Способ получения производных пиримидина - RU2019543C1
Код документа: RU2019543C1
Чертежи









Описание
Изобретение относится к способу по- лучения новых производных пиримидина, обладающих ценными фунгицидными свойствами, которые могут найти применение в сельском хозяйстве.
Известны такие соединения, обладающие фунгицидными свойствами, как манкозеб, карбоксин, пироксифур.
Недостатком этих известных средств является их сравнительно неширокий спектр фунгицидного действия.
Цель изобретения - способ получения новых производных пиримидина, обладающих более широким спектром фунгицидного действия.
Поставленная цель достигается
способом получения производных пиримидина общей
формулы I

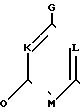

G - водород или галоген;
Х - водород, галоген, незамещенный С1-С4-алкил или замещенный галогеном гидроксилом или цианогруппой С2 -С4-алкенил, С2-С4 -алкинил, триметилсилилзамещенный С2-С4-алкенил, С2-С4-алкенилокси, С2-С4 -алкинилоксигруппа, фенил, циано-, тиоцианато- или нитрогруппа, группа -NR1R2, где R1 и R2 являются водородом или С1-С4-алкилом; С1-С4-алкилкарбониламино-, азидогруппа, С1-С4-алкоксил, фенокси-, бензилоксигруппа, С1-С4-алкоксикарбонилметил, С1-С4-алканоилокси-, С1-С4 -алкилсульфонилокси-, С1-С4-алкилтио-, С1-С4-алкилсульфинил-, С1-С4 -алкилсульфонилгруппа, формил, С1-С4-алканоил, гидроксимино (С1-С4)алкильная группа, карбамоил, тиокарбамоил или группа СН3О2 СС=СНОСН3;
Y - галоген, С1-С4-алкил, С1-С4-алкоксил, нитро-, ди(С1-С4)алкиламиногруппа или водород;
или Х и Y в случае, когда они находятся в орто-положении относительно друг друга, вместе с фенильным кольцом, к которому они присоединены, образуют нафталиновое кольцо;
А - галоген, водород, С1-С4-алкил или С1-С4-алкоксил.
Способ заключается в том, что соединение общей формулы II




П р и м е р 1. Получение /E/-метил-2-[2-(4-феноксипиримидин-2-илокси)-фенил]-3-метоксипропеноата (соединение 1 в табл.3).
К суспензии 0,3 г (6,85 ммоль, 50%-ная дисперсия в масле, предварительно промытая н-гексаном) гидрида натрия в 4 мл ДМФ прибавляют по каплям раствор 0,59 г (6,23 ммоль) фенола в 1 мл сухого ДМФ. Полученную смесь перемешивают в атмосфере азота до тех пор, пока не прекратится выделение пузырьков газа. Полученную смесь разбавляют 3 мл сухого ДМФ, а затем прибавляют по каплям к перемешиваемому раствору 1,0 г 4-хлор-2-метилтиопиримидина (6,23 ммоль) в 3 мл сухого ДМФ при 0оС. Происходит экзотермическая реакция и температура реакционной смеси повышается до 5оС. После перемешивания в атмосфере азота в течение 30 мин при 10оС ГХ-анализ указывает на образование одного продукта (98,8%). Реакционную смесь разбавляют 15 мл воды и экстрагируют 2 х 20 мл эфира. Объединенные эфирные экстракты промывают 2 х 15 мл 5% -ного раствора гидроксида натрия и 15 мл насыщенного раствора хлорида натрия, а затем сушат. Упаривание растворителя приводит к получению 1,40 г (чистота по ГХ 94%) 2-метилтио-4-феноксипиримидина в виде светло-желтого масла, которое используют непосредственно на следующей стадии. ПМР, δ: 2,37 (3Н, с) м.д.
К перемешиваемому раствору 1,00 г (4, 59 ммоль) 2-метил-тио-4-феноксипиримидина в 15 мл хлороформа при -15оС прибавляют 2,88 г (9,17 ммоль) м-хлорпербензойной кислоты в 35 мл хлороформа. Образуется белая мутная суспензия. Реакционной смеси дают нагреться до комнатной температуры и продолжают перемешивание в течение 4 ч. ГХ-анализ указывает на образование одного продукта (95% ). Реакционную смесь промывают 2 х 25 мл насыщенного водного раствора сульфита натрия, 2 х 25 мл насыщенного раствора карбоната натрия и 25 мл воды. Отделяют хлороформный раствор и сушат. Упаривают растворитель, получают бесцветное масло, которое кристаллизуется при охлаждении и затравке, получают 1,05 г 2-метансульфонил-4-феноксипиримидина в виде белого твердого продукта. Перекристаллизация из смеси хлороформа и н-гексана дает белый мелкокристаллический порошок, т.пл. 113-116оС, ПМР, δ: 3,17 (3Н, с), м.д. ИК-спектр (нуйол): ν 1133, 1315 см-1.
К раствору 200 мг 2-метансульфонил-4-феноксипиримидина (0,80 ммоль) в 2 мл сухого ДМФ при 0оС в атмосфере азота прибавляют 110 мг (0,80 ммоль) безводного карбоната калия. Раствор 166 мг (0,80 ммоль) /E/-метил-2-[2-оксифенил] -3-метоксипро- пеноата в 1 мл сухого ДМФ затем прибавляют по каплям при перемешивании. Реакционной смеси дают нагреться до комнатной температуры, а затем перемешивают в течение суток. Смесь разбавляют 15 мл воды, а затем экстрагируют 2 х 20 мл эфира. Объединенные эфирные экстракты промывают насыщенным солевым раствором, сушат и упаривают, получают желтое масло. Хроматография (элюент - эфир и н-гексан 5:1) дает светло-желтое мутное масло, которое тщательно растирают с эфиром, получают 0,10 г твердого белого цвета целевого продукта. Перекристаллизация из эфира и н-гексана дает 65 мг (выход 22%) твердого белого продукта, т.пл. 96-97оС; ПМР, δ: 3,57 (3Н, с), 3,70 (3Н, с), 6,48 (1Н, g), 7,12-7,45 (9Н, м), 7,42 (1Н, с), 8,29 (1Н, g) м.д.
ИК-спектр, ν: 1708, 1632 см-1.
П р и м е р 2. Получение /E/-метил-2-[2-(2-феноксипиримидин-4-илокси)фенил]-3-метоксипропеноата (соединение 1 в табл.2).
К перемешиваемому раствору 10,00 (62,3 ммоль) 4-хлор-2-метилтиопиримидина в 50 мл ледяной уксусной кислоты при 10-15оС прибавляют раствор 12,50 г (79,15 ммоль) перманганата калия в 100 мл воды. Реакционную смесь перемешивают всю ночь при комнатной температуре, охлаждают до 5оС, а затем обрабатывают газообразным сернистым ангидридом до тех пор, пока темный раствор не обесцветится. Прибавляют воду и смесь экстрагируют хлороформом. Объединенные органические слои промывают насыщенным водным раствором бикарбоната натрия, а затем водой и сушат. При выпаривании получают 10,84 г 4-хлор-2-метансульфонилпиримидина в виде белого твердого продукта, т.пл. 91-3оС. Обрабатывают 7,00 г (36,33 ммоль) 4-хлор-2-метансульфонилпиримидина феноксидом натрия (из 3,41 г (36,33 ммоль) фенола и 1,74 г (39,97 ммоль) 50% -ной дисперсии в масле гидрида натрия) в 100 мл сухого ДМФ при 0-5оС. Через 30 мин исходные соединения исчезают (ГХ-анализ). Реакционную смесь разбавляют водой, а затем два раза экстрагируют эфиром. Объединенные экстракты промывают 2 раза 5%-ным водным раствором гидроксида натрия и насыщенным солевым раствором, а затем сушат. При выпаривании растворителя получают 5,35 г очень светло-желтого подвижного масла. Хроматография (элюент - эфиры: н-гексан 2:3) с последующей кристаллизацией дает 3,50 г (84% чистого по ГХ) 4-хлор-2-феноксипиримидина в виде белого твердого продукта. Последующая хроматография дает чистый продукт (2,50 г, 33%), т.пл. 59-60оС.
К перемешиваемому раствору 2, 00 г (9,68 ммоль) 4-хлор-2-феноксипиримидина в 15 мл сухого ДМСО и 10 мл ДМФ при 10оС в атмосфере азота прибавляют по каплям суспензию 0,77 г (9,68 ммоль) метантиолата натрия в 15 мл сухого ДМСО и 5 мл ДМФ. После примерно часа при температуре ниже 15оС реакционную смесь разбавляют водой, а затем экстрагируют 3 раза эфиром. Объединенные экстракты промывают насыщенным солевым раствором, а затем сушат. Упаривание растворителя приводит к 2,00 г (87% чистого по ГХ) 4-метилтио-2-феноксипиримидина в виде плотного светло-желтого масла, которое используют на следующей стадии без дополнительной очистки.
Обрабатывают 2,00 г (7,96 ммоль) 4-метилтио-2-феноксипиримидина в 12 мл ледяной уксусной кислоты раствором 1,60 г (10,11 ммоль) перманганата калия в 20 мл воды, как описано для 4-хлор-2-метилтиопиримидина. После обработки получают светло-желтое масло, которое тщательно растирают с эфиром и н-гексаном, получая светло-желтый слегка плотный порошок (1,00 г). Перекристаллизация из четыреххлористого углерода (хлороформа (следы)) н-гексана дает 0,70 г (выход 35%) 4-метансульфонил-2-феноксипиримидин в виде белого порошка, т.пл. 86-87оС, ПМР, δ: 3,19 (3Н, с) м.д.
ИК-спектр (нуйол) ν: 1135, 1305 см-1.
К раствору 300 мг (1,20 ммоль) 4-метансульфонил-2-феноксипиримидина в 4 мл сухого ДМФ прибавляют 116 мг (1,20 ммоль) безводного карбоната калия. Раствор 0,250 г (1,20 ммоль) /E/-метил-2-(2-оксифенил)-3-метоксипропеноата в ДМФ прибавляют и реакционную смесь перемешивают всю ночь при комнатной температуре. Ее выливают в воду и экстрагируют эфиром. Эфирные экстракты промывают насыщенным солевым раствором, сушат и концентрируют, получая 0,48 г желтого масла. Хроматография (элюент-эфир и н-гексан 3:1) приводит к 0,34 г белого твердого продукта. Перекристаллизация из четыреххлористого углерода (дихлорметана (следы)) н-гексана приводит к целевому продукту в виде белого порошка (0,31 г, 69% выход); т.пл. 114-115оС, ПМР (270 МГц), δ: 3,60 (3Н, с), 3,74 (3Н, с), 6,43 (1Н, g), 7,11-7,42 (9Н, м), 7,46 (1Н, с), 8,28 (2Н, g) и м.д.
Масс-спектр м/е 378 (М+).
П р и м е р 3. Получение (Е)-метил-2-(2-)6-(2-цианофенокси)пиримидин-4-илокси)фе- нил)-3-метоксипропеноата (соединение 9 табл.1).
К раствору 0, 76 г (5,10 ммоль) 4,6-дихлорпиримидина в 4 мл сухого ДМФ при 0оС прибавляют 0,70 г (5,10 ммоль) безводного карбоната калия. Затем по каплям при перемешивании прибавляют раствор 0,53 г (2, 55 ммоль) /E/-метил-2-(2-оксифенил)-3-метоксипропеноата в 2 мл ДМФ. По окончании прибавления реакционной смеси дают нагреться до комнатной температуры и продолжат перемешивание в течение суток. Затем реакционную смесь разбавляют 15 мл воды и экстрагируют 3 х 20 мл эфира. Объединенные эфирные экстракты промывают насыщенным солевым раствором и сушат. После упаривания получают 1,10 г коричневой жидкости, которую хроматографируют (элюент - эфир и н-гексан, 3:2), получают /E/-метил-2-[2-(6-хлорпиримидин-4-илокси) фенил]-3-метоксипропеноата в виде тяжелого светло-желтого масла (0,58 г, 71% выход), которое кристаллизуется при стоянии. Перекристаллизация из эфира (дихлорметана (следы)) н-гексана при -78оС дает продукт в виде белого порошка 0,25 г, т. пл. 94-95оС. В другом случае получают 15 г продукта из 15,90 г 4,6-дихлорпиримидина, /E/-метил-2-(2-оксифенил)-3-метоксипропеноата (14,80 г) и 19,64 г безводного карбоната калия.
Нагревают в течение ночи 1,50 г (4,68 ммоль) /E/-метил-2-[2-(6-хлорпиримидин-4-илокси)фенил] -3-метоксипропеноата при 95-100оС с 0,61 г (5,15 ммоль) 2-цианофенола и 0,71 г (5,15 ммоль) карбоната калия в 35 мл ДМФ в присутствии каталитического количества однохлористой меди. Охлаждают реакционную смесь, разбавляют водой, а затем экстрагируют эфиром. Объединенные эфирные слои промывают 2 М раствором гидроксида натрия и насыщенным раствором соли, а затем сушат. Упаривание растворителя дает 1,52 г светло-желтого масла. Перекристаллизация из эфира (дихлорметана) н-гексана приводит к целевому продукту в виде светло-желтого порошка (1,20 г, выход 64%), т.пл. 110-111оС; ПМР, δ: 3,63 (3Н, с), 3,74 (3Н, с), 6,42 (1Н, с), 7,19-7,47 (6Н, м), 7,50 (1Н, с), 7,62-7,75 (2Н, м), 8, 40 (1Н, с) м.д. При повторном приготовлении целевого соединения перекристаллизация дает белый кристаллический продукт, т.пл. 118-119оС.
П р и м е р 4. Получение /E/-метил-2-[2-(6-(2-оксифенокси)пиримидин-4-илокси(фе- нил)-3- метоксипропеноата (соединение 26 табл.1).
Смесь 6,6 г (0,06 ммоль) катехола и 8,28 г (0,06 ммоль) безводного карбоната калия в 100 мл ДМФ нагревают 1 ч при 110оС. Затем прибавляют каталитическое количество (0,2 г) однохлористой меди, затем раствор 12,82 г (0,04 моль) /E/-метил-2-[2-(6-хлорпиримидин-4-илокси)фенил]-3-ме- токсипропеноата в 50 мл сухого ДМФ. Реакционную смесь нагревают 2 ч при 110оС, оставляют на ночь, а затем выливают в воду. Полученную смесь экстрагируют эфиром ("экстракт А"). Оставшийся водный слой подкисляют концентрированной соляной кислотой, а затем снова экстрагируют эфиром, затем оба эти экстракта промывают водой (х3), сушат и упаривают, получают 6,78 г коричневой смолы ("экстракт В"). Экстракт "А" промывают разбавленным раствором гидроксида натрия, полученную водную фазу подкисляют концентрированной соляной кислотой и экстрагируют этилацетатом, этот этилацетатный экстракт затем промывают водой, сушат и упаривают, получают 6,68 г коричневой смолы ("экстракт С"). Экстракты "В" и "С"объединяют и хроматографируют (элюент - эфир), получают 7,8 г (выход 49,5%) целевого соединения в виде желтого твердого продукта, который идентичен образцу, полученному ранее в малых масштабах, т.пл. 159-161оС, ИК-спектр, ν: 3100, 1712, 1642 см-1; ПМР, δ: 3,61 (3Н, с), 3,75 (3Н, с), 6,30 (1Н, с), 6,52 (1Н, с), 6,91-6,97 (1Н, м), 7,05-7,21 (4Н, м), 7,26-7,48 (3Н, м), 7,45 (1Н, с), 8, 44 (1Н, с) м.д.
П р и м е р 5. Получение /E/-метил-2-(2-)6-(2-метоксифенокси)пиримидин-4-илок- си[фенил]-3- метоксипропеноата (соединение 29 табл.1).
К перемешиваемому раствору 0,50 г (1,27 ммоль) /E/-метил-2-[2-](2-оксифенокси)пиримидин-4-илокси(фенил)-3-метоксип- ропеноата в 15 мл сухого ДМФ при 0оС прибавляют 0,17 г (1,27 ммоль) безводного карбоната калия и 0,22 г (1,52 ммоль) метилиодида. Реакционной смеси дают нагреться до комнатной температуры, перемешивают два часа, а затем оставляют стоять на сутки. Смесь разбавляют 20 мл воды, а затем экстрагируют 3 х 25 мл эфира. Объединенные эфирные экстракты промывают 2 х 20 мл разбавленного раствора гидроксида натрия и 20 мл насыщенного солевого раствора, а затем сушат. После упаривания получают 0,36 г светло-розовой пены, которую хроматографируют (элюент - эфир-гексан 7:1), получая целевое соединение в виде белой пены (0,21 г, 40% выход); ПМР, δ: 3,60 (3Н, с), 3,76 (3H, c), 3, 78 (3H, c), 6,25 (1Н, с), 6,95-7,52 (3Н, м), 7,49 (1Н, с), 8,42 (1Н, с) м.д.
В альтернативном приготовлении /E/-метил-2-[2-(6-хлорпиримидин-4-илокси)фе- нил]-3-метоксипропеноат (1, 00 г, 3,12 ммоль), полученный, как описано в примере 3, обрабатывают 1,09 г (15,60 ммоль) метантиолата натрия при комнатной температуре в 15 мл хлороформа и 10 мл воды в присутствии каталитического количества тетрабутиламмонийбромида. После перемешивания в течение ночи отделяют хлороформный слой и оставшийся водный слой затем экстрагируют хлороформом. Объединенные хлороформные слои промывают водой, сушат и упаривают, получая 1,56 г оранжевого масла. Хроматография (элюент - эфир-гексан 2: 1) приводит к 0,92 г (выход 89%) /E/-метил-2-[2-(6-метилтиопиримидин-4-илокси)фенил] -3-метоксипропеноата в виде светло-желтого масла; ПМР, δ: 2,25 (3Н, с), 3,59 (3Н, с), 3,73 (3Н, с), 6,55 (1Н, с), 7,17 (1Н, с), 7,20-7,55 (3Н, м), 7,45 (1Н, с), 8,57 (1Н, с) м.д.
Перемешивают 0,20 г (0,6 ммоль) по- лученного продукта и 0,38 г (55% чистоты) мета-хлорпербензойной кислоты в 25 мл хлороформа в течение ночи при комнатной температуре. Обработка дает 0,26 г соответствующего сульфона (чистота 94% по ГХ) в виде тяжелого бесцветного масла, которое непосредственно используют на следующей стадии без дополнительной очистки. ПМР, δ: 3,25 (SO2 CH3), 7,45 (олефиновый протон) м.д.
К перемешиваемому раствору 0,24 г сульфона в 6 мл сухого ДМФ прибавляют 0,091 г безводного карбоната калия и раствор 0,082 г 2-метоксифенола в 2 мл сухого ДМФ. Реакционную смесь перемешивают 4 ч, а затем всю ночь при комнатной температуре, разбавляют 15 мл воды, затем экстрагируют 3 х 20 мл эфира. Объединенные эфирные экстракты промывают 2 х 15 мл разбавленного раствора гидроксида натрия и 15 мл насыщенного солевого раствора, затем сушат. При упаривании получают 0,25 г плотного светло-желтого масла. Хроматография (элюент - эфир-гексан 7:1) приводит к целевому соединению в виде плотной белой пены (0,17 г, выход 63%). ПМР идентичен указанному ранее.
П р и м е р 6. Получение /E/-метил-2-[2-]6-(2-тиокарбоксамидофенокси) пиримидин-4-илокси[фенил]-3-метоксипропеноата (соединение 59 табл.1).
Избыток газообразного сероводорода барботируют через перемешиваемый раствор 2,09 г (15,19 ммоль) /E/-метил-2-[2-(6-(2-цианофенокси)пиримидин-4-илокси)фе- нил]-3-метоксипропеноата, полученного как описано в примере 3, и 0,52 г триэтиламина в 45 мл сухого пиридина при 50оС. В течение 4,5 ч при 50оС и в течение одной недели при комнатной температуре избыток сероводорода удаляют продувкой воздуха через реакционную смесь. Полученный в результате коричневый раствор упаривают и азеотропно сушат толуолом (2 х 50 мл), получают коричневое масло, которое тщательно растирают с 3 х 40 мл воды. Остаток хроматографируют (элюент - ацетон-гексан 2:3), получают 0,79 г светло-желтого масла. Тщательное растирание с гексаном приводит к целевому соединению в виде светло-оранжевого порошка (0,68 г, выход 30%), т.пл. 125-128оС. Образец, приготовленный после, имел т.пл. 131-133оС, ПМР, δ: 3,63 (3Н, с), 3,78 (3Н, с), 6,27 (1Н, с), 7,18 (1Н, с), 7,10-7,60 (6Н, с), 7,49 (1Н, с), 7,71 (1Н, с), 7,91 (1Н, с), 8,05 (1Н, с), 8,39 (1Н, с) м.д.
П р и м е р 7. Получение соединений


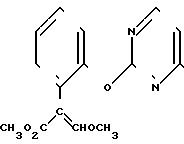

К перемешиваемой смеси 2,43 г /E/-метил-2-(2-оксифенил)-3-метоксипропеноата и 1,61 г безводного карбоната калия в 25 мл сухого ДМФ при 0оС прибавляют по каплям раствор 2,4,6-трихлорпиримидина в 5 мл сухого ДМФ. Реакционную смесь 30 мин перемешивают при 0оС и в течение суток при комнатной температуре, а затем выливают в воду и три раза экстрагируют эфиром. Объединенные эфирные экстракты промывают разбавленным раствором гидроксида натрия и трижды водой, затем сушат. После упаривания получают 2,62 г оранжевой смолы, которую хроматографируют (элюент смеси эфир-гексан), получают 0,65 г /E/-метил-2-2-(2,4-дихлорпиримидин-6-илокси)фе- нил/-3-метоксипропеноата в виде белого твердого продукта, т.пл. 88-90оС и 1,07 г смеси примерно 1:1, содержащей



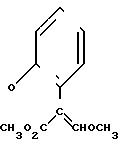
К перемешиваемому раствору части этой смеси (0,97 г) в 25 мл ТГФ прибавляют 0,11 г 5%-ного Pd/C катализатора, а затем в течение 5 мин по каплям прибавляют 0,405 г гипофосфита в 5 мл воды. После 2 ч перемешивания при комнатной температуре повышают температуру до 60оС и прибавляют дополнительные порции 0,41 г гипофосфита натрия в 5 мл воды (после дополнительных 30 мин) и 0,76 г карбоната калия и 0,11 г катализатора (после дополнительного часа). Когда исходный материал был израсходован (ГХ и ТСХ анализ), реакционную смесь фильтруют через цеолит, промывают на фильтре эфиром и водой. Разделяют слои фильтрата и водный слой экстрагируют более одного раза эфиром. Объединенные эфирные слои дважды промывают водой, сушат и упаривают, получают 0,78 г белой пены. Хроматография (элюент-эфир) дает элюированное первым соединение 123 табл.1 в виде белого твердого продукта 0,34 г, т.пл. 130-131оС, ИК-спектр, ν: 1705, 1693, 1636 см-1; ПМР, δ: 3,59 (6Н, с), 3,75 (6Н, с), 6,16 (1Н, с), 7,14-7,18 (2Н, м), 7,24-7,41 (6Н, м), 7,45 (2Н, с), 8,39 (1Н, с) м.д. и соединение 123 табл.2 в виде белой пены 0,23 г, т. пл. 60-70оС, ИК-спектр, ν: 1706, 1632 см-1, ПМР, δ: 3,56 (3Н, с), 3,70 (3Н, с), 3,74 (3Н, с), 6,34-6,37 (1Н, g), 7,15-7,35 (8Н, м), 7,44 (1Н, с), 7,47 (1Н, с), 8,21-8, 24 (1Н, с) м.д.
П р и м е р 8. Получение /E/-метил-2-[2-(4-фторпиримидин-6-илокси)фенил]-3-мето- ксипропеноата (промежуточного продукта).
Смесь 6,50 г 4, 6-дихлорпиримидина, 20,8 г тетрафторида серы и 35 мл Арктон 113 нагревают при 50оС при перемешивании в реакторе Монеля емкостью 100 мл в течение 3,3 ч. Повышают температуру до 100оС в течение 25 мин и выдерживают при 100оС в течение дополнительных 3 ч. Температуру повышают до 151оС в течение 20 мин и выдерживают при 151оС в течение 3 ч. Затем реактору дают остыть до комнатной температуры. Реакционную смесь выливают в насыщенный водный раствор бикарбоната натрия и экстрагируют дихлорметаном. На поверхности раздела фаз наблюдают плотный твердый продукт, который удаляют фильтрованием. Затем разделяют слои. Органический слой промывают водой, затем перегоняют при атмосферном давлении для удалении дихлорметана. Получают 400 мг 4, 6-дифторпиримидина перегонкой в вакууме 50оС/100 мм рт.ст. в виде светло-желтого масла (7,3% выход), ПМР, δ: 6,61 (1Н, с) и 8,69 (1Н, с) м.д.
К раствору 359 мг (1,724 ммоль) /E/-метил-2-(2-оксифенил)-3-метоксипропеноата в 3 мл сухого ДМФ при комнатной температуре прибавляют в один прием 376 мг (3,45 ммоль) безводного карбоната калия. Реакционную смесь 20 мин перемешивают при комнатной температуре, затем прибавляют раствор 200 мг 4,6-дифторпиримидина в 2 мл сухого ДМФ через шприц в течение ≈1 мин. Затем реакционную смесь перемешивают еще 20 мин при комнатной температуре, выливают в 20 мл воды и экстрагируют 4 х 30 мл этилацетата. Объединенные экстракты последовательно промывают 2 х 100 мл воды и 1 х 100 мл насыщенного солевого раствора, затем сушат и концентрируют, получая 464 мг целевого соединения в виде плотного желтого масла (выход 88%), ПМР, δ: 3,59 (3Н, с), 3,73 (3Н, с), 6,32 (1Н, с), 7, 16-7,43 (4Н, м), 7,45 (1Н, с), 8,51 (1Н, g) м. д.
Аналогично получают соединения табл.1 и 2.
Следующие примеры представляют собой примеры композиций, используемых для сельскохозяйственных и садоводческих целей, которые могут быть получены из предлагаемых соединений. Проценты в указанных примерах являются массовыми.
П р и м е р 9. Эмульгирующийся
концентрат готовят смешением и перемешиванием
ингредиентов до их полного растворения, %: Соединение 9 (см. табл. 1) 10 Бензиловый спирт 30 Додецилбензолсульфонат кальция 5 Нонилфенолэтоксилат (13
моль этиленоксида) 10 Алкилбензолы 45
П р
и м е р 10. Активный ингредиент растворяют в метилендихлориде и полученную жидкость распыляют на гранулы из аттапульгитовой глины. Затем дают
растворителю испариться, получают гранулированную
композицию, %: Соединение 9 (см. табл.1) 5 Аттапульгитовые гранулы 95
П р и м е р 11.Композицию, пригодную для использования для
протравливания семян, готовят путем измельчения и смешивания
трех ингредиентов, %: Соединение 9 (см. табл.1) 50 Минеральное масло 2 Китайская глина 48
П р и м е р 12. Дустовый порошок
готовят путем измельчения и смешивания активного ингредиента с
тальком, %: Соединение 9 (см. табл.1) 5 Тальк 95
П р и м е р 13. Суспензионный концентрат готовят путем измельчения в шаровой
мельнице ингредиентов с образованием водной суспензии
измельченной смеси в воде, %: Соединение 9 табл.1 40 Лигносульфонат натрия 10 Бентонитовая глина 1 Вода 49
Эта рецептура может быть
использована для распыления при разбавлении водой или
нанесена непосредственно на семена.
П р и м е р 14. Рецептуру смачивающегося порошка готовят смешением вместе и измельчением
ингредиентов до тех пор, пока все они не будут тщательно
перемещены, %: Соединение 9 (см. табл.1) 25 Лаурилсульфонат натрия 2 Лигносульфонат натрия 5 Оксид кремния 25 Китайская глина 43
П р
и м е р 15. Соединения испытывали против различных грибных
заболеваний листьев растений. Использовали следующую методику.
Растения выращивали в горшочном компосте Джон Иннес (N 1 или 2) в минигоршочках диаметром 4 см. Испытываемые соединения были формулированы или путем смешивания на шаровой мельнице с водным Дисперсолом Т или в виде раствора в ацетоне или ацетоне/этаноле, который затем разбавляют до желаемой концентрации непосредственно перед использованием. При заболеваниях листьев рецептуры (100 м.д. активного ингредиента) распыляют на листья и наносят на корни растений в почве. Распыляемые растворы наносят до максимального удерживания, а корни орошают до максимальной концентрации, эквивалентной примерно 40 м. д. в сухой почве. Прибавляют Твин-20 до получения конечной концентрации 0,05%, когда аэрозоль наносят на злаковые.
Для большинства испытаний соединение наносят на почву (корни) и на листья (распыление) за один-два дня до инокуляции растения патогеном. Исключением является испытание на Erysiphe graminis, в котором растения инокулируют за 24 ч перед обработкой.
Листовые патогены наносят распылением в виде суспензии спор на листья испытуемых растений. После инокуляции растения помещают в соответствующие условия окружающей среды, чтобы дать развиться инфекции, а затем инкубируют до тех пор, пока болезнь будет можно легко оценить. Период между инокуляцией и оценкой меняется от четырех до четырнадцати дней в зависимости от болезни и условий окружающей среды.
Оценивают контроль
заболевания по следующей шкале:
4 - нет заболевания;
3 - следы - 5% от заболевания на необработанных растениях;
2 - 6-25% от заболевания на необработанных растениях;
1 - 26-59% от заболевания на необработанных растениях;
0
- 60-100% от заболевания на необработанных растениях.
Результаты приведены в табл.3 и 4.
Сравнение широты спектра фунгицидного действия соединений формулы I, проведенное в аналогичных примеру 15 условиях для манкозеба, карбоксина, пироксифура, показывает (см. табл.5), что получаемые согласно предлагаемому способу по изобретению соединения обладают более широким спектром фунгицидного действия.
Реферат
Использование: в качестве фунгицидов в сельском хозяйстве. Сущность изобретения: продукт: производные пиримидина формулы I , где любые две группы из числа K, L и M являются азотами, а третья - группой CH=; C - водород или галоген; X - водород, галоген, незамещенный; C1-C4 - алкил или замещенный галогеном, гидроксилом или цианогруппой; C2-C4 -алкенил; C2-C4 -алкинил, триметилсилилзамещенный C2-C4 -алкенил, C2-C4 -алкенилокси C2-CH группа, фенил, цианотиоцианато нитрогруппа, группа- NR1R2 , где R1 и R2 являются водородом или C1-C4 -алкилом, C1-C4 -алкилкарбониламино-, азидогруппа, C1-C4 -алкокси, фенолкси-, бензилоксигруппа, C1-C4 -алкоксикарбонилметил, C1-C4 -алканоилокси-, C1-C4 -алкилсульфонилокси-, C1-C4 -алкилсульфонилгруппа, формил, C1-C4 -алканоил, гидроксии-мино- C1-C4 алкильная группа, карбамоил, тиокарбамоил или группа CH3O2CC=CHOCH3 , Y-галоген, C1-C4 -алкил, C1-C4 -алкоксил, нитро-, ди C1-C4 алкиламиногруппа или водород, или X и Y в случае, когда они находятся в орто-положении относительно друг друга, вместе с фенильным кольцом, к которому они присоединены, образуют нафталиновое кольцо; A-галоген, водород, C1-C4 -алкил, или C1-C4 -алкоксил. Реагент 1: соединение формулы II. Реагент 2:


Формула



где любые две группы из числа К, L и М являются азотами, а третья группой -СН=;
G - водород или галоген;
Х - водород, галоген, незамещенный С1 - С4-алкил или замещенный галогеном, гидроксилом, или цианогруппой С2 - С4-алкенил, С2 - С4-алкинил, триметилсилил замещенный С2 - С4-алкенил, С2 - С4 -алкенилоксигруппа, С2 - С4-алкинилоксигруппа, фенил, циано-, тиоцианато- или нитрогруппа, группа -NR1R2, где R1 и R2 - водород или С1- С4-алкил, С1 - С4-алкилкарбониламино-, азидогруппа, С1 - С4-алкоксил, фенокси-, бензилоксигруппа, С1 - С4 -алкоксикарбонил, С1 - С4-алкоксикарбонилметил, С1 - С4 -алканоилокси-, С1 - С4-алкилсульфонилокси-, С1 - С4 -алкилтио-, С1 - С4-алкилсульфинил-, С1 - С4 -алкилсульфонилгруппа, формил, С1 - С4-алканоил, гидроксиимино-(С1 - С4)-алкильная группа, карбамоил, тиокарбамоил или группа
CH3O2C

Y - галоген, С1 - С4-алкил, С1 - С4-алкоксил, нитро-, ди(С1 - С4)-алкиламиногруппа или водород,
или Х и У, в случае, когда они находятся в орто-положении относительно друг друга, вместе с фенильным кольцом, к которому они присоединены, образуют нафталиное кольцо;
А - галоген, водород, С1 - С4-алкил или С1 - С4-алкоксил,
отличающийся тем, что соединение общей формулы


где К, L, М и G имеют указанные значения,
подвергают взаимодействию с соединением общей формулы


где А, Х и У имеют указанные значения;
одна из групп Z1 или Z2 - уходящая группа из числа хлора или метансульфонила, когда другая группа является группой ТО, где Т - водород или катион щелочного металла,
в растворителе, таком как диметилформамид, при 0 - 110oС, возможно в присутствии катализатора, такого как хлорид одновалентной меди, в когда Т - водород, то в присутствии основания, такого как карбонат калия.
Комментарии