Катализаторы на основе оксида рутения для конверсии диоксида серы в триоксид серы - RU2422357C2
Код документа: RU2422357C2
Чертежи









Описание
Уровень техники, предшествующий данному изобретению
Данное изобретение относится в основном к катализаторам, содержащим оксид рутения, и к способам каталитического окисления и конверсии диоксида серы (SO2) в триоксид серы (SO3) при использовании таких катализаторов. Более конкретно, SO2 при низких концентрациях в потоках технологического газа может быть эффективным образом окислен до SO3 при сравнительно низких температурах при использовании катализаторов на основе оксида рутения по данному изобретению. Например, катализаторы, содержащие оксид рутения, особенно применимы для конверсии SO2 в SO3 на последней каталитической стадии многостадийного каталитического конвертера, используемого при производстве серной кислоты.
Обычный контактный способ производства серной кислоты включает каталитическое окисление в газовой фазе SO2 до SO3 на одной или нескольких стадиях каталитического окисления в конвертере, чтобы получить конвертированный газ, содержащий SO3, и абсорбцию SO3 водной серной кислотой, чтобы образовать продукт в виде дополнительного количества серной кислоты. Каталитическое окисление SO2 до SO3 протекает при допустимых расходах через катализатор в виде твердотельных частиц, который обычно содержит активные фазы, содержащие щелочной металл - ванадий или платину. Концентрации газообразного SO2 на входе первой каталитической стадии конвертера обычно находятся в интервале от примерно 4% до примерно 15%. При адиабатическом функционировании каждой стадии конвертера обычно требуется три или четыре каталитических стадии (или прохождения), чтобы достигнуть общих величин конверсии SO2 более 99,7% и обеспечить соответствие стандартам в отношении выпуска остаточного газа абсорбером. Обычно каждому прохождению через катализатор предшествуют внешние теплообменники, обеспечивая первое прохождение газового потока при охлаждении до желательной температуры на входе и прохождение четвертой стадии при температуре, обычно находящейся в интервале от примерно 360°C до примерно 415°C. Конверсию SO2, содержащегося в концентрации 99,7% на входе первой стадии, соответственно обеспечивают посредством четырехстадийного устройства с двойной абсорбцией, в котором SO3 удаляют из газового потока внутри абсорбционной колонны с орошением серной кислотой, которая расположена за второй каталитической стадией (конструкция с промежуточной абсорбцией (IPA) 2:2) или третьей каталитической стадией (конструкция IPA 3:1) конвертера. Конверсии от примерно 94% до примерно 95% SO2 обычно достигают на первых трех стадиях, при конвертировании остатка на четвертой, или последней, каталитической стадии конвертера перед прохождением через заключительную абсорбционную колонну для извлечения продукта в виде дополнительного количества серной кислоты.
Способы в соответствии с известным уровнем техники, такие как описанные в Патенте США №5264200 Felthouse et al., эффективным образом обеспечивают высокую суммарную конверсию SO2 и приемлемые уровни выпуска SO2 в остаточном газе абсорбера посредством контактирования газа, содержащего SO2, с монолитным катализатором с активной фазой, содержащей платину или щелочной металл - ванадий, в последовательности предварительных каталитических стадией перед промежуточной абсорбцией, за которой следует дополнительное прохождение через последнюю каталитическую стадию, включающую ванадиевый катализатор в виде частиц, содержащий цезий (т.е. катализатор на основе Cs-V). Посредством использования катализатора на основе Cs-V в виде частиц реакции на последней стадии может протекать до достижения термодинамического равновесия при низкой температуре газа на входе, находящейся в интервале от примерно 360°C до примерно 415°C, и данный температурный интервал способствует высокой степени конверсии SO2 в SO3.
Tomas Jirsak et al. в «Chemistry of SO2 on Ru(001): formation of SO3 и SO4», Surface Science 418 pp. 8-21 (1998), описывают воздействие кристаллов рутения (001) на SO2 и кислород, приводящее к диссоциации SO2, или разложению, или же диспропорционированию, вследствие чего образуются SO3 и SO4.
Вследствие стремления достичь экономии, обусловленной ростом масштабов производства, заводы для производства серной кислоты контактным способом часто строят при их производительности от 1500 до 2500 тонн в день (в расчете на 100% H2SO4). При таком уровне производства требуются каталитические конвертеры сравнительно большого диаметра (например, от 5 до 15 метров) с загрузкой катализатора от примерно 30 до примерно 50 литров на тонну (в расчете на 100% H2SO4) или более, в расчете на одну стадию. Повышенная каталитическая активность могла бы обеспечить использование меньших загрузок катализатора. Желательно, чтобы увеличенная эффективность конверсии SO2 и более низкий выпуск остаточных газов были достигнуты посредством использования катализатора последней стадии, имеющего повышенную активность при низкой температуре по сравнению с известными катализаторами окисления SO2. Поэтому имеется потребность в катализаторе окисления SO2, который обладает стабильностью и высокой активностью, посредством чего обеспечивает возможность использования меньших загрузок катализатора и более высоких скоростей газа и снижение сопутствующих капитальных затрат.
Сущность изобретения
Поэтому задачей данного изобретения является, в частности, создание катализатора окисления для использования в способах окисления SO2 до SO3; создание катализатора окисления, содержащего активную фазу оксида рутения; создание такого катализатора окисления, который обладает стабильностью и продолжительным сроком службы при функционировании в условиях кислой среды; создание такого катализатора окисления и способов, пригодных для эффективного каталитического окисления SO2 до SO3 в исходных газовых смесях при сравнительно низкой концентрации газообразного SO2 и при сравнительно низких рабочих температурах; и создание такого катализатора окисления, адаптированного для конверсии SO2 до SO3 на последней каталитической стадии конвертера, используемого в производстве серной кислоты контактным способом.
Следовательно, данное изобретение направлено на способы каталитического окисления SO2 до SO3. Более конкретно, SO2 при низких концентрациях в потоках технологического газа может быть эффективным образом окислен до SO3 при сравнительно низких температурах при использовании раскрытых здесь катализаторов на основе оксида рутения. В одном варианте осуществления способ включает контактирование исходной газовой смеси, содержащей SO2 и кислород, с катализатором окисления, содержащим активную фазу на основе оксида рутения, с получением в результате конвертированного газа, содержащего SO3.
Катализатор на основе оксида рутения по данному изобретению пригоден, в частности, для использования в качестве катализатора окисления для конверсии SO2 в SO3 на одной или нескольких каталитических стадиях многостадийного каталитического конвертера, используемого в производстве серной кислоты контактным способом. В одном таком варранте осуществления данное изобретение направлено на способ получения серной кислоты и/или олеума из исходного газа, содержащего SO2. Данный способ включает образование исходной газовой смеси для конверсии посредством объединения исходного газа с кислородсодержащим газом и введение такой исходной газовой смеси для конверсии в каталитический конвертер, имеющий несколько последовательных каталитических стадий. Каждая каталитическая стадия содержит катализатор окисления, эффективный для окисления SO2 до SO3. Исходная газовая смесь для конверсии при этом контактирует с катализатором окисления, находящимся по меньшей мере на первой каталитической стадии в последовательности таких стадией, чтобы образовать частично конвертированный газ, содержащий SO3 и остаточные SO2 и кислород. Частично конвертированный газ пропускается через по меньшей мере одну последующую каталитическую стадию в последовательности таких стадией, катализатор окисления, находящийся на данной стадии, включает активную фазу, содержащую оксид рутения, в результате чего остаточный SO2 в частично конвертированном газе окисляется до SO3 с образованием конвертированного газа, содержащего SO3 и остаточный SO2. Данный конвертированный газ контактирует с водным раствором, содержащим серную кислоту, для абсорбции находящегося в нем SO3 в зоне абсорбции SO3, чтобы получить дополнительное количество серной кислоты и/или олеума и газ, обедненный SO3, который содержит SO2.
Данное изобретение также направлено на катализатор окисления, применимый для окисления SO2 до SO3. В одном варианте осуществления катализатор окисления содержит кислотоустойчивый носитель и размещенную на нем активную фазу. Активная фаза содержит оксид рутения со средним размером кристаллитов менее примерно 500 Å.
В соответствии с другим вариантом осуществления катализатор окисления содержит кислотоустойчивый носитель и промотированную активную фазу на поверхности носителя, содержащую оксид рутения и промотор, содержащий дополнительный оксид металла в состоянии окисления +4 или +3.
В соответствии с еще одним вариантом осуществления катализатор окисления содержит носитель, включающий частицы микрофлюидизированного кремнезема и частицы коллоидного кремнезема, и активную фазу на поверхности носителя, содержащую оксид рутения. Микрофлюидизированный кремнезем характеризуется тем, что имеет средний размер частиц менее примерно 20 мкм, а коллоидный кремнезем характеризуется тем, что имеет средний размер частиц от примерно 10 нм до примерно 25 нм.
Данное изобретение также направлено на способы и методы изготовления катализатора окисления, включающего активную фазу, содержащую оксид рутения. В одном варианте осуществления способ изготовления катализатора окисления включает объединение раствора соли рутения и кислотоустойчивого носителя, чтобы образовать суспензию, и добавление основания к данной суспензии, чтобы образовать предшественник катализатора, имеющий покрытие из оксида рутения на поверхности носителя. Предшественник катализатора термообрабатывают при первой температуре от примерно 200°C до примерно 350°C в течение от примерно 0,1 часа до примерно 5 часов и при второй температуре от примерно 50°C до примерно 500°C в течение от примерно 0,1 часа до примерно 5 часов, чтобы преобразовать предшественник катализатора в катализатор окисления, включающий носитель и размещенную на нем активную фазу, содержащую оксид рутения.
В другом варианте осуществления способ изготовления катализатора на основе оксида рутения включает объединение кислотоустойчивого носителя, микрофлюидизированного кремнезема и коллоидного кремнезема, чтобы образовать первую суспензию. Микрофлюидизированный кремнезем характеризуется тем, что имеет средний размер частиц менее примерно 20 мкм, а коллоидный кремнезем характеризуется тем, что имеет средний размер частиц от примерно 10 нм до примерно 25 нанометров. Из первой суспензии формируют носитель с покрытием. С таким носителем с покрытием объединяют раствор соли рутения, чтобы образовать вторую суспензию. Ко второй суспензии добавляют основание, чтобы сформировать предшественник катализатора, имеющий покрытие из оксида рутения на поверхности указанного носителя с покрытием. После этого предшественник катализатора термообрабатывают.
Данное изобретение, кроме того, направлено на жидкую дисперсию, содержащую фазу жидкого носителя, суспензию микрофлюидизированного кремнезема и суспензию коллоидного кремнезема. Общее содержание кремнезема составляет более примерно 5 процентов в расчете на массу. Суспензия микрофлюидизированного кремнезема характеризуется тем, что имеет вязкость при примерно 24°C и при содержании твердотельных частиц примерно 15 массовых процентов, которая составляет менее примерно 50 сантипуаз, микрофлюидизированный кремнезем характеризуется тем, что имеет средний размер частиц менее примерно 20 мкм, а коллоидный кремнезем характеризуется тем, что имеет средний размер частиц от примерно 10 нм до примерно 50 нанометров.
Другие задачи и особенности данного изобретения будут частью очевидны и частью указаны здесь далее.
Краткое описание чертежей
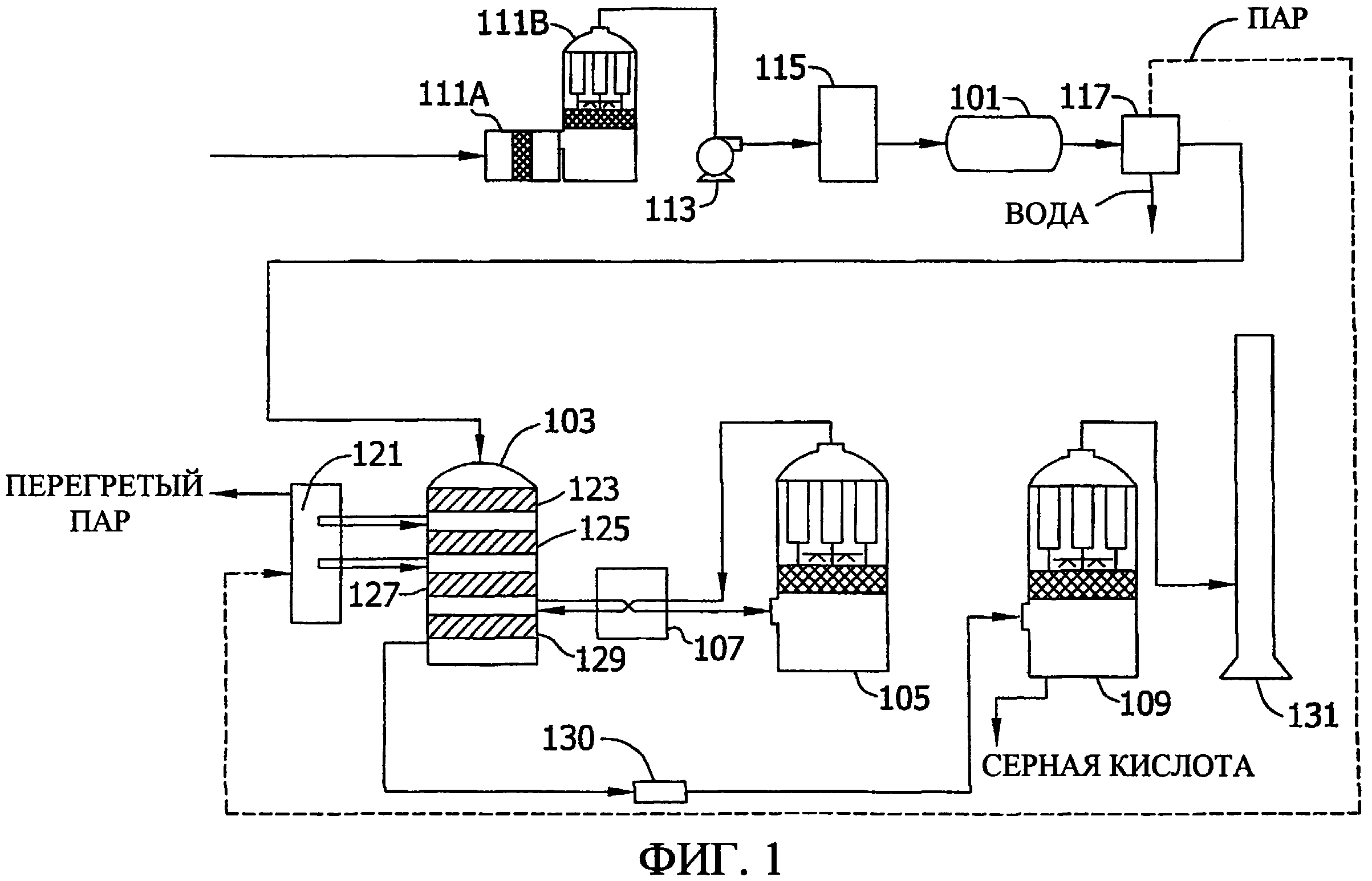
Фиг. 1 представляет схему обычного контактного процесса производства серной кислоты, в котором может быть предпочтительно использован катализатор на основе оксида рутения по данному изобретению.
Фиг. 2 представляет рентгеновские дифрактограммы для порошковых образцов гранул размером от 2,1 до 2,4 мкм катализатора на основе оксида рутения, изготовленного в Примере 6, после конверсии в SO3 в реакторной системе устройства для термического испытания катализатора на старение (TCAT) (что названо «после конверсии») (обозначено как 1); перед конверсией в SO3 (что названо «перед конверсией») (обозначено как 2); и «линейная рентгенограмма» для эталонного образца RuO2, взятая из PDF 40-1290 и представленная острыми пиками на оси «Два тета».
Фиг. 3 представляет рентгеновские дифрактограммы для монолитного катализатора, изготовленного в Примере 10 (оксид рутения/TEOS-Sylox-15/Sylox-15 на монолите с 200 ячеек на квадратный дюйм, разрезанном на кусочки примерно 5 мм × 5 мм), перед конверсией в SO3 (обозначено как 1) в сравнении с «линейной рентгенограммой» для эталонного образца RuO2, взятой из PDF 40-1290 и представленной острыми пиками на оси «Два тета».
Фиг. 4 представляет рентгеновские дифрактограммы для катализаторов, изготовленных в Примере 15, при этом верхняя дифрактограмма относится к порошковым образцам оксид рутения-гранулы 50 нм ZrO2/100 нм ZrO2/гранулы кремнезема (обозначено как 1), а нижняя дифрактограмма относится к порошковым образцам оксид рутения/гранулы кремнезема (обозначено как 2).
Фиг. 5 представляет изображение, полученное сканирующим просвечивающим электронным микроскопом (STEM) для образца, который получен из нарезанных кусочков монолитного катализатора (катализатора 15'), представляющего собой монолитный катализатор, изготовленный в Примере 12.
Фиг. 6 представляет использование рентгеновской спектроскопии с энергетической дисперсией (EDS) для анализа состава одной из ярких зон на Фиг. 5 при расположении луча, указанном кружком (обозначено как 1). Элементный анализ этой области приведен на Фиг. 7.
Фиг. 7 представляет выходной сигнал EDS для области, обозначенной как 1 на Фиг. 6, полученный при использовании прибора EDAX-TSL.
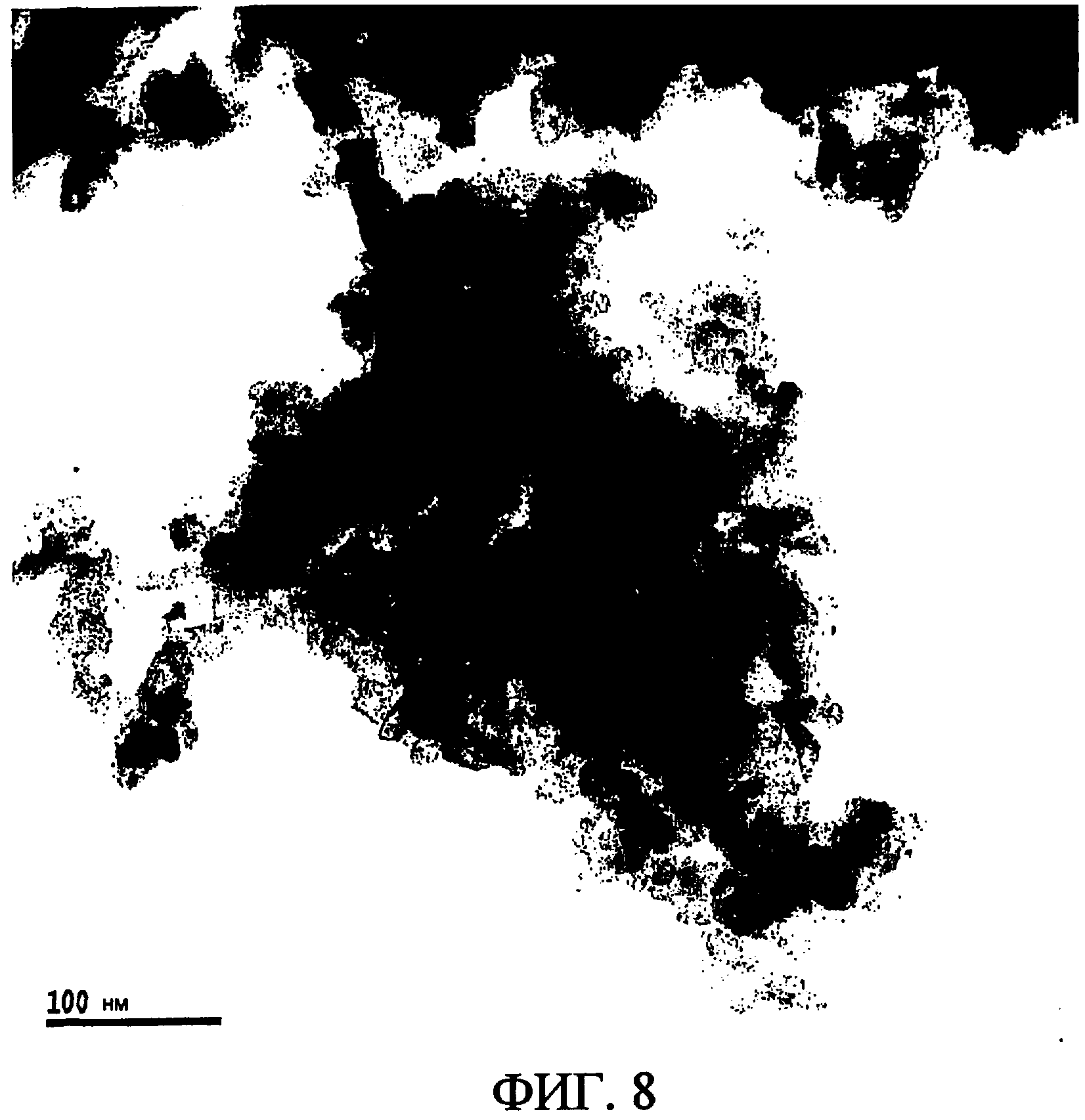
Фиг. 8 представляет изображение, полученное просвечивающей электронной микроскопией (TEM) для фазы диоксида рутения (темные области) на носителе из кремнезема (светлые области) образца монолитного катализатора (катализатор 15'), представляющего собой монолитный катализатор, изготовленный в Примере 12.
Фиг. 9 представляет собой увеличенное изображение, полученное TEM, на Фиг. 8.
Фиг. 10 представляет изображение STEM катализатора на основе оксида рутения на гранулированном носителе (катализатор 14), изготовленного в Примере 18 (после испытания в реакторе TCAT), квадратная область (обозначенная как 1) на котором показывает расположение луча при анализе состава этой области с помощью EDS. Элементный анализ этой области контактирован на Фиг. 11.
Фиг. 11 представляет выходной сигнал EDS для области, обозначенной как 1 на Фиг. 10, полученный при использовании прибора EDAX-TSL.
Фиг. 12 представляет собой типичное изображение TEM с высоким разрешением для катализатора на основе оксида рутения на гранулированном носителе (катализатор 14), изготовленного в Примере 18 (после испытания в реакторе TCAT).
Подробное описание изобретения
Катализатор по данному изобретению включает активную фазу, содержащую оксид рутения. Катализатор применим при окислении SO2 до SO3 при сравнительно низких температурах, например, менее примерно 400°C. Катализатор особенно пригоден для конверсии остаточного SO2 на последней каталитической стадии конвертера, используемого для производства в промышленных масштабах серной кислоты контактным способом.
Катализаторы на основе оксида рутения по данному изобретению обеспечивают улучшенную конверсию при низкой температуре SO2 до SO3 в газовых потоках со сравнительно низким содержанием SO2. Например, в Таблице 2 Примера 6 показано, что катализаторы на основе оксида рутения на носителе (катализаторы 2-5) обеспечивают значительно более высокую степень конверсии SO2 в газовом потоке, содержащем 0,5% SO2 и 7% кислорода, в температурном интервале от 250°C до 375°C по сравнению с обычным катализатором на носителе, содержащим смеси оксида цезия (Cs2O), оксида калия (K2O) и пятиокиси ванадия (V2O5).
Катализаторы на носителе по данному изобретению способны обеспечивать конверсию 98%, 99% или даже такую высокую, как 99,9% и практически 100% SO2, содержащегося в газовых потоках, в SO3 при низкой температуре. В частности, катализаторы на носителе по данному изобретению способны обеспечивать остаточное содержание SO2 менее 0,01% (менее 100 млн-1 по объему), менее 0,005% (менее 50 млн-1 по объему), менее 0,004% (менее 40 млн-1 по объему), менее 0,003% (менее 30 млн-1 по объему), менее 0,002% (менее 20 млн-1 по объему) или даже менее 0,001% (менее 10 млн-1 по объему) в газовых потоках, первоначально содержащих до примерно 1% SO2 или менее. Кроме того, катализаторы, раскрытые в данном изобретении, предусматривают увеличенную скорость газа и обладают повышенной химической и термической стабильностью.
Как описано более подробно ниже, катализаторы по данному изобретению могут включать активную фазу, содержащую оксид рутения, без носителя. Предпочтительно, однако, чтобы активная фаза катализатора присутствовала на носителе. Подходящие носители включают монолиты (например, носители с сотовой или иной структурой, имеющие сквозные отверстия, ячейки или каналы для протекания газа, содержащего SO2, при относительно высокой скорости и низком перепаде давления), а также носители меньшего размера для изготовления зерен катализатора, предназначенных для использования в неподвижном или уплотненном слое катализатора. В одном варианте осуществления катализатор содержит активную фазу на основе оксида рутения, характеризуемую средним размером кристаллитов менее примерно 500 Å, что придает катализатору повышенную активность и стабильность.
Катализаторы могут быть соответственно изготовлены способами осаждения из раствора, включающими растворение соединения, являющегося предшественником оксида рутения, в подходящем растворителе. Твердотельный предшественник катализатора может быть затем осажден из раствора, например, регулированием pH и/или нагреванием раствора. В тех вариантах осуществления, в которых активная фаза катализатора окисления, содержащая оксид рутения, присутствует на носителе, твердотельный предшественник катализатора может быть осажден из раствора на носитель катализатора. После осаждения предшественник катализатора на основе оксида рутения отделяют от раствора и опционально сушат перед преобразованием предшественника в оксид рутения и его активацией, например, прокаливанием предшественника в окислительной атмосфере. В качестве альтернативы, раствор, содержащий соединение, являющееся предшественником оксида рутения, может быть использован для увлажнения или пропитывания носителя катализатора с последующей опциональной сушкой и преобразованием соединения предшественника, чтобы образовать активную фазу, содержащую оксид рутения, на поверхности носителя катализатора. Носитель катализатора может опционально содержать тонкое грунтовочное покрытие с большой площадью поверхности, на котором формируют активную фазу, содержащую оксид рутения. В некоторых вариантах осуществления предшественник катализатора на основе оксида рутения может быть подвергнут восстановительной обработке.
Активная фаза на основе оксида рутения
В активной фазе катализатора, содержащей оксид рутения, по меньшей мере примерно 10%, в расчете на молярное содержание рутения, активного компонента рутения находится в форме оксида рутения. Предпочтительно, по меньшей мере 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98% и даже по меньшей мере 99% или более рутения присутствует в форме оксида рутения. Активная фаза, содержащая оксид рутения, может содержать рутений в различном состоянии окисления, например рутений в состоянии(ях) окисления +2, +3, +4 и/или +8 подходит для использования в данном изобретении. Предпочтительной является активная фаза на основе оксида рутения, содержащая оксид рутения с более низкой валентностью, например RuO, Ru2O3 и/или RuO2. Оксиды рутения и гидраты оксида рутения, в которых рутений имеет состояние окисления +4, например RuO2, особенно предпочтительны в активной фазе.
Активная фаза может, кроме того, содержать рутений, который не находится в форме оксида. Например, в активной фазе, содержащей оксид рутения, может присутствовать металлический рутений, гидроксид рутения (Ru(OH)3) или остаток соединения, являющегося предшественником оксида рутения, из которого образуют активную фазу (например, галоидной соли рутения, такой как RuCl3, или другого соединения, являющегося предшественником оксида рутения). Кроме того, как описано в деталях ниже, активная фаза на основе оксида рутения может включать один или несколько металлических промоторов, обычно присутствующих в форме оксида металла.
Было обнаружено, что размер кристаллитов оксида рутения в активной фазе влияет на каталитическую активность при низких температурах, а также на химическую и термическую стабильность. В частности, уменьшенный размер кристаллитов обеспечивает более высокую каталитическую активность и более продолжительный срок службы катализатора. Размер кристаллитов обычно измеряют при использовании рентгеновской дифракции (XRD) или электронной микроскопии, в частности просвечивающей электронной микроскопии высокого разрешения, посредством которой размер кристаллитов наблюдают непосредственным образом и определяют распределение частиц по размерам на основании совокупности проведенных наблюдений. Полагают, что средний размер кристаллитов оксида рутения менее примерно 500 Å повышает активность вследствие увеличения до максимума активной поверхности на единицу объема катализатора. Также полагают, что молекулы SO2 должны хемосорбироваться на поверхности катализатора, чтобы сделать возможным приближение атомов кислорода, которые будут принимать участие в образовании и десорбции SO3. Однако хемосорбированный SO2 может ослабить адгезию между активной фазой из оксида рутения и носителем, поддерживающим активную фазу, что приводит к нестабильности катализатора. Согласно одной теории, полагают, что катализаторы по данному изобретению обеспечивают повышенную стабильность катализатора вследствие увеличения площади каталитической поверхности и соответствующего увеличения активности, так что хемосорбция SO2 и десорбция SO3 происходят быстро, в результате чего ограничивается промежуток времени, в течение которого катализатор подвергается воздействию хемосорбированного SO2. Найдено, что катализаторы окисления SO2 в соответствии с данным изобретением, имеющие средний размер кристаллитов оксида рутения менее примерно 500 Å, проявляют повышенную активность и стабильность. Предпочтительно активная фаза катализатора, содержащая оксид рутения, имеет средний размер кристаллитов оксида рутения менее примерно 450 Å, менее примерно 400 Å, менее примерно 350 Å, менее примерно 300 Å, менее примерно 250 Å, менее примерно 200 Å, менее примерно 150 Å или даже менее примерно 100 Å. Предпочтительно активная фаза катализатора, содержащая оксид рутения, включает кристаллиты оксида рутения размером в интервале от примерно 10 Å до примерно 500 Å, более предпочтительно от примерно 20 Å до примерно 300 Å, еще более предпочтительно от примерно 30 Å до примерно 100 Å и даже еще более предпочтительно от примерно 50 Å до примерно 80 Å.
Различные способы изготовления катализаторов, включающих активную фазу, содержащую оксид рутения, без носителя или на носителе, включая методики контроля размера кристаллитов оксида рутения, изложены в деталях ниже.
Соединения, являющиеся предшественником оксида рутения, и их растворы
Способы осаждения из раствора, используемые для изготовления катализаторов по данному изобретению, включают растворение соединения, являющегося предшественником оксида рутения, в растворителе. Соединения, являющиеся предшественником оксида рутения, включают, например, следующие соединения, их гидраты и их смеси: оксид рутения; гидроксид рутения; галогениды рутения, такие как хлорид рутения, бромид рутения и йодид рутения; галогенсодержащие кислоты, такие как хлорорутениевая кислота, броморутениевая кислота и йодорутениевая кислота; оксикислоты, такие как рутениевая кислота, соли щелочных металлов и соли аммония хлорорутениевой кислоты и рутениевой кислоты, такие как хлорорутенат натрия и рутенат натрия, рутениевые соли неорганических кислот, такие как нитрат нитрозила рутения, нитрат рутения, ацетат рутения и сульфат рутения; 2,4-пентандионат рутения; и координационные комплексы, такие как галогениды тетрамминрутения, и трехъядерные карбоксилаты рутения, такие как тригидрат μ-оксоацетата рутения (III, III, III) и ацетат μ3-оксогексакис(μ-ацетато)триакватрирутения (Ru3O(O2CCH3)6(H2O)3(CH3CO2). Растворитель может быть водным, органическим или их смесью и выбирается таким образом, чтобы соединение, являющееся предшественником оксида рутения, легко растворялось в нем при первоначальных условиях стадии растворения. Подходящие органические растворители включают C1-4-спирты. Однако вода является предпочтительным растворителем при использовании совместно с водорастворимыми соединениями, являющимися предшественником оксида рутения, такими как гидрат трихлорида рутения (III) и гидрат нитрата нитрозила рутения и другие гидратированные галогениды или нитраты рутения.
Активная фаза на основе оксида рутения без носителя
Катализатор, включающий активную фазу на основе оксида рутения без носителя, может быть соответственно изготовлен посредством первого растворения соединения, являющегося предшественником оксида рутения, в растворителе. Соединение, являющееся предшественником оксида рутения, обычно растворяют в растворителе в концентрации от примерно 0,01 М до примерно 5 М, предпочтительно от примерно 0,1 М до примерно 5 М и более предпочтительно от примерно 0,1 М до примерно 3 М, в расчете на эффективную единицу количества рутения. Например, в случае использования трехъядерного комплекса рутения в качестве соединения, являющегося предшественником оксида рутения, молярность раствора вычисляется делением молекулярной массы комплекса на три, чтобы получить молекулярную массу моноядерных молекулярных частиц рутения.
Растворенное соединение, являющееся предшественником оксида рутения (т.е. растворенное вещество), осаждают из раствора предшественника, чтобы образовать суспензию, содержащую твердотельный предшественник катализатора на основе оксида рутения. Чтобы вызвать осаждение предшественника катализатора на основе оксида рутения из раствора, могут быть использованы различные способы и их комбинации, известные специалистам в данной области техники (например, способы перевода в нерастворимую форму или пересыщения), включающие регулирование pH, удаление растворителя (т.е. испарение), нагревание, снижение температуры охлаждением или «мгновенная кристаллизация» раствора предшественника и добавление второго растворителя или сорастворителя, в котором растворенное вещество имеет низкую растворимость. Независимо от используемого способа твердотельный предшественник катализатора на основе оксида рутения предпочтительно осаждают из раствора в течение периода времени, достаточного для обеспечения непрерывного и равномерного осаждения высокодиспергированных аморфных мелких частиц твердотельного предшественника катализатора на основе оксида рутения. А именно, предпочтительно избегают проведения быстрого, или по существу мгновенного, перевода в нерастворимую форму и/или осаждения, чтобы обеспечить получение в основном гомогенного твердотельного предшественника катализатора на основе оксида рутения с малым размером частиц.
В предпочтительном варианте осуществления твердотельный предшественник катализатора на основе оксида рутения является аморфным гидратом оксида рутения. Было найдено, что гидратированные твердотельные предшественники катализатора на основе оксида рутения обеспечивают изготовление катализаторов на основе оксида рутения с повышенной активностью, химической стабильностью и термической стабильностью. В случае водорастворимой соли рутения преобразование в гидрат оксида рутения происходит в ходе многостадийного процесса с привлечением гидроксидных ионов, который не понят полностью.
В одном варианте осуществления твердотельный предшественник катализатора на основе оксида рутения, содержащий гидрат оксида рутения, может быть осажден из раствора предшественника при использовании методики регулирования pH. Кислый водный раствор предшественника, содержащий растворенные в нем галоидную соль рутения или нитрат рутения, обрабатывают подходящим основанием, чтобы провести нейтрализацию (т.е. увеличение величины pH) и осаждение суспензии аморфного гидрата оксида рутения из раствора. Используемое основание может быть твердотельным, жидким или газообразным и предпочтительно выбрано из аммиака, гидроксида аммония, гидроксида натрия и гидроксида калия. Газообразный аммиак предпочтителен в тех случаях, когда полезно добавление газа, таких как объединение добавления аммиака с функционированием обезвоживающего устройства. В случае использования щелочных растворов для регулирования pH такие растворы предпочтительно имеют концентрацию основания более примерно 5% по массе на объем, более предпочтительно по меньшей мере 10% по массе на объем, 15% по массе на объем, 20% по массе на объем или даже 25% по массе на объем. Основание и раствор предшественника, содержащий соединение, являющееся предшественником оксида рутения, могут быть объединены при любом порядке добавления. В одном варианте осуществления раствор предшественника медленно добавляют к концентрированному раствору основания (например, раствору гидроксида аммония) при перемешивании в течение увеличенного периода времени, например в течение по меньшей мере 15 минут, примерно 30 минут, примерно 45 минут или по меньшей мере примерно 60 минут или более. Перемешивание суспензии продолжают после завершения добавления раствора предшественника в течение по меньшей мере примерно 15 минут, примерно 30 минут, примерно 45 минут, примерно 60 минут, примерно 75 минут, примерно 90 минут или более, чтобы обеспечить получение гомогенного осадка предшественника катализатора на основе оксида рутения. Температуру во время осаждения предпочтительно поддерживают ниже температуры кипения или температуры флегмы, предпочтительно в интервале от примерно 20°C до примерно 95°C.
В другом варианте осуществления способа изготовления осадка предшественника катализатора на основе оксида рутения раствор предшественника оксида рутения может быть нагрет до температуры, достаточно высокой для осаждения аморфного гидрата оксида рутения из раствора. Предпочтительно использование растворителя или композиции растворителей, содержащих воду. Например, водный раствор предшественника оксида рутения может быть нагрет до температуры от примерно 70°C до примерно 95°C, чтобы осадить гидрат оксида рутения. Скорость нагревания предпочтительно контролируют для того, чтобы селективным образом получить твердотельное вещество с высокой степенью диспергирования. Обычно следует избегать быстрого нагревания, чтобы предотвратить локализованное негомогенное осаждение. Некоторые водные растворы соединений, являющихся предшественником оксида рутения, таких как хлорид рутения, кислые и имеют величины pH примерно 1 или ниже. После осаждения, обусловленного нагреванием, величина pH может быть отрегулирована подходящим основанием, таким как газообразный аммиак, гидроксид аммония, гидроксид натрия или гидроксид калия.
После образования влажный осадок твердотельного предшественника катализатора на основе оксида рутения выделяют из суспензии любым способом разделения твердого тела и жидкости, известным в данной области техники, таким как фильтрация или центрифугирование. Примеси могут быть удалены из отделенного влажного осадка промывкой растворителем, таким как вода, или слабыми технологическими растворами. Влажный отделенный предшественник катализатора затем опционально сушат. Сушка может быть выполнена любым способом, известным в данной области техники, таким как сушка на лотке, сушка в псевдоожиженном слое, сушка распылением или сушка в вакуумном сушильном шкафу. Сушку предпочтительно проводят при температуре от примерно 100°C до примерно 200°C в вакуумном сушильном шкафу. Твердотельный предшественник катализатора на основе оксида рутения сушат до тех пор, пока не будет удалено достаточно влаги, чтобы получить в основном сыпучий порошок. Время сушки составляет обычно от по меньшей мере примерно 0,5 часа до примерно 5 часов или более.
Предшественник катализатора на основе оксида рутения в виде порошка или твердотельного материала может быть опционально спрессован в форме пластин или с приданием иной формы при использовании способов и устройств, известных специалистам в данной области техники, например пресса Carver, пригодного для изготовления образцов в лабораторных масштабах, или ротационного таблеточного пресса. Пластины или иные прессованные профили могут быть затем измельчены (например, при использовании молотковой мельницы, шаровой мельницы или другим способом уменьшения размеров частиц, известным в данной области техники) и просеяны, чтобы получить порошки или гранулы предшественника катализатора на основе оксида рутения с относительно равномерным распределением частиц по размеру. Например, могут быть приготовлены порошки с размером частиц в интервале от примерно 1 мкм до примерно 100 мкм или агрегированные частицы большего размера, например фракции, соответствующие размеру ячеек сита 10-12 меш.
Твердотельные предшественники катализатора на основе оксида рутения активируют термической обработкой (т.е. прокаливанием при высокой температуре) при температурах от примерно 200°C до примерно 600°C, более предпочтительно от примерно 300°C до примерно 500°C, в течение от примерно 0,5 до примерно 12 часов в атмосфере, содержащей кислород (например, прокаливанием на воздухе) и/или SO2. Активация при высокой температуре может быть выполнена в несколько этапов при разных температурах. Например, твердотельный предшественник катализатора на основе оксида рутения может быть термообработан вначале на первом этапе прокаливания при температуре от примерно 200°C до примерно 300°C и затем на втором этапе прокаливания при температуре от примерно 300°C до примерно 600°C. Вслед за каждым таким этапом может следовать стадия нагревания до температуры, поддерживаемой на последующем этапе.
Также найдено, что в некоторых случаях уменьшение размера твердотельных частиц предшественника оксида рутения приводит к получению активной фазы оксида рутения с желательными размером кристаллитов, активностью катализатора и/или сроком службы катализатора. Полагают, что такое уменьшение приводит к образованию высокодиспергированных и механически прочно сцепленных кристаллитов металлического рутения, которые, когда они подвергаются воздействию окислительной атмосферы при повышенных температурах, преобразуются в катализатор на основе оксида рутения, эффективный для окисления SO2. Однако проявление преимуществ, связанных с уменьшением размера твердотельных частиц предшественника оксида рутения, зависит от соединения предшественника, использованного для образования твердотельного предшественника. В частности, восстановительная обработка предоставляет преимущества, когда твердотельный предшественник оксид рутения образуют при использовании соли рутения, такой как хлорид рутения или нитрат нитрозила рутения. Подходящие восстановители включают, например, водород для газофазного восстановления или растворы борогидрида натрия, борогидрида лития, триацетилборогидрида калия, формальдегида, муравьиной кислоты, формиата натрия, гидразингидрохлорида, гидроксиламина, борана, борана-тетрагидрофурана, борана-пиридина, гидрида лития-алюминия, гидрида алюминия и фосфорноватистой кислоты для жидкофазного восстановления. Газофазное восстановление твердотельных частиц предшественника оксида рутения выполняют перед активацией катализатора, содержащего оксид рутения. Высушенный твердотельный предшественник катализатора на основе оксида рутения, выделенный из суспензии предшественника, может контактировать с восстановительным газом, таким как водород, при повышенной температуре. Концентрация восстановительного газа в восстановительной атмосфере составляет предпочтительно от примерно 1% до примерно 10%, более предпочтительно от примерно 1% до примерно 5% при остатке, состоящем в основном из подходящего инертного газа, например азота. В одном из предпочтительных вариантов осуществления восстановительная атмосфера содержит от примерно 2% до примерно 5% водорода при остатке, состоящем в основном из азота. Предпочтительные интервалы температур для газофазного восстановления зависят от соли рутения. В случае хлорида рутения предпочтительная температура восстановления находится в интервале от примерно 150°C до примерно 250°C при более предпочтительной температуре примерно 200°C. В случае нитрата рутения предпочтительная температура восстановления находится в интервале от примерно 125°C до примерно 175°C при более предпочтительной температуре примерно 150°C. В случае газофазного восстановления в присутствии водорода предшественника, образованного из хлорида рутения, продуктами восстановительной реакции являются металлический рутений и газообразная хлористоводородная кислота. В случае газофазного восстановления в водороде предшественника, образованного из нитрата рутения, продуктами восстановительной реакции являются металлический рутений и газообразная азотная кислота. Скорость образования побочного газообразного продукта может контролироваться, чтобы определить момент завершения в основном преобразования в металлический рутений. Время обработки с газофазным восстановлением обычно составляет менее примерно 24 часов, например, менее примерно 20 часов, менее примерно 16 часов, менее примерно 12 часов и, как правило, менее примерно 8 часов.
Площадь поверхности по БЭТ, обеспечиваемая прокаленным катализатором на основе оксида рутения после активации, обычно составляет по меньшей мере примерно 25 м2/г, предпочтительно по меньшей мере примерно 50 м2/г, по меньшей мере примерно 75 м2/г и даже по меньшей мере примерно 100 м2/г. Площадь поверхности по БЭТ относится к величине площади поверхности, определенной хорошо известным способом Брунауэра-Эмметта-Теллера, и, если не указано иное, все величины площади поверхности, приведенные в данном описании, относятся к площади поверхности по БЭТ.
Активированный катализатор на основе оксида рутения без носителя подходит для использования в качестве катализатора окисления при конверсии SO2 в SO3. Порошок или гранулы могут опционально объединены со связующими, известными специалистам в данной области техники, и затем таблетированы или сформованы иным образом с образованием различных форм желательного размера для использования в устройствах с неподвижным или уплотненным слоем катализатора, через который пропускают газ, содержащий SO2.
Пример изготовления катализатора на основе оксида рутения без носителя в соответствии с данным изобретением и его оценка при окислении SO2 до SO3 представлена в Примере 6, ниже.
Активная фаза на основе оксида рутения на носителе
В соответствии с предпочтительным вариантом осуществления катализатор окисления по данному изобретению включает активную фазу, содержащую оксид рутения, на поверхности носителя или опоры катализатора. Из соображений экономии использование катализатора на носителе предпочтительно, чтобы получить катализатор, в котором открыта внешнему воздействию более высокая доля оксида рутения на единичный объем активной фазы. Носители катализатора могут опционально содержать тонкое грунтовочное покрытие с большой площадью поверхности, наносимое на поверхность носителя.
Активная фаза, содержащая оксид рутения, обычно присутствует на носителе в количестве менее примерно 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3% или даже менее примерно 2% от массы катализатора, однако без ограничения этими величинами. Предпочтительные интервалы содержания активной фазы на основе оксида рутения составляют от примерно 0,1% до примерно 10%, от примерно 0,5% до примерно 10%, от примерно 0,5% до примерно 5%, от примерно 0,5% до примерно 4%, от примерно 0,5% до примерно 3% или даже от примерно 0,5% до примерно 2% от массы катализатора. Из расчета массы на объем катализатор на носителе по данному изобретению обычно содержит менее примерно 20 кг рутения на кубический метр объема катализатора, менее примерно 15 кг рутения на кубический метр, менее примерно 10 кг рутения на кубический метр, предпочтительно менее примерно 5 кг рутения на кубический метр, еще предпочтительнее менее примерно 4 кг рутения на кубический метр, еще предпочтительнее менее примерно 3 кг рутения на кубический метр и даже более предпочтительно менее примерно 2 кг рутения на кубический метр объема катализатора.
Предпочтительные носители обычно характеризуется тем, что они имеют большую площадь внешней поверхности, что предоставляет увеличенную долю оксида рутения, открытого внешнему воздействию, на единичный объем активной фазы и обеспечивает высокую эффективность реакции, низкий перепад давления и высокую стабильность каталитических свойств. В кислой окружающей среде, в которой функционируют катализаторы, используемые для каталитической конверсии SO2 в SO3, предпочтительны носители, изготовленные из кислотоустойчивых материалов или содержащие такие материалы, по причине их химической стабильности при таких условиях. Подходящие кислотоустойчивые материалы носителя включают, например, силикаты (т.е. соединения, содержащие кремний, кислород и один или несколько металлов с водородом или без него), муллит (т.е. силикат алюминия), кордиерит, диоксид циркония, гидроксид циркония, нержавеющую сталь, ферритные стали и никелевые сплавы, такие как инконель и хастеллой. Подходящие кислотоустойчивые носители могут также включать комбинацию кремнезема с одним или несколькими соединениями, выбранными из оксида циркония (ZrO2), оксида алюминия (Al2O3), диоксида титана (TiO2), диоксида олова (SnO2) и оксида лантана (La2O3). В одном варианте осуществления носители содержат комбинацию силикатов и одного или нескольких соединений из соединения циркония, соединения олова и соединения титана. В таком варианте осуществления соединение кремния обычно содержит по меньшей мере примерно 80 масс.%, примерно 85 масс.% или примерно 90 масс.% носителя.
Носитель для активной фазы, содержащей оксид рутения, может иметь разные размеры и формы, известные в данной области техники, включающие те, которые адаптированы для использования в неподвижных или уплотненных слоях катализатора, содержащих неупорядоченно распределенные частицы или зерна сравнительно небольшого размера, такие как, например, порошки, макрочастицы, гранулы, кольца (например, кольца Рашига и кольца Полла), зерна в виде колеса, зерна в виде седла, сферические или цилиндрические зерна, зерна с волнистым профилем, зерна в виде звезды, зерна в виде решеток и зерна в виде трилистника. Примеры подходящих носителей в виде частиц и формованных носителей включают те из них, которые изготовлены из SiO2, Zr(OH)4 и ZrO2.
Примеры подходящих порошковых носителей включают силикаты, такие как SiO2, и алюмосиликаты (например, цеолиты), имеющие сравнительно низкое содержание оксида алюминия (например, менее примерно 1% по массе), чтобы придать им достаточную устойчивость к кислотам, соединения циркония, такие как ZrO2 и Zr(OH)4, соединения олова, такие как SnO2, и соединения титана, такие как TiO2. Предпочтительный порошковый носитель содержит мезопористый диоксид циркония (ZrO2). Предпочтительно мезопоры имеют диаметр менее примерно 50 нм, 40 нм или даже 30 нм. Мезопоры могут иметь равномерное или неравномерное распределение. Предпочтительными являются порошковые материалы носителя со средним размером частиц от примерно 0,1 мкм до примерно 200 мкм, от примерно 0,5 мкм до примерно 100 мкм и даже примерно от 1 мкм до примерно 50 мкм. В одном варианте осуществления диаметр порошкового носителя составляет от примерно 2 мкм до примерно 10 мкм и в другом варианте осуществления - от примерно 2 мкм до примерно 5 мкм.
Найдено, что носители с большой площадью поверхности способствуют уменьшению размеру кристаллитов рутения и увеличению активности и срока службы катализатора. На основании экспериментальных данных, имеющихся на настоящий момент, полагают, что носители с большой площадью поверхности обеспечивают образование активных слоев, содержащих оксид рутения с высокой степенью диспергирования, которые химически и термически стабильны в присутствии реакционно-способных газов, таких как кислород, SO2 и SO3. Предпочтительны носители с площадью поверхностью по меньшей мере примерно 50 м2/г, например от примерно 50 м2/г до примерно 500 м2/г, более предпочтительно от примерно 100 м2/г до примерно 300 м2/г и еще более предпочтительно от примерно 150 м2/г до примерно 250 м2/г.
Пористость носителя катализатора может быть определена на основании распределения размера пор. В соответствии с одной схемой, предложенной ИЮПАК и использованной здесь, микропоры определяют как имеющие размер менее примерно 20 Å, мезопоры определяют как имеющие размер между примерно 20 Å и примерно 500 Å, и макропоры - как имеющие размер более примерно 500 Å. Носитель или тонкое грунтовочное покрытие обычно содержит комбинацию микропор, мезопор и макропор при их соотношении, изменяющемся в зависимости от площади поверхности и объема пор. Например, тонкие грунтовочные покрытия с большой площадью поверхности и большим объемом пор могут иметь распределение с отклонением в сторону интервала размеров микропор, в то время как носители катализатора со сравнительно малой площадью поверхности и малым объемом пор могут иметь распределение с отклонением в сторону интервала размеров мезопор и/или макропор.
Для пористых носителей и тонких грунтовочных покрытий предпочтительны материалы с объемом пор от примерно 0,1 см3/г до примерно 3,0 см3/г, более предпочтительно от примерно 0,3 см3/г до примерно 1,2 см3/г и еще более предпочтительно от примерно 0,6 см3/г до примерно 1,0 см3/г. Предпочтительными являются материалы, имеющие распределение пор по размерам, при котором по меньшей мере примерно 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9% или 10% объема пор относится к порам, имеющим диаметр менее примерно 20 Å (т.е. микропорам), по меньшей мере примерно 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70% или 75% объема пор относится к порам, имеющим диаметр между примерно 20 Å и примерно 500 Å (т.е. мезопорам), и по меньшей мере примерно 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9% или 10% объема пор относится к порам, имеющим диаметр более 500 Å (т.е. макропорам). В одном варианте осуществления материалы носителя или тонкого грунтовочного покрытия имеют распределение пор по размерам, при котором по меньшей мере примерно 5% объема пор относится к порам, имеющим диаметр менее примерно 20 Å, по меньшей мере примерно 50% объема пор относится к порам, имеющим диаметр между примерно 20 Å и примерно 500 Å, и по меньшей мере примерно 5% объема пор относится к порам, имеющим диаметр более 500 Å.
В соответствии с одним из предпочтительных вариантов осуществления данного изобретения носитель находится в форме монолита сравнительно большого размера, такого как, например, сотовая структура, имеющая сквозные отверстия, ячейки или каналы для протекания газа, содержащего SO2, через катализатор, и адаптированная для использования в слое катализатора, содержащем упорядоченную или структурированную сборку катализаторных монолитов. Носитель с сотовой структурой или монолит может иметь ячейки разного диаметра и поперечного сечения (например, квадратные ячейки, хотя выбор может варьироваться в зависимости от вида применения) и разную плотность ячеек, однако ячейки обычно достаточно велики, так что носитель не препятствует существенным образом протеканию газа, содержащего SO2, с высокой скоростью. Монолитные катализаторы предоставляют повышенную эффективность процесса посредством обеспечения высокой скорости потока газа, содержащего SO2, при сравнительно низком перепаде давления.
Материалы, подходящие для изготовления носителей в виде монолита с отверстиями, включают кордиерит (орторомбический метасиликат магния-алюминия, Mg2Al4O3(SiO3)5), муллит (3Al2O3·2SiO2), кремнезем, диоксид циркония (ZrO2) и α-оксид алюминия. Двумя предпочтительными материалами для использования в данном изобретении являются муллит и кремнезем. Кремнезем особенно предпочтителен.
Подходящие величины номинальной плотности ячеек в монолитном носителе с сотовой структурой составляют 9, 16, 25, 50, 100, 200, 300, 400, 500, 600, 700, 800 и 900 ячеек на квадратный дюйм (ячеек/кв. дюйм). Для данного изобретения предпочтительная плотность ячеек находится в интервале от примерно 100 до примерно 400 ячеек/кв. дюйм поперечного сечения, перпендикулярного направлению протекания газа через монолитный катализатор. Более предпочтительно плотность ячеек составляет от примерно 100 до примерно 300 ячеек/кв. дюйм. В одном варианте осуществления проницаемость носителя с отверстиями такова, что перепад давления газа, содержащего SO2, кислород и азот, который протекает со скоростью 600 стандартных футов в минуту (183 метра в минуту) через монолитный катализатор, содержащий такой носитель, составляет не более примерно 8 дюймов водяного столба на линейный фут (0,066 атмосферы на метр) в направлении потока. Обычно доля пустот в монолите, образованных отверстиями, находится в интервале между примерно 0,25 и примерно 0,75.
Предпочтительными являются монолитные носители, имеющие площадь поверхности по БЭТ по меньшей мере примерно 15 м2/г, например от примерно 15 м2/г до примерно 50 м2/г. В одном варианте осуществления активная фаза, содержащая оксид рутения, локализована на поверхности стенок отверстий, определяющих проходы или каналы через монолит и имеющих поверхностное покрытие с тонкими порами (часто микропорами), которое является интегральной частью монолитного носителя, образованной при изготовлении носителя, или сформировано впоследствии посредством формирования тонкого грунтовочного пленочного покрытия. Например, интегральное покрытие с большой площадью поверхности может быть образовано на поверхности стенок отверстий монолитного носителя во время изготовления носителя совместной экструзией оксида с большой площадью поверхности/высокой пористостью вместе с оксидом с меньшей площадью поверхности/меньшей пористостью. Подходящие носители для изготовления монолитных катализаторов по данному изобретению включают тонкостенные сотовые структуры. Типичные способы изготовления таких носителей раскрыты в Патентах США №№ 3790654, 4364888, 5175136 и 5264200, содержание которых включено здесь в прямой форме посредством ссылки.
Патент США № 5264200 описывает монолитные носители с сотовой структурой, в которых оксид с высокой пористостью объединен с оксидом с малой площадью поверхности, чтобы получить композиционный материал, обладающий проницаемостью, желательной для протекания газа, пористостью с тонкими порами, желательной для эффективной активности катализатора, и механической прочностью, обеспеченной оксидом с малой площадью поверхности. Типичные материалы для изготовления таких сотовых структур из кремнеземистых композитов включают порошок кремнезема с низкой плотностью и высокой пористостью, имеющий средний размер частиц менее примерно 20 микрон, и частицы кремнезема с низкой площадью поверхности, имеющие размер от примерно 20 до примерно 75 микрон. Пластицированную смесь (или «тесто»), подходящую для экструзии, приготавливают добавлением водной фазы, содержащей воду и низший спирт, такой как, например, изопропиловый спирт. Патент США № 5264200 описывает материал для монолитных катализаторов, такой как кремнезем, экструдированный с образованием номинально от 100 до 300 ячеек/кв. дюйм при квадратном профиле ячеек. Эти композиционные кремнеземистые носители имеют суммарные объемы пор от 0,25 до 0,50 см3/г при площади поверхности от 15 до 50 м2/г. Возможно получение более высоких величин объема пор (от 0,50 до 0,75 см3/г), однако получаемые при этом кремнеземистые монолитные носители могут иметь недостаточную механическую прочность. Механическая прочность является адекватной, когда модуль разрыва составляет более примерно 500 фунтов на квадратный дюйм (примерно 350000 кг/м2). Компонент кремнезема с высокой пористостью кремнеземистого композиционного монолитного носителя в Патенте США № 5264200 выбирали из нескольких порошков кремнезема с высокими величинами площади поверхности (от 100 до 500 м2/г) или кремнеземов с низкими величинами площади поверхности (менее 10 м2/г), однако высокими величинами объема пор, таких как диатомиты. Эти кремнеземистые сотовые структуры, описанные в Патенте США № 5264200, подходят для использования для поддержки активной фазы, содержащей оксид рутения, по данному изобретению. В Табл. I Патента США № 5264200, воспроизведенной ниже, представлен перечень типичных композиционных кремнеземистых сотовых структур. Эти сотовые структуры с 200 ячейками/кв. дюйм охарактеризованы представлением данных, полученных ртутной порометрией и абсорбцией воды. Сотовые структуры, обозначенные как 3 и 4, изготовлены при использовании 10 и 20% диатомита соответственно.
Композиционные кремнеземистые сотовые структуры Патента США № 5264200 демонстрируют очень широкий интервал площади поверхности для стенок отверстий носителя. В случае, когда для изготовления сотовой структуры был использован кремнезем с большой площадью поверхности/высокой пористостью, площадь поверхности находилась в интервале от 100 до 400 м2/г монолита при объеме пор от 0,5 до 2 см3/г. Когда был использован диатомит, то площадь поверхности составляла менее 2 м2/г при пористости примерно 1 см3/г.
Кремнеземистые сотовые структуры, описанные выше, представляют собой тип монолитного носителя активной фазы, содержащей оксид рутения, для использования при окислении SO2, особенно в случае изготовления при использовании диатомитового компонента для обеспечения пористости композиционного материала сотовой структуры. Эти цельнокремнеземистые монолитные носители являются возможными альтернативами сотовым структурам с тонким грунтовочным покрытием. В предпочтительном варианте осуществления очень эффективные носители для активной фазы, содержащей оксид рутения, в соответствии с данным изобретением получают при использовании тонкого грунтовочного покрытия, содержащего кремнезем, которое нанесено на монолитный носитель (например, сотовую структуру из муллита или кремнезема). Такие монолитные катализаторы с оксидом рутения и тонким грунтовочным покрытием обладают исключительной термической и химической стабильностью. Как описано более подробно ниже, такой катализатор изготавливают осаждением кремнеземистого порошка в виде тонкой пленки или тонкого грунтовочного покрытия на поверхности носителя с сотовой структурой. После сушки и прокаливания получают тонкое грунтовочное покрытие с большой площадью поверхности на поверхностях макропор керамического носителя с сотовой структурой. Тонкое грунтовочное покрытие может быть соединено с носителем посредством пленки, полученной прокаливанием суспензии, из которой осаждают тонкое грунтовочное покрытие. Большая площадь поверхности, образованной в носителе с тонким грунтовочном покрытием, способствует обеспечению термической стабильности активной фазы, содержащей оксид рутения.
Тонкое грунтовочное покрытие
Тонкое грунтовочное покрытие представляет собой тонкое, плотно соединенное покрытие из материала, осажденного на стенки и/или поверхность носителя (например, стенки, определяющие фораминальные ячейки, или газовые каналы, проходящие через монолитный носитель). Тонкие грунтовочные покрытия могут увеличивать площадь поверхности носителя, что обеспечивает осаждение предшественника катализатора на основе оксида рутения в высокодиспергированном состоянии и соответственно увеличенную активность, термическую стабильность и химическую стабильность. Тонкие грунтовочные покрытия могут также предоставлять поверхность носителя с высокой пористостью.
Тонкое грунтовочное покрытие обычно формируют из суспензии, раствора или коллоидного раствора («золя»), содержащих материал с большой площадью поверхности, такой как кремнезем, диоксид циркония, оксид олова, например диоксид олова (SnO2), диоксид титана или т.п., который наносят на стенки и/или поверхность носителя и затем сушат. Подходящие материалы тонкого грунтовочного покрытия включают коллоидный кремнезем (например, коллоидный кремнезем NYACOL 1440), цирконилхлорид (ZrOCl2), коллоиды оксида циркония (такие как коллоиды 50 нм и 100 нм, поставляемые Nyacol) и порошки, описанные в Патенте США № 5264200 и контактированные в Табл. II ниже.
После нанесения на носитель тонкое грунтовочное покрытие предпочтительно прокаливают, при этом кремнезем или другой материал с большой площадью поверхности прочно соединяется с поверхностями стенок носителя. В результате получают носитель, покрытый оксидом кремния, оксидом циркония, оксидом титана, оксидом олова, оксидом алюминия или их комбинацией, что предоставляет носитель, содержащий, например, в случае кремнеземистого носителя, SiO2-SiO2, ZrO2-SiO2, TiO2-SiO2, SnO2-SiO2 и/или Al2O5-SiO2.
В случае монолитного носителя нанесение композиции материала тонкого грунтовочного покрытия на стенки носителя предпочтительно выполняют окунанием носителя в данный материал. Оптимальной концентрацией суспензии является такая ее величина, которая обеспечивает максимальное нанесение материала тонкого грунтовочного покрытия с большой площадью поверхности за цикл нанесения покрытия окунанием, данный цикл обычно включает погружение носителя в приготовленную композицию материала тонкого грунтовочного покрытия и удаление суспензии материала из каналов сотовой структуры с помощью газового потока, пропускаемого через каналы. Предпочтительно суспензии, растворы и коллоидные растворы или золи материала тонкого грунтовочного покрытия содержат от примерно 5 масс.% до примерно 25 масс.%, более предпочтительно от примерно 10 масс.% до примерно 25 масс.% твердотельного материала. Чтобы увеличить количество нанесенного материала, цикл нанесения покрытия окунанием может быть при необходимости повторен. Между нанесениями материала покрытия окунанием может быть короткий период сушки на воздухе при комнатной температуре или при повышенной температуре (например, примерно 100°C). После формирования требуемого влажного тонкого грунтовочного покрытия повторением нанесения материала окунанием покрытый монолитный носитель обычно сушат при температуре от примерно 100°C до примерно 200°C в сушильном шкафу с нагнетанием воздуха и предпочтительно прокаливают при температуре от примерно 400°C до примерно 800°C, более предпочтительно от примерно 400°C до примерно 600°C и еще более предпочтительно от примерно 400°C до примерно 550°C.
В одном варианте осуществления композицию материала тонкого грунтовочного покрытия, эффективную в отношении адгезии к расположенному ниже монолитному носителю, предоставляют посредством включения пленкообразующего агента, такого как золь, содержащий кремнезем, диоксид циркония, оксид олова (например, SnO2), диоксид титана или их смеси, в тонкое грунтовочное покрытие. На основании экспериментальных данных, имеющихся к настоящему времени, и без намерения установления связи с какой-либо конкретной теорией полагают, что адгезия основана на связях, таких как -O-Si-O-, которые устойчивы к воздействию серной кислоты. Также полагают, что связи, образованные в материалах из золей, основаны на -O-Si-O- и поэтому обеспечивают повышенную стабильность в присутствии серной кислоты. Золи кремнистой кислоты, пригодные для использования в качестве компонента тонкого грунтовочного покрытия по данному изобретению, могут быть приготовлены различными способами, известными в данной области техники. Золь кремнистой кислоты, пригодный для нанесения тонкого грунтовочного покрытия, может быть получен, например, из силоксана, такого как частично гидролизованный тетраэтилортосиликат, Si(OC2H5)4 (TEOS), как описано S. Sakka, K. Kamiya, K. Makita и Y. Yamamoto в Journal of Non-Crystalline Solids, 63, 223-235 (1984) и включено здесь посредством ссылки. В конкретном способе, описанном в указанном источнике, воду, этанол и TEOS объединяют в молярном соотношении 8:4:1 и добавляют HNO3 в качестве кислоты таким образом, чтобы концентрация кислоты составляла 0,01 M. Циркониевые золи, которые подходят для нанесения тонкого грунтовочного покрытия, включают ZrOCl2, NYACOL Zr 10/20 и NYACOL Zircon.
Композиции материала тонкого грунтовочного покрытия, содержащие золь, могут быть приготовлены образованием суспензии, которая содержит: (1) кремнезем с большой площадью поверхности, например SYLOX 15, в концентрации от примерно 5 масс.% до примерно 30 масс.%, более предпочтительно от примерно 5 масс.% до примерно 25 масс.%, еще более предпочтительно от примерно 5 масс.% до примерно 20 масс.% и даже еще более предпочтительно от примерно 10 масс.% до примерно 20 масс.%; (2) золь, например, TEOS или коллоидного кремнезема в концентрации от примерно 5 масс.% до примерно 50 масс.%, более предпочтительно от примерно 10 масс.% до примерно 45 масс.% и еще более предпочтительно от примерно 10 масс.% до примерно 40 масс.%; (3) примерно от 0,01 масс.% до примерно 0,5 масс.% неорганической кислоты, например, азотной кислоты (HNO3); и (4) остаток, содержащий воду и водорастворимый растворитель, такой как низший спирт, при соотношении воды и водорастворимого растворителя, составляющем от примерно 2:1 до примерно 1:2. Золь может содержать дополнительные опциональные компоненты, такие как диспергирующие и смачивающие агенты, например поверхностно-активные вещества и диспергаторы. Эти агенты обычно уменьшают поверхностное натяжение и улучшают способность к формированию покрытия на носителе, на который наносят тонкое грунтовочное покрытие. Подходящие поверхностно-активные вещества включают неионогенные, катионактивные, анионактивные и амфотерные поверхностно-активные вещества. Предпочтительными являются неионогенные поверхностно-активные вещества, подходящим примером которых является TRITON CF-32 (конденсат амина и полигликоля, поставляемый Union Carbide).
Суспензию тонкого грунтовочного покрытия, содержащую золь, затем объединяют с подходящим носителем, таким как монолит, и обрабатывают, как описано выше. После сушки и прокаливания высушенный золь тонкого грунтовочного покрытия обеспечивает прочное соединение кремнеземистого тонкого грунтовочного покрытия с большой площадью поверхности и носителем посредством образования адгезионной пленки, которая приводит к формированию тонкого грунтовочного покрытия с большой площадью поверхности, прочно соединенного с носителем. В случае монолитов, после прокаливания конечный носитель с нанесенным тонким грунтовочным покрытием обычно имеет удельную площадь поверхности от примерно 15 м2/г до примерно 50 м2/г. Адгезия и большая площадь поверхности обеспечивают термическую стабильность активной фазы, содержащей оксид рутения, которая сформирована при высокой степени дисперсности на носителе с тонким грунтовочным покрытием.
В предпочтительном варианте осуществления для приготовления носителя активной фазы, содержащей оксид рутения, носитель, предпочтительно в форме монолита, покрывают композицией тонкого грунтовочного покрытия, содержащей коллоидный кремнезем и микрофлюидизированный кремнезем. Микрофлюидизированный кремнезем образует пористое покрытие с большой площадью поверхности, а коллоидный кремнезем служит в качестве адгезионного золя, чтобы эффективным образом связать кремнезем с большой площадью поверхности с носителем.
Коллоидный кремнезем обычно отличается тем, что содержит частицы кремнезема с максимальным размером в интервале от примерно 1 нм до примерно 1000 нм. Однако коммерческие продукты коллоидного кремнезема обычно доступны в виде материала, имеющего в основном более равномерное распределение частиц по размерам. Предпочтительно коллоидный кремнезем содержит частицы в интервале от примерно 5 нм до примерно 100 нм, более предпочтительно от примерно 10 нм до примерно 50 нм.
Микрофлюидизированный кремнезем обычно характеризуется тем, что содержит частицы кремнезема со средним размером примерно 10 мкм. Продажный порошок кремнезема с большой площадью поверхности обычно имеет размер частиц от примерно 10 мкм до примерно 20 мкм в высушенном состоянии. Кремнезем с большой площадью поверхности поставляется на рынок многими производителями, например это продукты SYLOX 15 и SYLOID 74 фирмы W.R. Grace and Company. Было найдено, что при суспендировании кремнезема с большой площадью поверхности в воде может происходить агломерация частиц, что приводит к размерам частиц от примерно 10 мкм до примерно 40 мкм или более. Поэтому в одном из вариантов осуществления суспендированные агломерированные частицы кремнезема с большой площадью поверхности деагломерируют или уменьшают в размере при использовании, например, микрофлюидизации, измельчения в шаровой мельнице и/или методов измельчения среды. Методы уменьшения размера частиц во влажном состоянии предпочтительны вследствие упрощения обработки и ее хорошей согласованности с остальными операциями нанесения тонкого грунтовочного покрытия влажным образом.
В одном из методов уменьшения размера частиц влажным способом микрофлюидизированный кремнезем приготавливают при использовании MICROFLUIZER, устройства для обработки текучей среды при приложении высоких срезающих усилий при высоком давлении, поставляемого Microfluidics Corporation (Newton, Massachusetts, USA). Суспензию, содержащую агломерированные частицы кремнезема, подают через ограничивающие каналы в указанном устройстве под высоким давлением, при этом частицы перемешиваются и подвергаются воздействию срезающих усилий с уменьшением размера до желательной величины. Полученные микрофлюидизированные частицы обычно имеют в основном мономодальное распределение по размерам от примерно 1 мкм до примерно 40 мкм, предпочтительно от примерно 5 мкм до примерно 20 мкм и более предпочтительно примерно 5 мкм до примерно 15 мкм.
Кремнеземы с большой площадью поверхности, известные в данной области техники, склонны к образованию плотных, часто агломерированных, суспензий в воде, которые обладают вязкостью от примерно 70 сантипуаз до примерно 80 сантипуаз при комнатной температуре при концентрации кремнезема от примерно 15 масс.% до примерно 20 масс.%. Суспензии, имеющие величины вязкости в этом интервале, обычно менее предпочтительны для носителей с тонким грунтовочным покрытием. В противоположность этому, суспензии микрофлюидизированного кремнезема сходного состава, используемые в данном изобретении, которые имеют уменьшенные размеры частиц с преимущественно мономодальным распределением по размерам, также предоставляют пониженную величину вязкости при комнатной температуре, составляющую менее примерно 50 сантипуаз, 40 сантипуаз, 30 сантипуаз, 20 сантипуаз, 10 сантипуаз или даже менее примерно 5 сантипуаз, что делает такие суспензии с низкой вязкостью особенно пригодными для использования при формировании тонкого грунтовочного покрытия на монолитном носителе с большой площадью поверхности вследствие облегчения доступа в поры и ячейки или каналы носителя без агрегирования. Кроме того, обычные золи, содержащие коллоидный кремнезем, как правило, требуют использования системы водного носителя, также содержащей горючий органический растворитель, например метиловый или этиловый спирт, чтобы уменьшить вязкость в степени, достаточной для обеспечения гомогенного осаждения золя на носитель. Однако использование микрофлюидизированного кремнезема в комбинации с коллоидным кремнеземом в соответствии с данным изобретением предоставляет композицию тонкого грунтовочного покрытия с достаточной низкой вязкостью, чтобы можно было устранить необходимость использования органического растворителя в водном носителе. Преимуществом при этом является то, что устранение горючих растворителей из водного носителя уменьшает неконтролируемое выделение в атмосферу летучих органических соединений (VOC) и обеспечивает устранение необходимости для капитального оборудования соответствовать классу II Электротехнических правил и норм и использовать дополнительные агрегаты для предотвращения неконтролируемого выделения в атмосферу летучих органических соединений.
Композицию микрофлюидизированного и коллоидного кремнезема для тонкого грунтовочного покрытия приготавливают объединением водной суспензии микрофлюидизированного кремнезема с водной суспензией коллоидного кремнезема. Суспензия микрофлюидизированного кремнезема обычно содержит от примерно 5 масс.% до примерно 30 масс.%, предпочтительно от примерно 5 масс.% до примерно 25 масс.% и более предпочтительно от примерно 5 масс.% до примерно 20 масс.% кремнезема, а суспензия коллоидного кремнезема обычно содержит от примерно 10 масс.% до примерно 50 масс.%, предпочтительно от примерно 15 масс.% до примерно 45 масс.% и более предпочтительно от примерно 20 масс.% до примерно 40 масс.% кремнезема. Массовое соотношение микрофлюидизированного кремнезема и коллоидного кремнезема в композиции тонкого грунтовочного покрытия обычно составляет от примерно 2:1 до примерно 1:2, предпочтительно, как правило, от примерно 1:1 до примерно 1:1,5. Композиция тонкого грунтовочного покрытия может содержать дополнительные опциональные компоненты, такие как неионогенные поверхностно-активные вещества, например Triton CF-32, Triton X-102 или Triton 770.
Носители, такие как монолиты, обычно покрывают композицией из микрофлюидизированного и коллоидного кремнезема нанесением окунанием с последующей сушкой при температуре от примерно 100°C до примерно 200°C. Как рассмотрено выше, повторяющиеся этапы нанесения материала покрытия и сушки могут выполняться последовательно для нанесения желательного количества материала. Носитель с нанесенным покрытием затем предпочтительно прокаливают при температуре от примерно 400°C до примерно 800°C, более предпочтительно от примерно 400°C до примерно 600°C и даже более предпочтительно от примерно 400°C до примерно 550°C, чтобы образовать на основе золя связи между носителем и микрофлюидизированным кремнеземом с большой площадью поверхности.
В одном варианте осуществления на прокаленный носитель с микрофлюидизированным кремнеземом, обладающим большой площадью поверхности, может быть опционально нанесено второе тонкое грунтовочное покрытие; при этом на поверхность носителя осаждают, например, цирконилхлорид (ZrOCl2) или коллоид оксида циркония (такой как коллоиды с размером частиц 50 нм и 100 нм, поставляемые Nyacol).
Формирование активной фазы из оксида рутения на носителе
Предшественники катализаторов на носителе по данному изобретению могут быть приготовлены объединением или контактированием с носителем, на котором опционально сформировано тонкое грунтовочное покрытие, с раствором соединения, являющегося предшественником оксида рутения, с последующей стадией осаждения, на которой твердотельный предшественник катализатора на основе оксида рутения, предпочтительно содержащий аморфный гидрат оксида рутения, осаждают на поверхностях носителя. После опциональной сушки предшественник катализатора может быть затем активирован нагреванием в подходящей окислительной атмосфере, содержащей кислород и/или SO2, чтобы образовать активную фазу, содержащую оксид рутения.
В альтернативном варианте осуществления предшественники катализатора на носителе могут быть приготовлены объединением или контактированием (например, пропитыванием) носителя с раствором соединения, являющегося предшественником оксида рутения, предпочтительно содержащем соль рутения, такую как хлорид рутения или нитрат нитрозила рутения, или трехъядерные карбоксилатные соединения рутения, такие как ацетат μ3-оксогексакис(μ-ацетато)триакватрирутения (Ru3O(O2CCH3)6(H2O)3(CH3CO2), посредством чего покрывают или увлажняют носитель и наносят на носитель соединение предшественника из раствора. В этом варианте осуществления скорее не осаждают предшественник катализатора на основе оксида рутения из раствора предшественника на поверхность носителя катализатора, а носитель катализатора, на который нанесено соединение, являющееся предшественником оксида рутения, отделяют от раствора предшественника и подвергают дальнейшей обработке, чтобы образовать активную фазу, содержащую оксид рутения. После опциональной сушки металлический рутений предшественника катализатора, размещенный на носителе, конвертируют, чтобы сформировать активную фазу, содержащую оксид рутения, нагреванием в подходящей окислительной атмосфере, содержащей кислород и/или SO2. В этом варианте осуществления раствор предшественника может также содержать кислоту, такую как серная кислота, при условии, что предшественник катализатора преобразовывается в гидрат оксида рутения нагреванием в атмосфере, содержащей влажный воздух и/или воздух и пар, и на последующем этапе нагревания формируется активная фаза, содержащая безводный оксид рутения. Без намерения установления связи с какой-либо конкретной теорией полагают, что когда водный раствор соединения, являющегося предшественником оксида рутения, такого как ацетат μ3-оксогексакис(μ-ацетато)триакватрирутения, приготавливают в присутствии серной кислоты, то превалирующими ионами в растворе становятся Ru3O(O2CCH3)6(H2O)3+, поскольку ацетатный лиганд протонируется в качестве уксусной кислоты. Посредством пропитывания носителя раствором с Ru3O(O2CCH3)6(H2O)3+ с последующей сушкой комплекс рутения на носителе равномерно распределяется в порах носителя. После сушки на воздухе (например, при температуре примерно 100-140°C) на носителе образуется сульфатная соль [Ru3O(O2CCH3)6(H2O)3]2SO4. При нагревании во влажном воздухе или смеси воздуха с паром (например, при температуре от примерно 200°C до примерно 250°C) ацетатные лиганды постепенно выделяются в виде уксусной кислоты (H2O+-O2CCH3→HO2CCH3+OH-). Остающийся «гидроксильный» лиганд образуется шесть раз для каждого трехъядерного комплекса, что приводит к образованию ионов Ru3O(OH)6+. Полагают, что агломерация на кремнеземистом носителе таких трехъядерных ионов приводит к образованию вытянутой матрицы из номинально водного оксида рутения (RuO2·xH2O). При нагревании такого водного оксида рутения (например, до температуры выше примерно 350-400°C) он преобразуется в безводный RuO2, а SO4 образует H2SO4, которая испаряется с образованием газа (H2O+SO3).
Подходящие растворы соединения, являющегося предшественником оксида рутения, используемые для формирования предшественников катализатора на носителе, в целом описаны здесь выше и приготавливаются растворением соединения, являющегося предшественником оксида рутения, в растворителе в концентрации от примерно 0,01 М до примерно 5 М, в расчете на эффективную единицу количества рутения. Оптимальные концентрации раствора предшественника зависят от площади поверхности и пористости носителя и могут быть легко определены. Если используется носитель, иной чем монолитный носитель (например, с сотовой структурой), такой как носитель с частицами в форме седла или колец, то концентрации предшественника оксида рутения в растворе обычно составляют от примерно 0,01 М до примерно 2 М, предпочтительно от примерно 0,01 М до примерно 1 М и часто от примерно 0,05 до 1 М, в расчете на эффективную единицу количества рутения. Если носитель находится в виде монолита, то концентрация предшественника оксида рутения в растворе обычно несколько выше и составляет, как правило, от примерно 0,1 М до примерно 5 М, предпочтительно от примерно 1,0 М до примерно 3 М, в расчете на эффективную единицу количества рутения.
Раствор соединения, являющегося предшественником оксида рутения, приводят в контакт или объединяют с носителем катализатора и тем самым размещают на нем и/или в нем указанное соединение при использовании любого из нескольких подходящих способов. Как описано выше, носитель может быть, например, порошком, гранулами или иметь другую форму, адаптированную для использования в неподвижных или уплотненных слоях катализатора, или быть монолитным и может опционально иметь тонкое грунтовочное покрытие. В одном варианте осуществления носитель, который способен к образованию теста или суспензии, такой как порошок или гранулы, первоначально объединяют с жидкостью, чтобы образовать суспензию с концентрацией носителя от примерно 1 масс.% до примерно 30 масс.%, предпочтительно от примерно 2 масс.% до примерно 20 масс.%. Предпочтительной является водная суспензия, которая содержит воду. Суспензию носителя и раствор соединения, являющегося предшественником оксида рутения, затем объединяют. В качестве альтернативы порошковый или подобного вида носитель может быть объединен непосредственно с раствором соединения, являющегося предшественником оксида рутения, чтобы образовать тесто или суспензию. Предпочтительная концентрация носителя в образованной суспензии составляет от примерно 5 масс.% до примерно 20 масс.%, более предпочтительно от примерно 10 масс.% до примерно 15 масс.%. Подходящие способы образования теста или суспензии известны специалистам в данной области техники и включают, например, перемешивание, измельчение в мельнице во влажном состоянии, встряхивание, взбалтывание и их комбинации. Носитель после размещения соединения, являющегося предшественником оксида рутения, может быть опционально отделен от раствора данного соединения, чтобы получить влажный носитель с размещенным предшественником.
В другом варианте осуществления монолитные носители, такие как сотовые структуры, и формованные носители катализатора увеличенного размера, такие как кольца и седла, опционально имеющие тонкое грунтовочное покрытие, могут быть погружены в раствор соединения, являющегося предшественником оксида рутения (например, для пропитывания или нанесения покрытия окунанием), что обеспечивает размещение указанного соединения на носителе. Независимо от вида используемого носителя способ и последовательность операций при объединении или контактировании носителя с раствором предшественника не являются критичными, и носитель, или его тесто, или суспензия могут быть добавлены к раствору соединения, являющегося предшественником оксида рутения, или же наоборот. Кроме того, носитель может контактировать с раствором соединения, являющегося предшественником оксида рутения, или пропитан им несколько раз, чтобы обеспечить размещение на носителе желаемого количества соединения. Например, монолитный носитель может быть последовательно несколько раз погружен в раствор предшественника. В случае более пористых носителей их оставляют погруженными в раствор соединения, суспендированными или приведенными в контакт иным образом с таким раствором в течение промежутка времени, достаточного для обеспечения в основном гомогенного нанесения, абсорбции и проникновения раствора предшественника на поверхность носителя и в его поры. Время контактирования носителя или его погружения может значительно варьироваться в зависимости от площади поверхности и пористости носителя и желательной глубины проникновения и обычно составляет по меньшей мере примерно 1 час, 2 часа, 3 часа, 4 часа, 5 часов, 6 часов и вплоть до примерно 24 часов или более.
Как указано выше, в одном варианте осуществления формирование активной фазы из оксида рутения на носителе включает осаждение твердотельного предшественника катализатора на основе оксида рутения, предпочтительно содержащего гидрат оксида рутения, из раствора предшественника на поверхность носителя катализатора. Подходящие способы осаждения описаны здесь выше и включают нагревание и/или регулирование pH раствора предшественника при контактировании с носителем. Как описано выше в связи с приготовлением активной фазы из оксида рутения без носителя, осаждение предшественников гидрата оксида рутения обычно обеспечивает получение катализаторов на основе оксида рутения, обладающих повышенной химической и термической стабильностью. В случае катализаторов на носителе осажденные предшественники гидрата оксида рутения обеспечивают образование прочных связей с расположенным ниже носителем или тонким грунтовочным покрытием, чтобы улучшить стабильность катализатора.
Осаждение предшественника гидрата оксида рутения нагреванием раствора предшественника может быть использовано, когда носитель контактирует с раствором предшественника в виде теста или суспензии, содержащим носитель, или погружением монолитного носителя в раствор предшественника. Предпочтительно использование растворителя или композиции растворителей, содержащих воду. Раствор предшественника, соприкасающийся с носителем, обычно нагревают до температуры от примерно 70°C до примерно 95°C, что приводит к осаждению аморфного гидрата оксида рутения на структуру носителя и/или внутри нее. Предпочтительно раствор предшественника, контактированный с носителем, который обладает способностью к образованию теста или суспензии, перемешивают во время нагревания. Как описано здесь выше, скорость нагревания предпочтительно контролируют, чтобы обеспечить непрерывное и равномерное осаждение высокодиспергированных аморфных частиц малого размера из гидрата оксида рутения, являющегося твердотельным предшественником катализатора. Обычно следует избегать быстрого нагревания, чтобы предотвратить локализованное негомогенное осаждение. Некоторые растворы рутениевого соединения предшественника, такого как хлорид рутения, кислые и имеют величины pH примерно 1 или ниже. После осаждения нагреванием величина pH растворов рутениевого соединения предшественника может быть отрегулирована подходящим основанием, таким как газообразный аммиак, гидроксид аммония, гидроксид натрия или гидроксид калия.
Осаждение предшественника гидрата оксида рутения регулированием pH раствора предшественника может быть использовано, когда носитель контактирует с раствором предшественника в виде теста или суспензии, содержащим носитель, погружением монолитного носителя в раствор предшественника или когда носитель с размещенным предшественником отделен от раствора соединения, являющегося предшественником оксида рутения. Кислый водный раствор предшественника может быть обработан подходящим основанием для нейтрализации (т.е. увеличения pH) и осаждения аморфного гидрата оксида рутения на структуру носителя и/или внутри нее. Как описано выше, используемое основание может быть твердотельным, жидким или газообразным и предпочтительно выбрано из аммиака, гидроксида аммония, гидроксида натрия и гидроксида калия. В случае использования щелочных растворов для регулирования pH такие растворы предпочтительно имеют концентрацию основания более примерно 5% по массе на объем, более предпочтительно по меньшей мере 10% по массе на объем, 15% по массе на объем, 20% по массе на объем или даже 25% по массе на объем. Основание и раствор, содержащий соединение, являющееся предшественником оксида рутения, при контактировании с носителем могут быть объединены при любом порядке добавления. Например, раствор соединения, являющегося предшественником оксида рутения, и носитель могут быть медленно добавлены к концентрированному раствору основания (например, раствору гидроксида аммония) при перемешивании в течение увеличенного промежутка времени, например в течение по меньшей мере 15 минут, примерно 30 минут, примерно 45 минут, примерно 60 минут или более. Перемешивание суспензии продолжают после завершения добавления раствора предшественника в течение по меньшей мере примерно 15 минут, примерно 30 минут, примерно 45 минут, примерно 60 минут, примерно 75 минут, примерно 90 минут или более, чтобы обеспечить осаждение гомогенного предшественника катализатора из гидрата оксида рутения. Температуру во время осаждения предпочтительно поддерживают ниже температуры кипения или температуры флегмы, предпочтительно в интервале от примерно 20°C до примерно 95°C. В другом варианте осуществления концентрированный раствор основания добавляют к раствору соединения, являющегося предшественником оксида рутения, при контактировании с носителем. В еще одном варианте проведения процесса кислая смесь раствора соединения, являющегося предшественников катализатора на основе оксида рутения, и носителя катализатора может быть отрегулирована до нейтральной или щелочной величины pH добавлением газообразного основания, такого как аммиак. Газообразное основание предпочтительно в тех случаях, когда добавление газа выгодно, например при совмещении добавления основания с функционированием обезвоживающего устройства. В еще одном варианте проведения процесса основа с размещенным предшественником может быть собрана в качестве влажного или сухого твердотельного материала, который затем обрабатывают газообразным аммиаком, посредством чего преобразуют рутениевое соединение предшественника в аморфный гидрат оксида рутения. Опционально основной раствор может быть затем пропущен через катализатор.
После осаждения твердотельного предшественника оксида рутения на носитель катализатора размещенный предшественник катализатора отделяют от смеси для осаждения, опционально промывают и затем сушат. По меньшей мере часть высушенного предшественника катализатора может быть в форме агломератов, что приводит к неравномерному распределению частиц по размерам. В этом случае агломераты могут быть разрушены, чтобы получить более однородное распределение частиц катализатора по размерам и тем самым оптимизировать его физические свойства, такие как текучесть, способность к уплотнению и объем пустот в уплотненном виде, а также каталитическую активность. Для деагломерации и уменьшения размера частиц подходят способы уменьшения размера частиц, известные в данной области техники, такие как измельчение в мельнице (например, шаровых мельницах, молотковых мельницах, ротационных мельницах, барабанных мельницах, вибрационных мельницах или струйных мельницах). Предшественник катализатора может быть затем отделен в соответствии с размером частиц, например, просеиванием или классификацией.
Предшественник катализатора на основе оксида рутения на носителе активируют прокаливанием в окислительной атмосфере, чтобы преобразовать данный предшественник и сформировать активную фазу, содержащую оксид рутения, которая обладает желательными свойствами в отношении размера кристаллитов, активности, химической стабильности и термической стабильности. Прокаливание, как описано выше, обычно проводят при температуре от примерно 200°C до примерно 600°C, более предпочтительно от примерно 300°C до примерно 500°C, в течение от примерно 0,5 до примерно 12 часов в атмосфере, содержащей кислород и/или SO2. Прокаливание предшественника катализатора на основе оксида рутения на носителе может опционально быть достигнуто при использовании методик, включающих несколько этапов, проводимых при разной температуре, при повышении температуры между этапами.
Независимо от того, сформирован ли предшественник катализатора на носителе при использовании методик осаждения или нанесения увлажнением, носитель с размещенным на нем предшественником может быть опционально подвергнут восстановительной обработке, как описано выше (например, нагреванием в восстановительной атмосфере или контактированием с восстановителем, например, раствором борогидрида натрия в случае жидкофазной обработки), чтобы способствовать образованию высокодиспергированных и механически прочно сцепленных кристаллитов металлического рутения, которые, когда они подвергаются воздействию окислительной атмосферы при повышенных температурах, преобразуются в катализатор на основе оксида рутения, эффективный для окисления SO2. Такая восстановительная обработка преобразует основную часть соединения рутения, размещенного на носителе, в металлический рутений, в результате чего получают предшественник катализатора на носителе, содержащий металлический рутений. Как указано выше, восстановительная обработка и сопутствующие ей преимущества в отношении размера кристаллитов оксида рутения, каталитической активности и/или срока службы катализатора зависят от соединения, являющегося предшественником оксида рутения, которое используется для формирования предшественника катализатора, и, в частности, такая обработка выгодна тогда, когда твердотельный предшественник оксида рутения сформирован при использовании соли рутения, такой как хлорид рутения или нитрат нитрозила рутения. В вариантах осуществления, в которых используются другие соединения, являющиеся предшественником оксида рутения (например, когда предшественник катализатора на носителе формируют увлажнением носителя раствором трехъядерного карбоксилатного соединения рутения), восстановительная обработка является ненужной и может оказывать неблагоприятное воздействие на эксплуатационные качества катализатора. Вслед за любой восстановительной обработкой металлический рутений предшественника катализатора преобразуют в оксид рутения, чтобы сформировать активную фазу, содержащую оксид рутения, нагреванием в подходящей окислительной атмосфере, содержащей кислород и/или SO2, как описано выше. Опционально нагревание носителя с размещенным предшественником в восстановительной атмосфере и нагревание в окислительной атмосфере могут быть объединены в процесс термообработки, чтобы восстановить осажденное соединение, являющееся предшественником оксида рутения, до металлического рутения и вместе с этим сформировать активную фазу, содержащую оксид рутения.
Промоторы
Промоторы могут быть включены в составы катализаторов, содержащих оксид рутения, по данному изобретению. Промоторы, как полагают, действуют таким образом, что уменьшают размер кристаллитов рутения и посредством этого повышают каталитическую активность и стабильность. Из теоретических соображений, без намерения установления связи с какой-либо конкретной теорией, полагают, что основность определенных активирующих металлов способствует образованию гидрата рутения, который, как, кроме того, полагают, благоприятствует формированию стабильного диоксида рутения с уменьшенным размером кристаллитов. Эти металлы могут, таким образом, действовать как промоторы, или синергисты каталитической активности, и увеличивать каталитическую активность, посредством чего обеспечивается возможность уменьшения количества рутения на катализаторе.
Предпочтительны промоторы на основе металлов с валентностью +4 или +3, более предпочтительно промоторы на основе металлов с валентностью +4. Подходящие металлы промоторов включают цирконий, олово, титан, гафний, свинец, церий, теллур, торий, уран, алюминий и лантан. Промоторы обычно присутствуют в виде оксидов, образованных из водорастворимых металлических солей, и включают ZrO2, SnO2, TiO2, HfO2, PbO2, CeO2, TeO2, ThO2, UO2, Al2O3 и La2O3 и их смеси. Диоксид циркония и оксид олова (например, SnO2) особенно предпочтительны. Любая растворимая соль металла промотора подходит для использования при изготовлении катализатора по данному изобретению, например оксиды, гидроксиды, галогениды, галогенсодержащие кислоты, оксикислоты, соли неорганических кислот и координационные комплексы, такие как тетраммингалогениды. Предпочтительные соединения металла промотора обычно имеют растворимость в воде по меньшей мере примерно 10% по массе на объем, более предпочтительно по меньшей мере примерно 25% по массе на объем и еще более предпочтительно по меньшей мере примерно 50% по массе на объем при pH менее примерно 3, например, 2 или даже 1. Подходящие водорастворимые соединения циркония включают гидроксид циркония (Zr(OH)4), цирконилхлорид (ZrOCl2), цирконилнитрат (ZrO(NO3)2), сульфат циркония (Zr(SO4)2), цирконилацетат (Zr(OH)2(C2H3O2)2) и их гидраты. Подходящие водорастворимые соединения олова включают диоксид олова (SnO2), хлорид двухвалентного олова (SnCl2), сульфат двухвалентного олова (SnSO4) и их гидраты.
Массовое соотношение оксида рутения и промотора (в виде оксида) в катализаторе составляет обычно от примерно 10:1 до примерно 1:10, предпочтительно от примерно 5:1 до примерно 1:10, от примерно 4:1 до примерно 1:10, от примерно 3:1 до примерно 1:10, от примерно 2:1 до примерно 1:10 или от примерно 1:1 до примерно 1:10.
Металл промотора может быть включен в катализатор на основе оксида рутения в составе носителя, в качестве тонкого грунтовочного покрытия и/или в качестве компонента активной фазы, содержащей оксид рутения.
В одном варианте осуществления носитель катализатора может быть импрегнирован раствором металлического промотора с концентрацией, например, от примерно 0,1 М до примерно 10 М, от примерно 0,5 М до примерно 5 М или от примерно 1 М до примерно 3 М. Раствор металлического промотора объединяют или приводят в контакт с носителем катализатора в течение времени, достаточного для проникновения раствора в носитель. Носитель с промотором затем отделяют и, при необходимости, выполняют нейтрализацию pH. Например, носитель катализатора может быть объединен с раствором ZrOCl2·8H2O концентрацией 1-3 М с последующей продувкой азотом для удаления избытка раствора и продувкой аммиаком для нейтрализации pH. В случае промоторов, имеющих пониженную растворимость при повышенной величине pH, адсорбция металлического промотора в носителе и/или осаждение на носитель в виде слоя или пленки были улучшены увеличением pH посредством добавления основания. Предпочтительные основания включают аммиак, гидроксид аммония, гидроксид натрия и гидроксид калия.
В другом варианте осуществления металлический промотор может быть нанесен на носитель совместно с композицией тонкого грунтовочного покрытия или в качестве ее компонента. В этом варианте осуществления приготавливают раствор промотора (как описано здесь выше). Раствор промотора затем объединяют с суспензией материала тонкого грунтовочного покрытия, такого как кремнезем с большой площадью поверхности. Промотор с материалом тонкого грунтовочного покрытия затем наносят на носитель, чтобы сформировать покрытие с большой площадью поверхности, содержащее равномерно распределенный в нем металлический промотор.
В другом варианте осуществления металлический промотор может быть нанесен на поверхность носителя с тонким грунтовочным покрытием (например, в качестве верхнего слоя покрытия). В этом варианте осуществления приготавливают раствор промотора с концентрацией от 0,1 М до 10 М, от 0,5 М до 5 М или от 1 М до 3 М. Раствор металлического промотора объединяют или приводят в контакт с носителем, имеющим тонкое грунтовочное покрытие, и выдерживают в течение времени, достаточного для проникновения раствора в слой пористого тонкого грунтовочного покрытия. При необходимости может быть выполнена нейтрализация pH.
В еще одном варианте осуществления раствор промотора (как описано здесь выше) может быть объединен с раствором рутениевого соединения предшественника (как описано здесь выше) и нанесен на носитель или носитель с тонким грунтовочным покрытием. В качестве альтернативы соединение металлического промотора может быть растворено в растворе соединения, являющегося предшественником оксида рутения. В этом варианте осуществления промотор и рутений совместно осаждают или наносят совместно иным образом на поверхность носителя или носителя с тонким грунтовочным покрытием.
Использование катализатора на основе оксида рутения
Катализаторы на основе оксида рутения в соответствии с данным изобретением обычно применимы в процессах каталитического окисления SO2 до SO3. Такие процессы включают контактирование исходной газовой смеси, содержащей SO2 и кислород, с описанным здесь катализатором на основе оксида рутения, чтобы получить конвертированный газ, содержащий SO3. Описанные здесь катализаторы на основе оксида рутения особенно подходят для окисления SO2 в исходной газовой смеси с концентрацией газообразного SO2 не более примерно 2%, предпочтительно не более примерно 1,5% и даже более предпочтительно не более примерно 1%, 0,9%, 0,8%, 0,7% или менее. Предпочтительно температура исходной газовой смеси, содержащей SO2, и концентрация газообразного SO2 таковы, что исходная газовая смесь приводится в контакт c катализатором на основе оксида рутения при температуре не более примерно 400°C, более предпочтительно от примерно 300°C до примерно 400°C, более предпочтительно от примерно 325°C до примерно 400°C и даже более предпочтительно от примерно 350°C до примерно 375°C. В частном варианте осуществления каталитическая конверсия SO2 в SO3 при использовании катализатора на основе оксида рутения является частью процесса производства серной кислоты контактным способом. Однако катализаторы на основе оксида рутения по данному изобретению, как правило, применимы в любых видах применения, в которых требуется каталитическое окисление SO2 до SO3, особенно в исходных газовых смесях с низкими концентрациями газообразного SO2, и там, где желательна низкая температура каталитического окисления.
Способы производства серной кислоты и/или олеума, известные в данной области техники, обычно включают сжигание сернистого сырья вместе с кислородсодержащим газом в серной печи для образования потока газообразных продуктов сгорания или исходной газовой смеси, содержащей SO2 и кислород, пропускание данного газового потока через конвертер, содержащий несколько каталитических стадий или проходов для нарастающей конверсии SO2 в SO3, рекуперацию тепла экзотермической конверсии в применимой форме посредством охлаждения газового потока, выходящего из каталитических стадий, и пропускание охлажденного газового потока от по меньшей мере одной стадии через зону абсорбции, в которой газовый поток приводят в контакт с водным раствором серной кислоты для удаления SO3 из газовой фазы, чтобы получить в качестве продукта серную кислоту и/или олеум. В конструкции с промежуточной абсорбцией, использующей каталитический конвертер с четырьмя стадиями, SO3 удаляют из газового потока посредством серной кислоты, орошающей абсорбционную колонну, которая расположена за второй каталитической стадией (конструкция IPA 2:2) или за третьей каталитической стадией (конструкция IPA 3:1) конвертера, и газовый поток из зоны промежуточной абсорбции возвращают на последующую стадию из нескольких каталитических стадий перед пропусканием через стадию заключительной абсорбции. На некоторых каталитических стадиях могут быть использованы катализаторы, содержащие активные фазы на основе платины или щелочного металла - ванадия. Газ, поступающий на последнюю каталитическую стадию конвертера, обычно имеет низкую концентрацию газообразного SO2 и температуру в интервале от примерно 360°C до примерно 415°C.
На Фиг. 1 приведена схема технологического процесса производства серной кислоты обычным контактным способом, включающего промежуточную абсорбцию и представляющего собой типичный промышленный вариант осуществления, в котором может быть эффективно использован катализатор на основе оксида рутения по данному изобретению. Невысушенный воздух для сжигания вводят в систему через фильтр 111A, сушилку 111B и компрессор 113. Температуру воздуха для сжигания увеличивают пропусканием через предварительный нагреватель, включающий теплообменник 115 с косвенным обогревом, в котором воздух нагревается непрямым образом, например, посредством передачи тепла от колонны для выпуска абсорбированной кислоты с рекуперацией тепла. Нагретый воздух затем используют для сжигания серы или другого сернистого сырья в серной печи 101. Таким образом, передача тепла в предварительном нагревателе воздуха способствует нагреванию газообразных продуктов сгорания, выпускаемых из серной печи 101. В качестве альтернативы поток SO2 может быть получен из таких источников как этап обжига руды при извлечении металла, и ссылка здесь на обжиг или сжигание сернистого сырья предназначена для включения таких этапов обжига руды или любого другого процесса, в котором сернистое сырье окисляется с образованием газа, содержащего SO2, из которого может быть получена серная кислота. Хотя процесс, представленный на Фиг. 1, включает сушилку для воздуха, используемого для сжигания, следует понимать, что катализатор по данному изобретению может быть использован в производстве серной кислоты при контактировании с влажным газом, в котором газообразные продукты сгорания, содержащие SO2, образованные при использовании невысушенного воздуха для сжигания и содержащие водяные пары в значительной концентрации, подают в конвертер, как это описано, например, в Патенте США № 5130112, все содержание которого включено здесь путем ссылки.
Газообразные продукты сгорания, выпускаемые из серной печи, пропускают через модуль 117 рекуперации отходящего тепла, предпочтительно паровой котел, в котором тепло от газообразных продуктов сгорания передают теплоносителю, например воде или пару, подаваемому в котел. Обычно газообразные продукты сгорания поступают в котел для рекуперации отходящего тепла при температуре примерно 1160°C и выходят из него при температуре выше точки росы. Пар предпочтительно образуют при давлении по меньшей мере примерно 25 бар (избыт.), обычно в интервале от 40 до 60 бар (избыт.). В представленной схеме технологического процесса теплота перегрева передается пару, образованному в котле для рекуперации отходящего тепла, пропусканием пара через пароперегреватели, включающие теплообменники 121 с косвенным теплообменом, в которых пару передается тепло от конвертированного газа, содержащего SO3, который образован в каталитическом конвертере 103.
SO2 в газообразных продуктах сгорания конвертируется в SO3 в конвертере 103, включающем первую 123, вторую 125, третью 127 и четвертую 129 каталитические стадии. В этой конструкции с промежуточной абсорбцией газ от третьей каталитической стадии конвертера направляется в абсорбционную колонну 105 с рекуперацией тепла. Абсорбция выполняется при высокой температуре в данной колонне с рекуперацией тепла, при этом производится серная кислота и выделяется теплота абсорбции. Выпускной газ из абсорбционной колонны с рекуперацией тепла направляется назад в конвертер 103, в котором остаточный SO2 конвертируется в SO3 на четвертой (т.е. последней) каталитической стадии 129. Газ от последней стадии конвертера направляется к последней абсорбционной колонне 109, в которой производится дополнительная серная кислота. Газ, выпускаемый из последней абсорбционной колонны, выводят из системы через вытяжную трубу 131.
Газообразные продукты сгорания, выходящие из модуля 117 рекуперации отходящего тепла и обычно содержащие от примерно 4% до примерно 15% SO2 вместе с исходным кислородом, (т.е. исходная газовая смесь для конверсии), поступают в конвертер и проходят по порядку через первые три каталитические стадии 123, 125 и 127 соответственно, на которых от примерно 94% до примерно 95% SO2 конвертируется в SO3 при использовании катализатора, содержащего, например, активную фазу на основе платины или щелочного металла - ванадия. Если в серную печь 101 не подается в достаточном избытке воздух для сжигания или другой кислородсодержащий газ, то дополнительный воздух или другой кислородсодержащий газ может быть смешан с газообразными продуктами сгорания, чтобы образовать исходную газовую смесь для конверсии. Более конкретно, газ, выходящий из модуля 117 рекуперации отходящего тепла, вводится в первую каталитическую стадию 123 конвертера 103. При конверсии SO2 в SO3 на стадии 123 вследствие экзотермичности реакции образуется значительное количество тепла, по меньшей мере часть которого рекуперируется в пароперегревателе 121, в котором тепло передается от конвертированного газа пару, образованному в модуле 117 рекуперации отходящего тепла, в виде его перегрева. Конвертированный газ, выходящий из пароперегревателя 121, поступает на вторую каталитическую стадию 125 конвертера, на которой дополнительное количество SO2 конвертируется в SO3. Как показано на Фиг. 1, горячий газ, выходящий из второй каталитической стадии, охлаждается в пароперегревателе 121, или же в качестве альтернативы он может быть охлажден передачей тепла газу, возвращаемому на четвертую каталитическую стадию конвертера от абсорбционной колонны 105 с рекуперацией тепла 105, в «горячем» теплообменнике, включающем теплообменник с косвенным обогревом. Охлажденный конвертированный газ от второй стадии проходит через третью каталитическую стадию 127 для дополнительной конверсии SO2 в SO3. Тепло, содержащееся в газе, выходящем от третьей стадии 127, рекуперируется в теплообменнике 107 с косвенным обогревом.
Серная кислоты, выводимая из абсорбционной колонны 105 с рекуперацией тепла, протекает к циркуляционному насосу и в конечном счете выпускается в качестве потока серной кислоты, содержащего основную часть ее произведенного количества. Газ, выпускаемый из абсорбционной колонны 105, вначале проходит через туманоуловитель внутри колонны 105 и затем выпускается из колонны и возвращается в конвертер для дополнительной конверсии SO2 в исходном газе на четвертой стадии, чтобы получить конвертированный газ, содержащий SO3, на последней каталитической стадии 129. Последняя стадия может функционировать по существу изотермическим образом при температуре не более примерно 375°C или адиабатическим образом при максимальной температуре контактирования газа на четвертой стадии с катализатором, предпочтительно не превышающей примерно 400°C.
Исходной газ на последней стадии обычно содержит не более примерно 5%, 4,5%, 4%, 3,5%, 3%, 2,5%, 2%, 1,5%, 1%, 0,9%, 0,8%, 0,7% или менее SO2. Конверсия по меньшей мере 75%, 80%, 85%, 90%, 95%, 99%, 99,5%, 99,6%, 99,7%, 99,8% и даже 99,9% или более SO2 в SO3 может быть достигнута при использовании катализатора на основе оксида рутения по данному изобретению на последней каталитической стадии 129 при температуре конверсии предпочтительно не более примерно 400°C, например от примерно 300°C до примерно 400°C, от примерно 325°C до примерно 400°C или даже от примерно 350°C до примерно 375°C. При этом легко может быть достигнута концентрация SO2 в конвертированном газе на выходе последней каталитической стадии, составляющая менее примерно 500 млн-1 по объему, примерно 400 млн-1 по объему, примерно 300 млн-1 по объему, примерно 200 млн-1 по объему, примерно 100 млн-1 по объему, примерно 90 млн-1 по объему, примерно 80 млн-1 по объему, примерно 70 млн-1 по объему, примерно 60 млн-1 по объему, примерно 50 млн-1 по объему, примерно 40 млн-1 по объему, примерно 30 млн-1 по объему, примерно 20 млн-1 по объему или даже примерно 10 млн-1 по объему. По причине благоприятного соотношения между скоростью конверсии и перепадом давления в предпочтительном варианте осуществления катализатор на основе оксида рутения по данному изобретению, используемый на последней каталитической стадии 129 конвертера 103, является монолитным катализатором, как это описано здесь. Использование монолитного катализатора на основе оксида рутения по данному изобретению обеспечивает существенно более высокую скорость протекания газа через последнюю каталитическую стадию и соответственно существенно меньший диаметр конвертера (в отношении последней каталитической стадии или всех каталитических стадий, если в них используются монолитные катализаторы).
В одном варианте осуществления четвертая каталитическая стадия 129 функционирует адиабатическим образом. В таком варианте осуществления нагревание исходного газа на четвертой стадии и содержание в нем SO2 предпочтительно таковы, что теплота экзотермической реакции при конверсии SO2 в SO3 не увеличивает температуру газа при контактировании с катализатором окисления выше примерно 400°C или даже примерно 375°C.
Найдено, что использование катализаторов на основе оксида рутения по данному изобретению на последней каталитической стадии 129 обеспечивает конверсию SO2 по меньшей мере примерно 99,7% от концентрации SO2 на входе первой стадии 123 при более низкой загрузке катализатора по сравнению с катализаторами четвертой стадии, известными в данной области техники. В частности, сравнение данных в Табл. 2 (ниже) показывает, что различные варианты осуществления катализатора на основе оксида рутения, изложенные здесь, обеспечивают достижение более высокой величины конверсии SO2 по сравнению со сравнительными катализаторами той же самой массы, содержащими цезий, калий и ванадий. Более высокая эффективность конверсии (т.е. активность), связанная с катализаторами на основе оксида рутения по данному изобретению, делает возможным уменьшение слоев катализатора четвертой стадии (т.е. уменьшенную загрузку катализатора), используемых для достижения требуемой конверсии на четвертой стадии, что приводит к снижению стоимости катализатора, повышению скорости газа и уменьшению капитальных затрат.
Конвертированный на четвертой стадии газ, выпускаемый из последней каталитической стадии 129, направляется в последнюю абсорбционную колонну 109 через другой теплообменник 130. Абсорбцию остаточного SO3 выполняют в последней абсорбционной колонне 109 противотоком серной кислоты и газа через зону насадочного абсорбера. Остаточный газ на выходе абсорбционной колонны 109 вначале проходит через туманоуловитель внутри колонны 109 и выпускается из системы через вытяжную трубу 131.
Примеры
Представленные ниже примеры предназначены только для дополнительной иллюстрации и объяснения данного изобретения. Поэтому данное изобретение не должно ограничиваться какими-либо деталями этих примеров.
Эксперименты были выполнены, чтобы оценить приготовление катализаторов на основе оксида рутения и их эффективность при конверсии SO2 в SO3.
В примерах, приведенных ниже, представлено несколько методик приготовления катализаторов, содержащих рутений, для окисления SO2 до SO3. Эти катализаторы могут быть без носителя или поддерживаться кислотоустойчивыми носителями, такими как диоксид циркония, кремнезем и смеси диоксида циркония и кремнезема. С носителем могут быть объединены другие активирующие и стабилизирующие элементы. Рутений может быть добавлен к носителю в виде водного раствора гидрата трихлорида рутения (III) или гидрата нитрата нитрозила рутения, при этом гидратная вода определяется элементным анализом соли рутения. В примерах описаны разные способы «фиксирования» рутения на катализаторе, включающие восстановление в водной среде в присутствии Zr(OH)4, нагревание раствора RuCl3 до температуры от 80 до 90°C в присутствии формованного или порошкового носителя из ZrO2, увлажнение носителя водным раствором RuCl3 с последующей обработкой влажного носителя с RuCl3 безводным аммиаком и обработку высушенного носителя с RuCl3 водородсодержащим газом при температуре от 150°C до 300°C до тех пор, пока в отходящих газах не будет практически полностью отсутствовать HCl. В случае нитрата нитрозила рутения высушенный носитель с RuNO(NO3)3 обрабатывают водородсодержащим газом при температуре от 150°C до 300°C до тех пор, пока в отходящих газах не будет практически полностью отсутствовать HNO3. Дальнейшая активация этих катализаторов может включать нагревание на воздухе или в воздушном потоке, содержащем SO2, до примерно 350°C перед использованием в качестве катализатора. Было найдено, что такие катализаторы обладают высокой активностью при низких температурах, особенно в газовых потоках с низкой концентрацией SO2, которые имеют низкие уровни содержания как SO2, так и O2. Было установлено, что ни ванадиевые, ни платиновые активные фазы, обычные для катализаторов предшествующего уровня техники, не являются особенно эффективными для конверсии с высокой степенью преобразования газовых потоков с низким содержанием SO2 при низких температурах на входе. Катализаторы, содержащие оксид рутения, представленные в описанных ниже примерах, демонстрируют высокие степени конверсии при низких температурах.
Оценка катализаторов проводилась в реакторной системе устройства для термического испытания катализатора на старение (TCAT). Реактор TCAT имеет восемь кварцевых реакторных труб, расположенных по окружности в обычной электропечи, и сконструирован для испытания разных образцов катализатора при одинаковых условиях для окисления SO2 при разных температурах на входе. Каждая труба реактора функционирует при практически изотермических условиях. Для оценки катализатора обычный подаваемый исходной газ смешивали и доставляли при одной и той же объемной скорости потока (100 стандартных кубических сантиметров в минуту (SCCM)) к каждому образцу посредством отдельных регуляторов массового расхода. Образцы газа на входе и выходе анализировали методом газовой хроматографии с калибровочными смесями и определяли конверсию SO2 во входящем газовом потоке. Этот анализ повторяли для каждого образца катализатора при заданной температуре на входе и затем температуру на входе повышали на заданную величину до новой температуры на входе. Температуру поддерживали при примерно 375°C в течение примерно 24 часов после завершения серии конверсий SO2 с приращением температуры на входе для всех образцов в их начальном состоянии. Такая термообработка имитировала короткий период приработки в рабочем режиме, которой катализаторы обычно подвергаются в конвертере при условиях протекания реакции. Температуру затем понижали до минимальной начальной температуры на входе и снова измеряли величины конверсии SO2 для всех образцов. Температуру снова увеличивали для создания набора таких же величин температуры, что и первоначальные температуры, использованные в цикле со свежими образцами, и вновь измеряли величины конверсии SO2. После завершения этого цикла для всех образцов величины конверсии SO2 в цикле с «состаренными» образцами сравнивали с величинами для образцов в начальном состоянии. Наиболее эффективно термически стабилизированными образцами катализатора являлись те из них, которые проявляли наименьшее снижение величины конверсии SO2 между циклами в начальном и состаренном состоянии при различных температурах на входе.
Способ газовой хроматографии (GC) использовали для обнаружения и количественного определения диоксида серы и кислорода во входном и выходном газовых потоках систем TCAT и интегрального реактора. Использовали двухканальный газовый микрохроматограф Agilent модель 200M с использованием гелия в качестве газа-носителя. Каждый из образцов газа на выходе реактора плюс образец на входе направляли в указанный газовый хроматограф через автоматический многопозиционный распределитель (Valco модель 2CSC4MWP). Канал A двухканального анализатора использовали для отделения и обнаружения кислорода, а канал B - для обнаружения диоксида серы. Анализируемые образцы разделяли внутри на два отдельных потока и направляли один из них в канал A, а другой - в канал B. Каждый канал включал инжекционный клапан, хроматографическую колонку и детектор по теплопроводности. Колонка для канала A (O2) имела длину 8 метров и была заполнена молекулярными ситами 5A. Данная колонка отделяла O2 от SO2 и N2 при 60°C, и O2 и N2 затем объединяли с N2, использованным в качестве внутреннего эталона. SO2 удерживали молекулярными ситами колонки до тех пор, пока температура колонки не повышалась до 150°C. Продувку O2 проводили один раз в сутки (в течение ночи). Колонку для канала B (SO2) длиной 4 метра с OV-1701 использовали для отделения SO2 от N2 и O2 при 45°C. SO2 и воздух (O2 и N2) объединяли и воздух использовали в качестве внутреннего эталона.
Для управления газовым хроматографом и определения площади пиков использовали настольный компьютер с программным обеспечением Ezchrom (версия 4.5). Анализатор калибровали при использовании четырех уровней концентрации калибровочного газа. Данные четыре уровня охватывали наибольшую и наименьшую концентрации O2 и SO2, которые ожидались для заданного набора условий реакции. Данные о площади пиков газовой хроматографии вводили в программу заказного программного обеспечения, которая рассчитывала содержание каждого компонента в мольных процентах и степень конверсии SO2 в процентах. Данные затем форматировали и выводили в виде заключительной сводки величин концентрации всех газов и конверсии реактора. Пакет заказного программного обеспечения также контролировал функционирование и синхронизацию многопозиционного распределителя образцов.
Анализ рутения и циркония выполняли способом рентгеновской люминесценции (XRF). Анализ проводили при использовании спектрометра Philips Minipal 2, модель PW 4025 с блоком для смены 12 образцов и системой продувки гелием области между источником энергии (мощность 9 ватт, напряжение в интервале от 1 до 4 кВ и ток в интервале от 1 мкА до 1 мА) и образцом. Для указанных систем компонентов в источнике энергии была использована родиевая рентгеновская трубка с 6 фильтрами для поглощения рентгеновских фотонов, поскольку абсорбция не была равномерной во всем спектральном диапазоне. Образцы предварительно измельчали в микромельнице в течение по меньшей мере 1 минуты, просеивали через сито 100 меш; просеянный порошок загружали в чашку для образца, соединенной одним концом с пропиленовой пленкой толщиной 4 мкм и закрытой на другом конце перед загрузкой чашки с образцом в одну из позиций блока для смены образцов. Для анализа рутениевых катализаторов на носителе были разработаны три системы компонентов, для каждой из которых использовали 30,0 кВ, 8 мкА, серебряный фильтр и время измерения 300 секунд.
Образцы катализатора, содержащие Ru, Si и Zr, анализировали с использованием ряда эталонов. Эталоны устанавливают линейную зависимость числа импульсов счета в секунду для линий Kα люминесценции анализируемого элемента от массового процентного содержания элемента в пределах всего интервала, охватываемого эталонами. Измеренные эталоны затем аппроксимировали прямой линией методом наименьших квадратов в пределах требуемого интервала величин массового процентного содержания элементов. В Табл. 1 приведены название системы компонентов, интервал в масс.% для анализируемого элемента, минимальная и максимальные величины для этого интервала и коэффициент корреляции для линейной аппроксимации методом наименьших квадратов.
Элементные анализы с определением содержания в масс.% в тех примерах, в которых оно выше величин, приведенных в таблице выше, проводили посредством измерения поглощенной массы.
Измерения площади поверхности и объема пор выполнялись компанией Porous Materials, Inc. (PMI), Ithaca, NY, способом Брунауэра-Эмметта-Теллера («БЭТ»). Теория БЭТ физической адсорбции была использована для измерения площадей поверхности одноточечным методом при использовании PMI BET Sorptometer, модель # CBET 201-A. Результаты сообщались в расчете на дегазированные образцы в м2 на грамм образца или в м2/г. Данные ртутной порометрии получали при использовании PMI Mercury/Nonmercury Porosimeter, модель # AMP-60K-A-1_NM. Объемы пор измеряли в кубических сантиметрах (см3) на грамм, см3/г. Распределение пор по объему измеряли в интервале от примерно 29 фунтов на квадратный дюйм абсолютного давления (фунтов на кв. дюйм абс. давл.) до примерно 60000 фунтов на кв. дюйм абс. давл., что соответствовало диаметру пор от 7,3 мкм до примерно 0,0035 мкм.
Размер кристаллитов измеряли способом порошковой рентгеновской дифрактометрии (XRD). Образцы в виде тонких порошков прессовали в чашках для образцов с пленкой Mylar, чтобы удержать порошок в чашках. Чашку с образцом размещали в дифрактометре Scintag PAD II с использованием излучения CuKα, детектора из высокочистого германия, поддерживаемого при 77 K (температуре кипения жидкого азота), и одноканального анализатора. Дебаеграмму порошка сравнивали с файлами порошковой дифракции, сохраненными в The International Centre for Diffraction Data® (ICDD®, http://www.icdd.com/). Материалам с уникальными кристаллическими фазами соответствует номер «файла порошковой дифракции» или PDF. Дебаеграммы материалов из PDF сравнивали с дебаеграммой катализатора, полученной порошковой рентгеновской дифрактометрией.
Исследования аналитической электронной микроскопией высокого разрешения проводили в Unit for Nanocharacterization при Hebrew University of Jerusalem, Израиль. Сканирующую просвечивающую электронную микроскопию (STEM) выполняли с использованием устройства Tecnai F20 G2 (FEI Company, США), функционировавшего при 200 кВ и оборудованного рентгеновским спектрометром с энергетической дисперсией (EDS с использованием прибора EDAX-TSL). Спектрометр EDS функционировал в режиме STEM. Съемку изображений STEM проводили с детектором STEM для получения темнопольных изображений в рассеянных электронах на высоких углах (HAADF), высокоэффективном для формирования изображений с контрастом по атомному номеру (Z).
ПРИМЕР 1
В сравнительном примере 1 оценивался известный катализатор для производства серной кислоты при уменьшенном числе проходов, который содержит смесь солей цезия, калия и ванадия на диатомитовом носителе и поставляется на рынок под торговым наименованием SCX-2000 компанией MECS, Inc. Типичный анализ этого катализатора с высвобождением летучих компонентов дал следующий примерный состав оксидов активной фазы: 11,4% Cs2O; 8,5% K2O и 7,3% V2O5. Этот катализатор является типичным представителем наиболее активных видов коммерчески доступных катализаторов на основе ванадия для производства серной кислоты. Экструдаты SCX-2000 измельчали для получения гранул размером от 2,1 до 2,4 мкм для сравнительного тестирования в реакторе TCAT при использовании 2,6 см3 гранул, масса которых составляла 1,58 г. Величину конверсии SO2 в % в газовом потоке, содержащем 0,5% SO2 и 7% O2, оценивали в реакторе TCAT при разных температурах для катализатора 1 (гранулы SCX-2000 - начальное состояние) и 1A (гранулы SCX-2000 - состаренное состояние), полученные результаты представлены в Табл. 2.
ПРИМЕР 2
45,0 г порошка Zr(OH)4 суспендировали в 400,5 г деионизированной воды. К этой суспензии добавляли 11,9 г RuCl3·1,79H2O, растворенного в 55 г воды. Суспензию нагревали до 84°C в течение 19 мин. По прошествии дополнительных 21 мин суспензию фильтровали через фильтровальную бумагу Whatman #50. При этом отфильтровывали твердотельный темно-серый материал. Влажный осадок размешивали примерно в 350 мл воды и нагревали до 84°C. Величина pH суспензии составляла <1, и ее увеличивали до pH 7,1 при 66°C концентрированным NH4OH. По прошествии 59 мин добавляли 17,7 мл 37%-ного формальдегида 10 порциями. Суспензию затем охлаждали при перемешивании в течение ночи. Суспензию затем повторно нагревали до 83°C при перемешивании. После этого суспензию фильтровали через фильтровальную бумагу Whatman #50. Влажный осадок сушили в течение ночи в вакуумной печи при 120°C. Суммарная масса полученного высушенного порошка составляла 42,6 г.
Высушенный порошок формовали в виде пластин при приложении давления 20 килофунтов/кв. дюйм. Пластины дробили и затем просеивали для получения частиц с размером в интервале между 2,1 и 2,4 мкм. Гранулы затем прокаливали на воздухе в муфельной печи при 200°C в течение 2,5 часов и затем повышали температуру до величины от 370 до 430°C в течение 90 мин. В результате получали зеленовато-серые гранулы общей массой 7,00 г, содержавшие, по данным рентгеновского люминесцентного анализа (XRF), 5,02 масс.% Ru. Порцию гранул объемом 2,6 см3, имевшую массу 3,31 г, загружали в одну из реакторных труб устройства для термического испытания катализатора на старение (TCAT). Величину конверсии SO2 в % в газовом потоке, содержащем 0,5% SO2 и 7% O2, оценивали при разных температурах в реакторе TCAT для катализатора 2 (катализатор примера 2 - начальное состояние) и 2A (катализатор примера 2 - состаренное состояние), полученные результаты представлены в Табл. 2.
ПРИМЕР 3
Четыре грамма гранул диоксида циркония размером 1/8 дюйма (Alfa Aesar # 43815, обычно с площадью поверхности по БЭТ 90 м2/г) погружали в водный 0,1 M раствор (примерно 25 мл) RuCl3·xH2O. Раствор нагревали до 80-90°C для осаждения тонкого покровного слоя водного оксида рутения на поверхности гранул диоксида циркония с проникновением покрытия из водного диоксида рутения в гранулы не более чем на 100 мкм. Покрытые (1Ч) гранулы тщательно промывали водой, после чего погружали в другие 25 мл водного 0,1 M раствора RuCl3·хH2O. Раствор снова нагревали до 80-90°C для осаждения второго тонкого слоя покрытия (2×) из водного диоксида рутения на поверхности гранул диоксида циркония, покрытой водным диоксидом рутения (1×). После промывания этих гранул водой повторяли процедуру нанесения покрытия, чтобы получить гранулы диоксида циркония с покрытием из трижды (3×) нанесенного водного диоксида рутения.
Трижды (3×) покрытые гранулы суспендировали при комнатной температуре в примерно 50 мл воды. Трижды (3×) покрытые гранулы обрабатывали при использовании избытка порошка NaBH4 в количестве примерно 0,2 г, добавляемого непосредственно в воду, в которую были погружены трижды (3×) покрытые гранулы. Покрытые гранулы интенсивно перемешивали в водном растворе NaBH4 и раствор затем декантировали для получения гранул, обработанных борогидридом. Гранулы тщательно промывали водой и сушили в сушильном шкафу с нагнетанием воздуха при температуре выше 100°C. Гранулы затем прокаливали на воздухе при 400°C в течение от 2 до 3 ч. При анализе XRF прокаленных гранул (обозначенных как 3x-RuO2/ZrO2) было найдено 0,454 масс.% Ru. Величину конверсии SO2 в % в газовом потоке, содержащем 0,5% SO2 и 7% O2, оценивали в реакторе TCAT при разных температурах для катализатора 3 (катализатор примера 3 - начальное состояние) и 3A (катализатор примера 3 - состаренное состояние), полученные результаты представлены в Табл. 2.
ПРИМЕР 4
Этот пример иллюстрирует нанесение покрытия из коллоидного кремнезема на частицы порошка Zr(OH)4 после осаждения рутениевой фазы. 45,0 г порошка Zr(OH)4 суспендировали и перемешивали в 439,5 г деионизированной воды в течение нескольких минут. В вибрационную мельницу на 2 л помещали 500 мл среды с диоксидом циркония 3/8 дюйма, к которой добавляли суспензию вместе с 48 г воды. В вибрационной мельнице измельчение шариками выполняли в течение 17 ч. Суспензию Zr(OH)4, измельченного в мельнице, смывали через ситовую сетку в лабораторный стакан на 1 л в суммарном объеме примерно 725 мл.
К суспензии частиц, измельченных в мельнице, добавляли 12,1 г RuCl3·1,79H2O и 50 мл воды. Суспензию нагревали до 90°C на масляной бане. Через промежуток времени в 2,5 ч суспензия становилась серо-зеленой. Суспензию затем фильтровали при примерно 70°C через тонкопористый фильтр из спеченного стекла (600 мл). Отфильтрованный влажный осадок промывали горячей водой для удаления избыточного водного раствора RuCl3.
Влажный осадок и моечную муть объединяли в смесителе с 38,6 г коллоидного кремнезема Nyacol 1440 (40% кремнезема) и получали примерно 200 мл суспензии. Суспензию смешивали в течение 12 мин, после чего перемещали в вакуумную печь и сушили при температуре выше 100°C в течение ночи.
Масса высушенного твердого остатка составляла 64,31 г. Агломераты просеивали для получения фракции с размером частиц от 10 до 12 меш, масса которой составляла примерно 16,8 г. Гранулы помещали в муфельную печь и нагревали на воздухе следующим образом: при температуре от 173 до 200°C в течение примерно 90 мин, затем при температуре от 355 до 450°C в течение примерно 2,0 часов, после чего охлаждали до комнатной температуры. Масса оливково-зеленых гранул составляла 15,9 г. Порцию гранул объемом 2,6 см3 (2,87 г, содержавших 4,67 масс.% Ru по XRF) загружали в реакторную трубу TCAT. Величину конверсии SO2 в % в газовом потоке, содержащем 0,5% SO2 и 7% O2, оценивали в реакторе TCAT при разных температурах для катализатора 4 (катализатор примера 4 - начальное состояние) и 4A (катализатор примера 4 - состаренное состояние), полученные результаты представлены в Табл. 2.
ПРИМЕР 5
К 98 мл воды в колбе Эрленмейера на 500 мл добавляли 2,40 г RuCl3·1,79H2O и несколько капель поверхностно-активного вещества Triton CF-32. 8,98 г мезопористого ZrO2 (приобретенного в Mesotech Modern Materials Inc.) добавляли к рутениевому раствору в виде 16 порций шпателем. Затем добавляли еще 83 мл воды с последующим добавлением нескольких капель поверхностно-активного вещества CF-32 для улучшения увлажнения порошка. Суспензию энергично перемешивали вручную каждые 20 мин в течение промежутка времени примерно 110 мин при нагревании при 90°C на масляной бане.
Суспензию фильтровали через бумагу Whatman #1 и получали остаток на фильтре, который затем промывали водой. Вторая фильтрация при использовании фильтра тонкой очистки не показала накопления материала. Порошок сушили при 115°C в вакуумной печи и получали порошок серо-кремового цвета массой 7,7 г. Этот порошок прокаливали на воздухе при использовании муфельной печи следующим образом: примерно при 200°C в течение 80 мин, затем при 420°C в течение 4,5 ч. Полученный таким образом прокаленный оливково-зеленый порошок прессовали для образования гранул размером от 10 до 12 меш объемом 2,6 см3 и массой 3,90 г. Анализ XRF прокаленного порошка показал содержание 1,50 масс.% Ru. Величину конверсии SO2 в % в газовом потоке, содержащем 0,5% SO2 и 7% O2, оценивали в реакторе TCAT при разных температурах для катализатора 5 (катализатор примера 5 - начальное состояние) и 5A (катализатор примера 5 - состаренное состояние), полученные результаты представлены в Табл. 2.
ПРИМЕР 6
Типичный пример оксида рутения приготавливали следующим образом. К 148 г воды добавляли 55,9 г RuCl3·1,79H2O (степень гидратации рассчитывали из процентного содержания Ru) и раствор перемешивали в течение примерно 20 мин до полного растворения соли. Раствор гидроксида аммония был приготовлен из 257 г концентрированного NH4OH (28,8% NH3 по химическому анализу), добавленного к 1006 г воды в лабораторном стакане на 2 литра. Раствор RuCl3 добавляли по каплям в течение часа в раствор NH4OH при энергичном перемешивании во время добавления. Раствор затем перемешивали в течение дополнительных 90 минут, после чего перемешивание прекращали. После 15 минут отстаивания раствор фильтровали через тонкопористый фильтр из спеченного стекла на 600 мл. Влажный осадок водного диоксида рутения дважды промывали водой, после чего его в количестве 121,0 г переносили в тигель и сушили в течение ночи в вакуумной печи при 135°C. Масса высушенного твердотельного материала составляла 28,9 г.
Твердотельный материал прессовали при использовании пресса Carver в виде пластин, которые затем дробили и просеивали. Масса фракции частиц с размером в интервале от 2,1 до 2,4 мкм составляла 8,43 г. Гранулы помещали в тигель и прокаливали на воздухе следующим образом: поддерживали при 200°C в течение 45 минут, при 350°C в течение 1 ч и при 400-460°C в течение 45 минут. Масса охлажденных гранул составляла 6,64 г. Порция в 2,6 см3 этих гранул имела массу 3,2 г. Площадь поверхности прокаленного диоксида рутения, определенная по БЭТ, составила 31,2 м2/г. Величину конверсии SO2 в % в газовом потоке, содержащем 0,5% SO2 и 7% O2, оценивали в реакторе TCAT при разных температурах для катализатора 6 (катализатор примера 6 - начальное состояние) и 6A (катализатор примера 6 - состаренное состояние), полученные результаты представлены в Табл. 2.
Данные в Табл. 2 показывают, что наиболее активные катализаторы на основе ванадия (катализаторы 1 и 1A) не проявляют степени конверсии выше 30% SO2 ниже температур от 350 до 375°C. В противоположность этому, катализаторы на основе рутениевой активной фазы (имеющие номера от 2 до 6) проявляют высокую активность при такой низкой температуре как 250°C для диоксида рутения, приготовленного в объемной форме, и при примерно от 300 до 325°C для катализаторов с рутениевой активной фазой на носителе, представленных в примерах с 2 по 5.
ПРИМЕР 7
Анализ порошковой рентгеновской дифрактометрией (XRD) катализатора 6 перед (Фиг. 2, график 2) и после (Фиг. 2, график 1) функционированием в реакторе TCAT показывает, что диоксид рутения наблюдается в виде кристаллической фазы как перед окислением SO2 в реакторе TCAT, так и после него. Для справки рентгеновская дифрактограмма представлена на Фиг. 2 наряду с «линейной рентгенограммой» RuO2, взятой из файла порошковой дифракции (PDF) для образца # 40-1290 (установленная дебаеграмма для RuO2).
Сравнительные дифрактограммы XRD на Фиг. 2 подтверждают, что RuO2 образует кристаллическую фазу, найденную в этом катализаторе для окисления SO2.
ПРИМЕР 8
Представленные ниже два примера иллюстрируют приготовление катализаторов на основе оксида рутения на носителе. Кольцеобразные таблетки размером 5 мм из кремнезема (Nikki Chemical, N601A3, площадь поверхности по БЭТ 264 м2/г) измельчали и просеивали, чтобы получить гранулы размером от 2,1 до 2,4 мкм. Гранулы кремнезема в суммарном количестве 3,4 г помещали в капельную воронку на 125 мл с запорными кранами на обоих концах и пробку из стекловаты над нижним запорным краном. Верхний запорный кран открывали и вакуумировали всю капельную воронку в целом с использованием централизованной вакуумной системы в течение 1,5 ч. Приготавливали раствор RuCl3 (15,1 г RuCl3·2,42H2O в 53,1 мл), содержащий 20 капель раствора поверхностно-активного вещества (приготовленного из 20 капель Triton CF-32, растворенных в 100 мл воды). Раствор RuCl3 втягивали посредством вакуума поверх гранул кремнезема в капельную воронку и оставляли раствор над гранулами на 2,3 ч. Нижний запорный кран открывали и удаляли избыток раствора продувкой азотом, подаваемым через верхний запорный кран. Газообразный азот затем заменяли безводным аммиаком. Нижний запорный кран удаляли и размещали бумагу для определения pH на нижнем выходе капельной воронки. В пределах 10 минут жидкость, удаляемая из гранул, изменяла цвет бумаги на синий (щелочная величина pH), что указывало на прохождение аммиака.
Гранулы извлекали в тигель и сушили в сушильном шкафу с нагнетанием воздуха при 120°C в течение ночи. Тигель затем перемещали в муфельную печь и прокаливали на воздухе при температуре от 200 до 265°C в течение 2 часов, после чего выдерживали между 380 и 440°C в течение дополнительных 2 часов. Порцию этих гранул объемом 2,6 см3 (1,2 г с содержанием 4,5 масс.% Ru по данным анализа XRF) загружали в реакторную трубу TCAT и оценивали в соответствии с установленной процедурой. Величину конверсии SO2 в % в газовом потоке, содержащем 0,5% SO2 и 7% O2, оценивали в реакторе TCAT при разных температурах для катализатора 8 (катализатор примера 8 - начальное состояние) и 8A (катализатор примера 8 - состаренное состояние), полученные результаты представлены в Табл. 3.
ПРИМЕР 9
В этом примере следовали обычной процедуре, использованной в примере 8. Гранулы кремнезема в суммарном количестве 3,4 г помещали в капельную воронку на 125 мл. Верхний запорный кран открывали и вакуумировали всю капельную воронку в целом с использованием централизованной вакуумной системы в течение 45 минут. Раствор ZrOCl2·8H2O (19,4 г в 50 мл воды), содержащий 20 капель раствора поверхностно-активного вещества CF-32 (приготовленного как описано в примере 8), втягивали через нижний запорный кран капельной воронки таким образом, чтобы покрыть им гранулы кремнезема. Гранулы пропитывали этим раствором в течение ночи. Раствор сливали с гранул и удаляли избыток раствора из воронки продувкой азотом, подаваемым через верхний запорный кран. Газообразный азот затем заменяли безводным аммиаком из баллона. Влажную бумагу для определения pH подвергали воздействию газа, выпускаемого через открытый нижний запорный кран. Когда цвет бумаги изменялся от цвета для кислой величины pH на цвет для щелочной величины pH, подачу аммиака прекращали и замещали продувкой азотом.
Приготавливали раствор RuCl3 (15,1 г RuCl3·2,42H2O в 51 мл), содержащий 20 капель раствора поверхностно-активного вещества Triton CF-32. Раствор RuCl3 втягивали посредством вакуума поверх гранул кремнезема в капельную воронку и оставляли раствор над гранулами на 1,2 ч. Нижний запорный кран открывали и удаляли избыток раствора продувкой азотом, подаваемым через верхний запорный кран.
Гранулы извлекали в тигель и сушили в сушильном шкафу с нагнетанием воздуха при 120°C в течение 1,2 ч. Тигель затем перемещали в муфельную печь и прокаливали на воздухе при температуре от 199 до 252°C в течение 1,2 часов, после чего выдерживали между 375 и 452°C в течение дополнительных 2 часов перед охлаждением до комнатной температуры. Порцию этих гранул объемом 2,6 см3 (1,4 г с содержанием 8,7 масс.% Ru по данным анализа XRF) загружали в реакторную трубу TCAT и оценивали. Результаты для этого катализатора приведены в Табл. 3 как для катализатора 9 (в начальном состоянии) и 9A (состаренного).
ПРИМЕР 10
Этот пример описывает приготовление монолитного носителя и катализатора, проявляющего активность при низкой температуре и стабильность. Кусочки монолитного носителя из кремнезема, имеющие примерно 200 ячеек на квадратный дюйм («ячеек/кв. дюйм») (примерно 31 ячейку на квадратный сантиметр), были изготовлены из комбинации: (1) 30 масс.% порошка кремнезема с большой площадью поверхности (Sylox 15, поставляемого W. R. Grace & Co., с площадью поверхности по БЭТ примерно 300 м2/г) и (2) 70 масс.% порошка кремнезема с малой площадью поверхности (площадью поверхности по БЭТ менее 1 м2/г, поставляемого Applied Ceramics, Inc.). Катализатор для оценки в лабораторном реакторе приготавливали при использовании цилиндрических кусочков монолита кремнезема с 200 ячеек/кв. дюйм, которые имели в среднем диаметр 2,3 см, длину 7,4 см и массу 21,6 г.
Кусочки монолита кремнезема с тонким грунтовочным покрытием приготавливали нанесением покрытия окунанием при использовании 15% суспензии (30,7 г) Sylox 15, добавленной к 70 г предварительно гидролизованного раствора тетраэтилортосиликата («TEOS»), этанолу (57,8 г), воде (47,3 г) и 0,14 г концентрированной азотной кислоты. Пять погружений было выполнено для каждого из трех образцов монолита, после чего избыток суспензии сдували струей воздуха. Монолиты со свеженанесенным покрытием сушили в сушильном шкафу с нагнетанием воздуха при 130°C по меньшей мере 2 часа. Высушенные образцы затем прокаливали на воздухе нагреванием до 200°C в течение одного часа, выдерживали по меньшей мере 30 минут, после чего температуру повышали до 550°C и выдерживали в течение 2 часов при этой температуре перед охлаждением до комнатной температуры в течение нескольких часов. Величины поглощения в масс.% для трех извлеченных образцов находились в интервале от 6,9 до 12,2% от массы необработанного монолита кремнезема.
На монолиты кремнезема с тонким грунтовочным покрытием затем размещали рутениевую активную фазу. Вначале образцы монолита помещали в первый стеклянный реакционный сосуд, закрытый сверху притертым стеклянным переходником. На каждой стороне реакционного сосуда находился тефлоновый запорный кран. При закрытом одном запорном кране образцы монолита с покрытием вакуумировали с использованием централизованной вакуумной системы для дегазации в течение 12 минут. Водный 2,95 M раствор RuCl3 приготавливали при использовании соли RuCl3·2,42H2O. Вакуумирование реакционного сосуда использовали для втягивания раствора RuCl3 поверх монолитов. Вакуумирование также использовали для дегазации образцов, погруженных в раствор. По прохождении примерно 10 минут вакуумирование прекращали и использовали газообразный азот для удаления избытка раствора RuCl3. Пропитанные монолиты перемещали в другой стеклянный реакционный сосуд и продолжали продувку азотом. Газообразный азот затем заменяли аммиаком при его расходе 150 стандартных кубических сантиметров в минуту. Подачу аммиака продолжали до тех пор, пока влажная бумага для определения pH на конце реакционного сосуда не становилась синей. Каждый из монолитов, обработанных аммиаком, подвешивали в стальном лабораторном стакане проволокой, обмотанной вокруг монолита. Избыток суспензии удаляли из каждого монолита с покрытием при использовании проволоки для освобождения суспензии из каналов. Влажные монолиты затем размещали в сушильном шкафу с нагнетанием воздуха при 130°C в течение по меньшей мере 2 часов. Высушенные монолиты имели пурпурно-черный цвет при их извлечении из сушильного шкафа. После этого их помещали в муфельную печь и прокаливали на воздухе при 200°C в течение примерно 45 минут и затем в интервале между 385 и 427°C в течение 3 часов.
Охлажденные монолиты с покрытием имели прирост массы в интервале между 16,4 и 17,5%. Один из монолитов с покрытием отбирали для измерений стабильности активности в реакторе TCAT. Данный монолит разрезали на прямоугольные кусочки размером примерно 5 мм по боковой стороне, которые имели примерно два (200 ячеек/кв. дюйм) канала на боковой стороне. Эти маленькие кусочки 5Ч5 мм суммарной массой 1,7 г загружали в объеме примерно 4,5 см3 в кварцевую реакторную трубу между пробками из кварцевой стекловаты. Величину конверсии SO2 в % в газовом потоке, содержащем 0,5% SO2 и 7% O2, оценивали в реакторе TCAT при разных температурах для кусочков катализатора, обозначенных как 10-1 в первом цикле в начальном состоянии, и 10-1A в первом цикле в состаренном состоянии. Данные о величине конверсии в зависимости от температуры для этого катализатора при проведении трех циклов в начальном состоянии (с 10-2 по 10-4) и трех циклов в состаренном состоянии (с 10-2A по 10-4A) представлены в Табл. 4 ниже. Данные о конверсии показывают, что, за исключением снижения активности (89% от активности в начальном состоянии) между первыми циклами в начальном состоянии и состаренном состоянии, кусочки катализатора проявляли стабильную активность в последующих циклах в начальном состоянии и состаренном состоянии.
ПРИМЕР 11
Анализ порошковой рентгеновской дифрактометрией (XRD) катализатора 10 перед функционированием в реакторе TCAT показывает, что диоксид рутения наблюдается в виде кристаллической фазы, образованной после обработки, описанной в Примере 10. Дебаеграмма порошка, представленная на Фиг. 3 (обозначена как 1), расположена над «линейной рентгенограммой» для RuO2.
Три наиболее интенсивных пика (соответствующих рефлексам 110, 101 и 211 при использовании системы обозначений индексами Миллера hkl) имеют ширину (полную ширину на половине высоты, FWHM) в среднем 0,934°, что дает средний размер кристаллитов 92 Å для диоксида рутения, сформированного на поверхности монолитного катализатора.
ПРИМЕР 12
Результаты в отношении активности и стабильности катализатора, полученные в Примере 10, были использованы для изготовления монолитных катализаторов из кремнезема большего размера. Монолиты кремнезема, полученные от Applied Ceramics, Inc., имели плотность ячеек в 200 ячеек/кв. дюйм и такой же состав, что и в Примере 10, и содержали кремнезем с большой и малой площадью поверхности. Для испытаний в интегральном реакторе использовали двенадцать монолитов. Эти 12 монолитов кремнезема имели в среднем диаметр 6,49 см, длину 75,0 см, объем 247,4 см3 и массу 109,8 г. Вследствие толщины стенки из кремнезема, окружающей каждый монолит, и присущей ей пористости монолит с внешней стороны был покрыт тефлоновой лентой перед нанесением тонкого грунтовочного покрытия. Суспензии материала тонкого грунтовочного покрытия были приготовлены в сосудах на 1 литр в виде порций, содержащих раствор для нанесения покрытия (108 г воды, 132 г этанола, 160 г TEOS и 0,32 г концентрированной азотной кислоты, смешанных за 1 час перед приготовлением суспензии) и порошковый кремнезем Sylox 15 (70,1 г). Суспензию помещали в лабораторный стакан для нанесения покрытия на 500 мл, содержащий стержневую магнитную мешалку размером 1,5 дюйма, и энергично перемешивали при погружении монолитов кремнезема с тефлоновой лентой в суспензию. Всего требовалось пять порций для нанесения покрытия на 12 монолитов. Избыток суспензии удаляли из каналов воздушной струей, после чего удаляли тефлоновую ленту. Монолиты сушили в сушильном шкафу с нагнетанием воздуха при температуре по меньшей мере 110°C.
Высушенные монолиты прокаливали в муфельной печи, запрограммированной на температуру 200°C, по меньшей мере 1 час, после чего повышали температуру до 550°C и выдерживали по меньшей мере 2 часа. Прокаленные монолиты в среднем поглощали 18,7 г кремнезема на монолит (среднее увеличение массы 14,5%).
Активную фазу рутения размещали в соответствии с обычными процедурами Примера 10. Монолиты с тонким грунтовочным покрытием из кремнезема были обернуты тефлоновой лентой вокруг внешней оболочки монолита, чтобы предотвратить непосредственное контактирование раствора активной фазы с внешними кремнеземными оболочками монолитов. Монолиты, обернутые лентой, размещали в большом реакционном сосуде, снабженном на каждом конце запорным краном. Сосуд вакуумировали при использовании централизованной вакуумной системы. Вакуумирование использовали для втягивания 3,1 M раствора RuCl3 в сосуд и пропитывали монолит в течение 4 минут этим раствором. Нижний запорный кран открывали и сливали раствор RuCl3 под действием силы тяжести с последующей продувкой азотом, подаваемым через верхний запорный кран на реакционном сосуде. Пропитанный монолит затем размещали в другом реакционном сосуде и пропускали газообразный аммиак через монолит до тех пор, пока влажная красная лакмусовая бумага на противоположном конце резервуара (на выходе запорного крана) не становилась синей от контактирования с парами аммиака. Слабозакрепленный осадок счищали с монолита и заливали монолит 4Н раствором NH4OH. Каналы очищали воздушной струей и, при необходимости, нихромовой проволокой. Затем удаляли тефлоновую пленку с оболочки монолита и каждый монолит укладывали боковой стороной в стальной лоток. Монолит размещали в сушильном шкафу с нагнетанием воздуха при 130°C по меньшей мере на 2 часа. Высушенные монолиты затем прокаливали в муфельной печи на воздухе первоначально при 200°C в течение примерно часа, а затем при 400°C в течение 2 часов. Каждый из 12 прокаленных монолитов проявлял среднее увеличение массы 21,4 г или 14,2%.
ПРИМЕР 13
12 активированных монолитных катализаторов, приготовленных в Примере 12, использовали для оценки в интегральном реакторе при использовании труб из нержавеющей стали диаметром 3 дюйма (7,6 см), которые имели длину 48 дюймов (121,9 см) и были закреплены одним концом на фланце из нержавеющей стали. Фланец предоставлял отверстия для отбора проб газа или регистрации температуры через каждые 3,5 дюйма (8,9 см) вдоль трубы, наряду с переходниками в 1 дюйм (2,5 см), которые соединяли трубу с системой газовых трубопроводов реактора, включавшую три регулятора массового расхода диоксида серы, воздуха (предварительно высушенного колонкой с молекулярными ситами) и азота. В реакторную трубу загружали каждый из 12 монолитных катализаторов при использовании предварительно прокаленной ленты из кремнезема, чтобы обвернуть с внешней стороны каждый из монолитов и обеспечить уплотнение между реакторной трубой (3 дюйма; 7,6 см) и монолитными катализаторами (2,55 дюйма; 9,0 см). Загруженный реактор опускали в трубу с вакуумной рубашкой, которая была нагрета с внешней стороны, чтобы минимизировать теплообмен между внутренним реактором и окружающей средой. Газ, направляемый в реактор, предварительно нагревали до заданной температуры газа на входе.
Монолитный катализатор затем приводили в такое состояние, чтобы моделировать 4-ое прохождение после промежуточной абсорбции в производстве серной кислоты, в котором сырье для первого прохождения содержало 11,7% SO2 и 9,3% O2. При этих условиях 4-е прохождение осуществлялось при подаче газа при 75 станд. линейных футов/мин, что для этого монолитного реактора соответствовало общему расходу газа 76 станд. литров/мин. Газ с расходом 76 станд. литров/мин содержал 0,704% SO2 и 4,54% O2, остальное - N2, и результаты прохождения интегрального реактора при температуре на входе 350°C приведены в Табл. 5.
По сравнению с этим, в случае, когда катализатор SCX-2000 загружали в трубу интегрального реактора 4 дюйма (10,2 см) и оценивали в реакторной системе, описанной в Примере 12, при использовании газа с линейной скоростью 76 станд. линейных футов/мин, содержащего после промежуточной абсорбции 0,689% SO2 и 4,88% O2 (что принято как конверсия 95,00% при первом прохождении газовой композиции с 11,5% SO2 и 9,55% O2) по данным газовой хроматографии, остальной газ - азот, кумулятивная конверсия в 99,67% имела место после 40 дюймов (101,6 см) катализатора, предоставляя 0,046% SO2 и 4,7% O2 по данным анализа газовой хроматографией или 460 млн-1 SO2 при такой высоте загрузки в 40 дюймов.
Результаты в Табл. 5 показывают, что монолитный катализатор обеспечивает выпуски менее 0,010% SO2 или 100 млн-1 на глубине 15 дюймов (38,1 см) монолитного катализатора (0,91 фунтов SO2/стандартных тонн в день (STPD) кислоты при условии производительности завода 2500 STPD), а на глубине 21 дюйм (53,3 см) эмиссия SO2 составляет 0,002% SO2 или 20 млн-1 (0,13 фунтов SO2/STPD кислоты).
ПРИМЕР 14
При использовании монолитных катализаторов по данному изобретению, имеющих рутениевую активную фазу, возможны увеличенные скорости протекания газа, как это показано в следующем примере. Катализатор, приготовленный в Примере 12 и оцененный в Примере 13, оценивали в таком же интегральном реакторе, который описан в Примере 13, однако скорость газа была увеличена от 75 станд. линейных футов/мин до 119 станд. линейных футов/мин. Температуру на входе поддерживали при 350°C при том же самом составе газа (0,7% SO2 и 4,5% O2, остальное - N2), что и в Примере 13. Результаты представлены в Табл. 6.
Результаты в Табл. 6 показывают, что монолитный катализатор обеспечивает выпуски менее 0,008% SO2 или 80 млн-1 на глубине 24 дюйма (61,0 см) монолитного катализатора (0,78 фунтов SO2/стандартных тонн в день (STPD) кислоты при условии производительности завода 2500 STPD), а на глубине 27 дюймов (68,6 см) эмиссия SO2 составляет 0,005% SO2 или 50 млн-1 (0,52 фунтов SO2/STPD кислоты), и, наконец, на глубине катализатора 36 дюймов (91,4 см) эмиссия SO2 достигает уровня 0,001% SO2 или 10 млн-1 (0,13 фунтов SO2/STPD кислоты).
ПРИМЕР 15
Способ нанесения покрытия из водного раствора был разработан для экструдированных керамических монолитов и применялся к группе монолитов кремнезема Sylox 15 c 200 ячеек/кв. дюйм, полученных от Applied Ceramics, Inc. Тонкое грунтовочное покрытие содержало исходный кремнезем с большой площадью поверхности, такой как порошок Sylox 15, полученный от W. R. Grace. В бутыль на 10 литров добавляли 1769,8 г порошка Sylox 15. В эту бутыль с порошком кремнезема добавляли 10031,8 г воды порциями по 1 кг. Суспензию помещали в вальцовую мельницу на несколько часов. Суспензию подавали через процессор Microfluidics модель M-110Y MICROFLUIDIZER с показанием датчика воздушного компрессора 7000 фунтов на квадратный дюйм (изб.). Два модуля размещали для обработки суспензии: вначале модуль с конфигурацией «Z» 200 мкм (модель номер H30Z) и затем модуль с конфигурацией «Y» 75 мкм (модель номер F20Y). Перед пропусканием через MICROFLUIDIZER суспензия 14,9% Sylox 15 имела вязкость примерно 75 сантипуаз (сП) при измерении вискозиметром Брукфильда с использованием шпинделя #1 при комнатной температуре (от 16°C до 22°C). После обработки в MICROFLUIDIZER вязкость понижалась до примерно 5 сП или менее. Измерения размера частиц, проведенные при использовании анализатора размера частиц Beckman Coulter LS 13320 для суспензии кремнезема с вязкостью 75 сП, показали бимодальное распределение частиц по размеру с основным пиком около 10 мкм и вторым пиком около 200 мкм. Средний размер частиц составлял 24,3 мкм. После обработки в MICROFLUIDIZER второй пик исчезал, и в распределении частиц по размеру наблюдался одиночный пик между 10 и 20 мкм при среднем размере частиц 11,3 мкм.
Суспензию с 14,9% микрофлюидизированного Sylox 15 в количестве 2469,6 г смешивали с 1270,2 г коллоидного кремнезема Ludox TMA (34% кремнезема в воде) и 12,3 г смеси поверхностно-активного вещества, состоящей из 20,0 г 95%-ного водного раствора Triton CF-32, 37,35 г Triton X-102, 60,02 г Triton 770 и 853,50 г деионизированной воды. Группу из 13 монолитных носителей Sylox 15 от Applied Ceramics со средним диаметром 6,50 см, средней длиной 7,62 см и средним объемом 248,3 кубических сантиметров (см3) обматывали тефлоновой лентой, используемой для уплотнения трубных резьбовых соединений, размером1/2 дюйма, закрывая внешнюю поверхность каждого монолита на протяжении всей его длины. Монолиты затем погружали для нанесения покрытия в смесь из Sylox 15, Ludox TMA и поверхностно-активного вещества, обдували струей воздуха и сушили в сушильном шкафу с нагнетанием воздуха при 120°C в течение по меньшей мере двух часов. На высушенные монолиты наносили покрытие окунанием второй раз и снова сушили при 120°C. Тефлоновую ленту удаляли и монолиты прокаливали на воздухе при нагревании до 550°C в течение шести часов с промежуточной выдержкой при 200°C перед подъемом температуры и поддержанием при 550°C в течение двух часов. После прокаливания было получено среднее увеличение массы кремнезема в 11,1 масс.% в расчете на конечную массу монолитного катализатора. Монолиты, покрытые кремнеземом, затем погружали в раствор (4 масс.%) ZrO2 с частицами 100 нм (при использовании 20% Nyacol Zr100/20), находящийся в вакуумном эксикаторе. Погруженные образцы обрабатывали при использовании централизованной вакуумной системы в течение по меньшей мере 10 минут, после чего очищали каналы при использовании струи воздуха. Монолиты, импрегнированные при вакуумировании, сушили при 120°C в сушильном шкафу с нагнетанием воздуха и затем прокаливали при нагревании до 400°C на воздухе. Увеличение массы (%) диоксида циркония в расчете на увеличение массы, наблюдавшееся после прокаливания, и конечной массы монолитного катализатора составляло 0,71%.
Приготавливали 0,75 M раствор RuCl3. Монолиты 6,50Ч7,62 см покрывали тонким грунтовочным покрытием из кремнезема и диоксида циркония. Наружную поверхность покрытых монолитов затем обматывали тефлоновой лентой, используемой для уплотнения трубных резьбовых соединений, размером1/2 дюйма, и после этого погружали в раствор RuCl3. Четыре погруженных монолита размещали в вакуумном эксикаторе и вакуумировали при использовании централизованной вакуумной системы в течение примерно 10 минут. Раствор сливали с монолитов, после чего выдували его из каналов при использовании струи воздуха. Образцы сушили при 120°C в течение по меньшей мере 2 часов. После сушки в сушильном шкафу с нагнетанием воздуха удаляли тефлоновую ленту.
Водный RuCl3 размещали нанесением покрытия окунанием при вакуумировании суммарно на 13 монолитах. Монолиты загружали попарно в проточный резервуар из нержавеющей стали при использовании предварительно прокаленной ленты из кремнезема, чтобы обеспечить уплотнение для монолитов при их загрузке в данный резервуар для обработки. По специальному заказу были изготовлены два резервуара для обработки, чтобы обрабатывать монолиты длиной 7,62 см (или катализаторы другой формы). Резервуары имели стандартные 3-дюймовые (7,6 см) трубные фитинги из нержавеющей стали сортамента 40 с одним резьбовым патрубком длиной 6 дюймов (15,2 см) и двумя резьбовыми заглушками. Внутренние размеры резервуара составляли 3,07 дюйма в диаметре × 8 дюймов по длине (7,8×20,3 см), что давало внутренний объем 970 см3. Две заглушки, а также один участок посередине вдоль длины патрубка были просверлены и имели внутреннюю трубную резьбу 1/8 дюйма (3,2 мм). В эти три места ввинчивали три соединителя Swagelok под трубку диаметром 1/16 дюйма и наружной трубной резьбой 1/8 дюйма (1,6×3,2 мм). Два оконечных фитинга использовали в качестве впускного и выпускного отверстий, а центральный фитинг был просверлен для термопары диаметром 1/16 дюйма (1,6 мм), используемой для измерения температуры газа внутри резервуара.
Резервуары, которые содержали расположенные последовательно два монолита из кремнезема, покрытые RuCl3/ZrO2-кремнеземом, размещали параллельным образом в муфельной печи с гибкими трубками 1/16 дюйма (1,6 мм) из нержавеющей стали, проходящими через стенку печи и соединенными с внешними системами для пропускания газа и промывки. Температуру регистрировали цифровым регистратором данных. Поток газообразного азота с расходом примерно 1 стандартный литр в минуту (SLPM) использовали для проверки резервуаров на утечку. Температуру печи устанавливали при 200°C и в оба резервуара подавали с расходом 1,2 станд. литров/мин газовую смесь из 2-3% водорода в газообразном азоте. Пропускание газа для обработки выполняли по меньшей мере 24 часа, после чего контролировали влажной бумагой для определения pH наличие HCl в отходящем газе (определяли кислую величину pH). После обработки в течение ночи газовый поток контролировали на исчезновение в нем HCl. После определения исчезновения HCl резервуары охлаждали и извлекали катализаторы. Извлеченные катализаторы затем размещали в сушильном шкафу с нагнетанием воздуха и нагревали при 200°C в течение ночи (>16 часов) на воздухе. Извлеченные монолиты показывали увеличение массы вследствие размещения фазы, содержащей рутений, по отношению к начальной массе монолитных катализаторов из кремнезема с покрытием из ZrO2-кремнезема. Среднее увеличение массы для этих образцов составляло 4,58%. Анализ XRF образцов катализатора, вырезанных из области каналов монолита (т.е. исключая внешнюю стенку монолита), показал 2,3 масс.% рутения (3,2 масс.% в расчете на RuO2) и 0,34 масс.% циркония (0,46 масс.% в расчете на ZrO2).
После обработки двенадцати монолитов (группами по четыре) с помощью трубчатых ячеек для обработки газом, содержащим водород, при 200°C данные монолитные катализаторы загружали в реактор, описанный в Примере 13. Группу монолитных катализаторов затем использовали для моделирования 4-го прохождения после промежуточной абсорбции в производстве серной кислоты, в котором сырье для первого прохождения содержало 11,7% SO2 и 9,5% O2. При этих условиях 4-ое прохождение осуществлялось при подаче газа при 98,7 станд. линейных футов/мин, что соответствовало общему расходу газа в реакторе 99,6 станд. литров/мин. Газ с расходом 99,6 станд. литров/мин содержал 0,702% SO2 и 4,77% O2, остальное - N2. Результаты испытаний в интегральном реакторе с температурой на входе 375°C представлены в Табл. 7. Выходной газ при высоте слоя 36 дюймов (91,4 см) имел содержание SO2 20 млн-1 (0,13 фунтов SO2/STPD кислоты).
ПРИМЕР 16
В этом примере оценено использование нитратных солей рутения для приготовления катализаторов по данному изобретению.
Для катализатора 11 приготавливали 100 мл водного 0,6 M раствора RuNO(NO3)3 растворением 19,038 г RuNO(NO3)3 от Alfa Aesar в 100 мл воды и добавлением 1 капли Triton CF-32. Кольцеобразные гранулы кремнезема Nikki (6,710 г, размер частиц от 2,1 до 2,4 мкм) помещали в лабораторный стакан на 100 мл, в который добавляли 0,6 M раствор RuNO(NO3)3 в количестве, достаточном, чтобы покрыть гранулы. Лабораторный стакан помещали в вакуумный эксикатор и вакуумировали с использованием централизованной вакуумной системы в течение 6 минут. Гранулы отделяли на стальном сите 20 меш от избытка раствора, после чего перемещали в сушильный шкаф с нагнетанием воздуха для сушки при 120°C.
Катализатор 12 приготавливали следующим образом. Кольцеобразные гранулы кремнезема Nikki с размещенным ZrO2 приготавливали погружением кольцеобразных гранул кремнезема Nikki (с размером частиц от 2,1 до 2,4 мкм) в коллоидный раствор 3 масс.% ZrO2 Nyacol с размером частиц 100 нм (приготовленный разбавлением коллоидного раствора 20 масс.% ZrO2 с размером частиц 100 нм). Кольцеобразные гранулы кремнезема Nikki с размещенным ZrO2 сушили при 120°C в сушильном шкафу с нагнетанием воздуха в течение по меньшей мере двух часов. К 25 мл 0,6 M раствора RuNO(NO3)3 добавляли еще 25 мл воды, получая 0,3 M раствор RuNO(NO3)3. В круглодонную колбу на 50 мл добавляли 7,471 г гранул из 100 нм ZrO2/кремнезема, высушенных при 120°C. Высушенные гранулы из 100 нм ZrO2/кремнезема затем последовательно покрывали вначале 23,438 г 3%-ного коллоидного раствора ZrO2 с размером частиц 50 нм, а затем 11,719 г 0,30 M раствора RuNO(NO3)3 при использовании роторного испарителя в течение двух часов на масляной бане при температуре 70°C. Гранулы отделяли на стальном сите 20 меш от избытка раствора, после чего перемещали в сушильный шкаф с нагнетанием воздуха для сушки при 120°C.
Катализатор 13 приготавливали следующим образом. Кольцеобразные гранулы кремнезема Nikki с размещенным ZrO2 приготавливали погружением кольцеобразных гранул кремнезема Nikki (с размером частиц от 2,1 до 2,4 мкм) в коллоидный раствор 3 масс.% ZrO2 Nyacol с размером частиц 50 нм (приготовленный разбавлением коллоидного раствора 12 масс.% ZrO2 с размером частиц 50 нм). Кольцеобразные гранулы кремнезема Nikki с размещенным ZrO2 сушили при 120°C в сушильном шкафу с нагнетанием воздуха в течение по меньшей мере двух часов. Высушенные гранулы из 50 нм ZrO2/кремнезема затем погружали в 0,3 M раствор RuNO(NO3)3 в вакуумном эксикаторе в течение 6 минут при использовании централизованной вакуумной системы. Гранулы отделяли на стальном сите 20 меш от избытка раствора, после чего перемещали в сушильный шкаф с нагнетанием воздуха для сушки при 120°C.
Катализатор 11, две порции катализатора 12 и катализатор 13 загружали в четыре проволочных корзины из нержавеющей стали 0,75 дюйма × 2 дюйма, каждую закрывали ватой из кварцевых волокон и после этого помещали в трубу с фланцами, имеющую фитинги Swagelok на обоих концах с фланцами. Гранулы первоначально нагревали при 150°C в азоте с 3% H2 в течение 5 часов. Катализаторы с 11 по 13 извлекали из трубы с фланцами и убеждались, что они имеют красно-коричневый цвет. Затем катализаторы возвращали в трубу с фланцами и нагревали до 205°C в азоте с 3% H2 в течение 6 часов. После охлаждения образцов в трубе с фланцами в течение ночи до комнатной температуры гранулы извлекали и находили, что они имеют матовый черный внешний вид при практически полном покрытии гранул этим черным продуктом.
Гранулы, обработанные в смеси 3% H2 с азотом, затем дополнительно активировали в газовом потоке, содержащем смесь азота с 2% SO2 и 5% O2. Гранулы обрабатывали при использовании отдельных проволочных корзин из нержавеющей стали в цилиндрической металлической корзине большего размера с дном в виде сита. Гранулы нагревали при примерно 350°C в течение двух часов.
Дальнейшие оценки проводили при использовании порций 2,6 см3 активированных образцов в реакторах TCAT. Результаты оценок в этих реакторах представлены в Табл. 8 ниже. Катализатор 11 в Табл. 8 является «гранулами RuO2/кремнезем», катализатор 12 является «гранулами RuO2-50 нм ZrO2/100 нм ZrO2/кремнезем», и катализатор 13 является «гранулами RuO2-50 нм ZrO2/кремнезем». Данные для состаренных образцов (375°C в течение 24 часов) с использованием реактора TCAT, полученные при указанных температурах, представлены в колонках, озаглавленных «11A», «12A» и «13A» для катализаторов 11, 12 и 13 соответственно.
Данные порошковой рентгеновской дифрактометрии для катализаторов 11 и 12 представлены в виде дифрактограмм на Фиг. 4. Нижняя дифрактограмма (обозначена как 2) представляет катализатор 11 (порошковые образцы гранул RuO2/кремнезем) и соответствует дебаеграмме порошка RuO2 с размером кристаллитов примерно 100 Å. Верхняя дифрактограмма (обозначена как 1) представляет катализатор 12 (порошковые образцы гранул RuO2-50 нм ZrO2/100 нм ZrO2/кремнезем) и интерпретирована как аморфные фазы, которые не соответствуют фазам RuO2 или ZrO2.
ПРИМЕР 17
Приготавливали водную суспензию с 15 массовыми процентами Sylox 15 и анализировали распределение частиц по размеру при использовании анализатора размера частиц Beckman Coulter LS. Результаты показывают средний размер частиц 21,2 микрона при 90% частиц менее 32 микрон и 50% частиц менее 10 микрон. Вязкость, измеренная при примерно 24°C, составляла примерно 75 сантипуаз.
Суспензию Sylox 15 пропускали при высоком давлении через устройство MICROFLUIDIZER для обработки текучей среды при приложении высоких срезающих усилий при высоком давлении, поставляемого Microfluidics Corporation (Newton, Massachusetts, USA). Результаты показывают средний размер частиц 11,3 микрона при 90% частиц менее 24 микрон и 50% частиц менее 9 микрон. Вязкость, измеренная при примерно 24°C, составляла примерно 5 сантипуаз.
Коллоидные кремнеземы, коммерчески доступные от W.R.Grace and Company (Ludox® TMA и A-30), анализировали в отношении размера частиц при использовании просвечивающей электронной микроскопии (TEM). Статистический анализ изображений образцов, взятых из ряда партий коллоидного кремнезема AS-30, показал средний размер частиц 12-14 нм и стандартное отклонение 3-4 нм. Статистический анализ изображений образцов, взятых из ряда партий коллоидного кремнезема TMA, показал средний размер частиц 22-24 нм и стандартное отклонение 5-6 нм.
ПРИМЕР 18
Альтернативные соединения, являющиеся предшественником оксида рутения, оценивали в отношении использования для приготовления катализаторов на носителе. Для носителя катализатора были выбраны гранулы DAVICAT SIZR 4700 (кремнезем с поверхностью, покрытой диоксидом циркония с содержанием примерно от 1 до 2 масс.% Zr по данным анализа XRF и площадью поверхности по БЭТ более 200 м2/г, полученные от W. R. Grace) размером от 2,1 до 2,4 мкм (гранулы, обеспыленные на ситовой сетке #20 меш после прохождения через ситовую сетку #10 меш). Исходный водный раствор, содержащий Ru3O(O2CCH3)6(H2O)3(CH3CO2) (полученный от Colonial Metals, Elkton, MD, продукт номер 8062, 38,6% Ru по данным химического анализа), который содержал 0,21 M рутения, 145 г 1 M серной кислоты и 0,5 мл Triton CF-32 (полученного от Sigma Aldrich). Исходный раствор разбавляли в четыре раза для получения 0,052 M раствора рутения. Этот разбавленный раствор использовали для пропитывания 15,515 г (примерно 30 см3) гранул DAVICAT SIZR 4700 при вакуумировании с использованием централизованной вакуумной системы в течение 13 минут. Пропитанные глянцевитые черные гранулы извлекали на сито # 20 меш. Гранулы сушили в сушильном шкафу с нагнетанием воздуха при 90°C в течение 17 часов, получая блестящие черные гранулы. Гранулы затем сушили дополнительно в течение промежутков времени по 2 часа при 110°C и 140°C.
Высушенные гранулы, содержащие комплекс Ru3O(O2CCH3)6(H2O)3+, затем загружали в трубчатый резервуар из нержавеющей стали (2,5 см диаметром и 20,3 см длиной), имеющий впускное и выпускное отверстия для обработки газом, между пробками из прокаленной стекловаты. Со стороны верхнего течения трубчатого резервуара подавали с расходом 2,6 станд. литров/мин воздух, увлажненный пропусканием через сосуд с водой при 73°C. Трубчатый резервуар, содержащий гранулы, помещали в печь и нагревали до 205°C и затем пропускали поток влажного воздуха через гранулы в течение периода в 2,5 часа, за который 108 г воды было преобразовано в пар. Гранулы с первоначальной массой 15,249 г извлекали и взвешивали, при этом конечная масса гранул составляла 14,689 г. Извлеченные гранулы были обозначены как катализатор 14. Анализ XRF этих гранул активированного катализатора показал содержание 0,80 масс.% Ru и 0,95 масс.% Zr.
Образец, высверленный из одного из монолитных катализаторов, приготовленных в Примере 12 при использовании гидрата трихлорида рутения в качестве соли, являющейся предшественником оксида рутения, был обозначен как катализатор 15. Данный высверленный монолит имел диаметр 1,56 см и длину 2,49 см и весил 1,93 г. Анализ XRF этого катализатора показал содержание 1,62 масс.% Ru и 0,31 масс.% Zr. Порцию в 1,079 г гранул активированного катализатора 14 и высверленный монолитный катализатор 15 массой 1,93 г загружали в два реактора реакторной системы TCAT. Более жесткие условия были использованы для газового потока (примерно 1% SO2 и 7% O2) и катализаторы подвергали старению при 425°C в течение 20,7 часов. Результаты определения конверсии в зависимости от температуры для этих катализаторов перед старением (14 и 15) и после него (14A и 15A) приведены в Табл. 9.
Сравнение относительных скоростей обработки катализаторами 14 и 15 при примерно 375°C показывает, что гранулы катализатора 14 с более низкой загрузкой Ru обеспечивают величину в 0,220 моля конвертированного SO2/минуту на моль Ru, в то время как высверленный монолитный катализатор 15 обеспечивает величину в 0,094 моля конвертированного SO2/минуту на моль Ru.
ПРИМЕР 19
Чтобы объяснить вариации в скоростях обработки катализаторами 14 и 15 в Примере 18, были выполнены микроскопические исследования образцов катализаторов 14 и 15. В случае катализаторов 14 для исследований электронной микроскопией имелись в распоряжении образцы, идентичные тем, которые были использованы при исследовании посредством TCAT, результаты которого приведены в Табл. 9. В случае катализатора 15 для исследований электронной микроскопией использовали катализатор, который являлся типичным примером монолитного катализатора, приготовленного в Примере 12, и был обозначен как катализатор 15'.
Катализатор 15' анализировали сканирующей просвечивающей электронной микроскопией (STEM) и рентгеновской спектрометрией с энергетической дисперсией (EDS), чтобы исследовать участки катализатора, содержащего диоксид рутения на носителе. Изображения монолитного катализатора 15' на Фиг. 5 и 6 показывают сравнительно большие области диоксида рутения, поддерживаемого на кремнеземе. Яркие участки представляют собой фазу диоксида рутения, поддерживаемую на частицах кремнезема. Элементный анализ EDS одного из ярких участков на Фиг. 5 при расположении луча, указанном кружком (обозначено как 1) на Фиг. 6, представлен на Фиг. 7. Эти данные позволяют сделать вывод о том, что фаза диоксида рутения поддерживается непосредственно на кремнеземе. Фаза диоксида циркония присутствует в малых количествах, позволяя предположить, что ее взаимодействие с диоксидом рутения происходит в межфазной области, и эта фаза не поддерживает диоксид рутения. Изображения монолитного катализатора 15' на Фиг. 8 и 9 представляют собой снимки, полученные просвечивающим электронным микроскопом (TEM), с фазой диоксида рутения (темные области), поддерживаемой на кремнеземе (светлые области). Анализ типичных изображений, таких как то, что представлено на Фиг. 9, позволяет оценить интервал размеров кристаллитов фазы диоксида рутения. Фаза диоксида рутения выглядит на этих изображениях как сравнительно большие вытянутые кристаллиты RuO2 размером в интервале от 200 Å до 1000 Å (от 20 нм до 100 нм).
Фиг. 10 представляет собой изображение STEM катализатора 14 на основе оксида рутения на гранулированном носителе, изготовленного в Примере 18 (после испытания в реакторе TCAT). Элементный анализ EDS для квадратной области (обозначена как 1) на Фиг. 10, выполненный при использовании прибора EDAX-TSL, представлен на Фиг. 11. На основании данного микроскопического анализа катализатор 14 представляется как содержащий высокодиспергированные кристаллиты RuO2, поддерживаемые на кремнеземе. При этом отсутствовали непосредственные свидетельства поддержки на фазе ZrO2. Изображение STEM на Фиг. 10 показывает поверхность, целиком покрытую небольшими кристаллитами RuO2. Элементный анализ EDS, представленный на Фиг. 11, показывает примерно 3 масс.% Zr, что указывает на обогащение поверхности поверхностной фазы, тогда как количество объемной фазы близко к 1-2%. Эти данные позволяют сделать вывод о том, что фаза диоксида рутения поддерживается в основном на кремнеземе. Фаза диоксида циркония присутствует в небольших количествах (3 масс.% Zr), позволяя предположить, что ее взаимодействие с диоксидом рутения происходит в межфазной области. Кроме того, анализ поверхности катализатора 14 (Фиг. 11) показывает, что присутствует примерно от 7 до 12 масс.% серы, по-видимому, в виде поверхностных сульфатных компонентов. Анализ XRF катализатора 14 показал, что он содержал 0,95 масс.% Zr. Фиг. 12 представляет собой типичное изображение TEM с высоким разрешением для катализатора 14, изготовленного в Примере 18 (после испытания в реакторе TCAT). Анализ нескольких таких изображений TEM высокого разрешения позволяет сделать вывод о том, что поверхность покрыта небольшими неупорядоченно ориентированными кристаллитами диоксида рутения размером от 50 Å до 100 Å (от 5 нм до 10 нм) на очень пористой поверхности.
На основании этих данных полагают, что использование комплекса предшественника катализатора Ru3O(O2CCH3)6(H2O)3+ для катализатора 14 приводит не только к меньшему размеру кристаллитов по сравнению с катализатором на основе RuO2, приготовленным с использованием RuCl3·xH2O (катализаторы 15 и 15'), но также и к получению катализатора с повышенной активностью и стабильностью посредством прямой активации воздухом с паром.
Данное изобретение не ограничено представленными выше вариантами осуществления и может быть модифицировано различным образом. Приведенное выше описание предпочтительных вариантов осуществления, включая примеры, предназначено лишь для ознакомления специалистов в данной области техники с изобретением, его принципами и его практическим использованием, так, чтобы специалисты в данной области техники могли адаптировать и применить данное изобретение в его различных видах, которые могут наилучшим образом отвечать требованиям конкретного вида применения.
При ссылке с использованием слова (слов) «содержит/включает» или «содержащий/включающий» в этом описании в целом (включая формулу изобретения ниже), если из контекста не следует иное, эти слова используются на основании и при ясном понимании того, что они должны быть интерпретированы во включительном, а не в исключительном смысле, и что каждое из этих слов следует интерпретировать таким образом при истолковывании этого описания в целом.
Реферат
Изобретения относятся к области химии. Исходную газовую смесь, содержащую диоксид серы и кислород, контактируют с катализатором окисления, содержащим активную фазу на основе оксида рутения, при температуре газа не выше, чем примерно 400°С. При этом получат конвертированный газ, содержащий триоксид серы. Для получения серной кислоты и/или олеума объединяют исходный газ с кислородсодержащим газом, вводят газовую смесь в каталитический конвертер, имеющий несколько последовательных каталитических стадий. Образованный частично конвертированный газ, содержащий триоксид серы и остаточные диоксид серы и кислород, пропускают через по меньшей мере одну последующую каталитическую стадию с катализатором окисления, включающим активную фазу, содержащую оксид рутения с образованием конвертированного газа, содержащего триоксид серы и остаточный диоксид серы. Контактируют данный конвертированный газ с водным раствором, содержащим серную кислоту, для абсорбции находящегося в нем триоксида серы в зоне абсорбции триоксида серы, чтобы получить дополнительное количество серной кислоты и/или олеума и газ, обедненный триоксидом серы, который содержит диоксид серы. Изобретения позволяют повысить эффективность процессов за счет снижения температуры окисления диоксида серы в триоксид. 2 н. и 42 з.п. ф-лы, 12 ил., 11 табл.
Формула
образование исходной газовой смеси для конверсии посредством объединения исходного газа с кислородсодержащим газом;
введение такой исходной газовой смеси для конверсии в каталитический конвертер, имеющий несколько последовательных каталитических стадий, каждая каталитическая стадия содержит катализатор окисления, эффективный для окисления диоксида серы до триоксида серы, и контактирование посредством этого исходной газовой смеси для конверсии с катализатором окисления, содержащимся по меньшей мере на первой каталитической стадии в указанной последовательности таких стадий, чтобы образовать частично конвертированный газ, содержащий триоксид серы и остаточные диоксид серы и кислород;
пропускание частично конвертированного газа через по меньшей мере одну последующую каталитическую стадию в последовательности таких стадий, катализатор окисления, находящийся на данной стадии, включает активную фазу, содержащую оксид рутения, в результате чего остаточный диоксид серы в частично конвертированном газе окисляется до триоксида серы с образованием конвертированного газа, содержащего триоксид серы и остаточный диоксид серы; и
контактирование данного конвертированного газа с водным раствором, содержащим серную кислоту, для абсорбции находящегося в нем триоксида серы в зоне абсорбции триоксида серы, чтобы получить дополнительное количество серной кислоты и/или олеума и газ, обедненный триоксидом серы, который содержит диоксид серы.
Комментарии