Замещенные производные пиразола, промежуточные длязамещенные производные пиразола, промежуточные для их получения, гербицидная композиция и способ бор их получения, гербицидная композиция и способ борьбы с сорняками ьбы с сорняками - RU2137771C1
Код документа: RU2137771C1
Чертежи















































Описание
Настоящее изобретение относится к новым замещенным производным пиразола, способам их получения, к промежуточным соединениям, и к использованию указанных производных в качестве гербицидов.
Известно, что 1-фенилпиразолы обладают гербицидной активностью (EP 154115).
Гербицидно-активные 1-фенилпиразолы известны из EP-A-167028, так же как и в журнале Гетероциклическая химия, ч.26, 1989, стр. 893-898 уже описаны пиразолилпиразола общей формулы I, а именно, 1-(1,5-диметил-3-пиразолил)-3,5-диметилпиразол, замещенные гербицидно-активные пиразолилпиразолы, отличающиеся заместителем R6, описаны в EP-A-0542388.
Однако из-за недостаточно высокой гербицидной активности или селективности этих соединений могут возникать определенные проблемы при использовании их в целях защиты важных сельскохозяйственных культур.
Задачей настоящего изобретения является создание новых соединений, обладающих улучшенными биологическими свойствами по сравнению с известными соединениями.
Обнаружено, что замещенные производные пиразола общей формулы I

где R1 представляет собой C1-C4-алкил;
R2 представляет собой C1-C4-алкил; C1-C4-алкилтио; C1-C4-алкокси, каждый из которых является необязательно замещенным одним или несколькими атомами галогена;
R1 и R2, взятые вместе, образуют группу -(CH2)m;
R3 представляет собой водород или галоген:
R4 представляет собой водород или C1-C4-алкил;
R5 представляет собой водород, нитро, циано, или группу -COOCR7, -C(= x)NR8R9 или -C(=x)R10;
R6 представляет собой водород, галоген, циано, C1-C4-алкил (необязательно замещенный одним или несколькими атомами галогена или гидроксигруппами), фенил (необязательно замещенный одним или несколькими заместителями, такими как галоген, нитро, циано, C1-C4-алкил, C1-C4-алкокси или галогено-C1 -C4-алкил), пирролил, либо R6 представляет собой C2-C8-алкильную, C3-C8-алкенильную, C3-C8-алкинильную или C3 -C8-алкокcигруппу, каждая из которых прерывается одним или несколькими атомами кислорода; либо К6 представляет собой группу:

R7, R8 и R9 могут быть одинаковыми или различными, и представляют собой водород, или C1-C4-алкил; либо
R8 и R9, взятые вместе с атомом азота, с которым они связаны, образуют 5- или 6-членное насыщенное карбоциклическое кольцо;
R10 представляет собой водород, или C1-C4-алкил, необязательно замещенный одним или несколькими атомами галогена;
R11 представляет собой водород, C1 -C4-алкил, C2-C6-алкенил, C3-C6-алкинил, или фенил(каждый из которых является необязательно замещенным одним или несколькими атомами галогена), C3-C2-циклоалкил, цианометил, или группу R21CO-;
R12 представляет собой C1-C6-алкил, C2-C6-алкенил, C3-C6-алкинил, или фенил (каждый из которых является необязательно замещенным одним или несколькими атомами галогена), C3-C8-циклоалкил, цианометил, C1-C4-алкокси-C1-C6-алкил, ди-C3-C6-алкиламино-C1-C4-алкил, тетрагидрофурфурилметил,
C3-C6-алкинилокси-C1-C4-алкил, бензил (необязательно замещенный одним или несколькими заместителями, такими как галоген, нитро, циано, C1-C4 -алкил, C1-C4-алкокси, или галогено-C1-C4-алкил), либо R12 представляет собой группу -C(= x)R21, -(CH2)a -(O)d-R28, - (CH2)a-O-(CH2)b-R28, или -(CH2)a-X-R34, R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют 3-, 5- или 6-членное насыщенное карбоциклическое или ароматическое кольцо, в котором атом углерода является необязательно замещенным атомом кислорода;
R13 представляет собой водород, C1-C4-алкил; C2-C6-алкенил или C3-C6-алкинил; либо R13 и R14, взятые вместе, образуют группу -(CH2)p;
R14 и R15 могут быть одинаковыми или различными и представляют собой C1-C4-алкил, C2-C6-алкенил, C3-C6-алкинил, или фенил (каждый из которых является необязательно замещенным одним или несколькими атомами галогена), водород, C3-C6-циклоалкил или группы -XR18 или -NR19R20;
R16 представляет собой водород, C1-C6-алкил, C2-C6 -алкенил, C3-C6-алкинил, C1-C4-алкилкарбонил, циано-C1-C3-алкил, C1-C4 -алкоксикарбонил-C1-C4-алкил, ди-C1-C4-алкоксикарбонил-C1-C4-алкил, бензил, C1-C4-алкокси-C1 -C4-алкинил; либо группу -(CH2)a-R33, -(CH2)a-X-R30, -(CH2)a-X-(CH2)b-R30 или -(CH2)a -X-(CH2)b-X-(CH2)c -R30;
R17представляет собой водород, C1-C4-алкил, C2-C6 -алкенил, C3-C6-алкинил, циано-C1-C3-алкил, C1-C4-алкилкарбонил-C1-C3 -алкил или фенил;
R18 представляет собой C1-C4-алкил, необязательно замещенный одним или несколькими атомами галогена;
R19 и R20 могут быть одинаковыми или различными и представляют собой водород или C1-C4-алкил;
R21 представляет собой C1-C4 -алкокси-C1-C4-алкил, C1-C4-алкилтио-C1-C4-алкил, фенил, (замещенный одним или несколькими атомами нитро-, циано-, C1 -C4-алкильной, C1-C4- алкокси-, или галогено-C1-C4-алкильной группами), или группу -NR31R32 или -(CH2)a-(O)d-R33;
R22 представляет собой C3-C6-алкоксикарбонил или карбокси;
R23 представляет собой хлорметил, цианметил, C3-C6-циклоалкил (необязательно прерываемый одним или несколькими атомами кислорода) или C1-C4-алкоксикарбонил-C1-C4 -алкил;
R24 представляет собой гидроксигруппу или группу формулы -NR25R26;
A представляет собой -NR25R26, или -S(O)n-R27;
R25 и R26 могут быть одинаковыми или различными и представляют собой водород или C1-C4-алкил;
R27 представляет собой C1-C4-алкил, C1-C4 -алкоксикарбонил-C1-C4-алкил или карбокси;
R28 представляет собой водород, гидрокси, галоген, C1-C4-алкил (необязательно замещенный одной или несколькими C1-C4-алкоксигруппами), C3-C6-циклоалкил (необязательно прерываемый одним или несколькими атомами кислорода и необязательно замещенный диметилом), фурил, тиенил или - C(= O)R29;
R29 и R30 могут быть одинаковыми или различными и представляют собой C1-C4-алкил, или C1-C4-алкокси;
R31 и R32 могут быть одинаковыми или различными и представляют собой C1-C4-алкил или фенил;
R33 представляет собой C3-C6-циклоалкил (необязательно прерываемый одним или несколькими атомами кислорода и необязательно замещенный диметилом), фурил, тиенил или группу -C(=O)R29;
R34 представляет собой C1-C4-алкил;
а, b и с равны 1, 2 или 3;
d равно 0 или 1;
m равно 3 или 4;
n равно 0, 1 или 2;
p равно 2 или 3; и
x является кислородом или серой;
обладает лучшими гербицидными свойствами, чем ранее упоминаемые известные соединения родственной структуры.
Особенно активными являются производные пиразола общей формулы I,
определенные выше, в которых:
R1
является метилом;
R2 является метилтио или
дифторметокси (а особенно дифторметокси); либо
R1 и R2, взятые вместе, образуют группу -(CH2
)4;
R3 является водородом,
хлор- или бромгруппой;
R4 является водородом;
R5 является водородом, нитро, циано или -C(=X)R10.
В особенно предпочтительной группе вышеописанных соединений R6 является водородом, галогеном, циано, C1-C4- алкилом, C1-C4-алкилтио или -NR11R12, при этом предпочтительно, если R11 и R12 являются водородом, C1-C4-алкилом или C1-C4-алкоксикарбонилом.
Термин "галоген" означает фтор, хлор, бром и иод.
Термины "алкил", "алкенил" и "алкинил" означают углеводородные группы с разветвленной или прямой цепью.
Настоящее изобретение также
относится к промежуточным соединениям общей формулы Ik:
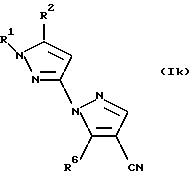
где R1, R2 и R6 имеют значения, определенные в общей формуле 1;
и к промежуточным соединениям общей формулы I:

где R1, R2, R3 и R6 имеют значения, определенные в общей формуле I.
Соединения настоящего
изобретения общей формулы I и промежуточные соединения, в которых R6 = NH2
, могут быть получены способом, в котором:
A) соединение общей формулы II:

где R1, R2 и R3 имеют значения, определенные в общей формуле I, подвергают реакции с соединением общей формулы III:

где R4 и R5 имеют значения, определенные в общей формуле I, a Y представляет собой C1-C6-алкокси, гидрокси или галоген, или, если является водородом, то:
В) соединение общей формулы II:

где R1, R2 и R3 имеют значения, определенные в общей формуле I, подвергают реакции с 2-галогенакрилнитрилом формулы IIIa:

либо с 2, 3-дигалогенпропионитрилом формулы III:

где Hal является галогеном; либо, если R3 является галогеном:
C) соединение общей формулы Ia:

где, R1, R2 , R5, R11 и R12 имеют значения, определенные в общей формуле I, сначала подвергают реакции с галогенирующим агентом, в результате чего получают соединение формулы Ib:

где R1, R2, R5, R11 и R12 имеют значения, определенные в общей формуле I; a Hal является галогеном, а затем, после обработки получают целевое соединение;
либо, если R16 является -OR16, то:
E) соединение общей формулы Id:

где R1, R2, R3, R4 и R5 имеют значения, определенные в общей формуле I, сначала подвергают диазотированию с получением соединения формула Ie:

где R1, R2 , R3, R4 и R5 имеют значения, определенные в общей формуле I, а затем нагревают, в результате чего получают соединение формулы If;

где R1, R2, R3, R4 и R5 имеют значения, определенные в общей формуле I, которое, в свою очередь, подвергают реакции с соединением общей формулы IV: QR16 (IV), где R16 имеет значения, определенные в общей формуле I, a Q является уходящей группой: либо, если R5 является нитрогруппой, а R6является SR17, то:
F) соединение общей формулы Ig:

где R1, R2, R3 и R4 имеют значения, определенные в общей формуле I, a Hal является галогеном, подвергают реакции с нуклеофилом общей формулы V;
⊖SR17 (V)
где R17 имеет значения, определенные в общей формуле I; либо, если R5 является нитрогруппой, a R6 является -S(O)nR17, где n равно 1 или 2, то:
G ) соединение общей формулы Ih:

где R1, R2, R3, R4 и R17 имеют значения, определенные в общей формуле I, подвергают постадийному окислению с использованием m-хлорпербензойной кислоты либо, если R5 является циано, то:
Н) соединение общей формулы IIa:

где R1 и R2 имеют значения, определенные в общей формуле I, подвергают реакции с соединением общей формулы Illc:
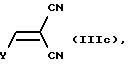
где Y является C1-C6-алкокси, гидрокси или галогеном; либо, если R16 является галогеном, то:
К) соединение общей формулы II:
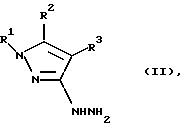
где R1, R2 и R3 имеют значения, определенные в общей формуле I, подвергают реакции с соединением общей формулы IIIc

где Y является C1-C6-алкокcи, диметиламино или галогеном, в результате чего получают соединение формулы Il
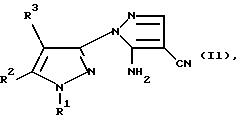
где R1, R2 и R3 имеют значения, определенные в общей формуле I, которое затем подвергают диазотированию известным способом с использованием нитрита натрия, и превращают в соответствующий галогенид; либо
L) соединение общей формулы lk:

где R1, R2 и R6 имеют значения, определенные в общей формулы I, обрабатывают галогенирующим агентом; либо
М) соединение общей формулы Im:

где R1, R2 и R3 имеют значения, определенные в общей формуле I, а R6представляет собой C1-C4 -алкил (необязательно замещенный одним или несколькими атомами галогена), или С2-С8-алкил (прерываемый одним или несколькими атомами кислорода), превращают известным способом в нитрил общей формулы I; либо, если R6 представляет собой группу -NR11R12, то
N) соединение общей формулы In:

где R1, R2 и R3 имеют значения, определенные в общей формуле I, подвергают реакции с амином в присутствии растворителя; либо, если R6 представляет собой группу -NR11R12, где R11 является водородом, a R12 является C1-C6-алкилом, то:
O) соединение общей формулы Il;

где R1, R2 и R3 имеют значения, определенные в общей формуле I, подвергают реакции со сложным триалкиловым орто-эфиром, с последующим восстановлением; либо
Р) соединение общей формулы Io:

где, R1, R2 и R3 имеют значения, определенные в общей формуле I, а R12 является C1-C6-алкилом, подвергают реакции с основанием и алкилирующим агентом или с хлорангидридом; либо, если R6 является -NR11R12, где R11 и R12 представляют собой C1-C6-алкил, то
Q) соединение общей формулы Il:

где R1, R2 и R3 имеют значения, определенные в общей формуле I, подвергают взаимодействию приблизительно с 2 молями основания и 2 молями соответствующего алкилирующего агента; либо
R ) соединение общей формулы Il:

где R1, R2 и R3 имеют значения, определенные в общей формуле I, подвергают взаимодействию с основанием, или в отсутствии основания, и c соответствующим хлорангидридом; либо
S) соединение общей формулы Ip:

где R1, R2, R3 и R21имеют значения, определенные в общей формуле I, подвергают реакции c основанием и c соответствующим алкилирующим агентом; либо
Т) соединение общей формулы In:

где R1, R2 и R3имеют значения, определенные в общей формуле I, а R5является пиано- или нитрогруппой, подвергают реакции с нуклеофилом кислорода, азота, серы или углерода; либо, если R6является замещенным метилом, то U ) соединение общей формулы Iq:
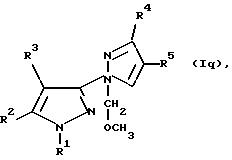
где R1, R2, R3, R4 и R5 имеют значения, определенные в общей формуле I, подвергают реакции с кислотой Льюиса; либо
V) соединение общей формулы Ir:

где R1, R2, R3, R4 и R5 имеют значения, определенные в общей формуле I, обрабатывают галогенирущим агентом; либо
W) соединение общей формулы Is:

где R1, R2, R3, R4 и R5 имеют значения, определенные в общей формуле 1, подвергают реакции с кислородным, азотным, серным или углеродным нуклеофилом; либо, если R6 является меркаптогруппой, то:
X) соединение общей формулы It:

где R1, R2, R3 и R4 имеют значения, определенные в общей формуле I, обрабатывают бисульфидом натрия; либо
Y) соединение общей формулы Iu:

где R1, R2, R3 и R4 имеют значения, определенные в общей формуле I, обрабатывают соответствующим алкилирующим агентом; либо
Z) соединение общей формулы Iv:
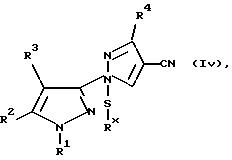
где R1, R2, R3 и R4 имеют значения, определенные в общей формуле I, a Rx является C1-C4-алкилом, подвергают постадийному окислению.
Соединения настоящего изобретения, имеющие общую формулу I, в которой R5 является нитрогруппой, - a R5 является галогеном, могут быть также получены способом, описанным в DE 3501323.
Соединения настоящего изобретения, имеющие общую формулу I, в которой R6 является группой -NR11R12, могут быть также получены известными способами, описанными в DE 3707686, DE 3543034, EP 224831, DE 3543035, JP 57167972, DE 2747531.
Соединения настоящего изобретения, имеющие общую формулу I, в которой R14 является группой -OR18 или -NR19R20, могут быть получены из незаявленных соединений общей формулы I, в которых R6 является аминогруппой, известными способами, описанными в Chem.Soc.Rev. 4, 231-50 (1975) и J.March, Advanced Organic Chemistry, 1985, p.370.
Соединения настоящего изобретения, имеющие общую формулу 1, в которой R5 является циано- или нитрогруппой, а R6 -C1-C4-алкилом, могут быть получены известными способами (J.Heterocyclic Chem. 24, 1669 (1987), там же, 24, 739 (1987)).
Реакции соединений формул II, IIa или III обычно осуществляют в подходящем растворителе, при температуре от -30 до 150oC, а предпочтительно при комнатной температуре.
В качестве галогенирующего агента могут быть использованы, например, сульфурилхлорид, гипохлорит натрия, N-хлорсукцинимид, N-бромсукцинимид, бром или хлор.
Удаляемыми группами в варианте способа E являются хлор- или бромгруппы.
Реакцию соединений общей формулы II обычно осуществляют методом, описанным в работе J.March, Advanced Organic Chemistry 1985, p. 647.
В варианте способа L), реакцию осуществляют в подходящем растворителе, предпочтительно, в ацетонитриле или в дихлорметане, при температуре от -10oC до 80oC.
Вариант способа (М) обычно осуществляют в соответствии с процедурой, описанной в Tetrahedron Letters, 1977, р. 1813.
Вариант способа О) обычно осуществляют известными методами (J.March, Advanced Organic Chemistry 1985, p. 798-800, и работы, цитируемые в настоящем описании).
В вариантах способа Р), Q), R) и S), подходящими основаниями являются, например, гидроксиды щелочных металлов и щелочноземельных металлов, метилат натрия, гидриды щелочных металлов, карбонаты щелочных и щелочноземельных металлов, третичные алифатические и ароматические амины, такие как триэтиламин и пиридин, а также гетероциклические основания.
Способ варианта Т) осуществляют, в основном, в соответствии c методами, описанными в J. Heterocyclic Chem. 25, 555 (1988).
Получение соединений может быть осуществлено с использованием или без использования растворителя. Если необходимо, то могут быть использованы растворители или разбавители, являющиеся инертными по отношению к реагентам. Примерами таких растворителей или разбавителей являются алифатические, алициклические и ароматические углеводороды, каждый из которых может быть необязательно хлорирован, например, такие как гексан, циклогексан, петролейный эфир, нафта, бензол, толуол, ксилол, метиленхлорид, хлороформ, тетрахлорметан, дихлорэтан, трихлорэтан и хлорбензол; простые эфиры, например, такие как диэтиловый эфир, метиловый эфир, метиловый т-бутилэфир, диизопропиловый эфир, дибутиловый эфир, диоксан и тетрагидрофуран; кетоны, например, такие как ацетон, метилэтилкетон, метилизопропилкетон и метилизобутилкетон; нитрилы, например, такие как ацетонитрил и пропионитрил; спирты, например, такие как метанол, этанол, изопропанол, бутанол, трет-бутанол, трет-амиловый спирт и этиленгликоль; сложные эфиры, например, такие как этилацетат и амилацетат; амиды, например, такие как диметилформамид и диметилацетамид; сульфоксиды, например, такие как диметилсульфоксид; сульфоны, например, такие как сульфолан; основания, например, такие как пиридин и триэтиламин; карбоновые кислоты, например, такие как уксусная кислота; и минеральные кислоты, например, такие как серная кислота и соляная кислота.
Соединения настоящего изобретения могут быть обработаны в соответствии со стандартными процедурами. Например, очистка может быть осуществлена посредством кристаллизации или колоночной хроматографии.
Соединения настоящего изобретения являются, как правило, бесцветными или слегка желтоватыми кристаллами или жидкостями, либо веществами, которые очень хорошо растворяются в галогенированных углеводородах таких, как метиленхлорид или хлороформ; простых эфирах таких, как диэтиловый эфир или тетрагидрофуран; спиртах таких, как метанол или этанол; кетонах таких, как ацетон или бутанон; амидах таких, как диметилформамид; и в сульфоксидах таких, как диметилсульфоксид.
Промежуточные соединения общей формулы II:

где R1, R2 и R3 имеют значения, определенные в общей формуле I, могут быть получены известным методом (см., например, JP 62158 260) из соединений общей формулы VI:

где R1, R2 и R3 имеют значения, определенные в общей формуле I.
Соединения
общей формулы II, где R1 и R2, взятые вместе,
образуют группу -(CH2)m-, а R3является водородом, могут быть получены путем обработки соединения
общей формулы IIIc;
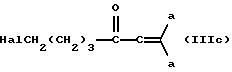
гидразином с добавлением основания. Соединение общей формулы IIIc может быть получено посредством реакции соединения общей формулы IIId

с 1,1-дигалогенэтиленом. Соединения общей формулы VI, где R1 и R2 имеют значения, определенные в общей формуле I, а R3 является галогеном, могут быть получены посредством реакции соединения общей формулы VI, в которой R3 является водородом, с галогенирующим агентом.
В качестве исходных материалов для получения соединений общей формулы VI используются
соединения общей формулы VII:

где R1 определен в общей формуле I, которые могут быть получены, например, способом, где в случае, если R2 является C1-C4-алкилом, необязательно замещенным галогеном,
а) соединение общей формулы VIII, VIIIa или IX:


где R2 является C1 -C4-алкилом, необязательно замещенным галогеном, подвергают реакции с соединением общей формулы X:
R1-NHNH2
где R1определен в общей формуле I, необязательно в присутствии растворителя; либо, если R2 является C1-C4-алкилтиогруппой, необязательно замещенной одним или несколькими атомами галогена, то:
b) соединение общей формулы XI:

где R35 является цианогруппой или группой -COOR36, в которой R36 представляет собой C1-C4-алкил, подвергают реакции с соединением общей формулы I необязательно в присутствии растворителя, например, в воде, и получают соединение общей формулы XII:

где R1 определен в общей формуле I, a R36 имеет вышеуказанные значения, которое затем подвергают реакции с соединением общей формулы XIII:
R37Q (XIII)
где R37 является C1-C4 -алкилом, необязательно замещенным одним или несколькими атомами галогена, а Q является уходящей группой, и полученное в результате соединение общей формулы XIV:

подвергают омылению и декарбоксилированию в соответствии с известными методами (см., например, Zeitschrift fur Chemie 420, (1968)); либо
c) соединение общей формулы XV:

где R35является цианогруппой или группой -COOR36, в которой R36 представляет собой C1 -C4-алкил, a R37 является C1 -C4-алкилом, необязательно замещенным одним или несколькими атомами галогена, подвергают реакции с соединением общей формулы X необязательно в присутствии растворителя, например, воды, в результате чего получают соединение общей формулы XIV; либо, если R2 является C1-C4-алкоксигруппой, необязательно замещенной одним или несколькими атомами галогена, то
d) соединение общей формулы XVI:

где R1 определен в общей формуле I, подвергают реакции с соединением общей формулы XIII в присутствии основания; либо
h) соединение общей формулы XVII:
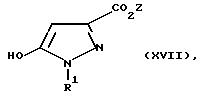
где R1 определен в общей формуле I, а Z является C1 -C4-алкилом, подвергают реакции, в присутствии основания, с соединением общей формулы XIII:
R37Q (XIII)
где R37 является C1-C4 -алкилом, необязательно замещенным одним или несколькими атомами галогена, а Q является удаляемой группой, и полученное в результате этой реакции соединение общей формулы XVIII:

где R1 определен в общей формуле I, R37 является C1-C4 -алкилом, необязательно замещенным одним или несколькими атомами галогена, а Z является C1-C4-алкилом, подвергают реакции с аммиаком, в результате чего получают соединение общей формулы XIX:
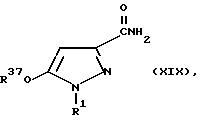
где R1 определен в общей формуле I, а R37 является C1-C4-алкилом, необязательно замещенным одним или несколькими атомами галогена, которое подвергают реакции с гидроксидом натрия и галогеном; либо, если в общей формуле I, R3 является галогеном, то :
f) соединение общей формулы XVIII или XIX:


где R1 определен в общей формуле I, R37 является C1-C4 -алкилом, необязательно замещенным одним или несколькими галогенами, а Z является C1-C4-алкилом, подвергают реакции с галогенирующим агентом, в результате чего получают соединение общей формулы XVIIIa и XIX:


где R1, R37 и Z имеют значения, определенные в общих формулах XVIII и XIX: либо
g) соединение общей формулы XIXa:

где R1 имеет значения, определенные в общей формуле I, R37 является C1-C4-алкилом, необязательно замещенным одним или несколькими атомами галогена, а Hal является галогеном, подвергают реакции с гидроксидом натрия и бромом, в результате чего получают соединение общей формулы XX:

где R1, R37 и Hal имеют значения, определенные в формуле XIXa; либо, если R1 и R2, взятые вместе, образуют три- или тетраметиленовую группу, то
h) соединение общей формулы XXI:
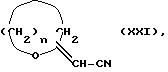
где n равно 2 или 3, подвергают реакции с гидразином, в результате чего получают 3(5)-амино-5(3)-гидроксиалкилпиразол общей формулы XXII

где n равно 2 или 3, который затем подвергают реакции с гексан-2,5-дионом, ангидридом фталевой кислоты или с ангидридом тетрагидрофталевой кислоты, в соответствии с известными методами, описанными в литературе (Bull. Chem. Soc. JP., 44, 2856-2858 (1971) или EP 305826), и получают соединение общей формулы XXIII:

где n равно 2 или 3, а Q является аминозащитной группой, например, такой, как Q1, Q2 или Q3:


которое подвергают циклизации по метод у Митсунобу (Synthesis I, (1981)) и получают соединение общей формулы XXIV:
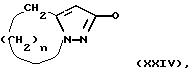
где n равно 2 или 3, а затем, в случае, если Q является Q1, это соединение обрабатывают гидроксиламином, как описано в J. Org.Chem., 49, 1224-1227 (1984), а в случае, если Q является Q2 или Q3, указанное соединение обрабатывают гидразином в соответствии с известными процедурами, описанными в литературе (Org. Synthesis, Coll. Vol. 3, 148 (1955)).
Исходные материалы общей формулы XXI могут быть получены известными методами (Chem. Ber., 109(1), 253-260, 1976).
Промежуточные соединения общей формулы Ik могут быть получены посредством реакции
соединения общей формулы
IIb

где R1 и R2 имеют значения, определенные в общей формуле IIb, которую осуществляют способом, описанным выше.
Промежуточные соединения общей формулы Im, где R6 является C1-C4-алкилом,
необязательно замещенным
одним или несколькими атомами галогена; или C2-C8-алкилом, прерываемым одним или несколькими атомами кислорода, могут быть получены известным способом, а
именно, путем
превращения соединения общей формулы Iq:

где R1, R2 и R3 имеют значения, определенные в общей формуле I, R6 является C1-C4-алкилом, необязательно замещенным одним или несколькими атомами галогена, или C2-C8-алкилом, прерываемым одним или несколькими атомами кислорода, а R7 является C1-C4-алкилом, в амид.
Соединения общей формулы Iq могут быть получены известным способом (J. Heterocyclic Chem. 24, 1669 (1987), там же, 24, 739 (1987)).
Получение промежуточных соединений может быть осуществлено в присутствии растворителя. Если возникает необходимость проведения реакции в присутствии растворителя, то для этой цели может быть использован один из растворителей, указанных выше.
Вышеуказанные исходные материалы являются либо известными соединениями, либо они могут быть получены известными методами.
Соединения настоящего изобретения обладают хорошей гербицидной активностью против широколистных сорняков и трав. Кроме того, соединения настоящего изобретения обладают избирательным действием в отношении различных сельскохозяйственных культур, например, таких как рапс, свекла, соевые культуры, хлопчатник, рис, ячмень, пшеница и другие злаковые культуры. Отдельные активные соединения являются особенно эффективными гербицидами избирательного действия в отношении таких культур, как свекла, хлопчатник, соя, кукуруза и зерновые. Однако соединения настоящего изобретения могут быть использованы для борьбы с сорняками в целях защиты монокультур, таких как лесные насаждения, декоративные растения, плодовые деревья, виноградники, цитрусовые деревья, ореховые культуры, бананы, кофейные деревья, чайные плантации, каучуковые деревья, масличные пальмы, ягодные культуры и плантации хмеля.
Так,
например, соединения настоящего изобретения могут быть использованы против следующих видов растений:
Двудольные сорняки вида: Siparis,
Lepidium, Galium, Stellaria, Matricaria, Anthemis,
Galinsoga, Chenopodium, Brassica, Urtica, Senecio, Amaranthus, Portulaca, Xanthium, Convolvulus, Ipomoea, Polygonum, Sesbania, Ambrosia, Cirsium,
Carduus, Sonchus, Solanum, Rorippa, Lamium, Veronica,
Abutilon, Datura, Viola, Galeopsis, Papaver, Centauera и Chrysanthemum.
Однодольные сорняки вида: Avena, Alopecurus, Echinochloa, Setaria, Panicum, Digitaria, Poa, Eleusine, Вrachiaria, Lolium, Bromus, Cyperus, Agropyron, Sagittaria, Monocharia, Fimbristylis, Eleocharis, Ischaemum и Apera.
Доза используемых соединений варьируется в зависимости от того, является ли данная обработка довсходовой или послевсходовой и, в основном, составляет от 0.001 до 5 кг/га.
Соединения настоящего изобретения могут быть также использованы в качестве дефолиантов, десикантов и гербицидов неизбирательного действия.
Соединения настоящего изобретения могут быть использованы как отдельно, так и в сочетании с другим соединением настоящего изобретения либо с другими активными агентами. В зависимости от целей обработки могут быть добавлены, но необязательно, и другие средства защиты растений или пестициды. В случае необходимости расширить спектр защитного действия, могут быть также добавлены другие гербициды. Примерами подходящих для этой цели активных гербицидов могут служить активные агенты, перечисленные в Weed Abstracts, Vol. 40, N 1, 1991 под заголовком "Lists of common names and abbreviations employed for currently used herbicides and plant growth regulators in Weed Abstracts".
Повышение эффективности и скорости действия активного ингредиента может быть достигнуто, например, путем добавления соответствующих адъювантов, таких как органические растворители, смачивающие агенты и масла. Указанные добавки позволяют уменьшить дозу активного соединения.
Вышеуказанные активные ингредиенты или их смеси могут быть изготовлены, например, в виде порошков, дустов, гранулированных препаратов, растворов, эмульсий или суспензий с добавлением жидких и/или твердых носителей, и/или разбавителей, и необязательно, связующих, смачивающих, эмульгирующих и/или диспергирующих добавок.
Подходящими жидкими носителями являются, например, алифатические и ароматические углеводороды, такие как бензол, толуол, ксилол, циклогексанон, изофорон, диметилсульфоксид, диметилформамид и другие нефтяные фракции, а также растительные масла.
Подходящими твердыми носителями являются измельченные минералы, например, бентонит, силикагель, тальк, каолин, аттапульгит, известняк, кремниевая кислота и растительные продукты, например мука.
В качестве поверхностно-активных агентов могут быть использованы, например, лигносульфонат кальция, полиоксиэтиленалкилфениловые простые эфиры, нафталинсульфоновые кислоты и их соли, фенолсульфоновые кислоты и их соли, конденсаты формальдегида, сульфаты жирных спиртов, а также замещенные бензолсульфоновые кислоты и их соли.
Процентные содержания активных ингредиентов в различных препаратах могут варьироваться в широких пределах. Так, например, композиции могут содержать от около 10 до 90 мас.% активного ингредиента и от около 90 до 10 мас.% жидких или твердых носителей, а также необязательно до 20 мас.% поверхностно-активных веществ.
Указанные агенты могут быть нанесены традиционным способом, например, в виде смеси для опрыскивания в объеме приблизительно от 100 до 1000 л/га, где в качестве носителя используют воду. Эти агенты могут быть внесены путем малообъемного или ультрамалообъемного опрыскивания, либо в виде так называемых микрогранул.
Изготовление вышеуказанных препаратов может быть осуществлено известными способами, например, путем размалывания или смешивания. Отдельные компоненты могут быть, но необязательно, смешаны непосредственно перед применением с помощью обычно используемого метода так называемого смешивания в резервуаре.
С использованием нижеуказанных ингредиентов могут быть получены
следующие препараты:
A) Смачивающийся
порошок
20 мас.% активного ингредиента
35 мас.% минерала-наполнителя
8 мас.% лигносульфоната кальция
2 мас.%
натриевой соли
N-метил-N-олеилтаурина
25
мас.% кремниевой кислоты
B) Паста
45 мас.% активного ингредиента
5 мас.% алюмосиликата натрия
15 мас.%
цетилполигликоля с 8 М
этиленоксида
2 мас.%
веретенного масла
10 мас.% полиэтиленгликоля
23 мас.% воды
C) Концентрат эмульсии
20 мас.% активного
ингредиента
75 мас.%
изофорона
5 мас. %
смеси натриевой соли N-метил-N-олеилтаурина и лигносульфоната кальция.
Нижеследующие примеры иллюстрируют получение соединений настоящего изобретения.
Пример 1.2
N-[1-(3-Хлор-4,5,6,7-тетрагидропиразол[1,5-a] пиридин-2-ил)-4-нитро-5- пиразолил]пропионамид
8.72 г (29.7 мМ)
N-[1-(3-Хлор-4,5,6,7-тетрагидропиразол[1,
5-a]пиридин-2-ил)-5- пиразолил]пропионамида суспендируют в 33 мл уксусной кислоты. К полученной суспензии при температуре 0-5oC добавляют 3.31 г
(32.5 мМ) уксусного ангидрида, охлаждая
при
этом льдом. Затем к раствору по капле добавляют 1.93 г (31 мМ) дымящей азотной кислоты. После перемешивания в течение 6 часов при комнатной температуре
смесь концентрируют. Образовавшийся
остаток
растворяют в дихлорметане, нейтрализуют водным раствором бикарбоната натрия, а затем промывают водным раствором хлорида натрия. Органическую фазу осушают
сульфатом магния и концентрируют.
Полученный
остаток очищают с помощью хроматографии на силикагеле, используя в качестве элюента смесь гексана/этилацетата (1:1).
Выход: 6.03 г = 60% от теоретич.
Т.пл.: 46-49oC.
Пример 2.0
N-[1-(4-Хлор-5-дифторметокси-1-метил-3-пиразолил)-4-нитро-5-пиразолил] - 2,2,2-трифорацетамид
0.79
г (2.1 мМ)
N-[1-(5-дифторметокси-1-метил-3-пиразолил)-4-нитро-5-пиразолил] - 2,2,2-трифторацетамида суспендируют в 35 мл дихлорметана и обрабатывают 0.17 мл сульфурилхлорида. Полученную смесь
перемешивают в
течение часа при комнатной температуре, а затем концентрируют.
Выход: 0.77 г = 89.5% от теоретич.
Т.пл.: 136-139oC.
Пример
2.1
N- [1- (4-Хлор-5-дифторметокси-1-метил-3-пиразолил)-4-нитро-5-пиразолил] ацетамид
1.3 г (5.0 мМ) 5-Амино-1-(4-хлор-5-дифторметокси-1-метил-3-пиразолил) пиразола растворяют в 20
мл уксусной
кислоты и обрабатывают 0.55 г (5.4 мМ) уксусного ангидрида. После 2-часового перемешивания при комнатной температуре, реакционный раствор охлаждают до 0oC, а затем добавляют
0.4 г
(6.4.мМ)
концентрированной азотной кислоты. После 8-часового перемешивания при комнатной температуре, реакционную смесь выливают в ледяную воду и экстрагируют этилацетатом. Органическую фазу
осушают
сульфатом
магния и концентрируют. Полученный остаток очищают с помощью хроматографии на колонке с силикагелем, используя в качестве элюента смесь гексана и этилацетата (1:1).
Выход: 1.4 г = 81.5% от теоретич.
Т.пл.: 132oC.
Пример 4.1
1- (3-Хлор-2, 4, 5, 6,7-тетрагидропиразол[1,
5-а]пиридин-2-ил)- 5-диэтиламино-4-пиразолкарбонитрил
10.45 г (0.35 М) 80% гидрида натрия добавляют к 100 мл тетрагидрофурана и полученную смесь охлаждают до 0oC. К этой смеси в
атмосфере азота по капле добавляют суспензию 43.6 г (0.17
М) 5-амино-1-(3-хлор-4,5,6,7-тетрагидропиразол[1,5-а] пиридин-2-ил) -4-пиразолкарбонитрила в 500 мл тетрагидрофурана. Полученную смесь
перемешивают в течение 1.5 часа, а затем при температуре 15oC по капле добавляют 31.4 мл (0.38 М) иодоэтана в 20 мл тетрагидрофурана. После перемешивания в течение 3 часов при температуре
15oC смесь охлаждают. К этой смеси по капле добавляют
воду, а затем экстрагируют этилацетатом. Органическую фазу отделяют, осушают, а затем концентрируют. Полученный остаток
перекристаллизовывают из этилацетата.
Выход: 47.3 г = 89,4% от теоретич.
Т.пл.: 68-70oC.
Пример 4.2
1-(3-Хлор-4,5,6,
7-тетрагидропиразол[1,5-а] пиридин-2-ил)-5-(этилметиламино)-4-пиразолкарбонитрил
23.3
г (88.7 мМ) 5-Амино-1-(3-хлор- 4,5,6,7-тетрагидропиразол[1,5-a]пиридин-2-ил)-4-пиразолкарбонитрила, 202
мл (1.21 мМ) триэтилортоформата и 10 капель трифторуксусной кислоты нагревают в течение 5 часов
в водяной бане при температуре 150oC. После этого воду удаляют, а реакционный раствор
концентрируют. Полученный в результате остаток суспендируют в 250 мл этанола, после чего порциями
обрабатывают 4.2 г (106.4 мМ) боргидрида натрия, охлаждая, при этом полученную смесь нагревают с
обратным холодильником до тех пор, пока не прекращается выделение газа. После этого смесь концентрируют
и полученный остаток осторожно добавляют к смеси льда и воды. Полученную таким образом смесь
три раза экстрагируют метиленхлоридом, после чего экстракты осушают. Органическую фазу концентрируют. После
этого при температуре 0oC к смеси добавляют 2,61 г (87.1 мМ) 80% гидрида натрия
в 150 мл тетрагидрофурана, а затем по капле добавляют 24.1 г (87.1 мМ) предварительно полученного 1-(3-хлор-4,
5,6,7-тетрагидропиразол [1,5-a] пиридин-2-ил)-5-метиламино-4-пиразолкарбонитрила в 500
мл
тетрагидрофурана. После перемешивания в течение часа при комнатной температуре к смеси добавляют 7.82 мл (95.8
мМ) иодэтана и нагревают при температуре 70oC в течение 3 часов. После
добавления по капле воды смесь три раза экстрагируют этилацетатом. Органическую фазу отделяют, осушают и концентрируют,
а полученный в результате остаток перекристаллизовывают из этилацетата.
Выход: 18.97 г = 71% от теоретич.
Т.пл.: 68-69oC.
Пример 4.3
5-Бром-1-(4-хлор-5-дифторметокси-1-метил-3-пиразолил)
-4-пиразолкарбонитрил
5.68 г (19.7 мМ) 5-Амино-1-(4-хлор-5-дифторметокси-1-метил-3-пиразолил)-4- пиразолкарбонитрила растворяют в 66.3
мл 47% бромводородной кислоты и полученную смесь
охлаждают до -6oC. После этого, к смеси по капле в атмосфере азота добавляют 2.36 г (34.2 мМ) нитрита натрия в 5.9 мл воды. Затем эту смесь
перемешивают в течение 15 минут при той же
температуре, после чего нагревают до комнатной температуры. После добавления 200 мл воды смесь 4 раза экстрагируют метиленхлоридом. Органическую фазу
промывают насыщенным водным раствором
бикарбоната
натрия, осушают сульфатом магния и концентрируют.
Выход: 6.94 г = 99.5% от теоретич.
Т.пл.: 78oC.
Получение исходных
материалов
1.
5-Амино-1- (4-хлор-5-дифторметокси-1-метил-3-пиразолил)-4-пиразолкарбонитрил
5.0 г (19.7 мМ)
5-Амино-1- (5-дифторметокси-1-метил-3- пиразолил)-4-пиразолкарбонитрила
растворяют в 180 мл
ацетонитрила и добавляют по капле 2.65 г (19.7 мМ) сульфурилхлорида. Полученную смесь перемешивают в течение
часа при комнатной температуре и концентрируют.
Выход: 5.68 г = 99.5% от теоретич.
Т.пл.: 140-142oC.
2.
5-Амино-1-(5-дифторметокси-1-метил-3-пиразолил)-4-пиразолкарбонитрил
22.5 г (0.13 М)
5-Дифторметокси-3-гидразино-1-метилпиразола растворяют в 310 мл этанола и обрабатывают 15.4 г (0.13 М)
этоксиметиленмалононитрила. Полученную смесь нагревают с обратным холодильником в течение часа,
а затем охлаждают. Образовавшийся осадок подвергают вакуумной фильтрации, а затем промывают небольшим
количеством этанола.
Выход: 19.28 г = 60% от теоретич.
Т.пл.: 141-143oC.
3. 5-Дифторметокси-3-гидразино-1-метилпиразол
39.8 г (0.25
М) 3-Амино-5-дифторметокси-1-метилпиразола растворяют в 225 мл воды и 450 мл
концентрированной соляной кислоты, К полученной смеси по капле и при температуре -10oC добавляют 18.55 г (0.27
М) нитрита натрия в 80 мл воды. После перемешивания в течение часа при
температуре -10oC к смеси по капле и при той же температуре добавляют 137.6 г хлорида олова (II), растворенного в 180
мл концентрированной соляной кислоты. После перемешивания еще в
течение часа при температуре -10oC к реакционной смеси по капле и при комнатной температуре добавляют 805 мл 32%-ного
гидроксида натрия. Затем эту смесь 8 раз перемешивают путем
встряхивания с этилацетатом, после чего объединенные органические фазы промывают насыщенным водным раствором хлорида натрия, осушают
сульфатом магния и концентрируют.
Выход: 42.24 г = 97.2% от теоретич.
4. З-Амино-6-дифторметокси-1-метилпиразол
71.7г (1.79 М) Гидроксида натрия добавляют к
600 мл воды и полученную смесь охлаждают до -5oC. При
той
же температуре к смеси по капле добавляют 57.3 г (0.36 М) брома так, чтобы температура не поднималась выше 0oC. После
этого к смеси порциями при 0oC добавляют 57.1 г (0.3
М)
3-карбамоил-5-дифторометокси-1-метилпиразола. Реакционную смесь перемешивают в течение часа при 80oC, а затем насыщают
хлоридом натрия. Образовавшийся в результате реакции осадок
подвергают вакуумной фильтрации, после чего фильтрат 6 раз перемешивают путем встряхивания с этилацетатом. Органическую фазу осушают
сульфатом магния и концентрируют. Ранее удаленный осадок
растворяют в 500 мл воды и полученный раствор нагревают до кипения в течение часа. Затем этот реакционный раствор насыщают хлоридом натрия и 6
раз встряхивают с этилацетатом. Органическую фазу
осушают сульфатом магния и концентрируют.
Выход: 34.2 г = 70.5% от теоретич.
Т.пл.: 57oC.
5. 3-Карбамоил-5-дифторметокси-1-метилпиразол
80.6 г (0.39 М) 3-Метоксикарбонил-5-дифторметокси-1-метилпиразола и 300 мл 33% водного раствора аммиака перемешивают в течение часа при
нагревании с обратным холодильником. После этого
реакционный раствор охлаждают. Образовавшийся осадок подвергают вакуумной фильтрации, а затем, промывают водой и диизопропиловым эфиром.
Выход: 58.9 г = 78.8% от теоретич.
Т.пл.: 154oC.
6. 5-Дифторметокси-3-метоксикарбонил-3-метилпиразол
67.6 г (0.43 М)
5-Гидрокси-3-метоксикарбонил-1-метилпиразола и 299.2 г (2.17 М)
карбоната кадия растворяют в 1500 мл диметилформамида и нагревают до 70oC. При той же температуре в смесь в течение 2 часов
вводят хлордифторметан. Полученную смесь перемешивают в течение
полутора часов при температуре 80oC. После добавления воды реакционную смесь 6 раз экстрагируют этилацетатом. Объединенные
органические фазы промывают насыщенным водным раствором хлорида
натрия и осушают сульфатом магния. Полученный реакционный раствор концентрируют.
Выход: 80.6 г = 90.3% от теоретич.
7. 5-Гидрокси-3-метоксикарбонил-1-метилпиразол
102.3 г (0.72 М) Диметилацетилендикарбоксилата добавляют к 1000 мл эфира и полученную смесь охлаждают до -5oC в бане из
льда/метанола. К этой смеси по капле добавляют 33 г (0.72 М)
метилгидразина в 100 мл эфира так, чтобы внутренняя температура не поднималась выше 0oC. После этого смесь перемешивают один час
при 0oC, в результате чего образовывается
осадок. Этот осадок подвергают вакуумной фильтрации, а затем промывают эфиром и осушают в вакууме при температуре 40oC. Промежуточное
соединение помещают в масляную баню, нагретую до
120oC. Полученный реакционный продукт перекристаллизовывают из метанола.
Выход: 67.6 г = 60.1% от теоретич.
Т.пл.: 197oC.
8.
4-Хлор-5-дифторметокси-3-метоксикарбонил-1-метилпиразол
2.1 г (10 мМ) 5-Дифторметокси-3-метоксикарбонил-1-метилпиразола, растворенного в 30 мл
метиленхлорида, обрабатывают 1.35 г (10 мМ)
сульфурилхлорида и полученную смесь перемешивают при комнатной температуре в течение 10 минут. После концентрирования полученный остаток
перекристаллизовывают из диизопропилового
эфира/этилацетата.
Выход: 1.8 г = 74.8% от теоретич.
Т.пл.: 51oC.
Пример 4.4
1-(4-Хлор-5-дифторметокси-1-метил-3-пиразолил)- 5-метил-4-пиразолкарбонитрил
0.57 г (2.25 мМ) 1-(5-Дифторметокси-1-метил-3-пиразолил) -5-метил-4-пиразолкарбонитрила, растворенного в 30 мл
метиленхлорида, при комнатной температуре обрабатывают 0.30 г (2.25 М) сульфурилхлорида. Полученную смесь перемешивают в течение часа, а затем концентрируют.
Выход: 0.65 г = 99.8% от теоретич.
Т.пл.: 69-70oC.
Получение исходных материалов
1. 1-(5-Дифторметокси-1-метил-3-пиразолил)-5-метил-4-пиразолкарбонитрил
Смесь
0.79
г (2.91 мМ) 1-(5-дифторметокси-1- метил-3-пиразолил)-5-метил-4-пиразолкарбоксамида, 0.46 г (5.85 мМ) пиридина и 20 мл 1,4-диоксана охлаждают до 5oC, а затем по капле добавляют 0.74 г
(3.51
мМ) ангидрида трифторуксусной кислоты. Полученную смесь перемешивают в течение 3 часов при комнатной температуре, а затем добавляют 100 мл воды и четыре раза экстрагируют этилацетатом.
Органическую
фазу осушают сульфатом магния и концентрируют.
Выход: 0.74 г = 99.8% от теоретич.
Т.пл.: 106-107oC.
2.
1-(5-Дифторметокси-1-метил-3-пиразолил)-5-метил-4-пиразолкарбоксамид
0.98 г (3.38 мМ) 1-(5-Дифторметокси-1-метил-3-пиразолил)- 5-метил-4-пиразолкарбонилхлорида растворяют в 20 мл
тетрагидрофурана и при перемешивании добавляют 50 мл водного раствора аммиака (33%). После перемешивания в течение 3 часов при комнатной температуре полученную смесь концентрируют до половины объема
и
подкисляют разбавленной соляной кислотой. Образовавшийся осадок отфильтровывают в вакууме, промывают небольшим количеством воды и осушают.
Выход: 0.27 г = 73% от теоретич.
Т.пл.: 116-118oC.
3. 1-(5-Дифторметокси-1-метил-3-пиразолил)-5-метил-4-пиразолкарбонилхлорид
0.2 г (3.8 мМ)
1-(5-Дифторметокси-1-метил-3-пиразолил)-5-метил-4-пиразолкарбоновой кислоты суспендируют в 30 мл 1,2-дихлороэтана. К полученной суспензии по капле и при комнатной температуре добавляют 1.19 г (10.0
мМ) тионилхлорида. После этого смесь нагревают с обратным холодильником в течение часа, а затем концентрируют.
Выход: 0.98 г = 100% от теоретич.
4.
1-(5-Дифторметокси-1-метил-3-пиразолил)-5-метил-4-пиразолкарбоновая кислота
Смесь 1.25 г (4.16 мМ) Этил 1-(5-дифторметокси-1-метил- 3-пиразолил)-5-метил-4-пиразолкарбоксилата, 20 мл этанола
и
0.97 мл 45% водного раствора гидроксида натрия перемешивают один час при температуре 80oC. Полученный реакционный раствор концентрируют до половины объема и подкисляют 37% соляной
кислотой.
Образовавшийся осадок отфильтровывают в вакууме, промывают водой, а затем осушают.
Выход: 1.05 г = 93% от теоретич.
Т.пл.: 205-207oC.
5.
Этил 1-(5-дифторметокси-1-метил-3-пиразолил)-5-метил-4- пиразолкарбоксилат
3.0 г (16.8 мМ) 5-Дифторметокси-3-гидразино-1-метилпиразолила добавляют в 25 мл этанола и полученную
смесь по капле
обрабатывают 2.96 г (16.0 мМ) этилдиметиламинометиленацетата, растворенного в 25 мл этанола. Затем смесь нагревают с обратным холодильником в течение 2 часов. После охлаждения осадок
отфильтровывают в
вакууме.
Выход: 2.52 г = 53% от теоретич.
Т.пл.: 110oC.
Получение используемых исходных материалов описано ниже.
1.1,1,
7-Трихлор-1-гептан-3-он
К 78.53 г (0.589 мМ) хлорида алюминия в 150 мл метиленхлорида, при комнатной температуре, по капле добавляют 100 г (0.62 М) 5-хлорвалероилхлорида.
После перемешивания
в течение часа к смеси по капле добавляют 45 мл (0.558 М) 1,1-дихлорэтилена в 25 мл метиленхлорида. Затем к смеси по капле и при охлаждении льдом добавляют 100 мл воды. Твердый
материал подвергают
вакуумной фильтрации через слой Целита. Фильтрат промывают водой, а органическую фазу осушают и концентрируют. Образовавшийся остаток подвергают дистилляции на роторном
испарителе.
Выход: 112.76 г = 93.8% от теоретич.
Т.кип.: 125oC/0.4 мбар.
2. 2-Гидразино-4,5,6,7-тетрагидропиразоло[1,5-а]пиридин
261.9 мл (5.4 М) гидрата
гидразина по капле и при температуре -2oC (баня из сухого льда/ацетона) добавляют к 116.6 г (0.54 М) 1,1,7-трихлор-1-гептан-3-она в 2000 мл 2-пропанола. После
12-часового перемешивания при
комнатной температуре добавляют 60.6 г (1.08 мМ) гидроксида калия и полученную смесь нагревают с обратным холодильником в течение 5 часов. Затем реакционную смесь
выпаривают досуха и полученный
остаток обрабатывают 100 мл воды и 100 мл солевого раствора. После экстрагирования (9 раз) этилацетатом органическую фазу промывают солевым раствором, осушают
сульфатом
натрия и концентрируют.
Выход: 29.29 г = 35.6% от теоретич.
Полученный продукт представляет собой желтое маслообразное вещество.
3.
5-Амино-4-циано-1-(1-метил-5-метилмеркапто-3-пиразолил)-пиразол
Смесь 2.0 г (13.1 мМ) 3-гидразино-1-метил-5-метилмеркаптопиразола и 1.8 г (14.4 мМ) этоксиметиленмалононитрила в 25 мл
этанола
перемешивают в течение 30 минут при комнатной температуре и нагревают до кипения в течение 3 часов. Реакционную смесь концентрируют и полученный остаток хроматографируют на силикагеле
(элюент:
гексан/этилацетат, 1:1).
Выход: 2.8 г = 91% от теоретич.
Т.пл.: 165-166oC.
4. 3-Гидразино-1-метил-5-метилмеркаптопиразол
1,1 г
(15.8 мМ) нитрита натрия в 4 мл воды по капле и при температуре 0oC добавляют к 1.9 г (13.1 мМ) 3-амино-1-метил-5-метилмеркаптопиразола в 28 мл концентрированной соляной
кислоты и
полученную смесь размешивают в течение 2 часов при 0oC. К этой смеси при температуре -30oC по капле добавляют 7.4 г (32.8 мМ) SnCl2•2H2O
в 5.5 мл
концентрированной соляной кислоты. После этого полученную смесь перемешивают в течение 3 часов при той же температуре. Затем реакционную смесь подщелачивают путем добавления 32% гидроксида
натрия и
экстрагируют метиленхлоридом. Органическую фазу осушают сульфатом натрия и концентрируют. Таким образом получают 2.0 г продукта, который используют без дополнительной очистки.
5.
3-Амино-1-метил-5-метилмеркаптопиразол
5.55 г (33.0 мМ) 3-Амино-4-циано-1-метил-5-метилмеркаптопиразола нагревают до кипения в течение 24 часов вместе с 50 мл 32% гидроксида
натрия.
Полученную реакционную смесь охлаждают, слегка подкисляют водным раствором бифосфата натрия, нагревают в течение 8 часов при температуре 50oC, а затем экстрагируют этилацетатом.
Органическую фазу осушают сульфатом натрия и концентрируют, в результате чего получают остаток. Этот остаток очищают с помощью хроматографии на силикагеле (элюент: гексан/этилацетат, 1:1).
Выход: 19 г = 39.8% от теоретич.
Т.пл.: 164-168oC.
6. 3-Амино-4-циано-1-метил-5-метилмеркаптопиразол
9.63 г (56.6 мМ)
[Бис(метилмеркапто)метилен]малононитрила суспендируют в 50 мл воды и обрабатывают 3.7 мл (67.9 мМ) метилгидразина. Полученную смесь нагревают до кипения в течение часа, после чего реакционный
раствор
охлаждают и образовавшийся осадок подвергают вакуумной фильтрации, а затем перекристаллизовывают из этанола.
Выход: 6.5 г = 68.0% от теоретич.
Т.пл.: 120-121oC.
7. 5-Амино-1-( 4,5,6,7-тетрагидропиразол[1,5-а] пиридин-2-ил)- 4-пиразолкарбоновая кислота и 2-гидразино-4,5,6,7-тетрагидропиразол [1,5-а]пиридин
Нижеследующие
соединения получают в соответствии с известными способами.
а) 2-Амино-4,5,6,7-тетрагидропиразол[1,5-a] пиридин
Раствор 8.19 г (146 мМ) гидроксида калия в 122
мл воды и 122
мл
этанола добавляют к 19.19 г (292 мМ) гидрохлорида гидроксиламина в 200 мл этанола. Полученную смесь перемешивают в течение 15 минут, а затем добавляют 12.5 г (58 мМ) 2-(2,
5-диметил-1-пирролил)-4,5,
6,
7-тетрагидропиразол[1,5-a] -пиридина. После этого смесь нагревают с обратным холодильником в течение 30 часов. После дистилляции этанола смесь обрабатывают этилацетатом,
твердые вещества
отфильтровывают, а водную фазу насыщают хлоридом натрия и экстрагируют этилацетатом. Органическую фазу промывают насыщенным водным раствором хлорида натрия, а затем осушают
сульфатом натрия и
концентрируют. Неочищенный продукт очищают с помощью хроматографии на силикагеле (элюент: этилацетат/метанол).
Выход: 6.12 г = 77% от теоретич.
1H-ЯМР (CDCl3, 300 МГц): δ 1.75-1.85 (м, 2H), 1.95-2.05 (м, 2H), 2,68 (т, 2H, J = 7.51), 3.5 (шир.с., 2H), 3.92 (т, 2H, J = 7.5 Гц), 5.33 (с, 1H).
b)
2-(2,
5-Диметил-1-пирролил)-4,5,6,7-тетрагидропиразоло-[1,5-a] пиридин
К 19.7 г (84 мМ) 3(5)-(4-гидроксибутил)-5(3)-(2,5-диметил-1-пирролил)пиразола и 22.1 г (84 мМ) трифенилфосфина в 300 мл
тетрагидрофурана по капле и при охлаждении льдом добавляют 16 г (92 мМ) диэтилазодикарбоксилата. Полученную смесь перемешивают в течение 4 часов при комнатной температуре, а затем концентрируют.
Полученный остаток очищают с помощью хроматографии на силикагеле (элюент: гексан/этилацетат).
Выход: 14.27 г = 97% от теоретич.
nD20: 1.5630.
c) 3 (5)-(4-Гидроксибутил)-5(3)-(2,5-диметил-1-пирролил)-пиразол
Смесь 18 г (116 мМ) 3(5)-амино-5(3)-(гидроксибутил)-пиразола, 14.6 г (128 мМ) 2,5-гександиона и 3.2
мл уксусной кислоты в 100 мл толуола нагревают с обратным холодильником в течение 8 часов, удаляя при этом воду. Полученный осадок подвергают вакуумной фильтрации, промывают толуолом и осушают.
Выход: 19.7 г = 72% от теоретич.
Т.пл.: 147-148oC.
d) 3 (5)- Амино-5(3)-(гидроксибутил)пиразол
4.8 мл Моногидрата гидразина при
комнатной температуре добавляют к раствору 12.3 г (0.1 М) тетрагидро-2H-пиран-2-илиденацетонитрила в 100 мл толуола. Полученную смесь нагревают с обратным холодильником в течение пяти часов.
Образовавшееся темно-желтое маслообразное вещество отделяют, а затем реакционную смесь концентрируют. Остаток очищают с помощью хроматографии на силикагеле (элюент: этилацетат/ метанол).
Выход: 11 г = 71% от теоретич.
Нижеследующие соединения (см. таблицы) получают способами, аналогичными способам, описанным в предыдущих Примерах.
Реферат
Описываются новые замещенные производные пиразола общей формулы I, где значения R1, R2, R3, R4, R5 и R6 указаны в 1 пункте формулы изобретения. Новые соединения обладают гербицидной активностью. Описывается гербицидная композиция на основе соединения формулы I, способ борьбы с сорняками, а также промежуточные для получения соединений формулы I. 5 с. и 3 з.п.ф-лы, 4 табл.

Формула

где R1 представляет собой C1-C4-алкил;
R2 представляет собой C1-C4-алкилтио, C1-C4-алкокси, каждый из которых является необязательно замещенным одним или несколькими атомами галогена;
R1 и R2, взятые вместе, образуют группу -(CH2)m;
R3 представляет собой водород или галоген;
R4 представляет собой водород или C1-C4-алкил;
R5 представляет собой водород, нитро, циано;
R6 представляет собой водород, галоген, циано, C1-C4-алкил, необязательно замещенный одним или несколькими атомами галогена или гидроксигруппами, фенил, необязательно замещенный заместителями как нитро, пирролил; либо
R6 представляет собой C2-C8-алкильную, C3-C8 -алкенильную, C3-C8-алкинильную или C3-C8-алкоксигруппу, каждая из которых прерывается одним или несколькими атомами кислорода; -NR11R12, либо
R6 представляет собой группу

R11 представляет собой водород, C1-C4-алкил, C2-C6-алкенил, C3-C6-алкинил, или группу R21CO-;
R12 представляет собой C1-C6-алкил, C2-C6-алкенил, C3-C6-алкинил или фенил, необязательно замещенный атомом галогена, C3 -C8-циклоалкил, цианометил, C1-C4-алкокси-C1-C6-алкил, ди-C1-C4-алкиламино-C1-C4-алкил, бензил, группу -C(= O)R21, -(CH2 )a-(O)d-R28, -(CH2)2-O(CH2)b-R28 или -(CH2)a-S'-R34,
R11 и R12, взятые вместе с атомом азота, с которым они связаны, образуют пирролидинильную, пиперидинильную, морфолинильную или азиридинильную группу;
R13 представляет собой водород;
R14 могут быть OR18, -NR19R20;
R15 представляет собой группу OR18;
R16 представляет собой C1-C6-алкил, C3-C6-алкинил, C1-C4-алкоксикарбонил-C1-C4-алкил, ди-C1 -C4 -алкокси-карбонил-C1-C4-алкил, бензил, либо группу -(CH2)a-R33, -(CH2 )a-O-R30, -(CH2 )a -O-(CH2)b-R30,
R17 представляет собой C1-C4-алкил, C3-C6-алкинил, C1-C4 -алкилкарбонил-C1-C3-алкил или фенил;
R18 представляет собой C1-C4-алкил,
R19 и R20 могут быть одинаковыми или различными и представляют собой водород или C1-C4-алкил;
R21 представляет собой C1-C4алкил, необязательно замещенный одним или несколькими атомами галогена, C1-C4-алкокси-C1-C4-алкил, фенил, необязательно замещенный галогеном, нитрогруппой или группой NR31R32;
R22 представляет собой C1-C4-алкоксикарбонил или карбокси;
R23 представляет собой C3-C6-циклоалкил, необязательно прерываемый одним или несколькими атомами кислорода;
R24 представляет собой гидрокси-группу или группу формулы - NR25R26;
А представляет собой - NR25R26 или -S(O)n-R27;
R25 и R26 могут быть одинаковыми или различными и представляют собой водород или C1-C4-алкил;
R27 представляет собой C1-C4-алкил, C1-C4-алкоксикарбонил-C1-C4-алкил или карбокси;
R28 представляет собой гидрокси, галоген, C1-C4-алкил, замещенный двумя C1 -C4-алкокси-группами, C3-C6 -циклоалкил, необязательно прерываемый одним или несколькими атомами кислорода и необязательно замещенный 1-2 метилом, фурил, тиенил или - С(=O)R29;
R29 и R30 могут быть одинаковыми или различными и представляют собой C1-C4-алкил или C1-C4-алкокси;
R31 и R32 могут быть одинаковыми или различными и представляют собой C1-C4-алкил или фенил;
R33 представляет собой C3-C6-циклоалкил, прерываемый одним или двумя атомами кислорода и необязательно замещенный 1-2 метилом, фурил, тиенил или группу -C(=O)R29;
R34 представляет собой C1-C4-алкил;
а, b равны 1, 2 или 3;
d равно 0 или 1;
m равно 3 или 4;
n равен 0,1 или 2;
Х представляет собой кислород или серу.
R1 представляет собой метил;
R2 представляет собой метилтио или дифторметокси, предпочтительно, дифторметокси, либо
R1 и R2, взятые вместе, образуют группу - (CH2)4;
R3 представляет собой водород, хлор или бром;
R4 представляет собой водород;
R5 представляет собой водород, нитро, циано.

в которых R1, R2 и R6 имеют значения, определенные в общей формуле I в п. 1, в качестве промежуточных соединений для получения замещенных производных пиразола по п.1, где R3 - означает галоген.

в которых R1, R2 и R3 имеют значения, определенные в общей формуле I в п. 1, а R6 - представляет собой C1-C4 алкил, необязательно замещенный одним или несколькими атомами галогена, или C2-C3-алкильную группу, прерываемую одним или несколькими атомами кислорода, в качестве промежуточных соединений для получения замещенных производных пиразола по п.1, в которой R1, R2, R3 и R6 имеют вышеопределенные значения, а R5 - обозначает циан.
Комментарии