0,100 г, 0,28 ммоль) и 4-фторфенилбороновую кислоту (57) (69 мг, 0,49 ммоль) суспендировали в смеси толуол/EtOH (5 мл/2 мл) и добавляли водный раствор K2CO3 (2 M, 1 мл). Перемешиваемую смесь продували N2 и затем обрабатывали Pd(dppf)Cl2 (5 мг, 6,83 мкмоль) и нагревали при кипячении с обратным холодильником в атмосфере N2 в течение 20 мин. Образовавшуюся смесь разбавляли водой (10 мл) и экстрагировали EtOAc (3×15 мл). Высушенные (MgSO4) органические слои адсорбировали на силикагеле и хроматографировали на силикагеле, элюируя EtOAc. Растирание продукта в Et2O давало 5 (90 мг, 86%): т. пл. 194-196°С;1H ЯМР [(CD3)2SO] δ 8,60 (д, J=1,7 Гц, 1 H), 8,14-8,10 (м, 2 H), 8,03 (с, 1 H), 7,95 (дд, J=8,2, 0,6 Гц, 1 H), 7,81 (дд, J=8,2, 2,3 Гц, 1 H), 7,31 (ушир. т, J=8,9 Гц, 2 H), 4,75 (д, J=12,2 Гц, 1 H), 4,71-4,68 (м, 2 H), 4,49 (д, J=11,7 Гц, 1 H), 4,31-4,26 (м, 3 H).Анализ (C18H15FN4O4. 1,5 Н2О) C, N, F. H: вычислено 4,57; найдено 3,87. Чистота по ВЭЖХ: 99,4%.
G. Синтез (6S)-6-({6-[3-фтор-4-(трифторметокси)фенил]-3-пиридинил}метокси)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (21) способом схемы 4
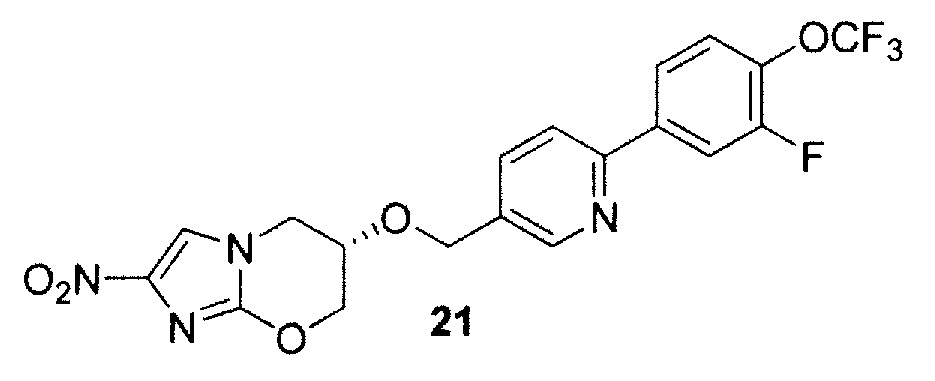
Перемешиваемую смесь бромида 52 (см. пример 2F) (502 мг, 1,41 ммоль), 3-фтор-4-(трифторметокси)фенилбороновой кислоты (56) (450 мг, 2,01 ммоль) и Pd(dppf)Cl2(130 мг, 0,178 ммоль) в толуоле (20 мл) и EtOH (10 мл) дегазировали в течение 12 мин (вакуумный насос) и затем вводили N2. Посредством шприца добавляли водный раствор 2 М Na2CO3 (3,8 мл, 7,6 ммоль) и перемешиваемую смесь снова дегазировали в течение 15 мин и затем вводили N2. Образовавшуюся смесь перемешивали при 90°С в течение 2 час и затем охлаждали, разбавляли водным NaHCO3 (100 мл) и экстрагировали CH2Cl2 (6×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-0,5% МеОН/CH2Cl2 сначала давало предварительные фракции, и затем дальнейшее элюирование смесью 0,5% МеОН/CH2Cl2 давало 21 (573 мг, 89%) в виде кремового твердого вещества: т. пл. (CH2Cl2/пентан) 187-189°С;1H ЯМР (CDCl3) δ 8,62 (д, J=1,5 Гц, 1 H), 7,90 (дд, J=11,3, 2,1 Гц, 1 H), 7,78 (ддд, J=8,6, 2,0, 1,3 Гц, 1 H), 7,75 (дд, J=8,2, 2,2 Гц, 1 H), 7,71 (дд, J=8,2, 0,8 Гц, 1 H), 7,41 (с, 1 H), 7,40 (ддкв, J=8,7, 7,6, 1,2 Гц, 1 H), 4,80 (д, J=12,0 Гц, 1 H), 4,70 (д, J=11,8 Гц, 1 H), 4,68 (ддд, J=12,2, 3,5, 2,3 Гц, 1 H), 4,40 (дд, J=12,2, 1,1 Гц, 1 H), 4,25 (дд, J=13,3, 4,5 Гц, 1 H), 4,22-4,15 (м, 2 H). Анализ (C19H14F4N4O5) C, H, N.
Н. Синтез (6S)-6-({6-[3-хлор-4-(трифторметокси)фенил]-3-пиридинил}метокси)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (28) способом схемы 4

Охлажденную льдом смесь 98% H2SO4 (0,75 мл) и воды (2,25 мл) добавляли к 3-хлор-4-(трифторметокси)анилину (53) (1,00 г, 4,73 ммоль) и образовавшуюся соль дробили (с применением стеклянного стержня) и охлаждали на ледяной бане. По каплям добавляли раствор NaNO2 (359 мг, 5,20 ммоль) в холодной воде (0,75 мл, затем 0,25 мл) и смесь перемешивали при 0°С в течение 12 мин. Добавляли раствор мочевины (42,6 мг, 0,709 ммоль) в холодной воде (0,25 мл) и смесь перемешивали при 0°С в течение 3 мин. Наконец, медленно добавляли раствор KI (1,65 г, 9,94 ммоль) в холодной воде (1,6 мл, затем 0,2 мл) и смесь перемешивали при комнатной температуре в течение 10 мин и затем при 52°С в течение 2 час. Образовавшуюся охлажденную смесь разбавляли смесью лед-вода (45 мл) и экстрагировали CH2Cl2 (4×50 мл). Экстракты последовательно промывали водным раствором Na2SO3 (30 мл 0,5% раствора) и затем водой (40 мл) и в заключение осторожно концентрировали при пониженном давлении и при 17°С. Образовавшееся масло хроматографировали на силикагеле с элюированием пентаном, получая при этом 2-хлор-4-иод-1-(трифторметокси)бензол (54) (1,24 г, 81%) в виде бесцветного масла (белого твердого вещества при замораживании);1H ЯМР (CDCl3) δ 7,82 (д, J=2,1 Гц, 1 H), 7,61 (дд, J=8,6, 2,1 Гц, 1 H), 7,05 (дкв, J=8,6, 2,0 Гц, 1 H). HRAPCIMS, вычислено для C7H3ClF3IO m/z (М+) 323,8834, 321,8864, найдено 323,8834, 321,8861.
Триизопропилборат (0,76 мл, 3,29 ммоль) и иодид 54 (815 мг, 2,53 ммоль) последовательно добавляли с помощью шприца к смеси безводного толуола (4 мл) и безводного перегнанного ТГФ (1 мл) в атмосфере N2 и смесь охлаждали до -78°С. На протяжении 75 мин по каплям добавляли н-бутиллитий (1,08 мл 2,5 M раствора в гексанах, 2,70 ммоль) к перемешиваемому раствору (при -78°С) и смесь перемешивали при -78°С в течение дополнительных 3 час и затем медленно нагревали до -20°С (на протяжении 1,5 час). Добавляли 2 н. HCl (2,6 мл) и смесь перемешивали при комнатной температуре в течение 30 мин и затем разбавляли водой (40 мл) и экстрагировали EtOAc (5×50 мл). Экстракты промывали насыщенным раствором соли (50 мл) и затем упаривали досуха. Остаток растирали в пентане (~ 3-4 мл), охлаждали до -78°С и быстро фильтровали в холодном состоянии (промывание пентаном, охлажденным до -78°С), получая при этом 3-хлор-4-(трифторметокси)фенилбороновую кислоту (55) (459 мг, 76%) в виде белого твердого вещества (смесь 1:1 тримерного бороксина и бороновой кислоты по данным ЯМР): т. пл. 202-204°С;1H ЯМР (CDCl3) δ 8,26 (д, J=1,5 Гц, 3 H, бороксин), 8,12 (дд, J=8,2, 1,5 Гц, 3 H, бороксин), 7,85 (д, J=1,5 Гц, 1 H, борная кислота), 7,65 (дд, J=8,2, 1,6 Гц, 1 H, борная кислота), 7,48 (дкв, J=8,2, 1,5 Гц, 3 H, бороксин), 7,35 (дкв, J=8,2, 1,5 Гц, 1 H, борная кислота), 4,57 (с, 2 H, борная кислота).
Сочетание Сузуки бромида 52 и бороновой кислоты 55, как в примере 2G, с последующей хроматографией продукта на силикагеле с элюированием смесью 0-0,5% MeOH/CH2Cl2 (предварительные фракции) и затем смесью 0,5% MeOH/CH2Cl2 давало 28 (90%) в виде кремового твердого вещества: т. пл. (CH2Cl2/пентан) 169-171°С;1H ЯМР (CDCl3) δ 8,63 (ушир. д, J=1,3 Гц, 1 H), 8,15 (д, J=2,2 Гц, 1 H), 7,91 (дд, J=8,6, 2,2 Гц, 1 H), 7,75 (дд, J=8,2, 2,1 Гц, 1 H), 7,71 (дд, J=8,2, 0,8 Гц, 1 H), 7,44-7,39 (м, 2 H), 4,80 (д, J=12,0 Гц, 1 H), 4,70 (д, J=1 1,8 Гц, 1 H), 4,68 (ддд, J=12,3, 3,5, 2,2 Гц, 1 H), 4,39 (дд, J=12,2, 1,2 Гц, 1 Н), 4,25 (дд, J=13,3, 4,5 Гц, 1 H), 4,22-4,15 (м, 2 H).Анализ (C19H14ClF3N4O5) C, H, N.
I. Синтез (6S)-2-нитро-6-({5-[4-(трифторметокси)фенил]-2-пиридинил}метокси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (7) способом схемы 5

NaH (0,525 г, 13,1 ммоль, 60% в минеральном масле) добавляли к раствору спирта 41 (1,872, 10,1 ммоль) и 5-бром-2-(хлорметил)пиридина (58) (получен хлорированием (5-бром-2-пиридинил)метанола, как описано van den Heuvel et al., 2004) (2,5 г, 12,1 ммоль) в безводном ДМФА (40 мл) при 5°С. Образовавшуюся смесь перемешивали при комнатной температуре в течение 2 час и затем гасили водой (300 мл). Осадок отделяли фильтрованием, промывали водой и сушили, получая при этом (6S)-6-[(5-бром-2-пиридинил)метокси]-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (59) (3,087 г, 86%) в виде бледно-коричневого твердого вещества: т. пл. 171-173°С;1H ЯМР [(CD3)2SO] δ 8,65 (дд, J=2,3, 0,4 Гц, 1 H), 8,04 (дд, J=8,4, 2,4 Гц, 1 H), 8,02 (с, 1 H), 7,35 (дд, J=8,4, 0,4 Гц, 1 H), 4,72-4,66 (м, 3 H), 4,49 (ушир. д, J=12,0 Гц, 1 H), 4,35-4,21 (м, 3 H). Анализ (C12H11BrN4O4) C, H, N. Чистота по ВЭЖХ: 99,4%.
Бромид 59 (0,100 г, 0,28 ммоль) и 4-(трифторметокси)фенилбороновую кислоту (44) (0,075 г, 0,366 ммоль) суспендировали в смеси толуол/EtOH (5 мл/2 мл) и добавляли водный раствор K2CO3 (1 мл; 2 M). Перемешиваемую смесь продували N2 и затем обрабатывали Pd(dppf)Cl2 (5 мг, 6,83 мкмоль) и нагревали при кипячении с обратным холодильником в атмосфере N2 в течение 30 мин. Образовавшуюся смесь разбавляли водой (10 мл) и экстрагировали EtOAc (3×15 мл). Высушенные (MgSO4) органические слои абсорбировали на силикагеле и хроматографировали на силикагеле, элюируя смесью 5% MeOH/EtOAc. Растирание продукта в Et2O давало 7 (97 мг, 79%): т. пл. 157-159°С;1H ЯМР [(CD3)2SO] δ 8,85 (д, J=2,0 Гц, 1 H), 8,11 (дд, J=8,1, 2,4 Гц, 1 H), 8,03 (с, 1 H), 7,85 (дт, J=8,8, 2,0 Гц, 2 H), 7,48 (т, J=7,7 Гц, 3 H), 4,82 (д, J=13,2 Гц, 1 H), 4,78 (д, J=13,2 Гц, 1 H), 4,72 (дт, J=12,0, 2,6 Гц, 1 H), 4,51 (д, J=12,0 Гц, 1 H), 4,37-4,24 (м, 3 H).Анализ (C19H15F3N4O5) C, H, N, F. Чистота по ВЭЖХ: 100%.
J. Синтез (6S)-2-нитро-6-({4-[5-(трифторметил)-2-пиридинил]бензил}окси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (8) способом схемы 6

Реакция спиртового оксазина 41 (5,00 г, 27,0 ммоль) с 4-бромбензилбромидом (60) (7,62 г, 30,5 ммоль) и NaH (60% масс./масс., 1,40 г, 35,0 ммоль) в ДМФА (100 мл) в течение 2 час при комнатной температуре давала (6S)-6-[(4-бромбензил)окси]-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (61) (8,368 г, 88%) в виде бледно-желтого твердого вещества: т. пл. (Et2O) 188-190°С;1H ЯМР [(CD3)2SO] δ 8,01 (с, 1 H), 7,54 (дт, J=8,4, 2,2 Гц, 2 H), 7,13 (дт, J=8,5, 2,2 Гц, 2 H), 4,67-4,62 (м, 2 H), 4,61 (д, J=12,2 Гц, 1 H), 4,46 (д, J=12,0 Гц, 1 H), 4,28-4,19 (м, 3 H). Анализ (C13H12BrN3O4) C, H, N.
Смесь бромида 61 (2,00 г, 5,65 ммоль), бис(пинаколято)диборона (1,59 г, 6,29 ммоль) и KOAc (3,40 г, 34,7 ммоль) в ДМСО (40 мл) продували N2. Добавляли Pd(dppf)Cl2 (0,14 г, 0,17 ммоль) и смесь продували N2 при нагревании до 90°С. Спустя 1 час реакционную смесь распределяли между EtOAc и водой и органический экстракт очищали хроматографией на силикагеле, элюируя EtOAc. Продукт растирали в Et2O, получая при этом (6S)-2-нитро-6-{[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил]окси}-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (62) (1,158 г, 51%): т. пл. 150-153°С;1H ЯМР [(CD3)2SO] δ 8,01 (с, 1 H), 7,65 (д, J=8,0 Гц, 2 H), 7,32 (д, J=8,0 Гц, 2 H), 4,70 (д, J=12,5 Гц, 1 H), 4,67-4,63 (м, 2 H), 4,46 (д, J=11,9 Гц, 1 H), 4,29-4,20 (м, 3 H), 1,29 (с, 12 H).Анализ (C19H24BN3O6) C, H, N.
Смесь боронатного эфира 62 (0,094 г, 0,23 ммоль) и 2-хлор-5-(трифторметил)пиридина (53 мг, 0,29 ммоль) в толуоле (5 мл), EtOH (3 мл) и водного K2CO3 (2 M, 1 мл, 2 ммоль) продували N2. Добавляли Pd(dppf)Cl2 (8 мг, 0,01 ммоль) и смесь кипятили с обратным холодильником в атмосфере N2 в течение 0,5 час, затем распределяли между EtOAc и водой. Органический слой сушили и упаривали, и затем колоночная хроматография остатка на силикагеле с применением градиентного элюирования (смесь 1:1 гексаны:EtOAc, затем EtOAc) давала 8 (60 мг, 62%) в виде белого твердого вещества: т. пл. (после растирания в Et2O) 252-254°С;1H ЯМР [(CD3)2SO] δ 9,03 (ушир. с, 1 H), 8,27 (дд, J=8,5, 2,1 Гц, 1 H), 8,18 (д, J=8,4 Гц, 1 H), 8,15 (д, J=8,3 Гц, 2 H), 8,03 (с, 1 H), 7,48 (д, J=8,3 Гц, 2 H), 4,66-4,78 (м, 3 H), 4,49 (д, J=11,8 Гц, 1 H), 4,23-4,33 (м, 3 H).Анализ (C19H15F3N4O4) C, H, N.
K. Синтез (6S)-2-нитро-6-({4-[6-(трифторметил)-3-пиридинил]бензил}окси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (9) способом схемы 6

Реакция боронатного эфира 62 (см. пример 2J) (0,157 г, 0,391 ммоль) и 5-бром-2-(трифторметил)пиридина (0,110 г, 0,487 ммоль), как в примере 2J, давала 9 (0,105 г, 64%) в виде белого твердого вещества: т. пл. (после растирания в Et2O) 221-222°С;1H ЯМР [(CD3)2SO] δ 9,08 (д, J=2,1 Гц, 1 H), 8,35 (дд, J=8,1, 1,9 Гц, 1 H), 8,03 (с, 1 H), 7,97 (д, J=8,1 Гц, 1 H), 7,81 (д, J=8,3 Гц, 2 H), 7,48 (д, J=8,3 Гц, 2 H), 4,67-4,77 (м, 3 H), 4,49 (д, J=11,8 Гц, 1 H), 4,22-4,33 (м, 3 H).Анализ (C19H15F3N4O4) C, H, N.
L. Синтез (6S)-2-нитро-6-({1-[4-(трифторметокси)фенил]-1H-пиразол-3-ил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (10) способом схемы 7

Тетрафторборат 4-трифторметоксибензолдиазония (63) (4,33 г, 15,7 ммоль) добавляли к раствору этил-2-хлорацетоацетата (64) (2,35 г, 14,3 ммоль) в пиридине (6 мл) и воде (6 мл) при -5°С. Смесь перемешивали при -5°С в течение 0,5 час и осадок отделяли фильтрованием и промывали охлажденной льдом водой. Перекристаллизация из смеси EtOH/вода давала этил-2-хлор-{[4-(трифторметокси)фенил]гидразоно}этаноат (65) (3,977 г, 82%) в виде бледно-оранжевых игл: т. пл. 128-130°С;1H ЯМР [(CD3)2SO] δ 10,68 (с, 1 H), 7,43 (д, J=9,2 Гц, 2 H), 7,34 (д, J=9,2 Гц, 2 H), 4,30 (т, J=7,1 Гц, 2 H), 1,30 (кв, J=7,1 Гц, 3 H). MSAPCI (химическая ионизация при атмосферном давлении) m/z 309, 311 [M-H]-.
Перемешиваемую смесь гидразоноилхлорида 65 (1,55 г, 4,99 ммоль), бицикло[2.2.1]гепта-2,5-диена (1,25 мл, 24,6 ммоль) и Et3N (2,0 мл, 14,3 ммоль) в толуоле (10 мл) нагревали до 70°С в течение 1 час. Смесь охлаждали и фильтровали, фильтровальный осадок промывали толуолом (10 мл) и органические фракции объединяли и упаривали. Остаток кипятили с обратным холодильником в ксилолах (30 мл) в течение 2 час. Колоночная хроматография на силикагеле с элюированием гексанами сначала давала ксилолы, и затем дальнейшее элюирование CH2Cl2 давало этил-1-[4-(трифторметокси)фенил]-1H-пиразол-3-карбоксилат (66) (1,176 г, 79%) в виде белого твердого вещества: т. пл. 76-78°С;1Н ЯМР (CDCl3) δ 7,91 (д, J=2,5 Гц, 1 H), 7,79 (д, J=8,9 Гц, 2 H), 7,33 (д, J=8,9 Гц, 2 H), 7,00 (д, J=2,5 Гц, 1 H), 4,44 (кв, J=7,1 Гц, 2 H), 1,43 (т, J=7,1 Гц, 3 H). МСAPCI m/z 301 [M+H]+.
LiAlH4 (0,137 г, 3,61 ммоль) добавляли к раствору сложного эфира 66 (1,081 г, 3,60 ммоль) в Et2O (20 мл) при 0°С и перемешиваемую смесь нагревали до комнатной температуры в течение 1 час, затем охлаждали до 0°С и гасили льдом. Смесь разбавляли Et2O (100 мл) и насыщенным водным тартратом натрия-калия (100 мл) и затем фильтровали через целит. Органический слой сушили и хроматографировали на силикагеле с элюированием смесью CH2Cl2:EtOAc (95:5), получая при этом {1-[4-(трифторметокси)фенил]-1Н-пиразол-3-ил}метанол (67) (0,888 г, 96%) в виде белого твердого вещества: т. пл. 53-54°С;1H ЯМР [(CD3)2SO] δ 8,44 (д, J=2,5 Гц, 1 H), 7,92 (д, J=8,5 Гц, 2 H), 7,48 (д, J=8,5 Гц, 2 H), 6,50 (д, J=2,5 Гц, 1 H), 5,15 (т, J=5,8 Гц, 1 H), 4,51 (д, J=5,8 Гц, 2 H). МСAPCI m/z 259 [M+H]+.
PBr3 (0,312 мл, 3,32 ммоль) добавляли к раствору спирта 67 (0,858 г, 3,32 ммоль) в простом эфире (15 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 17 час, затем охлаждали до 0°С, гасили льдом и распределяли между CH2Cl2 и водой. Колоночная хроматография органической части на силикагеле (элюирование CH2Cl2) давала 3-(бромметил)-1-[4-(трифторметокси)фенил]-1H-пиразол (68) (0,952 г, 89%) в виде белого твердого вещества: т. пл. 71-73°С;1H ЯМР (CDCl3) δ 7,84 (д, J=2,5 Гц, 1 H), 7,69 (д, J=9,1 Гц, 2 H), 7,31 (д, J=9,1 Гц, 2 H), 6,54 (д, J=2,5 Гц, 1 H), 4,56 (с, 2 H). МСAPCI m/z 321, 323 [M+H]+.
NaH (60% масс./масс., 160 мг, 4,00 ммоль) добавляли к раствору спиртового оксазина 41 (0,473 г, 2,55 ммоль) и бромида 68 (0,913 г, 2,84 ммоль) в ДМФА (50 мл) при 0°С. Смесь перемешивали при 0°С в течение 2 час и затем гасили льдом и распределяли между EtOAc и водой. Органическую фракцию сушили и упаривали и затем подвергали колоночной хроматографии на силикагеле с элюированием с градиентом от смеси 1:1 гексаны:EtOAc до EtOAc, получая при этом 10 (0,844 г, 78%) в виде белого твердого вещества: т. пл. 103-105°С;1H ЯМР [(CD3)2SO] δ 8,50 (д, J=2,5 Гц, 1 H), 8,01 (с, 1 H), 7,93 (д, J=9,1 Гц, 2 H), 7,49 (д, J=9,1 Гц, 2 H), 6,54 (д, J=2,5 Гц, 1 H), 4,68-4,74 (м, 2 H), 4,65 (дт, J=12,3, 2,4 Гц, 1 H), 4,47 (д, J=11,8 Гц, 1 H), 4,20-4,31 (м, 3 H). Анализ (C17H14F3N5O5) C, H, N.
M. Синтез (6S)-6-({1-метил-3-[4-(трифторметокси)фенил]-1Н-пиразол-5-ил}метокси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (11) способом схемы 8

Раствор 2-(2-пропинилокси)тетрагидро-2Н-пирана (69) (0,758 г, 5,41 ммоль), CuI (17 мг, 0,09 ммоль) и PdCl2(PPh3)2 (0,158 г, 0,023 ммоль) в ТГФ (15 мл) продували N2. Добавляли 1-иод-4-(трифторметокси)бензол (70) (1,30 г, 4,51 ммоль) в ТГФ (10 мл) с последующим добавлением раствора сульфата метилгидразина (1,95 г, 13,5 ммоль) и NaHCO3(2,27 г, 27 ммоль) в воде (25 мл). Смесь промывали струей монооксида углерода и затем перемешивали при комнатной температуре в течение 2 дней в атмосфере монооксида углерода. Образовавшуюся смесь распределяли между CH2Cl2 и водой, фракцию CH2Cl2 сушили и растворитель выпаривали. Колоночная хроматография остатка на силикагеле (элюирование CH2Cl2) давала 1-метил-5-[(тетрагидро-2Н-пиран-2-илокси)метил]-3-[4-(трифторметокси)фенил]-1H-пиразол (71) (1,034 г, 64%) в виде коричневого твердого вещества: т. пл. 40-42°С;1H ЯМР (CDCl3) δ 7,78 (д, J=8,8 Гц, 2 H), 7,21 (д, J=8,0 Гц, 2 H), 6,51 (с, 1 H), 4,75 (д, J=12,8 Гц, 1 H), 4,69 (т, J=3,3 Гц, 1 H), 4,57 (д, J=12,8 Гц, 1 H), 3,94 (с, 3 H), 3,84-3,91 (м, 1 H), 3,53-3,60 (м, 1 H), 1,68-1,88 (м, 2 H), 1,50-1,66 (м, 4 H). МСAPCI (химическая ионизация при атмосферном давлении) m/z 357 [M+H]+.
Перемешиваемый раствор простого эфира THP 71 (0,968 г, 2,72 ммоль) в HCl (4 М, 10 мл) и ТГФ (10 мл) нагревали до 80°С в течение 16 час. ТГФ выпаривали и остаток распределяли между EtOAc и водным NaHCO3. Органический слой сушили и упаривали и остаток перекристаллизовывали (iPr2O), получая при этом {1-метил-3-[4-(трифторметокси)фенил]-1H-пиразол-5-ил}метанол (72) (0,278 г, 38%) в виде белого твердого вещества: т. пл. 91-93°С.1H ЯМР [(CD3)2SO] δ 7,86 (д, J=8,9 Гц, 2 H), 7,36 (д, J=8,9 Гц, 2 H), 6,64 (с, 1 H), 5,30 (т, J=5,2 Гц, 1 H), 4,52 (д, J=5,2 Гц, 2 H), 3,84 (с, 3 H). МСAPCI m/z 273 [M+H]+.
PBr3 (0,15 мл, 1,60 ммоль) добавляли к раствору спирта 72 (0,205 г, 0,75 ммоль) в Et2O (10 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 16 час, охлаждали до 0°С, гасили льдом и разбавляли Et2O (100 мл). Хроматография органической части на силикагеле (элюирование CH2Cl2) давала 5-(бромметил)-1-метил-3-[4-(трифторметокси)фенил]-1H-пиразол (73) (0,212 г, 85%) в виде белого твердого вещества: т. пл. 70-71°С;1H ЯМР (CDCl3) δ 7,76 (д, J=8,9 Гц, 2 H), 7,22 (д, J=8,9 Гц, 2 H), 6,55 (с, 1 H), 4,50 (с, 2 H), 3,94 (с, 3 H). МСAPCI m/z 335,337 [M+H]+.
NaH (95% масс./масс., 25 мг, 0,99 ммоль) добавляли к раствору спирта 41 (0,113 г, 0,61 ммоль) и брома 73 (0,207 г, 0,62 ммоль) в ДМФА (6 мл) при 0°С. Смесь перемешивали при 0°С в течение 2 час, затем гасили льдом и распределяли между EtOAc и водой. Органический слой сушили и растворитель выпаривали. Колоночная хроматография остатка на силикагеле при элюировании с градиентом от смеси 1:1 гексаны:EtOAc до EtOAc давала 11 (0,130 г, 48%) в виде белого твердого вещества: т. пл. 178-179°С;1H ЯМР [(CD3)2SO] δ 8,02 (с, 1 H), 7,85 (д, J=8,9 Гц, 2 H), 7,36 (д, J=8,9 Гц, 2 H), 6,76 (с, 1 H), 4,77 (д, J=12,6 Гц, 1 H), 4,72 (д, J=12,6 Гц, 1 H), 4,69 (дт, J=12,1, 2,3 Гц, 1 H), 4,48 (д, J=11,8 Гц, 1 H), 4,21-4,32 (м, 3 H), 3,79 (с, 3 H). Анализ (C18H16F3N5O5) C, H, N.
N. Синтез (6S)-6-{[3-фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (12) способом схемы 9.

Раствор спирта 41 (1,403 г, 7,58 ммоль) и 4-бром-1-(бромметил)-2-фторбензола (74) (2,66 г, 9,93 ммоль) в безводном ДМФА (30 мл) в атмосфере N2 при 0°С обрабатывали 60% NaH (427 мг, 10,7 ммоль), затем быстро дегазировали и снова вводили N2. После перемешивания при комнатной температуре в течение 3 час реакционную смесь охлаждали (СО2/ацетон), гасили смесью лед/водный NaHCO3 (20 мл), добавляли к насыщенному раствору соли (150 мл) и экстрагировали CH2Cl2 (4×80 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% EtOAc/CH2Cl2 сначала давало предварительные фракции, и затем элюирование смесью 3-5% EtOAc/CH2Cl2 давало (6S)-6-[(4-бром-2-фторбензил)окси]-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (75) (2,633 г, 93%) в виде бледно-желтого твердого вещества: т. пл. (MeOH/CH2Cl2/гексан) 171-173°С;1H ЯМР [(CD3)2SO] δ 8,01 (с, 1 H), 7,54 (дд, J=9,7, 1,8 Гц, 1 H), 7,42 (дд, J=8,2, 1,8 Гц, 1 H), 7,37 (дд, J=8,1, 7,7 Гц, 1 H), 4,72-4,62 (м, 3 H), 4,47 (ушир. д, J=11,9 Гц, 1 H), 4,30-4,19 (м, 3 H). Анализ (C13H11BrFN3O4) C, H, N.
Перемешиваемую смесь бромида 75 (475 мг, 1,28 ммоль), 4-(трифторметокси)фенилбороновой кислоты (44) (395 мг, 1,92 ммоль) и Pd(dppf)Cl2(143 мг, 0,195 ммоль) в толуоле (18 мл) и EtOH (7 мл) дегазировали в течение 8 мин (вакуумный насос) и затем вводили N2. При помощи шприца добавляли 2 М водный раствор Na2CO3 (3,5 мл, 7,0 ммоль) и перемешиваемую смесь снова дегазировали в течение 8 мин и затем вводили N2. Образовавшуюся смесь перемешивали при 85°С в течение 70 мин и затем охлаждали, разбавляли водным NaHCO3 (50 мл) и экстрагировали CH2Cl2 (6×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало предварительные фракции, и затем дополнительное элюирование смесью 1-2% EtOAc/CH2Cl2 давало 12 (539 мг, 93%) в виде бледно-желтого твердого вещества: т. пл. (CH2Cl2/пентан) 160-162°С;1H ЯМР (CDCl3) δ 7,57 (дт, J=8,8, 2,5 Гц, 2 H), 7,42 (т, J=7,7 Гц, 1 H), 7,39 (с, 1 H), 7,35 (дд, J=7,9, 1,7 Гц, 1 H), 7,33-7,23 (м, 3 H), 4,81-4,73 (м, 2 H), 4,65 (ддд, J=12,2, 3,6, 2,0 Гц, 1 Н), 4,38 (ушир. д, J=12,1 Гц, 1 H), 4,25-4,13 (м, 3 H). Анализ (C20H15F4N3O5) C, H, N.
O. Синтез (6S)-6-({2-фтор-4-[6-(трифторметил)-3-пиридинил]бензил}окси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (23) способом схемы 9

Перемешиваемую смесь бромида 75 (см. пример 2N) (503 мг, 1,35 ммоль), 6-(трифторметил)-3-пиридинилбороновой кислоты (386 мг, 2,02 ммоль) и Pd(dppf)Cl2 (148 мг, 0,202 ммоль) в толуоле (20 мл) и EtOH (10 мл) дегазировали в течение 12 мин (вакуумный насос) и затем вводили N2. При помощи шприца добавляли 2 М водный раствор Na2CO3 (3,5 мл, 7,0 ммоль) и перемешиваемую смесь снова дегазировали в течение 12 мин и затем вводили N2. Образовавшуюся смесь перемешивали при 90°С в течение 3 час и затем охлаждали, разбавляли водным NaHCO3 (100 мл) и экстрагировали CH2Cl2 (6×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-3% EtOAc/CH2Cl2 сначала давало предварительные фракции, и затем дальнейшее элюирование смесью 3-4% EtOAc/CH2Cl2 давало 23 (530 мг, 90%) в виде кремового твердого вещества: т. пл. (МеОН/CH2Cl2/пентан) 195-198°С;1H ЯМР [(CD3)2SO] δ 9,12 (д, J=2,1 Гц, 1 H), 8,40 (дд, J=8,1, 1,9 Гц, 1 H), 8,03 (с, 1 H), 7,99 (д, J=8,1 Гц, 1 H), 7,75 (дд, J=11,3, 1,7 Гц, 1 H), 7,68 (дд, J=7,9, 1,8 Гц, 1 H), 7,57 (т, J=7,8 Гц, 1 H), 4,80 (ушир. д, J=13,0 Гц, 1 H), 4,76 (ушир. д, J=13,3 Гц, 1 H), 4,69 (дт, J=12,0, 2,5 Гц, 1 H), 4,50 (ушир. д, J=11,7 Гц, 1 H), 4,35-4,22 (м, 3 H). Анализ (C19H14F4N4O4) C, H, N.
Р. Синтез (6S)-6-({2-фтор-4-[5-(трифторметил)-2-пиридинил]бензил}окси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (14) способом схемы 9

Перемешиваемую смесь бромида 75 (см. пример 2N) (1,601 г, 4,30 ммоль), бис(пинаколято)диборона (1,179 г, 4,64 ммоль), Pd(dppf)Cl2 (0,473 г, 0,646 ммоль) и KOAc (1,497 г, 15,3 ммоль) в безводном ДМСО (24 мл) дегазировали в течение 35 мин (вакуумный насос) и затем вводили N2. Смесь перемешивали при 89°С в течение 5 час и затем охлаждали, добавляли к смеси лед-вода (150 мл) и экстрагировали EtOAc (5×100 мл). Экстракты промывали водой (2×100 мл), упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 50% EtOAc/петролейный эфир сначала давало предварительные фракции, и затем дальнейшее элюирование смесью 50-67% EtOAc/петролейный эфир давало (6S)-6-{[2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил]окси}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (76) (1,186 г, 66%) в виде кремового твердого вещества: т. пл. (CH2Cl2/Et2O/пентан) 147-149°С;1Н ЯМР (CDCl3) δ 7,58 (дд, J=7,5, 0,9 Гц, 1 H), 7,49 (ушир. д, J=10,3 Гц, 1 H), 7,38 (с, 1 H), 7,35 (т, J=7,3 Гц, 1 H), 4,76 (д, J=12,9 Гц, 1 H), 4,73 (д, J=12,7 Гц, 1 H), 4,59 (ддд, J=12,1, 3,8, 2,0 Гц, 1 H), 4,34 (дд, J=12,0, 1,5 Гц, 1 H), 4,20-4,07 (м, 3 H), 1,34 (с, 12 H). HRFABMS (масс-спектр высокого разрешения с бомбардировкой ускоренными атомами), вычислено для C19H23BFN3O6 m/z [М+Н]+ 420,1742, 419,1779, найдено 420,1733, 419,1763.
Перемешиваемую смесь боронатного эфира 76 (602 мг, 1,43 ммоль), 2-хлор-5-трифторметилпиридина (1,08 г, 5,96 ммоль) и Pd(dppf)Cl2 (0,232 г, 0,317 ммоль) в толуоле (18 мл) и EtOH (9 мл) дегазировали в течение 12 мин (вакуумный насос) и затем вводили N2. При помощи шприца добавляли 2 М водный раствор Na2CO3 (3,8 мл, 7,6 ммоль) и перемешиваемую смесь снова дегазировали в течение 12 час и затем снова вводили N2. Образовавшуюся смесь перемешивали при 90°С в течение 120 мин и затем охлаждали, разбавляли водным NaHCO3 (100 мл) и экстрагировали CH2Cl2 (6×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-2% EtOAc/CH2Cl2 сначала давало предварительные фракции, и затем дополнительное элюирование смесью 2-6% EtOAc/CH2Cl2 давало 14 (523 мг, 83%) в виде бледно-желтого твердого вещества: т. пл. (CH2Cl2/гексан) 233-235°С;1H ЯМР (CDCl3) δ 8,95 (м, 1 H), 8,01 (дд, J=8,3, 2,3 Гц, 1 H), 7,86-7,79 (м, 3 H), 7,49 (т, J=7,8 Гц, 1 H), 7,40 (с, 1 H), 4,82 (ушир. д, J=13,1 Гц, 1 H), 4,78 (ушир. д, J=13,3 Гц, 1 H), 4,66 (ддд, J=12,2, 3,5, 2,0 Гц, 1 H), 4,39 (дд, J=12,1, 1,4 Гц, 1 H), 4,26-4,14 (м, 3 H). Анализ (C19H14F4N4O4) C, H, N.
Q. Синтез (6S)-2-нитро-6-{[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (13) способом схемы 10
Смесь (S)-глицидного спирта (77) (20 г, 0,27 моль), бензилового спирта (27,9 мл, 0,27 моль) и CsF (0,82 г, 5,40 ммоль) нагревали с перемешиванием при 120°С в течение 16 час. Непрореагировавший бензиловый спирт удаляли с применением роторного испарителя, присоединенного к линии высокого вакуума. Продукт распределяли между EtOAc и водой и органический экстракт упаривали и хроматографировали на диоксиде кремния. Элюирование петролейным эфиром давало предварительные фракции, и затем дальнейшее элюирование смесью EtOAc/петролейный эфир (3:7) давало (2S)-3-(бензилокси)-1,2-пропандиол (78) (9,52 г, 19%) в виде вязкого масла: [α]19 -3,64° (c, 6,59, CHCl3);1H ЯМР (CDCl3) δ 7,38-7,28 (м, 5 H), 4,56 (с, 2 H), 3,92-3,87 (м, 1 H), 3,71 (дд, J=11,4, 3,9 Гц, 1 H), 3,64 (дд, J=11,4, 5,4 Гц, 1 H), 3,61-3,57 (м, 2 H), 2,60 (ушир., 1 H), 2,22 (ушир., 1 H). МСAPCI m/z 183 [M+H]+.
Хлор(триизопропил)силан (12,2 мл, 0,057 моль) добавляли по каплям при 20°С к перемешиваемому раствору диола 78 (9,52 г, 0,052 моль) и имидазола (5,33 г, 0,078 моль) в ДМФА (150 мл) и перемешивание продолжали в течение 16 час. Большую часть ДМФА удаляли при пониженном давлении и остаток распределяли между EtOAc и водой. Органический экстракт хорошо промывали водой, затем насыщенным раствором соли и упаривали, получая при этом масло, которое хроматографировали на диоксиде кремния. Элюирование петролейным эфиром давало предварительные фракции, и затем дальнейшее элюирование смесью EtOAc/петролейный эфир (1:19) давало (2R)-1-(бензилокси)-3-[(триизопропилсилил)окси]-2-пропанол (79) (13,80 г, 78%) в виде бесцветного масла: [α]19 -0,78° (с, 8,93, CHCl3);1H ЯМР (CDCl3) δ 7,41-7,27 (м, 5 H), 4,55 (с, 2 H), 3,90-3,84 (м, 1 H), 3,79-3,72 (м, 2 H), 3,59-3,51 (м, 2 H), 2,52 (д, J=5,1 Гц, 1 H), 1,13-1,03 (м, 21 H). МСAPCI m/z 339 [M+H]+.
1,1'-Диизопропилазодикарбоксилат (7,70 мл, 0,039 моль) добавляли по каплям при 5°С к раствору спирта 79 (12,40 г, 0,037 моль), 4'-(трифторметокси)[1,1'-бифенил]-4-ола (описан Edsall et al., 2003, получен посредством сочетания Сузуки 4-бромфенола и бороновой кислоты 44) (8,29 г, 0,033 моль) и трифенилфосфина (10,26 г, 0,039 моль) в безводном бензоле (25 мл) и раствор перемешивали при 20°С в течение 18 час. Продукт адсорбировали непосредственно на диоксиде кремния концентрированием при пониженном давлении, и хроматографирование на силикагеле с элюированием смесью EtOAc/петролейный эфир (1:19) давало 4-[((1S)-2-(бензилокси)-1-{[(триизопропилсилил)окси]метил}этил)окси]-4'-(трифторметокси)-1,1'-бифенил (80) (14,30 г, 69%) в виде бесцветного масла: [α]19 +5,9° (с, 6,95, CHCl3);1H ЯМР (CDCl3) δ 7,72 (д, J=8,8 Гц, 2 H), 7,58 (д, J=8,8 Гц, 2 H), 7,40 (д, J=8,8 Гц, 2 H), 7,35-7,24 (м, 5 H), 7,06 (д, J=8,8 Гц, 2 H), 4,64-4,57 (м, 1 H), 4,52 (с, 2 H), 3,98-3,87 (м, 2 H), 3,76-3,65 (м, 2 H), 1,08-0,98 (м, 21 H). МСAPCI m/z 576 [M+H]+.
Смесь бензилового эфира 80 (10,79 г, 0,019 моль) и 5% Pd-C (500 мг) в смеси 1:1 EtOAc/EtOH (250 мл) гидрировали при 413685,6 Па (60 фунт/кв. дюйм) в течение 4 час. Катализатор удаляли фильтрованием через целит и фильтрат концентрировали при пониженном давлении, получая при этом (2S)-2-{[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}-3-[(триизопропилсилил)окси]-1-пропанол (81) в виде вязкого масла, достаточно чистого для применения на следующей стадии. Иод (6,03 г, 0,024 моль) добавляли порциями при 20°С к энергично перемешиваемому раствору сырого спирта 81, трифенилфосфина (6,23 г, 0,024 моль) и имидазола (2,49 г, 0,036 моль) в бензоле (100 мл) и перемешивание продолжали в течение 1 час. После разбавления EtOAc смесь промывали водой, 2 н. Na2SO3 и снова водой. Экстракт упаривали и хроматографировали на силикагеле с элюированием смесью EtOAc/петролейный эфир (1:19), получая при этом 4-[((1R)-2-иод-1-{[(триизопропилсилил)окси]метил}этил)окси]-4'-(трифторметокси)-1,1'-бифенил (82) (9,08 г, общий выход 81%) в виде бесцветного масла;1H ЯМР (CDCl3) δ 7,54 (д, J=8,8 Гц, 2 H), 7,48 (д, J=8,8 Гц, 2 H), 7,26 (ушир. д, J=8,8 Гц, 2 H), 7,02 (д, J=8,8 Гц, 2 H), 4,31-4,25 (м, 1 H), 4,03 (дд, J=10,4, 4,8 Гц, 1 H), 3,93 (дд, J=10,4, 5,6 Гц, 1 H), 3,55 (дд, J=10,5, 5,6 Гц, 1 H), 3,45 (дд, J=10,5, 4,8 Гц, 1 H), 1,15-1,06 (м, 21 H). МС APCI m/z 595 [M+H]+.
Смесь 2-бром-4(5)-нитроимидазола (0,73 г, 3,82 ммоль), иодида 82 (2,50 г, 4,20 ммоль) и K2CO3 (0,63 г, 4,58 ммоль) в ДМФА (30 мл) перемешивали при 87°С в течение 20 час. Образовавшуюся смесь распределяли между EtOAc и насыщенным раствором соли и экстракт хорошо промывали насыщенным раствором соли. Упаривание давало масло, которое хроматографировали на силикагеле с элюированием смесью EtOAc/петролейный эфир (1:9), получая при этом 2-бром-4-нитро-1-{(2S)-2-{[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}-3-[(триизопропилсилил)окси]пропил}-1Н-имидазол (83) (1,05 г, 42%) в виде масла;1H ЯМР (CDCl3) δ 7,95 (с, 1 H), 7,51 (д, J=8,8 Гц, 2 H), 7,45 (д, J=8,8 Гц, 2 H), 7,25 (ушир. д, J=8,8 Гц, 2 H), 6,89 (д, J=8,8 Гц, 2 H), 4,63-4,56 (м, 2 H), 4,37-4,29 (м, 1 H), 4,02 (дд, J=10,7, 3,4 Гц, 1 H), 3,84 (дд, J=10,7, 6,8 Гц, 1 H), 1,17-1,07 (м, 21 H). МС APCI m/z 660, 658 [M+H]+.
Фторид тетра-н-бутиламмония (3,18 мл 1 M раствора в ТГФ, 3,18 ммоль) добавляли при 20°С к раствору силилового простого эфира 83 (1,05 г, 1,59 ммоль) в ТГФ (40 мл) и раствор перемешивали при комнатной температуре в течение 1 час. После разбавления EtOAc раствор промывали насыщенным водным раствором NaHCO3, затем водой и затем упаривали, получая при этом масло, которое хроматографировали на диоксиде кремния. Элюирование смесью EtOAc/петролейный эфир (1:1) давало предварительные фракции, и затем дальнейшее элюирование EtOAc давало спирт с удаленной защитной группой. Это вещество сразу растворяли в ДМФА (20 мл) и раствор охлаждали до 5°С и обрабатывали NaH (0,19 г 60% дисперсии в минеральном масле, 4,77 ммоль). Охлаждающую баню удаляли и смесь перемешивали при 20°С в течение 30 мин. Добавляли воду, смесь экстрагировали EtOAc и экстракт упаривали, получая при этом масло, которое хроматографировали на диоксиде кремния. Элюирование смесью EtOAc/петролейный эфир (1:1) давало предварительные фракции, и затем дальнейшее элюирование смесью EtOAc/петролейный эфир (2:1) давало 13 (175 мг, 26%) в виде белого твердого вещества: т. пл. 210°С; [α]19 -9,5° (c, 0,84, ацетон);1H ЯМР [(CD3)2SO] δ 8,07 (с, 1 H), 7,75 (д, J=8,8 Гц, 2 H), 7,66 (д, J=8,8 Гц, 2 H), 7,41 (ушир. д, J=8,8 Гц, 2 H), 7,15 (д, J=8,8 Гц, 2 H), 5,31-5,27 (м, 1 H), 4,71-4,63 (м, 2 H), 4,42 (дд, J=13,8, 3,2 Гц, 1 H), 4,34 (ушир. д, J=13,8 Гц, 1 H). Анализ (C19H14F3N3O5) C, H, N. Анализ хиральной ВЭЖХ показал, что данный продукт содержит 70% ее-изомера.
R. Синтез (6S)-6-{[2-фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (15) способом схемы 11

Суспензию 4-бром-3-фторбензойной кислоты (84) (1,61 г, 7,35 ммоль) в безводном ТГФ (10 мл, затем 4×3 мл для споласкивания) в атмосфере N2 добавляли по каплям (на протяжении 40 мин) к суспензии борогидрида натрия (400 мг, 10,6 ммоль) в безводном ТГФ (15 мл) в атмосфере N2 и затем смесь охлаждали на ледяной бане. Раствор иода (1,008 г, 3,97 ммоль) в безводном ТГФ (10 мл, затем 2×3 мл) по каплям добавляли (на протяжении 35 мин) к перемешиваемому раствору и затем смесь перемешивали при комнатной температуре в течение 14 час. Смесь концентрировали при пониженном давлении и затем последовательно обрабатывали водой (20 мл), 10% HCl (3,4 мл) и водой (20 мл) и экстрагировали CH2Cl2 (4×50 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле с элюированием смесью 50% CH2Cl2/петролейный эфир, получая при этом (4-бром-3-фторфенил)метанол (85) (1,096 г, 73%) в виде белого твердого вещества: т. пл. (CH2Cl2/петролейный эфир) 39-40°С;1H ЯМР (CDCl3) δ 7,52 (дд, J=8,0, 7,2 Гц, 1 H), 7,16 (дд, J=9,3, 1,8 Гц, 1 H), 7,02 (дд, J=8,2, 1,8 Гц, 1 H), 4,67 (д, J=5,9 Гц, 2 H), 1,75 (т, J=5,9 Гц, 1 H). HREIMS, вычислено для C7H6BrFO m/z (М+) 205,9567, 203,9586, найдено 205,9566, 203,9580.
Бромирование спирта 85, как в примере 2A, в течение 20 час давало 1-бром-4-(бромметил)-2-фторбензол (86) (100%) в виде белого твердого вещества: т. пл. (пентан) 39-41°С;1H ЯМР (CDCl3) δ 7,52 (дд, J=8,1, 7,1 Гц, 1 H), 7,17 (дд, J=9,0, 2,0 Гц, 1 H), 7,06 (дд, J=8,2, 1,8 Гц, 1 H), 4,41 (д, 2 H). HREIMS, вычислено для C7H5Br2F m/z (М+) 269,8701, 267,8722, 265,8742, найдено 269,8692, 267,8713, 265,8726.
Реакция бромида 86 (1,29 эквив.) со спиртовым производным оксазина 41, как в примере 2N, с последующей хроматографией продукта на силикагеле с элюированием смесью 0-2% EtOAc/CH2Cl2 (предварительные фракции) и затем смесью 2-4% EtOAc/CH2Cl2 давала (6S)-6-[(4-бром-3-фторбензил)окси]-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (87) (89%) в виде бледно-желтого твердого вещества: т. пл. (MeOH/CH2Cl2/гексан) 181-183°С;1H ЯМР [(CD3)2SO] δ 8,01 (с, 1 H), 7,68 (дд, J=8,0, 7,5 Гц, 1 H), 7,30 (дд, J=9,8, 1,9 Гц, 1 H), 7,12 (дд, J=8,2, 1,5 Гц, 1 H), 4,70-4,60 (м, 3 H), 4,47 (ушир. д, J=1 1,7 Гц, 1 H), 4,31-4,18 (м, 3 H). Анализ (C13H11BrFN3O4) C, H, N.
Перемешиваемую смесь бромида 87 (503 мг, 1,35 ммоль), 4-(трифторметокси)фенилбороновой кислоты (44) (500 мг, 2,43 ммоль) и Pd(dppf)Cl2(304 мг, 0,415 ммоль) в толуоле (16 мл) и EtOH (8 мл) дегазировали в течение 12 мин (вакуумный насос) и затем вводили N2. При помощи шприца добавляли водный раствор 2 M Na2CO3 (3,6 мл, 7,2 ммоль) и перемешиваемую смесь снова дегазировали в течение 12 мин и затем вводили N2. Образовавшуюся смесь перемешивали при 90°С в течение 6 час и затем охлаждали, разбавляли водным NaHCO3 (100 мл) и экстрагировали CH2Cl2 (6×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 0-1% EtOAc/CH2Cl2 сначала давало предварительные фракции, и затем дополнительное элюирование смесью 2% EtOAc/CH2Cl2 давало 15 (478 мг, 78%) в виде бледно-желтого твердого вещества: т. пл. (CH2Cl2/пентан) 181-183°С;1Н ЯМР (CDCl3) δ 7,55 (дтд, J=8,8, 2,4, 1,5 Гц, 2 H), 7,42 (т, J=7,9 Гц, 1 H), 7,40 (с, 1 H), 7,29 (ушир. дд, J=8,8, 0,9 Гц, 2 H), 7,17 (дд, J=7,8, 1,6 Гц, 1 H), 7,13 (дд, J=1 1,0, 1,4 Гц, 1 H), 4,77 (д, J=12,2 Гц, 1 H), 4,68-4,62 (м, 2 H), 4,38 (дд, J=12,2, 1,5 Гц, 1 H), 4,26-4,14 (м, 3 H). Анализ (C20H15F4N3O5) C, H, N.
S. Синтез (6S)-2-нитро-6-({2-[4-(трифторметокси)фенил]-5-пиримидинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (16) способом схемы 12

Смесь 5-бром-2-иодпиримидина (88) (1,50 г, 5,27 ммоль), 4-(трифторметокси)фенилбороновой кислоты (44) (1,185 г, 5,75 ммоль) и Na2CO3 (1,11 г, 10,5 ммоль) в толуоле (120 мл) и воде (15 мл) продували N2. Добавляли Pd(PPh3)4 (60 мг, 0,05 ммоль) и смесь кипятили с обратным холодильником в атмосфере N2 в течение 17,5 час, затем распределяли между EtOAc и водой. Колоночная хроматография органической части на силикагеле (элюирование смесью 4:1 гексаны/CH2Cl2) давала 5-бром-2-[4-(трифторметокси)фенил]пиримидин (89) (1,264 г, 75%) в виде белого твердого вещества: т. пл. 107-108°С;1H ЯМР (CDCl3) δ 8,83 (с, 2 H), 8,46 (д, J=9,0 Гц, 2 H), 7,31 (д, J=9,0 Гц, 2 H). МС APCI m/z 319, 321 [M+H]+.
n-BuLi (2,5 M, 1,88 мл, 4,7 ммоль) добавляли к раствору бромида 89 (1,252 г, 3,92 ммоль) в ТГФ (40 мл) при -95°С. Раствор перемешивали в течение 30 сек и затем добавляли ДМФА (5 мл). Реакционную смесь перемешивали при -90°С в течение 20 мин и затем гасили водным NH4Cl. Образовавшуюся смесь распределяли между EtOAc и водой и затем колоночной хроматографией органической части на силикагеле (элюирование смесью 1:7 EtOAc/гексаны) получали 2-[4-(трифторметокси)фенил]-5-пиримидинкарбальдегид (90) (0,778 г, 74%) в виде белого твердого вещества: т. пл. 114-115°С;1H ЯМР (CDCl3) δ 10,16 (с, 1 H), 9,22 (с, 2 H), 8,62 (д, J=9,0 Гц, 2 H), 7,36 (д, J=9,0 Гц, 2 H). МС APCI m/z 269 [M+H]+, 301 [M+H+MeOH]+.
NaBH4 (0,22 г, 5,82 ммоль) добавляли к раствору 90 (0,776 г, 2,89 ммоль) в MeOH (100 мл) при 0°С. Раствор перемешивали при 0°С в течение 1 час, затем гасили насыщенным раствором соли и распределяли между EtOAc и водой. Колоночная хроматография органической части на силикагеле (элюирование смесью 1:1 EtOAc/гексаны) давала {2-[4-(трифторметокси)фенил]-5-пиримидинил}метанол (91) (0,657 г, 84%) в виде белого твердого вещества: т. пл. 84-85°С;1H ЯМР [(CD3)2SO] δ 8,86 (с, 2 H), 8,50 (д, J=8,9 Гц, 2 H), 7,51 (д, J=8,9 Гц, 2 H), 5,46 (т, J=5,5 Гц, 1 H), 4,60 (д, J=5,5 Гц, 2 H). МС APCI m/z 271 [M+H]+.
Мезилхлорид (0,55 мл, 7,0 ммоль) добавляли к раствору спирта 91 (0,951 г, 3,52 ммоль) и Et3N (1,5 мл, 10,8 ммоль) в ТГФ (40 мл) при 0°С и смесь перемешивали при 0°С в течение 1 час. Смесь распределяли между EtOAc и водой и органический слой сушили и упаривали, получая при этом масло, которое растворяли в ацетоне (100 мл). Добавляли LiBr (6,10 г, 70,2 ммоль) и смесь кипятили с обратным холодильником в атмосфере N2 в течение 1 час, затем фильтровали и упаривали. Остаток распределяли между EtOAc и водой и органический слой сушили и упаривали. Колоночная хроматография на силикагеле (элюирование CH2Cl2) давала 5-(бромметил)-2-[4-(трифторметокси)фенил]пиримидин (92) (1,097 г, 94%) в виде белого твердого вещества: т. пл. 80-81°С;1H ЯМР (CDCl3) δ 8,82 (с, 2 H), 8,50 (д, J=9,0 Гц, 2 H), 7,32 (д, J=9,0 Гц, 2 H), 4,48 (с, 2 H). МС APCI m/z 333, 335 [M+H]+.
NaH (60% масс./масс., 0,125 г, 3,1 ммоль) добавляли к раствору спиртового оксазина 41 (0,375 г, 2,03 ммоль) и бромида 92 (0,709 г, 2,13 ммоль) в ДМФА (30 мл) при -78°С. Перемешиваемую смесь нагревали до 0°С в течение 1 час, гасили водой и распределяли между EtOAc и водой. Колоночная хроматография органической части на силикагеле (элюирование смесью 19:1 EtOAc/MeOH) давала 16 (0,512 г, 58%) в виде белого твердого вещества: т. пл. 227-230°С (MeOH);1H ЯМР [(CD3)2SO] δ 8,88 (с, 2 H), 8,49 (д, J=8,9 Гц, 2 H), 8,03 (с, 1 H), 7,51 (д, J=8,9 Гц, 2 H), 4,79 (д, J=12,6 Гц, 1 H), 4,76 (д, J=12,6 Гц, 1 H), 4,70 (дт, J=12,0, 2,5 Гц, 1 H), 4,49 (ушир. д, J=12,0 Гц, 1 H), 4,29-4,35 (м, 2 H), 4,25 (дд, J=13,3, 3,1 Гц, 1 H). Анализ (C18H14F3N5O5) C, H, N.
T. Синтез (6S)-2-нитро-6-({4-[4-(трифторметокси)бензил]бензил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (17) способом схемы 13

Раствор метил-4-(бромметил)бензоата (93) (0,23 мл, 1,0 ммоль) и 4-(трифторметокси)фенилбороновой кислоты (44) (0,24 г, 1,1 ммоль) в DME (3 мл) и 2 M водном K2CO3 (1 мл) дегазировали, затем обрабатывали тетракис(трифенилфосфин)палладием (58 мг, 50 мкмоль). Реакционную смесь перемешивали в атмосфере N2 при 105°С в течение 24 час и затем добавляли EtOAc (250 мл). Органический слой промывали водой, водный слой снова экстрагировали EtOAc (100 мл) и объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле с элюированием смесью петролейный эфир/EtOAc (9:1), получая при этом метил-4-[4-(трифторметокси)бензил]бензоат (94) (215 мг, 68%) в виде бесцветного масла;1H ЯМР (CDCl3) δ 7,98-7,94 (м, 2 H), 7,26-7,22 (м, 2 H), 7,20-7,12 (м, 4 H), 4,03 (с, 2 H), 3,90 (с, 3 H). HREIMS, вычислено для C16H13F3O3 m/z (М+) 310,0817, найдено 310,0815.
LiAlH4 (55 мг, 1,45 ммоль) добавляли к раствору сложного эфира 94 (203 мг, 0,65 ммоль) в простом эфире (5 мл) и смесь перемешивали при комнатной температуре в течение 3 час, затем добавляли EtOAc (150 мл). Органический слой промывали водой, водный слой снова экстрагировали EtOAc (100 мл) и объединенные органические слои промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали при пониженном давлении, получая при этом {4-[4-(трифторметокси)бензил]фенил}метанол (95) (185 мг, колич. выход) в виде белого твердого вещества: т. пл. (EtOAc/гексан) 60-61°С;1H ЯМР (CDCl3) δ 7,32-7,27 (м, 2 H), 7,20-7,14 (м, 4 H), 7,13-7,09 (м, 2 H), 4,67 (с, 2 H), 3,98 (с, 2 H). HREIMS, вычислено для C15H13F3O2 m/z (М+) 282,0868, найдено 282,0866.
Раствор спирта 95 (0,18 г, 0,64 ммоль) в CH2Cl2 (4 мл) обрабатывали PBr3 (115 мкл, 1,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 час, затем добавляли EtOAc (150 мл). Органический слой промывали водой (100 мл), водный слой снова экстрагировали EtOAc (100 мл) и объединенные органические слои промывали насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле с элюированием смесью петролейный эфир/EtOAc (9:1), получая при этом 1-(бромметил)-4-[4-(трифторметокси)бензил]бензол (96) (0,14 г, 64%) в виде белого твердого вещества: т. пл. (EtOAc/гексан) 33-35°С;1H ЯМР (CDCl3) δ 7,34-7,30 (м, 2 H), 7,20-7,10 (м, 6 H), 4,48 (с, 2 H), 3,97 (с, 2 H). HREIMS, вычислено для C15H1279BrF3O m/z (М+) 344,0024, найдено 344,0033; вычислено для C15H1281BrF3O m/z (М+) 346,0003, найдено 346,0011.
Раствор бромида 96 (0,12 г, 0,35 ммоль) и спирта 41 (54 мг, 0,29 ммоль) в ДМФА (2 мл) обрабатывали NaH (60% в масле, 17 мг, 0,43 ммоль) и смесь перемешивали при комнатной температуре в течение 3 час, затем добавляли EtOAc (150 мл). Органический слой промывали водой (100 мл), водный слой снова экстрагировали EtOAc (100 мл) и объединенные органические слои промывали насыщенным раствором соли (100 мл), сушили (Na2SO4) и концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле с элюированием смесью 0-3% МеОН/CH2Cl2, получая при этом 17 (95 мг, 73%) в виде бледно-желтого твердого вещества: т. пл. (CH2Cl2/MeOH) 132-133°С;1H ЯМР [(CD3)2SO] δ 8,00 (с, 1 H), 7,35-7,30 (м, 2 H), 7,28-7,19 (м, 6 H), 4,63 (дт, J=11,9, 2,3 Гц, I H), 4,61 (д, J=11,8 Гц, 1 H), 4,57 (д, J=11,8 Гц, 1 H), 4,45 (д, J=11,9 Гц, 1 H), 4,27-4,17 (м, 3 H), 3,96 (с, 2 H). Анализ (C21H18F3N3O5) C, H, N.
U. Синтез (6S)-2-нитро-6-[(5-{[4-(трифторметокси)фенил]этинил}-2-пиридинил)метокси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (18) способом схемы 14

Смесь бромида 59 (см. пример 21) (0,310 г, 0,873 ммоль), PdCl2(PPh3)2 (33 мг, 0,047 ммоль) и иодида меди (18 мг, 0,095 ммоль) в ДМФА (4 мл) и Et3N (4 мл) продували N2. Добавляли этинилтриметилсилан (0,61 мл, 4,3 ммоль) и смесь перемешивали в герметизированной трубке при 50°С в течение 18 час и затем распределяли между EtOAc и водой. Остаток растворяли в ТГФ (20 мл), охлаждали до 0°С и обрабатывали TBAF (1 М в ТГФ, 1,8 мл) и затем раствор перемешивали в течение 2 час. Удаление растворителя давало остаток, который распределяли между EtOAc и водой. Колоночная хроматография органической части на силикагеле с применением градиентного элюирования (0-5% МеОН:EtOAc) давала (6S)-6-[(5-этинил-2-пиридинил)метокси]-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазин (97) (0,178 г, 68%) в виде рыжевато-коричневого твердого вещества: т. пл. 135-136°С;1H ЯМР [(CD3)2SO] δ 8,61 (д, J=1,6 Гц, 1 H), 8,02 (с, 1 H), 7,90 (дд, J=8,0, 2,2 Гц, 1 H), 7,38 (д, J=8,0 Гц, 1 H), 4,78 (д, J=13,8 Гц, 1 H), 4,74 (д, J=13,8 Гц, 1 H), 4,69 (дт, J=12,0, 2,6 Гц, 1 H), 4,49 (д, J=12,0 Гц, 1 H), 4,40 (с, 1 H), 4,30-4,35 (м, 2 H), 4,25 (дд, J=13,7, 3,5 Гц, 1 H). Анализ (C14H12N4O4) C, H, N.
Смесь алкина 97 (0,075 г, 0,25 ммоль), 1-иод-4-(трифторметокси)бензола (70) (0,088 г, 0,30 ммоль) и иодида меди (5 мг, 0,03 ммоль) в ДМФА (2 мл) и Et3N (2 мл) продували N2. Добавляли PdCl2(PPh3)2 (9 мг, 0,01 ммоль) и смесь перемешивали при комнатной температуре в течение 0,5 час и затем распределяли между EtOAc и водой. Колоночная хроматография органической части на силикагеле с применением градиентного элюирования (0-5% MeOH:EtOAc) давала 18 (0,084 г, 73%) в виде белого твердого вещества: т. пл. 207-208°С;1H ЯМР [(CD3)2SO] δ 8,71 (д, J=1,7 Гц, 1 H), 8,03 (с, 1 H), 7,99 (дд, J=8,1, 2,2 Гц, 1 H), 7,73 (д, J=8,9 Гц, 2 H), 7,42-7,47 (м, 3 H), 4,81 (д, J=13,9 Гц, 1 H), 4,77 (д, J=13,9 Гц, 1 H), 4,71 (дт, J=12,0, 2,5 Гц, 1 H), 4,51 (д, J=1 1,9 Гц, 1 H), 4,31-4,37 (м, 2 H), 4,26 (дд, J=13,7, 3,5 Гц, 1 H). Анализ (C21H15F3N4O5) C, H, N.
V. Синтез (6S)-2-нитро-6-[(5-{[6-(трифторметил)-3-пиридинил]этинил}-2-пиридинил)метокси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (25) способом схемы 14

Сочетание Соногашира алкина 97 (см. пример 2U) (0,075 г, 0,25 ммоль) и 5-бром-2-(трифторметил)пиридина (98) (0,068 г, 0,30 ммоль), как в примере 2U, при 50°С в течение 0,5 час давало 25 (0,086 г, 77%) в виде белого твердого вещества: т. пл. 226-227°С;1H ЯМР [(CD3)2SO] δ 8,97 (д, J=1,3 Гц, 1 H), 8,78 (д, J=1,4 Гц, 1 H), 8,30 (дд, J=8,0, 1,4 Гц, 1 H), 8,06 (дд, J=8,1, 2,2 Гц, 1 H), 8,04 (с, 1 H), 8,00 (д, J=8,2 Гц, 1 H), 7,43 (д, J=8,2 Гц, 1 H), 4,83 (д, J=13,9 Гц, 1 H), 4,79 (д, J=13,9 Гц, 1 H), 4,72 (дт, J=12,0, 2,5 Гц, 1 H), 4,51 (д, J=11,9 Гц, 1 H), 4,32-4,38 (м, 2 H), 4,26 (дд, J=13,8, 3,5 Гц, 1 H). Анализ (C20H14F3N5O4) C, H, N.
W. Синтез (6S)-2-нитро-6-({(2Е])-3-[4'-(трифторметокси)[1,1'-бифенил]-4-ил]-2-пропенил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (19) способом схемы 15

Раствор метил-(Е)-3-(4-бромфенил)-2-пропеноата (99) (0,500 г, 2,07 ммоль) и 4-(трифторметокси)фенилбороновой кислоты (44) (0,612 г, 2,97 ммоль) в диоксане (40 мл) и водном K2CO3 (2 M, 10 мл, 20 ммоль) продували N2. Добавляли Pd(dppf)Cl2 (0,050 г, 0,06 ммоль) и раствор кипятили с обратным холодильником в атмосфере N2 в течение 1 час. Диоксан удаляли и остаток экстрагировали EtOAc, органическую фракцию сушили и растворитель удаляли. Колоночная хроматография остатка на силикагеле с применением градиентного элюирования (от гексанов до CH2Cl2) давала метил-(2E)-3-[4'-(трифторметокси)[1,1'-бифенил]-4-ил]-2-пропеноат (100) (0,567 г, 85%) в виде белого твердого вещества: т. пл. 98-100°С;1H ЯМР (CDCl3) δ 7,73 (д, J=16,0 Гц, 1 H), 7,56-7,63 (м, 6 H), 7,30 (дд, J=8,8, 0,9 Гц, 2 H), 6,48 (д, J=16,0 Гц, 1 H), 3,82 (с, 3 H). МС APCI m/z 323 [M+H]+.
DIBAL-H (20% масс./масс. в толуоле, 2 мл, 2,39 ммоль) добавляли к суспензии сложного эфира 100 (0,396 г, 1,23 ммоль) в толуоле (12 мл) при -78°С. Смесь нагревали до комнатной температуры, перемешивали в течение 1 час и затем выливали на охлажденный льдом раствор NH4Cl (50 мл). Смесь разбавляли CH2Cl2 (100 мл), фильтровали через целит и органический слой сушили и упаривали. Колоночная хроматография остатка на силикагеле с применением градиентного элюирования (от CH2Cl2 до смеси 95:5 CH2Cl2:Et0Ac) давала (2Е)-3-[4'-(трифторметокси)[1,1'-бифенил]-4-ил]-2-пропен-1-ол (101) (0,195 г, 54%) в виде белого твердого вещества: т. пл. 121-123°С;1H ЯМР (CDCl3) δ 7,60 (д, J=8,7 Гц, 2 H), 7,53 (д, J=8,4 Гц, 2 H), 7,47 (д, J=8,4 Гц, 2 H), 7,28 (д, J=8,1 Гц, 2 H), 6,66 (д, J=15,9 Гц, 1 H), 6,42 (дт, J=15,9, 5,7 Гц, 1 H), 4,36 (дд, J=5,9, 5,7 Гц, 2 H), 1,44 (т, J=5,9 Гц, 1 H). МС APCI m/z 307 [M-Н-Н2О+MeOH]-.
PBr3 (26 мкл, 0,28 ммоль) добавляли к раствору спирта 101 (0,159 г, 0,540 ммоль) в Et2O (10 мл) при 0°С. Смесь нагревали до комнатной температуры и перемешивали в течение 1 час, затем гасили льдом и экстрагировали Et2O. Органическую фракцию сушили и упаривали и затем колоночной хроматографией остатка на силикагеле (элюирование CH2Cl2) получали 4-[(1Е)-3-бром-1-пропенил]-4'-(трифторметокси)-1,1'-бифенил (102) (0,123 г, 71%) в виде белого твердого вещества: т. пл. 121-123°С;1Н ЯМР (CDCl3) δ 7,60 (д, J=8,8 Гц, 2 H), 7,53 (д, J=8,4 Гц, 2 H), 7,46 (д, J=8,4 Гц, 2 H), 7,28 (д, J=8,8 Гц, 2 H), 6,69 (д, J=15,6 Гц, 1 H), 6,45 (дт, J=15,6, 7,8 Гц, 1 H), 4,18 (дд, J=7,8, 0,9 Гц, 2 H). МС APCI m/z 277 [M+H-HBr]+.
NaH (60% масс./масс., 0,016 г, 0,40 ммоль) добавляли к раствору спиртового оксазина 41 (0,050 г, 0,27 ммоль) и бромида 102 (0,100 г, 0,28 ммоль) в ДМФА (6 мл) при -78°С. Смесь перемешивали при 0°С в течение 1 час, затем гасили льдом и распределяли между EtOAc и водой. Органическую фракцию сушили и упаривали и затем колоночной хроматографией остатка на силикагеле с применением градиентного элюирования (от смеси 1:1 гексаны:EtOAc до EtOAc) получали 19 (0,079 г, 63%) в виде белого твердого вещества: т. пл. 220-221°С;1H ЯМР [(CD3)2SO] δ 8,04 (с, 1 H), 7,80 (д, J=8,8 Гц, 2 H), 7,66 (д, J=8,4 Гц, 2 H), 7,55 (д, J=8,4 Гц, 2 H), 7,44 (д, J=8,8 Гц, 2 H), 6,66 (д, J=16,0 Гц, 1 H), 6,43 (дт, J=16,0, 5,9 Гц, 1 H), 4,65 (д, J=11,9 Гц, 1 H), 4,48 (д, J=11,9 Гц, 1 H), 4,21-4,35 (м, 5 H). Анализ (C22H18F3N3O5) C, H, N.
Х. Синтез (6S)-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин-6-ил-4-[4-(трифторметокси)фенил]-1-пиперазинкарбоксилата (20) способом схемы 16

Трифосген (0,80 г, 2,70 ммоль) добавляли порциями при перемешивании к охлаждаемой ледяной баней суспензии спиртового оксазина 41 (1,00 г, 5,40 ммоль) и Et3N (1,12 мл, 8,10 ммоль) в безводном ТГФ (30 мл). Спустя 15 мин ледяную баню удаляли и суспензию перемешивали при комнатной температуре в течение 90 мин, получая при этом раствор сырого карбонилхлорида 103. Затем добавляли раствор 1-[4-(трифторметокси)фенил]пиперазина (104) (1,40 г, 5,67 ммоль) в ТГФ (10 мл) и перемешивание продолжали в течение 2 час. Добавляли воду и смесь экстрагировали EtOAc. Упаривание данного экстракта давало маслянистое твердое вещество, которое хроматографировали на диоксиде кремния. Элюирование смесью EtOAc/петролейный эфир (1:1) давало предварительные фракции, и затем дальнейшее элюирование посредством EtOAc давало 20 (1,53 г, 62%) в виде желтого порошка после растирания с простым эфиром: т. пл. 166-168°С;1H ЯМР [(CD3)2SO] δ 8,06 (с, 1 H), 7,19 (д, J=9,0 Гц, 2 H), 6,99 (д, J=9,0 Гц, 2 H), 5,32 (ушир. с, 1 H), 4,62-4,55 (м, 2 H), 4,39 (дд, J=13,9, 3,5 Гц, 1 H), 4,27 (ушир. д, J=13,9 Гц, 1 H), 3,45 (ушир. м, 4 H), 3,13 (ушир. м, 4 H). Анализ (C18H18F3N5O6) C, H, N.
Y. Синтез (6S)-6-({5-[3-фтор-4-(трифторметокси)фенил]-2-пиридинил}метокси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (22) способом схемы 17

Перемешиваемую смесь (5-бром-2-пиридинил)метанола (105) (753 мг, 4,00 ммоль), 3-фтор-4-(трифторметокси)фенилбороновой кислоты (56) (см. пример 2G) (1,165 г, 5,20 ммоль) и Pd(dppf)Cl2(366 мг, 0,50 ммоль) в толуоле (40 мл) и EtOH (20 мл) дегазировали в течение 15 мин (вакуумный насос) и затем вводили N2. При помощи шприца добавляли 2 М водный раствор Na2CO3 (10 мл, 20,0 ммоль) и перемешиваемую смесь снова дегазировали в течение 15 мин и затем вводили N2. Образовавшуюся смесь перемешивали при 89°С в течение 2 час и затем охлаждали, разбавляли водным NaHCO3 (120 мл) и экстрагировали CH2Cl2 (6×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование смесью 50-75% CH2Cl2/петролейный эфир сначала давало предварительные фракции, и затем дальнейшее элюирование смесью 75% CH2Cl2/петролейный эфир и смесью 0-0,5% МеОН/CH2Cl2 давало {5-[3-фтор-4-(трифторметокси)фенил]-2-пиридинил}метанол (106) (687 мг, 60%) в виде бледно-желто-коричневого твердого вещества: т. пл. 51-53°С;1H ЯМР (CDCl3) δ 8,76 (д, J=1,9 Гц, 1 H), 7,84 (дд, J=8,1, 2,3 Гц, 1 H), 7,46-7,34 (м, 4 H), 4,83 (д, J=5,1 Гц, 2 H), 3,47 (т, J=5,2 Гц, 1 H). HRESIMS, вычислено для C13H10F4NO2 m/z (М+H)+ 288,0642, найдено 288,0641.
Раствор спирта 106 (678 мг, 2,36 ммоль) и трифенилфосфина (746 мг, 2,84 ммоль) в безводном CH2Cl2(30 мл) осторожно обрабатывали перекристаллизованным N-бромсукцинимидом (507 мг, 2,85 ммоль) (охлаждение водяной баней) и смесь перемешивали при комнатной температуре в течение 3 час. Образовавшийся раствор концентрировали и затем добавляли избыток петролейного эфира у верха колонки силикагеля (25 г в петролейном эфире), промывая минимальным дополнительным количеством CH2Cl2. Сначала элюирование петролейным эфиром давало предварительные фракции, и затем дополнительное элюирование смесью 10-20% Et2O/пентан давало 2-(бромметил)-5-[3-фтор-4-(трифторметокси)фенил]пиридин (107) (616 мг, 75%) в виде белого твердого вещества, которое применяли непосредственно в следующей стадии;1H ЯМР (CDCl3) δ 8,76 (дд, J=2,4, 0,6 Гц, 1 H), 7,84 (дд, J=8,1, 2,4 Гц, 1 H), 7,54 (дд, J=8,0, 0,6 Гц, 1 H), 7,46-7,33 (м, 3 H), 4,60 (с, 2 H). HRESIMS, вычислено для C13H9BrF4NO m/z [М+Н]+ 351,9778, 349,9798, найдено 351,9778, 349,9798.
Раствор спиртового оксазина 41 (311 мг, 1,68 ммоль) и бромида 107 (614 мг, 1,75 ммоль) в безводном ДМФА (6,5 мл) в атмосфере N2 при 0°С обрабатывали 60% NaH (88,5 мг, 2,21 ммоль), затем быстро дегазировали и снова вводили N2. После перемешивания при комнатной температуре в течение 2,5 час реакционную смесь охлаждали (СО2/ацетон), гасили смесью лед/водный NaHCO3 (20 мл), добавляли к насыщенному раствору соли (40 мл) и экстрагировали CH2Cl2 (8×50 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Сначала элюирование смесью 0-0,75% MeOH/CH2Cl2 давало предварительные фракции, и затем дальнейшее элюирование смесью 0,75-1,5% МеОН/CH2Cl2 давало 22 (676 мг, 89%) в виде бледно-желтого твердого вещества: т. пл. (CH2Cl2/пентан) 182-184°С;1H ЯМР (CDCl3) δ 8,74 (дд, J=2,3, 0,7 Гц, 1 H), 7,86 (дд, J=8,1, 2,4 Гц, 1 H), 7,48-7,38 (м, 4 H), 7,35 (ддд, J=8,4, 2,2, 1,0 Гц, 1 H), 4,87 (д, J=13,0 Гц, 1 H), 4,81 (д, J=13,0 Гц, 1 H), 4,70 (ддд, J=12,2, 3,5, 1,5 Гц, 1 H), 4,40 (дд, J=12,2, 1,4 Гц, 1 H), 4,33-4,20 (м, 3 H). Анализ (C19H14F4N4O5) C, H, N.
Z. Синтез (6S)-2-нитро-6-({6-[4-(трифторметокси)фенил]-3-пиридазинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (24) способом схемы 18

NaH (60% масс./масс., 0,304 г, 7,60 ммоль) добавляли к раствору спиртового оксазина 41 (0,893 г, 4,82 ммоль) в ДМФА (20 мл) при 0°С. Образовавшийся раствор охлаждали до -42°С и добавляли раствор 3-(бромметил)-6-хлорпиридазина (108) (полученного посредством свободно-радикального бромирования 3-хлор-6-метилпиридазина, как описано в EP 1555259) (1,053 г, 5,08 ммоль) в ДМФА (5 мл). Смесь перемешивали при -42°С в течение 1 час и затем гасили льдом. Добавляли EtOAc (200 мл) и органический слой сушили (MgSO4) и затем концентрировали при пониженном давлении. Остаток хроматографировали на силикагеле, сначала элюируя смесью гексаны/EtOAc (1:1) для удаления непрореагировавшего 3-(бромметил)-6-хлорпиридазина и затем EtOAc для получения (6S)-6-[(6-хлор-3-пиридазинил)метокси]-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (109) (0,843 г, 56%) в виде белого твердого вещества: т. пл. 180-184°С;1H ЯМР [(CD3)2SO] δ 8,02 (с, 1 H), 7,93 (д, J=8,8 Гц, 1 H), 7,74 (д, J=8,8 Гц, 1 H), 4,97 (д, J=13,2 Гц, 1 H), 4,94 (д, J=13,2 Гц, 1 H), 4,69 (дт, J=12,0, 2,6 Гц, 1 H), 4,50 (д, J=12,1 Гц, 1 H), 4,30-4,39 (м, 2 H), 4,25 (дд, J=13,5, 3,3 Гц, 1 H). Анализ (C11H10ClN5O4) C, H, N.
Сочетание Сузуки хлорида 109 и 4-(трифторметокси)фенилбороновой кислоты (44), как в примере 2D, давало 24 (66%) в виде белого твердого вещества: т. пл. 194°С (разл.);1H ЯМР [(CD3)2SO] δ 8,25-8,30 (м, 3 H), 8,03 (с, 1 H), 7,76 (д, J=8,9 Гц, 1 H), 7,55 (д, J=8,9 Гц, 2 H), 5,04 (д, J=13,2 Гц, 1 H), 5,00 (д, J=13,2 Гц, 1 H), 4,74 (дт, J=12,0, 2,6 Гц, 1 H), 4,52 (д, J=11,9 Гц, 1 H), 4,33-4,43 (м, 2 H), 4,28 (дд, J=13,5, 3,3 Гц, 1 H). Анализ (C18H14F3N5O5) C, H, N.
AA. Синтез (6S)-6-{[4-(5-фтор-2-пиридинил)бензил]окси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (26) способом схемы 19

Перемешиваемую смесь 4-(гидроксиметил)фенилбороновой кислоты (34) (501 мг, 3,30 ммоль) и Pd(dppf)Cl2 (338 мг, 0,462 ммоль) в толуоле (36 мл) и EtOH (18 мл) дегазировали в течение 15 мин (вакуумный насос) и затем вводили N2. Посредством шприца добавляли 2 М водный раствор Na2CO3 (9 мл, 18 ммоль) и перемешиваемую смесь снова дегазировали в течение 15 мин и затем вводили N2. Посредством шприца добавляли 2-бром-5-фторпиридин (110) (1,44 г, 8,18 ммоль) и образовавшуюся смесь перемешивали при 89°С в течение 200 мин. Охлажденную смесь затем разбавляли водным NaHCO3 (100 мл) и экстрагировали CH2Cl2 (5×100 мл). Экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование сначала CH2Cl2 и смесью 0-25% Et2O/петролейный эфир давало предварительные фракции, и затем дальнейшее элюирование смесью 33-50% Et2O/петролейный эфир давало [4-(5-фтор-2-пиридинил)фенил]метанол (111) (307 мг, 46%) в виде кремового твердого вещества (после растирания с пентаном): т. пл. 100-101°С;1H ЯМР (CDCl3) δ 8,54 (д, J=2,9 Гц, 1 H), 7,94 (дт, J=8,4, 1,9 Гц, 2 H), 7,72 (ддд, J=8,8, 4,2, 0,5 Гц, 1 H), 7,50-7,43 (м, 3 H), 4,76 (д, J=6,0 Гц, 2 H), 1,69 (т, J=6,0 Гц, 1 H). HRESIMS, вычислено для C12H11FNO m/z [М+Н]+ 204,0819, найдено 204,0824.
Раствор спирта 111 (305 мг, 1,50 ммоль) и трифенилфосфина (474 мг, 1,81 ммоль) в безводном CH2Cl2 (12 мл) осторожно обрабатывали перекристаллизованным N-бромсукцинимидом (322 мг, 1,81 ммоль) (охлаждение водяной баней) и смесь перемешивали при комнатной температуре в течение 3 час. Образовавшийся раствор концентрировали и затем добавляли к избыточному пентану с верха колонки с силикагелем (20 г в пентане), промывая минимальным дополнительным CH2Cl2. Элюирование сначала пентаном давало предварительные фракции, и затем дальнейшее элюирование смесью 20-50% Et2O/пентан давало 2-[4-(бромметил)фенил]-5-фторпиридин (112) (348 мг, 87%) в виде белого твердого вещества, которое применяли непосредственно в следующей стадии;1H ЯМР (CDCl3) δ 8,54 (д, J=2,9 Гц, 1 H), 7,92 (дт, J=8,4, 1,9 Гц, 2 H), 7,72 (ддд, J=8,7, 4,3, 0,4 Гц, 1 H), 7,52-7,43 (м, 3 H), 4,54 (с, 2 H). HRESIMS, вычислено для C12H10BrFN m/z [М+Н]+ 267,9955, 265,9975, найдено 267,9959, 265,9979.
Раствор спиртового оксазина 41 (242 мг, 1,31 ммоль) и бромида 112 (346 мг, 1,30 ммоль) в безводном ДМФА (5 мл) в атмосфере N2 при 0°С обрабатывали 60% NaH (70 мг, 1,75 ммоль), затем быстро дегазировали и снова вводили N2. После перемешивания при комнатной температуре в течение 135 мин реакционную смесь охлаждали (СО2/ацетон), гасили смесью лед/водный NaHCO3 (20 мл), добавляли к насыщенному раствору соли (100 мл) и экстрагировали CH2Cl2 (9×100 мл). Объединенные экстракты упаривали досуха и остаток хроматографировали на силикагеле. Элюирование сначала смесью 0-6% EtOA/CH2Cl2 давало предварительные фракции, и затем дальнейшее элюирование смесью 7-10% EtOAc/CH2Cl2 давало сырой продукт, который далее хроматографировали на силикагеле. Элюирование сначала петролейным эфиром и смесью 50-67% EtOAc/петролейный эфир давало предварительные фракции, и затем дальнейшее элюирование смесью 30% EtOAc/CH2Cl2 давало 26 (357 мг, 74%) в виде кремового твердого вещества: т. пл. (CH2Cl2/пентан) 180-181°С;1H ЯМР (CDCl3) δ 8,54 (д, J=2,9 Гц, 1 H), 7,95 (дт, J=8,4, 1,9 Гц, 2 H), 7,72 (ддд, J=8,8, 4,2, 0,4 Гц, 1 H), 7,48 (ддд, J=8,7, 8,1, 2,9 Гц, 1 H), 7,41 (ушир. д, J=8,4 Гц, 2 H), 7,37 (с, 1 H), 4,79 (д, J=12,2 Гц, 1 H), 4,68 (д, J=12,2 Гц, 1 H), 4,61 (ддд, J=12,1, 3,7, 1,9 Гц, 1 H), 4,35 (дд, J=12,1, 1,5 Гц, 1 H), 4,20-4,09 (м, 3 H). Анализ (C18H15FN4O4) C, H, N.
BB. Синтез (6S)-2-нитро-6-({1-[4-(трифторметокси)фенил]-1H-пиразол-4-ил}метокси)-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (27) способом схемы 20.

Смесь этил-(2Е)-2-циано-3-этокси-2-пропеноата (113) (1,87 г, 11,1 ммоль), гидрохлорида 4-(трифторметокси)фенилгидразина (114) (2,286 г, 10,00 ммоль) и NaOAc (0,90 г, 11,0 ммоль) в AcOH (7,5 мл) и воде (2,5 мл) нагревали до 100°С в атмосфере N2 в течение 15 час. Смесь выливали на лед и образовавшийся осадок отделяли фильтрованием и перекристаллизовывали (МеОН/вода), получая при этом этил-5-амино-1-[4-(трифторметокси)фенил]-1H-пиразол-4-карбоксилат (115) (2,965 г, 94%) в виде белых хлопьев: т. пл. 102-103°С;1H ЯМР [(CD3)2SO] δ 7,73 (с, 1 H), 7,68 (д, J=9,1 Гц, 2 H), 7,53 (д, J=9,1 Гц, 2 H), 6,41 (ушир. с, 2 H), 4,22 (кв, J=7,1 Гц, 2 H), 1,27 (т, J=7,1 Гц, 3 H). МС APCI m/z 316 [M+H]+.
Раствор аминопиразола 115 (1,850 г, 5,87 ммоль) и изоамилнитрита (0,83 мл, 6,18 ммоль) в ТГФ (20 мл) кипятили с обратным холодильником в течение 14 час, затем добавляли дополнительный изоамилнитрит (0,83 мл, 6,18 ммоль) и раствор кипятили с обратным холодильником в течение 6 час. Растворитель удаляли при пониженном давлении, получая при этом твердое вещество, которое перекристаллизовывали (EtOH) с получением этил-1-[4-(трифторметокси)фенил]-1Н-пиразол-4-карбоксилата (116) (1,527 г, 87%) в виде белых хлопьев: т. пл. 114-116°С;1H ЯМР (CDCl3) δ 8,38 (д, J=0,5 Гц, 1 H), 8,10 (с, 1 H), 7,74 (д, J=9,1 Гц, 2 H), 7,34 (д, J=9,1 Гц, 2 H), 4,35 (кв, J=7,1 Гц, 2 H), 1,38 (т, J=7,1 Гц, 3 H). МС APCI m/z 301 [M+H]+.
Смесь сложного эфира 116 (0,730 г, 2,43 ммоль) и LiAlH4(0,200 г, 5,28 ммоль) в Et2O (20 мл) кипятили с обратным холодильником в течение 2 час. Смесь охлаждали до 0°С, гасили льдом, разбавляли Et2O (100 мл) и фильтровали через целит. Органический слой сушили (MgSO4), затем подвергали колоночной хроматографии на силикагеле (смесь 19:1 CH2Cl2/EtOAc), получая при этом {1-[4-(трифторметокси)фенил]-1Н-пиразол-4-ил}метанол (117) (0,520 г, 83%) в виде белого твердого вещества: т. пл. 73-74°С;1H ЯМР (CDCl3) δ 7,91 (с, 1 H), 7,67-7,73 (м, 3 H), 7,31 (д, J=8,4 Гц, 2 H), 4,69 (д, J=5,5 Гц, 2 H), 1,57 (т, J=5,5 Гц, 1 H). МС APCI m/z 259 [M+H]+.
PBr3 (76 мкл, 0,81 ммоль) добавляли к раствору спирта 117 (0,210 г, 0,813 ммоль) в простом эфире (10 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 2 час, затем охлаждали до 0°С, гасили льдом и распределяли между Et2O и водой. Колоночная хроматография органической порции на силикагеле (элюирование CH2Cl2) давала 4-(бромметил)-1-[4-(трифторметокси)фенил]-1Н-пиразол (118) (0,212 г, 81%) в виде белого твердого вещества: т. пл. 50-51°С;1H ЯМР (CDCl3) δ 7,94 (д, J=0,5 Гц, 1 H), 7,74 (с, 1 H), 7,69 (д, J=9,1 Гц, 2 H), 7,31 (д, J=9,1 Гц, 2 H), 4,50 (с, 2 H). МС APCI m/z 321, 323 [M+H]+.
NaH (60% масс./масс., 30 мг, 0,75 ммоль) добавляли к раствору спиртового оксазина 41 (0,091 г, 0,49 ммоль) и бромида 118 (0,157 г, 0,49 ммоль) в ДМФА (10 мл) при 0°С. Смесь перемешивали в течение 2 час, гасили льдом и распределяли между EtOAc и водой. Колоночная хроматография органической порции на силикагеле с элюированием с градиентом от смеси 1:1 гексаны:EtOAc до EtOAc давала 27 (0,148 г, 71%) в виде белого твердого вещества: т. пл. 150-151°С;1H ЯМР [(CD3)2SO] δ 8,53 (с, 1 H), 8,02 (с, 1 H), 7,93 (д, J=9,1 Гц, 2 H), 7,77 (с, 1 H), 7,50 (д, J=9,1 Гц, 2 H), 4,56-4,66 (м, 3 H), 4,46 (д, J=11,8 Гц, 1 H), 4,20-4,26 (м, 3 H). Анализ (C17H14F3N5O5) C, H, N.
CC. Синтез (6S)-2-нитро-6-({5-[4-(трифторметокси)фенил]-2-пиримидинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (29) способом схемы 21

Смесь 5-бром-2-метилпиримидина (119) (1,34 г, 7,75 ммоль), N-бромсукцинимида (1,40 г, 7,87 ммоль) и AIBN (0,13 г, 0,79 ммоль) в CCl4 (15 мл) перемешивали при 60°С в течение 3 час. Образовавшуюся смесь фильтровали, фильтровальный осадок промывали Et2O (100 мл) и объединенные фильтраты концентрировали при пониженном давлении. Колоночная хроматография осадка с элюированием смесью 2:1 EtOAc:гексаны давала 5-бром-2-(бромметил)пиримидин (120) (0,214 г, 11%) в виде белого твердого вещества: т. пл. 55-57°С;1H ЯМР (CDCl3) δ 8,79 (с, 2 H), 4,57 (с, 2 H). Анализ (C5H4Br2N2) C, H, N.
NaH (60% масс./масс., 0,170 г, 4,25 ммоль) добавляли к раствору бромида 120 (0,879 г, 3,49 ммоль) и спирта 41 (0,520 г, 2,81 ммоль) в безводном ДМФА (10 мл) при -78°С. Смесь перемешивали при 0°С в течение 0,5 час и затем гасили льдом и экстрагировали EtOAc (200 мл). Органический слой сушили (MgSO4) и упаривали и затем остаток подвергали колоночной хроматографии с применением градиентного элюирования (смесь 0-5% МеОН/EtOAc), получая при этом (6S)-6-[(5-бром-2-пиримидинил)метокси]-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (121) (0,510 г, 51%) в виде бледно-коричневого твердого вещества: т. пл. >290°С;1H ЯМР [(CD3)2SO] δ 8,98 (с, 2 H), 8,03 (с, 1 H), 4,83 (д, J=13,2 Гц, 1 H), 4,80 (д, J=13,2 Гц, 1 H), 4,68 (дт, J=12,0, 2,6 Гц, 1 H), 4,48 (ушир. д, J=11,9 Гц, 1 H), 4,40-4,36 (м, 1 H), 4,31 (дт, J=13,5, 2,1 Гц, 1 H), 4,23 (дд, J=13,5, 3,3 Гц, 1 H). Анализ (C11H10BrN5O4) C, H. N: вычисл. 19,67; найдено 19,17.
Сочетание Сузуки бромида 121 и 4-(трифторметокси)фенилбороновой кислоты (44), как в примере 2D, давало 29 (89%) в виде белого твердого вещества: т. пл. 223-226°С;1H ЯМР [(CD3)2SO] δ 9,14 (с, 2 H), 8,05 (с, 1 H), 7,93 (д, J=8,8 Гц, 2 H), 7,53 (д, J=8,8 Гц, 2 H), 4,91 (д, J=14,5 Гц, 1 H), 4,87 (д, J=14,5 Гц, 1 H), 4,72 (дт, J=11,9, 2,6 Гц, 1 H), 4,51 (ушир. д, J=12,0 Гц, 1 H), 4,45-4,42 (м, 1 H), 4,35 (дт, J=13,5, 2,1 Гц, 1 H), 4,27 (дд, J=13,5, 3,3 Гц, 1 Н). Анализ (C18H14F3N5O5) C, H, N.
DD. Синтез (6S)-2-нитро-6-({3-[4'-(трифторметокси)[1,1'-бифенил]-4-ил]-2-пропинил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (30) способом схемы 22

NaH (60% масс./масс., 0,280 г, 7,0 ммоль) добавляли к раствору спиртового оксазина 41 (1,00 г, 5,40 ммоль) и 1-бром-4-(3-бром-1-пропинил)бензола (122) (получен двумя стадиями из 1-бром-4-иодбензола и пропаргилового спирта, как описано в WO 9524400) (1,57 г, 5,73 ммоль) в ДМФА (25 мл) при 0°С. Смесь перемешивали при 0°С в течение 1 час и затем гасили водой и экстрагировали EtOAc. Органическую фракцию сушили и растворитель удаляли и затем остаток подвергали колоночной хроматографии на силикагеле с применением градиентного элюирования (от смеси 1:1 гексаны:EtOAc до EtOAc), получая при этом (6S)-6-{[3-(4-бромфенил)-2-пропинил]окси}-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (123) (1,58 г, 77%) в виде белого твердого вещества: т. пл. 160-162°С;1H ЯМР [(CD3)2SO] δ 8,03 (с, 1 H), 7,60 (д, J=8,6 Гц, 2 H), 7,42 (д, J=8,6 Гц, 2 H), 4,66 (дт, J=12,1, 2,4 Гц, 1 H), 4,57 (с, 2 H), 4,49 (д, J=12,1 Гц, 1 H), 4,37-4,40 (м, 1 H), 4,30 (дт, J=13,7, 2,0 Гц, 1 H), 4,25 (дд, J=13,7, 3,2 Гц, 1 H). Анализ (C15H12BrN3O4) C, H, N.
Сочетание Сузуки бромида 123 и 4-(трифторметокси)фенилбороновой кислоты (44), как в примере 2D, с последующей колоночной хроматографией продукта на силикагеле с применением градиентного элюирования (от смеси 1:1 гексаны:EtOAc до EtOAc) давало 30 (72%) в виде белого твердого вещества: т. пл. 192-194°С;1Н ЯМР [(CD3)2SO] δ 8,04 (с, 1 H), 7,83 (д, J=8,8 Гц, 2 H), 7,72 (д, J=8,4 Гц, 2 H), 7,58 (д, J=8,4 Гц, 2 H), 7,46 (д, J=8,8 Гц, 2 H), 4,68 (дт, J=12,1, 2,4 Гц, 1 H), 4,61 (с, 2 H), 4,51 (д, J=12,0 Гц, 1 H), 4,39-4,42 (м, 1 H), 4,32 (дт, J=13,6, 2,0 Гц, 1 H), 4,27 (дд, J=13,6, 3,2 Гц, 1 H). Анализ (C22H16F3N3O5) C, H, N.
EE. Синтез (6S)-2-нитро-6-[(4-{(Е)-2-[4-(трифторметокси)фенил]этенил}бензил)окси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (31) способом схемы 23

4-(Трифторметокси)бензальдегид (125) (0,928 г, 4,88 ммоль), K2CO3 (2,8 г, 20 ммоль) и 18-краун-6 (0,04 г, 0,15 ммоль) добавляли к раствору бромида [4-(метоксикарбонил)бензил]трифенилфосфония (124) (2,00 г, 4,07 ммоль) в ТГФ (60 мл) и CH2Cl2 (40 мл). Смесь кипятили с обратным холодильником в атмосфере N2 в течение 18 час и затем распределяли между EtOAc и водой. Органический слой промывали насыщенным раствором соли и растворитель удаляли. Колоночная хроматография остатка на силикагеле с элюированием смесью 19:1 гексаны:EtOAc давала сырой продукт, который перекристаллизовывали из гексанов, получая при этом метил-4-{(Е)-2-[4-(трифторметокси)фенил]этенил}бензоат (126) (0,549 г, 42%) в виде белых хлопьев: т. пл. (гексаны) 120-122°С;1H ЯМР (CDCl3) δ 8,03 (д, J=8,4 Гц, 2 H), 7,51-7,58 (м, 4 H), 7,16-7,24 (м, 3 H), 7,09 (д, J=16,3 Гц, 1 H), 3,93 (с, 3 H). МС APCI m/z 323 [M+H]+.
LiAH4 (0,039 г, 1,03 ммоль) добавляли к раствору сложного эфира 126 (0,166 г, 0,515 ммоль) в Et2O (10 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 0,5 час и затем охлаждали до 0°С, гасили льдом и фильтровали через целит. Органическую фракцию сушили и упаривали и затем колоночной хроматографией остатка с элюированием смесью 95:5 CH2Cl2:MeOH получали (4-{(E)-2-[4-(трифторметокси)фенил]этенил}фенил)метанол (127) (0,182 г, 82%) в виде белого твердого вещества: т. пл. 161-163°С;1H ЯМР [(CD3)2SO] δ 7,78 (д, J=8,8 Гц, 2 H), 7,57 (д, J=8,2 Гц, 2 H), 7,31-7,38 (м, 4 H), 7,27 (с, 2 H), 5,16 (т, J=5,7 Гц, 1 H), 4,51 (д, J=5,7 Гц, 2 H). МС APCI m/z 277 [M+H -Н2О]+.
PBr3 (56 мкл, 0,60 ммоль) добавляли к раствору спирта 127 (0,177 г, 0,601 ммоль) в безводном CH2Cl2 (10 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 1 час и затем охлаждали до 0°С, гасили льдом и экстрагировали CH2Cl2. Органическую фракцию сушили и упаривали и затем колоночной хроматографией остатка при элюировании CH2Cl2 получали 1-{(Е)-2-[4-(бромметил)фенил]этенил}-4-(трифторметокси)бензол (128) (0,125 г, 58%) в виде белого твердого вещества: т. пл. 100-102°С;1H ЯМР (CDCl3) δ 7,52 (д, J=8,6 Гц, 2 H), 7,48 (д, J=8,3 Гц, 2 H), 7,39 (д, J=8,3 Гц, 2 H), 7,20 (ушир. д, J=8,1 Гц, 2 H), 7,10 (д, J=16,3 Гц, 1 H), 7,05 (д, J=16,3 Гц, 1 H), 4,51 (с, 2 H). МС APCI m/z 277 [M+H-HBr]+.
NaH (60% масс./масс., 0,010 г, 0,25 ммоль) добавляли к раствору спирта 41 (0,024 г, 0,13 ммоль) и бромида 128 (0,056 г, 0,16 ммоль) в безводном ДМФА (5 мл) при -78°С. Смесь затем перемешивали при 0°С в течение 1 час, гасили водой и экстрагировали EtOAc. Органическую фракцию сушили и упаривали и затем остаток подвергали колоночной хроматографии с применением градиентного элюирования (от смеси 1:1 гексаны:EtOAc до EtOAc), получая при этом 31 (0,043 г, 72%) в виде белого твердого вещества: т. пл. 228-230°С;1H ЯМР [(CD3)2SO] δ 8,02 (с, 1 H), 7,72 (д, J=8,8 Гц, 2 H), 7,59 (д, J=8,2 Гц, 2 H), 7,27-7,38 (м, 6 H), 4,61-4,70 (м, 3 H), 4,47 (д, J=11,9 Гц, 1 H), 4,20-4,31 (м, 3 H). Анализ (C22H18F3N3O5) C, H, N.
FF. Синтез (6S)-2-нитро-6-[(4-{[4-(трифторметокси)фенил]этинил}бензил)окси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина (32) способом схемы 24

Смесь иодида 43 (см. пример 2C) (1,00 г, 2,49 ммоль) и иодида меди (51 мг, 0,27 ммоль) в ДМФА (10 мл) и Et3N (10 мл) продували N2. Добавляли этинилтриметилсилан (1,0 мл, 7,1 ммоль) и PdCl2(PPh3)2 (93 мг, 0,13 ммоль) и смесь перемешивали в атмосфере N2 в течение 0,5 час. Образовавшуюся смесь распределяли между EtOAc и водой, органическую фракцию сушили и растворитель удаляли. Остаток растворяли в ТГФ (50 мл) и добавляли фторид тетра-н-бутиламмония (5 мл 1 M раствора в ТГФ, 5 ммоль). Раствор перемешивали в течение 2 час и затем концентрировали. Остаток распределяли между EtOAc и водой и органическую фракцию сушили и концентрировали. Колоночная хроматография остатка на силикагеле с применением градиентного элюирования (от смеси 1:1 гексаны:EtOAc до EtOAc) давала (6S)-6-[(4-этинилбензил)окси]-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (129) (0,530 г, 71%) в виде белого твердого вещества: т. пл. 162-164°С;1H ЯМР [(CD3)2SO] δ 8,01 (с, 1 H), 7,45 (д, J=8,3 Гц, 2 H), 7,32 (д, J=8,3 Гц, 2 H), 4,62-4,71 (м, 3 H), 4,47 (д, J=11,9 Гц, 1 H), 4,20-4,30 (м, 3 H), 4,14 (с, 1 H). Анализ (C15H13N3O4) C, H, N.
Сочетание Соногашира алкина 129 и 1-иод-4-(трифторметокси)бензола (70), как в примере 2U, с последующей колоночной хроматографией продукта на силикагеле с применением градиентного элюирования (от смеси 1:1 гексаны:EtOAc до EtOAc) давало 32 (72%) в виде белого твердого вещества: т. пл. 233-236°С;1H ЯМР [(CD3)2SO] δ 8,03 (с, 1 H), 7,69 (д, J=8,9 Гц, 2 H), 7,55 (д, J=8,3 Гц, 2 H), 7,42 (д, J=8,9 Гц, 2 H), 7,37 (д, J=8,3 Гц, 2 H), 4,62-4,73 (м, 3 H), 4,48 (д, J=11,9 Гц, 1 H), 4,22-4,32 (м, 3 H). Анализ (C22H16F3N3O5) C, H, N.
GG. Синтез (6S)-2-нитро-6-[(6-{[4-(трифторметокси)фенил]этинил}-3-пиридинил)метокси]-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазина (33) способом схемы 24

Сочетание Соногашира бромида 52 (см. пример 2F) (0,310 г, 0,873 ммоль) и этинилтриметилсилана (0,61 мл, 4,3 ммоль) при комнатной температуре в течение 18 час с последующим десилилированием посредством TBAF, как в примере 2U, давало (6S)-6-[(6-этинил-3-пиридинил)метокси]-2-нитро-6,7-дигидро-5H-имидазо[2,1-b][1,3]оксазин (130) (0,150 г, 57%) в виде белого твердого вещества: т. пл. 168-170°С;1H ЯМР [(CD3)2SO] δ 8,51 (д, J=1,6 Гц, 1 H), 8,02 (с, 1 H), 7,74 (дд, J=8,0, 2,2 Гц, 1 H), 7,54 (дд, J=8,0, 0,6 Гц, 1 H), 4,73 (д, J=12,6 Гц, 1 H), 4,70 (д, J=12,6 Гц, 1 H), 4,67 (дт, J=12,3, 2,3 Гц, 1 H), 4,47 (д, J=11,9 Гц, 1 H), 4,29 (с, 1 H), 4,20-4,28 (м, 3 H). МС APCI m/z 301 [M+H]+.
Сочетание Соногашира алкина 130 и 1-иод-4-(трифторметокси)бензола (70), как в примере 2U, давало 33 (55%) в виде белого твердого вещества: т. пл. 235-238°С;1H ЯМР [(CD3)2SO] δ 8,56 (д, J=1,7 Гц, 1 H), 8,03 (с, 1 H), 7,72-7,82 (м, 3 H), 7,65 (д, J=7,9 Гц, 1 H), 7,45 (д, J=8,0 Гц, 2 H), 4,76 (д, J=12,7 Гц, 1 H), 4,72 (д, J=12,7 Гц, 1 H), 4,69 (дт, J=12,0, 2,3 Гц, 1 H), 4,49 (д, J=11,9 Гц, 1 H), 4,22-4,32 (м, 3 H). Анализ (C21H15F3N4O5) C, H, N.
Пример 3. Физико-химические свойства, стабильность и биологическая активность
Физико-химические свойства соединений изобретения оценивали, как указано ниже. Результаты показаны ниже в таблице 2.
(а) Вычисленная липофильность (CLOGP). Липофильность вычисляли с применением программного обеспечения расчета LogP/log D от ACD/Labs (version 8.0, Advanced Chemistry Development, Inc., Toronto, Ontario, Canada).
(b) Растворимость в воде. Образец твердого соединения смешивали с водой (достаточной для получения 2 мМ раствора) в пробирке Эппендорфа и суспензию подвергали действию ультразвука в течение 15 мин и затем центрифугировали при 13000 об/мин в течение 6 мин. Аликвоту прозрачного супернатанта 2-кратно разбавляли водой и затем проводили ВЭЖХ. Растворимость вычисляли сравнением полученной площади пика с площадью пика стандартного раствора соединения в ДМСО (после принятия во внимание изменяющихся факторов разбавления и объемов ввода).
Оценивали также микросомальную стабильность и биологическую активность in vitro соединений изобретения с результатами, показанными в таблице 2.
(а) Минимальные ингибирующие концентрации (MIC). Соединения оценивали на их активность против репликации Mycobacterium tuberculosis 8-дневным анализом на микропланшетах с применением реагента Alamar синего (добавленного в день 7) для определения роста (MABA) (Collins et al., 1997; Falzari et al., 2005). Самую низкую концентрацию соединения, дающую >90% ингибирования, считают MIC. В скрининге на активность соединений против бактерий в нереплицирующемся состоянии, которое моделирует клиническую персистенцию, применяли 11-дневный анализ на основе люминесценции с низкой регенерацией кислорода (LORA) и высокой пропускной способностью, где бактерии М. tuberculosis, содержащие плазмиду с промотором ацетамидазы, запускающим ген бактериальной люциферазы, сначала адаптировали к условиям низкого содержания кислорода размноженной культуры (Cho et al., 2007).
(b) Стабильность соединений к действию микросом человека и мыши. Испытуемые соединения (1 мкМ) инкубировали при 37°С с препаратами пула микросом печени человека или мыши CD-1 (конечная концентрация белка 0,5 мг/мл) и системой регенерации NADPH (MgCl2, 3,3 мМ; G6P, 3,3 мМ; G6PD, 0,4 Е/мл; NADP+, 1,3 мМ) в фосфатном буфере (75 мМ, pH 7,4) с конечным объемом 200 мкл. Соединения растворяли в ДМСО, так чтобы конечная концентрация ДМСО была 0,5%. Реакции останавливали при времени 0 и 60 мин добавлением MeCN (100 мкл), содержащего 0,2 мкМ метопролола в качестве внутреннего стандарта. Образцы разводили в 10 раз и центрифугировали перед анализом ЖХ-МС/МС с применением ионизации электрораспылением и мониторинга SRM с применением метода градиентной ЖХ. Площади пиков суммировали и выражали как отношения площадей пиков анализируемое вещество/IS (внутренний стандарт) (PAR) и среднюю величину для каждой временной точки вычисляли из двух повторностей. Процентную величину остатка (микросом) вычисляли как остаток в % = 100×(среднее PART60/среднее PART0).
Биологическую активность соединений изобретения оценивали в двух анализах и определяли также фармакокинетические параметры с результатами, показанными ниже в таблице 3.
а) Анализ инфицирования мыши in vivo острой ТВ (туберкулезной бациллой). Мышей BALB/c инфицировали посредством аэрозоля с суспензией ~2×106 колониеобразующих «единиц» (CFU) M. tuberculosis Эрдман/мл (Falzari et al., 2005). Каждое соединение вводили перорально группе из 7 или 8 мышей при 100 мг/кг ежедневно 5 дней в неделю в течение 3 недель начиная с 11 дня после инфицирования. Соединения вводили в виде суспензии в смеси 0,5% СМС/0,08% твина 80 в воде. Мышей умерщвляли на 31 день и числа CFU в легких определяли и сравнивали с числом CFU для мышей, обработанных только наполнителем, в это время. РА-824 применяли в качестве положительного контроля в каждом эксперименте и результаты регистрировали как отношение среднее уменьшение CFU у мышей, обработанных соединением/среднее уменьшение CFU у мышей, обработанных РА-824. В этом анализе РА-824 вызывал уменьшения CFU до 2,5-3 log.
(b) Анализ инфицирования in vivo мыши хронической ТВ. Соединения вводили перорально, как в (а), но обработку начинали через ~70 дней после инфицирования. В этом анализе РА-824 вызывал уменьшения CFU ~2 log.
с) Фармакокинетика in vivo. Соединения вводили перорально мышам CD-1 при дозе 40 мг/кг в виде суспензии в смеси 0,5% карбоксиметилцеллюлозы/0,08% твина 80 в воде. Образцы, отобранные из плазмы и легких, анализировали ЖХ-МС/МС для получения требуемых фармакокинетических параметров.
Цитированные ссылки
Содержание каждого из документов, перечисленных ниже, таким образом, включено в контекст путем ссылки.
Патентные документы США
Патент США № 5668127.
Патент США № 6087358.
Международные патентные документы
EP 1555259.
WO 95/24400.
WO 2007/075872.
Непатентные публикации
Anderson et al., Org. Biomol. Chem. 6, 1973-1980 (2008).
Cho et al., Antimicrob. Agents Chemother. 51, 1380-1385 (2007).
Collins et al., Antimicrob. Agents Chemother. 41, 1004-1009 (1997).
deSolms et al., J Med. Chem. 46, 2973-2984 (2003).
Edsall et al., Bioorg. Med. Chem. 11, 3457-3474 (2003).
Falzari et al., Antimicrob. Agents Chemother. 49, 1447-1454 (2005).
Ferrara et al., Lancet 367, 1328-1334 (2006).
Kiener et al., Synlett 10, 814-816 (1994).
Kim et al., J. Med. Chem. 52, 1317-1328 и 1329-1344 (2009).
Li et al., Bioorg. Med. Chem. Lett. 18, 2256-2262 (2008).
Manjunatha et al., Proc. Natl. Acad. Sci. USA 103, 431-436 (2006).
Sasaki et al., J. Med. Chem. 49, 7854-7860 (2006).
Schubert et al., Synlett 3, 342-344 (1999).
Singh et al., Science 322, 1392-1395 (2008).
Stover et al., Nature 405, 962-966 (2000).
Tyagi et al., Antimicrob. Agents Chemother. 49, 2289-2293 (2005).
van den Heuvel et al., J. Org. Chem. 69, 250-262 (2004).
Реферат
Изобретение относится к области органической химии, а именно к производным нитроимидазооксазинам формулы I, где Х представляет собой О, ОСН, ОСНСН=СН или ОСНС≡С; Y представляет собой группу любой из формул IIa-IIc, гдеозначает место присоединения к X, и Z в формулах IIa-IIc представляет собой СН, СН=СН, С≡С или прямую связь, числа 2, 3 и 4 являются положениями концевого кольца, имеющего Rв качестве заместителя, концевое кольцо формулы I содержит С, СН или один атом азота в каждом положении кольца, и каждый из Rи Rв формулах I и IIa представляет собой один или два заместителя, расположенные в любом доступном положении кольца и независимо представляет собой Н, F, Cl, CF, OCFH, OCFили их комбинации. Также изобретение относится к фармацевтической композиции на основе соединения формулы I, способу лечения микробной инфекции, конкретным соединениям. Технический результат: получены новые соединения, полезные при лечении микробных инфекций, в т.ч. и при лечении заболеваний, вызванных Mycobacterium tuberculosis. 5 н. и 3 з.п. ф-лы, 28 ил., 3 табл., 3 пр.
Формула

где Х представляет собой О, ОСН2, ОСН2СН=СН или ОСН2С≡С;
Y представляет собой группу любой из формул IIa-IIc

где

Z в формулах IIa-IIc представляет собой СН2, СН=СН, С≡С или прямую связь,
числа 2, 3 и 4 являются положениями концевого кольца, имеющего R1 в качестве заместителя,
концевое кольцо формулы I содержит С, СН или один атом азота в каждом положении кольца, и
каждый из R1 и R2 в формулах I и IIa представляет собой один или два заместителя, расположенные в любом доступном положении кольца и независимо представляет собой Н, F, Cl, CF3, OCF2H, OCF3 или их комбинации.
Х представляет собой О, ОСН2, ОСН2СН=СН или ОСН2С≡С;
Y представляет собой группу любой из формул IIa-IIc

где

Z в формулах IIa-IIc представляет собой СН2, СН=СН, С≡С или прямую связь;
числа 2, 3 и 4 являются положениями концевого кольца, имеющего R1 в качестве заместителя,
концевое кольцо формулы I содержит С или СН в каждом положении кольца, или содержит атом азота во 2-положении и С или СН в каждом оставшемся положении кольца, или содержит атом азота в 3-положении и С или СН в каждом оставшемся положении кольца,
R1 в формуле I представляет собой любой один или два заместителя из F, расположенного в положении 4 кольца, OCF3, расположенного в положении 4 кольца, Cl, расположенного в положении 2 кольца, Cl, расположенного в положении 3 кольца, F, расположенного в положении 3 кольца, CF3, расположенного в положении 4 кольца, или их комбинации, и
R2 в формуле IIa представляет собой любой один или два заместителя из Н или F в любом доступном положении.
A. (6S)-6-{[2'-хлор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
B. (6S)-6-{[3'-фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
С. (6S)-2-нитро-6-{[4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
D. (6S)-2-нитро-6-({4-[5-(трифторметил)-2-пиридинил]бензил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
Е. (6S)-2-нитро-6-({4-[6-(трифторметил)-3-пиридинил]бензил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
F. (6S)-6-{[3-Фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
G. (6S)-2-нитро-6-{[4'-(трифторметокси)[1,1'-бифенил]-4-ил]окси}-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
Н. (6S)-6-({2-фтор-4-[5-(трифторметил)-2-пиридинил]бензил}окси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
I. (6S)-6-{[2-фтор-4'-(трифторметокси)[1,1'-бифенил]-4-ил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
J. (6S)-2-нитро-6-({4-[4-(трифторметокси)бензил]бензил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
K. (6S)-2-нитро-6-({(2Е)-3-[4'-(трифторметокси)[1,1'-бифенил]-4-ил]-2-пропенил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
L. (6S)-6-({2-фтор-4-[6-(трифторметил)-3-пиридинил]бензил}окси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
M. (6S)-6-{[4-(5-фтор-2-пиридинил)бензил]окси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
N. (6S)-2-нитро-6-({3-[4'-(трифторметокси)[1,1'-бифенил]-4-ил]-2-пропинил}окси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
O. (6S)-2-нитро-6-[(4-{(Е)-2-[4-(трифторметокси) фенил]этенил}бензил)окси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
Р. (6S)-2-нитро-6-[(4-{[4-(трифторметокси)фенил]этинил}бензил)окси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина и
их смесей, оптических или геометрических изомеров, фармакологически приемлемых производных в виде солей.
A. (6S)-6-{[6-(4-фторфенил)-3-пиридинил]метокси}-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
В. (6S)-2-нитро-6-({6-[4-(трифторметокси)фенил]-3-пиридинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
С. (6S)-2-нитро-6({5-[4-(трифторметокси)фенил]-2-пиридинил}метокси)-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
D. (6S)-2-нитро-6-[(5-{[4-(трифторметокси)фенил]этинил}-2-пиридинил)метокси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
Е. (6S)-6-({6-[3-фтор-4-(трифторметокси)фенил]-3-пиридинил}метокси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
F. (6S)-6-({5-[3-фтор-4-(трифторметокси)фенил]-2-пиридинил}метокси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
G. (6S)-2-нитро-6-[(5-{[6-(трифторметил)-3-пиридинил]этинил}-2-пиридинил)метокси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
Н. (6S)-6-({6-[3-хлор-4-(трифторметокси)фенил]-3-пиридинил}метокси)-2-нитро-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина;
I. (6S)-2-нитро-6-[(6-{[4-(трифторметокси)фенил]этинил}-3-пиридинил)метокси]-6,7-дигидро-5Н-имидазо[2,1-b][1,3]оксазина и
их смесей, оптических или геометрических изомеров, фармакологически приемлемых производных в виде солей.
Комментарии