Соединения амина, имеющие противовоспалительную, противогрибковую, противопаразитарную и противораковую активность - RU2677229C2
Код документа: RU2677229C2
Описание
УРОВЕНЬ ТЕХНИКИ
Большинство ядерных эукариотических клеток, в одноклеточных организмах или в качестве составляющих многоклеточного организма, включая человека, содержат подкисленные вакуоли, которые являются критическими для поддержки жизни и функционирования клеток. В клетках млекопитающих эти вакуоли включают лизосомы и другие эндосомальные везикулярные органеллы. Внутренний рН лизосом обычно составляет приблизительно от 4,5 до 5 и поддерживается вакуолярными АТФ-зависимыми протонными насосами, а также эффектами равновесия Доннана. Лизосомы участвуют в буферизации цитозоля, защите клетки от кислых сред, а также являются первичными местами разложения и рециркуляции компонентов старения или поврежденных органелл, таких как митохондрия, процесса, известного как аутофагия. Существует несколько важных патологических состояний, в которых лизосомальные характеристики изменяются и способствуют патогенезу заболевания, представляя потенциальную мишень для фармакологической терапии.
Растущий набор данных показывает, что общим фенотипическим изменением в инвазивных раковых клетках является перенацеливание лизосом на участие в деструкции окружающих клеток через экзоцитоз кислого содержимого, включая ферменты. Протеолитические ферменты, обычно находящиеся в лизосомах, но секретируемые раковыми клетками, такие как катепсины, могут разлагать белки внеклеточного матрикса, облегчая инвазию и метастазирование опухоли. Кроме того, лизосомы и другие кислые вакуолярные органеллы часто увеличиваются в раковых клетках, что способствует буферизации рН; многие солидные опухоли генерируют кислую внеклеточную среду, способствуя инвазии, которая требует, чтобы раковые клетки адаптировались как к продукции, так и к толерантности по отношению к низкому внеклеточному рН. Раковые клетки, выбранные in vitro для инвазивного потенциала, имеют более крупные, более кислые лизосомы, чем менее агрессивные клетки. Раковые клетки, подвергнутые действию ионизирующего облучения, дают защитную реакцию, включающую увеличение и подкисление лизосом. Родственной защитной реакцией, через которую раковые клетки приобретают преимущества для выживания, является активация аутофагии, которая включает слияние аутофагосом, содержащих поврежденные органеллы или другой клеточный детрит, с лизосомами; прерывание аутофагии может ослабить жизнеспособность раковой клетки. Некоторые раковые клетки также секвестрируют химиотерапевтические агенты в лизосомах в рамках механизма лекарственной резистентности. Хлорохин, противомалярийное лекарственное средство, которое аккумулируется в лизосомах млекопитающих, потенцирует, или восстанавливает чувствительность к ней, противораковую активность нескольких классов химиотерапевтических средств и терапий рака с использованием нацеленных малых молекул и антител. Лизосомотропные флуоресцентные красители, такие как акридиновый оранжевый, могут использоваться, чтобы визуально дифференцировать опухоли in situ от окружающих тканей, указывая потенциальное резкое отличие для определенных нацеливаемых на лизосомы цитотоксических средств, которые селективно уничтожают раковые клетки.
Лизосомальные изменения также являются важными признаками обычных воспалительных заболеваний, особенно таких, которые включают активированные макрофаги, где экзоцитоз лизосомальных ферментов, цитокинов и некоторых воспалительных медиаторов, таких как HMBG1, которые процессируются и высвобождаются через лизосомы, может участвовать в повреждении тканей и как в местном, так и в системном воспалении. Трансдукция сигналов глюкокортикоидов также связана с лизосомами, так, что ухудшение функционирования лизосом может усилить противовоспалительные пути, опосредующие эффекты глюкокортикоидов.
Большинство грибов имеет кислые вакуоли, подобные лизосомам. Эти кислые вакуоли являются критическими для гомеостаза ионов и рН, хранения аминокислот, аутофагии и для процессинга некоторых белков. Вакуоли подкисляются через протонный насос, вакуолярную Н+-АТФазу, или "V-АТФ-азу", и известно, что грибы с инактивацией мутаций субъединиц V-АТФ-азы, которые приводят к ослабленному подкислению вакуолей, также теряют вирулентность и плохо растут. Эргостерин, специфический грибковый стероид, аналогичный холестерину в клетках млекопитающих, как главный мембранный компонент является критическим для конформации и активности V-АТФ-азы, и дисфункция V-АТФ-азы, по-видимому, является главным механизмом противогрибковой активности ингибиторов синтеза эргостерина, которые включают несколько классов существующих противогрибковых средств. Противогрибковые средства, которые действуют через связывание с определенными белками, например, ингибиторами ферментов, являются неотъемлемо уязвимыми для развития лекарственной резистентности через отдельные мутации в генах, кодирующих целевые белки. Средства, нацеливаемые на грибы через адекватно специфическое нацеливание и разрушение грибковых кислых вакуолей улавливанием катионов, могут быть менее подвержены развитию резистентности через точечные мутации, чем лекарственные средства, действующие через связывание со специфическими белками-мишенями, вследствие ослабленной жизнеспособности и вирулентности, когда ослаблено подкисление вакуолей.
Известны клинически важные противомалярийные лекарственные средства, которые аккумулируются в кислых вакуолях и лизосомах, и их биологическая активность в значительной степени установлена через их концентрацию в кислых вакуолях, не только при малярии, но и при воспалительных заболеваниях, некоторых видах рака и немалярийных инфекций, вызываемых грибами и одноклеточными и простейшими паразитами. Противомалярийные лекарственные средства, являющиеся аналогами хинолина, нацеливаются на малярийные плазмодии через улавливание катионов в кислых пищеварительных вакуолях, где они могут накапливаться в концентрации на несколько порядков выше, чем во внеклеточных пространствах. Большая молярная фракция хлорохина, мефлохина, акрихина и нескольких из родственных им соединений является незаряженной при обычном внеклеточном рН приблизительно 7,4 и цитоплазматическом рН 7,1 и может таким образом проходить через клеточную мембрану и мембраны органелл. В кислой среде, такой как внутренняя среда лизосом или кислых вакуолей грибов, эти противомалярийные средства являются преимущественно катионными и таким образом не могут свободно проходить до вакуолярной мембраны. Противомалярийные средства, такие как хлорохин, ослабляют процессинг восстановительного плазмодиями после аккумуляции в питательных вакуолях, что составляет большую часть их специфической токсичности к плазмодиям. Однако хлорохин и подобные противомалярийные средства-аналоги хинолина могут аккумулироваться в лизосомах млекопитающих и кислых вакуолях грибов и ослаблять функцию вакуолей в степени, достаточной для того, чтобы обеспечить некоторую клиническую выгоду, даже только за счет частичного снижения кислотности вакуоли. Хлорохин используется для лечения в случае хронических аутоиммунных и воспалительных заболеваний, таких как системная красная волчанка или ревматоидный артрит, с умеренной эффективностью. О степени противогрибковой активности сообщалось для таких противомалярийных средств, как хлорохин или акрихин, в обоих случаях в виде индивидуальных средств или в комбинации с другими классами противогрибковых средств, таких как флуконазол, особенно в животных моделях системного криптококкоза. Однако их активность является субоптимальной, приводя к неполному торможению роста грибов. В недавней работе также была продемонстрирована умеренная подавляющая рост активность хлорохина, мефлохина и других слабокатионных лекарственных средств, таких как сирамесин, в животных моделях рака. Существующие лизосомотропные средства, такие как противомалярийные соединения хинолона, могут, таким образом, демонстрировать некоторую терапевтически релевантную активность при заболеваниях, в патогенезе которых участвуют кислые вакуоли. Однако активность и потенциал противомалярийных средств при таких заболеваниях ограничены, поскольку клетки-мишени могут переносить аккумуляцию относительно высоких концентраций противомалярийных средств; специфический летальный эффект соединений хинолина в случае малярии в значительной степени приписывается прерыванию процессинга восстановительного гематина в пределах пищеварительных вакуолей плазмодия, механизму цитотоксичности, не применимому в областях воспалительного заболевания, рака или грибковых инфекций. Несмотря на набор данных, показывающий высокий потенциал нацеливания на лизосомы для лечения рака, существующие средства не показали адекватную активность или терапевтический индекс для того, чтобы эффективно лечить рак у человека.
Сообщалось, что “лизосомотропные детергенты”, включая слабокатионные гетероциклические группы, несущие единственную алкильную цепь приблизительно с 10-14 атомами углерода, являются высоко цитотоксичными для клеток млекопитающих и показывают противогрибковую активность широкого спектра in vitro. Этот класс средств аккумулируется в лизосомах и кислых вакуолях через тот же самый тип процесса улавливания катионов, через который концентрируются противомалярийные средства, и когда они достигают критической мицеллярной концентрации в вакуоли, они ведут себя как детергенты, повреждая вакуолярные мембраны. Они демонстрируют характерную сигмовидную кривую доза-ответ, как следствие формирования ими мицеллярных микроструктур. Однако нет никакой информации об активности или безопасности этого класса средств in vivo в животных моделях релевантных заболеваний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Это изобретение относится к соединению, представленному Формулой I, или к его фармацевтически приемлемой соли
G-NH-A-Q-X-Y-ZI
в которой
G обозначает моноциклическое, бициклическое или трициклическое ароматическое кольцо, имеющее один, два или три атома азота в гетероцикле. G может быть незамещенным, или он может быть замещен на кольцевом атоме углерода группами амино, диметиламино, гидрокси, галогеном, метилом, перфторметилом или алкилом, имеющим от 1 до 16 атомов углерода, причем алкил является незамещенным или замещенным группами гидрокси или алкокси, имеющими от 1 до 12 атомов углерода, или ацетокси. Или он может быть замещен на атоме азота в гетероцикле алкилом, имеющим от 1 до 16 атомов углерода, причем алкил является незамещенным или замещенным группами гидрокси или алкокси, имеющим от 1 до 8 атомов углерода. N обозначает азот, Н обозначает водород, и NH отсутствует или присутствует. A отсутствует или присутствует и обозначает алкил, имеющий от 1 до 12 атомов углерода, при условии, что, если A имеет 1 атом углерода, Q должен отсутствовать;
Q отсутствует или присутствует и обозначает O, NHC(O) или NH, при условии, что, если A отсутствует, Q должен отсутствовать, и если и X, и Y отсутствуют, Q не может обозначать O или NH. X отсутствует или присутствует и обозначает алкил, имеющий от 1 до 5 атомов углерода, при условии, что, если Y отсутствует и Z обозначает алкокси или фенокси, X должен иметь более 1 атома углерода. Y отсутствует или присутствует и обозначает фенил, незамещенный или замещенный галогеном, или моноциклическое или бициклическое ароматическое кольцо, имеющее один или два атома азота. Z отсутствует или присутствует и обозначает водород, алкил, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный одной фенильной или феноксигруппой, алкокси, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный одной фенильной или феноксигруппой, фенил, фенокси или NHC(O)R6 или C(O)NHR6, или C(O)OR6, где R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, при условии, что, если все A, Q, X и Y отсутствуют, тогда Z должен обозначать алкил, имеющий от 6 до 12 атомов углерода.
Это изобретение также относится к применению или способу для лечения или профилактики состояния у млекопитающего; причем состояние выбрано из группы, состоящей из воспалительного заболевания, грибковой инфекции, инфекции, вызванной одноклеточным паразитом, и опухолевого заболевания; включающему введение пациенту эффективного количества соединения или соли по изобретению. Оно также относится к композициям, включающим эти соединения или соль. И оно относится к способу ингибирования грибов ex vivo, включающему контакт поверхности или гриба с соединением или солью.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Вне связи с теорией, это изобретение относится к соединениям и их применению для лечения заболеваний, характеризующихся патогенными клетками, показывающими лизосомы или другие кислые вакуоли со связанными с заболеванием изменениями, предрасполагающими их к аккумуляции соединений по изобретению, которые затем селективно инактивируют или удаляют такие патогенные клетки. Соединения по изобретению, многие из которых являются производными аминохинолина и аминохиназолина, демонстрируют значительное повышение потенциала и активности по сравнению с известными лекарственными средствами на основе аминохинолина, такими как хлорохин, как следствие структурных групп, которые сильно нарушают целостность лизосомальных или вакуолярных мембран, когда соединения аккумулируются в кислых вакуолях в клетках. Заболевания, которые являются по меньшей мере умеренно чувствительными к противомалярийным производным хинолина и аналогам, в целом более эффективно лечатся соединениями по изобретению. Такие заболевания в общем включают воспалительные заболевания, опухолевые заболевания, включая как гематологический рак, так и солидные опухоли, и инфекции эукариотическими патогенами, включая грибы и несколько классов простейших или других одноклеточных паразитов.
ОПРЕДЕЛЕНИЯ
В рамках изобретения термин "алкил" означает алкильную группу с прямой или разветвленной цепью или циклическую алкильную группу. Алкильная группа, идентифицированная как имеющая определенное число атомов углерода, подразумевает любую алкильную группу, имеющую указанное число атомов углерода. Например, алкил, имеющий три атома углерода, может быть пропилом или изопропилом; и алкил, имеющий четыре атома углерода, может быть н-бутилом, 1-метилпропилом, 2-метилпропилом или трет-бутилом.
В рамках изобретения термин "галоген" относится к одному или более атомов фтора, хлора, брома и йода.
В рамках изобретения термин “перфтор” как в перфторметиле означает, что рассматриваемая группа имеет атомы фтора вместо всех атомов водорода.
Некоторые химические соединения названы здесь их химическим названием или двухбуквенным кодом, показанным ниже. Следующее представляет собой соединения по изобретению.
CH N-[8-(Гексилокси)октил]хинолин-4-амин
CI N-(8-Бутоксиоктил)хинолин-4-амин
CJ N-(8-Метоксиоктил)хинолин-4-амин
CK N-[6-(Гексилокси)гексил]хинолин-4-амин
CL N-(6-Бутоксигексил)хинолин-4-амин
АL N-[10-(Гексилокси)децил]хинолин-4-амин
AM N-(10-Бутоксидецил)хинолин-4-амин
СМ N-(5-Метоксипентил)хинолин-4-амин
AV N-[8-(Гексилокси)октил]-2-метилхинолин-4-амин
AW 7-Хлор-N-[8-(гексилокси)октил]хинолин-4-амин
AX 8-Хлор-N-[8-(гексилокси)октил]хинолин-4-амин
AY N-[8-(Гексилокси)октил]-7-(трифторметил)хинолин-4-амин
CN N-[8-(Гексилокси)октил]-8-(трифторметил)хинолин-4-амин
BB N-{5-[3-(Гексилокси)пропокси]пентил}хинолин-4-амин
BC N-{3-[5-(Гексилокси)пентилокси]пропил}хинолин-4-амин
AJ N-[8-(3-Этоксипропокси)октил]хинолин-4-амин
BD N-[8-(2-Пропоксиэтокси)октил]хинолин-4-амин
CO N-[8-(Бензилокси)октил]хинолин-4-амин
AR N-(6-Феноксигексил)хинолин-4-амин
AN N-(8-Феноксиоктил)хинолин-4-амин
CP N-{2-[2-(Гексилокси)фенокси]этил}хинолин-4-амин
CQ N-{3-[2-(Гексилокси)фенокси]пропил}хинолин-4-амин
CR N-{4-[2-(Гексилокси)фенокси]бутил}хинолин-4-амин
CS N-[3-(2-Этоксифенокси)пропил]хинолин-4-амин
CT N-[3-(2-Метоксифенокси)пропил]хинолин-4-амин
CU N-{3-[2-(Бензилокси)фенокси]пропил}хинолин-4-амин
BH N-[8-(3-Метоксифенокси)октил]хинолин-4-амин
CV N-{4-[3-(Гексилокси)фенокси]бутил}хинолин-4-амин
AZ N-{3-[3-(Гексилокси)фенокси]пропил}хинолин-4-амин
CW N-{2-[3-(Гексилокси)фенокси]этил}хинолин-4-амин
AD N-[8-(4-Метоксифенокси)октил]хинолин-4-амин
CX N-[6-(4-Метоксифенокси)гексил]хинолин-4-амин
BA N-{2-[4-(Гексилокси)фенокси]этил}хинолин-4-амин
CY N-{3-[4-(Гексилокси)фенокси]пропил}хинолин-4-амин
CZ N-{4-[4-(Гексилокси)фенокси]бутил}хинолин-4-амин
BE N-[8-(м-Толилокси)октил]хинолин-4-амин
BF N-[8-(п-Толилокси)октил]хинолин-4-амин
BG N-[8-(o-Толилокси)октил]хинолин-4-амин
DA N-[8-(4-трет-Бутилфенокси)октил]хинолин-4-амин
BJ N-[8-(4-Фторфенокси)октил]хинолин-4-амин
BI N-[8-(3-Фторфенокси)октил]хинолин-4-амин
DB N-[8-(2-Фторфенокси)октил]хинолин-4-амин
DC N-(Бифенил-4-ил)хинолин-4-амин
AO N-(4-Гексилфенил)хинолин-4-амин
AP Гексил-4-(хинолин-4-иламино)бензоат
DD N-(4-Феноксифенил)хинолин-4-амин
DE N-(3-Феноксифенил)хинолин-4-амин
DF N-(2-Феноксифенил)хинолин-4-амин
DG N-[4-(Хинолин-4-иламино)фенил]гексанамид
DH N-[3-(Хинолин-4-иламино)фенил]гексанамид
AQ N-Гексил-4-(хинолин-4-иламино)бензамид
BV N-Гексил-3-(хинолин-4-иламино)бензамид
DI N-(4-Метоксифенил)хинолин-4-амин
DJ N-[4-(Бензилокси)фенил]хинолин-4-амин
DK N-(4-Бутоксифенил)хинолин-4-амин
DL N-[4-(Гексилокси)фенил]хинолин-4-амин
DM N-[3-(Бензилокси)фенил]хинолин-4-амин
DN N-[3-(Гексилокси)фенил]хинолин-4-амин
DO N-[2-(Бензилокси)фенил]хинолин-4-амин
DP N-[2-(Гексилокси)фенил]хинолин-4-амин
BL N-[2-Фтор-4-(гексилокси)фенил]хинолин-4-амин
DQ N-Бензилхинолин-4-амин
DR N-Фенэтилхинолин-4-амин
AA N-[4-(Гексилокси)бензил]хинолин-4-амин
AC N-[3-(Гексилокси)бензил]хинолин-4-амин
DS N-[2-(Гексилокси)бензил]хинолин-4-амин
BK N-[3-Фтор-4-(гексилокси)бензил]хинолин-4-амин
DT N-[4-(Децилокси)бензил]хинолин-4-амин
DU N-[3-(Децилокси)бензил]хинолин-4-амин
AF N-(3-Феноксибензил)хинолин-4-амин
BU N-[3-(Бензилокси)бензил]хинолин-4-амин
DV N-(3-Фенэтоксибензил)хинолин-4-амин
DW N-[4-(Хинолин-4-иламино)бутил]бензамид
DX N-[6-(Хинолин-4-иламино)гексил]бензамид
DY N-[8-(Хинолин-4-иламино)октил]бензамид
DZ 3-Метокси-N-[8-(хинолин-4-иламино)октил]бензамид
EA 4-Метокси-N-[8-(хинолин-4-иламино)октил]бензамид
EB 2-(Гексилокси)-N-[2-(хинолин-4-иламино)этил]бензамид
EC 2-(Гексилокси)-N-[3-(хинолин-4-иламино)пропил]бензамид
ED 2-(Гексилокси)-N-[4-(хинолин-4-иламино)бутил]бензамид
EE N-[8-(Хинолин-4-иламино)октил]пиколинамид
EF N-[8-(Хинолин-4-иламино)октил]никотинамид
EG N-[8-(Хинолин-4-иламино)октил]изоникотинамид
BZ N-(Пиридин-4-илметил)хинолин-4-амин
BY N-(Пиридин-3-илметил)хинолин-4-амин
EH N-(Пиридин-2-илметил)хинолин-4-амин
EI N-Гексилхинолин-4-амин
AG N-(Децил)хинолин-4-амин
EJ N-(Додецил)хинолин-4-амин
AI N1,N8-ди(хинолин-4-ил)октан-1,8-диамин
EK N-[8-(Гексилокси)октил]хинолин-6-амин
EL N-[8-(Гексилокси)октил]хинолин-3-амин
EM N-[8-(Гексилокси)октил]хинолин-8-амин
EN N-[8-(Гексилокси)октил]-2-(трифторметил)хинолин-4-амин
EO 7-Хлор-N-децилхинолин-4-амин
EP 7-Хлор-N-додецилхинолин-4-амин
AH N-(Децил)хиназолин-4-амин
EQ N-Додецилхиназолин-4-амин
ER N-Децил-7-фторхиназолин-4-амин
ES N-Додецил-7-фторхиназолин-4-амин
ET 7-Хлор-N-децилхиназолин-4-амин
EU 7-Хлор-N-додецилхиназолин-4-амин
EV N-(6-Бутоксигексил)хиназолин-4-амин
EW N-[8-(Гексилокси)октил]хиназолин-4-амин
AE N-[8-(4-Метоксифенокси)октил]хиназолин-4-амин
EX N-{2-[2-(Гексилокси)фенокси]этил}хиназолин-4-амин
EY N-{3-[2-(Гексилокси)фенокси]пропил}хиназолин-4-амин
EZ N-{4-[2-(Гексилокси)фенокси]бутил}хиназолин-4-амин
FA N-[8-(Хиназолин-4-иламино)октил]никотинамид
AK N-[3-(Гексилокси)бензил]хиназолин-4-амин
CG N-[3-(Децилокси)бензил]хиназолин-4-амин
BM N-(3-Феноксибензил)хиназолин-4-амин
BN N-[4-(Децилокси)бензил]хиназолин-4-амин
AB N-[4-(Гексилокси)бензил]хиназолин-4-амин
FB 1-[2-(Этоксиметил)-1H-имидазо[4,5-c]хинолин-1-ил]-2-метилпропан-2-ол
FC 1-(4-Амино-1-изобутил-1H-имидазо[4,5-c]хинолин-2-ил)пентил ацетат
FD 1-Изобутил-2-пентадецил-1H-имидазо[4,5-c]хинолин-4-ол
BP 1-Октил-1H-имидазо[4,5-c]хинолин
FE 1-гексадецил-1H-имидазо[4,5-c]хинолин
FF 1-гексадецил-1H-имидазо[4,5-c]хинолин-4-амин
FG 1-[2-(Додецилокси)этил]-1H-имидазо[4,5-c]хинолин
FH 1-[2-(Додецилокси)этил]-N,N-диметил-1H-имидазо[4,5-c]хинолин-4-амин
FI 1-[6-(Октилокси)гексил]-1H-имидазо[4,5-c]хинолин
CD 1-(8-Этоксиоктил)-1H-имидазо[4,5-c]хинолин
CE 1-(8-Метоксиоктил)-1H-имидазо[4,5-c]хинолин
BQ 1-(8-Бутоксиоктил)-1H-имидазо[4,5-c]хинолин
FJ 1-[9-(Гексилокси)нонил]-1H-имидазо[4,5-c]хинолин
FK 1-(10-Бутоксидецил)-1H-имидазо[4,5-c]хинолин
BO 4-Амино-1-[8-(гексилокси)октил]пиридиниевые соли
FL 4-(8-Метоксиоктиламино)-1-метилпиридиний йодид
AS 1-[8-(Гексилокси)октил]-1H-имидазо[4,5-c]пиридин
FM 1-гексадецил-1H-имидазо[4,5-c]пиридин
AT 1-(10-Бутоксидецил)-1H-имидазо[4,5-c]пиридин
FN N-(8-Метоксиоктил)пиридин-4-амин
FO N-[8-(Гексилокси)октил]пиридин-3-амин
FP N-[8-(Гексилокси)октил]пиридин-2-амин
AU N-[8-(Гексилокси)октил]пиримидин-4-амин
FQ N-[8-Гексилокси)октил)пиримидин-2-амин
FR 1-[8-(Гексилокси)октил]-4-фенил-1H-имидазол
FS N-[8-(Гексилокси)октил]изохинолин-1-амин
FT N-[8-(Гексилокси)октил]изохинолин-5-амин
FU N-[8-(Гексилокси)октил]хиноксалин-2-амин
CC 1-[8-(Гексилокси)октил]-1H-бензимидазол
FV N-[8-(Гексилокси)октил]пиразин-2-амин
FW 1-[8-(Гексилокси)октил]-1H-индол
FX 3-[8-(Гексилокси)октил]-3H-имидазо[4,5-b]пиридин
FY 1-Додецил-1H-имидазо[4,5-c]хинолин
FZ 1-[3-(Децилокси)пропил]-1H-имидазо[4,5-c]хинолин
GA 1-[4-(Децилокси)бутил]-1H-имидазо[4,5-c]хинолин
GB 1-[8-(Гексилокси)октил]-1H-имидазо[4,5-c]хинолин
GC 1-{5-[3-(Гексилокси)пропокси]пентил}-1H-имидазо[4,5-c]хинолин
GD 1-{3-[3-(Гексилокси)фенокси]пропил}-1H-имидазо[4,5-c]хинолин
Следующие соединения были менее активны в примере(ах) биологической активности, в котором(ых) они были протестированы.
BR N-(2-Метоксиэтил)хинолин-4-амин
BS N-[2-(Морфолин-4-ил)этил]хинолин-4-амин
BT N-[3-(Хинолин-4-иламино)пропил]бензамид
BW N-(2-диэтиламиноэтил)-4-(хинолин-4-иламино)бензамид BX N-(4-диметиламинобензил)хинолин-4-амин
CA N-(Пиридин-4-илметил)-8-(гексилокси)октанамид
CB N-(Хинолин-6-ил)-8-(гексилокси)октанамид
CF 1-{3-[(5-(Гексилокси)пентокси]пропил}1H-имидазо[4,5-c]хинолин
В рамках изобретения переходный термин "включающий" является открытым. Пункт формулы изобретения, в котором используется этот термин, может включать элементы в добавление к указанным в таком пункте.
В формуле изобретения слово "или" означает "и/или", за исключением случаев, когда такое прочтение в контексте является бессмысленным. Так, например, когда в отношении Формулы I отмечено, что переменная G может быть замещена на кольцевом атоме углерода "или" азота в гетероцикле, она может быть замещена на кольцевом атоме углерода, азота в гетероцикле, или и на кольцевом атоме углерода, и на атоме азота в гетероцикле.
Следующие сокращения используются в примерах химического синтеза и в других местах в этом описании:
DCM дихлорметан
DIEA N,N-диизопропилэтиламин
DMA N,N-диметилацетамид
DMAP 4-(N,N-диметиламино)пиридин
DME 1,2-диметоксиэтан
DMF N,N-диметилформамид
ДМСО диметил сульфоксид
ЕА этилацетат
Et2O простой диэтиловый эфир
EtOH этанол
FC флэш-хроматография
Hex гексаны
IPA 2-пропанол
LAH тетрагидридоалюминат лития
MeOH метанол
Т.пл. температура плавления
NMP N-метилпирролидинон
ЯМР спектрометрия ядерного магнитного резонанса
SPE твердофазная экстракция
ТЕА триэтиламин
THF тетрагидрофуран
TLC тонкослойная хроматография
СОЕДИНЕНИЯ
В одном варианте соединения или соли Формулы I, G выбраны из группы, состоящей из замещенного или незамещенного хинолила, замещенного или незамещенного хиназолила, незамещенного изохинолила, незамещенного хиноксалила, незамещенного бензимидазолила, незамещенного пиридила, незамещенного пиразинила, незамещенного индолила, замещенного или незамещенного имидазохинолила, замещенного пиридиния, незамещенного имидазопиридина, незамещенного пиримидила и замещенного имидазолила. В другом варианте соединения или соли Формулы I A-Q-X-Y-Z выбраны из группы, состоящей из алкоксифенилалкила, алкоксифенила, алкоксифеноксиалкила, алкоксиалкила, алкоксиалкоксиалкила, феноксифенила, феноксифенилалкила, фенилалкоксифенилалкила, феноксиалкила, фенилалкоксиалкила, алкилфеноксиалкила, алкила, (галогенфенокси)алкила, бифенила, алкилфенила, алкоксикарбонилфенила, N-алкилкарбамоилфенила, алкокси(галогенфенила), фенилалкила, алкокси(галогенфенил)алкила, (алкоксибензамидо)алкила, пиколинамидоалкила, никотинамидоалкила, изоникотинамидоалкила, N-(хинолиламино)алкила, N-(хиназолиламино)алкила, фенилалкоксифеноксиалкила, алкилалкоксифенила, фенилалкоксифенила, пиридилалкила и гидроксиалкила.
Некоторые из соединений по изобретению, в которых G обозначает незамещенный или замещенный хинолил, могут быть представлены Формулой IA

в которой A отсутствует или присутствует и алкил имеет от 1 до 12 атомов углерода, при условии, что, если A имеет 1 атом углерода, Q должен отсутствовать. Q отсутствует или присутствует и обозначает O, NHC(O) или NH, при условии, что, если A отсутствует, Q должен отсутствовать, и если и X и Y отсутствуют, Q не может быть O или NH.
X отсутствует или присутствует и алкил имеет от 1 до 5 атомов углерода, при условии, что, если Y отсутствует и Z обозначает алкокси или фенокси, X должен иметь больше чем 1 атом углерода. Y отсутствует или присутствует и обозначает фенил, незамещенный или замещенный галоген или моноциклическое или бициклическое ароматическое кольцо, имеющее один или два атома азота. Z отсутствует или присутствует и обозначает водород, алкил, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный одним фенилом или феноксигруппой, алкокси, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный одним фенилом или феноксигруппой, фенил, фенокси или NHC(O)R6 или C(O)NHR6 или C(O)OR6, где R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, при условии, что, если все A, Q, X и Y отсутствуют, тогда Z должен обозначать алкил, имеющий 6-12 атомов углерода. Один из R1 и R2 обозначает водород, и другой выбран из группы, состоящей из водорода, галогена, метила и перфторметила. В варианте осуществления изобретения и R1, и R2 обозначают водород. В варианте Формулы IA, A-Q-X-Y-Z выбран из группы, состоящей из алкоксифенилалкила, алкоксифенила, алкоксифеноксиалкила, алкоксиалкила, алкоксиалкоксиалкила, феноксифенила, феноксифенилалкила, фенилалкоксифенилалкила, феноксиалкила, фенилалкоксиалкила, алкилфеноксиалкила, алкила, (галогенфенокси)алкила, бифенила, алкилфенила, алкоксикарбонилфенила, N-алкилкарбамоилфенила, алкокси(галогенфенила), фенилалкила, алкокси(галогенфенил)алкила, (алкоксибензамидо)алкила, пиколинамидоалкила, никотинамидоалкила, изоникотинамидоалкила, фенилалкоксифеноксиалкила, алкилалкоксифенила, фенилалкоксифенила, пиридилалкила и N-(хинолиламино)алкила.
Более конкретный вариант соединений, в которых G обозначает хинолил, может быть представлен Формулой IA1

в которой n=0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12, при условии, что, если p=1 затем n не должен означать 0 или 1, p=0 или 1; и q=0 или 1, Один из R1 и R2 обозначает водород, и другой выбран из группы, состоящей из водорода, галогена, метила и перфторметила. R3 может обозначать алкил, имеющий от 1 до 10 атомов углерода, незамещенный или замещенный: a)фенилом или моноциклическим или бициклическим ароматическим кольцом, имеющим один или два атома азота, или фенокси, незамещенным или замещенным фенокси или алкокси, имеющим от 1 до 6 атомов углерода, или b) алкокси, имеющим от 1 до 6 атомов углерода, при условии, что, если R3 обозначает алкил, замещенный алкокси, тогда алкил должен иметь больше чем 1 атом углерода. Альтернативно R3 может быть фенилом, незамещенным или замещенным галогеном и незамещенным или замещенным: a) алкилом, имеющим от 1 до 6 атомов углерода, незамещенным или замещенным фенилом или фенокси, b) алкокси, имеющим от 1 до 10 атомов углерода, незамещенным или замещенным фенилом или фенокси, при условии, что когда он замещен фенокси, алкокси должен иметь больше чем один атом углерода, c)фенилом, d)фенокси или e) C(O)OR6, C(O)NHR6 или NHC(O)R6, в которых R6 обозначает алкил, имеющий от 1 до 6 атомов углерода.
В варианте соединений Формулы IA1, R1 обозначает водород, и R2 обозначает водород. В более конкретном варианте осуществления n=2, 3, 4, 5, 6, 7, 8, 9 или 10; p=1; и R3 обозначает алкил, имеющий от 1 до 6 атомов углерода. Примеры таких соединений включают N-[8-(гексилокси)октил]хинолин-4-амин, N-(8-Бутоксиоктил)хинолин-4-амин, N-(8-Метоксиоктил)хинолин-4-амин, N-[6-(гексилокси)гексил]хинолин-4-амин, N-(6-Бутоксигексил)хинолин-4-амин, N-[10-(гексилокси)децил]хинолин-4-амин, N-(10-Бутоксидецил)хинолин-4-амин, N-(5-Метоксипентил)хинолин-4-амин.
В другом варианте соединений Формулы IA1, n=2, 3, 4, 5, 6, 7, 8, 9 или 10; p=1; один из R1 и R2 обозначает водород, и другой выбран из группы, состоящей из галогена, метила и перфторметила; и R3 обозначает алкил, имеющий от 1 до 6 атомов углерода. Примеры таких соединений включают N-[8-(гексилокси)октил]-2-метилхинолин-4-амин, 7-Хлор-N-[8-(гексилокси)октил]хинолин-4-амин, 8-Хлор-N-[8-(гексилокси)октил]хинолин-4-амин, N-[8-(гексилокси)октил]-7-(трифторметил)хинолин-4-амин, N-[8-(гексилокси)октил]-8-(трифторметил)хинолин-4-амин.
В другом варианте соединений Формулы IA1, в которой R1 обозначает водород и R2 обозначает водород: n=2, 3, 4, 5, 6, 7, 8, 9 или 10; p=1; R3обозначает алкил, имеющий от 2 до 5 атомов углерода, замещенный алкокси, имеющим от 1 до 6 атомов углерода. Примеры таких соединений включают
N-{5-[3-(Гексилокси)пропокси]пентил}хинолин-4-амин, N-{3-[5-(Гексилокси)пентилокси)пропил}хинолин-4-амин, N-[8-(3-Этоксипропокси)октил]хинолин-4-амин, N-[8-(2-Пропоксиэтокси)октил]хинолин-4-амин.
Подгруппа соединений Формулы IA1 может быть представлена Формулой IA1a

в которой n=0, 1, 2, 3, 4, 5, 6, 7 или 8; p=0 или 1; q=0 или 1, при условии, что, если p=1, тогда n не должен означать 0 или 1, Один из R1 и R2 обозначает водород, и другой выбран из группы, состоящей из водорода, галогена, метила и перфторметила. R4 обозначает водород или галоген. R5 выбран из группы, состоящей из водорода; галогена; неразветвленного или разветвленного алкила, имеющего от 1 до 6 атомов углерода, незамещенного или замещенного фенилом или фенокси; алкокси, имеющего от 1 до 10 атомов углерода, незамещенного или замещенного фенилом или фенокси, при условии, что когда он замещен фенокси, алкокси должен иметь больше чем один атом углерода; фенила; фенокси; C(O)OR6; C(O)NHR6; или NHC(O)R6, в которых R6 обозначает алкил, имеющий от 1 до 6 атомов углерода. В варианте Формулы IA1a R1 обозначает водород, и R2 обозначает водород. В более конкретном варианте осуществления p=1, и R4 обозначает водород. В еще более конкретном варианте осуществления R5 обозначает водород. Примеры таких соединений включают N-[8-(бензилокси)октил]хинолин-4-амин, N-(6-Феноксигексил)хинолин-4-амин, N-(8-Феноксиоктил)хинолин-4-амин.
В другом варианте Формулы IA1a, и R1, и R2 обозначают водород, q=0, и R5 обозначает алкокси, имеющий от 1 до 6 атомов углерода, незамещенный или замещенный фенилом. В более конкретном варианте осуществления R5 находится в орто-положении. Примеры таких соединений включают N-{2-[2-(гексилокси)фенокси]этил}хинолин-4-амин, N-{3-[2-(гексилокси)фенокси)пропил}хинолин-4-амин, N-{4-[2-(гексилокси)фенокси]бутил}хинолин-4-амин, N-[3-(2-Этоксифенокси)пропил]хинолин-4-амин, N-[3-(2-Метоксифенокси)пропил]хинолин-4-амин, N-{3-[2-(бензилокси)фенокси)пропил}хинолин-4-амин. Альтернативно R5 находится в мета-положении. Примеры таких соединений включают N-[8-(3-Метоксифенокси)октил]хинолин-4-амин, N-{4-[3-(гексилокси)фенокси]бутил}хинолин-4-амин, N-{3-[3-(гексилокси)фенокси)пропил}хинолин-4-амин, N-{2-[3-(гексилокси)фенокси]этил}хинолин-4-амин. Альтернативно R5 находится в пара-положении. Примеры таких соединений включают N-[8-(4-Метоксифенокси)октил]хинолин-4-амин, N-[6-(4-Метоксифенокси)гексил]хинолин-4-амин, N-{2-[4-(гексилокси)фенокси]этил}хинолин-4-амин, N-{3-[4-(гексилокси)фенокси)пропил}хинолин-4-амин, N-{4-[4-(гексилокси)фенокси]бутил}хинолин-4-амин.
В другом варианте Формулы IA1a, R1 обозначает водород, и R2 обозначает водород, p=1, R4 обозначает водород, и R5 обозначает неразветвленный или разветвленный алкил, имеющий от 1 до 6 атомов углерода. Примеры таких соединений включают N-[8-(м-толилокси)октил]хинолин-4-амин, N-[8-(п-Толилокси)октил]хинолин-4-амин, N-[8-(o-Толилокси)октил]хинолин-4-амин, N-[8-(4-трет-Бутилфенокси)октил]хинолин-4-амин. Альтернативно R5 обозначает фтор. Примеры таких соединений включают N-[8-(4-Фторфенокси)октил]хинолин-4-амин, N-[8-(3-Фторфенокси)октил]хинолин-4-амин, N-[8-(2-Фторфенокси)октил]хинолин-4-амин.
В другом варианте Формулы IA1a, R1 обозначает водород, и R2 обозначает водород, и p=0. В более конкретном варианте осуществления q=0. В еще более конкретном варианте осуществления n=0. Примеры такого соединения включают N-(бифенил-4-ил)хинолин-4-амин, N-(4-Гексилфенил)хинолин-4-амин, Гексил-4-(хинолин-4-иламино)бензоат, N-(4-Феноксифенил)хинолин-4-амин, N-(3-Феноксифенил)хинолин-4-амин, N-(2-Феноксифенил)хинолин-4-амин, N-[4-(хинолин-4-иламино)фенил]гексанамид, N-[3-(хинолин-4-иламино)фенил]гексанамид, N-Гексил-4-(хинолин-4-иламино)бензамид, N-Гексил-3-(хинолин-4-иламино)бензамид. Альтернативно R5 обозначает алкокси, имеющий от 1 до 10 атомов углерода, незамещенный или замещенный фенилом. Примеры таких соединений включают N-(4-Метоксифенил)хинолин-4-амин, N-[4-(бензилокси)фенил]хинолин-4-амин, N-(4-Бутоксифенил)хинолин-4-амин, N-[4-(гексилокси)фенил]хинолин-4-амин, N-[3-(бензилокси)фенил]хинолин-4-амин, N-[3-(гексилокси)фенил]хинолин-4-амин, N-[2-(бензилокси)фенил]хинолин-4-амин, N-[2-(гексилокси)фенил]хинолин-4-амин, N-[2-Фтор-4-(гексилокси)фенил]хинолин-4-амин. В другом варианте осуществления Формулы IA1a, R1 обозначает водород, и R2 обозначает водород, p=0, q=0, и n=1 или 2. Примеры таких соединений включают N-Бензилхинолин-4-амин и N-Фенэтилхинолин-4-амин.
В другом варианте Формулы IA1a, R1 обозначает водород, и R2 обозначает водород, p=0, и q=1. В более конкретном варианте осуществления R5 обозначает алкокси, имеющий от 1 до 10 атомов углерода. Примеры таких соединений включают N-[4-(гексилокси)бензил]хинолин-4-амин, N-[3-(гексилокси)бензил]хинолин-4-амин, N-[2-(гексилокси)бензил]хинолин-4-амин, N-[3-Фтор-4-(гексилокси)бензил]хинолин-4-амин, N-[4-(децилокси)бензил]хинолин-4-амин, N-[3-(децилокси)бензил]хинолин-4-амин. Альтернативно R5 обозначает фенокси или алкокси, имеющий от 1 до 10 атомов углерода, замещенный фенилом. Примеры таких соединений включают N-(3-Феноксибензил)хинолин-4-амин, N-[3-(бензилокси)бензил]хинолин-4-амин, N-(3-Фенэтоксибензил)хинолин-4-амин.
Другой более конкретный вариант осуществления соединений, в которых G обозначает хинолил, может быть представлен Формулой IA2

в которой n=2, 3, 4, 5, 6, 7 или 8, R13 обозначает фенил, незамещенный или замещенный алкокси, имеющим от 1 до 6 атомов углерода; или 2-, 3- или 4-пиридил. В одном варианте осуществления R13 обозначает незамещенный фенил. Примеры таких соединений включают N-[4-(хинолин-4-иламино)бутил]бензамид, N-[6-(хинолин-4-иламино)гексил]бензамид, N-[8-(хинолин-4-иламино)октил]бензамид. В другом варианте осуществления R13 обозначает фенил, замещенный алкокси, имеющим от 1 до 6 атомов углерода. Примеры таких соединений включают 3-Метокси-N-[8-(хинолин-4-иламино)октил]бензамид, 4-Метокси-N-[8-(хинолин-4-иламино)октил]бензамид, 2-(Гексилокси)-N-[2-(хинолин-4-иламино)этил]бензамид, 2-(Гексилокси)-N-[3-(хинолин-4-иламино)пропил]бензамид, 2-(Гексилокси)-N-[4-(хинолин-4-иламино)бутил]бензамид. Альтернативно R13 обозначает 2-пиридил, 3-пиридил или 4-пиридил. Примеры таких соединений включают N-[8-(хинолин-4-иламино)октил]пиколинамид, N-[8-(хинолин-4-иламино)октил]никотинамид, N-[8-(хинолин-4-иламино)октил]изоникотинамид.
Другие примеры соединений Формулы IA включают N-(Пиридин-4-илметил)хинолин-4-амин, N-(Пиридин-3-илметил)хинолин-4-амин, N-(Пиридин-2-илметил)хинолин-4-амин, N-Гексилхинолин-4-амин, N-(Децил)хинолин-4-амин, N-(Додецил)хинолин-4-амин, N1,N8-ди(хинолин-4-ил)октан-1,8-диамин. Другие примеры соединений Формулы I, в которой G обозначает хинолил, включают N-[8-(гексилокси)октил]хинолин-6-амин, N-[8-(гексилокси)октил]хинолин-3-амин, N-[8-(гексилокси)октил]хинолин-8-амин, N-[8-(гексилокси)октил]-2-(трифторметил)хинолин-4-амин, 7-Хлор-N-децилхинолин-4-амин, 7-Хлор-N-додецилхинолин-4-амин.
Некоторые из соединений по изобретению, в которых G обозначает незамещенный или замещенный хиназолил, могут быть представлены Формулой IB

в которой A отсутствует или присутствует и обозначает алкил, имеющий от 1 до 12 атомов углерода, при условии, что, если A имеет 1 атом углерода, Q должен отсутствовать. Q отсутствует или присутствует и обозначает O, NHC(O) или NH, при условии, что, если A отсутствует, Q должен отсутствовать, и если и X, и Y отсутствуют, Q не может быть O или NH.
X отсутствует или присутствует и обозначает алкил, имеющий от 1 до 5 атомов углерода, при условии, что, если Y отсутствует и Z обозначает алкокси или фенокси, X должен иметь больше чем 1 атом углерода. Y отсутствует или присутствует и обозначает фенил, незамещенный или замещенный галогеном, или обозначает моноциклическое или бициклическое ароматическое кольцо, имеющее один или два атома азота. Z отсутствует или присутствует и обозначает водород, алкил, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный одним фенилом или феноксигруппой, алкокси, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный одним фенилом или феноксигруппой, фенил, фенокси или NHC(O)R6 или C(O)NHR6 или C(O)OR6, где R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, при условии, что, если все A, Q, X и Y отсутствуют, тогда Z должен обозначать алкил, имеющий 6-12 атомов углерода. R1 выбран из группы, состоящей из водорода, галогена, метила и перфторметила.
В варианте осуществления Формулы IB, R1 обозначает водород. В другом варианте осуществления, A-Q-X-Y-Z выбран из группы, состоящей из алкоксифенилалкила, алкоксифенила, алкоксифеноксиалкила, алкоксиалкила, алкоксиалкоксиалкила, феноксифенила, феноксифенилалкила, фенилалкоксифенилалкила, феноксиалкила, фенилалкоксиалкила, алкилфеноксиалкила, алкила, (галогенфенокси)алкила, бифенила, алкилфенила, алкоксикарбонилфенила, N-алкилкарбамоилфенила, алкокси(галогенфенила), фенилалкила, алкокси(галогенфенил)алкила, (алкоксибензамидо)алкила, пиколинамидоалкила, никотинамидоалкила, изоникотинамидоалкила, фенилалкоксифеноксиалкила, алкилалкоксифенила, фенилалкоксифенила, пиридилалкила, N-(хиназолиламино)алкила и N-(хинолиламино)алкила.
Подгруппа соединений Формулы IB может быть представлена Формулой IB1

в которой n=0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12; Q отсутствует или присутствует и обозначает O или NHC(O), при условии, что, если Q присутствует, n не может означать 0 или 1; и при условии, что, если Q отсутствует, то (CH2)nR7 должен иметь больше чем 5 атомов углерода. R1 обозначает водород или галоген. R7 выбран из группы, состоящей из: водорода; алкила, имеющего от 1 до 6 атомов углерода; и фенила или моноциклического ароматического кольца, имеющего один атом азота, незамещенный или замещенный, алкилом, имеющим от 1 до 6 атомов углерода, или алкокси, имеющим от 1 до 10 атомов углерода, или фенилом или фенокси. В варианте осуществления Q отсутствует. Примеры таких соединений включают N-(Децил)хиназолин-4-амин,N-Додецилхиназолин-4-амин, N-Децил-7-фторхиназолин-4-амин, N-Додецил-7-фторхиназолин-4-амин, 7-Хлор-N-децилхиназолин-4-амин, 7-Хлор-N-додецилхиназолин-4-амин. В другом варианте осуществления Q обозначает O или NHC(O). Примеры таких соединений включают N-(6-Бутоксигексил)хиназолин-4-амин, N-[8-(гексилокси)октил]хиназолин-4-амин, N-[8-(4-Метоксифенокси)октил]хиназолин-4-амин, N-{2-[2-(гексилокси)фенокси]этил}хиназолин-4-амин, N-{3-[2-(гексилокси)фенокси)пропил}хиназолин-4-амин, N-{4-[2-(гексилокси)фенокси]бутил}хиназолин-4-амин, N-[8-октил(Хиназолин-4-иламино)]никотинамид. В варианте Формулы IB1, n=1, Q отсутствует, и R7 обозначает фенил, замещенный алкокси, имеющим от 1 до 10 атомов углерода, или фенокси. Примеры таких соединений включают N-[3-(гексилокси)бензил]хиназолин-4-амин, N-[3-(децилокси)бензил]хиназолин-4-амин, N-(3-Феноксибензил)хиназолин-4-амин, N-[4-(децилокси)бензил]хиназолин-4-амин, N-[4-(гексилокси)бензил]хиназолин-4-амин.
Некоторые из соединений по изобретению, в котором G обозначает незамещенный или замещенный имидазохинолил, могут быть представлены Формулой IC

в которой R1 обозначает водород, ОН, NH2, или N(CH3)2; R2 выбран из группы, состоящей из водорода, галогена, метила и перфторметила; R8 обозначает водород или алкил, имеющий от 1 до 15 атомов углерода, незамещенный или замещенный алкокси, имеющим 1 или 2 атома углерода, или ацетокси; и R9 обозначает разветвленный или неразветвленный алкил, имеющий от 1 до 16 атомов углерода, незамещенный или замещенный гидрокси или алкокси, имеющим от 1 до 12 атомов углерода, при условии, что если он замещен гидрокси или алкокси, R9 должен иметь больше чем 1 атом углерода. В варианте осуществления R2 обозначает водород. Примеры таких соединений включают 1-[2-(Этоксиметил)-1H-имидазо[4,5-c]хинолин-1-ил]-2-метилпропан-2-ол, 1-(4-Амино-1-изобутил-1H-имидазо[4,5-c]хинолин-2-ил)пентил ацетат, 1-Изобутил-2-пентадецил-1H-имидазо[4,5-c]хинолин-4-ол, 1-Октил-1H-имидазо[4,5-c]хинолин, 1-гексадецил-1H-имидазо[4,5-c]хинолин, 1-гексадецил-1H-имидазо[4,5-c]хинолин-4-амин, 1-Додецил-1H-имидазо[4,5-c]хинолин, 1-{5-[3-(Гексилокси)пропокси]пентил}-1H-имидазо[4,5-c]хинолин, 1-{3-[3-(гексилокси)фенокси)пропил}-1H-имидазо[4,5-c]хинолин. В другом варианте Формулы IC, R2 обозначает водород, и R9 обозначает неразветвленный алкил, имеющий от 2 до 10 атомов углерода, замещенный алкокси, имеющим от 1 до 12 атомов углерода. Примеры таких соединений включают 1-[2-(додецилокси)этил]-1H-имидазо[4,5-c]хинолин, 1-[2-(додецилокси)этил]-N,N-диметил-1H-имидазо[4,5-c]хинолин-4-амин, 1-[6-(октилокси)гексил]-1H-имидазо[4,5-c]хинолин, 1-(8-Этоксиоктил)-1H-имидазо[4,5-c]хинолин, 1-(8-Метоксиоктил)-1H-имидазо[4,5-c]хинолин, 1-(8-Бутоксиоктил)-1H-имидазо[4,5-c]хинолин, 1-[9-(гексилокси)нонил]-1H-имидазо[4,5-c]хинолин, 1-(10-Бутоксидецил)-1H-имидазо[4,5-c]хинолин, 1-[3-(децилокси)пропил]-1H-имидазо[4,5-c]хинолин, 1-[4-(децилокси)бутил]-1H-имидазо[4,5-c]хинолин, 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]хинолин.
Некоторые из соединений по изобретению, в которых G обозначает замещенный пиридиний, могут быть представлены в соответствии с Формулой ID

в которой R10 обозначает алкил, имеющий от 1 до 8 атомов углерода, незамещенных или замещенных, алкокси, имеющим от 1 до 6 атомов углерода, при условии, что если замещено алкокси, R10 должен иметь больше чем 1 атом углерода. R11обозначает водород; или алкил, имеющий от 1 до 8 атомов углерода, незамещенный или замещенный алкокси, имеющим от 1 до 3 атомов углерода, при условии, что если он замещен алкокси, R11 должен иметь больше чем 1 атом углерода. X- обозначает противоион. Примеры таких соединений включают 4-Амино-1-[8-(гексилокси)октил]пиридиниевую соль и 4-(8-Метоксиоктиламино)-1-метилпиридиний йодид.
В варианте осуществления этого изобретения G обозначает 1H-имидазо[4,5-c]пиридин. Некоторые из этих соединений могут быть представлены Формулой IE

в которой R12 обозначает алкил, имеющий от 2 до 16 атомов углерода, незамещенный или замещенный алкокси, имеющим от 4 до 6 атомов углерода. Примеры таких соединений включают 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридин, 1-гексадецил-1H-имидазо[4,5-c]пиридин, 1-(10-Бутоксидецил)-1H-имидазо[4,5-c]пиридин.
Примеры этого изобретения, в котором G обозначает пиридил, включают N-(8-Метоксиоктил)пиридин-4-амин, N-[8-(гексилокси)октил]пиридин-3-амин и N-[8-(гексилокси)октил]пиридин-2-амин.
Примеры этого изобретения, в котором G обозначает пиримидил, включают N-[8-(гексилокси)октил]пиримидин-4-амин и N-[8-Гексилокси)октил)пиримидин-2-амин. В варианте осуществления этого изобретения G обозначает 5-арил-1H-имидазолил. Примеры таких соединений включают 1-[8-(гексилокси)октил]-4-фенил-1H-имидазол. Примеры соединений по изобретению, в котором G обозначает изохинолил, включают N-[8-(гексилокси)октил]изохинолин-1-амин, N-[8-(гексилокси)октил]изохинолин-5-амин. Примеры соединений, в которых G обозначает хиноксалил, включают N-[8-(гексилокси)октил]хиноксалин-2-амин. Примеры соединений, в которых G обозначает бензимидазолил, включают 1-[8-(гексилокси)октил]-1Н-бензимидазол. Примеры соединений, в которых G обозначает пиразинил, включают N-[8-(гексилокси)октил]пиразин-2-амин. Примеры соединений, в которых G обозначает индолил, включают 1-[8-(гексилокси)октил]-1Н-индол. В варианте осуществления этого изобретения G обозначает 3H-имидазо[4,5-b]пиридин. Примеры таких соединений включают 3-[8-(гексилокси)октил]-3H-имидазо[4,5-b]пиридин.
В некоторых вариантах осуществления этого изобретения, один или более из следующих соединений исключены: имихимод; 4-(н-дециламино)хинолин [58911-14-1]; 4-дециламинохиназолин [22754-12-7].
В варианте соединения по изобретению, соединение находится в в основном (по меньшей мере 98%) чистой форме. Это изобретение относится к пролекарствам соединений и солей, описанных выше, и к их применениям, как описано здесь. Всякий раз, когда фенильное кольцо замещено, замещение может быть в орто-, мета- или пара-положении.
РЕАКЦИОННЫЕ СХЕМЫ
Соединения согласно настоящему изобретению могут быть получены в соответствии со следующими реакционными схемами.
Соединение формулы I, в которых G обозначает моноциклическое или бициклическое ароматическое кольцо, имеющее один или два атома атома азота в гетероцикле, незамещенное или замещенное на кольцевом углероде галогеном, метилом или перфторметилом;
N обозначает азот, Н обозначает водород;
A отсутствует или присутствует и обозначает алкил, имеющий от 1 до 12 атомов углерода, при условии, что, если A имеет 1 атом углерода, Q должен отсутствовать;
Q отсутствует или присутствует и обозначает O, NHC(O) или NH, при условии, что, если A отсутствует, Q должен отсутствовать, и если и X, и Y отсутствуют, Q не может быть O или NH;
X отсутствует или присутствует и обозначает алкил, имеющий от 1 до 5 атомов углерода, при условии, что, если Y отсутствует и Z обозначает алкокси или фенокси, X должен иметь больше чем 1 атом углерода;
Y отсутствует или присутствует и обозначает фенил, незамещенный или замещенный галогеном, или обозначает моноциклическое или бициклическое ароматическое кольцо, имеющее один атом азота;
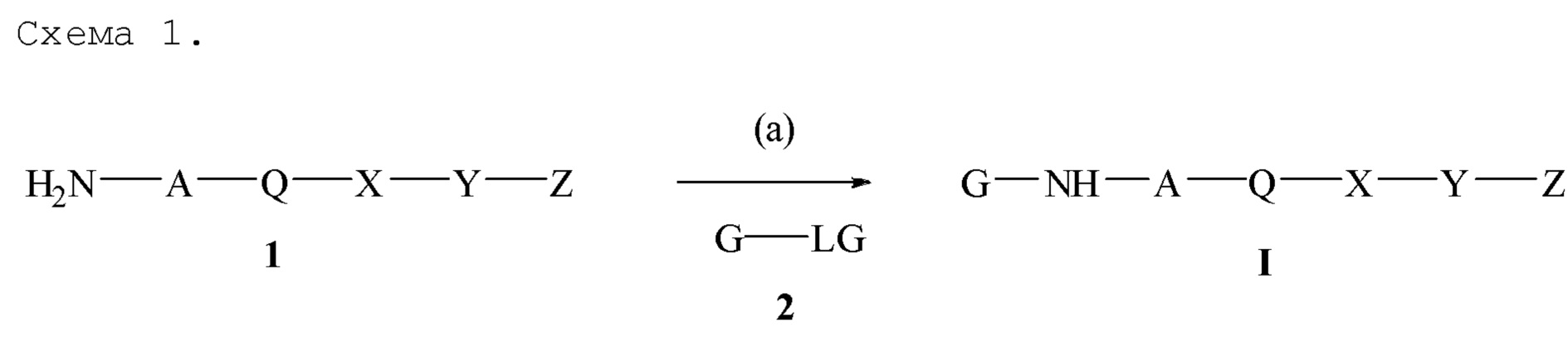
Z отсутствует или присутствует и обозначает: a) водород, b) алкил, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный одним фенилом или феноксигруппой, c) алкокси, имеющий от 1 до 10 атомов углерода, незамещенный или замещенный одним фенилом или феноксигруппой, d) фенил, e) фенокси, или f) NHC(O)R6 или C(O)NHR6 или C(O)OR6, где R6 обозначает алкил с 1-6 атомами углерода, за исключением случая, если и X, и Y отсутствуют, при условии, что, если все A, Q, X и Y отсутствуют, тогда Z должен обозначать алкил, имеющий 6-12 атомов углерода, может быть получено реакцией соединения формулы 1 с соединением формулы 2, где LG обозначает уходящую группу, такую как галоген, сульфонилокси, силокси или борат, через реакционную схему на Схеме 1. Если LG расположена в положении на ароматическом кольце, которое активировано атомом азота, реакция на стадии (a) может протекать термически без использования катализатора, и LG обозначает галоген, предпочтительно, и LG обозначает хлор, наиболее предпочтительно. G предпочтительно выбран из группы соединений, состоящих из незамещенного или замещенного 4-хинолила, 4-хиназолила, 2-хинолила, 2-хиназолила, 1-изохинолила, 3-изохинолила, 2-хиноксалила, 1-фталазила, 2-пиридила, 4-пиридила, 2-пиримидила, 4-пиримидила и 2-пиразинила. Соединение формулы 1 и соединение формулы 2 и подходящее основание, такое как триэтиламин, трипропиламин, N-метилморфолин или диизопропилэтиламин, нагревают в подходящем растворителе, таком как 1-пентанол, 1-бутанол, 2-пропанол, диметилформамид, N-метилпирролидинон, или смеси подходящих растворителей. Если LG не расположен в положении на ароматическом кольце, которое активировано атомом азота, реакция может проходить с использованием катализатора, такого как комплекс переходного металла, такой как комплекс палладия или комплекс никеля.
Соединение формулы 7, где T обозначает CH, и R2 присутствует, или T обозначает N, и R2 отсутствует и где также: a) n=2-12, и p=1; или b) n=0 или 1, и p=0; и где q=0 или 1, и один из R1 и R2 обозначает водород, и другой выбран из группы, состоящей из водорода, галогена, метила и перфторметила, и R3 обозначает алкил, имеющий от 1 до 10 атомов углерода, незамещенный или замещенный: a) моноциклическим или бициклическим ароматическим кольцом, имеющим один или два атома азота, или фенилом, незамещенным или замещенным алкокси, имеющим от 1 до 6 атомов углерода, или b) алкокси, имеющим от 1 до 6 углерода, при условии, что, если R3 обозначает алкил, замещенный алкокси, тогда алкил не может иметь 1 атом углерода; фенилом, незамещенным или замещенным галогеном и незамещенным или замещенным: a) алкилом, имеющим от 1 до 6 атомов углерода, b) алкокси, имеющим от 1 до 10 атомов углерода, незамещенным или замещенным фенилом или фенокси, при условии, что когда он замещен фенокси, алкокси должен иметь больше чем один атом углерода, c) фенилом, d) фенокси или e) C(O)OR6, C(O)NHR6 или NHC(O)R6, в которых R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, может быть получено исходя из соединения формулы 3 или исходя из соединения формулы 6 через реакционную схему на Схеме 2.
Некоторые соединения формулы 3 и некоторые соединения формулы 6 коммерчески доступны. Соединение формулы 3 вводят в реакцию с соединением формулы 4, получая соединение формулы 5 через реакцию стадии (a): соединение формулы 3 обрабатывают подходящим основанием и затем вводят в реакцию с соединением формулы 4. Селективность реакции для замещения только одного из бромидов соединения формулы 4 может быть увеличена при использовании стехиометрического избытка соединения формулы 3. Если n=1, любое основание, которое обычно используется для превращения спирта в алкоголят, является подходящим, такое как гидрид натрия или затрудненный алкоголят щелочного металла, такой как изопропилат натрия. Если n=1, основание должно быть полностью введено в реакцию с соединением формулы 3 до добавления соединения формулы 4. Если n=0, любое основание, которое обычно используется для превращения фенола в простой эфир фенола, является подходящим, такое как карбонат калия или карбонат натрия. Если n=0, соединение формулы 4 может присутствовать, когда основание вводят в реакцию с соединением формулы 3.
Соединение формулы 5 превращают в соединение формулы 6 через реакции стадии (b), синтез Габриэля первичных аминов. Соединение формулы 5 вводят в реакцию с фталимидом калия в обычно используемых условиях, получая промежуточное соединение фталимида, которое превращают в соединение формулы 6 в обычно используемых условиях, таких как гидразин моногидрат в этаноле при нагревании с обратным холодильником. Может использоваться любой способ расщепления фталимидов.
Соединение формулы 6 превращают в соединение формулы 7 через стадию (c): соединение формулы 6 вводят в реакцию с соединением формулы 7 в присутствии третичного аминового основания, такого как триэтиламин, диизопропилэтиламин или трипропиламин, при повышенной температуре в подходящем растворителе, таком как 2-пропанол при нагревании с обратным холодильником, если T обозначает N, или 1-пентанол при нагревании с обратным холодильником или диметилформамид или N-метилпирролидинон при 130-150°C, если T обозначает CH.

Соединение формулы 3, где q=0 или 1, и R3 обозначает алкил, имеющий от 1 до 10 атомов углерода, замещенный алкокси, имеющим от 1 до 12 атомов углерода, при условии, что, если R3 обозначает алкил, замещенный алкокси, тогда алкил не может иметь один атом углерода, может быть получено через реакционную схему на Схеме 3. На стадии (a) соединение формулы 9, где n=2-11, обрабатывают любым основанием, которое обычно используется для превращения спирта в алкоголят, таким как гидрид натрия или затрудненный алкоголят щелочного металла, такой как изопропилат натрия. Затем добавляют соединение формулы 10, где R6 обозначает алкил, имеющий 1 до 12 атомов углерода. Селективность реакции для алкилирования только одного из гидроксилов соединения формулы 9 может быть увеличена при использовании стехиометрического избытка соединения формулы 9.

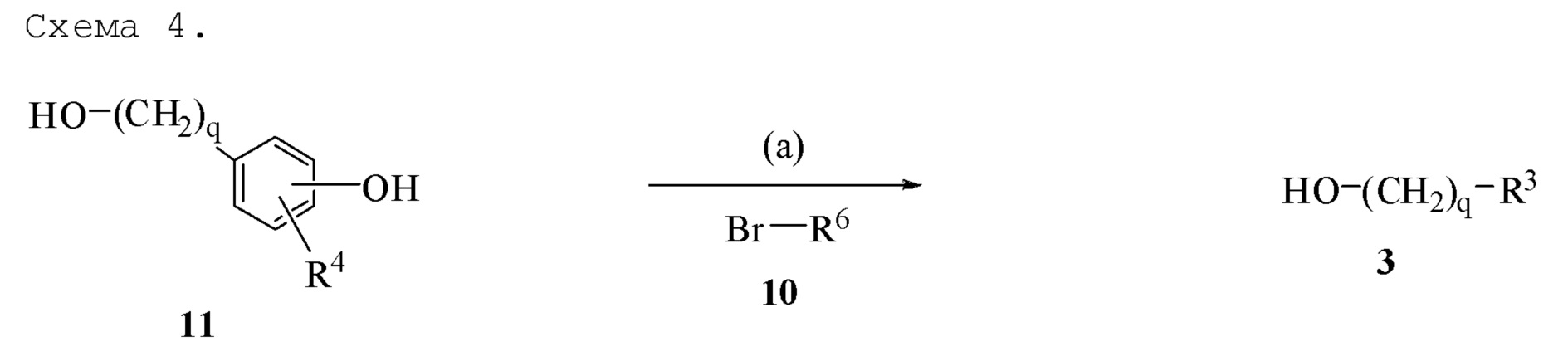
Соединение формулы 3, где q=0 или 1, и R3 обозначает фенил, замещенный галогеном, алкокси, имеющим от 1 до 10 атомов углерода, незамещенным или замещенным фенилом или фенокси, может быть получено из соединения формулы 11, где q=0 или 1, и R4 обозначает водород или галоген, через реакционную схему на Схеме 4. Соединение формулы 11 обрабатывают подходящим основанием, таким как карбонат калия или карбонат натрия, и вводят в реакцию с соединением формулы 10, где R6 обозначает алкил, имеющий от 1 до 10 атомов углерода, незамещенный или замещенный фенилом или фенокси. Используя карбонатные основания с соединением формулы 11, в котором q=1, ароматический гидроксил вводят селективно в реакцию с соединением формулы 10, несмотря на наличие алифатического гидроксила. Если n=0, то использование стехиометрического избытка соединения формулы 11 минимизирует количество диалкилированного побочного продукта.
Соединение формулы 6, где n=0, p=0, q=0, и R3 обозначает фенил, незамещенный или замещенный галогеном, C(O)OR6, в котором R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, может быть получено, исходя из соединения формулы 12, где R4 обозначает водород или галоген, и соединения формулы 13, где R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, через реакционную схему на Схеме 5. Соединение формулы 12 может быть коммерчески доступным или может быть получено из карбоновой кислоты с использованием обычных способов. Соединение формулы 14, где R4 обозначает водород или галоген, и R5 обозначает C(O)OR6, в котором R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, получают реакцией соединения формулы 12 с соединением формулы 13 в присутствии основания, такого как пиридин или триэтиламин, через стадию (a). Любой из обычных способов получения карбоновых сложных эфиров из карбоновых кислот или их производных и спиртов может использоваться для получения соединения формулы 14. Если соединение формулы 13 заменено аминным аналогом, то схема реакции приведет к соединению формулы 6, где R3 замещен C(O)NHR6. Соединение формулы 14 восстанавливают, чтобы сформировать соединение формулы 6 каталитическим восстановлением, используя водород и палладий на катализаторе на основе активированного угля через стадию (b). Любой из обычных способов для селективного восстановления нитрогрупп до аминогрупп в присутствии карбоновых сложноэфирных групп может использоваться на стадии (b).

Соединение формулы 6, где n=0, p=0, q=0, и R3 обозначает фенил, незамещенный или замещенный галогеном, NHC(O)R6, в котором R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, может быть получено, исходя из соединения формулы 15, где R4 обозначает водород или галоген, и соединения формулы 16, где R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, через реакционную схему на Схеме 5. Соединение формулы 15 и соединение формулы 16 могут реагировать с образованием соединения формулы 14, где R4 обозначает водород или галоген, и R5 обозначает NHC(O)R6, в котором R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, через реакцию стадии (a) в любых обычных условиях для получения карбоксамидов реакцией аминов с хлорангидридами карбоновой кислоты. Соединение формулы 14 восстанавливают, чтобы сформировать соединение формулы 6 каталитическим восстановлением, используя водород и палладий на катализаторе на основе активированного угля через стадию (b). Любой из обычных способов восстановления нитрогрупп до аминогрупп может использоваться на стадии (b).

Соединение формулы 6, где n=0, p=0, q=0, и R3 обозначает фенил, незамещенный или замещенный галогеном, алкокси, имеющим от 1 до 12 атомов углерода, незамещенным или замещенным одним фенилом или феноксигруппой, может быть получено, исходя из соединения формулы 17, где R4 обозначает водород или галоген, и соединения формулы 10, где R6 обозначает алкил, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный фенилом или фенокси, через реакционную схему на Схеме 7. Смесь соединения формулы 17 и соединения формулы 10 вводят в реакцию в присутствии подходящего основания, такого как карбонат калия или карбонат натрия, и подходящего растворителя, такого как диметилформамид, получая соединение формулы 14, где R4 обозначает водород или галоген, и R5 обозначает алкокси, имеющий от 1 до 12 атомов углерода, незамещенный или замещенный одним фенилом или феноксигруппой. Соединение формулы 14 восстанавливают, чтобы сформировать соединение формулы 6 каталитическим восстановлением, используя водород и палладий на катализаторе на основе активированного угля, через стадию (b). Любой из обычных способов восстановления нитрогрупп до аминогрупп может использоваться на стадии (b).

Соединение формулы 6, где n=0, p=0, q=1, и R3 обозначает либо фенил, либо моноциклическое или бициклическое ароматическое кольцо, имеющее один или два атома азота, которое является незамещенным или замещенным галогеном и: a) алкилом, имеющим от 1 до 12 атомов углерода, b) алкокси, имеющим от 1 до 10 атомов углерода, незамещенным или замещенным одним фенилом или феноксигруппой, c) фенилом, d) фенокси, или e) NHC(O)R6 или C(O)NHR6 или C(O)OR6, где R6 обозначает алкил, имеющий 1 до 6 атомов углерода, может быть получено, исходя из соединения формулы 3, где q=1, и R3 обозначает либо фенил, либо моноциклическое или бициклическое ароматическое кольцо, имеющее один или два атома азота, которое является незамещенным или замещенным галогеном и: a) алкилом, имеющим от 1 до 12 атомов углерода, b) алкокси, имеющим от 1 до 10 атомов углерода, незамещенным или замещенным одним фенилом или феноксигруппой, c) фенилом, d) фенокси, или e) NHC(O)R6 или C(O)NHR6 или C(O)OR6, где R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, через реакционную схему на Схеме 8. Соединение формулы 3 превращают в соединение формулы 18 через реакцию стадии (a) обработкой тионил хлоридом. Любой из реактивов и реакций, которые обычно используются для превращения спирта и особенно бензилового спирта в галогенид и особенно бензил галогенид, может использоваться на стадии (a). Альтернативно, соединение формулы 3 превращают в соединение формулы 19 через реакцию стадии (b) обработкой метансульфонил хлоридом и триэтиламином. На стадии (b) любой сульфонилирующий реактив, который традиционно используется для превращения гидроксила в уходящую группу, можно использовать вместо метансульфонил хлорида, и любое подходящее основание может использоваться вместо триэтиламина. Соединение формулы 18 или соединение формулы 19 превращают в соединение формулы 6 через реакции стадии (c), синтез Габриэля первичных аминов. Соединение формулы 18 или соединение формулы 19 вводят в реакцию с фталимидом калия в обычно используемых условиях, получая промежуточное соединение фталимида, которое превращают в соединение формулы 6 в обычно используемых условиях, таких как гидразин моногидрат в этаноле при нагревании с обратным холодильником. Может использоваться любой способ расщепления фталимидов.

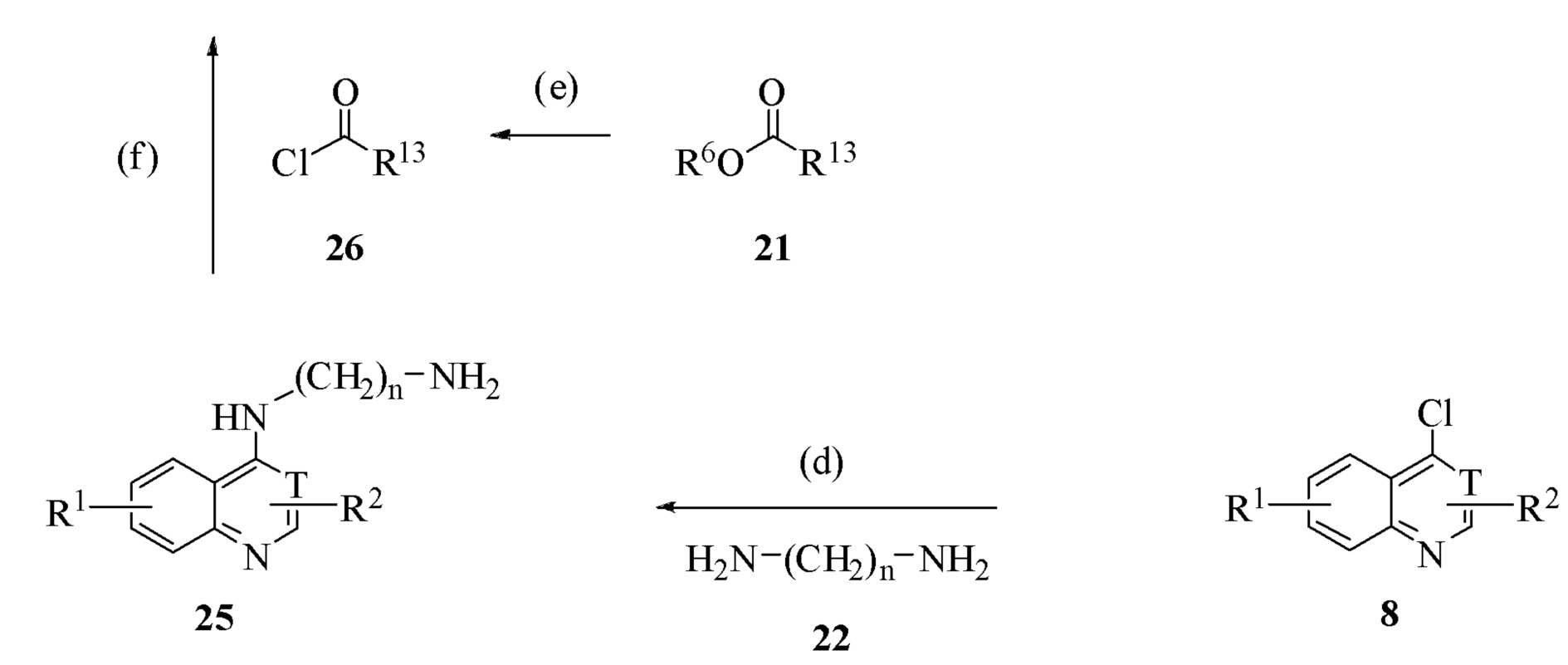
Соединение формулы 24, где T обозначает CH, и R2 присутствует, или T обозначает N, и R2 отсутствует, и в котором n=2, 3, 4, 5, 6, 7 или 8; R1 и R2 обозначают водород; и R13 обозначает фенил, 2-, 3- или 4-пиридил, незамещенный или замещенный: a) алкилом, имеющим от 1 до 12 атомов углерода, незамещенным или замещенным одним фенилом или феноксигруппой, b) алкокси, имеющим от 1 до 12 атомов углерода, незамещенным или замещенным одним фенилом или феноксигруппой, c) фенилом или d) фенокси, может быть получено, исходя из соединения формулы 20, где R6 обозначает алкил, имеющий 1-6 атомов углерода, или, если оно коммерчески доступно, исходя из соединения формулы 21, где R6 обозначает алкил, имеющий 1-6 атомов углерода, и R13 обозначает фенил, 2-, 3- или 4-пиридил, незамещенный или замещенный: a) алкилом, имеющим от 1 до 12 атомов углерода, незамещенным или замещенным одним фенилом или феноксигруппой, b) алкокси, имеющим от 1 до 12 атомов углерода, незамещенным или замещенным одним фенилом или феноксигруппой, c) фенилом или d) фенокси, через реакционную схему на Схеме 9. Соединение формулы 20 вводят в реакцию с соединением формулы 10, где R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, в присутствии подходящего основания, такого как карбонат калия, через реакцию стадии (a). Производное бензойной кислоты соединения формулы 20 может использоваться как исходный материал, также, если используются два эквивалента соединения формулы 10 и два эквивалента подходящего основания. Соединение формулы 21 может быть введено в реакцию с соединением формулы 22, где n=2-8, с получением соединения формулы 23 через реакцию стадии (b). Стадия (b) может быть осуществлена в отсутствие растворителя при температуре 100-130°C. Селективность ацилирования только одной из аминогрупп соединения формулы 22 может быть увеличена при использовании стехиометрического избытка соединения формулы 22. Соединение формулы 23 может быть введено в реакцию с соединением формулы 8 с получением соединения формулы 24 через реакцию стадии (c). Смесь соединения формулы 23 и соединения формулы 7, где T обозначает CH, и R1 и R2 обозначают водород, нагревают в 1-пентаноле с обратным холодильником или диметилформамиде или N-метилпирролидиноне, или их смеси при 130-160°C в присутствии подходящего основания, такого как триэтиламин, трипропиламин, N-метилморфолин или диизопропилэтиламин, получая соединение формулы 24, где T обозначает CH. Смесь соединения формулы 23 и соединения формулы 7, где T обозначает N, и R1 и R2 обозначают водород, нагревают в 2-пропаноле с обратным холодильником в присутствии подходящего основания, такого как триэтиламин или диизопропилэтиламин, получая соединение формулы 24, где T обозначает N. Как альтернативное получение соединения формулы 24, соединение формулы 8, где T обозначает CH и R2присутствует, или T обозначает N, и R2 отсутствует, может быть введено в реакцию с соединением формулы 22, с получением соединения формулы 25, где T обозначает CH, и R2 присутствует, или T обозначает N, и R2 отсутствует, через реакцию стадии (d). Стадию (d) проводят, используя тот же самый растворитель, температуру и основание, как описано для стадии (c). Соединение формулы 21 может быть превращено в соединение формулы 26 через реакции стадии (e). Любой обычный способ превращения карбонового сложного эфира в хлорангидрид карбоновой кислоты может использоваться для стадии (e); например, основное омыление и затем реакция с тионил хлоридом, оксалил хлоридом, фосфорил хлоридом или хлоридом фосфора (V). Соединение формулы 25, где T обозначает CH или N и где R1 и R2 обозначают водород, и соединение формулы 26 могут быть введены в реакцию с получением соединения формулы 24, где T обозначает CH или N, через реакцию стадии (f) с использованием любого из обычных способов для формирования карбоксамидов из хлорангидридов карбоновой кислоты и аминов.
Схема 9.

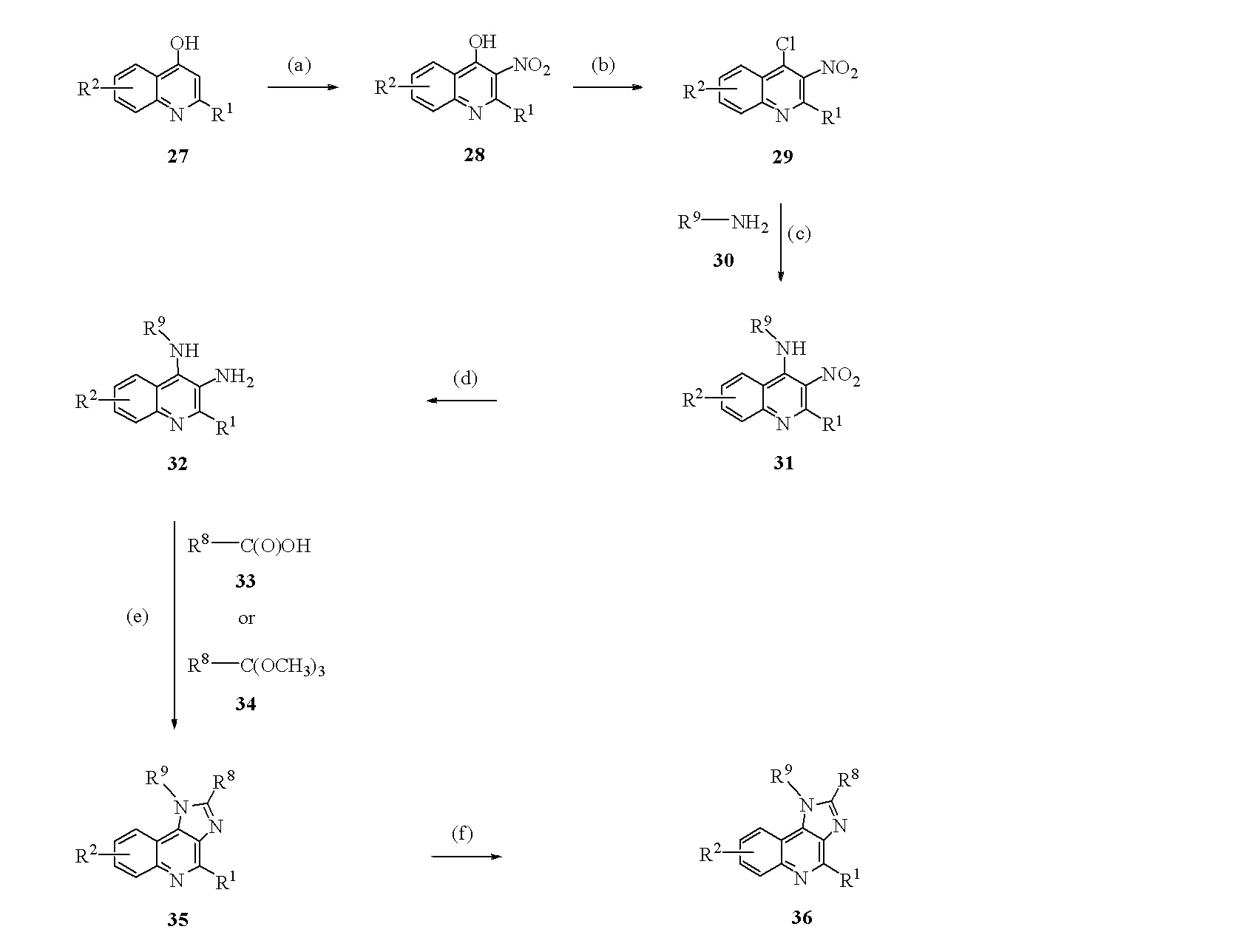
Соединение формулы I, в которой G обозначает имидазохинолил, незамещенный или замещенный на кольцевом углероде галогеном, метилом или перфторметилом; NH отсутствует; R1 обозначает водород, ОН, NH2 или N(CH3)2; и также: a) AQXYZ представлен R8, и R9 обозначает разветвленный или неразветвленный алкил, имеющий от 1 до 16 атомов углерода, незамещенный или замещенный гидрокси или алкокси, имеющим от 1 до 12 атомов углерода, при условии, что если он замещен гидрокси или алкокси, R9 не может иметь 1 атом углерода, или b) AQXYZ представлен R9, и R8 обозначает водород или алкил, имеющий от 1 до 15 атомов углерода, незамещенный или замещенный алкокси, имеющим 1 или 2 атома углерода, или ацетокси, может быть получено, исходя из соединения формулы 27, где R1 обозначает водород или гидрокси, и R2 обозначает водород, галоген, метил или перфторметил, через реакционную схему на Схеме 10. На стадии (a) соединение формулы 27, где R1 обозначает водород или гидрокси, нитрируют, получая соединение формулы 28, используя азотную кислоту в горячей уксусной кислоте или пропионовой кислоте. На стадии (b) соединение формулы 28 обрабатывают хлорирующим средством, таким как фосфорил хлорид, индивидуально или в комбинации с хлоридом фосфора (V), или дихлоридом фенилфосфоновой кислоты, получая соединение формулы 29, где R1 обозначает хлор, если соединение формулы 28 имело гидроксигруппу в качестве R1. На стадии (c) соединение формулы 29 вводят в реакцию с соединением формулы 30 в присутствии третичного аминового основания, такого как триэтиламин, в инертном растворителе, таком как дихлорметан, с мягким нагреванием, получая соединение формулы 31. Из литературы известно, что 4-хлор соединения формулы 29, где R1 обозначает хлор, более реакционоспособен с аминами. Любой из аминов, описанных в изобретении, может использоваться на стадии (c). Было обнаружено, что, если соединение формулы 29, где R1 обозначает хлор, перемешивают с соединением формулы 30 сначала в смеси диметилформамида и дихлорметана, и затем дихлорметан заменяют толуолом, и смесь нагревают с обратным холодильником, формируется соединение формулы 31, где R1 обозначает N(CH3)2. На стадии (d) нитрогруппу соединения формулы 31 восстанавливают любым из множества способов. Если R1 обозначает водород или хлор, гидрирование с использованием 5% или 10% Pd-C или восстановление с использованием цинковой пыли и соляной кислоты приводит к соединению формулы 32, где R1 обозначает водород. Если R1 обозначает хлор, то гидрирование с использованием 10% Pt-C приводит к соединению формулы 32, где R1 обозначает хлор. Если R1 обозначает диметиламино, все эти способы оставляют R1 неизменным. На стадии (e) орто-диамин соединения формулы 32 нагревают с соединением карбоновой кислоты формулы 33 или соединением формулы 34, сложным орто-эфиром соединения 33, получая соединение формулы 35. Может использоваться любой орто-эфирный аналог соединения формулы 33. На стадии (f), если соединение формулы 35, где R1 обозначает хлор, обрабатывают в гидролитических условиях, получают соединение формулы 36, где R1 обозначает гидрокси. На стадии (f), если соединение формулы 35, где R1 обозначает хлор, обрабатывают аммиаком или первичным амином, получают производное R1-амино соединения формулы 36. На стадии (f), если соединение формулы 35, где R1 обозначает хлор, обрабатывают цинковой пылью и соляной кислотой, получают соединение формулы 36, где R1 обозначает водород. Соединение формулы 35, где R1 и R2 и R8 обозначают водород, и R9 является стабильным по отношению к органолитиевым основаниям, может быть введено в реакцию с органолитиевым основанием и затем алкилировано органогалогенидом или альдегидом с получением соединения формулы 36, где R8 содержит производное реактива алкилирования.
Схема 10.
Если соединение формулы 33 или соединение формулы 34, или соединение формулы 37, в котором n=0-12, при условии, что, если p=1, тогда n не должен означать 0 или 1; p=0 или 1; q=0 или 1; R3 выбран из группы, состоящей из: алкила, имеющего от 1 до 10 атомов углерода, незамещенного или замещенного: a) моноциклическим или бициклическим ароматическим кольцом, имеющим один или два атома азота, незамещенным или замещенным алкокси, имеющим от 1 до 6 атомов углерода, или b) алкокси, имеющим от 1 до 6 атомов углерода, при условии, что, если R3 обозначает алкил, замещенный алкокси, тогда алкил должен иметь больше чем 1 атом углерода; и фенила, незамещенного или замещенного галогеном и незамещенного или замещенного: a) алкилом, имеющим от 1 до 6 атомов углерода, b) алкокси, имеющим от 1 до 10 атомов углерода, незамещенным или замещенным фенилом или фенокси, при условии, что когда он замещен фенокси, алкокси должен иметь больше чем один атом углерода, c) фенилом, d) фенокси или e) C(O)OR6, C(O)NHR6, или NHC(O)R6, в которых R6 обозначает алкил, имеющий от 1 до 6 атомов углерода, не доступно коммерчески или как промежуточное соединение синтеза, соединение формулы 5 может быть превращено в соединение формулы 37, и следовательно, в соединение формулы 33, или соединение формулы 5 может быть превращено в соединение формулы 34 через реакцию Пиннера в соответствии со схемой, показанной на Схеме 11. На стадии (a) соединение формулы 5 вводят в реакцию с солью щелочного металла уксусной кислоты, такой как ацетат калия или ацетат натрия, или ацетат лития, в подходящем растворителе, таком как диметилформамид. Затем ацетатный сложный эфир гидролизуют при умеренно основном рН, получая соединение формулы 37. Соединение формулы 37, первичный спирт, может быть окислено до соединения карбоновой кислоты формулы 33 через реакцию стадии (b) с использованием любого из многочисленных подходящих способов для окисления спиртов до кислот, таким как окисление Джонса. Альтернативно, соединение формулы 5 вводят в реакцию с цианидом щелочного металла, таким как цианид натрия или цианид калия, в подходящем растворителе, таком как диметилформамид, с получением соединения формулы 38 через реакцию стадии (c). На стадии (d) соединение формулы 38 обрабатывают спиртом, таким как метанол, и кислотным катализатором, таким как соляная кислота, чтобы сформировать соединение формулы 34.

Соединение формулы ID, в котором R10 обозначает алкил, имеющий от 1 до 8 атомов углерода, незамещенный или замещенный алкокси, имеющим от 1 до 6 атомов углерода, при условии, что если он замещен алкокси, R10 должен иметь больше чем 1 атом углерода; R11 обозначает водород или алкил, имеющий от 1 до 8 атомов углерода, незамещенный или замещенный алкокси, имеющим от 1 до 3 атомов углерода, при условии, что если он замещен алкокси, R11 должен иметь больше чем 1 атом углерода; и X- обозначает противоион, может быть получено в соответствии со схемой, показанной на Схеме 12. Если соединение формулы 41 не является коммерчески доступным, соединение 39, 4-хлорпиридин гидрохлорид, может использоваться для его получения через реакцию стадии (a). Соединение 39 нагревают при 130-140°C в затрудненном спирте, таком как 2-пропанол, в присутствии третичного аминового основания, такого как триэтиламин, с соединением формулы 40, получая соединение формулы 41. Через реакцию стадии (b) соединение формулы 41 вводят в реакцию с алкил сульфонатом, таким как соединение формулы 42, в подходящем растворителе, таком как ацетон, получая соединение формулы ID, где X- обозначает противоион, такой как метансульфонат, йодид, бромид или хлорид. Любой алкил йодидное или алкил бромидное или алкил сульфонатное производное R10 может использоваться в реакции стадии (b).
Схема 12.

Соединение формулы 48, где R12 обозначает алкил, имеющий от 2 до 16 атомов углерода, незамещенный или замещенный алкокси, имеющим от 4 до 6 углерода, может быть получено, исходя из соединения 43, 4-гидрокси-3-нитропиридина, в соответствии со схемой, показанной на Схеме 13. Соединение 43 вводят в реакцию с подходящим галогенирующим реагентом, таким как дихлорид фенилфосфоновой кислоты, получая соединение 44, 4-хлор-3-нитропиридин, через реакцию стадии (a). Соединение 44 вводят в реакцию с соединением формулы 45 в присутствии подходящего основания, такого как триэтиламин, в подходящем растворителе, таком как пиридин, получая соединение формулы 46 через реакцию стадии (b). Любой из аминов, описанных в изобретении, может использоваться на стадии (b). Нитрогруппу соединения формулы 46 восстанавливают до аминогруппы соединения формулы 47 каталитическим гидрированием через реакцию стадии (c). Соединение формулы 47 нагревают в триэтоксиметане, получая соединение формулы 48 через реакцию стадии (d). Используя те же самые стадии (b), (c) и (d), но исходя из коммерчески доступного соединения 49, 2-хлор-3-нитропиридина, получают соединение формулы 52.

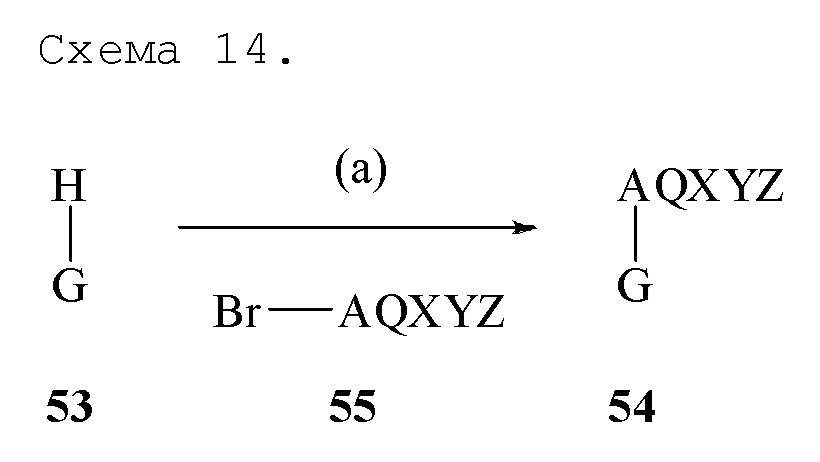
Любое соединение формулы 53, где G обозначает моноциклическое, бициклическое или трициклическое ароматическое кольцо, имеющее один, два или три атомов атома азота в гетероцикле, где атом азота в гетероцикле связан с водородом, может реагировать с соединением формулы 55, где Br-AQXYZ является первичным алкил бромидом, с получением соединения формулы 54, где AQXYZ приведен в пункте 1 формулы изобретения для соединения формулы I, в соответствии со схемой, показанной на Схеме 14. Соединение формулы 53 обрабатывают с сильным основанием, таким как трет-бутоксид натрия, в подходящем растворителе, таком как диметилформамид, и полученный анион амида обрабатывают соединением формулы 55, получая соединение формулы 54 через реакцию стадии (a). Если анион амида находится в резонансе с соседним азотом, алкилирование соединением формулы 55 происходит селективно на менее затрудненном азоте. Первичный алкил йодид, хлорид, алкансульфонат или арилсульфонат AQXYZ может использоваться вместо соединения формулы 55 для реакции стадии (a).
Любое соединение формулы 58, где G обозначает моноциклическое, бициклическое или трициклическое ароматическое кольцо, имеющее один, два или три атома азота в гетероцикле, как определено в пункте 1 формулы изобретения, где кольцевой атом углерода связан с группой NH2, может подвергаться процедуре алкилирования с получением соединения с формулой 59, где A, Q, X, Y и Z имеют значения, определенные в пункте 1 формулы изобретения, исходя из соединения формулы 56, где (AQXYZ) обозначает радикал, который оканчивается группой первичного спирта, в соответствии со схемой, показанной на Схеме 15. Многие соединения формулы 58 доступны коммерчески. Соединение формулы 56, где радикал (AQXYZ) заканчивается функциональной группой первичного спирта и где (AQXYZ) не содержит другую спиртовую группу или аминогруппу, может подвергаться окислению любым из множества обычных способов, таких как окисление Сверна или окисление тетрапропиламмоний перрутенатом/N-метилморфолин N-оксидом, получая соединение формулы 57 через реакцию стадии (a). Соединение формулы 58 может подвергаться гидроалкилированию соединением формулы 57 через реакцию стадии (b) с использованием любого обычного способа для гидроалкилирования амина, такого как цианоборгидрид натрия в тетрагидрофуране. Альтернативно, соединение формулы 58 может подвергаться ацилированию радикалом карбоновой кислоты (AQXYZ) через реакцию стадии (d) с использованием любого обычного способа для формирования амида, такого как конденсация карбодиимида или ацилирование смешанного ангидрида, с использованием изопропил хлорформиата. Кроме того, стадия (d) может быть осуществлена с использованием производного хлорангидрида кислоты соединения формулы 60, которое может быть получено с использованием любого обычного реактива для получения хлорангидридов кислоты, такого как тионил хлорид или оксалил хлорид. Соединение формулы 60 может быть получено из соединения формулы 56 через реакцию стадии (c) с использованием любого подходящего обычного реактива для окисления спиртов, такого как реактив Джонса. Амидная группа соединения формулы 61, где (AQXYZ) не содержит сложный эфир или другую амидную группу, может быть восстановлена до аминогруппы соединения формулы 59 через реакцию стадии (e) с использованием подходящего восстановителя, такого как литий-алюминийгидрид.
Схема 15.

Любое соединение формулы 58, где G обозначает моноциклическое, бициклическое или трициклическое ароматическое кольцо, имеющее один, два или три атома азота в гетероцикле, как определено в пункте 1 формулы изобретения, где кольцевой атом углерода связан с группой NH2, может подвергаться процедуре алкилирования с получением соединения с формулой 59, где A, Q, X, Y и Z имеют значения, определенные в пункте 1 формулы изобретения, исходя из соединения формулы 56, где (AQXYZ) обозначает радикал, который заканчивается группой первичного спирта, в соответствии со схемой, показанной на Схеме 16. Многие соединения формулы 58 доступны коммерчески. Соединение формулы 56, где радикал (AQXYZ) заканчивается функциональной группой первичного спирта и где (AQXYZ) не содержит другой спирт или аминогруппу, может подвергаться реакции сульфонилирования с использованием метансульфонил хлорида и аминного основания, такого как пиридин или триэтиламин, с получением соединения формулы 62 через реакцию стадии (a). Соединение формулы 58 может подвергаться замещающему алкилированию соединением формулы 62 с получением соединения формулы 59 через реакцию стадии (b) с использованием любого обычного способа алкилирования амина, такого как нагревание смеси в тетрагидрофуране или диметилформамиде в отсутствие или в присутствии основания, такого как триэтиламин, диизопропиламин или N-метилморфолин. Аналоги соединения формулы 62, где метансульфонатная группа заменена обычной уходящей группой, такой как йодид, бромид, хлорид или другая сульфонатная группа, могут использоваться на стадии (b).

ПРИМЕНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ
Это изобретение относится к некоторым соединениям, описанным ниже, для лечения заболеваний, характеризующихся патогенными клетками, демонстрирующими лизосомы или другие кислые вакуоли со связанными с заболеванием изменениями, предрасполагающими их к аккумуляции соединений по изобретению, которые затем селективно инактивируют или удаляют такие патогенные клетки. Соединения по изобретению, многие из которых являются производными аминохинолина и аминохиназолина, показывают значительные увеличения потенциала и активности по сравнению с известными аминохинолиновыми лекарственными средствами, такими как хлорохин, как следствие структурных групп, которые эффективно разрушают целостность лизосомальной или вакуолярной мембраны, когда соединения аккумулируются в клетках в кислых вакуолях. Заболевания, которые являются по меньшей мере умеренно чувствительными к противомалярийным производным и аналогам хинолина, в целом более эффективно лечатся соединениями по изобретению. Такие заболевания в общем включают воспалительные заболевания, опухолевые заболевания, включая как гематологический рак, так и солидные опухоли, и инфекции эукариотическими патогенами, включая грибы и несколько классов простейших или других одноклеточных паразитов.
ПРОТИВОВОСПАЛИТЕЛЬНОЕ ПРИМЕНЕНИЕ
Важным действием соединений по изобретению является противовоспалительная активность, позволяющая осуществлять лечение или профилактику заболеваний или симптомов, связанных с чрезмерным воспалением ткани. Это изобретение также относится к композициям, содержащим соединение согласно настоящему изобретению, а также к применению соединения согласно настоящему изобретению для получения лекарственного средства для лечения или профилактики воспалительных заболеваний. Соединения по изобретению показывают селективность в отношении подавления или инактивации макрофагов, которые стимулировались в провоспалительном состоянии, с меньшим действием на нестимулированные макрофаги. Активированные провоспалительные макрофаги участвуют в патогенезе большого разнообразия воспалительных и аутоиммунных заболеваний. Макрофаги являются как антигенпредставляющими клетками, так и эффекторами для повреждения ткани, направляемого аутореактивными Т-клетками, и участвуют в повреждении и дисфункции тканей при заболеваниях, включая, но не ограничиваясь ими, ревматоидный артрит, системную красную волчанку, псориаз, воспалительное заболевание кишечника и атопический дерматит. Воспалительные макрофаги участвуют во многих системных заболеваниях, включая аутоиммунные заболевания, сердечно-сосудистые нарушения и нарушения обмена веществ, и нейродегенеративные состояния. Активированные макрофаги играют первичную роль в повреждении ткани при нестабильности атеросклеротических бляшек, с последовательным риском разрушения и тромбической окклюзии сосуда. Активированные макрофаги в жировой ткани участвуют в метаболических патологиях, включая инсулинорезистентность, диабет типа 2 и другие последствия ожирения. Остеокласты представляют собой макрофагоподобные клетки, которые опосредуют дегенерацию кости при остеопорозе и участвуют в деструкции костей и “костной боли” при раке, возникающем в костях или метастазирующем в кости. Композиции по изобретению могут быть использованы для лечения этих и других нарушений, при которых активированные макрофаги участвуют в патогенезе воспалительного заболевания.
Несколько классов топических средств используются для лечения воспалительных заболеваний кожи, таких как атопический дерматит, экзема или псориаз. Кортикостероиды широко используются, но имеют потенциал как для местной, так и для системной токсичности, особенно при длительном применении. Они могут вызывать местную атрофию или истончение кожи, которая может привести к разрушению кожи, а также телеангиэктазии. Кроме того, топические кортикостероиды могут системно абсорбироваться в количествах, достаточных для того, чтобы вызвать системные побочные эффекты. Второй класс средств для лечения атопического дерматита составляют Т-клеточные иммунодепрессанты, такие как ингибиторы кальцинеурина такролимус и пимекролимус. Их местные и системные иммуносупрессивные эффекты вызывают вопросы об угнетении иммунного надзора рака, включая меланомы и лимфомы.
Аналоги витамина D, особенно кальципотриол, известны как средства для топического лечения псориаза. Кальципотриол действует, ингибируя чрезмерную пролиферацию кератиноцитов. Нанесение на нормальную кожу противопоказано вследствие отбеливающего эффекта, и существует также возможность побочных эффектов вследствие системной абсорбции. Кожное раздражение или зуд известно как побочный эффект кальципотриола. Соединения по изобретению особенно активны против макрофагальных предшественников, которые были активированы экспонированием к действию витамина D3. Возможно, что лечение псориаза кальципотриолом, обеспечивая некоторые улучшения за счет ингибирования пролиферации кератиноцитов, может также направлять местные макрофаги к провоспалительному состоянию, способствуя известным побочным эффектам, таким как раздражение, и ограничивая чистый терапевтический эффект. Способность соединений по изобретению инактивировать примируемые витамином D3 предшественники провоспалительных макрофагов, как показано в нескольких Примерах ниже, показывает, что комбинированное топическое лечение соединениями по изобретению и аналогами витамина D может обеспечить неожиданные преимущества при псориазе и псориатическом дерматите, как в лечении воспалительной эпидермальной гиперпролиферации, так и в уменьшении раздражения или зуда как побочных эффектов аналогов витамина D.
Соединения по изобретению могут быть использованы для лечения воспаления глаз, включая кератит, вызванный инфекцией (грибковой, бактериальной, амебной) или неинфекционными триггерами, такими как повреждение роговицы или контактные линзы. Соединения по изобретению являются особенно подходящими в случае грибкового кератита, противодействуя как инфекционным грибам, так и параллельному воспалительному повреждению. Соединения по изобретению ингибируют ангиогенез роговицы и другие воспалительные изменения в ответ на механическое или химическое повреждение.
Соединения по изобретению могут быть использованы для лечения различных воспалительных или гиперпролиферативных состояний или поражений кожи, включая, но не ограничиваясь ими, экзему, атопический дерматит, псориаз и импетиго. Импетиго представляет собой поверхностную бактериальную инфекцию кожи с воспалительным повреждением эпидермы; соединения по изобретению и подавляют воспаление, и имеют прямые подавляющие или бактерицидные эффекты на грамположительные бактерии, включая, но не ограничиваясь ими, Staphylococcus aureus и Staphylococcus pyogenes, первичные организмы, ответственные за импетиго. Соединения по изобретению также ингибируют предопухолевые и опухолевые изменения кожи, которые часто показывают характеристики как воспаления, так и неоплазии, включая, но не ограничиваясь ими, актиничный кератоз, себорейный кератоз и бородавки.
Примеры E и F демонстрируют эффективность соединений по изобретению для лечения воспаления кожи и псориатического дерматита в стандартных мышиных моделях человеческих нарушений кожи.
Макрофаги и родственные типы клеток участвуют в патогенезе аутоиммунных заболеваний, в которых задействованы адаптивная иммунная система, и как антигенпредставляющие клетки, и как эффекторы разрушения тканей после неадекватной стимуляции Т-клетками, которые секретируют гамма интерферон и другие воспалительные медиаторы, которые рекрутируют и активируют макрофаги. Соединения по изобретению разрывают представление антигена макрофагами и дендритными клетками, а также инактивируют провоспалительные эффекторные макрофаги, которые повреждают ткани. Общий принцип состоит в том, что соединения по изобретению могут быть использованы для лечения хронических или эпизодических аутоиммунных заболеваний, где хлорохин, гидроксихлорохин или другие противомалярийные аналоги хинолина показывают активность у человека или в релевантных моделях животных, и в целом являются более мощными и активными, чем противомалярийные средства, при воспалительных и немалярийных инфекционных заболеваниях. Такие заболевания включают, но не ограничены ими, ревматоидный артрит, системную и дисковидную красную волчанку, псориатический артрит, васкулит, синдром Съегрена, склеродермию, аутоиммунный гепатит и рассеянный склероз.
Синдром активации макрофагов (MAS) представляет собой острое осложнение нескольких аутоиммунных заболеваний, особенно при состояниях, возникающих в детском возрасте, таких как идиопатический ювенильный артрит, где он поражает более 10% пациентов, а также при воспалительных заболеваниях кишечника. При MAS макрофаги сверхактивируются, вызывая повреждения гематопоэтической системы и системное воспаление; MAS иногда летален. Соединения по изобретению могут быть использованы для лечения MAS и опционно доставляться перорально или внутривенной инъекцией или инфузией.
Пример G демонстрирует полезную активность соединений по изобретению при пероральном введении мышам в модели рассеянного склероза, аутоиммунного заболевания.
Для лечения хронических аутоиммунных нарушений, соединения по изобретению вводят системно, предпочтительно перорально. Для лечения острых воспалительных состояний или обострений аутоиммунных заболеваний, внутривенное лечение соединениями по изобретению является дополнительным подходящим путем доставки.
Для перорального или внутривенного лечения аутоиммунных или воспалительных заболеваний, соединения по изобретению обычно вводят в дозах в пределах от 1 до 1000 миллиграмм в сутки, предпочтительно от 100 до 600 миллиграмм в сутки, в единственной дозе или разделенной на две или три дозы в сутки.
ПРОТИВОГРИБКОВЫЕ И ПРОТИВОПАРАЗИТАРНЫЕ ПРИМЕНЕНИЯ
Соединения согласно настоящему изобретению полезны в ингибировании роста грибов, как и in vivo, так и ex vivo. Соответственно, это изобретение также относится к способам и применениям для ингибирования роста грибков у пациента-млекопитающего, например, человека. Эти способы могут использоваться для лечения и профилактики грибковой инфекции. Ex vivo, полезно обрабатывать поверхности соединением согласно настоящему изобретению, чтобы ингибировать или предотвратить рост грибка, или в сельском хозяйстве или садоводстве, чтобы предотвратить или вылечить грибковые заболевания, которые поражают ценные растения. Это изобретение также относится к композициям, содержащим соединение согласно настоящему изобретению, а также к применениям соединения согласно настоящему изобретению для получения лекарственного средства для ингибирования роста грибов.
Это изобретение основано, частично, на открытии, что соединения согласно настоящему изобретению являются эффективными в ингибировании роста разнообразных видов грибов, как показано ниже в примерах биологической активности. Вне связи с теорией, считается, что соединения согласно этому раскрытию используют уязвимость кислой вакуоли грибов. Они, как считается, аккумулируются в кислых вакуолях через захват катионов, и кроме того, проявляют противогрибковую активность, разрывая структуру и функцию кислых вакуолей.
В соответствии с этим изобретением, рост грибов в целом ингибируется. Примеры грибов, которые могут ингибироваться, включают, но не ограничены ими, Candida, Saccharomyces, Trichophyton, Cryptococcus, Aspergillus и Rhizopus. В более конкретных вариантах осуществления этого изобретения грибы представляют собой Candida albicans; Candida glabrata; Saccharomyces cerevisiae; Trichophyton rubrum; Cryptococcus neoformans, например, Cryptococcus neoformans серотипов D и A; и Aspergillus fumigatus.
Это изобретение также относится к способам лечения и профилактики паразитарных инфекций. Вследствие способности соединений по изобретению проникать и аккумулироваться в кислых вакуолях в клетках, они могут быть использованы для лечения инфекций, вызываемых паразитическими микроорганизмами, которые находятся внутри кислых вакуолей в макрофагах и других типах клеток. Туберкулез (микобактерии), листерия или стафилококк (грамположительные бактерии), cryptococcus (грибы) и лейшмании и трипаносомы (амебы), Coxiella burnetii (грамотрицательные бактерии) и плазмодии (некоторые из которых вызывают малярию) являются неограничивающими примерами таких важных инфекционных организмов, для которых нахождение в пределах макрофагов может защитить организмы от клеточного или гуморального иммунитета или снизить эффективность фармакотерапий.
Соединения по изобретению, которые несут липофильные группы и обычно являются частично нейтральными при физиологическом рН (7,3), могут свободно проходить в кислые вакуоли, содержащие паразиты и концентрируются и захватываются там вследствие ионизации в кислой среде (рН 4-6,5). Эти соединения разрывают структуру и функцию кислых вакуолей, являющихся убежищами для паразитов, а также имеют прямую противопаразитарную активность вследствие кислых вакуолей во многих паразитарных организмах.
Паразиты, жизнеспособность или вирулентность которых зависят от целостности и функции кислой вакуоли, также уязвимы для соединений по изобретению, подобно основанию для их противогрибковой активности. Кислая вакуоль малярийных плазмодиев обеспечивает среду для концентрации соединений по изобретению. Точно так же трипаносомы имеют большую кислую вакуоль, которая является необходимой для использования питательных веществ. Соединения по изобретению пригодны для лечения или профилактики малярии и трипаносомных инфекций. Более широко, простейшие паразиты в целом используют подкисленные пищеварительные вакуоли для захвата и переваривания пищи и поэтому могут подвергаться противопаразитарному действию соединений по изобретению.
Сообщалось, что противомалярийное лекарственное средство хлорохин имеет противопаразитарную активность против различных организмов, содержащихся в кислых вакуолях в клетках-хозяевах, или которые имеют кислые вакуоли непосредственно, включая, но не ограничиваясь ими, туберкулезные микобактерии, криптоспоридии, лейшмании и криптококки. В целом, хлорохин действует, аккумулируясь в кислых вакуолях через захват катионов. Активность хлорохина является, таким образом, индикатором вероятной активности соединений по изобретению (многие из которых включают аминохинолин или другой гетероцикл, подобный таковому хлорохина, для нацеливания на кислые вакуоли), с тем отличием, что соединения по изобретению являются в основном более мощными и активными, чем хлорохин, как показано на Cryptococcus neoformans в Примере K, где хлорохин вызвал меньше чем 50%-ое ингибирование роста в концентрации 100 микромоль, тогда как многие соединений по изобретению вызвали 100%-ое ингибирование роста в намного более низких концентрациях. Хлорохин, несмотря на опубликованные отчеты, показывающие, что он может улучшить выживание в животных моделях криптококкоза, показывает потолок приблизительно 40% ингибирования роста C. neoformans in vitro, тогда как соединения по изобретению в основном являются более мощными, чем хлорохин, и могут вызывать 100%-ое ингибирование роста Cryptococcus, вследствие более эффективного разрушения мембран кислых вакуолей, в которых аккумулируются соответствующие лекарственные средства.
Для лечения грибковых или паразитарных инфекций, соединения по изобретению вводят в таких носителях и такими путями введения, которые адекватны природе и локализации инфекции. Для кожной или ногтевой инфекции соединение по изобретению наносят в топическом составе, который может быть лосьоном, мазью, раствором, суспензией или спреем. Для глазных грибковых инфекций, соединения по изобретению составляют в глазных каплях. Для системных инфекций, соединения по изобретению вводят перорально в таблетках, капсулах, драже, растворах или суспензиях, или вводят системно инъекцией в солевом растворе, липидных эмульсиях, липосомах или других стандартных парентеральных носителях. Инфекции легкого, особенно включающие организмы, находящиеся в альвеолярных макрофагах, могут подвергаться лечению ингаляционной доставкой соединений по изобретению и подходящих эксципиентов, известных как приемлемые для ингаляционного применения лекарственного средства. Для внутривенного или перорального введения для лечения системных инфекций, соединения по изобретению вводят в дозах в интервале от 10 до 2000 миллиграмм в сутки, предпочтительно от 200 до 1000 миллиграмм в сутки.
Другие классы противогрибковых средств в клиническом использовании включают ингибиторы синтеза эргостерина (“азольные” противогрибковые средства, включая, но не ограничиваясь ими, флуконазол, кетоконазол, вориконазол, и аллиламины, включая, но не ограничиваясь им, тербинафин), полиеновые противогрибковые средства, который действуют, связываясь с компонентами грибковых мембран, особенно эргостерин (включая, но не ограничиваясь ими, амфотерицин B или нистатин), эхинокандиновые ингибиторы синтеза глуоана (включая, но не ограничиваясь им, каспофунгин), и другие средства, известными в медицинской практике как активные противогрибковые средства. Соединения по изобретению действуют через другой механизм действия по сравнению с существующими клинически важными противогрибковыми средствами, и они могут быть введены совместно с одним или более другими противогрибковыми средствами, чтобы улучшить общее противогрибковое лечение. Соединения по изобретению совместно вводят как отдельные фармацевтические составы, или они могут быть составлены в единый комбинированный лекарственный продукт. Комбинация соединений по изобретению с азольными противогрибковыми средствами особенно предпочтительна для полностью перорального режима для использования против криптококкозов, которое иначе потребовало бы инъекций или инфузий амфотерицина B для начальной индукции. Соединения по изобретению также могут быть введены совместно с амфотерицином B. Один состав амфотерицина B включает его включение в липиды, включающие мембраны липосом. Поскольку многие из соединений по изобретению несут липофильные группы, которые встраиваются в липидные мембраны, их предпочтительно включают в липосомы, либо как единственные средства, либо в комбинации с амфотерицином B или другими известными полиеновыми противогрибковыми средствами.
ПРОТИВОРАКОВЫЕ ПРИМЕНЕНИЯ
Это изобретение относится к соединениям, которые являются полезными для системного лечения рака, основанного на последовательных лизосомальных изменениях, характеризующих инвазивный рак. Лизосомальные изменения при раке, включая их расширение и подкисление, способствуют выживание раковых клеток в кислых внеклеточных средах, а также увеличивает способность раковых клеток к инвазии в окружающие ткани, через экзоцитоз лизосомального содержимого, включая протеазы и полисахаридазы, которые могут разлагать компоненты внеклеточного матрикса. Однако эти стереотипные изменения в лизосомальных свойствах могут сделать раковые клетки уязвимыми для разрывающих лизосомы средств с адекватными физико-химическими свойствами для того, чтобы селективно аккумулироваться в лизосомах и повреждать их в раковых клетках по сравнению с нормальными тканями.
Соединения по изобретению аккумулируются в лизосомах в раковых клетках и нарушают их целостность, таким образом демонстрируя мощную селективную цитотоксическую активность против раковых клеток in vivo и in vitro.
Поскольку одним главным механизмом для резистентности раковой клетки к различным химиотерапевтическим средствам является их изоляция в лизосомах и других кислых вакуолярных камерах, соединения по изобретению могут восстановить или усилить чувствительность раковых клеток к различным классам противораковых средств, включая антиметаболиты, ингибиторы тирозин киназы, противораковые антитела к рецепторам фактора роста, антрациклины, соединения платины, алкилирующие агенты и антитела. Соединения по изобретению обычно не показывают токсичности, перекрывающей ограничивающую дозу токсичность большинства противораковых средств, что позволяет использовать комбинацию соединений по изобретению с другими классами противоопухолевых лекарственных средств с чистым улучшением эффективности и терапевтического индекса.
Раковые клетки, подвергнутые действию сублетальных доз ионизирующего облучения, переносят защитный ответ, который увеличивает их резистентность к последующему облучению. Компонентом этого защитного ответа является формирование увеличенных лизосом или других подкисленных вакуолярных органелл; ингибирование вакуолярной АТФ-азы, ответственной за подкисление лизосом бафиломицином А, препятствует защитному ответу в сублетально облученных клетках и сенсибилизирует раковые клетки к ионизирующему облучению. Повреждение лизосом является значительным медиатором вызываемой облучением гибели в раковых клетках. Разрывая целостность лизосомальных мембран, соединения по изобретению полезны для уменьшения резистентности раковых клеток к терапевтическому ионизирующему облучению и для потенцирования противораковой эффективности ионизирующей радиотерапии. Соединения по изобретению могут быть введены до ионизирующей радиотерапии рака (с внешним облучением или введением нацеливаемых антителами радиоизотопов) как радиосенсибилизаторы, или их можно дать после облучения, чтобы воздействовать на выжившие раковые клетки, переносящие защитные ответы на нелетальное облучение, включающие продукцию или расширение кислых вакуолей.
Одним механизмом, придающим преимущества селективного выживания и пролиферации в некоторых видах рака, является апрегуляция аутофагии, процесса, через который поврежденные органеллы или другой клеточный детрит захватываются аутофагосомами, которые соединяются с лизосомами для переваривания и переработки составляющих молекул. Концентрируясь в лизосомах и разрывая их, соединения по изобретению ослабляют аутофагию в раковых клетках, таким образом уменьшая их жизнеспособность и резистентность к другим противораковым лечениям.
Для лечения рака, соединения по изобретению вводят пероральным или внутривенным введением в дозах от 10 до 2000 миллиграмм в сутки. Соединения по изобретению вводят как единственные средства или в комбинации с другими лечениями рака, адекватными специфическому типу рака, и в целом в дозах, когда такие средства используют индивидуально, поскольку соединения по изобретению обычно не будут иметь перекрывающуюся токсичность с другими классами противораковых средств, что потребовало бы существенного сокращения дозы.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Это изобретение относится к фармацевтической композиции, включающей биологически активное средство, как описано здесь, и фармацевтически приемлемый носитель. Другие варианты фармацевтической композиции согласно настоящему изобретению включают любой из вариантов биологически активных средств, описанных выше. Во избежание ненужного дублирования, каждое такое средство и группы средств не повторяются, но они включены в это описание фармацевтических композиций, как если бы они были повторены.
Предпочтительно композиция адаптирована для перорального введения, например, в форме таблетки, таблетки, покрытой оболочкой, драже, твердой или мягкой желатиновой капсулы, раствора, эмульсии или суспензии. В целом пероральная композиция включает от 10 до 1000 мг соединения согласно настоящему изобретению. Для пациента удобно проглатывать одну или две таблетки, таблетки, покрытых оболочкой, драже или желатиновых капсулы в сутки. Однако композиция может также быть адаптирована для введения любыми другими обычными средствами системного введения, включая ректальное, например, в форме суппозиториев, парентеральное, например, в форме растворов для инъекции, или назальное.
Биологически активные соединения могут быть обработаны с фармацевтически инертными, неорганическими или органическими носителями для получения фармацевтических композиций. Лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли и т.п. могут использоваться, например, в качестве таких носителей для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие многоатомные спирты и т.п. В зависимости от природы активного ингредиента, носители, однако, обычно не требуются в случае мягких желатиновых капсул, кроме непосредственно мягкого желатина. Подходящими носителями для получения растворов и сиропов являются, например, вода, многоатомные спирты, глицерин, растительные масла и т.п. Подходящими носителями для суппозиториев являются, например, натуральные или гидрированные масла, воски, жиры, полужидкие или жидкие многоатомные спирты и т.п.
Фармацевтические композиции могут, кроме того, содержать консерванты, солюбилизаторы, стабилизаторы, смачивающие вещества, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферы, средства для создания покрытий или антиоксиданты. Они могут также содержать другие терапевтически ценные вещества, особенно противовоспалительные или противогрибковые средства (в зависимости от того, нацелено ли лечение на воспалительное заболевание или грибковую инфекцию или рак у пациента), которые действуют через механизмы, отличные от тех, которые лежат в основе эффектов соединений по изобретению.
Для лечения рака, предпочтительные дополнительные лекарственные средства, которые можно использовать совместно или составлять совместно с соединением по изобретению, включают перорально активные противораковые средства. Поскольку соединения по изобретению действуют через уникальный механизм, не разделяемый другими противораковыми лекарственными средствами, они совместимы с большим разнообразием параллельных терапий, включая антиметаболиты, антрациклины, ингибиторы тирозин киназы, платиновые препараты или алкилирующие агенты. Такие средства, когда они являются перорально активными, вводят или составляют для доставки количеств лекарственных средств, определенных в предыдущих клинических испытаниях как эффективные и адекватно переносимые.
Для системного лечения заболеваний, включая некоторые виды рака, воспалительные состояния и грибковые или протозойные инфекции, соединения по изобретению могут вводиться внутривенной инъекцией или инфузией. Для внутривенного введения, соединения по изобретению растворяют в подходящих внутривенных составах как растворы или в липидных эмульсиях, используя стандартные эксципиенты, известные в данной области техники как хорошо переносимые ингредиенты внутривенных составов и композиций. Подходящие объемы и концентрации выбирают для доставки 10-2000 миллиграмм соединений по изобретению в сутки, в зависимости от определенных требований для соединения и болезненного состояния, как определено в клинических испытаниях.
Соединения по изобретению могут быть включены в липосомальные составы. Липофильные группы соединений по изобретению обеспечивают их прямое включение в липидные слои липосом. Липосомы выгодны в некоторых состояниях для внутривенного введения вследствие улучшенной эффективности и более умеренным инфузионным реакциям по сравнению с нелипосомальными составами. Липосомы являются также подходящими для ингаляционной доставки для лечения грибковых или паразитарных инфекций легких, или воспаления легких и дыхательных путей. В некоторых вариантах осуществления, соединения по изобретению включают в липосомальные составы для доставки с другими лекарственными средствами, включая, но не ограничиваясь ими, противогрибковые средства, такие как липосомальный амфотерицин B, или противораковые средства, такие как липосомальный доксорубицин.
Для лечения воспалительных состояний кожи или грибковых инфекций кожи или ногтей, или носовых путей, соединения по изобретению наносят топически в фармацевтически приемлемом составе. Топическая композиция может быть в различных формах, включая, но не ограничиваясь ими, раствор, спрей, гель, гидрогель, лосьон, крем, мазь, пасту или эмульсию в форме жидкой суспензии, лосьона или крема. Композиция может также быть нанесена через кожный пластырь или бандаж, который может быть нанесен на пораженную область, при необходимости, обеспечивая расширенное экспонирование кожи к лечению; в таких составах подходящие стандартные топические эксципиенты лекарственного средства и носители являются подходящими для доставки соединений по изобретению. Стандартные компоненты для топических составов известны в данной области техники и являются подходящими в качестве носителей для соединений по изобретению. Основы мази могут включать один или более углеводородов (парафин, мягкий парафин, микрокристаллический воск или церезин), абсорбционные основы (ланолин или пчелиный воск), макроголи (полиэтиленгликоль) или растительные масла. Лосьоны и кремы представляют собой эмульсии вода в масле или масло в воде; масляные компоненты могут включать длинноцепочечные жирные кислоты, спирты или сложные эфиры, и дополнительно содержат биологически совместимые неионогенные поверхностно-активные вещества. Соединения по изобретению включают в топические носители в концентрациях в диапазоне от 0,01% до 5%, предпочтительно от 0,02 до 1%. Соединения по изобретению наносят на поражения кожи от одного до трех раз в сутки на время, зависящее от скорости разрешения состояния.
Для лечения некоторых инфекций легкого, включая грибковые инфекции или паразитов, находящихся в альвеолярных макрофагах, ингаляционные составы соединений по изобретению являются подходящими. Эксципиенты и ингаляционные устройства для использования лекарственного средства известны в данной области техники и могут быть использованы для доставки соединений по изобретению для лечения инфекций легкого, включая криптококкоз и туберкулез.
Соединения по изобретению предпочтительно составляют с другими противогрибковыми или противовоспалительными средствами для топического или системного введения, особенно когда оба лекарственных средства вводят тем же самым путем и по одной и той же схеме. Соединения по изобретению совместимы со стандартными составами и эксципиентами, используемыми для других топических или системных противогрибковых или противовоспалительных средств, включая, но не ограничиваясь ими, мази и таблетки или капсулы. Предпочтительные категории лекарственных средств для комбинации в топических противовоспалительных составах включают кортикостероиды, ингибиторы кальциневрина и аналоги витамина D, и другие средства, известные как имеющие независимую терапевтическую активность в случае воспалительных состояний кожи.
Изобретение будет более понятно со ссылкой на следующие примеры, которые иллюстрируют, но не ограничивают изобретение, описанное здесь.
ПРИМЕРЫ
ПРИМЕРЫ ХИМИЧЕСКОГО СИНТЕЗА
Пример 1: N-[8-(гексилокси)октил]хинолин-4-амин
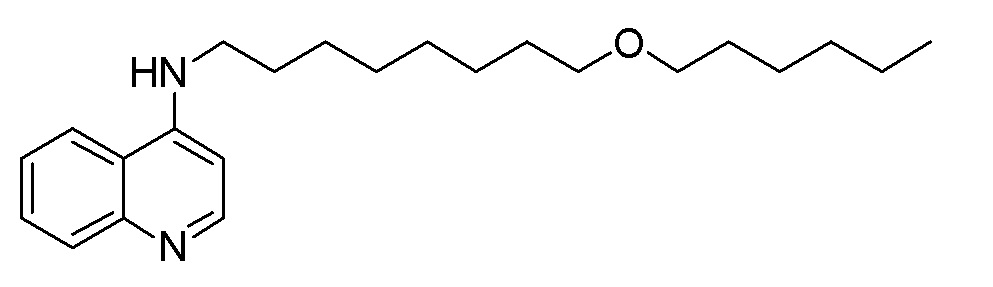
Смесь 4-хлорхинолина (300 мг, 1,84 ммоль), 8-(гексилокси)октан-1-амина (558 мг, 2,44 ммоль) и DMAP (260 мг, 2,13 ммоль) нагревали при 135°C в течение 3 часов. Смесь охлаждали и разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над Na2SO4 и концентрировали. FC (градиент стадии 10%, 12%, 14% MeOH/DCM) дала 279 мг продукта в форме твердого вещества. Rf 0,26 (10% MeOH/DCM); Т.пл. 64,0-65,5°C (из ЕА/Гексан);1H ЯМР (CDCl3) δ 8,51 (д, 1H, J=5,2 Гц), 7,94 (д, 1H, J=8,4 Гц), 7,74 (д, 1H, J=8,4 Гц), 7,57 (м, 1H), 7,37 (м, 1H), 6,37 (д, 1H, J=5,5 Гц), 5,24 (шир. с, 1H, NH), 3,39-3,34 (м, 4H), 3,25 (м, 2H), 1,73-1,26 (м, 20H), 0,84 (м, 3H).
Пример 2: N-(8-Бутоксиоктил)хинолин-4-амин

8-Бутоксиоктан-1-ол 60% Гидрид натрия в минеральном масле (3,5 г, 87,5 ммоль) промывали дважды 20 мл гексанов. Добавляли безводный DMF (300 мл), смесь охлаждали ванной со льдом и добавляли 1,8-октандиол (51,2 г, 351 ммоль). Через 1,5 часа медленно добавляли 1-бромбутан (6 г, 43,8 ммоль). Смесь нагревали до комнатной температуры. Через 24 часа смесь концентрировали. Остаток забирали в Et2O (500 мл) и промывали насыщенным NaHCO3 и H2O (400 мл каждый). Водные фазы экстрагировали Et2O (3×400 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая 3,9 г бесцветного масла. Rf 0,4 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,6 (т, 2H), 3,4-3,3 (м, 4H), 1,6-1,4 (м, 6H), 1,4-1,2 (м, 10H), 0,9 (т, 3H).
8-Бутоксиоктил метансульфонат Смесь 8-бутоксиоктан-1-ола (3,99 г, 20,2 ммоль) и ТЕА (3,4 мл, 24,2 ммоль) в 70 мл DCM охлаждали, используя ванну со льдом. Затем добавляли метансульфонил хлорид (1,87 мл, 24,1 ммоль). Через 2 часа смесь промывали H2O, насыщенным NaHCO3, H2O, 1M HCl и H2O (50 мл каждый). Органическую фазу высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 1,3 г бесцветного масла.
1-Бутокси-8-йодоктан Смесь 8-бутоксиоктил метансульфоната (1,3 г, 6,6 ммоль) и йодида натрия (1,0 г, 6,7 ммоль) в 100 мл ацетона нагревали с обратным холодильником в течение 2 часов. Смесь охлаждали, фильтровали и концентрировали. Остаток забирали в ЕА (400 мл) и промывали насыщенным NaHCO3 и солевым раствором (100 мл каждый). Органическую фазу высушивали над Na2SO4, фильтровали и концентрировали, получая 1,3 г желтой жидкости.
N-(8-Бутоксиоктил)фталимид 1-Бутокси-8-йодоктан (6,2 г, 20,2 ммоль) и фталимид калия (3,73 г, 20,2 ммоль) в 50 мл DMF перемешивали при 60-80°C в течение 12 часов. Охлажденную смесь концентрировали, и остаток разделяли между ЕА (3×300 мл) и 5% Na2S2O3, H2O и солевым раствором (100 мл каждый). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая 5,2 г твердого вещества.1H ЯМР (CDCl3) δ 7,8 и 7,7 (м, 4H, AA’BB’), 3,6 (т, 2H), 3,4-3,3 (м, 4H), 1,7-1,2 (м, 16H), 0,9 (т, 3H).
8-Бутоксиоктан-1-амин Гидразин моногидрат (0,92 мл, 19 ммоль) добавляли к смеси N-(8-бутоксиоктил)фталимида (5,2 г, 15,9 ммоль) и 80 мл EtOH. Смесь нагревали с обратным холодильником в течение 2 часов. Затем смесь охлаждали ванной со льдом и перемешивали энергично, добавляя 200 мл Et2O. Осадок фильтровали и промывали Et2O, и органические фазы концентрировали, получая 3,9 г янтарного масла.1H ЯМР (CD3OD) 3,5-3,4 (м, 4H), 2,9 (т, 2H), 1,7-1,3 (м, 16H), 0,9 (т, 3H).
N-(8-Бутоксиоктил)хинолин-4-амин Смесь 8-бутоксиоктан-1-амина (0,569 мг, 2,89 ммоль), 4-хлорхинолина (710 мг, 4,33 ммоль), ТЕА (5 мл, 36 ммоль) и 0,5 мл NMP закупоривали в толстостенной стеклянной трубке и перемешивали при 130°C в течение 4 дней. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали. Очистка FC (60% ЕА/Гексан +2% ТЕА) дала 244 мг масла.1H ЯМР (CDCl3) δ 8,9 (м, 1H, NH), 8,7 (д, 1H), 8,2-8,1 (м, 2H), 7,6 (м, 1H), 7,4 (м, 1H), 6,4 (д, 1H), 3,5 (м, 2H), 3,4-3,3 (м, 4H), 1,8 (м, 2H), 1,7-1,3 (м, 14H), 0,9 (т, 3H).
Пример 3: N-(8-Метоксиоктил)хинолин-4-амин

8-(Бензилокси)октан-1-ол 60%-ую дисперсию гидрида натрия в минеральном масле (5,38 г, 134 ммоль) промывали гексанами, чтобы удалить масло. Охлаждая ванной со льдом, медленно добавляли смесь 1,8-октандиол (24,49 г, 168 ммоль) в 300 мл DMF. Смеси давали нагреться до комнатной температуры. Через 1 час, смесь бензил хлорида (7,70 мл, 66,7 ммоль) в 30 мл DME добавляли по каплям. Через 2 часа добавляли дополнительное количество бензил хлорида (1,00 мл, 8,7 ммоль), и смесь перемешивали в течение ночи. Затем добавляли 2 мл концентрированного NH4OH. Через 1 час выпаривали летучие компоненты. Остаток забирали в Et2O и трижды промывали 1M HCl и однократно солевым раствором. Органическую фазу высушивали над безводным MgSO4 и упаривали на силикагель. SPE с промывкой 5% ЕА/Гексан и с последующим элюированием 20% ЕА/Гексан дал 12,19 г продукта в форме бесцветного масла. (Элюирование с ЕА дало 12,19 г рекуперируемого 1,8-октандиола после перекристаллизации из ЕА/Гексан.) Rf 0,55 (20% ЕА/Гексан).
[(8-Метоксиоктилокси) метил]бензол 60%-ую дисперсию гидрида натрия в минеральном масле (2,1 г, 52 ммоль) промывали гексанами, чтобы удалить масло. Охлаждая ванной со льдом, медленно добавляли смесь 8-(бензилокси)октан-1-ол (9,9 г, 42 ммоль) в 25 мл DMF. Смеси давали нагреться до комнатной температуры. Через 1 час добавляли диметил сульфат (4,0 мл, 42 ммоль), и смесь перемешивали в течение ночи. Смесь разбавляли Et2O, промывали 1М HCl, дважды 0,1М HCl и солевым раствором, высушивали над MgSO4 и концентрировали. SPE с промывкой 1% ЕА/Гексан и последующим элюированием 10% Et2O/Hex дал 8,63 г продукта в форме масла. Rf 0,62 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,36-7,24 (м, 5H), 4,49 (с, 2H), 3,45 (т, 2H, J=6,7 Гц), 3,35 (т, 2H, J=6,7 Гц), 3,32 (с, 3H), 1,62-1,50 (м, 4H), 1,40-1,25 (м, 8H).
8-Метоксиоктан-1-ол Смесь [(8-метоксиоктилокси)метил]бензола (8,60 г, 34,4 ммоль) и 860 мг 5%-ого Pd-C в 80 мл THF перемешивали в атмосфере водорода в течение 40 часов. Смесь помещали в атмосферу аргона и фильтровали через слой Целита, промывая дополнительным THF. Аликвоту упаривали досуха для спектроскопии. Rf 0,26 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,59 (т, 2H, J=6,7 Гц), 3,33 (т, 2H, J=6,4 Гц), 3,29 (с, 3H), 1,84 (с, 1H, OH), 1,60-1,45 (м, 4H), 1,40-1,25 (м, 8H).
8-Метоксиоктил метансульфонат Смесь 8-метоксиоктан-1-ола (34,3 ммоль) в 100 мл THF охлаждали ванной со льдом. Добавляли метансульфонил хлорид (4,50 мл, 57,5 ммоль) и ТЕА (8,30 мл, 59,2 ммоль), и быстро образовывался белый осадок. Через 2 часа смесь разбавляли ЕА и промывали H2O, насыщенным NaHCO3, солевым раствором, 1M HCl и солевым раствором, и органическую фазу высушивали над MgSO4 и концентрировали. SPE с промывкой 10% ЕА/Гексан и последующим элюированием 30% ЕА/Гексан дал 7,34 г масла, содержащего 8-метоксиоктил метансульфонат и 8-метоксиоктан-1-ол в молярном отношении 9:1, как определено ЯМР. 8-Метоксиоктил метансульфонат имел Rf 0,31 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 4,19 (т, 2H, J=6,7 Гц), 3,34 (т, 2H, J=6,5 Гц), 3,30 (с, 3H), 2,98 (с, 3H), 1,72 (м, 2H), 1,52 (м, 2H), 1,40-1,25 (м, 8H).
N-(8-Метоксиоктил)фталимид 9:1 смесь 8-метоксиоктил метансульфоната и 8-метоксиоктан-1-ола (4,10 г) забирали в 80 мл DMF и добавляли фталимид калия (4,4 г, 24 ммоль). Смесь нагревали при 80-100°C в течение 4 часов. Затем смесь охлаждали, разбавляли ЕА и промывали H2O, дважды 0,1M HCl и солевым раствором. Органическую фазу высушивали над MgSO4 и концентрировали на силикагеле. SPE с элюированием 30% ЕА/Гексан дал 4,32 г продукта в форме твердого вещества. Rf 0,50 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,81 и 7,67 (м, 4H, AA’BB’), 3,64 (т, 2H, J=7,3 Гц), 3,32 (т, 2H, J=6,7 Гц), 3,29 (с, 3H), 1,62 (м, 2H), 1,50 (м, 2H), 1,40-1,20 (м, 8H).
8-Метоксиоктан-1-амин Гидразин моногидрат (1,00 мл, 20,6 ммоль) добавляли к смеси N-(8-метоксиоктил)фталимида (4,32 г, 14,9 ммоль) в 100 мл EtOH, и смесь нагревали с обратным холодильником в течение 6 часов, в течение которых формировался белый осадок. Затем смесь охлаждали, добавляли 4 мл 6M HCl, большую часть летучих компонентов упаривали, добавляли 100 мл 0,1M HCl, и смесь оставляли на 30 мин. Осадок фильтровали и промывали дважды 50 мл 0,1M HCl. Объединенный фильтрат промывали трижды 50 мл Et2O. Фильтрат подщелачивали до рН более 10, добавляя твердый NaOH, охлаждая ванной со льдом. Фильтрат экстрагировали DCM (150 мл, 2×100 мл). Органические фазы высушивали над безводным Na2SO4 и концентрировали, получая 2,17 г масла.1H ЯМР (CDCl3) δ 3,30 (т, 2H, J=6,6 Гц), 3,27 (с, 3H), 2,62 (м, 2H), 1,53-1,24 (м, 12H), 1,41 (с, 2H, NH2).
N-(8-Метоксиоктил)хинолин-4-амин Смесь 4-хлорхинолина (3,00 ммоль), 8-метоксиоктан-1-амина (233 мг, 1,46 ммоль), DIEA (0,52 мл, 3,00) и 4 мл IPA нагревали при 135°C в течение 16 часов в герметизированной трубке. Смесь обрабатывали дополнительным количеством 8-метоксиоктан-1-амина (343 мг, 2,16 ммоль) и нагревали в течение дополнительных 64 часов. Затем смесь обрабатывали дополнительным количеством 8-метоксиоктан-1-амина (140 мг, 0,88 ммоль) и нагревали в течение дополнительных 48 часов. Смесь охлаждали, и летучие компоненты выпаривали. Остаток разделяли между ЕА и 5%-ым Na2CO3, и органические фазы промывали солевым раствором, высушивали над безводным Na2SO4 и концентрировали. Продукт очищали, используя FC, элюируя 10% и затем 15% MeOH/DCM. Содержащие продукт фракции концентрировали, и остаток забирали в DCM, промывали 5%-ым Na2CO3, высушивали над безводным Na2SO4 и упаривали, получая 694 мг продукта в форме твердого вещества. Rf 0,26 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,41 (д, 1H, J=5,7 Гц), 7,93 (м, 1H), 7,52 (м, 1H), 7,30 (м, 1H), 6,33 (д, 1H, J=5,7 Гц), 6,09 (шир. с, 1H, NH), 3,31-3,23 (м, 7H), 1,65, (м, 2H), 1,48 (м, 2H), 1,33-1,25 (м, 8H).
Пример 4: N-[6-(гексилокси)гексил]хинолин-4-амин

6-(Гексилокси)гексан-1-амин получали, исходя из 1,6-гександиола в соответствии со способом получения 10-(гексилокси)декан-1-амин.
6-(Гексилокси)гексан-1-ол Rf 0,16 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,59 (м, 2H), 3,36 (т, 2H, J=6,7 Гц), 3,35 (т, 2H, J=6,8 Гц), 1,87 (с, 1H, OH), 1,56-1,47 (м, 6H), 1,36-1,25 (м, 10H), 0,85 (м, 3H).
6-(гексилокси)гексил метансульфонат Rf 0,16 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 4,21 (т, 2H, J=6,6 Гц), 3,38 (т, 2H, 6,4 Гц), 3,37 (т, 2H, J=6,7 Гц), 2,98 (с, 3H), 1,74 (м, 2H), 1,61-1,46 (м, 4H), 1,40-1,37 (м, 4H), 1,35-1,24 (м, 6H), 0,87 (т, 3H, J=6,8 Гц).
N-[6-(гексилокси)гексил]фталимид Rf 0,40 (20% ЕА/Гексан).
6-(Гексилокси) гексан-1-амин1H ЯМР (CDCl3) δ 3,36 (м, 2H), 3,35 (т, 2H, J=6,8 Гц), 2,67 (м, 2H), 2,10 (шир. с, 2H, NH2), 1,78-1,19 (м, 16H), 0,85 (т, 3H, J=6,8 Гц).
Смесь 6-(гексилокси)гексан-1-амина (234 мг, 1,16 ммоль), 4-хлорхинолина (235 мг, 1,44 ммоль) и ТЕА (0,50 мл, 3,56 ммоль) в 1 мл NMP нагревали при 160°C в течение 16 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали. SPE с промывкой 40% ЕА/Гексан и 4%-ым MeOH/DCM и с элюированием 8% MeOH/DCM дал 137 мг продукта в форме твердого вещества. Rf 0,42 (7,5% MeOH/DCM); Т.пл. 41-44°C (из ЕА/Гексан);1H ЯМР (CDCl3) δ 8,45 (д, 1H, J=5,5 Гц), 7,92 (д, 1H, J=8,4 Гц), 7,86 (д, 1H, J=8,4 Гц), 7,55 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,33 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 6,35 (шир. с, 1H, NH), 3,37-3,22 (м, 6H), 1,72-1,19 (м, 16H), 0,83 (м, 3H).
Пример 5: N-(6-Бутоксигексил)хинолин-4-амин

6-Бутоксигексан-1-ол 60% Гидрид натрия в минеральном масле (3,56 г, 89 ммоль) промывали дважды 20 мл гексанов. Добавляли безводный DMF (250 мл), смесь охлаждали ванной со льдом и добавляли 1,6-гександиол (41,4 г, 351 ммоль). Через 1,5 часа медленно добавляли 1-бромбутан (4,71 мл, 43,7 ммоль). Смесь нагревали до комнатной температуры. Через 24 часа смесь концентрировали. Остаток забирали в Et2O (500 мл) и промывали насыщенным NaHCO3 и H2O (400 мл каждый). Водные фазы экстрагировали Et2O (3×400 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая 6,55 г бесцветного масла. Rf 0,4 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,6 (т, 2H), 3,4-3,3 (м, 4H), 1,6-1,4 (м, 6H), 1,4-1,2 (м, 6H), 0,8 (т, 3H).
6-Бутоксигексил метансульфонат Смесь 6-бутоксигексан-1-ола (6,55 г, 37,6 ммоль) и ТЕА (5,51 мл, 39,5 ммоль) в 100 мл DCM охлаждали, используя ванну со льдом. Затем добавляли метансульфонил хлорид (3,06 мл, 39,5 ммоль). Через 1,5 часа смесь промывали H2O, насыщенным NaHCO3, H2O, 1M HCl и H2O (50 мл каждый). Органическую фазу высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 9,24 г бесцветного масла.1H ЯМР (CDCl3) δ 4,2 (т, 2H), 3,4-3,3 (м, 4H), 2,9 (с, 3H), 1,7 (м, 2H), 1,6-1,2 (м, 10H), 0,8 (т, 3H).
1-Бутокси-6-йодгексан Смесь 6-бутоксигексил метансульфоната (9,23 г, 36,6 ммоль) и йодида натрия (5,5 г, 36,6 ммоль) в 300 мл ацетона нагревали с обратным холодильником в течение 3 часов. Смесь охлаждали, фильтровали и концентрировали. Остаток забирали в ЕА (400 мл) и промывали насыщенным NaHCO3 и солевым раствором (100 мл каждый). Органическую фазу высушивали над Na2SO4, фильтровали и концентрировали, получая 10,4 г желтой жидкости.
N-(6-Бутоксигексил)фталимид 1-Бутокси-6-йодгексан (10,4 г, 36,6 ммоль) и фталимид калия (6,78 г, 36,6 ммоль) в 300 мл DMF перемешивали при 60-80°C в течение 12 часов. Охлажденную смесь концентрировали, и остаток разделяли между ЕА (3×300 мл) и 5% Na2S2O3, H2O и солевым раствором (100 мл каждый). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая 7,2 г твердого вещества.1H ЯМР (CDCl3) δ 7,8 и 7,7 (м, 4H, AA’BB’), 3,6 (т, 2H), 3,4-3,3 (м, 4H), 1,7-1,2 (м, 12H), 0,8 (т, 3H).
6-Бутоксигексан-1-амин Гидразин моногидрат (1,3 мл, 27 ммоль) добавляли к смеси N-(6-бутоксигексил)фталимида (6,72 г, 22,2 ммоль) и 100 мл EtOH. Смесь нагревали с обратным холодильником в течение 16 часов. Затем смесь охлаждали ванной со льдом и перемешивали энергично, добавляя 200 мл Et2O. Осадок фильтровали и промывали Et2O, и органические фазы концентрировали, получая 4,2 г янтарного масла.1H ЯМР (CD3OD) 3,5-3,4 (м, 4H), 2,9 (т, 2H), 1,7-1,3 (м, 12H), 0,9 (т, 3H).
N-(6-Бутоксигексил)хинолин-4-амин Смесь 6-бутоксигексан-1-амина (0,5 г, 2,9 ммоль), 4-хлорхинолина (711 мг, 4,4 ммоль), ТЕА (5 мл, 36 ммоль) и 0,5 мл NMP закупоривали в толстостенной стеклянной трубке и перемешивали при 130°C в течение 4 дней. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали. Очистка FC (60% ЕА/Гексан +2% ТЕА) дала 220 мг янтарного масла.1H ЯМР (CDCl3) δ 8,4 (д, 1H), 8,3-8,1 (м, 3H), 7,6 (м, 1H), 7,4 (м, 1H), 6,4 (д, 1H), 3,5 (м, 2H), 3,4-3,3 (м, 4H), 1,8 (м, 2H), 1,7-1,3 (м, 10H), 0,9 (т, 3H).
Альтернативный Синтез
6-Бутоксигексан-1-ол 60% Дисперсию гидрида натрия в минеральном масле (14 г, 350 ммоль) промывали двумя частями по 50 мл гексана, и затем высушивали в вакууме. Охлаждая ванной со льдом, осторожно добавляли IPA (50 мл) и 1,6-гександиол (200 г, 1700 ммоль), с наблюдаемым выделением газа. Смеси давали нагреться до комнатной температуры и добавляли 1-бромбутан (25,0 мл, 234 ммоль). Смесь нагревали при 45°C в течение 3 дней. Затем добавляли 6,6 мл уксусной кислоты и осуществляли перегонку летучих компонентов до достижения температуры кипения 90°C. Остаток загружали на силикагель. Два раунда SPE (50% ЕА/Гексан) дали 36,7 г светло-желтой жидкости. Rf 0,40 (50% ЕА/Гексан).
6-Бутоксигексил метансульфонат 6-Бутоксигексан-1-ол (36,7 г, 211 ммоль) забирали в 600 мл Et2O, охлажденного ванной со льдом. Добавляли метансульфонил хлорид (19,8 мл, 253 ммоль) и ТЕА (35,5 мл, 253 ммоль), что сопровождалось немедленным формированием осадка. Через 1,5 часа добавляли 100 мл H2O, и фазы разделяли. Водную фазу экстрагировали ЕА (2×150 мл), и органические фазы промывали насыщенным NaHCO3, H2O, 1M HCl, H2O и солевым раствором (100 мл каждый). Органические фазы высушивали над безводным Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 52,2 г светло-желтой жидкости. Rf 0,55;1H ЯМР (CDCl3) δ 4,19 (м, 2H), 3,65-3,34 (м, 4H), 2,97 (с, 3H), 1,72 (м, 2H), 1,56-1,50 (м, 4H), 1,50-1,30 (м, 6H), 0,88 (т, 3H);13C ЯМР (CDCl3) δ 70,8, 70,7, 70,2, 37,4, 32,0, 29,7, 29,2, 25,8, 25,4, 19,5, 14,0.
1-Бутокси-6-йодгексан Смесь 6-бутоксигексил метансульфоната (52,2 г, 207 ммоль) и йодида натрия (40 г, 267 ммоль) в 400 мл ацетона нагревали с обратным холодильником в течение 1 часа. Смесь охлаждали, концентрировали и разделяли между ЕА (3×300 мл) и H2O, 5% Na2S2O3, H2O и солевым раствором (150 мл каждый). Органические фазы высушивали над Na2SO4 и концентрировали, получая продукт в форме желтой жидкости, которая содержала 13 мол.% исходного материала.1H ЯМР (CDCl3) δ 3,38-3,35 (м, 4H), 3,16 (т, 2H, J=7,0 Гц), 1,80 (м, 2H), 1,58-1,48 (м, 4H), 1,40-1,30 (м, 6H), 0,88 (т, 3H, J=7,3 Гц);13C ЯМР (CDCl3) δ 70,8, 70,7, 33,6, 32,0, 30,5, 29,7, 25,3, 19,5, 14,1, 7,2.
N-(6-Бутоксигексил)фталимид Сырой 1-бутокси-6-йодгексан и фталимид калия (46 г, 249 ммоль) в 300 мл DMF перемешивали при комнатной температуре в течение 41 часа и при 60-80°C в течение 24 часов. Охлажденную смесь концентрировали, и остаток разделяли между ЕА (3×350 мл) и H2O, 5% Na2S2O3, H2O и солевым раствором (100 мл каждый). Объединенные органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. SPE (10% ЕА/Гексан) дал 51,6 г бесцветной жидкости. Rf 0,38 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,77 и 7,65 (м, 4H, AA’BB’), 3,62 (т, 2H, J=7,3 Гц), 3,34-3,31 (м, 4H), 1,63 (м, 2H), 1,52-1,44 (м, 4H), 1,35-1,25 (м, 6H), 0,85 (м, 3H);13C ЯМР (CDCl3) δ 168,5, 133,9, 132,3, 123,2, 70,8, 70,7, 38,0, 31,9, 29,7, 28,7, 26,8, 25,9, 19,4, 14,0.
6-Бутоксигексан-1-амин Гидразин моногидрат (9,1 мл, 187 ммоль) добавляли к смеси N-(6-бутоксигексил)фталимида (51,6 г, 170 ммоль) и 900 мл EtOH. Смесь нагревали с обратным холодильником в течение 12 часов и оставляли при комнатной температуре в течение 3 дней. Затем 250 мл летучего материала удаляли перегонкой. 1M HCl (200 мл) добавляли к все еще теплому остатку. После охлаждения до комнатной температуры, осадок удаляли фильтрацией, промывали тремя частями по 200 мл 50%-ого водного раствора EtOH. Фильтрат подщелачивали до рН 10, добавляя гранулы NaOH, концентрировали и забирали в 800 мл DCM. Водную фазу отделяли, и органическую фазу высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой DCM и 5% MeOH/DCM и с элюированием 8% MeOH/DCM +3% NH4OH, дал фракции продукта нингидрина (+). Фракции продукта концентрировали и забирали в DCM. Органическую фазу отделяли, высушивали над безводным Na2SO4 и концентрировали, получая 29,1 г желтой жидкости. Rf 0,09 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 3,26 (т, 2H, J=6,6 Гц), 3,25 (т, 2H, J=6,6 Гц), 2,55 (т, 2H, J=6,9 Гц), 1,46-1,38 (м, 4H), 1,32 (м, 2H), 1,34 (шир. с, 2H, NH2), 1,26-1,20 (м, 6H), 0,78 (т, 3H, J=7,4 Гц);13C ЯМР (CDCl3) δ 70,7, 70,6, 42,1, 33,6, 31,8, 29,7, 26,7, 26,0, 19,3, 13,8.
N-(6-Бутоксигексил)хинолин-4-амин 6-Бутоксигексан-1-амин (6,05 г, 34,6 ммоль) забирали в 150 мл 1-пентанола и 15 мл удаляли перегонкой. Добавляли трипропиламин (15,8 мл, 82,9 ммоль) и 4-хлорхинолин (8,20 г, 50,3 ммоль), и смесь нагревали с обратным холодильником в течение 25 часов и оставляли при комнатной температуре в течение 2 дней. Затем большую часть летучих компонентов упаривали и добавляли 30 мл 1н. NaOH и 60 мл 5%-ого Na2CO3. Смесь экстрагировали DCM (3×150 мл), и органические фазы высушивали над Na2SO4 и упаривали на силикагеле. SPE, с промывкой 50% ЕА/Гексан и с элюированием 5% MeOH/DCM +2% ТЕА, дал коричневое масло. После охлаждения ниже 0°C, масло затвердевало. Твердое вещество промывали холодной смесью 10% ЕА/Гексан и высушивали в вакууме, получая 6,62 г бесцветного твердого вещества. Rf 0,07 (50% ЕА/Гексан) 0,35 (10% MeOH/DCM); Т.пл. 62,5-65,0°C;1H ЯМР (CDCl3) δ 8,52 (д, 1H, J=5,5 Гц), 7,99 (дд, 1H, J=0,7, 8,4 Гц), 7,77 (дд, 1H, J=0,7, 8,4 Гц), 7,62 (ддд, 1H, J=1,5, 7,0, 8,4 Гц), 7,42 (ддд, 1H, J=1,4, 6,9, 8,4 Гц), 6,42 (д, 1H, J=5,5 Гц), 5,26 (шир. с, 1H, NH), 3,41 (т, 2H, J=6,6 Гц), 3,40 (т, 2H, J=6,6 Гц), 3,33 (м, 2H), 1,78 (м, 2H), 1,64-1,31 (м, 10H), 0,91 (т, 3H, J=7,3 Гц);13C ЯМР (CDCl3) δ 150,5, 150,3, 147,8, 129,5, 129,4, 124,9, 119,6, 118,8, 98,9, 70,9, 70,8, 43,4, 32,0, 29,9, 29,1, 27,2, 26,2, 19,6, 14,1.
Пример 6: N-[10-(гексилокси)децил]хинолин-4-амин

10-(Гексилокси)декан-1-ол 60% дисперсию гидрида натрия в минеральном масле (1,08 г, 27 ммоль) промывали гексаном. Добавляли 2-пропанол (150 мл), сначала медленно. Затем добавляли 1,10-деканeдиол (31,3 г, 180 ммоль), и смесь немного нагревали, чтобы достигнуть гомогенности. 1-Бромгексан (2,50 мл, 17,9 ммоль) добавляли по каплям. При перемешивании при комнатной температуре в течение ночи, смесь нагревали с обратным холодильником в течение 2 часов, и затем 100 мл летучих компонентов удаляли перегонкой. Добавляли 1M HCl (10 мл), и затем остаток растворителя удаляли перегонкой. Очистка твердофазной экстракцией с элюированием 12% ЕА/Гексан дала 1,20 г 10-(гексилокси)декан-1-ола в форме бесцветной жидкости. Rf 0,22 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,63 (м, 2H), 3,40-3,35 (м, 4H), 1,65-1,55 (м, 6H), 1,40-1,20 (м, 18H), 0,87 (м, 3H).
10-(Гексилокси)декан-1-амин Метансульфонил хлорид (0,50 мл, 6,39 ммоль) добавляли к смеси 10-(гексилокси)декан-1-ола (1,20 г, 4,65 ммоль) и триэтиламина (0,98 мл, 6,99 ммоль) в 100 мл DME, охлажденного ванной со льдом. Через 1 час смесь разделяли между ЕА (3×100 мл) и H2O, насыщенным NaHCO3, H2O, 0,1M HCl и солевым раствором (50 мл каждый), и органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. Остаток забирали в 150 мл ацетона, добавляли йодид натрия (1,27 г, 8,47 ммоль), и смесь нагревали с обратным холодильником в течение 3 часов. Затем смесь охлаждали, растворитель выпаривали, и остаток разделяли между ЕА (3×100 мл) и 5% Na2S2O3 и H2O (50 мл каждого), и органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. Остаток забирали в 20 мл NMP и добавляли фталимид калия (1,66 г, 8,97 ммоль). После того, как йодид был израсходован, что наблюдается с помощью TLC, смесь разделяли между ЕА (3×100 мл) и 0,1M HCl и солевым раствором (50 мл каждого), и органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. Остаток забирали в 30 мл этанола, добавляли гидразин моногидрат (0,60 мл, 12,5 ммоль), и смесь нагревали с обратным холодильником в течение 8 часов. Затем летучие компоненты выпаривали, остаток разделяли между DCM (3×60 мл) и 5% Na2CO3 (50 мл), и органические фазы высушивали над Na2SO4 и концентрировали, получая 964 мг 10-(гексилокси)декан-1-амина в форме масла, которое затвердевало при отстаивании.1H ЯМР (CD3OD) δ 3,45-3,36 (м, 4H), 2,72 (м, 2H), 1,65-1,45 (м, 6H), 1,45-1,25 (м, 18H), 0,89 (м, 3H).
N-[10-(гексилокси)децил]хинолин-4-амин Смесь 10-(гексилокси)декан-1-амина (256 мг, 1,00 ммоль), 4-хлорхинолина (240 мг, 1,47 ммоль) и частицу гранулированного DMAP в 1,5 мл DIEA нагревали при 150°C в герметизированной трубке в течение 24 часов. Охлажденную смесь разделяли между DCM (3×60 мл) и 5% Na2CO3 (50 мл), и органические фазы высушивали над Na2SO4 и концентрировали. Очистка твердофазной экстракцией, промывка 50% ЕА/Гексан и затем элюирование продукта 50% ЕА/Гексан +2% ТЕА дала 175 мг продукта в форме твердого вещества. Rf 0,42 (50% ЕА/Гексан +0,5% ТЕА);1H ЯМР (CDCl3) δ 8,51 (д, 1H, J=5,2 Гц), 7,94 (дд, 1H, J=1,0, 8,4 Гц), 7,74 (д, 1H, J=8,2 Гц), 7,57 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,36 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 6,37 (д, 1H, J=5,4 Гц), 5,23 (шир. с, 1H, NH), 3,36 (т, 4H, J=6,7 Гц), 3,25 (м, 2H), 1,70 (м, 2H), 1,56-1,26 (м, 22H), 0,85 (м, 3H).
Пример 7: N-(10-Бутоксидецил)хинолин-4-амин
1-Бром-10-бутоксидекан 60% дисперсию гидрида натрия в минеральном масле (1,7 г, 42 ммоль) промывали гексаном. Охлаждая ванной со льдом, добавляли смесь 1-бутанола (10 мл, 109 ммоль) и DMF (40 мл), сначала медленно. После того, как выделение газа прекратилось, смесь 1,10-дибромдекана (47,1 г, 157 ммоль) и 100 мл DCM и 40 мл DMF добавляли в одной части. Смеси давали нагреться до комнатной температуры в течение ночи. Затем DCM выпаривали, и остаток разделяли между ЕА (3×250 мл) и 0,1M HCl и солевым раствором (100 мл каждый), и органические фазы высушивали над Na2SO4 и концентрировали. Очистка SPE, с промывкой Гексаном, чтобы рекуперировать избыток дибромида, и последующим элюированием 10% ЕА/Гексан дала 10,7 г 1-бром-10-бутоксидекана, загрязненного 1,10-дибутоксидеканом. Rf 0,39 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,40-3,36 (м, 6H), 1,82 (м, 2H), 1,57-1,47 (м, 4H), 1,41-1,26 (м, 14H), 0,89 (м, 3H).
10-Бутоксидекан-1-амин Смесь 1-бром-10-бутоксидекана (21,1 г, 72 ммоль) и азида натрия (5,1 г, 78 ммоль) в 30 мл DMF перемешивали при комнатной температуре, пока бромид не был израсходован, что наблюдается с помощью TLC. Смесь разделяли между ЕА (3×350 мл) и H2O (3×100 мл) и солевым раствором (100 мл), и органические фазы высушивали над Na2SO4 и концентрировали. Очистка SPE с использованием 10% ЕА/гексан дала 19,6 г продукта азида. Азид забирали в 40 мл ЕА, и добавляли 40 мл MeOH под аргоном, 2,0 г 5% Pd/C, и смесь перемешивали в атмосфере водорода, пока азид не был израсходован, что наблюдается с помощью TLC. Катализатор удаляли фильтрацией, и летучие компоненты выпаривали. Очистка SPE, с промывкой 50% ЕА/Гексан и последующим элюированием 15%-ым MeOH/DCM +2% ТЕА, дала 7,0 г 10-бутоксидекан-1-амина в форме бесцветного твердого вещества.1H ЯМР (CDCl3) δ 3,40-3,34 (м, 4H), 2,55 (м, 2H), 2,1 (шир. с, 2H, NH2), 1,58-1,26 (м, 20H), 0,90 (м, 3H).
N-(10-Бутоксидецил)хинолин-4-амин Смесь 10-бутоксидекан-1-амина (312 мг, 1,36 ммоль), 4-хлорхинолина (375 мг, 2,30 ммоль) и DIEA (0,50 мл, 2,87 ммоль) в 3 мл 2-пропанола нагревали при 130°C в течение 3 дней и 160°C в течение 1 дня. Летучие компоненты выпаривали. Смесь разделяли между DCM (3×60 мл) и 5% Na2CO3 (50 мл), и органические фазы высушивали над Na2SO4 и концентрировали. Очистка FC на длинной колонке (10% MeOH/DCM) дала N-(10-бутоксидецил)хинолин-4-амин. Rf 0,34 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,52 (д, 1H, J=5,4 Гц), 7,96 (д, 1H, J=8,4 Гц), 7,75 (д, 1H, J=8,4 Гц), 7,60 (дд, 1H, J=7,0, 8,2 Гц), 7,39 (дд, 1H, J=6,9, 8,4 Гц), 6,39 (д, 1H, J=5,2 Гц), 5,20 (шир. с, 1H, NH), 3,41-3,35 (м, 4H), 3,28 (м, 2H), 1,73 (м, 2H), 1,59-1,28 (м, 18H), 0,89 (м, 3H).
Пример 8: N-(5-Метоксипентил)хинолин-4-амин

1-Бром-5-метоксипентан MeOH (20 мл) добавляли по каплям к промытому гексаном гидриду натрия (61,8 ммоль), охлаждая ванной со льдом. Смесь добавляли по каплям к смеси 1,5-дибромпентана (99,44 г, 0,432 моль) и 100 мл 1:1 MeOH и THF. Через 42 часа большую часть растворителя удаляли перегонкой при комнатном давлении. Затем аккуратная перегонка в вакууме дала приблизительно 20 мл жидкости, которая состояла из 1:1 смеси 1,5-дибромпентана и 1-бром-5-метоксипентана. Резервуар разделяли между DCM и H2O, и органическую фазу высушивали над MgSO4 и концентрировали перегонкой при комнатном давлении, получая 96 г 2,1:1 смеси 1,5-дибромпентана и DCM. Дибромид повторно обрабатывали метилатом натрия. Сырые смеси 1-бром-5-метоксипентана объединяли и разделяли SPE, промывая пентаном, получая 1,5-дибромпентан, и элюируя 10% Et2O/пентан, получая 8,40 г бесцветной жидкости после концентрации перегонкой. Rf 0,53 (5% ЕА/Гексан) 0,44 (10% Et2O/Hex);1H ЯМР (CDCl3) δ 3,4-3,3 (м, 4H), 3,31 (с, 3H), 1,86 (м, 2H), 1,6 (м, 2H), 1,3 (м, 2H).
1-Азидо-5-метоксипентан Смесь 1-бром-5-метоксипентана (2,76 г, 15,2 ммоль) и азида натрия (1,14 г, 17,5 ммоль) в 10 мл DMF перемешивали при комнатной температуре в течение 16 часов. Затем смесь разделяли между Et2O (3×70 мл) и H2O (3×50 мл) и солевым раствором. Органические фазы высушивали над Na2SO4, и перемешивание продолжали. Rf 0,36 (10% Et2O/Hex).
5-Метоксипентан-1-амин Смесь 1-азидо-5-метоксипентана в Et2O и 286 мг 5% Pd-C перемешивали в атмосфере водорода в течение 24 часов. Смесь продували аргоном и фильтровали через слой Целита. Большую часть Et2O удаляли перегонкой при атмосферном давлении.1H ЯМР (CDCl3) δ 3,35 (т, 2H), 3,3 (с, 3H), 2,6 (м, 2H), 1,6-1,3 (м, 6H).
N-(5-Метоксипентил)хинолин-4-амин Смесь 5-метоксипентан-1-амина, 4-хлорхинолина (900 мг, 5,52 ммоль) и DIEA (0,50 мл, 2,87 ммоль) нагревали при 130°C в герметизированной трубке в течение 24 часов. Смесь охлаждали и разделяли между ЕА и 5% Na2CO3 и солевым раствором. Органические фазы высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой 40% ЕА/Гексан +2% ТЕА и с элюированием 80% ЕА/Гексан +2% ТЕА, дал твердое вещество. Rf 0,20 (80% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,46 (д, 1H, J=5,2 Гц), 7,90 (дд, 1H, J=1,0, 8,4 Гц), 7,77 (м, 1H), 7,51 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,28 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 6,31 (д, 1H, J=5,4 Гц), 5,55 (м, 1H, NH), 3,30 (т, 2H, J=6,2 Гц), 3,25 (с, 3H), 3,20 (м, 2H), 1,65 (п, 2H, J=7 Гц), 1,57-1,42 (м, 4H).
Пример 9: N-[8-(гексилокси)октил]-2-метилхинолин-4-амин

N-[8-(гексилокси)октил]-2-метилхинолин-4-амин Смесь 8-(гексилокси)октан-1-амина (479 мг, 2,09 ммоль), 4-хлорхиналдина (575 мг, 3,25 ммоль) и DIEA (1,00 мл, 5,74 ммоль) нагревали при 140°C в герметизированной пробирке в течение 4 дней. Затем летучий материал выпаривали, и остаток очищали FC (7% MeOH/DCM), получая 217 мг N-[8-(гексилокси)октил]-2-метилхинолин-4-амина.1H ЯМР (CDCl3) δ 7,87 (д, 1H, J=8,4 Гц), 7,67 (д, 1H, J=8,0 Гц), 7,53 (м, 1H), 7,29 (м, 1H), 6,26 (с, 1H), 5,10 (м, 1H, NH), 3,35 (т, 4H, J=6,5 Гц), 3,21 (м, 2H), 2,57 (с, 3H), 1,73-1,21 (м, 20H), 0,85 (м, 3H).
Пример 10: 7-Хлор-N-[8-(гексилокси)октил]хинолин-4-амин

7-Хлор-N-[8-(гексилокси)октил]хинолин-4-амин Смесь 8-(гексилокси)октан-1-амина (537 мг, 2,34 ммоль), 4,7-дихлорхинолина (565 мг, 2,85 ммоль), DIEA (0,50 мл, 2,87 ммоль) и 1 мл NMP нагревали при 140°C в герметизированной пробирке в течение 24 часов. Затем летучий материал выпаривали, и остаток очищали SPE (5% MeOH/DCM и затем 30% ЕА/Гексан +2% ТЕА), получая 358 мг 7-хлор-N-[8-(гексилокси)октил]хинолин-4-амина. Rf 0,20 (5% MeOH/DCM), 0,31 (30% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,43 (д, 1H, J=5,4 Гц), 7,87 (д, 1H, J=2,0 Гц), 7,68 (д, 1H, J=8,9 Гц), 7,22 (дд, 1H, J=2,2, 8,9 Гц), 6,30 d, 1H, J=5,4 Гц), 5,46 (т, 1H, J=4,8 Гц, NH), 3,33 (т, 4H, J=6,7 Гц), 3,19 (м, 2H), 1,70-1,23 (м, 20H), 0,82 (м, 3H).
Пример 11: 8-Хлор-N-[8-(гексилокси)октил]хинолин-4-амин
8-Хлор-N-[8-(гексилокси)октил]хинолин-4-амин Смесь 8-(гексилокси)октан-1-амина (456 мг, 1,99 ммоль), 4,8-дихлорхинолина (480 мг, 2,42 ммоль), DIEA (0,43 мл, 2,47 ммоль) и 1 мл NMP нагревали при 140°C в герметизированной пробирке в течение 24 часов. Затем летучий материал выпаривали, и остаток очищали SPE (5% MeOH/DCM и затем 30% ЕА/Гексан +2% ТЕА), получая 338 мг 8-хлор-N-[8-(гексилокси)октил]хинолин-4-амин. Rf 0,28 (5% MeOH/DCM), 0,38 (30% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,61 (д, 1H, J=5,5 Гц), 7,72-7,64 (м, 2H), 7,26 (м, 1H), 6,41 (д, 1H, J=5,4 Гц), 5,19 (т, 2H, J=4,7 Гц, NH), 3,38-3,33 (м, 4H), 3,26 (м, 2H), 1,76 (м, 20H), 0,85 (м, 3H).
Пример 12: N-[8-(гексилокси)октил]-7-(трифторметил)хинолин-4-амин

Смесь 8-(гексилокси)октан-1-амина (546 мг, 2,38 ммоль), 4-хлор-7-трифторметилхинолина (711 мг, 3,06 ммоль), DIEA (0,50 мл, 2,87 ммоль) и 1 мл NMP нагревали при 140-150°C в герметизированной пробирке в течение 24 часов. Затем остаток разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, и органические фазы высушивали над Na2SO4 и концентрировали. Очистка SPE не удалась, но FC (25% ЕА/Гексан) дала 626 мг желтого масла, которое затвердевало при отстаивании. Rf 0,10 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,53 (д, 1, J=5,4 Гц), 8,19 (с, 1), 7,87 (д, 1, J=8,9 Гц), 7,47 (дд, 1, J=1,7, 8,9 Гц), 6,42 (д, 1, J=5,5 Гц), 5,47 (м, 1), 3,36-3,32 (м, 4), 3,25 (м, 2), 1,81-1,17 (м, 20), 0,83 (м, 3).
Пример 13: N-[8-(гексилокси)октил]-8-(трифторметил)хинолин-4-амин

N-[8-(гексилокси)октил]-8-(трифторметил)хинолин-4-амин Смесь 8-(гексилокси)октан-1-амина (590 мг, 2,58 ммоль), 4-хлор-8-(трифторметил)хинолина (780 мг, 3,36 ммоль) и DIEA (0,50 мл, 2,86 ммоль) в 1 мл NMP нагревали в толстостенной герметизированной пробирке при 140-150°C в течение 48 часов. Затем остаток разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, и органические фазы высушивали над Na2SO4 и концентрировали. FC (20% ЕА/Гексан) дала 793 мг желтого масла. Rf 0,28 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,60 (д, 1, J=5,4 Гц), 7,94 (д, 1, J=8,6 Гц), 7,91 (д, 1, J=7,4 Гц), 7,35 (м, 1), 6,42 (д, 1, J=5,4 Гц), 5,23 (м, 1, NH), 3,36 (т, 4, J=6,6 Гц), 3,23 (м, 2), 1,74-1,25 (м, 20), 0,85 (м, 3).
Пример 14: N-{5-[3-(Гексилокси)пропокси]пентил}хинолин-4-амин

3-(Гексилокси)пропан-1-ол Один моль металлического натрия добавляли частями к 250 г 1,3-пропандиола, охлажденного ванной со льдом и продутого аргоном. После того, как металл растворился, 0,466 моль 1-йодгексана, смешанного в 100 мл DMF, добавляли по каплям. Смеси давали нагреться до комнатной температуры в течение ночи. Затем смесь нагревали до 60°C в течение 2 часов. Затем смесь охлаждали до комнатной температуры и обрабатывали 10 мл концентрированного NH4OH в течение 1 часа. Затем смесь разделяли между ЕА (3×250 мл) и 1,5 L H2O+H3PO4 (pH~10), H2O, 1M HCl, 2×0,1M HCl и солевым раствором. Органические фазы высушивали над MgSO4 и концентрировали. Очистка SPE с промывкой 10% ЕА/Гексан и элюированием 30% ЕА/Гексан, дала 44,2 г 3-(гексилокси)пропан-1-ол в форме светло-желтой жидкости. Rf 0,28 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,74 (т, 2H), 3,60 (т, 2H, J=5,7 Гц), 3,39 (т, 2H), 2,66 (с, 1H, OH), 1,80 (м, 2H), 1,53 (м, 2H), 1,56-1,20 (м, 6H), 0,85 (м, 3H).
3-(Гексилокси)пропил метансульфонат получали способом, используемым для получения 3-феноксибензил метансульфоната, используя 44,2 г 3-(гексилокси)пропан-1-ол, 43 мл ТЕА и 24 мл метансульфонил хлорида в 540 мл DCM. Сырой материал забирали в 450 мл ацетона и вводили в реакцию с 55,7 г йодида натрия при нагревании с обратным холодильником в течение 4 часов. Затем смесь охлаждали и разбавляли 1 объемом гексанов. Твердое вещество отфильтровывали, и фильтрат концентрировали. Остаток забирали в 350 мл DCM и промывали 5%-ым Na2S2O3 (чтобы удалить окраску) и H2O. Органическую фазу высушивали над Na2SO4 и концентрировали, получая сырой 1-(3-йодпропокси)гексан.
1,5-пентанeдиол (230 мл) продували аргоном и добавляли частями 22,6 г металлического калия. Экзотермическое выделение газа сдерживали, охлаждая ванной со льдом. Затем при комнатной температуре добавляли по каплям смесь сырого 1-(3-йодпропокси)гексана и 100 мл DMA. При перемешивании в течение ночи, непрореагировавший йодид наблюдали с помощью TLC. Гидрид натрия (7,4 г) добавляли 2-граммовыми частями с охлаждением ванной со льдом. Смесь перемешивали при комнатной температуре в течение 60 часов. Затем смесь охлаждали ванной со льдом и нейтрализовали добавлением концентрированного HCl. Смесь разделяли между ЕА и H2O, и органические фазы промывали 5%-ым Na2S2O3 (чтобы удалить окраску) и солевым раствором, высушивали над Na2SO4 и концентрировали. Очистка SPE, с промывкой 5% ЕА/Гексан и последующим элюированием 30% ЕА/Гексан, дала 39,0 г 5-[3-(гексилокси)пропокси]пентан-1-ола в форме бесцветного масла. Rf 0,19 (30% ЕА/Гексан), 0,31 (40% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,60 (т, 2H, J=6,6 Гц), 3,48-3,34 (м, 8H), 1,8 (м, 2H), 1,6-1,5 (м, 4H), 1,5-1,2 (м, 10H), 0,85 (т, 3H, J=6,7 Гц).
5-[3-(Гексилокси)пропокси]пентил метансульфонат (51,0 г) получали способом, используемым для 3-(гексилокси)пропил метансульфоната, используя 39,0 г 5-[3-(гексилокси)пропокси]пентан-1-ола, 24,4 мл ТЕА, 13,6 мл метансульфонил хлорида и 420 мл DCM. Rf 0,38 (40% ЕА/Гексан);1H ЯМР (CDCl3) δ 4,23 (т, 2H, J=6,4 Гц), 3,5-3,3 (м, 8H), 2,98 (с, 3H), 1,8-1,7 (м, 4H), 1,7-1,4 (м, 6H), 1,4-1,2 (м, 6H), 0,9 (т, 3H).
5-Азидопентил-3-(гексилокси)пропиловый эфир (29,3 г) получали реакцией 5-[3-(гексилокси)пропокси]пентил метансульфоната (51 г) и азида натрия (11,3 г) в 80 мл DMF при комнатной температуре согласно способу, используемому для 8-(3-этоксипропокси)октан-1-амина. Rf 0,20 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,4-3,3 (м, 8H), 3,22 (т, 2H), 1,7 (м, 2H), 1,6-1,2 (м, 14H), 0,84 (т, 3H).
5-[3-(Гексилокси)пропокси]пентан-1-амин (26,4 г) получали из 5-азидопентил-3-(гексилокси)пропилового эфира, используя LAH, способом, используемым для получения [4-(гексилокси)фенил]метанамина.1H ЯМР (CDCl3) δ 3,5-3,3 (м, 8H), 2,65 (т, 2H, J=6,4 Гц), 1,8 (м, 2H), 1,7-1,2 (м, 14H), 0,84 (т, 3, J=6,8 Гц).
N-{5-[3-(Гексилокси)пропокси]пентил}хинолин-4-амин Смесь 5-[3-(гексилокси)пропокси]пентан-1-амина (482 мг, 1,97 ммоль), 4-хлорхинолина (345 мг, 2,12 ммоль), DIEA (0,80 мл, 4,59 ммоль) и 2 мл NMP нагревали при 160°C в течение 3 дней в герметизированной пробирке. Затем смесь охлаждали, летучий материал выпаривали, остаток разделяли между DCM и 5%-ым Na2CO3, и органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 50% ЕА/Гексан и последующим элюированием 60% ЕА/Гексан +2% ТЕА, дал 502 мг N-{5-[3-(гексилокси)пропокси]пентил}хинолин-4-амина в форме янтарного масла. Rf 0,20 (60% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,48 (д, 1H, J=5,4 Гц), 7,91 (дд, 1H, 1,2, 8,4 Гц), 7,76 (м, 1H), 7,54 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,32 (ддд, 1H, J=1,2, 6,9, 8,2 Гц), 6,34 (д, 1H, J=5,4 Гц), 5,42 (т, 1H, J=5,0 Гц), 3,46-3,20 (м, 10H), 1,83-1,39 (м, 10H), 1,31-1,15 (м, 6H), 0,81 (м, 3H).
Пример 15: N-{3-[5-(Гексилокси)пентилокси)пропил}хинолин-4-амин
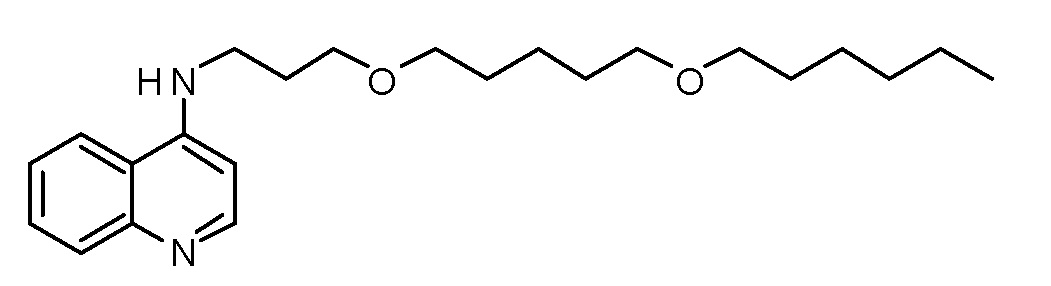
N-{3-[5-(Гексилокси)пентилокси)пропил}хинолин-4-амин (426 мг) получали способом, аналогичным используемому для получения N-{5-[3-(гексилокси)пропокси]пентил}хинолин-4-амина, но оба диола вводили в реакцию в обратной последовательности. Rf 0,18 (60% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,47 (д, 1H, J=5,5 Гц), 7,90 (дд, 1H, J=0,7, 8,4 Гц), 7,70 (м, 1H), 7,54 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,32 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 6,30 (д, 1H, J=5,4 Гц), 6,19 (м, 1H), 3,57 (м, 2H), 3,44-3,24 (м, 8H), 1,96 (м, 2H), 1,86-1,16 (м, 14H), 0,81 (м, 3H).
Пример 16: N-[8-(3-Этоксипропокси)октил]хинолин-4-амин

1-Бром-8-(3-этоксипропокси)октан 60% Дисперсию гидрида натрия в минеральном масле (1,4 г, 35 ммоль) промывали дважды 20 мл гексана. Добавляли безводный NMP (50 мл) и DME (50 мл), смесь охлаждали ванной со льдом и добавляли 3-этокси-1-пропанол (2,00 мл, 17,4 ммоль). После того, как выделение газа прекратилось, 1,8-дибромоктан (25,7 мл, 139 ммоль) добавляли в одной части. Через 16 часов при комнатной температуре, смесь нагревали с обратным холодильником в течение 1,5 часов. Затем летучие компоненты выпаривали, и остаток разбавляли 150 мл H2O и экстрагировали DCM (2×25 мл). Объединенные органические фазы промывали 0,05M HCl, высушивали над безводным MgSO4 и концентрировали. SPE, с промывкой гексаном с получением 1,8-дибромоктана и последующим элюированием 10% ЕА/Гексан, дал 4,15 г 1-бром-8-(3-этоксипропокси)октана. Rf 0,28 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,50-3,31 (м, 10H), 1,88-1,77 (м, 4H), 1,56-1,38 (м, 10H), 1,17 (т, 3H, J=6,9 Гц).
1-Азидо-8-(3-этоксипропокси)октан 1-Бром-8-(3-этоксипропокси)октан (4,15 г, 14,1 ммоль) забирали в 50 мл DMF и добавляли азид натрия (1,09 г, 16,8 ммоль) и каталитический йодид натрия. Через 88 часов смесь разделяли между ЕА (150 мл) и H2O (50 мл), и органическую фазу промывали солевым раствором (50 мл), высушивали над Na2SO4 и концентрировали. FC (5% ЕА/Гексан) дала 2,55 г бесцветной жидкости. Rf 0,37 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,50-3,42 (м, 6H), 3,38 (т, 2H, J=6,7 Гц), 3,24 (т, 2H, J=6,9 Гц), 1,82 (м, 2H), 1,64-1,49 (м, 4H), 1,31 (шир. м, 8H), 1,18 (т, 3H, J=6,9 Гц).
8-(3-Этоксипропокси)октан-1-амин 1-Азидо-8-(3-этоксипропокси)октан (2,55 г, 9,84 ммоль) забирали в 100 мл ЕА. Смесь помещали в атмосферу аргона, добавляли 10% Pd/C (200 мг) и аргон заменяли водородом. Когда исходный материал был израсходован, что наблюдается с помощью TLC, водород заменяли аргоном, и смесь фильтровали через Целит, с промывкой ЕА. Фильтрат концентрировали, получая 1,0 г желтого масла.1H ЯМР (CDCl3) δ 3,6-3,3 (м, 8H), 2,6 (м, 1H), 2,4 (м, 1H), 1,8 (м, 2H), 1,7-1,1 (м, 15H).
N-[8-(3-Этоксипропокси)октил]хинолин-4-амин Смесь 8-(3-этоксипропокси)октан-1-амина (1,0 г, 4,4 ммоль), 4-хлорхинолина (1,46 г, 9,0 ммоль), ТЕА (4,0 мл, 28 ммоль) и 0,2 мл NMP закупоривали в толстостенной стеклянной трубке и перемешивали при 130°C в течение 4 дней. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали. Очистка FC (60% ЕА/Гексан +2% ТЕА) дала 147 мг янтарного масла.1H ЯМР (CDCl3) δ 8,4 (д, 1H), 8,1-7,9 (м, 2H), 7,6 (м, 1H), 7,4 (м, 1H), 6,4 (д, 1H), 6,2 (шир. с, 1H, NH), 3,6-3,3 (м, 10H), 1,9-1,7 (м, 6H), 1,6-1,2 (м, 8H), 1,2 (м, 3H).
Пример 17: N-[8-(2-Пропоксиэтокси)октил]хинолин-4-амин

N-[8-(2-Пропоксиэтокси)октил]хинолин-4-амин (550 мг) получали, используя этиленгликоль монопропиловый эфир (2,00 мл, 17,5 ммоль), 1,8-дибромоктан (25,7 мл, 139 ммоль) и 4-хлорхинолин (1,42 г), с использованием способа для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.
1-Бром-8-(2-пропоксиэтокси)октан: Rf 0,29 (10% ЕА/Гексан); 3,55 (шир. с, 4H, A2B2), 3,46-3,34 (м, 6H), 1,81 (м, 2H), 1,65-1,52 (м, 4H), 1,42-1,30 (м, 8H), 0,88 (т, 3H, J=7,4 Гц).
1-Азидо-8-(2-пропоксиэтокси)октан: Rf 0,37 (10% ЕА/Гексан); 3,55 (шир. с, 4H, A2B2), 3,43 (т, 2H, J=6,7 Гц), 3,40 (т, 2H, J=6,8 Гц), 3,22 (м, 2H, J=6,9 Гц), 1,65-1,52 (м, 6H), 1,29-1,20 (м, 8H), 0,88 (т, 3H, J=7,4 Гц).
N-[8-(2-Пропоксиэтокси)октил]хинолин-4-амин:1H ЯМР (CDCl3) δ 8,3 (м, 2H), 8,1 (д, 1H), 7,6 (м, 1H), 7,4 (м, 2H), 6,4 (д, 1H), 3,55 (шир. с, 4H, A2B2), 3,45-3,35 (м, 6H), 1,8 (м, 2H), 1,6-1,2 (м, 12H), 0,9 (т, 3H).
Пример 18: N-[8-(бензилокси)октил]хинолин-4-амин

8-(Бензилокси)октан-1-амин (880 мг) получали из 8-(бензилокси)октан-1-ола (4,23 г) согласно способу, используемому в получении 10-(гексилокси)декан-1-амина.
Смесь 8-(бензилокси)октан-1-амина (235 мг, 1,00 ммоль), 4-хлорхинолина (201 мг, 1,23 ммоль), DIEA (0,50 мл, 2,87 ммоль) и 2 мл IPA нагревали в толстостенной стеклянной трубке при 150°C в течение 4 дней. Смесь охлаждали и разделяли между DCM и 5%-ым Na2CO3, и органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 3% MeOH/DCM и с элюированием 8% MeOH/DCM, дал 150 мг продукта в форме желтого масла. Rf 0,13 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 8,49 (д, 1H, J=5,4 Гц), 7,97 (д, 1H, J=8,4 Гц), 7,86 (д, 1H, J=8,4 Гц), 7,58 (ддд, 1H, J=1,2, 7,0, 8,5 Гц), 7,40-7,21 (м, 6H), 6,38 (д, 1H, J=5,4 Гц), 5,68 (м, 1H), 4,48 (с, 2H), 3,44 (т, 2H, J=6 Гц), 3,27 (м, 2H), 1,75-1,52 (м, 4H), 1,37-1,32 (м, 8H).
Пример 19: N-(6-Феноксигексил)хинолин-4-амин

N-(6-Феноксигексил)хинолин-4-амин (188 мг) получали, исходя из 1,6-дибромгексана (4,25 мл) и фенола (326 мг), согласно способу, используемому для получения N-(8-феноксиоктил)хинолин-4-амина.
(6-Бромгексилокси)бензол (409 мг): Rf 0,46 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,3 (м, 2H), 6,9 (м, 3H), 4,0 (м, 2H), 3,4 (м, 2H), 2,0-1,7 (м, 4H), 1,6-1,4 (м, 4H).
(6-Азидогексилокси)бензол (344 мг):1H ЯМР (CDCl3) δ 7,3 (м, 2H), 6,9 (м, 3H), 4,0 (м, 2H), 3,28 (т, 2H, J=6,8 Гц), 1,8 (м, 2H), 1,7-1,4 (м, 6H).
6-Феноксигексан-1-амин (224 мг):1H ЯМР (CDCl3) δ 7,3 (м, 2H), 6,9 (м, 3H), 3,91 (т, 2H, J=6,4 Гц), 2,6 (м, 2H), 1,8-1,3 (м, 8H).
N-(6-Феноксигексил)хинолин-4-амин: Rf 0,15 (50% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,53 (д, 1H, J=5,2 Гц), 7,97 (м, 1H), 7,75 (м, 1H), 7,60 (ддд, 1H, J=1,2, 6,9, 8,2 Гц), 7,38 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 7,30-7,22 (м, 2H), 6,95-6,86 (м, 3H), 6,39 (д, 1H, J=5,5 Гц), 5,22 (т, 1H, J=4,7 Гц), 3,94 (т, 2H, J=6 Гц), 3,29 (м, 2H), 1,81-1,44 (м, 8H).
Пример 20: N-(8-Феноксиоктил)хинолин-4-амин
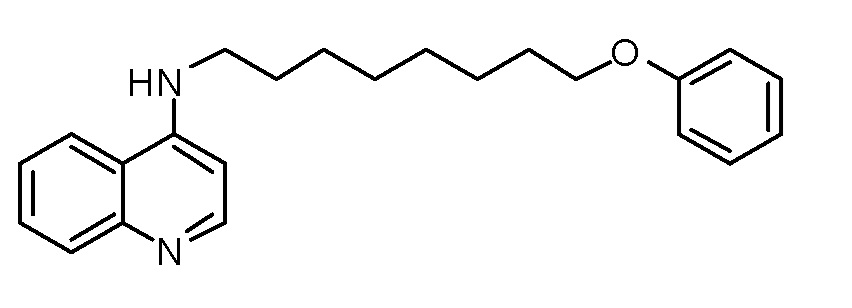
(8-Бромоктилокси)бензол Смесь фенола (321 мг, 3,41 ммоль), 1,8-дибромоктана (5,00 мл, 27,0 ммоль) и K2CO3 (1,41 г, 10,2 ммоль) в 6 мл DMF и 6 мл 1,2-диметоксиэтана нагревали при 90°C в течение 24 часов. Смесь охлаждали и разделяли между простым эфиром (3×175 мл) и 0,1н. NaOH (75 мл) и 1:1 0,1M HCl/солевой раствор (75 мл). Органические фазы высушивали над MgSO4 и концентрировали. Очистка FC (5% ЕА/Гексан) дала 533 мг (8-бромоктилокси)бензола в форме бесцветного масла. Rf 0,50 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,31-7,24 (м, 2H), 6,95-6,88 (м, 3H), 3,95 (т, 2H, J=6,5 Гц), 3,41 (т, 2H, J=6,8 Гц), 1,91-1,73 (м, 4H), 1,47-1,27 (м, 8H).
(8-Азидооктилокси)бензол (460 мг бесцветного масла) и затем 8-феноксиоктан-1-амин (339 мг бесцветного твердого вещества) получали согласно способу, используемому для получения 10-бутоксидекан-1-амина, с использованием 533 мг (8-бромоктилокси)бензола и 170 мг азида натрия.
(8-Азидооктилокси)бензол:1H ЯМР (CDCl3) δ 7,33-7,25 (м, 2H), 6,97-6,88 (м, 3H), 3,96 (м, 2H), 3,26 (т, 2H, J=7,0 Гц), 1,80 (м, 2H), 1,60 (м, 2H), 1,50-1,38 (м, 8H).
8-Феноксиоктан-1-амин:1H ЯМР (CDCl3) δ 7,26-7,20 (м, 2H), 6,91-6,84 (м, 3H), 3,90 (т, 2H, J=6,4 Гц), 2,63 (м, 2H), 1,74 (м, 2H), 1,5-1,2 (м, 10H).
N-(8-Феноксиоктил)хинолин-4-амин Смесь 8-феноксиоктан-1-амина (339 мг, 1,53 ммоль), 4-хлорхинолина (328 мг, 2,01 ммоль) и ТЕА (0,50 мл, 3,56 ммоль) в 1 мл NMP нагревали при 160°C в течение 24 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали. Очистка FC (50% ЕА/Гексан +2% ТЕА) дала 431 мг N-(8-феноксиоктил)хинолин-4-амин. Rf 0,18 (50% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,53 (д, 1H, J=5,4 Гц), 7,97 (дд, 1H, J=1,0, 8,4 Гц), 7,74 (м, 1H), 7,60 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,39 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,30-7,22 (м, 2H), 6,95-6,86 (м, 3H), 6,39 (д, 1H, J=5,4 Гц), 5,17 (шир. с, 1H, NH), 3,93 (т, 2H, J=6,5 Гц), 3,27 (м, 2H), 1,82-1,68 (м, 4H), 1,47-1,40 (м, 8H).
Пример 21: N-{2-[2-(гексилокси)фенокси]этил}хинолин-4-амин

2-[2-(гексилокси)фенокси]этанол Смесь 2-(гексилокси)фенола (9,10 г, 46,9 ммоль), этилен карбоната (6,4 г, 72,7 ммоль) и K2CO3 (10,0 г, 72,5 ммоль) в 50 мл DMF нагревали при 70-75°C в течение 17 часов и затем при 90°C в течение 6 часов. Смесь охлаждали, частично нейтрализовали 1M HCl и разделяли между ЕА и 1M HCl, H2O (2x) и солевым раствором. Органические фазы высушивали над MgSO4, фильтровали через слой силикагеля и концентрировали до коричневого масла. SPE, с промывкой 10% ЕА/Гексан и последующим элюированием 37% ЕА/Гексан, дал 10,73 г светло-желтой жидкости. Rf 0,15 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 6,99-6,94 (м, 2H), 6,92-6,87 (м, 2H), 4,12 (м, 2H), 4,00 (т, 2H), 3,88 (м, 2H), 2,80 (с, 1H, OH), 1,82 (м, 2H), 1,46 (м, 2H), 1,38-1,31 (м, 4H), 0,90 (м, 3H);13C ЯМР (CDCl3) δ 150,2, 148,6, 122,8, 121,3, 117,2, 113,9, 72,5, 69,3, 61,5, 31,8, 29,4, 25,9, 22,8, 14,2.
2-[2-(гексилокси)фенокси]этил метансульфонат Сырой 2-[2-(гексилокси)фенокси]этанол (10,73 г, 45,1 ммоль) забирали в 170 мл 1,2-диметоксиэтана и охлаждали ванной со льдом. Добавляли метансульфонил хлорид (4,90 мл, 62,6 ммоль) и затем ТЕА (9,40 мл 67,0 ммоль). Через 2 часа добавляли 5 мл H2O, и летучие компоненты выпаривали. Остаток разделяли между ЕА и H2O, насыщенным NaHCO3, H2O, 1M HCl, H2O (2x) и солевым раствором. Органические фазы высушивали над MgSO4 и концентрировали, получая 13,67 г бесцветного твердого вещества. Rf 0,37 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 6,99-6,86 (м, 4H), 4,60 (м, 2H), 4,25 (м, 2H), 3,98 (м, 2H), 3,16 (с, 3H), 1,78 (м, 2H), 1,46 (м, 2H), 1,38-1,30 (м, 4H), 0,90 (м, 3H);13C ЯМР (CDCl3) δ 149,7, 147,9, 122,8, 121,1, 115,5, 113,7, 69,1, 69,0, 67,6, 38,1, 31,8, 29,5, 25,9, 22,8, 14,2.
[2-(Гексилокси)этил]фталимид Смесь 2-[2-(гексилокси)фенокси]этил метансульфоната (13,67 г, 43,2 ммоль), фталимида калия (15,5 г, 84 ммоль) и йодида натрия (610 мг) в 50 мл DMF нагревали при 90°C в течение 24 часов. Охлажденную смесь разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали, и остаток фильтровали через слой силикагеля в 30% ЕА/гексан и упаривали, получая твердое вещество. Перекристаллизация из EtOH дала 10,4 г бесцветного твердого вещества.1H ЯМР (CDCl3) δ 7,85 и 7,72 (м, 4H, AA’BB’), 6,94-6,82 (м, 4H), 4,26 и 4,12 (м, 4H, A2B2), 3,88 (т, 2H), 1,71 (м, 2H), 1,42-1,27 (м, 6H), 0,90 (м, 3H);13C ЯМР (CDCl3) δ 168,3, 149,8, 148,6, 134,1, 132,4, 123,5, 122,3, 121,1, 115,6, 114,3, 69,3, 66,4, 37,7, 31,8, 29,4, 25,8, 22,8, 14,2.
2-[2-(гексилокси)фенокси]этанамин N-[2-(Гексилокси)этил]фталимид (10,4 г, 28,3 ммоль) забирали в 130 мл EtOH и добавляли гидразин моногидрат (2,0 мл, 41 ммоль). Смесь нагревали с обратным холодильником в течение 16 часов. После прекращения нагревания, 140 мл 1M HCl добавляли к еще теплой смеси, и смесь перемешивали энергично в течение охлаждения. Осадок фильтровали и промывали EtOH. Фильтрат концентрировали. SPE, с промывкой 7%-ым MeOH/DCM и затем 7% MeOH/DCM +2% ТЕА, дал фракции, содержащие 6,80 г масляно-твердого продукта нингидрина (+). Rf 0,40 (5% MeOH/DCM +2% ТЕА);1H ЯМР (CDCl3) δ 6,94-6,82 (м, 4H), 4,00 (т, 2H, J=5,2 Гц), 3,97 (т, 2H, J=6,7 Гц), 3,05 (т, 2H, J=5,2 Гц), 1,80 (м, 2H), 1,54 (шир. с, 2H, NH2), 1,50-1,28 (м, 6H), 0,89 (м, 3H).
N-{2-[2-(гексилокси)фенокси]этил}хинолин-4-амин Сырой 2-[2-(гексилокси)фенокси]этанамин (6,8 г, 28,7 ммоль) забирали в 30 мл DMA, и 25 мл выпаривали в вакууме. Остаток разбавляли 5 мл NMP и добавляли 4-хлорхинолин (4,20 г, 25,8 ммоль) и DIEA (10,0 мл, ммоль). Смесь нагревали в герметизированной пробирке при 160°C в течение 24 часов. Затем смесь охлаждали, разделяли между ЕА и 5%-ым Na2CO3 (3x) и солевым раствором. Органическую фазу высушивали над Na2SO4 и концентрировали, получая твердое вещество. Растирание с Et2O и высушивание дало 3,11 г бесцветного твердого вещества. Rf 0,31 (10% MeOH/DCM); Т.пл. 104,5-106,0°C;1H ЯМР (CDCl3) δ 8,55 (д, 1H, J=5,5 Гц), 8,04 (м, 1H), 7,85 (д, 1H, J=8,4 Гц), 7,66 (ддд, 1H, J=1,4, 6,9, 8,4 Гц), 7,44 (м, 1H), 7,02-6,97 (м, 2H), 6,95-6,89 (м, 2H), 6,50 (д, 1H, J=5,5 Гц), 6,00 (шир. с, 1H, NH), 4,37 (т, 2H, J=5,1 Гц), 4,02 (т, 2H, J=6,9 Гц), 3,71 (м, 2H), 1,79 (м, 2H), 1,40 (м, 2H), 1,28-1,20 (м, 4H), 0,83 (м, 3H).
Пример 22: N-{3-[2-(гексилокси)фенокси)пропил}хинолин-4-амин
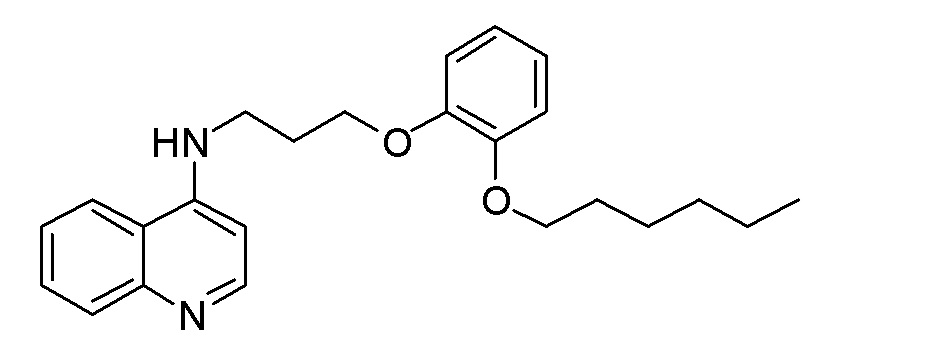
2-(Гексилокси)фенол Смесь катехола (28,9 г, 263 ммоль), K2CO3 (37 г, 268 ммоль) и 1-бромгексана (29,0 мл, 207 ммоль) в 130 мл DMA вводили в реакцию при комнатной температуре в течение 20 часов при помощи механического перемешивания. TLC аликвоты показала наличие существенного количества катехола. Смесь нагревали при 80°C, и TLC аликвоты показала хороший прогресс реакции. Добавляли 1-Бромгексан (5,9 мл, 42 ммоль) и K2CO3 (6 г, 43 ммоль), и нагревание продолжали в течение 10 часов. Затем смесь охлаждали, и большую часть летучих компонентов упаривали. Остаток разделяли между ЕА (3×250 мл) и H2O, 5% Na2CO3 (2x), H2O, 0,1M HCl и солевым раствором (200 мл каждый). Объединенные органические фазы высушивали над MgSO4 и концентрировали. SPE (5% ЕА/Гексан) дал 34,8 г 4:1 смеси 2-(гексилокси)фенола и 1,2-бис-(гексилокси)бензола, как определено 1H ЯМР. Образец очищали SPE, с промывкой Гексаном, получая диэфир, и затем элюируя 2-(гексилокси)фенолом, используя 5% ЕА/гексан. Rf 0,38 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,0-6,8 (м, 4H), 5,7 (с, 1H), 4,0 (т, 2H), 1,9 (м, 2H), 1,5 (м, 2H), 1,4-1,3 (м, 4H), 1,9 (т, 3H).
N-{3-[2-(гексилокси)фенокси)пропил}фталимид Смесь 2-(гексилокси)фенола, которая содержала 1,2-бис-(гексилокси)бензол (чистый на 90 мол.%, 61,8 г), K2CO3 (43,6 г, 316 ммоль) и N-(3-бромпропил)фталимида (76,9 г, 287 ммоль) в 150 мл DMF, нагревали при 60°C в течение 24 часов при помощи механического перемешивания. TLC (5% ЕА, 45% толуол, 50% Гексан) аликвоты показала, что осталось существенное количество исходного бромида, поэтому температуру повышали до 100°C. Через 16 часов реакция была закончена, как показала TLC. Затем смесь охлаждали, и большую часть летучих компонентов упаривали. Остаток разделяли между ЕА (3×250 мл) и H2O, нейтрализовали, используя H3PO4, 0,1M HCl, H2O и солевой раствор (200 мл каждый). Объединенные органические фазы высушивали над MgSO4 и концентрировали, получая 83 г продукта в форме твердого вещества светлого желто-коричневого цвета, которое содержало 2-(гексилокси)фенол и 1,2-бис-(гексилокси)бензол, как показал 1H ЯМР. Rf 0,21 (1:9:10 ЕА/толуол/Гексан) 0,19 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ и 7,71 (м, 4H, AA’BB’), 6,93-6,82 (м, 4H), 4,06 (т, 2H), 3,96-3,88 (м, 4H), 2,19 (м, 2H), 1,76 (м, 2H), 1,46-1,24 (м, 6H), 0,87 (м, 3H).
3-[2-(гексилокси)фенокси)пропан-1-амин Сырой N-{3-[2-(гексилокси)фенокси)пропил}фталимид растворяли в 450 мл теплого IPA и добавляли гидразин моногидрат (24,8 мл, 327 ммоль). Смесь нагревали при 80°C в течение 12 часов при помощи механического перемешивания, и затем смесь оставляли при комнатной температуре на 48 часов. Твердое вещество разбивали, разбавляли 400 мл Et2O и перемешивали в течение 1 часа. Осадок фильтровали и промывали 50% MeOH/Et2O (2×200 мл). Объединенные фильтраты концентрировали, получая 73 г янтарной жидкости. Жидкость забирали в 400 мл DCM и промывали 1н. NaOH и H2O (100 мл каждый). Органическую фазу концентрировали. Смесь разделяли SPE. Элюирование с 1% MeOH/DCM дало 20 г смеси 2-(гексилокси)фенола и 1,2-бис-(гексилокси)бензола. Затем элюирование 7% MeOH/DCM +2% NH4OH дало продукт. Частично концентрированные фракции промывали 200 мл H2O, водную фазу экстрагировали 150 мл DCM, и объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая 33,6 г янтарной жидкости. Rf 0,06 (5% MeOH/DCM, нингидрин (+));1H ЯМР (CDCl3) δ 6,91-6,87 (м, 4H), 4,09 (т, 2H), 3,98 (т, 2H, J=6,6 Гц), 2,93 (т, 2H), 1,95 (кв., 2H), 1,80 (м, 2H), 1,50-1,31 (м, 6H), 0,90 (м, 3H);13C ЯМР (CDCl3) δ 121,5, 121,2, 114,4, 114,1, 69,3, 67,9, 40,0, 33,4, 31,8, 29,5, 25,9, 22,8, 14,2.
N-{3-[2-(гексилокси)фенокси)пропил}хинолин-4-амин 3-[2-(гексилокси)фенокси)пропан-1-амин (28,4 г, 113 ммоль) забирали в 230 мл 1-пентанола, и 70 мл летучего материала удаляли перегонкой, чтобы обеспечить безводные условия. Смеси давали охладиться ниже температуры кипения и добавляли трипропиламин (43 мл, 226 ммоль) и 4-хлорхинолин (23,9 г, 147 ммоль). Нагревание с обратным холодильником возобновляли. Через 15 часов TLC аликвоты не указала наличия исходного нингидрина (+). После перемешивания при комнатной температуре в течение 48 часов, 120 мл летучего материала удаляли перегонкой. Охлажденную смесь разбавляли 350 мл DCM и промывали 2н. NaOH, H2O и 5% Na2CO3 (100 мл каждый). Водные фазы экстрагировали в свою очередь 350 мл DCM. Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали. Очистка FC, с элюированием с градиентом стадии 40, 50 и 60% ЕА/Гексан +2% ТЕА, дала чистые фракции продукта, как показали TLC и ЯМР. Смесь продукта концентрировали, забирали в ЕА, промывали 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали, получая желтое масло. Отстаивание под Et2O и охлаждение с использованием ванны со льдом дали бесцветный осадок. Осадок собирали фильтрацией и промывали ледяным Et2O, получая 33,9 г продукта после высушивания в вакууме. Т.пл. 61,0-62,0°C;1H ЯМР (CDCl3) δ 8,55 (д, 1H, J=5,1 Гц), 7,95 (дд, 1H, J=0,8, 8,5 Гц), 7,84 (дд, 1H, J=1,1, 8,4 Гц), 7,60 (м, 1H), 7,35 (м, 1H), 6,98-6,87 (м, 4H), 6,44 (д, 1H, J=5,5 Гц), 5,98 (т, 1H, J=4,4 Гц, NH), 4,21 (т, 1H, J=5,5 Гц), 4,02 (т, 2H), 3,58 (м, 2H), 2,27 (м, 2H), 1,75 (м, 2H), 1,40 (м, 2H), 1,27-1,21 (м, 4H), 0,84 (м, 3H);13C ЯМР (CDCl3) δ 151,2, 150,1, 149,6, 148,7, 148,6, 130,0, 129,0, 124,5, 122,3, 121,1, 120,2, 119,2, 115,2, 113,8, 98,7, 69,2, 69,2, 42,1, 31,6, 29,3, 28,5, 25,8, 22,7, 14,1.
Пример 23: N-{4-[2-(гексилокси)фенокси]бутил}хинолин-4-амин

N-(4-Бромбутил)фталимид Смесь 1,4-дибромбутана (22 мл, 185 ммоль) и фталимида калия (11,35 г, 61,4 ммоль) в 60 мл DMF перемешивали при комнатной температуре в течение 1 дня. Затем реакционную смесь экстрагировали гексаном (3×150 мл). Фракции гексана высушивали над MgSO4, фильтровали и концентрировали, получая 30 г 1:2,2 молярной смеси рекуперируемых 1,4-дибромбутана и DMF. Эту смесь разбавляли 30 мл DMF и повторно обрабатывали фталимидом калия (4,80 г, 26 ммоль) при комнатной температуре в течение 1 дня. Эти две реакционных смеси в DMF разделяли между 1:1 ЕА/Гексан (3×150 мл) и H2O (2×100 мл), 0,1M HCl (100 мл) и солевым раствором (100 мл). Органические фазы высушивали над MgSO4 и концентрировали. SPE, с элюированием 0% и 10% ЕА/Гексан, дал 17,3 г бесцветного твердого вещества. Rf 0,55 (40% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,86-7,81 (м, 2H), 7,73-7,69 (м, 2H), 3,71 (т, 2H), 3,43 (т, 2H), 1,94-1,80 (м, 4H);13C ЯМР (CDCl3) δ 168,5, 134,2, 132,3, 123,5, 37,2, 32,9, 30,1, 27,4.
N-{4-[2-(гексилокси)фенокси]бутил} фталимид Смесь N-(4-бромбутил)фталимида (17,3 г, 61,3 ммоль), 2-(гексилокси)фенола (14,9 г, 61 ммоль) и K2CO3 (9,5 г, 69 ммоль) в 80 мл DMF нагревали при 80°C в течение 20 часов. Затем смесь охлаждали, разделяли между 40% ЕА/Гексан (3×300 мл) и 0,25M HCl (340 мл), H2O, 0,1M HCl и солевым раствором (150 мл каждый), высушивали над MgSO4, концентрировали, фильтровали через слой силикагеля с 40% ЕА/Гексан и концентрировали, получая 25,7 г твердого вещества светло-желтого цвета.
4-[2-(гексилокси)фенокси]бутан-1-амин Сырой N-{4-[2-(гексилокси)фенокси]бутил}фталимид забирали в 400 мл IPA и добавляли гидразин моногидрат (4,40 мл, 91 ммоль). Смесь нагревали при 80°C в течение 12 часов. Затем смесь охлаждали, приводя к осаждению. Добавляли Et2O (400 мл), и гетерогенную смесь энергично перемешивали. Осадок удаляли фильтрацией через Целит, и осадок промывали Et2O (4×150 мл). Летучие компоненты выпаривали, получая 14,2 г бесцветного твердого вещества.1H ЯМР (CDCl3) δ 6,88-6,83 (м, 4H), 3,98 (т, 2H, J=6,2 Гц), 3,96 (т, 2H, J=6,7 Гц), 2,77 (т, 2H, J=6,9 Гц), 2,17 (шир. с, 2H), 1,89-1,74 (м, 4H), 1,64 (м, 2H), 1,50-1,23 (м, 6H), 0,89 (м, 3H).
N-{4-[2-(гексилокси)фенокси]бутил}хинолин-4-амин Сырой 4-[2-(гексилокси)фенокси]бутан-1-амин (14,2 г, 53,6 ммоль) забирали в 400 мл 1-пентанола, и 100 мл удаляли перегонкой. Смесь охлаждали до температуры ниже температуры кипения и добавляли трипропиламин (15 мл, 78,7 ммоль) и 4-хлорхинолин (8,75 г, 53,7 ммоль). Нагревание с обратным холодильником продолжали в течение 18 часов. Затем смесь концентрировали перегонкой. SPE, с промывкой 50% ЕА/Гексан и последующим элюированием 10%-ым MeOH/DCM, дал после концентрации коричневое масло. Масло забирали в DCM и промывали 5%-ым Na2CO3, высушивали над Na2SO4 и концентрировали. Очистка FC (60% ЕА/Гексан +2% ТЕА), выпаривание растворителей из фракций продукта и затем выпаривание MeOH и высушивание дали 3,7 г продукта в форме бесцветного твердого вещества.1H ЯМР (CDCl3) δ 8,53 (д, 1H, J=5,5 Гц), 7,95 (дд, 1H, J=0,7, 8,4 Гц), 7,74 (м, 1H), 7,59 (ддд, 1H, J=1,1, 7,0, 8,1 Гц), 7,33 (м, 1H), 6,97-6,88 (м, 4H), 6,43 (д, 1H, J=5,2 Гц), 5,63 (т, 1H, NH), 4,11 (т, 1H), 4,00 (т, 2H), 3,49 (м, 2H), 2,01-1,94 (м, 4H), 1,74 (м, 2H), 1,39 (м, 2H), 1,23-1,16 (м, 4H), 0,80 (м, 3H);13C ЯМР (CDCl3) δ 151,3, 150,0, 149,5, 148,8, 148,8, 130,1, 129,1, 124,6, 121,8, 121,1, 119,8, 119,1, 114,4, 113,7, 98,8, 69,2, 69,2, 42,8, 31,7, 29,4, 26,8, 25,9, 25,8, 22,8, 14,1.
Пример 24: N-[3-(2-Этоксифенокси)пропил]хинолин-4-амин

N-[3-(2-Этоксифенокси)пропил]хинолин-4-амин (217 мг) получали согласно способу, используемому для получения N-{3-[4-(гексилокси)фенокси)пропил}хинолин-4-амина, исходя из 2-этоксифенола (1,5 г) и N-(3-бромпропил)фталимида (2,91 г).
N-[3-(2-Этоксифенокси)пропил]фталимид (2,57 г):1H ЯМР (CDCl3) δ 7,85 и 7,75 (м, 4H, AA’BB’), 6,95-6,80 (м, 4H), 4,1-4,0 (м, 4H), 3,9 (т, 2H), 2,2 (м, 2H), 1,4 (т, 3H).
3-(2-Этоксифенокси)пропан-1-амин (0,76 г):1H ЯМР (CDCl3) δ 6,9 (м, 4H), 4,1-4,0 (м, 4H), 2,95 (т, 2H), 1,95 (м, 2H), 1,5 (шир. с, 2H, NH2), 1,4 (т, 3H).
N-[3-(2-Этоксифенокси)пропил]хинолин-4-амин:1H ЯМР (CDCl3) δ 8,8 (шир. с, 1H, NH), 8,5 (м, 1H), 8,4 (м, 1H), 8,2 (д, 1H), 7,7 (м, 1H), 7,5 (м, 1H), 7,0-6,8 (м, 4H), 6,6 (д, 1H), 4,2 (м,2H), 4,1 (м,2H), 3,8 (кв., 2H), 2,4 (м,2H), 1,4 (т, 3H).
Пример 25: N-[3-(2-Метоксифенокси)пропил]хинолин-4-амин

3-(2-Метоксифенокси)пропан-1-амин получали согласно способу, используемому для получения 3-[4-(гексилокси)фенокси)пропан-1-амина, исходя из 2-метоксифенола (1,5 г) и N-(3-бромпропил)фталимида (3,2 г).
N-[3-(2-Метоксифенокси)пропил]фталимид (3,19 г):1H ЯМР (CDCl3) δ 7,8 и 7,7 (м, 4H, AA’BB’), 6,9-6,8 (м, 4H), 4,1 (т, 2H), 3,9 (т, 2H), 3,7 (с, 3H), 2,2 (м, 2H).
3-(2-Метоксифенокси)пропан-1-амин (770 мг):1H ЯМР (CDCl3) δ 6,9-6,8 (м, 4H), 4,1 (т, 2H), 3,8 (с, 3H), 2,9 (т, 2H), 2,0 (м, 2H), 1,5 (шир. с, 2H, NH2).
N-[3-(2-Метоксифенокси)пропил]хинолин-4-амин Смесь 3-(2-метоксифенокси)пропан-1-амина (770 мг, 3,95 ммоль), 4-хлорхинолина (777 мг, 4,77 ммоль), 0,15 мл NMP и 2 мл ТЕА нагревали при 130°C в герметизированной пробирке в течение 5 дней. Затем смесь охлаждали и концентрировали в вакууме. Очистка препаративной TLC (5% MeOH/DCM) дала продукт.1H ЯМР (CDCl3) δ 8,4 (д, 1H), 8,2 (д, 1H), 8,1 (д, 1H), 7,7 (м, 1H), 7,4 (м, 1H), 7,1 (шир. с, 1H, NH), 7,0-6,9 (м, 4H), 6,5 (д, 1H), 4,3 (т, 2H), 3,9 (с, 3H), 3,7 (м, 2H), 2,3 (м, 2H).
Пример 26: N-{3-[2-(бензилокси)фенокси)пропил}хинолин-4-амин
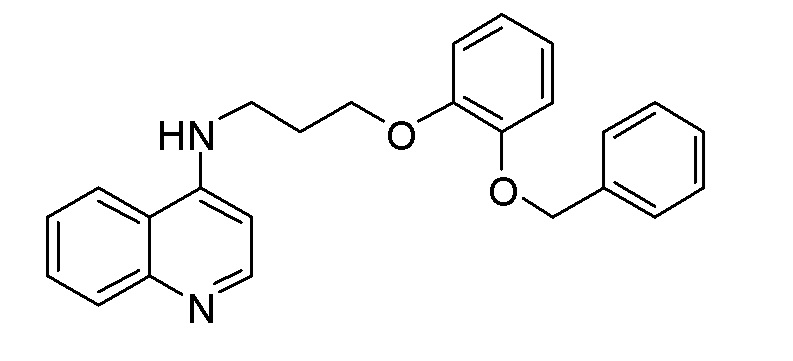
N-{3-[2-(бензилокси)фенокси)пропил}хинолин-4-амин получали согласно способу, используемому для получения N-{3-[4-(гексилокси)фенокси)пропил}хинолин-4-амина, исходя из 2-(бензилокси)фенола (2,0 г) и N-(3-бромпропил)фталимида (2,68 г).
N-{3-[2-(бензилокси)фенокси)пропил}фталимид (3,6 г):1H ЯМР (CDCl3) δ 7,8 и 7,7 (м, 4H, AA’BB’), 7,5-7,3 (м, 4H), 7,0-6,8 (м, 5H), 5,1 (с, 2H), 4,1 (т, 2H), 3,9 (т, 2H), 2,2 (м, 2H).
3-[2-(бензилокси)фенокси)пропан-1-амин (1,92 г):1H ЯМР (CDCl3) δ 7,5-7,3 (м, 5H), 6,9-6,8 (м, 4H), 5,1 (с, 2H), 4,1 (т, 2H), 2,9 (т, 2H), 2,0 (м, 2H).
N-{3-[2-(бензилокси)фенокси)пропил}хинолин-4-амин:1H ЯМР (CDCl3) δ 8,5 (д, 1H), 7,9 (д, 1H), 7,8 (д, 1H), 7,5 (м, 1H), 7,4-7,2 (м, 6H), 7,0-6,9 (м, 4H), 6,4 (д, 1H), 6,0 (шир. с, 1H, NH), 5,1 (с, 2H), 4,2 (т, 2H), 3,6 (м, 2H), 2,3 (м, 2H).
Пример 27: N-[8-(3-Метоксифенокси)октил]хинолин-4-амин
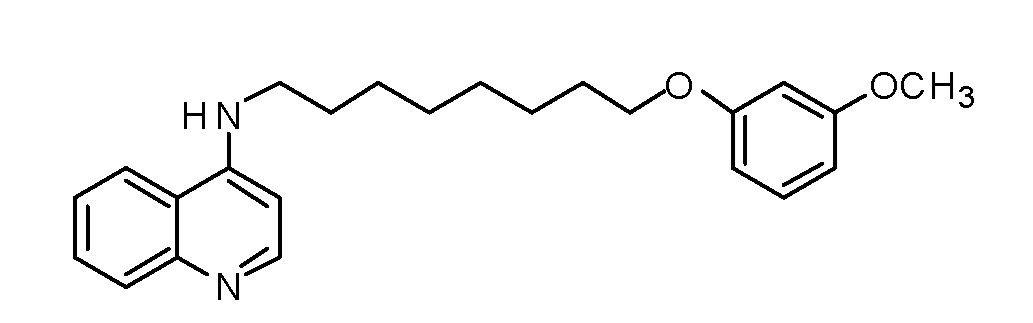
1-(8-Бромоктилокси)-3-метоксибензол (1,28 г) получали тем же самым способом, используемым для 1-(8-бромоктилокси)-3-метилбензола с использованием 3-метоксифенола (638 мг, 5,14 ммоль), 1,8-дибромоктана (14,3 г, 53 ммоль) и K2CO3 (852 мг, 6,17 ммоль) в 14 мл NMP и 7 мл DME, с нагреванием в течение 24 часов.1H ЯМР (CDCl3) δ 7,2 (м, 1H), 6,46 (м, 3H), 3,9 (т, 2H), 3,4 (т, 2H, J=6,9 Гц), 1,9-1,7 (м, 4H), 1,6-1,2 (м, 8H).
1-(8-Йодоктилокси)-3-метоксибензол (1,47 г) получали из 1-(8-бромоктилокси)-3-метоксибензола (1,28 г, 6,78 ммоль) и йодида натрия (601 мг) в 50 мл ацетона согласно способу, используемому в получении 10-(гексилокси)декан-1-амина.
N-[8-(3-Метоксифенокси)октил]фталимид (1,0 г) получали из 1-(8-йодоктилокси)-3-метоксибензола (1,47 г, 4,06 ммоль) и фталимида калия (1,13 г) в 50 мл DMF при 60-80°C в течение 12 часов согласно способу, используемому для получения N-[8-(гексилокси)октил]фталимида.1H ЯМР (CDCl3) δ 7,85 (м, 2H), 7,7 (м, 2H), 7,2 (м, 1H), 6,7-6,5 (м, 3H), 3,9 (м, 2H), 3,8 (с, 3H), 3,65 (м, 2H), 1,8-1,6 (м, 4H), 1,5-1,3 (м, 8H).
8-(3-Метоксифенокси)октан-1-амин (438 мг, 1,74 ммоль) получали из N-[8-(3-метоксифенокси)октил]фталимида (1,0 г, 2,6 ммоль) с использованием гидразин моногидрата (0,20 мл) в EtOH (50 мл) согласно способу, используемому для получения [3-(гексилокси)фенил]метанамина.1H ЯМР (CD3OD) δ 7,1 (м, 1H), 6,5-6,4 (м, 3H), 3,9 (т, 2H), 3,7 (с, 3H), 2,7 (т, 2H), 1,8 (м, 2H), 1,6-1,4 (м, 10H).
N-[8-(3-Метоксифенокси)октил]хинолин-4-амин (200 мг) получали из 8-(3-метоксифенокси)октан-1-амина (438 мг, 1,74 ммоль), 4-хлорхинолина (572 мг), ТЕА (2 мл) и NMP (0,2 мл) согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,5 (д, 1H), 8,0 (д, 1H), 7,75 (д, 1H), 7,6 (м, 1H), 7,4 (м, 1H), 7,15 (м, 1H), 6,5-6,4 (м, 4H), 5,1 (шир. с, 1H, NH), 3,9 (т, 2H), 3,3 (м, 2H), 1,8 (м, 4H), 1,6-1,3 (м, 8H).
Пример 28: N-{4-[3-(гексилокси)фенокси]бутил}хинолин-4-амин

1-(4-Бромбутокси)-3-(гексилокси)бензол Смесь 3-(гексилокси)фенола (1,21 г, 6,26 ммоль), 1,4-дибромбутана (7,00 мл, 59 ммоль) и K2CO3 (950 мг, 6,88 ммоль) в 14 мл 1:1 NMP/1,2-диметоксиэтан аккуратно нагревали до кипения в течение 40 часов. Смесь охлаждали и разделяли между DCM и 1M HCl. Органическую фазу высушивали над MgSO4 и концентрировали в вакууме с нагреванием, чтобы удалить избыток дибромида. Остаток разделяли SPE, с промывкой Гексаном и последующим элюированием продукта 5% ЕА/Гексан, получая 1-(4-бромбутокси)-3-(гексилокси)бензол (1,42 г). Rf 0,40 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,15 (м, 1H), 6,51-6,43 (м, 3H), 3,99-3,90 (м, 4H), 3,48 (т, 2H, J=6,6 Гц), 2,11 (м, 2H), 1,93 (м, 2H), 1,81 (м, 2H), 1,50-1,29 (м, 6H), 0,92 (м, 3H).
N-{4-[3-(гексилокси)фенокси]бутил}фталимид 1-(4-Бромбутокси)-3-(гексилокси)бензол (1,40 г, 4,26 ммоль), фталимид калия (1,18 г, 6,38 ммоль) и DMF (5 мл) перемешивали при комнатной температуре, пока бромид не был израсходован, что наблюдали с помощью TLC аликвоты. Смесь разделяли между ЕА и H2O и солевым раствором, и органическую фазу высушивали над MgSO4 и концентрировали. SPE (15% ЕА/Гексан) дал 1,60 г продукта. Rf 0,40 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,83 и 7,70 (м, 4H, AA’BB’), 7,12 (м, 1H), 6,48-6,42 (м, 3H), 3,98-3,88 (м, 4H), 3,76 (т, 2H, J=6,8 Гц), 1,92-1,70 (м, 6H), 1,49-1,25 (м, 6H), 0,89 (м, 3H).
4-[3-(гексилокси)фенокси]бутан-1-амин Смесь N-{4-[3-(гексилокси)фенокси]бутил}фталимида (1,60 г, 4,05 ммоль), гидразин моногидрата (0,30 мл, 6,3 ммоль) и 15 мл EtOH нагревали с обратным холодильником в течение 8 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым K2CO3 и солевым раствором, и органические фазы высушивали над Na2SO4 и концентрировали. SPE, с промывкой 5% MeOH/DCM и с элюированием 10% MeOH/DCM +2% ТЕА, дал 1,05 г амина в форме бесцветного твердого вещества.1H ЯМР (CD3OD + CDCl3) δ 7,01 (т, 1H, J=7,8 Гц), 6,37-6,32 (м, 3H), 3,83-3,76 (м, 4H), 2,66 (т, 2H), 1,74-1,50 (м, 6H), 1,34-1,17 (м, 6H), 0,77 (м, 3H).
N-{4-[3-(гексилокси)фенокси]бутил}хинолин-4-амин Смесь 4-[3-(гексилокси)фенокси]бутан-1-амина (300 мг, 1,20 ммоль), 4-хлорхинолина (283 мг, 1,74 ммоль), DIEA (0,50 мл, 2,87 ммоль) и 1,5 мл IPA закупоривали в толстостенной стеклянной трубке и перемешивали при 180°C в течение 3 дней. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4 и концентрировали. SPE, с промывкой 3% MeOH/DCM и с элюированием 10% MeOH/DCM, дал 293 мг продукта в форме твердого вещества. Rf 0,26 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,52 (д, 1, J=5,2 Гц), 7,97 (д, 1, J=8,4 Гц), 7,72 (д, 1, J=8,4 Гц), 7,61 (м, 1H), 7,37 (м, 1H), 7,17 (т, 1, J=8 Гц), 6,53-6,47 (м, 3), 6,42 (д, 1, J=5,5 Гц), 5,35 (шир. с, 1H, NH), 4,03 (м, 2H), 3,91 (м, 2H), 3,40 (м, 2H), 1,96-1,95 (м, 4), 1,75 (м, 2H), 1,46-1,31 (м, 6), 0,89 (м, 3).
Пример 29: N-{3-[3-(гексилокси)фенокси)пропил}хинолин-4-амин

3-(Гексилокси)фенол Смесь резорцина (7,1 г), K2CO3 (1,13 г) и 1-бромгексана (1,0 мл) в 60 мл NMP вводили в реакцию при 50-60°C в течение 20 часов при помощи механического перемешивания. Затем смесь охлаждали, и большую часть летучих компонентов выпаривали. Остаток разделяли между ЕА (3×250 мл) и H2O, 5% Na2CO3 (2x), H2O, 0,1M HCl и солевым раствором (200 мл каждый). Объединенные органические фазы высушивали над MgSO4 и концентрировали. SPE (5% ЕА/Гексан) дал 1,29 г 3-(гексилокси)фенола.1H ЯМР (CDCl3) δ 7,10 (м, 1H), 6,48 (м, 1H), 6,42-6,38 (м, 2H), 3,91 (т, 2H, J=6,7 Гц), 1,75 (м, 2H), 1,48-1,31 (м, 6H), 0,89 (м, 3H).
N-{3-[3-(гексилокси)фенокси)пропил}фталимид Смесь 3-(гексилокси)фенола (9,8 г), K2CO3 (9,8 г) и N-(3-бромпропил)фталимид (15,5 г) в 150 мл 2-бутанон нагревали с обратным холодильником в течение 24 часов при помощи механического перемешивания. Затем смесь охлаждали, и большую часть летучих компонентов упаривали. Остаток разделяли между ЕА (3×250 мл) и H2O, нейтрализовали, используя H3PO4, 0,1M HCl, H2O и солевой раствор (200 мл каждый). Объединенные органические фазы высушивали над MgSO4 и концентрировали, получая 7,58 г продукта.1H ЯМР (CDCl3) δ 7,81 и 7,68 (м, 4H, AA’BB’), 7,09 (т, 1H, J=8,2 Гц), 6,45 (ддд, 1H, J=1,0, 2,5, 8,4 Гц), 6,39-6,32 (м, 2H), 3,99 (т, 2H, J=6,0 Гц), 3,91-3,83 (м, 4H), 2,16 (м, 2H), 1,73 (м, 2H), 1,45-1,21 (м, 6H), 0,90 (м, 3H).
3-[3-(гексилокси)фенокси)пропан-1-амин Сырой N-{3-[3-(гексилокси)фенокси)пропил}фталимид (1,20 г) растворяли в 50 мл EtOH и добавляли гидразин моногидрат (0,22 мл). Смесь нагревали с обратным холодильником в течение 12 часов, и затем смесь оставляли при комнатной температуре на 48 часов. Твердое вещество разбивали, разбавляли 50 мл простого эфира и перемешивали в течение 1 часа. Осадок фильтровали и промывали 50% MeOH/простой эфир (2×40 мл). Объединенные фильтраты концентрировали. Жидкость забирали в 100 мл DCM и промывали 1н. NaOH и H2O (10 мл каждый). Органическую фазу концентрировали. SPE, с промывкой 1% MeOH/DCM и последующим элюированием 7% MeOH/DCM +2% NH4OH, дал продукт. Частично сконцентрированные фракции промывали 20 мл H2O, водную фазу экстрагировали 40 мл DCM, и объединенные органические фазы высушивали над Na2SO4 и концентрировали, получая 763 мг янтарной жидкости.1H ЯМР (CDCl3) δ 7,13 (м, 1H), 6,49-6,43 (м, 3H), 4,00 (т, 2H, J=6,1 Гц), 3,90 (т, 2H), 2,89 (т, 2H, J=6,7 Гц), 1,96-1,84 (м, 4H), 1,74 (м, 2H), 1,48-1,28 (м, 6H), 0,89 (м, 3H).
N-{3-[3-(гексилокси)фенокси)пропил}хинолин-4-амин Смесь 3-[3-(гексилокси)фенокси)пропан-1-амина (763 мг, 3,04 ммоль), 4-хлорхинолина (746 мг, 4,58 ммоль), DIEA (1,0 мл, 5,74 ммоль) и 0,1 мл DMF герметизировали в толстостенной стеклянной трубке и нагревали при 130°C в течение 4 дней. Смесь охлаждали. SPE, с промывкой 50% ЕА/Гексан и с элюированием 10% MeOH/DCM, дал продукт, загрязненный нингидрином (+). FC (8%-9% MeOH/DCM) привел к частичной очистке. SPE (60% ЕА/Гексан +1% ТЕА) дал 389 мг продукта в форме масла, которое затвердевало при отстаивании. Rf 0,25 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,52 (д, 1H, J=5,2 Гц), 7,96 (дд, 1H, J=0,8, 8,4 Гц), 7,77 (дд, 1H, J=1,0, 8,4 Гц), 7,61 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,40 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,17 (м, 1H), 6,53-6,48 (м, 3), 6,42 (д, 1H, J=5,4 Гц), 5,74 (шир. с, 1H, NH), 4,14 (м, 2H), 3,90 (м, 2H), 3,54 (м, 2H), 2,23 (м, 2H), 1,76 (м, 2H), 1,49-1,24 (м, 6), 0,89 (м, 3).
Пример 30: N-{2-[3-(гексилокси)фенокси]этил}хинолин-4-амин
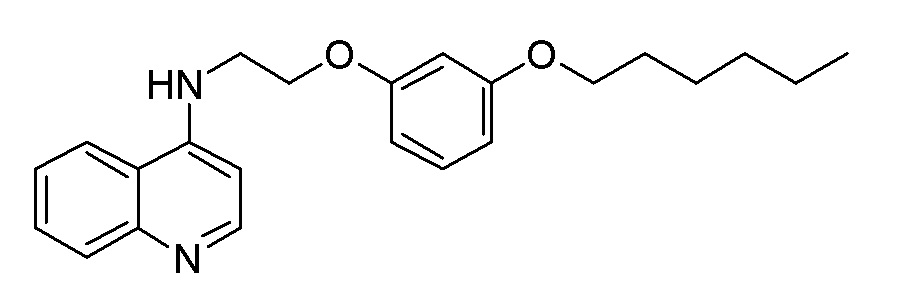
3-(Гексилокси)фенол (2,5 г), N-(2-бромэтил)фталимид (3,27 г), и K2CO3 (1,95 г) в ацетоне (50 мл) при нагревании с обратным холодильником и с последующей обработкой гидразин моногидратом (3,5 мл) в EtOH (24 мл) при нагревании с обратным холодильником дали 226 мг нингидрин (+) 2-[3-(гексилокси)фенокси]этан-1-амина.1H ЯМР (CDCl3) δ 7,10 (м, 1H), 6,55-6,40 (м, 3H), 4,00-3,80 (м, 4H), 3,00 (шир. с, 2H), 1,90-1,70 (м, 4H), 1,50-1,30 (м, 6H), 0,90 (м, 3H).
N-{2-[3-(гексилокси)фенокси]этил}хинолин-4-амин Смесь 2-[3-(гексилокси)фенокси]этан-1-амина (226 мг, 0,95 ммоль), 4-хлорхинолина (233 мг, 1,43 ммоль), DIEA (1,0 мл, 5,74 ммоль) и 0,15 мл DMF герметизировали в толстостенной стеклянной трубке и перемешивали при 140°C и перемешивали в течение 5 дней. Охлажденную смесь концентрировали и разделяли FC (7% MeOH/DCM), получая 150 мг продукта в форме твердого вещества розового цвета. Rf 0,32 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,50 (д, 1H, J=5,5 Гц), 7,99 (д, 1H, J=8,2 Гц), 7,93 (д, 1H, J=8,1 Гц), 7,62 (м, 1H), 7,42 (м, 1H), 7,16 (м, 1H), 6,54-6,47 (м, 4), 6,21 (шир. с, 1H, NH), 4,28 (т, 2H, J=5,2 Гц), 3,92 (м, 2H), 3,75 (м, 2H), 1,75 (м, 2H), 1,48-1,24 (м, 6), 0,88 (т, 3, J=6,7 Гц).
Пример 31: N-[8-(4-Метоксифенокси)октил]хинолин-4-амин

1-(8-Бромоктилокси)-4-метоксибензол Смесь 4-метоксифенола (5,08 г, 41,0 ммоль) и K2CO3 (6,12 г, 44,3 ммоль) в 40 мл DMF перемешивали в течение 1,25 часов. Затем добавляли смесь 1,8-дибромоктана (86,0 г, 316 ммоль) в 40 мл DMF. Смесь перемешивали в течение 24 часов и затем оставляли на 6 дней. Смесь разделяли между 1:1 ЕА/Гексан и H2O (3x), 0,1M HCl и солевым раствором, и органические фазы высушивали над Na2SO4, фильтровали и концентрировали. Остаток в 10% ЕА/Гексан фильтровали через слой силикагеля, и затем большую часть растворителей выпаривали. Осуществляли перегонку в вакууме, чтобы удалить большую часть избытка дибромида, и остаток в резервуаре состоял из почти бесцветного твердого вещества и небольшого количества жидкости. Резервуар промывали дважды Гексаном, и твердое вещество высушивали в вакууме. Rf 0,42 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 6,82 (с, 4H), 3,89 (т, 2H), 3,76 (с, 3H), 3,40 (т, 2H, J=6,8 Гц), 1,90-1,70 (м, 4H), 1,48-1,33 (м, 8H).
N-[8-(4-Метоксифенокси)октил]фталимид Смесь сырого 1-(8-бромоктилокси)-4-метоксибензола и фталимида калия (7,59 г, 41,0 ммоль) в 60 мл NMP перемешивали при комнатной температуре, пока бромид не был израсходован, как показал анализ TLC аликвоты. Затем добавляли 30 мл H2O, и большую часть летучего материала выпаривали в вакууме. Остаток разделяли между 1:1 ЕА/Гексан и H2O и солевым раствором. Органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая 14,88 г бесцветного твердого вещества. Rf 0,11 (10% ЕА/Гексан).
8-(4-Метоксифенокси)октан-1-амин Гидразин моногидрат (4,00 мл, 84 ммоль) добавляли к смеси N-[8-(4-метоксифенокси)октил]фталимида (14,8 г, 38,8 ммоль) и 125 мл денатурированного EtOH с использованием механического перемешивания. Смесь нагревали с обратным холодильником в течение 15 часов, в течение какого времени формировался бесцветный осадок. Смесь концентрировали упариванием, и остаток разделяли между изопропил ацетатом (300, 2×125 мл) и 5% Na2CO3 (200, 3×100 мл) и солевым раствором (100 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая 8,63 г твердого вещества белого цвета после высушивания в вакууме.1H ЯМР (CDCl3) δ 6,79 (с, 4H), 4,66 (с, 3H), 3,86 (т, 2H, J=6,4 Гц), 3,72 (с, 3H), 2,72 (т, 2H, J=7,4 Гц), 1,71 (м, 2H), 1,55-1,33 (м, 10H).
N-[8-(4-Метоксифенокси)октил]хинолин-4-амин 8-(4-Метоксифенокси)октан-1-амин (4,60 г, 18,3 ммоль) забирали в 100 мл 1-пентанола, и 30 мл летучего материала удаляли перегонкой. Смесь охлаждали до температуры ниже температуры кипения и добавляли трипропиламин (7,00 мл, 36,7 ммоль) и 4-хлорхинолин (3,28 г, 20,1 ммоль). Нагревание с обратным холодильником продолжали. Через 26,25 часов, смесь охлаждали и добавляли 20 мл 1н. NaOH. Летучий материал удаляли выпариванием. Смесь разбавляли DCM (350 мл) и промывали 5%-ым Na2CO3 (50 мл). Водную фазу экстрагировали DCM (100 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали. SPE, с промывкой 50% ЕА/Гексан и последующим элюированием 50% ЕА/Гексан +2% ТЕА, дал фракции продукта, которые объединяли и концентрировали. Остаток разделяли между DCM и 5%-ым Na2CO3. Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая твердое вещество желтого цвета. Твердое вещество растирали с ледяной смесью 20% Et2O/Hex и высушивали в вакууме. Твердое вещество имело Т.пл. 141,0-144,0°C. Твердое вещество растворяли в минимальном горячем бутаноне, и затем смеси давали охладиться до комнатной температуры. После охлаждения в ванне со льдом в течение 2 часов, осадок собирали и промывали ледяным бутаноном, получая 3,98 г твердого вещества желто-коричневого цвета. Rf 0,23 (5% MeOH/DCM +2% ТЕА); Т.пл. 143,0-145,5°C;1H ЯМР (CDCl3) δ 8,56 (д, 1H, J=5,1 Гц), 7,98 (дд, 1H, J=0,7, 8,5 Гц), 7,72 (м, 1H), 7,62 (м, 1H), 7,42 (м, 1H), 6,85-6,80 (м, 4H, AA’BB’), 6,42 (д, 1H, J=5,5 Гц), 4,97 (шир. с, 1H, NH), 3,90 (т, 2H, J=6,6 Гц), 3,76 (с, 3H), 3,31 (м, 2H), 1,80-1,73 (м, 4H), 1,48-1,39 (м, 8H);13C ЯМР (CDCl3) δ 153,9, 153,5, 151,3, 149,8, 148,7, 130,3, 129,1, 124,8, 119,3, 118,9, 115,6, 114,8, 99,0, 68,8, 56,0, 43,4, 29,6, 29,5, 29,5, 29,2, 27,3, 26,2.
Пример 32: N-[6-(4-Метоксифенокси)гексил]хинолин-4-амин

1-(6-Бромгексилокси)-4-метоксибензол Смесь 1,6-дибромгексана (2,4 мл, 15,7 ммоль), 4-метоксифенола (243 мг, 1,96 ммоль) и K2CO3 (550 мг, 3,99 ммоль) в 4 мл DMF и 3 мл DME перемешивали 16 часов при комнатной температуре, 4 часа при 80°C и 64 часа при комнатной температуре. Смесь разбавляли ЕА и промывали H2O, 5%-ым Na2CO3, H2O, 0,1M HCl и солевым раствором. Органическую фазу высушивали над безводным Na2SO4, фильтровали через слой силикагеля и концентрировали. SPE, с промывкой Гексаном и последующим элюированием 15% ЕА/Гексан, дал 623 мг продукта в форме бесцветного твердого вещества. Rf 0,29 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 6,82 (с, 4H, AA’BB’), 3,90 (т, 2H, J=6,3 Гц), 3,76 (с, 3H), 3,41 (м, 2H, AB), 1,88 (м, 2H), 1,76 (м, 2H), 1,56-1,39 (м, 4H).
1-(6-Азидогексилокси)-4-метоксибензол Смесь 1-(6-бромгексилокси)-4-метоксибензола (623 мг, 2,17 ммоль) и азида натрия (210 мг, 3,23 ммоль) в 5 мл DMF перемешивали при комнатной температуре в течение 48 часов. Затем смесь разбавляли ЕА и промывали H2O и солевым раствором. Органическую фазу высушивали над MgSO4 и концентрировали, получая 500 мг масляного твердого вещества. Rf 0,50 (15% Et2O/Hex);1H ЯМР (CDCl3) δ 6,82 (с, 4H, AA’BB’), 3,89 (т, 2H, J=6,5 Гц), 3,74 (с, 3H), 3,25 (т, 2H, J=6,9 Гц), 1,76 (м, 2H), 1,62 (м, 2H), 1,55-1,36 (м, 4H).
6-(4-Метоксифенокси) гексан-1-амин Смесь 1-(6-азидогексилокси)-4-метоксибензола (500 мг) и 65 мг 5%-ого Pd-C в 25 мл MeOH перемешивали в атмосфере водорода в течение 16 часов. Смесь продували аргоном и фильтровали через слой Целита. Фильтрат концентрировали, получая 448 мг масла.1H ЯМР (CDCl3) δ 6,77 (с, 4H, AA’BB’), 3,84 (м, 2H), 3,70 (с, 3H), 2,64 и 2,56 (м, 2H, AB), 1,71 (м, 2H), 1,51-1,31 (м, 6H).
N-[6-(4-Метоксифенокси)гексил]хинолин-4-амин Четыре мл пиридина выпаривали из 6-(4-метоксифенокси)гексан-1-амина (448 мг, 2,01 ммоль). Затем смесь амина, 4-хлорхинолина (424 мг, 2,60 ммоль), DIEA (0,80 мл, 4,59 ммоль) и 1,5 мл NMP нагревали при 160°C в герметизированной пробирке в течение 24 часов. Смесь охлаждали и разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над безводным Na2SO4 и концентрировали. FC (50% ЕА/Гексан +2% ТЕА) дала масло, которое содержало остаточный NMP, что наблюдали с помощью ЯМР. Разбавление EtOH и упариванием под высоким вакуумом повторяли, пока NMP не переставал выявляться в ЯМР. Rf 0,12 (50% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,52 (д, 1H, J=5,2 Гц), 7,96 (д, 1H, J=8,4 Гц), 7,74 (д, 1H, J=8,4 Гц), 7,59 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,37 (ддд, 1H, J=1,2, 6,9, 8,2 Гц), 6,82-6,80 (м, 4H), 6,39 (д, 1H, J=5,4 Гц), 5,20 (м, 1H, NH), 3,89 (т, 2H, J=6,3 Гц), 3,74 (с, 3H), 3,31 (м, 2H), 1,78-1,75 (м, 4H), 1,52-1,49 (м, 4H).
Пример 33: N-{2-[4-(гексилокси)фенокси]этил}хинолин-4-амин

4-(Гексилокси)фенол получали способами, подобными используемому для получения 3-(гексилокси)фенола. 4-(Бензилокси)фенол (11,45 г), K2CO3 (8,68 г), 1-бромгексан (10,4 мл) и DMF (50 мл) при 80-100°C дали 1-(бензилокси)-4-(гексилокси)бензол (12,97 г). Rf 0,68 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,44-7,28 (м, 5H), 6,91-6,76 (м, 4H), 5,00 (с, 2H), 3,89 (т, 2H, J=6,6 Гц), 1,74 (м, 2H), 1,49-1,24 (м, 6H), 0,89 (м, 3H).
4-(гексилокси)фенол Смесь 1-(бензилокси)-4-(гексилокси)бензола (12,97 г) и 5% Pd/C (1,2 г) в 200 мл 1:1 MeOH/EA перемешивали под водородом в течение 16 часов. Исходный материал был израсходован, что наблюдали анализом TLC. Реакционную смесь фильтровали через Целит, растворители обменивали на 12% ЕА/Гексан, и смесь фильтровали через слой силикагеля и концентрировали, получая 8,84 г 4-(гексилокси)фенола. Rf 0,21 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 6,80-6,72 (м, 4H), 3,88 (т, 2H, J=6,7 Гц), 1,79-1,68 (м, 2H), 1,48-1,30 (м, 6H), 0,91-0,86 (м, 3H).
2-[4-(гексилокси)фенокси]этанол Смесь 4-(гексилокси)фенола (11,0 г, 56,7 ммоль), этилен карбоната (7,5 г, 85 ммоль) и K2CO3 (11,7 г, 85 ммоль) в 60 мл DMF нагревали при 60°C в течение 16 часов. Смесь разделяли между ЕА и H2O, 0,1M HCl, H2O и солевым раствором. Органические фазы высушивали над MgSO4 и концентрировали. SPE, с промывкой 10% ЕА/Гексан (которая дала 5,8 г рекуперируемого стартового фенола) и с элюированием 37% ЕА/Гексан, дал продукт в форме бесцветного твердого вещества. Рекуперированный исходный материал повторно обрабатывали реагентами. Объединенный выход продукта составил 11,4 г бесцветного твердого вещества. Rf 0,20 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 6,83-6,81 (м, 4H, AA’BB’), 4,03 и 3,93 (м, 4H, A2B2), 3,90 (т, 2H, J=6,6 Гц), 1,79-1,72 (м, 2H), 1,45 (м, 2H), 1,36-1,30 (м, 4H), 0,90 (м, 3H);13C ЯМР (CDCl3) δ 153,9, 152,9, 115,8, 115,7, 70,2, 68,9, 61,8, 31,8, 29,6, 25,9, 22,8, 14,2.
2-[4-(гексилокси)фенокси]этанамин получали способом, используемым для получения [3-(гексилокси)фенил]метанамина.
2-[4-(гексилокси)фенокси]этанол (11,4 г), метансульфонил хлорид (5,60 мл), ТЕА (11,0 мл) и DCM (150 мл) при 0°C дали 2-[4-(гексилокси)фенокси]этил метансульфонат (13,9 г).1H ЯМР (CDCl3) δ 6,85-6,81 (м, 4H, AA’BB’), 4,54 и 4,19 (м, 4H, A2B2), 3,90 (т, 2H, J=6,6 Гц), 3,08 (с, 3H), 1,76 (м, 2H), 1,44 (м, 2H), 1,36-1,30 (м, 4H), 0,90 (м, 3H);13C ЯМР (CDCl3) δ 154,3, 152,2, 116,0, 115,8, 68,9, 68,4, 66,9, 38,0, 31,8, 29,5, 25,9, 22,8, 14,2.
2-[4-(гексилокси)фенокси]этил метансульфонат (13,9 г), фталимид калия (8,57 г) и DMF (40 мл) при 60°C дали N-{2-[4-(гексилокси)фенокси]этил}фталимид (11,58 г после перекристаллизации из EtOH/H2O). Rf 0,40 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,85 и 7,71 (м, 4H, AA’BB’), 6,79 (м, 4H, AA’BB’), 4,18 и 4,08 (м, 4H, A2B2), 3,86 (т, 2H, J=6,6 Гц), 1,73 (м, 2H), 1,42 (м, 2H), 1,34-1,28 (м, 4H), 0,89 (м, 3H);13C ЯМР (CDCl3) δ 168,4, 153,9, 152,6, 134,2, 132,3, 123,5, 115,9, 115,6, 68,8, 65,7, 37,7, 31,8, 29,5 25,9, 22,8, 14,2.
2-[4-(гексилокси)фенокси]этанамин N-{2-[4-(гексилокси)фенокси]этил}фталимид (11,6 г), гидразин моногидрат (2,25 мл), IPA (125 мл) и EtOH (50 мл) при нагревании с обратным холодильником дали бесцветное твердое вещество (7,50 г).1H ЯМР (CDCl3) δ 6,73 (с, 4H, AA’BB’), 3,80 (т, 2H, J=5,2 Гц), 3,79 (т, 2H, J=6,7 Гц), 2,93 (т, 2H), 1,66 (м, 2H), 1,41-1,21 (м, 6H), 0,85-0,80 (м, 3H).
N-{2-[4-(гексилокси)фенокси]этил}хинолин-4-амин Сырой 2-[4-(гексилокси)фенокси]этанамин (7,40 г, 31,2 ммоль) забирали в 30 мл DMA, и затем 25 мл выпаривали. Остаток переносили в толстостенную герметизированную пробирку и добавляли 5 мл NMP, 4-хлорхинолин (5,09 г, 31,2 ммоль) и DIEA (10,8 мл, 62 ммоль). Смесь нагревали при 160°C в течение 16 часов. После охлаждения, разбавление смеси 5%-ым Na2CO3 привело к формированию осадка. Осадок фильтровали и промывали H2O. Осадок перекристаллизовывали из MeOH/H2O и затем из MeOH, получая 7,50 г бесцветного твердого вещества. Rf 0,20 (5% MeOH/DCM); Т.пл. 131,5-132,0°C;1H ЯМР (CDCl3) δ 8,58 (д, 1H, J=5,2 Гц), 8,00 (дд, 1H, J=0,8, 8,4 Гц), 7,79 (дд, 1H, J=0,8, 8,4 Гц), 7,66-7,62 (м, 1H), 7,44 (ддд, 1H, J=1,5, 7,0, 8,5 Гц), 6,86 (м, 4H, AA’BB’), 6,49 (д, 1H, J=5,5 Гц), 5,60 (шир. с, 1H, NH), 4,25 (т, 2H), 3,90 (т, 2H, J=6,6 Гц), 3,70 (м, 2H), 1,74 (м, 2H), 1,45 (м, 2H), 1,36-1,30 (м, 4H), 0,90 (м, 3H);13C ЯМР (CDCl3) δ 154,2, 152,6, 151,0, 149,9, 148,5, 130,0, 129,4, 125,1, 119,7, 119,1, 115,9, 115,8, 99,2, 68,9, 66,9, 42,9, 31,8, 29,5, 25,9, 22,8, 14,2.
Пример 34: N-{3-[4-(гексилокси)фенокси)пропил}хинолин-4-амин

N-{3-[4-(гексилокси)фенокси)пропил} фталимид Смесь 4-(гексилокси)фенола (1,04 г, 5,36 ммоль), N-(3-бромпропил)фталимида (1,44 г, 5,37 ммоль), K2CO3 (1,12 г, 8,12 ммоль) и 10 мл DMF вводили в реакцию в течение 26 часов. Затем смесь разбавляли ЕА и промывали H2O, 0,1M HCl и солевым раствором, высушивали над безводным Na2SO4 и концентрировали. Остаток фильтровали через слой силикагеля, используя 20% ЕА/гексан, и фильтрат концентрировали, получая 1,96 г твердого вещества светло-желтого цвета. Rf 0,20 (15% ЕА/Гексан), 0,38 20% ЕА/гексан +2% DIEA);1H ЯМР (CDCl3) δ 7,83 и 7,69 (м, 4H, AA’BB’), 6,79-6,71 (м, 4H, AA’BB’), 3,96 (т, 2H, J=6,2 Гц), 3,91-3,81 (м, 4H), 2,14 (м, 2H), 1,73 (м, 2H), 1,48-1,28 (м, 6H), 0,89 (м, 3H).
3-[4-(гексилокси)фенокси)пропан-1-амин Смесь N-{3-[4-(гексилокси)фенокси)пропил}фталимида (1,96 г) и гидразин моногидрата (0,40 мл, 8,24 ммоль) в 40 мл EtOH нагревали с обратным холодильником в течение 20 часов. Затем летучие компоненты выпаривали. SPE, с промывкой 5% MeOH/DCM и последующим элюированием 5% MeOH/DCM +2% ТЕА, дал 632 мг бесцветного твердого вещества. Rf 0,21 (5% MeOH/DCM +25 DIEA);1H ЯМР (CDCl3) δ 6,75 (шир. с, 4H), 3,92 (т, 2H, J=6,0 Гц), 3,83 (т, 2H, J=6,7 Гц), 3,00 (шир. м, 2H, NH2), 2,82 (т, 2H, J=6,8 Гц), 1,87 (м, 2H), 1,68 (м, 2H), 1,43-1,23 (м, 6H), 0,83 (м, 3H).
N-{3-[4-(гексилокси)фенокси)пропил}хинолин-4-амин Смесь 3-[4-(гексилокси)фенокси)пропан-1-амина (476 мг, 1,90 ммоль), 4-хлорхинолина (416 мг, 2,55 ммоль) и DIEA (0,50 мл, 2,86 ммоль) в 1 мл NMP нагревали при 150°C в герметизированной пробирке в течение 18 часов. Затем смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 2,5% MeOH/DCM и последующим элюированием 7% MeOH/DCM, дал 633 мг твердого вещества. Rf 0,28 (10% MeOH/DCM); Т.пл. 84,5-86,0°C (из ЕА/Гексан);1H ЯМР (CDCl3) δ 8,51 (д, 1H, J=5,4 Гц), 7,95 (дд, 1H, J=1,0, 8,5 Гц), 7,79 (м, 1H), 7,57 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,35 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 6,82 (шир. с, 4H, AA’BB’), 6,38 (д, 1H, J=5,4 Гц), 5,97 (м, 1H, NH), 4,03 (т, 2H, J=5,4 Гц), 3,86 (т, 2H, J=6,4 Гц), 3,47 (м, 2H), 2,15 (м, 2H), 1,73 (м, 2H), 1,47-1,25 (м, 6H), 0,88 (м, 3H).
Пример 35: N-{4-[4-(гексилокси)фенокси]бутил}хинолин-4-амин
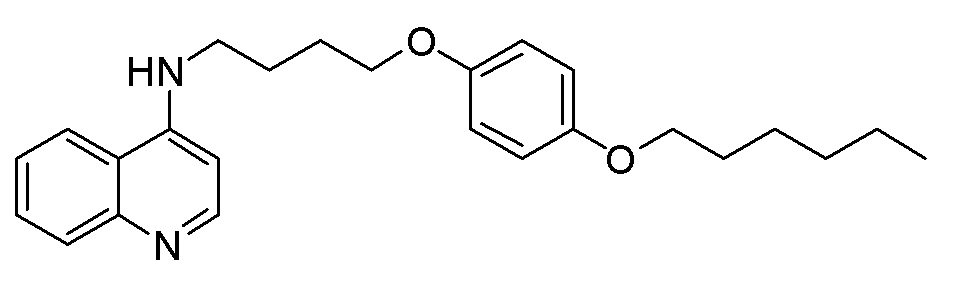
1-(4-Бромбутокси)-4-(гексилокси)бензол 4-(гексилокси)фенол (1,52 г, 7,84 ммоль), 1,4-дибромбутан (7,4 мл, 62 ммоль) и K2CO3 (1,22 г, 8,84 ммоль) в 8 мл DMF перемешивали в течение 16 часов. Смесь разделяли между ЕА и 0,1M HCl и солевым раствором, и органические фазы высушивали над MgSO4, фильтровали и концентрировали. SPE с промывкой 1% ЕА/Гексан и последующим элюированием 5% ЕА/Гексан дал 2,36 г бесцветного твердого вещества. Rf 0,59 (15% ЕА/Гексан);1H ЯМР (CDCl3) δ 6,80 (шир. с, 4H, AA’BB’), 3,93 (т, 2H, J=6,0 Гц), 3,88 (т, 2H, J=6,7 Гц), 3,48 (м, 2H), 2,05 (м, 2H), 1,90 (м, 2H), 1,74 (м, 2H), 1,48-1,28 (м, 6H), 0,89 (м, 3H).
N-{4-[4-(гексилокси)фенокси]бутил} фталимид 1-(4-Бромбутокси)-4-(гексилокси)бензол (2,36 г, 7,17 ммоль) и фталимид калия (2,0 г, 10,8 ммоль) в 12 мл DMF перемешивали в течение 60 часов. Смесь разделяли между ЕА и 0,1M HCl и солевым раствором, и органические фазы высушивали над MgSO4, фильтровали и концентрировали. SPE с промывкой 5% ЕА/Гексан и последующим элюированием 15% ЕА/Гексан дал 2,64 г бесцветного твердого вещества. Rf 0,31 (15% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,83 и 7,70 (м, 4H, AA’BB’), 6,78 (шир. с, 4H, AA’BB’), 3,92 (т, 2H, J=6,1 Гц), 3,87 (т, 2H, J=6,7 Гц), 3,75 (т, 2H, J=7,0 Гц), 1,92-1,68 (м, 6H), 1,48-1,22 (м, 6H), 0,89 (м, 3H).
4-[4-(гексилокси)фенокси]бутан-1-амин Смесь N-{4-[4-(гексилокси)фенокси]бутил}фталимида (2,64 г, 6,68 ммоль) и гидразин моногидрата (0,65 мл, 13,4 ммоль) в 60 мл EtOH нагревали с обратным холодильником в течение 20 часов. Смесь охлаждали, концентрировали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4, фильтровали и концентрировали. SPE с промывкой 4% MeOH/DM и последующим элюированием 6% MeOH/DCM +2% DIEA дал содержащие продукт фракции. Эти фракции концентрировали, забирали в DCM и промывали 5%-ым Na2CO3, высушивали над Na2SO4, фильтровали и концентрировали, получая 1,69 г бесцветного твердого вещества. Rf 0,20 (5% MeOH/DCM +2% DIEA, нингидрин (+));1H ЯМР (CDCl3) δ 6,80 (шир. с, 4H, AA’BB’), 3,93-3,85 (м, 4H), 2,75 (т, 2H, J=7 Гц), 1,87-1,26 (м, 14H), 0,89 (м, 3H).
N-{4-[4-(гексилокси)фенокси]бутил}хинолин-4-амин Смесь 4-[4-(гексилокси)фенокси]бутан-1-амина (499 мг, 1,88 ммоль), 4-хлорхинолина (3999 мг, 2,45 ммоль) и DIEA (0,50 мл, 2,86 ммоль) в 1 мл NMP нагревали при 150°C в герметизированной пробирке в течение 18 часов. Затем смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 2,5% MeOH/DCM и последующим элюированием 7% MeOH/DCM, дал 633 мг твердого вещества. Rf 0,25 (10% MeOH/DCM); Т.пл. 113,0-114,0°C (из ЕА/Гексан);1H ЯМР (CDCl3) δ 8,53 (д, 1H, J=5,2 Гц), 7,95 (м, 1H), 7,70 (д, 1H, J=7,6 Гц), 7,58 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,34 (ддд, 1H, J=1,2, 6,9, 8,2 Гц), 6,82 (шир. с, 4H, AA’BB’), 6,40 (д, 1H, J=5,4 Гц), 5,38 (шир. т, 1H, NH), 3,96 (т, 2H, J=5,6 Гц), 3,88 (т, 2H, J=6,5 Гц), 3,36 (шир. м, 2H), 1,92-1,90 (м, 4H), 1,74 (м, 2H), 1,48-1,28 (м, 6H), 0,89 (м, 3H).
Пример 36: N-[8-(м-толилокси)октил]хинолин-4-амин

1-(8-Бромоктилокси)-3-метилбензол Смесь м-крезола (1,00 мл, 9,54 ммоль), 1,8-дибромоктана (15,0 мл, 81 ммоль) и K2CO3 (2,6 г, 18,8 ммоль) в 20 мл NMP и 10 мл DME нагревали с обратным холодильником в течение 66 часов. Затем смесь охлаждали, разбавляли DCM (20 мл) и экстрагировали 0,05н. NaOH (150, 100 мл) и 1M HCl (100 мл). Водные фазы экстрагировали DCM (20 мл), и объединенные органические фазы высушивали над MgSO4 и концентрировали. SPE, с промывкой Гексаном для рекуперации дибромида и последующим элюированием 3% ЕА/Гексан, дал 1,7 г 1-(8-бромоктилокси)-3-метилбензола. Rf 0,39 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,15 (т, 1H), 6,8-6,65 (м, 3H), 3,95 (т, 2H), 3,4 (т, 2H), 3,3 (с, 3H), 1,9-1,7 (м, 4H), 1,5-1,2 (м, 8H).
1-(8-Азидооктилокси)-3-метилбензол (1,7 г) получали из 1-(8-бромоктилокси)-3-метилбензола (1,7 г, 5,69 ммоль) и азида натрия (740 мг, 11,4 ммоль) в 50 мл DMF согласно способу, используемому для получения 10-бутоксидекан-1-амина.
8-(м-толилокси)октан-1-амин (0,6 г) получали из 1-(8-азидооктилокси)-3-метилбензола (1,7 г) способом, используемым для получения 10-бутоксидекан-1-амина.1H ЯМР (CDCl3) δ 7,1 (м, 1H), 6,6 (м, 3H), 3,9 (м, 2H), 2,7 (т, 1H), 2,3 (м, 4H), 1,8-1,6 (м, 4H), 1,5-1,3 (м, 8H).
N-[8-(м-толилокси)октил]хинолин-4-амин (166 мг) получали из 8-(м-толилокси)октан-1-амина (0,6 г), 4-хлорхинолина (840 мг), ТЕА (2 мл) и NMP (0,2 мл) согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,6 (м, 2H), 8,05 (м, 2H), 7,6 (т, 1H), 7,4 (т, 1H), 7,1 (т, 1H), 6,8-6,6 (м, 3H), 6,4 (д, 1H), 3,9 (т, 2H), 3,5 (м, 2H), 2,3 (с, 3H), 1,9-1,7 (м, 4H), 1,5-1,3 (м, 8H).
Пример 37: N-[8-(п-Толилокси)октил]хинолин-4-амин
1-(8-Бромоктилокси)-4-метилбензол (1,9 г) получали тем же самым способом, используемым для 1-(8-бромоктилокси)-3-метилбензола с использованием п-крезола (1,00 мл, 9,54 ммоль), 1,8-дибромоктана (15,0 мл, 51 ммоль) и K2CO3 (2,6 г, 18,8 ммоль) в 20 мл NMP и 10 мл DME, нагреваемых в течение 66 часов.1H ЯМР (CDCl3) δ 7,0 (д, 2H), 6,8 (д, 2H), 3,9 (т, 2H), 3,4 (т, 2H), 2,3 (с, 3H), 1,9-1,7 (м, 4H), 1,5-1,2 (м, 8H).
1-(8-Азидооктилокси)-4-метилбензол (1,9 г) получали из 1-(8-бромоктилокси)-4-метилбензола (1,9 г, 6,36 ммоль) и азида натрия (830 мг, 12,7 ммоль) в 50 мл DMF согласно способу, используемому для получения 10-бутоксидекан-1-амина.
8-(п-Толилокси)октан-1-амин (0,6 г) получали 1-(8-азидооктилокси)-4-метилбензола (1,9 г) способом, используемым для получения 10-бутоксидекан-1-амина.1H ЯМР (CDCl3) δ 7,05 (д, 2H), 6,75 (д, 2H), 3,9 (м, 2H), 2,7 (м, 1H), 2,35 (т, 1H), 2,3 (с, 3H), 1,8-1,2 (м, 12H).
N-[8-(п-Толилокси)октил]хинолин-4-амин (161 мг) получали из 8-(п-толилокси)октан-1-амина (0,6 г), 4-хлорхинолина (840 мг), ТЕА (2 мл) и NMP (0,2 мл) согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,5 (д, 1H), 8,0 (д, 1H), 7,85 (д, 1H), 7,6 (т, 1H), 7,4 (т, 1H), 7,1 (м, 3H), 6,8 (м, 3H), 6,4 (д, 1H), 3,9 (т, 2H), 3,4 (м, 2H), 2,3 (с, 3H), 1,9-1,7 (м, 4H), 1,5-1,3 (м, 8H).
Пример 38: N-[8-(o-Толилокси)октил]хинолин-4-амин

1-(8-Бромоктилокси)-2-метилбензол (1,3 г) получали тем же самым способом, используемым для 1-(8-бромоктилокси)-3-метилбензола с использованием o-крезола (696 мг, 6,44 ммоль), 1,8-дибромоктана (14 г, 81 ммоль) и K2CO3 (1,00 г, 7,25 ммоль) в 12 мл NMP и 12 мл DME, нагреваемых в течение 16 часов.
1-(8-Йодоктилокси)-2-метилбензол (1,3 г) получали из 1-(8-бромоктилокси)-2-метилбензола (1,3 г, 4,35 ммоль) и йодида натрия (652 мг, 4,35 ммоль) в 50 мл ацетона согласно способу, используемому в получении 10-(гексилокси)декан-1-амина.
N-[8-(o-Толилокси)октил]фталимид (1,3 г) получали из 1-(8-йодоктилокси)-2-метилбензола (1,3 г) и фталимида калия (1,0 г, 5,4 ммоль) в 50 мл DMF согласно способу, используемому для получения N-[8-(гексилокси)октил]фталимида.1H ЯМР (CDCl3) δ 7,85 (м, 2H), 7,7 (м, 2H), 7,15 (м, 2H), 6,8 (м, 2H), 3,95 (м, 2H), 3,7 (м, 2H), 2,2 (м, 3H), 1,9-1,6 (м, 4H), 1,6-1,25 (м, 8H).
8-(o-Толилокси)октан-1-амин (390 мг) получали из N-[8-(o-толилокси)октил]фталимида (1,0 г, 2,74 ммоль) с использованием гидразин моногидрата (0,2 мл) в EtOH (50 мл) согласно способу, используемому для получения [3-(гексилокси)фенил]метанамина.1H ЯМР (DMSO-d6) δ 7,1 (м, 2H), 6,9-6,75 (м, 2H), 3,9 (т, 2H), 2,5 (м, 2H), 2,15 (с, 3H), 1,75 (м, 2H), 1,5-1,2 (м, 10H).
N-[8-(o-Толилокси)октил]хинолин-4-амин (300 мг) получали из 8-(o-толилокси)октан-1-амина (390 мг), 4-хлорхинолина (544 мг), ТЕА (2 мл) и NMP (0,2 мл) согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,55 (д, 1H), 8,0 (д, 1H), 7,75 (д, 1H), 7,65 (м, 1H), 7,45 (м, 1H), 7,15 (м, 2H), 6,8 (м, 2H), 6,4 (д, 1H), 3,95 (т, 2H), 3,35 (м, 2H), 2,3 (с, 3H), 1,8 (м, 4H), 1,6-1,3 (м, 8H).
Пример 39: N-[8-(4-трет-Бутилфенокси)октил]хинолин-4-амин

1-(8-Бромоктилокси)-4-трет-бутилбензол (900 мг) получали тем же самым способом, используемым для 1-(8-бромоктилокси)-3-метилбензола с использованием 4-трет-бутилфенола (647 мг, 4,31 ммоль), 1,8-дибромоктана (11,7 г, 43 ммоль) и K2CO3 (714 мг, 5,17 ммоль) в 12 мл NMP и 6 мл DME, нагреваемых в течение 24 часов.1H ЯМР (CDCl3) δ 7,28 и 6,82 (м, 4H, AA’BB’), 3,93 (м, 2H), 3,40 (т, 2H, J=6,8 Гц), 1,90-1,71 (м, 4H), 1,46-1,22 (м, 8H), 1,29 (с, 9H).
1-трет-Бутил-4-(8-йодоктилокси)бензол (900 мг) получали из 1-(8-бромоктилокси)-4-трет-бутилбензола (900 мг) и йодида натрия (400 мг) в 50 мл ацетона согласно способу, используемому для получения 10-(гексилокси)декан-1-амина.
N-[8-(4-трет-Бутилфенокси)октил]фталимид (1,3 г) получали из 1-трет-бутил-4-(8-йодоктилокси)бензола (900 мг) и фталимида калия (860 мг) в 50 мл DMF согласно способу, используемому для получения N-[8-(гексилокси)октил]фталимида.1H ЯМР (CDCl3) δ 7,85 и 7,70 (м, 4H, AA’BB’), 7,3 и 6,8 (м, 4H, AA’BB’), 3,9 (т, 2H), 3,65 (м, 2H), 1,8-1,6 (м, 4H), 1,6-1,3 (м, 17H).
8-(4-трет-Бутилфенокси)октан-1-амин (590 мг) получали из N-[8-(4-трет-бутилфенокси)октил]фталимида (900 мг) и гидразин моногидрата (0,17 мл) в 50 мл EtOH согласно способу, используемому для получения [3-(гексилокси)фенил]метанамина.1H ЯМР (DMSO-d6) δ 7,25 и 6,80 (м, 4H, AA’BB’), 3,9 (т, 2H), 2,5 (м, 2H), 1,68 (м, 2H), 1,5-1,2 (м, 19H).
N-[8-(4-трет-Бутилфенокси)октил]хинолин-4-амин Смесь 8-(4-трет-бутилфенокси)октан-1-амина (510 мг, 1,84 ммоль), 4-хлорхинолина (604 мг, 3,70 ммоль), ТЕА (4,0 мл, 28 ммоль) и 0,4 мл NMP нагревали в толстостенной стеклянной трубке при 130°C в течение 4 дней. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали. Очистка FC (60% ЕА/Гексан +2% ТЕА) дала 320 мг твердого вещества. Т.пл. 108-110°C (из MeOH);1H ЯМР (CDCl3) δ 8,4 (д, 1H), 8,0 (д, 1H), 7,8 (д, 1H), 7,6 (м, 1H), 7,4 (м, 1H), 7,3 и 6,8 (м, 4H, AA’BB’), 6,4 (д, 1H), 5,2 (шир. с, 1H, NH), 3,9 (м, 2H), 3,3 (м, 2H), 1,8-1,6 (м, 4H), 1,6-1,3 (м, 8H), 1,3 (с, 9H).
Пример 40: N-[8-(4-Фторфенокси)октил]хинолин-4-амин

1-(8-Бромоктилокси)-4-фторбензол (2,75 г) получали тем же самым способом, используемым для 1-(8-бромоктилокси)-3-метилбензола с использованием 4-фторфенола (1,33 г, 12,1 ммоль), 1,8-дибромоктана (20 мл, 108 ммоль) и K2CO3 (1,77 г, 14,3 ммоль) в 20 мл NMP и 10 мл DME, нагреваемых в течение 24 часов.1H ЯМР (CDCl3) δ 7,0-6,9 (м, 2H), 6,8 (м, 2H), 3,89 (т, 2H, J=6,4 Гц), 3,40 (т, 2H, J=6,8 Гц), 1,9-1,7 (м, 4H), 1,6-1,2 (м, 8H).
1-Фтор-4-(8-йодоктилокси)бензол получали из 1-(8-бромоктилокси)-4-фторбензола (2,75 г, 9,08 ммоль) и йодида натрия (1,63 г, 10,9 ммоль) в 70 мл ацетона согласно способу, используемому в получении 10-(гексилокси)декан-1-амина.
N-[8-(4-Фторфенокси)октил]фталимид (2,19 г) получали из 1-фтор-4-(8-йодоктилокси)бензола и фталимида калия (2,52 г, 13,6 ммоль) в 50 мл DMF при 60-80°C в течение 12 часов согласно способу, используемому для получения N-[8-(гексилокси)октил]фталимида.1H ЯМР (CDCl3) δ 7,85 (м, 2H), 7,7 (м, 2H), 6,9 (м, 2H), 6,8 (м, 2H), 3,9 (т, 2H), 3,7 (т, 2H), 1,8-1,6 (м, 4H), 1,5-1,3 (м, 8H).
8-(4-Фторфенокси)октан-1-амин (657 мг, 2,75 ммоль) получали из N-[8-(4-фторфенокси)октил]фталимида (2,19 г, 5,94 ммоль) с использованием гидразин моногидрата (0,43 мл) в EtOH (50 мл) согласно способу, используемому для получения [3-(гексилокси)фенил]метанамина.1H ЯМР (CD3OD) δ 7,0-6,8 (м, 4H), 3,9 (т, 2H), 2,7 (т, 2H), 1,75 (м, 2H), 1,6-1,3 (м, 10H).
N-[8-(4-Фторфенокси)октил]хинолин-4-амин получали из 8-(4-фторфенокси)октан-1-амина (657 мг, 2,75 ммоль), 4-хлорхинолина (676 мг), ТЕА (2 мл) и NMP (0,2 мл) при 130°C в герметизированной пробирке в течение 5 дней согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,5 (д, 1H), 8,0 (д, 1H), 7,9 (д, 1H), 7,65 (м, 1H), 7,4 (м, 1H), 7,1-6,8 (м, 4H), 6,4 (д, 1H), 5,6 (шир. с, 1H, NH), 4,0 (т, 2H), 3,35 (м, 2H), 1,8 (м, 2H), 1,7-1,2 (м, 10H).
Пример 41: N-[8-(3-Фторфенокси)октил]хинолин-4-амин

1-(8-Бромоктилокси)-3-фторбензол (2,06 г) получали тем же самым способом, используемым для 1-(8-бромоктилокси)-3-метилбензола с использованием 3-фторфенола (1,60 г, 14,3 ммоль), 1,8-дибромоктана (25 мл, 135 ммоль) и K2CO3 (2,56 г, 18,5 ммоль) в 25 мл NMP и 12 мл DME, нагреваемых в течение 24 часов. Rf 0,42 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,2 (м, 1H), 6,7-6,6 (м, 3H), 3,9 (т, 2H), 3,4 (т, 2H), 1,9-1,7 (м, 4H), 1,6-1,2 (м, 8H).
1-Фтор-3-(8-йодоктилокси)бензол получали из 1-(8-бромоктилокси)-3-фторбензола (2,06 г, 6,78 ммоль) и йодида натрия (1,22 г, 8,13 ммоль) в 60 мл ацетона согласно способу, используемому в получении 10-(гексилокси)декан-1-амина.
N-[8-(3-Фторфенокси)октил]фталимид (1,85 г) получали из 1-фтор-3-(8-йодоктилокси)бензола и фталимида калия (1,9 г, 10,3 ммоль) в 50 мл DMF при 60-80°C в течение 12 часов согласно способу, используемому для получения N-[8-(гексилокси)октил]фталимида.1H ЯМР (CDCl3) δ 7,85 (м, 2H), 7,7 (м, 2H), 7,2 (м, 1H), 6,7-6,5 (м, 3H), 3,9 (т, 2H), 3,7 (т, 2H), 1,8-1,6 (м, 4H), 1,5-1,3 (м, 8H).
8-(3-Фторфенокси)октан-1-амин (874 мг, 3,66 ммоль) получали из N-[8-(3-фторфенокси)октил]фталимида (1,85 г, 5,01 ммоль) с использованием гидразин моногидрата (0,36 мл) в EtOH (50 мл) согласно способу, используемому для получения [3-(гексилокси)фенил]метанамина.1H ЯМР (CD3OD) δ 7,25 (м, 1H), 6,8-6,6 (м, 3H), 3,9 (т, 2H), 2,7 (т, 2H), 1,8 (м, 2H), 1,6-1,3 (м, 10H).
N-[8-(3-Фторфенокси)октил]хинолин-4-амин получали из 8-(3-фторфенокси)октан-1-амина (874 мг, 3,66 ммоль), 4-хлорхинолина (900 мг), ТЕА (2 мл) и NMP (1 мл) при 130°C в герметизированной пробирке в течение 5 дней согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,5 (д, 1H), 8,0 (д, 1H), 7,85 (д, 1H), 7,65 (м, 1H), 7,4 (м, 1H), 7,15 (м, 1H), 6,7-6,5 (м, 3H), 6,5 (д, 1H), 5,6 (шир. с, 1H, NH), 3,9 (т, 2H), 3,35 (м, 2H), 1,8 (м, 4H), 1,6-1,3 (м, 8H).
Пример 42: N-[8-(2-Фторфенокси)октил]хинолин-4-амин

1-(8-Бромоктилокси)-2-фторбензол (2,97 г) получали тем же самым способом, используемым для 1-(8-бромоктилокси)-3-метилбензола с использованием 2-фторфенола (1,69 г, 15,1 ммоль), 1,8-дибромоктана (38,3 г, 141 ммоль) и K2CO3 (2,76 г, 20 ммоль) в 25 мл NMP и 20 мл DME, нагреваемых в течение 24 часов. Rf 0,33 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,10-6,83 (м, 4H), 4,0 (м, 2H), 3,38 (т, 2H, J=6,9 Гц), 1,91-1,76 (м, 4H), 1,47-1,32 (м, 8H).
1-Фтор-2-(8-йодоктилокси)бензол (3,43 г) получали из 1-(8-бромоктилокси)-2-фторбензола (2,97 г, 9,80 ммоль) и йодида натрия (1,76 г, 11,7 ммоль) в 70 мл ацетона согласно способу, используемому в получении 10-(гексилокси)декан-1-амина.
N-[8-(2-Фторфенокси)октил]фталимид (2,84 г) получали из 1-фтор-2-(8-йодоктилокси)бензола (3,43 г) и фталимида калия (2,72 г, 14,7 ммоль) в DMF при 60-80°C в течение 12 часов согласно способу, используемому для получения N-[8-(гексилокси)октил]фталимида.1H ЯМР (CDCl3) δ 7,85 и 7,70 (м, 4H, AA’BB’), 7,10-6,80 (м, 4H), 4,00 (т, 2H), 3,70 (т, 2H), 1,90-1,60 (м, 4H), 1,55-1,25 (м, 8H).
8-(2-Фторфенокси)октан-1-амин (1,27 г, 5,32 ммоль) получали из N-[8-(2-фторфенокси)октил]фталимида (2,84 г, 7,70 ммоль) с использованием гидразин моногидрата (0,50 мл) в EtOH (50 мл) согласно способу, используемому для получения [3-(гексилокси)фенил]метанамина.
N-[8-(2-Фторфенокси)октил]хинолин-4-амин (100 мг) получали из 8-(2-фторфенокси)октан-1-амина (1,27 г, 5,32 ммоль), 4-хлорхинолина (1,3 г, 7,98 ммоль), ТЕА (2 мл) и NMP (1 мл) при 130°C в герметизированной пробирке в течение 5 дней согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,4 (д, 1H), 8,0 (д, 1H), 7,9 (д, 1H), 7,6 (м, 1H), 7,4 (м, 1H), 7,0-6,7 (м, 4H), 6,4 (д, 1H), 5,9 (шир. с, 1H, NH), 3,9 (т, 2H), 3,3 (м, 2H), 1,9-1,2 (м, 12H).
Пример 43: N-(бифенил-4-ил)хинолин-4-амин

Смесь 4-бифениламина (200 мг, 1,18 ммоль), 4-хлорхинолина (228 мг) и DIEA (0,25 мл, 1,43 ммоль) в 1 мл NMP нагревали при 150°C в герметизированной пробирке в течение 24 часов. Охлажденную смесь разбавляли ЕА, промывали 5%-ым Na2CO3 (2x) и солевым раствором, высушивали над безводным Na2SO4 и концентрировали. SPE, с элюированием градиентом стадии 1%, 3% и 5% MeOH/DCM, дал фракции, которые концентрировали, получая твердое вещество коричневого цвета. Твердое вещество промывали MeOH и высушивали в вакууме. Rf 0,21 (5% MeOH/DCM); Т.пл. 222-226°C;1H ЯМР (20% CD3OD/CDCl3) δ 8,38 (д, 1H, J=5,7 Гц), 8,06 (м, 1H), 7,91 (м, 1H), 7,67-7,26 (м, 11H), 6,98 (д, 1H, J=5,5 Гц).
Пример 44: N-(4-Гексилфенил)хинолин-4-амин

Смесь 4-гексиланилина (197 мг, 1,11 ммоль), 4-хлорхинолина (210 мг) и DIEA (0,24 мл) в 1 мл NMP нагревали при 150°C в герметизированной пробирке в течение 24 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали. Очистка SPE (градиент стадии 1, 2, 3, 5, 6% MeOH/DCM) дала фракции, приводящие к твердому веществу желтого цвета. Перекристаллизация из MeOH дала 229 мг бесцветного твердого вещества. Rf 0,14 (5% MeOH/DCM); Т.пл. 132,5-133,0°C;1H ЯМР (CDCl3) δ 8,52 (д, 1H, J=5,7 Гц), 8,03 (дд, 1H, J=0,7, 8,4 Гц), 7,85 (д, 1H, J=7,6 Гц), 7,64 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,44 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 6,88-6,81 (м, 4H), 6,50 (д, 1H, J=5,7 Гц), 5,92 (шир. с, 1H, NH), 4,26 (т, 2H, J=5 Гц), 3,89 (т, 2H, J=6 Гц), 3,73 (кв., 2H, J=5,2 Гц), 1,74 (м, 2H), 1,48-1,28 (м, 6H), 0,89 (м, 3H).
Пример 45: Гексил-4-(хинолин-4-иламино)бензоат

Гексил-4-аминобензоат (282 мг), полученный из 1-гексанола и 4-нитробензоил хлорида в две обычные стадии, вводили в реакцию с 4-хлорхинолином (322 мг) и DIEA (0,50 мл) в 2 мл NMP, нагретого при 160°C в герметизированной пробирке в течение 16 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали. Очистка SPE, с промывкой 20% ЕА/Гексан и последующим элюированием 55% ЕА/Гексан, дала твердое вещество желтого цвета. Перекристаллизация из ЕА/Гексан дала бесцветное твердое вещество. Rf 0,14 (50% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,61 (д, 1H, J=5,2 Гц), 8,09-8,03 (м, 4H), 7,70 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,52 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,34-7,31 (м, 2H), 7,19 (д, 1H, J=5,2 Гц), 4,30 (т, 2H, J=6,6 Гц), 1,76 (м, 2H), 1,47-1,24 (м, 6H), 0,89 (м, 3H).
Пример 46: N-(4-Феноксифенил)хинолин-4-амин
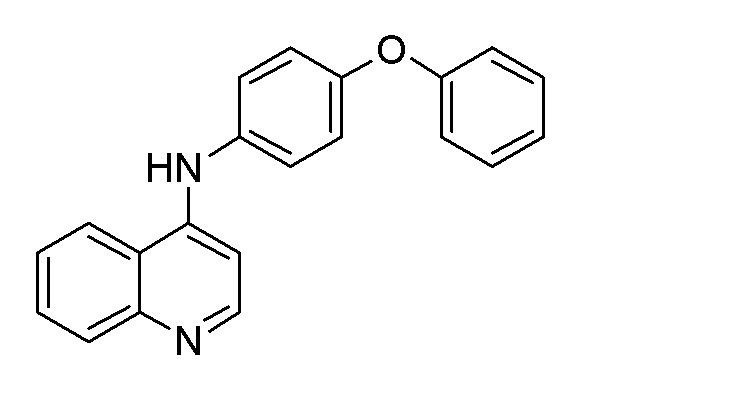
Смесь 4-феноксианилина (182 мг, 0,98 ммоль), 4-хлорхинолина (175 мг, 1,07 ммоль) и DIEA (0,50 мл, 2,87 ммоль) в 1 мл NMP нагревали при 140-150°C в герметизированной пробирке в течение 24 часов. Затем смесь охлаждали и разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 50% ЕА/Гексан и с элюированием 5% MeOH/DCM, дал твердое вещество. Перекристаллизация из ЕА/Гексан дала 111 мг твердого вещества желто-коричневого цвета. Второй выход твердого вещества светлого желто-коричневого цвета на 111 мг получали из MeOH. Эти два выхода имели сопоставимые спектры ЯМР. Rf 0,19 (5% MeOH/DCM); Т.пл. 170-172°C (из MeOH)1H ЯМР (CDCl3) δ 8,51 (д, 1H, J=5,5 Гц), 8,05 (д, 1H, J=8,7 Гц), 7,99 (д, 1H, J=8,4 Гц), 7,68 (ддд, 1H, J=1,3, 6,9, 8,2 Гц), 7,50 (ддд, 1H, J=1,3, 6,9, 8,2 Гц), 7,40-7,25 (м, 5H), 7,22-6,99 (м, 5H), 6,83 (д, 1H, J=5,4 Гц).
Пример 47: N-(3-Феноксифенил)хинолин-4-амин

Смесь 3-феноксианилина (307 мг, 1,66 ммоль), 4-хлорхинолина (296 мг, 1,82 ммоль) и DIEA (0,32 мл, 1,84 ммоль) в 1 мл NMP нагревали при 140-150°C в герметизированной пробирке в течение 24 часов. Затем смесь охлаждали и разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 20% ЕА/Гексан, 20% ЕА/Гексан +2% ТЕА и 35% ЕА/Гексан +2% ТЕА, затем с элюированием 50% ЕА/Гексан +2% ТЕА, дал 208 мг твердого вещества желтого цвета. Rf 0,26 (7,5% MeOH/DCM); Т.пл. 189-192°C (из MeOH);1H ЯМР (CDCl3) δ 8,40 (д, 1H, J=5,2 Гц), 7,98-7,91 (м, 2H), 7,62 (м, 1H), 7,45 (м, 1H), 7,34-7,26 (м, 3H), 7,10-6,98 (м, 6H), 6,90 (т, 1H, J=2,2 Гц), 6,75 (дд, 1H, J=2,5, 8,1 Гц).
Пример 48: N-(2-Феноксифенил)хинолин-4-амин
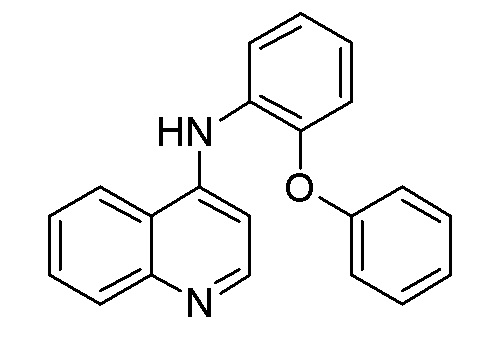
Смесь 2-феноксианилина (286 мг, 1,54 ммоль), 4-хлорхинолина (278 мг, 1,70 ммоль) и 4-метилморфолина (0,19 мл, 1,73 ммоль) в 0,5 мл NMP нагревали в толстостенной герметизированной пробирке при 130°C в течение 20 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали. FC (7,5% MeOH/DCM) дала темное масло, которое содержало остаток 4-метилморфолина. Масло фильтровали через слой силикагеля, используя 30% ЕА/гексан +2% ТЕА, получая 402 мг твердого вещества. Rf 0,10 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 8,61 (д, 1H, J=5,2 Гц), 8,03 (дд, 1H, J=0,7, 8,4 Гц), 7,85-7,81 (м, 1H), 7,64 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,59 (м, 1H), 7,43 (м, 1H), 7,34-7,24 (м, 2H), 7,19-6,98 (м, 8H).
Пример 49: N-[4-(хинолин-4-иламино)фенил]гексанамид

N-(4-Нитрофенил)гексанамид Гексаноил хлорид ((0,81 мл, 5,8 ммоль) медленно добавляли к смеси 4-нитроанилина ((800 мг, 5,79 ммоль) в 5 мл пиридина и 15 мл DMF, охлажденных ванной со льдом. Через 30 минут, смесь нагревали до комнатной температуры. Через дополнительных 2 часа выпаривали летучие компоненты. Остаток забирали в ЕА (100 мл) и промывали насыщенным NaHCO3 (2×75 мл), H2O (2×50 мл), 0,1н. HCl (2×25 мл) и H2O. Органическую фазу концентрировали в вакууме, получая 1,50 г продукта.1H ЯМР (CDCl3) δ 8,2 (м, 2H), 7,7 (м, 2H), 7,4 (шир. с, 1H, NH), 2,4 (м, 2H), 1,8 (м, 2H), 1,4-1,3 (м, 4H), 0,9 (м, 3H).
N-(4-Аминофенил)гексанамид Смесь N-(4-нитрофенил)гексанамида (1,50 г), 10% Pd-C (200 мг) и 75 мл MeOH перемешивали в атмосфере водорода до потребления исходного материала, что наблюдали с помощью аналитической TLC. Затем атмосферу продували аргоном, и смесь фильтровали через слой Целита. Выпаривание растворителя дало 1,22 г продукта.1H ЯМР (CDCl3) δ 7,2 (м, 3H), 7,0 (шир. с, 1H, NH), 6,6 (м, 2H), 3,6 (шир. с, 2H, NH2), 2,3 (м, 2H), 1,7 (м, 2H), 1,4-1,2 (м, 4H), 0,9 (м, 3H).
N-[4-(хинолин-4-иламино)фенил]гексанамид Смесь 4-хлорхинолина (358 мг, 2,20 ммоль), N-(4-аминофенил)гексанамида (300 мг, 1,46 ммоль) и ТЕА (1 мл) нагревали при 130°C в герметизированной пробирке в течение 5 дней. Затем летучие компоненты выпаривали. Остаток очищали препаративной TLC (10% MeOH/DCM), получая 329 мг продукта. Rf 0,3 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,56 (д, 1H, J=5,5 Гц), 8,04 (д, 2H, J=8,9 Гц), 8,05-7,99 (м, 2H), 7,69 (ддд, 1H, J=1,2, 6,9, 8,2 Гц), 7,51 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,30 (д, 2H, J=8,9 Гц), 7,18 (д, 1H, J=5,4 Гц), 4,35 (кв., 2H, J=7 Гц), 1,38 (т, 3H, J=7 Гц).
Пример 50: N-[3-(хинолин-4-иламино)фенил]гексанамид

N-[3-(хинолин-4-иламино)фенил]гексанамид получали согласно способу, используемому для получения N-[4-(хинолин-4-иламино)фенил]гексанамида, исходя из 3-нитроанилина (800 мг) и гексаноил хлорида (0,81 мл) и используя 4-хлорхинолин (358 мг).
N-(4-Нитрофенил)гексанамид (1,50 г):1H ЯМР (CDCl3) δ 8,4 (м, 1H), 8,0-7,9 (м, 2H), 7,8 (шир. с, 1H, NH), 7,5 (м, 1H), 2,4 (м, 2H), 1,8 (м, 2H), 1,4-1,2 (м, 4H), 0,9 (м, 3H).
N-(4-Аминофенил)гексанамид (1,34 г):1H ЯМР (CDCl3) δ 7,4 (шир. с, 1H, NH), 7,2 (шир. с, 1H), 7,0 (т, 1H), 6,7 (д, 1H), 6,4 (д, 1H), 3,5 (шир. с, 2H, NH2), 2,3 (т, 2H), 1,7 (м, 2H), 1,4-1,2 (м, 4H), 0,9 (м, 3H).
N-[3-(хинолин-4-иламино)фенил]гексанамид: Rf 0,2 (10% MeOH/DCM);1H ЯМР (CD3OD) δ 8,5 (д, 1H), 8,4 (д, 1H), 8,0-7,8 (м, 3H), 7,7 (м, 1H), 7,5-7,3 (м, 2H), 7,1 (м, 1H), 7,0 (д, 1H), 2,4 (т, 2H), 1,7 (м, 2H), 1,4-1,2 (м, 4H), 0,9 (м, 3H).
Пример 51: N-Гексил-4-(хинолин-4-иламино)бензамид

N-Гексил-4-(хинолин-4-иламино)бензамид 4-Амино-N-гексилбензамид (220 мг), полученный из 1-аминогексана (0,70 мл) и 4-нитробензоил хлорида (450 мг) в две обычные стадии, вводили в реакцию с 4-хлорхинолином (239 мг) и DIEA (0,50 мл) в 1 мл IPA, нагретыми при 130-180°C в герметизированной пробирке в течение 8 дней. Смесь охлаждали и разделяли между DCM и 5%-ым Na2CO3. Органические фазы высушивали над Na2SO4 и концентрировали. Очистка SPE, с промывкой 3% MeOH/DCM и последующим элюированием 15% MeOH/DCM, дала 105 мг твердого вещества. Rf 0,08 (5% MeOH/DCM);1H ЯМР (20% CD3OD/CDCl3) δ 8,39 (д, 1H, J=5,4 Гц), 8,15 (дд, 1H, J=0,7, 8,4 Гц), 7,89 (дд, 1H, J=0,7, 8,4 Гц), 7,80-7,75 (м, 2H), 7,65 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,47 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,36-7,30 (м, 2H), 7,07 (д, 1H, J=5,5 Гц), 3,35 (м, 2H, AB), 1,57 (м, 2H), 1,32-1,21 (м, 6H), 0,84 (т, 3H, J=6 Гц).
N-Гексил-4-нитробензамид (467 мг):1H ЯМР (CDCl3) δ 8,17 (д, 2H, J=8,7 Гц), 7,91 (д, 2H, J=8,7 Гц), 7,00 (шир. с, 1H, NH), 3,39 (м, 2H), 1,56 (м, 2H), 1,4-1,1 (м, 6H), 0,81 (м, 3H).
4-Амино-N-гексилбензамид: Rf 0,22 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 7,56 (м, 2H), 6,58 (м, 2H), 6,56 (шир. с, 1H, NH), 4,12 (шир. с, 2H, NH2), 3,57 (м, 2H), 1,53 (м, 2H), 1,47-1,22 (м, 6H), 0,84 (м, 3H).
Пример 52: N-Гексил-3-(хинолин-4-иламино)бензамид
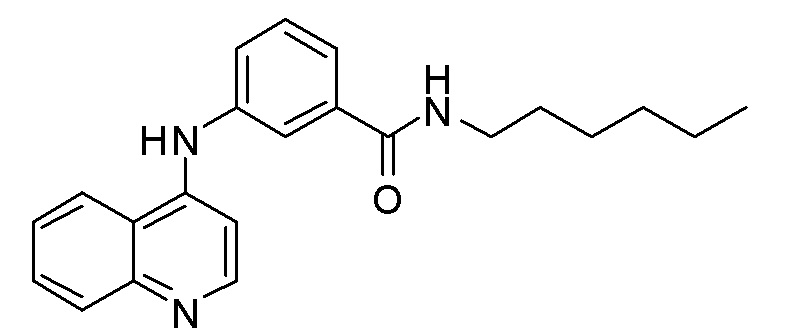
N-Гексил-3-(хинолин-4-иламино)бензамид (117 мг) получали согласно способу, используемому для получения N-гексил-4-(хинолин-4-иламино)бензамида, исходя из 3-нитробензойной кислоты (1,17 г) и 1-гексиламина (1,02 мл) и используя 4-хлорхинолин (225 мг).
N-Гексил-3-нитробензамид:1H ЯМР (CDCl3) δ 8,56 (м, 1H), 8,28 (м, 1H), 8,13 (ддд, 1H, J=1,2, 1,7, 7,7 Гц), 7,58 (т, 1H, J=7,9 Гц), 6,84 (шир. с, 1H, NH), 3,44 (м, 2H), 1,60 (м, 2H), 1,39-1,23 (м, 6H), 0,84 (т, 3H, J=7,0 Гц).
3-Амино-N-гексилбензамид (1,47 г): Rf 0,25 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 7,14-7,00 (м, 3H), 6,71 (м, 1H), 6,42 (шир. с, 1H, NH), 3,80 (шир. с, 2H, NH2), 3,34 (м, 2H), 1,53 (м, 2H), 1,48-1,21 (м, 6H), 0,84 (м, 3H).
N-Гексил-3-(хинолин-4-иламино)бензамид: Rf 0,05 (5% MeOH/DCM);1H ЯМР (20% CD3OD/CDCl3) δ 8,34 (д, 1H, J=5,6 Гц), 8,18 (дд, 1H, J=0,7, 8,4 Гц), 7,91-7,88 (м, 1H), 7,70-7,64 (м, 2H), 7,53-7,38 (м, 4H), 6,93 (д, 1H, J=5,7 Гц), 3,35 (м, 2H), 1,57 (м, 2H), 1,32-1,20 (м, 6H), 0,84 (м, 3H).
Пример 53: N-(4-Метоксифенил)хинолин-4-амин

Смесь п-анизидина (138 мг, 1,12 ммоль), 4-хлорхинолина (235 мг, 1,44 ммоль) и DIEA (0,50 мл, ммоль) нагревали при 130°C в герметизированной пробирке в течение 40 часов. Охлажденную смесь разделяли между ЕА (3x) и 5%-ым Na2CO3 (3x) и солевым раствором, и органические фазы высушивали над безводным Na2SO4 и концентрировали, получая 385 мг коричневого масла. Очистка препаративной TLC (10% MeOH/DCM) дала 294 мг коричневого масла, которое затвердевало при отстаивании.1H ЯМР (CDCl3) δ 8,48 (д, 1H, J=5,4 Гц), 7,99 (д, 1H, J=8,4 Гц), 7,96 (д, 1H, J=8,4 Гц), 7,64 (ддд, 1H, J=1,3, 7,0, 8,5 Гц), 7,45 (м, 1H), 7,21 (м, 2H), 6,93 (м, 2H), 6,68 (д, 1H, J=5,2 Гц), 3,82 (с, 3H).
Пример 54: N-[4-(бензилокси)фенил]хинолин-4-амин

Смесь 4-(бензилокси)анилина (197 мг, 0,99 ммоль), 4-хлорхинолина (169 мг, 1,04 ммоль) и DIEA (0,18 мл, 1,03 ммоль) в 1 мл NMP нагревали при 150°C в герметизированной пробирке в течение 24 часов. Затем смесь охлаждали и разделяли между ЕА (2x) и 5%-ым Na2CO3 (2x) и солевым раствором. Органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 1% MeOH/DCM и с элюированием 5% MeOH/DCM с отсечением фракций, дал 152 мг бесцветного твердого вещества. Rf 0,18 (5% MeOH/DCM); Т.пл. 201-202°C (из MeOH);1H ЯМР (CDCl3) δ 8,49 (д, 1H, J=5,4 Гц), 8,02 (дд, 1H, J=1,0, 8,6 Гц), 7,91 (дд, 1H, J=0,7, 8,4 Гц), 7,66 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,51-7,31 (м, 6H), 7,26-7,20 (м, 2H), 7,06-6,98 (м, 2H), 6,71 (д, 2H, J=5,2 Гц), 5,09 (с, 2H).
Пример 55: N-(4-Бутоксифенил)хинолин-4-амин

Смесь 4-бутоксианилина (236 мг, 1,43 ммоль), 4-хлорхинолина (236 мг, 1,45 ммоль) и DIEA (0,26 мл, 1,49 ммоль) в 1 мл NMP нагревали при 150°C в герметизированной пробирке в течение 24 часов. Охлажденную смесь разделяли между ЕА (2x) и 5%-ым Na2CO3 (2x) и солевым раствором, и органические фазы высушивали над безводным Na2SO4 и концентрировали, получая твердое вещество. SPE, с промывкой 1% MeOH/DCM и с элюированием 5% MeOH/DCM, дал фракции, приводящие к твердому веществу после концентрации. Перекристаллизация из MeOH дала 177 мг. Rf 0,18 (5% MeOH/DCM); Т.пл. 181-185°C;1H ЯМР (CDCl3) δ 8,45 (д, 1H, J=5,4 Гц), 8,03 (дд, 1H, J=1,0, 8,7 Гц), 7,97 (д, 1H, J=8,4 Гц), 7,67 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 7,48 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,22 и 6,95 (м, 4H, AA’BB’), 6,67 (д, 1H, J=5,4 Гц), 3,98 (т, 2H, J=6,5 Гц), 1,79 (м, 2H), 1,51 (м, 2H), 0,99 (т, 3H, J=7,3 Гц).
Пример 56: N-[4-(гексилокси)фенил]хинолин-4-амин

1-(Гексилокси)-4-нитробензол Смесь 4-нитрофенола (480 мг, 3,45 ммоль), 1-бромгексана (0,43 мл, 3,08 ммоль), K2CO3 (481 мг, 3,57 ммоль) и йодида натрия на 20 мг в 5 мл DMF нагревали при 60°C в течение 18 часов. Охлажденную смесь разбавляли Et2O и промывали 5%-ым Na2CO3 и солевым раствором, поочередно, пока водная фаза не обесцветилась. Органическую фазу высушивали над MgSO4 и концентрировали, получая 532 мг желтого масла. Rf 0,21 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,19-8,13 (м, 2H, AA’BB’), 6,94-6,88 (м, 2H, AA’BB’), 4,02 (т, 2H), 1,80 (м, 2H), 1,50-1,29 (м, 6H), 0,89 (м, 3H).
4-(гексилокси)анилин Смесь 1-(гексилокси)-4-нитробензола (532 мг, 2,38 ммоль) и 5% Pd/C (60 мг) в 20 мл MeOH перемешивали в атмосфере водорода в течение 3 часов. Затем смесь фильтровали через слой Целита и концентрировали, получая 458 мг масла.1H ЯМР (CDCl3) δ 6,78-6,72 (м, 2H, AA’BB’), 6,65-6,59 (м, 2H, AA’BB’), 3,88 (т, 2H), 3,44 (шир. с, 2H, NH2), 1,75 (м, 2H), 1,50-1,28 (м, 6H), 0,92 (м, 3H).
N-[4-(гексилокси)фенил]хинолин-4-амин Смесь 4-(гексилокси)анилина (430 мг, 2,23 ммоль), 4-хлорхинолина (431 мг, 2,64 ммоль) и DIEA (1,0 мл, 5,74 ммоль) в 1 мл NMP нагревали в толстостенной герметизированной пробирке при 160°C в течение 24 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали, получая твердое вещество, которое перекристаллизовывали из EtOH, получая бесцветное твердое вещество.1H ЯМР (CDCl3) δ 8,49 (д, 1, J=5,2 Гц), 8,02 (дд, 1, J=0,7, 8,4 Гц), 7,91 (д, 1, J=8,4 Гц), 7,67 (ддд, 1, J=1,5, 6,9, 8,4 Гц), 7,48 (ддд, 1, J=1,5, 6,9, 8,4 Гц), 7,25-7,18 (м, 2H), 6,98-6,92 (м, 2H), 6,69 (д, 1, J=5,5 Гц), 6,64 (шир. с, 1H), 3,97 (т, 2H, J=6 Гц), 1,80 (м, 2H), 1,50-1,30 (м, 6), 0,92 (м, 3).
Пример 57: N-[3-(бензилокси)фенил]хинолин-4-амин
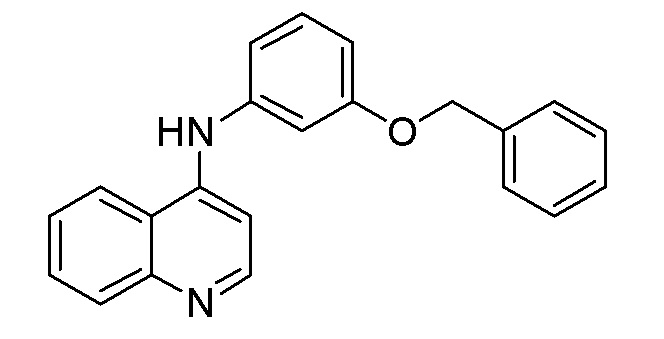
Смесь 3-(бензилокси)анилина (312 мг, 1,57 ммоль), 4-хлорхинолин (280 мг, 1,72 ммоль) и DIEA (0,30 мл, 1,72 ммоль) в 1 мл NMP нагревали при 150°C в герметизированной пробирке в течение 24 часов. Затем смесь охлаждали и разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 20% ЕА/Гексан, 20% ЕА/Гексан +2% ТЕА и 35% ЕА/Гексан +2% ТЕА, затем с элюированием 50% ЕА/Гексан +2% ТЕА, дал 528 мг твердого вещества желтого цвета. Перекристаллизация из MeOH дала 390 мг твердого вещества светло-желтого цвета. Rf 0,26 (7,5% MeOH/DCM); Т.пл. 77-80°C (от MeOH);1H ЯМР (CDCl3) δ 8,45 (д, 1H, J=5,5 Гц), 8,04 (д, 1H, J=8,4 Гц), 7,98 (д, 1H, J=8,4 Гц), 7,67 (м, 1H), 7,53-7,24 (м, 8H), 6,94-6,79 (м, 4H), 5,08 (с, 2H).
Пример 58: N-[3-(гексилокси)фенил]хинолин-4-амин

1-(Гексилокси)-3-нитробензол Смесь 3-нитрофенола (553 мг, 3,98 ммоль), 1-бромгексана (0,50 мл, 3,58 ммоль) и K2CO3 (618 мг, 4,48 ммоль) в 5 мл DMF нагревали при 60-80°C в течение 12 часов. Охлажденную смесь разбавляли Et2O и промывали 5%-ым Na2CO3 и солевым раствором, поочередно, пока водная фаза не обесцветилась, и затем 0,1M HCl и солевым раствором. Органическую фазу высушивали над MgSO4 и концентрировали, получая 756 мг масла.1H ЯМР (CDCl3) δ 7,78 (ддд, 1H, J=1,0, 2,0, 7,9 Гц), 7,70 (м, 1H), 7,39 (м, 1H), 7,19 (ддд, 1H, J=1,0, 2,4, 8,1 Гц), 4,01 (т, 2H, J=6,6 Гц), 1,80 (м, 2H), 1,58-1,30 (м, 6H), 0,89 (м, 3H).
3-(гексилокси)анилин Смесь 1-(гексилокси)-3-нитробензола (756 мг, 3,39 ммоль) и 5% Pd/C (90 мг) в 20 мл MeOH перемешивали в атмосфере водорода в течение 3 часов. Затем смесь фильтровали через слой Целита и концентрировали, получая 660 мг светло-оранжевого масла.1H ЯМР (CDCl3) δ 7,04 (м, 1H), 6,34-6,23 (м, 3H), 3,90 (т, 2H), 3,62 (шир. с, 2H, NH2), 1,75 (м, 2H), 1,49-1,26 (м, 6H), 0,90 (м, 3H).
N-[3-(гексилокси)фенил]хинолин-4-амин Безводный пиридин (4 мл) выпаривали из сырого 3-(гексилокси)анилина (406 мг, 2,10 ммоль), затем добавляли 4-хлорхинолин (420 мг, 2,58 ммоль), DIEA (0,80 мл, 4,59 ммоль) и 1,5 мл NMP, и смесь нагревали при 160°C в толстостенной герметизированной пробирке в течение 24 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали. SPE, с промывкой 20% ЕА/Гексан и последующим элюированием 50% ЕА/Гексан +2% ТЕА, дал продукт в форме коричневого масла, которое содержало остаточный NMP. Кристаллизация из ЕА/Гексан дала 410 мг твердого вещества светлого желто-коричневого цвета. Rf 0,32 (50% 50% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,55 (д, 1, J=5,2 Гц), 8,03-7,96 (м, 2H), 7,63 (ддд, 1, J=1,2, 6,9, 8,4 Гц), 7,43 (ддд, 1, J=1,2, 6,7, 8,2 Гц), 7,26 (м, 1H), 7,14 (шир. с, 1H), 7,04 (д, 1, J=5,5 Гц), 6,87-6,83 (м, 2H), 6,69 (м, 1H), 3,90 (т, 2H, J=6 Гц), 1,75 (м, 2H), 1,45-1,30 (м, 6), 0,89 (м, 3).
Пример 59: N-[2-(бензилокси)фенил]хинолин-4-амин

Смесь 2-(бензилокси)анилина (301 мг, 1,51 ммоль), 4-хлорхинолина (268 мг, 1,64 ммоль) и 4-метилморфолина (0,18 мл, 1,64 ммоль) в 0,5 мл NMP нагревали в толстостенной герметизированной пробирке при 130°C в течение 20 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали. FC (7,5% MeOH/DCM) дал темное масло, которое содержало остаток 4-метилморфолина. Масло фильтровали через слой силикагеля, используя 30% ЕА/гексан +2% ТЕА, получая 268 мг твердого вещества желто-коричневого цвета. Rf 0,12 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 8,60 (д, 1H, J=5,4 Гц), 8,05 (дд, 1H, 1,0, 8,4 Гц), 7,88 (дд, 1H, J=0,8, 8,4 Гц), 7,66 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,53-7,40 (м, 2H), 7,37-7,29 (м, 5H), 7,15 (д, 1H, J=5,2 Гц), 7,07-6,98 (м, 3H), 5,17-5,10 (м, 2H, AB).
Пример 60: N-[2-(гексилокси)фенил]хинолин-4-амин

1-(Гексилокси)-2-нитробензол 2-нитрофенол (1,38 г, 9,93 ммоль), 1-бромгексан (1,30 мл, 9,30 ммоль) и K2CO3 (1,38 г, 10,0 ммоль) в 6 мл DMF перемешивали при комнатной температуре в течение 3 дней. Смесь разбавляли Et2O и промывали 0,25н. NaOH, пока водная фаза не обесцветилась, и затем солевым раствором. Органическую фазу высушивали над MgSO4 и концентрировали. Rf 0,39 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,78 (дд, 1H, J=1,7, 8,2 Гц), 7,48 (ддд, 1H, J=1,8, 7,3, 8,9 Гц), 7,04 (дд, 1H, J=1,0, 8,5 Гц), 6,97 (ддд, 1H, 1,2, 7,4, 8,2 Гц), 4,07 (т, 2H, J=6,4 Гц), 1,80 (м, 2H), 1,51-1,28 (м, 6H), 0,90 (м, 3H).
2-(гексилокси)анилин Смесь 1-(гексилокси)-2-нитробензола и 5% Pd/C (94 мг) в 15 мл MeOH и 15 мл ЕА перемешивали в атмосфере водорода в течение 5 часов. Затем смесь фильтровали через слой Целита и концентрировали. Остаток фильтровали через силикагель, используя 30% ЕА/гексан, получая 1,51 г коричневого масла, которое содержало остаток 1-бромгексана, как показал анализ ЯМР. SPE с промывкой гексаном и с элюированием 30% ЕА/Гексан дал 1,38 г красно-коричневого масла. Rf 0,26 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 6,81-6,68 (м, 4H), 3,98 (т, 2H, J=6,4 Гц), 3,76 (шир. с, 2H, NH2), 1,81 (м, 2H), 1,53-1,23 (м, 6H), 0,91 (м, 3H).
N-[2-(гексилокси)фенил]хинолин-4-амин Смесь 2-(гексилокси)анилина (282 мг, 1,46 ммоль), 4-хлорхинолина (258 мг, 1,58 ммоль) и 4-метилморфолина (0,18 мл, 1,64 ммоль) в 0,5 мл NMP нагревали в толстостенной герметизированной пробирке при 130°C в течение 20 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали. FC (7,5% MeOH/DCM) дала темное масло, которое содержало остаток 4-метилморфолинв. Масло фильтровали через слой силикагеля, используя 30% ЕА/гексан +2% ТЕА, получая 416 мг твердого вещества желто-коричневого цвета. Rf 0,13 (5% MeOH/DCM) 0,50 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,59 (дд, 1H, J=6,3, 11,5 Гц), 8,05 (м, 1H), 7,95 (м, 1H), 7,65 (ддд, 1H, J=1,3, 6,7, 9,7 Гц), 7,50-7,44 (м, 2H), 7,19-7,13 (м, 2H), 7,06-6,91 (м, 3H), 3,99 (т, 2H, J=6,4 Гц), 1,75 (м, 2H), 1,45-1,17 (м, 6H), 0,83 (м, 3H).
Пример 61: N-[2-Фтор-4-(гексилокси)фенил]хинолин-4-амин

2-Фтор-4-(гексилокси)-1-нитробензол (2,6 г) получали из 3-фтор-4-нитрофенола (5,0 г, 31,5 ммоль), 60% гидрида натрия (1,9 г), 1-бромгексана (4,75 мл) и 30 мл DMF согласно способу, используемому для получения 1-(8-бромоктилокси)-3-метилбензола.1H ЯМР (CDCl3) δ 8,05 (т, 1H), 6,7 (м, 2H), 4,0 (т, 2H), 1,8 (м, 2H), 1,6-1,3 (м, 6H), 0,9 (м, 3H).
2-Фтор-4-(гексилокси)анилин (1,6 г) получали из 2-фтор-4-(гексилокси)-1-нитробензолаа (2,6 г) согласно способу, используемому для получения 8-(3-этоксипропокси)октан-1-амина.1H ЯМР (CDCl3) δ 6,75-6,5 (м, 3H), 3,85 (т, 2H), 3,4 (шир. с, 2H, NH2), 1,75 (м, 2H), 1,5-1,2 (м, 6H), 0,9 (м, 3H).
N-[2-Фтор-4-(гексилокси)фенил]хинолин-4-амин (114 мг) получали из 2-фтор-4-(гексилокси)анилина (1,6 г), 4-хлорхинолина (1,33 г), ТЕА (5 мл) и NMP (0,5 мл) при 130°C в герметизированной пробирке в течение 5 дней согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,55 (д, 1H), 8,05 (д, 1H), 7,95 (д, 1H), 7,7 (м, 1H), 7,5 (м, 1H), 7,3 (м, 1H), 6,75 (м, 2H), 6,65 (д, 1H), 6,4 (шир. с, 1H, NH), 3,95 (т, 2H), 1,8 (м, 2H), 1,6-1,3 (м, 6H), 0,9 (м, 3H).
Пример 62: N-Бензилхинолин-4-амин
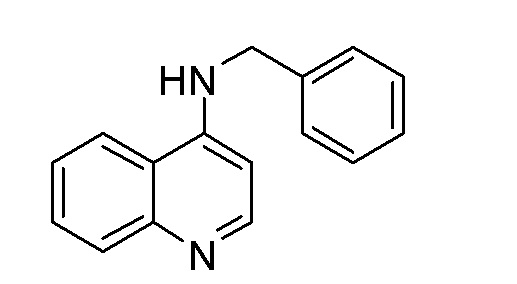
Смесь бензиламина (166 мг, 1,55 ммоль), 4-хлорхинолина (268 мг, 1,64 ммоль) и DIEA (0,50 мл, 2,87 ммоль) нагревали в толстостенной герметизированной пробирке при 130°C в течение 40 часов. Смесь охлаждали, добавляли смесь EtOH и H2O, и герметизированную смесь нагревали в течение 16 часов. Затем смесь охлаждали и разделяли между ЕА (3x) и 5%-ым Na2CO3 (3x) и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали, получая 385 мг масла. Очистка препаративной TLC (10% MeOH/DCM) дала 294 мг коричневого масла. Rf 0,33 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,49 (д, 1H, J=5,2 Гц), 7,98 (дд, 1H, J=0,8, 8,4 Гц), 7,82 (д, 1H, J=8,4 Гц), 7,61 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,42-7,27 (м, 6H), 6,41 (д, 1H, J=5,4 Гц), 5,76 (шир. с, 1H), 4,51 (м, 2H, AB).
Пример 63: N-Фенэтилхинолин-4-амин
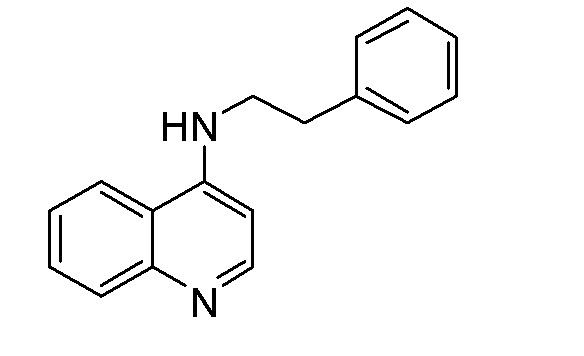
Смесь 2-фенэтиламина (177 мг, 1,46 ммоль), 4-хлорхинолина (258 мг, 1,58 ммоль) и DIEA (0,50 мл, 2,87 ммоль) нагревали при 130°C в герметизированной пробирке в течение 40 часов. Охлажденную смесь разделяли между ЕА (3x) и 5%-ым Na2CO3 (3x) и солевым раствором, и органические фазы высушивали над безводным Na2SO4 и концентрировали, получая твердое вещество. Промывка Et2O дала 230 мг твердого вещества красного цвета.1H ЯМР (CDCl3) δ 8,55 (д, 1H, J=5,4 Гц), 7,98 (м, 1H), 7,64-7,58 (м, 2H), 7,42-7,24 (м, 6H), 6,48 (д, 1H, J=5,4 Гц), 5,17 (шир. с, 1H, NH), 3,60 (м, 2H), 3,06 (т, 2H, J=6,9 Гц).
Пример 64: N-[4-(гексилокси)бензил]хинолин-4-амин
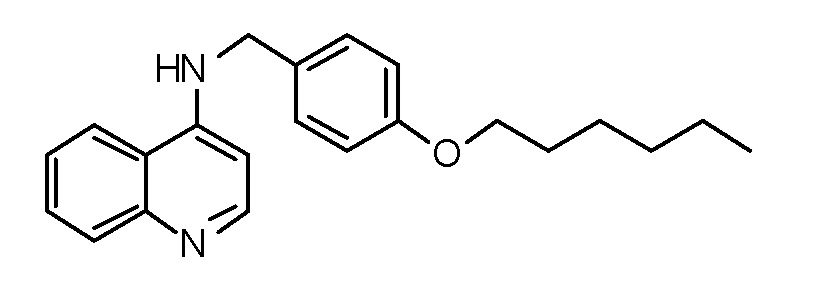
4-(Гексилокси)бензонитрил Смесь 4-цианофенола (25,2 г, 212 ммоль), K2CO3 (24,7 г, 233 ммоль) и 1-бромгексана (29,6 мл, 212 ммоль) в 150 мл DMF перемешивали при комнатной температуре в течение 24 часов и затем при 55°C в течение 24 часов. 4-Цианофенол оставался, как показала TLC. Na2CO3 (7,0 г, 66 ммоль), и добавляли 1-бромгексанe (3,0 мл, 21 ммоль), и через 24 часа температуру понижали до 40°C, и добавляли дополнительное количество Na2CO3 (12,4 г, 117 ммоль) и 1-бромгексана (10,0 мл, 72 ммоль). Однако через 24 часа не наблюдалось потребления оставшегося 4-цианофенола. Смесь охлаждали до комнатной температуры и добавляли 6 мл концентрированного NH4OH. После отстаивания в течение 3 дней смесь разделяли между ЕА (3×250 мл) и H2O (300 и 200 мл), 1M HCl (100 мл) и солевым раствором (150 мл). Объединенные органические фазы высушивали над MgSO4 и концентрировали. SPE (10% ЕА/Гексан) дал 35,8 г бесцветного масла, которое затвердевало при отстаивании. Rf 0,63 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,55 и 6,92 (м, 4H, AA’BB’), 3,98 (т, 2H, J=6,6 Гц), 1,78 (м, 2H), 1,43 (м, 2H), 1,35-1,30 (м, 4H), 0,89 (м, 3H);13C ЯМР (CDCl3) δ 162,6, 134,1, 119,5, 115,4, 103,8, 68,6, 31,7, 29,1, 25,8, 22,7, 14,2.
[4-(гексилокси)фенил]метанамин 4-(гексилокси)бензонитрил (35,8 г, 176 ммоль) забирали в 350 мл THF, и смесь охлаждали ванной со льдом. LAH (7 г, 184 ммоль) добавляли осторожно частями. Через 1 час смесь нагревали с обратным холодильником. Через 15 часов смесь охлаждали ванной со льдом. Осторожно, с полным перемешиванием, частями и в этой последовательности, 7 мл H2O, 7 мл 15%-ого NaOH и 21 мл H2O добавляли к ледяной смеси. Полученную гетерогенную смесь разбавляли 350 мл IPA. Смесь фильтровали через слой Целита, и твердые частицы промывали 200 мл IPA. Фильтрат концентрировали, получая 34,4 г продукта, который содержал остаточный IPA. Rf 0,25 (5% MeOH/DCM +2% ТЕА, нингидрин (+));1H ЯМР (CDCl3) δ 7,17 и 6,83 (м, 4H, AA’BB’), 3,90 (т, 2H, J=6,7 Гц), 3,74 (с, 2H), 2,00 (шир. с, 2H, NH2), 1,78 (м, 2H), 1,48-1,27 (м, 6H), 0,88 (м, 3H).
N-[4-(гексилокси)бензил]хинолин-4-амин [4-(гексилокси)фенил]метанамин (166 ммоль) забирали в 400 мл 1-пентанола, и 150 мл летучего материала удаляли перегонкой, чтобы обеспечить безводные условия. Смеси давали охладиться до 70°C и добавляли трипропиламин (63 мл, 330 ммоль) и 4-хлорхинолин (28 г, 172 ммоль). Нагревание с обратным холодильником продолжали. Через 16 часов TLC аликвоты показала сохранение очень небольшого количества исходного материала нингидрина (+). Летучий материал удаляли перегонкой и упариванием. Охлажденную смесь разбавляли 1:2 DCM/EA и промывали 3н. NaOH (60 мл), H2O и солевым раствором. Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали. SPE, с элюированием 50% ЕА/Гексан и затем 15% EtOH/DCM, дал коричневое масло. Масло забирали в ЕА и промывали 5%-ым Na2CO3 и солевым раствором. Органическую фазу высушивали над Na2SO4, фильтровали и концентрировали. К остатку добавляли ЕА (10 мл) и затем гексаны (20 мл). Получали осадок. Бесцветный осадок собирали фильтрацией и промывали 100 мл 50% ЕА/гексан и затем 50 мл 30% ЕА/гексан. Второй выход получали из объединенных фильтратов. Выходы объединяли и высушивали в вакууме, получая 38,4 г. Rf 0,25 (5% MeOH/DCM); Т.пл. 103,5-104,0°C;1H ЯМР (CDCl3) δ 8,55 (д, 1H, J=5,5 Гц) 8,00 (д, 1H, J=0,7 Гц), 7,98 (д, 1H, J=0,7 Гц), 7,74 (м, 1H), 7,65-7,61 (м, 1H), 7,41 (м, 1H), 7,30 и 6,90 (м, 4H, AA’BB’), 6,46 (д, 1H, J=5,1 Гц), 5,33 (м, 1H), 4,43 (м, 2H, AB), 3,96 (т, 2H, J=6,6 Гц), 1,79 (м, 2H), 1,46 (м, 2H), 1,39-1,30 (м, 4H), 0,90 (м, 3H);13C ЯМР (CDCl3) δ 159,2, 151,4, 149,6, 148,7, 130,3, 129,5, 129,2, 129,1, 124,9, 119,5, 119,0, 115,2, 99,5, 68,4, 47,4, 31,8, 29,4, 25,9, 22,8, 14,2.
Пример 65: N-[3-(гексилокси)бензил]хинолин-4-амин

3-(гексилокси)бензальдегид Смесь 3-гидроксибензальдегид (10,3 г, 84,4 ммоль), K2CO3 (13,9 г, 100,7 ммоль) и 1-бромгексан (11,2 мл, 80,0 ммоль) в 90 мл DMF нагревали при 60°C в течение 12 часов. Смесь охлаждали до комнатной температуры, вливали в 30% ЕА/гексан и промывали H2O, 5%-ым Na2CO3, H2O, 0,1M HCl и солевым раствором. Органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 15,8 г коричневого масла. Rf 0,56 (20% ЕА/Гексан).1H ЯМР (CDCl3) δ 9,94 (с, 1H), 7,43-7,36 (м, 3H), 7,14 (м, 1H), 3,99 (т, 2H, J=6,6 Гц), 1,79 (м, 2H), 1,45 (м, 2H), 1,37-1,28 (м, 4H), 0,89 (м, 3H);13C ЯМР (CDCl3) δ 192,4, 159,9, 137,9, 130,1, 123,4, 122,1, 113,0, 68,4, 31,7, 29,2, 25,8, 22,7, 14,2.
[3-(гексилокси)фенил]метанол 3-(гексилокси)бензальдегид забирали в 160 мл MeOH, и смесь охлаждали, используя ванну со льдом. NaBH4 (3,17 г, 83 ммоль) добавляли в трех частях, в течение которых из смеси выделялся газ. Спустя три часа после заключительного добавления, добавляли 10 мл ацетона, и смесь оставляли на 3 дня. Затем летучий материал выпаривали, и остаток разделяли между 1:1 ЕА/Гексан и H2O, 5% Na2CO3 (2x), H2O, 0,1M HCl (2x) и солевым раствором. Органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 15,3 г светло-коричневого масла. Rf 0,28 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,16 (м, 1H), 7,83-7,81 (м, 2H), 7,73 (м, 1H), 5,55 (с, 2H), 4,86 (т, 2H, J=6,6 Гц), 2,86 (шир. с, 1H, OH), 2,69 (м, 2H), 2,37 (м, 2H), 2,27-2,23 (м, 4H), 1,82 (т, 3H, J=7,0 Гц);13C ЯМР (CDCl3) δ 159,6, 142,7, 129,7, 119,1, 114,0, 113,1, 69,2, 65,4, 31,8, 29,4, 25,9, 22,8, 14,2.
3-(гексилокси)бензил метансульфонат [3-(гексилокси)фенил]метанол забирали в 180 мл THF и 100 мл ЕА и охлаждали с использованием ванны со льдом. Добавляли ТЕА (12,4 мл, 88 ммоль) и затем метансульфонил хлорид (6,30 мл, 80 ммоль). Быстро образовывался белый осадок. Через 2 часа добавляли 5 мл H2O, и летучие компоненты выпаривали. Остаток разделяли между ЕА (3×300 мл) и H2O, насыщенным NaHCO3, H2O, 0,1M HCl и солевым раствором (100 мл каждый). Объединенные органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 20,75 г светло-коричневого масла. Rf 0,50 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,3 (м, 1H), 6,9-6,8 (м, 3H), 5,2 (с, 2H), 4,0 (т, 2H, J=6,6 Гц), 2,9 (2s, 3H), 1,8 (м, 2H), 1,4 (м, 2H), 1,4-1,3 (м, 4H), 0,9 (м, 3H);13C ЯМР (CDCl3) δ 159,7, 134,9, 130,1, 120,9, 115,7, 114,9, 71,7, 68,3, 38,6, 31,7, 29,4, 25,9, 22,8, 14,2.
N-[3-(гексилокси)бензил]фталимид Смесь 3-(гексилокси)бензил метансульфоната и фталимида калия (15,4 г, 83,2 ммоль) в 200 мл DMF перемешивали, используя приводную мешалку, при комнатной температуре в течение 4 часов и затем при 50°C в течение 4 часов. Затем добавляли H2O (100 мл), и летучий материал выпаривали. Остаток разделяли между ЕА и 5%-ым Na2CO3 (2x), H2O, 0,1M HCl и солевым раствором. Органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. Кристаллизация от IPA дала 20,74 г бесцветного твердого вещества. Rf 0,56 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,9 и 7,7 (м, 4H, AA’BB’), 7,2 (м, 1H), 7,0-6,9 (м, 2H), 6,8 (м, 1H), 4,8 (с, 2H), 3,9 (т, 2H, J=6,6 Гц), 1,8 (м, 2H), 1,5 (м, 2H), 1,3-1,2 (м, 4H), 0,9 (м, 3H);13C ЯМР (CDCl3) δ 168,2, 159,6, 138,0, 134,2, 132,3, 129,8, 123,6, 120,8, 114,8, 114,1, 68,1, 41,8, 31,8, 29,4, 25,9, 22,8, 14,2.
[3-(гексилокси)фенил]метанамин Гидразин моногидрат (2,20 мл, 45,3 ммоль) добавляли к смеси N-[3-(гексилокси)бензил]фталимида (10,1 г, 30,0 ммоль) и 90 мл денатурированного EtOH с механическим перемешиванием. Смесь нагревали с обратным холодильником в течение 15 часов, в течение какого времени формировался бесцветный осадок. Смесь концентрировали упариванием, и остаток разделяли между DCM (150, 2×80 мл) и 5% Na2CO3 (2×100 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали. SPE, с промывкой 50% изопропил ацетат/Гексан и последующим элюированием 3% MeOH/DCM +2% ТЕА дал 4,40 г продукта в форме светло-желтой жидкости, которую использовали без дополнительного высушивания. Rf 0,26 (10% MeOH/DCM, нингидрин (+));1H ЯМР (CDCl3) δ 7,22 (м, 1H), 6,87-6,84 (м, 2H), 6,76 (дд, 1H, J=2,4, 8,0 Гц), 3,94 (т, 2H, J=6,6 Гц), 3,82 (шир. с, 2H, AB), 1,76 (м, 2H), 1,59 (шир. с, 2H, NH2), 1,47-1,29 (м, 6H), 0,89 (т, 3H, J=6,8 Гц).
N-[3-(гексилокси)бензил]хинолин-4-амин [3-(гексилокси)фенил]метанамин (7,20 г, 34,8 ммоль) забирали в 100 мл 1-пентанола, и затем 25 мл летучего материала удаляли перегонкой. Смесь охлаждали до температуры ниже температуры кипения и добавляли трипропиламин (10,0 мл, 52,4 ммоль) и 4-хлорхинолин (5,67 г, 34,8 ммоль). Нагревание с обратным холодильником продолжали. Через 26 часов летучий материал удаляли выпариванием. Смесь разбавляли DCM (350 мл) и промывали 1н. NaOH (50 мл) и 5% Na2CO3 (50 мл). Водные фазы экстрагировали DCM (100 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали. SPE, с промывкой 50% ЕА/Гексан и последующим элюированием 50% ЕА/Гексан +2% ТЕА, дал фракции продукта, которые объединяли и концентрировали. Остаток разделяли между ЕА (400, 175 мл) и 5% Na2CO3 и солевым раствором (50 мл каждый). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали приблизительно до 50 мл, после чего образовывался осадок. Осадок перекристаллизовывали нагреванием и охлаждением, в конце чего добавляли 20 мл гексанов. При отстаивании в течение ночи, бесцветный осадок собирали фильтрацией и промывали 30% ЕА/Гексан. (Маточный раствор содержал приблизительно 2,4 г материала, но его дополнительно не обрабатывали.) Высушивание в вакууме дало 4,05 г. Rf 0,20 (10% MeOH/DCM); Т.пл. 109,5-110,0°C;1H ЯМР (CDCl3) δ 8,55 (д, 1H, J=5,1 Гц), 8,00 (дд, 1H, J=0,7, 8,4 Гц), 7,76 (дд, 1H, J=1,1, 8,5 Гц), 7,65 (ддд, 1H, J=1,4, 6,9, 8,4 Гц), 7,44 (м, 1H), 7,29 (т, 1H), 6,98-6,94 (м, 2H), 6,86 (дд, 1H, J=1,8, 8,1 Гц), 6,46 (д, 1H, J=5,2 Гц), 5,34 (т, 1H, NH), 4,50 (м, 2H, AB), 3,94 (т, 2H, J=6,6 Гц), 1,80-1,73 (м, 2H), 1,46-1,40 (м, 2H), 1,35-1,30 (м, 4H), 0,91-0,87 (м, 3H);13C ЯМР (CDCl3) δ 160,0, 151,4, 149,6, 148,8, 139,4, 130,4, 130,2, 129,2, 125,0, 119,8, 119,5, 119,1, 114,2, 113,9, 99,7, 68,3, 47,9, 31,8, 29,5, 25,9, 22,8, 14,2.
Пример 66: N-[2-(гексилокси)бензил]хинолин-4-амин

[2-(гексилокси)фенил]метанол Смесь 3-гидроксибензилового спирта (3,06 г, 24,7 ммоль), 1-бромгексана (3,20 мл, 22,9 ммоль), K2CO3 (3,50 г, 25,4 ммоль) и 10 мл DMF вводили в реакцию в течение 40 часов. Смесь разделяли между ЕА и H2O, 5%-ым Na2CO3, H2O, 0,1M HCl и солевым раствором. Органические фазы высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой 5% ЕА/Гексан и с элюированием 15% ЕА/Гексан, дал 2,86 г продукта. Rf 0,31 (15% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,27-7,22 (м, 2H), 6,95-6,85 (м, 2H), 4,69 (с, 2H), 4,01 (т, 2H, J=6,5 Гц), 2,45 (шир. с, 1H, OH), 1,81 (м, 2H), 1,52-1,32 (м, 6H), 0,91 (м, 3H).
N-[2-(гексилокси)бензил]фталимид DIEA (4,90 мл, 28,1 ммоль) добавляли к смеси [2-(гексилокси)фенил]метанола (2,86 г, 13,8 ммоль) и метансульфонил хлорида (2,10 мл, 26,8 ммоль) в 25 мл диоксана и 10 мл ЕА, охлажденных ванной со льдом. Через 2 часа смесь разделяли между ЕА и H2O, насыщенным NaHCO3, H2O, 0,1M HCl и солевым раствором. Органические фазы высушивали над безводным Na2SO4 и концентрировали. Остаток фильтровали через слой силикагеля, используя 50% ЕА/гексан, и фильтрат концентрировали, получая сырой 2-(гексилокси)бензилов метансульфонат. Сырой 2-(гексилокси)бензилов метансульфонат забирали в 150 мл ацетона, добавляли йодид натрия (3,1 г, 21 ммоль), и смесь нагревали с обратным холодильником в течение 1,5 часов. Затем растворитель выпаривали, и твердый остаток разделяли между ЕА и H2O. Органическую фазу обесцвечивали водным раствором Na2S2O3 и промывали H2O и солевым раствором, высушивали над безводным MgSO4 и концентрировали. Остаток фильтровали через слой силикагеля, используя 25% ЕА/гексан, и фильтрат концентрировали, получая сырой 1-(гексилокси)-2-(йодметил)бензол. Смесь сырого 1-(гексилокси)-2-(йодметил)бензола и фталимида калия (3,8 г, 20 ммоль) в 12 мл DMF вводили в реакцию при комнатной температуре в течение 24 часов. Смесь разделяли между ЕА и H2O, водным Na2S2O3, H2O, 5%-ым Na2CO3, H2O, 0,1M HCl и солевым раствором, и органические фазы высушивали над безводным MgSO4 и концентрировали. SPE, с промывкой 5% ЕА/Гексан и с элюированием 15% ЕА/Гексан, дал 2,30 г масла. Аккуратная TLC (с избеганием перегрузки и с использованием более длинного планшета) показала, что продукт содержал почти совместно мигрирующие примеси. Rf 0,37 (15% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,84 и 7,71 (м, 4H, AA’BB’), 7,27-7,14 (м, 2H), 6,88-6,81 (м, 2H), 4,91 (с, 2H), 3,96 (т, 2H, J=6,5 Гц), 1,77 (п, 2H, J=6,7 Гц), 1,46-1,22 (м, 6H), 0,88 (м, 3H).
[2-(гексилокси)фенил]метанамин Гидразин моногидрат добавляли к смеси N-[2-(гексилокси)бензил]фталимида и 80 мл EtOH, и смесь нагревали с обратным холодильником в течение 20 часов. Смесь охлаждали, и летучие компоненты выпаривали. Остаток разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой 18% ЕА/Гексан, затем 4% MeOH/DCM и с элюированием 6%-ым MeOH/DCM +2% ТЕА, дал продукт нингидрин (+). Rf 0,61 (5% MeOH/DCM +2% ТЕА).
N-[2-(гексилокси)бензил]хинолин-4-амин Смесь [2-(гексилокси)фенил]метанамина (417 мг, 2,01 ммоль), 4-хлорхинолина (430 мг, 2,64 ммоль) и DIEA (0,50 мл, 2,86 ммоль) в 1 мл NMP нагревали при 150°C в герметизированной пробирке в течение 18 часов. Затем смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 2,5% MeOH/DCM и последующим элюированием 7% MeOH/DCM, дал 545 мг твердого вещества. Rf 0,20 (10% MeOH/DCM); Т.пл. 90-91°C (из ЕА/Гексан);1H ЯМР (CDCl3) δ1H ЯМР (CDCl3) δ 8,52 (д, 1H, J=5,5 Гц), 7,98 (дд, 1H, J=0,7, 8,4 Гц), 7,77 (дд, 1H, J=1,0, 8,4 Гц), 7,61 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,39 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 7,31-7,23 (м, 2H), 6,92-6,87 (м, 2H), 6,48 (д, 1H, J=5,2 Гц), 5,71 (шир. т, 1H, J=5,2 Гц, NH), 4,54 (м, 2H, AB), 4,02 (т, 2H, J=6,4 Гц), 1,84-1,74 (м, 2H), 1,50-1,17 (м, 6H), 0,87-0,81 (м, 3H).
Пример 67: N-[3-Фтор-4-(гексилокси)бензил]хинолин-4-амин
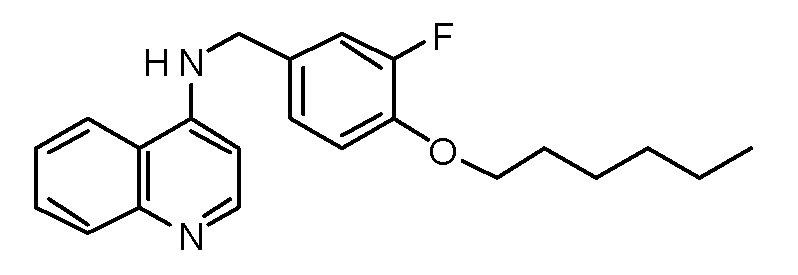
3-Фтор-4-(гексилокси)бензонитрил (721 мг) получали из 3-фтор-4-гидроксибензoнитрила (1,5 г, 10,9 ммоль), 60% гидрида натрия (654 мг), 1-бромгексана (1,30 мл), и 10 мл DMF согласно способу, используемому для получения 1-(8-бромоктилокси)-3-метилбензола.1H ЯМР (CDCl3) δ 7,5 (т, 1H), 6,8-6,6 (м, 2H), 3,95 (т, 2H), 1,8 (м, 2H), 1,5-1,2 (м, 6H), 0,9 (м, 3H).
[3-Фтор-4-(гексилокси)фенил]метанамин (212 мг, 0,9 ммоль) получали из 3-фтор-(4-гексилокси)бензонитрила (721 мг, 3,3 ммоль) и LAH (6,6 ммоль) в THF (50 мл) при 0°C в течение 4 часов и при комнатной температуре в течение 12 часов согласно способу, используемому для получения [4-(гексилокси)фенил]метанамина.1H ЯМР (CDCl3) δ 7,15 (т, 1H), 6,7-6,5 (м, 2H), 3,9 (т, 2H), 3,75 (с, 2H), 1,75 (м, 2H), 1,6-1,2 (м, 8H), 0,9 (м, 3H).
N-[3-Фтор-4-(гексилокси)бензил]хинолин-4-амин (325 мг) получали из [3-фтор-4-(гексилокси)фенил]метанамина (486 мг, 2,2 ммоль), 4-хлорхинолина (541 мг, 3,3 ммоль), ТЕА (4 мл) и NMP (0,5 мл) при 130°C в герметизированной пробирке в течение 5 дней согласно способу, используемому для получения N-[8-(3-этоксипропокси)октил]хинолин-4-амина.1H ЯМР (CDCl3) δ 8,5 (д, 1H), 8,0 (д, 1H), 7,8 (д, 1H), 7,6 (м, 1H), 7,4 (м, 1H), 7,25 (т, 1H), 6,6 (м, 2H), 6,45 (д, 1H), 5,8 (шир. с, 1H, NH), 4,5 (м, 2H, AB), 3,9 (т, 2H), 1,8 (м, 2H), 1,6-1,2 (м, 6H), 0,9 (м, 3H).
Пример 68: N-[4-(децилокси)бензил]хинолин-4-амин

4-(децилокси)бензонитрил Смесь 4-гидроксибензoнитрила (4,32 г, 36,3 ммоль), 1-бромдекана (6,80 мл, 32,9 ммоль) и K2CO3 (6,61 г, 47,8 ммоль) в 20 мл DMF вводили в реакцию в течение 2 дней. Растворитель выпаривали в вакууме. Остаток разделяли между 50% ЕА/Гексан (3×150 мл) и 5% Na2CO3 (3×80 мл), H2O (40 мл), 0,1M HCl (40 мл) и солевым раствором (80 мл). Органические фазы высушивали над безводным Na2SO4 и концентрировали, получая 8,30 г бесцветного масла, которое затвердевало при отстаивании.1H ЯМР (CDCl3) δ 7,54 и 6,90 (м, 4H, AA’BB’), 3,97 (т, 2H, J=6,6 Гц), 1,78 (м, 2H), 1,42 (м, 2H), 1,34-1,25 (м, 12H), 0,86 (м, 3H);13C ЯМР (CDCl3) δ 162,6, 134,0, 119,4, 115,3, 103,7, 68,5, 32,0, 29,6, 29,4, 29,4, 29,1, 26,0, 22,8, 14,2.
[4-(децилокси)фенил]метанамин Литий-алюминийгидрид (2,0 г, 53 ммоль) добавляли частями к смеси 4-(децилокси)бензонитрила (8,30 г, 32,0 ммоль) и 80 мл THF, охлажденного ванной со льдом. Затем смеси давали нагреться до комнатной температуры. Через 2 часа смесь охлаждали ванной со льдом, и добавляли последовательно и осторожно 2 мл H2O, 2 мл 15% NaOH и 6 мл H2O. Полученные твердые частицы отфильтровывали, и твердые частицы промывали 5% MeOH/DCM +1% ТЕА. Фильтрат концентрировали, затем забирали в DCM и промывали 5%-ым Na2CO3. Органическую фазу высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой 40% изопропил ацетат/Гексан и с элюированием 3% MeOH/DCM +2% ТЕА, дал фракции нингидрина (+). Эти фракции концентрировали, и остаток забирали в DCM, промывали 5%-ым Na2CO3, высушивали над безводным Na2SO4 и концентрировали, получая 7,61 г бесцветного твердого вещества. Rf 0,11 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 7,18 и 6,83 (м, 4H, AA’BB’), 3,90 (т, 2H, J=6,6 Гц), 3,76 (с, 2H), 1,75 (м, 2H), 1,56 (шир. с, 2H, NH2), 1,43 (м, 2H), 1,39-1,26 (м, 12H), 0,87 (т, 3H, J=6,9 Гц);13C ЯМР (CDCl3) δ 158,1, 135,4, 128,2, 114,5, 68,0, 46,0, 32,0, 29,6, 29,6, 29,5, 29,4, 26,1, 22,7, 14,2.
N-[4-(децилокси)бензил]хинолин-4-амин [4-(децилокси)фенил]метанамин (5,90 г, 22,4 ммоль) забирали в 100 мл 1-пентанола, и 25 мл удаляли перегонкой. Смесь немного охлаждали и добавляли трипропиламин (6,50 мл, 34,1 ммоль) и 4-хлорхинолин (3,63 г, 22,3 ммоль). Нагревание с обратным холодильником продолжали в течение 24 часов. Затем летучие компоненты выпаривали, и остаток разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над безводным Na2SO4 и концентрировали на силикагеле. SPE, с промывкой 50% ЕА/Гексан и последующим элюированием 10% MeOH/DCM, дал твердое вещество. Твердое вещество забирали в DCM, промывали 5% Na2CO3, высушивали над безводным Na2SO4 и концентрировали, получая твердое вещество. Перекристаллизация из ЕА/Гексан дала 3,70 г бесцветного твердого вещества. Rf 0,13 (10% MeOH/DCM); Т.пл. 96,5-97,0°C;1H ЯМР (CDCl3) δ 8,55 (д, 1H, J=5,2 Гц), 7,99 (д, 1H, J=8,5 Гц), 7,74 (д, 1H, J=8,4 Гц), 7,63 (м, 1H), 7,42 (м, 1H), 7,30 и 6,90 (м, 4H, AA’BB’), 6,47 (д, 1H, J=5,1 Гц), 5,30 (шир. с, 1H, NH), 4,44 (м, 2H, AB), 3,95 (м, 2H), 1,79 (м, 2H), 1,46 (м, 2H), 1,32-1,27 (м, 10H), 0,88 (м, 3H);13C ЯМР (CDCl3) δ 159,1, 151,3, 149,6, 148,7, 130,2, 129,4, 129,2, 129,2, 124,9, 119,5, 118,9, 115,1, 99,5, 68,3, 47,3, 32,1, 29,8, 29,8, 29,6, 29,5, 29,5, 26,2, 22,9, 14,3.
Пример 69: N-[3-(децилокси)бензил]хинолин-4-амин

3-(децилокси)бензальдегид 1-Бромдекан (15,0 мл, 72,6 ммоль) добавляли к смеси 3-гидроксибензальдегида (9,75 г, 79,9 ммоль) и K2CO3 (12,2 г, 88,4 ммоль) в 80 мл DMF, нагретого при 50°C, используя механическое перемешивание. Через 22 часа смесь разбавляли H2O (100 мл) и экстрагировали ЕА (3×100 мл), и органические фазы промывали 5%-ым Na2CO3 и H2O (100 мл каждый), 0,1M HCl (2×100 мл) и солевым раствором (100 мл), и высушивали над безводным Na2SO4. Выпаривание летучих компонентов привело к 18,74 г продукта в форме коричневого масла. Rf 0,54 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 9,96 (с, 1H), 7,44-7,37 (м, 3H), 7,18 (м, 1H), 4,00 (т, 2H, J=6,6 Гц), 1,80 (м, 2H), 1,46 (м, 2H), 1,36-1,23 (м, 12H), 0,88 (м, 3H);13C ЯМР (CDCl3) δ 192,4, 159,9, 138,0, 130,2, 123,5, 122,2, 113,0, 68,5, 32,1, 29,8, 29,7, 29,6, 29,5, 29,3, 26,2, 22,9, 14,3.
[3-(децилокси)фенил]метанол Боргидрид натрия (2,63 г, 69,2 ммоль) добавляли к смеси 3-(децилокси)бензальдегида (18,74 г) и 160 мл MeOH, охлажденного ванной со льдом. Через 1 час, остаточный гидрид гасили, добавляя H2O, и медленно добавляли 80 мл 1M HCl, приводя к осаждению. Летучие компоненты выпаривали, и остаток разделяли между 50% ЕА/Гексан и H2O, 5% Na2CO3 (2x), H2O и солевым раствором. Органические фазы высушивали над безводным Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 21,05 г продукта в форме твердого вещества светло-коричневого цвета. Rf 0,11 (10% ЕА/Гексан) 0,28 (1:4:5 ЕА/толуол/Гексан);1H ЯМР (CDCl3) δ 7,24 (м, 1H), 6,90-6,88 (м, 2H), 6,81 (м, 1H), 4,60 (шир. с, 2H, AB), 3,94 (т, 2H, J=6,6 Гц), 2,55 (шир. с, 1H, OH), 1,78 (м, 2H), 1,46 (м, 2H), 1,38-1,24 (м, 12H), 0,91 (м, 3H);13C ЯМР (CDCl3) δ 159,5, 142,7, 129,6, 119,0, 113,8, 113,0, 68,1, 65,2, 32,0, 29,8, 29,7, 29,6, 29,5, 29,4, 26,2, 22,8, 14,3.
3-(децилокси)бензил метансульфонат Триэтиламин (11,8 мл, 84,4 ммоль) добавляли к смеси [3-(децилокси)фенил]метанамина (21,05 г, ммоль) и метансульфонил хлорида (6,60 мл, 84,4 ммоль) в 120 мл THF, охлажденного ванной со льдом. Быстро образовывался осадок. Через 1 час добавляли 5 мл H2O, и летучие компоненты выпаривали. Остаток разделяли между ЕА и H2O, насыщенным NaHCO3, H2O, 0,1M HCl и солевым раствором. Органические фазы высушивали над безводным Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 23,53 г 3-(децилокси)бензил метансульфоната в форме янтарного масла, которое затвердевало при отстаивании. Rf 0,45 (1:4:5 ЕА/толуол/Гексан) 0,35 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,29 (м, 1H), 6,98-6,90 (м, 3H), 5,19 (м, 2H, AB), 3,95 (т, 2H, J=6,6 Гц), 2,90 (с, 3H), 1,78 (м, 2H), 1,43 (м, 2H), 1,36-1,28 (м, 12H), 0,88 (м, 3H);13C ЯМР (CDCl3) δ 159,6, 134,9, 130,1, 120,8, 115,6, 114,8, 71,7, 68,2, 38,4, 32,0, 29,7, 29,7, 29,5, 29,4, 29,4, 26,2, 22,8, 14,3.
N-[3-(децилокси)бензил]фталимид Смесь 3-(децилокси)бензил метансульфоната (23,53 г, 68,8 ммоль) и фталимида калия (14,00 г, 75,7 ммоль) в 90 мл DMF вводили в реакцию при комнатной температуре в течение 16 часов и при 50-60°C в течение 3 часов. Смесь охлаждали, разбавляли 350 мл H2O и экстрагировали ЕА (3×400 мл). Органические фазы промывали H2O (3×200 мл) и солевым раствором (2×200 мл), высушивали над безводным Na2SO4 и концентрировали, получая бесцветное твердое вещество. Твердое вещество разбивали и промывали 10% ЕА/Гексан, получая 11,40 г твердого вещества в форме бесцветного твердого вещества. Смывы частично концентрировали, получая дополнительные 6,95 г бесцветного твердого вещества. Rf 0,50 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,84 и 7,70 (м, 4H, AA’BB’), 7,21 (м, 1H), 7,00-6,96 (м, 2H), 6,79 (м, 1H), 4,81 (с, 2H, AB), 3,92 (т, 2H, J=6,6 Гц), 1,74 (м, 2H), 1,43 (м, 2H), 1,30-1,26 (м, 12H), 0,88 (м, 3H);13C ЯМР (CDCl3) δ 168,2, 159,6, 137,9, 134,2, 132,4, 129,9, 123,6, 120,8, 114,8, 114,1, 68,2, 41,8, 32,1, 29,8, 29,8, 29,6, 29,5, 29,5, 29,5, 26,2, 22,9, 14,3.
[3-(децилокси)фенил]метанамин Гидразин моногидрат (3,90 мл, 80,3 ммоль) добавляли в трех частях к смеси N-[3-(децилокси)бензил]фталимида (5,12 г, 13,0 ммоль) и IPA при нагревании с обратным холодильником. После того, как исходный материал был израсходован, что наблюдали с помощью TLC (30 часов), смесь охлаждали и концентрировали. Остаток разделяли между изопропил ацетатом и 5%-ым Na2CO3 и солевым раствором, и органические фазы высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой 50% изопропил ацетат/Гексан и с последующим элюированием 3% MeOH/DCM +2% ТЕА, дал материал нингидрина (+). Частичная концентрация и промывка фильтрата 5%-ым Na2CO3 и высушивание над Na2SO4 дали 3,25 г желтого масла после высушивания в вакууме.
N-[3-(децилокси)бензил]хинолин-4-амин Смесь [3-(децилокси)фенил]метанамина (2,54 г, 9,66 ммоль), 4-хлорхинолина (1,73 г, 10,62 ммоль) и трипропиламина (4,00 мл, 21,0 ммоль) в 65 мл 1-пентанол нагревали с обратным холодильником в течение 16 часов. Аналитическая TLC показала существенное количество непрореагировавшего [3-(децилокси)фенил]метанамина. Добавляли 4-Хлорхинолин (0,85 г, 5,21 ммоль) и трипропиламин (2,00 мл, 10,5 ммоль). Через 24 часа смесь охлаждали и добавляли 15 мл 1н. NaOH. Летучие компоненты выпаривали, остаток забирали в DCM и промывали 5%-ым Na2CO3, и органическую фазу высушивали над безводным Na2SO4 и упаривали на силикагеле. SPE, с промывкой 70% ЕА/Гексан и с элюированием 50% ЕА/Гексан +2% ТЕА, дал 2,62 г твердого вещества белого цвета после кристаллизации из IPA. Перекристаллизация из 30% ЕА/гексан дала 2,00 г N-[3-(децилокси)бензил]хинолин-4-амина в форме порошкообразного твердого вещества белого цвета. Rf 0,24 (50% ЕА/Гексан +2% ТЕА) 0,40 (10% MeOH/DCM); Т.пл. 71,0-72,0°C;1H ЯМР (CDCl3) δ 8,55 (д, 1H, J=5,1 Гц), 8,00 (м, 1H), 7,77 (м, 1H), 7,64 (ддд, 1H, J=1,5, 7,0, 8,5 Гц), 7,43 (ддд, 1H, J=1,5, 7,0, 8,5 Гц), 7,28 (м, 1H), 6,97-6,93 (м, 2H), 6,85 (дд, 1H, J=1,8, 8,1 Гц), 6,45 (д, 1H, J=5,5 Гц), 5,38 (м, 1H, NH), 4,49 (м, 2H, AB), 3,94 (м, 2H), 1,77 (м, 2H), 1,42 (м, 2H), 1,34-1,26 (м, 10H), 0,87 (м, 3H); 159,9, 151,4, 149,6, 148,7, 139,3, 130,3, 130,2, 129,2, 125,0, 119,7, 119,5, 18,95, 114,1, 113,8, 99,6, 68,3, 47,8, 32,1, 29,8, 29,8, 29,6, 29,5, 29,5, 26,3, 22,9, 14,3.
Пример 70: N-(3-Феноксибензил)хинолин-4-амин

3-Феноксибензил метансульфонат Смесь 3-феноксибензилового спирта (15,44 г, 77,2 ммоль) и ТЕА (13,1 мл, 93,4 ммоль) в 180 мл THF и 100 мл ЕА охлаждали, используя ванну со льдом. Затем добавляли метансульфонил хлорид (6,60 мл, 84,4 ммоль). Быстро образовывался белый осадок. Через 2 часа добавляли 5 мл H2O, и летучие компоненты выпаривали. Остаток разделяли между ЕА (3×300 мл) и H2O, насыщенным NaHCO3, H2O, 0,1M HCl и солевым раствором (100 мл каждый). Объединенные органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 22,02 г бесцветного масла. Rf 0,38 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,4-7,3 (м, 3H), 7,2-7,1 (м, 2H), 7,1-7,0 (м, 4H), 5,2 (м, 2H, AB), 2,9 (с, 3H);13C ЯМР (CDCl3) δ 158,0, 156,7, 135,5, 130,4, 130,1, 124,0, 123,4, 119,5, 119,4, 118,8, 71,0, 38,4.
N-(3-Феноксибензил)фталимид Смесь 3-феноксибензил метансульфоната (22,5 г, 80,9 ммоль) и фталимида калия (16,4 г, 88,6 ммоль) в 200 мл NMP перемешивали при 50°C в течение 17 часов, используя приводную мешалку. Затем добавляли H2O (100 мл), и летучий материал выпаривали. Остаток разделяли между ЕА и 5%-ым Na2CO3 (2x), H2O, 0,1M HCl и солевым раствором. Органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. Кристаллизация из IPA дала 23,55 г бесцветного твердого вещества. Rf 0,53 (30% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,85 и 7,73 (м, 4H, AA’BB’), 7,34-7,24 (м, 3H), 7,15-7,07 (м, 3H), 6,99-6,97 (м, 2H), 6,88-6,85 (м, 1H), 4,82 (м, 2H, AB);13C ЯМР (CDCl3) δ 168,1, 157,6, 157,1, 138,4, 134,5, 134,2, 132,2, 130,2, 129,9, 123,8, 123,6, 123,6, 123,2, 119,1, 119,1, 118,1, 41,4.
(3-Феноксифенил)метанамин Гидразин моногидрат (3,50 мл, 72,1 ммоль) добавляли к смеси N-(3-феноксибензил)фталимида (6,28 г, 19,1 ммоль) и 200 мл IPA, используя механическое перемешивание. Смесь нагревали с обратным холодильником в течение 7 часов. При отстаивании в течение ночи, образовывался осадок. Смесь концентрировали упариванием, и остаток разделяли между изопропил ацетатом и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4, фильтровали и концентрировали. SPE, с промывкой 50% изопропил ацетат/Гексан и с последующим элюированием 3% MeOH/DCM +2% ТЕА дал фракции, которые содержали продукт нингидрина (+). Объединенные фракции продукта промывали 5%-ым Na2CO3, высушивали над Na2SO4, фильтровали и концентрировали, получая 3,25 г желтого масла. Rf 0,28 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 7,36-7,25 (м, 3H), 7,12-6,95 (м, 5H), 6,87 (ддд, 1H, J=1,0, 2,5, 8,2 Гц), 3,82 (шир. с, 2H), 2,15 (шир. с, 2H, NH2).
N-(3-Феноксибензил)хинолин-4-амин (3-Феноксифенил)метанамин (2,02 г, 10,2 ммоль) забирали в 60 мл 1-пентанола, и затем 15 мл летучего материала удаляли перегонкой. Смесь охлаждали до температуры ниже температуры кипения и добавляли трипропиламин (3,80 мл, 19,9 ммоль) и 4-хлорхинолин (1,65 г, 10,2 ммоль). Нагревание с обратным холодильником продолжали. Через 66 часов летучий материал удаляли выпариванием. Смесь разделяли между DCM (150, 100 мл) и 5% Na2CO3 (80 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая твердое вещество. Перекристаллизация из ЕА/Гексан дала 2,08 г бесцветного твердого вещества. Rf 0,34 (10% MeOH/DCM); Т.пл. 163,0-164,0°C;1H ЯМР (CDCl3) δ 8,54 (д, 1H, J=5,5 Гц), 8,00 (м, 1H), 7,76 (д, 1H, J=8,1 Гц), 7,64 (м, 1H), 7,43 (м, 1H), 7,34-7,29 (м, 3H), 7,11 (м, 1H), 7,05 (с, 1H), 7,02-6,99 (м, 2H), 6,94 (дд, 1H, J=2,2, 8,0 Гц), 6,42 (д, 1H, J=5,5 Гц), 5,46 (шир. с, 1H, NH) 4,51 (м, 2H, AB);13C ЯМР (CDCl3) δ 158,2, 156,9, 151,3, 149,5, 148,7, 139,9, 130,5, 130,3, 130,0, 129,3, 125,0, 123,8, 122,2, 119,5, 119,3, 118,9, 118,0, 117,8, 99,7, 47,4.
Пример 71: N-[3-(бензилокси)бензил]хинолин-4-амин
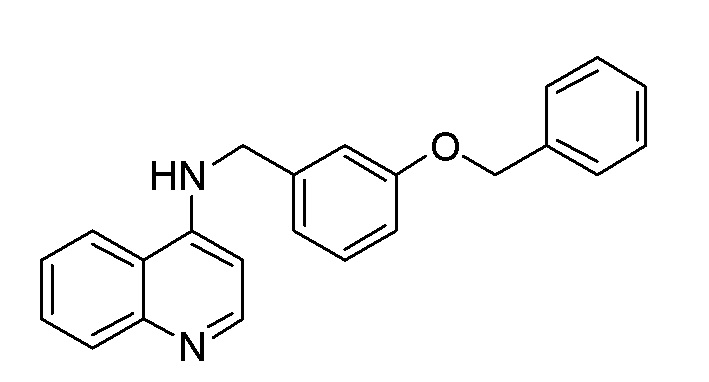
3-(бензилокси)бензонитрил Смесь 3-гидроксибензoнитрила (504 мг, 4,24 ммоль), бензил хлорида (607 мг, 4,78 ммоль) и K2CO3 (605 мг, 4,38 ммоль) в 2 мл DMF вводили в реакцию в течение 42 часов. Смесь разбавляли 50% ЕА/Гексан и промывали 5%-ым Na2CO3 (2x) и солевым раствором, подкисленным 1M HCl. Органическую фазу высушивали над безводным MgSO4 и концентрировали. FC (15% ЕА/Гексан) дал 780 мг бесцветного масла. Rf 0,50 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,43-7,31 (м, 6H), 7,26-7,17 (м, 3H), 5,08 (м, 2H, AB).
[3-(бензилокси)фенил]метанамин Смесь 3-(бензилокси)бензонитрила и 30 мл THF охлаждали ванной со льдом. Добавляли LAH (195 мг и затем 190 мг). Смеси давали нагреться до комнатной температуры. Через 24 часа смесь охлаждали ванной со льдом, и последовательно добавляли 0,40 мл H2O, 0,40 мл 15% NaOH и 1,2 мл H2O. Гетерогенную смесь разбавляли 5% MeOH/DCM и предварительно загружали на силикагеле. SPE с промывкой 5%-ым MeOH и с элюированием 10% MeOH/DCM +2% ТЕА дал 672 мг бесцветного масла, которое затвердевало при отстаивании.1H ЯМР (CDCl3) δ 7,48-7,23 (м, 6H), 6,98-6,83 (м, 3H), 5,07 (м, 2H, AB), 3,83 (м, 2H, AB).
N-[3-(бензилокси)бензил]хинолин-4-амин (600 мг) получали из [3-(бензилокси)фенил]метанамина (670 мг, 3,14 ммоль), 4-хлорхинолина (767 мг, 4,70 ммоль) и DIEA (1,20 мл, 6,88 ммоль) в 0,5 мл DMF, нагретого в герметизированной пробирке. FC (7% MeOH/DCM) дала 600 мг продукта. Rf 0,38 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,43 (д, 1H, J=5,4 Гц), 8,01-7,96 (м, 2H), 7,62-7,56 (м, 1H), 7,40-7,22 (м, 7H), 6,99-6,88 (м, 3), 6,53 (шир. с, 1H, NH), 6,34 (д, 1H, J=5,5 Гц), 4,99 (с, 2H), 4,48 (м, 2H, AB).
Пример 72: N-(3-Фенэтоксибензил)хинолин-4-амин

N-(3-Фенэтоксибензил)хинолин-4-амин получали способом, используемым для получения N-[3-(бензилокси)бензил]хинолин-4-амина, исходя из 3-гидроксибензoнитрила (561 мг, 4,71 ммоль), 2-бромэтилбензола (1,34 г, 7,24 ммоль) и K2CO3 (1,00 г, 7,25 ммоль) в 2 мл DMF, нагретом при 60°C.
3-(Фенэтокси)бензонитрил (454 мг): Rf 0,46 (20% ЕА/Гексан):1H ЯМР (CDCl3) δ 7,38-7,20 (м, 7H), 7,10 (м, 2H), 4,18 (т, 2H, J=6,9 Гц), 3,11 (т, 2H, J=6,9 Гц).
(3-(Фенэтоксифенил)метанамин (480 мг):1H ЯМР (CDCl3) δ 7,36-7,20 (м, 6H), 6,87 (м, 2H), 6,78 (м, 1H), 4,18 (т, 2H, J=7,2 Гц), 3,82 (м, 2H, AB), 3,10 (т, 2H, J=7,2 Гц), 2,16 (шир. с, 2H, NH2).
N-(3-Фенэтоксибензил)хинолин-4-амин (358 мг): Rf 0,12 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 8,39 (д, 1H, J=5,4 Гц), 7,96 (д, 1H, J=8,4 Гц), 7,91 (д, 1H, J=8,4 Гц), 7,59 (м, 1H), 7,38 (м, 1H), 7,31-7,18 (м, 6H), 6,94-6,90 (м, 2H), 6,80 (дд, 1H, J=2,4, 8,1 Гц), 6,35 (д, 1H, J=5,5 Гц), 6,26 (шир. с, 1H), 4,48 (м, 2H, AB), 4,12 (т, 2H, J=7,0 Гц), 3,05 (м, 2H).
Пример 73: N-[4-(хинолин-4-иламино)бутил]бензамид

N1-(Хинолин-4-ил)бутан-1,4-диамин Смесь 1,4-бутандиамина (1,54 г, 17,5 ммоль), 4-хлорхинолина (357 мг, 2,19 ммоль) и DIEA (0,50 мл, 2,87 ммоль) нагревали при 130°C в герметизированной пробирке в течение 24 часов. Смесь охлаждали, забирали в ЕА и промывали 5%-ым Na2CO3 (3x) и солевым раствором. Органическую фазу высушивали над Na2SO4 и концентрировали.1H ЯМР (20% CD3OD/CDCl3) δ 8,33 (д, 1H, J=5,5 Гц), 7,86 (ддд, 1H, J=0,5, 1,5, 8,4 Гц), 7,81 (ддд, 1H, J=0,5, 1,2, 8,4 Гц), 7,53 (ддд, 1H, J=1,3, 6,7, 8,4 Гц), 7,33 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 6,29 (д, 1H, J=5,5 Гц), 3,20 (м, 2H), 2,66 (т, 2H, J=6,9 Гц), 1,69 (м, 2H), 1,51 (м, 2H).
N-[4-(хинолин-4-иламино)бутил]бензамид N1-(Хинолин-4-ил)бутан-1,4-диамин (185 мг, 0,86 ммоль) забирали в 5 мл пиридина, и смесь концентрировали. Остаток забирали в 10 мл DCM, охлаждали ванной со льдом и добавляли ТЕА (0,49 мл, 3,5 ммоль) и затем бензоил хлорид (0,40 мл, 3,43 ммоль). Смеси давали нагреться до комнатной температуры. Через 2 часа добавляли 3,43 мл 1н. NaOH, и летучие компоненты удаляли перегонкой. Остаток разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали. SPE, с промывкой 5% MeOH/DCM и с элюированием 15%-ым MeOH/DCM, дал масляное твердое вещество. Повторная очистка препаративной TLC (15% MeOH/DCM) дала продукт в форме твердого вещества. Rf 0,21 (15% MeOH/DCM);1H ЯМР (CDCl3) δ 8,31 (д, 1H, J=5,7 Гц), 8,10 (м, 1H), 7,80-7,77 (м, 3H), 7,62 (ддд, 1H, J=1,2, 6,6, 8,4 Гц), 7,55-7,39 (м, 4H), 6,51 (д, 1H, J=5,5 Гц), 3,45 (кв., 2H, J=7 Гц), 1,86-1,76 (м, 4H).
Пример 74: N-[6-(хинолин-4-иламино)гексил]бензамид

N1-(Хинолин-4-ил)гексан-1,6-диамин Смесь 1,6-гександиамина (2,05 г, 17,7 ммоль) и 4-хлорхинолина (297 мг, 1,82 ммоль) нагревали при 130°C в герметизированной пробирке в течение 24 часов. Смесь охлаждали, разделяли между ЕА (3x) и 5%-ым Na2CO3 (3x) и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали.1H ЯМР (20% CD3OD/CDCl3) δ 8,39 (д, 1H, J=5,4 Гц), 7,87 (д, 1H, J=8,1 Гц), 7,75 (д, 1H, J=8,4 Гц), 7,56 (ддд, 1H, J=1,3, 6,9, 8,4 Гц), 7,36 (м, 1H), 6,35 (д, 1H, J=5,4 Гц), 3,26 (м, 2H), 2,63 (м, 2H), 1,71 (м, 2H), 1,49-1,38 (м, 6H).
N-[6-(хинолин-4-иламино)гексил]бензамид N1-(Хинолин-4-ил)гексан-1,6-диамин (230 мг, 0,946 ммоль) забирали в 5 мл пиридина, и смесь концентрировали. Остаток забирали в 10 мл DCM, охлаждали ванной со льдом и добавляли ТЕА (0,53 мл, 3,8 ммоль) и затем бензоил хлорид (0,44 мл, 3,78 ммоль). Смеси давали нагреться до комнатной температуры. Через 2 часа добавляли 3,78 мл 1н. NaOH. Смесь разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над Na2SO4 и концентрировали. Очистка препаративной TLC (15% MeOH/DCM) дала продукт. Остаток от концентрации экстракта из адсорбента забирали в DCM, промывали 5%-ым Na2CO3, высушивали над Na2SO4 и концентрировали, получая продукт. Rf 0,23 (15% MeOH/DCM);1H ЯМР (CDCl3) δ 8,30 (д, 1H, J=6,0 Гц), 8,09 (д, 1H, J=8,4 Гц), 7,91 (д, 1H, J=8,4 Гц), 7,82-7,78 (м, 2H), 7,55 (м, 1H), 7,45-7,30 (м, 4H), 6,94 (т, 1H, J=6 Гц), 6,81 (шир. с, 1H), 6,24 (д, 1H, J=6,2 Гц), 3,40 (м, 2H), 3,25 (м, 2H), 1,68-1,54 (м, 8H).
Пример 75: N-[8-(хинолин-4-иламино)октил]бензамид

N-(8-Аминооктил)бензамид Смесь 1,8-октандиамина (3,27 г, 22,7 ммоль) и метил бензоата (0,40 мл, 3,20 ммоль) нагревали при 115°C в течение 24 часов. Смесь охлаждали и разделяли между ЕА и H2O. Органическую фазу, которая содержала 1:1 молярное отношение диамина и моноамида, концентрировали. SPE с обратной фазой с промывкой 20% MeOH/H2O и с элюированием MeOH дал фракцию продукта, которую концентрировали, забирали в DCM, промывали 5%-ым Na2CO3, высушивали над Na2SO4 и концентрировали, получая 698 мг продукта.1H ЯМР (20% CD3OD/CDCl3) δ 7,64-7,59 (м, 2H), 7,43 (шир. с, 1H, NH), 7,32-7,18 (м, 3H), 3,19 (м, 2H), 2,45 (м, 2H), 1,42 (м, 2H), 1,27-1,04 (м, 10H).
N-[8-(хинолин-4-иламино)октил]бензамид Смесь N-(8-аминооктил)бензамида (357 мг, 1,44 ммоль), 4-хлорхинолина (312 мг, 1,91 ммоль) и DIEA (0,50 мл, 2,87 ммоль) в 1 мл NMP нагревали при 160°C в герметизированной пробирке в течение 24 часов. Смесь охлаждали, разбавляли DCM и промывали 5%-ым Na2CO3. Органическую фазу высушивали над Na2SO4 и концентрировали. SPE, с промывкой 5%-ым MeOH/DCM и с элюированием 2,5% MeOH/DCM +2% ТЕА, дал продукт в форме масла, которое кристаллизовали из EtOH. Rf 0,33 (50% ЕА/Гексан +2% ТЕА);1H ЯМР (CDCl3) δ 8,33 (д, 1H, J=5,7 Гц), 7,87 (дд, 1H, J=0,7, 8,4 Гц), 7,80 (д, 1H, J=8,7 Гц), 7,74-7,71 (м, 2H), 7,58 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,48-7,34 (м, 4H), 6,38 (д, 1H, J=5,7 Гц), 3,38-3,26 (м, 4H), 1,74-1,35 (м, 12H).
Пример 76: 3-Метокси-N-[8-(хинолин-4-иламино)октил]бензамид
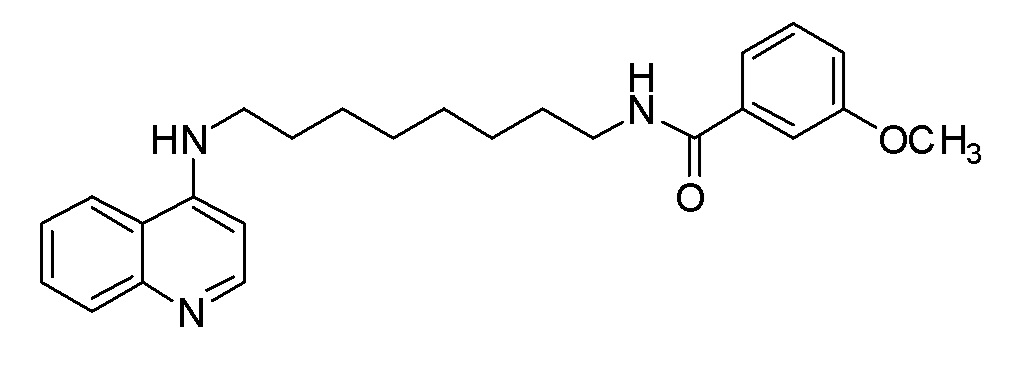
N-(8-Аминооктил)-3-метоксибензамид Смесь метил-3-метоксибензоата (863 мг, 5,20 ммоль) и 1,8-октандиамина (6,90 г) нагревали при 110-120°C в течение 24 часов. Смесь охлаждали и разделяли между ЕА (3×60 мл) и H2O, 2,5% Na2CO3 (3x) и солевым раствором (60 мл каждый). Органические фазы высушивали над безводным Na2SO4 и концентрировали. ЯМР показал, что остаток состоял из амида и диамина в отношении 2,3:1, SPE с обратной фазой (ODS-силикагель) с промывкой 20% MeOH/H2O и с последующим элюированием MeOH, дал 1,43 г желтого масла. ЯМР показал, что масло состояло из амида и диамина в отношении 7,3:1.
3-Метокси-N-[8-(хинолин-4-иламино)октил]бензамид Смесь N-(8-аминооктил)-3-метоксибензамида (540 мг, 1,94 ммоль), 4-хлорхинолина (340 мг, 2,08 ммоль) и DIEA (0,80 мл, 4,59 ммоль) в 2,5 мл NMP нагревали при 160°C в герметизированной пробирке в течение 3 дней. Смесь охлаждали, разбавляли ЕА, промывали 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4 и концентрировали. SPE, с промывкой 1% MeOH/DCM и последующим элюированием 7,5% MeOH/DCM +2% ТЕА, дал продукт в форме твердого вещества. Rf 0,19 (ЕА+2% ТЕА); Т.пл. 162-165°C (из MeOH);1H ЯМР (20% CD3OD/CDCl3) δ 8,38 (д, 1H, J=5,7 Гц), 8,04 (д, 1H, J=8,4 Гц), 7,92 (д, 1H, J=8,4 Гц), 7,57 (м, 1H), 7,40-7,21 (м, 4H), 6,95 (ддд, 1H, J=1,2, 2,7, 8,1 Гц), 6,85 (м, 1H), 6,36 (д, 1H, J=5,7 Гц), 6,31 (шир. с, 1H, NH), 3,75 (с, 3H), 3,41-3,25 (м, 4H), 1,72-1,16 (м, 12H).
Пример 77: 4-Метокси-N-[8-(хинолин-4-иламино)октил]бензамид

N-(8-Аминооктил)-4-метоксибензамид Смесь метил-4-метоксибензоата (874 мг, 5,26 ммоль) и 1,8-октандиамина (6,18 г) нагревали при 110-120°C в течение 4 дней. Смесь охлаждали и разделяли между ЕА (3×60 мл) и H2O, 2,5% Na2CO3 (3x) и солевым раствором (60 мл каждый). Органические фазы высушивали над безводным Na2SO4 и концентрировали. SPE с обратной фазой (ODS-силикагель), с промывкой 20% MeOH/H2O и последующим элюированием MeOH, дал масло. Масло забирали в DCM и промывали 5%-ым Na2CO3, высушивали над Na2SO4 и концентрировали, получая 533 мг клейкого твердого вещества желтого цвета.1H ЯМР (CD3OD) δ 7,77 и 6,96 (м, 4H, AA’BB’), 4,88 (с, 3H), 3,34 (м, 2H), 3,13 (м, 1H, NH), 2,60 (м, 2H), 1,91 (2xs, 2H, NH2), 1,62-1,33 (м, 12H).
4-Метокси-N-[8-(хинолин-4-иламино)октил]бензамид Смесь N-(8-аминооктил)-4-метоксибензамида (533 мг, 1,92 ммоль) и 7,5 мл безводного пиридина упаривали досуха. Затем добавляли 4-хлорхинолин (335 мг, 2,08 ммоль) и DIEA (0,80 мл, 4,59 ммоль) в 2,5 мл NMP, и смесь нагревали при 160°C в герметизированной пробирке в течение 3 дней. Смесь охлаждали, разбавляли ЕА, промывали 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4 и концентрировали. SPE, с промывкой 1% MeOH/DCM и последующим элюированием 7,5% MeOH/DCM +2% ТЕА, дал продукт в форме твердого вещества. Rf 0,00 (5% MeOH/DCM) 0,20 (ЕА+2% ТЕА);1H ЯМР (20% CD3OD/CDCl3) δ 8,30 (д, 1H, J=5,7), 7,82-7,76 (м, 2H), 7,65 и 6,82 (м, 4H, AA’BB’), 7,53 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,33 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 6,32 (д, 1H, J=5,5 Гц), 3,74 (с, 3H), 3,32-3,19 (м, 4H), 1,70-1,25 (м, 12H).
Пример 78: 2-(Гексилокси)-N-[2-(хинолин-4-иламино)этил]бензамид

Метил-2-(гексилокси)бензоат Смесь метил салицилата (7,76 г, 51,1 ммоль), K2CO3 (8,8 г, 64 ммоль) и 1-бромгексана (8,60 мл, 61,5 ммоль) в 30 мл DMF нагревали при 50°C в течение 20,5 часов. Смесь разделяли между 1:1 ЕА/Гексан (3×150 мл) и 0,2M HCl, 0,1M HCl и солевым раствором (50 мл из каждого). Органические фазы высушивали над Na2SO4 и концентрировали. SPE, с промывкой Гексаном и с элюированием 20% ЕА/Гексан, дал 11,7 г бесцветную жидкость.
N-(2-Аминоэтил)-2-(гексилокси)бензамид Смесь метил-2-(гексилокси)бензоата (2,11 г, 8,94 ммоль) и 1,2-этандиамина (5,40 мл, 81,0 ммоль) нагревали при 115°C в герметизированной пробирке в течение 72 часов. Затем летучие компоненты выпаривали в вакууме. Остаток забирали в 10 мл MeOH и упаривали в вакууме, получая 2,34 г янтарной жидкости.1H ЯМР (CD3OD) δ 7,84 (м, 1H), 7,45 (ддд, 1H, J=1,9, 7,4, 9,2 Гц), 7,09 (д, 1H, J=8,1 Гц), 7,02 (м, 1H), 4,13 (т, 2H, J=6,5 Гц), 3,47 (м, 2H), 2,84 (м, 2H), 1,86 (м, 2H), 1,49 (м, 2H), 1,39-1,34 (м, 4H), 0,93 (м, 3H);13C ЯМР (CD3OD) δ 169,0, 158,5, 134,1, 132,0, 123,7, 122,0, 114,0, 70,4, 43,5, 42,3, 32,9, 30,4, 27,2, 23,9, 14,6.
2-(Гексилокси)-N-[2-(хинолин-4-иламино)этил]бензамид N-(2-Аминоэтил)-2-(гексилокси)бензамид (2,34 г, 8,86 ммоль) забирали в 65 мл 1-пентанола, и 15 мл удаляли перегонкой. Смесь немного охлаждали и добавляли трипропиламин 3,40 мл, 17,8 ммоль) и 4-хлорхинолин (1,60 г, 9,82 ммоль). Смесь нагревали с обратным холодильником в течение 63 часов. Затем смесь концентрировали в вакууме. Остаток разделяли между DCM и 5%-ым Na2CO3, и органическую фазу высушивали над Na2SO4 и концентрировали. FC (5% MeOH/DCM +2% ТЕА) дала 1,84 г коричневого сиропа, который затвердевал при отстаивании. Твердое вещество промывали 20%, 33% и 50% Et2O/Hex и высушивали в вакууме, получая 1,67 г твердого вещества. Rf 0,30 (5% MeOH/DCM +2% ТЕА);1H ЯМР (CDCl3) δ 8,56-8,51 (м, 2H), 8,28 (дд, 1H, J=1,8, 8,1 Гц), 7,92 (д, 1H, J=8,8 Гц), 7,60 (м, 1H), 7,46-7,41 (м, 2H), 7,08 (м, 1H), 6,93 (д, 1H, J=8,0 Гц), 6,77 (шир. с, 1H, NH), 6,33 (д, 1H, J=5,1 Гц), 4,06 (т, 2H, J=6,6 Гц), 3,90 (м, 2H), 3,50 (м, 2H), 1,77 (м, 2H), 1,42-1,23 (м, 6H), 0,87 (т, 3H, J=7 Гц);13C ЯМР (CDCl3) δ 168,1, 157,3, 151,2, 150,3, 148,6, 133,5, 132,5, 129,8, 129,1, 124,9, 121,5, 120,9, 120,6, 119,1, 112,5, 98,1, 69,3, 46,1, 39,1, 31,5, 29,2, 26,0, 22,7, 14,2.
Пример 79: 2-(Гексилокси)-N-[3-(хинолин-4-иламино)пропил]бензамид

2-(Гексилокси)-N-[3-(хинолин-4-иламино)пропил]бензамид (1,6 г) получали способом, используемым для получения 2-(гексилокси)-N-[4-(хинолин-4-иламино)бутил]бензамида, исходя из метил-2-(гексилокси)бензоата (2,13 г) и 1,3-диаминопропана (6,00 мл) и используя 4-хлорхинолин (1,70 г).
N-(3-Аминопропил)-2-(гексилокси)бензамид:1H ЯМР (CDCl3) δ 7,85 (дд, 1H, J=1,8, 7,7 Гц), 7,44 (ддд, 1H, J=1,8, 7,3, 9,2 Гц), 7,10 (д, 1H, J=8,4 Гц), 7,02 (м, 1H), 4,14 (м, 2H), 3,48 (м, 2H), 3,30 (м, 2H), 2,72 (м, 2H), 1,86 (м, 2H), 1,75 (м, 2H), 1,40-1,35 (м, 4H), 0,93 (м, 3H);13C ЯМР (CDCl3) δ 168,8, 158,5, 134,1, 132,0, 123,6, 122,0, 114,0, 70,4, 40,0, 38,2, 33,9, 32,9, 30,4, 27,2, 23,9, 14,6.
2-(Гексилокси)-N-[3-(хинолин-4-иламино)пропил]бензамид: Rf 0,08 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 8,50 (д, 1H, J=5,5 Гц), 8,25 (дд, 1H, J=1,8, 7,7 Гц), 8,24-8,20 (м, 1H), 8,01-7,98 (м, 1H), 7,93 (дд, 1H, J=0,7, 8,4 Гц), 7,58 (ддд, 1H, J=1,1, 7,0, 8,1 Гц), 7,44-7,36 (м, 2H), 7,10-7,06 (м, 1H), 6,92 (д, 1H, J=8,1 Гц), 6,49-6,46 (т, 1H, J=6 Гц, NH), 6,39 (д, 1H, J=5,5 Гц), 4,03 (т, 2H), 3,63-3,59 (м, 2H), 3,46-3,42 (м, 2H), 2,64 (шир. с, 1H, NH), 1,95-1,89 (м, 2H), 1,81-1,74 (м, 2H), 1,45-1,27 (м, 6H), 0,89-0,86 (м, 3H);13C ЯМР (CDCl3) δ 166,8, 157,2, 151,0, 150,0, 148,7, 133,1, 132,4, 129,7, 129,1, 124,7, 121,4, 21,3, 120,4, 119,3, 112,4, 98,3, 69,2, 39,6, 39,6, 36,8, 31,6, 29,3, 28,7, 26,0, 22,7, 14,1.
Пример 80: 2-(Гексилокси)-N-[4-(хинолин-4-иламино)бутил]бензамид

N-(4-Аминобутил)-2-(гексилокси)бензамид Смесь 1,4-диаминобутана (5,37 г, 61 ммоль) и метил-2-(гексилокси)бензоата (1,80 г, 7,63 ммоль) нагревали при 110°C в герметизированной пробирке в течение 48 часов. Смесь разделяли между изопропил ацетатом (3×125 мл) и H2O (100 мл), 5% Na2CO3 (2×100 мл) и солевым раствором (100 мл). Органические фазы высушивали над безводным Na2SO4 и концентрировали, получая 2,10 г бесцветного сиропа.1H ЯМР (CDCl3) δ 8,15 (дд, 1H, J=7,7, 1,8 Гц), 8,01 (шир. с, 1H), 7,33 (ддд, 1H, J=9,2, 7,3, 1,8 Гц), 6,98 (м, 1H), 6,88 (д, 1H, J=8,4 Гц), 4,04 (м, 2H), 3,41 (м, 2H), 2,68 (м, 2H), 1,80 (м, 2H), 1,59 (м, 2H), 1,52-1,40 (м, 4H), 1,32-1,25 (м, 4H), 1,12 (шир. с, 2H), 0,86 (м, 3H);13C ЯМР (CDCl3) δ 165,3, 157,0, 132,6, 132,2, 121,6, 121,1, 112,2, 69,0, 42,0, 39,6, 31,6, 31,3, 29,3, 27,1, 26,0, 22,6, 14,0.
2-(Гексилокси)-N-[4-(хинолин-4-иламино)бутил]бензамид N-(4-Аминобутил)-2-(гексилокси)бензамид забирали в 60 мл 1-пентанола, и 15 мл летучей жидкости удаляли перегонкой. Смесь немного охлаждали и добавляли трипропиламин (2,70 мл, 14,2 ммоль) и 4-хлорхинолин (1,29 г, 7,91 ммоль). Нагревание с обратным холодильником продолжали в течение 42 часов. Охлажденную смесь концентрировали и разделяли между DCM и 5%-ым Na2CO3, и органическую фазу высушивали над безводным Na2SO4 и концентрировали. Остаток забирали в ЕА и затем снова концентрировали. Полученное масло затвердевало при отстаивании. Твердое вещество разбивали и промывали 20%, 50% и 100% Et2O/Hex. Высушивание в вакууме дало 1,53 г твердого вещества желто-серого цвета. Rf 0,21 (5% MeOH/DCM +2% ТЕА);1H ЯМР (CD3OD) δ 8,53 (д, 1H, J=5,5 Гц), 8,24 (дд, 1H, J=1,9, 7,7 Гц), 8,16 (м, 1H, NH), 7,95 (д, 1H, J=8,4 Гц), 7,85 (д, 1H, J=8,4 Гц), 7,61 (м, 1H), 7,44-7,38 (м, 2H), 7,07 (м, 1H), 6,94 (д, 1H, J=8,4 Гц), 6,41 (д, 1H, J=5,1 Гц), 5,44 (шир. с, 1H, NH), 4,08 (м, 2H), 3,57 (м, 2H), 3,39 (м, 2H), 1,91-1,75 (м, 6H), 1,44 (м, 2H), 1,34-1,27 (м, 4H), 0,86 (м, 3H);13C ЯМР (CDCl3) δ 165,9, 157,2, 151,2, 149,9, 148,7, 133,0, 132,5, 130,1, 129,1, 124,8, 121,5, 121,4, 119,8, 119,0, 112,4, 98,9, 69,2, 43,2, 39,3, 31,7, 29,4, 28,0, 26,2, 26,1, 22,8, 14,2.
Пример 81: N-[8-(хинолин-4-иламино)октил]пиколинамид

N-(8-Аминооктил)пиколинамид Смесь 1,8-октандиамина (8,19 г, 56,9 ммоль) и метил пиколинaта (970 мг, 7,08 ммоль) нагревали при 130°C в течение 60 часов. Смесь охлаждали, забирали в метанол и упаривали на силикагеле. Предварительно загруженный силикагель загружали на вершину колонки для флэш-хроматографии и элюировали с использованием 15% MeOH/DCM +2% ТЕА. Концентрация содержащих продукт фракций дала 1,28 г жидкости. Rf 0,23 (15% MeOH/DCM +2% ТЕА);1H ЯМР (20% CD3OD/CDCl3) δ 8,5 (ддд, 1H, J=1,0, 1,7, 4,9 Гц), 8,2 (м, 1H), 8,0 (шир. с, 1H, NH), 7,8 (м, 1H), 7,4 (ддд, 1H, J=1,5, 4,9, 7,7 Гц), 3,43 (м, 2H), 2,66 (м, 2H), 2,17 (шир. с, 2H, NH2), 1,65-1,28 (м, 12H).
N-[8-(хинолин-4-иламино)октил]пиколинамид Смесь N-(8-аминооктил)пиколинамида (557 мг, 2,24 ммоль), 4-хлорхинолина (544 мг, 3,34 ммоль), DIEA (1 мл, 6 ммоль) и 0,5 мл DMF нагревали при 140°C в герметизированной пробирке в течение 89 часов. Затем летучие компоненты выпаривали, и остаток очищали FC (8% MeOH/DCM), получая 520 мг продукта. Rf 0,38 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,6 (д, 1H), 8,4 (д, 1H), 8,1 (д, 1H), 8,1-7,9 (м, 3H), 7,7 (м, 1H), 7,5 (м, 1H), 7,30 (м, 1H), 6,3 (д, 1H), 3,4-3,3 (м, 4H), 1,7 (м, 2H), 1,5 (м, 2H), 1,3-1,0 (м, 8H).
Пример 82: N-[8-(хинолин-4-иламино)октил]никотинамид

N-(8-Аминооктил)никотинамид Смесь 1,8-диаминооктана (9,78 г, 67,0 ммоль) и метил никотинaта (1,50 г, 10,9 ммоль) нагревали при 84°C в течение 16 часов и при 110-120°C в течение дополнительных 56 часов. Охлажденную смесь разделяли SPE с промывкой 5% MeOH/DCM +2% ТЕА, чтобы удалить октан-1,8-бис(амид) и остаточный метил никотинaт, и затем 15% MeOH/DCM +2% ТЕА, чтобы элюировать фракции продукта нингидрина (+). Фракции продукта концентрировали, забирали в DCM, промывали 5%-ым Na2CO3, высушивали над Na2SO4, фильтровали и высушивали, получая 2,07 г твердого вещества светло-желтого цвета. Rf 0,10 (15% MeOH/DCM +2% ТЕА);1H ЯМР (CD3OD) δ 8,95 (дд, 1H, J=0,8, 2,2 Гц), 8,67 (м, 1H), 8,23 (м, 1H), 7,53 (м, 1H), 3,38 (т, 2H, J=7,3 Гц), 2,60 (т, 2H), 1,61 (м, 2H), 1,47-1,33 (м, 10H);13C ЯМР (CD3OD) δ 167,8, 152,7, 149,2, 137,1, 132,4, 125,3, 42,8, 41,3, 34,1, 30,7, 30,6, 28,2, 28,2, 22,2.
N-[8-(хинолин-4-иламино)октил]никотинамид N-(8-аминооктил)никотинамид (5,66 г, 22,7 ммоль) забирали в 100 мл 1-пентанола, и затем 50 мл летучего материала удаляли перегонкой. Смесь охлаждали до температуры ниже температуры кипения и добавляли трипропиламин (9,50 мл, 49,8 ммоль) и 4-хлорхинолин (4,08 г, 25,0 ммоль). Нагревание с обратным холодильником продолжали. Через 22 часа летучий материал удаляли выпариванием. Смесь разделяли между DCM (175, 2×100 мл) и комбинацией 25 мл 1н. NaOH и 25 мл 5%-ого Na2CO3. Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая темный сироп. Две кристаллизации из MeOH/H2O и высушивание в вакууме над P2O5 дали 2,31 г твердого вещества желто-коричневого цвета. Rf 0,56 (15% MeOH/DCM +2% ТЕА); Т.пл. 139,5-141,0°C;1H ЯМР (DMSO-d6) δ 8,97 (м, 1H), 8,66 (м, 1H), 8,61 (т, 1H, J=5,5 Гц), 8,35 (д, 1H, J=5,1 Гц), 8,19 (д, 1H, J=8,8 Гц), 8,14 (ддд, 1H, J=1,4, 2,2, 7,7 Гц), 7,74 (дд, 1H, J=1,1, 8,5 Гц), 7,57 (м, 1H), 7,46 (м, 1H), 7,38 (ддд, 1H, J=1,4, 7,0, 8,4 Гц), 7,16 (т, 1H, J=5 Гц), 6,40 (д, 1H, J=5,5 Гц), 3,27-3,22 (м, 4H), 1,65 (м, 2H), 1,44 (м, 2H), 1,30 (м, 8H);13C ЯМР (DMSO-d6) δ 164,6, 151,6, 150,4, 150,,2, 148,3, 148,0, 134,8, 130,1, 128,7, 123,7, 123,4, 121,7, 118,8, 98,0, 42,4, 39,2, 29,0, 28,8, 28,7, 27,8, 26,6, 26,4.
Пример 83: N-[8-(хинолин-4-иламино)октил]изоникотинамид

N-(8-Аминооктил)изоникотинамид Смесь 1,8-диаминооктана (7,66 г, 53 ммоль) и метил изоникотинaта (910 мг, 6,64 ммоль) нагревали при 130°C в течение 60 часов. Охлажденную смесь разделяли между DCM и 5% Na2CO3, и органическую фазу высушивали над безводным Na2SO4 и концентрировали. FC (15% MeOH/DCM +2% ТЕА) дала 539 мг масляного твердого вещества. Rf 0,15 (15% MeOH/DCM +2% ТЕА);1H ЯМР (20% CD3OD/CDCl3) δ 8,59 и 7,66 (м, 4H, AA’BB’), 3,33 (м, 2H), 3,10 (м, 1H, NH), 2,78 (м, 2H), 1,85 (с, 2H, NH2), 1,57-1,24 (м, 12H).
N-[8-(хинолин-4-иламино)октил]изоникотинамид Смесь N-(8-аминооктил)изоникотинамида (539 мг, 2,16 ммоль), 4-хлорхинолина (536 мг, 3,29 ммоль), DIEA (2 мл, 12 ммоль) и 0,5 мл DMF нагревали при 140°C в герметизированной пробирке в течение 89 часов. Затем летучие компоненты выпаривали, и остаток очищали FC (8% MeOH/DCM), получая 113 мг продукта. Rf 0,13 (10% MeOH/DCM);1H ЯМР (20% CD3OD/CDCl3) δ 8,58 и 7,62 (м, 4H, AA’BB’), 8,35 (д, 1H, J=5,4 Гц), 7,83 (дд, 1H, J=0,7, 8,4 Гц), 7,71 (м, 1H), 7,55 (ддд, 1H, J=1,3, 7,0, 8,2 Гц), 7,35 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 6,34 (д, 1H, J=5,5 Гц), 3,37-3,21 (м, 4H), 1,70-1,22 (м, 12H).
Пример 84: N-(Пиридин-4-илметил)хинолин-4-амин
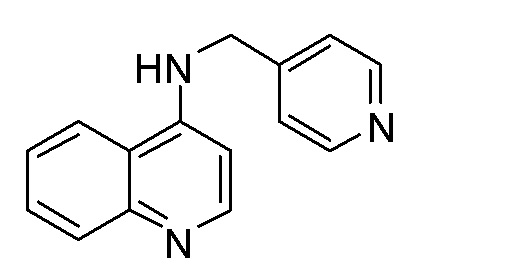
N-(Пиридин-4-илметил)хинолин-4-амин получали согласно способу, используемому для получения N-(пиридин-2-илметил)хинолин-4-амина. Rf 0,29 (5% MeOH/DCM +2% ТЕА);1H ЯМР (CDCl3) δ 8,51-8,47 (м, 2H), 8,39 (д, 1H, J=5,4 Гц), 8,03-8,00 (м, 1H), 7,95 (дд, 1H, J=1,0, 8,4 Гц), 7,59 (ддд, 1, J=1,2, 6,9, 8,4 Гц), 7,40 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,28-7,22 (м, 2H), 6,61 (шир. с, 1H), 6,19 (д, 1H, J=5,4 Гц), 4,56 (шир. с, 2H).
Пример 85: N-(Пиридин-3-илметил)хинолин-4-амин

N-(Пиридин-3-илметил)хинолин-4-амин получали согласно способу, используемому для получения N-(пиридин-2-илметил)хинолин-4-амина. Rf 0,36 (5% MeOH/DCM +2% ТЕА);1H ЯМР (CDCl3) δ 8,56 (д, 1H, J=2,0 Гц), 8,45 (дд, 1H, J=1,7, 5,0 Гц), 8,41 (д, 1H, J=5,2 Гц), 7,98 (д, 1H, J=8,4 Гц), 7,91 (дд, 1H, J=1,0, 8,4 Гц), 7,61 (ддд, 1H, J=1,7, 2,0, 7,9 Гц), 7,54 (ддд, 1H, J=1,2, 6,9, 8,2 Гц), 7,33 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,17 (дд, 1H, J=5,0, 7,9 Гц), 6,61 (шир. с, 1H), 6,29 (д, 1H, J=5,5 Гц), 4,50 (м, 2H, AB).
Пример 86: N-(Пиридин-2-илметил)хинолин-4-амин

Смесь 4-хлорхинолина (228 мг, 1,40 ммоль), 2-(аминометил)пиридина (144 мг, 1,33 ммоль) и DIEA (0,50 мл) нагревали при 130°C в герметизированной пробирке в течение 48 часов. Затем смесь охлаждали, разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, высушивали над Na2SO4 и концентрировали. FC (3% MeOH/DCM +2% ТЕА) дала содержащие продукт фракции, которые концентрировали. Остаток забирали в DCM и промывали 5%-ым Na2CO3, высушивали над Na2SO4 и концентрировали, получая продукт. Rf 0,54 (5% MeOH/DCM +2% ТЕА);1H ЯМР (CDCl3) δ 8,57-8,54 (м, 1H), 8,46 (д, 1H, J=5,4 Гц), 7,99-7,91 (м, 2H), 7,62-7,52 (м, 2H), 7,37 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 7,26-7,23 (м, 1H), 7,18-7,13 (м, 1H), 7,03 (шир. с, 1H), 6,32 (д, 1H, J=5,4 Гц), 4,52 (м, 2H, AB).
Пример 87: N-Гексилхинолин-4-амин

Смесь 4-хлорхинолина (248 мг, 1,52 ммоль) и 1-гексиламина (2 мл, 15 ммоль) нагревали в герметизированной пробирке при 100°C в течение 2 дней, при 120-130°C в течение 2 дней и при 150°C в течение 1 дня. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, и органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. SPE, с промывкой 25% ЕА/Гексан и с элюированием 12% MeOH/DCM с последующей повторной очисткой препаративной TLC (10% MeOH/DCM), дала продукт в форме масла. Rf 0,16 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 8,48 (д, 1H, J=5,4 Гц), 7,97 (дд, 1H, J=1,0, 8,4 Гц), 7,87 (д, 1H, J=8,4 Гц), 7,60 (ддд, 1H, J=1,5, 6,9, 8,4 Гц), 7,40 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 6,40 (д, 1H, J=5,7 Гц), 5,66 (шир. с, 1H, NH), 3,32 (м, 2H), 1,75 (м, 2H), 1,46-1,26 (м, 6H), 0,89 (м, 3H).
Пример 88: N-(Децил)хинолин-4-амин

Смесь 1-аминодекана (4,36 г, 27,8 ммоль), трипропиламина (8,00 мл, 42,0 ммоль) и 4-хлорхинолина (4,55 г, 27,9 ммоль) в 25 мл 1-пентанола нагревали с обратным холодильником в течение 3 дней. Затем летучие компоненты выпаривали. Остаток забирали в DCM (150 мл) и промывали 5%-ым Na2CO3 (100 мл). Водную фазу экстрагировали DCM (100 мл), и объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая темную жидкость. SPE, с элюированием 1% и затем 5% MeOH/DCM +2% ТЕА, дал фракции продукта, которые концентрировали, разделяли между DCM (150, 100 мл) и 5% Na2CO3 (100 мл), высушивали над Na2SO4, фильтровали и концентрировали. Перекристаллизация из ЕА/Гексан дала 4,14 г бесцветного твердого вещества. Rf 0,30 (5% MeOH/DCM +2% ТЕА); Т.пл. 79,0-80,0°C;1H ЯМР (CDCl3) δ 8,56 (д, 1H, J=5,5 Гц), 7,97 (дд, 1H, J=1,1, 8,4 Гц), 7,72 (м, 1H), 7,62 (ддд, 1H, J=1,4, 7,0, 8,4 Гц), 7,41 (м, 1H), 6,43 (д, 1H, J=5,5 Гц), 4,97 (шир. с, 1H, NH), 3,31 (м, 2H), 1,76 (м, 2H), 1,46 (м, 2H), 1,39-1,27 (м, 12H), 0,88 (м, 3H);13C ЯМР (CDCl3) δ 152,2, 149,9, 149,6, 129,2, 128,2, 125,0, 122,7, 121,0, 102,4, 62,0, 51,8, 32,6, 28,0, 25,7, 22,4, 14,0.
Пример 89: N-(Додецил)хинолин-4-амин
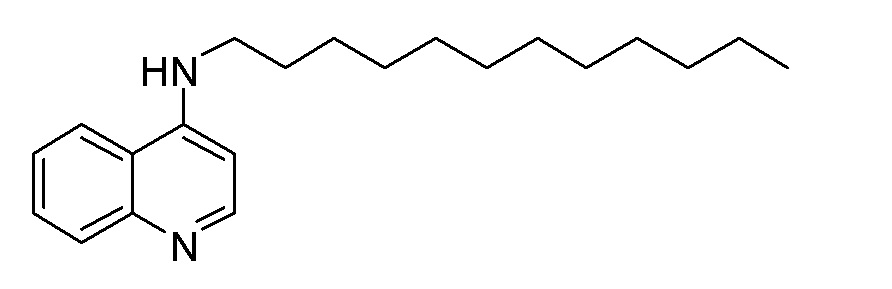
Смесь 4-хлорхинолина (3,25 г, 19,9 ммоль), 1-додециламина (3,80 г, 20,5 ммоль) и трипропиламина (5,90 мл, 30,9 ммоль) в 30 мл 1-пентанола нагревали с обратным холодильником в течение 16,5 часов. Затем летучие компоненты выпаривали в вакууме. Остаток разделяли между DCM (150, 100 мл) и смесью 1н. NaOH и 5% Na2CO3 (20 мл каждый). Органические фазы высушивали над Na2SO4 и концентрировали. Кристаллизация из ледяной смеси 10% ЕА/гексан, промывка собранного твердого вещества ледяной смесью 20% Et2O/Hex дала 4,95 г бесцветного твердого вещества (Т.пл. 81,5-82,0°C). LC/MS (230 нм) показала наличие примеси в количестве 5-10%. SPE (1% ТЕА/ЕА) отделила примесь с преимущественно арильными атомами водорода (ЯМР). Продукт перекристаллизовывали из ледяной смеси 10% ЕА/гексан, получая 4,70 г бесцветного твердого вещества. Rf 0,12 (10% MeOH/DCM); Т.пл. 80,5-81,5°C;1H ЯМР (CDCl3) δ 8,56 (д, 1H, J=5,1 Гц), 7,97 (дд, 1H, J=1,1, 8,4 Гц), 7,72 (м, 1H), 7,62 (ддд, 1H, J=1,5, 7,0, 8,5 Гц), 7,42 (ддд, 1H, J=1,5, 7,0, 8,5 Гц), 6,42 (д, 1H, J=5,5 Гц), 4,98 (шир. с, 1H, NH), 3,31 (м, 2H), 1,76 (п, 2H, J=7,3 Гц), 1,47 (м, 2H), 1,38-1,26 (м, 16H), 0,88 (т, 3H, J=6,8 Гц);13C ЯМР (CDCl3) δ 151,3, 149,8, 148,7, 130,3, 129,1, 124,7, 119,3, 118,9, 99,0, 43,5, 32,1, 29,8, 29,8, 29,8, 29,8, 29,6, 29,5, 29,2, 27,4, 22,9, 14,3.
Пример 90: N1,N8-ди(хинолин-4-ил)октан-1,8-диамин

Смесь 1,8-октандиамина и избытка 4-хлорхинолина и DIEA в NMP нагревали при 160°C в герметизированной пробирке в течение 3 дней. Смесь охлаждали и очищали SPE, с промывкой 1% MeOH/DCM и последующим элюированием 7,5% MeOH/DCM +2% ТЕА, получая продукт в форме твердого вещества. Rf 0,05 (ЕА+2% ТЕА);1H ЯМР (20% CD3OD/CDCl3) δ 8,32 (д, 2H, J=5,7 Гц), 7,85-7,80 (м, 4), 7,58 (ддд, 2H, J=1,2, 6,9, 8,2 Гц), 7,38 (ддд, 2H, J=1,2, 6,9, 8,4 Гц), 6,37 (д, 2H, J=5,7 Гц), 3,38-3,25 (м, 4H), 1,73-1,24 (м, 12H).
Пример 91: N-[8-(гексилокси)октил]хинолин-6-амин

8-(Гексилокси)октановая кислота Приблизительно 6,0 мл реагента Джонса добавляли к смеси 8-(гексилокси)октан-1-ола (2,1 г, 9,1 ммоль) и 50 мл DCM, охлажденной ванной со льдом, после чего зеленый цвет смеси исчез. Затем смесь промывали H2O и 0,1M HCl, и органическую фазу высушивали над MgSO4, разбавляли 5 мл MeOH, фильтровали через слой силикагеля, промывая этот слой 5% MeOH/DCM, и концентрировали. FC (5% MeOH/DCM) дала 1,6 г продукта. Rf 0,3 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 3,4 (т, 4H), 2,3 (м, 2H), 1,7-1,4 (м, 6H), 1,4-1,2 (м, 12H), 0,9 (м, 3H).
8-(Гексилокси)-N-(хинолин-6-ил)октанамид Смесь 6-аминохинолина (0,5 г, 3,5 ммоль), 8-(гексилокси)октановой кислоты (847 мг, 3,47 ммоль), 1-гидроксибензoтриазола (469 мг, 3,47 ммоль), 4-диметиламинопиридина (42 мг, 0,3 ммоль) и EDC (663 мг, 3,47 ммоль) в 20 мл DCM вводили в реакцию, пока исходный материал не был израсходован, что наблюдали с помощью TLC. Затем летучие компоненты выпаривали, и остаток разделяли между ЕА и H2O, 5%-ым Na2CO3, H2O и солевым раствором, и органические фазы высушивали над Na2SO4 и концентрировали. FC (50% ЕА/Гексан) дала 225 мг продукта. Rf 0,4 (50% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,8 (м, 1H), 8,4 (м, 1H), 8,15 (м, 1H), 8,05 (м, 1H), 7,9 (шир. с, 1H, NH), 7,6 (м, 1H), 7,4 (м, 1H), 3,4 (т, 4H), 2,4 (т, 2H), 1,7 (м, 2H), 1,6-1,4 (м, 4H), 1,4-1,2 (м, 12H), 0,85 (м, 3H).
N-[8-(гексилокси)октил]хинолин-6-амин Смесь 8-(гексилокси)-N-(хинолин-6-ил)октанамида (171 мг, 0,46 ммоль) и 20 мл THF охлаждали ванной со льдом, после чего добавляли 70 мг литий-алюминийгидрида. Смеси давали медленно нагреться до комнатной температуры в течение ночи. Затем смесь повторно охлаждали и осторожно добавляли 0,7 мл H2O, 0,7 мл 15%-ого NaOH и 2,1 мл H2O. Смесь фильтровали через слой Целита, промывая 5% MeOH/DCM, и фильтрат концентрировали. Остаток разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, и органическую фазу высушивали над Na2SO4 и концентрировали. FC (50% ЕА/Гексан) дала 100 мг продукта. Rf 0,3 (50% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,6 (м, 1H), 7,95-7,85 (м, 2H), 7,3 (м, 1H), 7,1 (м, 1H), 7,7 (м, 1H), 3,4 (т, 4H), 3,2 (т, 2H), 1,8-1,2 (м, 20H), 0,85 (т, 3H).
Пример 92: N-[8-(гексилокси)октил]хинолин-3-амин

N-[8-(гексилокси)октил]хинолин-3-амин (66 мг) получали согласно способу, используемому для получения N-[8-(гексилокси)октил]хинолин-6-амина, исходя из 3-аминохинолина (728 мг).
8-(Гексилокси)-N-(хинолин-3-ил)октанамид:1H ЯМР (CDCl3) δ 9,05 (шир. с, 1H), 8,95 (шир. с, 1H), 8,5 (шир. с, 1H, NH), 8,1 (д, 1H), 7,8 (д, 1H), 7,7-7,5 (м, 2H), 3,4 (м, 4H), 2,5 (т, 2H), 1,8 (м, 2H), 1,7-1,2 (м, 16H), 0,85 (т, 3H).
206-181 N-[8-(гексилокси)октил]хинолин-3-амин1H ЯМР (CDCl3) δ 8,6 (д, 1H), 8,0 (д, 1H), 7,6 (д, 1H), 7,5-7,3 (м, 2H), 7,0 (м, 1H), 4,3 (шир. с, 1H, NH), 3,5-3,3 (м, 4H), 3,2 (м, 2H), 1,8-1,2 (м, 20H), 0,9 (м, 3H).
Пример 93: N-[8-(гексилокси)октил]хинолин-8-амин

N-[8-(гексилокси)октил]хинолин-8-амин (58 мг) получали согласно способу, используемому для получения N-[8-(гексилокси)октил]хинолин-6-амина, исходя из 8-аминохинолина (472 мг).
8-(Гексилокси)-N-(хинолин-8-ил)октанамид: Rf 0,7 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 9,8 (шир. с, 1H, NH), 8,85-8,75 (м, 2H), 8,2 (м, 1H), 7,6-7,4 (м, 3H), 3,4 (м, 4H), 2,6 (т, 2H), 1,8 (м, 2H), 1,7-1,2 (м, 16H), 0,9 (м, 3H).
N-[8-(гексилокси)октил]хинолин-8-амин: Rf 0,6 (50% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,7 (д, 1H), 8,1 (шир. с, 1H), 7,5-7,3 (м, 2H), 7,0 (д, 1H), 6,7 (д, 1H), 3,5-3,3 (м, 4H), 3,3 (м, 2H), 1,8 (м, 2H), 1,7-1,2 (м, 18H), 0,9 (м, 3H).
Пример 94: N-[8-(гексилокси)октил]-2-(трифторметил)хинолин-4-амин

Смесь 8-(гексилокси)октан-1-амина (350 мг, 1,53 ммоль), 4-хлор-2-трифторметилхинолина (420 мг, 1,81 ммоль) и ТЕА (0,32 мл, 1,84 ммоль) в 1 мл NMP нагревали при 150°C в течение 16 часов. Смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали. Очистка препаративной TLC дала продукт. Rf 0,38 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,01 (м, 1H), 7,75 (д, 1H, J=8,4 Гц), 7,62 (ддд, 1H, J=1,2, 6,9, 8,4 Гц), 7,42 (ддд, 1H, J=1,2, 7,0, 8,4 Гц), 6,65 (с, 1H), 5,45 (м, 1H, NH), 3,38-3,34 (м, 4H), 3,27 (м, 2H), 1,76-1,18 (м, 20H), 0,85 (м, 3H).
Пример 95: 7-Хлор-N-децилхинолин-4-амин

7-Хлор-N-децилхинолин-4-амин (8,10 г) получали согласно способу, используемому для получения 7-хлор-N-додецилхинолин-4-амина, исходя из 5,18 г 1-дециламина и 6,53 г 4,7-дихлорхинолина. Т.пл. 102,5-103,0°C (ЕА/Гексан);1H ЯМР (CDCl3) δ 88,5 (д, 1H, J=5,5 Гц), 7,9 (д, 1H, J=1,9 Гц), 7,6 (д, 1H, J=8,8 Гц), 7,3 (м, 1H), 6,4 (д, 1H, J=5,5 Гц), 5,1 (шир. м, 1H, NH), 3,3 (м, 2H), 1,7 (м, 2H), 1,5-1,3 (м, 14H), 0,8 (м, 3H);13C ЯМР (CDCl3) δ 152,2, 149,9, 149,4, 134,9, 129,0, 125,4, 121,1, 117,3, 99,2, 43,5, 32,1, 29,7, 29,7, 29,6, 29,5, 29,1, 27,3, 22,9, 14,3.
Пример 96: 7-Хлор-N-додецилхинолин-4-амин

Смесь 1-додециламина (4,57 г, 24,7 ммоль), трипропиламина (9,4 мл, 49 ммоль), 4,7-дихлорхинолина (4,89 г, 24,7 ммоль) и 50 мл 1-пентанола нагревали с обратным холодильником в течение 22 часов. Затем летучие компоненты выпаривали. Остаток разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, и органическую фазу высушивали над Na2SO4 и концентрировали. SPE (50% ЕА/Гексан) дал продукт в форме твердого вещества желтого цвета. Продукт забирали в DCM, промывали 5%-ым Na2CO3, высушивали над Na2SO4 и концентрировали. Продукт кристаллизовали из ледяной всмеси 20% ЕА/гексан, получая 7,50 г бесцветного твердого вещества. Rf 0,30 (50% ЕА/Гексан); Т.пл. 95,0-97,0°C;1H ЯМР (CDCl3) δ 8,5 (д, 1H, J=5,1 Гц), 7,9 (д, 1H, J=1,9 Гц), 7,6 (д, 1H, J=8,8 Гц), 7,3 (м, 1H), 6,39 (д, 1H, J=5,5 Гц), 5,0 (шир. м, 1H, NH), 3,3 (м, 2H), 1,8 (м, 2H), 1,5-1,2 (м, 20H, 0,9 (м, 3H);13C ЯМР (CDCl3) δ 152,3, 149,9, 149,4, 135,0, 129,1, 125,4, 121,0, 117,3, 99,3, 43,5, 32,1, 29,8, 29,8, 29,8, 29,7, 29,6, 29,5, 29,1, 27,3, 22,9, 14,3.
Пример 97: N-(Децил)хиназолин-4-амин

Смесь 4-хлорхиназолина (6,90 г, 42,1 ммоль), 1-дециламина (10,8 мл, 54,3 ммоль) и ТЕА (8,90 мл, 62,7 ммоль) в 50 мл IPA нагревали с обратным холодильником в течение 6 часов, затем оставляли стоять в течение ночи. Затем летучие компоненты выпаривали, и остаток забирали в DCM и промывали смесью 20 мл 1н. NaOH и 20 мл 5%-ого Na2CO3. Органическую фазу высушивали над безводным Na2SO4 и фильтровали через слой силикагеля, промывая 5% MeOH/DCM. Фильтрат концентрировали, получая твердое вещество. Твердое вещество промывали частями по 10 мл и по 25 мл 20% Et2O/Hex, затем высушивали в вакууме, получая 11,22 г бесцветного твердого вещества. Rf 0,41 (10% MeOH/DCM); Т.пл. 72,5-73,0°C;1H ЯМР (CDCl3) δ 8,66 (с, 1H), 7,82 (дд, 1H, J=1,1, 8,8 Гц), 7,73-7,69 (м, 2H), 7,44 (м, 1H), 5,83 (шир. с, 1H, NH), 3,65 (м, 2H), 1,72 (м, 2H), 1,46-1,25 (м, 14H), 0,86 (т, 3H, J=7,0 Гц);13C ЯМР (CDCl3) δ 159,7, 155,7, 149,6, 132,7, 128,8, 126,1, 120,6, 115,2, 41,6, 32,1, 29,8, 29,7, 29,6, 29,5, 27,6, 22,9, 14,3.
Пример 98: N-Додецилхиназолин-4-амин

1-Додециламин (4,20 г, 22,7 ммоль) забирали в 45 мл IPA, и 10 мл удаляли перегонкой. Затем смесь немного охлаждали и добавляли ТЕА (6,5 мл, 46 ммоль) и 4-хлорхиназолин (3,72 г, 22,7 ммоль). Смесь нагревали с обратным холодильником в течение 7 часов. Затем большую часть летучих компонентов удаляли перегонкой. Остаток разделяли между DCM (150, 100 мл) и смесью 1н. NaOH и 5% Na2CO3 (20 мл каждый). Органические фазы высушивали над Na2SO4 и концентрировали. SPE (градиент стадии 30, 50 и 60% ЕА/гексан) дал содержащие продукт фракции, которые концентрировали, забирали в DCM, промывали 5%-ым Na2CO3, высушивали над Na2SO4 и концентрировали, получая сироп. Кристаллизация из ледяной смеси 30% ЕА/гексан дала 6,05 г бесцветного твердого вещества. Rf 0,20 (50% ЕА/Гексан); Т.пл. 74,0-75,0°C;1H ЯМР (CDCl3) δ 866 (с, 1H), 7,82 (м, 1H), 7,74-7,69 (м, 2H), 7,45 (м, 1H), 5,76 (шир. с, 1H, NH), 3,65 (м, 2H), 1,72 (м, 2H), 1,46-1,25 (м, 18H), 0,87 (м, 3H);13C ЯМР (CDCl3) δ 159,6, 155,7, 149,6, 132,7, 128,9, 126,1, 120,6, 115,1, 41,6, 32,1, 29,8, 29,8, 29,8, 29,8, 29,6, 29,6, 29,5, 27,3, 22,9, 14,3.
Пример 99: N-Децил-7-фторхиназолин-4-амин
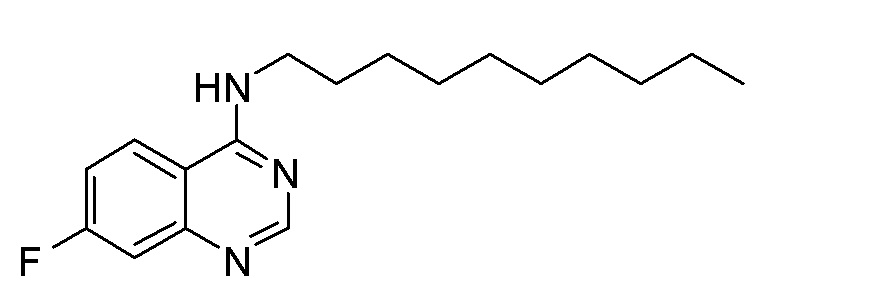
Смесь 1-дециламина (1,2 мл, 6,0 ммоль), 4-хлор-7-фторхиназолина (1,1 г, 6,0 ммоль) и ТЕА (1,3 мл, 9,3 ммоль) в 10 мл IPA нагревали с обратным холодильником в течение 6 часов. Затем летучие компоненты выпаривали, и остаток разделяли между DCM (400, 300 мл) и 5% Na2CO3 (400 мл). Органические фазы высушивали над безводным Na2SO4, фильтровали через слой силикагеля, промывая 10% MeOH/DCM, и концентрировали. Продукт кристаллизовали из ЕА/Гексан.
Пример 100: N-Додецил-7-фторхиназолин-4-амин
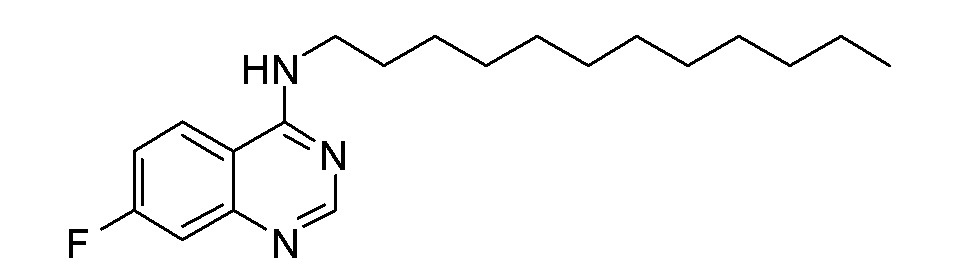
N-Додецил-7-фторхиназолин-4-амин получали из 1-додециламина (1,2 мл, 5,2 ммоль), 4-хлор-7-фторхиназолина (1,0 г, 5,5 ммоль), и ТЕА (1,2 мл, 8,6 ммоль) в 10 мл IPA согласно способу, используемому для получения N-децил-7-фторхиназолин-4-амина.
Пример 101: 7-Хлор-N-децилхиназолин-4-амин

7-хлор-N-децилхиназолин-4-амин получали из 1-дециламина (1,5 мл, 7,0 ммоль), 4,7-дихлорхиназолина (1,4 г, 7,0 ммоль) и ТЕА (2,0 мл, 14 ммоль) в 15 мл IPA согласно способу, используемому для получения N-децил-7-фторхиназолин-4-амина.
Пример 102: 7-Хлор-N-додецилхиназолин-4-амин

7-хлор-N-додецилхиназолин-4-амин получали из 1-додециламина (1,3 г, 7,0 ммоль), 4,7-дихлорхиназолина (1,4 г, 7,0 ммоль) и ТЕА (2,0 мл, 14 ммоль) в 15 мл IPA согласно способу, используемому для получения N-децил-7-фторхиназолин-4-амина.
Пример 103: N-(6-Бутоксигексил)хиназолин-4-амин

6-Бутоксигексан-1-амин (7,20 г, 41,1 ммоль) забирали в 200 мл, и 50 мл удаляли перегонкой. Смесь немного охлаждали и добавляли ТЕА (17,4 мл, 124 ммоль) и 4-хлорхиназолин (11,11 г, 67,7 ммоль). Смесь нагревали с обратным холодильником в течение 38 часов, затем оставляли стоять при комнатной температуре в течение 3 дней. Летучие компоненты выпаривали. Остаток разделяли между DCM (150, 2×50 мл) и смесью 40 мл 1н. NaOH и 40 мл 5%-ого Na2CO3. Органические фазы высушивали над безводным Na2SO4 и упаривали на силикагеле. SPE, с промывкой 30% ЕА/Гексан и с элюированием 60% ЕА/Гексан, дал желтый сироп, который кристаллизовали из 10% ЕА/гексан при -20°C, получая 4,64 г бесцветного твердого вещества. Rf 0,25 (50% ЕА/Гексан); Т.пл. 40-46°C;1H ЯМР (CDCl3) δ 8,64 (с, 1H), 7,84 (д, 1H, J=8,4 Гц), 7,78-7,70 (м, 2H), 7,46 (ддд, 1H, J=1,4, 7,3, 8,4 Гц), 6,12 (шир. с, 1H, NH), 3,66 (м, 2H), 3,41-3,37 (м, 4H), 1,74 (м, 2H), 1,62-1,30 (м, 10H), 0,90 (т, 3H, J=7,3 Гц);13C ЯМР (CDCl3) δ 159,8, 155,2, 148,6, 133,0, 128,1, 126,3, 120,9, 115,0, 70,9, 70,9, 41,6, 32,0, 29,8, 29,4, 27,1, 26,2, 19,6, 14,1.
Пример 104: N-[8-(гексилокси)октил]хиназолин-4-амин

8-(Гексилокси)октан-1-ол 1,8-Октандиол (201,4 г, 1,38 моль) забирали в 1,3 л IPA, и 250 мл летучего материала удаляли перегонкой. Смеси позволяли охладиться до температуры ниже температуры кипения, и металл натрия (6,9 г, 0,30 моль) добавляли частями, поддерживая атмосферу аргона. После завершения добавления смесь кипятили в течение одного часа и затем перемешивали при комнатной температуре в течение ночи. 1-Бромгексан (32,2 мл, 0,23 моль) добавляли в медленном потоке. Через 25 часов смесь мягко нагревали. Начал формироваться осадок. После 2 дней нагревания, смесь нагревали для перегонки 400 мл летучего материала. Затем нагревание прекращали и добавляли 16 г NH4Cl в 48 мл H2O. Через 1 час перегонку возобновляли и собирали 450 мл дистиллята. Нагревание останавливали, и 214 г силикагеля добавляли к горячей смеси. Теплую смесь тщательно перемешивали и охлаждали. Избыток диола удаляли SPE с использованием 30% ЕА/гексан, получая 25,9 г светло-желтого масла, содержащего желаемый продукт. Rf 0,19 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,63-3,58 (м, 2H), 3,37 (т, 4H, J=6,7 Гц), 1,66 (шир. с, 1H, OH), 1,57-1,50 (м, 6H), 1,30-1,28 (м, 14H), 0,87 (т, 3H, J=6,6 Гц). 1,8-Октандиол рекуперировали, элюируя 5%- MeOH/DCM, выпаривали растворитель и кристаллизовли три выхода из смеси ЕА/Гексан, получая 182,4 г бесцветного твердого вещества.
8-(гексилокси)октил метансульфонат 8-(Гексилокси)октан-1-ол забирали в 250 мл DCM и охлаждали с использованием ванны со льдом. Добавляли по очереди ТЕА (21,0 мл, 150 ммоль) и метансульфонил хлорид (10,5 мл, 134 ммоль). Через 1,25 часа добавляли 20 г кубиков льда. Большую часть летучего материала выпаривали. Остаток разделяли между 1:1 ЕА/Гексан (3×300 мл) и H2O, насыщенным NaHCO3, H2O, 1M HCl, H2O и солевым раствором (100 мл каждый). Объединенные органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. Rf 0,28 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 4,21 (т, 2H, J=6,6 Гц), 3,38 (т, 2H, J=6,4 Гц), 3,37 (т, 2H, J=6,7 Гц), 2,98 (с, 3H), 1,72 (м, 2H), 1,61-1,46 (м, 4H), 1,40-1,24 (м, 14H), 0,87 (т, 3H, J=6,8 Гц).
N-[8-(гексилокси)октил]фталимид Толуол (100 мл) смешивали с сырым 8-(гексилокси)октил метансульфонатом и затем упаривали. Остаток забирали в 120 мл DMF и 60 мл NMP. Добавляли фталимид калия (25,0 г, 135 ммоль). После смешивания в течение 21,5 часов, добавляли 50 мл H2O, и летучий материал выпаривали. Остаток разделяли между ЕА (3×300 мл) и H2O (150 мл), насыщенным NaHCO3 (150 мл) и солевым раствором (2×150 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. Rf 0,50 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,81 и 7,68 (м, 4H, AA’BB’), 3,65 (т, 2H, J=7,3 Гц), 3,36 (т, 2H, J=6,7 Гц), 3,35 (т, 2H, J=6,7 Гц), 1,67-1,48 (м, 6H), 1,29-1,22 (м, 14H), 0,86 (т, 3H, J=6,8 Гц).
8-(Гексилокси)октан-1-амин IPA (100 мл) смешивали с сырым N-[8-(гексилокси)октил]фталимидом и затем упаривали. Остаток забирали в 450 мл EtOH, добавляли гидразин моногидрат (6,60 мл, 136 ммоль), и смесь нагревали с обратным холодильником в течение ночи. Смесь концентрировали перегонкой 300 мл летучего материала. Нагревание останавливали, 150 мл 1M, HCl добавляли к горячей смеси, и смеси давали охладиться. Осадок удаляли фильтрацией и промывали 1:1 EtOH/H2O (2×100 мл). Фильтрат концентрировали до 100 мл и подщелачивали до рН>10 с использованием гранул NaOH. Смесь экстрагировали DCM (3×250 мл), и объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая 27,6 г мутной жидкости.1H ЯМР (CDCl3) δ 3,36 (т, 4H, J=6,7 Гц), 2,66 (т, 2H, J=6,9 Гц), 1,52 (м, 2H), 1,44-1,28 (м, 18H), 0,86 (м, 3H).
N-[8-(гексилокси)октил]хиназолин-4-амин Сырой 8-(гексилокси)октан-1-амин забирали в 400 мл IPA, и 250 мл летучего материала удаляли перегонкой. Смесь охлаждали и добавляли ТЕА (16,8 мл, 120 ммоль) и 4-хлорхиназолин (9,8 г, 60 ммоль). Смесь нагревали с обратным холодильником в течение 4 часов. TLC аликвоты показала существенное количество остаточного материала нингидрина (+). Добавляли ТЕА (11,2 мл, 80 ммоль) и 4-хлорхиназолин (6,5 г, 38 ммоль). После дополнительного нагревания в течение 5 часов смеси давали охладиться и перемешивали 12 часов. Затем летучие компоненты выпаривали, и остаток разделяли между DCM (300, 2×150 мл) и 1н. NaOH и 5% Na2CO3 (100 мл каждый). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали. SPE, с элюированием 20%, 30% и 50% ЕА/Гексан, дал фракции продукта, которые объединяли и концентрировали. Остаток забирали в 300 мл ЕА, фильтровался и концентрировали. Полученное твердое вещество желтого цвета перекристаллизовано дважды из 10% ЕА/гексан, получая 30,3 г твердого вещества светло-желтого цвета. Rf 0,11 (40% ЕА/Гексан); Т.пл. 67,0-67,5°C;1H ЯМР (CDCl3) δ 8,66 (с, 1H), 7,83 (д, 1H, J=7,8 Гц), 7,75-7,70 (м, 2H), 7,46 (м, 1H), 5,81 (шир. с, 1H, NH), 3,65 (дт, 2H, J=5,5, 7,4 Гц), 3,38 (т, 4H), 1,73 (м, 2H), 1,59-1,52 (м, 4H), 1,46-1,24 (м, 14H), 0,87 (т, 3H, J=6,9 Гц);13C ЯМР (CDCl3) δ 159,7, 155,6, 149,4, 132,8, 128,7, 126,2, 120,6, 115,1, 71,2, 71,1, 41,7, 41,5, 31,9, 30,0, 29,6, 29,6, 29,5, 27,2, 26,4, 26,1, 22,8, 14,3.
Пример 105: N-[8-(4-Метоксифенокси)октил]хиназолин-4-амин
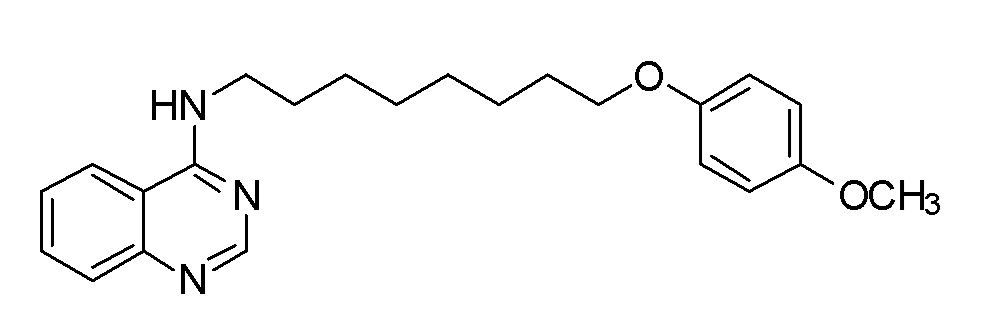
8-(4-Метоксифенокси)октан-1-амин (4,03 г, 16,1 мм) забирали в 125 мл IPA, и 50 мл летучих компонентов удаляли перегонкой. Смесь немного охлаждали и добавляли ТЕА (4,50 мл, 32,1 ммоль) и 4-хлорхиназолин (2,92 г, 17,7 ммоль). Нагревание с обратным холодильником продолжали. Через 24 часа, смеси давали охладиться и добавляли 15 мл 1н. NaOH. Летучие компоненты выпаривали. Остаток разбавляли DCM, промывали 5%-ым Na2CO3, высушивали над безводным Na2SO4 и концентрировали на силикагеле. SPE, с промывкой 50% ЕА/Гексан и с элюированием 40% ЕА/Гексан +2% ТЕА, дал содержащие продукт фракции, которые концентрировали, забирали в DCM, промывали 5%-ым Na2CO3, высушивали над безводным Na2SO4 и концентрировали, получая твердое вещество желтого цвета. Перекристаллизация из ЕА/Гексан дала 3,93 г твердого вещества белого цвета. Rf 0,41 (50% ЕА/Гексан +2% ТЕА); Т.пл. 97,0-98,0°C;1H ЯМР (CDCl3) δ 8,66 (с, 1H), 7,81 (дд, 1H, J=0,7, 8,4 Гц), 7,51 (м, 1H), 7,69 (ддд, 1H, J=1,5, 7,0, 8,5 Гц), 7,41 (ддд, 1H, J=1,5, 7,0, 8,4 Гц), 6,83-6,78 (м, 4H, AA’BB’), 6,09 (м, 1H, NH), 3,87 (т, 2H, J=6,6 Гц), 3,74 (с, 3H), 3,67 (м, 2H), 1,76-1,66 (м, 4H), 1,46-1,33 (м, 8H);13C ЯМР (CDCl3) δ 159,7, 155,6, 153,8, 153,4, 149,5, 132,6, 128,6, 126,0, 120,8, 115,6, 115,2, 114,8, 68,7, 55,9, 41,5, 29,5, 29,4, 29,4, 27,1, 26,1.
Пример 106: N-{2-[2-(гексилокси)фенокси]этил}хиназолин-4-амин

2-[2-(гексилокси)фенокси]этанамин (15,32 г, 64,6 ммоль) забирали в 350 мл IPA, и 50 мл удаляли перегонкой. Смесь немного охлаждали и добавляли ТЕА (18,0 мл, 128 ммоль) и 4-хлорхиназолин (11,0 г, 67,1 ммоль). Смесь нагревали с обратным холодильником в течение 16 часов. Затем летучие компоненты выпаривали, и остаток разделяли между DCM и 5%-ым Na2CO3 (500 мл каждого). Органическую фазу высушивали над Na2SO4 и концентрировали. Твердое вещество перекристаллизовано из ЕА/Гексан, получая 16,0 г твердого вещества.1H ЯМР (CDCl3) δ 8,6 (с, 1H), 7,9-7,7 (м, 3H), 7,4 (м, 1H), 7,0-6,8 (м, 4H), 6,6 (шир. с, 1H, NH), 4,3 (м, 2H), 4,1-4,0 (м, 4H), 1,8 (м, 2H), 1,4 (м, 2H), 1,3-1,2 (м, 4H), 0,8 (м, 3H).
Пример 107: N-{3-[2-(гексилокси)фенокси)пропил}хиназолин-4-амин
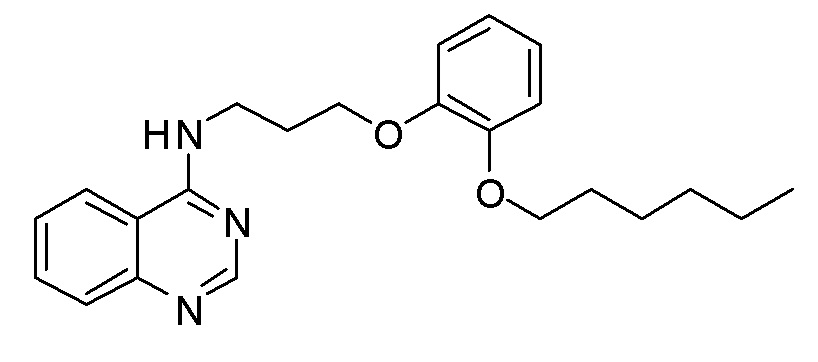
2-(Гексилокси)фенол Смесь катехола (47,5 г, 432 ммоль), 1-бромгексана (71,2 г, 432 ммоль) и K2CO3 (71,5 г, 518 ммоль) в 120 мл NMP и 240 мл DMF нагревали при 60°C в течение 24 часов. Затем летучие компоненты выпаривали, и суспензию разделяли между ЕА (600, 2×250 мл) и H2O, 5% Na2CO3 (2x), H2O, 0,1M HCl и солевым раствором (150 мл каждый). Органические фазы высушивали над Na2SO4 и упаривали на силикагеле. SPE (10% ЕА/Гексан) дал 75,5 г бесцветной жидкости, которая содержала в молярном отношении 2,5:1 2-(гексилокси)фенол и 1,2-бис-(гексилокси)бензол, как было вычислено из спектра ЯМР. Реакцию повторяли, используя катехол (71,68 г, 652 ммоль), 1-бромгексан (91,0 мл, 651 ммоль) и K2CO3 (108 г, 783 ммоль) в 240 мл DMF при комнатной температуре. Реакция дала 96,3 г светло-желтой жидкости, которая содержала в молярном отношении 1:1 2-(гексилокси)фенол и 1,2-бис-(гексилокси)бензол.
N-{3-[2-(гексилокси)фенокси)пропил}фталимид 1:1 смесь 2-(гексилокси)фенола и 1,2-бис-(гексилокси)бензола (47,2 г, 100 ммоль фенола), K2CO3 (18,7 г, 136 ммоль) и N-(3-бромпропил)фталимида (26,8 г, 100 ммоль) в 100 мл DMF нагревали при 55°C в течение 24 часов. Затем смесь охлаждали, и большую часть летучих компонентов выпаривали. Остаток разделяли между ЕА (3×250 мл) и H2O (3×200 мл), 0,05M HCl (2×150 мл) и солевым раствором (150 мл). Объединенные органические фазы высушивали над Na2SO4 и концентрировали. SPE с промывкой 5% ЕА/Гексан для элюирования остаточных исходных материалов и затем элюированием продукта смесью 20% ЕА/Гексан дали 29,8 г твердого вещества белого цвета. Rf 0,41 (20% ЕА/Гексан).
3-[2-(гексилокси)фенокси)пропан-1-амин Смесь N-{3-[2-(гексилокси)фенокси)пропил}фталимида (29,8 г, 78,2 ммоль) и гидразин моногидрата (4,80 мл, 101 ммоль) в 300 мл EtOH нагревали с обратным холодильником в течение 16 часов. Затем нагревание останавливали и добавляли 50 мл 2M HCl. Суспензию смешивали в течение 2 часов, затем фильтровали через слой Целита, промывая 100 мл 10%-ого водного раствора EtOH. Фильтрат подщелачивали до рН 10 с использованием гранул NaOH и концентрировали. SPE, с промывкой 3% MeOH/DCM и с элюированием 8% MeOH/DCM +2% ТЕА, дал 15,5 г желтого масла.
3-[2-(гексилокси)фенокси)пропан-1-амин (15,5 г, 61,8 ммоль) забирали в 250 мл IPA, и 50 мл удаляли перегонкой. Смесь немного охлаждали и добавляли ТЕА (10,5 мл, 74,8 ммоль) и 4-хлорхиназолин (11,1 г, 67,6 ммоль). Смесь нагревали с обратным холодильником в течение 16 часов. Затем большую часть летучих компонентов выпаривали, и остаток разделяли между ЕА (300, 2×250 мл) и 5% Na2CO3 и солевым раствором (150 мл каждый). Органические фазы высушивали над безводным Na2SO4 и концентрировали, получая темную жидкость. Растирание с двумя частями ледяной смеси 50% Et2O/Hex дало 14,9 г твердого вещества светлого желто-коричневого цвета. Rf 0,20 (50% ЕА/Гексан +2% ТЕА) 0,28 (5% MeOH/DCM +2% ТЕА); Т.пл. 67,0-67,5°C;1H ЯМР (CDCl3) δ 8,65 (с, 1H), 7,85-7,81 (м, 2H), 7,70 (ддд, 1H, J=1,5, 7,0, 8,4 Гц), 7,38 (ддд, 1H, J=1,1, 6,9, 8,0 Гц), 7,11 (шир. с, 1H, NH), 7,00-6,89 (м, 4H), 4,24 (м, 2H), 4,04 (м, 2H), 3,93 (м, 2H), 2,24 (м, 2H), 1,71 (м, 2H), 1,37 (м, 2H), 1,23-1,17 (м, 4H), 0,81 (м, 3H);13C ЯМР (CDCl3) δ 159,7, 155,5, 149,5, 149,2, 148,6, 132,6, 128,3, 126,0, 122,5, 121,6, 121,3, 115,5, 115,3, 113,8, 70,5, 69,2, 40,9, 31,6, 29,2, 28,5, 25,8, 22,7, 14,1.
Пример 108: N-{4-[2-(гексилокси)фенокси]бутил}хиназолин-4-амин

4-[2-(гексилокси)фенокси]бутан-1-амин (13,82 г, 52,2 ммоль) забирали в 300 мл IPA, и 50 мл удаляли перегонкой. Затем смесь немного охлаждали и добавляли ТЕА (15 мл, 107 ммоль) и 4-хлорхиназолин (8,6 г, 52 ммоль). Смесь нагревали с обратным холодильником в течение 16 часов. Затем летучие компоненты выпаривали, и остаток разделяли между DCM и 5%-ым Na2CO3 (500 мл из каждого). Органическую фазу высушивали над Na2SO4 и концентрировали. Твердое вещество перекристаллизовано из ЕА/Гексан, получая 8,3 г бесцветного твердого вещества.
Пример 109: N-[8-октил(Хиназолин-4-иламино)]никотинамид

N-(8-Аминооктил)никотинамид (2,60 г, 10,4 ммоль) забирали в 65 мл IPA, и 30 мл летучих компонентов удаляли перегонкой. Смесь охлаждали и добавляли ТЕА (2,90 мл, 20,7 ммоль) и 4-хлорхиназолин (1,88 г, 11,5 ммоль). Смесь нагревали с обратным холодильником в течение 6 часов. Затем летучие компоненты выпаривали, и остаток разделяли между DCM и смесью 20 мл 1н. NaOH и 20 мл 5%-ого Na2CO3. Темную водную фазу экстрагировали 40 мл 1-бутанола. Объединенные органические фазы концентрировали. Остаток забирали в 10% MeOH/DCM +2% ТЕА и фильтровали через слой силикагеля. Фильтрат концентрировали, получая темное твердое вещество. Твердое вещество перекристаллизовывали из 10%-ого водного раствора MeOH, что частично удалило окраску. Перекристаллизация из EtOH дала два выхода твердого вещества светлого желто-коричневого цвета с сопоставимыми спектрами1H ЯМР; выходы объединяли, получая 2,08 г с Т.пл. 173-176°C и 67% чистотой LC (230 нм). FC (градиент стадии 10%-12% MeOH/DCM) и перекристаллизация из IPA/H2O дала 1,52 г твердого вещества светло-желтого цвета, 89% чистота LC (230 нм). Растирание с ледяным Et2O и затем 30% ЕА/Гексан при комнатной температуре дало твердое вещество с Т.пл. 172,5-176,0°C и 90% чистотой LC (230 нм).1H ЯМР (40oC, DMSO-d6) δ 8,96 (д, 1H, J=1,5 Гц), 8,66 (д, 1H, J=3,3 Гц), 8,56 (шир. с, 1H), 8,42 (с, 1H), 8,21-8,13 (м, 3H), 7,72 (м, 1H), 7,63 (м, 1H), 7,48-7,44 (м, 2H), 3,51 (м, 2H), 3,23 (м, 2H), 1,62 (м, 2H), 1,51 (м, 2H), 1,4-1,2 (м, 8H);13C ЯМР (DMSO-d6) δ 164,6, 159,3, 155,1, 151,6, 149,0, 148,3, 134,8, 132,3, 130,1, 127,4, 125,4, 123,4, 122,6, 114,9, 40,4, 39,2, 29,0, 28,8, 28,7, 28,5, 26,5, 26,4.
Пример 110: N-[3-(гексилокси)бензил]хиназолин-4-амин

[3-(гексилокси)фенил]метанамин (18,5 г 89,3 ммоль) забирали в 300 мл IPA, и 100 мл летучего материала удаляли перегонкой. Смесь охлаждали и добавляли ТЕА (25,3 мл, 180 ммоль) и 4-хлорхиназолин (16,1 г, 98,3 ммоль). Смесь нагревали с обратным холодильником в течение 5 часов и затем перемешивали при комнатной температуре в течение ночи. Затем летучие компоненты выпаривали, и остаток забирали в DCM (200 мл) и промывали 1н. NaOH (100 мл). Водную фазу экстрагировали DCM (100 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали, получая твердое вещество красно-коричневого цвета. SPE, с элюированием 20%, 30% и 50% ЕА/Гексан, дал фракции продукта, которые объединяли и концентрировали, получая твердое вещество коричневого цвета. Перекристаллизация из смеси ЕА/Гексан дала 21,8 г продукта в форме бесцветного твердого вещества. Rf 0,21 (50% ЕА/Гексан); Т.пл. 106,0-107,0°C;1H ЯМР (CDCl3) δ 8,69 (с, 1H), 7,84 (д, 1H), 7,74-7,71 (м, 2H), 7,44 (м, 1H), 7,25 (м, 1H), 6,96-6,93 (м, 2H), 6,83 (дд, 1H, J=2,2, 8,5 Гц), 6,18 (шир. с, 1H), 4,83 (м, 2H, AB), 3,92 (т, 2H, J=6,6 Гц), 1,75 (м, 2H), 1,42 (м, 2H), 1,33-1,28 (м, 4H), 0,89 (м, 3H);13C ЯМР (CDCl3) δ 159,8, 159,5, 155,8, 149,6, 139,7, 132,9, 130,1, 128,8, 126,3, 120,8, 120,2, 115,0, 114,5, 113,8, 68,2, 45,5, 31,8, 29,4, 25,9, 22,8, 14,2.
Пример 111: N-[3-(децилокси)бензил]хиназолин-4-амин
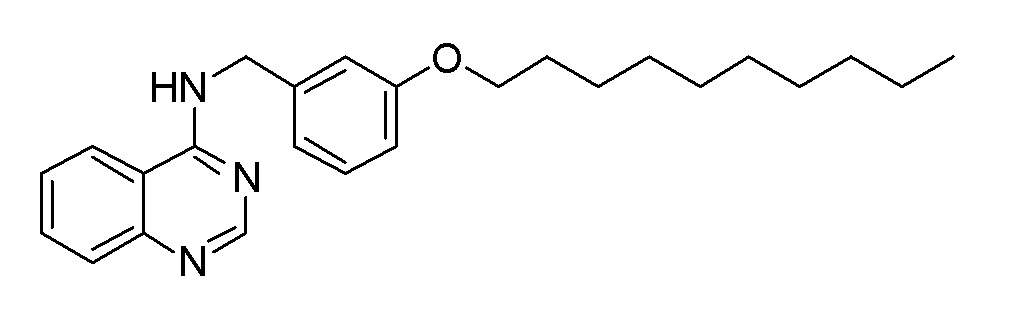
(3-(децилокси)фенил)метанол Смесь 3-гидроксибензилового спирта (36,2 г, 292 ммоль), 1-бромдекана (55,5 мл, 269 ммоль) и K2CO3 (44,3 г, 321 ммоль) в 60 мл NMP и 120 мл DMF перемешивали при 60°C в течение 2 дней при помощи приводной мешалки. Затем летучие компоненты удаляли в вакууме. Полученную суспензию разделяли между 50% ЕА/Гексан (300, 2×250 мл) и H2O (400 мл), 0,2N NaOH (150 мл), H2O (150 мл), 2M HCl (150 мл), H2O (150 мл) и солевым раствором (150 мл). Органические фазы высушивали над безводным Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 67,8 г янтарного масла. Масло затвердевало экзотермически. ЯМР показал наличие остаточного 1-бромдекана и ЕА.1H ЯМР (CDCl3) δ 7,2 (м, 1H), 6,9 (м, 2H), 6,8 (м, 1H), 3,9 (шир. с, 2H, AB), 3,9 (т, 2H, J=6,6 Гц), 2,6 (шир. с, 1H, OH), 1,8 (м, 2H), 1,5 (м, 2H), 1,4-1,2 (м, 12H), 0,9 (м, 3H);13C ЯМР (CDCl3) δ 159,5, 142,7, 129,6, 119,0, 113,8, 113,0, 68,1, 65,2, 32,0, 29,8, 29,7, 29,6, 29,5, 29,4, 26,2, 22,8, 14,3.
1-(Хлорметил)-3-(децилокси)бензол Смесь [3-(децилокси)фенил]метанола (58,4 г, 221 ммоль) и 150 мл толуола добавляли по каплям к смеси тионил хлорида (19,4 мл, 266 ммоль) и 50 мл толуола. В ходе добавления наблюдалось выделение газа. Через 16 часов смесь нагревали с обратным холодильником. Через 1 час 150 мл летучего материала удаляли перегонкой. Затем оставшиеся летучие компоненты выпаривали в вакууме.
N-[3-(децилокси)бензил]фталимид Остаток забирали в 120 мл DMF и 60 мл NMP, добавляли фталимид калия (49,2 г, 266 ммоль), и смесь нагревали при 60°C в течение 24 часов. Затем смесь охлаждали и разделяли между 50% ЕА/Гексан и H2O (2x), 0,1M HCl и солевым раствором. Органические фазы высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали, получая 90,4 г янтарного масла.1H ЯМР (CDCl3) δ 7,8 и 7,7 (м, 4H, AA’BB’), 7,2 (м, 1H), 7,0 (м, 2H), 6,8 (м, 1H), 4,8 (с, 2H), 3,9 (т, 2H, J=6,6 Гц), 1,7 (м, 2H), 1,4 (м, 2H), 1,4-1,2 (м, 12H), 0,9 (м, 3H);13C ЯМР (CDCl3) δ 168,2, 159,6, 137,9, 134,2, 132,3, 129,9, 123,6, 120,8, 114,8, 114,1, 68,2, 41,8, 32,1, 29,8, 29,8, 29,6, 29,5, 29,5, 26,2, 22,9, 14,3.
[3-(децилокси)фенил]метанамин IPA (50 мл) смешивали с остатком и затем упаривали, чтобы удалить остаточный ЕА. Остаток забирали в 400 мл EtOH, добавляли гидразин моногидрат (14,5 мл, 299 ммоль), и смесь нагревали с обратным холодильником. Через 6 часов смесь охлаждали и добавляли 150 мл 2M HCl. Твердый осадок дробили, формируя суспензию, которую фильтровали и промывали 20%-ым водным IPA. Фильтрат подщелачивали до рН 10, добавляя гранулы NaOH. Затем смесь концентрировали. Полученную жидкость разделяли между DCM и 5%-ым Na2CO3, и органическую фазу высушивали над безводным Na2SO4 и концентрировали.
N-[3-(децилокси)бензил]хиназолин-4-амин Сырой [3-(децилокси)фенил]метанамин забирали в 400 мл IPA, и 100 мл летучих компонентов удаляли перегонкой. Смеси давали немного охладиться. Добавляли ТЕА (39 мл, 278 ммоль) и 4-хлорхиназолин 22,4 г, 136 ммоль). Смесь нагревали с обратным холодильником в течение 20 часов. Затем смеси давали охладиться, и летучие компоненты выпаривали. Смесь разделяли между DCM (350, 2×100 мл) и 2н. NaOH (150 мл). Органические фазы высушивали над безводным Na2SO4, добавляли 150 мл MeOH, и смесь фильтровали через слой силикагеля. Фильтрат концентрировали, получая твердое вещество розового цвета. Твердое вещество перекристаллизовывали из смеси ЕА/Гексан, получая слегка окрашенное твердое вещество. Твердое вещество перекристаллизовывали из IPA, получая 43,4 г бесцветного твердого вещества. Rf 0,47 (10% MeOH/DCM); Т.пл. 93,0-95,5°C;1H ЯМР (CDCl3) δ 8,71 (с, 1H), 7,86 (д, 1H, J=8,4 Гц), 7,76-7,68 (м, 2H), 7,46 (м, 1H), 7,27 (м, 1H), 6,98-6,94 (м, 2H), 6,84 (м, 1H), 5,95 (шир. с, 1H, NH), 4,84 (м, 2H, AB), 3,94 (т, 2H, J=6,6 Гц), 1,77 (м, 2H), 1,43 (м, 2H), 1,29-1,26 (м, 12H), 0,87 (м, 3H);13C ЯМР (CDCl3) δ 159,8, 159,4, 155,6, 149,8, 139,8, 132,9, 130,1, 128,9, 126,3, 120,7, 120,2, 115,0, 114,6, 113,8, 68,3, 45,6, 32,1, 29,8, 29,8, 29,6, 29,5, 29,5, 26,3, 22,9, 14,3.
Пример 112: N-(3-Феноксибензил)хиназолин-4-амин

(3-Феноксифенил)метанамин (1,55 г, 7,79 ммоль) забирали в 60 мл IPA, и 15 мл летучего материала удаляли перегонкой. Смесь охлаждали и добавляли ТЕА (1,50 мл, 10,7 ммоль) и 4-хлорхиназолин (1,20 г, 7,32 ммоль) в 15 мл IPA. Смесь нагревали с обратным холодильником в течение 5,5 часов и затем перемешивали при комнатной температуре в течение ночи. Затем летучие компоненты выпаривали, и остаток разделяли между DCM (3×70 мл) и 5% Na2CO3 (40 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и концентрировали. SPE, с элюированием 25% и затем 55% ЕА/Гексан, дал фракции продукта, которые объединяли и концентрировали, получая к твердое вещество оранжевого цвета. Перекристаллизация из смеси ЕА/Гексан дала твердое вещество розового цвета, и затем из MeOH дала 1,29 г твердого вещества светло-розового цвета. Rf 0,19 (50% ЕА/Гексан); Т.пл. 146,5-148,0°C;1H ЯМР (CDCl3) δ 8,66 (с, 1H), 7,83 (д, 1H, J=8,5 Гц), 7,77 (д, 1H, J=8,1 Гц), 7,71 (м, 1H), 7,42 (м, 1H), 7,30 (м, 3H), 7,10 (м, 2H), 7,04 (шир. с, 1H), 6,99 (м, 2H), 6,90 (м, 1H), 6,44 (м, 1H, NH), 4,84 (м, 2H, AB);13C ЯМР (CDCl3) δ 159,5, 157,9, 157,0, 155,5, 149,6, 140,4, 132,9, 130,3, 130,0, 128,7, 126,3, 123,7, 122,6, 120,9, 119,2, 118,3, 117,9, 115,1, 45,1.
Пример 113: N-[4-(децилокси)бензил]хиназолин-4-амин

4-(децилокси)бензонитрил Смесь 4-гидроксибензoнитрила (4,32 г, 36,3 ммоль), 1-бромдекана (6,80 мл, 32,9 ммоль) и K2CO3 (6,61 г, 47,8 ммоль) в 20 мл DMF вводили в реакцию в течение 2 дней. Растворитель выпаривали в вакууме. Остаток разделяли между 50% ЕА/Гексан (3×150 мл) и 5% Na2CO3 (3×80 мл), H2O (40 мл), 0,1M HCl (40 мл) и солевым раствором (80 мл). Органические фазы высушивали над безводным Na2SO4 и концентрировали, получая 8,30 г бесцветного масла, которое затвердевало при отстаивании.1H ЯМР (CDCl3) δ 7,54 и 6,90 (м, 4H, AA’BB’), 3,97 (т, 2H, J=6,6 Гц), 1,78 (м, 2H), 1,42 (м, 2H), 1,34-1,25 (м, 12H), 0,86 (м, 3H);13C ЯМР (CDCl3) δ 162,6, 134,0, 119,4, 115,3, 103,7, 68,5, 32,0, 29,6, 29,4, 29,4, 29,1, 26,0, 22,8, 14,2.
[4-(децилокси)фенил]метанамин (7,61 г) получали в форме бесцветного твердого вещества способом, используемым для [4-(гексилокси)фенил]метанамина, обрабатывая 4-(децилокси)бензoнитрил 2 г LAH.1H ЯМР (CDCl3) δ 7,2 (м, 2H), 6,8 (м, 2H), 3,90 (т, 2H, J=6,6 Гц), 3,76 (с, 2H), 1,75 (м, 2H), 1,55 (м, 2H), 1,43 (м, 2H), 1,4-1,2 (м, 10H), 0,87 (м, 3H);13C ЯМР (CDCl3) δ 158,1, 135,4, 128,3, 114,5, 68,0, 46,0, 32,0, 29,6, 29,6, 29,5, 29,4, 29,4, 28,1, 26,1, 22,7, 14,2.
N-[4-(децилокси)бензил]хиназолин-4-амин (3,77 г) получали из [4-(децилокси)фенил]метанамина (3,04 г, 11,6 ммоль), 4-хлорхиназолина (2,60 г, 15,8 ммоль), ТЕА (3,40 мл, 24,2 ммоль) и IPA (50 мл) с использованием способа, используемого для N-(3-феноксибензил)хиназолин-4-амина. Продукт перекристаллизовывали из 30% ЕА/гексан. Rf 0,24 (5% MeOH/DCM); Т.пл. 103,0-104,5°C;1H ЯМР (CDCl3) δ 8,71 (с, 1H), 7,85 (дд, 1H, J=0,7, 8,4 Гц), 7,74 (дд, 1H, J=1,5, 6,9 Гц), 7,69 (м, 1H), 7,44 (ддд, 1H, J=1,1, 7,0, 8,1 Гц), 7,31 (м, 2H), 6,88 (м, 2H), 5,90 (шир. с, 1H, NH), 4,78 (м, 2H, AB), 3,95 (т, 2H, J=6,6 Гц), 1,77 (м, 2H), 1,45 (м, 2H), 1,4-1,2 (м, 12H), 0,88 (м, 3H);13C ЯМР (CDCl3) δ 159,6, 159,1, 155,7, 149,7, 132,8, 130,0, 129,7, 128,9, 126,2, 120,8, 115,0, 68,3, 45,2, 32,1, 29,8, 29,8, 29,6, 29,5, 29,4, 26,2, 22,9, 14,3.
Пример 114: N-[4-(гексилокси)бензил]хиназолин-4-амин

N-[4-(гексилокси)бензил]хиназолин-4-амин (31,9 г) получали из [4-(гексилокси)фенил]метанамина (32 г), 4-хлорхиназолина (19 г), ТЕА (32,5 мл) и IPA (250 мл) согласно способу, используемому для получения N-(3-феноксибензил)хиназолин-4-амина. Т.пл. 109,0-111,0°C (из IPA);1H ЯМР (CDCl3) δ 8,68 (с, 1H), 7,82 (м, 1H), 7,71 (м, 2H), 7,41 (м, 1H), 7,29 (м, 2H, J=2,9, 4,8, 9,5 Гц, AA’BB’), 6,87 (м, 2H, J=2,9, 5,1, 9,5 Гц, AA’BB’), 6,11 (шир. с, 1H, NH), 4,77 (м, 2H, AB), 3,93 (т, 2H, J=6,6 Гц), 1,76 (м, 2H), 1,5 (м, 2H), 1,4-1,3 (м, 4H), 0,89 (м, 3H);13C ЯМР (CDCl3) δ 159,4, 150,0, 155,6, 149,6, 132,8, 130,0, 129,6, 128,7, 126,2, 120,8, 115,0, 115,0, 68,3, 45,1, 31,8, 29,4, 25,9, 22,8, 14,2.
Пример 115: 1-[2-(Этоксиметил)-1H-имидазо[4,5-c]хинолин-1-ил]-2-метилпропан-2-ол

3-Нитрохинолин-4-ол 70% водный раствор азотной кислоты (6,1 мл) добавляли по каплям к смеси 4-гидроксихинолина (10 г, 69 ммоль) и 100 мл уксусной кислоты при нагревании с обратным холодильником. Через 15 минут, смеси давали охладиться до комнатной температуры. Разбавление EtOH привело к формированию осадка, который фильтровали и промывали последовательно EtOH, H2O и EtOH. Высушивание фильтрата в вакууме дало 4,62 г светло-желтого порошка.1H ЯМР (DMSO-d6) δ 9,2 (с, 1H), 8,3 (д, 1H), 7,9-7,7 (м, 2H), 7,5 (м, 1H).
4-Хлор-3-нитрохинолин Оксихлорид фосфора (2,5 мл, 27 ммоль) добавляли по каплям к смеси 3-нитрохинолин-4-ола (4,6 г, 24 ммоль) и 100 мл DMF. Смесь нагревали при 100°C в течение 15 минут и затем лили на перемешиваемый лед. Суспензию нейтрализовали твердым NaHCO3, и осадок фильтровали и промывали насыщенным NaHCO3 и H2O. Фильтрат забирали в DCM, высушивали над безводным Na2SO4 и концентрировали, получая 2,3 г твердого вещества.
2-Метил-1-(3-нитрохинолин-4-ил) пропан-2-ол Смесь 4-хлор-3-нитрохинолина (2,3 г, 11 ммоль), 1-амино-2-метилпропан-2-ола (1,0 г, 11 ммоль), ТЕА (9,3 мл) и 100 мл DCM нагревали с обратным холодильником, пока исходный материал не был израсходован. Смеси давали охладиться, промывали насыщенным NaHCO3 и H2O, высушивали над безводным Na2SO4 и концентрировали, получая 1,01 г продукта.1H ЯМР (DMSO-d6) δ 9,9 (шир. с, 1H, NH), 9,2 (с, 1H), 8,5 (д, 1H), 7,9-7,8 (м, 2H), 7,6 (м, 1H), 5,1 (с, 1H, OH), 3,8 (м, 2H, ABX), 1,2 (с, 6H).
1-(3-Аминохинолин-4-иламино)-2-метилпропан-2-ол 2-Метил-1-(3-нитрохинолин-4-ил)пропан-2-ол (1,01 г, ммоль), 10% Pd-C (200 мг) и 20 мл толуола перемешивали в атмосфере водорода, пока исходный материал не был израсходован. Водород заменяли аргоном, и смесь фильтровали через слой Целита и концентрировали упариванием, получая 586 мг продукта.1H ЯМР (CD3OD) δ 8,3 (с, 1H), 8,1 (м, 1H), 7,8 (м, 1H), 7,5-7,4 (м, 2H), 7,2-7,0 (м, 2H, ABX), 1,2 (с, 6H).
1-[2-(Этоксиметил)-1H-имидазо[4,5-c]хинолин-1-ил]-2-метилпропан-2-ол Смесь 1-(3-аминохинолин-4-иламино)-2-метилпропан-2-ола (586 мг, 2,54 ммоль) и 0,4 мл этоксиуксусной кислоты нагревали при 130°C в течение 3 часов. Охлажденную смесь лили в 5 мл H2O и подщелачивали 6н. NaOH. Полученное твердое вещество собирали фильтрацией, промывали H2O и высушивали в вакууме, получая 655 мг продукта.1H ЯМР (CDCl3) δ 9,1 (с, 1H), 8,3 (м, 1H), 8,1 (м, 1H), 7,7-7,5 (м, 2H), 4,9 (шир. с, 2H), 4,8 (шир. с, 2H), 3,6 (кв., 2H), 1,3 (с, 6H), 1,2 (т, 3H).
Пример 116: 1-(4-Амино-1-изобутил-1H-имидазо[4,5-c]хинолин-2-ил)пентил ацетат

N-Изобутил-3-нитрохинолин-4-амин 4-Хлор-3-нитрохинолин получали из 3-нитрохинолин-4-ола (5,5 г, 28,8 ммоль). Изобутиламин (3,2 мл, 32 ммоль) медленно добавляли к смеси 4-хлор-3-нитрохинолина, ТЕА (24 мл, 170 ммоль) и 40 мл DCM. Смесь нагревали с обратным холодильником в течение 30 мин. Затем летучие компоненты выпаривали, и остаток забирали в водный раствор кислоты и фильтровали. Фильтрат подщелачивали до рН 8-9, добавляя концентрированный NH4OH, и полученное твердое вещество отфильтровывали и промывали H2O. Высушивание в вакууме дало 6,49 г продукта.1H ЯМР (CDCl3) δ 9,8 (шир. с, 1H, NH), 9,3 (с, 1H), 8,3 (м, 1H), 8,0 (м, 1H), 7,8 (м, 1H), 7,4 (м, 1H), 3,8 (м, 2H), 2,1 (м, 1H), 1,1 (д, 6H).
N4-Изобутилхинолин-3,4-диамин Смесь N-изобутил-3-нитрохинолин-4-амина (19,0 г, 77,6 ммоль) и 10% Pd-C (700 мг) в 200 мл ЕА вводили в реакцию в атмосфере водорода при 42 psi, пока исходный материал не был израсходован. Затем водород заменяли аргоном, и смесь фильтровали через слой Целита. Фильтрат концентрировали, получая 15,2 г продукта.1H ЯМР (CDCl3) δ 8,4 (с, 1H), 7,9 (м, 1H), 7,8 (м, 1H), 7,5-7,4 (м, 2H), 3,9-3,6 (шир. м, 3H, NH), 3,0 (д, 2H), 1,9 (м, 1H), 1,0 (д, 6H).
1-Изобутил-1H-имидазо[4,5-c]хинолин Смесь N4-изобутилхинолин-3,4-диамина (2,33 г, 10,8 ммоль) и 17 мл муравьиной кислоты нагревали при 100°C в течение 3 часов. Летучие компоненты выпаривали в вакууме. Остаток разбавляли H2O, подщелачивали с использованием концентрированного NH4OH и экстрагировали DCM. Органический растворитель заменяли Et2O, обрабатывали активированным углем, фильтровали через слой Целита и концентрировали. ЯМР показал наличие исходного материала. Сырой продукт смешивали с триэтоксиметаном, нагревали при 100°C в течение 3 часов и обрабатывали как перед этим, получая 1,4 г продукта.1H ЯМР (CDCl3) δ 9,3 (с, 1H), 8,3 (м, 1H), 8,1 (м, 1H), 7,9 (с, 1H), 7,7-7,5 (м, 2H), 4,3 (д, 2H), 2,3 (м, 1H), 1,0 (д, 6H).
1-(1-Изобутил-1H-имидазо[4,5-c]хинолин-2-ил)пентан-1-ол N-бутиллитий (1,5M в гексанах, 3,6 мл) добавляли к смеси 1-изобутил-1H-имидазо[4,5-c]хинолина (1,4 г, 4,9 ммоль) и 25 мл THF, охлажденной ванной со смесью сухой лед/IPA. Через 15 минут добавляли валериановый альдегид (0,80 мл, 7,5 ммоль). Смеси давали нагреться до комнатной температуры. Через 3 часа добавляли H2O и Et2O, и органическую фазу отделяли, высушивали над безводным MgSO4 и концентрировали. FC, с элюированием ЕА, дала 990 мг продукта.1H ЯМР (CDCl3) δ 9,2 (с, 1H), 8,1 (м, 1H), 7,9 (м, 1H), 7,7-7,5 (м, 2H), 4,95 (м, 1H), 4,5 (м, 1H), 4,3 (м, 1H), 2,3 (м, 2H), 1,6-1,3 (м, 4H), 1,1 (д, 3H), 1,0-0,8 (м, 6H).
1-(1-Изобутил-1H-имидазо[4,5-c]хинолин-2-ил)пентил ацетат Уксусный ангидрид (0,400 мл, 4,24 ммоль) и ТЕА (0,510 мл, 3,64 ммоль) добавляли последовательно к смеси 1-(1-изобутил-1H-имидазо[4,5-c]хинолин-2-ил)пентан-1-ола (818 мг, 2,75 ммоль) и 20 мл DCM. Через 16 часов смесь разбавляли 1 объемом DCM и промывали H2O и насыщенным NaHCO3. Органическую фазу высушивали над безводным MgSO4 и концентрировали, получая 1,00 г продукта.1H ЯМР (CDCl3) δ 9,3 (с, 1H), 8,25 (м, 1H), 8,1 (м, 1H), 7,75-7,55 (м, 2H), 6,1 (м, 1H), 4,5 (м, 2H, ABX), 2,3 (м, 2H), 2,1 (с, 3H), 1,5-1,3 (м, 4H), 1,1 (д, 3H), 1,0-0,8 (м, 6H).
2-(1-Ацетоксипентил)-1-изобутил-1H-имидазо[4,5-c]хинолин-5-оксид Смесь 1-(1-изобутил-1H-имидазо[4,5-c]хинолин-2-ил)пентил ацетата (980 мг, 2,91 ммоль) и 32% перуксусной кислоты (0,22 мл, 3,2 ммоль) в 20 мл ЕА нагревали с обратным холодильником в течение 1 часа и перемешивали при комнатной температуре в течение ночи. Летучие компоненты выпаривали в вакууме, и остаток разделяли между DCM и насыщенным NaHCO3 и H2O. Органическую фазу высушивали над безводным Na2SO4 и концентрировали, получая твердое вещество. Твердое вещество суспендировали в холодном ацетоне, фильтровали, и высушивали, получая 750 мг продукта.1H ЯМР (CDCl3) δ 9,3 (с, 1H), 9,0 (м, 1H), 8,5 (шир. с, 2H, NH2), 8,15 (м, 1H), 7,85-7,75 (м, 2H), 6,0 (дд, 1H), 4,5 (м, 2H, ABX), 2,3 (м, 2H), 2,1 (с, 3H), 1,5-1,3 (м, 4H), 1,1 (д, 3H), 0,95 (д, 3H), 0,9 (м, 3H).
1-(4-Амино-1-изобутил-1H-имидазо[4,5-c]хинолин-2-ил)пентил ацетат Смесь 4-толуолсульфонил хлорида (447 мг, 2,34 ммоль) и 15 мл DCM медленно добавляли к смеси 2-(1-ацетоксипентил)-1-изобутил-1H-имидазо[4,5-c]хинолин-5-оксида (750 мг, 2,13 ммоль) и 8 мл концентрированного NH4OH, охлажденной ванной со льдом. Смеси давали нагреться до комнатной температуры в течение ночи. Смесь разбавляли DCM и промывали насыщенным NaHCO3, и органическую фазу высушивали над безводным Na2SO4 и концентрировали, получая 650 мг бесцветного твердого вещества.1H ЯМР (CDCl3) δ 7,9 (д, 1H), 7,7 (д, 1H), 7,5 (м, 1H), 7,3 (м, 1H), 6,1 (дд, 1H), 5,5 (шир. с, 2H, NH2), 4,4 (м, 2H, ABX), 2,3 (м, 2H), 2,15 (м, 1H), 2,1 (с, 3), 1,5-1,3 (м, 4H), 1,1 (д, 3H), 1,0-0,8 (м, 6H).
Пример 117: 1-Изобутил-2-пентадецил-1H-имидазо[4,5-c]хинолин-4-ол

2-Хлор-N-изобутил-3-нитрохинолин-4-амин Смесь изобутиламина (10,0 мл, 101 ммоль) и ТЕА (15,6 мл, 111 ммоль) в 10 мл 1:1 DMF/DCM медленно добавляли к 2,4-дихлор-3-нитрохинолину (26,94 г, 111 ммоль) в 100 мл 4:1 DMF/DCM, охлажденной ванной со льдом. Смеси давали нагреться до комнатной температуры в течение ночи. Затем летучие компоненты выпаривали, и остаток разделяли между ЕА и насыщенным NaHCO3 и солевым раствором, высушивали над Na2SO4 и концентрировали. FC (15% ЕА/Гексан) дала продукт в форме твердого вещества оранжевого цвета. Перекристаллизация из смеси ЕА/Гексан дала 3 выхода продукта (17,97 г) в форме твердого вещества светло-оранжевого цвета.
2-Хлор-N4-изобутилхинолин-3,4-диамин Смесь 2-хлор-N-изобутил-3-нитрохинолин-4-амина (996 мг, 3,57 ммоль) и 35 мг 5% Pt-C в 15 мл MeOH перемешивали под 2 атмосферами водорода в течение 90 мин. Затем смесь продували аргоном, фильтровали через слой Целита и концентрировали досуха.
4-Хлор-1-изобутил-2-пентадецил-1H-имидазо[4,5-c]хинолин Смесь сырого 2-хлор-N4-изобутилхинолин-3,4-диамина и пальмитиновой кислоты (3,66 г, 14,3 ммоль) нагревали при 180°C в течение 4 часов. Затем смесь частично охлаждали и разбавляли при перемешивании 400 мл ЕА и 10 мл 1M NaOH и 40 мл 5%-ого Na2CO3. Теплую смесь охлаждали ванной со льдом и получали твердое вещество (предположительно, пальмитат натрия). Жидкость декантировали от твердого вещества, слои разделяли, и водный слой экстрагировали ЕА (2×150 мл). Органические фазы промывали 5%-ым Na2CO3 (3×50 мл) и солевым раствором, высушивали над Na2SO4 и концентрировали. FC (4% MeOH/DCM) дала фракции, которые содержали продукт, что наблюдали с помощью TLC. Фракции концентрировали, и два выхода продукта (1,14 г) кристаллизовали из DCM/Hex. Rf 0,27 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 7,8 (м, 2H), 7,4 (м, 1H), 7,3 (м, 1H), 4,2 (д, 2H, ABX), 2,9 (м, 2H), 2,3 (м, 1H), 1,9 (м, 2H), 1,5-1,2 (м, 24H), 1,0 (д, 6H), 0,85 (т, 3H).
1-Изобутил-2-пентадецил-1H-имидазо[4,5-c]хинолин-4-ол Смесь 4-хлор-1-изобутил-2-пентадецил-1H-имидазо[4,5-c]хинолина (165 мг, 0,35 ммоль) в 5 мл 50% концентрированного NH4OH/MeOH нагревали при 160°C в течение 72 часов. Затем смесь охлаждали и упаривали, получая твердое вещество. Твердое вещество промывали насыщенным NaHCO3 и H2O и высушивали в вакууме, получая твердое вещество светло-серого цвета в количестве 160 мг. Rf 0,29 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 12,1 (шир. с, 1H, OH), 7,8 (м, 2H), 7,4 (м, 1H), 7,3 (м, 1H), 4,2 (д, 2H, ABX), 2,9 (м, 2H), 2,3 (м, 1H), 1,9 (м, 2H), 1,5-1,2 (м, 24H), 1,0 (д, 6H), 0,85 (т, 3H).
Пример 118: 1-Октил-1H-имидазо[4,5-c]хинолин
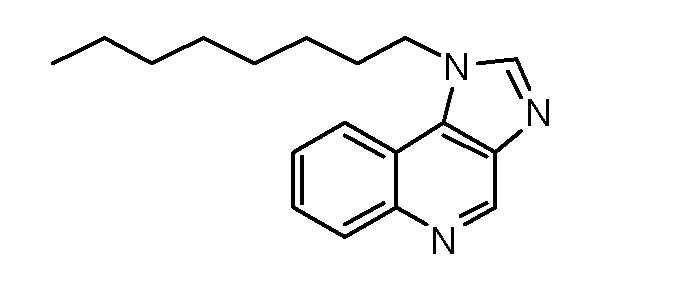
2,4-дигидрокси-3-нитрохинолин Концентрированную азотную кислоту (12,4 мл) добавляли к механически перемешиваемой смеси 2,4-дигидроксихинолина (20,2 г, 125 ммоль) в 160 мл уксусной кислоты при нагревании с обратным холодильником. Через 20 минут нагревание останавливали. Через еще 15 минут добавляли 3 объема кубиков льда, и смесь перемешивали 30 мин. Осадок фильтровали и промывали четыре раза с 1 объемом ледяной H2O. После высушивания в вакууме, получали 23,0 г твердого вещества оранжевого цвета.
2,4-дихлор-3-нитрохинолин Смесь 2,4-дигидрокси-3-нитрохинолина (5,08 г, 24,7 ммоль) и дихлорида фенилфосфоновой кислоты (13,9 мл, 98,4 ммоль) нагревали при 140°C в течение 3 часов. После того, как смесь несколько охладилась, ее добавляли к 18,5 г NaHCO3 в 150 мл ледяного H2O. Величина рН составила по меньшей мере 6, Твердое вещество отфильтровывали и промывали дважды с H2O. После высушивания в вакууме, получали 5,09 г твердого вещества желто-коричневого цвета.
2-Хлор-3-нитро-N-октилхинолин-4-амин Смесь 2,4-дихлор-3-нитрохинолина (1,0 г, 4,1 ммоль), 1-октиламина (0,75 мл), ТЕА (3,5 мл) и 20 мл DCM нагревали с обратным холодильником в течение 1 часа. Затем летучий материал выпаривали, остаток забирали в H2O и подщелачивали до рН 8-9 концентрированной HCl и концентрированным NH4OH. Осадок собирали и промывали H2O. После высушивания в вакууме, получали 1,65 г твердого вещества.
N4-Октилхинолин-3,4-диамин (515 мг) получали, обрабатывая 2-хлор-3-нитро-N-октилхинолин-4-амин (1,33 г) в условиях, используемых для получения N-[8-(гексилокси)октил]пиримидин-4-амина.1H ЯМР (CDCl3) δ 8,5 (с, 1H), 8,05 (д, 1H), 7,9 (д, 1H), 7,5 (м, 1H), 7,35 (м, 1H), 4,1 (шир. с, 2H, NH2), 3,5 (м, 2H), 1,75 (м, 2H), 1,6-1,1 (м, 10H), 0,85 (м, 3H).
1-Октил-1H-имидазо[4,5-c]хинолин (400 мг) получали, обрабатывая N4-октилхинолин-3,4-диамин (515 мг) в условиях, используемых для получения 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридина.1H ЯМР (CDCl3) δ 9,35 (с, 1H), 8,6 (м, 1H), 8,2 (д, 1H), 8,0 (с, 1H), 7,75 (м, 2H), 4,6 (т, 2H), 2,0 (м, 2H), 1,5-1,1 (м, 10H), 0,9 (м, 3H).
Пример 119: 1-гексадецил-1H-имидазо[4,5-c]хинолин

2-Хлор-3-нитро-N-октилхинолин-4-амин Смесь 2,4-дихлор-3-нитрохинолина (1,0 г, 4,1 ммоль), 1-октиламина (0,75 мл), ТЕА (3,5 мл) и 20 мл DCM нагревали с обратным холодильником в течение 1 часа. Затем летучий материал выпаривали, остаток забирали в H2O и подщелачивали до рН 8-9 концентрированной HCl и концентрированным NH4OH. Осадок собирали и промывали H2O. После высушивания в вакууме, получали 1,65 г твердого вещества.
N4-Октилхинолин-3,4-диамин (515 мг) получали, обрабатывая 2-хлор-3-нитро-N-октилхинолин-4-амин (1,33 г) в условиях, используемых для получения N-[8-(гексилокси)октил]пиримидин-4-амина.1H ЯМР (CDCl3) δ 8,5 (с, 1H), 8,05 (д, 1H), 7,9 (д, 1H), 7,5 (м, 1H), 7,35 (м, 1H), 4,1 (шир. с, 2H, NH2), 3,5 (м, 2H), 1,75 (м, 2H), 1,6-1,1 (м, 10H), 0,85 (м, 3H).
1-Октил-1H-имидазо[4,5-c]хинолин (400 мг) получали, обрабатывая N4-октилхинолин-3,4-диамин (515 мг) в условиях, используемых для получения 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридина.1H ЯМР (CDCl3) δ 9,35 (с, 1H), 8,6 (м, 1H), 8,2 (д, 1H), 8,0 (с, 1H), 7,75 (м, 2H), 4,6 (т, 2H), 2,0 (м, 2H), 1,5-1,1 (м, 10H), 0,9 (м, 3H).
Пример 120: 1-гексадецил-1H-имидазо[4,5-c]хинолин-4-амин

1-гексадецил-1H-имидазо[4,5-c]хинолин-4-амин получали согласно способу, используемому для получения 1-изобутил-2-пентадецил-1H-имидазо[4,5-c]хинолин-4-ола, используя 2,4-дихлор-3-нитрохинолин (1,00 г), 1-гексадециламин (1,00 г), 8 мл триэтоксиметана при нагревании с обратным холодильником для циклизации имидазола, и раствора 1 мл безводного NH3 в 8 мл безводного IPA в заключительной реакции. При финальной очистке использовали FC (5% MeOH/DCM, Rf 0,17).1H ЯМР (CDCl3) δ 7,9 (м, 1H), 7,8 (м, 1H), 7,75 (с, 1H), 7,5 (м, 1H), 7,3 (м, 1H), 5,6 (шир. с, 1H, NH), 4,5 (т, 2H), 2,0 (м, 2H), 1,5-1,2 (м, 26H), 0,85 (т, 3H).
Пример 121: 1-[2-(додецилокси)этил]-1H-имидазо[4,5-c]хинолин

2-(Додецилокси)этанол 60% Дисперсию гидрида натрия в минеральном масле (8,3 г, 208 ммоль) промывали в Гексане (2x). Затем медленно добавляли смесь этиленгликоля (17,4 мл, 312 ммоль) в 250 мл DMF и 25 мл DCM, охлаждая ванной со льдом. Через 1 час добавляли 1-йоддодекан (104 ммоль). Смеси давали нагреться до комнатной температуры. Через 24 часа выпаривали летучие компоненты, и остаток разделяли между ЕА и 100 мл 1M HCl, затем 0,1M HCl и 5% Na2S2O3, затем 0,1M HCl, затем солевым раствором, и органические фазы высушивали над MgSO4 и концентрировали. SPE, с промывкой 5% ЕА/Гексан и с элюированием 40% ЕА/Гексан, дал 10,15 г продукта. Rf 0,48 (40% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,7 (м, 2H), 3,55-3,40 (м, 4H), 2,1 (шир. с, 1H, OH), 1,6 (м, 2H), 1,4-1,2 (м, 18H), 0,85 (т, 3H).
2-(додецилокси)этил метансульфонат как сырой материал получали из 2-(додецилокси)этанола (10,15 г, 44,1 ммоль), метансульфонил хлорида (4,3 мл, 53 ммоль) и триэтиламина (7,5 мл, 53 ммоль) в 200 мл THF, и использовали. Rf 0,56 (40% ЕА/Гексан).
1-(2-Йодэтокси)додекан (14,9 г) получали из 2-(додецилокси)этил метансульфоната и 12,9 г йодида натрия реакцией Finkelstein. Rf 0,94 (40% ЕА/Гексан) 0,46 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,7 (т, 2H), 3,45 (т, 2H), 3,25 (т, 2H), 1,6 (м, 2H), 1,4-1,2 (м, 18H), 0,85 (т, 3H).
1-(2-Азидоэтокси)додекан как сырой продукт получали из 1-(2-йодэтокси)додекана (14,9 г, 43,8 ммоль) и азида натрия (2,85 г, 43,8 ммоль) в 33 мл DMF. Rf 0,28 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,6 (т, 2H), 3,45 (т, 2H), 3,35 (т, 2H), 1,6 (м, 2H), 1,4-1,2 (м, 18H), 0,85 (т, 3H).
2-(Додецилокси)этанамин получали каталитическим гидрированием сырого 1-(2-азидоэтокси)додекана, используя 1,5 г 5%-ого Pd-C в 150 мл MeOH. SPE, с промывкой 50% ЕА/Гексан и с элюированием 15% MeOH/DCM +2% ТЕА, дал 8,0 г продукта.
1-[2-(додецилокси)этил]-1H-имидазо[4,5-c]хинолин (103 мг) получали способом, используемым для получения 1-гексадецил-1H-имидазо[4,5-c]хинолин-4-амина, исходя из 2-(додецилокси)этанамина (2,73 г, 11,9 ммоль) и 2,4-дихлор-3-нитрохинолина (2,94 г, 12,1 ммоль), используя восстановление как нитро, так и арил хлорида с использованием цинк/HCl, и образование кольца имидазола с использованием 7 мл триэтоксиметана при нагревании с обратным холодильником. Финальная очистка представляла собой FC (5% MeOH/DCM, Rf 0,10).1H ЯМР (CDCl3) δ 9,3 (с, 1H), 8,2 (д, 1H), 8,1 (д, 1H), 7,95 (с, 1H), 7,7-7,5 (м, 2H), 4,7 (м, 2H), 3,85 (м, 2H), 3,3 (м, 2H), 1,4 (м, 2H), 1,3-1,1 (м, 18H), 0,8 (м, 3H).
Пример 122: 1-[2-(додецилокси)этил]-N,N-диметил-1H-имидазо[4,5-c]хинолин-4-амин

N4-[2-(додецилокси)этил]-N2,N2-диметил-3-нитрохинолин-2,4-диамин Стехиометрический избыток 2-(додецилокси)этанамина и 2,4-дихлор-3-нитрохинолина (486 мг, 2,0 ммоль) и DIEA (0,38 мл, 2,18 ммоль) в 10 мл DMF и 10 мл DCM смешивали при комнатной температуре в течение 2 дней. Никакой реакции не наблюдалось при помощи TLC. DCM выпаривали и заменяли толуолом, и смесь нагревали с обратным холодильником в течение 6 часов. Затем реакционную смесь охлаждали, разделяли между ЕА и насыщенным NaHCO3 и солевым раствором, и органическую фазу высушивали над Na2SO4 и концентрировали. FC (градиенту стадии от 10% до 20% ЕА/гексан) дал 306 мг N4-[2-(додецилокси)этил]-N2,N2-диметил-3-нитрохинолин-2,4-диамина в форме оранжевого масла, а также 376 мг N2,N4-бис[2-(додецилокси)этил]-3-нитрохинолин-2,4-диамина в форме оранжевого масла.1H ЯМР (CDCl3) δ 7,9 (м, 2H), 7,6-7,55 (м, 2H), 7,1 (м, 1H), 3,8 (м, 2H), 3,5-3,4 (м, 4H), 3,0 (с, 6H), 1,6 (м, 2H), 1,4-1,2 (м, 18H), 0,85 (т, 3H).
1-[2-(додецилокси)этил]-N,N-диметил-1H-имидазо[4,5-c]хинолин-4-амин Нитрогруппу N4-[2-(додецилокси)этил]-N2,N2-диметил-3-нитрохинолин-2,4-диамина (306 мг, 0,70 ммоль) восстанавливали, используя цинк/HCl, и орто-диамин вводили в реакцию с триэтоксиметаном при нагревании с обратным холодильником, получая 197 мг продукта после FC (5%MeOH/DCM). Rf 0,15 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 7,9 (м, 2H), 7,8 (с, 1H), 7,45 (м, 1H), 7,2 (м, 1H), 4,6 (т, 2H), 3,85 (т, 2H), 3,6 (с, 6H), 3,3 (т, 2H), 1,5 (м, 2H), 1,3-1,1 (м, 18H), 0,85 (т, 3H).
Пример 123: 1-[6-(октилокси)гексил]-1H-имидазо[4,5-c]хинолин

6-(Октилокси)гексан-1-ол Гидрид натрия (6,38 г, 266 ммоль) осторожно добавляли к смеси 1,6-гександиола (47,2 г, 400 ммоль) и 120 мл DMF, охлажденной ванной со льдом. Через 15 минут добавляли смесь 1-йодоктана (31,9 г, 133 ммоль) в 120 мл DCM. Смеси давали нагреться до комнатной температуры в течение ночи. Затем летучие компоненты выпаривали, и остаток разделяли между ЕА и 0,1M HCl, 5%-ым Na2S2O3, H2O и солевым раствором. Органические фазы высушивали над безводным MgSO4 и концентрировали. SPE, с промывкой 2% ЕА/Гексан и с элюированием 40% ЕА/Гексан, дал 13,0 г бесцветного масла. Rf 0,40 (50% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,59 (т, 2H, J=6,7 Гц), 3,36 (т, 2H, J=6,7 Гц), 3,35 (т, 2H, J=6,7 Гц), 2,02 (шир. с, 1H, OH), 1,56-1,47 (м, 6H), 1,40-1,20 (м, 14H), 0,84 (м, 3H).
2-Хлор-3-нитро-N-[6-(октилокси)гексил]хинолин-4-амин ТЕА (8,40 мл, 59,9 ммоль) добавляли к смеси 6-(октилокси)гексан-1-ола (7,60 г, 33,0 ммоль) и метансульфонил хлорида (4,56 мл, 58,3 ммоль) в 190 мл DME, охлажденной ванной со льдом. Смеси давали нагреться до комнатной температуры. Через 4 часа добавляли 5 мл H2O, и летучие компоненты выпаривали. Остаток разделяли между ЕА (3×150 мл) и H2O, насыщенным NaHCO3, H2O, 1M HCl, H2O и солевым раствором (100 мл каждый). Органические фазы высушивали над MgSO4 и концентрировали, получая бесцветное масло. Масло забирали в 250 мл ацетона, добавляли йодид натрия (9,9 г, 66 ммоль), и смесь нагревали с обратным холодильником в течение 2 часов. Летучие компоненты выпаривали, и остаток разделяли между ЕА и H2O, 5%-ым Na2S2O3, H2O и солевым раствором. Органические фазы высушивали над MgSO4 и концентрировали. SPE (5% ЕА/Гексан) дал фиолетовое масло. Масло забирали в 25 мл DMF и 10 мл толуола, добавляли фталимид калия (5,55 г, 30 ммоль), и смесь нагревали с обратным холодильником в течение 4 часов. Затем смесь охлаждали и разделяли между ЕА и 0,1M HCl, 5%-ым Na2S2O3, H2O и солевым раствором. Органические фазы высушивали над MgSO4 и концентрировали. SPE, с промывкой 5% ЕА/Гексан и с элюированием 7,5% ЕА/Гексан, дал 10,05 г бесцветного масла. Масло забирали в 500 мл 5% IPA/EtOH, добавляли гидразин моногидрат (2,0 мл, 41 ммоль), и смесь нагревали с обратным холодильником в течение 4 часов. Смесь охлаждали и концентрировали. Остаток разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой 50% ЕА/Гексан и с элюированием 15% MeOH/DCM +2% ТЕА, дал 1,91 г бесцветного масла. Масло забирали в смеси 9 мл DMA и 9 мл толуола и добавляли 2,4-дихлор-3-нитрохинолин (2,16 г, 8,87 ммоль) и DIEA (1,45 мл, 8,32 ммоль). Смесь вводили в реакцию при комнатной температуре в течение 88 часов и при нагревании с обратным холодильником в течение 2 дней. Смесь охлаждали, летучие компоненты выпаривали, и остаток разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором. Органические фазы высушивали над Na2SO4 и концентрировали. SPE (20% ЕА/Гексан) дал содержащие продукт фракции с примесями. FC (20% ЕА/Гексан) дал 2,06 г желтого масла, которое затвердевало при отстаивании. Твердое вещество перекристаллизовывали из смеси ЕА/Гексан, получая 1,70 г твердого вещества желтого цвета. Rf 0,22 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,84 (д, 1H, J=7,9 Гц), 7,76 (дд, 1H, J=1,2, 8,4 Гц), 7,63 (ддд, 1H, J=1,2, 6,9, 8,1 Гц), 7,42 (ддд, 1H, J=1,3, 7,0, 8,4 Гц), 5,98 (т, 1H, J=4,7 Гц, NH), 3,38-3,29 (м, 6H), 1,66 (м, 2H), 1,56-1,42 (м, 4H), 1,36-1,34 (м, 4H), 1,2-1,1 (м, 10H), 0,8 (м, 3H).
1-[6-(октилокси)гексил]-1H-имидазо[4,5-c]хинолин Четыре мл смеси 1:3 концентрированной HCl и MeOH медленно добавляли к смеси 2-хлор-3-нитро-N-[6-(октилокси)гексил]хинолин-4-амина (357 мг, 0,82 ммоль), цинковой пыли (320 мг) и 20 мл DCM, охлажденной ванной со льдом. Смеси давали нагреться до комнатной температуры. Через 16 часов выпаривали летучие компоненты, остаток разбавляли 75 мл DCM и подщелачивали до рН>8 с использованием 5% Na2CO3. Органическую фазу отделяли, высушивали над безводным Na2SO4 и концентрировали. Триэтоксиметан (5 мл) добавляли к сырому продукту, и смесь нагревали при 130°C в течение 6 часов. Затем смесь охлаждали и концентрировали. Остаток разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над Na2SO4 и концентрировали. FC (градиент стадии 3% и 5% MeOH/DCM) дал 101 мг коричневого масла. Rf 0,21 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 9,31 (с, 1H), 8,26 (м, 1H), 8,12 (м, 1H), 7,92 (с, 1H), 7,70-7,58 (м, 2H), 4,54 (т, 2H, J=7,2 Гц), 3,34 (т, 2H, J=6,2 Гц), 3,33 (т, 2H, J=6,7 Гц), 2,00 (м, 2H), 1,56-1,39 (м, 6H), 1,3-1,1 (м, 12H), 0,83 (м, 3H).
Пример 124: 1-(8-Этоксиоктил)-1H-имидазо[4,5-c]хинолин

1-(8-Этоксиоктил)-1H-имидазо[4,5-c]хинолин получали способом, используемым для получения 1-октил-1H-имидазо[4,5-c]хинолина, заменяя 1-октиламин 8-этоксиоктан-1-амином. 8-Этоксиоктан-1-амин получали способом, используемым для получения 8-(гексилокси)октан-1-амина, используя йодэтан и 1,8-октандиол как исходные материалы.
Пример 125: 1-(8-Метоксиоктил)-1H-имидазо[4,5-c]хинолин

1-(8-Метоксиоктил)-1H-имидазо[4,5-c]хинолин получали способом, используемым для получения 1-октил-1H-имидазо[4,5-c]хинолина, заменяя 1-октиламин 8-метоксиоктан-1-амином.
Пример 126: 1-(8-Бутоксиоктил)-1H-имидазо[4,5-c]хинолин

1-(8-Бутоксиоктил)-1H-имидазо[4,5-c]хинолин получали способом, используемым для получения 1-октил-1H-имидазо[4,5-c]хинолина, заменяя 1-октиламин 8-бутоксиоктан-1-амином. 8-Бутоксиоктан-1-амин получали способом, используемым для получения 10-(гексилокси)декан-1-амина, используя 1-бромбутан и 1,8-октандиол как исходные материалы.
Пример 127: 1-[9-(гексилокси)нонил]-1H-имидазо[4,5-c]хинолин

9-(Бензилокси)нонан-1-ол, как 8,79 г бесцветного масла, получали способом, используемым для получения 8-(бензилокси)октан-1-ола, используя 27,1 г 1,9-нонанeдиол, 7,85 мл бензил хлорида в 20 мл DME, 1,80 г гидрида натрия, 60% дисперсию в минеральном масле, и 300 мл DMF. Rf 0,12 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,37-7,22 (м, 5H), 4,49 (с, 2H), 3,61 (т, 2H, J=6,6 Гц), 3,45 (т, 2H, J=6,7 Гц), 1,65-1,49 (м, 4H), 1,36-1,21 (м, 10H).
{[9-(Гексилокси)нонилокси]метил}бензол Гидрид натрия (920 мг, 38,3 ммоль) добавляли к смеси 9-(бензилокси)нонан-1-ола (8,79 г, 35,2 ммоль) и 200 мл DME. Через 1 час добавляли 1-йодгексан (10,6 г, 50 ммоль). Через 40 часов анализ TLC показал небольшое превращение. Добавляли другую часть гидрида натрия. Через 8 часов добавляли другую часть гидрида натрия и 1-бромгексан (7,0 мл, 50 ммоль). Смесь перемешивали 48 часов, затем оставляли на несколько недель. Затем осторожно добавляли 6 мл концентрированного NH4OH. Через 16 часов выпаривали летучие компоненты. Остаток разделяли между ЕА (3×250 мл) и H2O (100 мл), 5% Na2S2O3 (100 мл), H2O (100 мл), 0,1M HCl (2×100 мл) и солевым раствором (100 мл). Органические фазы высушивали над безводным Na2SO4 и концентрировали. SPE (5% ЕА/Гексан) дал 8,47 г бесцветного масла. Rf 0,75 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,34-7,23 (м, 5H), 4,49 (с, 2H), 3,48-3,36 (м, 6H), 1,68-1,51 (м, 6H), 1,5-1,2 (м, 16H), 0,88 (т, 3H, J+6,8 Гц).
1-(Гексилокси)-9-йоднонан Смесь {[9-(гексилокси)нонилокси]метил}бензола (8,47 г, 25,4 ммоль), хлортриметилсилана (20 мл, 158 ммоль) и йодида натрия (23,7 г, 158 ммоль) в 150 мл DCM нагревали с обратным холодильником в течение 60 часов, затем перемешивали при комнатной температуре в течение 48 часов. Затем летучие компоненты выпаривали. Остаток разделяли между ЕА (3×250 мл) и насыщенным NaHCO3 (100 мл), 5% Na2S2O3 (100 мл), H2O (100 мл) и солевым раствором (100 мл). Органические фазы высушивали над безводным MgSO4 и концентрировали. Анализ TLC показал наличие 9-(гексилокси)нонан-1-ола с низким Rf. Смесь забирали в 25 мл толуола и затем концентрировали. Фиолетовое масло забирали в других 25 мл толуола, добавляли 5 мл оксихлорида фосфора, и смесь нагревали с обратным холодильником, пока предполагаемый спирт не был израсходован, что наблюдали анализом TLC. Смесь охлаждали ванной со льдом и медленно добавляли насыщенный раствор NaHCO3, что сопровождалось выделением газа. Смесь экстрагировали ЕА (3×250 мл), и органические фазы промывали H2O, 0,1M HCl и солевым раствором (100 мл каждый), высушивали над MgSO4 и концентрировали. SPE (2% ЕА/Гексан), с исключением ранних фракций, которые содержали бензилгалогениды, дал 3,76 г продукта в форме янтарного масла. Rf 0,53 (5% ЕА/Гексан);1H ЯМР (CDCl3) δ 3,37 (т, 4H, J=6,7 Гц), 3,16 (м, 2H), 1,80 (м, 2H), 1,57-1,49 (м, 4H), 1,4-1,2 (м, 16H), 0,87 (м, 3H).
N-[9-(гексилокси)нонил]фталимид Смесь 1-(гексилокси)-9-йоднонана (3,80 г, 14,4 ммоль) и фталимида калия (2,70 г, 14,6 ммоль) в 8 мл DMF нагревали при 100°C в течение 5 часов. Смесь охлаждали и разделяли между ЕА (3×250 мл) и 5% Na2CO3, H2O, 5% Na2S2O3, H2O, 0,1M HCl и солевым раствором (100 мл каждый). Органические фазы высушивали над безводным MgSO4 и концентрировали. SPE, с промывкой 5% ЕА/Гексан и с элюированием 7,5% ЕА/Гексан, дал 3,30 г продукта в форме твердого вещества. Rf 0,26 (10% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,80 и 7,67 (м, 4H, AA’BB’), 3,64 (м, 2H), 3,35 (т, 2H, J=6,7 Гц), 3,34 (т, 2H, J=6,7 Гц), 1,77-1,47 (м, 6H), 1,28-1,22 (м, 16H), 0,86 (м, 3H).
9-(Гексилокси)нонан-1-амин Смесь N-[9-(гексилокси)нонил]фталимид (3,05 г, 8,18 ммоль) и гидразин моногидрат (0,58 мл, 12 ммоль) в 50 мл 5% IPA/EtOH нагревали с обратным холодильником в течение 4 часов. Смесь охлаждали и концентрировали. Остаток разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой 50% ЕА/Гексан и с элюированием 15% MeOH/DCM +2% ТЕА, дал 1,08 г смеси 9-(гексилокси)нонан-1-амина и фталгидразида. Rf 0,11 (15% MeOH/DCM +2% ТЕА);1H ЯМР (CDCl3) δ 4,6 (шир. с, 2H, NH2), 3,4-3,3 (м, 4H), 2,7 (т, 2H), 1,7-1,1 (м, 22H), 0,8 (м, 3H).
2-Хлор-N-[9-(гексилокси)нонил]-3-нитрохинолин-4-амин Смесь 9-(гексилокси)нонан-1-амина и фталгидразида вводили в реакцию с 2,4-дихлор-3-нитрохинолином (1,11 г, 4,56 ммоль) и ТЕА (0,63 мл, 4,49 ммоль) в 9 мл DMF и 16 мл толуола, при нагревании с обратным холодильником. Через 24 часа смесь охлаждали, разделяли между ЕА и H2O, 5%-ым Na2CO3 и солевым раствором, высушивали над безводным Na2SO4 и концентрировали. FC, с элюированием 15% и затем 20% ЕА/Гексан, дал 1,35 г желтого продукта в форме масла, которое затвердевало при отстаивании. Перекристаллизация из холодной смеси ЕА/гексан дала 650 мг твердого вещества желтого цвета. Rf 0,18 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,87 (д, 1H, J=8,6 Гц), 7,78 (дд, 1H, J=1,3, 9,5 Гц), 7,67,65 (м, 1H), 7,45 (м, 1H), 5,99 (т, 1H, J=4,7 Гц, NH), 3,39-3,31 (м, 6H), 1,66 (м, 2H), 1,53-1,45 (м, 4H), 1,4-1,1 (м, 16H), 0,82 (м, 3H).
1-[9-(гексилокси)нонил]-1H-имидазо[4,5-c]хинолин Шесть мл смеси 1:3 концентрированной HCl и MeOH медленно добавляли к смеси 2-хлор-N-[9-(гексилокси)нонил]-3-нитрохинолин-4-амина (674 мг, 1,50 ммоль), цинковой пыли (585 мг) и 25 мл DCM, охлажденной ванной со льдом. Смеси давали нагреться до комнатной температуры. Через 1 час выпаривали летучие компоненты, остаток разбавляли 75 мл DCM и подщелачивали до рН>8 с использованием 5% Na2CO3. Органическую фазу отделяли, высушивали над безводным Na2SO4 и концентрировали. Rf 0,41 (15% MeOH/DCM) Триэтоксиметан (4 мл) добавляли к сырому продукту, и смесь нагревали при 130°C в течение 6 часов. Затем смесь охлаждали и концентрировали. FC (3% и 5% градиент стадии MeOH/DCM) дала 273 мг коричневого масла. Rf 0,27 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 9,22 (с, 1H), 8,16 (м, 1H),7,98 (м, 1H), 7,60-7,47 (м, 2H), 4,38 (т, 2H, J=7,1 Гц), 3,27 (т, 2H, J=6,7 Гц), 3,26 (т, 2H, J=6,7 Гц), 1,86 (м, 2H), 1,45-1,41 (м, 4H), 1,4-1,1 (м, 16H), 0,78 (м, 3H).
Пример 128: 1-(10-Бутоксидецил)-1H-имидазо[4,5-c]хинолин

1-(10-Бутоксидецил)-1H-имидазо[4,5-c]хинолин получали способом, используемым для получения 1-октил-1H-имидазо[4,5-c]хинолина, заменяя 1-октиламин 10-бутоксидекан-1-амином. 10-Бутоксидекан-1-амин получали способом, используемым для получения 10-(гексилокси)декан-1-амина, используя 1-бромбутан и 1,10-деканeдиол как исходные материалы. Rf 0,23 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 9,32 (с, 1H), 8,27 (м, 1H), 8,12 (м, 1H), 7,93 (с, 1H), 7,66 (м, 2H), 4,54 (т, 2H, J=7,2 Гц), 3,36 (т, 2H, J=6,5 Гц), 3,35 (т, 2H, J=6,5 Гц), 1,99 (м, 2H), 1,57-1,13 (м, 18H), 0,88 (т, 3H, J=7,3 Гц).
Пример 129: 4-Амино-1-[8-(гексилокси)октил]пиридиниевая соль

Смесь 8-(гексилокси)октилметансульфоната (0,5 г, 1,62 ммоль) и 4-аминопиридина (450 мг) в 20 мл THF нагревали с обратным холодильником в течение 18 часов. Смесь концентрировали и очищали FC (5% MeOH/DCM), получая 396 мг масляного твердого вещества. Перекристаллизация из MeOH дала твердое вещество. Т.пл. 108-110°C;1H ЯМР (CDCl3) δ 8,4 (шир. с, 1,4H), 7,8 (д, 2H), 7,2 (д, 2H), 4,1 (м, 2H), 3,35 (м, 4H), 2,4 (шир. с, 4,5H), 1,8 (м, 2H), 1,6 (м, 4H), 1,4-1,2 (м, 14H), 0,8 (м, 3H).
Пример 130: 4-(8-Метоксиоктиламино)-1-метилпиридиний йодид

Смесь N-(8-метоксиоктил)пиридин-4-амина (176 мг, 0,74 ммоль) и йодметана (0,5 мл, 8 ммоль) в 4 мл ацетона нагревали при 80°C в герметизированной пробирке в течение 1,5 часов, затем оставляли стоять при комнатной температуре в течение 2 дней, в течение которых формировался осадок. Летучие компоненты выпаривали из осажденного продукта.1H ЯМР (CDCl3) δ 8,47 (м, 1H), 7,99 (м, 2H), 7,57 (м, 1H), 6,59 (м, 1H), 4,04 (с, 3H), 3,35-3,21 (м, 4H), 3,29 (с, 3H), 1,71 (м, 2H), 1,54-1,28 (м, 10H).
Пример 131: 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридин

N-[8-(гексилокси)октил]-3-нитропиридин-4-амин Смесь 3-нитропиридин-4-ола (510 мг, 3,64 моль) в 1 мл дихлорида фенилфосфоновой кислоты нагревали при 170-140°C в течение 3 часов. Затем смесь охлаждали и разделяли между ЕА и насыщенным NaHCO3. Органическую фазу промывали солевым раствором, высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали, получая сырой 4-хлор-3-нитропиридин. 8-(Гексилокси)октан-1-амин забирали в 10 мл пиридина, и 5 мл летучего материала выпаривали из смеси. Смесь охлаждали ванной со льдом, добавляли ТЕА (0,44 мл, 3,14 моль), и затем добавляли смесь хлорпиридина, полученного выше, и 10 мл DCM. Смеси давали нагреться до комнатной температуры в течение ночи. Затем реакционную смесь концентрировали упариванием, и остаток разделяли между ЕА и насыщенным NaHCO3. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали. Очистка FC (50% ЕА/Гексан) дала 405 мг N-[8-(гексилокси)октил]-3-нитропиридин-4-амина в форме желтого масла. Rf 0,28 (50% ЕА/Гексан);1H ЯМР (CDCl3) δ 9,16 (с, 1H), 8,24 д, 1H, J=6,2 Гц), 8,12 (шир. с, 1H), 6,66 (д, 1H, J=6,2 Гц), 3,38-3,25 (м, 6H), 1,70 (м, 2H), 1,52-1,47 (м, 4H), 1,39-1,18 (м, 14H), 0,84 (т, 3H, J=6,7 Гц).
N4-[8-(гексилокси)октил]пиридин-3,4-диамин Смесь N-[8-(гексилокси)октил]-3-нитропиридин-4-амина (405 мг, 1,15 моль) и 45 мг 10% Pd/C в 30 мл MeOH перемешивали в атмосфере водорода в течение 5 часов. Затем катализатор удаляли фильтрацией через Целит, и фильтрат концентрировали. Очистка SPE, с промывкой 10% MeOH/DCM и последующим элюированием 15% MeOH/DCM +2% ТЕА, дала 216 мг N4-[8-(гексилокси)октил]пиридин-3,4-диамина. Rf 0,05 (15% MeOH/DCM, нингидрин (+));1H ЯМР (CDCl3) δ 7,86 (д, 1H, J=5,4 Гц), 7,79 (с, 1H), 6,38 (д, 1H, J=5,4 Гц), 4,53 (шир. с, 1H), 3,62 (шир. с, 2H), 3,34 (т, 4H, J=6,7 Гц), 3,08 (м, 2H), 1,62-1,46 (м, 6H), 1,27-1,24 (м, 14H), 0,83 (т, 3H, J=6,8 Гц).
1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридин Смесь N4-[8-(гексилокси)октил]пиридин-3,4-диамина (216 мг, 0,67 моль) в 2 мл триэтоксиметана нагревали с обратным холодильником в течение 6 часов. Затем летучий материал удаляли выпариванием, и остаток разделяли между ЕА и насыщенным NaHCO3. Органические фазы промывали солевым раствором, высушивали над Na2SO4 и концентрировали. Очистка FC (7% MeOH/DCM) дала 217 мг 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридина в форме янтарного масла. Rf 0,11 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 9,02 (с, 1H), 8,34 (д, 1H, J=5,7 Гц), 7,86 (с, 1H), 7,25 (м, 1H), 4,08 (т, 2H, J=7,0 Гц), 3,30-3,25 (м, 4H), 1,78 (м, 2H), 1,45-1,43 (м, 4H), 1,22-1,19 (м, 14H), 0,78 (т, 3H, J=6,7 Гц).
Пример 132: 1-гексадецил-1H-имидазо[4,5-c]пиридин

N-гексадецил-3-нитропиридин-4-амин 1-гексадециламин забирали в 10 мл пиридина, и 6 мл летучих компонентов удаляли перегонкой. Смесь охлаждали и добавляли смесь 4-хлор-3-нитропиридина в 10 мл DCM и 10 мл DMF. Затем добавляли ТЕА (0,46 мл, 3,28 ммоль), и смесь мягко нагревали с обратным холодильником. Через 16 часов охлажденную смесь забирали в ЕА и промывали насыщенным NaHCO3, H2O, и солевым раствором. Органическую фазу высушивали над безводным Na2SO4 и концентрировали. SPE, с промывкой 10% ЕА/Гексан и с элюированием 20% ЕА/Гексан, дал 626 мг твердого вещества. Rf 0,34 (50% ЕА/Гексан);1H ЯМР (CDCl3) δ 9,19 (с, 1H), 8,26 (д, 1H, J=6,1 Гц), 8,15 (шир. с, 1H, NH), 6,68 (д, 1H, J=6,2 Гц), 3,30 (м, 2H), 1,72 (м, 2H), 1,42-1,17 (м, 26H), 0,86 (м, 3H).
1-гексадецил-1H-имидазо[4,5-c]пиридин Смесь N-гексадецил-3-нитропиридин-4-амина (626 мг, 1,79 ммоль) и 65 мг 10%-ого Pd-C в 25 мл 1:1 EA/MeOH перемешивали в атмосфере водорода в течение 40 часов. Атмосферу водорода заменяли аргоном, и смесь фильтровали через слой Целита и концентрировали. SPE, с промывкой 10% MeOH/DCM и с элюированием 10% MeOH/DCM +2% ТЕА, дал 540 мг бесцветного твердого вещества. Твердое вещество забирали в 8 мл триэтоксиметана и нагревали с обратным холодильником в течение 4 часов. Затем летучие компоненты выпаривали. Остаток забирали в свежей 8 мл части триэтоксиметана и нагревали с обратным холодильником в течение 6 часов. Летучие компоненты выпаривали. FC остатка (5% MeOH/DCM) дала 375 мг твердого вещества желто-коричневого цвета. Rf 0,10 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 9,06 (с, 1H), 8,39 (д, 1H, J=5,7 Гц), 7,92 (с, 1H), 7,31 (дд, 1H, J=1,0, 5,7 Гц), 4,12 (м, 2H), 1,82 (м, 2H), 1,26-1,18 (м, 26H), 0,81 (т, 3H, J=6,6 Гц).
Пример 133: 1-(10-Бутоксидецил)-1H-имидазо[4,5-c]пиридин

1-(10-Бутоксидецил)-1H-имидазо[4,5-c]пиридин (231 мг) в форме янтарного масла получали согласно способу, используемому для получения 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридина, используя 492 мг 4-гидрокси-3-нитропиридина и 535 мг 10-бутоксидекан-1-амина.
N-(10-Бутоксидецил)-3-нитропиридин-4-амин: Rf 0,30 (50% ЕА/Гексан);1H ЯМР (CDCl3) δ 9,18 (с, 1H), 8,25 (д, 1H, J=6,0 Гц), 8,14 (шир. с, 1H, NH), 6,68 (д, 1H, J=6,2 Гц), 3,39-3,26 (м, 6H), 1,71 (м, 2H), 1,57-1,47 (м, 4H), 1,40-1,27 (м, 14H), 0,88 (т, 3H, J=7,2 Гц).
N4-(10-Бутоксидецил)пиридин-3,4-диамин: Rf 0,08 (15% MeOH/DCM);1H ЯМР (CDCl3) δ 7,89 (д, 1H, J=6,4 Гц), 7,83 (с, 1H), 6,41 (д, 1H, J=6,4 Гц), 4,41 (шир. с, 1H, NH), 3,58 (шир. с, 2H, NH2), 3,39-3,33 (м, 4H), 3,11-3,10 (шир. м, 2H), 1,66-1,47 (м, 6H), 1,40-1,26 (м, 14H), 0,88 (т, 3H, J=7,2 Гц).
1-(10-Бутоксидецил)-1H-имидазо[4,5-c]пиридин: Rf 0,15 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 9,06 (с, 1H), 8,38 (д, 1H, J=5,7 Гц), 7,88 (д, 1H), 7,28 (д, 1H, J=5,4 Гц), 4,12 (м, 2H), 3,35-3,29 (м, 4H), 1,82 (м, 2H), 1,53-1,43 (м, 4H), 1,36-1,20 (м, 14H), 0,84 (м, 3H).
Пример 134: N-(8-Метоксиоктил)пиридин-4-амин

Смесь 4-хлорпиридин гидрохлорида (1,50 г, 10,0 ммоль), 8-метоксиоктан-1-амина (894 мг, 5,62 ммоль), ТЕА (1,80 мл, 10,4 ммоль) и 4 мл IPA нагревали при 130-140°C в герметизированной пробирке в течение 48 часов. Затем смесь охлаждали, и летучие компоненты выпаривали. Остаток разделяли между DCM и 5%-ым Na2CO3, и органическую фазу высушивали над Na2SO4 и концентрировали. FC (градиент стадии 1% ТЕА+0%, 2%, 3% MeOH/DCM) дал 176 мг твердого вещества. Rf 0,13 (10% MeOH/DCM);1H ЯМР (CDCl3) δ 8,6 (м, 1H), 7,8 (м, 2H), 6,9 (м, 2H), 3,3 (м, 5H), 3,2 (м, 2H), 1,7 (м, 2H), 1,5 (м, 2H), 1,4-1,2 (м, 8H).
Пример 135: N-[8-(гексилокси)октил]пиридин-3-амин

8-(Гексилокси)октанал (1,12 г, 4,91 ммоль), полученный окислением Swern 8-(гексилокси)октан-1-ола, смешивали с 3-аминопиридином (500 мг, 5,32 ммоль) в 5 мл ацетонитрила и 0,4 мл 1M HCl. Затем добавляли 0,37 мл 1M натрий цианоборгидрида в THF. Через 20 часов смесь разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, и органическую фазу высушивали над Na2SO4 и концентрировали. FC (70% ЕА/Гексан) дала 160 мг продукта.1H ЯМР (CDCl3) δ 8,0 (м, 1H), 7,9 (м, 1H), 7,1 (м, 1H), 6,9 (м, 1H), 3,4 (т, 4H), 3,1 (т, 2H), 1,7-1,5 (м, 6H), 1,5-1,2 (м, 14H), 0,85 (м, 3H).
Пример 136: N-[8-(гексилокси)октил]пиридин-2-амин

Смесь 2-аминопиридина (458 мг, 4,8 ммоль) и 8-(гексилокси)октилметансульфоната (0,5 г, 1,6 ммоль) в 20 мл THF нагревали с обратным холодильником в течение 3 часов. Затем реакционную смесь охлаждали и обрабатывали в соответствии с процедурой, используемой для N-[8-(гексилокси)октил]пиридин-3-амина, получая 100 мг продукта.1H ЯМР (CDCl3) δ 8,0 (м, 1H), 7,4 (м, 1H), 6,55 (м, 1H), 6,35 (м, 1H), 4,6 (шир. с, 1H, NH), 3,4 (т, 4H), 3,2 (м, 2H), 1,7-1,5 (м, 6H), 1,5-1,2 (м, 14H), 0,85 (м, 3H).
Пример 137: N-[8-(гексилокси)октил]пиримидин-4-амин

6-Хлор-N-[8-(гексилокси)октил]пиримидин-4-амин 8-(Гексилокси)октан-1-амин (636 мг, 2,78 ммоль) забирали в 15 мл пиридина, и затем 10 мл летучего материала удаляли перегонкой. Смесь охлаждали до комнатной температуры и последовательно добавляли 15 мл DCM, 4,6-дихлорпиримидин (621 мг, 4,17 ммоль) и ТЕА (0,47 мл, 3,35 ммоль). При перемешивании в течение ночи, TLC показала наличие исходного материала амина, поэтому добавляли второе количество 4,6-дихлорпиримидина, и смесь нагревали с обратным холодильником в течение 3 часов. Затем смесь охлаждали, летучий материал выпаривали, и остаток разделяли между ЕА и 5%-ым Na2CO3. Органические фазы промывали солевым раствором, высушивали над Na2SO4, фильтровали через слой силикагеля и концентрировали. Очистка FC (30% ЕА/Гексан) дала 767 мг 6-хлор-N-[8-(гексилокси)октил]пиримидин-4-амина в форме твердого вещества желто-коричневого цвета. Rf 0,18 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,30 (с, 1H), 6,30 (д, 1H, J=1,0 Гц), 5,36 (шир. с, 1H, NH), 3,37 (т, 4H, J=6,9 Гц), 3,24 (м, 2H, AB), 1,6-1,5 (м, 6H), 1,3-1,2 (м, 14H), 0,87 (м, 3H).
N-[8-(гексилокси)октил]пиримидин-4-амин Смесь 6-хлор-N-[8-(гексилокси)октил]пиримидин-4-амина (767 мг, 2,25 ммоль) в 30 мл DCM и 6,8 мл 2M HCl/IPA охлаждали, используя ванну со льдом. Затем добавляли 876 мг цинковой пыли. Через 45 минут смеси давали нагреться до комнатной температуры. При перемешивании в течение ночи, смесь разделяли между DCM и 5%-ым Na2CO3. Органическую фазу высушивали над Na2SO4 и концентрировали. Очистка FC (5%MeOH/DCM) дала 229 мг N-[8-(гексилокси)октил]пиримидин-4-амина в форме бесцветного твердого вещества. Rf 0,21 (5% MeOH/DCM);1H ЯМР (CDCl3) δ 8,46 (с, 1H), 8,08 (д, 1H, J=5,7 Гц), 6,25 (дд, 1H, J=1,2, 5,9 Гц), 5,59 (шир. с, 1H), 3,33 (т, 4H, J=6,7 Гц), 3,21 (м, 2H, AB), 1,58-1,45 (м, 6H), 1,26-1,17 (м, 14H), 0,83 (м, 3H).
Пример 138: N-[8-Гексилокси)октил)пиримидин-2-амин

Смесь 2-хлорпиримидина (272 мг, 2,39 ммоль), 8-(гексилокси)октан-1-амина (548 мг, 2,39 ммоль) и ТЕА (0,34 мл, 2,42 ммоль) в 10 мл DMF нагревали при 80-90°C в течение 2 часов. Затем смесь разделяли между ЕА и 5%-ым Na2CO3 (2x) и солевым раствором, и органическую фазу высушивали над Na2SO4 и концентрировали. FC (50% ЕА/Гексан) дала 227 мг продукта в форме твердого вещества желтого цвета.1H ЯМР (CDCl3) δ 8,2 (д, 2H), 6,4 (д, 2H), 5,6 (шир. с, 1H, NH), 3,3 (м, 4H), 1,6-1,4 (м, 6H), 1,4-1,2 (м, 14H), 0,8 (м, 3H).
Пример 139: 1-[8-(гексилокси)октил]-4-фенил-1H-имидазол

4-Фенилимидазол (1,0 г, 6,9 ммоль) добавляли к смеси трет-бутоксида натрия (7,9 ммоль) в 20 мл DMF, охлажденной ванной со льдом. Через 30 минут добавляли 8-(гексилокси)октилметансульфонат (2,14 г, 6,95 ммоль), и смеси давали нагреться до комнатной температуры. Через 6 часов выпаривали летучие компоненты. Остаток забирали в ЕА и промывали насыщенным NaHCO3, 0,1M HCl и H2O. Органическую фазу высушивали над безводным Na2SO4 и концентрировали. FC (70% ЕА/Гексан) дала 2,5 г 1-[8-(гексилокси)октил]-4-фенил-1H-имидазол.1H ЯМР (CDCl3) δ 7,8 (м, 2H), 7,6 (с, 1H), 7,4 (м, 2H), 7,2 (м, 2H), 3,9 (т, 2H), 3,4 (м, 4H), 1,8 (м, 2H), 1,6-1,5 (м, 4H), 1,4-1,2 (м, 14H), 0,9 (м, 3H).
Пример 140: N-[8-(гексилокси)октил]изохинолин-1-амин

1-Хлоризохинолин (390 мг, 2,38 ммоль), 8-(гексилокси)октан-1-амин (360 мг, 1,57 ммоль) и триэтиламин (0,22 мл, 1,57 ммоль) в 2 мл DMA нагревали при 80°C в течение 24 часов. Затем смесь охлаждали и разделяли между ЕА и 5%-ым Na2CO3 и солевым раствором, и органическую фазу высушивали над Na2SO4 и концентрировали. FC (20% ЕА/Гексан) дала 87 мг продукта. Rf 0,25 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,97 (д, 1, J=6,0 Гц), 7,76-7,73 (м, 1), 7,67-7,64 (м, 1), 7,59-7,53 (м, 1), 7,47-7,41 (м, 1), 6,89 (д, 1, J=5,9 Гц), 5,25 (шир. с, 1), 3,62-3,55 (м, 2), 3,38 (т, 4, J=6,7 Гц), 1,77-1,67 (м, 2), 1,58-1,24 (м, 18), 0,89-0,84 (м, 4).
Пример 141: N-[8-(гексилокси)октил]изохинолин-5-амин

N-[8-(гексилокси)октил]изохинолин-5-амин (123 мг) получали согласно способу, используемому для получения N-[8-(гексилокси)октил]хинолин-6-амина, исходя из 8-(гексилокси)октановой кислоты (300 мг, 123 ммоль) и 5-аминоизохинолина (174 мг, 1,21 ммоль).1H ЯМР (CDCl3) δ 9,14 (д, 1, J=0,7 Гц), 8,44 (д, 1, J=6,1 Гц), 7,57-7,54 (м, 1), 7,45 (т, 1, J=7,9 Гц), 7,30-7,25 (м, 1), 6,74 (дд, 1, J=0,7, 7,7 Гц), 4,35 (шир. с, 1), 3,41-3,35 (м, 4), 3,27-3,22 (м, 2), 1,80-1,70 (м, 2), 1,57-1,21 (м, 18), 0,89-0,84 (м, 3).
Пример 142: N-[8-(гексилокси)октил]хиноксалин-2-амин

N-[8-(гексилокси)октил]хиноксалин-2-амин (238 мг) получали согласно способу, используемому для получения N-[8-(гексилокси)октил]изохинолин-1-амина, исходя из 8-(гексилокси)октан-1-амина (380 мг, 1,66 ммоль) и 2-хлорхиноксалина (413 мг, 2,50 ммоль), но реакцию осуществляли при комнатной температуре в течение 4 дней. Rf 0,20 (20% ЕА/Гексан);1H ЯМР (CDCl3) δ 8,14 (с, 1), 7,80 (дд, 1, J=1,2, 8,1 Гц), 7,64 (м, 1), 7,50 (м, 1), 7,29 (м, 1), 5,24 (шир. т, 1), 3,46 (м, 2), 3,37-3,32 (м, 4), 1,66-1,47 (м, 6), 1,31-1,25 (м, 14), 0,84 (м, 3).
Пример 143: 1-[8-(гексилокси)октил]-1Н-бензимидазол

8-(гексилокси)октил метансульфонат (9,4 г, 31 ммоль) добавляли к смеси бензимидазола (4,0 г, 31 ммоль) и трет-бутоксида натрия (31 ммоль) в 100 мл DMF. Через 6 часов выпаривали летучие компоненты, и остаток разделяли между ЕА и насыщенным NaHCO3, 0,1M HCl и H2O, и органические фазы высушивали над Na2SO4 и концентрировали. FC (70% ЕА/Гексан) дала 7,4 г продукта.1H ЯМР (CDCl3) δ 7,9 (с, 1H), 7,8 (м, 1H), 7,4 (м, 1H), 7,2 (м, 2H), 4,1 (т, 2H), 3,3 (м, 4H), 1,9 (м, 2H), 1,7-1,5 (м, 4H), 1,4-1,2 (м, 14H), 0,9 (м, 3H).
Пример 144: N-[8-(гексилокси)октил]пиразин-2-амин

N-[8-(гексилокси)октил]пиразин-2-амин (102 мг) получали согласно способу, используемому для получения N-[8-(гексилокси)октил]изохинолин-1-амина, исходя из 8-(гексилокси)октан-1-амина (583 мг, 2,54 ммоль) и 2-хлорпиразина (0,25 мл, 2,81 ммоль) и при нагревании при 70°C в течение 5 дней. Rf 0,26 (40% ЕА/Гексан);1H ЯМР (CDCl3) δ 7,9 (м, 1H), 7,8 (м, 1H), 7,7 (м, 1H), 4,8 (шир. с, 1H, NH), 3,4-3,2 (м, 6H), 1,6-1,4 (м, 6H), 1,4-1,2 (м, 14H), 0,8 (м, 3H).
Пример 145: 1-[8-(гексилокси)октил]-1Н-индол

1-[8-(гексилокси)октил]-1Н-индол (1,0 г) получали согласно способу, используемому для получения 1-[8-(гексилокси)октил]-1Н-бензимидазола, исходя из индола (836 мг, 7,1 ммоль), 8-(гексилокси)октилметансульфоната (1,1 г, 3,6 ммоль) и 7,1 ммоль трет-бутоксида натрия.1H ЯМР (CDCl3) δ 7,6 (д, 1H), 7,3 (д, 1H), 7,2 (м, 1H), 7,1 (м, 2H), 6,5 (д, 1H), 4,1 (т, 2H), 3,4 (м, 4H), 1,8 (м, 2H), 1,7-1,5 (м, 4H), 1,4-1,2 (м, 14H), 0,9 (м, 3H).
Пример 146: 3-[8-(гексилокси)октил]-3H-имидазо[4,5-b]пиридин

3-[8-(гексилокси)октил]-3H-имидазо[4,5-b]пиридин получали согласно способу, используемому для получения 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридина, исходя из 2-хлор-3-нитропиридина (479 мг, 3,0 ммоль) и 8-(гексилокси)октан-1-амина (0,69 г, 3,0 ммоль). Так как 2-хлор-3-нитропиридин был коммерчески доступен, первую стадию в получении 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]пиридина (хлорирование с использованием дихлорида фенилфосфоновой кислоты) не осуществляли. Rf 0,31 (5% MeOH/DCCM);1H ЯМР (CDCl3) δ 8,21 (дд, 1, J=1,5, 4,7 Гц), 7,89 (с, 1), 7,87 (м, 1), 7,02 (дд, 1, J=4,7, 7,9 Гц), 4,09 (м, 2), 3,21-3,15 (м, 4), 1,74 (м, 2), 1,36-1,32 (м, 4), 1,14-1,10 (м, 14), 0,69 (м, 3).
Пример 147: 1-Додецил-1H-имидазо[4,5-c]хинолин

1-Додецил-1H-имидазо[4,5-c]хинолин (510 мг) получали согласно способу, используемому для получения 1-октил-1H-имидазо[4,5-c]хинолина, исходя из 2,4-дихлор-3-нитрохинолина (1,0 г, 4,1 ммоль) и 1-додециламина (1,0 г, 4,5 ммоль).1H ЯМР (CDCl3) δ 8,5 (с, 1H), 8,15 (д, 1H), 8,05 (д, 1H), 7,5 (м, 1H), 7,3 (м, 1H), 3,7 (т, 2H), 1,8 (м, 2H), 1,5-1,1 (м, 18H), 0,8 (м, 3H).
Пример 148: 1-[3-(децилокси)пропил]-1H-имидазо[4,5-c]хинолин

3-(Децилокси)пропан-1-амин (7,17 г твердого вещества) получали согласно способу, используемому для получения 8-бутоксиоктан-1-амина, исходя из 1,3-пропанeдиола (26,3 мл, 363 ммоль) и 1-йоддекана (121 ммоль), смешанных в 240 мл 1:1 DCM/DMF.
1-[3-(децилокси)пропил]-1H-имидазо[4,5-c]хинолин (127 мг) получали согласно способу, используемому для получения 1-октил-1H-имидазо[4,5-c]хинолина, исходя из 2,4-дихлор-3-нитрохинолина (1,94 г, 7,99 ммоль) и 3-(децилокси)пропан-1-амина (1,72 г, 7,99 ммоль).1H ЯМР (CDCl3) δ 8,9,3 (с, 1H), 8,3 (м, 2H), 7,95 (с, 1H), 7,7-7,5 (m 2H), 4,7 (т, 2H), 3,5-3,3 (м, 4H), 2,2 (м, 2H), 1,6 (м, 2H), 1,4-1,2 (м, 14H), 0,8 (т, 3H).
Пример 149: 1-[4-(децилокси)бутил]-1H-имидазо[4,5-c]хинолин

4-(Децилокси)бутан-1-амин (2,42 г, 7,28 ммоль) получали восстановлением с помощью литий-алюминийгидрида 4-(децилокси)бутиронитрила, который получали с недостаточным выходом из алкоголята натрия 1-деканола и 4-бромбутиронитрила.
1-[4-(децилокси)бутил]-1H-имидазо[4,5-c]хинолин (78 мг) получали согласно способу, используемому для получения 1-октил-1H-имидазо[4,5-c]хинолина, исходя из 2,4-дихлор-3-нитрохинолина (1,77 г, 7,28 ммоль) и 4-(децилокси)бутан-1-амина (2,42 г, 7,28 ммоль).1H ЯМР (CDCl3) δ 9,3 (с, 1H), 8,25 (м, 1H), 8,15 (м, 1H), 7,95 (с, 1H), 7,7-7,5 (м, 2H), 4,6 (т, 2H), 3,5-3,3 (м, 4H), 2,1 (м, 2H), 1,7 (м, 2H), 1,5 (м, 2H), 1,4-1,1 (м, 14H), 0,8 (т, 3H).
Пример 150: 1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]хинолин

1-[8-(гексилокси)октил]-1H-имидазо[4,5-c]хинолин получали способом, используемым для получения 1-октил-1H-имидазо[4,5-c]хинолина, заменяя 1-октиламин 8-(гексилокси)октан-1-амином.
Пример 151: 1-{5-[3-(Гексилокси)пропокси]пентил}-1H-имидазо[4,5-c]хинолин

1-{5-[3-(Гексилокси)пропокси]пентил}-1H-имидазо[4,5-c]хинолин (2,75 г коричневого масла) получали способом, используемым для получения 1-октил-1H-имидазо[4,5-c]хинолина, исходя из 2,4-дихлор-3-нитрохинолина (5,35 г, 22 ммоль) и 5-[3-(гексилокси)пропокси]пентан-1-амина (4,90 г, 20 ммоль).1H ЯМР (CDCl3) δ 9,3 (с, 1H), 8,25 (м, 1H), 8,1 (м, 1H), 7,9 (с, 1H), 7,7-7,5 (м, 2H), 4,5 (т, 2H), 3,5-3,3 (м, 8H), 2,0 (м, 2H), 1,8 (м, 2H), 1,7-1,4 (м, 6H), 1,4-1,2 (м, 6H), 0,8 (м, 3H).
Пример 152: 1-{3-[3-(гексилокси)фенокси)пропил}-1H-имидазо[4,5-c]хинолин

1-{3-[3-(гексилокси)фенокси)пропил}-1H-имидазо[4,5-c]хинолин (1,33 г коричневого масла) получали способом, используемым для получения 1-октил-1H-имидазо[4,5-c]хинолина, исходя из 2,4-дихлор-3-нитрохинолина (4,33 г, 17,8 ммоль) и 3-[2-(гексилокси)фенокси)пропан-1-амина (4,37 г, 17,8 ммоль).1H ЯМР (CDCl3) δ 9,3 (с, 1H), 8,3-8,1 (м, 2H), 7,9 (с, 1H), 7,7-7,5 (м, 2H), 7,1 (м, 1H), 6,6-6,4 (м, 3H), 4,7 (т, 2H), 3,95-3,80 (м, 4H), 2,4 (м, 2H), 1,7 (м, 2H), 1,5-1,2 (м, 6H), 0,8 (м, 3H).
ПРИМЕРЫ БИОЛОГИЧЕСКОЙ АКТИВНОСТИ
ПРОТИВОВОСПАЛИТЕЛЬНЫЕ ПРИМЕРЫ
ПРИМЕР A: Селективное уничтожение ЛПС-активируемых воспалительных макрофагов Соединением АС.
Краткое описание: THP-1 представляет собой человеческую линию клеток AML, которая может быть индуцирована в макрофагоподобную клетку обработкой 0,2 мкМ витамина-D3 (vit-D3) в течение 3-5 дней. В отсутствие воспалительного активатора (ЛПС; бактериальный эндотоксин), АС оказывает небольшой эффект на жизнеспособность клеток в клетках THP-1 за период 6 часов. Точно так же ЛПС в отсутствие АС индуцировал только низкий уровень гибели клеток. Напротив, когда оба компонента, ЛПС и АС, добавляли к активированным vit-D3 клеткам THP-1, массовая цитотоксичность наблюдалась в течение 6 часов. Эти наблюдения показывают, что стимулируемые макрофаги, участвующие в воспалительной реакции, могут являться специфичемкой мишенью для дезактивации с использованием АС.
Краткий обзор эксперимента:
1. Активированные Vit-D3 клетки THP-1 переносили в лунки планшета с 24 лунками
2. Добавляли Соединение АС, ЛПС от E.coli 0111:B4 или оба компонента
3. После 6 часов при 37C подсчет жизнеспособных клеток в лунках осуществляли с помощью FACS
Экспериментальные процедуры
Клеточная культура:
Клетки THP-1 (ATCC), обработанные 0,2 мкМ витамина-D3 (EMD Biosciences) в течение 4 дней до дня 0 переносили в лунки планшета с 24 лунками (1×106 клеток в 1 мл cRPMI [RPMI (ATCC)+10% ΔFBS (ATCC)]. ЛПС от E.coli 0111:B4 (Sigma-Aldrich) и соединение АС добавляли в соответствующие лунки, и планшеты помещали в 37C термостат. Через 6 часов лунки обрабатывали для теста апоптоза, вызываемого Аннексином V.
Подсчет клеток FACS и тест жизнеспособности: через 6 часов 500 мкл суспензии клеток из каждой лунки переносили в пробирки FACS на 3 мл, и 50 мкл шариков CountBright (Invitrogen) добавляли в каждую пробирку. Образцы перемешивали в вортексе, добавляли 2 мкл пропидий йодида (150 мМ) (Sigma-Aldrich), затем помещали на FACSCalibur.
Результаты:
Как показано в Таблице 1, в отсутствие второго провоспалительного сигнала (ЛПС), АС оказывал небольшой эффект на жизнеспособность клеток в клетках THP-1 за период 6 часов. Точно так же ЛПС в отсутствие АС индуцировал только низкий уровень гибели клеток. Напротив, когда и ЛПС, и АС добавляли к активированным vit-D3 клеткам THP-1, массовая цитотоксичность наблюдалась в течение 6 часов. Цитотоксичность увеличивалась зависимым от дозы АС образом.
Как показано в Таблице 2, в отсутствие второго сигнала (ЛПС), АС, в диапазоне концентраций от 0,1 до 2 мкМ, оказывал небольшой эффект на жизнеспособность клеток в клетках THP-1 за период 6 часов. Точно так же ЛПС в отсутствие АС индуцировал низкий уровень гибели клетки, который увеличивался дозозависимым образом. Напротив, когда оба компонента, ЛПС и АС, добавляли к активированным vit-D3 клеткам THP-1, массовая цитотоксичность наблюдалась в течение 6 часов. Цитотоксичность, по-видимому, достигала максимального уровня с самой низкой использованной дозой ЛПС (1 нг/мл).
Заключение:
АС селективно уменьшает жизнеспособность провоспалительных ЛПС-активированных макрофагов, с относительным сохранением нестимулированных макрофагов. Очень низкая доза ЛПС (1 нг/мл) обеспечила достаточную активацию макрофагов, чтобы сделать их чувствительными к АС.
ПРИМЕР B: Относительный потенциал соединения АС и хлорохина для инактивации воспалительных макрофагов
Предпосылки: THP-1 представляет собой человеческую линию клеток AML, которая может быть индуцирована в макрофагоподобную клетку витамином-D3 (vit-D3), затем активирована в воспалительное состояние стимуляцией ЛПС (бактериальный эндотоксин). В макрофаге связывание ЛПС с толл-подобным рецептором 4 (TLR-4) приводит к активации NF-κB и секреции воспалительных цитокинов, что может привести к повреждению ткани при воспалительных заболеваниях.
Соединения по изобретению инактивируют воспалительные макрофаги, аккумулируясь в кислых вакуолях и разрушая их структуру и функцию, ингибируя высвобождение везикулярных воспалительных медиаторов и индуцируя цитозольные изменения, которые вызывают гибель или дисфункцию макрофагов, включая ингибирование аутофагии; аутофагия важна для дифференцировки моноцитов в макрофаги. Цель этого исследования состояла в том, чтобы сравнить относительный потенциал соединения по изобретению, АС, с хлорохином. Как АС, так и хлорохин представляют собой производные 4-аминохинолина, и известно, что хлорохин может быть использован для лечения нескольких клинических воспалительных заболеваний.
В этом эксперименте проверяли жизнеспособность клеток, и захват и аккумуляцию акридинового оранжевого, лизосомотропного флуоресцентного красителя, использовали, чтобы оценить подкисление и целостность лизосом. Краситель JC-1 использовали, чтобы измерить эффекты тестируемых соединений на потенциал митохондриальной мембраны (ММP); восстановление ММP является признаком апоптотической гибели клетки.
Экспериментальные процедуры:
1. Vit-D3 активированные клетки THP-1 (0,5×106 клеток в 2 мл) переносили в лунки планшета с 24 лунками
2. Соединение АС добавляли в концентрации 0,5 мкМ, 1,0 мкМ или 5,0 мкМ
3. Хлорохин добавляли в концентрации 25,0 мкМ, 50,0 мкМ или 100,0 мкМ
4. ЛПС от E.coli 0111:B4 (конечная концентрация 1 нг/мл) добавляли в некоторые лунки
5. Через 5 часов количество жизнеспособных клеток, захват Акридинового оранжевого (A.O). и митохондриальную загрузку JC-1 определяли сортировкой активированных флюоресценцией клеток (FACS)
Информация о линии клеток:
THP-1: ATCC TIB-202 Организм: Человек, мужского пола, годовалый младенец
Орган: Периферическая кровь Заболевание: Острый Моноцитарный Лейкоз (AML)
Тип клетки: Моноцит Свойства роста: Суспензия в RPMI плюс 10% FBS
Клеточная культура:
Клетки THP-1 (p39), обработанные 0,1 мкМ vit-D3 (100 мкМ) в ДМСО в течение 3 дней, подсчитывали, осаждали, повторно суспендировали в бессывороточном RPMI (Lonza 12-115F) и переносили в лунки двух планшетов с 24 лунками (0,5×106 клеток в 2 мл). Добавляли Соединение АС (в трех экземплярах) в 0,1 мкМ, 0,5 мкМ и 1,0 мкМ, добавляли хлорохин дифосфат (в трех экземплярах) в 10,0 мкМ, 50,0 мкМ и 100,0 мкМ. Сырой ЛПС от E.coli 0111:B4 добавляли к некоторым лункам (конечная концентрация 1 нг/мл), и планшеты помещали в 37C термостат. 1 мкл Baf A1 (конечная концентрация 50 нМ) добавляли к одной лунке (без ЛПС) в час T=4 и использовали в качестве компенсационного контроля для загрузки Акридинового оранжевого. Через 5 часов, 500 мкл аликвот клеток переносили в пробирки FACS, и количество жизнеспособных клеток, загрузку A.O. и аккумуляцию JC-1 определяли с помощью FACS.
Тест захвата Акридинового оранжевого (A.O). и подсчет жизнеспособных клеток - момент времени 5 часов:
Образцы перемешивали в вортексе, добавляли 2 мкл 50 мкг/мл сток-раствора A.O. (конечная концентрация 200 нг/мл), и пробирки инкубировали при 37C в течение 15 минут. Пробирки промывали дважды в DPBS, повторно суспендировали в 500 мкл DPBS и помещали на FACSCalibur. Акридиновый оранжевый вызывает сильную флюоресценцию как в FL-1 (зеленый - связывание РНК), так и в FL-3 (дальнекрасный - кислые лизосомы).
Результаты:
Как показано в Таблице 3 ниже, в отсутствие ЛПС, низкие дозы АС имели низкие прямые цитотоксические эффекты, которые увеличивались зависимым от концентрации образом в острый/(5-часовой) момент времени. Хлорохин демонстрировал подобную тенденцию, хотя он требовал в 100 раз больше лекарственного средства против АС; 100 мкМ Хлорохина были приблизительно эквивалентны 1 мкМ АС.
В присутствии низкой дозы ЛПС (1 нг/мл), цитотоксичность возрастала с добавлением 0,1 мкМ (100 нM) АС. Добавление 10 мкМ, 50 мкМ или 100 мкМ Хлорохина имело меньший эффект на ЛПС-индуцированную гибель клеток, чем 1 мкМ АС, что указывает на приблизительно 100x более высокий потенциал АС, чем хлорохина для инактивации стимулируемых ЛПС, а также базовых клеток THP-1.
Как АС, так и хлорохин уменьшали флюоресценцию акридинового оранжевого в клетках THP-1, инициированных витамином D3 и активированных 0,1 нг/мл ЛПС (Таблица 4), что указывает на устранение кислотности или нарушение целостности лизосом. АС был приблизительно 50x более активным чем хлорохин в отношении снижения флюоресценции акридинового оранжевого.
Обработка АС приводила к дозозависимому сокращению деполяризации митохондрий, приводя к уменьшению аккумуляции в митохондриях накопление димеров красного JC-1.
ЛПС индивидуально (1 нг/мл) не имел никакого эффекта на целостность митохондрий, но потенцировал АС-индуцированную деполяризацию митохондрий. Напротив, Хлорохин оказывал небольшой прямой эффект или не оказывал эффекта на целостность митохондрий в концентрациях до 100 мкМ в отсутствие или в присутствии ЛПС.
Заключение:
АС показывает селективность в отношении инактивации ЛПС-активированных макрофагов против нестимулированных клеток. АС также уменьшал аккумуляцию акридинового оранжевого в лизосомах, что указывает на то, что он вызвал разрушение лизосом. АС является приблизительно в 100 раз более активным, чем хлорохин, в отношении инактивации макрофагов и приблизительно в 50 раз более активным, чем хлорохин, в отношени нарушения целостности лизосом, что измеряется на основе аккумуляции акридинового оранжевого.
ПРИМЕР C: Скрининг соединений по изобретению на противовоспалительную активность in vitro
Предпосылки: THP-1 представляет собой человеческую линию клеток острого миелогенного лейкоза (AML), которая может быть индуцирована в макрофагоподобную клетку витамином-D3 (vit-D3). В макрофаге ЛПС (липополисахарид; эндотоксин), стимуляция толл-подобного рецептора 4 (TLR-4) приводит к активации NF-κB и секреции воспалительных цитокинов, а также к инициации запрограммированных путей гибели через RIP и Каспазу 8, Баланс этой сложной регуляторной сети зависит от высокоспецифичных киназ, ферментов, которые требуют АТФ. Нарушение цитозольного рН или доступности/энергетического уровня АТФ нарушает эту контрольную сеть и может вызывать макрофагальный сдвиг от продукции воспалительных цитокинов к запрограммированному событию гибели, который имеет чистый эффект в отношении ограничения воспалительного повреждения.
Было показано, что соединения по изобретению быстро (в течение 5-6 часов) инактивируют макрофаги, когда макрофаги были помещены в провоспалительное состояние, активированное ЛПС.
Больше чем 200 соединений по изобретению были скринированы в отношении противовоспалительной активности в системе THP-1 для оценки их относительного потенциала и активности in vitro.
Краткое описание:
Добавление ЛПС к обработанным соединением макрофагам привело к острой/5-часовой гибели клеток; эта активность увеличивалась зависимым от концентрации образом. Обработка только тестируемыми соединениями показала только низкий уровень острой цитотоксичности.
Большинство протестированных соединений показало значительную способность инактивировать провоспалительные клетки THP-1 в соответствии с предложенным механизмом действия, включающим разрушение лизосомы, которое не зависит от связывания со специфической белковой мишенью. Из протестированных соединений семь демонстрировали более высокую активность, чем активное эталонное соединение АС: CJ, AM, AG, CX, AF, BM и AH.
В самой низкой протестированной концентрации (0,1 мкМ) все семь протестированных соединений были более активны, чем АС, в индукции гибели клеток, обработанных ЛПС. В концентрациях 0,5 мкМ и выше, все соединения, включая АС, достигали максимального порога активности.
Результаты:
Добавление ЛПС к обработанным тестируемым соединением макрофагам вызывало массовую острую/5-часовую гибель клеток; эта активность увеличивалась зависимым от концентрации образом (Таблица 6). Обработка только соединениями без провоспалительной активации макрофагов ЛПС показала только низкий уровень острой цитотоксичности.
В самой низкой протестированной концентрации (0,1 мкМ), семь соединений были более активны, чем АС, в приведении клеток в услвоия для ЛПС-индуцированной гибели клеток. В концентрациях 0,5 мкМ и выше, все восемь соединений, включая АС, достигали максимального порога активности.
Соединение СХ было самым эффективным цитотоксическим соединением в острый/5-часовой момент времени, за которым следовала группа умеренной активности, включая CJ, AF, 30006 и BM. AG и AM оказывали самый низкий эффект на цитоплазматическую адаптацию, хотя и больший, чем тот, который показывает АС.
ПРИМЕР D: Противовоспалительная активность соединений по изобретению
Было показано, что соединения по изобретению непосредственно ингибируют NF-κB, повреждают внутриклеточные кислые лизосомы, приводя к утечке протонов и подкислению цитоплазмы, а также повреждают митохондрии, снижая энергетический уровень клетки. Вместе эти действия приводят к прямой гибели клеток в некоторых уязвимых типах клеток в течение приблизительно 48 часов. Дополнительно в макрофаге подкисление цитоплазмы и истощение энергии соединениями по изобретению примируют клетку для инактивации под действием низких концентраций ЛПС, приводя к острому (5-часовому) событию гибели клетки через комбинацию Каспаза-управляемого апоптоза и RIP-управляемого некроза.
Соединения по изобретению тестировали в количестве 0,1 мкМ против АС в тесте вызываемой ЛПС гибели клеток THP-1. Оценивали как острую/5-часовую, так и хроническую/48-часовую фазы гибели клеток. Соединения скринировали партиями с ДМСО в качестве отрицательного контроля и АС в качестве высокоактивного контроля. Соединения тестировали в низкой концентрации 0,1 мкМ с целью идентификации средств, более мощных, чем эталонное средство АС; в более высоких концентрациях, например, 1 мкМ, большинство соединений по изобретению активно в стимулировании гибели клетки в этом тесте, что делает дифференциацию от АС менее чистой, чем при в 10 раз более низкой концентрации лекарственного средства.
Результаты/Краткое описание:
Семь из соединений по изобретению не только демонстрировали активность, эквивалентную АС, в острый/5-часовой момент времени (подготовка клеток), но были также более активны, чем АС в хронический/48-часовой момент времени (удержание): CJ, AM, AG, CX, AF, BM и AH.
Другие 15 протестированных соединений демонстрировали активность, эквивалентную АС, как в 5-часовой, так и в 48-часовой моменты времени: CI, CL, AL, AR, AN, AD, BH, CV, AJ, BD, BU, BK, EW, AK и AE.
Остальные 187 соединений показали более низкую противовоспалительную активность, чем АС, в тестируемой концентрации 0,1 мкМ. Однако это скрининг проводили в подоптимальной концентрации, чтобы обнаружить самые мощные соединения в библиотеке; низкая активность в концентрации 0,1 мМ в контексте этого теста все еще совместима со значительной и мощной противовоспалительной активностью по сравнению с хлорохином или другими противомалярийными средствами.
ПРИМЕР E: Противовоспалительные свойства соединения АС в модели воспаления кожи
Цель: Оценить противовоспалительные свойства соединений по изобретению в мышиной модели индуцированного 12-O-тетрадеканoилфорбол-13-ацетатом (TPA) хронического воспаления кожи. Нанесенные топически сложные эфиры форбола, такие как TPA, индуцируют воспаление кожи, включающее отек, инфильтрацию макрофагов и Т-клеток и эпидермальную гиперплазию (Alford et al., 1992), и эту систему использовали как модель животных для дерматита, имитируя аспекты человеческих воспалительных нарушений кожи. TPA также известен как соканцерогенный фактор, так, что средства, которые ингибируют гиперпролиферативные или ангиогенные действия TPA, могут, по-видимому, ингибировать промотирование опухоли.
Способы
Составы лекарственного средства: Соединение АС растворяли в смеси изопропил миристат:пропилен гликоль (1:1)+0,9% ДМСО в указанных концентрациях. TPA растворяли в смеси ацетон:вода (99:1). Дексаметазон (0,06%) растворяли в нормальном солевом растворе.
Мыши: В этом эксперименте использовались самки мышей HSD-ICR (CD-1R) в возрасте 8-10 недель.
Схема эксперимента: Мышей размещали в шести группах по 10 мышей в каждой. 20 мл 0,01% TPA наносили в каждое ухо в дни 0, 2, 4, 7, 9, 11, 13, 15, 18, 20 и 22, 20 мл АС в различных концентрациях или 20 мл раствора дексаметазона наносили в уши ежедневно, начиная с дня 7, после того, как воспалительные изменения в толще уха были установлены. Толщину уха измеряли толщиномерами каждые три дня.
Результаты
Лечение соединением АС предотвращало воспалительное утолщение ушей мышей, обработанных TPA. Гистология показала, что TPA-индуцированный отек и эпидермальная гиперплазия были уменьшены АС, как и ангиогенез. Потенциал АС был сопоставим с таковым дексаметазона, причем значительная активность наблюдалась в самой низкой дозе 12,5 микрограммов АС в ухо в сутки.
Ссылка
Alford JG, Stanley PL, Todderud G, Tramposch KM. (1992) Temporal infiltration of leukocyte subsets into mouse skin inflamed with phorbol ester. Agents Actions. 37(3-4): 260-7
ПРИМЕР F: Противовоспалительные эффекты соединений по изобретению на псориазиформный дерматит у мышей
Топический имихимод (IMQ), агонист толл-подобного рецептора, был принят как модель воспалительных кожных заболеваний, включая псориаз и атопический дерматит. Кожные воспалительные изменения и экспрессия генов у мышей, обработанных топическим имихимодом, имитировали псориаз и дерматит у человека (van der Fits et al., 2009; Swindell et al., 2011). Эффект ряда соединений по изобретению проверяли на мышиной модели имихимод-индуцированного дерматита, используя для сравнения топический такролимус и дексаметазон для оценки безопасности и эффективности относительно стандартных средств, используемых для лечения дерматита у человека.
Соединения, тестируемые на противовоспалительную активность, индивидуально растворяли в этаноле в концентрации 0,6% и затем смешивали с 9 объемами вазелина (расплавленного на горячей водяной бане при 50 градусах С), получая мази, содержащие активное лекарственное средство в количестве 0,06%. Мазь дексаметазона получали точно так же, но в конечной концентрации 0,03%, потому что дексаметазон в концентрации 0,06%, нанесенный топически, в предварительных экспериментах вызвал значительное снижение массы вследствие системной абсорбции. Коммерческая мазь с 0,1% такролимуса (ProTopic™; Novartis) также использовалась как активный компаратор. Вазелин, содержащий 10% этанола, использовался в качестве контроля.
Самок мышей Balb/C (8 недель) рандомизировали и разделяли на группы по 5 животных в каждой. Воротники из полиэтилена надевали на мышей, чтобы предотвратить возможность легко царапать уши.
5% имихимода наносили на оба уха каждой мыши (20 микролитров на ухо) ежедневно в течение 5 дней, и затем через день в течение всей продолжительности исследования. Воспалительные изменения, включая удвоение толщины уха, были очевидны в день 5. В день 7 после инициирования имихимода было начато лечение топическими средствами. Оба уха каждой мыши обрабатывали тестируемыми мазями, используя одно соединениее на одну мышь.
Толщину уха и исследования PASI (Psoriasis Area and Severity Index, стандартная система оценки псориаза) регистрировали дважды в неделю в течение исследования. Оценка PASI включает сумму оценок набухания, эритемы и шелушения от 0 до 4; максимальный бал PASI составляет 12, и минимум, в незатронутой коже, - 0).
Результаты
Лечение имихимодом привело к значительным воспалительным изменениям, включая увеличение толщины уха и изменения в оценках PASI; уши в контрольной группе достигли максимального возможного значения в системе оценки PASI с серьезными утолщением, эритемой и шелушением. Соединения по изобретению, нанесенные топически в основе мази, уменьшали имихимод-индуцированное воспалительное повреждение ушей мыши, что было оценено путем измерений толщины с использованием толщиномера и оценкой PASI внешнего вида. Сравнительные лекарственные средства такролимус и дексаметазон также уменьшали толщину уха и оценку PASI. Особенно AF превосходила коммерческую клиническую форму топических 0,1% такролимуса (протопическая мазь) в уменьшении толщины уха и оценки PASI. Противовоспалительная активность дексаметазона сопровождалась значительным снижением массы тела, указывая на системную токсичность вследствие абсорбции дексаметазона. Ни соединения по изобретению, ни такролимус не влияли на массу тела. В добавление к стимулированию воспаления ушей, перенос имихимода от ушей на скальпы мышей привел к потере волос и псориазиформному дерматиту на голове в области от участка между ушей до носа. У мышей, получающих лечение дексаметазоном эта область оставалась лысой после лечения в конце эксперимента; напротив, рост волос сохранялся в этой области в течение ежедневного лечения AF, указывая, что AF ингибировал патологическое воспаление, также не ослабляя нормальное обеспечение жизнедеятельности тканей. Известным побочным эффектом лечения дексаметазоном и другими топическими кортикостероидами является истончение и ослабление подвергнутых лечению областей; отсутствие возобновления роста волос может отражать клиническую проблему атрофии кожи, известную как побочный эффект топического дексаметазона. AF был одинаково эффективен в концентрациях 0,6% и 0,06% в основе мази, отражая широкое терапевтическое окно. Все протестированные соединения по изобретению уменьшали IMQ-индуцированные изменения в толщине уха, таким образом демонстрируя их противовоспалительную активность in vivo.
Библиография
Swindell WR, Johnston A, Carbajal S, Han G, Wohn C, Lu J, Xing X, Nair RP.
Voorhees JJ, Elder JT, Wang XJ, Sano S, Prens EP, DiGiovanni J, Pittelkow MR.
Ward NL, Gudjonsson JE. (2011) Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PLoS One. 6(4):e18266
van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, Cornelissen
F, Mus AM, Florencia E, Prens EP, Lubberts E. (2009) Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 182(9):5836-45
ПРИМЕР G: Эффекты соединений по изобретению на мышиной модели рассеянного склероза
Рассеянный склероз (MS) представляет собой аутоиммунное заболевание опосредуемое, в том числе, деструкцией иммунной системой миелиновых оболочек, окружающих аксоны нейрона в мозге. Принятой моделью животных для этого заболевания является Экспериментальный Аутоиммунный Энцефалит (EAE), индуцируемый иммунизацией мышей белками или пептидами, которые вызывают иммунный ответ на миелин-специфические белки.
В этом эксперименте, EAE был индуцирован иммунизацией мышей пептидом от протеолипидного белка (PLP), известной антигенной мишени в MS. Несколько соединений по изобретению вводили перорально, чтобы оценить их эффект на течение EAE, с получением, как результат, количественной оценки симптомов заболевания. Линомид, малая молекула-иммуномодулятор с известной активностью в моделях EAE, использовался как лекарственное средство для сравнения.
Материалы и Методы
41 мышь получали подкожные инъекции 90 мкг PLP139-151 в 200 мл PBS в День 0.
PLP получали в неполном адъюванте Фройнда (IFA), смешивая 10 мл IFA с 40 мг M. tuberculosis H37Ra (конечная концентрация 4 мг/мл M. tuberculosis). Полученная смесь представляла собой полный адъювант Фройнда (CFA).
Для инъекции эмульсию PLP139-151 и CFA получали, смешивая 1 мл сток-раствора с 1 мл CFA при интенсивном перемешивании в течение 15 минут, формируя эмульсию.
Мыши получали носитель или тестируемое соединение (60 мкмоль/кг; суспендированное в 1%-ом водном растворе гидроксипропилметилцеллюлозы) пероральным скармливанием, три раза в неделю в течение 2 недель, затем один раз в сутки в течение 4 дополнительных недель, начиная с Дня 14, Ампулы с носителем и с соединениями кодировали буквами (A-E), чтобы получить слепую оценку серьезности заболевания.
Группа 1 (n=7) Носитель
Группа 2 (n=6): AZ
Группа 3 (n=7): CZ
Группа 4 (n=7): CP
Группа 5 (n=7): CQ
Группа 6 (n=7) Линомид
Мышей тестировали через день в отношении развития клинических симптомов согласно системе сортировки, приведенной ниже.
Результаты:
Мыши во всех группах показывали сопоставимые умеренные симптомы заболевания EAE в день 14 после инъекции PLP, и в этот момент времени инициировали пероральное лечение тестируемыми средствами. В завершении исследования, в День 46, получающие носитель мыши показали более тяжелые баллы оценки симптома заболевания, чем мыши в группе, получающей лечение. Соединения по изобретению показали защитную активность, сопоставимую с активностью соединения для положительного контроля, линомида.
ПРИМЕРЫ ПРОТИВОГРИБКОВОЙ И ПРОТИВОПАРАЗИТАРНОЙ АКТИВНОСТИ
ПРИМЕР H: Антикандидозная Активность Соединений по Изобретению

Краткий обзор эксперимента:
Отдельную колонию Candida Albicans выращивали в 50 мл бульона в течение ночи (19 часов). Клетки промывали PBS и 3,5×104 КОЕ/мл C. Albicans (144 мл/лунка) в среде YPD высевали в 96-луночные планшеты. Тестируемые соединения затем добавляли в каждую лунку в концентрации от 5 до 40 мкМ как конечные концентрации. Планшеты инкубировали при 30°C в течение ночи (24 часа), и OD при 600 нм считывали в конце инкубации как индекс плотности дрожжевых клеток.
Результаты:
Большинство протестированных соединений показали ингибирование роста Candida. На основании кривых ингибирования вычисляли значения IC50 (50%-ое ингибирование роста грибов) и MIC (99% ингибирования роста грибов) соединений, используя Xlfit, и они представлены в следующей таблице. Соединения с более высокой противогрибковой активностью имеют более низкие числовые обозначения.
Таблица 21
Значение 50% ингибирующей (IC50) и максимальной ингибирующей концентрации (MIC)

*MIC не может быть вычислен для этих соединений вследствие недостатка точечных данных.
Процедура: часть I: Получение клеток Candida albicans
1. За день до получения инокулума берут отдельную колонию Candida albicans, штамм 3153 (партия# 61794) из планшета с агаром Сабуро с декстрозой, используя петлю для инокулума, и затравливают в колбу на 250 мл, содержащую 50 мл среды для выращивания YPD
2. Инкубируют при 30°C с перемешиванием при 150 об./мин. в течение по меньшей мере 18 часов с приоткрытой крышкой, чтобы позволить воздуху входить и облегчать рост.
3. Исследуют аликвоту культуры под микроскопом на морфологию клеток Candida и отсутствие бактериального заражения; >95% клеток Candida должны быть бластоконидиями.
4. Переносят 25 мл ночной культуры в пластмассовую пробирку для центрифугирования на 50 мл и центрифугируют при 1000×g в течение 20 мин.
5. Отбрасывают супернатант и промывают осадок с использованием 4 мл PBS три раза. перемешивают в вортексе и центрифугируют при 1000×g в течение 10 мин.
6. После третьей промывки распределяют осадок в 2 мл PBS и перемешивают в вортексе.
7. Готовят три 1:10 серийных разведения в стерильном PBS (10-1, 10-2, 10-3) из 2 мл суспензии клетки, используя 15 мл культуральные пробирки. Конечный объем в каждой пробирке составляет 5 мл.
8. Подсчитывают число клеток в суспензии клеток из пробирки с разведением 10-3 на гемоцитометре.
Для вычисления концентрации клеток в мл:
Среднее число клеток в одном большом квадрате x фактор разведения ×104
104 = переводной коэффициент для преобразования 10-4 мл в 1 мл число клеток в 50-кратном разведении 10-3 составило: 14×104 КОЕ/мл
9. Получают 1:4 разведение в среде YPD от 50-кратного разведения суспензий клеток 10-3 для тестирования соединений.
Конечная концентрация клеток C. albicans для теста: 3,5×104 КОЕ/мл
10. Высевают 144 мкл/лунка вышеупомянутого разведения клеток на планшеты с 96 лунками.
часть II: C. Тестирование ингибирования роста Albicans соединениями
1. Из 10 мМ сток-растворов ДМСО готовят серийные разведения соединений до растворов 0,13, 0,25, 0,40, 0,55, 0,75 и 1,0 мМ
2. Добавляют 6 мкл каждого из разбавленных растворов соединения на лунку в двух экземплярах. Конечные концентрации составили 0, 5, 10, 16, 22, 30 и 40 микромоль.
3. Инкубируют все планшеты при 30C в течение ночи (~24 часа).
4. Считывают поглощение света при OD600 для каждого планшета.
6. Вычисляюти % ингибирования каждого соединения против клеток, обработанных ДМСО.
ПРИМЕР I: Оценка Активности Соединений против Saccharomyces cerevisiae

Краткий обзор эксперимента:
Ночную культуру S. cereviseae разбавляли в бульоне YPD в концентрации 40000/мл, и 150 мл/лунка высевали в 96-луночные планшеты. Соединения затем добавляли в каждую лунку в концентрации от 4 до 50 мкМ как конечная концентрация. Планшеты инокулировали при 30°C в течение ночи с перемешиванием при 220 об./мин., и поглощение света при 600 нм считывали после инкубации в течение 18 часов.
Результаты:
Среди всех эффектов соединений против S. cerevisiae, соединения AL, BG и AW были самыми эффективными. Соединение AI генерировало более низкую IC50 от вычисления XLfit, даже при том, что оно не могло достигнуть близкого к 100% уничтожения в высокой концентрации, как другие соединения. Хлорохин (C.Q). не показывал ингибирования роста дрожжей до 50 мкM. Следующие перечисленные значения IC50 (50%-ое ингибирование роста грибов) и MIC (99% ингибирования роста грибов) соединений (вычисленные с использованием XLfit) основаны на кривых ингибирования.
Таблица 22
Анти-S. cerevisiae - Значение 50% ингибирующей (IC50) и максимальной ингибирующей концентрации (MIC)

*MIC не может быть вычислен для этих соединений вследствие недостатка точечных данных.
Процедура: часть I: Получение дрожжевых клеток
1. За день до получения инокулума берут отдельную колонию S. cereviseae из планшета с агаром Сабуро с декстрозой, используя петлю для инокулума, и затравливают в пробирку на 50 мл, содержащую 10 мл среды для выращивания YPD
2. Инкубируют при 30°C с перемешиванием при 220 об./мин. в течение 24 часов с приоткрытой крышкой, чтобы позволить воздуху входить и облегчать рост.
3. Исследуют аликвоту культуры под микроскопом на морфологию дрожжевых клеток и отсутствие бактериального заражения.
4. Разбавляют ночную культуру средой YPD в разведении 1:30 (70 мкл на 2,1 мл) и подсчитывают число клеток как 4230000/мл.
5. Смешивают 620 мл 1:30 разведения и 64,4 мл YPD, чтобы получить конечную концентрацию 40000/мл клеток
6. Высевают 144 мл/лунка в четырех планшетах с 96 лунками. часть II: Тестирование ингибирования роста дрожжей
1. Из 10 мМ сток-растворов ДМСО готовят серийные разведения соединений до растворов 0,1, 0,2, 0,3, 0,63 и 1,25 мМ
2. Добавляют 6 мкл каждого из разбавленных растворов соединения на лунку в двух экземплярах. Конечные концентрации составили 0, 4, 8, 12, 25 и 50 микромоль.
3. Инкубируют все планшеты при 30C в течение ночи (~18 часов) с перемешиванием при 220 об./мин.
4. Считывают поглощение света при OD600 для каждого планшета на спектрофотометре для считывания планшетов Spectra Max Plus.
5. Вычисляют % ингибирования каждого соединения против клеток, обработанных ДМСО, и выстраивают график.
ПРИМЕР J: Активность соединений по изобретению против Trichophyton
Tricophyton rubrum представляет собой один из первичных грибков, ответственных за персистирующие резистентные к лечению инфекции ногтя на пальце ноги.

Краткий обзор эксперимента:
Trichophyton, выращенные на двух агаровых пластинках, собирали, соскребая в 10 мл солевого раствора, и фильтровали через 8 мкМ фильтры. Отфильтрованный раствор разбавляли (1:75) и высевали в 96-луночные планшеты, и обрабатывали выбранными соединениями по изобретению.
Результаты:
Этот эксперимент включал некоторые активные соединения из предыдущего эксперимента, к которым добавили несколько неиспытанных соединений. Культура, обработанная соединениями AW, AX, AT, AE или AH, не показывала видимого роста грибов даже при самой низкой протестированной концентрацией (6 мкМ), что демонстрирует их сильнейшее ингибирующее действие против роста trichophyton. Большинство остальных соединений также ингибировали грибковый рост при более высокой концентрации (12-18 мкМ). AO, AP, AF, BL, AQ and BO показали только частичное или отсутствующее ингибирование на рост грибов при самой высокой протестированной концентрации (40 мкМ). В следующей таблице представлена максимальная ингибирующая концентрация (MIC), основанная на визуальной оценке.
Процедура: часть I: Получение клеток Trichophyton rubrum
Соскребают замороженную культуру Trichophyton из ампулы ATCC и суспендируют в 100 мл PDB, и затем высевают на планшете PDA. Инкубируют планшет при 30C в течение 4 дней.
Планшет накрывают почти полностью. Соскребают колонии с двух планшетов в 10 мл солевого раствора и фильтруют через 8 мкМ фильтр в 24-луночный прозрачный планшет (используя 2 лунки). Измеряют OD собранного раствора при 520 нм и 600 нм:
520 нм = 0,13; 600 нм = 0,092 1x без разбавления
520 нм = 0,061; 600 нм = 0,037 разбавление 1:2,5
Получают 90 мл разведения 1:75 в бульоне PDB от отфильтрованной суспензии клеток, смешивая 1,2 мл раствора клеток с 88,8 мл PDB и разделяют на аликвоты по 144 мл/лунка в 5×96-луночные планшеты. часть II: Тестирование ингибирования роста Trichophyton соединениями
1. Из 10 мМ сток-растворов ДМСО готовят серийные разведения соединений до растворов 0,15, 0,3, 0,45, 0,63 и 1 мМ
2. Добавляют 6 мкл каждого из разбавленных растворов соединения на лунку в трех экземплярах. Конечные концентрации составили 0, 6, 12, 18, 25 и 40 микромоль.
3. Оборачивают планшеты парапленками и инкубируют все планшеты при 30°C в течение 6 дней.
Делают снимок планшетов на блоке для формирования изображений KODAK с 17 фиксациями 1,5 сек./фиксация с общей экпозицией 25,5 секунд.
ПРИМЕР K: Активность против Cryptococcus соединений по изобретению

Краткий обзор эксперимента:
Cryptococcus neoformans (серотип D) высевали в 96-луночные планшеты с 144 мкл/лунка 8×105 КОЕ/мл в среде для выращивания YM. Разбавленные соединения затем добавляли в каждую лунку в концентрации от 4 до 60 мкМ как конечная концентрация в двух экземплярах. Планшеты инокулировали при 37°C в течение всего 48 часов. Два измерения OD при 600 нм осуществляли после обработки в течение 30 и 48 часов.
Результаты:
Большинство соединений, протестированных в этом тесте, ингибировало рост Cryptococcus, причем соединения AL, AG, AW, AX, AA, AE, AH, AK BM, и BN были наиболее эффективные. Примечательно, что соединения AA и АС были весьма активными против Cryptococcus по сравнению с их относительными слабыми активностями в отношении Candida и S. cereviseae. В целом кажется, что Cryptococcus более чувствительны к соединениям по изобретению, чем другие протестированные грибы. Хлорохин имел очень слабую активность против Cryptococcus с максимальным ингибированием роста 40% в концентрации 100 микромоль, таким образом, его IC50 была выше этой концентрации. IC50 (концентрация для 50% ингибирования) и MIC (концентрация для максимальных 99% ингибирования) вычисляли, используя XLfit, на основании OD после 48 часов, и они представлены в следующей таблице.
Таблица 24

Процедура: часть I: Получение грибковых клеток
1. Собирают отдельную колонию Cryptococcus с агаровой пластинки YM, используя затравку кристаллизации, используя петлю для инокулума, и затравливают в колбу на 125 мл, содержащую 25 мл среды для выращивания YM.
2. Инкубируют при 37°C с перемешиванием при 220 об./мин. в течение 24 часов с приоткрытой крышкой, чтобы позволить воздуху входить и облегчать рост.
3. Исследуют аликвоту культуры под микроскопом на морфологию дрожжевых клеток и отсутствие бактериального заражения.
4. Разбавляют ночную культуру средой YM в разведении 1:100 и подсчитывают число клеток как 1×106 кое/мл.
5. Получают конечную концентрацию суспензии клеток 8×105 кое/мл в среде YM.
6. Высевают 144 мл/лунка 8×105 кое/мл суспензии клетки на планшеты с 96 лунками. часть II: Тестирование ингибирования роста Cryptococcus соединениями
1. Из 10 мМ сток-растворов ДМСО готовят серийные разведения соединений до растворов 0,1, 0,2, 0,3, 0,5, 1,0 и 1,5 мМ
2. Добавляют 6 мкл каждого из разбавленных растворов соединения на лунку в двух экземплярах. Конечные концентрации составили 0, 4, 8, 12, 20, 40 и 60 микромоль.
3. Инкубируют все планшеты при 37°C в течение ночи (30 часов) с перемешиванием при 150 об./мин.
4. Считывают поглощение света при OD600 для каждого планшета.
5. Оставляют планшеты в термостате при 37°C в течение еще одного дня и снова считывают поглощение света при OD600 в момент времени 48 часов, чтобы обеспечить ингибирующее действие соединений.
6. Вычисляют% ингибирования и IC50 каждого соединения против необработанных клеток.
ПРИМЕР L: Активность соединений по изобретению против Cryptococcus (серотип A)

Краткий обзор эксперимента:
Cryptococcus neoformans (серотип A) высевали в 96-луночные планшеты с 144 мл/лунка 5×105 КОЕ/мл в среде для выращивания YPD. Разбавленные соединения затем добавляли в каждую лунку с концентрацией от 0,05 до 10 мМ как конечная концентрация, в двух экземплярах. Планшеты инокулировали при 30°C. Два измерения OD при 600 нм осуществляли после обработки в течение 18 часов и 48 часов.
Результаты:
Большинство соединений, протестированных в этом тесте, ингибировало рост Cryptococcus (серотип A), причем AX, AK, BM, AE and AH были наиболее эффективными. C12-имидазол имел относительно слабую активность против серотипа А Cryptococcus в низкой концентрации. Выстроенные данные были основаны на считывании в момент времени 26 часов, поскольку считывания на 18 часах были слишком низкими. IC50 (концентрация для 50% ингибирования) и MIC (концентрация для максимума - 99% ингибирования) вычисляли, используя XLfit, на основании OD в момент времени 26 часов, и они перечислены в следующей таблице.
Таблица 25
Значение 50% ингибирующей (IC50) и максимальной ингибирующей концентрации (MIC)

Следует отметить, что C. neoformans (серотип A) является самым чувствительным к соединениям грибком по сравнению с другими протестированными видами, включая C. аlbicans, S. cerevisiae, Trichophyton rubrum и Cryptococcus серотип D
Процедура: часть I: Получение грибковых клеток
1. Собирают отдельную колонию Cryptococcus с агаровой пластинки Сабуро с декстрозой, используя петлю для инокулума, и затравливают в колбу на 125 мл, содержащую 25 мл среды для выращивания YPD
2. Инкубируют при 30°C с перемешиванием при 220 об./мин. в течение 24 часов с приоткрытой крышкой, чтобы позволить воздуху входить и облегчать рост.
3. Исследуют аликвоту культуры под микроскопом на морфологию дрожжевых клеток и отсутствие бактериального заражения.
4. Разбавляют ночную культуру средой YPD в разведении 1:100 и подсчитывают число клеток как 8×106 кое/мл.
5. Получают конечную концентрацию суспензии клеток 5×105 кое/мл в среде YPD после хранения стоковой культуры при 4°C в течение 3 дней.
6. Высевают 144 мл/лунка 5×105 кое/мл суспензии клетки на планшеты с 96 лунками. часть II: Тестирование ингибирования роста Cryptococcus соединениями
1. Из 10 мМ сток-растворов ДМСО готовят серийные разведения соединений до растворов 0,0013, 0,0025, 0,0125, 0,025, 0,05, 0,125 и 0,25 мМ
2. Добавляют 6 мкл каждого из разбавленных растворов соединения на лунку в двух экземплярах. Конечные концентрации составили 0, 0,05, 0,1, 0,5, 1,0, 2,0, 5,0 и 10 микромоль.
3. Инкубируют все планшеты при 30°C в течение ночи с перемешиванием при 175 об./мин.
4. Считывают поглощение света при OD600 после 18 и 26 часов для каждого планшета.
5. Вычисляют % ингибирования и IC50 каждого соединения против необработанной клетки.
ПРИМЕР М: Эффекты соединений по изобретению на противогрибковую активность макрофагов, происходящих из THP-1; Развитие противогрибкового скрининга подвергнутого фагоцитозу Cryptococcus neoformans
Предпосылки: В предыдущих примерах было показано, что соединения обладают непосредственной противогрибковой активностью против Cryptococcus neoformans в концентрациях менее 5 мкМ. Соединения, будучи слабыми основаниями, являются лизосомотропными, концентрируясь в кислом лизосомальном компартменте макрофагов. Некоторые патогенные грибы, такие как Cryptococcus neoformans, содержатся в кислых лизосомах макрофагов, чтобы избежать иммунной системы хозяина (Srikanta et al., 2011).
Было показано, что другое лизосомотропное лекарственное средство, хлорохин, который имеет некоторую непосредственную противогрибковую активность в намного более высокой концентрации 100 мкМ в отношении C. neoformans, усиливает противогрибковую активность макрофагов против C. neoformans при тестировании в концентрации всего лишь 10 мкМ. Было показано, что этот эффект является следствием концентрации лекарственного средства в лизосомах, в которых содержатся дрожжи (Harrison et al., 2000), поэтому существует потенциальная возможность того, что соединения по изобретению будут вести себя подобно хлорохину для того, чтобы воздействовать на Cryptococcus или другие организмы, находящиеся в макрофагах, но в намного более низких концентрациях.
Результаты/Резюме:
Все протестированные соединения (АМ, BM, АН и АС) показали чистое дозозависимое ингибирование роста грибов после фагоцитоза и лизиса. АН показал самый высокий потенциал с приблизительно 100%-ым ингибированием роста грибов при 2мкМ.
Значения IC50 после макрофагального фагоцитоза были сопоставимы со значениями IC50 для прямого ингибирования роста грибов в отсутствие макрофагов, приведенных в более раннем исследовании.
Соединения были способны к уничтожению C. neoformans (серотип A), даже когда грибы находились внутри живых макрофагов.
Библиография:
1: A sensitive high-throughput assay for evaluating host-pathogen interactions in Cryptococcus neoformans infection Srikanta, D et al (2011) PLoS ONE 6(7): e22773
2: Conditional lethality of the diprotic weak bases Chloroquine and Quinacrine against Cryptococcus neoformans Harrison, T. S et al (2000) J Infect Disease 182: p283-289
Результаты:
Две концентрации макрофагов (1×105 и 2×105/лунка) и высокую концентрацию C. neoformans (4×106/лунка) (значения MOI 40 и 20, соответственно) тестировали в этом эксперименте.
Все протестированные соединения показали чистое дозозависимое ингибирование роста грибов после фагоцитоза и лизиса. Фагоцитоз макрофагами не защищал клетки грибов от противогрибковой активности соединений по изобретению.
Значения IC50 после макрофагального фагоцитоза были сопоставимы со значениями IC50 для прямого ингибирования роста грибов в отсутствие макрофагов.
Экспериментальные процедуры:
Краткий обзор эксперимента для развития теста планшет #4:
Клетки THP-1 разводили до 5×105/мл или 1×106/мл в cRPMI+PMA
200 мкл переносили в плоскодонную чашку с 96 лунками (1×105 и 2×105/лунка) (48 ч при 37°C)
Среду удаляли и добавленный свежий cRPMI+PMA (еще 24 ч при 37°C)
Клетки C. neoformans в DPBS опсонизировали человеческой сывороткой (60 мин. при 30C)
Опсонизированные дрожжи промывали (DPBS) и повторно суспендировали в количестве 1×107/мл или 2×107/мл в cRPMI.
100 мкл добавляли на планшет с макрофагами (1×106 и 2×106/лунка) (4 ч при 37°C), промытый X4 с использованием DPBS
100 мкл cRPMI добавляли в каждую лунку (18 ч при 37°C), соединение АС добавляли в некоторые лунки. Среду удаляли без промывки, добавляли 25 мкл 0,05% Triton X-100, чтобы лизировать клетки (3 минуты взбалтывания при комнатной температуре)
Добавляли 125 мкл бульона YPD, и планшет инкубировали (24 ч при 30°C, затем 24 ч при 37°C)
Рост C. neoformans определяли на Спектрофотометре (600 нм) через 24 и 48 часов
Информация о линии клеток:
THP-1: ATCC TIB-202 Организм: Человек, мужского пола, младенец в возрасте одного года
Орган: Периферическая кровь Заболевание: Острый Моноцитарный Лейкоз (AМЛ)
Тип клеток: Моноцит Свойства роста: Суспензия в RPMI плюс 10% FBS
Протокол дифференцировки макрофагов, происходящих из THP-1 (PMA):
Клетки THP-1 (p15), выращенные в cRPMI [RPMI (Lonza 12-115F) плюс 10% ΔFBS (Lonza DE14-701F)] подсчитывали на гемацитометре. Клетки центрифугировали при 1 800 об./мин. при комнатной температуре в течение 5 минут, аспирировали супернатант, осадок после центрифугирования разрушали, затем разводили до 5,0×105/мл и 1,0×106/мл в cRPMI, дополненном 0,2 мкг/мл форбол 12-миристат 13-ацетат (PMA) (1 мг/мл в ДМСО Sigma P8139). Аликвоты по 200 мкл каждой концентрации клеток переносили в 42 лунки (половина планшета) плоскодонного планшета с 96 лунками (1×105 и 2×105/лунка) и помещали в термостат, настроенный на 37°C, на 48 часов, затем среду удаляли и добавляли 200 мкл свежего cRPMI+PMA. Планшет инкубировали в течение дополнительных 24 часов при 37°C, затем обрабатывали для сбора дрожжей.
Информация о дрожжевом штамме:
Cryptococcus neoformans: ATCC MYA-1017 Обозначение: CDC21
Выделение: Получены из штамма H99 от пациента с болезнью Ходжкина, Нью-Йорк
Антигенные свойства: Серотип-A Свойства роста: Суспензия в бульоне YEPD 25C
Опсонизация клеток Cryptococcus neoformans (только человеческая сыворотка):
Параллельно получению макрофагов, клетки C. neoformans выращивали из единственной колонии в 20мл бульона YPD при 30C в течение ночи. Измерение поглощения света в разведении 1:10 ночной (ON) культуры дало 0,89 OD при 600 нм. Предполагаемая концентрация этого стока составила 4×108 клеток/мл (2×108 клеток/мл дали OD 600 нм 0,426 в более раннем исследовании проявления планшета с макрофагами Cryptococcus 3 МL113012). Клетки промывали однократно DPBS и повторно суспендировали в 2 мл DPBS. 230 мкл этого стока (~60×107 клеток) разбавляли до 500 мкл с DPBS в пробирке Eppendorf. Для опсонизации добавляли 500 мкл человеческой сыворотки (SIGMA S7023), и пробирку инкубировали при 30 C в течение 60 минут с планетарным перемешиванием. Опсонизированные грибковые клетки промывали три раза с 800 мкл DPBS (1100 г в течение 2 минут) и повторно суспендировали в 800 мкл DPBS. 1:200 разведение клеток подсчитывали (4,25×106/мл), эквивалент 8,5×108/мл для 1X стока. 470 мкл 1X стока забирали в 10 мл с cRPMI для конечной концентрации 4×107/мл.
Тест макрофаг-опосредованной противогрибковой активности:
Среду аспирировали с полученного макрофагального планшета и в лунки добавляли 100 мкл опсонизированной суспензии грибковых клеток. Среду без дрожжей добавляли в три лунки для каждой концентрации макрофагов, чтобы обеспечить ретроспективные считывания. Три пустых лунки (без макрофагов) засевали грибами, и они служили промытым контролем. Планшеты затем инкубировали при 37°C в течение 4 часов, затем промывали 4 раза с DPBS (планшеты коротко встряхивали после добавления DPBS, чтобы увеличить моющую эффективность). 144 мкл cRPMI добавляли в каждую лунку и 6 мкл, 12,5 мкМ, 25 мкМ и 50 мкМ стока соединений AM, BM, AH и AC добавляли в трех экземплярах для конечных концентраций 0,5 мкМ, 1мкМ или 2 мкМ. Планшет инкубировали при 37°C, 5% CO2 в течение 18 часов. Среду удаляли, 25 мкл 0,05% Triton X-100 (SIGMA T-9284) в DPBS добавляли в каждую лунку, и планшет взбалтывали при комнатной температуре в течение 3 минут для того, чтобы лизировать клетки. 125 мкл бульона YPD (KD Medical YLF-3260) добавляли в каждую лунку, и планшет помещали в термостат при 30°C. Рост клеток C. neoformans определяли, измеряя поглощение света при 600 нм на Спектрофотометре (Spectra Max Plus с использование программы SoftMax Pro) через 30 часов.
ПРИМЕР N: Противогрибковая активность, определяемая по минимальной ингибирующей и фунгицидной концентрациям
ЦЕЛЬ
Цель этого исследования состояла в определении противогрибковой активности восьми экспериментальных соединений против репрезентативной группы грибковых изолятов, включая Candida albicans, C. glabrata, Cryptococcus neoformans, Trichophyton rubrum, Aspergillus fumigatus и Rhizopus spp. Противогрибковую активность измеряли по минимальной ингибирующей концентрации (MIC) и минимальной фунгицидной концентрации (MFC).
МАТЕРИАЛЫ
Изоляты
Тестировали три недавних клинических штамма каждого вида, взятых из коллекции культур в Центре Медицинской Микологии, Case Western University.
Противогрибковые средства
Соединения в порошковой форме растворяли в ДМСО. Серийные разведения каждого соединения получали в RPMI-1640 в диапазоне 0,125-64 мг/мл.
МЕТОДЫ
Тестирование MIC проводили согласно стандартам CLSI M27-A3 и M38-A2 для тестирования чувствительности дрожжей и нитчатых грибов, соответственно (1, 2). Тестируемые изоляты субкультивировали из замороженных скошенных агаров на агаровые пластинки с картофельной декстрозой (Trichophyton rubrum субкультивировали на овсяных планшетах для продукции конидий) и проверяли на чистоту. Инокуляты затем получали в RPMI-1640 (YNB для Cryptococcus) в концентрации 0,5-2,5×103 колониеобразующих единиц (КОЕ)/мл или 0,4-5×104 конидий/мл для дрожжей и нитчатых грибов, соответственно. Результаты MIC считывали при 50%-ом и 100%-ом ингибировании по сравнению с контролем, и в моменты времени 24 и 48 часов (C. neoformans инкубировали в течение 72 часов, и штаммы T. rubrum инкубировали в течение 96 часов).
Определения MFC проводили согласно модификациям, предварительно описанным Canton et al. and Ghannoum and Isham. (3, 4) В частности, полное содержимое каждой чистой лунки из теста MIC субкультивировали на агаре с картофельной декстрозой. Чтобы избежать противогрибкового переноса, аликвотам давали впитаться в агар и затем насекали для выделения после высыхания, таким образом удаляя клетки из источника лекарственного средства. Фунгицидная активность была определена как сокращение на ≥99,9% количества колониеобразующих единиц (КОЕ)/мл от исходного показателя инокулума, причем соединения определяли как «цидные», если MFC падал в ходе 4 разведений MIC.
РЕЗУЛЬТАТЫ
Данные показывают, что все восемь соединений демонстрировали противогрибковую активность против протестированных штаммов, хотя результаты MIC и MFC были штаммспецифичными. Как можно заметить в Таблице 27, соединение АС показало самые низкие значения MIC против штаммов C. albicans как при 50%-ом, так и при 100%-ом ингибировании в момент времени 24 часа (<0,12-0,25 и <0,12-1 мг/мл, соответственно) и 48 часов (<0,12-1 и 0,5-2 мг/мл, соответственно). Важно, что соединение АС было «цидным» против 2 из 3 протестированных штаммов C. albicans. Соединение AG демонстрировало схожие значения MIC и MFC против штаммов C. albicans.
Таблица 28 показывает, что соединения AG и АС были также наиболее активными против протестированных штаммов C. glabrata. Через 24 часа MIC при 50% для соединения AG составила 0,25-1 мг/мл и 0,5-2 при 100%. Через 48 часов, соответствующие значения для соединения AG составили в обоих случаях 0,5-2 мг/мл. Через 24 часа MIC при 50% для соединения АС составила 0,5-1 мг/мл и 1-2 при 100%. Через 48 часов, соответствующие значения соединения АС составили 1-2 (50%) и 2-4 мг/мл (100%). Оба соединения AG и АС были «цидными» против всех протестированных штаммов C. glabrata.
Как можно заметить в Таблице 29, соединения AX и AH демонстрировали самую большую противогрибковую активность против протестированных штаммов Cryptococcus neoformans. Соединение АХ имело значения MIC 0,12-2 и 0,5-4 мг/мл при 50%-ом и 100%-ом ингибировании, соответственно, в то время как соединение АН имело соответствующие значения 0,004-2 и 0,25-2 мг/мл. Оба соединения были «цидными» против всех 3 изолятов neoformans.
Таблица 30 показывает значения MIC и MFC восьми соединений против штаммов Aspergillus fumigatus. Соединения AE, AH и AC показали эквивалентную ингибирующую активность, причем соединение АЕ демонстрировало значения MIC<0,12-0,5 и <0,12-1 мг/мл при 50%-ом и 100%-ом ингибировании, соответственно, через 24 часа. Через 48 часов, соответствующие значения для соединения АЕ были 0,5-2 и 1-4 мг/мл. Соединение АН демонстрировало значения MIC<0,12 и 0,25-0,5 мг/мл при 50%-ом и 100%-ом ингибировании, соответственно, через 24 часа. Через 48 часов, соответствующие значения для соединения АН были 0,25-1 и 0,25-4 мг/мл. Для соединения АС значения MIC в 24 часа были <0,12 и 0,25-0,5 мг/мл для 50%-ого и 100%-ого ингибирования, соответственно, в то время как соответствующие значения в 48 часов были 0,5-1 и 1 мг/мл. Однако только соединения AL, AM и AG были «цидными» против одного из штаммов A. fumigatus (MRL 28397).
В Таблице 31 можно видеть, что соединения АЕ АН и АС были наиболее активными против штаммов Rhizopus. В 24 часа соединение АЕ показало значения MIC<0,12 и 1-2 мг/мл для 50%-ого и 100%-ого ингибирования, соответственно, с соответствующими значениями в 48 часов 1-2 и 1-4 мг/мл. Соединение АН показало значения MIC 0,25-0,5 и 2 мг/мл для 50%-ого и 100%-ого ингибирования, соответственно, в 24 часа и 2 мг/мл для обоих конечных считываний в 48 часов. В 24 часа соединение АС показало значения MIC<0,12-0,25 и 0,5 мг/мл для 50%-ого и 100%-ого ингибирования, соответственно, с соответствующими значениями в 48 часов 0,5 и 0,5-1 мг/мл. В целом, никакой «цидной» активности не было продемонстрировано против протестированных штаммов Rhizopus.
Наконец, Таблица 32 показывает значения MIC и MFC этих восьми соединений против T. rubrum. При 50%-ом конечном ингибировании соединения AG, AX, AE, AH и AC показали эквивалентную активность (<0,12-4 мг/мл в целом). При 100%-ом конечном ингибировании соединения AG, AH и AC были эквивалентными (0,25-4 мг/мл в целом), при этом соединения АХ и АЕ располагались немного выше (0,25-16 мг/мл). В пределах определения «цидности» (MFC в пределах 4 разведений MIC) все соединения считались «цидными» против штаммов T. rubrum, хотя для некоторых штаммов MFC были высокими (8-16 мг/мл).
В целом, соединения АЕ, АН и АС, по-видимому, продемонстрировали самую высокую ингибирующую активность против большинства протестированных грибковых штаммов.
Библиография для Примера N
1. CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard - Second Edition. CLSI document M27-A2 (ISBN 1-56238-469-4). CLSI, 940 West Valley Road, Suite 1400, Wayne, PA 19087-1898 USA, 2002.
2. CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi; Approved Standard- Second Edition. CLSI document M38-A2 [ISBN 1-56238-668-9]. CLSI, 940 West Valley Road, Suite 1400, Wayne, PA 19087-1898 USA, 2008.
3. Canton E, Peman J, Viudes A, Quindos G, Gobernado M, Espinel-Ingroff A. 2003. Minimum fungicidal concentrations of amphotericin B for bloodstream Candida species. Diagn Microbiol Infect Dis. 45:203-6.
4. Ghannoum MA, Isham N. 2007, Voriconazole and Caspofungin Cidality Against Non-Albicans Candida Species. Infectious Diseases in Clinical Practice. 15(4):250-253.
ПРИМЕРЫ ПРОТИВОРАКОВОЙ АКТИВНОСТИ
ПРИМЕР O: Соединения по изобретению ингибируют рост сингенного рака молочной железы у мышей
Модели рака на мышах обычно включают либо сингенные мышиные опухоли в организме иммунокомпетентных мышей, либо ксенотрансплантаты человеческих опухолей у иммунокомпрометированных мышей. Важным аспектом использования мышиных опухолей у мышей является то, что опухоль и хозяин имеют намного более близкое генетическое сходство, чем в случае человеческих ксенотрансплантатов у мышей, и поэтому они могут рассматриваться как очень строгий тест селективности средств для ингибирования пролиферации раковых клеток против нормальных тканей. 4T1 представляет собой линию клеток рака молочной железы, обычно используемую в качестве модели сингенного рака. Тестируемые соединения выбирали на основании их способности селективно уничтожать клетки рака молочной железы 4T1 мышей относительно нормальной линии клеток молочной железы мышей in vitro.
Самок мышей Balb/C рандомизировали на группы лечения, и 103 клеток 4T1 вводили в жировой слой молочных желез каждой мыши в 0,1 мл PBS 4/28/10 (день 0). Мыши получали тестируемые соединения пероральным скармливанием в 1% гидроксипропилметилцеллюлозы со дня 2 до дня 30. Рост опухоли оценивали измерениями с помощью толщиномера два раза в неделю, и массу опухоли после вскрытия и массу тела также мониторили.
Группы лечения были следующими:
1. Носитель (1% гидроксипропилметилцеллюлоза; HPMC)
2. CI (120 мкмоль/кг/сутки)
3. ВА (120 мкмоль/кг/сутки)
4. СР (120 мкмоль/кг/сутки)
5. CQ (120 мкмоль/кг/сутки)
6. AA (120 мкмоль/кг/сутки)
7. АС (120 мкмоль/кг/сутки)
Соединения по изобретению уменьшали рост опухоли по сравнению с получавшими носитель мышами после ежедневного перорального приема в дозе 120 мкМ/кг/сутки в течение 33 дней, с приемлемой токсичностью (менее 10% снижения массы тела). CQ было самым активным из соединений, протестированных в этом эксперименте на модели рака молочной железы 4T1. Соединения выбирали для тестирования in vivo на основании их способности селективно уничтожать клетки рака молочной железы 4T1 мышей относительно нормальной линии клеток молочной железы мышей in vitro, что указывает на соответствие между цитотоксичностью соединений по изобретению в отношении линии раковых клеток in vitro и их противораковой активностью in vivo.
ПРИМЕР P: Эффекты соединения АС у мышей, несущих ксенотрансплантаты человеческого гормоннезависимого рака предстательной железы
Экспериментальная процедура
В стандартных моделях рака предстательной железы используют подкожные ксенотрансплантаты человеческой линии раковых клеток предстательной железы. Местные измеримые опухоли продуцируются в месте инъекции клеток и метастазируют в критические ткани, такие как кости, легкие и печень. Летальность в этой модели является следствием метастаз, ослабляющих функцию ткани. Соединения по изобретению оценивали в отношении ингибирования роста опухоли и снижения или задержки летальности на модели рака предстательной железы РС-3, имитирующей продвинутую андрогеннезависимую стадию рака предстательной железы.
10 самок голых мышей (самки Hsd:бестимусные голые-Foxn1nu) получали клетки РС-3 (5×106 на мышь в 0,1 мл PBS) подкожной инъекцией в заднюю часть правого бока. Через 8 дней опухоли были осязаемы, и мышей разделяли на две группы с приблизительно равными средними размерами опухоли. Мыши получали АС или носитель (солевой раствор) путем внутрибрюшинной (i.p). инъекции один раз в сутки до дня 79.
1. Носитель (солевой раствор 0,9%): Средний объем опухоли до лечения 55,7 мм3; масса тела 26,6±0,9 г)
2. Соединение АС: 120 ммоль/кг/сутки. Средний объем опухоли до лечения 59,6 мм3; масса тела 26,8±0,5 г)
Опухоли измеряли толщиномерами дважды в неделю, и массу тела и летальность также мониторили.
Результаты
Все 5 получавших носитель мышей умерли ко дню 35 (Индивидуальные дни смерти 20, 24, 24, 26 и 35). Одна мышь в леченной АС группе умерла в день 65, и остальные 4 оставались в живых до завершения исследования в день 79.
У прожившей дольше всех получавшей носитель мыши объем опухоли был на 3007% больше ко времени смерти в день 35, чем в начале лечения; все другие получавшие носитель животные умерли от метастатического заболевания с меньшими первичными размерами опухоли. Среди мышей, получавших АС, опухоли увеличились до среднего значения 949% от начального размера в день 77; две из мышей, оставшихся в живых до конца исследования, не имели к тому моменту никаких обнаружимых опухолей и считались достигшими полной регрессии, и одна регрессировала больше чем на 50% от начального размера опухоли. Леченные АС мыши имели в среднем массу тела 28,9±1,3 г в конце исследования; увеличение, а не снижение массы тела от начальной массы тела группы 26,8±0,5 г показывает, что лечение хорошо переносилось. Ежедневные инъекции АС поэтому заметно улучшили выживание и уменьшили размер опухоли, включая полную и частичную регрессии у мышей, несущих гормоннезависимый рак предстательной железы.
ПРИМЕР Q: Эффекты соединений по изобретению на мышиной модели метастазов в печень колоректального рака человека
Основной причиной осложненного течения и летальности у больных с колоректальным раком является метастазирование опухоли в печень; колоректальный рак может часто успешно резецироваться от первичного очага, но метастазы в печень намного менее доступны для хирургического лечения. Мышиную модель метастаза колоректального рака в печень получали, используя клетки аденокарциномы толстой кишки HCT-116, введенные в селезенку безтимусовых (голых) мышей. Раковые клетки HCT-116 спонтанно распространялись от селезенки в печень через кровообращение, и они формируют опухоли в печени (Ishizu, K., Sunose, N., Yamazaki, K., Tsuruo, T., Sadahiro, S., Makuuchi, H., and Yamori, T. Development and Characterization of a Model of Liver Metastasis Using Human Colon Cancer HCT-116, Biol. Pharm. Bull. 2007, 30(9):1779-1783).
Соединения CQ и AA тестировали на противоопухолевую активность на модели HCT-116 метастазирующего колоректального рака.
Методы:
Мышей (самки Hsd:бестимусовые голые-Foxn1nu) анестезировали внутрибрюшинной инъекцией ксилазина/кетамина с последующим разрезом приблизительно 10 мм на левой подреберной области (область дезинфицировали этанолом) для оголения брюшины. Брюшину открывали приблизительно на 8 мм около селезенки, и 2,5×106 клеток в 50 мл PBS инъецировали в селезенку, используя иглу 30G. Селезенку устанавливали на место, и хирургическую область закрывали, используя швы и зажимы.
В день после получения клеток, мышей рандомизировали в группы по пять на основании массы тела, получая группы с приблизительно эквивалентной средней массой тела. Мыши получали один раз в сутки пероральную дозу тестируемого соединения или носителя (1% гидроксипропилметилцеллюлоза), начиная с 48 часов после инъекции клеток в селезенку.
В завершении исследования спустя 28 дней после инъекции клеток HCT-116, регистрировали массу тела, и селезенку и печень удаляли, взвешивали и фиксировали в 10%-ом водном растворе формальдегида. Печень нарезали на секции и окрашивали; относительные области нормальной и опухолевой тканей определяли количественно в гистологических секциях с помощью программного обеспечения для количественной планиметрии.
Результаты:
Опухоли в контрольной группе носителя занимали 14% печени по оценке количественной планиметрией в гистологических секциях. Оба соединения, CQ и АС, заметно уменьшали область печени, в которую происходила инвазия метастазирующих раковых клеток. Группа носителя имела на 12% более высокое отношение масса печени/масса тела, чем группы, получавшие CQ или АС, что подтверждало результаты измерения планиметрии гистологии, указывающие, что опухоли увеличивали полную массу печени в группе носителя. Масса тела не различалась достоверно между группами мышей, получавших носитель или тестируемые соединения, что указывает, что соединения по изобретению хорошо переносились в дозе 240 ммоль/кг/сутки в течение 28 дней.
ПРИМЕР R: Эффекты соединений по изобретению, сорафениба и комбинаций на мышиной модели гепатоцеллюлярного рака человека
Гепатоцеллюлярный рак (HCC) является одной из самых распространенных и летальных форм рака во всем мире, обычно развиваясь как следствие хронической инфекции вирусами гепатита B или C. Ингибитор тирозин киназы сорафениб представляет собой мультикиназный ингибитор, используемый для лечения продвинутого HCC, и имеет как прямые противоопухолевые, так и антиангиогенные свойства. Соединения по изобретению действуют через другие механизм действия, чем сорафениб или другие ингибиторы киназы; поэтому возможно, что соединения по изобретению, в добавление к демонстрации активности единственного средства, могут также усиливать эффективность сорафениба или других стандартных видов лечения в случае HCC и других видов рака.
Линия клетки гепатоцеллюлярного рака Hep3B является человеческой по происхождению, содержит генетические следы вируса гепатита B и может быть введена в печень бестимусных иммунокомпрометированных мышей как модель первичного HCC. Пероральный сорафениб активен в этой модели и использовался и как положительный контроль, и как партнер для комбинированной терапии с выбором соединений по изобретению. Все тестируемые соединения вводили перорально.
Методы:
Тестируемые соединения по изобретению суспендировали в 1% гидроксипропилметилцеллюлозе (HPMC) с использованием соникатора, оборудованного микронаконечником, чтобы минимизировать величину частиц и максимизировать однородность суспензии. Сорафениб растворяли в 1:1 смеси Кремофора EL и этанола, нагревая до 60°C в течение 1 минуты и затем обрабатывая ультразвуком в течение 10 минут, чтобы полностью суспендировать.
Самок голых мыши (Hsd:бестимусные голые-Foxn1nu) весом приблизительно 25 г анестезировали смесью кетамин/ксилазин, оставляли лежать на спине и делали поперечный разрез на 1 см через кожу и брюшину левого верхнего отдела брюшной полости. Срединную долю печени экспонировали, применяя мягкое давление на брюшную полость. 1,5-2×106 клеток Hep3B в объеме 20 мл бессывороточного матригель:EMEM (1:1) медленно имплантировали подсерозной инъекцией в печень, используя иглу 27 калибра на Гамильтонском шприце. Печени давали вернуться на место, и брюшину закрывали швами и раневыми зажимами.
Мышей разделяли на 8 групп мышей после инъекции клеток; группа носитель/носитель включала 12 мышей, и другие группы включали 8 или 9, Мыши начинали получать тестируемые лекарственные средства перорально через 48 ч после инъекции клеток.
Тестируемые соединения, сорафениб и носители вводили пероральным скармливанием. Сорафениб или его кремофор-содержащий носитель давали утром, и соединения по изобретению или их носитель HPMC вводили во второй половине дня каждый день; все животные получали два введения лекарственных средств или подходящих носителей в день. В группе с сорафенибом в качестве единственного активного тестируемого средства, суточная доза составляла 30 мг/кг; в комбинации с соединениями по изобретению доза сорафениба была уменьшена до 20 мг/кг, потому что толерантность комбинации была неизвестна, а также потому что возможная улучшенная противораковая активность соединений по изобретению, скомбинированных с более низкой дозой сорафениба по сравнению с более высокой дозой сорафениба, используемого индивидуально, будет более ясно демонстрировать выгодную активность соединений по изобретению.
Мышей умерщвляли в день 35 после того, как 2 из начальных 12 получавших носитель мышей умерли от прогрессии опухоли; печень удаляли и фотографировали, и опухоли вырезали для измерения и взвешивания.
Результаты
У всех получавших носитель мышей развивались опухоли со средней массой приблизительно 2 грамма в момент умерщвления. Сорафениб (30 мг/кг/день) как единственное средство уменьшил размер опухоли на более чем 50%. Соединения АС и AB индивидуально также уменьшали размер опухоли на более чем 50%; AK индивидуально приводил к численному, но не статистически значимому сокращению размера опухоли по сравнению с носителем. Добавление сорафениба (20 мг/кг/день) к соединениям по изобретению привело к лучшему ингибированию роста опухоли, чем которое было достигнуто с сорафенибом индивидуально 30 мг/кг/день. Комбинации DD или AB с сорафенибом приводили к более полным регрессиям (отсутствие жизнеспособных опухолей, обнаруживаемых во время вскрытия), чем единственное средство. Все лечения, включая комбинации, хорошо переносились, на что указывает поддержание постоянной массы тела на протяжении всего исследования.
ПРИМЕР S: In vitro скрининг противораковой активности против мышиного рака молочной железы 4T1 и рака предстательной железы человека РС-3
Соединения по изобретению скринировали в отношении способности уничтожать или ингибировать пролиферацию линий раковых клеток in vitro, в дополнение к исследованиям in vivo подгруппы соединений, демонстрирующих противораковую эффективность in vivo, в дозах, которые хорошо переносились после перорального или внутрибрюшинного введения.
Противораковую активность против мышиных раковых клеток рака молочной железы 4T1 оценивали in vitro, высевая 1×104 клеток/лунка в плоскодонные культуральные планшеты, затем обрабатывая выбранными концентрациями 1 мкМ или 5 мкМ соединений в течение 18 ч после высевания. Затем добавляли 10 мкл красящего реагента Wst1, индикатора окраски тетразолия для гибели клеток, на лунку и инкубировали приблизительно 2 ч, после чего тестировали на устройстве для считывания микропланшетов Biotek EL800 Universal (450 нм, референсная длина волны 630 нм).
Активность против человеческих раковых клеток предстательной железы PC3 in vitro оценивали подобным же способом.
Раковые клетки предстательной железы РС-3 высевали в количестве 2×104 клеток/лунка в 96-луночные плоскодонные планшеты для культуры тканей и инкубировали в течение приблизительно 20 часов с носителем или тестируемым соединением в концентрациях 0,4, 0,5 или 2,5 мкМ, как указано для конкретных соединений в правом столбце Таблицы 37. Добавляли 1/10 объема краски Wst1/лунка и инкубировали в течение двух часов в термостате для клеточной культуры. Образцы анализировали в трех экземплярах на устройстве для считывания микропланшетов EL800 Universal при 450 нм, референсная длина волны 630 нм. числовые обозначения в Таблице 37 представляют процент выживания раковых клеток относительно обработанных носителем клеток в указанных концентрациях, причем значения ниже 100 указывают на противораковую цитотоксическую активность при протестированных концентрациях лекарственных средств.
ПРИМЕР T: Эффекты соединений по изобретению на резистентность человеческих раковых клеток предстательной железы к цитотоксическим химиотерапевтическим средствам in vitro
Терапия рака осложняется врожденной или приобретенной резистентностью опухоли к отдельным цитотоксическим противораковым средствам. Один механизм резистентности раковых клеток к химиотерапевтическим средствам, таким как антрациклины, соединения платины, алкалоиды барвинка, таксаны и некоторые ингибиторы тирозин киназы, состоит в захвате противораковых средств в лизосомах или родственных кислых вакуолях. Соединения по изобретению тестировали in vitro на их способность увеличить чувствительность к нескольким другим классам противораковых средств, к которым раковые клетки предстательной железы РС-3 являются относительно резистентными in vitro и in vivo.
Раковые клетки предстательной железы РС-3 высевали в количестве 2×104 клеток/лунка в 96-луночные плоскодонные планшеты для культуры и инкубировали приблизительно 20 часов. Клетки обрабатывали с ожидаемой подоптимальной концентрацией тестируемых соединений для уничтожения клеток в качестве единственного средства в течение приблизительно 30 минут. Добавляли химиотерапевтические средства (доксорубицин, оксалиплатин, паклитаксел или винкристин в концентрациях, субоптимальных для уничтожения клеток РС-3), и клетки РС-3 инкубировали в течение дополнительных 72 часов, после чего тестировали с использованием реагента Wst1. Добавляли 1/10 объема краски Wst1/лунка и инкубировали в течение двух часов в термостате для клеточной культуры. Образцы анализировали в трех экземплярах на устройстве для считывания микропланшетов EL800 Universal при 450 нм, референсная длина волны 630 нм.
В Таблице 38, числовые обозначения в столбце, озаглавленном “Без химиотерапевтических средств”, представляют процент выживания клеток после воздействия соединений по изобретению в концентрациях, обозначенных налево от этого столбца. В столбцах, озаглавленных по названиям четырех химиотерапевтических средств, значения ниже соответствующих значений “Без химиотерапевтических средств” указывают лучшую противораковую активность конкретной комбинации цитотоксического средства в комбинации с соединением по изобретению, чем была получена с любым классом соединения индивидуально. В указанных концентрациях минимальная активность химиотерапевтических средств индивидуально в течение 72 часов экспонирования была подвергнута нормализации к 100% для ясности в различении синергических или аддитивных эффектов соединений по изобретению. Результаты показывают, что в протестированных концентрациях широкий диапазон соединений по изобретению увеличивал чувствительность раковых клеток к одному или более протестированных цитотоксических химиотерапевтических средств доксорубицину, оксалиплатину, паклитакселу или винкристину.
Реферат
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы IA1a или его фармацевтически приемлемой соли, где n равно 2-8; p равно 1; q равно 0; Rобозначает водород, и Rобозначает водород; Rобозначает водород; и Rвыбран из группы, состоящей из галогена; алкокси, имеющего от 1 до 10 атомов углерода, незамещенного или замещенного фенилом; или где n равно 0; p равно 0; q равно 1; Rобозначает водород, и Rобозначает водород; Rобозначает водород или галоген; и Rобозначает алкокси, имеющий от 1 до 10 атомов углерода, фенокси или алкокси, имеющий от 1 до 10 атомов углерода, замещенный фенилом. Также изобретение относится к способу лечения, основанному на использовании соединения формулы IА1а, применению соединения формулы IА1а и композиции на его основе. Технический результат: получены новые гетероциклические соединения, полезные при лечении воспалительного заболевания, грибковой инфекции, одноклеточной паразитарной инфекции и опухолевого заболевания. 7 н. и 43 з.п. ф-лы, 38 табл., 171 пр.
Формула

Комментарии