Пиразол или его соль и способ получения, гербицидная композиция и ее применение - RU2707086C1
Код документа: RU2707086C1
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к области пестицидов, в частности, относится к соединению пиразола или его соли, к способу ее получения, к гербицидной композиции и ее применению.
Уровень техники
Просо куриное является наиболее распространенным сорняком в рисовом поле; в особенности с распространением технологии посева семенами, борьба с просом куриным стала ключевой задачей для увеличения урожайности. Разработано большое количество гербицидов для борьбы с просом куриным, например, ингибиторы ALS (например, фенокссулам, триафамон и т. п.) и ингибиторы ACCase (например, цигалофоп-бутил, метамифоп, феноксапроп-п-этил, клефоксидим и т. п.), однако вследствие массового нанесения этих гербицидов устойчивость проса куриного по отношению к ним стала все более важной. Сообщают, что в настоящее время у множества биотипов проса куриного развилась устойчивость к основным гербицидам проса куриного. Поэтому настоятельно необходима разработка гербицида, не приводящего к перекрестной устойчивости по отношению к главным современным гербицидам проса куриного.
Содержание изобретения
Для решения указанной выше задачи предшествующего уровня техники в настоящем изобретении предложены пиразол или его соли, способ ее получения, гербицидная композиция и ее применение. Неожиданно было установлено, что пиразол не только оказывает сильное воздействие на просо куриное, но и безопасен для риса при послевсходовом нанесении. Также неожиданно было установлено, что он оказывает сильное воздействие на просо куриное, устойчивое по отношению к основным гербицидам, таким как фенокссулам, хинклорак, цигалофоп-бутил и пропанил и т. п.
Для решения указанной выше задачи в настоящем изобретении предложено следующее техническое решение:
Соединение пиразола формулы (I) или его соль:

в которой
R означает

R1 означает C1-C3 алкил;
R2 означает водород или C1-C4 алкил;
R3 означает водород или C1-C6 алкил, необязательно замещенный фенил, необязательно замещенный пиридил, необязательно замещенный алкенил, необязательно замещенный алкинил, C1-C6 алкилкарбонил, C1-C6 алкоксилкарбонил, C1-C6 алкилкарбонилметил, C1-C6 алкоксилкарбонилметил, C1-C4 алкилсульфонил, C1-C4 галогенированный алкилсульфонил, фенилсульфонил или фенилсульфонил, замещенный алкилом, алкоксилом или галогеном, бензоил или бензоил, замещенный галогеном, нитрогруппой, алкилом или алкоксилом, феноксилкарбонил или феноксилкарбонил, замещенный галогеном, нитрогруппой, алкилом или алкоксилом, бензоилметил или бензоилметил, замещенный галогеном, нитрогруппой, алкилом или алкоксилом, феноксилкарбонилметил или феноксилкарбонилметил, замещенный галогеном, нитрогруппой, алкилом или алкоксилом.
Предпочтительно, если R', R'' и R''' означают водород, метил, метоксил, фтор метил или хлор, где R', R'', R''' могут быть одинаковыми или разными.
R1 означает метил, этил или изопропил;
R2 означает водород, метил, этил или циклопропил;
R3 означает водород или C1-C6 алкил, необязательно замещенный фенил, необязательно замещенный пиридил, необязательно замещенный алкенил, необязательно замещенный алкинил, C1-C6 алкилкарбонил, C1-C6 алкоксилкарбонил, C1-C6 алкилкарбонилметил, C1-C6 алкоксилкарбонилметил, C1-C4 алкилсульфонил, C1-C4 галогенированный алкилсульфонил, фенилсульфонил или фенилсульфонил, замещенный алкилом, алкоксилом или галогеном, бензоил или бензоил, замещенный галогеном, нитрогруппой, алкилом или алкоксилом, феноксилкарбонил или феноксилкарбонил, замещенный галогеном, нитрогруппой, алкилом или алкоксилом, бензоилметил или бензоилметил, замещенный галогеном, нитрогруппой, алкилом или алкоксилом, феноксилкарбонилметил или феноксилкарбонилметил, замещенный галогеном, нитрогруппой, алкилом или алкоксилом.
Соединение формулы (III):

в которой
R', R'' и R''' означают водород, C1-C4 алкил, C1-C4 галогенированный алкил, C1-C4 алкоксил или галоген; R', R'' и R''' могут быть одинаковыми или разными.
Предпочтительно, если R', R'' и Rʺ' означают водород, метил, метоксил, или хлор; R', R'' и R''' могут быть одинаковыми или разными.
Соединение формулы (V):

в которой
R', R'' и R''' означают водород, C1-C4 алкил, C1-C4 галогенированный алкил, C1-C4 алкоксил или галоген; R', R'' и R''' могут быть одинаковыми или разными.
R1 означает C1-C3 алкил;
R2 означает водород или C1-C4 алкил;
Предпочтительно, если R', R'' и R''' означают водород, метил, метоксил, или хлор; R', R'' и R''' могут быть одинаковыми или разными.
R1 означает метил, этил или изопропил;
R2 означает водород, метил, этил или циклопропил.
В определении указанного выше соединения термины, использующиеся по отдельности или в комбинации с другими терминами, означают следующие группы:
галоген означает фтор, хлор, бром, или йод;
алкил означает обладающий линейной цепью алкил или обладающий разветвленной цепью алкил;
галогенированный алкил означает линейный или разветвленный алкил, в котором все или часть атомов водорода, замещены атомами галогенов;
алкоксил означает функциональную группу, образованную путем связывания алкила с кислородом.
При необходимости соединение формулы (I) можно превратить в соответствующую его соль по обычным методикам. Соль может быть в любой форме при условии, что она является сельскохозяйственно приемлемой, например, соль щелочного металла (например, соль натрия или соль калия), соль щелочноземельного металла (например, соль магния или соль кальция ), или соль аммония (например, соль диметиламина или соль триэтиламина).
Соединение, предлагаемое в настоящем изобретении, может находиться в форме одного или нескольких стереоизомеров. Стереоизомеры включают энантиомеры, диастереоизомеры и геометрические изомеры. Все эти стереоизомеры и их смесь входят в объем настоящего изобретения.
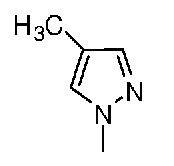
В настоящем изобретении также раскрыт способ получения соединения пиразола формулы (I) или его соли, в котором 2-хлор-3-бромметил-4-метилсульфонилбензойную кислоту используют в качестве исходного вещества.
Также раскрыт способ получения соединения пиразола формулы (I-1) или его соли, включающий следующие стадии:
(1) взаимодействия 2-хлор-3-бромметил-4-метилсульфонилбензойную кислоты вводят с соединением формулы (II) с получением соединения формулы (III);
(2) взаимодействие соединения формулы (III) с соединением формулы (IV) с получением соединения формулы (V);
(3) подвергание соединения формулы (V) перегруппировке с получением соединения формулы (I), в котором R3 означает водород(а именно, формулы (I-1)); соединение пиразола формулы (I-1) или его соль можно получить по следующему пути реакции:

Указанную выше реакцию алкилирования необходимо проводить в присутствии растворителя. Использующийся растворитель инертен по отношению к реакционной смеси. Такой растворитель обычно представляет собой апротонный полярный растворитель, такой как ацетонитрил, ДМФ (диметилформамид), ДМСО (диметилсульфоксид), или смешанный растворитель, предпочтительно ацетонитрил.
Указанную выше реакцию N-алкилирования необходимо проводить в присутствии щелочи, которой обычно является обычно гидрид металла, такой как гидрид натрия, гидрид калия, предпочтительно гидрид натрия.
Температура, при которой проводят указанную выше реакцию N-алкилирования обычно равна -10-30°C, предпочтительно 0-10°C; продолжительность реакции составляет 0,5-48 ч, предпочтительно 1-12 ч.
Для уменьшения продолжительности реакции и повышения скорости реакции этерификации указанная выше реакция этерификации обычно включает 2 стадии: на первой стадии соединение формулы (III) превращают в соответствующий ацилхлорид; на второй стадии ацилхлорид проводят реакцию с соединением формулы (IV) с получением соединения формулы (V). Таким образом, соединение формулы (V) получают реакцией соединения формулы (III') с соединением формулы (IV). Соединение формулы (III') описывается следующей формулой:

где R', R'', R''' являются такими, как определено выше.
Соединение формулы (III) можно превратить в соединение формулы (III') следующим путем:

где R', R'', R''' являются такими, как определено выше.
Указанную выше реакцию этерификации необходимо проводить в присутствии растворителя. Использующийся растворитель инертен по отношению к реакционной смеси. Растворителем обычно является апротонный растворитель, который является полярным или неполярным. Например, ацетонитрил, метилбензол, диметилбензол, дихлорметан, дихлорэтан, тетрагидрофуран, или ацетон и т. п., предпочтительно 1,2-дихлорэтан.
Вторую стадию реакции этерификации необходимо проводить в присутствии реагента, снижающего кислотность; использующимся реагентом, снижающим кислотность, является обычная щелочь, неорганическая или органическая. Одну или большее количество таких щелочей можно выбрать для использования из числа следующих: карбонаты (например, карбонат натрия, карбонат калия), бикарбонаты (например, бикарбонат натрия, бикарбонат калия), амины (например, диметиламин, триэтиламин, N,N-диизопропилэтиламин) и пиридины (например, пиридин, 4-диметиламинопиридин), предпочтительно триэтиламин.
Температура, при которой проводят реакцию этерификации обычно равна от -10 до 50°C, предпочтительно от 0 до 10°C; продолжительность реакции составляет от 0,5 до 24 ч, предпочтительно от 1 до 6 ч.
Реакцию перегруппировки проводят в реакционной системе реакции этерификации. Или, скорее, реакционную жидкость можно использовать прямо для реакции перегруппировки без какой-либо обработки после реакции этерификации. Следовательно, растворителем, необходимым для реакции перегруппировки, является тот же, который используют для указанной выше реакции этерификации.
Указанная выше реакция перегруппировки включает 2 стадии. Первая стадия, перегруппировка соединения формулы (V); вторая, регулирование pH системы. А именно, необходимое количество воды добавляют в систему после первой стадии и систему подкисляют. Обычно кислотой, использующейся для регулирования pH, является хлористоводородная кислота.
Температура, при которой проводят первую стадию реакции перегруппировки, обычно равна от 0 до 100°C, предпочтительно от 40 до 60°C; продолжительность реакции составляет от 0,5 до 24 ч, предпочтительно от 1 до 6 ч. Температура реакции, при которой проводят стадию регулирования pH, равна от 10 до 50°C, предпочтительно от 10 до 25°C.
При необходимости до проведения реакции перегруппировки добавляют соответствующее количество катализатора. В настоящем изобретении катализатором предпочтительно является циангидрин ацетона.
Соединение формулы (III) и формулы (V), участвующие в пути реакции, являются новыми промежуточными продуктами и их можно использовать для получения соединения, предлагаемого в настоящем изобретении.
Для увеличения разнообразия соединений структуру соединения формулы (I), в котором R3 означает водород (т. е. формулы (I-1)), соответствующим образом модифицируют по методике конструирования молекул, таким образом получают соединение формулы (I) с не являющейся водородом группой R3 (т. е. формулы (I')). Соединение пиразола формулы (I'), относящийся к настоящему изобретению получают по реакции соединения формулы (I-1) с соединением формулы (VI). Путь реакции описывается следующим уравнением:

где Y означает галоген, предпочтительно хлор, бром или йод.
Получение соединения указанной выше формулы (I') необходимо проводить в присутствии растворителя. Использующийся растворитель должен быть инертным по отношению к реакционной смеси. Такой растворитель обычно является непротонным растворителем, полярным или неполярным, например, ацетонитрил, метилбензол, диметилбензол, дихлорметан, дихлорэтан, тетрагидрофуран или ацетон, предпочтительно ацетонитрил или дихлорметан.
Способ получения соединения формулы (I') необходимо проводить в присутствии реагента, снижающего кислотность; использующимся реагентом, снижающим кислотность, является обычная щелочь, неорганическая или органическая. Одну или большее количество таких щелочей можно выбрать для использования из числа следующих: карбонаты (например, карбонат натрия, карбонат калия), бикарбонаты (например, бикарбонат натрия, бикарбонат калия), амины (например, диметиламин, триэтиламин, N,N-диизопропилэтиламин) и пиридины (например, пиридин, 4-диметиламинопиридин), предпочтительно триэтиламин или карбонат калия.
Температура, при которой проводят реакцию получения соединения формулы (I') обычно равна от -10 до 50°C, предпочтительно от 0 до 20°C; продолжительность реакции обычно составляет от 0,1 до 12 ч, предпочтительно от 0,5 до 3 ч.
Здесь также раскрывается гербицидная композиция, которая содержит гербицидно эффективное количество по меньшей мере одного соединения пиразола или его соли.
Гербицидная композиция кроме того содержит вспомогательное вещество, использующееся для ее приготовления.
Описывается также способ борьбы с вредным растением, который включает стадию нанесения гербицидно эффективного количества по меньшей мере одного соединения пиразола или его соли, или гербицидной композиции на растение или участок, на котором произрастает вредное растение.
Применение по меньшей мере одного соединения пиразола или его соли или гербицидной композиции для борьбы с вредным растением, предпочтительно, если соединение пиразола или его соль наносят для борьбы с вредным растением в желательной сельскохозяйственной культуре, предпочтительно, если желательной сельскохозяйственной культурой является генетически модифицированная сельскохозяйственная культура или сельскохозяйственная культура, обработанная по методике редактирования генома.
Соединения формулы (I), предлагаемые в настоящем изобретении, обладают чрезвычайно высокой гербицидной активностью по отношению к широкому спектру экономически важных однодольных и двудольных вредных растений. Активные соединения также эффективно действуют на многолетние сорняки, которые образуют побеги из ризом, корневищ или других многолетних органов и с которыми трудно бороться. В этом контексте обычно несущественно, наносят ли соединения до высевания, до всходов или после всходов. В частности, можно отметить примеры некоторых представителей однодольной и двудольной сорной флоры, с которыми можно бороться с помощью соединений, предлагаемых в настоящем изобретении, и они не ограничиваются определенными видами. Примерами видов сорняков, на которые эффективно действуют активные соединения, из числа однодольных являются виды Avena, Lolium, Alopecurus, Phalaris, Echinochloa, Digitaria, Setaria и также виды Cyperus из числа однолетних и из числа многолетних виды Agropyron, Cynodon, Imperata и Sorghum и также многолетние виды Cyperus.
В случае двудольных видов сорняков, спектр воздействия распространяется на такие виды, как, например, Galium, Viola, Veronica, Lamium, Stellaria, Amaranthus, Sinapis, Ipomoea, Sida, Matricaria и Abutilon из числа однолетних, и Convolvulus, Cirsium, Rumex и Artemisia из числа многолетних сорняков. Активные соединения, предлагаемые в настоящем изобретении, также обеспечивают превосходную борьбу с вредными растениями, которые появляются при особых условиях выращивания риса, такими как, например, Echinochloa, Sagittaria, Alisma, Eleocharis, Scirpus и Cyperus. Если соединения, предлагаемые в настоящем изобретении, наносят на поверхность почвы до всходов, то полностью предупреждается появление проростков сорняков или сорняки растут до стадии семядоли, но затем их рост останавливается и, в конечном счете через 3-4 недели они полностью гибнут. В частности, соединения, предлагаемые в настоящем изобретении, обладают превосходной активностью по отношению к видам Apera spica venti, Chenopodium album, Lamium purpureum, Polygonum convulvulus, Stellaria media, Veronica hederifolia, Veronica persica, Viola tricolor и по отношению к видам Amaranthus, Galium и Kochia.
Хотя соединения, предлагаемые в настоящем изобретении, обладают превосходной гербицидной активностью по отношению к однодольным и двудольным сорнякам, растения экономически важных сельскохозяйственных культур, такие как, например, пшеница, ячмень, рожь, рис, кукуруза, сахарная свекла, хлопчатник и соя, не повреждаются совсем или повреждаются лишь в незначительной степени. В частности, они обладают превосходной совместимостью со злаками, такими как пшеница, ячмень и кукуруза, в особенности пшеница. Поэтому соединения, предлагаемые в настоящем изобретении, являются весьма подходящими для селективной борьбы с ростом вредного растения в посевах сельскохозяйственного использования или в посевах декоративных растения.
Благодаря их гербицидной способности, эти активные соединения также можно использовать для борьбы с вредными растениями в известных сельскохозяйственных культурах или в разрабатываемых с помощью генетической инженерии растениях. Трансгенные растения обычно обладают особенно благоприятными характеристиками, например, устойчивостью по отношению к некоторым пестицидам, в частности, некоторым гербицидам, устойчивостью по отношению к болезням растений или возбудителям болезней растений, таким как некоторые насекомые или микроорганизмы, такие как грибы, бактерии или вирусы. Другие конкретные характеристики относятся, например, к количеству, качеству, стабильности при хранении, составу и к особым ингредиентам собранного продукта. Так, известны трансгенные растения, обладающие повышенным содержанием крахмала или крахмалом измененного качества, или обладающие иным составом жирных кислот в собранном продукте.
Применение соединений формулы (I), предлагаемых в настоящем изобретении, или их солей для экономически важных трансгенных полезных сельскохозяйственных культур и декоративных растений, например, злаков, таких как пшеница, ячмень, рожь, овес, просо, рис, маниок и кукуруза или даже культур сахарной свеклы, хлопчатника, сои, рапса, картофеля, томата, гороха и других видов овощей является предпочтительным. Соединения формулы (I) предпочтительно можно использовать в качестве гербицидов в культурах полезных растений, которые устойчивы или которым с помощью генной инженерии придана устойчивость по отношению к фитотоксическим воздействиям гербицидов.
Обычные пути получения новых растений, характеристики которых изменены по сравнению с характеристиками известных растений, включают, например, традиционные методики скрещивания и генерации мутантов. Альтернативно, новые растения, обладающие измененными характеристиками, можно получить по методикам генной инженерии (см., например, EP-A 0221044, EP-A 0131624). Например, описаны различные случаи:
введенных с помощью генной инженерии изменений в сельскохозяйственных растениях для изменения крахмала, синтезируемого в растениях (например, WO 92/11376, WO 92/14827, WO 91/19806),
трансгенных сельскохозяйственных растений, которые устойчивы по отношению к некоторым гербицидам типа глуфосината (см., например, EP-A 0242236, EP-A 0242246) или глифосата (WO 92/00377), или типа сульфонилмочевины (EP-A 0257993, патент U.S. № 5013659),
трансгенных сельскохозяйственных растений, например, хлопчатника, обладающих способностью продуцировать токсины Bacillus thuringiensis (Bt-токсины), которые придают устойчивость по отношению к некоторым вредителям растений (EP-A 0142924, EP-A 0193259), трансгенных сельскохозяйственных растений, обладающих измененным составом жирных кислот (WO 91/13972).
В принципе, известны многочисленные биологические методики, которые позволяют получить новые трансгенные растения, обладающие измененными характеристиками; см., например, Sambrook et al., 1989, Molecular Cloning, A Laboratory Manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.; или Winnacker "Gene und Klone" [Genes and Clones], VCH Weinheim, 2nd edition 1996, или Christou, "Trends in Plant Science" 1 (1996) 423-431). Для проведения таких генно-инженерных операций можно вводить молекулы нуклеиновых кислот в плазмиды, что делает возможным протекание мутагенеза или изменение в последовательности путем рекомбинации последовательностей ДНК. С помощью указанных выше стандартных процедур можно, например, заменить основания, для удаления частичных последовательностей или добавить природные или синтетические последовательности. Для связывания фрагментов ДНК друг с другом, к фрагментам можно добавить адапторы или линкеры.
Клетки растений, обладающие уменьшенной активностью генного продукта, можно получить, например, путем экспрессирования по меньшей мере одной подходящей антисмысловой РНК, смысловой РНК для обеспечения эффекта совместной супрессии, или путем экспрессирования по меньшей мере одной соответствующим образом сконструированной рибозимы, которая специфически расщепляет транскрипты указанного выше генного продукта.
Для этой цели можно использовать обе молекулы ДНК, которые включают полную кодирующую последовательность генного продукта, включая любые фланкирующие последовательности, которые могут содержаться, и молекулы ДНК, которые включают только части кодирующей последовательности, необходимо, чтобы эти части были достаточно длинными для оказания антисмыслового воздействия в клетках. Также можно использовать последовательности ДНК, которые обладают высокой степенью гомологии с кодирующими последовательностями генного продукта, но не полностью идентичны.
При экспрессировании молекул нуклеиновой кислоты в растениях синтезированный белок может быть локализован в любом желательном компартменте клеток растений. Однако для обеспечения локализации в некотором компартменте, например, можно связать кодирующую область с последовательностями ДНК, которые обеспечивают локализацию в определенном компартменте. Такие последовательности известны специалисту в данной области техники (см., например, Braun et al., EMBO J. 11 (1992), 3219-3227; Wolter et al., Proc. Natl. Acad. Sci. USA 85 (1988), 846-850; Sonnewald et al., Plant J. 1 (1991), 95-106).
Клетки трансгенного растения можно регенерировать в целые растения по известным методикам. Трансгенные растения, в принципе, могут представлять собой растения любого желательного вида растений, т. е. и однодольные, и двудольные растения. Таким образом можно получить трансгенные растения, которые обладают измененными характеристиками, путем сверхэкспрессии, супрессии или ингибирования гомологичных (=натуральных) генов или последовательностей генов, или путем экспрессии гетерологичных (=посторонних) генов или последовательностей генов.
При использовании активных соединений, предлагаемых в настоящем изобретении, в трансгенных сельскохозяйственных культурах в дополнение к воздействиям на вредные растения, которое можно наблюдать в случае других сельскохозяйственных культур, часто наблюдаются эффекты, которые специфичны для воздействия на соответствующую трансгенную сельскохозяйственную культуру, например, измененный или специфически расширенный спектр сорняков, с которыми можно бороться, измененные нормы расхода, которые можно использовать при нанесении, предпочтительно хорошая сочетаемость с гербицидами, по отношению к которым обладают устойчивостью трансгенные сельскохозяйственные культуры, и влияние на рост и урожайность трансгенных культурных растений. Поэтому настоящее изобретение также относится к применению соединений, предлагаемых в настоящем изобретении, в качестве гербицидов для борьбы с вредными растениями в трансгенных культурных растениях.
Кроме того, соединения, предлагаемые в настоящем изобретении, обладают чрезвычайно высокой способностью к регуляции культурных растений. Они реглирующим образом участвуют в метаболизме растения и поэтому их можно использовать для направленного воздействия на компоненты растения и для облегчения сбора урожая, например, путем обеспечения высыхания и задержки роста. Кроме того, они также являются подходящими для обычного регулирования и подавления нежелательного вегетативного роста без одновременного разрушения растений. Подавление вегетативного роста играет важную роль для многих однодольных и двудольных сельскохозяйственных культур, поскольку таким образом можно уменьшить или полностью предотвратить полегание.
Соединения, предлагаемые в настоящем изобретении, можно наносить в виде обычных препаратов в форме смачивающихся порошков, эмульгирующихся концентратов, растворов для опрыскивания, дустов или гранул. Поэтому настоящее изобретение также относится к гербицидным композициям, включающим соединения формулы (I). Соединения формулы (I) можно приготовить в разных формах в зависимости от преобладающих биологических и/или физико-химических параметров. Примерами подходящих препаратов являются: смачивающиеся порошки (WP), растворимые в воде порошки (SP), растворимые в воде концентраты, эмульгирующиеся концентраты (EC), эмульсии (EW), такие как эмульсии типа масло-в-воде и вода-в-масле, растворы для опрыскивания, концентраты суспензии (SC), дисперсии на масляной или водной основе, смешивающиеся с маслом растворы, дусты (DP), суспензии капсул (CS), композиции для протравливания семян, гранулы для внесения вразброс и в почву, гранулы (GR) в форме микрогранул, гранулы для опрыскивания, гранулы с покрытием и адсорбирующиеся гранулы, диспергирующиеся в воде гранулы (WG), растворимые в воде гранулы (SG), сверхмалообъемные препараты, микрокапсулы и воска. Эти отдельные типы препаратов, в принципе, известны и описаны, например, в публикациях Winnacker-Kuhler, "Chemische Technologie" [Chemical Technology], Volume 7, C. Hauser Verlag Munich, 4th. Edition 1986; Wade van Valkenburg, "Pesticide Formulations", Marcel Dekker, N.Y., 1973; K. Martens, "Spray Drying" Handbook, 3rd Ed. 1979, G. Goodwin Ltd. London.
Необходимые вспомогательные вещества, использующиеся для приготовления препаратов, такие как инертные вещества, поверхностно-активные вещества, растворители и другие добавки, также известны и описаны, например, в публикациях Watkins, "Handbook of Insecticide Dust Diluents and Carriers", 2nd Ed., Darland Books, Caldwell N.J., H. v. Olphen, "Introduction to Clay Colloid Chemistry"; 2nd Ed., J. Wiley & Sons, N.Y.; C. Marsden, "Solvents Guide"; 2nd Ed., Interscience, N.Y. 1963; McCutcheon's "Detergents and Emulsifiers Annual", MC Publ. Corp., Ridgewood N.J.; Sisley and Wood, "Encyclopedia of Surface Active Agents", Chem. Publ. Co. Inc., N.Y. 1964; Schonfeldt, "Grenzflchenaktive thylenoxidaddukte" [Surface-active ethylene oxide adducts], Wiss. Verlagsgesell., Stuttgart 1976; Winnacker-Kuchler, "Chemische Technologie" [Chemical Technology], Volume 7, C. Hauser Verlag Munich, 4th Edition 1986.
Смачивающиеся порошки являются препаратами, которые равномерно диспергируются в воде и которые в дополнение к активному соединению также содержат разбавитель или инертные вещества, поверхностно-активные вещества ионогенного и/или неионогенного типа (смачивающие агенты, диспергирующие средства), например, полиэтоксилированные алкилфенолы, полиэтоксилированные жирные спирты, полиэтоксилированные жирные амины, сульфаты простых эфиров жирного спирта и полигликоля, алкансульфонаты, алкилбензолсульфонаты, лигнинсульфонат натрия, 2,2'-динафтилметан-6,6'-дисульфонат натрия, дибутилнафталилсульфонат натрия или даже олеоилметилтауринат натрия. Для получения смачивающихся порошков используются гербицидно активные соединения, тонкоизмельченные, например, в обычной аппаратуре, такой как молотковые мельницы, вентиляторные мельницы и воздухоструйные мельницы, и их смешивают, одновременно или последовательно со вспомогательными веществами, использующимися для их приготовления.
Эмульгирующиеся концентраты получают растворением активного соединения в органическом растворителе, например, бутаноле, циклогексаноне, диметилформамиде, ксилоле или даже в относительно высококипящих ароматических соединениях или углеводородах или смесях растворителей, путем добавления одного или большего количества поверхностно-активных веществ ионогенного и/или неионогенного типа (эмульгаторы). Примерами эмульгаторов, которые можно использовать, являются алкиларилсульфонаты кальция, такие как додецилбензолсульфонат Ca, или неионогенные эмульгаторы, такие как эфиры полигликоля с жирными кислотами, алкилариловые эфиры полигликоля, эфиры жирного спирта с полигликолем, продукты конденсации пропиленоксид-этиленоксид, алкиловые простые полиэфиры, сорбитановые сложные эфиры, например, сорбитановые эфиры жирных кислот или полиоксиэтиленсорбитановые сложные эфир, например, полиоксиэтиленсорбитановые эфиры жирных кислот.
Дусты получают путем размола активного соединения с тонкоизмельченными твердыми веществами, такими как тальк, природные длины глины, такие как каолин, бентонит и пирофиллит, или диатомовая земля. Концентраты суспензии могут готовиться на водной или масляной основе. Их можно получить, например, путем мокрого размола с использованием обычных имеющихся в продаже шаровых мельниц с добавлением или без добавления поверхностно-активных веществ, как уже отмечено выше, например, в случае препаратов других типов.
Эмульсии, например, эмульсии типа масло-в-воде (EW), можно получить например, с помощью смесителей, коллоидных мельниц и/или статических смесителей использованием водно-органических растворителей и при желании поверхностно-активных веществ, как уже отмечено выше, например, в случае препаратов других типов.
Гранулы можно получить путем разбрызгивания активного соединения на адсорбирующий гранулированный инертный материал или путем нанесения концентратов активного соединения на поверхность носителей, таких как песок, каолиниты или гранулированный инертный материал, с использованием адгезивных связующих, таких как поливиниловый спирт, полиакрилат натрия или даже минеральные масла. Подходящие активные соединения также можно гранулировать по методике, обычно использующейся для получения гранул удобрений, при желании в виде смеси с удобрениями. Диспергирующиеся в воде гранулы обычно получают по обычным методикам, таким как распылительная сушка, гранулирование в псевдоожиженном слое, дисковое гранулирование, перемешивание с использованием высокоскоростных смесителей и экструзия без использования твердого инертного материала.
Методики получения гранул с помощью дискового, проводимого с помощью экструдера и разбрызгивания гранулирования описаны, например, в публикациях "Spray-Drying Handbook" 3rd ed. 1979, G. Goodwin Ltd., London; J. E. Browning, "Agglomeration", Chemical and Engineering 1967, pages 147 ff.; "Perry's Chemical Engineer's Handbook", 5th Ed., McGraw-Hill, New York 1973, pp. 8-57. Дополнительные подробности приготовления средст защиты растений описаны, например, в публикациях G. C. Klingman, "Weed Control as a Science", John Wiley and Sons Inc., New York, 1961, pages 81-96 и J. D. Freyer, S. A. Evans, "Weed Control Handbook", 5th Ed., Blackwell Scientific Publications, Oxford, 1968, pages 101-103.
Агрохимические препараты обычно содержат от 0,1 до 99 мас.%, в частности, от 0,1 до 95 мас.% активного соединения формулы (I). В смачивающихся порошках концентрация активного соединения, например, равна примерно от 10 до 90 мас.%, остальное до 100 мас.% представляет собой обычные компоненты препарата. В эмульгирующихся концентратах концентрация активного соединения может равняться примерно от 1 до 90%, предпочтительно от 5 до 80 мас.%. Препараты в форме дустов содержат от 1 до 30 мас.% активного соединения, чаще всего предпочтительно от 5 до 20 мас.% активного соединения, а растворы для опрыскивания содержат примерно от 0,05 до 80%, предпочтительно от 2 до 50 мас.% активного соединения. В случае диспергирующихся в воде гранул содержание активного соединения частично зависит от того, находится ли активное соединение в жидкой или твердой форме, и от использующихся вспомогательных веществ для гранулирования, наполнителей и т. п. В диспергирующихся в воде гранулах содержание активного соединения, например, равно от 1 до 95 мас.%, предпочтительно от 10 до 80 мас.%.
Кроме того, указанные препараты активного соединения могут включать агенты, придающие липкость, смачивающие агенты, диспергирующие средства, эмульгаторы, средства, обеспечивающие проницаемость, консерванты, антифризные агенты, растворители, наполнители, носители, красители, противовспениватели, средства, препятствующие испарению, и регуляторы pH и вязкости, которые в каждом случае являются обычными.
На основе этих препаратов также можно приготовить комбинации с другими пестицидно активными соединениями, например, инсектицидами, акарицидами, гербицидами и фунгицидами, и также с антидотами, удобрениями и/или регуляторами роста, например, в форме готовой к применению смеси или баковой смеси.
Подходящими активными соединениями, которые можно объединять с активными соединениями, предлагаемыми в настоящем изобретении, в смешанных препаратах или баковой смеси, являются, например, известные активные соединения, описанные, например, в публикации World Herbicide New Product Technology Handbook, China Agricultural Science and Farming Techniques Press, 2010.9 и в цитированной в ней литературе. Например, следующие активные соединения можно отметить в качестве гербицидов, которые можно объединять с соединениями формулы (I) (примечание: соединения называют с использованием "обычного названия" в соответствии с нормативами Международной организации стандартизации (ISO) или химических названий, при необходимости вместе с обычным кодовым номером): ацетохлор, бутахлор, алахлор, пропизохлор, метолахлор, s-метолахлор, претилахлор, пропахлор, этахлор, напропамид, R-левовращающий напропамид, пропанил, мефенацет, дифенамид, дифлуфеникан, этапрохлор, бефлубутамид, бромобутид, диметенамид, диметенамид-P, этобензанид, флуфенацет, тенилхлор, метазахлор, изоксабен, флампроп-M-метил, флампроп-M-пропил, аллидохлор, пентоксамид, хлоранокрил, ципразин, мефлуидид, моналид, делахлор, принахлор, тербухлор, ксилахлор, диметахлор, цисанилид, тримексахлор, кломепроп, пропизамид, пентанохлор, карбетамид, бензоилпроп-этил, ципразол, бутенахлор, тебутам, бензипрам, 1379, дихлофлуанид, напроанилид, диэтатилэтил, напталам, флуфенацет, бензадокс, хлортиамид, хлорфталимид, изокарбамид, пиколинафен, атразин, симазин, прометрин, цианатрин, симетрин, аметрин, пропазин, дипропетрин, SSH-108, тербутрин, тербутилазин, триазифлам, ципразин, проглиназин, триэтазин, прометон, симетон, азипротрин, десметрин, диметаметрин, проциазин, мезопразин, себутилазин, секбуметон, тербуметон, метопротрин, цианатрин, ипазин, хлоразин, атратон, пендиметалин, эглиназин, циануровая кислота, индазифлам, хлорсульфурон, метсульфурон-метил, бенсульфурон метил, хлоримурон-этил, трибенурон-метил, тифенсульфурон-метил, пиразосульфурон-этил, мезосульфурон, йодосульфурон-метилнатрий, форамсульфурон, циносульфурон, триасульфурон, сульфометурон метил, никосульфурон, этаметсульфурон-метил, амидосульфурон, этоксисульфурон, циклосульфамурон, римсульфурон, азимсульфурон, флазасульфурон, моносульфурон, моносульфурон-сложный эфир, флукарбазон-натрий, флупирсульфурон-метил, галосульфурон-метил, оксасульфурон, имазосульфурон, примисульфурон, пропоксикарбазон, просульфурон, сульфосульфурон, трифлоксисульфурон, трифлусульфурон-метил, тритосульфурон, натрий-метсульфурон-метил, флуцетосульфурон, HNPC-C, ортосульфамурон, пропирисульфурон, метазосульфурон, ацифлюорфен, фомесафен, лактофен, фторгликофен, оксифлуорфен, хлорнитрофен, аклонифен, этоксифен-этил, бифенокс, нитрофторфен, хлометоксифен, фтордифен, фторнитрофен, фурилоксифен, нитрофен, TOPE, DMNP, PPG1013, AKH-7088, галосафен, хлортолурон, изопротурон, линурон, диурон, димрон, флуометурон, бензтиазурон, метабензтиазурон, кумилурон, этидимурон, изоурон, тебутиурон, бутурон, хлорбромурон, метилдимрон, фенобензурон, SK-85, метобромурон, метоксурон, афесин, монурон, сидурон, фенурон, флуотиурон, небурон, хлороксурон, норурон, изонорурон, 3-циклооктил-1, тиазфлурон, тебутиурон, дифеноксурон, парафлурон, метиламин трибунил, карбутилат, триметурон, димефурон, монисоурон, анисурон, метиурон, хлорэтурон, тетрафлурон, фенмедифам, фенмедифам-этил, десмедифам, асулам, тербукарб, барбан, профам, хлорпрофам, роумат, свеп, хлорбуфам, карбоксазол, хлорпрокарб, фенасулам, BCPC, CPPC, карбасулам, бутилат, бентиокарб, вернолат, молинат, триаллат, димепиперат, эспрокарб, пирибутикарб, циклоат, авадекс, EPTC, этиолат, орбенкарб, пебулат, просульфокарб, тиокарбазил, CDEC, димексано, изополинат, метиобенкарб, бутиловый эфир 2,4-D, MCPA-Na, изооктиловый эфир 2,4-D, изооктиловый эфир MCPA, натриевая соль 2,4-D, соль 2,4-D с диметиламином, MCPA-тиоэтил, MCPA, 2,4-D-пропионовая кислота, соль 2,4-D с пропионовой кислотой в высокой концентрации, 2,4-D масляная кислота, MCPA-пропионовая кислота, Соль MCPA с пропионовой кислотой, MCPA-масляная кислота, 2,4,5-D, 2,4,5-D-пропионовая кислота, 2,4,5-D-масляная кислота, Соль MCPA с амином, дикамба, эрбон, хлорфенак, сайзон, TBA, хлорамбен, метокси-TBA, диклофоп-метил, флуазифоп-бутил, флуазифоп-п-бутил, галоксифоп-метил, галоксифоп-P, хизалофоп-этил, хизалофоп-п-этил, феноксапроп-этил, феноксапроп-п-этил, пропахизафоп, цигалофоп-бутил, метамифоп, клодинафоп-пропаргил, фентиапроп-этил, хлоразифоп-пропинил, поппенат-метил, трифопсим, изоксапирифоп, паракват, дикват, оризалин, эталфлуралин, изопропалин, нитралин, профлуралин, продинамин, бенфлуралин, флухлоралин, динитрамин, дипропалин, хлорнидин, металпропалин, динопроп, глифосат, анилофос, глуфосинат аммоний, амипрофос-метил, сульфосат, пиперофос, биалафос-натрий, бенсулид, бутамифос, фокарб, 2,4-DEP, H-9201, зитрон, имазапир, имазетапир, имазахин, имазамокс, аммониевая соль имазамокса, имазапик, имазаметабенз-метил, флуроксипир, изооктиловый эфир флуроксипира, клопиралид, пиклорам, трихлопир, дитиопир, галоксидин, 3,5,6-трихлор-2-пиридинол, тиазопир, флуридон, аминопиралид, дифлубензопир, триклопир-бутотил, Клиодинат, сетоксидим, клетодим, циклоксидим, аллоксидим, клефоксидим, бутроксидим, тралкоксидим, тепралоксидим, бутидазол, метрибузин, гексазинон, метамитрон, этиозин, аметридион, амбузин, бромоксинил, бромоксинилоктаноат, иоксинилоктаноат, иоксинил, дихлобенил, diphenatrile, пираклонил, хлороксинил, йодобонил, флуметсулам, флорасулам, фенокссулам, метосулам, клорансулам-метил, диклосулам, пироксулам, бенфуресат, биспирибак-натрий, пирибензоксим, пирифталид, пириминобак-метил, пиритиобак-натрий, бензобициклон, мезотрион, сулкотрион, темботрион, тефурилтрион, бициклопирон, кетодпирадокс, изоксафлутол, кломазон, феноксасульфон, метиозолин, флуазолат, пирафлуфен-этил, пиразолинат, дифензокват, пиразоксифен, бензофенап, нипираклофен, пирасульфотол, топрамезон, пироксасульфон, кафенстрол, флупоксам, аминотриазол, амикарбазон, азафенидин, карфентразон-этил, сульфентразон, бенкарбазон, бензфендизон, бутафенацил, бромацил, изоцил, ленацил, тербацил, флупропацил, цинидон-этил, флумиклорак-пентил, флумиоксазин, пропизамид, MK-129, флумезин, пентахлорфенол, диносеб, динотерб, ацетат динотерба, диносам, DNOC, хлорнитрофен, меацетат динотерба, динофенат, оксадиаргил, оксадиазон, пентоксазон, флуфенацет, флутиацет-метил, фентразамид, флуфенпир-этил, пиразон, бромпиразон, метфлуразон, кузакира, димидазон, оксапиразон, норфлуразон, пиридафол, хинклорак, хинмерак, бентазон, пиридат, оксазикломефон, беназолин, кломазон, цинметилин, ZJ0702, пирибамбенз-пропил, инданофан, хлорат натрия, далапон, трихлоруксусная кислота, монохлоруксусная кислота, гексахлорацетон, флупропанат, циперкват, бромфеноксим, эпроназ, метазол, флуртамон, бенфуресат, этофумезат, тиоклорим, хлортал, фторхлоридон, таврон, акролеин, бентранил, тридифан, хлорфенпропметил, тидиаризонамин, фенизофам, бузоксинон, метоксифенон, сафлуфенацил, клацифос, хлоропон, алорак, диэтамкват, этнипромид, ипримидам, ипфенкарбазон, тиенкарбазон-метил, пиримисульфан, хлорфлуразол, трипропиндан, сулгликапин, просульфалин, камбендихлор, аминоциклопирахлор, родетанил, беноксакор, фенклорим, флуразол, фенхлоразол-этил, клохинтоцет-мексил, оксабетринил, MG/91, циометринил, DKA-24, мефенпир-диэтил, фурилазол, флуксофеним, изоксадифен-этил, дихлормид, галауксифен-метил, DOW848, UBH-509, D489, LS 82-556, KPP-300, NC-324, NC-330, KH-218, DPX-N8189, SC-0744, DOWCO535, DK-8910, V-53482, PP-600, MBH-001, KIH-9201, ET-751, KIH-6127 и KIH-2023.
Для использования препараты, которые в настоящее время имеются в продаже, при необходимости разбавляют обычным образом, например, водой в случае смачивающихся порошков, эмульгирующихся концентратов, дисперсий и диспергирующихся в воде гранул. Продукты в форме дустов, гранул для внесения в почву или вразброс и растворы для опрыскивания обычно до использования дополнительно не разбавляют другими инертными веществами. Норма расхода соединений формулы (I) меняется в зависимости от внешних условий, таких как температура, влажность, тип использующегося гербицида и т. п. Она может меняться в широких пределах, например, от 0,001 до 1,0 кг/га или более активного соединения, но предпочтительно равна от 0,005 до 750 г/га, более предпочтительно от 0,005 до 250 г/га.
Вследствие экономических причин, разнообразия и биологической активности соединения, мы предпочли синтезировать некоторые соединения, часть из которых выбрана и представлена в приведенной ниже таблице. Структура и соответствующая информация для некоторых соединений приведены в таблице 1. Соединение, указанные в таблице 1, предназначены для разъяснения настоящего изобретения и их не следует считать каким-либо образом ограничивающими настоящее изобретение. Специалисты в данной области техники не должны интерпретировать объект настоящего изобретения, как ограниченный указанными ниже соединениями. Данные по физическим характеристикам, относящиеся к настоящему изобретению, не были калиброваны.
Таблица 1. Структура соединений и данные1H ЯМР


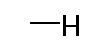


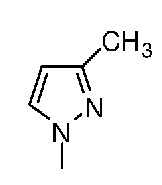






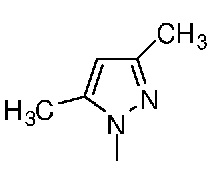


















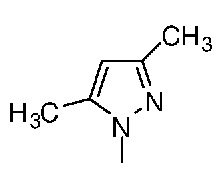












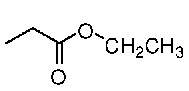




Конкретная методика осуществления изобретения
Приведенное выше содержание настоящего изобретения дополнительно пояснено с помощью следующих вариантов осуществления и специалисты в данной области техники не должны считать их ограниченными вариантами осуществления: любые методики, разработанные на основе содержания настоящего изобретения следует включать в объем настоящего изобретения. Технологические параметры и выходы продуктов представлены в вариантах осуществления без оптимизации.
Пример 1
Способ получения соединения 01 в таблице 1 описан в варианте осуществления
Стадия 1: синтез промежуточного продукта (a)

50 мл Ацетонитрила добавляли в трехгорлую колбу объемом 250 мл. Колбу помещали в баню из воды со льдом и температуру устанавливали равной от 5 до 10°C. 3,0 г (0,075 моля) NaH отвешивали и медленно добавляли в трехгорлую колбу. Температуру поддерживали равной ниже 10°C. Затем 3 г (0,036 моля) 4-метилпиразола растворяли в небольшом количестве ацетонитрила, раствор помещали в капельную воронку и по каплям добавляли к системе примерно при 0°C. После добавления полученную смесь перемешивали в бане из воды со льдом. Когда температура системы становилась постоянной, 10 г (0,030 моля) 2-хлор-3-бромметил-4-метилсульфонилбензойной кислоты отвешивали и порциями медленно добавляли в систему при регулируемой температуре, равной не выше 10°C. Систему перемешивали в бане из воды со льдом. За протеканием реакции следили с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) до полного израсходования вещества. Ацетонитрил удаляли путем выпаривания в роторном испарителе. К остатку добавляли 200 мл воды. По каплям добавляли HCl, полученную смесь перемешивали при комнатной температуре для осаждения твердых частиц. Твердые частицы собирали фильтрованием с отсасыванием и получали почти белое твердое вещество, которое было промежуточным продуктом (a). Промежуточный продукт сушили в сушильном шкафу для последующего использования.
Стадия 2: синтез промежуточного продукта (b)

10 г (0,030 моля) Промежуточного продукта (a) отвешивали и добавляли в колбу объемом 250 мл, затем добавляли 50 мл дихлорэтана и небольшое количество ДМФ в качестве катализатора. Затем 5 г (0,039 моля) оксалилхлорида растворяли в небольшом количестве дихлорэтана. Раствор помещали в капельную воронку и по каплям добавляли в систему при комнатной температуре. После добавления по каплям систему продолжали перемешивать в течение примерно 2 ч при комнатной температуре и получали раствор реакционной смеси, содержащий промежуточный продукт (b). Раствор реакционной смеси сразу использовали в следующей реакции без какой-либо обработки.
Стадия 3: синтез соединения 01

4,0 г (0,036 моля) 1,3-диметил-5-пиразолола отвешивали и добавляли в трехгорлую колбу объемом 250 мл. Для растворения добавляли 50 мл 1,2-дихлорэтана. 12 г (0,12 моля) Триэтиламина отвешивали и добавляли в систему. Раствор реакционной смеси, содержащий промежуточный продукт (b) (0,030 моля), по каплям добавляли в систему в бане из воды со льдом и в защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч. Раствор реакционной смеси, содержащий промежуточный продукт (c), получали после полного израсходования сырья. 3,0 г (0,030 моля) Триэтиламина и 0,5 мл циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (c), в защитной атмосфере аргона при регулируемой температуре, равной от 50 до 60°C, и реакцию проводили в течение 2 ч. Для слежения за протеканием реакции использовали ВЭЖХ. После завершения реакции добавляли 100 мл воды, затем медленно по каплям при перемешивании при комнатной температуре добавляли HCl до установления pH, равного примерно 3. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, органический растворитель удаляли путем выпаривания в роторном испарителе и получали 8,1 г бледно-коричневого порошкообразного твердого вещества, т. е. соединения 01. Содержание, определенное с помощью ВЭЖХ равнялось 93,9%, и выход равнялся 67,8%.
Данные1H ЯМР см. в таблице 1.
В примерах 2-4 раскрыт синтез соединения 02 с преобразованием в соединение 04 соответственно, методики синтеза которого аналогичны методикам примера 1, поэтому их описание не приведено.
Пример 5
В этом примере раскрыт синтез соединения 05, приведенного в таблице 1.
Стадия 1: синтез промежуточного продукта (d)

50 мл Ацетонитрила отвешивали и добавляли в трехгорлую колбу объемом 250 мл и помещали в баню из воды со льдом при регулируемой температуре, равной от 5 до 10°C. 4,4 г (0,11 моля) NaH отвешивали и медленно добавляли в колбу при регулируемой температуре, равной не выше 10°C. 4,6 г (0,045 моля) 4-Хлорпиразола отвешивали и растворяли в небольшом количестве ацетонитрила, раствор помещали в капельную воронку и по каплям добавляли с охлаждением системы примерно до 0°C. После добавления полученный реакционный раствор перемешивали в бане из воды со льдом. Когда температура системы становилась постоянной, 10 г (0,030 моля) 2-хлор-3-бромметил-4-метилсульфонилбензойной кислоты отвешивали и порциями добавляли в систему при регулируемой температуре, равной не выше 10°C, и перемешивали в бане из воды со льдом. За протеканием реакции следили с помощью ВЭЖХ до полного израсходования сырья. Ацетонитрил удаляли путем выпаривания в роторном испарителе. К остатку добавляли 200 мл воды, затем медленно по каплям добавляли HCl и перемешивали при комнатной температуре для осаждения твердых частиц. Твердые частицы собирали фильтрованием с отсасыванием и получали почти белое твердое вещество, т. е. промежуточный продукт (d). Промежуточный продукт сушили в сушильном шкафу для последующего использования.
Стадия 2: синтез промежуточного продукта (e)

10,5 г (0,030 моля) Промежуточного продукта (d) отвешивали и добавляли в колбу объемом 250 мл и добавляли 50 мл дихлорэтана и небольшое количестве ДМФ в качестве катализатора. Затем 5 г (0,039 моля) оксалилхлорида отвешивали и растворяли в небольшом количестве дихлорэтана. Раствор помещали в капельную воронку и по каплям добавляли в систему при комнатной температуре. Систему перемешивали в течение примерно 2 ч при комнатной температуре после добавления и получали раствор реакционной смеси, содержащий промежуточный продукт (e). Раствор реакционной смеси сразу использовали в следующей реакции без какой-либо обработки.
Стадия 3: синтез соединения 05

4,5 г (0,036 моля) 1-Метил-3-этил-5-пиразолола отвешивали и добавляли в трехгорлую колбу объемом 250 мл и для растворения добавляли 50 мл 1,2-дихлорэтана. 12 г (0,012 моля) Триэтиламина отвешивали и вводили в систему. Раствор реакционной смеси, содержащий промежуточный продукт (e) (0,030 моля), по каплям добавляли в систему в бане из воды со льдом в защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч, раствор реакционной смеси, содержащий промежуточный продукт (f), получали после полного израсходования сырья. 3,0 г (0,030 моля) Триэтиламина и 0,5 мл циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (f), в защитной атмосфере аргона при регулируемой температуре, равной от 40 до 50°C, и реакцию проводили в течение 2 ч. За протеканием реакции следили с помощью ВЭЖХ. После завершения реакции добавляли 100 мл воды, затем при перемешивании по каплям медленно добавляли HCl при комнатной температуре до установления pH, равного примерно 3. Водный слой удаляли путем экстракции и органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, органический растворитель удаляли путем выпаривания в роторном испарителе и получали 6,7 г темно-коричневого порошкообразного твердого вещества, т. е. соединения 05. Содержание, определенное с помощью ВЭЖХ равнялось 86,8%, и выход равнялся 42,4%.
Данные1H ЯМР см. в таблице 1.
В примере 6 раскрыт синтез соединения 6, методика синтеза которого аналогична методике примера 5, поэтому его описание не приведено.
Пример 7
В этом примере раскрыт синтез соединения 07, приведенного в таблице 1.
Стадия 1: синтез промежуточного продукта (a)
См. пример 1.
Стадия 2: синтез промежуточного продукта (b)
См. пример 1.
Стадия 3: синтез соединения 07

3,2 г (0,033 моля) 1-Метил-5-пиразолола отвешивали и добавляли в трехгорлую колбу объемом 250 мл и для растворения добавляли 50 мл 1,2-дихлорэтана. 10 г (0,010 моля) Триэтиламина добавляли в систему. Раствор реакционной смеси (0,030 моля), содержащий промежуточный продукт (b), по каплям добавляли в бане из воды со льдом и в защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч, раствор реакционной смеси, содержащий промежуточный продукт (g), получали после полного израсходования сырья. 3,0 г (0,030 моля) Триэтиламина и 0,5 мл циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (g), в защитной атмосфере аргона при регулируемой температуре, равной от 50 до 60°C. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 2 ч. После завершения реакции добавляли 100 мл воды, затем медленно по каплям при перемешивании при комнатной температуре добавляли HCl до установления pH, равного примерно 3. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали 8,3 г бледно-коричневого порошкообразного твердого вещества, т. е. соединения 07. Содержание, определенное с помощью ВЭЖХ равнялось 96,5%, и выход равнялся 72,4%.
Данные1H ЯМР см. в таблице 1.
В примерах 8-10 раскрыт синтез соединения 08 с преобразованием в соединение 10, приведенное в таблице 1 соответственно, методики синтеза которого аналогичны методикам примера 7, поэтому их описание не приведено.
Пример 11
В этом примере раскрыт синтез соединения 11, приведенного в таблице 1.
Стадия 1: синтез промежуточного продукта (d)
См. пример 5.
Стадия 2: синтез промежуточного продукта (e)
См. пример 5.
Стадия 3: синтез соединения 11

4,6 г (0,033 моля) 1-Метил-3-циклопропил-5-пиразолола отвешивали и добавляли в трехгорлую колбу объемом 250 мл и для растворения добавляли 50 мл 1,2-дихлорэтана. 12 г (0,012 моля) Триэтиламина добавляли в систему. Раствор реакционной смеси, содержащий промежуточный продукт (e) (0,030 моля), по каплям добавляли в систему в бане из воды со льдом ив защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч. Раствор реакционной смеси, содержащий промежуточный продукт (h), получали после полного израсходования сырья. 3,0 г (0,030 моля) Триэтиламина и 0,5 мл циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (h), в защитной атмосфере аргона при регулируемой температуре, равной от 40 до 50°C. После проведения реакции в течение 2 ч за протеканием реакции следили с помощью ВЭЖХ. После завершения реакции добавляли 100 мл воды, затем по каплям при перемешивании медленно добавляли HCl при комнатной температуре до установления pH, равного примерно 3. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали 6,6 г темно-коричневого порошкообразного твердого вещества, т. е. соединения 11. Содержание, определенное с помощью ВЭЖХ равнялось 83,1%, и выход равнялся 56,5%.
Данные1H ЯМР см. в таблице 1.
В примерах 12-14 раскрыт синтез соединения 12 с преобразованием в соединение 14, приведенное в таблице 1 соответственно, методики синтеза которого аналогичны методикам примера 11, поэтому их описание не приведено.
Пример 15
В этом примере раскрыт синтез соединения 15, приведенного в таблице 1.
Стадия 1: синтез промежуточного продукта (i)

50 мл Ацетонитрила отвешивали и добавляли в трехгорлую колбу объемом 250 мл. Колбу помещали в баню из воды со льдом при регулируемой температуре, равной от 5 до 10°C. 3,0 г (0,075 моля) NaH медленно добавляли в колбу при регулируемой температуре, равной 10°C. Затем 2,4 г (0,036 моля) пиразола растворяли в небольшом количестве ацетонитрила, полученный раствор помещали в капельную воронку и по каплям добавляли когда температура системы снижалась примерно до 0°C. После добавления по каплям систему перемешивали в бане из воды со льдом. Когда температура системы становилась постоянной, 10 г (0,030 моля) 2-хлор-3-бромметил-4-метилсульфонилбензойной кислоты отвешивали и медленно добавляли в систему порциями при регулируемой температуре, равной не выше 10°C, и перемешивали в бане из воды со льдом. За протеканием реакции следили с помощью ВЭЖХ до полного израсходования сырья. Ацетонитрил удаляли путем выпаривания в роторном испарителе, затем добавляли 200 мл воды. Медленно по каплям добавляли HCl и перемешивали при комнатной температуре для осаждения твердых частиц. Частицы собирали фильтрованием с отсасыванием и получали почти белое твердое вещество, т. е. промежуточный продукт (i). Промежуточный продукт сушили в сушильном шкафу для последующего использования.
Стадия 2: синтез промежуточного продукта (j)
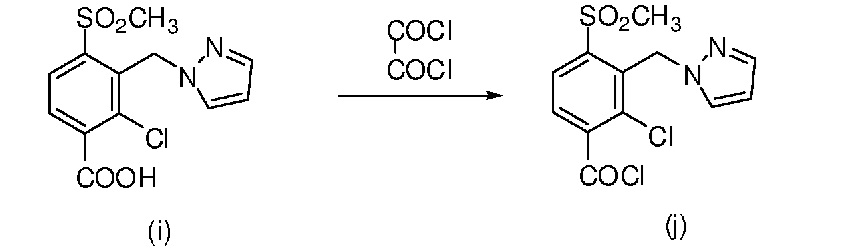
10 г (0,030 моля) Промежуточного продукта (i) отвешивали и добавляли в колбу объемом 250 мл и добавляли 50 мл дихлорэтана. Небольшое количество ДМФ по каплям добавляли в качестве катализатора. Затем 5 г (0,039 моля) оксалилхлорида отвешивали и растворяли в небольшом количестве дихлорэтана, полученный раствор помещали в капельную воронку и по каплям добавляли к системе при комнатной температуре. После добавления по каплям систему перемешивали в течение примерно 2 ч при комнатной температуре и получали раствор реакционной смеси, содержащий промежуточный продукт (j). Раствор реакционной смеси сразу использовали в следующей реакции без какой-либо обработки.
Стадия 3: синтез соединения 15

1,9 г (0,015 моля) 1-Метил-3-циклопропил-5-пиразолола отвешивали и добавляли в трехгорлую перегонную колбу объемом 250 мл и для растворения добавляли 50 мл 1,2-дихлорэтана. 4,0 г (0,040 моля) Триэтиламина отвешивали и добавляли в систему. Раствор 1,2-дихлорэтана (содержащий 0,010 моля (j)), содержащий промежуточный продукт (j), по каплям добавляли в систему в бане из воды со льдом и в защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч. Раствор реакционной смеси, содержащий промежуточный продукт (k), получали после полного израсходования сырья. 1,0 г (0,010 моля) Триэтиламина и несколько капель циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (k), в защитной атмосфере аргона при регулируемой температуре, равной от 50 до 60°C, и реакцию проводили в течение 2 ч. За протеканием реакции следили с помощью ВЭЖХ. После завершения реакции добавляли 100 мл воды, и по каплям медленно при перемешивании добавляли HCl при комнатной температуре до установления pH, равного примерно 3. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали 3,6 г бледно-коричневого порошкообразного твердого вещества, т. е. соединения 15. Содержание, определенное с помощью ВЭЖХ равнялось 95,6%, и выход равнялся 81,5%.
Данные1H ЯМР см. в таблице 1.
В примерах 16-17 раскрыт синтез соединения 16 с преобразованием в соединение 17, приведенное в таблице 1 соответственно, методики синтеза которого аналогичны методикам примера 15, поэтому их описание не приведено.
Пример 18
В этом примере раскрыт синтез соединения 18, приведенного в таблице 1.
Стадия 1: синтез промежуточного продукта (i)
См. пример 15.
Стадия 2: синтез промежуточного продукта (j)
См. пример 15.
Стадия 3: синтез соединения 18

1,7 г (0,015 моля) 1-Этил-5-пиразолола отвешивали и добавляли в трехгорлую колбу объемом 250 мл и для растворения добавляли 50 мл 1,2-дихлорэтана. 4,0 г (0,040 моля) Триэтиламина отвешивали и добавляли в систему. Раствор 1,2-дихлорэтана (содержащий 0,010 моля (j)), содержащий промежуточный продукт (j), по каплям добавляли в систему в бане из воды со льдом и в защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч. Раствор реакционной смеси, содержащий промежуточный продукт (l), получали после полного израсходования сырья. 1,0 г (0,010 моля) Триэтиламина и несколько капель циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (l), при регулируемой температуре, равной от 50 до 60°C, в защитной атмосфере аргона и реакцию проводили в течение 2 ч. За протеканием реакции следили с помощью ВЭЖХ. После завершения реакции добавляли 100 мл воды, затем медленно при перемешивании при комнатной температуре по каплям добавляли HCl до установления pH, равного примерно 3. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали 3,5 г бледно-коричневого порошкообразного твердого вещества, т. е. соединения 18. Содержание, определенное с помощью ВЭЖХ равнялось 94,9%, и выход равнялся 81,3%.
Данные1H ЯМР см. в таблице 1.
В примерах 19-20 раскрыт синтез соединения 19 с преобразованием в соединение 20, приведенное в таблице 1 соответственно, методики синтеза которого аналогичны методикам примера 18, поэтому их описание не приведено.
Пример 21
В этом примере раскрыт синтез соединения 21, приведенного в таблице 1.
Стадия 1: синтез промежуточного продукта (i)
См. пример 15.
Стадия 2: синтез промежуточного продукта (j)
См. пример 15.
Стадия 3: синтез соединения 21

1,8 г (0,012 моля) 1-Метил-3-циклопропил-5-пиразолола отвешивали и добавляли в трехгорлую колбу объемом 250 мл и для растворения добавляли 50 мл 1,2-дихлорэтана. 3,0 г (0,040 моля) Триэтиламина добавляли в систему. Раствор 1,2-дихлорэтана (содержащий 0,010 моля (j)), содержащий промежуточный продукт (j), по каплям добавляли в систему в бане из воды со льдом и в защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч. Раствор реакционной смеси, содержащий промежуточный продукт (m), получали после полного израсходования сырья. 1,0 г (0,010 моля) Триэтиламина и несколько капель циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (m), при регулируемой температуре, равной от 50 до 60°C, в защитной атмосфере аргона и реакцию проводили в течение 2 ч. За протеканием реакции следили с помощью ВЭЖХ. После завершения реакции добавляли 100 мл воды, и медленно при перемешивании при комнатной температуре по каплям добавляли HCl до установления pH, равного примерно 3. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали 3,9 г бледно-коричневого порошкообразного твердого вещества, т. е. соединения 21. Содержание, определенное с помощью ВЭЖХ равнялось 93,6%, и выход равнялся 81,4%.
Данные1H ЯМР см. в таблице 1.
В примерах 22-23 раскрыт синтез соединения 22 с преобразованием в соединение 23, приведенное в таблице 1 соответственно, методики синтеза которого аналогичны методикам примера 21, поэтому их описание не приведено.
Пример 24
В этом примере раскрыта специальная методика синтеза соединения 24, приведенного в таблице 1.
Стадия 1: синтез промежуточного продукта (i)
См. пример 15.
Стадия 1: синтез промежуточного продукта (j)
См. пример 15.
Стадия 3: синтез соединения 24

1,7 г (0,012 моля) 1,3-Диэтил-5-пиразолола отвешивали и добавляли в трехгорлую колбу объемом 250 мл, и для растворения добавляли 50 мл 1,2-дихлорэтана. 3,0 г (0,030 моля) Триэтиламина отвешивали и добавляли в систему. Раствор 1,2-дихлорэтана (содержащий 0,010 моля (j)), содержащий промежуточный продукт (j), по каплям добавляли в систему в бане из воды со льдом и в защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч. Раствор реакционной смеси, содержащий промежуточный продукт (n), получали после полного израсходования сырья. 1,0 г (0,010 моля) Триэтиламина и несколько капель циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (n), при регулируемой температуре, равной от 50 до 60°C, в защитной атмосфере аргона и реакцию проводили в течение 2 ч. За протеканием реакции следили с помощью ВЭЖХ. После завершения реакции добавляли 100 мл воды, затем по каплям при перемешивании при комнатной температуре медленно добавляли HCl до установления pH, равного примерно 3. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали 3,9 г бледно-коричневого порошкообразного твердого вещества, т. е. соединения 24. Содержание, определенное с помощью ВЭЖХ равнялось 92,1%, и выход равнялся 82,3%.
Данные1H ЯМР см. в таблице 1.
В примерах 25-26 раскрыт синтез соединения 25 с преобразованием в соединение 26, приведенное в таблице 1 соответственно, методики синтеза которого аналогичны методикам примера 24, поэтому их описание не приведено.
Пример 27
В этом примере раскрыт синтез соединения 24, приведенного в таблице 1.
Стадия 1: синтез промежуточного продукта (i)
См. пример 15.
Стадия 1: синтез промежуточного продукта (j)
См. пример 15.
Стадия 3: синтез соединения 27
В этом примере раскрыт синтез соединения 27, приведенного в таблице 1. Соединение 27 можно синтезировать следующим путем:

2,0 г (0,012 моля) 1-Изопропил-3-циклопропил-5-пиразолола отвешивали и добавляли в трехгорлую колбу объемом 250 мл и для растворения добавляли 50 мл 1,2-дихлорэтана. 3,0 г (0,030 моля) Триэтиламина отвешивали и добавляли в систему. Раствор 1,2-дихлорэтана (содержащий 0,010 моля (j)), содержащий промежуточный продукт (j) добавляли в систему в бане из воды со льдом и в защитной атмосфере аргона. За протеканием реакции следили с помощью ВЭЖХ после проведения реакции в течение 1 ч. Раствор реакционной смеси, содержащий промежуточный продукт (o), получали после полного израсходования сырья. 1,0 г (0,010 моля) Триэтиламина и несколько капель циангидрина ацетона добавляли в раствор реакционной смеси, содержащий промежуточный продукт (o), при регулируемой температуре, равной от 50 до 60°C, в защитной атмосфере аргона и реакцию проводили в течение 2 ч. За протеканием реакции следили с помощью ВЭЖХ. После завершения реакции добавляли 100 мл воды, затем по каплям при перемешивании при комнатной температуре медленно добавляли HCl до установления pH, равного примерно 3. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 200 мл воды, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали 4,0 г бледно-коричневого порошкообразного твердого вещества, т. е. соединения 27. Содержание, определенное с помощью ВЭЖХ равнялось 91,8%, и выход равнялся 79,3%.
Данные1H ЯМР см. в таблице 1.
В примерах 28-30 раскрыт синтез соединения 28 с преобразованием в соединение 30, приведенное в таблице 1 соответственно, методики синтеза которого аналогичны методикам примера 27, поэтому их описание не приведено.
Пример 31
В этом примере раскрыт синтез соединения 31, приведенного в таблице 1. Соединение 31 можно синтезировать следующим путем:
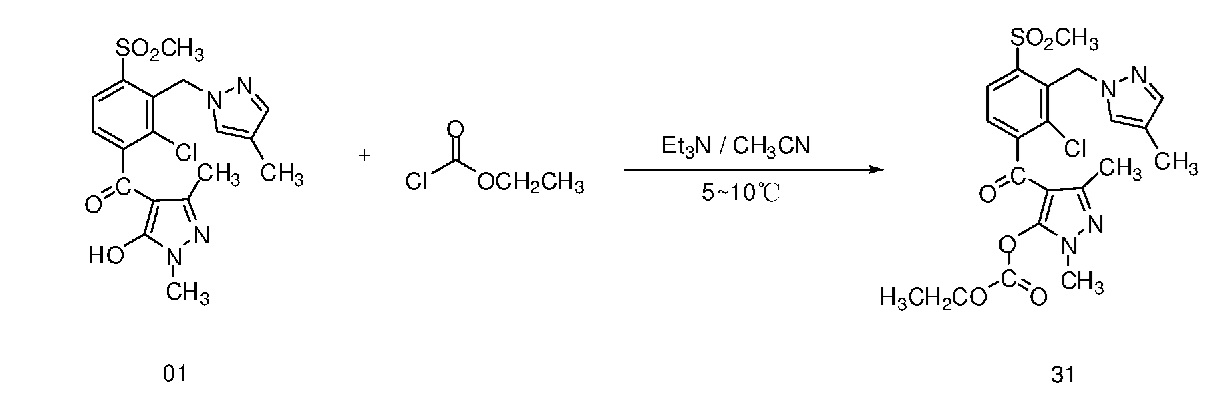
2,2 г (0,005 моля) Соединения 01 отвешивали и добавляли в колбу объемом 100 мл, добавляли 15 мл ацетонитрила и 1,0 г (0,010 моля) триэтиламина и перемешивали в бане из воды со льдом. 0,9 г (0,007 моля) Этилхлорформиата растворяли в 10 мл ацетонитрила и помещали в капельную воронку, и по каплям добавляли в бане из воды со льдом. После добавления по каплям смесь вводили в реакцию в гомотермических условиях в бане из воды со льдом. За протеканием реакции следили с помощью ВЭЖХ до полного израсходования соединения 01. После завершения реакции добавляли 100 мл воды и 100 мл этилацетата. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 100 мл насыщенного раствора соли, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали бледно-коричневое порошкообразное твердое вещество. 1,4 г Бледно-желтого порошка, т. е. соединения 31, получали после перекристаллизации из 95% этилового спирта. Содержание, определенное с помощью ВЭЖХ равнялось 92,9%, и выход равнялся 61,8%.
Данные1H ЯМР см. в таблице 1.
Пример 32
В этом примере раскрыт синтез соединения 32, приведенного в таблице 1. Соединение 32 синтезировали следующим путем:

2,2 г (0,005 моля) Соединения 01 отвешивали и добавляли в колбу объемом 100 мл, добавляли 15 мл ацетонитрила и 1,4 г (0,010 моля) карбоната калия, и перемешивали в бане из воды со льдом. 0,8 г (0,006 моля) Этансульфонилхлорида растворяли в 10 мл ацетонитрила и помещали в капельную воронку, и по каплям добавляли в бане из воды со льдом. После добавления по каплям смесь вводили в реакцию в гомотермических условиях в бане из воды со льдом. За протеканием реакции следили с помощью ВЭЖХ до полного израсходования соединения 01. После завершения реакции добавляли 100 мл воды и 100 мл этилацетата. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 100 мл насыщенного раствора соли, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали желтое порошкообразное твердое вещество. 1,6 г Бледно-желтого порошка, т. е. соединения 32, получали после перекристаллизации из 95% этилового спирта. Содержание, определенное с помощью ВЭЖХ равнялось 95,1%, и выход равнялся 65,3%.
Данные1H ЯМР см. в таблице 1.
Пример 33
В этом примере раскрыт синтез соединения 33, приведенного в таблице 1. Соединение 33 синтезировали следующим путем:

2,20 г (0,005 моля) Соединения 04 отвешивали и добавляли в колбу объемом 100 мл, добавляли 20 мл ацетонитрила и 1,40 г (0,010 моля) карбоната калия и перемешивали в бане из воды со льдом. 0,95 г (0,005 моля) Толуолсульфонилхлорида растворяли в 10 мл ацетонитрила и помещали в капельную воронку, и по каплям добавляли в бане из воды со льдом. После добавления по каплям смесь вводили в реакцию в гомотермических условиях в бане из воды со льдом. За протеканием реакции следили с помощью ВЭЖХ до полного израсходования соединения 04. После завершения реакции добавляли 100 мл воды и 100 мл этилацетата. Водный слой удаляли путем экстракции. Органический слой промывали 2 раза с помощью 100 мл насыщенного раствора соли, сушили над безводным сульфатом натрия, концентрировали путем выпаривания в роторном испарителе и получали коричневое порошкообразное твердое вещество. 1,5 г Светло-коричневого порошка, т. е. соединения 29, получали после перекристаллизации из 95% этилового спирта. Содержание, определенное с помощью ВЭЖХ равнялось 95,5%, и выход равнялся 48,5%.
Данные1H ЯМР см. в таблице 1.
В примерах 34-40 раскрыт синтез соединения 34 с преобразованием в соединение 40, приведенное в таблице 1, методики синтеза которого аналогичны методикам примера 33, поэтому их описание не приведено.
Исследование биологической активности:
Стандарт уровней активности при повреждении вредного растения (т. е. степень подавления роста) является следующим:
Уровень 10: полностью погибают;
Уровень 9: степень подавления роста более 90%;
Уровень 8: степень подавления роста более 80%;
Уровень 7: степень подавления роста более 70%;
Уровень 6: степень подавления роста более 60%;
Уровень 5: степень подавления роста более 50%;
Уровень 4: степень подавления роста более 30%;
Уровень 3: степень подавления роста более 30%;
Уровень 2: степень подавления роста более 20%;
Уровень 1: степень подавления роста более 1-10%;
Уровень 0: отсутствие эффекта.
Приведенные выше степени подавления роста являются степенями подавления роста для массы растений в сыром виде.
Семена однодольных и двудольных сорняков и семена основной сельскохозяйственной культуры (т. е. пшеницы, кукурузы, риса, сои, хлопчатника, масличной культуры, проса и сорго) помещали в пластмассовый горшок, заполненный почвой. Затем их закрывали слоем почвы толщиной 0,5-2 см, семенам предоставляли возможность расти в теплице в благоприятной среде. Исследуемые растения обрабатывали на стадии 2-3 листьев через 2-3 недели после высевания. Исследуемые соединения, предлагаемые в настоящем изобретении, растворяли в ацетоне, затем добавляли Tween 80 и разбавляли некоторым количеством воды до определенной концентрации. Растения опрыскивали этим раствором с помощью опрыскивателя. Затем растения выращивали в теплице в течение 3 недель. Эксперимент, приводивший к борьбе с сорняками через 3 недели, описан в таблице 2.
Таблица 2: Эксперимент по борьбе с сорняками на послевсходовой стадии
*му - китайская единица измерения площади - 1/15 га
В таблице 2 показано, что многие соединения, предлагаемые в настоящем изобретении, при нанесении после всходов безопасны для риса и обладают высокой эффективностью при борьбе с просом куриным. Кроме того, большинство соединений также безопасны для кукурузы и пшеницы и их можно использовать в кукурузе и пшенице для борьбы с травянистыми и широколиственными сорняками.
Исследование безопасности для пересаженного риса и исследование борьбы с сорняками в рисовом поле:
Почву рисового поля помещали в горшок площадью 1/1000000 га. Семена проса, камыша ситниковидного, череды трехраздельной и стрелолиста трехлистного высевали и закрывали тонким слоем почвы, затем выдерживали в теплице под слоем воды глубиной 0,5-1 см. Клубень стрелолиста трехлистного высевали на следующий день или через 2 дня. Затем его выдерживали под слоем воды глубиной 3-4 см. Сорняки равномерно обрабатывали по капельной методике с помощью разбавленной в воде WP или SC, полученной обычным способом получения соединений, предлагаемых в настоящем изобретении, с помощью пипетки с обеспечением заданного эффективного количества, когда просо, камыш ситниковидный и череда трехраздельная достигали стадии 0,5 листа и стрелолист трехлистный достигал момента времени, соответствующего стадии первичного листа.
Кроме того, почву рисового поля, которую помещали в горшок площадью 1/1000000 га, выравнивали, так чтобы образовывался слой воды глубиной 3-4 см. На следующий день рис на стадии 3 листьев (японский рис) пересаживали на глубину 3 см. Через 5 дней после пересадки таким же образом обрабатывали соединением, предлагаемым в настоящем изобретении.
Визуально исследовали продуктивность проса, камыша ситниковидного, череды трехраздельной и стрелолиста трехлистного через 14 дней после обработки соединением, предлагаемым в настоящем изобретении, и продуктивность риса через 21 день после обработки соединением, предлагаемым в настоящем изобретении соответственно. Исследовали борьбу с сорняками для стандартных уровней 1-10 активности и результаты приведены в таблице 3.
Таблица 3: Результаты эксперимента по борьбе с сорняками в пересаженном рисовом поле (500 г активного ингредиента/га)
Примечание: Семена проса куриного, осокообразного камыша, стрелолиста и череды собирали в Heilongjing Province of China. Исследования показали, что сорняки устойчивы по отношению к пиразосульфурон-этилу при обычных нормах расхода.
Одновременно, после нескольких исследований установлено, что соединение, предлагаемое в настоящем изобретении, обладает хорошей селективностью по отношению ко многим злаковым травянистым растениям, таким как цойсия японская, бермудская трава, овсяница тростниковая, мятлик, плевел и паспалум влагалищный и т. п., и может обеспечить борьбу со многими травянистыми сорняками и широколиственными сорняками. Соединение также обладает превосходной селективностью и коммерческой ценностью по данным исследований на сое, хлопчатнике, подсолнечнике, картофеле, фруктовых садах и овощах при использовании различных методик нанесения гербицида.
Реферат
Изобретение относится к области органической химии, а именно к соединениям пиразола формулы (I), в которой R означает,где R', R'', R''' означают водород, C1-C4 алкил, C1-C4 галогенированный алкил или галоген, R', R'', R''' могут быть одинаковыми или разными; Rозначает C1-C3 алкил; Rозначает водород или C1-C4 алкил; Rозначает водород, C1-C6 алкилкарбонил, C1-C6 алкоксилкарбонил, C1-C6 алкоксилкарбонилметил, C1-C4 алкилсульфонил, или фенилсульфонил, замещенный алкилом, или бензоил, или его соль. Также раскрываются конкретные соединения пиразола формулы (I), промежуточные сеодиенения формулы (III) и формулы (V), способ получения соединений пиразола формулы (I), гербицидная композиция на основе соединений пиразола формулы (I), способ борьбы с сорняками и применение соединений пиразола формулы (I) для борьбы с сорняками. Технический результат: получены новые соединения пиразола, которые используются в качестве гербицидов для борьбы с сорняками. 9 н. и 4 з.п. ф-лы, 12 пр., 3 табл.
Формула


























































Документы, цитированные в отчёте о поиске
Производные пиразола или их соли с основаниями
Комментарии