2-амидо-4-арилокси-1-карбонилпирролидиновые производные в качестве ингибиторов серинпротеаз, в частности протеазы ns3-ns4a hcv - RU2440368C2
Код документа: RU2440368C2
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к соединениям, ингибирующим серинпротеазную активность, в частности активность протеазы NS3-NS4A вируса гепатита С. Как таковые, они действуют, препятствуя жизненному циклу вируса гепатита С, и также применимы в качестве противовирусных средств. Изобретение также относится к способам получения таких соединений. Изобретение также относится к композициям, содержащим указанные соединения, или для применения ex vivo, или для введения пациенту, страдающему от заражения HCV. Изобретение также относится к способам лечения инфекции HCV у пациента путем введения композиции, содержащей соединение данного изобретения.
Предпосылки создания изобретения
Заражение вирусом гепатита С ("HCV") является непреодолимой медицинской проблемой человечества. HCV признают причинным фактором большинства случаев "не-А, не-В" гепатита, с оценкой распространенности в сывортке 3% в мировом масштабе [A. Alberti et al., "Natural History of Hepatitis C", J. Hepatology, 31., (Suppl.1), pp.17-24 (1999)]. Приблизительно четыре миллиона индивидуумов могут быть инфицированы вирусным гепатитом только в Соединенных Штатах [M.J. Alter et al., "The Epidemiology of Viral Hepatitis in the United States", Gastroenterol. Clin. North Am., 23, pp.437-455 (1994); M.J. Alter "Hepatitis C Virus Infection in the United States", J. Hepatology, 31., (Suppl.1), pp.88-91 (1999)].
После первого воздействия HCV только примерно у 20% инфицированных индивидуумов развивается острый клинический гепатит, в то время как у других, как оказывается, инфекция устраняется спонтанно. Однако почти в 70% случаев вирус вызывает хроническое инфекционное заболевание, которое длится десятилетиями [S. Iwarson, "The Natural Course of Chronic Hepatitis", FEMS Micrbiology Reviews, 14, pp.201-204 (1994); D. Lavanchy, "Global Surveillance and Control of Hepatitis C", J. Viral Hepatitis, 6, pp.35-47 (1999)]. Оно, как правило, приводит к рецидивирующему и непрерывно ухудшающемуся воспалению печени, которое часто ведет к более тяжелым болезненным состояниям, таким как цирроз и печеночно-клеточный рак [M.C. Kew, "Hepatitis C and Hepatocellular Carcinoma", FEMS Micrbiology Reviews, 14, pp.211-220 (1994); I. Saito et al., "Hepatitis C Virus Infection is Associated with the Development of Hepatocellular Carcinoma", Proc. Natl. Acad. Sci. USA, 87, pp.6547-6549 (1990)]. К несчастью не существует высокоэффективных способов лечения для ослабления развития хронического HCV.
Геном HCV кодирует полипротеин в 3010-3033 аминокислоты [Q.L. Choo et al., "Genetic Organization and Diversity of the Hepatitis C Virus", Proc. Natl. Acad. Sci. USA, 88, pp.2451-2455 (1991); N. Kato et al., "Molecular Cloning of the Human Hepatitis C Virus Genome From Japanese Patients with Non-A, Non B Hepatitis", Proc. Natl. Acad. Sci. USA, 87, pp.9524-9528 (1990); A. Takamizawa et al., "Structure and Organization Hepatitis C Virus Genome Isolated From Human Carriers", J. Virol., 65, pp.1105-1113 (1991)]. Полагают, что неструктурные (NS) белки HCV обеспечивают необходимый каталитический механизм репликации вируса. NS белки образуются при протеолитическом расщеплении полипротеина [R Bartenschlager et. al., "Nonstructural Protein 3 of the Hepatitis C Virus Encodes a Serine-Type Proteinase Required for Cleavage at the NS3/4 and NS4/5 Junctions", J. Virol., 67, pp.3835-3844 (1993); A. Grakoui et. al., "Characterization of the Hepatitis C Virus-Encoded Serine Proteinase: Determination of Proteinase-Dependent Polyprotein Cleavage Sites", J. Virol., 67, pp.2832-2843 (1993); A. Grakoui et. al., "Expression and Indentification of Hepatitis C Virus Polyprotein Cleavage Products", J. Virol., 67, pp.1385-1395 (1993); L. Tomei et. al., "NS3 is a serine protease required for processing of hepatitis C virus polyprotein", J. Virol., 67, pp.4017-4026 (1993)].
NS белок 3 HCV (NS3) содержит серинпротеазную активность, которая способствует процессированию большинства вирусных ферментов, и, таким образом, считается необходимым для репликации и инфекционности вируса. Известно что протеаза NS3 вируса желтой лихорадки понижает инфекционность вируса [Chambers T.J. et al., "Evidence that the N-terminal Domain of Nonstructural Protein NS3 From Yellow Fever Virus is a Serine Protease Responsible for Site-Specific Cleavage in the Viral Polyprotein", Proc. Natl. Acad. Sci. USA, 87, pp.8898-8902 (1990)]. Показано что первые 181 аминокислот NS3 (остатки 1027-1207 вирусного полипротеина) содержат домен серинпротеазы NS3, которая процессирует все четыре сайта в прямом направлении полипротеина HCV [C. Lin et al., "Hepatitis C Virus NS3 Serine Proteinase: Trans-Cleavage Requirements and Processing Kinetics", J. Virol., 68, pp.8147-8157 (1994)].
Серинпротеаза NS3 HCV и ее ассоциативный кофактор NS4A способствуют процессированию всех вирусных ферментов и, таким образом, считаются необходимыми для репликации вируса. Оказывается что такой процессинг аналогичен процессингу, осуществляемому аспартилпротеазой вируса иммунодефицита человека, которая также вовлекается в процессинг вирусных ферментов. Ингибиторы HIV-протеазы, подавляющие процессинг вирусных белков, являются сильными противовирусными средствами для людей, указывая, что прерывание данной стадии жизненного цикла вируса является результатом действия терапевтически активных средств. Следовательно, это является привлекательной мишенью для разработки лекарственных средств.
Описаны некоторые потенциальные ингибиторы HCV-протеазы [публикации РСТ №№ WO 02/18369, WO 02/08244, WO 00/09558, WO 00/09543, WO 99/64442, WO 99/07733, WO 99/07734, WO 99/50230, WO 98/46630, WO 98/17679 и WO 97/43310, патент США 5990276, M. Llinas-Brunet et al., Bioorg.Med. Chem. Lett., 8, pp.1713-18 (1998); W. Han et al., Bioorg. Med. Chem. Lett., 10, 711-13 (2000); R. Dunsdon et al., Bioorg. Med. Chem. Lett., 10, pp.1571-79 (2000); M. Llinas-Brunet et al., Bioorg. Med. Chem. Lett., 10, pp.2267-70 (2000); и S. LaPlante et al., Bioorg. Med. Chem. Lett., 10, pp.2271-74 (2000)].
В настоящее время не существует каких-либо удовлетворительных средств против HCV или способов лечения. Единственной общепринятой терапией в случае заболевания HCV является лечение интерфероном. Однако интерфероны оказывают существенное побочное действие [M.A. Wlaker et al., "Hepatitis C Virus: An Overview of Current Approaches and Progress", DDT, 4, pp.518-29 (1999); D. Moradopour et al., "Current and Evolving Therapies for Hepatitis C", Eur. J. Gastroenterol. Hepatol., 11, pp.1199-1202 (1999); H.L.A. Janssen et al., "Suicide Associated with Alfa-Interferon Therapy for Chronic Viral Hepatitis", J. Hepatol., 21, pp.241-243 (1994); P.F. Renault et al., "Side Effects of Alfa Interferon", Seminars in Liver Disease, 9, pp.273-277 (1989)] и вызывают длительную ремиссию только в части (~25%) случаев [O. Weilland, "Interferon Therapy in Chronic Hepatitis C Virus Infection", FEMS Microbiol. Rev., 14, pp.279-288 (1994)]. Более того, перспективы в отношении эффективных вакцин против HCV остаются неопределенными.
Таким образом, существует потребность в соединениях, применимых в анти-HCV терапии. Такие соединения должны обладать лечебным потенциалом как ингибиторы протеаз, в частности как ингибиторы серинпротеаз, и в особенности ингибиторы серинпротеазы NS3 HCV. Такие соединения могли бы применяться как анти-HCV средства. Существует особая потребность в соединениях с улучшенным ингибированием ферментов или клеточной активностью.
Сущность изобретения
Настоящее изобретение направлено на указанные потребности и относится к соединению формулы I:

где переменные имеют значения, указанные в данном описании.
Изобретение также относится к композициям, содержащим вышеуказанные соединения, и их применению. Такие композиции можно применять для предварительной обработки инвазивных устройств для внедрения пациенту, для обработки биологических образцов, таких как кровь, перед введением пациенту и для прямого введения пациенту. В каждом случае композиция будет использоваться для ингибирования репликации HCV и уменьшения опасности и тяжести заражения HCV.
Изобретение также относится к получению соединений формулы I.
Подробное описание изобретения
Настоящее изобретение относится к соединению формулы I:

где
Ar представляет собой 5-10-членное ароматическое кольцо, содержащее до 4 гетероатомов, выбранных из О, S, N(H), SO и SO2, где 1-3 атома кольца, необязательно и независимо, замещены J;
R1 и R2 представляют собой независимо:
(С1-С12)-алифатический радикал,
(С3-С10)-циклоалкил- или -циклоалкенил-,
[(C3-С10)-циклоалкил- или -циклоалкенил]-(С1-С12)-алифатический радикал,
(С6-С10)-арил-(С1-С12)-алифатический радикал,
(С6-С10)-гетероарил-(С1-С12)-алифатический радикал,
где до 3 алифатических атомов углерода в R1 и R2 могут быть заменены гетероатомом, выбранным из О, N, S, SO или SO2, в химически устойчивой структуре;
где каждый из R1 и R2, независимо и необязательно, замещен заместителями, до 3, выбранными, независимо, из J;
R3 и R3' представляют собой, независимо, водород или (С1-С12)-алифатический радикал, где любой атом водорода, необязательно, заменен галогеном; где любой концевой атом углерода R3, необязательно, замещен сульфгидрилом или гидрокси; или R3 представляет собой фенил или -СН2-фенил, где указанная фенильная группа, необязательно, замещена заместителями, до 3, выбранными, независимо, из J; или
R3 и R3' вместе с атомом, с которым они связаны, представляют собой 3-6-членное кольцо, содержащее до 2 гетероатомов, выбранных из N, NH, О, SO или SO2; где кольцо имеет до 2 заместителей, выбранных, независимо, из J;
R4 и R4' представляют собой, независимо,
водород-,
(С1-С12)-алифатический радикал,
(С3-С10)-циклоалкил- или -циклоалкенил-,
(С3-С10)-циклоалкил-(С1-С12)-алифатический радикал,
(С6-С10)-арил-,
(С3-С10)-гетероциклил- или
(С5-С10)-гетероарил-;
где до двух алифатических атомов углерода в R4 и R4' могут быть заменены гетероатомом, выбранным из О, N, S, SO или SO2;
где каждый из R4 и R4', независимо и необязательно, замещен заместителями, до 3, выбранными, независимо, из J;
W представляет собой:

где каждый R6 представляет собой независимо:
водород-,
(С1-С12)-алифатический радикал,
(С6-С10)-арил-,
(С6-С10)-арил-(С1-С12)-алифатический радикал,
(С3-С10)-циклоалкил- или -циклоалкенил-,
[(C3-С10)-циклоалкил- или -циклоалкенил]-(С1-С12)-алифатический радикал,
(С3-С10)-гетероциклил-,
(С3-С10)-гетероциклил-(С1-С12)-алифатический радикал,
(С5-С10)-гетероарил- или
(С5-С10)-гетероарил-(С1-С12)-алифатический радикал, или
две группы R6, связанные с одним и тем же атомом азота, вместе с указанным атомом азота образуют (С3-С10)-гетероциклическое кольцо;
где R6, необязательно, замещен заместителями J, до 3;
где каждый R8, независимо, представляет собой -OR'; или группы R8 вместе с атомом бора представляют собой (С3-С10)-гетероциклическое кольцо, содержащее, кроме атома бора, до 3 других гетероатомов, выбранных из N, NH, О, SO и SO2;
Т представляет собой:
(С1-С12)-алифатический радикал,
(С6-С10)-арил-,
(С6-С10)-арил-(С1-С12)-алифатический радикал,
(С5-С10)-гетероарил- или
(С5-С10)-гетероарил-(С1-С12)-алифатический радикал,
где каждый Т, необязательно, замещен заместителями J, до 3;
J представляет собой галоген, -OR', -NO2, -CN, -CF3, -OCF3, -R', оксо, тиоксо, 1,2-метилендиокси, 1,2-этилендиокси, =N(R'), =N(OR'), -N(R')2, -SR', -SOR', -SO2R', -SO2N(R')2, -SO3R',
-C(O)R', -C(O)C(O)R', -C(O)CH2C(O)R', -C(S)R', -C(S)OR',
-C(O)OR', -C(O)C(O)OR', -C(O)C(O)N(R')2, -OC(O)R', -C(O)N(R')2,
-OC(O)N(R')2, -C(S)N(R')2, -(CH2)0-2NHC(O)R', -N(R')N(R')COR',
-N(R')N(R')C(O)OR', -N(R')N(R')CON(R')2, -N(R')SO2R',
-N(R')SO2N(R')2, -N(R')C(O)OR', -N(R')C(O)R', -N(R')C(S)R',
-N(R')C(O)N(R')2, -N(R')C(S)N(R')2, -N(COR')COR', -N(OR')R',
-C(=NH)N(R')2, -C(O)N(OR')R', -C(=NOR')R', -OP(O)(OR')2,
-P(O)(R')2, -P(O)(OR')2 или -P(O)(H)(OR');
R' выбирают, независимо, из:
водорода-,
(С1-С12)-алифатического радикала,
(С3-С10)-циклоалкила- или -циклоалкенила-,
[(C3-С10)-циклоалкил- или -циклоалкенил]-(С1-С12)-алифатического радикала,
(С6-С10)-арила-,
(С6-С10)-арил-(С1-С12)-алифатического радикала,
(С3-С10)-гетероциклила-,
(С6-С10)-гетероциклил-(С1-С12)-алифатического радикала,
(С5-С10)-гетероарила- или
(С5-С10)-гетероарил-(С1-С12)-алифатического радикала;
где R', необязательно, замещен группами J, до 3;
где две группы R', связанные с одним и тем же атомом, образуют 3-10-членное ароматическое или неароматическое кольцо, содержащее до 3 гетероатомов, выбранных, независимо, из N, О, S, SO или SO2, где указанное кольцо, необязательно, конденсировано с (С6-С10)-арилом, (С5-С10)-гетероарилом, (C3-С10)-циклоалкилом или (С3-С10)-гетероциклилом, где любой цикл содержит до 3 заместителей, выбранных, независимо, из J.
Указанные соединения решают изложенные выше проблемы, будучи соединениями с улучшенной активностью в отношении ферментов и/или клеточной активностью. Например, соединения данного изобретения, в частности предпочтительные соединения, показывают лучшее ингибирование ферментов, чем соединения WO 98/17679. Соединения данного изобретения также имеют более хорошие результаты в отношении клеток, чем другие соединения, о которых сообщается в литературе (см. документы, цитированные в данном описании).
В одном воплощении, если R1 представляет собой циклогексил, R2 представляет собой трет-бутил, R3' представляет собой Н, R3 представляет собой н-пропил, W представляет собой -С(О)С(О)N(H)-циклопропил, и Т представляет собой:

тогда Ar представляет собой 4-хиназолинил или 5-хлор-2-пиридил.
Настоящее изобретение также относится к соединению формулы I:

где
n равен 0 или 1;
Ar представляет собой 5-10-членное ароматическое кольцо, содержащее до 4 гетероатомов, выбранных из О, S, N(H), SO и SO2, где 1-3 атома кольца, необязательно и независимо, замещены J;
R1, R2, R12 и R13 представляют собой независимо:
(С1-С12)-алифатический радикал,
(С3-С10)-циклоалкил- или -циклоалкенил-,
[(C3-С10)-циклоалкил- или -циклоалкенил]-(С1-С12)-алифатический радикал,
(С6-С10)-арил-(С1-С12)-алифатический радикал,
(С6-С10)-гетероарил-(С1-С12)-алифатический радикал,
где до 3 алифатических атомов углерода в R1 и R2 могут быть заменены гетероатомами, выбранными из О, N, NH, S, SO или SO2, в химически устойчивой структуре;
где каждый из R1 и R2, независимо и необязательно, замещен в каждом поддающемся замещению положении заместителями, до 3, выбранными, независимо, из J;
R3 и R3' представляют собой, независимо, водород или (С1-С12)-алифатический радикал, где любой атом водорода, необязательно, заменен галогеном; где любой концевой атом углерода R3, необязательно, замещен сульфгидрилом или гидрокси; или R3 представляет собой фенил или -СН2-фенил, где указанная фенильная группа, необязательно, замещена заместителями, до 3, выбранными, независимо, из J; или
R3 и R3' вместе с атомом, с которым они связаны, представляют собой 3-6-членное кольцо, содержащее до 2 гетероатомов, выбранных из N, NH, О, SO и SO2; где кольцо имеет до 2 заместителей, выбранных, независимо, из J;
R4 и R4' представляют собой независимо:
водород-,
(С1-С12)-алифатический радикал,
(С3-С10)-циклоалкил- или -циклоалкенил-,
(С3-С10)-циклоалкил-(С1-С12)-алифатический радикал,
(С6-С10)-арил-,
(С3-С10)-гетероциклил- или
(С5-С10)-гетероарил-;
где до двух алифатических атомов углерода в R4 и R4' могут быть заменены гетероатомами, выбранными из О, N, S, SO и SO2;
где каждый из R4 и R4', независимо и необязательно, замещен заместителями, до 3, выбранными, независимо, из J;
W представляет собой:

где
Y представляет собой -СО2Н, производное группы -СО2Н или биоизостеру группы
-СО2Н;
каждый R6 представляет собой независимо:
водород-,
(С1-С12)-алифатический радикал,
(С6-С10)-арил-,
(С6-С10)-арил-(С1-С12)-алифатический радикал,
(С3-С10)-циклоалкил- или -циклоалкенил-,
[(C3-С10)-циклоалкил- или -циклоалкенил]-(С1-С12)-алифатический радикал,
(С3-С10)-гетероциклил-,
(С3-С10)-гетероциклил-(С1-С12)-алифатический радикал,
(С5-С10)-гетероарил- или
(С5-С10)-гетероарил-(С1-С12)-алифатический радикал, или
две группы R6, связанные с одним и тем же атомом азота, вместе с указанным атомом азота образуют (С3-С10)-гетероциклическое кольцо;
где R6, необязательно, замещен заместителями J, до 3;
где каждый R8, независимо, представляет собой -OR'; или группы R8 вместе с атомом бора представляют собой (С3-С10)-гетероциклическое кольцо, содержащее, кроме атома бора, до 3 других гетероатомов, выбранных из числа N, NH, О, SO и SO2;
Т представляет собой
(С1-С12)-алифатический радикал,
(С6-С10)-арил-,
(С6-С10)-арил-(С1-С12)-алифатический радикал,
(С5-С10)-гетероарил- или
(С5-С10)-гетероарил-(С1-С12)-алифатический радикал,
где каждый Т, необязательно, замещен заместителями J, до 3; и
где до 3 алифатических атомов углерода в Т могут быть заменены гетероатомами, выбранными из О, N, NH, S, SO или SO2, в химически устойчивой структуре;
при условии, что если Т представляет собой пиррол, пиррол не является замещенным J в положении 3, причем J представляет собой -C(O)R', -C(O)C(O)R', -C(O)CH2C(O)R', -C(S)R', -C(S)OR',
-C(O)OR', -C(O)C(O)OR', -C(O)C(O)N(R')2, -C(O)N(R')2,
-C(S)N(R')2, -C(=NH)N(R')2, -C(O)N(OR')R' или -C(=NOR')R';
J представляет собой галоген, -OR', -NO2, -CN, -CF3, -OCF3, -R', оксо, тиоксо, 1,2-метилендиокси, 1,2-этилендиокси, =N(R'), =N(OR'), -N(R')2, -SR', -SOR', -SO2R', -SO2N(R')2, -SO3R',
-C(O)R', -C(O)C(O)R', -C(O)CH2C(O)R', -C(S)R', -C(S)OR',
-C(O)OR', -C(O)C(O)OR', -C(O)C(O)N(R')2, -OC(O)R', -C(O)N(R')2,
-OC(O)N(R')2, -C(S)N(R')2, -(CH2)0-2NHC(O)R', -N(R')N(R')COR',
-N(R')N(R')C(O)OR', -N(R')N(R')CON(R')2, -N(R')SO2R',
-N(R')SO2N(R')2, -N(R')C(O)OR', -N(R')C(O)R', -N(R')C(S)R',
-N(R')C(O)N(R')2, -N(R')C(S)N(R')2, -N(COR')COR', -N(OR')R',
-C(=NH)N(R')2, -C(O)N(OR')R', -C(=NOR')R', -OP(O)(OR')2,
-P(O)(R')2, -P(O)(OR')2 или -P(O)(H)(OR');
R' представляет собой:
водород-,
(С1-С12)-алифатический радикал,
(С3-С10)-циклоалкил- или -циклоалкенил-,
[(C3-С10)-циклоалкил- или -циклоалкенил]-(С1-С12)-алифатический радикал,
(С6-С10)-арил-,
(С6-С10)-арил-(С1-С12)-алифатический радикал,
(С3-С10)-гетероциклил-,
(С6-С10)-гетероциклил-(С1-С12)-алифатический радикал,
(С5-С10)-гетероарил- или
(С5-С10)-гетероарил-(С1-С12)-алифатический радикал;
где R', необязательно, замещен группами J, до 3;
где две группы R', связанные с одним и тем же атомом, образуют 3-10-членное ароматическое или неароматическое кольцо, содержащее до 3 гетероатомов, выбранных, независимо, из N, О, S, SO или SO2, где указанное кольцо, необязательно, конденсировано с (С6-С10)-арилом, (С5-С10)-гетероарилом, (C3-С10)-циклоалкилом или (С3-С10)-гетероциклилом, где любое кольцо содержит до 3 заместителей, выбранных, независимо, из J.
В одном воплощении, если R1 представляет собой циклогексил, R2 представляет собой трет-бутил, R3' представляет собой Н, R3 представляет собой н-пропил, W представляет собой -С(О)С(О)N(H)-циклопропил, и Т представляет собой:

тогда Ar не является 4-хиназолинилом или 5-хлор-2-пиридилом.
В другом воплощении, если Т представляет собой пиррол, пиррол не является замещенным J, но, необязательно, конденсирован с 5-членным или 6-членным арильным или гетероарильным кольцом.
В другом воплощении, если Т представляет собой пиррол, пиррол не является замещенным J в положении 3, но, необязательно, конденсирован с 5-членным или 6-членным арильным или гетероарильным кольцом.
В другом воплощении Т не является:

В другом воплощении Т не является пирролом.
В более конкретном воплощении данное изобретение относится к соединению, где n равен 0, и соединение имеет формулу I-А:

где переменные имеют значения, указанные в любом из воплощений, раскрытых в данном описании. В соединении формулы А W представляет собой предпочтительно:

В другом конкретном воплощении данное изобретение относится к соединению, где n равен 1, и соединение имеет формулу I-В:

где переменные имеют значения, указанные в любом из воплощений, раскрытых в данном описании. В соединении формулы I-B W представляет собой предпочтительно:

Определения
Термин "арил", используемый в данном описании, обозначает моноциклическую или бициклическую карбоциклическую ароматическую кольцевую систему. Фенил является примером моноциклической ароматической кольцевой системы. Бициклические ароматические кольцевые системы включают системы, в которых оба кольца являются ароматическими, например нафтил, и системы, в которых только одно кольцо из двух является ароматическим, как, например, в тетралине.
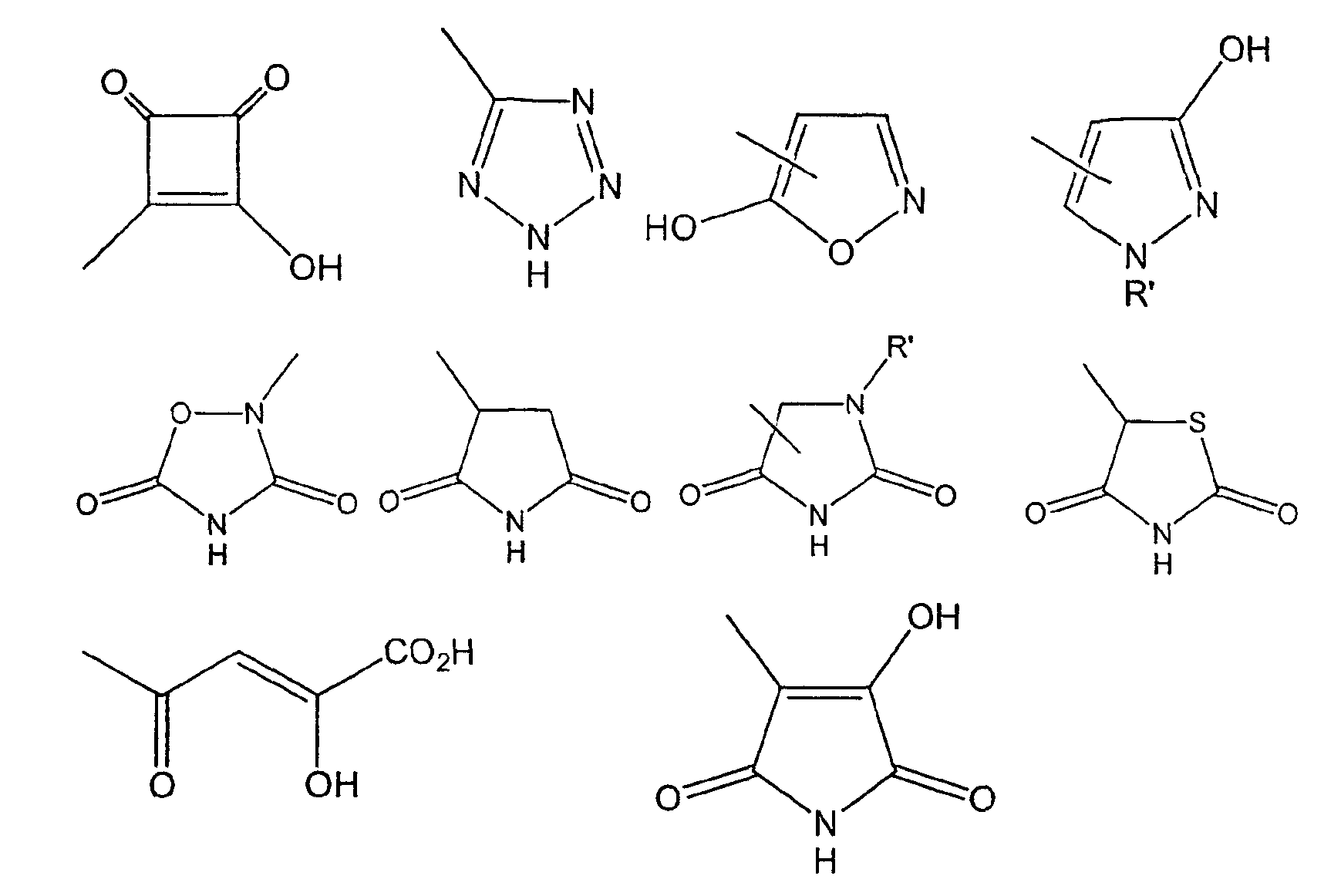
Термин "биоизостера" группы -СО2Н, используемый в данном описании, относится к химической группе, которая может замещать карбоксильную группу в биологически активной молекуле. Примеры таких групп раскрываются в Christopher A. Lipinski, "Bioisosteres in Drug Design", Annual Reports in Medicinal Chemistry, 21, pp.286-88 (1986), и в C.W. Thornber, "Isosterism and Molecular Modification in Drug Design", Chemical Society Reviews, pp.563-580 (1979). Примерами таких групп являются, но не ограничиваются ими, -СОСН2ОН, -CONHOH, SO2NHR', -SO3H,
-PO(OH)NH2, -CONHCN, -OSO3H, -CONHSO2R', -PO(OH)2,
-PO(OH)(OR'), -PO(OH)(R'), -OPO(OH)2, -OPO(OH)(OR'),
-OPO(OH)(R'), HNPO(OH)2, -NHPO(OH)(OR'), -NHPO(OH)(R'),

Термин "гетероциклил", используемый в данном описании, обозначает моноциклическую или бициклическую неароматическую кольцевую систему с 1-3 гетероатомами или группами гетероатомов в каждом кольце, выбранными из О, N, NH, S, SO или SO2, в химически устойчивой структуре. В воплощении бициклической неароматической кольцевой системы "гетероциклила" одно кольцо или оба кольца могут содержать указанные гетероатомы или группы гетероатомов.
Примерами гетероциклических колец являются 3-1Н-бензимидазол-2-он, 3-(1-алкил)-бензимидазол-2-он, 2-тетрагидрофуранил, 3-тетрагидрофуранил, 2-тетрагидротиофенил, 3-тетрагидротиофенил, 2-морфолино, 3-морфолино, 4-морфолино, 2-тиоморфолино, 3-тиоморфолино, 4-тиоморфолино, 1-пирролидинил, 2-пирролидинил, 3-пирролидинил, 1-тетрагидропиперазинил, 2-тетрагидропиперазинил, 3-тетрагидропиперазинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 1-пиразолинил, 3-пиразолинил, 4-пиразолинил, 5-пиразолинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-пиперидинил, 2-тиазолидинил, 3-тиазолидинил, 4-тиазолидинил, 1-имидазолидинил, 2-имидазолидинил, 4-имидазолидинил, 5-имидазолидинил, индолинил, тетрагидрохинолинил, тетрагидроизохинолинил, бензотиолан, бензодитиан и 1,3-дигидроимидазол-2-он.
Термин "гетероарил", используемый в данном описании, обозначает моноциклическую или бициклическую ароматическую кольцевую систему с 1-3 гетероатомами или группами гетероатомов в каждом кольце, выбранными из О, N, NH или S, в химически устойчивой структуре. В воплощении такой бициклической ароматической кольцевой системы "гетероарила":
- одно или оба кольца могут быть ароматическими; и
- одно или оба кольца могут содержать указанные гетроатомы или группы гетероатомов.
Примерами гетероарильных колец являются 2-фуранил, 3-фуранил, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, бензимидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, N-пирролил, 2-пирролил, 3-пирролил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пиридазинил (например, 3-пиридазинил), 2-тиазолил, 4-тиазолил, 5-тиазолил, тетразолил (например, 5-тетразолил), триазолил (например, 2-триазолил и 5-триазолил), 2-тиенил, 3-тиенил, бензофурил, бензотиофенил, индолил (например, 2-индолил), пиразолил (например, 2-пиразолил), изотиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-триазолил, 1,2,3-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил, пуринил, пиразинил, 1,3,5-триазинил, хинолинил (например, 2-хинолинил, 3-хинолинил, 4-хинолинил) и изохинолинил (например, 1-изохинолинил, 3-изохинолинил или 4-изохинолинил).
Термин "алифатический радикал", используемый в данном описании, обозначает линейный или разветвленный алкил, алкенил или алкинил. Подразумевается, что воплощения алкенила или алкинила требуют, по меньшей мере, два атома углерода в алифатической цепи.
Термин "циклоалкил или циклоалкенил" относится к моноциклической или конденсированной или мостиковой бициклической карбоциклической кольцевой системе, которая не является ароматической. Циклоалкенильные кольца содержат одну или несколько единиц ненасыщенности. Предпочтительными циклоалкильными группами являются циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, циклогептил, циклогептенил, норнборнил, адамантил и декалинил.
В данном описании у обозначения атома углерода могут иметься указанное целое число и любое промежуточное целое число. Например, число атомов углерода в (С1-С4)-алкильной группе равно 1, 2, 3 или 4. Следует иметь в виду, что такое обозначение относится к общему числу атомов в соответствующей группе. Например, в (С3-С10)-гетероциклиле общее число атомов углерода и гетероатомов равно 3 (как в азиридине), 4, 5, 6 (как в морфолине), 7, 8, 9 или 10.
Выражение "химически устойчивая структура", используемое в данном описании, относится к структуре соединения, которая придает соединению достаточную устойчивость для возможности получения и введения млекопитающему способами, известными в технике. Типично такие соединения устойчивы при температуре 40°С или ниже в отсутствие влаги или других условий химического воздействия в течение, по меньшей мере, недели.
Предпочтительные воплощения
В другом воплощении данного изобретения Ar представляет собой фенил, пиридил, хинолинил, пиримидинил или нафтил, где каждая группа, необязательно, замещена 1, 2 или 3 группами J.
В другом воплощении данного изобретения Ar представляет собой:


В еще одном воплощении данного изобретения Ar представляет собой:

В особенно предпочтительном воплощении Ar представляет собой:

В другом воплощении данного изобретения Ar представляет собой 6- или 10-членное ароматическое кольцо, содержащее 0, 1 или 2 гетероатома азота, где 1, 2, или 3 атома кольца, необязательно и независимо, имеют заместителей J.
В любом воплощении данного изобретения каждая группа J в Ar представляет собой, независимо, OR', NO2, CN, CF3, OCF3, R', COR', C(O)OR', C(O)N(R')2, SO2R', SO2N(R')2, 1,2-метилендиокси, 1,2-этилендиокси, NR'C(O)OR' или NR'SO2R'.
В любом воплощении данного изобретения каждая группа J в Ar представляет собой, предпочтительно и независимо, OR', галоген, CN, CF3, R' или COR'. Предпочтительнее такой J представляет собой галоген (в частности, хлор).
В других воплощениях данного изобретения каждая группа J в Ar представляет собой, независимо, галоген, трифторметил, метил или NO2.
Согласно любому предпочтительному воплощению Т представляет собой (С6-С10)-арил- или (С5-С10)-гетероарил-, где каждый Т, необязательно, замещен 1, 2 или 3 заместителями J.
В предпочтительном воплощении Т представляет собой 6-членную или 10-членную арильную группу. В другом предпочтительном воплощении Т представляет собой 6-членную гетероарильную группу, которая, необязательно, конденсирована с другой 5- или 6-членной арильной или гетероарильной группой.
Более предпочтительными являются воплощения, где Т представляет собой:

В более предпочтительном воплощении Т представляет собой:

В некоторых воплощениях данного изобретения любая группа Т, необязательно, конденсирована с 5-членной или 6-членной арильной или гетероарильной группой.
Соответственно, одно из воплощений данного изобретения относится к соединениям, в которых Т представляет собой:
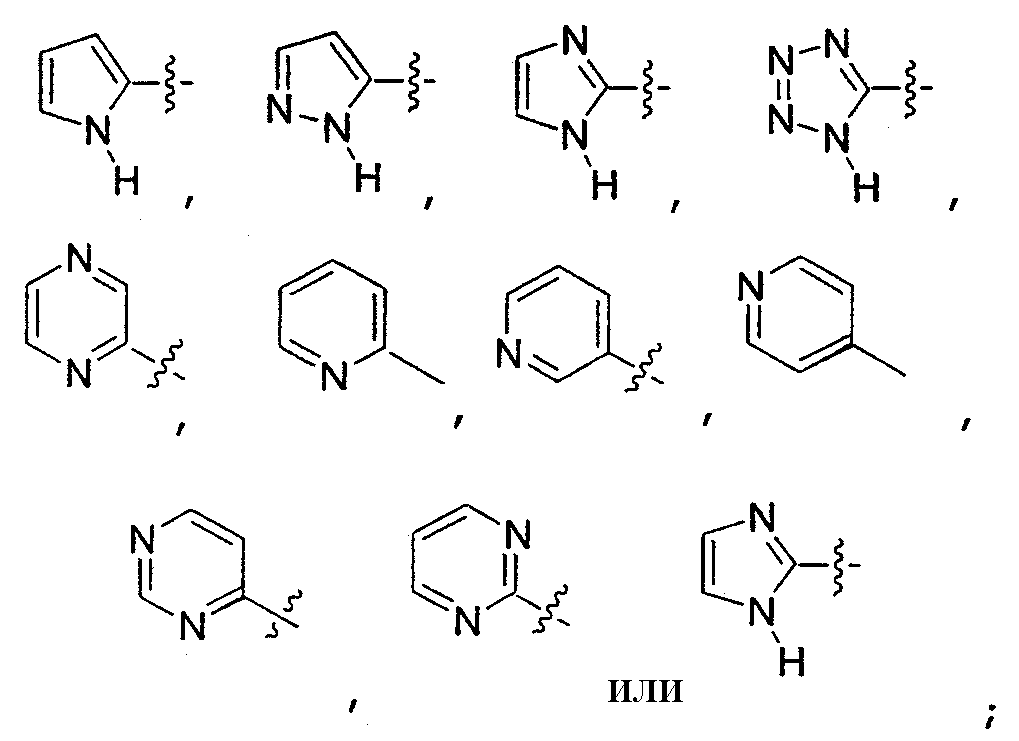
где каждая группа Т, необязательно, конденсирована с 5-членной или 6-членной арильной или гетероарильной группой.
В другом воплощении Т представляет собой:

где Т, необязательно, конденсирован с 5-членной или 6-членной арильной или гетероарильной группой.
В некоторых воплощениях Т не является конденсированным с другим кольцом.
В любом воплощении данного изобретения каждая арильная или гетероарильная группа Т, необязательно и независимо, замещена 1, 2 или 3 группами, выбранными из
-СН3, -Н2СН3, галогена, ацетила, -СО2Н, -((С1-С6)-алкил)-СО2Н или -O2R'.
В некоторых воплощениях 1 алифатический атом углерода в Т заменен на гетероатом, выбранный из O, N, NH, S, SO или SO2, в химически устойчивой структуре. Гетероатомом, предпочтительно, является О или NH. В другом воплощении алифатический атом углерода в Т не заменяют.
В некоторых воплощениях данного изобретения Т с остальной частью молекулы соединяет алифатическая группа. Как правило, в таких алифатических линкерах замена алифатического атома углерода предпочтительна в соединениях, где n равен 0. В соединениях, где n равен 1, замену на гетероатом в такой алифатической линкерной группе, предпочтительно, не производят. Более того, соединения, где n равен 1, предпочтительно, не содержат алифатической линкерной группы.
Согласно предпочтительному воплощению W представляет собой -C(O)-C(O)-R6. Предпочтительно R6 представляет собой фенил, пиридил, (С3-С6)-алкил, (С3-С6)-циклоалкил, -ОН, -О-(С1-С6)-алкил, -N(Н)-(C3-C6)-циклоалкил, -N(Н)-С(Н)(СН3)-(С6-С10)-арил, -N(Н)-С(Н)(СН3)-(С3-С10)-гетероциклил или -N(Н)-С(Н)(СН3)-(С5-С10)-гетероарил, где каждый арил, гетероциклил и гетероарил, необязательно, замещен атомом галогена. Предпочтительные воплощения выбирают из:

Предпочтительнее R6 представляет собой изопропил.
Согласно одному воплощению W представляет собой -C(O)-C(O)-OR6. В таком воплощении R6, предпочтительно, представляет собой водород, (С1-С12)-алифатический радикал (предпочтительнее, (С1-С6)-алкил), (С6-С10)-арил, (С3-С10)-циклоалкил или -циклоалкенил, (С3-С10)-гетероциклил, (С5-С10)-гетероарил или (С3-С6)-циклоалкил-(С1-С3)-алкил, где циклоалкил представляет собой, предпочтительно, циклопропильную группу. Арильная группа, необязательно, замещена группами J, до 3, где J представляет собой галоген, предпочтительно хлор или фтор. Предпочтительнее R6 представляет собой Н или метил.
Согласно другому воплощению W представляет собой -C(O)-C(O)-N(R6)2, где R6 представляет собой водород, (С1-С6)-алкил, (С1-С6)-алкенил, (С6-С10)-арил-(С1-С6)-алкил или (С6-С10)-гетероарил-(С1-С6)-алкил, где R6, необязательно, замещен группами J, до 3. Предпочтительно R6 представляет собой водород, (С3-С10)-циклоалкил или -циклоалкенил или (С3-С10)-гетероциклил. С другой стороны, один R6 представляет собой водород, а другой R6 представляет собой (С6-С10)-арил-(С1-С6)-алкил, где алкил, необязательно, замещен СО2Н; (С3-С6)-циклоалкил-; (С5)-гетероциклил-(С1-С3)-алкил-; (С3-С6)-алкенил-; или каждый R6 представляет собой (С1-С6)-алкил-. С другой стороны, каждый R6 представляет собой (С1-С3)-алкил-.
Наиболее предпочтительно -NHR6 в W представляет собой:

Предпочтительные заместители J на алкильных и арильных группах в таком воплощении представляют собой галоген, карбокси и гетероарил. Более предпочтительными заместителями на арильных группах являются галоген (предпочтительно, хлор или фтор), и более предпочтительными заместителями J на алкильных группах являются карбокси и гетероарил.
Согласно еще другим предпочтительным воплощениям соединений формулы I, W представляет собой:

где NR6R6 представляет собой -NH-((С1-С6)-алифатический радикал), -NH-((С3-С6)-циклоалкил), -NH-СН(СН3)-арил или -NH-СН(СН3)-гетероарил, где указанный арил или указанный гетероарил, необязательно, замещен атомами галогена, до 3.
В любом предпочтительном воплощении W NR6R6 представляет собой:

В любом предпочтительном воплощении W NR6R6 представляет собой:

В любом более предпочтительном воплощении W NR6R6 представляет собой:

В любом более предпочтительном воплощении W NR6R6 представляет собой:

Согласно одному воплощению R1 выбирают из:

Согласно предпочтительному воплощению R1 выбирают из:

Согласно другому воплощению R1 выбирают из:

Согласно другому воплощению R1 выбирают из:
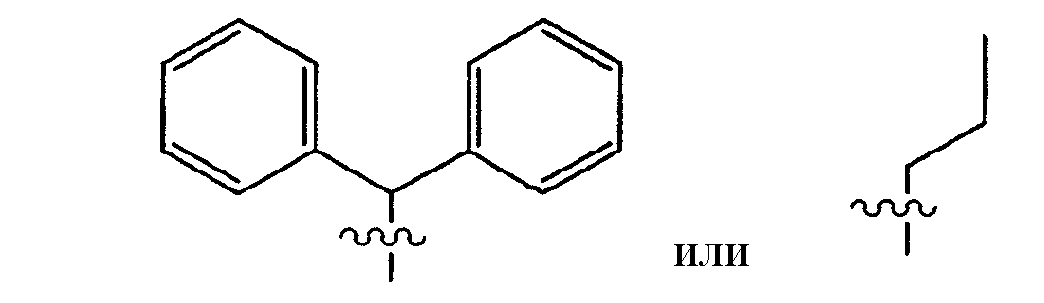
Наиболее предпочтительно R1 представляет собой циклогексил.
Согласно одному воплощению R2 представляет собой:

Согласно другому воплощению R2 представляет собой:

Согласно любым предпочтительным воплощениям R2 представляет собой:

Согласно любым более предпочтительным воплощениям R2 представляет собой:

В любых наиболее предпочтительных воплощениях R2 представляет собой трет-бутил.
Согласно одному воплощению R3 представляет собой:

Согласно предпочтительному воплощению R3 представляет собой:

Предпочтительнее R3 представляет собой пропил (предпочтительно, н-пропил).
В любом предпочтительном воплощении данного изобретения R3' представляет собой Н.
Согласно другому воплощению R3 и R3' вместе с атомом, с которым они связаны, образуют кольцевую систему:

Согласно другому воплощению R3' представляет собой водород, и R3 представляет собой:

Согласно предпочтительным воплощениям R3' представляет собой водород, и R3 представляет собой:

В еще других предпочтительных воплощениях данного изобретения R3' представляет собой водород, и R3 представляет собой:

Согласно одному воплощению один из R4 или R4' представляет собой водород.
Согласно другому воплощению один из R4 или R4' представляет собой (С1-С6)-алкил.
Согласно предпочтительному воплощению R4 и R4' представляют собой атомы водорода.
В некоторых воплощениях 1 или 2 атома углерода в R1, R2, R3 или R4, необязательно и независимо, заменяют на N, NH, O или S.
Соответственно, одно воплощение данного изобретения относится к соединению, где R1 представляет собой циклогексил, где 1 или 2 атома углерода, необязательно, заменены на N, NH, O или S, и где каждый атом, необязательно и независимо, замещен 1, 2 или 3 заместителями из групп J, где J представляет собой галоген, ОН, OR', NH, N(R')2 (и R' представляет собой, предпочтительно, (С1-С6)-алкил).
В некоторых других воплощениях ни один из атомов углерода в R1, R2, R3 или R4 не заменяют на N, NH, O или S. В других воплощениях указанные группы не имеют заместителей J.
Соединения данного изобретения могут содержать один или несколько асимметричных атомов углерода и, таким образом, могут существовать в виде рацематов и рацемических смесей, отдельных энантиомеров, смесей диастереомеров и отдельных диастереомеров. Все такие изомерные формы указанных соединений определенно входят в настоящее изобретение. Каждый стереогенный атом углерода может находиться в R- или S-конфигурации.
Предпочтительно соединения данного изобретения имеют структуру и стереохимию, отображенные в соединениях 1-76.
Любое из предпочтительных воплощений, перечисленных выше, включая такие воплощения в указанных выше видах, может одно определять формулу I или входить в сочетание для получения предпочтительного воплощения данного изобретения.
Аббревиатуры, которые используются на схемах, в описаниях получения и примерах, перечислены далее.
ТГФ - тетрагидрофуран,
ДМФА - N,N-диметилформамид,
EtOAc - этилацетат,
АсОН - уксусная кислота,
HOBt - гидрат 1-гидроксибензотриазола,
EDC - гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида,
NMM - N-метилмофролин,
NMP - N-метилпирролидинон,
EtOH - этанол,
t-BuOH - трет-бутанол,
Et2O - диэтиловый эфир,
ВОС - трет-бутилоксикарбонил,
ВОС2О - ди-трет-бутилдикарбонат,
Cbz - бензилоксикарбонил,
Chg - циклогексилглицин,
TbG - трет-бутилглицин,
Fmoc - 9-флуоренилметоксикарбонил,
ДМСО - диметилсульфоксид,
ТФК - трифторуксусная кислота,
DCCA - дихлоруксусная кислота,
DCE - дихлорэтан,
DIEA - диизопропилэтиламин,
MeCN - ацетонитрил,
PyBrOp - гексафторфосфат трис(пирролидино)бромфосфония,
TBTU или HATU - тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония,
DMAP - 4-диметиламинопиридин,
PPTS - п-толуолсульфонат пиридиния,
IBX - периодбензойная кислота,
AIBN - 2,2'-азобисизобутиронитрил,
ТЕМРО - 2,2,6,6-тетраметил-1-пиперидинилокси (свободный радикал),
rt или RT - комнатная температура,
ON - в течение ночи,
ND - не определено,
MC - масс-спектроматрия,
ЖХ - жидкостная хроматография.
Общая методология синтеза
Соединения данного изобретения можно получить, как правило, способами, известными специалистам в данной области техники. Приведенные ниже схемы 1-7 иллюстрируют пути синтеза соединений настоящего изобретения. С другой стороны, другие равноценные схемы, которые будут очевидны для специалиста в области органической химии, можно использовать для синтеза различных частей молекулы, как иллюстрирует приведенная ниже общая схема и приведенные далее препаративные примеры.
Схема 1



Выше на схеме 1 приводится общий путь синтеза для получения соединений формулы I с различными группами Ar, где Т представляет собой пиразин, R1 представляет собой циклогексил, R2 представляет собой трет-бутил, и W представляет собой -C(O)C(O)-N(H)-циклопропил. Как будет понятно специалистам-практикам, соединения формулы I, где T, R1 и R2 иные, чем указаны выше, можно получить, варьируя способы синтеза.
Например, соединения формулы I, где Т иной, чем пиразин, можно получить взаимодействием соединения 5 (после удаления группы Z) с соединением формулы Т-С(О)-ОН в подходящих условиях сочетания.
Подобным образом, замещение другим производным аминокислоты вместо Z-Tbg-OH при превращении соединения 3 в соединение 4, или другим производным аминокислоты вместо Z-Chg-OH при превращении соединения 4 в соединение 5 будет приводить к соединениям с другими группами R2 и R1, соответственно.
Схема 2

Схема 2, приведенная выше, показывает общий путь синтеза для получения соединения 59.
Схема 3

Другой подход к получению соединений данного изобретения отбражен на схеме 3. В данном подходе производное 4-гидроксипролина 13 вводят во взаимодействие с соединением 14, и получают соединение 15. На схеме 3 Р1 представляет собой водород или соответствующую аминозащитную группу, Р2 представляет собой водород или соответствующую карбоксизащитную группу, Ar имеет значения, указанные в данном описании, и Х представляет собой соответствующую удаляемую группу. В одном воплощении набор следующий: Р1 представляет собой трет-бутоксикарбонил, Р2 представляет собой водород, Ar представляет собой 4-хлор-2-пиридин, Х представляет собой Cl, и соединения 13 и 14 вводят во взаимодействие в присутствии tBuOK, ДМСО и ТГФ.
Как могут понять специалисты-практики, соединение 15 затем можно превратить в соединения формулы I обычными способами. Один такой способ отображен ниже на схеме 4.
Схема 4

Схема 5

Схема 5 отображает другой подход к получению соединения данного изобретения (59). Стадии, выполняемые по схеме 5, можно модифицировать, например, используя другие реагенты или осуществляя взаимодействия в другом порядке.
Схема 6

Схема 6 отображает подход к получению соединения 28. В таком воплощении соединение 27 превращают в соединение 28 удалением бензилоксикарбонильной защитной группы в условиях гидрогенолиза. Данную схему 6 можно модифицировать с использованием методов, известных специалистам-практикам, и получить соединение 28.
Схема 7

Схема 7 отображает другой подход к получению соединений данного изобретения. На схеме 7 переменные имеют значения, указанные в данном описании.
Соответственно, одно воплощение данного изобретения относится к способу получения соединения формулы I, определение которому дается в любом из приведенных в данном описании воплощений, включающему стадию взаимодействия соединения формулы II в присутствии соединения формулы III с образованием соединения формулы IV:

где в формулах:
R10 представляет собой аминозащитную группу, остаток Р3 ингибитора HCV-протеазы, раскрытого в данном описании, или остаток Р4-Р3 ингибитора HCV-протеазы, раскрытого в данном описании, и где остатки Р3 и Р4-Р3, необязательно, защищены аминоконцевой защитной группой;
R11 представляет собой карбоксизащитную группу или остаток Р1 ингибитора HCV-протеазы, раскрытого в данном описании, где остаток Р1, необязательно, защищен карбоксиконцевой защитной группой или W. Ar имеет значения, указанные в любом из воплощений, раскрытых в данном описании. Х представляет собой подходящую удаляемую группу. Как могут понять специалисты-практики, подходящую удаляемую группу можно получить in situ.
В другом воплощении 4-гидроксигруппу в формуле II можно превратить в удаляемую группу. В таком воплощении Х представляет собой нуклеофильный атом кислорода, который реагирует с II с образованием IV.
Используемые в данном описании обозначения Р1, Р3 и Р4 относятся к остаткам ингибиторов HCV-протеазы, раскрытых в данном описании, и хорошо известных специалистам-практикам.
Соединение формулы IV можно превратить в соединение формулы I согласно способам, раскрытым в данном описании.
Хотя ниже отображены и описаны некоторые примеры воплощений, следует иметь в виду, что соединения данного изобретения можно получить согласно способам, описанным, в целом, выше, с использованием соответствующих исходных веществ, как правило, доступных рядовому специалисту в данной области техники.
Другое воплощение данного изобретения относится к композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль. Согласно предпочтительному воплощению в композиции присутствует соединение формулы I в количестве, эффективном для снижения вирусной нагрузки в образце или у пациента, где указанный вирус кодирует серинпротеазу, необходимую для жизненного цикла вируса, и фармацевтически приемлемый носитель.
Если в таких композициях используют фармацевтически приелемые соли соединений данного изобретения, такие соли, предпочтительно, получают с неорганическими или органическими кислотами и основаниями. К числу таких солей присоединения кислот относятся ацетаты, адипаты, альгинаты, аспартаты, бензоаты, бензолсульфонаты, бисульфаты, бутираты, цитраты, камфораты, камфорсульфонаты, циклопентанпропионаты, диглюконаты, додецилсульфаты, этансульфонаты, фумараты, глюкогептаноаты, глицерофосфаты, гемисульфаты, гептаноаты, гексаноаты, гидрохлориды, гидробромиды, гидроиодиды, 2-гидроксиэтансульфонаты, лактаты, малеаты, метансульфонаты, 2-нафталинсульфонаты, никотинаты, оксалаты, памоаты, пектинаты, персульфаты, 3-фенилпропионаты, пикраты, пивалаты, пропионаты, сукцинаты, тартраты, тиоцианаты, тозилаты и ундеканоаты. Соли присоединения оснований включают аммониевые соли, соли щелочных металлов, такие как натриевые и калиевые соли, соли щелочноземельных металлов, такие как кальциевые и магниевые соли, соли с органическими основаниями, такие как соли дициклогексиламина, N-метил-D-глюкамина, и соли, образованные с аминокислотами, такими как аргинин, лизин и т.д.
Также основные азотсодержащие группы могут быть кватернизованы такими агентами, как низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлорид, -бромид и -иодид; диалкилсульфаты, такие как диметил-, диэтил-, дибутил- и диамилсульфаты; длинноцепные галогениды, такие как децил-, лаурил-, миристил- и стеарилхлорид, -бромид и -иодид; аралкилгалогениды, такие как бензил- и фенетилбромид, и другие. Посредством этого получают водо- или маслорастворимые или диспергируемые продукты.
Соединения, используемые в композициях и способах данного изобретения, также можно модифицировать добавлением подходящих функциональных групп для усиления избирательных биологических свойств. Такие модификации известны в данной области техники и включают изменения, повышающие биологическое проникание в данную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему), повышают оральную доступность, повышают растворимость для возможности введения путем инъекции, изменяют метаболизм и изменяют скорость выведения.
Фармацевтически приемлемые носители, которые можно использовать в указанных композициях, включают, но не ограничиваются перечисленным, иониты, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных жирных кислот растительных масел, воду, соли или электролиты, такие как протаминсульфат, динатрийгидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрийкарбоксиметилцеллюлозу, полиакрилаты, воски, полиэтилен-полиоксипропиленовые блоксополимеры, полиэтиленгликоль и ланолин.
Согласно предпочтительному воплощению композиции данного изобретения получают для введения млекопитающему, предпочтительно человеку, как фармацевтическое средство.
Такие фармацевтические композиции настоящего изобретения можно вводить орально, парентерально, ингаляцией, местно, ректально, назально, буккально, вагинально или с помощью имплантированного резервуара. Термин "парентеральный", используемый в данном описании, включает методы подкожной, внутривенной, внутримышечной, интраартикулярной, интрасиновиальной, интрастернальной, интратекальной, внутрипеченочной, внутриочаговой и интракраниальной инъекции или инфузии. Предпочтительно композиции вводят орально или внутривенно.
Стерильные инъецируемые формы композиций данного изобретения могут представлять собой водные или масляные суспензии. Такие суспензии можно получить согласно методам, известным в технике, с использованием подходящих диспергаторов или смачивающих веществ и суспендирующих веществ. Стерильный препарат для инъекций также может представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. К числу приемлемых сред и растворителей, которые можно использовать, относятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют нелетучие масла. Для такой цели можно использовать любое не вызывающее раздражения нелетучее масло, в том числе синтетические моно- или диглицериды. При получении препаратов для инъекций применимы жирные кислоты, такие как олеиновая кислота и ее глицериды, а также природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, в особенности в их полиоксиэтилированных формах. Такие масляные растворы или суспензии также могут содержать длинноцепочечный спирт как разбавитель, или диспергатор, такой как карбоксиметилцеллюлоза или подобные диспергаторы, которые обычно используют при получении фармацевтически приемлемых дозированных форм, в том числе эмульсий и суспензий. Также в целях получения препаратов можно использовать другие обычно используемые поверхностно-активные вещества, такие как твины, спаны, и другие эмульгаторы или усилители биодоступности, которые обычно используют при получении фармацевтически приемлемых жидких, твердых или других дозированных форм.
Уровни дозировки от примерно 0,01 до примерно 100 мг/кг массы тела в сутки, предпочтительно от примерно 0,5 до примерно 75 мг/кг веса тела в сутки, соединений ингибиторов протеаз, раскрытых в данном описании, применимы для предупреждения и лечения в монотерапии против вирусов, в частности заболеваний, опосредуемых HCV. Типично фармацевтические композиции данного изобретения следует вводить от примерно 1 до примерно 5 раз в сутки или, с другой стороны, в виде непрерывной инфузии. Такое введение можно использовать как при длительном лечении, так и при неотложной помощи. Количество активного ингредиента, которое можно объединять с носителями для получения единичной дозированной формы, будет изменяться в зависимости от реципиента, которого лечат, и конкретного способа введения. Типичный препарат будет содержать от примерно 5% до примерно 95% активного соединения (мас./мас.). Предпочтительно такие препараты содержат от примерно 20% до примерно 80% активного соединения. Как признано специалистами-практиками, дозировки интерферона, типично, измеряют в МЕ (например, от примерно 4 миллиона МЕ до примерно 12 миллионов МЕ).
Когда композиции данного изобретения содержат комбинацию соединения формулы I и одного или нескольких других лечебных или профилактических средств, как данное соединение, так и другое средство должны присутствовать в количествах от примерно 10 до 100%, предпочтительнее от примерно 10 до 80%, от дозировки, обычно вводимой по лечебной схеме монотерапии.
Фармацевтические композиции данного изобретения можно вводить орально в орально приемлемой дозированной форме, включая капсулы, таблетки, водные суспензии или растворы и другие формы. В случае таблеток для орального применения носители, которые обычно используют, включают лактозу и кукурузный крахмал. Также типично используют лубриканты, такие как стеарат магния. Для орального введения в форме капсул применимыми разбавителями являются лактоза и сухой кукурузный крахмал. Когда для орального применения требуются водные суспензии, активный ингредиент объединяют с эмульгатором и суспендирующим веществом. При необходимости также можно добавлять подслащивающие вещества, корригенты или красители.
С другой стороны, фармацевтические композиции данного изобретения можно вводить в форме суппозиториев для ректального применения. Их можно получить, смешивая вещество с подходящим нераздражающим эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре, и, следовательно, будет плавиться в прямой кишке, высвобождая лекарственное средство. К таким веществам относятся масло какао, пчелиный воск и полиэтиленгликоль.
Фармацевтические композиции данного изобретения также можно вводить местно, в особенности когда мишень для лечения включает участки или органы, легко доступные для местного применения, в том числе при заболеваниях глаз, кожи или нижнего отдела кишечника. Подходящие композиции для местного применения к каждому такому участку или органу получают легко.
Местное применение к нижнему отделу кишечника можно осуществить с композицией - ректальным суппозиторием (см. выше) или подходящим составом для клизмы. Также для местного применения можно использовать трансдермальные пластыри.
Для местных применений композиции можно получить в форме подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или нескольких носителях. К носителям для местного применения соединений данного изобретения относятся, но не ограничиваясь ими, минеральное масло, вазелиновое масло, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгирующий воск и вода. С другой стороны, фармацевтические композиции можно получить в форме подходящего лосьона или крема, содержащих активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Подходящими носителями являются, но не ограничиваются ими, минеральное масло, моностеарат сорбита, полисорбат 60, воск цетилэфиров, цетиловый спирт, 2-октилдодеканол, бензиловый спирт и вода.
Для офтальмического применения фармацевтические композиции можно получить в виде тонких суспензий в изотоническом, с отрегулированным рН, стерильном физиологическом растворе или, предпочтительно, в виде растворов в изотоническом, с отрегулированным рН, стерильном физиологическом растворе, или без консерванта или с консервантом, таким как хлорид бензалкония. С другой стороны, фармацевтические композиции для офтальмических применений можно получить в форме мазей, таких как на вазелиновой основе.
Фармацевтические композиции данного изобретения также можно вводить с помощью назального аэрозоля или ингаляции. Такие композиции получают согласно методам, хорошо известным в технике фармацевтических препаратов, и получают в виде растворов в физиологическом растворе, с использованием бензилового спирта или других подходящих консервантов, промоторов абсорбции для усиления биодоступности, фтороуглеводородов и/или других обычных солюбилизаторов или диспергаторов.
Наиболее предпочтительными являются фармацевтические композиции, полученные для орального применения.
В другом воплощении композиции данного изобретения также содержат другое противовирусное средство, предпочтительно средство против HCV. К таким противовирусным средствам относятся, включая, но не ограничиваясь ими, иммуномодуляторы, такие как α-, β- и γ-интерфероны, пэгилированные производные α-интерферона и тимозин; другие противовирусные средства, такие как рибаварин, амантадин и тельбувидин; другие ингибиторы протеаз вируса гепатита С (ингибиторы NS2-NS3 и ингибиторы NS3-NS4); ингибиторы других мишеней в жизненном цикле HCV, в том числе ингибиторы геликазы и полимеразы; ингибиторы внутреннего ввода рибосом; ингибиторы вирусов широкого спектра, такие как ингибиторы IMPDH (например, соединения по патентам США 5807876, 6498178, 6344465, 6054472, WO 97/40028, WO 98/40381, WO 00/56331 и микофеноловая кислота и ее производные, и, включая, но не ограничиваясь ими, VX-497, VX-148 и/или VX-944); или их любые комбинации. См. также W. Markland et al., Antimicrobial & Antiviral Chemotherapy, 44, p.859 (2000); и патент США 6541496.

В данном описании используются приведенные далее определения (с торговыми знаками, относящимся к продуктам, доступным на дату подачи данной заявки).
"Пэгинтрон" обозначает ПЭГ-интрон® - пэгинтерферон-альфа-2b, доступный от Schering Corporation, Kenilworth, NJ;
"интрон" обозначает интрон-А® - интерферон-альфа-2b, доступный от Schering Corporation, Kenilworth, NJ;
"рибаварин" обозначает рибаварин (1-бета-D-рибофуранозил-1Н-1,2,4-триазол-3-карбоксамид, доступный от ICN Pharmaceuticals, Inc., Costa Mesa, CA; описан в Merck Index, запись 8365, двенадцатое издание; также доступен как ребетол® от Schering Corporation, Kenilworth, NJ, или как копегус® от Hoffmann-La Roche, Nutley, NJ;
"пегасис" обозначает пегасис® - пэгинтерферон-альфа-2а, доступный от Hoffmann-La Roche, Nutley, NJ;
"роферон" обозначает роферон® - рекомбинантный интерферон-альфа-2а, доступный от Hoffmann-La Roche, Nutley, NJ;
"берефор" обозначает берефор® - интерферон-альфа-2а, доступный от Boehringer Ingelheim Pharmaceutical, Inc., Ridgelfield, CT;
Сумиферон® - очищенная смесь природных альфа-интерферонов, такая как сумиферон, доступный от Sumitomo, Япония;
велферн® - интерферон-альфа-n1, доступен от Glaxo-Wellcome Ltd., Великобритания;
альферон® - смесь природных альфа-интерферонов, производимая Interferon Sciences и доступная от Purdue Frederick Co., CT.
Термин "интерферон", используемый в данном описании, обозначает член семейства высокогомологичных видоспецифических белков, которые ингибируют репликацию вирусов и клеточную пролиферацию и модулируют иммунную реакцию, такой как интерферон-альфа, интерферон-бета или интерферон-гамма. Merck Index, запись 5015, двенадцатое издание.
Согласно одному воплощению настоящего изобретения интерферон представляет собой α-интерферон. Согласно другому воплощению в терапевтической комбинации по настоящему изобретению используют природный альфа-интерферон-2а, или в терапевтической комбинации по настоящему изобретению используют природный альфа-интерферон-2b. В другом воплощении в терапевтической комбинации по настоящему изобретению используют рекомбинантный альфа-интерферон-2а или 2b. В еще одном воплощении интерферон представляет собой пэгилированный альфа-интерферон-2а или 2b. К интерферонам, подходящим для настоящего изобретения, относятся
(а) интрон (интерферон-альфа-2В, Schering Plough);
(b) пэг-интерферон;
(с) пегасис;
(d) роферон;
(е) берофор;
(f) сумиферон;
(g) велферон;
(h) консенсус-альфа-интерферон, доступный от Amgen, Inc., Newbury Park, CA;
(i) альферон;
(j) вираферон®;
(k) инферген®.
Как признано специалистами-практиками, ингибитор протеазы можно было бы, предпочтительно, вводить орально. Интерферон типично не вводят орально. Тем не менее, в данном случае ничто не ограничивает способы или комбинации данного изобретения какими-либо характерными формами или схемой применения. Так, каждый компонент комбинации по данному изобретению можно вводить по отдельности, их можно вводить вместе или в любой их комбинации.
В одном воплощении ингибитор протеазы и интерферон вводят в отдельных дозированных формах. В одном воплощении любое другое средство вводят как часть одной дозированной формы с ингибитором протеазы или как отдельную дозированную форму. Так как данное изобретение включает комбинацию соединений, конкретные количества каждого соединения могут зависеть от конкретного количества каждого другого соединения в комбинации. Как признано специалистами-практиками, дозировки интерферона типично определяют в МЕ (например, от примерно 4 миллионов МЕ до примерно 12 миллионов МЕ).
Соответственно, средства (действующие как иммономодуляторы или иначе), которые можно использовать в комбинации с соединением данного изобретения, включают интерферон-альфа-2В (интрон А, Schering Plough); ребатрон (Schering Plough, интерферон-альфа-2В + рибаварин); пэгилированный интерферон-альфа (Reddy K.R. et al., "Efficacy and Safety of Pegylated (40-kd) interferon alpha-2a compared with interferon alpha-2a in noncirrhotic patients with chronic hepatitis C" (Hepatology, 33, pp.433-438 (2001)); консенсусный интерферон (Kao, J.H., et al., "Efficacy of Consensus in the Treatement of Chronic Hepatitis" J. Gastroenterol. Hepatol. 15, pp.1418-1423 (2000)), интерферон-альфа 2A (Роферон A; Roche), лимфобластоидный или природный интерферон; интерферон тау (Clayette, P. et al., "IFN-tau, A Nev Interferon Type I with Antiretroviral activity" Pathol. Biol. (Paris) 47, pp.553-559 (1999)); интерлейкин 2 (Davis, G.L. et al., "Future Option for the Management of Hepatitis C." Seminars in Liver Disease, 19, pp.103-112 (1999)); интерлейкин 6 (Davis et al. "Future Option for the Management of Hepatitis C." Seminars in Liver Disease, 19, pp.103-112 (1999)); интерлейкин 12 (Davis, G.L. et al., "Future Options for the Management of Hepatitis C", Seminars in Liver Disease, 19, pp.103-112 (1999)); рибавирин; и соединения, которые усиливают развитие Т-хелперных клеток типа 1 (Davis et al., "Future Option for the Management of Hepatitis C", Seminars in Liver Disease, 19, pp.103-112 (1999)). Интерфероны могут уменьшать интенсивность вирусных инфекций, проявляя прямое противовирусное действие и/или модифицируя иммунную реакцию на заражение. Противовирусное действие интерферонов часто передается через ингибирование проникновения вирусов или декапсидацию вирусов, синтез вирусной РНК, трансляцию вирусных белков и/или сборку и высвобождение вирусов.
Соединения, стимулирующие синтез интерферона в клетках (Tazulakhova E.B. et al., "Russian Experience in Screening, analysis, and Clinical Application of Novel Interferon Inducers", J. Interferon Cytokine Res., 21, pp.65-73), включают, но не ограничиваются ими, двухцепочечную РНК, одну или в комбинации с тобрамицином, и имиквимод (3М Pharmaceuticals; Sauder D.N., "Immunomodulatory and Pharmacologic Properties of Imiquimod", J. Am. Acad. Dermatol., 43, pp.S6-11 (2000)).
Можно использовать другие неиммуномодулирующие или иммуномодулирующие соединения в комбинации с соединением данного изобретения, но не ограничиваясь ими, описанные в WO 02/18369, включенной в данное описание посредством ссылки (см., например, с.273, строки 9-22, и от с.274, строка 4, до с.276, строка 11), и другие соединения.
Данное изобретение также может включать введение ингибитора цитохром-450-монооксигеназы. Ингибиторы CYP могут быть пригодны при повышении концентраций в печени и/или повышении содержания в крови соединений, которые ингибируются CYP.
Если воплощение данного изобретения включает ингибитор CYP, в способе данного изобретения можно использовать любой ингибитор CYP, который улучшает фармакокинетику релевантной протеазы NS3/4A. К таким ингибиторам CYP относятся, но не ограничиваются ими, ритонавир (WO 94/14436), кетоконазол, тролеандомицин, 4-метилпиразол, циклоспорин, клометиазол, циметидин, итраконазол, флуконазол, миконазол, флувоксамин, флуоксетин, нефазодон, сертралин, индинавир, нелфинавир, ампренавир, фозампренавир, саквинавир, лопинавир, делавирдин, эритромицин, VX-944 и VX-497. Предпочтительными ингибиторами CYP являются ритонавир, кетоконазол, тролеандомицин, 4-метилпиразол, циклоспорин и клометиазол. В отношении предпочтительных дозированных форм ритонавира см. патент США 6037157 и документы, цитированные в нем: патент США 5484801, заявку на патент США 08/402690 и заявки на Международный патент WO 95/07696 и WO 95/09614.
Способы измерения способности соединения ингибировать активность цитохром-450-монооксигеназы известны (см. US 6037157, и Yun et al., Drug Metabolism & Disposition, vol.21, pp.403-407 (1993)).
После улучшения состояния пациента, при необходимости, можно вводить поддерживающую дозу соединения, композиции или комбинации данного изобретения. Затем можно уменьшить дозировку или частоту введения или то и другое, как функцию симптомов, до уровня, при котором улучшенное состояние сохраняется, и когда симптомы ослаблены до нужного уровня, лечение следует прекратить. Однако пациентам может потребоваться периодическое лечение в течение длительного времени после рецидива симптомов заболевания.
Следует также иметь в виду, что конкретная дозировка и схема лечения для определенного пациента будут зависеть от ряда факторов, включая активность конкретного используемого соединения, возраст, массу тела, общее состояние здоровья, пол, питание пациента, время введения, скорость выведения, комбинацию лекарственных средств и оценку лечащего врача и тяжесть определенного заболевания, от которого лечат. Количество активных ингредиентов также будет зависеть от определенного выписанного соединения и отсутствия или наличия и природы другого противовирусного средства в композиции.
Согласно другому воплощению изобретение относится к способу лечения пациента, зараженного вирусом, характеризующимся кодированной вирусом серинпротеазы, необходимой для жизненного цикла вируса, путем введения указанному пациенту фармацевтически приемлемой композиции данного изобретения. Предпочтительно способы данного изобретения применяют для лечения пациента, страдающего от заражения HCV. Такое лечение может полностью устранить вирусную инфекцию или уменьшить ее тяжесть. Предпочтительнее пациентом является человек.
В другом воплощении способы данного изобретения дополнительно включают стадию введения указанному пациенту противовирусного средства, предпочтительно средства против HCV. К таким противовирусным средствам относятся, но не ограничиваются ими, иммуномодуляторы, такие как α-, β- и γ-интерфероны, пэгилированные производные α-интерферона и тимозин; другие противовирусные средства, такие как рибаварин и амантадин; другие ингибиторы протеаз вируса гепатита С (ингибиторы NS2-NS3 и ингибиторы NS3-NS4); ингибиторы других мишеней в жизненном цикле HCV, в том числе ингибиторы геликазы и полимеразы; ингибиторы внутреннего ввода рибосом; ингибиторы вирусов широкого спектра, такие как ингибиторы IMPDH (ингибиторы IMPDH, раскрытые в патенте США 5807876, микофеноловая кислота и ее производные); или любые комбинации средств, перечисленных выше.
Такое другое средство можно вводить пациенту в одной дозированной форме, содержащей как соединение данного изобретения, так и другое противовирусное средство. С другой стороны, другое средство можно вводить отдельно от соединения данного изобретения как часть сложной дозированной формы, где указанное другое средство вводят до, вместе с или после композиции, содержащей соединение данного изобретения.
В еще одном воплощении настоящее изобретение относится к предварительной обработке биологического вещества, предназначенного для введения пациенту, включающей стадию контактирования указанного биологического вещества с фармацевтически приемлемой композицией данного изобретения. Такими биологическими веществами являются, но не ограничиваются ими, кровь и ее компоненты, такие как плазма, тромбоциты, субпопуляции клеток крови и т.п.; органы, такие как почки, печень, сердце, легкие и т.д.; сперма и зрелые яйцеклетки; костный мозг и его компоненты, и другие жидкости, которые вливают пациенту, такие как физиологический раствор, декстроза и т.д.
Согласно другому воплощению изобретение относится к способам обработки материалов, которые потенциально могут контактировать с вирусом, характеризуемым кодированной вирусом серинпротеазой, необходимой для жизненного цикла вируса. Данный способ включает стадию контактирования указанного материала с соединением по изобретению. К таким материалам относятся, но не ограничиваются ими, хирургические инструменты и предметы одежды (например, пеленки, перчатки, фартуки, халаты, маски, очки, обувь и т.д.); лабораторные инструменты и предметы одежды (например, пеленки, перчатки, фартуки, халаты, маски, очки, обувь и т.д.); приспособления и материалы для взятия крови и инвазивные устройства, такие как шунты, стенты и т.д.
В другом воплощении соединения данного изобретения можно применять в качестве лабораторных инструментов, помогающих при выделении кодированной вирусом серинпротеазы. Такой способ включает стадии предоставления соединения данного изобретения, присоединенного к твердому носителю; приведения в контакт указанного твердого носителя с образцом, содержащим вирусную серинпротеазу, в условиях, вынуждающих указанную протеазу связываться с указанным твердым носителем; и элюирования указанной серинпротеазы из указанного твердого носителя. Предпочтительно вирусная серинпротеаза, выделяемая таким способом, представляет собой протеазу NS3-NS4A HCV.
Для того чтобы данное изобретение понималось полнее, далее приводятся препаративные примеры и примеры испытаний. Указанные примеры приводятся только в целях пояснения и никоим образом не должны рассматриваться как ограничивающие объем изобретения.
Примеры
Спектры1Н ЯМР регистрируют при 500 МГц с использованием прибора Brucker AMX 500. Образцы для масс-спектров анализируют на масс-спектрометре MicroMass ZQ или Quattro II, работающем в одном режиме с ионизацией электронным распылением. Образцы вводят в масс-спектрометр с использованием впрыскивания (FIA) или хроматографии. Подвижная фаза для всех масс-спектрографических анализов состоит из смесей ацетонитрил-вода с 0,2% муравьиной кислоты как модификатора.
Используемые в данном описании обозначения "Rt(мин)" или "RT" относятся ко времени удерживания при ВЭЖХ, в минутах, связанному с соединением. Приведенные времена удерживания при ВЭЖХ получают из масс-спектрографических данных или с использованием способа, описанного далее (способ В).
Прибор: Hewlett Packard HP-1050;
Колонка: YMC C18 (кат. №326289С46);
Градиент/время в градиенте: 10-90% CH3CN/H2O в течение 9 минут, затем 100% CH3CN в течение 2 минут;
Скорость потока: 0,8 мл/мин;
Длина волны детекции: 215 нм и 245 нм.
Химические названия выбранных соединений в данном описании получают с использованием программы наименований, предоставленной CambridgeSoft Corporations ChemDraw Ultra®, версия 7.0.1.
Пример 1
Получение соединения 59
2-трет-Бутиловый эфир, 1-бензиловый эфир 4-гидроксипирролидин-1,2-дикарбоновой кислоты (3)
Коммерчески доступный (Bachem) Z-гидроксипролин (1) (10 г, 42,51 ммоль) растворяют в 90 мл ТГФ (тетрагидрофуран) и охлаждают до 0°С на бане со смесью воды со льдом. К полученному раствору через капельную воронку в течение 30 минут добавляют предварительно полученный трет-бутил-N,N'-диизопропилимидокарбамат (2) (27 мл, 135 ммоль). По окончании добавления ледяную баню убирают, и реакционную смесь перемешивают при температуре окружающей среды в течение 24 часов. Объем реакционной смеси уменьшают, и затем добавляют диэтиловый эфир перед промыванием насыщенным раствором бикарбоната натрия, затем 0,5 М соляной кислотой, затем водой, и, наконец, рассолом. Органический слой сушат над сульфатом натрия и концентрируют в вакууме, и получают 15 г сырого вещества. Вещество пропускают через слой SiO2, элюируют смесью 45% EtOAc-гексан, и получают 2-трет-бутиловый эфир, 1-бензиловый эфир 4-гидроксипирролидин-1,2-дикарбоновой кислоты 3 в виде бесцветного масла 11,0 г (81%).
1Н ЯМР (CDCl3, м.д.) δ 7,35 (м, 5Н), 5,2 (м, 2Н), 4,3 (м, 2Н), 4,65 (м, 3Н), 2,35 (м, 1Н), 2,1 (т, 1Н), 1,35, 1,55 (ротамеры, 1,45, 9Н).
трет-Бутиловый эфир 1-(2-{2-циклогексил-2-[(пиразин-2-карбонил)амино]ацетиламино}-3,3-диметилбутирил)-4-гидроксипирролидин-2-карбоновой кислоты (6)
Перемешивают соединение (3) в EtOH, и добавляют каталитическое количество 10% Pd-на-угле, затем смесь перемешивают при давлении 1 атмосфера водорода с использованием баллона. Через 12 часов ТСХ показывает, что реакция завершилась, и катализатор отфильтровывают и промывают EtOH. Фильтрат концентрируют, остаток сушат в высоком вакууме, и получают амин в виде желтого твердого вещества, которое переносят на следующую стадию. Z-Tbg-OH (8,3 г, 31,1 ммоль) растворяют в NMP, и к раствору добавляют EDC (6,0 г, 31,1 ммоль), НОВТ (4,2 г, 31,1 ммоль) и DMAP (340 мг, 2,8 ммоль), и смесь охлаждают до 0°С с использованием бани со смесью воды со льдом. К полученной смеси добавляют амин в виде раствора в NMP, и реакционную смесь перемешивают в течение 2 суток. Реакционную смесь выливают на лед и подкисляют 0,5 н соляной кислотой до рН 5 и затем экстрагируют EtOAc. Органические экстракты промывают насыщенным раствором бикарбоната натрия, затем водой и, наконец, рассолом. Органический слой сушат над сульфатом натрия и концентрируют в вакууме, и получают 14,8 г сырого вещества. Осуществляют очистку с использованием хроматографии на SiO2 с элюированием смесью 50% EtOAc-гексан. Концентрирование однородных фракций дает 10,5 г соединения 4 в виде бесцветной пены (85%), которое используют как оно есть на следующей стадии.
К смеси соединения 4 (10,5 г, 24,16 ммоль) в EtOH добавляют каталитическое количество 10% Pd-на-угле, и затем смесь перемешивают при давлении 1 атмосфера водорода с использованием баллона. Через 12 часов ТСХ показывает, что реакция завершилась, и катализатор отфильтровывают и промывают EtOH. Фильтрат концентрируют, остаток сушат в высоком вакууме, и получают амин в виде желтого твердого вещества, которое передают на следующую стадию. Z-Chg-OH (7,7 г, 26,6 ммоль) растворяют в NMP, и к раствору добавляют EDC (5,1 г, 26,7 ммоль) и НОВТ (3,6 г, 26,6 ммоль), и смесь охлаждают до 0°С с использованием бани со смесью воды со льдом. К полученной смеси добавляют полученный ранее амин в виде раствора в NMP, и реакционную смесь перемешивают в течение 2 суток. Реакционную смесь выливают на лед с рассолом и затем экстрагируют EtOAc. Органические экстракты промывают 0,5 н соляной кислотой, насыщенным раствором бикарбоната натрия, водой и, наконец, рассолом. Органический экстракт сушат над сульфатом натрия и концентрируют в вакууме, и получают 15,31 г соединения 5 в виде неочищенного вещества, которое используют как оно есть на следующей стадии.
К раствору соединения 5 (5,6 г, 9,76 ммоль) в EtOH добавляют каталитическое количество 10% Pd-на-угле, затем смесь перемешивают при давлении 1 атмосфера водорода с использованием баллона. Через 12 часов ТСХ показывает, что реакция завершилась, и катализатор отфильтровывают и промывают EtOH. Фильтрат концентрируют, остаток сушат в высоком вакууме, и получают амин в виде аморфного твердого вещества, которое передают на следующую стадию. Пиразин-2-карбоновую кислоту (1,45 г, 11,7 ммоль) растворяют в NMP, и к раствору добавляют EDC (2,24 г, 11,7 ммоль) и НОВТ (1,34 г, 11,7 ммоль), и смесь охлаждают до 0°С с использованием ледяной бани. К полученной смеси добавляют полученный ранее амин в виде раствора в NMP, и реакционную смесь перемешивают в течение 2 суток. Реакционную смесь выливают на лед с рассолом и затем экстрагируют EtOAc. Органические экстракты промывают 0,5 н соляной кислотой, насыщенным раствором бикарбоната натрия, затем водой и, наконец, рассолом. Органический экстракт сушат над сульфатом натрия и концентрируют в вакууме, и получают 5,3 г (99%) соединения 6 в виде бесцветной пены, которую используют как она есть на следующей стадии.
({1-[4-(5-Хлорпиридин-2-илокси)-2-(1-циклопропиламинооксалилбутилкарбамоил)пирролидин-1-карбонил]-2,2-диметилпропилкарбамоил}циклогексилметил)амид пиразин-2-карбоновой кислоты (59)
К раствору 6 (0,15 г, 0,28 ммоль) в безводном ТГФ добавляют трифенилфосфин (0,131 г, 0,5 ммоль), 2-гидрокси-4-хлорпиридин (65 мг, 0,5 ммоль), и последним добавляют диэтилазодикарбоксилат (0,100 мл, 1,85 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 18 часов или до тех пор, пока ВЭЖХ не покажет отсутствие соединения 6. ТГФ удаляют из реакционной смеси, и затем вещество растворяют в EtOAc, и раствор промывают 0,1 н NaOH, 0,5 н соляной кислотой, водой и, наконец, рассолом. Органический экстракт сушат над сульфатом натрия и концентрируют в вакууме, и получают сырой трет-бутиловый эфир. трет-Бутиловый эфир гидролизуют до карбоновой кислоты обработкой 50% трифторуксусной кислотой в дихлорметане в течение 3 часов. Растворитель удаляют в вакууме, и затем остаток растворяют в 0,1 н NaOH и промывают EtOAc. Водную фазу подкисляют 5% лимонной кислотой и затем экстрагируют EtOAc. Полученную органическую фазу промывают водой и рассолом, затем органический экстракт сушат над сульфатом натрия и концентрируют в вакууме, и получают 4-(5-хлорпиридин-2-илокси)-2-1-(2-{2-циклогексил-2-[(пиразин-2-карбонил)амино]ацетиламино}-3,3-диметилбутирил)пирролидин-2-карбоновую кислоту 7а в виде бесцветной пены, которую используют как она есть на следующей стадии.
К раствору соединения 7а в 2 мл диметилформамида добавляют TBTU (0,15 г, 0,47 ммоль) и DIEA (0,15 мл, 1,1 ммоль), реакционную смесь перемешивают в течение 1,5 часов, и затем к смеси добавляют амин 10 [U. Schoellkogf et al., Justus Liebigs Ann. Chem. GE, pp.183-202 (1976); и J. Semple et al., Org. Letts., 2, pp.2769-2772 (2000)] и затем 4-метилморфолин (0,2 мл, 1,82 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 12 часов и затем выливают в воду и экстрагируют EtOAc. Органический экстракт сушат над сульфатом натрия и концентрируют в вакууме, и получают [(1-{4-(5-хлорпиридин-2-илокси)-2-[1-(циклопропилкарбамоилгидроксиметил)-бутилкарбамоил]пирролидин-1-карбонил}-2,2-диметилпропилкарбамоил)циклогексилметил]амид пиразин-2-карбоновой кислоты 11а (40 мг) в виде бесцветной пены, которую используют как она есть на следующей стадии. К раствору 11а (40 мг) в дихлорметане (4 мл) добавляют трет-бутанол (0,1 мл) и периодинан Десс-Мартина (40 мг, 0,086 ммоль), и затем смесь перемешивают при температуре окружающей среды в течение 6 часов. К реакционной смеси добавляют 1 мл смеси 1 н раствора тиосульфата натрия и насыщенного раствора бикарбоната натрия (1:1). Через 15 минут реакционную смесь экстрагируют EtOAc, и затем растворитель удаляют в вакууме. Очистку осуществляют с использованием хроматографии на SiO2 с элюированием смесью 50% EtOAc-гексан. Концентрирование однородных фракций дает 0,095 г соединения 59 в виде бесцветной пены (4,5% относительно 0,28 ммоль 6).1Н ЯМР (CDCl3, м.д.) δ 9,39 (с, 1Н), 8,76 (с, 1Н), 8,56 (с, 1Н), 8,24 (д, 1Н, J=9,6 Гц), 8,08 (с, 1Н), 7,8 (д, 1Н, 6,4 Гц), 7,69 (с, 1Н), 7,47-7,40 (м, 2Н), 7,47 (д, 1Н, J=8,75 Гц), 5,63 (с, 1Н), 5,60-5,50 (м, 1Н), 4,90 (м, 1Н), 4,70 (м, 1Н), 4,11 (д, 1Н, J=11,6 Гц), 4,0 (м, 1Н), 2,90 (м, 1Н) 2,60 (м, 1Н), 2,25 (м, 1Н), 2,0 (м), 1,90-1,4 (м, 1Н), 1,25-0,8 (м, 18Н), 0,73 (м, 2Н); ЖХ/МС: RT = 3,61 мин, 4,15 мин (10-90% CH3CN/7 мин); МН+ = 767,3, М- = 765,5.
Пример 2
Другой способ получения соединения 59
Вос-Pro-(4(R)-5-Хлорпиридин-2-илокси)ОН (15а)
Вос-Нур-ОН (130 г, 562,16 ммоль) растворяют в безводном ДМСО (1,6 л), и к полученному раствору добавляют 1 М раствор трет-бутоксида калия в ТГФ (1,4 л, 140 ммоль), поддерживая температуру смеси ниже 25°С. После перемешивания раствора в течение 1,5 часов при RT добавляют 2,5-дихлорпиридин (90,0 г, 608,15 ммоль), и реакционную смесь перемешивают в течение 18 часов при RT. Смесь выливают в воду и экстрагируют диэтиловым эфиром (1 л) для удаления избытка 2,5-дихлорпиридина. Затем водный слой подкисляют 1 н HCl (0,8 л) и два раза экстрагируют этилацетатом (2,5 л). Органические слои объединяют и промывают рассолом. Этилацетатный слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, и получают коричневое масло - 210 г сырого вещества.
Вос-Pro-(4(R)-5-Хлорпиридин-2-илокси)аллиловый эфир (16)
Вос-Pro-(4(R)-5-Хлорпиридин-2-илокси)ОН (15а) (~210 г, 557 ммоль) растворяют в безводном ацетонитриле (1,5 л), последовательно добавляют 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (DBU) (0,13 л, 869,28 ммоль) и аллилбромид (81,5 г, 673,66 ммоль), и реакционную смесь перемешивают в течение 18 часов при RT. Смесь концентрируют, полученное масло разбавляют этилацетатом (2 л) и последовательно промывают два раза водой (500 мл) и рассолом (500 мл). Этилацетатный слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, и получают коричневое масло, которое вносят в колонку с диоксидом кремния с метиленхлоридом и элюируют смесью 25% этилацетата в гексане, и получают желтое масло, 181 г, 472,77 ммоль, 84% относительно двух стадий.
Pro-(4(R)-5-Хлорпиридин-2-илокси)аллиловый эфир (17)
Вос-Pro-(4(R)-5-Хлорпиридин-2-илокси)аллиловый эфир (16) (181 г, 472,77 ммоль) обрабатывают 440 мл охлажденной до 0°С смеси трифторуксусной кислоты и метиленхлорида (90:10). Смесь оставляют нагреваться до RT и перемешивают в течение 3 час. Через 3 часа к смеси добавляют 400 мл толуола, смесь концентрируют при пониженном давлении, и получают сырую соль (17) трифторуксусной кислоты.
Вос-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллиловый эфир (18)
Неочищенную соль трифторуксусной кислоты Pro-(4(R)-5-хлорпиридин-2-илокси)аллилового эфира (17) (187 г, теор.) соединяют с охлажденным до 0°С раствором Вос-Tbg-ОН (110 г, 475,56 ммоль), NMM (155 мл, 1,410 ммоль), EDC (99 г, 518,32 ммоль) и HOBt (70 г, 518,32 ммоль) в 400 мл метиленхлорида. Смесь оставляют нагреваться до RT и перемешивают в течение 18 час. Смесь концентрируют, полученное масло разбавляют этилацетатом (2 л) и последовательно промывают два раза 0,5 н HCl (500 мл), водой (500 мл) и рассолом (500 мл). Этилацетатный слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, и получают коричневое масло, которое вносят в колонку с диоксидом кремния с метиленхлоридом и элюируют смесью 15% этилацетата в гексане, и получают бледно-желтую пену, 180 г, 362,83 ммоль, 77%.
Tbg-Pro-(4(R)-5-Хлорпиридин-2-илокси)аллиловый эфир (19)
Вос-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллиловый эфир (18) (180 г, 362,83 ммоль) обрабатывают 330 мл охлажденной до 0°С смеси трифторуксусной кислоты и метиленхлорида (90:10). Смесь оставляют нагреваться до RT и перемешивают в течение 3 час. Через 3 часа к смеси добавляют толуол (200 мл), смесь концентрируют при пониженном давлении, и получают желтое масло, к которому последовательно добавляют метиленхлорид (100 мл) и диэтиловый эфир (1,5 л). Смесь перемешивают, и добавляют 4 н раствор HCl в диоксане, продолжают перемешивать в течение 1 часа, сырой гидрохлорид дипептида отфильтровывают и промывают этиловым эфиром, охлажденным до 0°С, и получают 142,5 г вещества (323,86 ммоль, 91%).
Вос-Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллиловый эфир (20)
Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллиловый эфир (19) (142,5 г, 323,86 ммоль) соединяют с охлажденным до 0°С раствором Вос-Chg-ОН (94 г, 365,29 ммоль), NMM (109 мл, 994,18 ммоль), EDC (69 г, 361,26 ммоль) и HOBt (48,77 г, 361,26 ммоль) в 400 мл метиленхлорида. Смесь оставляют нагреваться до RT и перемешивают в течение 18 час. Смесь концентрируют, полученное масло разбавляют этилацетатом (2 л) и последовательно промывают два раза 0,5 н HCl (500 мл), водой (500 мл) и рассолом (500 мл). Этилацетатный слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, и получают коричневое масло, которое вносят в колонку с диоксидом кремния с метиленхлоридом и элюируют смесью 25% этилацетата в гексане, и получают бледно-желтую пену, 180 г, 362,83 ммоль, 77%.
Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллиловый эфир (21)
Вос-Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллиловый эфир (20) (65 г, 102,33 ммоль) обрабатывают 250 мл охлажденной до 0°С смеси трифторуксусной кислоты и метиленхлорида (90:10). Смесь оставляют нагреваться до RT и перемешивают в течение 3 час. Через 3 часа к смеси добавляют толуол (200 мл), смесь концентрируют при пониженном давлении, и получают желтое масло, к которому последовательно при перемешивании добавляют метиленхлорид (200 мл) и диэтиловый эфир (1,5 л), сырую соль ТФК трипептида отфильтровывают и промывают диэтиловым эфиром, охлажденным до 0°С, и получают 66 г вещества, 101,53 ммоль, 98%.
Пиразин-2-карбонил-Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллиловый эфир (22)
Неочищенную соль ТФК Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллилового эфира (21) (66 г, 101,53 ммоль) соединяют с охлажденным до 0°С раствором пиразин-2-карбоновой кислоты (13,6 г, 109,69 ммоль), NMM (44 мл, 400,19 ммоль), EDC (21 г, 109,95 ммоль) и HOBt (14,85 г, 109,95 ммоль) в 500 мл метиленхлорида. Смесь оставляют нагреваться до RT и перемешивают в течение 18 час. Смесь концентрируют, полученное масло разбавляют этилацетатом (2 л) и последовательно промывают два раза 0,5 н HCl (500 мл), водой (500 мл) и рассолом (500 мл). Этилацетатный слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, и получают коричневое масло, которое вносят в колонку с диоксидом кремния с метиленхлоридом и элюируют смесью 45% этилацетата в гексане, и получают бледно-желтую пену, 64,66 г, 100,85 ммоль, 99%.
Пиразин-2-карбонил-Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)ОН (23)
Пиразин-2-карбонил-Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)аллиловый эфир (22) (64,66 г, 100,85 ммоль) растворяют в 250 мл безводной смеси ацетонитрила и метиленхлорида (50:50). Добавляют катализатор тетракис(трифенилфосфин)палладий (0) (1,5 г, 1,30 ммоль), а затем пирролидин (8,55 мл, 102,44 ммоль). Реакционную смесь перемешивают при RT в течение 18 час. Через 18 час растворитель выпаривают. Масло растворяют в этилацетате (2 л) и экстрагируют два раза 10% лимонной кислотой (250 мл) и рассолом (250 мл). Этилацетатный слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, и получают бледно-желтое твердое вещество, 58,5 г, 97,32 ммоль, 96%.
Циклопропиламид 3-амино-2-гидроксигексановой кислоты (24)
Бензиловый эфир [1-(циклопропилкарбамоилгидроксиметил)бутил]карбаминовой кислоты (48 г, 149,82 ммоль) растворяют в метаноле (1 л) и дегазируют азотом в течение пяти минут, добавляют палладий, 10 мас.%, на активированном угле (2,5 г), и затем пропускают водород в течение 18 час. Через 18 час источник водорода удаляют, реакционную смесь дегазируют азотом и фильтруют, полученный фильтрат упаривают, остаток сушат в высоком вакууме, и получают белое твердое вещество, 26,9 г, 144,42 ммоль, 97%.
Пиразин-2-карбонил-Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)-Nva-гидроксициклопропиламид (25)
Циклопропиламид 3-амино-2-гидроксигексановой кислоты (24) (20 г, 107,37 ммоль) соединяют с пиразин-2-карбонил-Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)ОН (23) (58,5 г, 97,32 ммоль), NMM (11,76 мл, 106,96 ммоль), EDC (20,45 г, 107,07 ммоль) и HOBt (14,45 г, 107,07 ммоль) в 250 мл метиленхлорида. Смесь перемешивают в течение 18 час. Смесь концентрируют, полученное масло разбавляют этилацетатом (2 л) и последовательно промывают два раза 0,5 н HCl (500 мл), водой (500 мл) и рассолом (500 мл). Этилацетатный слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении, и получают коричневое масло, которое вносят в колонку с диоксидом кремния с метиленхлоридом и элюируют смесью 2% метанола в этилацетате, и получают бледно-желтую пену, 61,5 г, 79,94 ммоль, 74%.
({1-[4-(5-Хлорпиридин-2-илокси)-2-(1-циклопропиламинооксалилбутилкарбамоил)пирролидин-1-карбонил]-2,2-диметилпропилкарбамоил}циклогексилметил)амид пиразин-2-карбоновой кислоты (59)
В 4-л круглодонную колбу, снабженную подвесной мешалкой, термопарой и вводом для азота, добавляют EDC (229,0 г, 151,83 ммоль), а затем безводный этилацетат (1230 мл). Начинают перемешивание для образования густой взвеси. К полученной взвеси добавляют пиразин-2-карбонил-Chg-Tbg-Pro-(4(R)-5-хлорпиридин-2-илокси)-Nva-гидроксициклопропиламид (25) (61,5 г, 79,94 ммоль), растворенный в этилацетате (250 мл), а затем безводный ДМСО (460 мл). Применяют охлаждающую баню для достижения температуры в колбе 7°С. Добавляют охлажденный до 7°С раствор дихлоруксусной кислоты (65,94 мл, 787,20 ммоль) в этилацетате (100 мл) так, что температура в колбе сохраняется на уровне 12-25°С. Охлаждающую баню убирают, и тонкую взвесь перемешивают в течение 1 часа. Применяют охлаждающую баню, и гасят реакцию 1 н HCl (1,230 мл), в то время как температура сохраняется на уровне 15-25°С. Органический слой отделяют и промывают три раза водой (200 мл) и рассолом (200 мл). Этилацетатный слой сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении на бане при температуре не выше 40°С, и получают коричневое масло, которое вносят в колонку с диоксидом кремния с метиленхлоридом и элюируют смесью 90% этилацетата в гексане, и получают бледно-желтую пену, 44,0 г, 57,34 ммоль, 72%.1Н ЯМР (CDCl3) 9,39 1Н(с), 8,76 1Н(с), 8,55 1Н(с), 8,22 1Н(д), 8,08 1Н(с), 7,47 1Н(NH), 7,45 1Н(д), 7,35 1Н(NH), 7,01 1Н(NH), 6,49 1Н(д), 5,63 1Н(м), 5,53 1Н(м), 4,48 1Н(м), 4,70 1Н(д), 4,67 1Н(м), 4,11 1Н(м), 4,01 1Н(м), 2,86 1Н(м), 2,57 1Н(м), 2,25 1Н(м), 1,96 1Н(м), 1,80 1Н(м), 1,70 6Н(м), 1,60 2Н(м), 1,50 2Н(м), 1,25 3Н(м), 0,96 12Н(м), 0,91 2Н(м), 0,72 2Н(м).
Пример 3
Соединения 1-72 и 74-76 получают, по существу, так, как описано в данном описании. Результаты анализа указанных соединений согласуются с раскрытыми структурами соединений. Другие выбранные результаты приводятся ниже.









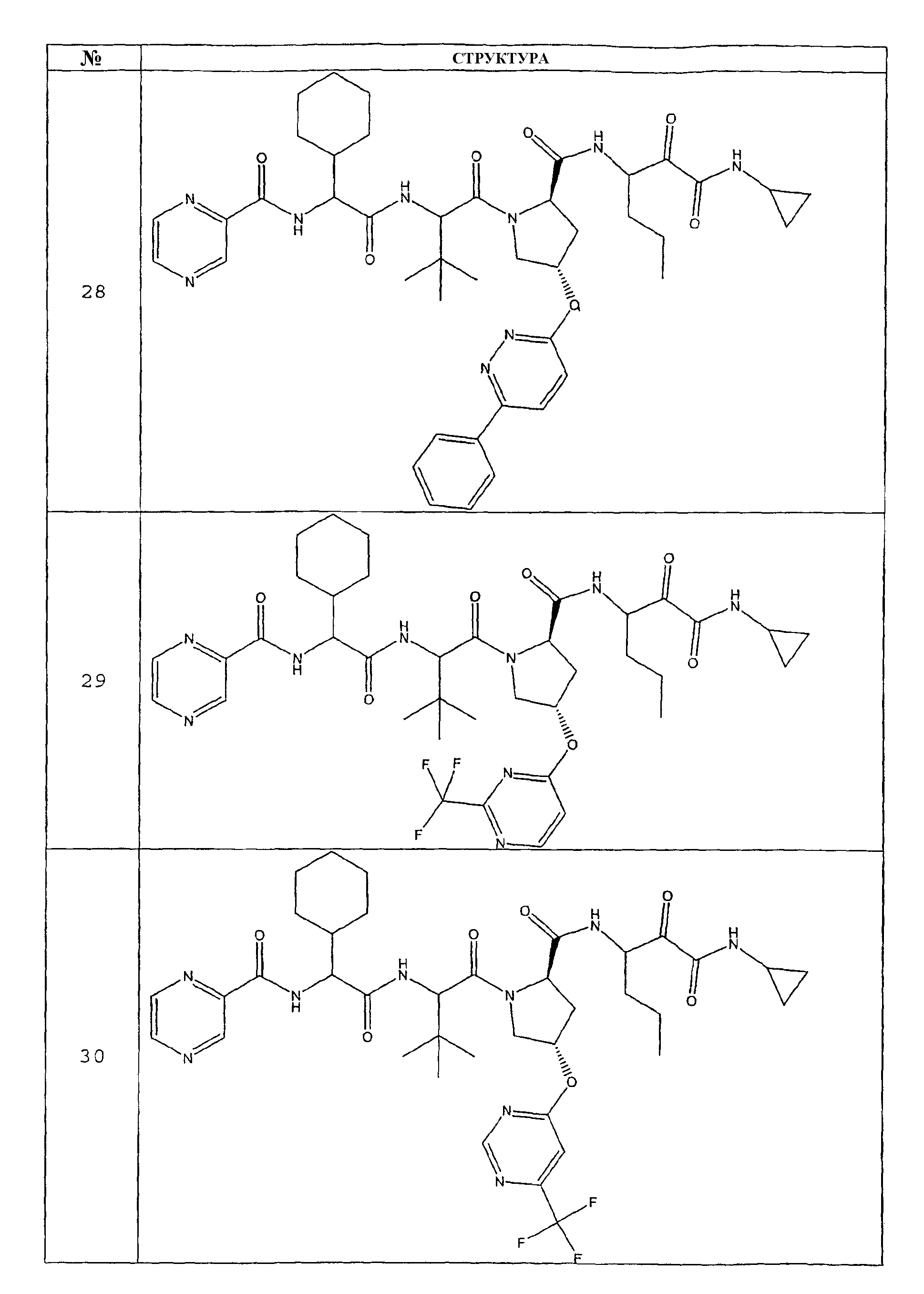











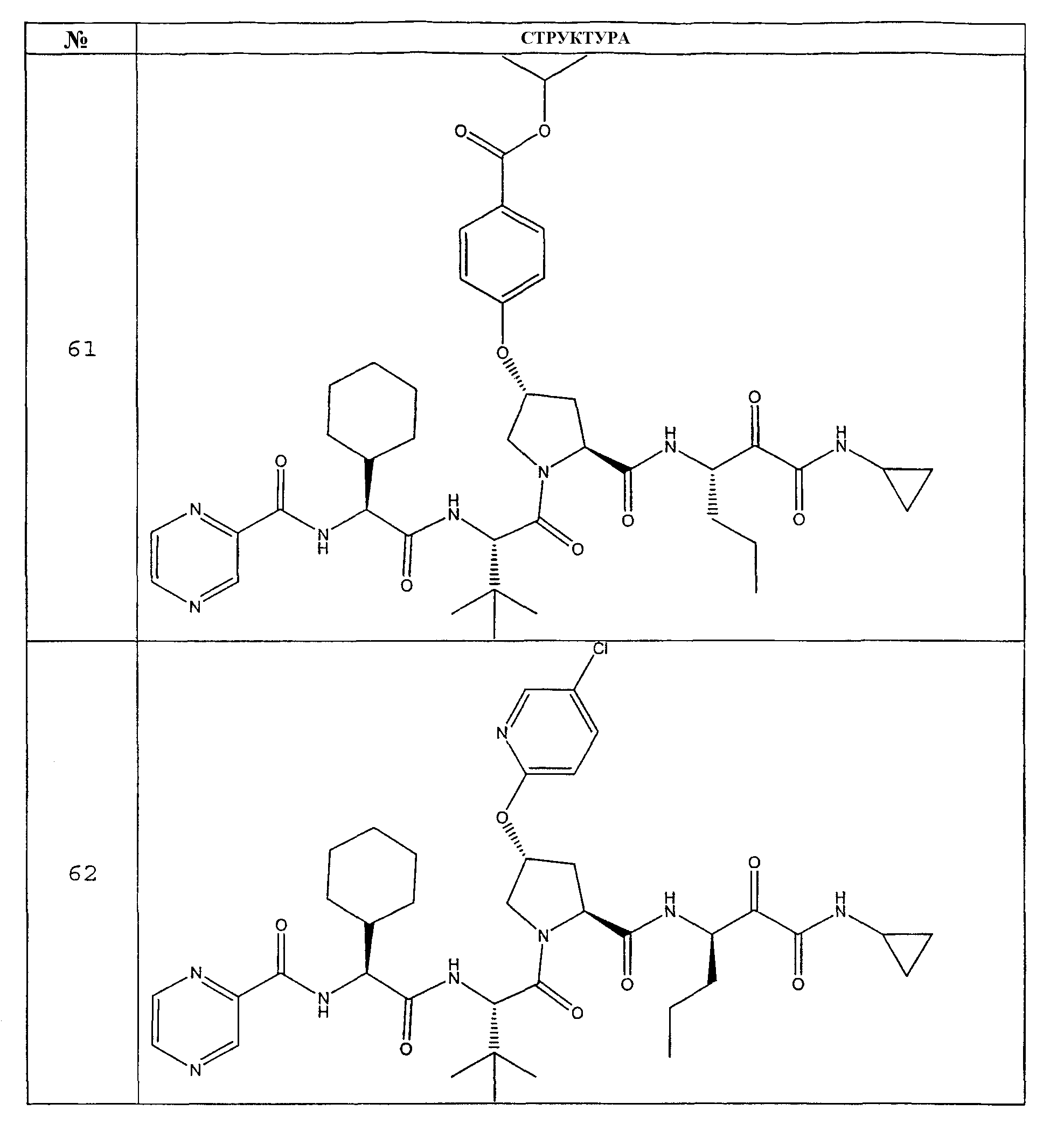






Пример 4
Протокол анализа репликона HVC в клетках
Клетки, содержащие репликон вируса гепатита С (HCV), поддерживают в среде DMEM, содержащей 10% сыворотки плода коровы (FBS) и 0,25 мг на мл G418 с соответствующими добавками (среда А).
В день 1, монослой репликонсодержащих клеток обрабатывают смесью трипсин:ЭДТК, удаляют ее, и затем разбавляют средами А, а именно до конечной концентрации 100000 клеток на мл. Высевают по 10000 клеток в 100 мкл в каждую лунку 96-луночного планшета для культивирования тканей и культивируют в течение ночи в камере для культивирования тканей при 37°С.
В день 2, соединения (в 100% ДМСО) разводят серийно в DMEM, содержащей 2% FBS и 0,5% ДМСО с соответствующими добавками (среда В). В серийных разведениях поддерживают конечную концентрацию ДМСО 0,5%.
Среды на монослое репликонсодержащих клеток удаляют, и затем добавляют среды В, содержащие различные концентрации соединения. В другие лунки добавляют среды В без какого-либо соединения как контроль в отсутствие соединения.
Клетки инкубируют с соединением или 0,5% ДМСО в средах В в течение 48 часов в камере для культивирования тканей при 37°С. По окончании 48-часовой инкубации среды удаляют, и монослой репликонсодержащих клеток промывают один раз PBS и хранят при -80°С до экстрагирования РНК.
Культуральные планшеты с обработанными монослоями репликонсодержащих клеток оттаивают, и в каждую лунку добавляют фиксированное количество другого РНК-вируса, такого как вирус вирусной диареи крупного рогатого скота (BVDV). Сразу же к клеткам добавляют реагенты для экстрагирования РНК (такие как реагенты из наборов RNeasy) для того, чтобы избежать разложения РНК. Полную РНК экстрагируют согласно инструкции изготовителя с изменениями для улучшения эффективности и воспроизводимости экстракции. Наконец, полную клеточную РНК, включая РНК репликона HCV, элюируют и хранят при -80°С до последующей обработки.
Проводят количественный анализ методом Tacqman ОТ-ПЦР (RT-PCR) в режиме реального времени с двумя наборами специфических праймеров и зонда. Один предназначен для HCV, а другой для BVDV. Экстракты полной РНК из обработанных репликонсодержащих клеток добавляют в реакционные смеси для ПЦР реакций для определения количества РНК как HVC, так и BVDV в одной и той же ПЦР-лунке. Ошибку эксперимента уменьшают или устраняют на основании уровня РНК BVDV в каждой лунке. Уровень РНК HVC в каждой лунке вычисляют согласно стандартной кривой по результатам из того же планшета для ПЦР. Процент ингибирования или снижения уровня РНК HVC вследствие обработки соединением вычисляют, принимая контроль ДМСО или в отсутствие соединения за 0% ингибирования. IC50 (концентрация, при которой наблюдают 50% снижение уровня РНК HVC) вычисляют из титрационной кривой для любого взятого соединения.
Пример 5
Протокол анализа Ki HCV
Метод микроколоночной ВЭЖХ для разделения субстрата 5АВ и продуктов
Субстрат:
NH2-Glu-Asp-Val-Val-(альфа)Abu-Cys-Ser-Met-Ser-Tyr-COOH
Получают исходный раствор 20 мМ 5АВ (или концентрации по выбору) в ДМСО (мас./0,2 М DTT). Полученный раствор хранят в аликвотах при -20°С.
Буфер: 50 мМ HEPES, pH 7,8; 20% глицерина; 100 мМ NaCl.
Общий объем смеси для анализа 100 мкл.
Буфер, КК4А, DTT и tNS3 объединяют и распределяют каждые 78 мкл в лунки 96-луночного планшета. Планшет инкубируют при 30°С в течение ~5-10 мин.
Растворяют 2,5 мкл раствора испытываемого соединения соответствующей концентрации в ДМСО (только ДМСО для контроля) и добавляют в каждую лунку. Все инкубируют при комнатной температуре в течение 15 мин.
Инициируют реакцию, добавляя 20 мкл 250 мкМ раствора субстрата 5АВ (концентрация 25 мкМ эквивалентна или несколько ниже Km для 5АВ).
Проводят инкубацию в течение 20 мин при 30°С.
Реакцию обрывают, добавляя 25 мкл 10% ТФК.
Переносят 120-мкл аликвоты в сосуды для ВЭЖХ.
Продукт SMSY отделяют от субстрата и КК4А способом, описанным далее.
Метод микроколоночного разделения
Приборы: Agilent 1100;
Дегазатор G1322A;
Сдвоенный насос G1312A;
Автодозатор G1313A;
Камера термостатирования колонки G1316A;
Детектор с диодной матрицей G1315A.
Колонка:
Phenomenex Jupiter; 5 микрон С18; 300 Å; 150×2 мм; Р/О 00F-4053-B0;
Термостат колонки: 40оС;
Объем впрыска: 100 мкл;
Растворитель А = вода для ВЭЖХ + 0,1% ТФК;
Растворитель В = ацетонитрил для ВЭЖХ + 0,1% ТФК.
Время остановки: 17 мин.
Время после выполнения: 10 мин.
Соединения данного изобретения испытывали в анализах примера 4 и/или примера 5, и видно, что они обладают активностью ингибирования протеазы NS3-NS4 HCV. Некоторые предпочтительные соединения данного изобретения показывают сравнимые результаты с клетками (пример 4) и с ферментами (пример 5). Соединения 2, 7, 12, 13, 14, 18, 22, 24, 26, 34, 45, 50, 53, 54, 55, 57, 59, 61, 65 и 66 показывают сравнимые результаты с клетками и с ферментами. Более предпочтительными соединениями являются соединения 24, 45, 53, 54, 59 и 61, имеющие сравнимые результаты с клетками и с ферментами, и причем оба типа результатов подпадают под категорию А.
Соединения данного изобретения испытывали согласно примеру 5 (ферменты), и обнаружено, что по своим величинам Ki, соответствующим Ki <0,1 мкМ (категория А); 0,1-0,3 мкМ (категория В) и >0,3 мкМ (категория С), они распределяются следующим образом:
категория А: 15, 19, 20, 24, 32, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 49, 51, 52, 53, 54, 56, 58, 59, 61, 64, 67 и 69;
категория В: 1, 2, 3, 4, 5, 7, 8, 10, 12, 13, 14, 16, 18, 22, 23, 25, 30, 31, 33, 26, 34, 48, 50, 55, 57, 60, 65, 66, 70, 71 и 76;
категория С: 6, 9, 11, 17, 21, 27, 28, 29, 47, 74, 75 и 76.
Соединения данного изобретения испытывали согласно примеру 4 (клетки), и обнаружено, что по своим величинам IC50, соответствующим IC50 <0,5 мкМ (категория А); 0,5-1,0 мкМ (категория В) и >1,0 мкМ (категория С), они распределяются следующим образом:
категория А: 7, 12, 13, 14, 15, 18, 19, 22, 24, 34, 35, 36, 38, 39, 40, 42, 43, 44, 45, 46, 47, 48, 50, 53, 54, 55, 56, 26, 57, 58, 59, 61, 64, 65, 66 и 67;
категория В: 1, 2, 3, 4, 5, 8, 10, 16, 20 и 70;
категория С: 6, 11, 41, 46, 47, 48, 49, 51, 60 и 71.
Хотя в данном описании раскрыт ряд воплощений данного изобретения, очевидно, что приведенные базовые примеры можно изменять и получить другие воплощения, в которых используются соединения и способы данного изобретения. Поэтому следует иметь в виду, что объем данного изобретения определяется прилагаемой формулой изобретения, а не конкретными воплощениями, представленными в примерах, описанных выше. Все цитированные документы включены в данное описание посредством ссылок.
Реферат
Настоящее изобретение относится к соединениям формулы I-A и I-B, которые ингибируют серинпротеазную активность, в частности активность протеазы NS3-NS4A вируса гепатита С. Соединения действуют, препятствуя жизненному циклу вируса гепатита С, и также применимы в качестве противовирусных средств. Изобретение также относится к композициям, содержащим указанные соединения, или для применения in vivo, или для введения пациенту, страдающему от заражения HCV. 6 н. и 29 з.п. ф-лы.
Формула

где Ar представляет собой 6- или 10-членное ароматическое кольцо, содержащее 0, 1 или 2 атома азота в качестве гетероатома, где 1, 2 или 3 атома кольца необязательно и независимо замещены J;
R1 и R2 представляют собой, независимо:
(С1-С12)-алифатический радикал или (С3-С10)-циклоалкил-;
R3 и R3' представляют собой, независимо, водород или (С1-С12)-
алифатический радикал, где любой атом водорода, необязательно, заменен галогеном;
R4 и R4' представляют собой, независимо: водород;
W представляет собой:

где NR6R6 представляет собой

Т представляет собой

J представляет собой галоген, -OR', -NO2, -CN, -CF3, -OCF3, -R', -SO2N(R')2, -C(O)R', -C(O)OR', -C(O)N(R')2 или -N(R')C(O)R';
R' представляет собой:
водород,
(С1-С12)-алифатический радикал, (С6-С10)-арил- или (С3-С10)-гетероциклил-;
где R', необязательно, замещен группами J, до 3;
где две группы R', связанные с одним и тем же атомом, образуют 3-10-членное неароматическое кольцо, содержащее до 3 атомов азота в качестве гетероатома, где любое кольцо содержит до 3 заместителей, выбранных, независимо, из J.

где n равен 0 или 1;
Ar представляет собой 6- или 10-членное ароматическое кольцо, содержащее 0, 1 или 2 атома азота в качестве гетероатома, где 1, 2 или 3 атома кольца, необязательно и независимо, замещены J;
R1 и R2 представляют собой, независимо:
(С1-С12)-алифатический радикал или
(С3-С10)-циклоалкил-;
R3 и R3' представляют собой, независимо, водород или (С1-С12)-алифатический радикал, где любой атом водорода, необязательно, заменен галогеном;
R4 и R4' представляют собой, независимо:
водород-;
W представляет собой
где NR6R6 представляет собой
Т представляет собой

J представляет собой галоген, -OR', -NO2, -CN, -CF3, -OCF3, -R', -SO2N(R')2, -C(O)R', -C(O)OR', -C(O)N(R')2 или -N(R')C(O)R';
R' представляет собой:
водород-,
(С1-С12)-алифатический радикал,
(С6-С10)-арил- или
(С3-С10)-гетероциклил-;
где R', необязательно, замещен группами J, до 3;
где две группы R', связанные с одним и тем же атомом, образуют 3-10-членное неароматическое кольцо, содержащее до 3 атомов азота в качестве гетероатома, где любое кольцо содержит до 3 заместителей, выбранных, независимо, из J.


R3 представляет собой

R3 представляет собой
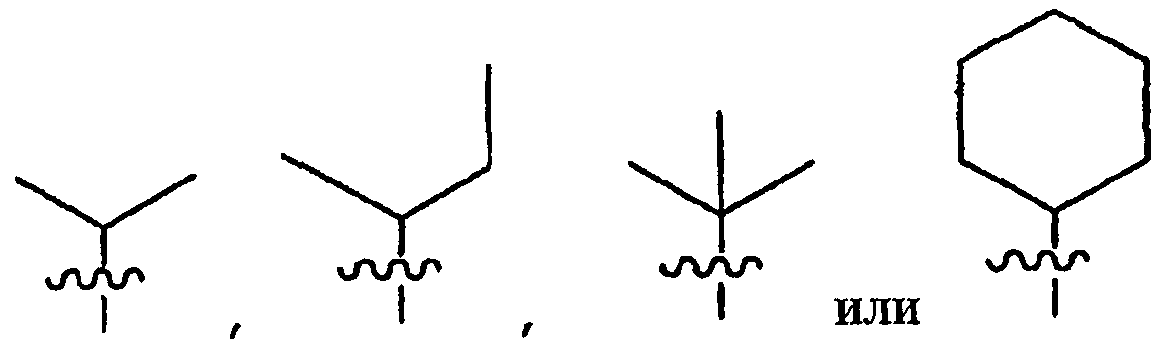

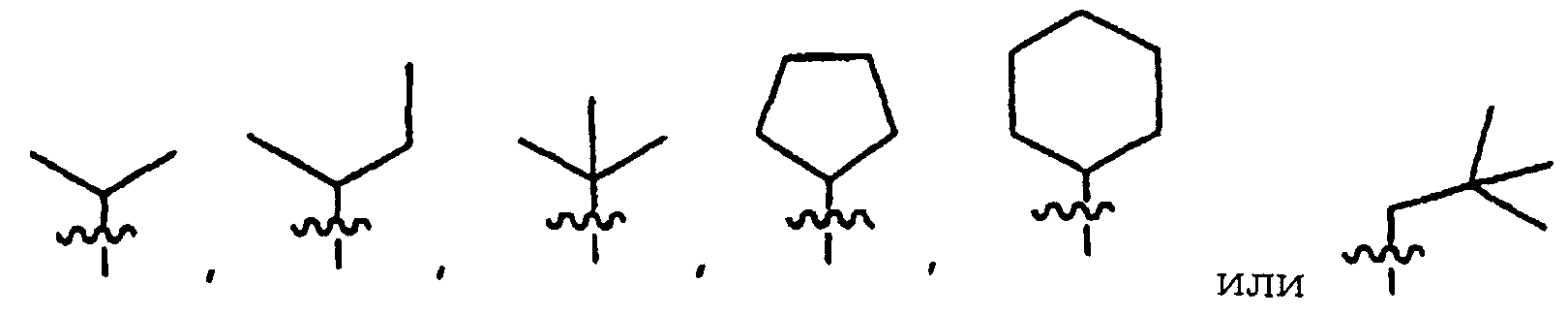












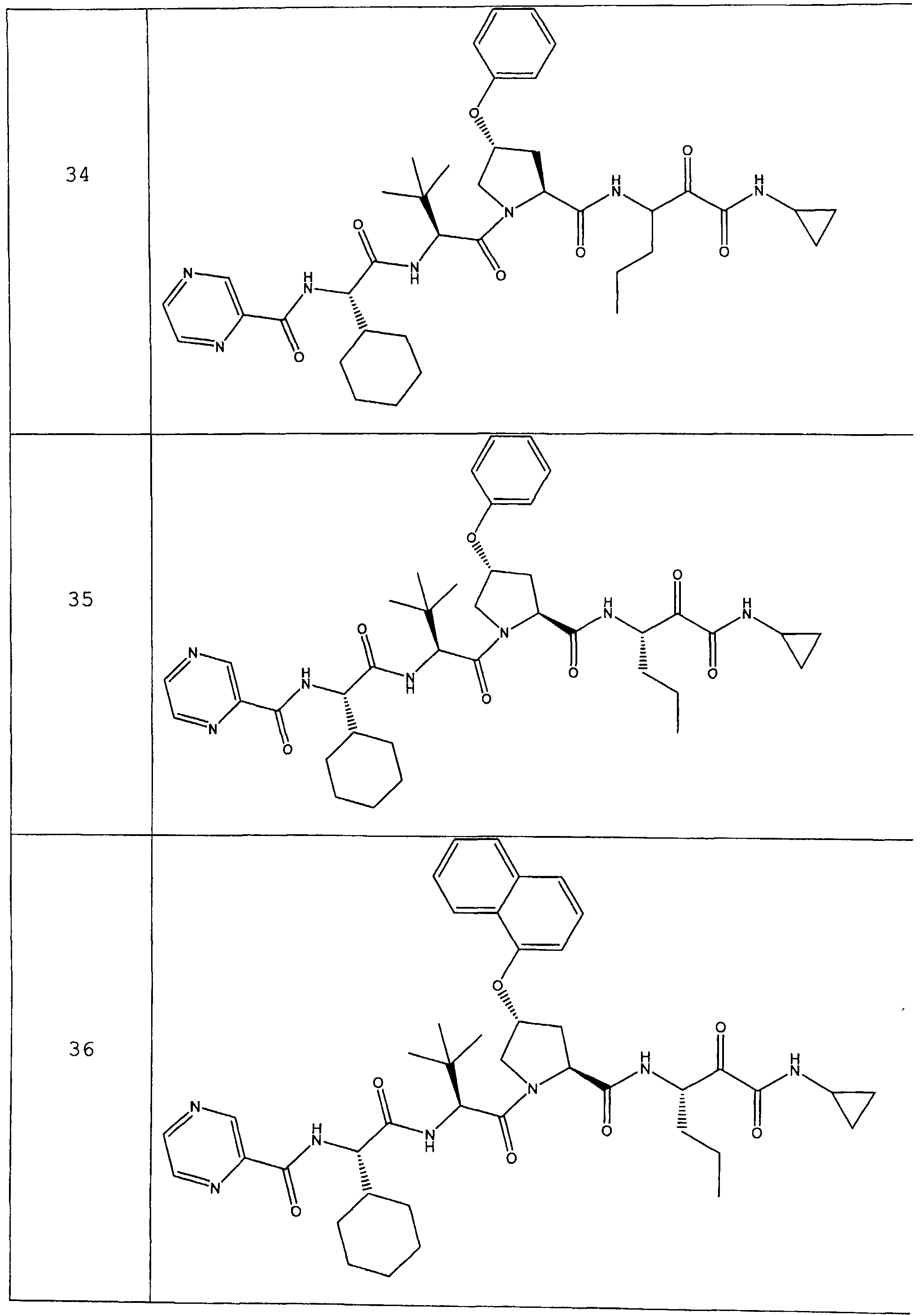









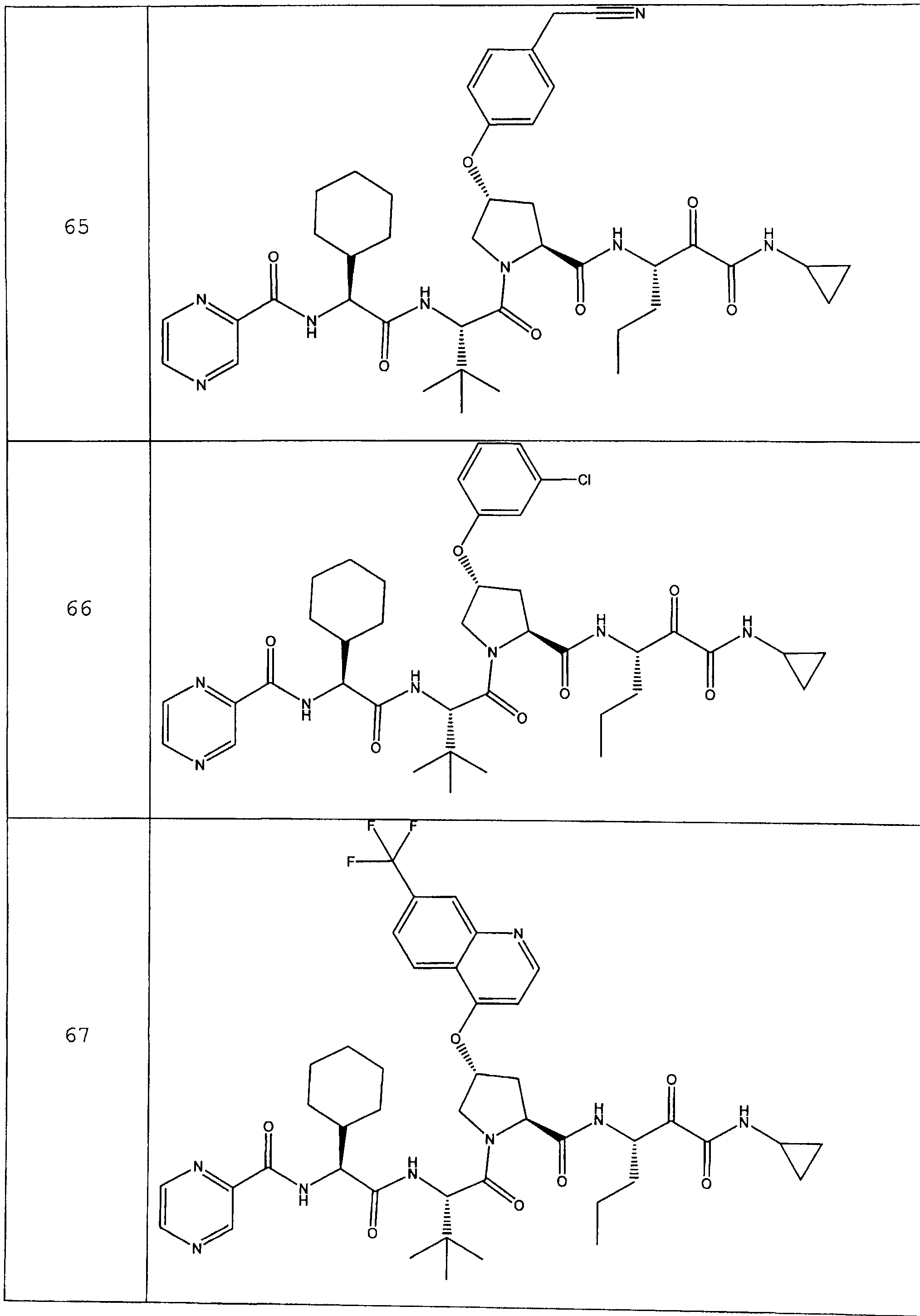


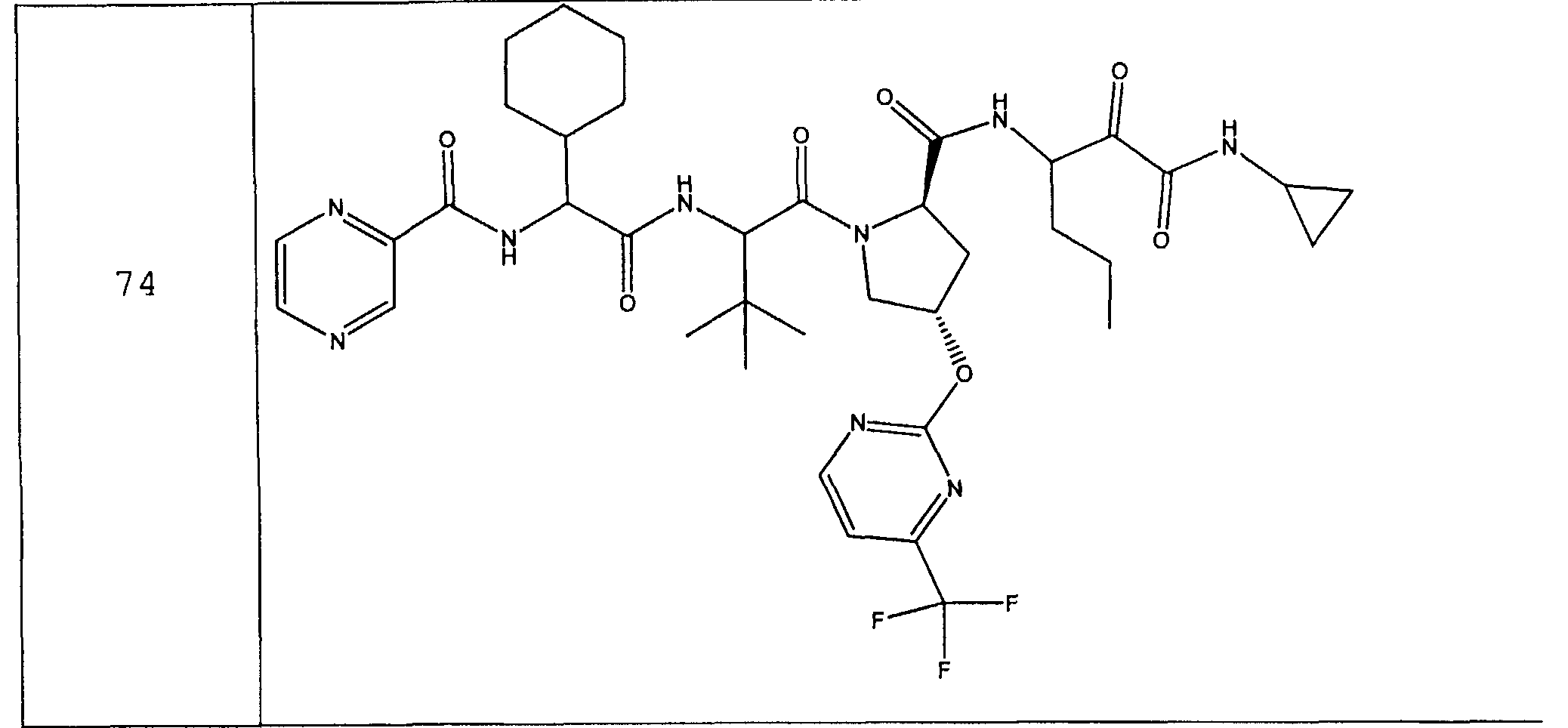
второго ингибитора протеазы HCV, ингибитора другой мишени в жизненном цикле HCV или их комбинации, где указанное дополнительное средство представляет часть указанной композиции или отдельную дозированную форму.








Комментарии