Способ получения нитрата металла на подложке - RU2516467C2
Код документа: RU2516467C2
Чертежи



Описание
Настоящее изобретение относится к получению нитратов металлов на подложке, пригодных для использования в качестве предшественников катализаторов или сорбентов.
Нитраты металлов представляют собой пригодные для использования предшественники катализаторов или сорбентов благодаря их относительно низкой стоимости и простоте получения. При получении катализатора или сорбента, как правило, один или несколько растворимых нитратов металлов импрегнируются в соответствующий материал подложки и сушатся для удаления растворителя. Затем импрегнированная подложка, как правило, нагревается на воздухе, на стадии, часто называемой кальцинированием, до повышенной температуры, равной температуре разложения нитрата металла или превышающей ее, для получения оксида металла. Однако такой способ не всегда приводит к получению удовлетворительных оксидных материалов. В частности, когда оксид металла представляет собой восстанавливаемый оксид металла, диспергирование и распределение кристаллитов оксида металла, а, следовательно, и восстановленного металла, получаемого с помощью этих способов, часто плохое.
Опубликованы меры для улучшения кальцинирования. WO 2007/071899 описывает процедуру кальцинирования для преобразования нитратов металлов в соответствующие оксиды металла посредством нагрева нитрата металла для осуществления его разложения в атмосфере газовой смеси, содержащей оксид азота (NO) и имеющей содержание кислорода <5% об. WO 2008/029177 описывает идентичный способ с использованием закиси азота (N2O). Хотя эти документы рассматривают высокотемпературное превращение нитрата в оксид, они не рассматривают эффекта выдерживания нитрата металла на подложке в газах при температурах ниже, чем те, при которых образуется оксид.
Авторы обнаружили, что посредством использования низкотемпературной термической обработки высушенного или невысушенного нитрата металла может достигаться улучшенное диспергирование металла. Повышенная дисперсность металла является желательной, поскольку каталитическая активность или сорбция часто позитивно связана с площадью поверхности получаемых соединений металлов на подложке.
Соответственно, настоящее изобретение предусматривает способ получения нитрата металла на подложке, пригодного для использования в качестве предшественника для катализатора или сорбента, включающий стадии:
(i) импрегнирования материала подложки нитратом металла, и
(ii) выдерживания импрегнированного материала в газовой смеси, содержащей оксид азота, при температуре в пределах 0-150°C, с образованием диспергированного на подложке нитрата металла.
Кроме того, настоящее изобретение предусматривает нитрат металла на подложке, который может получаться с помощью указанного выше способа.
Таким образом, в отличие от способов, описанных в указанных выше WO 2007/071899 и WO 2008/029177, способ по настоящему изобретению действует, стабилизируя нитрат и уменьшая его тенденцию к агломерации на поверхности подложки.
Нитрат металла может наноситься на подложку с помощью ряда способов, включая импрегнирование расплавленным нитратом, то есть импрегнирования с использованием расплавленного нитрата металла, или посредством импрегнирования с использованием соответствующего раствора нитрата металла. Например, нитрат металла может импрегнироваться на материале подложки из водного или неводного раствора, например, раствора в этаноле или ацетоне, которые могут содержать другие растворители. В растворе могут присутствовать один или несколько нитратов металлов. Может осуществляться одна или несколько стадий импрегнирования для увеличения нагрузки металла или для создания последовательных слоев различных нитратов металлов перед сушкой. Импрегнирование может осуществляться с использованием любого из способов, известных специалистам в области получения катализаторов или сорбентов, но предпочтительно осуществляется посредством так называемого 'сухого' импрегнирования или импрегнирования с помощью 'пропитки по влагоемкости', поскольку это сводит к минимуму количество растворителя, который используется и должен удаляться при последующей сушке. Импрегнирование с помощью пропитки по влагоемкости является особенно пригодным для пористых материалов подложек и включает смешивание материала подложки с таким количеством жидкости, которое достаточно только для заполнения пор подложки.
Как правило, импрегнирование осуществляют до тех пор, пока содержание металла для импрегнированного материала (вычисляемое по отношению к сухому веществу) не будет находиться в пределах 1-30% масс.
В настоящем изобретении, импрегнированный материал выдерживается в газовой смеси, содержащей оксид азота, при температуре в пределах 0-150°C, предпочтительно 10-120°C, более предпочтительно 25-75°C, с получением диспергированного на подложке нитрата металла.
Таким образом, в одном из вариантов осуществления это выдерживание приводит к удалению растворителя из импрегнированного материала, то есть обработка служит для сушки и одновременной стабилизации нитрата металла на подложке. В альтернативном варианте осуществления, если это желательно, может осуществляться дополнительная стадия сушки при низкой температуре перед выдерживанием в газовой смеси, содержащей NO, с целью удаления растворителя. Если осуществляется эта дополнительная стадия, она должна осуществляться при температуре ниже 60°C, например, в пределах от 0 до 60°C, в вакууме или на воздухе или в атмосфере инертного газа, такого как азот, с тем, чтобы не вызывать агломерации нитрата металла. Соответственно, импрегнированный материал, желательно, должен поддерживаться при температуре примерно ниже 60°C перед выдерживанием в газовой смеси, содержащей NO.
Импрегнированный материал, следовательно, может сушиться в атмосфере NO или, альтернативно, сушиться при низкой температуре, примерно ниже 60°C, в вакууме, на воздухе или в инертной атмосфере для удаления растворителя, а затем нагреваться в атмосфере оксида азота для удаления любого оставшегося растворителя и получения стабилизированного нитрата металла.
Может быть желательным, в частности, когда термическую обработку осуществляют при выше примерно 20°C, чтобы атмосфера, в которой нитрат металла на подложке выдерживают во время нагрева, содержала очень мало свободного кислорода или вообще не содержала его, поскольку он, как обнаружено, является источником дисперсности восстановленного оксида металла в материалах, полученных из нитратов. Следовательно, содержание кислорода (O2) в газовом потоке предпочтительно ≤5%, более предпочтительно ≤1%, наиболее предпочтительно ≤0,1 % об.
Газовый поток, в котором выдерживают нитрат металла, может представлять собой любой газовый поток, который содержит оксид азота. Предпочтительно газовый поток содержит оксид азота и один или несколько газов, выбранных из монооксида углерода, диоксида углерода или инертного газа. Предпочтительно инертный газ представляет собой один или несколько газов, выбранных из азота, гелия или аргона. Предпочтительно газовый поток, в котором выдерживают оксид металла на подложке, состоит из одного или нескольких инертных газов и оксида азота.
Газовая смесь, в которой выдерживают нитрат металла на подложке, может находиться при атмосферном давлении или при чуть более высоком давлении, как правило, примерно до 10 бар абс. Могут использоваться различные способы, известные в области осуществления стадии нагрева. Когда стадия нагрева осуществляется посредством прохождения газовой смеси через слой нитрата металла на подложке, объемная часовая скорость газа (GHSV) для газовой смеси предпочтительно находится в пределах от 100 до 600000 час-1.
Концентрация оксида азота в газовом потоке предпочтительно находится в пределах 0,001-15% об., более предпочтительно 0,1-15% об., наиболее предпочтительно 1-10% об., для достижения желаемого воздействия при масштабируемой объемной скорости и сведения в то же время к минимуму требований к скруббингу.
Нитрат металла на подложке включает один или несколько нитратов металлов на поверхности и/или в порах подложки. Нитрат металла может представлять собой любой нитрат металла, но предпочтительно представляет собой нитрат металла, используемого при получении катализаторов, предшественников катализаторов или сорбентов. Нитрат металла может представлять собой нитрат щелочного, щелочноземельного металла или переходного металла. Предпочтительно нитрат металла представляет собой нитрат переходного металла, то есть нитрат металлов, выбранных из Групп 3-12 Периодической таблицы элементов, включительно. Соответствующие легкодоступные нитраты металлов для получения катализатора, предшественника катализатора или сорбента включают нитраты La, Ce, Zr, Cr, Mn, Fe, Ru, Co, Rh, Ir, Ni, Pd, Pt, Cu и Zn, более предпочтительно нитраты Cr, Mn, Fe, Ru, Co, Rh, Ir, Ni, Pd, Pt, Cu и Zn. Могут присутствовать один или несколько нитратов металлов. Под термином "нитраты металлов" авторы включают соединения нитратов металлов формулы M(NO3)x·(H2O)a, где x представляет собой валентность металла M, и 'a' может представлять собой 0 или целое число ≥1.
Однако настоящее изобретение, как обнаружено, является особенно пригодным для использования, когда получаемый продукт нитрата металла на подложке содержит нитрат металла формулы Mx(OH)y(NO3)z, в которой x, y и z представляют собой целые числа ≥1 и M представляет собой переходной металл, предпочтительно железо, рутений, кобальт, родий, иридий, никель, палладий, платина, медь, или их смесь, более предпочтительно медь, никель или кобальт, в особенности, медь. Таким образом, раствор нитрата металла предпочтительно содержит нитрат железа, рутения, кобальта, родия, иридия, никеля, палладия, платины, меди или их смесь, более предпочтительно нитрат меди, никеля или кобальта, в особенности, меди. Могут присутствовать и другие нитраты металлов.
Подложка, на которую может наноситься нитрат металла, может представлять собой металл, углерод, оксид металла, смешанный оксид металла или твердую полимерную подложку.
Углеродные подложки, такие как активированные угли, графиты с высокой площадью поверхности, углеродные нановолокна и фуллерены в форме порошка, таблеток или гранул и имеющие соответствующую пористость, например, выше 0,1 мл/г, могут использоваться в качестве подложек для настоящего изобретения, предпочтительно, когда газовый поток содержит ≤0,1% об. кислорода.
Предпочтительно подложка представляет собой оксидную подложку, которая может представлять собой материал одинарного или смешанного оксида металла, включая керамику, цеолиты, перовскиты, шпинели и тому подобное. Оксидная подложка может также находиться в форме реактивной грунтовки на керамической, металлической, углеродной или полимерной подложке.
Подложка может находиться в форме порошка, имеющего усредненный по поверхности диаметр D[3, 2] в пределах от 1 до 200 микрон. Термин усредненный по поверхности диаметр D[3,2], иным образом определяемый как средний диаметр Саутера, определяется M. Alderliesten в статье "A Nomenclature for Mean Particle Diameters"; Anal. Proc, vol 21, May 1984, pages 167-172, и вычисляется из анализа размеров частиц, который удобно осуществлять с помощью дифракции лазерного света, например, с использованием Malvern Mastersizer. Агломераты таких порошков, имеющие размеры частиц в пределах от 200 микрон до 1 мм, также могут использоваться в качестве подложки. Альтернативно, подложка может находиться в форме формованных единиц, таких как таблетки, экструдаты или гранулы, как правило, имеющие размеры частиц в пределах от 1 до 25 мм и аспектное отношение меньше чем 2. Под размером частиц авторы имеют в виду самый маленький размер частиц, такой как ширина, длина или диаметр. Альтернативно, подложка может находиться в форме монолита, например, сотовой структуры, или такого ячеистого материала, как пена с открытой структурой ячеек.
Подложка предпочтительно выбирается из оксида алюминия, алюмината металла, диоксида кремния, алюмосиликата, оксида цинка, оксида титана, диоксида циркония или из их смесей, включая совместные гели, в любой форме из порошка, формованной единицы, монолита или ячеистой структуры.
Подложка может представлять собой подложку на основе диоксида кремния. Подложки на основе диоксида кремния могут формироваться из природных источников, таких, например, как кизельгур, могут представлять собой пирогенный или коллоидный диоксид кремния или могут представлять собой синтетический, например, преципитированный диоксид кремния или силикагель. В качестве подложек могут использоваться структурированные мезопористые диоксиды кремния, такие как SBA-15. Также могут использоваться преципитированные диоксиды кремния. Диоксид кремния может находиться в форме порошка или формованного материала, например, как кусочки диоксида кремния в форме экструдированных частиц, таблеток или гранул. Пригодные для использования порошкообразные диоксиды кремния, как правило, имеют частицы с усредненным по поверхности диаметром D[3, 2] в пределах от 3 до 100 мкм. Формованные диоксиды кремния могут иметь разнообразные формы и размеры частиц, в зависимости от формы или фильеры, используемой при их получении. Например, частицы могут иметь форму поперечного сечения, которая является круговой, дольчатой или другой формой, и длину примерно от 1 до более чем 10 мм. Площадь поверхности по БЭТ пригодных для использования порошкообразных или гранулированных диоксидов кремния, как правило, находится в пределах 10-500 м2/г, предпочтительно, 100-400 м2/г. Объем пор находится, как правило, в пределах примерно между 0,1 и 4 мл/г, предпочтительно, в пределах 0,2-2 мл/г, и средний диаметр пор предпочтительно находится в пределах от 0,4 примерно до 30 нм. Если это желательно, диоксид кремния может смешиваться с другими оксидами металла, такими как оксид титана или диоксид циркония. Альтернативно, диоксид кремния может присутствовать в качестве покрытия на формованной единице, которая предпочтительно состоит из оксида алюминия, как правило, как покрытия из 0,5-5 монослоев диоксида кремния на лежащей под ним подложке.
Подложка может представлять собой оксид цинка, который предпочтительно представляет собой материал с высокой площадью поверхности. Оксид цинка может также представлять собой часть смешанного оксида, например, титаната цинка.
Подложка может представлять собой подложку на основе оксида титана. Подложка на основе оксида титана предпочтительно является синтетической, например, содержит преципитированные оксиды титана. Оксид титана может необязательно содержать, например, до 20% масс. другого материала тугоплавкого оксида, как правило, диоксида кремния, оксида алюминия или диоксида циркония. Оксид титана альтернативно может присутствовать в виде покрытия на подложке, которая предпочтительно представляет собой диоксид кремния или оксид алюминия, например, в виде покрытия из 0,5-5 монослоев оксида титана на лежащей под ним подложке на основе оксида алюминия или оксида кремния. Площадь поверхности по БЭТ для соответствующего оксида титана, как правило, находится в пределах 10-500 м2/г, предпочтительно 100-400 м2/г. Объем пор оксида титана предпочтительно, как правило, находится в пределах между примерно 0,1 и 4 мл/г, более предпочтительно, 0,2-2 мл/г и средний диаметр пор предпочтительно находится в пределах от 2 примерно до 30 нм.
Подобным же образом, подложки на основе диоксида циркония могут быть синтетическими, например, содержать преципитированный диоксид циркония. Опять же, диоксид циркония может необязательно содержать, например, до 20% масс. другого материала тугоплавкого оксида, как правило, диоксида кремния, оксида алюминия или оксида титана. Альтернативно, диоксид циркония может представлять собой стабилизированный диоксид циркония, например, диоксид циркония, стабилизированный оксидом иттрия или оксидом церия. Диоксид циркония альтернативно может присутствовать в виде покрытия на подложке, которая предпочтительно представляет собой диоксид кремния или оксид алюминия, например, в виде покрытия из 0,5-5 монослоев диоксида циркония на лежащей под ним подложке на основе оксида алюминия или оксида кремния.
Подложка может представлять собой алюминат металла, например, алюминат кальция.
Материал подложки может представлять собой переходной оксид алюминия. Переходные оксиды алюминия определяются в "Ullmans Encyklopaedie der technischen Chemie", 4, neubearbeitete und erweiterte Auflage, Band 7 (1974), pp.298-299. Соответствующий переходной оксид алюминия может относиться к группе гамма-оксидов алюминия, например, представлять собой эта-оксид алюминия или хи-оксид алюминия. Эти материалы могут быть получены посредством кальцинирования гидроксидов алюминия при 400-750°C и, как правило, имеют площадь поверхности по БЭТ в пределах от 150 до 400 м2/г. Альтернативно, переходной оксид алюминия может принадлежать группе дельта-оксидов алюминия, которая содержит высокотемпературные формы, такие как дельта- и тета-оксид алюминия, которые могут формироваться посредством нагрева оксида алюминия гамма-группы до температуры выше примерно 800°C. Оксиды алюминия дельта-группы, как правило, имеют площадь поверхности по БЭТ в пределах от 50 до 150 м2/г. Альтернативно, переходной оксид алюминия может представлять собой альфа-оксид алюминия. Переходные оксиды алюминия содержат меньше чем 0,5 моль воды на моль Al2O3, реальное количество воды зависит от температур, до которых их нагревают. Пригодный для использования порошок переходного оксида алюминия, как правило, имеет усредненный по поверхности диаметр D [3, 2] в пределах от 1 до 200 мкм. В определенных применениях, таких как катализаторы, предназначенные для использования в реакциях в суспензии, преимущественным является использование очень мелкодисперсных частиц, которые в среднем предпочтительно меньше чем 20 мкм, например, имеют размер 10 мкм или меньше. Для других применений, например, в качестве катализатора для реакций, осуществляемых в псевдоожиженном слое, может быть желательным использование частиц больших размеров, предпочтительно, с размерами в пределах от 50 до 150 мкм. Предпочтительно, чтобы порошок оксида алюминия имел относительно большой средний диаметр пор, поскольку использование таких оксидов алюминия придает катализаторам особенно хорошую селективность. Предпочтительные оксиды алюминия имеют средний диаметр пор, по меньшей мере, 10 нм, в частности, диаметр в пределах от 15 до 30 нм. Под термином средний диаметр пор авторы подразумевают учетверенный объем пор, как измерено на ветви десорбции изотермы физической сорбции азота при относительном давлении 0,98, деленный на площадь поверхности по БЭТ. Предпочтительно материал на основе оксида алюминия представляет собой гамма-оксид алюминия или тета-оксид алюминия, более предпочтительно, тета-оксид алюминия, имеющий площадь поверхности по БЭТ от 90 до 120 м2/г и объем пор от 0,4 до 0,8 см3/г. Материал подложки на основе оксида алюминия может находиться в форме порошка, высушенного распылением, или формироваться в виде формованных единиц, таких как сферы, таблетки, цилиндры, кольца или таблетки с множеством отверстий, которые могут быть многодольчатыми или рифлеными, например, иметь поперечное сечение в форме клеверного листа, или находиться в форме экструдатов, известных специалистам в данной области. Подложка на основе оксида алюминия может выбираться преимущественно благодаря высокой фильтруемости и стойкости к истиранию.
Настоящее изобретение может использоваться для преобразования нитратов металлов на любом материале подложки, однако определенные сочетания нитрат металла/подложка являются более предпочтительными. Например, в зависимости от металла может быть или не быть желательным объединение нитрата металла с подложкой, которая способна, при условиях нагрева, используемых для разложения нитрата металла, образовывать соединения смешанных оксидов металлов с получаемым оксидом металла на подложке. Подложки с низкой активностью, такие как углерод или альфа-оксид алюминия, могут использоваться для уменьшения или предотвращения образования смешанного оксида металла с помощью подложки, когда это является нежелательным.
Нитраты металлов на подложке, получаемые с помощью способа по настоящему изобретению, содержат малые кристаллиты, как правило, размером ≤10 нм нитрата металла формулы M(NO3)x·(H2O)a, где x представляет собой валентность металла M и a представляет собой целое число ≥1, и/или предпочтительно Mx(OH)y(NO3)z, в которых x, y и z представляют собой целые числа ≥1. Как сформулировано выше, M предпочтительно представляет собой железо, рутений, кобальт, родий, иридий, никель, палладий, платину, медь или их смесь, предпочтительно медь, никель или кобальт, в особенности, медь.
Нитрат металла на подложке может преобразовываться в высокодисперсный оксид металла на подложке посредством кальцинирования нитрата металла на подложке. Это может осуществляться с использованием обычных способов кальцинирования на воздухе при температурах в пределах 200-1200°C, предпочтительно 200-800°C, более предпочтительно 250-450°C. Предпочтительно кальцинирование осуществляется с использованием способов кальцинирования оксида азота или закиси азота из WO 2007/071899 и WO 2008/029177, для лучшего сохранения дисперсии металла. Альтернативно, кальцинирование в присутствии водорода или монооксида углерода в условиях, когда восстановления не происходит, может также использоваться для получения материалов оксидов металла на подложке с низкими остаточными уровнями нитратов. Таким образом, предпочтительно, кальцинирование осуществляется в атмосфере газовой смеси, которая содержит оксид азота, закись азота, водород или монооксид углерода или их смесь и имеет содержание кислорода ≤5% об. для осуществления его разложения посредством нагрева до температуры его разложения, при которой он образует оксид металла, или, если это желательно, до более высокой температуры. Содержание кислорода (O2) в газовом потоке предпочтительно ≤1%, наиболее предпочтительно ≤0,1% об. Температура, до которой может доводиться нитрат металла для осуществления его разложения, может находиться в пределах 100-1200°C, но предпочтительно температура находится в пределах 200-600°C для обеспечения преобразования нитрата в оксид, в то же время сводя к минимуму спекание оксида. Обнаружено, что более мелкие кристаллиты оксида металла могут быть получены посредством кальцинирования при более низких температурах в этом же диапазоне, например, в пределах между 200 и 450°C. Однако, когда является желательным образование фаз оксида, шпинеля или перовскита, на подложке или с ее помощью, может быть желательным использование температур в пределах 500-1200°C. Время, в течение которого нитрат металла на подложке находится при температуре в этих пределах, предпочтительно ≤16 часов, более предпочтительно ≤8 часов. Короткое время кальцинирования, например, ≤4 часов, в частности, ≤2 часов, является наиболее предпочтительным. При использовании таких технологий, по меньшей мере, 90% масс., более предпочтительно, по меньшей мере, 95%, наиболее предпочтительно, по меньшей мере, 99% нитрата металла, желательно, преобразуется в соответствующий оксид металла.
Соответственно, настоящее изобретение дополнительно предусматривает оксид на подложке, который может быть получен посредством (i) импрегнирования материала подложки нитратом металла, (ii) выдерживания импрегнированного материала в газовой смеси, содержащей оксид азота, при температуре в пределах 0-150°C, с получением диспергированного на подложке нитрата металла, и (iii) кальцинирования нитрата металла для осуществления его разложения и образования оксида металла на подложке.
Оксиды металлов на подложке имеют меньшие размеры кристаллитов оксида металла и, следовательно, более высокую дисперсность оксида металла, чем для оксида металла, получаемого с использованием способов, известных из литературы. Это происходит благодаря стабилизации нитрата металла при низких температурах до превращения в оксид металла, например, во время сушки, и, в частности, это представляет собой случай, когда кальцинирование осуществляется с использованием газовых смесей оксида азота и/или закиси азота, посредством их кумулятивного воздействия. Оксиды металлов на подложке по настоящему изобретению, как обнаружено с помощью сканирующей трансмиссионной электронной микроскопии (STEM) и дифракции рентгеновского излучения (XRD), имеют размеры кристаллитов оксидов металлов ≤10 нанометров, предпочтительно, ≤6 нанометров, при получаемых нагрузках оксидов металлов на подложках до 30% масс.
Когда оксид металла представляет собой восстанавливаемый оксид металла, такой как оксид меди, никеля, железа или кобальта, способ может дополнительно включать стадию нагрева оксида металла на подложке в восстанавливающем газовом потоке, содержащем монооксид углерода и/или водород, для осуществления восстановления, по меньшей мере, части оксида металла.
Альтернативно, когда нитрат металла представляет собой восстанавливаемый нитрат металла, такой как нитрат меди, никеля, железа или кобальта, может быть необходимым или желательным не кальцинирование материала, а воздействие на него непосредственно стадии восстановления с помощью восстанавливающего газового потока, при так называемом непосредственном восстановлении. В настоящем изобретении более высокое диспергирование металлов, достигаемое благодаря низкотемпературной обработке оксидом азота, делает непосредственное восстановление особенно привлекательным. Таким образом, в предпочтительном варианте осуществления, когда нитрат металла представляет собой восстанавливаемый нитрат металла, способ дополнительно включает стадию нагрева нитрата металла на подложке в восстанавливающем газовом потоке, содержащем монооксид углерода и/или водород, для осуществления восстановления, по меньшей мере, части нитрата металла.
Соответственно настоящее изобретение дополнительно предусматривает восстановленный нитрат или оксид металла на подложке, который может быть получен посредством (i) импрегнирования материала подложки нитратом металла, (ii) выдерживания импрегнированного материала в газовой смеси, содержащей оксид азота, при температуре в пределах 0-150°C, с получением диспергированного на подложке нитрата металла, (iii) необязательного кальцинирования нитрата металла для осуществления его разложения и образования оксида металла на подложке и (iv) нагрева оксида металла на подложке или нитрата металла на подложке в восстанавливающем газовом потоке, содержащем монооксид углерода и/или водород, для осуществления восстановления, по меньшей мере, части нитрата или оксида металла.
Композиция восстановленного металла на подложке, полученная таким образом, будет содержать металл в элементарной форме и, возможно, малые количества невосстановленного оксида или нитрата металла на материале подложки. В дополнение к этому, и другие восстанавливаемые или невосстанавливаемые оксиды металлов могут присутствовать на подложке.
Стадия восстановления может осуществляться посредством прохождения газа, содержащего водород, такого как водород, синтез-газ или смесь водорода с азотом, метаном или другим инертным газом, над восстанавливаемым оксидом или нитратом металла на подложке при повышенной температуре, например, посредством прохождения газа, содержащего водород, над композицией при температурах в пределах 150-600°C, предпочтительно 300-500°C, в течение 0,1-24 часов, при атмосферном давлении или при более высоких давлениях, примерно до 25 бар. Оптимальные условия восстановления для оксида никеля, оксида кобальта, оксида меди или оксидов железа известны специалистам в данной области.
В восстановленном оксиде или нитрате металла на подложке, полученном способом по настоящему изобретению, предпочтительно, по меньшей мере, 70%, более предпочтительно >80% и наиболее предпочтительно >90% восстанавливаемого металла восстанавливается до элементарной активной формы. Восстановленные оксиды металлов с очень высокой дисперсностью металла, выраженной как площадь поверхности металла на грамм катализатора или грамм металла в восстановленном материале, могут быть получены способом по настоящему изобретению. Площади поверхности металла могут определяться с помощью хемосорбции (например, хемосорбции водорода) с использованием способов, известных специалистам в данной области.
Восстановленные оксиды или нитраты содержат высокодисперсный металл и по этой причине окисление путем выдерживания на воздухе могут приводить к нежелательному саморазогреву в результате экзотермических реакций окисления. Такой саморазогрев может приводить к возникновению высоких температур, превышающих 250°C, и к последующему спеканию металла и потере площади поверхности. Для предотвращения этого и для простоты обращения, желательным является пассивирование восстановленного материала после стадии восстановления посредством обработки газовыми смесями, содержащими воздух и/или диоксид углерода. Такие способы описаны, например, в патенте США 4090980, в патенте Великобритании GB 1319622 и WO 95/33644.
Оксиды металлов на подложке и восстановленные оксиды или нитраты металлов на подложке могут использоваться во многих областях техники. Такие области включают катализаторы, предшественники катализаторов, сорбенты, полупроводники, сверхпроводники, магнитные среды памяти, твердотельные среды памяти, пигменты и поглотители УФ-излучения. Предпочтительно оксиды металлов на подложке и восстановленные оксиды или нитраты металлов на подложке используются в качестве катализаторов, предшественников катализаторов или сорбентов. Под термином "сорбенты" авторы включают адсорбенты и поглотители.
В предпочтительных вариантах осуществления, оксиды металлов на подложке и восстановленные оксиды или нитраты металлов на подложке используются как предшественники катализаторов или катализаторы при синтезе метанола, конверсии водяного пара, в реакциях гидрирования, реакциях парового риформинга, реакциях метанирования и при синтезе углеводородов Фишера-Тропша. Например, восстановленные материалы на основе Cu на подложке, такие как Cu/ZnO/Al2O3, используют в качестве катализаторов синтеза метанола и катализаторов конверсии водяного пара.
Восстановленные оксиды Ni, Cu и Co на подложке могут использоваться сами по себе или в сочетании с другими оксидами металлов в качестве катализаторов для реакции гидрирования, а восстановленные оксиды Fe или Co могут использоваться в качестве катализаторов для синтеза углеводородов Фишера-Тропша. Катализаторы на основе Ni и Co находят применение при гидродесульфуризации. Катализаторы на основе восстановленного Fe могут использоваться в реакциях высокотемпературной конверсии водяного пара и при синтезе аммиака. Катализаторы на основе восстановленного Ni и благородных металлов находят применение в качестве катализаторов парового риформинга и в качестве катализаторов метанирования. Катализаторы на основе оксида Co находят применение в реакциях окисления, включая окисление аммиака и разрушение N2O. Катализаторы на основе оксида Ni могут использоваться для разложения гипохлорита в водных растворах.
Типичные реакции гидрирования включают гидрирование альдегидов и нитрилов до спиртов и аминов, соответственно, и гидрирование циклических ароматических соединений или ненасыщенных углеводородов. Катализаторы по настоящему изобретению являются особенно пригодными для гидрирования ненасыщенных органических соединений, в частности, масел, жиров, жирных кислот и производных жирных кислот, подобных нитрилам. Такие реакции гидрирования, как правило, осуществляют непрерывным или периодическим способом посредством обработки соединения, которое должно гидрироваться, газом, содержащим водород, под давлением в автоклаве, при температуре окружающей среды или при повышенной температуре в присутствии катализатора, например, гидрирование может осуществляться с помощью водорода при 80-250°C и при давлении в пределах 0,1-5,0×106 Па.
Синтез углеводородов Фишера-Тропша хорошо известен. Синтез Фишера-Тропша преобразует смесь монооксида углерода и водорода в углеводороды. Смесь монооксида углерода и водорода, как правило, представляет собой синтез-газ, имеющий отношение водород:монооксид углерода в пределах 1,7-2,5:1. Реакция может осуществляться непрерывным или периодическим способом с использованием одного или нескольких перемешиваемых реакторов в фазе суспензии, барботажных колонных реакторов, петлевых реакторов или реакторов с псевдоожиженным слоем. Способ может работать при давлениях в пределах 0,1-10 МПа и при температурах в пределах 150-350°C. Объемная часовая скорость газа (GHSV) для непрерывной работы находится в пределах 100-25000 час-1. Катализаторы по настоящему изобретению являются особенно пригодными для использования благодаря их высокой площади поверхности металла/г катализатора.
При паровом риформинге углеводородов, как правило, газ, содержащий метан, такой как природный газ, или нафта взаимодействует с паром и/или, где это возможно, с диоксидом углерода, над каталитически активным материалом с получением газа, содержащего водород, и оксидов углерода. Эти реакции являются сильно эндотермическими, и способ является особенно пригодным, когда они осуществляются с помощью внешнего нагрева, как при трубчатом паровом риформинге. Альтернативно, тепло может подаваться посредством нагрева реагентов и прохождения пара над катализатором в адиабатическом слое или в гибридном способе, в котором кислород представляет собой реагент, так что тепло, выделяющееся при окислении, поглощается эндотермическими реакциями. Гибридный способ может применяться к продукту трубчатого или адиабатического способа, то есть, при "вторичном риформинге", или к свежим исходным материалам ("каталитическое частичное окисление" или "аутотермический риформинг"). Обычно эти реакции сопровождаются реакцией конверсии водяного пара. Для получения синтез-газа, содержащего водород, выходная температура предпочтительно составляет, по меньшей мере, 600°C, для обеспечения низкого содержания метана. Хотя температура, как правило, находится в пределах 750-900°C для получения синтез-газа для производства аммиака или метанола, она может достигать 1100°C для получения металлургического восстанавливающего газа или опускаться до 700°C для получения бытового газа. Для гибридного способа с использованием кислорода температура может достигать 1300°C в самой горячей части слоя катализатора.
При предварительном риформинге смесь углеводород/пар подвергается воздействию стадии адиабатического низкотемпературного парового риформинга. При таком способе смесь углеводород/пар нагревается, как правило, до температуры в пределах 400-650°C, а затем проходит адиабатически через неподвижный слой соответствующего катализатора в виде частиц, обычно катализатора, имеющего высокое содержание никеля, например, свыше 40% масс. Катализаторы могут представлять собой простые цилиндры, имеющие дольчатую форму с множеством отверстий. Катализаторы предварительного риформинга, как правило, предусматриваются в предварительно восстановленной и пассивированной форме, хотя и оксидный катализатор может также устанавливаться. Во время такой стадии адиабатического низкотемпературного риформинга любые углеводороды более тяжелые, чем метан, взаимодействуют с паром с получением смеси метана, оксидов углерода и водорода. Использование такой стадии адиабатического риформинга, обычно называемой предварительным риформингом, является желательным для обеспечения того, чтобы исходные материалы для реактора парового риформинга не содержали углеводородов более тяжелых, чем метан, а также содержали значительное количество водорода. Это является желательным для сведения к минимуму риска образования углерода на катализаторе в следующем далее реакторе парового риформинга.
Для реакции парового риформинга катализатор обычно содержит металлический никель на подложке из оксида алюминия, диоксида циркония или алюмината кальция. Давление, как правило, находится в пределах 1-50 бар абс., но предполагаются и давления до 120 бар абс. Обычно используется избыток пара и/или диоксида углерода, в частности, в пределах 1,5-6, например 2,5-5, моль пара или диоксида углерода на грамм-атом углерода в исходных углеводородах.
Когда катализатор на основе Ni должен использоваться для метанирования, для удаления низких концентраций CO и CO2 (0,1-0,5% об.) из газа, содержащего водород, газ, содержащий водород, как правило, проходит через неподвижный слой частиц при температуре в пределах 230-450°C и при давлениях примерно от 50 бар абс. или выше и вплоть до 250 бар абс. В отличие от парового риформинга, катализатор предпочтительно представляет собой простые цилиндрические таблетки без отверстий, хотя и такие таблетки могут использоваться, если это желательно. Типичные диаметры таблеток находятся в пределах 2,5-5 мм, при этом высота находится в тех же пределах. Катализаторы могут предусматриваться в оксидной форме или в предварительно восстановленной и пассивированной форме.
Композиции сорбента, содержащие соединения Cu и/или Zn, могут использоваться при удалении соединений серы из газообразных или жидких потоков, в частности, из потоков углеводородов и потоков синтез-газа. Удаление соединений серы может осуществляться просто посредством прохождения потока, содержащего соединение серы, над неподвижным слоем сорбента в соответствующей емкости при температурах в пределах 0-300°C при атмосферном давлении или при повышенных давлениях, например, до 100 бар абс.
Композиции сорбентов, содержащие соединения серы и переходных металлов, в частности, композиции, содержащие соединения серы и меди, могут использоваться для удаления тяжелых металлов, таких как Hg или As, из загрязненных газообразных или жидких потоков. Удаление тяжелых металлов может осуществляться просто посредством прохождения потока, содержащего тяжелые металлы, над неподвижным слоем сорбента в соответствующих емкостях при температурах в пределах 0-100°C при атмосферном давлении или при повышенных давлениях, например, при давлении до 100 бар абс.
Стадии удаления соединений серы и соединений тяжелых металлов могут осуществляться последовательно или одновременно.
Далее настоящее изобретение иллюстрируется со ссылками на следующие далее примеры и со ссылками на фиг.1-4, в которых;
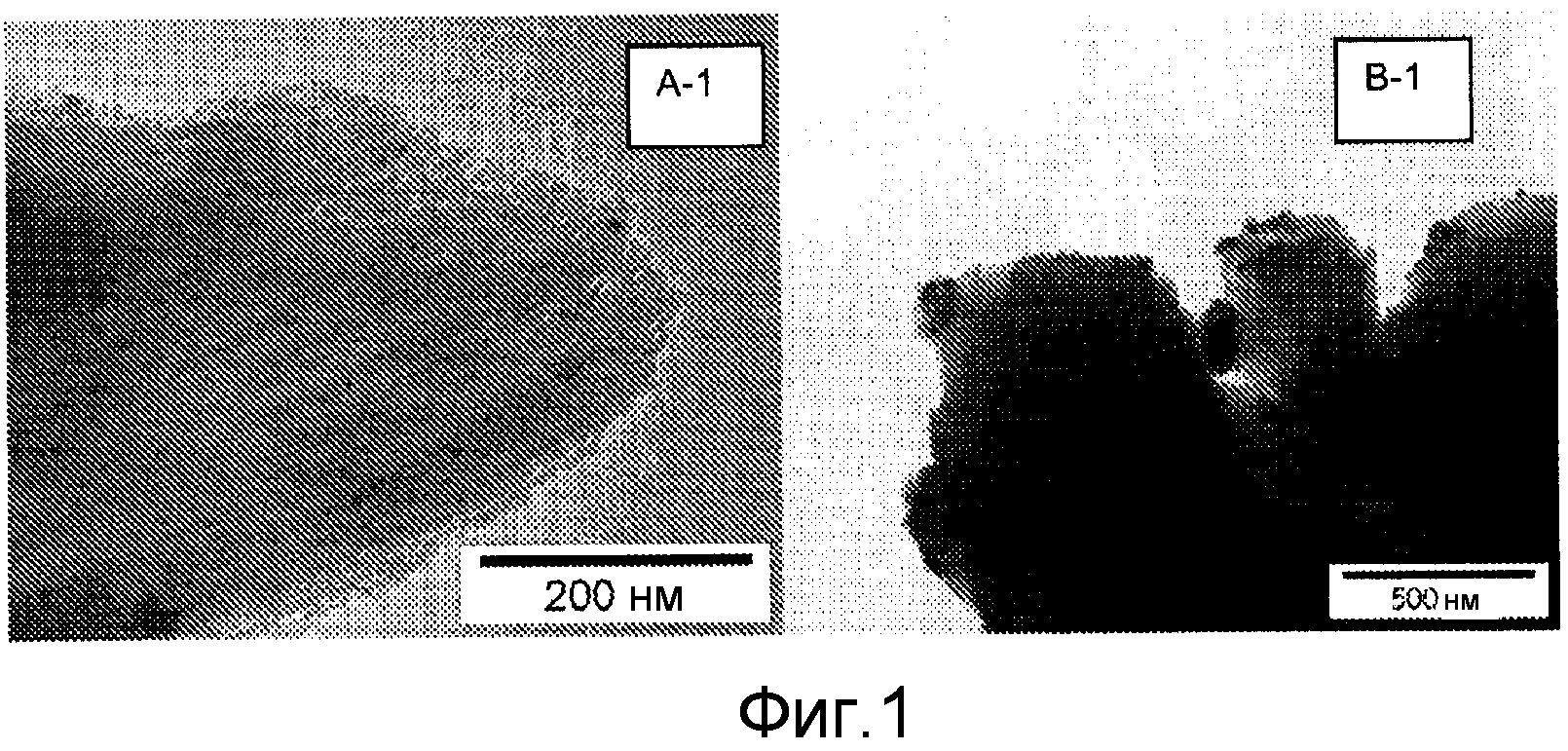
Фиг.1 представляет собой фотографии, сделанные с помощью трансмиссионного электронного микроскопа дляCuO на подложке из диоксида кремния (A-1, B-1).
Фиг.2 является изображением, полученным с помощью HAADF-STEM для нитрата меди на подложке из диоксида кремния (образец D).
Фиг.3 является изображением, полученным с помощью STEM в светлом поле, для CuO на подложке из диоксида кремния (D-1, D-2).
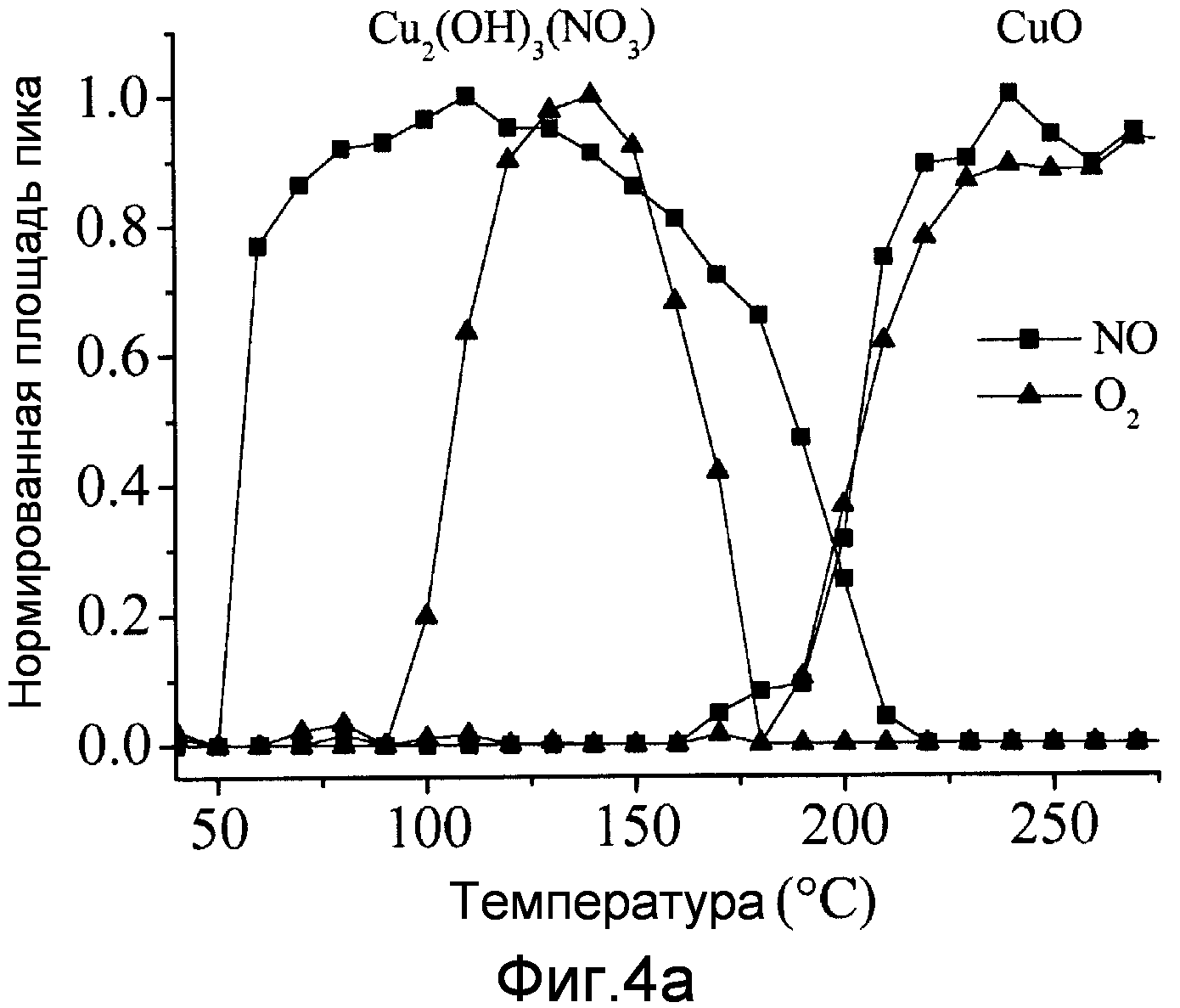
Фиг.4a изображает нормированную площадь пика Cu2(OH)3NO3 и линии дифракции для CuO, полученные при анализе XRD in-situ во время нагрева, как функцию температуры, и
Фиг.4b изображает линии дифракции XRD при 120°C для обеих термических обработок.
Пример 1: Оксид меди на положке из SBA-15
Стадию импрегнирования с помощью пропитки по влагоемкости осуществляют при 60 мбар с помощью 4,3 M водного раствора нитрата меди (II) (Cu(NO3)2·6H2O) на 0,25 г порошка SBA-15 (площадь поверхности по БЭТ=600 м2/г, общий объем пор=0,7 см3/г) с получением 16% масс. Cu/SiO2. После прохождения времени уравновешивания 15 минут, осуществляют два различных опыта. Первый образец непосредственно переносят в реактор с поршневым потоком (диаметр 1 см, длина 17 см) и подвергают комбинированной низкотемпературной и высокотемпературной термической обработке в 1% NO/Ar или на воздухе в соответствии с таблицей 1. Второй образец сначала сушат при 120°C в неподвижном воздухе в течение 12 часов перед воздействием на него таких же термических обработок в 1% NO/Ar или на воздухе. В каждом случае масса образца составляет 100 мг.
Образцы, обрабатываемые в 1% об. NO в Ar, обозначают как A-1 и B-1, в то время как образцы, обрабатываемые на воздухе, обозначают A-2 и B-2.
Образец A-1 получают в соответствии с настоящим изобретением с использованием низкотемпературной термической обработки в газе, содержащем NO, перед высокотемпературной термической обработкой. Образцы A-2 и B-2 представляют собой сравнительные образцы. Характеризацию осуществляют с помощью дифракции рентгеновского излучения на порошках (XRD) и трансмиссионной электронной микроскопии (TEM). Картины XRD получают при комнатной температуре при 2θ от 35° до 70° с помощью установки рентгеновского дифрактометра Bruker-Nonius D8 Advance с использованием излучения Co-Kα12 (λ=1,79026 Å). Средние размеры частиц оксида меди вычисляют в соответствии с уравнением Шерера, используя самую интенсивную линию дифракции при 45,3°.
Результаты анализа уширения линий приводятся в таблице 2. Образцы A-1 и A-2 получают низкотемпературную и высокотемпературную термическую обработку непосредственно после получения. A-1 имеет малый средний размер кристаллитов, то есть высокую дисперсность, которая видна как из XRD, так и из TEM (фиг.1). Образец, обработанный на воздухе (A-2), имеет плохую дисперсность. Важность низкотемпературной термической обработки показывается посредством низкой дисперсности, получаемой, если образец сначала сушат при 120°C на воздухе (образец B). Образец B-1 показывает, что термическая обработка NO (высокотемпературная и низкотемпературная) является гораздо менее эффективной после сушки на воздухе при 120°C (фиг.1). Хотя образец B1 выдерживают в NO/Ar, дисперсность нитрата меди уменьшается из-за стадии сушки на воздухе при 120°C. Малые частицы по-прежнему находятся внутри пор, но агломераты на наружной поверхности становятся гораздо больше (20-400 нм). Кроме того, средний размер кристаллита, вычисленный по XRD, гораздо больше.
Результаты показывают, что более высокая дисперсность получается, когда стадия сушки на воздухе заменяется низкотемпературной термической обработкой в NO.
Пример 2: Оксид меди на положке из SBA-15: низкотемпературная и высокотемпературная термическая обработка после сушки при комнатной температуре
Импрегнирование осуществляют, как описано в примере 1. После прохождения времени уравновешивания 15 минут, импрегнированный материал сушат в десикаторе, содержащем 4-Å молекулярные сита, при комнатной температуре (25°C) и при атмосферном давлении в течение 24 часов для удаления воды растворителя (как правило, удаляется 90% растворителя). Полученный высушенный материал обозначают как образец C. Малое количество высушенного материала (100 мг) подвергают комбинированной низкотемпературной и высокотемпературной термической обработке в 1% NO/Ar (обозначают как C-1) или на воздухе (обозначают как C-2) с использованием способа и устройства, описанных в примере 1 (см. таблицу 1 относительно условий).
Образцы характеризуются с помощью XRD. Анализ уширения линий показывает, что получаемые дисперсности сравнимы с примером 1, средний размер кристаллитов 4,5 нм, для материала, кальцинированного с помощью NO, и 23 нм, для материала, кальцинированного на воздухе (C-2). Средний размер кристаллита после термической обработки с помощью NO (низкотемпературная + высокотемпературная) несколько меньше, чем в примере 1, что может быть приписано более высокому содержанию воды для образца A.
Эти результаты показывают, что вода растворителя может удаляться перед низкотемпературной термической обработкой с помощью NO, поскольку она осуществляется при низких температурах.
Пример 3: Оксид меди на положке из SBA-15: низкотемпературная и высокотемпературная термическая обработка после сушки при 60°C
Импрегнирование осуществляют, как описано в примере 1. После прохождения 15 мин для уравновешивания, импрегнированный материал сушат в неподвижном воздухе при 60°C в течение 12 часов (образец D). После сушки, малое количество образца (100 мг) подвергают воздействию комбинированной низкотемпературной и высокотемпературной термической обработки в 1% NO/Ar (обозначают как D-1) или на воздухе (обозначают как D-2) с использованием устройства, описанного в примере 1 (см. таблицу 3 относительно условий).
Фиг.2 показывает, что образец может сушиться при 60°C, но что более низкие температуры могут быть предпочтительными, поскольку начинает происходить некоторая агломерация. Анализ с помощью XRD показывает средний размер кристаллитов 6,5 нм для D-1 и 23 нм для D2. TEM (фиг.3) показывает, что большая часть фазы CuO является сильно дисперсной после кальцинирования с помощью NO (dTEM=2-40 нм), тогда как кальцинирование на воздухе дает в основном очень большие агломераты (dTEM=10-500 нм).
Эти результаты показывают, что более высокие скорости нагрева и температуры сушки могут использоваться, но более низкие температуры и скорости нагрева являются предпочтительными.
Пример 4: Дисперсность Cu2(OH)3NO3 после сушки
Осуществляют эксперименты XRD in situ с помощью установки рентгеновского дифрактометра Bruker-AXS D8 Advance с использованием излучения CoKα12. 50 мг образца C из примера 2 нагревают при скорости нагрева 1°C/мин до 350°C в реакционной камере Anton-Paar XRK при потоке 10% O2/N2 или 10% NO/He. Условия приводятся в таблице 4. Образец, обрабатываемый с помощью 10% об. NO в He, обозначают E-1, а на воздухе, E-2.
Во время комбинированной низкотемпературной и высокотемпературной термической обработки в NO и O2 in situ наблюдают только две фазы, Cu2(OH)3NO3 и CuO. Фиг.4a изображает нормированную площадь пика Cu2(OH)3NO3 и линии дифракции CuO как функцию температуры, а фиг.4b линии дифракции при 120°C для обеих термических обработок. Анализ уширения линий по Шереру показывает размер кристаллического домена Cu2(OH)3NO3 9 нм для низкотемпературной обработки в NO и 23 нм для воздуха. После полного разложения (высокотемпературная термическая обработка) получают средние кристаллиты CuO 6 и 23 нм для термической обработки с помощью NO и на воздухе, соответственно.
Фиг.4a ясно показывает, что CuO не образуется при этих условиях в обоих случаях примерно до 175°C, однако преобразование гидроксинитрата меди заметно отличается, когда присутствует NO, по сравнению с O2. Когда присутствует NO, образование гидроксинитрата меди начинается примерно при 50°C и достигает пика примерно при 110°C, тогда как для O2, образование начинается примерно при 90°C и достигает максимума примерно при 130°C. Таким образом, видно, что NO вызывает образование гидроксинитрата меди при более низких температурах.
Эти эксперименты также показывают, что низкотемпературная термическая обработка с помощью NO приводит к образованию высокодисперсной фазы Cu2(OH)3NO3, тогда как термическая обработка на воздухе дает очень плохую дисперсность.
Кроме того, дисперсность этой фазы непосредственно определяет дисперсность CuO после высокотемпературной термической обработки для преобразования нитрата меди в оксид меди.
Пример 5: Оксид никеля на подложке из SBA-15
Импрегнирование с помощью пропитки по влагоемкости осуществляют с помощью 3,0 M водного раствора нитрата никеля (II) (Ni(NO3)2·6H2O) при 60 мбар на 0,25 г порошка SBA-15 (площадь поверхности по БЭТ=600 м2/г, общий объем пор=0,7 см3/г) с получением 15% масс. Ni/SiO2. После прохождения времени уравновешивания 15 минут, импрегнированный материал сушат при комнатной температуре (25°C) в десикаторе, как описано в примере 2. Для сравнения, другой образец сушат при 120°C, как описано в примере 1. Высушенный при комнатной температуре материал обозначают как образец F, а материал, высушенный при 120°C, как образец G. Малое количество (50 мг) высушенного образца подвергают воздействию комбинированной низкотемпературной и высокотемпературной термической обработки в 10% NO/He или 20% O2/N2, как описано в примере 4 (таблица 4). Образцы, обрабатываемые в 10% об. NO в He, обозначают как F-1, G-1, а на воздухе, как F-2, G-2.
Образцы F-1, F-2, G-1, G-2 анализируют с помощью XRD. Результаты анализа уширения линий для отражения при 50,8° приводятся в таблице 5.
Результаты показывают, что опять более мелкие кристаллиты (более высокую дисперсность) получают, когда стадия сушки на воздухе при 120°C заменяется низкотемпературной термической обработкой в NO.
Пример 6: Использование N2O вместо NO для низкотемпературной обработки
Образцы A (пример 1), F и G (пример 5) подвергают воздействию комбинированной низкотемпературной и высокотемпературной термической обработки в 1% об./об. N2O в He или в 1% NO/He. Термическую обработку осуществляют внутри HVC-DRP-3 Diffuse Reflectance Reaction Chamber (поставляется Harrick) как часть механистических ИК исследований. Ячейка сконструирована так, что она работает в условиях поршневого потока. Все обработки на воздухе и в NO, которые осуществляют, дают дисперсность, сходную с экспериментами ex situ. Как правило, загружают 10 мг образца и нагревают его при скорости 1°C/мин до 350°C в потоке 1% NO/He или 1% N2O/He. Детали термической обработки приведены в таблице 6.
ИК исследования показывают, что во время низкотемпературной термической обработки (25-120°C) в N2O образуется очень мало Cu2(OH)3NO3 (образец A) или Ni3(OH)4(NO3)2 (образец F) или они вообще не образуются, тогда, как в NO четко наблюдается образование этих соединений. Анализ с помощью XRD после высокотемпературной обработки показывает, что N2O не может предотвратить агломерацию, тогда как после обработки в NO получают высокую дисперсность (таблица 7). Образец G сушат на воздухе в течение 12 часов, и, таким образом, он уже частично разлагается на Ni3(OH)4(NO3)2. Таблица 7 показывает, что здесь N2O и NO дают сравнимые результаты.
Результаты показывают, что N2O является неэффективным при низкотемпературной термической обработке нитрата, как меди, так и никеля, тогда как NO является эффективным. Различие между двумя газами показывает различие между низкотемпературной и высокотемпературной термической обработкой.
Пример 7: Оксид кобальта на подложке из SBA: низкотемпературная термическая обработка в NO, объединенная с высокотемпературной термической обработкой на воздухе
Стадию импрегнирования с помощью пропитки по влагоемкости осуществляют с использованием 3,0 M водного раствора нитрата кобальт (II) (Co(NO3)2·6H2O) на 0,25 г порошка SBA-15 (площадь поверхности по БЭТ=600 м2/г, общий объем пор=0,7 см3/г) с получением 13% масс. Co/SiO2. После прохождения времени уравновешивания 15 минут, импрегнированный материал (обозначают как образец H) сушат в десикаторе при комнатной температуре в течение 24 часов, как описано в примере 2.
Малое количество (10 мг) образца H подвергают воздействию низкотемпературной термической обработки в 10% NO/He, с последующей высокотемпературной термической обработкой в 20% O2/N2 внутри HVC-DRP-3 Diffuse Reflectance Reaction Chamber, используемой для примера 6. Детали термической обработки приводятся в таблице 8. Термически обработанный материал обозначают как образец H-1.
Для сравнения, комбинированная низкотемпературная и высокотемпературная термическая обработка осуществляется в 10% NO/He (образец H-2) и 20% O/N2 (образец H-3) с использованием термической обработки, приведенной в таблице 4.
Результаты анализа уширения линий XRD для самой интенсивной линии дифракции полученной фазы Co3O4 (2θ=43,1°) приводятся в таблице 9.
Результаты демонстрируют, что низкотемпературная термическая обработка в NO приводит к получению более высокой дисперсности даже после высокотемпературного кальцинирования в O2/N2.
Пример 8: Оксид никеля на подложке из SBA: низкотемпературная термическая обработка в NO, объединенная с высокотемпературной термической обработкой в неподвижном воздухе
Стадию импрегнирования с помощью пропитки по влагоемкости осуществляют с использованием 4,3 M водного раствора нитрата никеля (II) (Ni(NO3)2·6H2O) на 1 г порошка SBA-15 (площадь поверхности по БЭТ=600 м2/г, общий объем пор=0,8 см3/г) с получением 15% масс. Ni/SiO2. После прохождения времени уравновешивания 15 минут импрегнированный материал (обозначают как образец I) разделяют на три части и подвергают воздействию различных низкотемпературных термических обработок. Первый образец переносят в керамический тигель и нагревают в муфельной печи в неподвижном воздухе до 150°C при скорости нагрева 2°C/мин и выдерживают при этой температуре в течение 16 часов (образец I-1). Другие два образца переносят в реактор с поршневым потоком (см. пример 1) и подвергают воздействию низкотемпературной термической обработки в N2 (образец I-2) или в 10% NO/He (образец I-3), как описано в таблице 10. После низкотемпературной термической обработки все образцы переносят в тигли для высокотемпературной термической обработки в неподвижном воздухе в муфельной печи (см. таблицу 10 относительно деталей).
Результаты анализа уширения линий XRD для самой интенсивной линии дифракции полученной фазы NiO (2θ=50,8°) приводятся в таблице 11.
Результаты демонстрируют, что низкотемпературная термическая обработка в NO приводит к получению более высокой дисперсности даже после высокотемпературного кальцинирования в неподвижном воздухе.
Реферат
Изобретение относится к области катализаОписанпособ получения оксида металла на подложке и восстановленного оксида металла на подложке, пригодного для использования в качестве предшественника для катализатора или сорбента, включающий стадии: (i) импрегнирования материала подложки раствором нитрата металла в растворителе, (ii) выдерживания импрегнированного материала в газовой смеси, содержащей оксид азота, при температуре в пределах 0-150°C для удаления растворителя из импрегнированного материала с одновременным высушиванием и стабилизацией нитрата металла на подложке, с получением диспергированного на подложке нитрата металла и (iii) кальцинирования диспергированного на подложке нитрата металла для осуществления его разложения и образования оксида металла на подложке, где кальцинирование осуществляют в газовой смеси, которая состоит из одного или нескольких инертных газов и оксида азота и концентрация оксида азота в газовой смеси находится в пределах 0,001-15% об. Технический результат - увеличение каталитической активности полученных продуктов. 4 н. и 8 з.п. ф-лы, 4 ил., 11 табл., 8 пр.
Формула
(i) импрегнирования материала подложки раствором нитрата металла в растворителе,
(ii) выдерживания импрегнированного материала в газовой смеси, содержащей оксид азота, при температуре в пределах 0-150°C для удаления растворителя из импрегнированного материала с одновременным высушиванием и стабилизацией нитрата металла на подложке, с получением диспергированного на подложке нитрата металла и
(iii) кальцинирования диспергированного на подложке нитрата металла для осуществления его разложения и образования оксида металла на подложке, где кальцинирование осуществляют в газовой смеси, которая состоит из одного или нескольких инертных газов и оксида азота и концентрация оксида азота в газовой смеси находится в пределах 0,001-15% об.
Комментарии