Поликристаллические абразивные материалы и способ их изготовления - RU2404021C2
Код документа: RU2404021C2
Чертежи





Описание
УРОВЕНЬ ТЕХНИКИ
Настоящее изобретение относится к поликристаллическим абразивным изделиям и материалам и к способу их изготовления.
Абразивные спрессованные элементы широко применяются для резания, размола, шлифования, сверления и других абразивных операций. Обычно они содержат сверхтвердые абразивные частицы, диспергированные в матрице второй фазы. Матрица может быть металлической или керамической, или керметной (металлокерамической). Сверхтвердые абразивные частицы могут представлять собой алмаз, кубический нитрид бора (cBN), карбид кремния, нитрид кремния и т.п. Эти частицы могут быть связаны друг с другом с помощью обычно применяющихся технологий уплотнения при высоком давлении и высокой температуре с образованием поликристаллической массы или могут быть связаны с помощью матрицы материала (материалов) второй фазы с образованием поликристаллической массы. Такие материалы обычно известны, как поликристаллический алмаз или поликристаллический кубический нитрид бора, если в качестве абразива они содержат алмаз или cBN соответственно.
Примеры абразивных спрессованных элементов из алмаза и кубического нитрида бора описаны в патентах US №№3745623; 3767371; 3743489; 4334928; 5466642 и 5328875.
Например, в патенте US №4334928 описан спеченный спрессованный элемент, предназначенный для применения в инструментах, в основном содержащий от 80 до 20 об.% образующейся при высоком давлении формы нитрида бора; и остальное представляет собой матрицу, образованную по меньшей мере одним связующим композиционным материалом, выбранным из группы, включающей карбиды, нитриды, карбонитриды, бориды и силициды переходных металлов групп IVa или Va периодической системы, их смеси и их твердые растворы. Матрица образует непрерывную связанную структуру в спеченном материале, представляющем собой образующуюся при высоком давлении форму нитрида бора, распределенную в непрерывной матрице. Все описанные в этом патенте методики включают объединение необходимых материалов с помощью технологий механического размола/перемешивания, таких как размол в шаровой мельнице, размол в ступке и т.п.
В патенте US №5466642 указано, что стойкий к истиранию режущий инструмент на основе cBN, обладающий превосходной ударной вязкостью, включает заданное количество по меньшей мере одного из следующих компонентов: карбид/нитрид Ti, соединение, включающее Ti и/или Al, карбид вольфрама, Al2O3 и остальное представляет собой cBN и случайные примеси. Описанная методика изготовления включает мокрое смешивание на шаровой мельнице. Случайные примеси могут представлять собой материал, образовавшийся вследствие истирания шаров и корпуса мельницы.
В патенте US №5328875 заявлена керамика PCBN, включающая композицию, содержащую в качестве компонентов связывающую фазу, диспергированную фазу и неизбежные примеси, представляющая собой высокопрочную керамику, предназначенную для режущих инструментов, обладающих высокой ударной вязкостью и высокой износостойкостью и стойкостью к скалыванию. Связывающая фаза содержит один или большее количество следующих компонентов: карбид алюминия и титана, нитриды и карбонитриды, включая кислород, и от 20 до 48 об.% разложившейся реакционной фазы кубического кристаллического нитрида бора. Диспергированная фаза содержит кубический кристаллический нитрид бора и разложившаяся реакционная фаза содержит один или большее количество следующих компонентов: карбид титана, нитрид титана и карбонитрид титана, и оксид алюминия и/или нитрид алюминия, а также борид титана. Указано, что размеры кристаллических зерен в связывающей фазе, содержащей разложившуюся реакционную фазу, и размеры кристаллических зерен в диспергированной фазе, содержащей кубический нитрид бора, все равны менее 1 мкм. Карбид титана и алюминия предпочтительно представляет собой Ti2-3AlC, нитрид титана и алюминия в основном представляет собой Ti3-3AlN и карбонитрид титана и алюминия в основном представляет собой Ti2-3AlCN. Разложившаяся реакционная фаза содержит один или большее количество следующих компонентов: TiC, TiN, TiCN, Al2O3, AlN и TiB2. Описанная методика изготовления включает мокрый размол и перемешивание необходимых измельченных компонентов в шаровой мельнице.
В технологиях предшествующего уровня техники возникает несколько значительных затруднений. Общие технологии, включающие процедуры механического размола и перемешивания для объединения необходимых исходных материалов, приводят к неизбежному измельчению и дроблению указанных компонентов. В свою очередь, это приводит к образованию широкого распределения по размерам частиц часто сложных и разных компонентов и обусловленной этим неоднородности компонентов. Вследствие этой неоднородности невозможно точно определять и регулировать фазовую структуру конечного материала после спекания и поэтому невозможно полностью использовать возможности материала в режущем инструменте. Такие материалы часто обладают неудовлетворительными рабочими характеристиками, что обусловлено плохой диспергированностью и неоднородностью компонентов.
Кроме того, эти технологии неприменимы, когда частицы необходимых исходных компонентов становятся мельче, в особенности в случае субмикрометровых измельченных материалов и тем более в случае нанометровых компонентов, что обусловлено значительной трудностью диспергирования. Поэтому применение таких технологий налагает ограничения на изготовление композиционных материалов, содержащих однородные субмикрометровые и нанометровые фазы.
Кроме того, сверхтвердые абразивные измельченные вещества невозможно размолоть без протекающего в той или иной степени истирания материала шаров, стержней и корпуса мельницы. Образовавшийся вследствие такого истирания материал обязательно загрязняет смесь необходимых компонентов или нежелательным материалом или, если этот материал можно рассматривать в качестве желательного, он вводится в нерегулируемом и неопределенном количестве. Это загрязнение особенно значительно, когда в попытках получения субмикрометровых и нанометровых исходных материалов используют интенсивный размол. За срок службы корпусов, шаров и стержней неизбежное истирание приводит к постепенному изменению размеров и текстуры поверхности этих элементов, что приводит к постепенному изменению их размалывающей, перемешивающей и измельчающей способности. Эти изменения приводят к дальнейшему изменению диспергирования, однородности и степени загрязнения объединяемых материалов и, поэтому, к изменению структуры, характеристик и свойств готовых композиционных материалов и инструментов. Кроме того, эти затруднения особенно характерны для материалов с зернами субмикрометрового и нанометрового размера и их трудно изготовить по таким технологиям.
Размол и перемешивание также склонны приводить к повреждению и разрушению волокон, нитевидных кристаллов и вообще измельченных материалов с большим аспектным отношением, которые могут прибавляться для изменения механических характеристик получаемого композиционного материала, обычно для увеличения ударной вязкости, что приводит к ухудшению характеристик изделия.
В предшествущем уровне техники имеются примеры того, когда размол и перемешивание не являются преимущественно использующимися технологиями. Например, в US 5211726 показано, что на гранулы cBN или алмаза в диапазоне размеров от мелких, примерно 0,1 мкм, до крупных, примерно 1 мм, можно нанести один или большее количество слоев активного покрытия и эти обладающие покрытием объекты спечь при давлении и температуре, достаточных для получения смешанных абразивных спрессованных элементов. Для нанесения покрытий на смешанные гранулы конкретного типа материала cBN, обладающие размером от примерно 50 мкм до примерно 1 мм, методики нанесения покрытия ограничиваются химическим осаждением из паровой фазы (ХПФ).
В ЕР 0577375 описана методика получения абразивных спрессованных элементов с использованием алмаза или cBN в качестве сверхтвердых компонентов, в которой огнеупорные оксиды, нитриды и карбиды осаждают на алмаз или cBN и покрытие спекают при температурах и давлениях, при которых алмаз и cBN предположительно являются термодинамически стабильными. Описанной методикой нанесения покрытия является химическое осаждение из паровой фазы и покрытие наносят на частицы алмаза или cBN размером от 20 до 40 мкм.
В US 5536485 описана методика, при которой спеченный материал из алмаза или cBN можно получить путем нанесения покрытия на частицы алмаза или cBN в газовой или паровой фазе с последующим спеканием указанных обладающих покрытием частиц при температурах и давлениях, при которых и алмаз, и cBN могут быть термодинамически стабильными или термодинамически метастабильными.
Большинство описанных в предшествущем уровне техники материалов, в которых сверхтвердым компонентом является cBN, зависит от реакций с металлами, такими как алюминий, титан и кремний, которые в расплавленном состоянии могут смачивать cBN, в значительной степени вступать в реакцию с cBN и приводить к его частичному разложению. Поэтому такие методики приводят к материалам, которые содержат разложившиеся фазы, включенные в сложную микроструктуру образовавшегося материала. Обязательно должны содержаться комплексные бориды, нитриды и боронитриды реагирующих металлов, часто с неоднородным распределением других включенных фаз. Это приводит к ограничению типов материалов, которые можно получить, теми, которые могут образоваться по соответствующим реакциям, и чрезмерно сложными структурами.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном варианте осуществления настоящего изобретения способ изготовления поликристаллического абразивного элемента включает стадии использования множества сверхтвердых абразивных частиц, обладающих витреофильными поверхностями, нанесения на сверхтвердые абразивные частицы покрытия из материала-предшественника матрицы, обработки обладающих покрытием сверхтвердых абразивных частиц, чтобы сделать их пригодными для спекания, предпочтительно превращения материала-предшественника матрицы в оксид, нитрид, карбид, оксинитрид, оксикарбид или карбонитрид материала-предшественника матрицы, или в элементную форму материала-предшественника матрицы, или в их комбинации, и объединения и спекания обладающих покрытием сверхтвердых абразивных частиц при давлении и температуре, при которых они являются кристаллографически или термодинамически стабильными.
Материал-предшественник матрицы предпочтительно представляет собой аморфный или нанокристаллический оксид, гидроксид или оксогидроксид.
Сверхтвердые абразивные частицы предпочтительно выбраны из группы, включающей алмаз, кубический нитрид бора, карбид кремния, нитрид кремния, карбид бора, субоксид бора (В6О) и т.п.
Предпочтительно, если сверхтвердые абразивные частицы представляют собой алмаз или кубический нитрид бора или комбинацию этих материалов, в этом случае частицы должны быть подвергнуты поверхностной обработке, чтобы сделать их поверхности витреофильными. Это образует другой объект настоящего изобретения, согласно которому находящиеся на поверхности химические частицы выбираются и генерируются путем соответствующей обработки для того, чтобы образовавшиеся таким образом на поверхности химические частицы могли быть совместимыми с последующими мокрыми химическими реакциями и средствами нанесения покрытия на сверхтвердые частицы и участвовали в них. Поверхностные химические частицы такого рода можно описать, как витреофильные, или склонные взаимодействовать со стеклом, в том отношении, что они могут образовывать связи с оксидными компонентами, типичными для стекла и стеклоподобных аморфных материалов. В этом случае материалы покрытия, вероятно, химически свяжутся с поверхностью сверхтвердых частиц.
Подвергнутые превращению материалы-предшественники матрицы обычно выбраны из числа обладающих зернами микрометрового, субмикрометрового или нанометрового размера оксидов, нитридов, карбидов, оксинитридов, оксикарбидов, карбонитридов или элементных форм материалов матрицы, или их комбинаций. Они обычно включают оксиды, нитриды, карбиды, оксинитриды, оксикарбиды и карбонитриды алюминия, титана, кремния, ванадия, циркония, ниобия, гафния, тантала, хрома, молибдена и вольфрама и любые подходящие комбинации этих материалов. Предпочтительно, если эти материалы-предшественники матрицы обладают зернами нанометрового размера. Предпочтительными элементными матрицами являются вольфрам, молибден или комбинации, или сплавы этих металлов, предпочтительно обладающие зернами нанометрового размера.
Материалы-предшественники матрицы предпочтительно наносят на сверхтвердые абразивные частицы с помощью так называемой золь-гелевой методики. Сверхтвердые частицы суспендируют в жидких средах, в которые введены подходящие химические реагенты, предпочтительно один или большее количество алкоксидов, так чтобы могли образоваться коллоидные частицы, которые связываются с поверхностями и включаются в покрытия, находящиеся на указанных частицах. Образованные таким образом покрытия преимущественно представляют собой микропористые оксиды, гидроксиды или оксогидроксиды указанных выше металлов или металлоидов.
Для удаления летучих веществ и нежелательных химических веществ, присоединенных к большим участкам поверхности микропористых аморфных покрытий, таких как гидроксилсодержащие частицы, в особенности -ОН, предпочтительно проводить нагревание на воздухе, в вакууме или инертном газе с регулированием температуры.
Для кристаллизации покрытий с образованием мелкозернистых или нанометровых оксидных керамик можно использовать дополнительную термическую обработку или прокаливание.
Поскольку некоторые оксидные керамики в некоторых температурных диапазонах подвергаются фазовым превращениям, выбор конкретных кристаллических фаз путем использования соответствующих температуры и длительности является другим объектом настоящего изобретения.
Некоторые из оксидных материалов покрытий не кристаллизуются в широких диапазонах температур и таким образом могут образовывать стекла и могут уплотняться по механизмам спекания стекол.
Реакции с регулированием температуры в реакционно-способных газах также можно использовать для превращения аморфных оксидов или кристаллических оксидных керамик в кристаллические неоксидные керамики. В частности, по реакции покрытий с аммиаком образуются нитриды. Карбиды можно получить по реакции покрытий со смесями углеродсодержащих газов с водородом, например со смесями метана или этана с водородом. Если некоторые оксидные покрытия восстанавливаются водородом, то их можно превратить в обладающие зернами микрометрового и нанометрового размера элементы или металлы.
Отличительной особенностью настоящего изобретения является то, что вследствие аморфного или микрокристаллического характера оксидных предшественников покрытий температуры, необходимые для их превращения в соответствующие керамики или металлы по реакции с газами, намного ниже температур, необходимых для обычных оксидных керамик, получаемых с помощью обычного прокаливания и плавления.
Обладающие покрытием сверхтвердые частицы предпочтительно объединяют, уплотняют, и покрытия спекают с помощью горячего прессования, предпочтительно при высоком давлении и температуре, такого как горячее прессование при подходящих температурах, при выбранных давлениях в течение выбранного периода времени. Выбираемые условия зависят от конкретных сверхтвердых частиц и конкретного спекаемого материала покрытия. Предпочтительное оборудование для горячего прессования включает устройства высокого давления, такие как ленточные прессы высокого давления и т.п., хорошо известные в данной области техники.
Нанесение покрытий на сверхтвердые абразивные частицы образует другой объект настоящего изобретения, как и обработка обладающих покрытием частиц.
Уплотнение и спекание обладающих покрытием и обработанных частиц при высоком давлении и температуре образуют еще один объект настоящего изобретения.
Поликристаллические абразивные элементы или спрессованные элементы, предлагаемые в настоящем изобретении, предпочтительно представляют собой композиционные материалы, включающие массу сверхтвердых измельченных материалов, обладающих любыми размерами или распределениями по размерам, меньшими, чем примерно несколько сотен микрометров, и включающие субмикрометровые, а также нанометровые частицы (частицы размером менее 0,1 мкм, т.е. 100 нм), которые хорошо диспергированы в непрерывной матрице, изготовленной из чрезвычайно мелкозернистой оксидной керамики, неоксидной керамики, керметов или комбинаций материалов этих классов.
Способ, предлагаемый в настоящем изобретении, также предоставляет возможность изготовления множества поликристаллических сверхтвердых абразивных элементов или композиционных материалов. Они включают поликристаллические сверхтвердые абразивные элементы, содержащие алмаз в матрице, выбранной из группы, включающей диоксид титана, TiO2, диоксид гафния, HfO2, диоксид кремния, SiO2, диоксид циркония, ZrO2, нитрид титана, TiN, нитрид ванадия, VN, нитрид гафния, HfN, нитриды ниобия, NbN, Nb2N, нитрид тантала, TaN, нитрид молибдена, Мо2N, нитрид вольфрама, W2N, карбид титана, TiC, карбид ванадия, VC, карбид гафния, HfC, карбид ниобия, NbC, карбид тантала, ТаС, карбид молибдена, Mo2C, карбиды вольфрама, W2C, WC, молибден, Мо, и вольфрам, W; поликристаллические сверхтвердые абразивные элементы, содержащие кубический нитрид бора в матрице, выбранной из группы, включающей оксид алюминия, Al2O3, диоксид титана, TiO2, диоксид гафния, HfO2, диоксид кремния, SiO2, диоксид циркония, ZrO2, нитрид ванадия, VN, нитрид гафния, HfN, нитриды ниобия, NbN, Nb2N, нитрид тантала, TaN, нитрид молибдена, Mo2N, нитрид вольфрама, W2N, карбид ванадия, VC, карбид гафния HfC, карбид ниобия, NbC, карбид тантала, ТаС, карбид молибдена, Мо2С, карбиды вольфрама, W2C, WC, молибден, Мо, и вольфрам, W; и поликристаллические сверхтвердые абразивные элементы, содержащие комбинации алмаза и кубического нитрида бора в матрице, выбранной из группы, включающей оксид алюминия, Al2O3, диоксид титана, TiO2, диоксид гафния, HfO2, диоксид кремния, SiO2, диоксид циркония, ZrO2, нитрид титана, TiN, нитрид ванадия, VN, нитрид гафния, HfN, нитриды ниобия, NbN, Nb2N, нитрид тантала, TaN, нитрид молибдена, Mo2N, нитрид вольфрама, W2N, карбид титана, TiC, карбид ванадия, VC, карбид гафния HfC, карбид ниобия, NbC, карбид тантала, ТаС, карбид молибдена, Mo2C, карбиды вольфрама, W2C, WC, молибден, Мо, и вольфрам, W. Другие специальные элементы или композиционные материалы, которые можно изготовить в соответствии с настоящим изобретением, включают алмаз в матрице из оксида алюминия, кубический нитрид бора в матрице из нитрида титана и кубический нитрид бора в матрице из карбида титана при условии, что зерна материала матрицы обладают нанометровым размером.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Настоящее изобретение ниже только в качестве примера будет более подробно описано со ссылкой на прилагаемые чертежи, на которых представлено следующее:
на фиг.1 приведена блок-схема стадий способа, предлагаемого в настоящем изобретении;
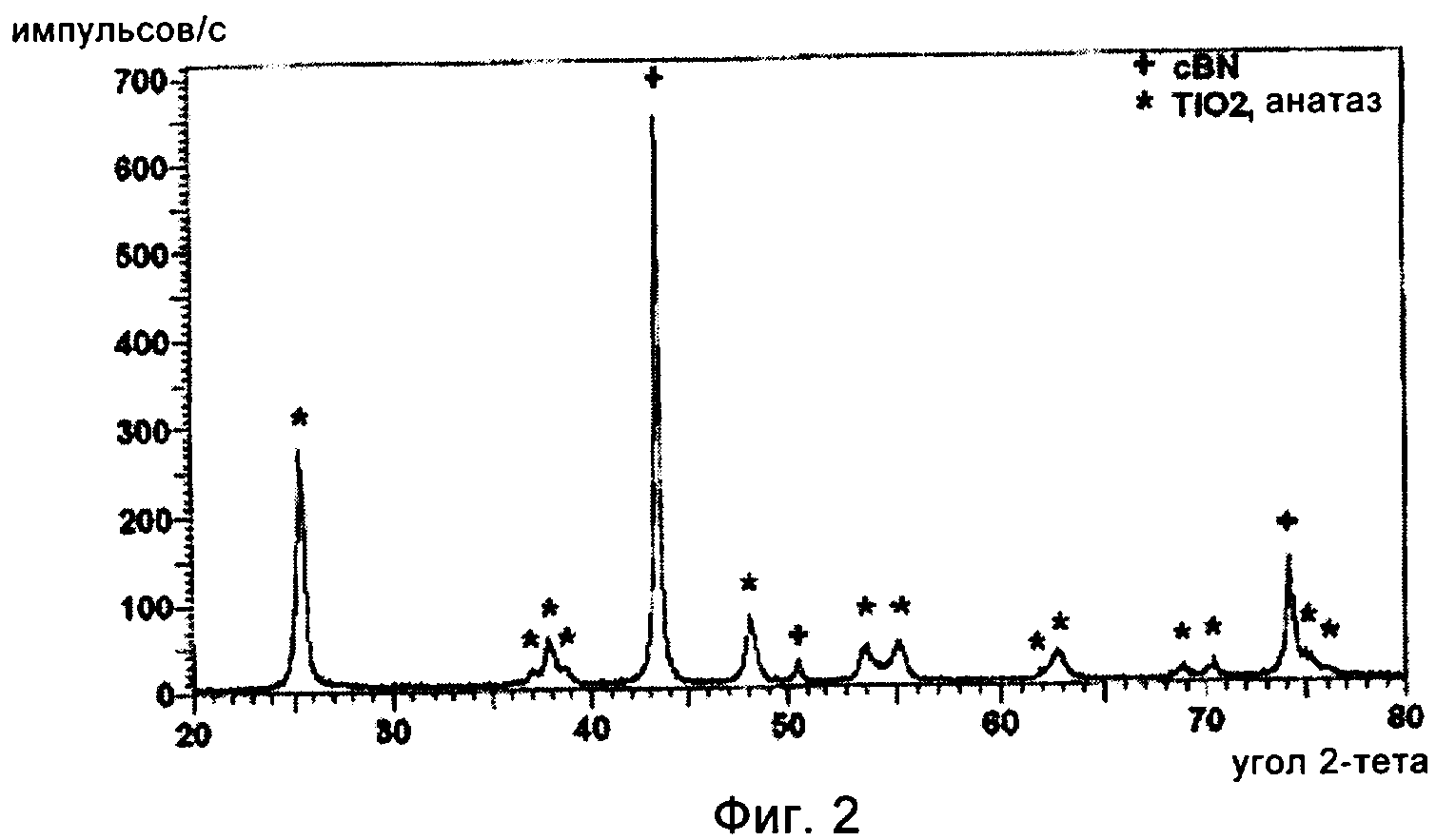
на фиг.2 приведена рентгенограмма частиц cBN, обладающих покрытием из диоксида титана, промежуточного материала в предпочтительном варианте осуществления способа, предлагаемого в настоящем изобретении;
на фиг.3 приведена рентгенограмма частиц cBN, обладающих покрытием из нитрида титана, полученного термической обработкой частиц cBN, обладающих покрытием из диоксида титана, охарактеризованных на фиг.2;
на фиг.4 приведена рентгенограмма спеченного материала, полученного из частиц cBN, обладающих покрытием из нитрида титана, охарактеризованных на фиг.3;
на фиг.5 приведена рентгенограмма частиц алмаза, обладающих покрытием из нитрида титана, полученных в соответствии с другим предпочтительным вариантом осуществления способа, предлагаемого в настоящем изобретении; и
на фиг.6 приведена рентгенограмма композиционного материала алмаз-оксид кремния, полученного в соответствии с еще одним предпочтительным вариантом осуществления способа, предлагаемого в настоящем изобретении.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Настоящее изобретение относится к поликристаллическим абразивным изделиям и материалам, также называющимся поликристаллическими абразивными элементами, предназначенными для применения в качестве режущих инструментов для обтачивания на токарном станке, размола и затачивания, сверления камня, керамики и металлов, изготовления износостойких деталей и т.п., и к способу их изготовления. Настоящее изобретение предпочтительно относится к улучшенным однородным композиционным материалам, в которых содержащиеся фазы материалов обладают зернами микрометрового, субмикрометрового и/или нанометрового размера, так что можно использовать ожидаемое улучшение характеристик и свойств при использовании этих материалов.
Эти материалы обладают улучшенными характеристиками, что приводит к улучшенным свойствам в различных случаях применения, в которых можно использовать композиционные абразивные изделия, предлагаемые в настоящем изобретении. С помощью настоящего изобретения устранены многие затруднения, проявляющиеся в предшествущем уровне техники вследствие неоднородности микроструктуры сложности образующих материалы фаз и введения примесей.
Композиционные абразивные изделия состоят из микрометровых, субмикрометровых или нанометровых сверхтвердых абразивных частиц, диспергированных в микрометровых, субмикрометровых или нанометровых матричных материалах.
В настоящем изобретении субмикрометровые частицы или зерна определяются, как обладающие наибольшим диаметром, равным от 1 мкм (1000 нм) до 0,1 мкм (100 нм), и нанометровые частицы или зерна определяются, как обладающие наибольшим диаметром, равным менее 0,1 мкм (100 нм).
Сверхтвердые абразивные частицы включают алмаз, кубический нитрид бора, карбид кремния, нитрид кремния, карбид бора, субоксид бора (B6O) и т.п. и любую комбинацию этих типов частиц. Предпочтительными сверхтвердыми частицами являются алмаз и кубический нитрид бора или комбинация этих материалов.
Матричные материалы включают, но не ограничиваются только ими, обладающие зернами микрометрового, субмикрометрового или нанометрового размера оксидные, нитридные, карбидные, оксинитридные, оксикарбидные и карбонитридные матрицы. Субмикрометровые или нанометровые матричные материалы включают оксиды, нитриды, карбиды, оксинитриды, оксикарбиды и карбонитриды алюминия, титана, кремния, ванадия, циркония, ниобия, гафния, тантала, хрома, молибдена и вольфрама и любые подходящие комбинации этих материалов. Предпочтительно, если эти матрицы являются обладающими зернами нанометрового размера соединениями алюминия, титана, тантала, кремния или циркония.
Композиционные абразивные изделия, предлагаемые в настоящем изобретении, включают композиции, в которых содержание диспергированных сверхтвердых частиц в материале матрицы составляет от примерно 25 до более 98 об.%, хотя они не ограничиваются этими содержаниями.
Основной особенностью способа, предлагаемого в настоящем изобретении, является то, что предшественники требующихся матричных материалов можно равномерно нанести на каждую отдельную сверхтвердую частицу с помощью коллоидной технологии, так что каждая частица будет обладать покрытием, обладающим такими же размерами и структурой, что и на других частицах. Это позволяет обеспечить значительную структурную однородность, которая намного лучше, чем обеспечиваемая в способах предшествующего уровня техники. Это можно осуществить для мелких сверхтвердых частиц микрометрового, субмикрометрового или даже нанометрового размеров, поскольку отличительной особенностью способа, предлагаемого в настоящем изобретении, является то, что материалы-предшественники покрытий и последующие конечные материалы матрицы, образующиеся с помощью выбранных методик термической обработки, могут обладать очень мелкими зернами нанометрового размера. В свою очередь, это позволяет обеспечить очень большое содержание сверхтвердых частиц, составляющее более 90 об.%, при высокой однородности.
Способ, предлагаемый в настоящем изобретении, обычно включает четыре технологические стадии, а именно 1) использование сверхтвердых абразивных частиц, обладающих витреофильными поверхностями, или, если это является целесообразным, химическую обработку поверхностей сверхтвердых абразивных частиц для придания им витреофильности; 2) использование методик коллоидных суспензионных реакций для нанесения на сверхтвердые частицы покрытия из материала-предшественника; 3) термическую обработку обладающих нанесенным таким образом покрытием сверхтвердых частиц в газовых средах, включая содержащие реакционно-способные газы, для превращения покрытия в выбранные оксиды, нитриды, карбиды, оксинитриды, оксикарбиды и/или карбонитриды; и 4) проводимое при высоком давлении и при высокой температуре уплотнение и обжиг с получением кусков не содержащего пор тонко структурированного и наноструктурированного композиционного материала.
На первой стадии на поверхности сверхтвердого измельченного материала проводят химические реакции для придания частицам витреофильной природы. Витреофильная природа, склонная взаимодействовать со стеклом, определяется, как обладающая такой природой, что легко может образовывать химические связи с оксидными материалами. Виды обработки, которые могут привести к образованию на поверхности химических соединений, необходимых для проявления витреофильности сверхтвердых частиц, включают, но не ограничиваются только ими, кипячение в кислотах-окислителях, таких как концентрированная азотная кислота, если это является подходящим, или обработка сильными окислительными реагентами, такими как растворы пероксида водорода, или нагревание на воздухе или в кислороде. Образованные таким образом поверхности обеспечивают образование и рост покрытий на основе оксидов или гидроксидов на измельченном материале и хорошую адгезию с образованными таким образом предшественниками покрытий на основе оксидов.
На второй стадии используется коллоидное суспензионное нанесение на сверхтвердые абразивные частицы покрытия из аморфных и/или обладающих зернами нанометрового размера гидратированных оксидных материалов-предшественников. Обнаружено, что модификация некоторых коллоидных методик позволяет аккуратно наносить покрытия на микрометровые, субмикрометровые и даже нанометровые частицы сверхтвердых материалов. Имеются две общие коллоидные методики, с помощью которых можно получить подходящие покрытия, в одной из которых используют водные растворы неорганических солей, а в другой используют металлоорганические соединения. Для этого предпочтительным подходом является указанная золь-гелевая методика, более предпочтительными - золь-гелевые методики с использованием гидролиза и поликонденсации алкоксидов или алкоголятов. Предшественники покрытий, сформированные по этой методике, являются микропористыми, аморфными или обладающими зернами нанометрового размера гидратированными оксидами с большой площадью поверхности. Золь-гелевые методики, в частности, являются весьма универсальными и пригодными для регулирования гетерогенного зародышеобразования и роста чрезвычайно правильных покрытий из гидратированных оксидных материалов-предшественников на поверхностях витреофильных суспендированных частиц, размер которых может составлять лишь 10 нм или даже менее.
Предпочтительной золь-гелевой методикой является медленное прибавление спиртового раствора алкоксида металла или комбинации алкоксидов металлов к суспензии частиц сверхтвердого материала в аликвоте раствора воды низкой концентрации в том же спирте. Алкоксиды металлов гидролизуются водой с образованием мономеров гидроксидов металлов, которые в свою очередь вступают в реакцию поликонденсации, которая постепенно приводит к образованию гидратированных микропористых оксидов, которые в настоящем изобретении называют оксидными материалами-предшественниками или покрытиями. Путем соответствующего выбора типа спирта, которые обычно содержат такие же алкильные группы, как и алкоксид(ы), концентрации суспендированных сверхтвердых частиц, концентрации раствора алкоксида в спирте, соотношения алкоксид/вода, температуры и наличия или отсутствия других реагентов, таких как кислоты или основания, можно регулировать образование покрытия из оксидного предшественника на суспендированных сверхтвердых частицах. Для нанесения на суспендированный сверхтвердый измельченный материал необходимого покрытия в случае каждого типа использованного алкоксида необходимы специальные условия.
Важной особенностью этого подхода является то, что побочными продуктами реакций гидролиза алкоксидов и поликонденсации являются вода, спирты и гидроксидные соединения, находящиеся на части свободных поверхностей покрытия. Все эти побочные продукты легко удаляются путем сушки и термической обработки при низкой температуре. Кроме того, сами алкоксиды легко доступны в виде продуктов высокой чистоты. Таким образом, золь-гелевая методика приводит к очень чистым незагрязненным оксидам. Поэтому можно получить весьма чистые конечные матричные материалы, что отличает данный подход от методик предшествующего уровня техники. Разумеется, в способе, предлагаемом в настоящем изобретении, отсутствуют примеси, всегда вводимые при использовании методик размола/перемешивания.
Еще одной очень важной особенностью способа, предлагаемого в настоящем изобретении, является то, что путем одновременного использования более одного типа алкоксида разных металлов можно получить большое количество смешанных оксидных материалов-предшественников. При этом подходе полученный таким образом оксидный материал-предшественник будет представлять собой смешанный оксид, в котором различные металлы распределены в молекулярном масштабе. Альтернативно, известно, что можно получить алкоксидные комплексы, содержащие более одного металла. Эти алкоксидные комплексы можно использовать в способе, предлагаемом в настоящем изобретении. Следовательно, оксиды, нитриды и карбиды, полученные при полном применении способа, предлагаемого в настоящем изобретении, могут включать смешанные и легированные фазы. Кроме того, известно, что можно получить смешанные структуры алкоксидов металлов. Использование таких смешанных алкоксидов металлов также приводит к смешанным предшественникам оксидов металлов и затем к содержащим металлы смешанным композиционным матричным фазам.
Применение смесей алкоксидов или смешанных алкоксидов также позволяет легировать матричные материалы-предшественники и последующие материалы агентами, модифицирующими спекание и структуру, такими как оксид иттрия, оксид магния и т.п. При получении композиционных материалов способом, предлагаемым в настоящем изобретении, можно использовать большое количество информации, имеющейся в области керамики, керметов и металлургии.
После извлечения из суспензии и промывки обладающие покрытием частицы медленно сушат, например, путем нагревания в вакууме при температуре ниже 100°С. Микропористые, аморфные или обладающие зернами нанометрового размера структуры материалов-предшественников покрытий делают их идеальными для реакционной термической обработки с программированием температуры в газообразных реагентах или средах с получением необходимых мелкозернистых и обладающих зернами нанометрового размера керамических и других материалов для использования в качестве компонентов композиционных материалов.
На третьей стадии реакционную термическую обработку с программированием температуры предшественников обладающих покрытием сверхтвердых частиц в выбранной газовой среде используют для частичного уплотнения покрытия и для его превращения в выбранный мелкозернистый или обладающий зернами нанометрового размера керамический материал. Термическую обработку на воздухе или в кислороде используют для прокаливания, частичного уплотнения покрытия, удаления всех оставшихся водных и спиртовых компонентов и кристаллизации покрытия в виде требующейся оксидной фазы. Выбор скорости нагрева, максимальной температуры и длительности нагревания при максимальной температуре зависит от структуры, фазы и типа требующегося оксида.
Если покрытие необходимо превратить в нитрид, то высушенный или прокаленный на воздухе обладающий покрытием материал можно нагреть в сухом аммиаке при температурах, обычно достигающих 1100°С, хотя в некоторых случаях может потребоваться использование температур, достигающих примерно 1400°С включительно. Обнаружено, что эта реакционная термическая обработка с программированием температуры приводит к постепенному восстановлению материала покрытия и может превратить оксидные основные покрытия в стехиометрические и нестехиометрические нитриды и оксинитриды. И в этом случае выбор скорости нагрева, скоростей потоков газов, максимальной температуры и длительности нагревания при максимальной температуре зависит от структуры, фазы и типа требующегося нитрида. Также обнаружено, что путем соответствующего выбора условий можно получить оксинитридные фазы.
Если покрытие необходимо превратить в карбид, то высушенный или прокаленный на воздухе обладающий покрытием материал можно нагреть в смеси углеродсодержащих газов, таких как метан или этан, с водородом при температурах, обычно ниже 1200°С, хотя в некоторых случаях может потребоваться использование температур, достигающих примерно 1500°С включительно. И в этом случае выбор скорости нагрева, скоростей потоков газов, максимальной температуры и длительности нагревания при максимальной температуре зависит от структуры, фазы и типа требующегося карбида. Также обнаружено, что путем соответствующего выбора условий можно получить оксикарбидные фазы. Альтернативно, обнаружено, что нитридные покрытия, полученные так, как описано выше, можно превратить в карбиды путем соответствующей термической обработки в смесях метана или этана с водородом. Путем соответствующего выбора условий можно получить карбонитридные фазы.
Некоторые оксидные покрытия можно легко восстановить в соответствующий элементарный металл путем восстановления в чистом водороде. Примерами таких покрытий являются оксиды вольфрама и молибдена, WO3 и МоО3, которые можно легко восстановить в металлы при низких температурах, обычно в диапазоне от 500 до 700°С.
Основной особенностью стадии реакции с программированием температуры способа, предлагаемого в настоящем изобретении, является то, что обнаружено, что размеры всех зерен полученных оксидных, нитридных, карбидных покрытий на сверхтвердых частицах обычно являются нанометровыми. Кроме того, другой важной особенностью этой термической обработки является то, что температуры и времена, необходимые для превращения, являются низкими и непродолжительными соответственно по сравнению с температурами и временами, необходимыми для аналогичных превращений обычных оксидных материалов, проводимых по методикам плавления или сплавления. В некоторых случаях в способе, предлагаемом в настоящем изобретении, температуры образования нитридов ниже температур образования нитридов обычных оксидных материалов на величину, достигающую 400°С. Кроме того, обладающие покрытием сверхтвердые частицы можно отделить в неагломерированном виде. Это способствует однородности структур, получаемых при последующих стадиях уплотнения при высоком давлении и высокой температуре и спекания.
На четвертой стадии высокотемпературное уплотнение и спекание проводят при температурах и давлениях, при которых сверхтвердые частицы материалов термодинамически и химически стабильны для получения не содержащего пор или почти не содержащего пор кусков монолитного микрометрового, субмикрометрового и нанометрового композиционного материала. Для предпочтительных сверхтвердых материалов алмаза и cBN эти условия соответствуют диапазонам давления от 2,5 до 8 ГПа и температуры от 600 до 1800°С. Полученные таким образом не содержащие пор композиционные материалы также можно связать с подложками из карбидов металлов in situ во время уплотнения и спекания. Используемый аппарат для обработки при высоком давлении и высокой температуре может быть любым из известных в данной области техники, в котором возможно создание соответствующих условий.
Указанные выше стадии способа будут подробнее описаны ниже со ссылкой на фиг.1.
1. Обработка поверхности сверхтвердых частиц для придания им витреофильности
В случае обладающего зернами микрометрового, субмикрометрового или нанометрового размера алмаза с помощью таких методик, как нагревание в концентрированных окисляющих кислотах, таких как смеси азотной и/или серной кислоты, можно сделать так, чтобы концевые поверхностные функциональные группы в основном представляли собой группы С-ОН, С-О-С, С=O и O=С-O-. Альтернативно, газовая термическая обработка в смеси 20% водород/аргон при 900°С для образования на поверхности концевых Н с последующей обработкой в смеси 20% кислород/аргон при 480°С приводит к тому, что на поверхности преобладают кислородсодержащие частицы. Также можно использовать другие методики образования кислородсодержащих функциональных групп, присоединенных к поверхности алмаза. Окисление поверхности алмаза делает ее витреофильной, т.е. способной к образованию химических связей с оксидами, включая, в частности, гидратированные оксидные структуры.
Предполагается, что в случае субмикрометрового cBN термическая обработка на воздухе при температуре выше 600°С приведет к увеличению концентрации борокислородных и азоткислородных частиц на поверхности и это можно обнаружить с помощью инфракрасной Фурье-спектроскопии отражения. Такая поверхность обладает витреофильностью при последующем коллоидном нанесении покрытия на оксиды, полученные по золь-гелевой методике. Многие другие хорошо известные сверхтвердые материалы, такие как карбид кремния и нитрид кремния и т.п., содержат на своей поверхности окисленные химические группы, что обычно делает их витреофильными и пригодными для использования в способе, предлагаемом в настоящем изобретении.
2. Коллоидное нанесение покрытия на частицы сверхтвердого материала
В части 2(а) блок-схемы используются обычные золь-гелевые методики получения предшественников гидратированных оксидных материалов для необходимых матричных материалов. Один пример такого подхода включает гидролиз растворов сульфата алюминия при повышенных температурах, таких как равная 100°С, в присутствии органических соединений, таких как мочевина, для нанесения покрытия на частицы в суспензии. Таким образом можно получить покрытия из водного оксида алюминия.
Однако предпочтительным более общим подходом является применение реакций гидролиза и поликонденсации алкоксидов металлов в спиртовых растворах. Алкоксиды или алкоголяты металлов обладают общей формулой вида Mn+[OR]n, где М обозначает металл валентности n, О обозначает кислород и R обозначает алкильную группу. Металл связан с алкильными группами через атомы кислорода. Большинство алкоксидов металлов растворимы в спиртах и могут легко гидролизоваться водой в спиртовом растворе с образованием гидроксидов:
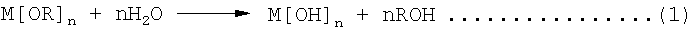
Затем можно провести реакции поликонденсации, такие как представленные приведенным ниже уравнением (2), и образовать связи М-О-М.
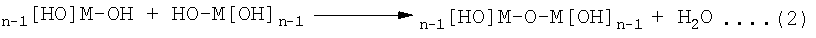
Последовательное проведение этих реакций приводит к трехмерной сетке -М-O-М-O-М-. Образовавшийся таким образом оксидный материал-предшественник обычно является аморфным или обладающим зернами нанометрового размера с очень большой площадью поверхности и является микропористым, содержащим в порах H2O и спирт. На поверхностях пористой структуры находятся концевые гидроксильные группы, ОН. Путем соответствующего выбора концентраций, соотношений спирт/вода, температуры, спирта-растворителя и введения других химикатов, таких как кислоты или основания, можно сделать так, чтобы в спиртовом растворе происходили зародышеобразование и рост пористого оксидного материала-предшественника. Необходимо подобрать подходящие концентрации суспендированных частиц, выступающих в качестве центров роста материала покрытия.
Раствор алкоксида (алкоксидов) металла получают в безводном спирте и затем в течение нескольких часов его при непрерывном перемешивании прибавляют к суспензии сверхтвердых частиц в аликвоте чистой воды обычно в том же спирте. Для стабилизации суспензии можно прибавить пептизирующий реагент, такой как кислоту или основание.
Альтернативно, если необходимо использовать особенно реакционно-способный алкоксидный реагент, лучшее регулирование образования покрытия можно обеспечить путем медленного прибавления аликвоты воды в спирте к суспензии сверхтвердых частиц в суспензии алкоксида в безводном спирте.
Побочные продукты реакции - воду и спирт - можно удалить путем сушки и термической обработки при низкой температуре 2(b). Аналогичным образом можно удалить поверхностные функциональные группы ОН. Обычно после фильтрования или центрифугирования суспензии с последующей промывкой свежим чистым спиртом и затем деионизированной водой обладающие покрытием частицы можно медленно высушить в течение примерно двух дней при температуре около 60°С в низком вакууме. Последующее удаление остаточной воды и спирта можно обеспечить путем нагревания примерно до 300°С на воздухе.
Многие элементы периодической системы могут образовывать алкоксиды. Алкоксиды, найденные пригодными для получения оксидных матриц способом, предлагаемым в настоящем изобретении, включают алкоксиды титана, алюминия, циркония, хрома, кремния, а алкоксиды кальция, магния, гафния, иттрия иногда пригодны в качестве добавок, включая комбинации этих алкоксидов. Алкоксиды, найденные пригодными для получения нитридных матриц способом, предлагаемым в настоящем изобретении, включают алкоксиды алюминия, титана, циркония, кремния, тантала, хрома, ниобия, гафния, ванадия, молибдена и вольфрама и их комбинации. Алкоксиды, найденные пригодными для получения карбидных матриц способом, предлагаемым в настоящем изобретении, включают алкоксиды титана, циркония, кремния, тантала, хрома, ниобия, гафния, ванадия, молибдена и вольфрама и их комбинации.
Алкильные группы R в общей формуле алкоксидов металлов, M[OR]n, могут включать метил, этил, н-пропил, н-бутил и любую группу общей формулы-СхН2х+1. Кроме того, включаются алкильные группы, в которых содержатся боковые алкильные группы, такие как изопропильная группа, -СН(СН3)2, или втор-бутильная группа, -СНСН2СН3СН3.
Скорость реакции гидролиза и время достижения точки гелеобразования для каждого алкоксида металла сильно зависят от длины цепи алкильной группы. Чем меньше длина цепи R, тем быстрее гидролиз и тем меньше время достижения точки гелеобразования оксидного материала-предшественника в покрытии сверхтвердых частиц. На характеристики покрытия для каждого типа требующегося гидратированного оксидного предшественника покрытия может сильно повлиять выбор R.
Спирты, применяющиеся в качестве растворителя для алкоксида и воды и в качестве суспендирующей жидкости для сверхтвердых частиц, можно выбрать из числа любых обычно имеющихся в продаже жидких растворителей. Предпочтительными спиртами являются этанол, метанол и изопропиловый спирт.
В таблице 1 приведен примерный, но не полный перечень некоторых алкоксидов, наиболее подходящих для способа, предлагаемого в настоящем изобретении.
После сушки/предварительной термической обработки обладающие покрытием частицы можно исследовать с помощью сканирующего электронного микроскопа и/или трансмиссионного электронного микроскопа.
3. Термическая обработка с программированием температуры (ТПТ)
Затем обладающие покрытием частицы подвергают термической обработке с программированием температуры. Это выполняют в выбранных газовых средах, при выбранных скоростях нагрева, при выбранных максимальных температурах, в течение выбранных периодов времени для регулирования удаления остаточных летучих примесей, уплотнения и спекания, перехода в другие структурные фазы и проведения химической реакции покрытия с газами, приводящей к другим типам материалов и фаз. Предпочтительным подходом является использование проточных газовых систем при тщательно подобранной и регулируемой скорости потока. Нагревание обладающего покрытием измельченного материала можно проводить в трубчатой печи, вращающейся трубчатой печи, приспособленной для медленного перемешивания частиц и тем самым предотвращения спекания или агломерации, или в любой конструкции печи, пригодной для регулируемого нагрева измельченных материалов в выбранных регулируемых газовых средах.
Как показано на схеме, приведенной на фиг.1, после предварительной сушки/термической обработки 2(b) существуют несколько возможных путей превращения обладающего покрытием материала в требующиеся материалы. (Сама предварительная сушка/термическая обработка 2(b) может представлять собой многостадийную процедуру, например сушку в вакууме при температуре ниже 100°С для удаления большей части свободной воды из микропор покрытия с последующим нагреванием, например примерно до 300°С в вакууме или на воздухе для удаления остаточных спиртов и абсорбированных гидроксильных функциональных групп с поверхности.)
Одним путем, путем А, является прокаливание обладающих покрытием частиц на воздухе или в кислороде для превращения покрытия в необходимый еще не полностью уплотненный оксид. В зависимости от конкретного используемого пористого оксидного материала-предшественника будет происходить частичное спекание или кристаллизация, включающая частичное уплотнение. Также могут происходить фазовые превращения в оксиды различной кристаллической структуры и их можно осуществить для получения требующихся оксидов. По этой методике обычно получают нанометровые оксиды. В каждом случае необходимые условия термической обработки определяют с помощью методик мониторинга и исследования реакции, таких как термогравиметрический анализ (ТГА), дифференциальный термический анализ (ДТА), рентгеноструктурный анализ (РСА) и т.п. Нагревание можно проводить в любом обычном оборудовании, пригодном для обработки тонкоизмельченного материала, хотя предпочтительными являются вращающиеся печи и печи с псевдоожиженным слоем.
Путь В используют для нагревания высушенных обладающих покрытием частиц, полученных на стадии 2(b), в аммиаке или смеси аммиака с инертным газом для превращения пористого оксидного предшественника покрытия в нитрид(ы) или оксинитрид(ы). Аммиак разлагается с образованием высокоактивных азот- и водородсодержащих частиц, которые постепенно восстанавливают и азотируют оксидный предшественник покрытия. Путем подбора условий можно получить различные оксинитридные и нитридные структуры. И в этом случае необходимые условия термической обработки определяют с помощью методик мониторинга и исследования реакции, таких как термогравиметрический анализ (ТГА), дифференциальный термический анализ (ДТА), рентгеноструктурный анализ (РСА) и т.п. Обычно образуются обладающие зернами нанометрового размера покрытия.
Путь С используют для нагревания высушенных обладающих покрытием частиц, полученных на стадии 2(b), в смесях углеродсодержащих газов с водородом для превращения пористого оксидного предшественника покрытия в карбид(ы) или оксикарбид(ы). Углеродсодержащим газом, в принципе, может быть любой газообразный углеводород, но предпочтительно - метан или этан. Смеси углеродсодержащий газ/водород можно разбавить инертным газом-носителем, таким как, например, аргон. Если активные газы составляют не более 20% от инертного газа-носителя, то маловероятно, что при утечке образуется взрывоопасная смесь газов с воздухом, так что улучшается безопасность. Типичные значения отношений количества метана или этана к количеству водорода составляют от 1/5 до 1/20. Необходимые условия термической обработки определяют с помощью методик мониторинга и исследования реакции, таких как термогравиметрический анализ (ТГА), дифференциальный термический анализ (ДТА), рентгеноструктурный анализ (РСА) и т.п.
Альтернативой превращения покрытий в оксинитриды и нитриды является использование пути А для выбранного оксида с последующим использованием пути D путем проведения термической обработки в содержащей аммиак среде с получением нитридов. Кроме того, при последующем использовании пути Е путем проведения обработки полученных таким образом нитридных покрытий в системах углеродсодержащий газ/водород можно получить другие карбидные микроструктуры, не такие как для пути С.
Кроме того, после получения оксидных структур с использованием пути А можно использовать путь F для получения карбидных микроструктур непосредственно из оксидных фаз.
Альтернативные комбинации путей допускают внесение изменений в содержание углерода, азота и кислорода в каждом карбиде, нитриде и оксиде. Например, посредством выбора пути и условий ТПТ можно получить оксинитридные материалы, материалы MNOx, в которых М обозначает металл, с выбором х в диапазоне от 0,5 до 0,05. В другом примере посредством выбора пути и условий ТПТ можно получить карбонитридные материалы, материалы MCNy, в которых у может находиться в диапазоне от 0 до 1.
Температуры нагрева, необходимые для получения кристаллических систем требующегося состава и структуры для материалов покрытий, являются относительно низкими. Это может привести к образованию низкотемпературных кристаллических систем, которые не образуются по более часто применяющимся твердофазным реакциям, обычно проводимым при более высоких температурах. В большей части случаев необходимые температуры ниже 1200°С, часто ниже 1000°С и в некоторых случаях составляют лишь 550°С.
Затем сверхтвердые частицы, по отдельности обладающие покрытием из требуемой оксидной нитридной или карбидной фазы необходимой микроструктуры, можно уплотнить в не содержащие пор или почти не содержащие пор обладающие зернами нанометрового размера куски композиционного материала путем обработки посредством горячего прессования, предпочтительно путем обработки при высоком давлении/высокой температуре.
4. Уплотнение при высокой температуре и спекание
По сравнению со спеканием без приложения давления горячее прессование обладает теми преимуществами, что при уплотнении обладающих зернами нанометрового размера материалов обеспечивается максимальная плотность и минимальный рост зерен. Примерами технологий горячего прессования, которые можно использовать, являются одноосное горячее прессование в углеродных штампах в специально сконструированных печах, горячее изостатическое прессование (ГИП), экструзия и технологии высокого давления. Предпочтительными сверхтвердыми частицами в настоящем изобретении являются алмаз и кубический нитрид бора, которые при продолжительном нагревании при высоких температурах, таких как 1600°С или выше, при нормальном давлении склонны к превращению в мягкие графитовые или гексагональные фазы. Вследствие этого проводимое при высоком давлении/высокой температуре горячее прессование является предпочтительной технологией для способа, предлагаемого в настоящем изобретении. Типичными условиями, которые можно использовать, но не ограничиваясь только ими, являются давления, равные от примерно 2,5 до примерно 8 ГПа, и температуры для каждого значения давления, определяющиеся термодинамической и/или химической стабильностью алмаза и cBN, обычно находящиеся в диапазоне от примерно 600 до примерно 1800°С. Типы оборудования для работы при высоком давлении/высокой температуре, которое можно использовать, включают поршневые и цилиндрические устройства, поршневые и опорные устройства, кубические опорные устройства и тороидальные и ленточные аппараты, и другие, хорошо известные в данной области техники.
Сверхтвердые частицы, по отдельности включенные в полученные, обычно не полностью уплотненные оксидные нитридные или карбидные покрытия, можно подвергать гранулированию, распылительной сушке, сушке вымораживанием, гранулированию в псевдоожиженном слое, все с использованием или без использования временных органических связующих материалов. Обычное холодное прессование также можно использовать для получения пористых, не полностью уплотненных полуфабрикатов изделий любой обычной формы с использованием сыпучего порошкообразного или гранулированного материала. В каждом случае значения давления/температуры/времени выбираются для проведения уплотнения и спекания материала покрытия, сведения к минимуму или регулирования роста зерен и получения кусков композиционного материала.
Полученные таким образом весьма однородные мелкозернистые и обладающие зернами нанометрового размера абразивные композиционные материалы характеризуются обусловленными их однородностью и сверхмелкозернистой микроструктурой улучшенными характеристиками по сравнению с аналогичными композиционными материалами, изготовленными по более распространенным технологиям, таким как размол и перемешивание отдельных порошкообразных исходных материалов. Обнаружено повышение ударной вязкости, но более примечательным является обнаруженное значительное повышение высокотемпературной прочности и твердости. Способом, предлагаемым в настоящем изобретении, также можно получить композиционные материалы, содержащие новые комбинации материалов и обладающие новыми составами и микроструктурой.
Полученные таким образом абразивные мелкозернистые композиционные материалы как в форме монолитов, так и в связанной с твердой металлической подложкой форме, можно применять для таких целей, как резание, размол, шлифование, сверление слишком твердых для обработки на станке материалов, включая сверление камня, и другие абразивные операции.
Способ, предлагаемый в настоящем изобретении, отличается от предшествующего уровня техники тем, что в нем используются мокрые коллоидные суспензионные технологии и поэтому он является весьма подходящим для эффективного нанесения покрытий на измельченные сверхтвердые частицы материала, обладающие размером от более 100 мкм до субмикрометрового и нанометрового включительно. В свою очередь это позволяет в дополнение к описанным в предшествующем уровне техники получить материалы новой структуры и состава. Имеются другие особенности настоящего изобретения, которые также отличают его от предшествующего уровня техники, включая специальные методики проведения химических реакций на поверхности измельченных сверхтвердых компонентов, коллоидное нанесение покрытий из микропористых основных оксидных предшественников, термическую обработку указанного обладающего покрытием материала с образованием выбранных структур и фаз с последующим использованием специальных методик уплотнения и спекания с получением улучшенных и новых композиционных структур, композиций и материалов.
Настоящее изобретение будет более подробно описано с помощью приведенных ниже неограничивающих примеров.
Пример 1
50 г субмикрометрового кубического нитрида бора, обладающего средним размером частиц, равным 0,7 мкм, в диапазоне размеров от 0,5 до 1,0 мкм обрабатывали в дымящей концентрированной серной кислоте, к которой прибавлен нитрат калия. После промывания и сушки субмикрометровый cBN дополнительно нагревали на воздухе при 600°С в течение 30 мин. Эта процедура приводила к тому, что в составе поверхности cBN преобладали кислородсодержащие функциональные группы и поэтому она стала витреофильной.
Затем 15 г этого субмикрометрового cBN с подвергнутой обработке поверхностью суспендировали в 865 мл чистого этанола в стакане, в который прибавляли 7,3 мл деионизированной воды. Суспензию энергично перемешивали лопастной мешалкой примерно при 100 оборотов/мин. 15,3 г жидкого изопропоксида титана, Ti(ОС3Н7)4 растворяли в 100 мл безводного этанола. Затем этот раствор по каплям в течение 1 ч при комнатной температуре (примерно 25°С) медленно прибавляли к суспензии cBN/этанол/вода, продолжая перемешивание. Перемешивание продолжали в течение еще 2 ч и содержимое стакана выдерживали в течение ночи. Полученные обладающие покрытием частицы извлекали из суспензии путем вакуумного фильтрования, трижды промывали этанолом и трижды деионизованной водой и затем сушили при 60°С в течение 2 дней в вакуумном сушильном шкафу. С помощью исследования на сканирующем электронном микроскопе (СЭМ) обнаружено, что каждая частица cBN была полностью покрыта соединением оксида титана, предположительно представляющим собой микропористый аморфный диоксид титана, TiO2.
Затем 10 г частиц cBN, обладающих покрытием из TiO2, подвергали термической обработке в потоке воздуха при 700°С в течение 3 ч. Скорость нагревания и скорость охлаждения поддерживали равными 5°С/мин. С помощью исследования на рентгеновском дифрактометре обнаружено, что покрытие закристаллизовалось в виде преимущественно анатазной фазы диоксида титана, как это показано на фиг.2, на котором приведена рентгенограмма, показывающая, что этот материал состоит только из диоксида титана и cBN. С помощью исследования этого измельченного материала на трансмиссионном электронном микроскопе, ТЭМ, обнаружено, что покрытие из диоксида титана закристаллизовалось в форме нанометровых кристаллитов размером примерно 30 нм.
5 г нагретых на воздухе субмикрометровых частиц cBN, обладающих покрытием из диоксида титана, дополнительно нагревали при 1100°С в течение 5 ч в трубчатой печи при пропускании потока сухого газообразного аммиака, NH3. Использовали скорость нагревания, равную 10°С/мин. Эта термическая обработка в аммиаке приводила к превращению обладающего зернами нанометрового размера покрытия из диоксида титана в обладающий зернами нанометрового размера нитрид титана, TiN. Исследование этого материала с помощью ТЭМ показало, что теперь покрытие состоит из кристаллитов нитрида титана размером примерно 40 нм. На фиг.3 приведена рентгенограмма, показывающая, что полученный порошок состоит только из cBN и нитрида титана со структурой осборнита.
Затем 2,5 г cBN, обладающего покрытием из обладающего зернами нанометрового размера TiN, обрабатывали при температуре, равной примерно 1300°С, при давлении, равном примерно 5,0 ГПа, в течение 20 мин при взаимодействии с подложкой из карбида вольфрама, WC, в аппарате высокого давления ленточного типа, хорошо известном в данной области техники. Таким образом получали не содержащий трещин поликристаллический cBN, содержащий примерно 78 мас.% cBN в сплошной матрице из TiN, связанный с подложкой или подкладкой из WC. Рентгенограмма спеченного материала приведена на фиг.4. Очевидно наличие следов фазы рутила диоксид титана. При исследовании с помощью СЭМ обнаружено, что полученный материал представляет собой просто дисперсию субмикрометровых частиц cBN в матрице из TiN. Это является одной особенностью, которая отличает настоящее изобретение от предшествующего уровня техники, в котором предполагается обнаружение таких фаз, как карбид вольфрама, WC, совместно с другими компонентами-металлами, образовавшимися из использованного размалывающего/перемешивающего оборудования. Кроме того, спектры дисперсии электронов, СДЭ, снятые на СЭМ, не приводят к обнаружению других элементов, кроме Ti, N и В. Полученные на СЭМ микрофотографии также показали, что расстояние между частицами cBN меняется в диапазоне примерно от 50 до 100 нм. Это означает, что размер зерен матрицы TiN самое большее равен этим значениям или меньше них. Это было подтверждено исследованием с помощью ТЭМ соответствующим образом утонченного образца материала, при котором наблюдались зерна TiN, обладающие размером от примерно 20 до 100 нм. Описанную выше процедуру повторяли несколько раз для получения материалов, предназначенных для исследований путем обработки на станке.
Материалы подвергали электроискровой обработке для получения образцов инструментов соответствующих размеров, предназначенных для исследований износа, включая обработку сталей, которые затруднительно обрабатывать на станках. Обнаружено, что материал, полученный так, как это описано выше, в качестве материала для токарных резцов обладает хорошими рабочими характеристиками при обработке этих сталей.
Пример 2
30 г порошкообразного cBN со средним размером частиц, равным 2 мкм, суспендировали в смешанном растворе 15% пероксида водорода, H2O2, и 15% гидроксида аммония, NH4OH, в воде, состава 1:1. Это приводило к гидролизу поверхностей частиц cBN и тем самым делало их витреофильными. Затем порошок cBN, обладающий частицами размером 2 мкм, извлекали из суспензии путем фильтрования и промывали деионизированной водой.
Затем 25,5 г полученного таким образом порошкообразного cBN суспендировали в 1440 мл этанола, в который прибавляли 13,1 мл деионизированной воды. Суспензию обрабатывали с помощью ультразвукового зонда в течение 15 мин для разрушения всех агломератов частиц cBN. 20,7 г изопропоксида титана растворяли в 100 мл безводного этанола. Затем этот раствор по каплям в течение 1 ч при комнатной температуре при энергичном перемешивании прибавляли к суспензии cBN в смеси этанол/вода. После прибавления суспензию перемешивали в течение еще 2 ч и затем выдерживали в течение ночи. Затем измельченный материал извлекали из суспензии путем фильтрования и трижды промывали чистым этанолом и затем трижды промывали деионизованной водой и затем сушили при 60°С в течение 2 дней в вакуумном сушильном шкафу. Исследование этого измельченного материала на электронном микроскопе с использованием устройства СДЭ показало, что на cBN имеется покрытие из соединения титана с кислородом. Все частицы были полностью покрыты в одинаковой степени.
Затем 20 г этого обладающего покрытием cBN прокаливали в трубчатой печи в потоке сухого воздуха при 450°С в течение 3 ч. Скорость нагревания и охлаждения поддерживали равными 5°С/мин. Исследование с помощью рентгеновского дифрактометра показало, что покрытие представляет собой диоксид титана, TiO2, со структурой анатаза.
8 г прокаленного cBN, обладающего покрытием из диоксида титана со структурой анатаза, нагревали в трубчатой печи при 1100°С в течение 5 ч в потоке сухого газообразного аммиака. С помощью исследования на рентгеновском дифрактометре показано, что покрытие из диоксида титана со структурой анатаза превратилось в нитрид титана, TiN.
Затем полученные таким образом частицы cBN размером 2 мкм, обладающие покрытием из TiN, обрабатывали при температуре, равной примерно 1300°С, при давлении, равном примерно 5,0 ГПа, в течение 20 мин. После отрезания и полировки сечения полученной таким образом поликристаллической массы материала показано, что покрытия из TiN подверглись спеканию с образованием непрерывной матрицы, в которой равномерно распределены частицы cBN. Другие фазы материала, кроме cBN и TiN, не обнаружены. Не обнаружены ни металлы, ни соединения металлов (кроме нитрида титана). Таким образом получен простой композиционный материал, содержащий только примерно 85 мас.% сверхтвердых частиц cBN, обладающих средним размером, равным 2 мкм, равномерно распределенных в непрерывной матрице TiN.
Пример 3
50 г алмаза, обладающего частицами микрометрового размера, полученного из синтетического алмаза с помощью дробления и сортировки, обладающего средним размером частиц, равным 1,0 мкм, в диапазоне размеров от 0,75 до 1,5 мкм, обрабатывали в дымящей концентрированной серной кислоте, к которой прибавлен нитрат калия. Эта очистка показала, что на поверхности алмаза не содержится металлов и неорганических загрязнений. Затем алмаз нагревали в потоке 20% кислорода в аргоне при 480°С в течение 1 ч. Эта процедура приводила к доведению до максимума количества кислородсодержащих функциональных групп, присоединенных к поверхностям алмаза, и делала их витреофильными.
15 г этого алмаза, обладающего частицами размером 1 мкм, с подвергнутой обработке поверхностью затем суспендировали в 865 мл чистого этанола в стакане, в который прибавляли 7,3 мл деионизированной воды. Суспензию энергично перемешивали лопастной мешалкой примерно при 100 оборотов/мин. 15,6 г жидкого изопропоксида титана, Ti(ОС3Н7)4, растворяли в 100 мл безводного этанола. Затем этот раствор по каплям в течение 1 ч при комнатной температуре (примерно 25°С) медленно прибавляли к суспензии алмаз/этанол/вода, продолжая перемешивание. Перемешивание продолжали в течение еще 2 ч и содержимое стакана выдерживали в течение ночи. Полученные обладающие покрытием частицы извлекали из суспензии путем вакуумного фильтрования, трижды промывали этанолом и трижды деионизованной водой и затем сушили при 60°С в течение 2 дней в вакуумном сушильном шкафу.
Затем 12 г высушенного обладающего покрытием алмаза нагревали на воздухе в статических условиях при 450°С в течение 2 ч. Использовали скорость нагревания, равную 5°С/мин. Затем материал исследовали с помощью СЭМ рентгеновской дифракции и обнаружили, что теперь на алмазе имеется покрытие из кристаллического диоксида титана со структурой анатаза, а другие фазы и соединения не обнаружены. Исследование с помощью ТЭМ подтвердило, что покрытие состоит из кристаллитов со структурой анатаза, обладающих размером, равным примерно от 10 до 20 нм.
Затем 5 г этого обладающего покрытием материала подвергали термической обработке в потоке сухого аммиака в течение 5 ч при 1100°С. Скорость потока аммиака составляла примерно 1 л/мин и использовали скорость нагревания, равную примерно 10°С/мин. Исследование с помощью СЭМ и ДРИ (дифракции рентгеновского излучения) показало, что теперь на алмазе имеется покрытие из нитрида титана. На фиг.5 приведена рентгенограмма, показывающая наличие алмаза и нитрида титана, а другие фазы и соединения не обнаружены. Исследование этого материала с помощью ТЭМ показало, что теперь покрытие состоит из кристаллитов нитрида титана размером примерно от 20 до 250 нм.
Путем обработки этого порошка при температуре, равной примерно 1350°С, и при давлении, равном примерно 5,5 ГПа, в ленточном аппарате высокого давления, в течение примерно 20 мин получали алмаз в виде композиционного материала в матрице из нитрида титана. Рентгеноструктурный анализ подтвердил, что полученный композиционный материал представляет собой алмаз в матрице из нитрида титана и что содержание нитрида титана соответствует формуле (TiN)0,88.
Пример 4
Методику, описанную выше в примере 3, можно проводить до получения порошкообразного алмаза, обладающего покрытием из кристаллического анатаза. Предполагается, что если этот порошок обрабатывать в потоке, состоящем из газовой смеси 10% метана в аргоне и 10% водорода в аргоне при соответствующем соотношении метан:водород и при температуре, равной примерно 1350°С, в течение нескольких часов (вероятно, более 5 ч), то покрытие из диоксида титана превратиться в карбид титана.
Вследствие этого предполагается, что множество этих частиц алмаза, обладающих покрытием из карбида титана, можно подвергнуть спеканию при высоких давлениях и температурах с получением композиционного материала, содержащего тонкоизмельченный алмаз в потенциально обладающей зернами нанометрового размера непрерывной матрице из карбида титана.
Пример 5
Алмаз, обладающий частицами размером 1 мкм (диапазон размеров от 0,75 до 1,5 мкм), подвергали кислотной очистке, как это описано в примере 3. 20 г подвергнутого кислотной очистке порошка обрабатывали смесью пероксида водорода (30 мас.%) и гидроксида аммония (25%) в объемном соотношении 1:1 путем кипячения с обратным холодильником при 60°С. Окисленные частицы алмаза обрабатывали по методике, описанной в примере 3, пока не получали порошкообразный алмаз, обладающий покрытием из кристаллического анатаза.
Этот порошкообразный алмаз, обладающий покрытием из диоксида титана, обрабатывали при температуре, равной 1350°С, и при давлении, равном 5,5 ГПа, в аппарате высокого давления ленточного типа. Таким образом получали матричный композиционный материал алмаз-диоксид титана.
Пример 6
Порошкообразный cBN, обладающий покрытием из диоксида титана, получали, как это описано в примере 1, путем термической обработки при температуре до 700°С с образованием cBN, обладающего покрытием преимущественно из анатаза. Этот порошок обрабатывали при температуре, равной 1350°С, и при давлении, равном 5,5 ГПа, в аппарате высокого давления ленточного типа. Таким образом получали композиционный материал, состоящий из cBN и диоксида титана в фазе рутила.
Пример 7
Синтетический порошкообразный алмаз, обладающий частицами размером 1 мкм с диапазоном размеров от 0,75 до 1,5 мкм, подвергали кислотной очистке, как это описано в примере 3. 20 г этого порошкообразного алмаза суспендировали в растворе, содержащем 258 мл чистого изопропанола и 175 мл деионизированной воды. Эту суспензию нагревали при 60°С в установке с обратным холодильником и механически перемешивали мешалкой лопастного типа примерно при 100 оборотов/мин. 24 г втор-бутоксида алюминия, обладающего химической формулой AlO3C12H27, растворяли в 100 мл безводного изопропанола и по каплям в течение 1 ч 45 мин при нагревании и перемешивании прибавляли к суспензии алмаза суспензию. После прибавления алкоксида суспензию перемешивали в течение 1 ч 15 мин при 60°С. Затем к нагретой суспензии прибавляли примерно 1 мл хлористоводородной кислоты (32%) и затем нагревали до 80°С и перемешивали в течение еще 1 ч, поддерживая указанную температуру. Затем суспензии давали охладиться до комнатной температуры и ее выдерживали при комнатной температуре в течение ночи. Затем суспензию сушили на роторном испарителе при температуре, равной 80°С, и в вакууме при давлении, равном 400 мбар.
Алмаз, обладающий покрытием из соединения алюминия, дополнительно сушили в вакуумном сушильном шкафу при 60°С в течение 2 дней. Исследование с помощью СЭМ показало, что частицы алмаза обладают покрытием из соединения оксида алюминия.
Затем этот порошок подвергали термической обработке при 400°С на воздухе в статических условиях в течение 3 ч. Использовали скорость нагревания, равную 5°С/мин. Рентгеноструктурный анализ показал, что после этой термической обработки покрытие на алмазе являлось преимущественно аморфным. Это было подтверждено исследованием с помощью ТЭМ. Затем подвергнутый термической обработке порошок подвергали горячему прессованию при взаимодействии с подложкой в виде диска из карбида вольфрама при температуре, равной примерно 1300°С, и при давлении, равном 5 ГПа, в течение примерно 20 мин при высоком давлении/высокой температуре в аппарате ленточного типа и получали композиционный материал алмаз-оксид алюминия.
Частицы алмаза, обладающие средним размером, равным 1 мкм, хорошо распределены в непрерывной матрице из оксида алюминия. Расстояние между частицами алмаза находилось в диапазоне примерно от 50 до 500 нм. В композиционный материал, находящийся рядом с подложкой из карбида вольфрама, частично приникал металлический кобальт с небольшим количеством вольфрама. Наличие этих металлов обнаружено с помощью спектрометрии дисперсии электронов (СДЭ) в приборе СЭМ. Композиционный материал алмаз-оксид алюминия очень прочно связан с подложкой из карбида вольфрама. Рентгеноструктурный анализ показал, что полученный спеченный материал в основном представляет собой алмаз в закристаллизованной непрерывной матрице из оксида алюминия.
Пример 8
Синтетический порошкообразный алмаз, обладающий частицами размером 1 мкм (с диапазоном размеров частиц 0,75-1,5 мкм), подвергали кислотной очистке, как это описано в примере 3. 30 г этого порошкообразного алмаза суспендировали в смеси 2,5 л чистого этанола, 500 мл деионизированной воды и 60 мл 25 об.% водного раствора гидроксида аммония. Эту суспензию обрабатывали ультразвуком с использованием ультразвукового зонда в течение 15 мин, а затем механически перемешивали мешалкой лопастного типа примерно при 100 оборотов/мин. 80 г тетраэтилортосиликата, обладающего химической формулой Si[C2H5O]4, растворяли в 100 мл чистого этанола. Суспензию тетраэтилортосиликата при энергичном перемешивании по каплям прибавляли к суспензии алмаза в течение 3 ч, продолжая механическое перемешивание. После прибавления алкоксида коллоидную суспензию перемешивали в течение еще 2 ч и ее выдерживали в течение 64 ч. Затем выдержанный раствор 6 раз промывали чистым этанолом и затем сушили на роторном испарителе при температурах от 70 до 80°С в вакууме при давлении, сниженном до 150 мбар. Порошок дополнительно сушили в вакуумном сушильном шкафу при 60°С в течение 2 дней.
Затем высушенный алмаз, обладающий покрытием из соединения кремния, подвергали термической обработке в трубчатой печи при скорости нагревания, равной 5°С/мин до 800°С в потоке аргона. Порошок обрабатывали при 800°С в течение 3 ч в потоке аргона. Рентгеноструктурный анализ подвергнутого термической обработке порошка показал, что покрытие из оксида кремния на алмазе все еще являлось преимущественно аморфным. Это было подтверждено исследованием с помощью ТЭМ.
Порошкообразный алмаз, обладающий покрытием из оксида кремния, обрабатывали при температуре, равной примерно 1300°С, и при давлении, равном примерно 5 ГПа, в течение 20 мин в аппарате ленточного типа и получали композиционный материал алмаз-оксид кремния. Исследование полированного образца с помощью СЭМ показало, что композиционный материал состоял из алмаза, обладающего средним размером частиц, равным 1 мкм, равномерно распределенного в непрерывной матрице. С помощью прибора СДЭ на микроскопе показано, что матрица представляет собой фазу оксида кремния, а исследование с помощью рентгеновского дифрактометра показало, что она представляет собой мелкозернистый кварц, как это показано на фиг.6, на котором приведена рентгенограмма этого материала.
Пример 9
Субмикрометровый кубический нитрид бора, обладающий размером частиц в диапазоне от 0,5 до 1 мкм (средний размер частиц равен 0,7 мкм), подвергали кислотной обработке, как это описано в примере 1. 34,04 г подвергнутого кислотной обработке порошкообразного cBN суспендировали в 2021 мл чистого этанола и 42 мл деионизированной воды. Эту суспензию cBN обрабатывали с помощью ультразвукового зонда в течение 20 мин для удаления агломератов, а затем энергично механически перемешивали лопастной мешалкой.
19,79 г н-пропоксида циркония(IV) (70 мас./мас.% в н-пропаноле), обладающего химической формулой Zr[O(СН2)2СН3]4, растворяли в 122 мл сухого этанола. Раствор алкоксида при перемешивании при комнатной температуре по каплям в течение 3 ч прибавляли к суспензии cBN и перемешивали в течение еще 1,5 ч после прибавления алкоксида. Суспензию обладающего покрытием cBN выдерживали при комнатной температуре в течение ночи. cBN, обладающий покрытием из оксида циркония, трижды промывали чистым этанолом и сушили на роторном испарителе в вакууме при давлении, равном от 600 до 390 мбар, и при температуре, равной от 70 до 80°С. Полученный порошок дополнительно сушили в вакуумном сушильном шкафу при 60°С в течение 2 дней. Высушенный порошок исследовали на сканирующем электронном микроскопе и обнаружено, что частицы cBN обладают хорошим покрытием.
Затем этот высушенный порошок подвергали термической обработке на воздухе в статических условиях при 600°С в течение 3 ч. Использовали скорость нагревания, равную 5°С/мин. Рентгеноструктурный анализ подвергнутого термической обработке порошка показал, что в нем содержатся фазы кубического нитрида бора и тетрагонального оксида циркония (ZrO2). Микрофотографии, полученные с помощью ТЭМ, показали, что покрытие из диоксида циркония состоит из кристаллитов размером не более 5 нм.
Порошкообразный cBN, обладающий покрытием из тетрагонального оксида циркония, подвергали горячему прессованию при взаимодействии с подложкой в виде диска из карбида вольфрама при температуре, равной примерно 1300°С, и при давлении, равном примерно 5 ГПа, в течение 20 мин в прессе ленточного типа для обработки при высоком давлении/высокой температуре. Таким образом получали композиционный материал, обладающий большим содержанием cBN в матрице из диоксида циркония, связанный с подложкой из карбида вольфрама. Содержание cBN найдено равным примерно 85 мас.%.
Исследование микроструктуры с помощью СЭМ показало, что зерна cBN хорошо распределены в непрерывной матрице и расстояние между частицами составляет не более 100 нм. Рентгеноструктурный анализ материала показал, что матрица состоит из чрезвычайно мелкозернистого диоксида циркония и содержатся и тетрагональная (ZrO1,88), и моноклинная (ZrO2) фазы. Ширина пиков ДРИ материала матрицы согласуется с обладающей зернами нанометрового размера ожидаемой структурой матрицы из диоксида циркония.
Пример 10
Субмикрометровый кубический нитрид бора, обладающий размером частиц в диапазоне от 0,5 до 1 мкм (средний размер частиц равен 0,7 мкм), подвергали кислотной обработке, как это описано в примере 1. 25 г этого порошка суспендировали в 1,5 л чистого этанола и 30 мл деионизированной воды и в течение 25 мин подвергали ультразвуковой обработке для удаления агломератов. В отдельном стакане 0,483 г гексагидрата нитрата иттрия, Y(NO3)3·6Н2О, растворяли в 50 мл чистого этанола, а затем прибавляли 13,9 г н-пропоксида циркония(IV), обладающего химической формулой Zr[O(СН2)2СН3]4, и еще 50 мл чистого этанола. Содержимое последнего стакана перемешивали стеклянной палочкой и дополнительно перемешивали путем встряхивания содержимого в делительной воронке. Раствор смеси гексагидрата нитрата иттрия с н-пропоксидом циркония(IV) при перемешивании по каплям при комнатной температуре в течение 2 ч прибавляли к суспензии cBN. После этого прибавления раствор дополнительно механически перемешивали в течение 1 ч 10 мин. Затем раствор выдерживали в течение ночи при комнатной температуре. Обнаружено, что после выдерживания в течение ночи полученное множество обладающих покрытием частиц образовало высоковязкий гель. После выдерживания всего в течение 48 ч золь-гель сушили на роторном испарителе в вакууме при давлении, равном 400 мбар, и при температуре, равной от 70 до 80°С.
Этот порошок дополнительно сушили в вакуумном сушильном шкафу при 60°С в течение 2 дней. Затем высушенный порошок cBN, обладающий покрытием из оксида циркония, подвергали термической обработке на воздухе в статических условиях при 600°С в течение 3 ч. Использовали скорость нагревания, равную 5°С/мин. Рентгеноструктурный анализ показал, что полученный порошок содержит фазы cBN и тетрагональную ZrO1,99. Полученные на ТЭМ микрофотографии показали, что зерна диоксида циркония обладают размером, равным от 4 до 5 нм.
Подвергнутый термической обработке порошок обрабатывали при температуре, равной примерно 1300°С, и при давлении, равном примерно 5 ГПа, в течение 20 мин при высоком давлении/высокой температуре в аппарате ленточного типа. Это приводило к получению композиционного материала cBN-оксид циркония. Содержание cBN найдено равным не менее 85 мас.%. Исследование микроструктуры с помощью СЭМ показало, что зерна cBN хорошо распределены в непрерывной матрице и расстояние между частицами составляет не более 100 нм. Рентгеноструктурный анализ материала показал, что матрица состоит из чрезвычайно мелкозернистого диоксида циркония и содержатся и тетрагональная (ZrO1,95), и моноклинная (ZrO2) фазы. Обнаружены следовые количества фазы ZrB2. Ширина пиков ДРИ материала матрицы согласуется с обладающей зернами нанометрового размера ожидаемой структурой матрицы из диоксида циркония.
Пример 11
10 г алмаза, обладающего частицами микрометрового размера, полученного из дробленого синтетического алмаза, со средним размером частиц, равным 2 мкм, подвергали кислотной очистке и подвергали термической обработке в кислороде, как это описано в примере 3. Это делало поверхности частиц витреофильными и способными образовывать связи с оксидными материалами.
Затем этот материал в стакане объемом 2 л суспендировали в 150 мл чистого этанола, в который прибавляли 5 мл деионизированной воды. Суспензию энергично перемешивали лопастной мешалкой примерно при 100 оборотов/мин. 20,25 г этоксида вольфрама формулы W(OC2H5)5 растворяли в 100 мл чистого безводного этанола. Затем этот раствор по каплям при перемешивании в течение 1,5 ч медленно прибавляли к суспензии алмаза в этаноле и воде. После завершения прибавления раствора алкоксида вольфрама суспензию перемешивали в течение еще 2 ч. Затем реакционную смесь выдерживали в течение примерно 16 ч. Измельченный материал оседал на дно стакана и надосадочная жидкость выглядела немного белесоватой. Надосадочную жидкость сливали и измельченный материал трижды промывали чистым этанолом, каждый раз перемешивая, давая отстояться и сливая. После последней декантации измельченный материал медленно сушили при условиях окружающей среды в течение 2 дней. Затем материал дополнительно сушили в вакуумном сушильном шкафу при 60°С в течение 24 ч. При исследовании с помощью СЭМ и блока СДЭ в указанном приборе обнаружено, что частицы алмаза размером 2 мкм обладают покрытием из соединения оксида вольфрама с отношением вольфрама:кислород, соответствующим оксиду вольфрама формулы WO3.
Затем обладающий покрытием материал нагревали на воздухе при 380°С в течение 3 ч для дополнительного высушивания и в особенности для удаления гидроксилсодержащих частиц.
Затем 5 г полученного обладающего покрытием измельченного материала нагревали в трубчатой печи в потоке смеси метана (СН4), водорода (Н2) и аргона. 90% объема газа составлял аргон, а остальные 10% - смесь метана с водородом. Объемное отношение метан:водород составляло 1:4. Полная использованная скорость потока газа равнялась примерно 50 мл/мин. Температуру печи повышали со скоростью 10°С/мин и максимальную постоянную температуру поддерживали в течение 10 ч. После охлаждения и последующего исследования с помощью СЭМ и ДРИ обнаружено, что измельченный алмаз теперь обладает покрытием из чрезвычайно мелкозернистого карбида вольфрама, WC.
Затем алмаз, обладающий покрытием из WC, подвергали спеканию в ленточном аппарате высокого давления при температуре, равной примерно 1400°С, и при давлении, равном примерно 5,5 ГПа. Получали композиционный материал, состоящий примерно из 50 мас.% алмаза, обладающего частицами размером 2 мкм, распределенный в чрезвычайно мелкозернистой спеченной матрице из карбида вольфрама. Предполагается, что зерна матрицы из карбида вольфрама в основном являются нанометровыми, т.е. наибольший размер зерен значительно меньше 100 нм.
5 г оставшегося алмаза, обладающего покрытием из оксида вольфрама, также нагревали в чистом водороде при 550°С в течение 3 ч. Таким образом оксидное покрытие восстанавливалось с образованием микрокристаллического металлического вольфрама.
Затем частицы алмаза, обладающего покрытием из металлического вольфрама, можно обработать при температуре, равной примерно 1400°С, и при давлении, равном примерно 5,5 ГПа. Предполагается, что будет получен композиционный материал, содержащий примерно 50 мас.% алмаза, обладающего частицами размером 2 мкм, распределенного в матрице из микрокристаллического металлического вольфрама.
Пример 12
Партию алмаза, обладающего частицами размером 2 мкм, очищали и поверхности частиц обрабатывали по той же методике, что и описанная в примере 1. cBN, обладающий частицами размером 2 мкм, также подвергали кислотной очистке и обрабатывали таким же образом, как это описано в примере 3. Эти порошкообразные алмаз и cBN можно смешать в спиртовой суспензии и нанести на них покрытие таким же образом, как это описано в примерах 2 и 3, в массовом отношении 1:1. Предполагается, что это приведет к порошкообразной системе алмаз/cBN, обладающей покрытием из аморфного диоксида титана, причем каждая частица будет обладать индивидуальным покрытием из диоксида титана. Затем этот порошок подвергали термической обработке на воздухе, а после этого - термической обработке в сухом аммиаке при 1100°С (как это подробно описано в примере 3), что приводит к порошкообразной системе алмаз/cBN, обладающей покрытием из кристаллического нитрида титана. Спекание этого порошка при высоком давлении/высокой температуре, как это описано в примере 2, приводит к равномерному распределению частиц и cBN, которые, в свою очередь, равномерно распределены в непрерывной микро/нанокристаллической матрице из нитрида титана.
Пример 13
Партии синтетического алмаза, обладающего частицами размером 1 мкм, подвергали кислотной очистке и поверхность обрабатывали таким же образом, как это описано в примере 3.
Алкоксиды циркония и иттрия, такие как н-пропоксид циркония(IV) и изопропоксид иттрия(III) можно смешать в безводном спирте, так чтобы в результате содержалось от 3 до 8 мол.% ZrO2. Эту смесь алкоксидов можно по каплям прибавить к спиртовой суспензии алмаза, как это описано в примере 10. Обладающий покрытием порошкообразный алмаз сушили и подвергали термической обработке, как это описано в примере 10, и получали частицы алмаза, обладающие покрытием из преимущественно нанометровой тетрагональной фазы диоксида циркония. Предполагается, что спекание этого порошка при высоком давлении/высокой температуре, как это подробно описано в примере 2, приведет к композиционному материалу алмаз-диоксид циркония.
Реферат
Изобретении относится к изготовлению поликристаллических абразивных элементов. На сверхтвердые абразивные частицы, обладающие поверхностями, склонными к образованию химических связей с оксидами, наносят коллоидное покрытие из материала-предшественника матрицы, содержащего гидратированный оксид или металл, или металлоид. Обладающие покрытием абразивные частицы подвергают термообработке в газовой среде, уплотняют и спекают при давлении и температуре, при которых они являются кристаллографически или термодинамически стабильными. Поликристаллический абразивный элемент содержит обладающие зернами микрометрового, субмикрометрового или нанометрового размера сверхтвердые абразивы, диспергированные в микрометровых, субмикрометровых или нанометровых матричных материалах. Полученный материал обладает однородной мелкозернистой структурой, а также высокими ударной вязкостью, высокотемпературной прочностью и твердостью. 5 н. и 30 з.п. ф-лы, 6 ил., 1 табл.
Комментарии