Гетероциклические соединения и фармацевтические композиции как ингибиторы катепсина s - RU2424239C2
Код документа: RU2424239C2
Описание
По настоящей заявке испрашивается приоритет по заявке на патент США 60/566990 от 30 апреля 2004, которая в полном объеме и во всех заявленных целях входит в предлагаемое изобретение в качестве ссылки.
ПРЕДПОСЫЛКА СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Область изобретения
Изобретение описывает соединения, фармацевтические композиции, включающие эти соединения, и методы применения данных соединений для лечения или предупреждения заболеваний или нарушений, опосредованных активностью катепсина S.
Предпосылка
Цистеиновые протеазы представляют собой энзиматический класс белков, которые катализируют гидролиз пептидной связи за счет нуклеофильной сульфгидрильной группы остатка цистеина в активном сайте фермента.
Катепсины являются подклассом цистеиновых протеаз, которые играют главную роль в лизосомальной, эндосомальной и внеклеточной протеиновой деградации и, таким образом, вовлечены во многие процессы заболевания. Было показано, что катепсин S [ЕС 3.4.22.27] нужен для должной презентации антигенов МНС класса II. В результате своей заметной роли в презентации антигенов МНС класса II катепсин S связывают с некоторыми нормальными и болезненными процессами у млекопитающих. Эти заболевания или нарушения включают, но не ограничиваются ими, остеопороз, остеоартрит, мышечную дистрофию, воспаление, инвазию опухоли, гломерулонефрит, малярию, периодонтальное заболевание, метахроматическую лейкодистрофию, периодонтальные заболевания, болезнь Педжета, атеросклероз, рассеянный склероз, ревматоидный артрит, юношеский начальный диабет, волчанку, астму, отторжение ткани, болезнь Альцгеймера, болезнь Паркинсона, нейрональную дегенерацию, шок, рак, невропатическую боль, хроническое обструктивное заболевание легких, воспалительное заболевание кишечника, аллергию, болезнь Шагаса, лейшманиоз, шистосомиаз и Африканскую сонную болезнь (трипаносомоз).
Новые соединения предлагаемого изобретения ингибируют активность катепсина S, и поэтому предполагается, что они будут применимы в лечении болезней, опосредованных катепсином S.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одной части изобретение предлагает соединения формулы I

в которых
R1 выбирают из формул (a) (b) или (с)



где n означает целое число 0, 1 и 2;
R6 и R7 независимо выбирают из водорода, С1-С6алкила, цианоС0-С6алкила, С3-С12циклоалкилС0-С4алкила и С6-С10арилС0-С4алкила; или R6 и R7 вместе с атомом углерода, к которому R6 и R7 присоединены, образуют С3-С8гетероциклоалкил или С3-С12циклоалкил;
при этом любой алкил в R6 и R7 может необязательно иметь метиленовую группу, замененную атомом или группой, выбранной из О или S(O)0-2; при этом любой арил, гетероциклоалкил или циклоалкил в R6 и R7 или образованный комбинацией R6 и R7 может быть необязательно замещен 1-3 радикалами, независимо выбранными из группы: галоид, гидрокси, нитро, циано, C1-С6алкил, C1-С6алкокси, галоидзамещенный C1-С6алкил, галоидзамещенный C1-С6алкокси, -XC(O)OR10, -XS(O)0-2R10, -XNRS(O)0-2R10 и -XS(O)0-2NR10R10; при этом Х означает связь или С1-С4алкилен; и R10 независимо выбран из водорода и C1-С6алкила;
R8 выбирают из водорода, С6-С10арилС0-С4алкила, С5-С10гетероарилС0-С4алкила, -C(O)OR10 и -C(0)NR10R11; при этом R10 выбирают из водорода и С1-С6алкила; и R11 выбирают из водорода, С1-С6алкила и -[CR12R13]m- R14; при этом m выбирают из 0, 1 и 2; R12 и R13 независимо выбирают из водорода и C1-С6алкила; и R14 выбирают из С6-С10арила, С5-С10гетероарила, С3-С12циклоалкила и С3-С8гетероциклоалкила;
при этом любой арил, гетероарил, циклоалкил или гетероциклоалкил в R8 и R14 может быть необязательно замещен 1-3 радикалами, независимо выбранными из группы: галоид, гидрокси, нитро, циано, C1-С6алкил, C1-С6алкокси, галоидзамещенный C1-С6алкил, галоидзамещенный C1-С6алкокси, С6-С10арил, С5-С10гетероарил, С3-С12циклоалкил и С3-С8гетероциклоалкил; и
R9 выбирают из C1-С6алкила, С6-С10арилС0-С4алкила и С3-С12циклоалкилС0-С4алкила;
R2 выбирают из водорода и C1-С6алкила;
R3 и R4 независимо выбирают из водорода, C1-С6алкила, С3-С12циклоалкилС0-С4алкила; и С6-С10арилС0-С4алкила; где любой алкил в R3 и R4 может необязательно иметь метиленовую группу, замененную атомом или группой, выбранной из О и S(O)0-2, при этом любой арил или циклоалкил в R3 и R4 может необязательно быть замещен 1-3 радикалами, независимо выбранными из группы: галоид, гидрокси, нитро, циано, С1-С6алкил, C1-С6алкокси, галоидзамещенный C1-С6алкил и галоидзамещенный C1-С6алкокси;
R5 выбирают из С3-С8гетероциклоалкила и NR12R13, где R12 и R13 независимо выбирают из водорода и C1-С6алкила; при этом любой гетероциклоалкил в R5 может необязательно быть замещен 1-3 радикалами, независимо выбранными из группы: галоид, гидрокси, нитро, циано, C1-С6алкил, C1-С6алкокси, галоидзамещенный С1-С6алкил и галоидзамещенный С1-С6алкокси, XC(O)OR10, -XS(O)0-2R10, -XNR10S(O)0-2R10 и -XS(O)0-2NR10R10; при этом Х означает связь или С1-С4алкилен; и R10 независимо выбирают из водорода и C1-С6алкила;
и их N-оксидные производные, пролекарственные производные, защищенные производные, индивидуальные изомеры и смеси изомеров; а также фармацевтически приемлемые соли и сольваты (например, гидраты) таких соединений.
Второй аспект настоящего изобретения предлагает фармацевтическую композицию, которая содержит соединение формулы I или N-оксидное производное, его индивидуальные изомеры и смеси изомеров и его фармацевтически приемлемую соль в смеси с одним или более наполнителем.
Третий аспект настоящего изобретения предлагает метод лечения заболевания у животного, при котором ингибирование активности катепсина S может предотвращать, ингибировать или ослаблять патологию и/или симптоматику заболевания; этот метод включает введение животному терапевтически эффективного количества соединения формулы I или N-оксидного производного, его индивидуальных изомеров и смеси изомеров и его фармацевтически приемлемой соли.
Четвертый аспект настоящего изобретения предлагает применение соединения формулы I в приготовлении лекарства для лечения заболевания у животного, при котором активность катепсина S вносит вклад в патологию и/или симптоматику заболевания.
В пятом аспекте настоящего изобретения соединения формулы I селективно ингибируют катепсин S относительно катепсинов К, L, В или их комбинаций.
Шестой аспект настоящего изобретения предлагает способ получения соединений формулы I и их N-оксидных производных, пролекарственных производных, защищенных производных, их индивидуальных изомеров и смеси изомеров и их фармацевтически приемлемых солей.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения.
«Алкил» означает группу или структурный элемент других групп, например, галоидзамещенного алкила или алкокси-группы, и может быть или прямоцепным или разветвленным. C1-С6алкокси включает метокси-, этокси-группы и так далее. Галоидзамещенный алкил включает трифторметил, пентафторэтил и так далее.
«Арил» означает моноциклический или сочлененный бициклический ароматический кольцевой ансамбль, включающий шесть-десять атомов углерода в кольце. Арил может означать, например, фенил или нафтил, преимущественно фенил. «Арилен» означает двухвалентный радикал, произведенный из арильной группы. «Гетероарил» означает то же самое, что определено для арила, и при этом один или два атома в кольце являются гетероатомами. Гетероарил включает, например, пиридил, индолил, индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил, бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензимидазолил, пиримидинил, фуранил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, тиенил и так далее. «С6-С10арилС0-С4алкил» означает арил, как было определено выше, присоединенный через алкиленовую группировку. С6-С10арилС0-С4алкил включает, например, фенетил, бензил и так далее. «Циклоалкил» означает насыщенный или частично ненасыщенный моноциклический, сочлененный бициклический, мостиковый полициклический кольцевой ансамбль или спиральный кольцевой ансамбль (где два кольца соединены через общий атом, например, спиро[5.5]ундекан и тому подобное), содержащий указанное число атомов в кольце. Например, С3-С10циклоалкил включает циклопропил, циклобутил, циклопентил, циклогексил и т.д.
«Гетероциклоалкил» означает циклоалкил, как определено выше, в котором один или более указанных атомов кольца заменены группами, выбранными из -O-, -N=, -NR-, -С(O)-, -S-, -S(O)- или -S(O)2-, где R означает водород, С1-С4алкил или азотную защитную группу. Так, например, термин С3-С8гетероциклоалкил, используемый для описания соединений предлагаемого изобретения, включает группы морфолино-, пирролидинил, пиперазинил, пиперидинил, пиперидинилон, 1,4-диокса-8-азаспиро[4.5]дец-8-ил и т.д.
«Галоген» (или гало) предпочтительно представлен хлором или фтором, но может также означать бром или йод.
Термины «лечить», «лечение» относятся к методам уменьшения или смягчения заболевания и/или его сопровождающих симптомов.
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ЧАСТЕЙ ИЗОБРЕТЕНИЯ
Настоящее изобретение предлагает соединения, композиции и методы лечения заболеваний, при которых ингибирование активности катепсина S может предотвращать, ингибировать или смягчать патологию и/или симптоматику заболеваний; метод включает введение животному терапевтически эффективного количества соединения формулы I.
Одна часть изобретения представляет соединения формулы Ia

в которых R1 выбирают из соединений формул (а), (b) или (с)



в которых n означает целое число 0, 1 или 2;
R6 выбирают из водорода, C1-С6алкила, цианоС0-С6алкила, С3-С12циклоалкилС0-C4алкила и С6-С10арилС0-C4алкила;
при этом любой алкил из R6 может необязательно иметь метиленовую группу, замененную атомом или группой, выбранной из О и S(O)0-2; при этом любой арил или циклоалкил из R6 может быть необязательно замещен 1-3 радикалами, независимо выбранными из группы: галоид, гидрокси, нитро, циано, C1-С6алкил, C1-С6алкокси, галоидзамещенный C1-С6алкил и галоидзамещенный C1-С6алкокси, -XC(O)OR10, -XS(O)0-2R10, -XNRS(O)0-2R10 и -XS(O)0-2NR10R10; при этом Х означает связь или С1-С4алкилен; и R10 независимо выбирают из водорода и C1-С6алкила;
R8 выбирают из водорода, С6-С10арилС0-С4алкила, С5-С10гетероарилС0-С4алкила, -C(O)OR10 и -C(O)NR10R11; при этом R10 выбирают из водорода и С1-С6алкила; и R11 выбирают из водорода, C1-С6алкила и -[CR12R13]m-R14; при этом m выбирают из 0, 1 и 2; R12 и R13 независимо выбирают из водорода и С1-С6алкила; и R14 выбирают из С6-С10арила, С5-С10гетероарила, С3-С12циклоалкила и С3-С8гетероциклоалкила;
при этом любой арил, гетероарил, циклоалкил или гетероциклоалкил в R8 и R14 может быть необязательно замещен 1-3 радикалами, независимо выбранными из группы: галоид, гидрокси, нитро, циано, C1-С6алкил, С1-С6алкокси, галоидзамещенный C1-С6алкил, галоидзамещенный C1-С6алкокси, С6-С10арил, С5-С10гетероарил, С3-С12циклоалкил и С3-С8гетероциклоалкил; и
R9 выбирают из C1-С6алкила, С6-С10арилС0-С4алкила и С3-С12циклоалкилС0-С4алкила;
R3 выбирают из водорода, C1-С6алкила, С3-С12циклоалкилС0-С4алкила и С6-С10арилС0-С4алкила; при этом любой алкил из R3 может необязательно иметь метилен, замененный атомом или группой, выбранной из О и S(O)0-2; при этом любой арил или циклоалкил из R3 может быть необязательно замещен 1-3 радикалами, независимо выбранными из группы: галоид, гидрокси, нитро, циано, C1-С6алкил, C1-С6алкокси, галоидзамещенный C1-С6алкил и галоидзамещенный C1-С6алкокси;
R5 выбирают из С3-С8гетероциклоалкила и NR12R13, при этом R12 и R13 независимо выбирают из водорода и С1-С6алкила; при этом любой гетероциклоалкил из R5 может быть необязательно замещен 1-3 радикалами, независимо выбранными из группы: галоид, гидрокси, нитро, циано, C1-С6алкил, C1-С6алкокси, галоидзамещенный C1-С6алкил и галоидзамещенный С1-С6алкокси, -XC(O)OR10, -XS(O)0-2R10, -XNR10S(O)0-2R10 и -XS(O)0-2NR10R10; при этом X означает связь или С1-С4алкилен; и R10 независимо выбирают из водорода и C1-С6алкила.
В другой части изобретения R2 и R4 оба означают водород; R6 выбирают из водорода, C1-С6алкила, цианоС0-С6алкила, С3-С12циклоалкилС0-С4алкила и С6-С10арилС0-С4алкила; при этом любой алкил из R6 может необязательно иметь метиленовую группу, замененную атомом или группой, выбранной из О и S(O)0-2; при этом любой арил или циклоалкил из R6 может быть необязательно замещен 1-3 радикалами, независимо выбранными из галоидов;
R7 означает водород;
R8 означает С5-С10гетероарил, необязательно замещенный 1-3 радикалами, независимо выбранными из группы: галоид, C1-С6алкил, галоидзамещенный C1-С6алкил, С6-С10арил, С5-С10гетероарил, С3-С12циклоалкил и С3-С8гетероциклоалкил; и
R9 означает C1-С6алкил;
R3 выбирают из C1-С6алкила, С3-С12циклоалкилС0-С4алкила и С6-С10арилС0-С4алкила; при этом любой алкил из R3 может необязательно иметь метиленовую группу, замененную атомом или группой, выбранной из О и S(O)0-2;
R5 выбирают из С3-С8гетероциклоалкила и NR12R13, при этом R12 и R13 независимо выбирают из водорода и C1-С6алкила.
В следующей части изобретения R6, выбирают из водорода, метила, этила, пропила, изопропила, циклопропила, цианометила, 2-хлорбензилоксиметила, бензилоксиметила, бензилоксиэтила, фенетила и бензила.
В другой части изобретения R8 означает бензоксазол-2-ил, бензотиазол-2-ил, [1,2,4]оксадиазол-3-ил, [1,2,4]оксадиазол-5-ил и оксазол[5,4-b]пиридин-2-ил; при этом любой гетероарил из R8 необязательно замещен 1-3 радикалами, независимо выбранными из галоида, этила, фенила, циклопропила и трифторметила.
В другой части изобретения R3 выбирают из циклогексилметила, циклопентилметила, бензилсульфонилметила, циклогексилэтила, фенила, изобутила, трет-бутилметила, циклогексила, бензила и фенетила; и R5 выбирают из группы: морфолино, диметиламино, пиперидинил и пирролидинил.
Предпочтительные соединения формулы I детализируются в примерах и в таблице I и отбираются из 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира 2-(бензоксазол-2-ил)-(1S)-циклопропил-2-оксоэтилкарбаминовой кислоты, 2-фенилметансульфонил-(1R)-(морфолин-4-карбонил)этилового эфира 2-(бензоксазол-2-ил)-(1S)-метил-2-оксоэтилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(5-этил[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(5-фенил[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира 4-оксотетрагидрофуран-(3S)-илкарбаминовой кислоты, (S)-1-трет-бутоксикарбонил-4-циано-4-(1-циклогексилметил-2-морфолин-4-ил-2-оксоэтоксикарбониламино)пиперидина, (S)-4-циано-4-(1-циклогексилметил-2-морфолин-4-ил-2-оксоэтоксикарбониламино)-1-метилпиперидина, (1S)-циклогексилметил-2-морфолин-4-ил-2-оксоэтилового эфира 2-(2-хлорбензилокси)-(1R)-цианоэтилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира 2-(бензоксазол-2-ил)-(1S)-метил-2-оксоэтилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(бензоксазол-2-карбонил)бутилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира 2-(5-фторбензоксазол-2-ил)-(1S)-метил-2-оксоэтилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира 2-(6-фторбензоксазол-2-ил)-(1S)-метил-2-оксоэтилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(бензоксазол-2-карбонил)-2-метилпропилкарбаминовой кислоты, 2-циклопентил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(бензотиазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(бензотиазол-2-карбонил)этилкарбаминовой кислоты, 2-фенилметансульфонил-(1R)-(морфолин-4-карбонил)этилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, 3-циклогексил-(1S)-(морфолин-4-карбонил)пропилового эфира (1S)-(бензоксазол-2-карбонил)этилкарбаминовой кислоты, 3-циклогексил-(1S)-(морфолин-4-карбонил)пропилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, (1S)-(морфолин-4-карбонил)-1-фенилметилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, 3,3-диметил-(1S)-(морфолин-4-карбонил)бутилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, (1S)-циклогексил-1-(морфолин-4-карбонил)метилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, (1S)-циклогексилметил-2-охо-2-(пиперидин-1-ил)этилового эфира (1S)-(бензоксазол-2-карбонил)этилкарбаминовой кислоты, (1S)-циклогексилметил-2-оксо-2-(пирролидин-1-ил)этилового эфира 2-(бензоксазол-2-ил)-(1S)-циклопропил-2-оксоэтилкарбаминовой кислоты, (1S)-циклогексилметил-2-оксо-2-(пирролидин-1-ил)этилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, (1S)-циклогексилметил-2-оксо-2-(пирролидин-1-ил)этилового эфира (1S)-(бензоксазол-2-карбонил)бутилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира 4-оксотетрагидрофуран-(3R)-илкарбаминовой кислоты, (1S)-циклогексилметил-2-(морфолин-4-ил)-2-оксоэтилового эфира (1S)-циано-2-метилпропилкарбаминовой кислоты, (1S)-циклогексилметил-2-(морфолин-4-ил)-2-оксоэтилового эфира (1S)-цианопропилкарбаминовой кислоты, (1S)-циклогексилметил-2-(морфолин-4-ил)-2-оксоэтилового эфира 2-бензилокси-(1R)-цианоэтилкарбаминовой кислоты, (1S)-циклогексилметил-2-(морфолин-4-ил)-2-оксоэтилового эфира 3-бензилокси-(1S)-цианопропилкарбаминовой кислоты, (1S)-циклогексилметил-2-(морфолин-4-ил)-2-оксоэтилового эфира (1S)-циано-3-фенилпропилкарбаминовой кислоты, (1S)-циклогексилметил-2-(морфолин-4-ил)-2-оксоэтилового эфира (1S)-2-дицианоэтилкарбаминовой кислоты, (1S)-циклогексилметил-2-(морфолин-4-ил)-2-оксоэтилового эфира (1S)-циано-2-фенилэтилкарбаминовой кислоты, (1S)-циклогексилметил-2-(морфолин-4-ил)-2-оксоэтилового эфира цианометилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(6-фторбензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклопентил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(6-фторбензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(оксазоло[5,4-b]пиридин-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-циклопропил-2-(6-фторбензоксазол-2-ил)-2-оксоэтилкарбаминовой кислоты, (1S)-циклогексил-1-(морфолин-4-карбонил)метилового эфира (1S)-(6-фторбензоксазол-2-карбонил)этилкарбаминовой кислоты, (1S)-циклогексил-1-(морфолин-4-карбонил)метилового эфира (1S)-(6-фторбензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-диметилкарбамоилэтилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(6-фторбензоксазол-2-карбонил)бутилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(5-циклопропил[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты, (1S)-циклогексил-1-(морфолин-4-карбонил)метилового эфира (1S)-(бензоксазол-2-карбонил)бутилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(6-метилбензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(5-хлорбензоксазол-2-карбонил)пропилкарбаминовой кислоты, (1S)-(морфолин-4-карбонил)-2-фенилэтилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, (1S)-(морфолин-4-карбонил)-3-фенилэтилового эфира (1S)-(бензоксазол-2-карбонил)пропилкарбаминовой кислоты, (1S)-циклогексилметил-2-оксо-2-(пирролидин-1-ил)этилового эфира (1S)-(7-фторбензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(5-трифторметил[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(7-фторбензоксазол-2-карбонил)пропилкарбаминовой кислоты, 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(5-фторбензоксазол-2-карбонил)бутилкарбаминовой кислоты; (1S)-циклогексилметил-2-оксо-2-(пирролидин-1-ил)этилового эфира (1S)-(6-фторбензоксазол-2-карбонил)пропилкарбаминовой кислоты; 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1S)-(3-циклопропил[1,2,4]оксадиазол-5-карбонил)пропилкарбаминовой кислоты; 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира (1R)-(5-циклопропил[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты; 3-метил-1-(морфолин-4-карбонил)бутилового эфира (S,S)-[1-(бензоксазол-2-карбонил)пропил]карбаминовой кислоты; 3-метил-1-(морфолин-4-карбонил)бутилового эфира (S,S)-(2-бензоксазол-2-ил-1-метил-2-оксоэтил)карбаминовой кислоты; 3,3-диметил-1-(пирролидин-1-карбонил)бутилового эфира (S,S)-[1-(5-циклопропил[1,2,4]оксадиазол-3-карбонил)пропил]карбаминовой кислоты; 3,3-диметил-1-(морфолин-4-карбонил)бутилового эфира (S,S)-[1-(5-циклопропил[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты.
Фармакология и полезность
Соединения предлагаемого изобретения ингибируют активность катепсина S, и поэтому полезны для лечения заболеваний или расстройств, в которых катепсин S способствует патологии и/или симптоматике заболевания. Предлагаемое изобретение далее представляет соединения этого изобретения для использования в получении лекарств для лечения заболеваний или расстройств, в которых катепсин S вносит вклад в патологию и/или симптоматику заболевания. Опосредованные катепсином S заболевания или состояния включают, но не ограничиваются ими: дистрофию мышц, воспаление, инвазию опухоли, гломерулонефрит, периодонтальное заболевание, невропатическую боль, хроническое обструктивное воспаление легких, воспалительное заболевание кишечника, аллергию, метахроматическую лейкодистрофию, остеопороз, остеоартрит (Inui Т., О. Ishibashi, J. Biol. Chem., 1997, 272, №13, cc.8109-8112; Saftig P., E. Hunziker, и др., Adv. Exp. Med. Biol., 2000 + ADs 2000, 477, cc.293-303; Saftig P., E. Hunziker, и др., Proc. Natl. Acad. Sci. USA, 1998, 95, №23, сс.13453-1358), периодонтальные заболевания, болезнь Педжета, атеросклероз (Jormsjo S., D.М.Wuttge, и др., Am. J. Pathol., 2002, 161, №3, cc.939-945), рассеянный склероз (Beck H., G. Schwarz, и др., Eur. J. Immunol., 2001, 31, №12, сс.3726-3736), ревматоидный артрит (Nakagawa Т.Y., W.H.Brissette, и др., Immunity, 1999, 10, №2, cc.207-217; Hou W.S., Z.Li, и др., Am. J. Pathol., 2001, 159, №6, сс.2167-2177), юношеский начальный диабет, волчанку, астму (Cimerman N., Р.М.Brguljan, и др., Pflugers Arch., 2001, 442, №6, Suppl. 1, сс.204-206), отторжение ткани, болезнь Альцгеймера (Lemere С.A., J.S.Munger, и др., Am. J. Pathol., 1995, 146, №4, сс.848-860), болезнь Паркинсона (Liu Y., L. Fallon, и др., Cell, 2002, 111, №2, сс.209-18), нейрональную дегенерацию, шок (Jaeschke H., М.A.Fisher, и др., J. Immunol., 1998, 160, №7, сс.3480-3486), рак (Fernandez P.L., X.Farre, и др., Int. J. Cancer, 2001, 95, №1, сс.51-55), малярию (Malhotra Р., Р.V.Dasaradhi, и др., Mol. Microbiol., 2002, 45, №5, cc.1245-1254), болезнь Шагаса (Eakin A.E., A.A.Mills, и др., J. Biol. Chem., 1992, 267, №11, сс.7411-20), лейшманиоз, шистосомиаз и Африканскую сонную болезнь (трипаносомоз) (Caffrey С.R., S.Scory, и др., Curr. Drug Targets, 2000, 1, №2, сс.155-162; Lalmanach G., A.Boulange, и др., Biol. Chem., 2002, 383, №5, сс.739-749).
Как упоминалось выше, настоящее изобретение предлагает метод предупреждения или лечения любого заболевания или нарушения, описанного выше, у пациента, нуждающегося в таком лечении; данный метод включает введение указанному пациенту терапевтически эффективного количества (см. ниже раздел «Введение и фармацевтические композиции») соединения формулы I или его фармацевтически приемлемой соли. Для любого случая применения, указанного выше, требуемая доза варьируется в зависимости от способа введения, индивидуального состояния пациента и желательного эффекта.
Введение и фармацевтические композиции
В общем случае, соединения предлагаемого изобретения вводятся в терапевтически эффективных количествах любым обычным и приемлемым способом, известным в этой области, или сами по себе или в комбинации с одним или более терапевтическими агентами. Терапевтически эффективное количество может варьироваться в широких пределах в зависимости от остроты заболевания, возраста и общего здоровья пациента, возможностей используемого соединения и других факторов. В общем случае удовлетворительные результаты достигаются систематически при дневных дозах от примерно 0,03 до 10,0 мг/кг веса тела. Показанная дневная доза для крупных млекопитающих, например, человека, находится в интервале от примерно 0,5 мг до примерно 100 мг, вводимых обычным путем, например в дробных дозах до четырех раз в день, или в форме продленного действия. Подходящие разовые дозовые формы для орального введения содержат от примерно 1 до 50 мг активного компонента.
Соединения предлагаемого изобретения могут быть введены в виде фармацевтических композиций любым подходящим способом, в частности энтерально, например, орально, например, в форме таблеток или капсул, или парэнтерально, например, в форме растворов или суспензий для инъекций, топикально, например, в форме примочек, гелей, мазей или кремов, или в нос, или в форме суппозитория. Фармацевтические композиции, содержащие соединение предлагаемого изобретения в свободной форме или в форме фармацевтически приемлемой соли, в ассоциации с по крайней мере одним фармацевтически приемлемым носителем или разбавителем могут быть получены подходящим способом смешения, гранулирования или покрытия. Оральные композиции, например, могут быть таблетками или желатиновыми капсулами, содержащими активные ингредиент вместе с а) разбавителями, например, лактозой, декстрозой, сахарозой, маннитолом, сорбитолом, целлюлозой и/или глицином; б) лубрикантами, например, кремнием, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток также с с) связующими, например, магнийалюминийсиликатом, крахмальной пастой, желатином, астрагалом, метилцеллюлозой, натрий карбоксиметилцеллюлозой и/или поливинилпирролидоном; если нужно с д) дезинтегрирующими веществами, например крахмалами, агаром, альгиновой кислотой или ее натриевой солью, или шипучими смесями; и/или с е) абсорбентами, красителями, отдушками и умягчителями. Композициями для инъекций могут быть водные изотонические растворы или суспензии, и суппозитории могут быть получены из жировых эмульсий или суспензий. Композии могут быть стерилизованы и/или содержать вспомогательные вещества, такие как консервирующие, стабилизирующие, увлажняющие или эмульгирующие агенты, активаторы раствора, соли для регулирования осмотического давления и/или буферы. Кроме того, они могут также содержать другие терапевтически подходящие вещества. Подходящие композиции для чрескожных аппликаций включают эффективное количество соединения предлагаемого изобретения с носителем. Носитель может включать абсорбируемые фармакологически приемлемые растворители, способствующие проникновению через кожу хозяина. Например, устройство для чрескожного введения существует в форме бандажа, содержащего деталь поддержки, резервуар, содержащий соединение необязательно с носителем, необязательно устройство, контролирующее скорость прохождения соединения к коже хозяина с контролируемой и предопределенной скоростью в течение пролонгированного отрезка времени, и способы крепления устройства к коже. Матриксные формы чрескожного введения также могут применяться. Подходящими формами для топикального применения, например, на кожу и глаза, предпочтительно являются водные растворы, мази, кремы или гели, хорошо известные специалистам. Они могут содержать солюбилизаторы, стабилизаторы, агенты, усиливающие тонус, буферы и консерванты.
Соединения предлагаемого изобретения могут вводиться в терапевтически эффективных количествах в комбинации с одним или более терапевтическими агентами (фармацевтические комбинации). Например, синергетические эффекты могут быть достигнуты с другими веществами, используемыми при лечении остеопороза, остеоартрита, мускульной дистрофии, воспаления, инвазии опухоли, гломерулонефрита, малярии, периодонтального заболевания, метахроматической лейкодистрофии, периодонтальных заболеваний, болезни Педжета, атеросклероза, рассеянного склероза, ревматоидного артрита, юношеского начального диабета, волчанки, астмы, отторжения ткани, болезни Альцгеймера, болезни Паркинсона, нейродегенерации, шока, рака, малярии, невропатической боли, хронического обструктивного воспаления легких, воспалительного заболевания кишечника, аллергии, болезни Шагаса, лейшманиоза, шистозомиаза, и/или африканского трипаносомоза (сонная болезнь). В тех случаях, когда соединения предлагаемого изобретения вводятся совместно с другими терапевтическими средствами, дозы совместно вводимых соединений, конечно, могут варьироваться в зависимости от типа совместно применяемых лекарств, от специфики применяемого лекарства, от условий применения и так далее.
Изобретение также предлагает фармацевтические комбинации, например, набор, содержащий а) первый агент, являющийся соединением предлагаемого изобретения, как здесь раскрывается, в свободной форме или в форме фармацевтически приемлемой соли, и б) по крайней мере один со-агент. Набор может содержать инструкции для его введения.
Термины «со-введение» или «совместное введение» или подобное, как используется здесь, означают введение выбранных терапевтических агентов отдельному пациенту и применение режимов лечения, при которых агенты вводят необязательно одним и тем же способом введения или не обязательно в одно и то же время.
Термин «фармацевтическая комбинация», как здесь используется, означает продукт, который получается из смешения или комбинации более чем одного активного ингредиента, и включает обе - фиксированную и нефиксированную комбинации активных ингредиентов. Термин «фиксированная комбинация» означает, что активные ингредиенты, например соединение формулы I и со-агент, вводят пациенту одновременно в форме одной дозы. Термин «нефиксированная комбинация» означает, что активные ингредиенты, например соединение формулы I и со-агент, вводят пациенту как отдельные агенты или одновременно, или последовательно без специфического ограничения времени, при этом такое введение обеспечивает терапевтически эффективные уровни обоих соединений в теле пациента. Последний также предлагает «коктейль»-терапию, например введение 3 или более активных ингредиентов.
Способы получения соединений предлагаемого изобретения
Предлагаемое изобретение включает также способы получения соединений изобретения. В описанных реакциях может возникнуть необходимость в защите реакционных функциональных групп, например, гидрокси-, амино-, имино-, тио- или карбоксильной группы, желательных в конечном продукте, для того, чтобы избежать их нежелательного участия в реакциях. Для этого могут быть использованы обычные защитные группы, стандартные в практике синтезов, см., например, T.W.Greene и P.G.M.Wuts в "Protective Groups in Organic Chemistry", John Wiley and Sons, 1991.
Следующие схемы иллюстрируют некоторые способы получения соединений предлагаемого изобретения. Специалистам должно быть понятно, что эти способы являются лишь презентативными и ни в коем случае не охватывают все способы получения соединений предлагаемого изобретения. Радикалы в схемах описаны в формуле I.
Схема 1

На схеме 1 проиллюстрирован синтез соединений предлагаемого изобретения, в которых R1 означает -CR7R6C(=O)R8, a R2, R4 и R7 означают водород. W.Kelly и др., Org. Lett., 2004, 6, с.497, приводят обзор различных способов синтеза α-гидроксикислот. α-Гидроксикислоту 1а вводят в реакцию с амином R5H в обычных условиях образования амида (например, DIC/HOBt, HATU, РуВОР и т.д., DIC - ?, HOBt - 1-гидроксибензотриазол, HATU - гексафторфосфат о-(7-азобензотриазол-1-ил)-1,1,3,3-тетраметилурония, PyBOP - гексафторфосфат бензотриазол-1-ил-оксотрипирролидинфосфония), получая соединение 1b. Обзоры реакций сочетания амидов приведены в Chamberlin и др., Chem. Rev., 1997, 97, с.2243 и M.Bodanszky и др., The practice of peptide synthesis, 2-е изд., Springer-Verlag 1994. Соединение 1b обрабатывают п-нитрофенилхлорформиатом в среде основания, получая смешанный карбонат 1с. Реакция 1с с NH2CHR6CH(OH)R8 приводит к карбамату 1е, который окисляют до 1f в стандартных условиях окисления. Предпочтительные способы окисления вторичных спиртов до соответствующих кетонов включают, но не ограничиваются ими, периодинан Десса-Мартина (D.В.Dess и др., J. Am. Chem. Soc., 1991, 113, с.7277 и J. Org. Chem., 1983, 48, c.4155), окисление по Шверну и его модификации (D.Swern и др., J. Org. Chem., 1978, 43, с.2480; Т.Т.Tidwell, Org. React., 1990, 39, c.297; M. Hudlicky, Oxidations in Organic Chemistry; ACS: Вашингтон, 1990), PCC (E.J.Corey и др., Tetrahedron Lett, 1975, c.2647; G. Piancatelli, Synthesis, 1982, c.245), PDC (E.J.Corey и др., Tetrahedron Lett., 1979, с.399) и катализируемое ТРАР (перрутенат тетра-н-пропиламмония) окисление (S.V.Ley и др., Synthesis, 1994, с.639).
Схема 2

Получение вицинального аминоспирта NH2CHR6CH(OH)R8, в котором R8 означает необязательно замещенный 0-3 Х бензоксазол, где Х означает заместители в R8, как указано в п.1, приведено в качестве примера на схеме 2. N-замещенную аминокислоту восстанавливают, используя или ВН3, или восстановление ее смешанного ангидрида с изобутилхлорформиатом действием NaBH4 [см. R.С.Larock, A guide to functional group preparations, cc.548-552, Wiley-VCH, 1989] для получения соединения 2b (Схема 2). Полученный спирт 2b может затем быть окислен до альдегида 2с. Предпочтительные способы окисления спиртов до соответствующих альдегидов включают, но не ограничиваются ими, периодинан Десса-Мартина (D.В.Dess и др., J. Am. Chem. Soc., 1991, 113, с.7277 и J. Org. Chem., 1983, 48, c.4155), окисление по Шверну и его модификации (D.Swern и др., J. Org. Chem., 1978, 43, с.2480; Т.Т.Tidwell, Org. React, 1990, 39, c.297; M.Hudlicky, Oxidations in Organic Chemistry; ACS: Вашингтон, 1990), ТЕМРО/трихлоризоциануровая кислота (TEMPO - 2,2,6,6-тетраметил-1-пиперидинилокси) (L. De Luca и др., Org. Lett, 2001, 3, с.3041), ТРАР катализируемое окисление (S.V.Ley и др., Synthesis, 1994, с.639). Добавление реагента Гриньяра, полученного из бензоксазола, к соединению 2-с приводит к соединению 2-d, которое после снятия защиты дает аминоспирт 2-е.
Предпочтительным способом получения замещенных бензоксазолов, используемых в предлагаемом изобретении и не являющихся коммерческими продуктами, является циклоконденсация о-аминофенола с триметилортоформиатом. Смотри ссылки A.R.Katritzky и др., Heterocycles, 1995, 41, с.345; J.H.Musser и др., J. Med. Chem., 1985, 28, c.1255; и K.R.Kunz и др., OPPI, 1990, 22, с.613.
Схема 3

Альтернативно, аминоспирт 2-d может быть получен последовательными реакциями, описанными в схеме 3 (смотри М.Е.McGrath и др., Biochemistry, 2003, 42, с.15018). Аминоальдегид 2-с превращают в циангидрин 3-a (С.Н.Heathcock и др., Org. Synth. Coll., т.7, с.381), который затем обрабатывают безводным этанолом и хлористым ацетилом, получая эфир имидата 3-b. Конденсация 3-b с о-аминофенолом приводит к аминоспирту 2-d.
Схема 4

Получение вицинального аминоспиртаtBOCNHCHR6CH(ОН)R8, где R8 означает 1,2,4-оксадиазол, необязательно замещенный 0-1 X, где Х означает заместители у R8, как определено в п.1, показано на схеме 4. Гидроксиламин добавляют к циангидрину 3-а в основной среде, получая гидроксиамидин 4-а, который ацилируют с помощью ХСООН (или соответствующего ей ацилхлорида или ангидрида), получая 4-b, 4-b нагревают в микроволновой печи, где он циклизуется в оксадиазол 4-c.
Схема 5

Иллюстрация синтеза соединений предлагаемого изобретения, в которых R1 означает -CHR6CN, и R2 и R4 оба означают водород, дана на схеме 5. α-Аминоамид 5-а добавляют к смешанному карбонату 1-c. Продукт 5-b дегидратируют цианурхлоридом, получая 5-с. Для детальных обсуждений получения коммерчески недоступных α-аминоамидов и их превращений в соответствующие α-аминонитрилы смотри Y.D.Ward и др., J. Med. Chem., 2002, 45, c.5471; P.D.Greenspan и др., J. Med. Chem., 2001, 44, с.4524.
Дополнительные способы получения соединений предлагаемого изобретения.
Соединение предлагаемого изобретения может быть получено в форме фармацевтически приемлемой кислотно-аддитивной соли реакцией свободной основной формы соединения с фармацевтически приемлемой неорганической или органической кислотой. Альтернативно, фармацевтически приемлемые основно-аддитивные соли соединения предлагаемого изобретения могут быть получены реакцией свободной кислотной формы соединения с фармацевтически приемлемым неорганическим или органическим основанием. Альтернативно, соединения предлагаемого изобретения в форме соли могут быть получены с использованием солей исходных материалов или промежуточных продуктов.
Свободные кислотные или основные формы соединений предлагаемого изобретения могут быть получены из соответствующей основно-аддитивной соли или кислотно-аддитивной соли, соответственно. Например, соединение предлагаемого изобретения в форме кислотно-аддитивной соли может быть превращено в соответствующую свободную основную форму обработкой подходящим основанием (например, раствором гидроксида аммония, гидроксида натрия и подобное). Соединение предлагаемого изобретения в форме основно-аддитивной соли может быть превращено в соответствующую кислотную свободную форму обработкой подходящей кислотой (например, хлористоводородной кислотой и т.д.).
Соединения предлагаемого изобретения в неокисленной форме могут быть получены из N-оксидов соединений предлагаемого изобретения обработкой их восстанавливающим агентом (например, серой, двуокисью серы, трифенилфосфином, борогидридом лития, борогидридом натрия, треххлористым фосфором, трехбромистым фосфором или подобными) в подходящем инертном органическом растворителе (например, ацетонитриле, этаноле, водном диоксане или подобных растворителях) при температуре от 0°С до 80°С.
Пролекарственные производные соединений предлагаемого изобретения могут быть получены способами, известными специалистам в этой области (для уточнения деталей смотри, например, Saulnier и др., Bioorganic and Medicinal Chemistry Letters, 1994, 4, с.1985). Например, подходящие пролекарства могут быть получены реакцией недериватизированного соединения предлагаемого изобретения с подходящим карбамилирующим агентом (например, 1,1-ацилоксиалкилкарбанохлоридатом, пара-нитрофенилкарбонатом или подобными).
Защищенные производные соединений предлагаемого изобретения могут быть получены способами, известными специалистам в этой области. Детальное описание техники, применяемой для введения защитных групп и их удаления, можно найти в Т.W.Greene, "Protecting Groups in Organic Chemistry", 3-е изд., John Wiley and Sons, Inc., 1999.
Соединения предлагаемого изобретения могут быть легко получены или могут образоваться во время процессов, описанных в предлагаемом изобретении, в виде сольватов (например, гидратов). Гидраты соединений предлагаемого изобретения могут быть легко получены при перекристаллизации из смеси вода/органический растворитель при использовании таких органических растворителей, как диоксан, тетрагидрофуран или метанол.
Соединения предлагаемого изобретения могут быть получены в виде их индивидуальных стереоизомеров реакцией рацемической смеси соединения с оптически активным разделяющим агентом для образования пары диастереомерных соединений, разделением диастереомеров и выделением оптически чистых энантиомеров. Поскольку разделение энантиомеров может быть проведено с использованием ковалентных диастереомерных производных соединений предлагаемого изобретения, предпочтительными являются диссоциирующие комплексы (например, кристаллические диастереомерные соли). Диастереомеры различаются по физическим свойствам (например, температурам плавления, температурам кипения, растворимости, реакционной способности и т.д.) и, обладая такими различиями, могут быть легко разделены. Диастереомеры могут быть разделены хроматографически или, предпочтительно, техникой разделения, основанной на различиях в растворимости. Оптически чистый энантиомер затем выделяют вместе с разделяющим агентом практическими способами, которые не приводят к рацемизации. Более детальное описание техники, пригодной к выделению стереоизомеров соединений из их рацемической смеси, можно найти в Jean Jacques, Andre Collet, Samuel H. Wilen "Enantiomers, Racemates and Resolutions", John Wiley и Sons, Inc., 1981.
Суммируя вышесказанное, можно сказать, что соединения формулы I могут быть приготовлены по схеме, включающей:
(a) реакцию, указанную на схемах 1, 2, 3, 4 и 5 и
(b) необязательное превращение соединения предлагаемого изобретения в фармацевтически приемлемую соль;
(c) необязательное превращение солевой формы соединения предлагаемого изобретения в не-солевую форму;
(d) необязательное превращение неокисленной формы соединения предлагаемого изобретения в фармацевтически приемлемый N-оксид;
(e) необязательное превращение N-оксидной формы соединения предлагаемого изобретения в его неокисленную форму;
(f) необязательное выделение индивидуального изомера соединения предлагаемого изобретения из смеси изомеров;
(g) необязательное превращение недериватизированного соединения предлагаемого изобретения в фармацевтически приемлемое пролекарственное производное; и
(h) необязательное превращение пролекарственного производного соединения предлагаемого изобретения в недериватизированную форму.
Там, где приготовление исходных материалов не описано специально, соединения являются хорошо известными веществами или могут быть получены аналогично известным методам или в соответствии с описанными здесь в Примерах.
Специалистам понятно, что приведенные выше превращения являются только презентативными способами приготовления соединений предлагаемого изобретения, и могут быть также использованы другие хорошо известные методы.
Примеры
Предлагаемое изобретение включает дальше примеры, которые не лимитируют весь объем, а иллюстрируют получение интермедиатов (указание 1) и соединений предлагаемого изобретения (указание 2).
Указание 1
Получение α-гидроксикислоты через диазотирование хиральной аминокислоты (презентативная процедура)
К перемешиваемой суспензии L-циклогексилаланина (4,00 г, 23,4 ммоль) в 0,5М H2SO4 (120 мл) при 0°С медленно по каплям добавляют водный раствор NaNO2 (12,1 г в 40 мл Н2О). Добавление заканчивают примерно через 1 час, и раствору дают нагреться до комнатной температуры. Через 16 час реакционную смесь экстрагируют эфиром (3×100 мл), и объединенные органические экстракты промывают 1М NaHSO4 (1×200 мл), насыщенным солевым раствором (1×100) и затем сушат над безводным Na2SO4. Растворитель удаляют в вакууме, и сырой продукт кристаллизуют из смеси Et2O/пентан (10 мл/100 мл), получая 2,1 г (52% выход) (S)-2-гидрокси-3-циклогексилпропионовой кислоты в виде блестящих белых игл.
Пример 1
2-Циклогексил-(1S)-(морфолин-4-карбонил)этиловый эфир 2-(бензоксазол-2-ил)-(1S)-циклопропил-2-оксоэтилкарбаминовой кислоты
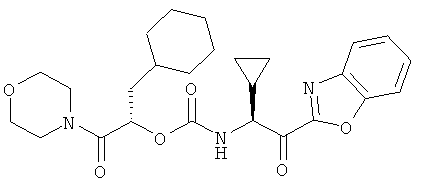
Схема 6

Стадия А: (S)-3-фенилмолочную кислоту (1,21 г, 126 ммоль) растворяют в дихлорметане (300 мл) и обрабатывают морфолином (55 г, 631 ммоль). Реакцию охлаждают на бане лед/вода и по каплям добавляют раствор PyBOP (72 г, 139 ммоль) в дихлорметане (200 мл) через уравнивающую давление воронку. Реакционную смесь перемешивают в течение ночи и оставляют нагреться до комнатной температуры. После добавления 110 мл 4М HCl реакционную смесь фильтруют, и отделяют органическую фазу. Водный слой дважды экстрагируют дихлорметаном. Объединенные органические растворы высушивают над MgSO4, и удаляют растворитель. Полученное вещество хроматографируют на силикагеле и элюируют этилацетатом, получая 20,65 г (68% выход) (S)-2-гидрокси-1-морфолин-4-ил-3-фенилпропан-1-она (2) в виде масла; ВЭЖХ-МС, рассчитанный для C13H17NO3 (М+Н+) 236,1; найденный 236,4.
Стадия В. Раствор соединения 2 (20,5 г, 87,1 ммоль) в смеси этанола (80 мл), воды (10 мл) и уксусной кислоты (10 мл) помещают в баллон Парра и обрабатывают 2 г 10% Rh/C. Баллон закручивают, вакуумируют и заполняют водородом до давления 1000 psi. Затем реакционную смесь нагревают при перемешивании при 50°С в течение ночи. После охлаждения до комнатной температуры и сброса давления баллон откачивают и снова дважды заполняют азотом. Реакционную смесь фильтруют через слой целита, и растворитель удаляют, выделяя 18,6 г (88,5%) (S)-3-циклогексил-2-гидрокси-1-морфолин-4-илпропан-1-она (3) в виде масла; ВЭЖХ-МС вычислено для C13H23NO3 (М+Н+) 242,2, найдено 242,4.
Стадия С. Раствор 4-нитрофенилхлорформиата (20,55 г, 102 ммоль) в диоксане (150 мл) обрабатывают пиридином (50 мл), получая суспензию, которую нагревают до 70°С, после чего в течение ~5 мин добавляют через капельную воронку с противодавлением раствор соединения 3 (12,30 г, 51,0 ммоль) в диоксане (50 мл). После перемешивания в течение еще 5 мин реакции дают охладиться до комнатной температуры, и растворитель удаляют упариванием на роторе. Оставшуюся массу распределяют между этилацетатом и водой. Органический слой собирают, водную фазу еще раз экстрагируют этилацетатом и удаляют. Объединенные органические слои высушивают над MgSO4, и растворитель удаляют. Полученный твердый остаток растворяют в минимальном количестве горячего дихлорэтана и обрабатывают дихлорметаном. Затем смесь помещают на ночь в холодильник при -4°С, затем фильтруют. Маточную жидкость концентрируют и очищают, используя 330 г силикагеля при градиенте этилацетата в гексане от 0 до 100%, выделяя 15,2 г (выход 73%) (S)-2-циклогексил-1-(морфолин-4-карбонил)этилового эфира 4-нитрофенилкарбоновой кислоты (4) в виде слегка желтоватого масла;1Н ЯМР (CDCl3, 400 МГц) δ 0,97-1,08 (м, 2Н), 1,09-1,36 (м, 3Н), 1,48-4,62 (м, 2Н), 1,65-1,68 (м, 4Н), 1,82-1,97 (м, 2Н), 3,41-3,60 (м, 3Н), 3,64-3,80 (м, 5Н), 5,29-5,36 (м, 1Н), 7,38-7,44 (м, 2Н), 8,24-8,30 (М, 2Н); ВЭЖХ-МС вычисленный для C20H26N2O7 (М+H+) 407,2, найденный 407,4.
Схема 7

Бензиловый эфир (S)-(1-циклопропил-2-гидроксиэтил)карбаминовой кислоты (5), указанный в схеме 7, получают следующим образом:
(i) Образец (S)-фенетил-(S)-циклопропилглицина (16,8 г, 76,7 ммоль, получен из циклопропанкарбоксальдегида, цианида калия и (S)-(-)-α-метилбензиламина, используя модифицированную методику, описанную в Daniel J. Bayson и др., патент США 6191306) обрабатывают ТГФ (200 мл), водой (100 мл) и 10% Pd/C (4,76 г). К перемешиваемой смеси добавляют муравьиную кислоту (17 мл) и перемешивают в течение ночи. Катализатор затем удаляют фильтрованием через слой целита, и растворитель удаляют упариванием на роторе. Остаток несколько раз обрабатывают метанолом, который упаривают, и остаток высушивают в вакууме, выделяя 4,75 г (выход 54%) (S)-аминоциклопропилуксусной кислоты в виде твердого вещества, используемого без дальнейшей очистки.
(ii) Продукт из предыдущей стадии (4,75 г, 41 ммоль) растворяют в 130 мл 1н. NaOH и обрабатывают при интенсивном перемешивании бензилхлорформиатом (5,92 г, 49,5 ммоль). Реакционную смесь перемешивают в течение ночи, затем дважды экстрагируют дихлорметаном. Органические слои удаляют, водную фазу подкисляют концентрированной HCl и трижды экстрагируют дихлорметаном. Объединенные органические слои высушивают над MgSO4, и растворитель удаляют, выделяя 7,38 г (выход 72%) (S)-бензилоксикарбониламиноциклопропилуксусной кислоты в виде белого твердого вещества.
(iii) Раствор (S)-бензилоксикарбониламиноциклопропилуксусной кислоты (3,2 г, 12,8 ммоль) в ТГФ (20 мл) охлаждают в бане лед/вода и обрабатывают 1М раствором ВН3 в ТГФ (16,7 мл, 16,7 ммоль). Реакционную смесь перемешивают 4 час и затем обрабатывают 1М HCl до прекращения выделения пузырьков. Смесь перемешивают в течение ночи, после чего органический растворитель упаривают на роторе. Остаток обрабатывают этилацетатом и переносят в делительную воронку. Водную фазу отбрасывают, органические фазы дважды промывают 1М NaOH, сушат над MgSO4, и растворитель удаляют. Остаток очищают, пропуская через силикагель, используя этилацетат в гексане с градиентом от 0 до 100%, и выделяют 1,5 г (выход 50%) спирта в виде белого твердого вещества.1Н ЯМР (CDCl3, 400 МГц) δ 0,26-0,37 (м, 1Н), 0,34-0,44 (м, 1Н), 0,47-0,61 (м, 2Н), 0,83-0,94 (м, 1Н), 2,95-3,04 (м, 1Н), 3,70 (дд 1Н, J1=5,8, h=11,1), 3,79-3,88 (м, 1Н), 5,00-5,12 (м, 1Н), 5,10 (с, 2Н), 7,29-7,31 (м, 5Н); ВЭЖХ-МС вычислено для C13H17NO3 (М+H+) 236,1, найдено 236,3.
Синтез 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира 2-(бензоксазол-2-ил)-(1S)-циклопропил-2-оксоэтилкарбаминовой кислоты (Схема 7).
Стадия А. Превращение проводят в соответствии с процедурами, описанными в М.Graupe и др., WO 02098850, ссылка на Пример 17(а), за исключением того, что применяют периодинан Десса-Мартина для превращения соединения 5 в соответствующий ему альдегид, получая бензиловый эфир 1-(S)-циклопропил-2-(бензоксазол-2-ил)-2-гидроксиэтилкарбаминовой кислоты (6) с общим выходом 45%; ВЭЖХ-МС вычислено для C20H20N2O4 (M+H+) 353,1; найдено 353,4.
Стадии В, С и D. Раствор соединения 6 (269 мг, 0,763 ммоль) в метаноле (8 мл) обрабатывают 20% Pd(OH)2 на угле (34 мг). Заменяют атмосферу реакции на водород, впрыскивая 3 мин раствор с помощью длинного шприца, и затем реакционную смесь перемешивают 2 часа под давлением 1 атмосферы водорода. Заменяют атмосферу вновь на азот, опять впрыскивая раствор через длинный шприц в течение 3 мин. Реакционную смесь фильтруют через целит и удаляют растворитель. Полученное масло обрабатывают раствором соединения 4 (310 мг, 0,763 ммоль) в этилацетате, и растворитель удаляют. Смесь растворяют затем в изопропиловом спирте (10 мл) и обрабатывают диизопропилэтиламином (148 мг, 1,15 ммоль). Реакцию выдерживают при температуре 50°С в течение ночи. Летучие вещества удаляют в вакууме и полученный остаток растворяют в этилацетате и промывают 5% раствором NaHSO4. Органические фазы высушивают над MgSO4 и растворитель удаляют. Полученное масло растворяют в дихлорметане (10 мл) и обрабатывают периодинаном Десса-Мартина (937 мг, 2,21 ммоль). После перемешивания в течение ночи реакцию обрабатывают насыщенным водным раствором NaHCO3 (~15 мл) и 1М Na2S2O3 (~15 мл) и перемешивают 20 мин. Смесь затем переносят в делительную воронку и отбирают органический слой. Водный слой дважды экстрагируют дихлорметаном и отбрасывают. Объединенные органические вытяжки сушат над MgSO4 и удаляют растворитель. Остаток очищают на силикагеле, используя градиент от 0 до 100% этилацетата в гексане, получая 114 мг (31%) 2-циклогексил-(1S)-(морфолин-4-карбонил)этилового эфира 2-(бензоксазол-2-ил)-(1S)-циклопропил-2-оксоэтилкарбаминовой кислоты в виде твердого вещества после лиофилизации.1Н ЯМР (CDCl3, 400 МГц) δ 0,51-0,65 (м, 3Н), 0,73-1,00 (м, 3Н), 1,04-1,29 (м, 4Н), 1,35-1,47 (м, 2Н), 1,58-1,84 (м, 6Н), 3,37-3,76 (м, 8Н), 4,88-4,93 (м, 1Н), 5,29 (дд 1Н, J1=2,7, h=10,1), 5,82 (д, 1Н, J=7,1), 7,44-7,50 (м, 1Н), 7,52-7,58 (м, 1Н), 7,67(д, 1Н, J=8,3), 7,90 (д, 1Н, J=8,0); ВЭЖХ-МС вычислено для C26H33N3O6 (М+H+) 484,2, найдено 484,5.
Пример 2
2-Фенилметансульфонил-(1R)-(морфолин-4-карбонил)этиловый эфир 2-(бензоксазол-2-ил)-(1S)-метил-2-оксоэтилкарбаминовой кислоты
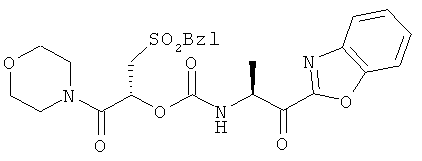
Схема 8

Стадия А: Эту реакцию проводят, как описано в Deechongkit S.; You S.-L.; Kelly J. W., Org. Lett, 2004, 6, с.497, используя (S)-метиловый эфир глициновой кислоты 1 и бензилмеркаптан. Метиловый эфир (R)-3-бензилсульфанил-2-гидроксипропионовой кислоты 2 (7,41 г, 31,41 ммоль, 92%) выделяют в виде вязкого масла: МС вычислено для C11H14O3S (М+Н+) 227,1, найдено 227,3.
Стадия В: Эту реакцию проводят, как ранее описано в Deechongkit S.; You S.-L.; Kelly J. W., Org. Lett., 2004, 6, с.497, используя метиловый эфир (R)-3-бензилсульфанил-2-гидроксипропионовой кислоты 2 и гидроксид лития. (R)-3-бензилсульфанил-2-гидроксипропионовую кислоту 3 (3,08 г, 14,51 ммоль, 46%) выделяют в виде вязкого масла: МС вычислено для C10H12O3S (М+Na+) 235,1, найдено 235,3.
Стадия С: Эту реакцию проводят, как описано в примере 1, используя (R)-3-бензилсульфанил-2-гидроксипропионовую кислоту 3. (R)-3-бензилсульфанил-2-гидрокси-1-морфолин-4-илпропан-1-он 4 (3,41 г, 11,87 ммоль, 67%) выделяют в виде вязкого масла: МС вычислено для C14H19NO3S (М+Н+) 282,1, найдено 282,4.
Стадия D: Оксон (2KHSO5×KHSO4×K2SO4, 10,55 г, 17,17 ммоль, 3,0 экв) растворяют в H2O (25 мл, 0,7 М) и добавляют к раствору (R)-3-бензилсульфанил-2-гидрокси-1-морфолин-4-илпропан-1-она 4 (1,61 г, 5,73 ммоль, 1,0 экв) в МеОН (25 мл, 0,3 М) в течение 30 мин при 0°С. Мониторинг реакции проводят совместно методами ЖХ/МС. После того, как реакция проходит полностью (около 12 час), МеОН удаляют в вакууме. Полученный раствор разбавляют водой (30 мл) и экстрагируют CH2Cl2 (3×50 мл). Органические экстракты объединяют, промывают водой (75 мл) и насыщенным раствором NaCl (50 мл). Органический слой высушивают над MgSO4 и фильтруют. Органический растворитель удаляют в вакууме, получая (R)-2-гидрокси-1-морфолин-4-ил-3-фенилметансульфонилпропан-1-он (5) в виде вязкого масла (1,60 г, 5,11 ммоль, 89%), который используют прямо без дальнейшей очистки: МС вычислено для C14H19NO5S (М+Н+) 314,1, найдено 314,3.
Стадия Е. Эту реакцию проводят так, как описано в примере 1, используя (R)-2-гидрокси-1-морфолин-4-ил-3-фенилметансульфонилпропан-1-он (5). (R)-1-(морфолин-4-карбонил)-2-фенилметансульфонилэтил(4-нитрофенил)карбонат 6 (1,98 г, 4,14 ммоль, 81%) выделяют в виде белого твердого вещества после хроматографии на колонке. МС вычислено для C21H22N2O9S (М+Н+) 479,1; найдено 479,3.
Это вещество затем используют для синтеза озаглавленного соединения, применяя процедуру, аналогичную описанной в примере 1.1Н ЯМР (CD3OD, 600 МГц) δ 1,54 (д, 3Н, J=6,6 Гц), 3,51-3,69 (м, 10Н), 4,51-4,58 (м, 2Н), 5,08-5,10 (м, 1Н), 5,74-5,77 (м, 1Н), 6,94-7,01 (м, 1Н), 7,28-7,58 (м, 6Н), 7,72-7,79 (м, 1Н), 7,93 (д, 1Н, J=8,4 Гц); ВЭЖХ-МС рассчитано для C25H27N3O8S (М+Н+) 530,2; найдено 530,4.
Пример 3
2-Циклогексил-(1S)-(морфолин-4-карбонил)этиловый эфир (1S)-(5-этил-[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты

Этот продукт получен с выходом 49% при использовании (S)-2-амино-1-(5-этил-1,2,4-оксадиазол-3-ил)бутан-1-ола и (S)-2-циклогексил-1-(морфолин-4-карбонил)этил(4-нитрофенил)карбоната, как описано в примере 1.1Н ЯМР (CDCl3, 400 МГц) δ 0,78-0,93 (м, 2Н), 0,97 (дд, 3Н, J1=J2=7,4), 1,05-1,30 (м, 3Н), 1,33-1,44 (м, 1Н), 1,43 (дд, 3Н, J1=J2=7,6), 1,60-1,83 (м, 8Н), 1,98-2,11 (м, 1Н), 3,00 (кв, 2Н, J=7,6), 3,38-3,48 (м, 1Н), 3,50-3,58 (м, 2Н), 3,60-3,76 (м, 5Н), 5,18 (дд 1Н, J1=4,8, J2=7,8, J3=12,6), 5,26-5,32 (м, 1Н), 5,67 (д, 1Н, J=8,2); ВЭЖХ-МС вычислено для C22H34N4O6 (М+H+) 451,3, найдено 451,5.
Пример 4
2-Циклогексил-(1S)-(морфолин-4-карбонил)этиловый эфир (1S)-(5-фенил-[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты.

Этот продукт получен с выходом 49% при использовании (S)-2-амино-1-(5-фенил-1,2,4-оксадиазол-3-ил)бутан-1-ола и (S)-2-циклогексил-1-(морфолин-4-карбонил)этил(4-нитрофенил)карбоната, как описано в примере 1.1Н ЯМР (CDCl3, 400 МГц) δ 0,80-1,03 (м, 2Н), 1,01 (дд, 3Н, J1=J2=7,4), 1,06-1,32 (м, 2Н), 1,39-1,89 (м, 9Н), 2,04-2,17 (м, 1Н), 3,39-3,78 (м, 8Н), 5,27 (ддд, 1Н, J1=4,9, J2=7,7, J3=12,6), 5,29-5,34 (м, 1Н), 5,68 (д, 1Н, J=8,2), 7,54-7,60 (м, 2Н), 7,62-7,68 (м, 1Н), 8,18-8,24 (м, 2Н); ВЭЖХ-МС вычислено для C26H34N4O6 (М+Н+) 499,3, найдено 499,4.
Пример 5
2-Циклогексил-(1S)-(морфолин-4-карбонил)этиловый эфир 4-оксотетрагидрофуран-(3S)-илкарбаминовой кислоты
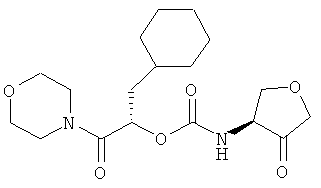
Используемый (4S)-аминотетрагидрофуран-(3R)-ол получают, как описано у E.N. Jacobsen и др., J. Am. Chem. Soc., 1995, 117, cc.5897-5898.
2-Циклогексил-(1S)-(морфолин-4-карбонил)этиловый эфир (4R)-гидрокситетрагидрофуран-(3S)-илкарбаминовой кислоты получают, как описано в примере 1.
Схема 9

Стадия А: 2-циклогексил-(1S)-(морфолин-4-карбонил)этиловый эфир (4R)-гидрокситетрагидрофуран-(3S)-илкарбаминовой кислоты (40 мг, 0,1079 ммоль) растворяют в сухом дихлорметане (5 мл) и охлаждают до 0°С, затем добавляют периодинан Десса-Мартина в сухом виде и перемешивают при этой температуре в течение получаса, прежде чем дать нагреться до комнатной температуры в течение 1,5 час. Летучие компоненты реакции удаляют упариванием на роторном испарителе, сырой продукт очищают с помощью препаративной ВЭЖХ, получая 15 мг, 37,5%. ВЭЖХ-МС вычислено для C18H28N2O6 (M+H+) 369,2, найдено 369,4.
Пример 6
(S)-1-трет-бутоксикарбонил-4-циано-4-(1-циклогексилметил-2-морфолин-4-ил-2-оксоэтоксикарбониламино)пиперидин
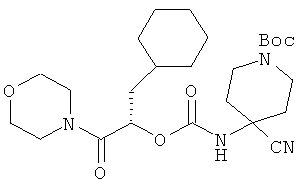
Схема 10

Стадия А: 1-Вос-4-пиперидон 1 (2 г, 10 ммоль) добавляют в колбу, содержащую KCN (3,256 г, 50 ммоль) и ацетат аммония (7,7 г, 100 ммоль). В колбу, закрытую резиновой пробкой, добавляют через шприц сухой метанол (50 мл). Реакционную смесь гомогенизируют ультразвуком в течение 15 мин для размельчения солей и затем перемешивают при комнатной температуре в течение ночи. Летучие вещества удаляют упариванием на роторном испарителе, остаток нейтрализуют насыщенным водным раствором бикарбоната натрия и экстрагируют этилацетатом. Органические слои высушивают над MgSO4, растворитель удаляют, выделяя с выходом 98% 2,21 г чистого трет-бутилового эфира 4-амино-4-цианопиперидин-1-карбоновой кислоты 2.1Н ЯМР (CDCl3, 400 МГц) δ 3,95 (с, 2Н), 3,18 (т. J=11,3 Гц, 2Н), 1,96 (д, J=13,2 Гц, 2Н), 1,82 (с, 2Н), 1,63 (т, J=12 Гц, 2Н), 1,45 (с, 9Н).
Стадия В: Образец 2 (192 мг, 0,852 ммоль) и лутидин (91,3 мг, 0,852 ммоль) растворяют в сухом ДХМ, добавляют по каплям к раствору трифосгена (278 мг, 2,81 ммоль) в сухом ДХМ и перемешивают 1,5 часа при комнатной температуре. Раствор (S)-3-циклогексил-2-гидрокси-1-морфолин-4-илпропан-1-она (полученного, как описано в примере 1, 205 мг, 0,852 ммоль) и лутидина (91,3 мг, 0,852 ммоль) добавляют по каплям в реакционную смесь в течение 10 мин и перемешивают 3 часа. Летучие продукты удаляют в вакууме. Полученный остаток очищают препаративной ВЭЖХ, выделяя 150 мг (S)-1-трет-бутоксикарбонил-4-циано-4-(1-циклогексилметил-2-морфолин-4-ил-2-оксоэтоксикарбониламино)пиперидина.1Н ЯМР (CD3OD, 400 МГц) δ 5,35 (д, J=9,2 Гц, 1Н), 3,81 (д, J=10,5 Гц, 2Н), 3,74-3,62 (м, 6Н), 3,56-3,48 (м, 2Н), 3,32 (м, 4Н), 2,23 (д, J=11,2 Гц, 2Н), 1,87 (м, 3Н), 1,71 (м, 5Н), 1,52-1,46 (м, 11Н), 1,29-1,20 (м, 3Н), 1,03-0,97 (м, 2Н). ВЭЖХ-МС вычислено для C25H40H4O6 (М+Н+) 493,3, найдено 493,3.
Пример 7
(S)-4-Циано-4-(1-циклогексилметил-2-морфолин-4-ил-2-оксоэтоксикарбониламино)-1-метилпиперидин

Образец титульного соединения примера 6 (125 мг, 0,254 ммоль) растворяют в ДХМ (1 мл) и ТФУ (1 мл) и перемешивают 1 час, летучие продукты удаляют упариванием на роторном испарителе и выделяют (S)-4-циано-4-(1-циклогексилметил-2-морфолин-4-ил-2-оксоэтоксикарбониламино)пиперидин с количественным выходом в виде соли ТФУ. Соответствующий свободный амин получают добавлением 1М NaOH и экстракцией ДХМ с последующим высушиванием и упариванием органического слоя на роторном испарителе. ВЭЖХ-МС вычислено для C20H32N4O4 (M+H+) 393,2, найдено 393,3.
Образец свободного амина, полученный в предыдущей стадии (40 мг, 0,102 ммоль), растворяют в МеОН (1 мл) и добавляют раствор формальдегида (6,06 мг, 0,202 ммоль) в метаноле (2 мл) с одной каплей уксусной кислоты. Затем в реакционную смесь добавляют твердый цианоборогидрид натрия (2,1 мг, 0,034 ммоль) и перемешивают в течение получаса. Растворитель удаляют в вакууме, продукт очищают препаративной ВЭЖХ, выделяя титульный продукт, 29,8 мг, выход 72%. ВЭЖХ-МС C21H34N4O4 (M+H+) 407,3, найдено 407,3.
Пример 8
(1S)-Циклогексилметил-2-морфолин-4-ил-2-оксоэтиловый эфир 2-(2-хлорбензилокси)-(1R)-цианоэтилкарбаминовой кислоты
Схема 11

(S)-2-Амино-3-(2-хлорбензилокси)пропионамид получают, как описано у P.D.Greenspan и др., J. Med. Chem., 2001, 44, сс.4524-4534.
Стадия А: (S)-2-Циклогексил-1-(морфолин-4-карбонил)этил(4-нитрофенил)карбонат (получение в примере 1, 132,8 мг, 0,326 ммоль) и (S)-2-амино-3-(2-хлорбензилокси)пропионамид (89,7 мг, 0,392 ммоль) помещают в сухую колбу с 2 мл безводного ДМФ, добавляют ДМАП (4-диметиламино)пиридин) (159 мг, 1,3 ммоль) в 0,5 мл сухого ДМФ и реакционную смесь перемешивают в течение ночи. После обработки продукт получают очисткой с помощью препаративной ВЭЖХ с выходом 39% в виде аморфного твердого вещества. ВЭЖХ-МС для C24H34ClN3O6 (М+1)=496,9.
Стадия В: Продукт стадии А (61 мг, 0,122 ммоль) растворяют в 0,5 мл сухого ДМФ и добавляют к раствору цианурхлорида (67 мг, 0,369 ммоль) в сухом ДМФ. Реакционную смесь перемешивают 2 часа, затем растворитель удаляют упариванием на роторном испарителе, сырой продукт очищают с помощью препаративной ВЭЖХ, получая 14,66 мг чистого аморфного твердого вещества.1Н ЯМР (CD3OD, 400 МГц) δ 7,59 (д, J=7,1 Гц, 1Н), 7,42 (д, J=7,3 Гц, 1Н), 7,34 (м, 2Н), 5,38 (д, J=9,8 Гц, 1Н), 4,86 (т, J=5,7 Гц, 1Н), 4,75 (д, J=1,8 Гц, 2Н), 3,83 (д J=5,6 Гц, 2Н), 3,7-3,67 (м, 6Н), 3,6-3,49 (м, 2Н), 1,87 (д, J=12,7 Гц, 1Н), 1,77-1,72 (м, 5Н), 1,58-1,49 (м, 2Н), 1,37-1,18 (м, 3Н), 1,09-0,92 (м, 2Н). ВЭЖХ-МС для C24H32ClN3O5 (М+1)=479,2.
Пример 45
2-Циклогексил-(1S)-(морфолин-4-карбонил)этиловый эфир (1S)-(5-циклопропил-[1,2,4]оксадиазол-3-карбонил)пропилкарбаминовой кислоты

Схема 12

Стадия А: Промежуточный продукт 2 получают из коммерчески доступного (S)-2-аминобутан-1-ола (1): соединение 1 (10,0 г, 112 ммоль, 1,0 экв) растворяют в 500 мл сухого ДХМ, Вос-ангидрид (26,93 г, 123,4 ммоль, 1,1 экв) растворяют в 200 мл сухого ДХМ и добавляют к соединению 1 через капельную воронку при температуре 0°С в течение 1 часа. После перемешивания реакционной смеси в течение ночи ее обрабатывают 25% NH4OH. Органический слой отделяют и высушивают над MgSO4 После упаривания растворителя получают чистое соединение 2 с количественным выходом.
Стадия В: Продукт 2 (1,02 г, 5,39 ммоль, 1 экв) растворяют в 60 мл сухого ДХМ и охлаждают до 0°С, добавляют трихлоризоциануровую кислоту (1,32 г, 5,65 ммоль, 1,05 экв) и перемешивают 10 мин, получая белую суспензию. TEMPO (8,8 мг, 0,056 ммоль, 0,01 экв) добавляют к охлажденной реакционной смеси, которая сразу же становится оранжевой, и образуется дополнительный осадок. Реакционный сосуд вынимают из охлаждающей бани, реакционную смесь перемешивают еще 45 мин, затем фильтруют через слой целита, промывают 5% лимонной кислотой, затем насыщенным раствором бикарбоната, высушивают над MgSO4, фильтруют и выделяют чистый продукт 3 (0,979 г, выход 97%), который сразу же используют в следующей стадии С.
Стадия С: Продукт 3 (0,99 г, 5,28 ммоль, 1 экв) растворяют в 20 мл диоксана и за 10 мин охлаждают до 0°С, затем добавляют NaHSO3 (2,75 г, 21,1 ммоль, 4 экв, растворенный в 10 мл воды). Реакционную смесь перемешивают 10 мин при 0°С и добавляют KCN (1,37 г, 21,1 ммоль, 4 экв, в 10 мл воды), после чего перемешивают в течение ночи. Реакционную смесь обрабатывают, разбавляя 150 мл этилацетата и промывая органический слой тремя порциями насыщенного раствора бикарбоната натрия. Органический слой высушивают над сульфатом натрия, фильтруют и концентрируют до сухости, получая чистый циангидрин с количественным выходом.
Стадия D: Продукт 4 (5,68 г, 26,5 ммоль, 1 экв) растворяют в 60 мл этанола и добавляют к раствору гидроксиламина (50% вес/объем) в воде (2,44 мл, 39,7 ммоль, 1,5 экв). Реакционную смесь нагревают в течение 2 часов при 60°С, затем удаляют летучие продукты, оставшуюся белую пену выдерживают в высоком вакууме 18 часов, получая с количественным выходом чистый продукт 5 в виде аморфного белого твердого вещества.
Стадия Е: Продукт 5 (9,7 г, 39,23 ммоль, 1 экв) растворяют в 50 мл сухого ДМФ, к нему при интенсивном перемешивании добавляют ангидрид циклопропанкарбоновой кислоты (6,148 г, 39,23 ммоль, 1 экв). Полученный раствор делят на пять сосудов по 20 мл для микроволновой печи, снабженных мешалками. Сосуды закрывают, каждый индивидуально нагревают при 200°С в течение 75 с. Содержимое сосудов затем объединяют, разбавляют 250 мл этилацетата, промывают водой, бикарбонатом и солевым раствором. Органический слой высушивают над сульфатом магния, фильтруют и концентрируют до сухости. Полученное масло хроматографируют на колонке с ISCO силикагелем с градиентом этилацетат/гексан от 20 до 80% в течение 40 мин. Выход 8,2 г (70%).
Стадии F и G: Продукт 7 (8 г, 26,9 ммоль, 1 экв) растворяют в 50 мл ДХМ, и медленно в течение 15 мин к перемешиваемому раствору добавляют 50 мл ТФУ. Реакционную смесь перемешивают 2 часа, затем упаривают до сухости. Полученное вязкое масло растворяют в 40 мл сухого ДМФ, туда добавляют DIPEA (диизопропилэтиламин) (17,35 г, 134,5 ммоль, 5 экв), затем раствор соединения 9 в 40 мл сухого ДМФ. Полученный желтый раствор перемешивают в течение ночи. Упариванием на роторном испарителе частично удаляют ДМФ до ~ 35 мл, и остаток разбавляют 500 мл этилацетата, промывают порциями 4×200 мл 25% Na2CO3. Органический слой промывают солевым раствором, концентрируют до сухости и помещают в метанол. Полученную смесь очищают с помощью широкошкальной обратимо-фазовой препаративной ВЭЖХ, фракции объединяют, удаляют ацетонитрил и экстрагируют этилацетатом, высушивают над MgSO4, фильтруют и упаривают, получая чистый продукт 10 (11,5 г, 24,7 ммоль, выход 92%).
Стадия Н: Продукт 10 (8,8 г, 18,94 ммоль, 1 экв) растворяют в 500 мл сухого ДХМ и охлаждают до 0°С, к нему при той же температуре добавляют в твердом виде периодинан Десса-Мартина (24,1 г, 56,8 ммоль, 3 экв), и реакционной массе дают нагреться до комнатной температуры, затем перемешивают в течение ночи и обрабатывают насыщенным раствором Na2S2O3 (400 мл), затем насыщенным раствором бикарбоната натрия. Органический слой высушивают над MgSO4, фильтруют и концентрируют до 40 мл. Суспензию фильтруют, раствор хроматографируют на двух 110 граммовых ISCO колонках с градиентом этилацетата от 0 до 100% за 30 мин, затем промывают 100% этилацетом в течение 15 мин, чтобы обеспечить выход из колонки всего продукта. ВЭЖХ фиксирует смесь гидрата, метилкеталя (образец был получен в метаноле) и желаемого продукта. Спектр1Н ЯМР ясный и определенный.
Получение 6: Циклопропанкарбоновую кислоту (29 г, 336,9 ммоль, 1 экв) растворяют в 500 мл сухого ДХМ, к раствору добавляют при 0°С по каплям за 0,5 часа ДСС (34,75 г, 168,5 ммоль, 0,5 экв) в виде раствора в 100 мл ДХМ. Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Полученную суспензию фильтруют через слой целита, упаривают и растворяют в 500 мл гексана, затем снова фильтруют через слой целита, чтобы удалить оставшиеся побочные продукты, и упаривают. Полученное масло перегоняют в вакууме, получая чистый ангидрид.
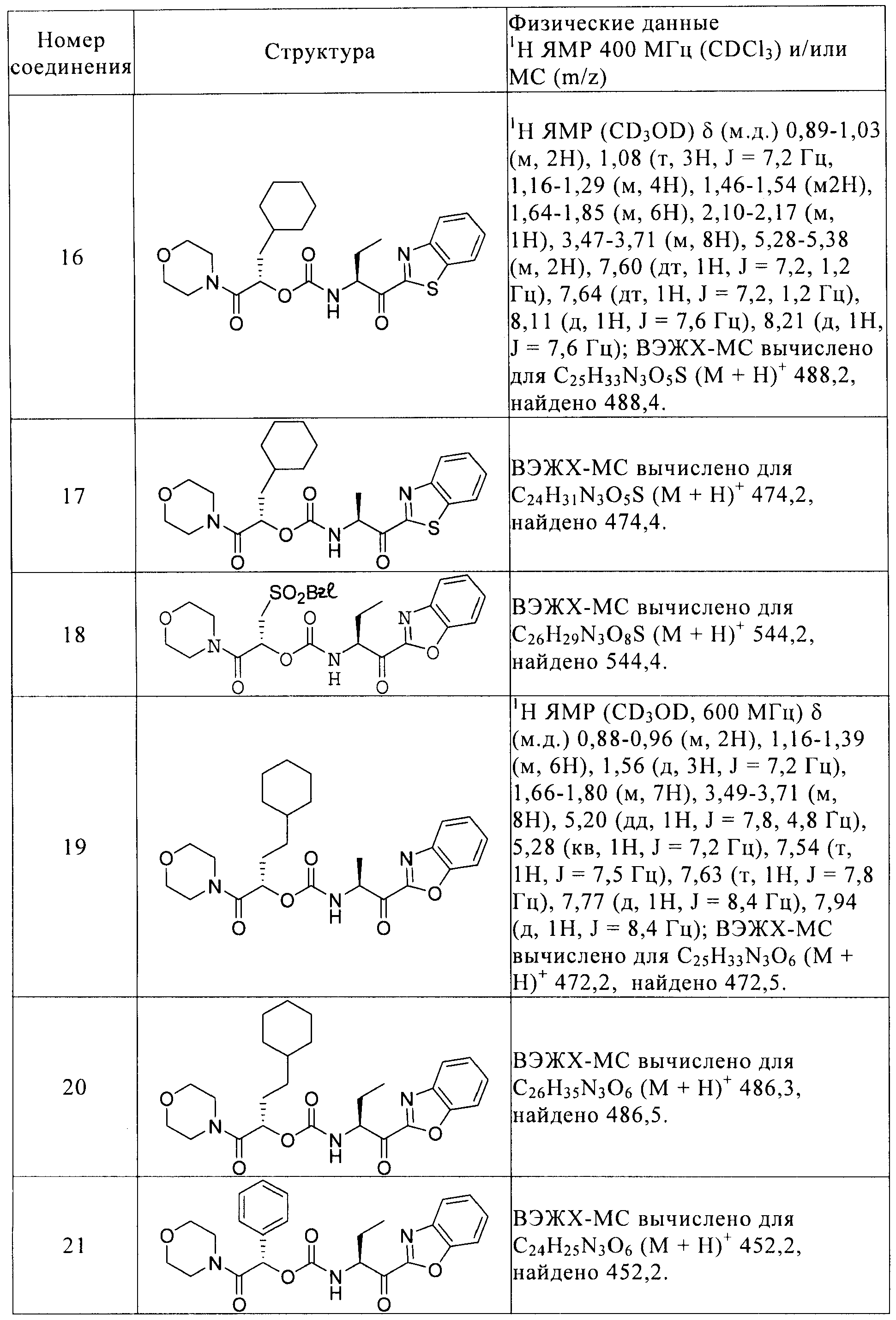
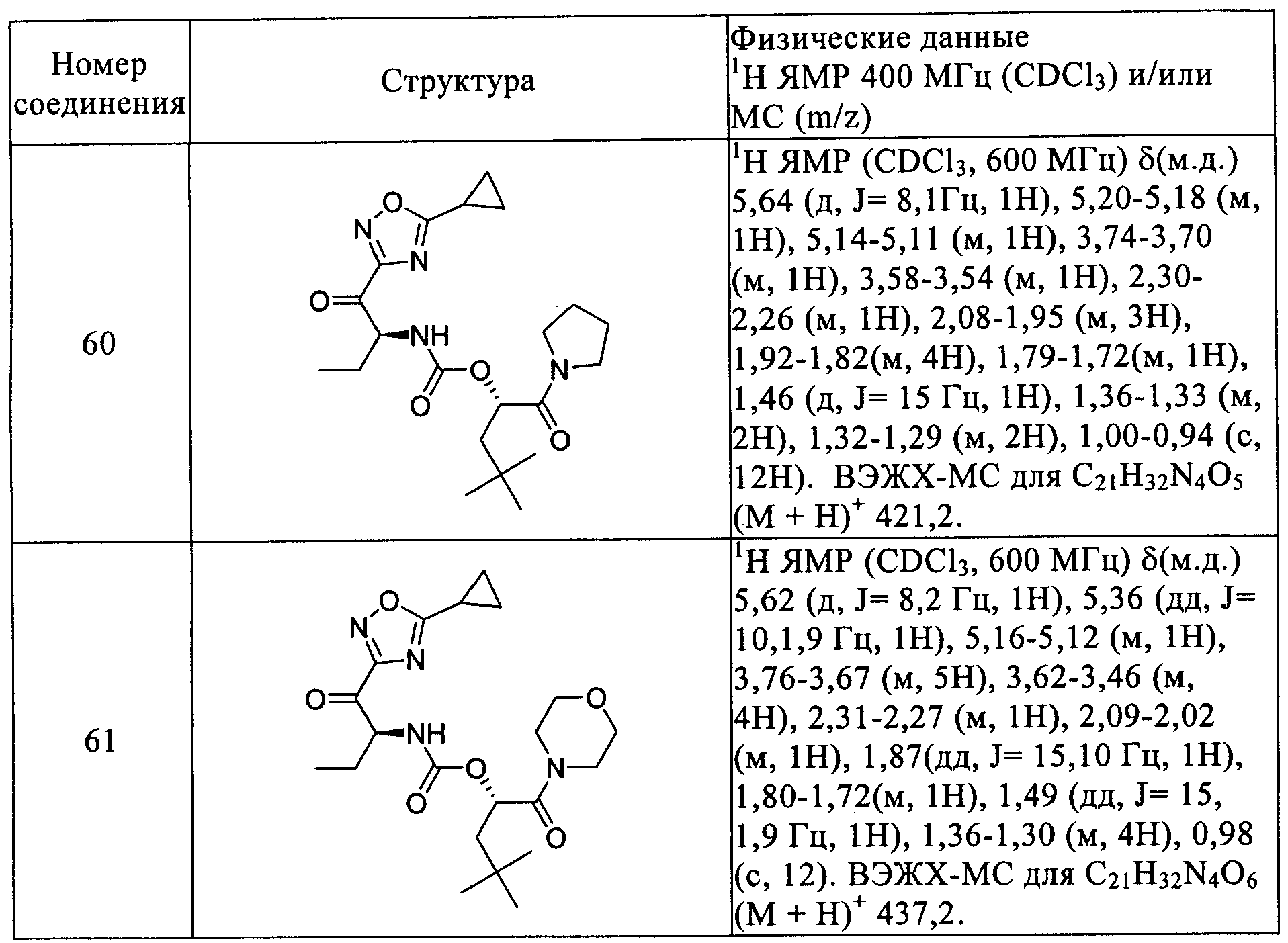
Повторением процессов, описанных в вышеприведенных примерах, с использованием подходящих исходных материалов получены следующие соединения формулы I, приведенные в таблице 1.









Опыт с катепсином S.
Кинетические измерения проводят в общем реакционном объеме 30 мкл на 384 луночных микротитрационных плашках. Катепсин S с конечной концентрацией 0,3-3 нМ (активный центр) инкубируют с соединением предлагаемого изобретения при двенадцати варьирующихся концентрациях в буфере, содержащем 100 мМ NaAc (pH 5,5), 1 мМ ЭДТА, 100 мМ NaCl. 0,01% Brij-35 (этоксилированный лауриловый спирт) при комнатной температуре в течение 20 мин. Контрольные реакции в отсутствие ингибитора осуществляют в 24 повторах. Реакции инициируют добавлением субстрата - ацетил-гистидин-пролин-валин-лизин-аминокарбамоилкумарина до конечной концентрации 50 мкл. Скорость гидролиза субстрата определяют, фиксируя повышение флуоресценции при длине волны возбуждения 380 нм и длине волны эмиссии 450 нм, которая возникает при расщеплении анилиновой связи в субстрате под воздействием фермента. Кажущиеся константы ингибирования для соединений определяют по кривым энзиматического развития и затем используют для расчета констант конкурентного ингибирования.
Опыт с катепсином К.
Кинетические измерения проводят в общем реакционном объеме 30 мкл на 384 луночных микротитрационных плашках. Катепсин К с конечной концентрацией 3,5 нМ (активный центр) инкубируют с соединением предлагаемого изобретения при двенадцати варьирующихся концентрациях в буфере, содержащем 100 мМ NaAc (pH 5,5), 1 мМ ЭДТА, 100 мМ NaCl. 0,01% Brij-35 при комнатной температуре в течение 20 мин. Контрольные реакции в отсутствие ингибитора осуществляют в 24 повторах. Реакции инициируют добавлением субстрата - ацетил-лизин-гистидин-пролин-лизин-аминокарбамоилкумарина до конечной концентрации 40 мкл. Скорость гидролиза субстрата определяют, фиксируя повышение флуоресценции при длине волны возбуждения 380 нм и длине волны эмиссии 450 нм, которая возникает при расщеплении анилиновой связи в субстрате под воздействием фермента. Кажущиеся константы ингибирования для соединений определяют по кривым энзиматического развития и затем используют для расчета констант конкурентного ингибирования.
Опыт с катепсином В.
Кинетические измерения проводят в общем реакционном объеме 30 мкл на 384 луночных микротитрационных плашках. Катепсин В с конечной концентрацией 1,5 нМ (активный центр) инкубируют с соединением предлагаемого изобретения при двенадцати варьирующихся концентрациях в буфере, содержащем 100 мМ NaAc (pH 5,5), 1 мМ ЭДТА, 100 мМ NaCl. 0,01% Brij-35 при комнатной температуре в течение 20 мин. Контрольные реакции в отсутствие ингибитора осуществляют в 24 повторах. Реакции инициируют добавлением субстрата - ацетил-гистидин-пролин-валин-лизин-аминокарбамоилкумарина до конечной концентрации 10 мкл. Скорость гидролиза субстрата определяют, мониторируя повышение флуоресценции при длине волны возбуждения 380 нм и длине волны эмиссии 450 нм, которая возникает при расщеплении анилиновой связи в субстрате под воздействием фермента. Кажущиеся константы ингибирования для соединений определяют по кривым энзиматического развития и затем используют для расчета констант конкурентного ингибирования.
Опыт с катепсином L.
Кинетические измерения проводят в общем реакционном объеме 30 мкл на 384 луночных микротитрационных плашках. Катепсин L с конечной концентрацией 0,1 нМ (активный центр) инкубируют с соединением предлагаемого изобретения при двенадцати варьирующихся концентрациях в буфере, содержащем 100 мМ NaAc (pH 5,5), 1 мМ ЭДТА, 100 мМ NaCl. 0,01% Brij-35 при комнатной температуре в течение 20 мин. Контрольные реакции в отсутствие ингибитора осуществляют в 24 повторах. Реакции инициируют добавлением субстрата - ацетил-гистидин-лизин-фенилаланин-аминокарбамоилкумарина до конечной концентрации 20 мкл. Скорость гидролиза субстрата определяют, мониторируя повышение флуоресценции при длине волны возбуждения 380 нм и длине волны эмиссии 450 нм, которая возникает при расщеплении анилиновой связи в субстрате под воздействием фермента. Кажущиеся константы ингибирования для соединений определяют по кривым энзиматического развития и затем используют для расчета констант конкурентного ингибирования.
Соединения формулы I в свободной форме или в форме фармацевтически приемлемых солей проявляют ценные фармакологические свойства, например, как показано в тестах in vitro, описанных в изобретении. Предпочтительные константы ингибирования катепсина S для соединений предлагаемого изобретения составляют менее 10 мкМ. Более предпочтительные константы ингибирования для соединений предлагаемого изобретения составляют менее 1,0 мкМ. Наиболее предпочтительные константы ингибирования для соединений предлагаемого изобретения составляют менее 0,1 мкМ. Селективность для катепсина S в присутствии изозимов катепсина определяют как соотношение константы ингибирования изозима катепсина для соединения предлагаемого изобретения к константе ингибирования катепсина S для того же соединения. Предпочтительные соединения предлагаемого изобретения, селективные к катепсину S, имеют значения этого соотношения больше, чем 10. Более предпочтительные соединения предлагаемого изобретения, селективные к катепсину S, имеют значения этого соотношения больше, чем 100. Наиболее предпочтительные соединения предлагаемого изобретения, селективные к катепсину S, имеют значения этого соотношения больше, чем 1000.
Так, например, 2-циклогексил-(1S)-(морфолин-4-карбонил)этиловый эфир 2-(бензооксазол-2-ил)-(1S)-циклопропил-2-оксоэтилкарбаминовой кислоты (Пример 1) имеет значение IC50, равное 6,6 нМ, и, по крайней мере, в 100 раз большую селективность для катепсина S, чем для катепсина К, В и L. Ниже примеры активности и селективности соединений предлагаемого изобретения детализированы в Таблице 2.
Является очевидным, что описанные примеры и разделы служат только иллюстративным целям, и специалистам могут быть предложены различные модификации или изменения, которые также включаются в предлагаемое изобретение и формулу изобретения. Все публикации, патенты и патентные приложения, цитированные в тексте, включены в виде ссылок во всей полноте.
Реферат
Настоящее изобретение относится к соединениям общей формулы (I), где R1 выбирают из формул (а), (b) и (с), n означает 0; R6 и R7 независимо выбирают из водорода, C1-С6алкила, цианоС1-С6алкила, С3-С6циклоалкилС0-С4алкила и С6арилС0-С4алкила; или R6 и R7 вместе с атомом углерода, к которому R6 и R7 присоединены, образуют 6-членный гетероциклоалкил с одним атомом азота; при этом любой алкил в R6 и R7 может необязательно включать метиленовую группу, замещенную атомом О; при этом любой арил в R6 и R7 или образованный комбинацией R6 и R7 может быть необязательно замещен 1 радикалом, независимо выбранным из группы: галоид, C1-С6алкил, -XC(O)OR10; при этом Х означает связь; R10 независимо выбирают из C1-С6алкила; R8 выбирают из С5-С9гетероарилС0-С4алкила, содержащего 2-3 гетероатома, независимо выбранных из N, О и S; при этом любой гетероарил в R8 может быть необязательно замещен 1 радикалом, независимо выбранным из группы: галоид, С1-С6алкил, С3-С6циклоалкил; R2 обозначает водород; R3 и R4 независимо выбирают из водорода, C1-С6алкила, С3-С6циклоалкилС0-С4алкила и С6арилС0-С4алкила; при этом любой алкил в R3 и R4 может необязательно включать метиленовую группу, замещенную группой S(O)2; R5 выбирают из С5-С6гетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, и NR12R13; где R12 и R13 независимо выбирают из C1-С6алкила; а также к их фармацевтически приемлемым солям и изомерам. Также изобретение относится к применению соединений формулы (I) для получения лекарственного средства и к фармацевтической композиции, обладающим свойством ингибитора катепсина S, содержащая терапевтически эффективное количество соединения формулы (I) в комбинации с фармацевтически приемлемым наполнителем. Технический рез
Формула

в котором R1 выбирают из формул (а), (b) и (с),

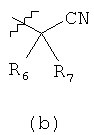

в которых n означает 0;
R6 и R7 независимо выбирают из водорода, C1-С6алкила, цианоС1-С6алкила, С3-С6циклоалкилС0-С4алкила и С6арилС0-С4алкила; или R6 и R7вместе с атомом углерода, к которому R6 и R7 присоединены, образуют 6-членный гетероциклоалкил с одним атомом азота;
при этом любой алкил в R6 и R7 может необязательно включать метиленовую группу, замещенную атомом О; при этом любой арил в R6 и R7 или образованный комбинацией R6 и R7 может быть необязательно замещен 1 радикалом, независимо выбранным из группы: галоид, C1-С6алкил, -ХС(O)OR10; при этом Х означает связь; и R10 независимо выбирают из C1-С6алкила;
R8 выбирают из С5-С9гетероарилС0-С4алкила, содержащего 2-3 гетероатома, независимо выбранных из N, О и S; при этом любой гетероарил в R8 может быть необязательно замещен 1 радикалом, независимо выбранным из группы: галоид, C1-С6алкил, С3-С6циклоалкил; и R2 обозначает водород;
R3 и R4 независимо выбирают из водорода, C1-С6алкила, С3-С6циклоалкилС0-С4алкила и С6арилС0-С4алкила; при этом любой алкил в R3 и R4 может необязательно включать метиленовую группу, замещенную группой S(O)2;
R5 выбирают из С5-С6гетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, и NR12R13; где R12 и R13 независимо выбирают из С1-С6алкила;
и их фармацевтически приемлемые соли и изомеры.

в котором R1 выбирают из формул (а), (b) и (с)

в которых n означает 0;
R6 выбирают из водорода, C1-С6алкила, циано-С1-С6алкила, С3-С6циклоалкилС0-С4алкила и С6арилС0-С4алкила;
при этом любой алкил из R6 может необязательно включать метиленовую группу, замененную атомом О; при этом арил в R6 может быть необязательно замещен 1 радикалом, независимо выбранными из группы: галоид, С1-С6алкил, -ХС(O)OR10; при этом Х означает связь; и R10независимо выбирают из C1-С6алкила;
R8 выбирают из С5-С9гетероарилС0-С4алкила, содержащего 2-3 гетероатома, независимо выбранных из N, О и S; при этом любой гетероарил в R8 может быть необязательно замещен 1 радикалом, независимо выбранным из группы: галоид, С1-С6алкил, С3-С6циклоалкил; и
R3 выбирают из водорода, C1-С6алкила, С3-С6циклоалкилС0-С4алкила и С6арилС0-С4алкила; при этом любой алкил из R3 может необязательно включать метиленовую группу, замененную группой S(O)2;
R5 выбирают из С5-Сбгетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, и NR12R13, при этом R12 и R13 независимо выбирают из C1-С6алкила.
R3 и R4 оба означают водород;
R6 выбирают из водорода, C1-С6алкила, цианоС1-С6алкила, С3-С6циклоалкилС0-С4алкила и С6арилС0-С4алкила; при этом любой алкил из R6 может необязательно включать метиленовую группу, замещенную атомом О; при этом любой арил в R6 может быть необязательно замещен 1 радикалом, независимо выбранными из группы галоидов;
R7 означает водород;
R8 выбирают из С5-С9гетероарилС0-С4алкила, содержащего 2-3 гетероатома, независимо выбранных из N, О и S; при этом любой гетероарил в R8 может быть необязательно замещен 1 радикалом, независимо выбранным из группы: галоид, C1-С6алкил, С3-С6циклоалкил; и
R3 выбирают из C1-С6алкила, С3-С6циклоалкилС0-С4алкила и С6арилС0-С4алкила; при этом любой алкил в R3 может необязательно включать метиленовую группу, замещенную группой S(O)2;
R5 выбирают из С5-С6гетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, и NR12R13, при этом R12 и R13 независимо выбирают
из C1-С6алкила.

и его фармацевтически приемлемые соли.
Документы, цитированные в отчёте о поиске
Производные амида, способ их получения и фармацевтическая композиция на их основе
Комментарии