Производные тетрагидрофурана, способ их получения и способ борьбы с грибками - RU2079274C1
Код документа: RU2079274C1
Чертежи



Описание
Изобретение относится к новым средствам защиты растений. В частности, изобретение относится к новым производным тетрагидрофурана, к способу их получения и к их использованию для борьбы с грибковыми заболеваниями растений.
Известны из заявки на европейский патент N 151084 производные тетрагидрофурана, обладающие фунгицидной активностью.
Задача заключается в разработке новых соединений более высокой эффективности и более широкого спектра действия. Эта задача решается
с помощью производных тетрагидрофурана формулы I:

в котором
X1 и X2 одинаковые или различные и обозначают водород, галоген, O-C1-C4-алкил, гидрокси, -C1-C4-алкил, ацил, замещенный галогеном, ди(C1-C4-алкил)амино, C1-C4-алкоксиимино; A фенил, замещенный двумя атомами галогена; R1, R2, R3, R4, R5 водород;
а также их оптических изомеров, рацематов и диастереомеров.
Изобретение относится также к солям соединений согласно изобретению. Солевые формы используют в сельском хозяйстве и среди них можно назвать: гидрат хлора, сульфат, оксалат, нитрат или арилсульфонат, а также аддитивные комплексы этих соединений с солями металлов, в частности солями железа, хрома, меди, марганца, цинка, кобальта, олова, магния и алюминия.
Изобретение относится также к способам получения соединений формулы I. Так, соединения формулы I, где X2 соответствует галогену, получают
по способу, заключающему в том, что галогенкетон формулы IIa:

где A фенил, замещенный 2 атомами галогена;
R5 водород, а
z означает атом галогена;
подвергают взаимодействию с металлоорганическим соединением формулы IIb:

где X1, R1 R4 имеют значения, указанные выше; а
M атом металла;
с последующим взаимодействием полученного соединения формулы:
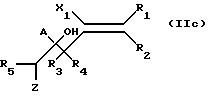
с незамещенным триазолом в среде минерального или органического основания, полученное соединение формулы:

где A, R1 R5, X и Tr принимают вышеуказанные значения, обрабатывают галогеном или смешанным галогеном в инертном растворителе и полученное соединение формулы 11e:

циклизуют в присутствии органического или минерального основания.
Первый этап этого способа, заключающийся во взаимодействии соединения формулы IIa с соединением формулы IIb, осуществляют в растворителе, предпочтительно выбранном из эфиров, например диэтилового эфира, тетрагидрофурана; алифатических, алициклических или ароматических углеводородов, например гексана, толуола, при температуре от -50oC до рефлюкса данного растворителя и при молярном соотношении IIa:IIb, предпочтительно составляющем 1,1 0,2.
Реакцию соединения формулы IIc с незамещенным триазолом осуществляют в присутствии органического или минерального основания, например пиридина, триэтиламина, едкого натра, едкого кали, карбонатов и бикарбонатов щелочных или щелочно-земельных металлов, и в соответствующем растворителе, например в спиртах, кетонах, амидах, нитрилах, ароматических углеводородах, при необходимости галогенированных, при температуре от 80oC до рефлюкса растворителя и при молярном соотношении IIc:триазол, предпочтительно составляющем 1,1 0,2, в результате чего получают соединение формулы IId.
Этап взаимодействия соединения IId с галогеном или со смешанным галогеном осуществляют при добавлении моля на моль используемых реагентов, а в качестве растворителя используют, например, насыщенные или ароматические, при необходимости галогенированные, углеводороды, в результате чего получают соединение IIe.
Циклизацию соединения IIe осуществляют предпочтительно при температуре окружающего воздуха в среде такого органического или минерального основания, которое используют на втором этапе способа, при молярном соотношении соединение IIe: основание, предпочтительно составляющем 1,1 0,66. Реакция может проходить в протонном или апротонном растворителе (вода, или галогенированный ароматический углеводород, диметилсульфоксид, амид, например диметилформамид).
Для получения соединений общей формулы 1, в которой X2 означает QR6, где R6 алкил(C1-C4), а Q означает атом кислорода или серы, а X1 отличается от галогена, полученное выше соединение формулы 1, где X2 означает галоген, подвергают взаимодействию с гетероатомным нуклеофильным соединением формулы R6 Q E, в которой E означает катион, например щелочного или щелочно-земельного металла или четвертичного аммония. Реакцию осуществляют в соответствующем растворителе в присутствии основания и, при необходимости, катализатора фазового перехода и при температуре реакции от -30oC до рефлюкса примененного растворителя, при молярном соотношении реагентов, предпочтительно составляющем 1,2 0,1.
Для получения соединений формулы 1, когда X2 соответствует гидроксильной группе, а X1 отличается от галогена, можно осуществить реакцию в присутствии инертного органического растворителя при температуре от -30oC до рефлюкса растворителя, соединений формулы 1, где X2 галоген, полученных как описано выше, с гидроперекисью, гидроокисью, окисью или перекисью щелочных, щелочно-земельных металлов, или металлов, в молярном соотношении реагентов, предпочтительно составляющем 1,1 0,2.
Соединения формулы I, где X2 означает S- или O-низший алкил, а X1 отличается от галогена, можно также получать путем
взаимодействия соединений формулы III:
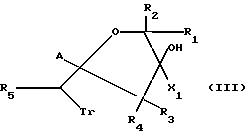
где X1 отличается от галогена, с алкилгалогенидом при молярном соотношении реагентов, предпочтительно составляющем 1,1 и 0,2, предпочтительно в присутствии органического или минерального основания.
Для получения
соединений формулы I, в которой X1 и X2 одновременно означают Q- R6, где R6 означает алкил(C1-C4), а Q означает кислород или серу,
осуществляют реакцию соединения формулы IVa или IV:

в которых X1 означает атом кислорода или серы, связанный двойной связью с тетрагидрофураном, с соединением формулы HQR6, при молярном соотношении, предпочтительно составляющем 1,1 0,2, в присутствии кислотного катализатора и в соответствующем растворителе, которым может быть спирт или сам тиол, или в инертном растворителе, например в углеводородах или спиртах. Если взято соединение формулы IVa, то полученное соединение затем сополимеризуют с триазольным соединением.
Соединения формулы IV или IVa, в которых X1 означает атом кислорода, получают путем окисления
соединения формулы III или IIIa:

где X1 означает атом водорода, известными методами.
Соединения формулы IV или IVa, где X1 означает атом серы, получают реакцией тионизации кетофункции, обрабатывая соединения формулы IV или IVa, где X 0, с помощью, например, H2S или P2S5, при молярном соотношении реагентов, предпочтительно составляющем 1,1 0,2, в присутствии инертного органического растворителя (например пиридина или углеводорода) при температуре от 20oC до рефлюкса.
Соединения формулы I, в которой X2 означает N-R7R8, где R7 R8 ди(C1-C4)-алкил, получают путем взаимодействия соединения формулы I, в которой H2 означает галоген, с амином формулы H-N-R7R8, при молярном соотношении между 1,1 и 0,2, при необходимости в присутствии другого органического или минерального основания.
Соединения формулы I, в которой X1 (или X2) означаетN-R9, где R9 (C1-C4)-алкокси, получают путем взаимодействия соединения формулы IV или IVa с амином формулы R9-NH2 или его солью, при молярном соотношении предпочтительно 1,1 0,2 в инертном органическом растворителе. В случае использования соединения IVa полученное соединение подвергают взаимодействию с триазольным соединением.
Настоящее изобретение относится также к применению соединений формулы I в качестве фунгицидов.
Соединения согласно изобретению могут применяться как для профилактики, так и для борьбы с грибками, в частности с базидиомицетами, аскомицетами, аделомицетами или fungi-imperfecti в особенности с ржавчиной, мучнистой росой, церкоспореллезом, фузариозом, гельминтоспориозом, септориозом, ризоктониозом растений и растительности целом и, в частности, пшеницы, ячменя, ржи, овса и их гибридов, а также риса и кукурузы. Соединения согласно изобретению особенно активно воздействуют на такие грибки, как базидиомицеты, аскомицеты, аделомицеты или fungi-imperfecti, например, Botrytis cinerea, Erysiphe graminis, Puccinia recondita, Piricularia oryzae, Cercospora beticola, Puccinia striiformis, Erysiphe cichoracearum, Fusarium oxysporum (melonis), Pyrenophora avenae, Septoria tritici, Venturia inaequalis, Whetzelinia sclerotiorum, Monilia laxa, Mycosphaerella fijiensis, Marssonina panettoniana, Alternaria solani, Aspergillus niger, Cercospora arachidicola, Cladosporium herbarum, Helminthosporium oryzae, Henicillium expansum, Pestalozzia sp. Phialophora cinerescens, Phoma betae, Phoma foveata, Phoma lingam, Ustilago maydis, Verticillium dahliae, Ascochita pisi, Guignardia bidwellii, Corticium rolfsii, Phomopsis viticola, Sclerotinia sclerotiorum, Sclerotinia minor, Coryneum cardinale, Rhizoctonia solani.
Кроме того, они активно воздействуют также на следующие грибки: Acrostalagmus koningi, Alternaria, Colletotrichum, Corticium rolfsii, Diplodia natalensis, Gaeumannomyces graminis, Gibberella fujikuori, Hormodendron cladosporioides, Lentinus degener или tigrinus, Lenzites quercina, Memnoniella echinata, Myrothecium verrucaria, Paecylomyces varioti, Pellicularia sasakii, Phellinus megaloporus, Polystictus sanguineus, Poria vaporaria, Sclerotium rolfsii, Stachybotris atra, Stereum, Stilbum sp. Trametes trabea, Trichderma pseudokoningi, Trichothecium roseum.
Соединения по изобретению особенно интересны из-за их широкого спектра воздействия при заболеваниях зерновых мучнистой росой, ржавчиной, церкоспореллезом, гельминтоспроиозом, септориозом, фузариозом. Они являются особенно активными в отношении серой плесневидной гнили винограда (Botrytis) и церкоспориозов и поэтому их можно применять для самых разнообразных культур, например виноградной лозы, овощных культур и древоводства и для тропических культур, например арахиса, бананов, кофе, плодов пекана и других.
Кроме вышеуказанных применений вещества согласно изобретению обнаруживают отличную биоцидную активность в отношении многих других разновидностей микроорганизмов, из которых
можно назвать, не ограничивая объема изобретения, следующие грибки:
Pullularia P.pullulans
Chaetomium C.globosum
Aspergillius Aspergillius niger
Coniophora
C.puteana
Вследствие их биоцидной активности вещества по изобретению позволяют эффективно бороться с микроорганизмами, размножение которых создает многочисленные проблемы в сельском хозяйстве
и промышленности. С этой целью они особенно пригодны для защиты растений или промышленных изделий, например древесины, кожи, красок, бумаги, такелажа, пластмассы, системы промышленного
водоснабжения.
Они особенно пригодны для защиты лигноцеллюлозных изделий, в частности деревянных, например мебельных материалов, строительного леса или древесины, подверженной атмосферным воздействиям, например оград, столбов на виноградниках, железнодорожных шпал.
Соединения согласно изобретению, применяемые отдельно или в виде композиций при обработке древесины, обычно используют с органическими растворителями и могут объединяться с одним или несколькими известными биоцидными веществами, например пентахлорфенолом, солями металлов, в частности меди, марганца, кобальта, хрома, цинка, производных неорганических или карбоновых кислот (гептановой, октановой, гексагидробензойной кислот); с органическими комплексными соединениями олова, 2-меркаптобензтиазолом, инсектицидами, например пиретроидами или хлорорганическими соединениями.
Кроме того, они имеют отличную избирательность относительно культур.
Они эффективно применяются в дозах 0,005 5 кг/га, в частности 0,11 1 кг/га.
Наиболее интересным представителем соединений согласно изобретению является соединение формулы:

в виде изомера или эквимолекулярной смеси, которое превосходит по фунгицидной активности известные аналоги.
Примеры I VII иллюстрируют частные способы приготовления соединений по изобретению, а также сами соединения.
Пример 1. Получение 1-[4-бром-2-(2, 4-дихлорфенил)-тетрагидро-2-фуранилметил]-1Н-1,2,4-триазола. Соединения N 1a, 1b и 1a + 1b.
Этап а). Получение 1-хлор-2-(2,4-дихлорфенил)-4-пентен-2-ола.
Готовят магнийорганическое соединение путем добавления раствора аллилбромида (110 мл) в этиловый эфир (700 мл) и тетрагидрофуран (200 мл) с магнием (110 г) при температуре 15 20oC в течение 3 часов. Нагревают с обратным холодильником в течение 30 минут, отстаивают и промывают остаток эфиром.
В эту органическую фазу добавляют при -30oC раствор альфа-трихлор-2, 4-ацетофенона (175 г) в тетрагидрофуране (250 г), нейтрализуют уксусной кислотой. Промывают водой, сушат над сульфатом натрия, концентрируют, затем отгоняют в вакууме. Получают бесцветное масло (205 г). Температура кипения (3•10-2 мм рт. ст.) 140 142oC.
Этап б). Получение 1-[2-(2,4-дихлорфенил)-2- гидрокси-4-пентенил]-1Н-1,2,4-триазола.
Смесь полученного на этапе а) продукта (106 г), триазола (55 г) и карбоната калия (160 г) нагревают в течение 4 часов при 120oC в диметилформамиде (600 мл). Нерастворимые вещества фильтруют, промывают диметилформамидом и реакционную смесь концентрируют в вакууме. Остаток, растворенный в метиленхлориде, промывают водой, затем концентрируют. Продукт получают кристаллизацией в этилацетате после разбавления с гептаном. Изолируют твердое вещество бледно-розового цвета с температурой плавления 101oC.
Этап с). Получение соединений N 1a и N 1b.
Полученное на этапе б) соединение (35 г) в хлороформе (200 мл) обрабатывают бромом при 0oC. После обесцвечивания растворитель испаряют и остаток снова растворяют в метаноле. Затем добавляют водный раствор едкого кали до получения основного pH. После испарения среды в вакууме остаток экстрагируют этилацетатом, промывают водой и концентрируют. Полученное масло (40 г) состоит из смеси двух диастереоизомеров в равном соотношении. Посредством хроматографии на двуокиси кремния последовательно выделяют наименее полярный изомер N 1a: белые кристаллы с температурой плавления 83oC, затем наиболее полярный изомер N 1b: белые кристаллы с температурой плавления 94oC. После перекристаллизации получают 1a с температурой плавления 96oC и 1b с температурой плавления 104oC. Смесь 50/50 1a и 1b имеет температуру плавления 74oC.
Аналогично готовят из соответствующих исходных материалов следующие соединения: 1-(4-бром-2-(4-хлорфенил)-тетрагидро-2- фуранил]-1Н-1,2,4-триазол 1c, т.пл. 74oC, 1d т.пл. 78oC, 1c + 1d т.пл. 69oC.
Пример II. Получение 1-[2-(2, 4-дихлорфенил)-4-гидрокситетрагидро-2-фуранилметил]-1Н-1,2,4 -триазола. Соединения N 2a и 2b.
10 г изомера 1a из примера I, являющегося наименее полярным изомером, растворенного в 30 мл хлорбензола, нагревают с обратным холодильником в течение 48 часов в присутствии 20 г бензоата натрия в 30 мл воды и 1 г катализатора фазового перехода "ADOGEN 464" метилтриалкиламмонийхлорида.
После разбавления эфиром органическую фазу промывают водой и удаляют в вакууме. Остаток обрабатывают при рефлюксе в течение 3 часов в метаноле (100 мл) в присутствии едкого кали (7 г). Охлаждают, разбавляют водой, экстрагируют этилацетатом, промывают до нейтрального состояния и очищают хроматографией полученный сырой продукт после концентрации в вакууме. Спирт 2a выделяют в виде белого порошка (2,8 г) с температурой плавления 193oC.
Производя аналогичные операции с наиболее полярным изомером 1b, полученным в примере I, получают оптически активный спирт 2b, который образуется в виде белого порошка с температурой плавления 162oC. Смесь 50/50 2a и 2b представляет собой масло.
Пример III. Получение 1-[2-(2, 4- дихлорфенил)-4-этокситетрагидро-2-фуранилметил]-1Н-1,2,4-триазола. Соединения N 3a и 3b.
Растворенный в диметилсульфоксиде (12 мл) спирт 2a (2,2 г) обрабатывают последовательно 80%-ным раствором гидрида натрия (0,42 г), затем этилиодидом (1,15 мл). Через 15 минут среду разбавляют водой и экстрагируют этилацетатом. После промывания водой растворитель испаряют и остаток очищают хроматографией на двуокиси кремния и получают бесцветное масло, которое представляет собой изомер 3a, который кристаллизуется растиранием в пентане с температурой плавления 90oC, аналогично из 2b получают изомер 3b в виде белого порошка с температурой плавления 63oC. Смесь 50/50 3a + 3b представляет собой масло.
Аналогично были получены соединения формулы VIII с использованием соответствующих спиртов, сведенных в следующую таблицу.
Пример IV. Получение 1-[1-(2,4-дихлорфенил)-4-этилтиотетрагидро-2-фуранилметил]-1,2,4-триазола. Соединения N 7a и 7b.
Растворенный в диметилсульфоксиде (38 мл) с водой (2 мл) бромил 1a (3,8 г) обрабатывают гидросульфидом натрия (2,8 г) в течение 2 часов. Затем добавляют едкий кали в порошке (3,3 г) и этилиодид (4 мл). Через 10 минут взбалтывания среда разбавляется водой и экстрагируется эфиром. После сушки и испарения получают изомер 7a, представляющий собой масло желтого цвета с температурой плавления (3,9 г) 88oC.
Производя аналогичные операции с 1b, получают 7b в виде порошка бледно-желтого цвета с температурой плавления 64oC. Смесь изомеров представляет собой масло.
Аналогичным образом были получены соединения формулы VIII с использованием соответствующих тиолов, сведенные в следующую таблицу.
Пример V. Получение 1-[7-(2,4-дихлорфенил)-1,4,6-триоксо-спиро[4,4] 7-нонанилметил]-1Н-1,2,4-триазола. Соединение N 13.
Этап а). Получение 1-хлор-2-(2,4-дихлорфенил)-3,4, 5-пентан-триола.
Полученный на этапе а) в примере 1 хлоргидрин (91 г) эпоксидируют в 1,2-дихлорэтане (125 мл) в присутствии ацетилацетоната ванадия (5 г) и перекиси третичного бутила 70% (200 мл) нагреванием с обратным холодильником в течение 48 часов. Охлажденную среду разбавляют водой, неоднократно промывают раствором бисульфита натрия и затем концентрируют. Остаток преобразуют в триол посредством нагревания в воде (200 мл) и диоксане (200 мл) в присутствии хлорной кислоты (5 мл) в течение 3 часов. После растворения в воде среду экстрагируют толуолом (300 мл), затем концентрируют.
Этап б). Получение 2-(2,4-дихлорфенил)-2-хлорметил-тетрагидро-4-фуранона.
Полученный на этапе а) маслянистый остаток нагревают в толуоле (100 мл) и бутаноле (200 мл) в присутствии паратолуолсульфокислоты (0,5 г) с разделением образовавшейся воды. После испарения реакционной среды остаток хроматографируют на двуокиси кремния (элюент этилацетат/гептан 40:60) и получают бесцветное масло (14,5 г), соответствующее смеси диастереоизомеров спирта, которая представляет собой 2-(2,4-дихлорфенил)-4-гидрокси-2-хлорметилтетрагидрофуран. Этот продукт окисляют непосредственно хромовым ангидридом в уксусной кислоте, получая после очищения хроматографией на двуокиси кремния фуранон в виде белых кристаллов с температурой плавления 99oC.
Этап c). Получение 7-хлорметил-7-(2,4-дихлорфенил)-1,4,6-триоксаспиро[4,4]нонана.
Полученный на этапе б) фуранон (4,2 г) в толуоле (50 мл) нагревают с обратным холодильником в присутствии этиленгликоля (6,5 мл) и толуолсульфокислоты (0,1 г) с отделением образовавшейся воды до исчезновения исходного продукта.
Среду промывают едким натром, затем разбавляют водой, экстрагируют эфиром и концентрируют. Получают твердое вещество белого цвета с температурой плавления 99oC.
Этап д). Получение соединения 13.
Галогенид этапа с) (5 г) в диметилсульфоксиде (20 мл) нагревают при 170oC в присутствии триазолилнатрия в течение 6 часов. Среду выливают в воду, экстрагируют этилацетатом, концентрируют и очищают хроматографией на двуокиси кремния. Выделяют бледно-желтые кристаллы (3,6 г) после перекристаллизации в смеси этилацетата и гептана с температурой плавления 123oC.
Пример VI. Получение 1-(4-хлор-2-(2,4-дихлорфенил)-тетрагидро-2-фуранилметил]-1Н-1,2,4-триазола. Соединения N 14a и 14b.
Этап а). Получение 2-(2, 4-дихлорфенил)-1,4,5-трихлор-2-пентанола.
Полученный на этапе а) примера 1 хлоргидрин в растворе с дихлорметаном (150 мл) обрабатывают газом хлора (13,4 г) при -15oC. Затем среду обрабатывают раствором бисульфита натрия 37% (15 мл), промывают водой, сушат и выпаривают. Получают сырой продукт в виде бесцветного масла (49,7 г), содержащего примерно 70% целевого продукта в виде смеси двух диастереоизомеров.
Этап б). Получение 1-(2,4- дихлорфенил)-1-(2,3-дихлор-1-пропанил)оксирана.
Первый способ заключается в получении раствора полученного на предыдущем этапе а) сырого хлоргидрида (10,3 г) в метаноле (30 мл) и обработке его раствором метаноловой гидроокиси калия с титром 2,55 10-3 молей/л (12 мл) при температуре окружающего воздуха. Осадок фильтруют и метаноловый раствор выпаривают. Остаток очищают хроматографией на двуокиси кремния. Получают бесцветное масло (7,4 г).
Второй способ заключается в получении раствора полученного на этапе а) примера 1 хлоргидрина (19,9 г) в метаноле (75 мл) и в обработке его раствором гидроокиси калия (4,9 г) в метаноле (20 мл) при температуре окружающего воздуха. После фильтрования нерастворимых веществ и выпаривания получают эпоксидную смолу (17,1 г) в виде масла желтого цвета. Эпоксидную смолу обрабатывают хлором до получения устойчивого желтого цвета (10,1 г) при -15oC. Затем среду промывают раствором бисульфита натрия, водой, затем выпаривают в вакууме. Получают масло желтого цвета (20,8 г), состоящее из смеси двух диастереоизомеров в соотношении 45/55.
Этап с). Получение 1-[4-хлор-2-(2,4-дихлорфенил)-тетрагидро-2-фуранилметил]-1H-1,2,4-триазола.
Полученный на этапе б) эпоксид (61,7 г) в бутаноле-1 (0,5 л) подвергают нагреванию при 90oC в течение 6 часов в присутствии триазолилнатрия (18,6 г). Минеральный осадок фильтруют и бутанол выпаривают. Остаток очищают хроматографией на двуокиси кремния (элюент 48% этилацетата, 48% гептана и 4% метанола) и последовательно получают первый диастереоизомер 14a с температурой плавления 113oC, затем второй диастереоизомер 14b с температурой плавления 97oC. Смесь 50/50 14a и 14b имеет температуру плавления 90oC.
Пример VII. Получение 1-[4-оксо-2(2, 4-дихлорфенил)-тетрагидро-2-фуранилметил]-1H-1,2,4-триазола. Соединение 15.
Спирт 2a (37,7 г) добавляют при -60oC в раствор диметилсульфоксида (17 мл) в дихлорметане (120 мл), обрабатывают при -60oC раствором трифторуксусного ангидрида (25,4 мл) в дихлорметане (60 мл). После 30 минут при -60oC доводят температуру до температуры окружающей среды, затем добавляют триэтиламин (48 мл). Среду выливают в воду, экстрагируют дихлорметаном и выпаривают. Кристаллизацией в эфире получают порошок белого цвета с температурой плавления 91oC.
Пример VIII. Получение 1-[4,4- дихлор-2-(2,4-дихлорфенил)-тетрагидро-2-фуранилметил]-1H-1,2,4-триазола. Соединение N20.
Смесь кетона 15 (3,1 г), пентахлорида фосфора (2,3 г), дихлорметана (30 мл), содержащего хлорид триэтилбензиламмоний (0,25 г), перемешивают в течение 2 часов при температуре окружающего воздуха до исчезновения исходного продукта. Среду выпаривают, разбавляют водой (100 мл), нейтрализуют бикарбонатом натрия и экстрагируют эфиром. После сушки и выпаривания маслянистый остаток рекристаллизуют в простом изопропиловом эфире (2 раза). Получают порошок белого цвета (0,6 г) с температурой плавления 138oC.
Пример IX. Получение 1-[4-диметиламино-2-(2,4-дихлорфенил)тетрагидро-2-фуранилметил]-1H-1,2,4 -триазола. Соединение N 16.
В раствор гидроокиси калия (0,24 г) и хлоргидрата диметиламина (1,05 г) в метаноле последовательно добавляют кетон из примера III, затем цианоборгидрид натрия (0,24 г). Через 15 часов среду разбавляют водой, экстрагируют эфиром. Целевой продукт экстрагируют из органической фазы хлористоводородной кислотой 6 N (3 x 20 мл). После нейтрализации, экстракции и хроматографии на двуокиси кремния выделяют целевой продукт (1,6 г) в виде масла бледно-желтого цвета (смесь 50/50 двух изомеров).
Пример X. Получение 1-[4-метоксиимино-2-(2, 4-дихлорфенил)тетрагидро-2-фуранилметил]-1H-1,2,4 -триазола. Соединение 17.
Кетон из примера III (2 г) в этаноле (30 мл) нагревают с обратным холодильником в присутствии хлоргидрата метоксиламина (5,8 мл в 25%-ном водном растворе) в течение 2 часов. Среду разбавляют водой, экстрагируют дихлорметаном и выпаривают. Продукт выделяют кристаллизацией из смеси простого диизопропилового эфира и гептана в виде порошка белого цвета с температурой плавления 108oC (смесь двух геометрических изомеров).
Аналогично приготавливают соединения, где R11 H
(4-гидроксимино), т.пл. 195oC 18

Примеры XI и следующие иллюстрируют применение соединений по изобретению в качестве фунгицидов.
В этих примерах разбрызгивание растворов или суспензий действующих веществ осуществляется в таких условиях, чтобы разбрызгивание раствора или суспензии в концентрации, равной 1 г/л, в среднем соответствовало нанесению приблизительно 2 микрограмм действующего вещества на см2 листьев растения.
В условиях примера XI и последующих соединения не проявляли фитотоксичности.
В этих примерах считается, что продукт обеспечивает полную защиту от грибковых заболеваний, когда защита составляет по меньшей мере 95% защита считается хорошей, когда она составляет по меньшей мере 80% (но меньше 95%), как довольно хорошая, когда она составляет по меньшей мере 70% (но меньше 80%), средней, когда она составляет по меньшей мере 50% (но меньше 70%).
В настоящем описании, за исключением противоположных указаний и указаний, касающихся эффективности, проценты представляют собой весовые проценты. Когда проценты выражены относительно стехиометрии, речь идет о молярных процентах. Что касается концентраций, некоторые из них выражены в ppm (часть на миллион), что соответствует мг/л.
Пример XI. Испытание в естественных условиях на Botrytis cinerea на томате.
Посредством тонкого измельчения готовят
водную эмульсию исследуемого действующего вещества следующего состава:
исследуемое действующее вещество 60 г
-Tween 80 (поверхностно-активное вещество), состоящий из олеата
полиоксиэтиленированного производного сорбитана, разбавленный до 10%-ной концентрации водой 0,3 мл
добавляют до 60 мл воды
Затем эту водную эмульсию разбавляют водой для получения
необходимой концентрации.
Разводимые в теплице томаты сорта Марманд в возрасте 30 40 дней обрабатывают разбрызгиванием водных эмульсий (называемые пульпой), описанных выше и имеющих различные концентрации испытываемого соединения. Испытание проводят два раза с каждой концентрацией.
Через 24 и 48 часов листья срезают и помещают в две чашки Петри (диаметр 14 см), на дне которых предварительно уложены влажные фильтрующие бумажные диски (5 листочков в каждой чашке).
Затем инокулят вводят с помощью шприца и наносят по три капли на листочек споровой суспензии. Споровую суспензию Botrytis cinerea получают из культуры в возрасте 15 дней, которую суспендируют в питательном растворе (100000 единиц/см3).
Контроль осуществляют через 3 6 дней после заражения путем сравнения с необработанным контрольным образцом.
В этих условиях наблюдают при дозе 1 г/л хорошую или полную защиту с соединениями 1a, 1b, 1a + 1b, 3b, 5b, 6b, 8b, 9b, 12b, 14a, 14b, 14a + 14b, 17.
Пример XII. Испытание в естественных условиях на Erysiphe graminis на ячмене (мучнистая роса ячменя).
Ячмень в чашках, высеянный в перегнойную почву, обрабатывают на стадии 10 см высотой посредством разбрызгивания водной эмульсии (называемой пульпой) с нижеуказанной концентрацией. Испытание повторяют два раза. Через 24 часа опудривают растения ячменя спорами Erysiphe graminis, причем опудривание осуществляют с помощью больных растений.
Проверку осуществляют через 8 14 дней после заражения. В этих условиях наблюдают следующие результаты: при дозе 1 г/л хорошая или полная защита с соединениями 1a, 1b, 1a + 1b, 2a, 2b, 3a, 3b, 3a + 3b, 4a, 4b, 5a, 5b, 6a, 6b, 7a + 7b, 8a, 8b, 13, 14a, 14b, 14a + 14b, 15, 17, 19, 20.
Пример XIII. Испытание в естественных условиях на Puccina recondita, вызывающую ржавчину пшеницы.
Пшеницу в чашках, высеянную в перегнойную почву, обрабатывают на стадии 10 см высотой посредством разбрызгивания водной эмульсии (называемой пульпой) такого же соединения, как и описанного в примере XI, и с разными концентрациями испытываемого соединения. Испытание проводят два раза с каждой концентрацией.
Через 24 часа водную споровую суспензию (50000 сп/см3) разбрызгивают на пшеницу; эту суспензию получают из зараженных растений. Затем пшеницу выдерживают в течение 48 часов в инкубационной камере при температуре приблизительно 18oC и 100%-ной относительной влажности.
Через эти два дня относительную влажность снижают до 60% Проверку состояния растений осуществляют в промежутке от 11-го до 15-го дня после заражения путем сравнения с необработанным контрольным образцом.
При дозе 1 г/л защита хорошая или полная с соединениями 1a, 1b, 1a + 1b, 3a, 3b, 3a + 3b, 5b, 6b, 7a, 7a + 7b, 8b, 12b, 13, 14a, 14b, 14a + 14b, 17, 19, 20.
Пример XIV. Испытание в естественных условиях на Piricularia oryzae, вызывающий пирикуляриозис риса (Rice Blast).
Рис в чашках, высеянный в смесь 50/50 торфа, обогащенного пуццоланом, обрабатывают на стадии 10 см высотой посредством разбрызгивания вышеуказанной водной эмульсией (называемой пульпой) с концентрацией, указанной ниже. Испытание проводят два раза. Через 48 часов производят обработку путем нанесения на листья суспензии спор, полученных в виде чистой культуры.
Проверку осуществляют через 8 дней после заражения. В этих условиях наблюдают следующие результаты: при дозе 1 г/л наблюдается хорошая или полная защита с соединениями 1a, 1b, 1a + 1b, 3a, 3b, 6a, 6b, 8a, 8b, 9a, 9b, 10b, 11a, 11b, 12a, 12b, 14a, 14b, 14a + 14b, 15, 20.
Пример XV. Испытание в лабораторных условиях на семенных и почвенных грибках.
Исследуют действие соединений согласно изобретению на следующие грибки, вызывающие заболевания
зерновых культур и других растений:
1) Pyrenophorae avenae
2) Septoria nodorum
3) Helminthosporium teres
4) Fusarium roseum
5) Fusarium nivale
6)
Fusarium culmorum
7) Rhizoctonia cerealis
8) Septoria tritici
9) Botrytis cinerea, чувствительный по отношению к карбеназиму и циклическим имидам
10) Botrytis
cinerea, обладающий стойкостью к карбеназиду и циклическим имидам
11) Pseudocercosporella herpotrichoides
12) Fusarium oxysporum F. sp. melonis
13) Rhizoctonia solani
14) Helminthosporium gramineum
Цифры перед названиями будут использованы для обозначения этих грибков в таблице (II).
При каждом испытании осуществляют следующие операции: питательную среду, состоящую из картофеля, глюкозы и агара (среда PDA), вводят в переохлажденном виде в серию чашек Петри (20 мл в чашке) после стерилизации в автоклаве при 120o C.
При заполнении чашек в переохлажденную среду вводят ацетоновый раствор действующего вещества для получения необходимой конечной концентрации.
В качестве контрольных образцов применяют аналогичные предшествующим чашки Петри, в которые выливают аналогичные количества питательной среды, не содержащей действующего вещества.
Через 24 или 48 часов в каждую чашку помещают фрагмент мицелия из указанной выше культуры одного и того же грибка.
Чашки хранятся в течение 2 10 дней (в зависимости от испытываемого грибка) при температуре 22oC и затем сравнивают рост грибков в чашках, содержащих испытываемое действующее вещество, с ростом того же грибка в чашке контрольном образце.
Таким образом, для каждого испытываемого соединения определяют степень подавления соответствующего грибка при дозе 30 ppm. Результаты указаны в нижеследующей таблице 3.
Реферат
Использование: в качестве фунгицидов как для профилактики, так и для борьбы с грибками. Сущность изобретения:
производные тетрагидрофурана формулы

где X1 и X2 - одинаковые или различные и обозначают H, галоген, O-(C1-C4)-алкил, гидрокси, -S-(C1-C4)-алкил, гидрокси, -S-(C1-C4)-алкил, ацил, замещенный галогеном, ди-(C1-C4-алкил)амино, (C1-C4)-алкоксимино, A - фенил, замещенный 2 атомами галогена, R1-R5 - водород. Способ борьбы с грибками путем обработки растений или их биотопа соединением формулы I в виде изомера с температурой пл. 96oC или эквимолекулярной смеси изомеров в количестве 0,11 - 1 кг/га. 3с. и 1 з.п. ф-лы, 3 табл.
Формула

где Х1 и Х2 одинаковые или различные и обозначают водород, галоген, О-(С1-С4)-алкил, гидрокси, S-(С1-С4)-алкил, ацил, замещенный галогеном, ди (С1-С4-алкил)-амино, (С1-С4)- алкоксиимино;
А фенил, замещенный 2 атомами галогена;
R1 R5 водород, а также их оптические изомеры, рацематы и диастереомеры.

где А фенил, замещенный 2 атомами галогена,
R5 водород,
Z атом галогена,
подвергают взаимодействию с металлоорганическим соединением формулы IIв

где Х1, R1 R4 имеют указанные значения;
М атом металла,
с последующим взаимодействием полученного соединения формулы IIc

с незамещенным триазолом в среде минерального или органического основания, полученное соединение формулы IId

где А, R1 R5, Х1 и Тr имеют указанные значения,
обрабатывают галогеном или смешанным галогеном в инертном растворителе и полученное соединение формулы IIе

циклизуют в присутствии органического или минерального основания.

в виде изомера с температурой плавления 96oС или эквимолекулярной смеси изомеров в количестве 0, 11 1 кг/га.
Комментарии